 Amanda L. Miller
Amanda L. Miller Shingo Bessho
Shingo Bessho Kaitlyn Grando
Kaitlyn Grando Çagla Tükel
Çagla Tükel- Department of Microbiology and Immunology, Lewis Katz School of Medicine, Temple University, Philadelphia, PA, United States
The human microbiota is the community of microorganisms that live upon or within their human host. The microbiota consists of various microorganisms including bacteria, fungi, viruses, and archaea; the gut microbiota is comprised mostly of bacteria. Many bacterial species within the gut microbiome grow as biofilms, which are multicellular communities embedded in an extracellular matrix. Studies have shown that the relative abundances of bacterial species, and therefore biofilms and bacterial byproducts, change during progression of a variety of human diseases including gastrointestinal, autoimmune, neurodegenerative, and cancer. Studies have shown the location and proximity of the biofilms within the gastrointestinal tract might impact disease outcome. Gram-negative enteric bacteria secrete the amyloid curli, which makes up as much as 85% of the extracellular matrix of enteric biofilms. Curli mediates cell-cell attachment and attachment to various surfaces including extracellular matrix components such as fibronectin and laminin. Structurally, curli is strikingly similar to pathological and immunomodulatory human amyloids such as amyloid-β, which has been implicated in Alzheimer's disease, α-synuclein, which is involved in Parkinson's disease, and serum amyloid A, which is secreted during the acute phase of inflammation. The immune system recognizes both bacterial amyloid curli and human amyloids utilizing the same receptors, so curli also induces inflammation. Moreover, recent work indicates that curli can participate in the self-assembly process of pathological human amyloids. Curli is found within biofilms of commensal enteric bacteria as well as invasive pathogens; therefore, evidence suggests that curli contributes to complex human diseases. In this review, we summarize the recent findings on how bacterial biofilms containing curli participate in the pathological and immunological processes in gastrointestinal diseases, systemic autoimmune diseases, and neurodegenerative diseases.
Introduction
The community of microorganisms that live upon or within a host are referred to as the microbiota. The human microbiota includes bacteria, fungi, viruses, and archaea that colonize the surface or deep layers of the skin (skin microbiota), the mouth (oral microbiota), the vagina (vaginal microbiota), and the digestive tract (gut microbiota) (1). The human microbiota has received increasing attention in numerous research fields over the last 15 years. The gut microbiota is of interest as numerous studies have reported that there are changes in the gut microbiota during obesity, diabetes, liver diseases, cancer, and neurodegenerative diseases (2–7).
Studies of the diversity of the human microbiota started as early as the mid-seventeenth century with Antonie van Leeuwenhoek who compared the oral and the fecal microbiota. He observed differences between the microbes in these two locations and also between samples from healthy vs. diseased individuals (8). In 2001, Joshua Lederberg coined the term microbiome to refer to the “the ecological community of commensal, symbiotic, and pathogenic microorganisms that literally share our body space” (9). Many fundamental questions concerning the human microbiota have been difficult or impossible to address until recently. In 2017, ~4,000 publications focusing on the study of the gut microbiota were published, accounting for 80% of the publications on the subject since 1977 (2). The advancement of laboratory techniques and “omics” technologies have allowed researchers to characterize the composition of the microbiota and its functions in human health and disease (10).
The coevolution of humans and their microbial symbionts have selected for a specialized community of microorganisms that thrive in the gut (11). Bacteria comprise the bulk of the gut microbiota with archaea, eukaryotes, and viruses present in much smaller numbers (10). In a healthy human adult, the gut microbiota is dominated by two phyla: Firmicutes and Bacteroidetes (12). Other phyla, including Actinobacteria, Proteobacteria, Verrucomicrobia, and Euryarchaeota, are found in lower abundance (12). The gut microbiota is unlike any free-living microbial communities found in the environment due to the unique environment of the digestive tract (13). The microbiota plays a crucial role in maintaining immune and metabolic homeostasis and protecting the host against pathogens through microbial crosstalk with the mucosal immune system through integrated signaling pathways and gene regulatory networks (13–15). The interactions between the host immune system and the colonizing gut microbiota initiate at birth and are important for host immune system development and homeostasis (16–18). A variety of genetic and environmental factors influence the composition and the function of the gut microbiota including host diet, genetics, age, location, and medication use, especially antibiotics (19). When this homeostatic relationship is disrupted, it can lead to dysbiosis or “an imbalance in the composition and metabolic capacity of our microbiota” (20). Growing evidence indicates that dysbiosis shifts the microbiota in ways that increase inflammation and accelerate the onset or contribute to the pathogenesis of chronic diseases (20) such as cardiovascular disease, obesity, diabetes, cancer, asthma, and inflammatory bowel disease (20–26).
Many species of bacteria that colonize the gut live in biofilms. A bacterial biofilm is a group of bacteria that are encapsulated in a three-dimensional, self-produced extracellular matrix that is adhered to a biotic or abiotic surface (27, 28). The biofilm provides a layer of protection to microorganisms that grow in stressful environments where nutrients are scarce and during changes in temperature, osmolarity, and oxygen availability (29–31). Furthermore, the biofilm blocks access by toxic agents such as antibiotics and the host's immune system (32).
Biofilms can be composed of a single species of bacteria or a consortium of multiple species of bacteria. Biofilms can be formed by a variety of bacterial species including Gram-positive (e.g., Bacillus spp, Listeria monocytogenes, Staphylococcus spp, and lactic acid bacteria including Lactobacillus plantarum and Lactococcus lactis) and Gram-negative species (e.g., Escherichia coli, Salmonella enterica, and Pseudomonas aeruginosa) (33). The Proteobacteria phylum, which makes up ~0.1% of the gut microbiota in a healthy individual, expands during inflammation. The Gammaproteobacteria class includes several medically and scientifically important families including Enterobacteriaceae, Vibrionaceae, and Pseudomonadaceae. Enterobacteriaceae is a large family of Gram-negative bacteria that includes many harmless enteric commensals as well as pathogens such as Salmonella, E. coli, Yersinia pestis, Klebsiella, and Shigella. All these bacteria have the ability to make biofilms (30, 34–37).
Bacterial biofilms form throughout the human oro-gastrointestinal tract and mixed species biofilms have been observed in dental and gastric infections as well as in intestinal diseases, chronic gut wounds, and colon cancer (38). Biofilms are also formed by species necessary for a healthy gut mucosa, and these biofilms may benefit the host by fortifying host defenses, enhancing the exchange of nutrients between the microbiota and the host, and interfering with colonization by pathogenic bacteria (38). Therefore, biofilms within the human gut can be both beneficial and detrimental to the host depending on whether they are produced by the commensal microbiota or enteric pathogens (38).
Only 10% of the biomass of the biofilm are actual bacterial cells (29, 39). The composition and the structure of the biofilm is dependent upon the bacteria within it and the environment in which it is formed (27, 28). The bacterial cells within a biofilm are physiologically distinct from their planktonic counterparts (40). In a biofilm, cells are embedded within an extracellular matrix (ECM) composed of extracellular polymeric substances such as lipids, polysaccharides, proteins, and DNA (41–43). The ECM accounts for 90% of the total biomass of the biofilm. In E. coli biofilms, the major proteinaceous component, which comprises 85% of the ECM, is the amyloid curli. Curli encapsulates individual bacterial cells and forms an interwoven mesh that supports the ECM (44–46). Curli expression is triggered when enteric bacteria are grown under stressful environmental conditions that favor biofilm formation over planktonic cell growth. Curli is responsible for the overall development of the biofilm architecture (47–50) as curli-deficient bacteria do not form mature three-dimensional biofilms and only grow in a single cell layer.
Curli proteins form thin amyloid fibers on the surface of enteric bacterial cells (51–53). These fibers range from 4 to 10 nm in width and have a β-sheet-rich structure in which the β-sheet strands are orientated perpendicular to the axis of the fiber (54). Human amyloids also have a cross-beta structure and share a strikingly similar quaternary structure with bacterial amyloids, including the pathological and immunomodulatory human amyloids such as amyloid-β (Aβ), which is involved in Alzheimer's disease (AD), α-synuclein (αSyn), which is implicated in Parkinson's disease (PD), and serum amyloid A (55–59).
Like other amyloids, curli is a conserved molecular pattern that causes the activation of toll-like receptors (TLR) 1 and 2 as well as intracellular NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome (60–62). Studies have shown that the immune system recognizes both bacterial amyloid curli and human amyloids through the same receptors that facilitate the inflammatory processes (61). Recent studies have demonstrated that curli and curli-associated biofilms in the gut participate in the pathogenesis of human diseases, including colorectal cancer, systemic lupus erythematosus (SLE), and PD (Table 1). In this review, we summarize the recent findings that suggest how bacterial biofilms containing curli participate in pathological and immunological processes of these diseases by direct interactions such as cross-seeding of human amyloids and by indirect interactions that trigger inflammation.
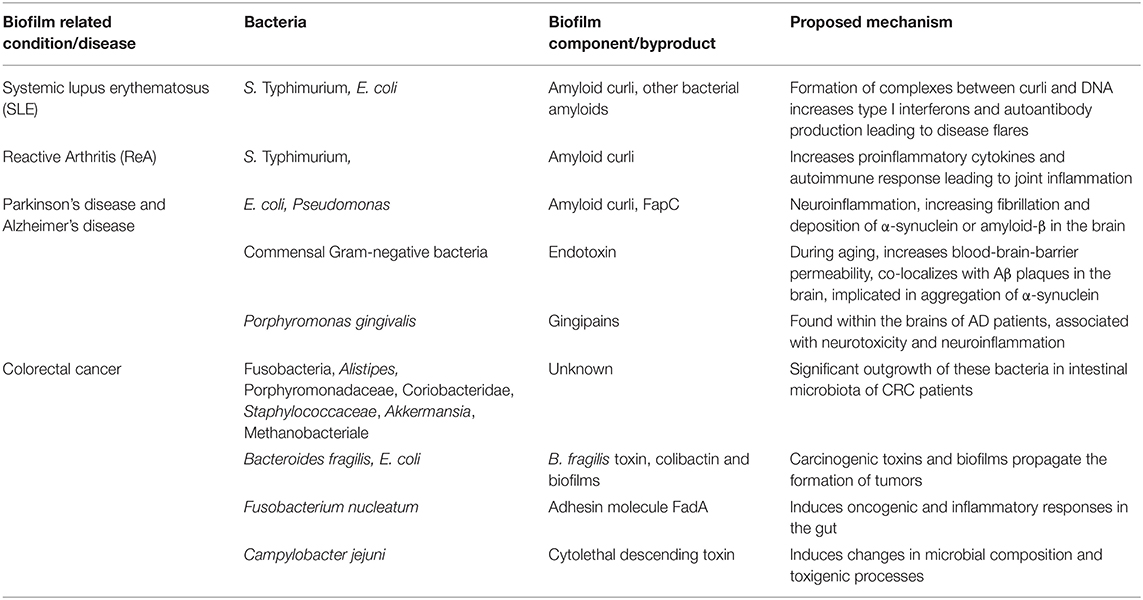
Table 1. Human diseases exaggerated by bacterial biofilms or biofilm by-products.
Curli Containing Bacteria in The Gastrointestinal Tract and The Urinary Tract
Biofilms can occur throughout the entire length of the gastrointestinal tract (38) and can be disease-linked or important for health. Two driver species of pathogenic biofilm formation in the gut are Salmonella and E. coli. These enteric bacteria thrive in a pro-inflammatory environment, conditions in which they outcompete the commensal microbiota. An outgrowth of enteric bacteria is common during inflammatory states that are associated with many gut disorders (63). It was highly debated whether or not enteric bacteria expressed curli and grow as biofilms in the gut. However, there was indirect evidence in support of this: for example, patients recovering from E. coli-induced sepsis harbor antibodies against curli (64). Similarly, antibodies against curli are detected after infection of mice with Salmonella enterica serovar Typhimurium (65). A recent study showed direct evidence for curli synthesis in the intestinal tract by S. Typhimurium during infection (66). Curli is recognized as a PAMP by the mucosal immune system via TLR2/TLR1 heterocomplex (60, 62) leading to the activation of NF-κB, eliciting the production of proinflammatory chemokines and cytokines including, IL-6, IL-8, and IL-17A (61, 67). The detection of curli by a healthy gut mucosa leads to reinforcement of the gut barrier preventing the leakage of bacteria and possibly the pathological bacterial amyloids (Figure 1) (68, 69).
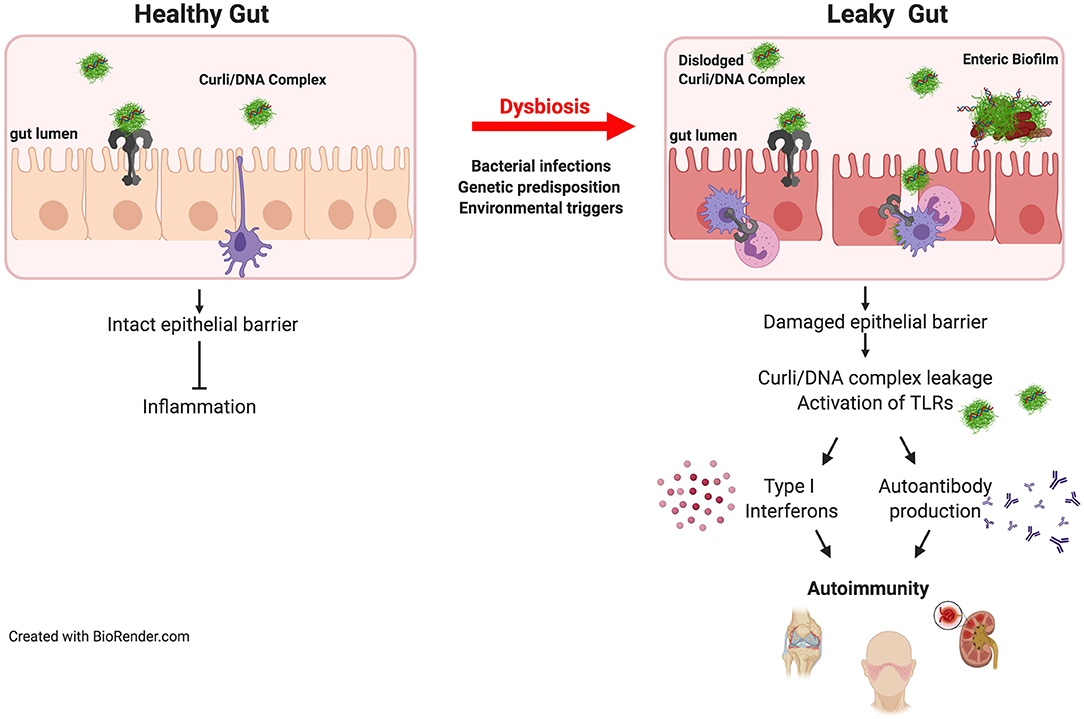
Figure 1. Bacterial amyloids and autoimmunity. Amyloids are produced by the members of gut microbiome. Curli/DNA complexes produced by commensal bacteria are recognized by TLR2/TLR1 heterocomplex which dampens inflammation in healthy intestinal tract. However, when the epithelial barrier is damaged during invasive infections or by other environmental factors or diseases, dislodged curli/DNA complexes from the biofilm activates TLR2/TLR1 heterocomplex and TLR9 leading to the generation of type I interferons and autoantibodies resulting in initiation and exacerbation of autoimmunity. In genetically predisposed individuals, the autoimmune effects of the curli/DNA complexes will likely to be exacerbated.
The urinary tract microbiome is just beginning to be characterized. Uropathogenic E. coli (UPEC), the most common cause of urinary tract infections (UTIs) (70, 71), is a member of the family Enterobacteriaceae and has the ability to form a curli-containing biofilm (72). UPEC is a frequent colonizer of medical devices and the primary cause of recurrent urogenital infections (73). UPEC forms curli-containing biofilms that are difficult to treat and eradicate with antibiotics leading to multidrug resistance (73). UTI infections can become persistent and result in bacteriuria which can lead to sepsis (71). Expression of csg gene cluster, genes that encode curli, is upregulated in UPEC isolated from urine from patients suffering from UTIs, whereas no csg expression is detected in urine from healthy controls (74, 75). It was reported that curli promotes colonization and immune induction by enhancing bacterial adherence and invasion into the uroepithelium during early stages of UTI (74).
The Involvement of Bacterial Amyloids in Autoimmune Diseases
The primary function of the immune system is to protect the host from possibly harmful substances and pathogens. The first line of defense against non-self-pathogens is the innate immune response. It immediately prevents the spread of foreign pathogens throughout the body. The second line of defense is the adaptive immune response, which is specific to the pathogen presented. This response is long lasting and highly specific. The exact etiologies for autoimmune disorders such as rheumatoid arthritis, SLE, and inflammatory bowel disease remain unknown, but various genetic and environmental factors contribute to their development (76). Furthermore, in those individuals who are predisposed, self-tolerance, the ability of the immune system recognizing self-produced antigens as non-threatening, becomes disrupted. The immune system begins to recognize self-antigens as foreign, leading to the bodies inability to “tolerate” self tissues as it attacks itself causing systemic and organ-specific damage (76–78). Numerous microorganisms use molecular mimicry or mimotopes to avoid detection by the immune system, which in turn could amplify the autoimmune response.
Infections and exposure to pathogens or opportunistic organisms may initiate or exacerbate autoimmune disorders. In addition to true autoimmune diseases mentioned above, a small group of patients experience autoimmune symptoms months and sometimes years after an infection is cleared. These autoimmune sequelae are observed following infections with human pathogens such as E. coli, Borrelia burgdorferi, S. Typhimurium, Mycobacterium tuberculosis, P. aeruginosa, Group A streptococci, and Staphylococcus aureus (76, 79–87). Joints are affected in many cases and post-infectious arthritis is observed. Most interestingly, all these bacteria express curli-like amyloids and form biofilms during infections. One of best understood examples of post-infectious arthritis is reactive arthritis (ReA). ReA is an inflammatory arthritis that develops in 5–10% of the patients following gastrointestinal infections with Salmonella, Shigella, Yersinia, or Campylobacter or following genital infections with Chlamydia trachomatis (79). Symptoms usually start 1–4 weeks post-infection and can last more than 5 years (79). Histocompatibility leukocyte antigen (HLA) B27 allele is a risk factor for ReA. About 90% of individuals who develop ReA following Salmonella infection carry the HLA-B27 genotype (88). ReA patients are not responsive to antibiotic treatment and cultures of joint fluids yield no bacterial growth. However, one study that employed immunohistochemical staining and mass spectrometry reported the presence of bacterial byproducts in synovial fluid (89–91). A recent study showed that in a mouse model of Salmonella infection curli is synthesized in the gastrointestinal tract and leads to increased anti-double-stranded DNA autoantibodies and to synoviocyte proliferation coupled with bone resorption in the knee joints (66). Infection with a curli mutant or a non-invasive strain did not cause such responses suggesting that the presence of curli during invasive infection with S. Typhimurium causes ReA (66).
Several mechanisms have been suggested to underlie the development of ReA. T cell-mediated immune responses clearly play a large role in autoimmune diseases. In rheumatoid arthritis, another autoimmune disease that affects joints, the functions of certain subsets of CD4+ T cells with regulatory capacity such as CD25+ regulatory T cells and Th2 cells are severely impaired. As ReA is a rare condition the role of these cells have not been elucidated, but it was proposed that CD4+ T cells that produce IL-17 and generate a type 17-mediated inflammatory response contribute to joint damage (92). Curli binds to and activates TLR2, leading to the production of pro-inflammatory cytokines and chemokines including IL-6, IL-8, TNFα, and IL-17 (60, 61, 67, 93). Therefore, TLR2-mediated IL-17 production is a plausible mechanism for the curli-driven development of ReA. However, recent studies have also shown that curli binds to DNA in the ECM forming highly immunogenic curli/DNA complexes. Curli amyloid acts as a carrier to bring DNA into endosomes where the DNA is recognized by TLR9 and activates type I interferons (94). Curli/DNA complexes also trigger the generation of anti-dsDNA and anti-chromatin autoantibodies following translocation into systemic sites from the gut (Figure 1) (41, 66, 94). In autoimmune diseases where anti-dsDNA autoantibodies are observed, DNA seems to be the key component. Nevertheless, it is not known whether the autoantibodies generated during S. Typhimurium infection that recognize curli alone or curli/DNA complexes facilitate joint damage directly. Additional studies are needed to assess the role of anti-dsDNA autoantibodies and to determine whether curli without DNA can elicit joint inflammation and damage.
As curli is also be produced commensal strains from the Enterobacteriaceae family (69), dissecting the mechanisms by which curli-producing bacteria trigger arthritogenic processes in autoimmune diseases is critical. Studies have shown that bacteria from the normal gut microbiota can also be arthritogenic and cause experimental arthritis in animals (95). Antibiotic treatment can prevent and suppress arthritis in murine models prone to arthritis. Additionally, germ-free animals do not develop arthritis (95). Overall, these studies suggest that amyloids from the gut microbiota contribute to the autoimmune processes. Phylogenetic analysis suggests that the curli assembly machinery is widespread, as homologs of the csg genes, which encode curli and are responsible for its biosynthesis and secretion, are found within four phylum, Bacteroidetes, Proteobacteria, Firmicutes, and Thermodesulfobacteria (30, 96, 97). As these members of these bacterial phyla are found in the gut microbiota and biofilms are observed in the intestinal tract, it is likely that the gut microbiota harbors amyloids.
SLE is a classical autoimmune disease in which the immune system causes widespread inflammation and tissue damage in joints, skin, brain, lungs, kidneys, and blood vessels. Bacterial infections are a major cause of morbidity and mortality in patients leading to and exacerbating SLE flares. Epidemiological studies suggest that bacterial infections promote SLE disease in predisposed individuals, but the underlying mechanisms remain unknown. SLE patients are more susceptible to infections, particularly bacterial infections, involving the upper respiratory tract, skin, and urinary tract than subjects without SLE (98–101). SLE patients produce autoantibodies against a wide variety of cellular antigens including double-stranded DNA and nuclear proteins (41). Curli/DNA complexes are powerful immune stimulators (41). When given systemically, curli/DNA complexes trigger immune activation and production of type I interferons as well as autoantibodies in SLE-prone and wild-type mice (41). It was also found that infection of SLE-prone mice with curli-producing bacteria triggers higher autoantibody titers than do curli-deficient bacteria (41). Furthermore, clinical studies of SLE patients with persistent bacteriuria and E. coli within their urine were positive for anti-curli/DNA antibodies (102). The levels of anti-curli/DNA IgG correlated with both bacteriuria and flares in the SLE cohort, further suggesting a link between curli/DNA complexes and increased disease severity in SLE (102). These data suggest that enteric bacteria production of curli potentiates disease pathogenesis in individuals predisposed to autoimmune disorders.
Neurological Diseases
Neurodegenerative diseases such as Alzheimer's Disease (AD) and Parkinson's Disease (PD) are characterized by proteins such as Aβ, hyperphosphorylated tau, and αSyn misfolded into pathological amyloid aggregates in and around neurons; these aggregates are associated with elevated inflammation (103–105). The gut microbiota impacts the nervous system through the gut-brain axis, a bidirectional “highway” for immune, metabolic, endocrine, and neural signals (Figure 2) (106, 107). Gut bacteria can synthesize neurotransmitters and bacterial metabolites, like indoles and short-chain fatty acids that can bypass the blood-brain-barrier and impact the brain (108–110). One hypothesis is that these signals and microbial products are transmitted not through the circulatory system, where they would need to navigate the blood-brain-barrier, but via the vagus nerve: the longest cranial nerve in the body, connecting the enteric nervous system to the brain stem, and containing both afferent and efferent fibers (111–113). Thus, it is similarly possible that aggregation of human amyloidogenic proteins may be seeded or indirectly induced by bacterial amyloids that originate in the gut.
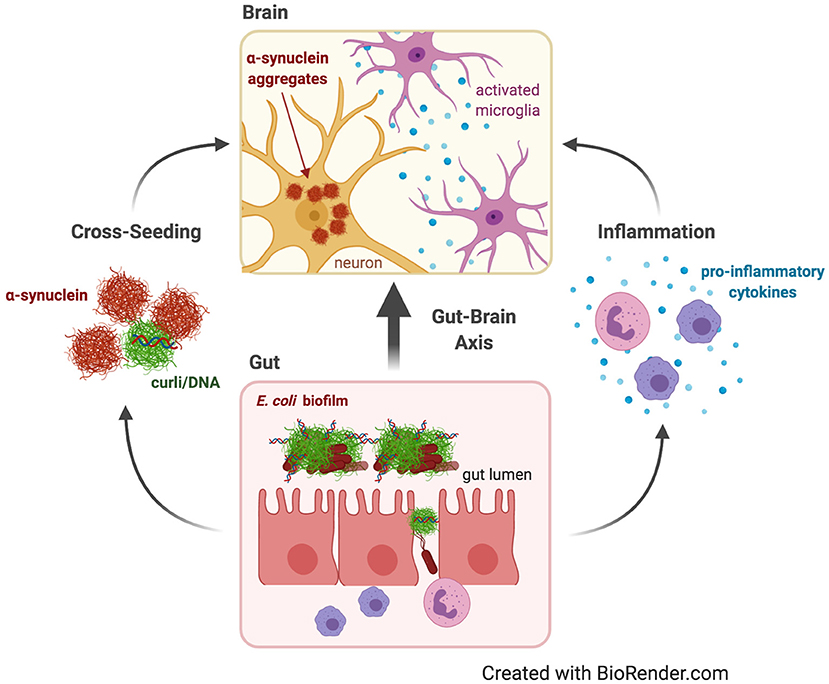
Figure 2. Parkinson's disease. Dysbiosis and leaky gut often occur with age, allowing contents of the gut lumen, such as curli from E. coli biofilms, to escape and induce changes in the brain through the gut-brain axis. Curli may contribute to Parkinson's Disease by increasing systemic inflammation and neuroinflammation in the form of activated microglia and astrocytes and elevated pro-inflammatory cytokines. Curli may also cross-seed fibrillation and aggregation of α-synuclein, which is capable of prion-like propagation from the gut to the brain to cause neurodegeneration.
There are two predominant theories about how biofilm-forming gut bacteria might affect neurodegenerative disease: indirectly by provoking neuroinflammation or directly due to cross-seeding of aggregation of human amyloids by bacterial amyloids (Figure 2). Neuroinflammation is emerging as a critical component of neurodegenerative diseases. It involves chronic activation of microglia and astrocytes, elevated pro-inflammatory cytokines and chemokines, and accumulation of Aβ and αSyn (104, 105). Dysbiosis of the gut microbiota may cause neuroinflammation by increasing pro-inflammatory cytokines, systemic inflammation, and weakening of the gut barrier by decreasing immune regulatory function (114, 115). Aging is a major risk factor for neurodegenerative diseases, and aging has been shown to change the microbiome composition and increase inflammation (116, 117). Aging also disrupts blood-brain-barrier function, often as a result of dysbiosis, allowing passage into the brain of bacterial cells or inflammatory metabolites that may exacerbate neuroinflammation (118–120).
Bacterial amyloids such as curli share structural and physical properties with human pathogenic amyloids (76, 121). Curli activates the same TLRs that recognize Aβ and αSyn (62, 122, 123). Curli induces elevated pro-inflammatory cytokines such as TNF-α, IL-6, and IL-1β, which have been shown to impair blood-brain-barrier integrity (64, 124–126). Therefore, it has been proposed that bacterial amyloids cause neuroinflammation and induce protein aggregation, indirectly leading to neurodegeneration (123, 127, 128). In addition, Aβ, tau, and αSyn have antimicrobial properties and so aggregation may be triggered by inflammation and dysbiosis (129–133).
PD is characterized by a loss of dopaminergic neurons in the nigrostriatal pathway. This loss is caused by Lewy bodies and Lewy neurites composed of intraneuronal αSyn aggregates (Figure 2) (103). Braak hypothesized that PD begins in the enteric nervous system and spreads up the vagus nerve to the brain (134). The earliest symptoms of PD, which occur up to 20 years before diagnosis in more than 65% of patients, include delayed gastric emptying, gastroparesis, constipation, and other gastrointestinal dysfunctions (135, 136). PD patients were recently demonstrated to have an altered gut microbiome compared to healthy controls (5). Elimination of gut bacteria in transgenic mouse models expressing human αSyn, either by using germ-free mice or treating with antibiotics, resulted in decreased αSyn deposition, neuroinflammation, and motor deficits; an effect that was reversed by recolonizing mice with microbiota from PD patients but not with healthy human microbiota (137). Furthermore, colonization of Fischer 344 rats with E. coli led to increased αSyn deposition in gut and brain neurons, increased gliosis, and increased inflammatory cytokines; effects replicated in C. elegans, but not observed with E. coli incapable of producing curli (Table 1) (127). A recent study corroborated these results: colonization of αSyn-overexpressing mice with wild-type E. coli, and not with curli-deficient E. coli, resulted in elevated αSyn pathology in the gut and brain and exacerbated cognitive, intestinal, and motor deficits and increased neuroinflammation. Interestingly, treatment of these mice with epigallocatechin gallate, an amyloid inhibitor restricted to the gut, prevented the increase in PD pathology and behavioral deficits. The same study confirmed in vitro that purified curli accelerated αSyn aggregation (138). Additionally, FapC, a functional amyloid produced by Pseudomonas biofilms, contributed to αSyn fibrillation, whereas a mutated FapC inhibited αSyn fibrillation (Table 1) (139). Thus, these studies also support the second theory: that bacterial amyloids directly cross-seed the aggregation of human pathological amyloids in the gut, which spread to the brain.
Human amyloids Aβ and αSyn have proven capable of cross-seeding tau (140, 141). Curli homologs from different bacteria strains are also capable of cross-seeding, even between E. coli and Shewanella onedensis (142). The first paper to propose the idea of cross-seeding by non-mammalian proteins showed that E. coli curli accelerated the fibrillation of serum amyloid A in a secondary amyloidosis mouse model (143). Data from in vitro studies also suggest that cross-seeding of Aβ aggregation by bacterial amyloids could initiate AD (Table 1) (127, 144). Curli is capable of cross-seeding Aβ fibrillation, and treatment of S. Typhimurium with D-enantiomeric peptides, known to inhibit Aβ fibrillation, inhibits curli fibrillation and reduces biofilm formation (121). Infection of pulmonary microvascular endothelial cells with clinical P. aeruginosa, which produce the bacterial amyloid FapC, induces production of Aβ and tau, capable of prion-like propagation to naïve cells (145). Evidence from animal models will be required to determine whether bacterial amyloids are capable of directly seeding Aβ or tau aggregation in a manner relevant to neurodegeneration.
Numerous recent studies confirmed that αSyn aggregates are capable of propagating from the gut to the brain, causing cognitive and motor deficits. Mice injected with αSyn preformed fibrils into the duodenal or gastric wall developed Lewy body-like αSyn aggregates in the dorsal motor nucleus of the vagus nerve, which spread to the brain and resulted in motor deficits and neurodegeneration (146, 147). The retrograde transport of αSyn pathology to the brain was corroborated in rat models (112, 148). However, studies by one group found that the pathology failed to progress past the dorsal motor nucleus (149, 150). Another group injected baboons with patient-derived Lewy body extracts and found that pathology spread to the central nervous system through circulation rather than via the vagus nerve (151). Regardless, there is mounting evidence indicating that bacterial amyloids such as curli, present at high levels in biofilms in the gut, are capable of cross-seeding αSyn aggregation and that αSyn aggregates are capable of spreading from the gut to the brain as a possible initiating event in PD (112, 127, 134, 139, 146–148, 151).
Meanwhile, similar observations are being made in AD, with some important differences from PD. AD is characterized by progressive accumulation of both extracellular Aβ plaques and intraneuronal hyperphosphorylated tau aggregates called neurofibrillary tangles (103). Evidence is inconclusive whether AD pathology begins in the intestines. Some samples from AD patients show increased Aβ deposits in the intestines (152), and several AD mouse models corroborate increased Aβ plaques and hyperphosphorylated tau overexpression in the intestines and enteric nervous system as well as impaired gut motility and function and increased inflammation (152–155). However, one study found no difference in gut motility and absorption between AD and control mice, though they detected intestinal Aβ and tau deposits in AD mice and AD patient samples (156). Another did not find evidence of Aβ in enteric neurons of an AD mouse model (157). Elimination of the gut bacteria by antibiotic treatment of APP/PS1 mice led to reduced Aβ plaque load and associated gliosis, altered cytokine profile, and increased regulatory T cell levels (158, 159). AD patients, and AD mouse models, show altered gut microbiota composition, favoring pro-inflammatory species, compared to healthy patients or wild-type mice (160–162). Microbiome composition affected cognition in APP/PS1 transgenic mice (162). Germ-free APP/PS1 transgenic mice showed reduction in Aβ deposition and pro-inflammatory cytokines in the brain (160). A recent study showed that fecal microbiota transplant from wild-type mice reduced Aβ plaques and tau tangles, gut permeability, systemic and neuroinflammation, and cognitive deficits in an AD mouse model (163). However, there is no in vivo evidence directly linking biofilm-forming bacteria and bacterial amyloids in the gut to AD pathology.
Rather than cross-seeding, AD microbiome studies point to a pattern of systemic inflammation and gut leakage that lead to AD pathology and cognitive deficits. Recent studies established that AD neuroinflammation involves NLRP3 inflammasome activation in microglia (164–166). Systemic inflammation induced by endotoxin or by fecal microbiota transplant from AD patients exacerbated microgliosis via the NLRP3 inflammasome in a mouse model (167, 168). Interestingly, curli activates the NLRP3 inflammasome in macrophages in vitro, suggesting a mechanism of microbiota-induced neuroinflammation (124). The endotoxin hypothesis also posits that microbiota-induced neuroinflammation underlies AD (169). Endotoxin, also known as lipopolysaccharide, produced by Gram-negative bacteria, is elevated in the serum of AD patients and during aging, has been shown to increase blood-brain-barrier permeability, and has even colocalized with Aβ plaques in the brain (Table 1) (169, 170). Endotoxin has also been implicated in the aggregation of a strain of αSyn in PD (171).
Another interesting possibility is direct infection of the brain in AD patients. Some have suggested that there is a separate microbiome within the brain, dominated by proteobacteria as well as fungal species (170, 172–174). If true, this would support the antimicrobial hypothesis of Aβ, wherein Aβ is secreted in response to infection as an antimicrobial peptide (129, 130). Still other studies implicate Porphyromonas gingivalis, another biofilm-forming bacteria, which causes chronic periodontitis in the mouth. P. gingivalis has been found in the brains of AD patients and is associated with neurotoxicity and neuroinflammation (Table 1) (175). Such studies are controversial due to contamination concerns during autopsy (176) and require further investigation. Regardless of how exactly the microbiome affects AD, antibiotics and probiotics have been proposed as treatment options (177–179).
Biofilms in Colorectal Cancer and Other Diseases of The Gastrointestinal Tract
Cancer results from uncontrolled, malignant cell proliferation caused by accumulated genetic and epigenetic mutations. The triggers for these mutations are multifactorial in origin and remain elusive in many cases, but genetics play a critical role. Accumulating evidence also supports the involvement of infectious agents in the development of cancer, especially in those organs that are exposed to microorganisms. Approximately 20% of cancers around the world have been estimated to be caused by microbes (180). For example, human papillomaviruses and the bacterium Helicobacter pylori cause cervical and gastric cancers, respectively (181, 182). Studies using Helicobacter have demonstrated that the protein encoded by cytotoxin-associated gene (cagA) induces DNA damage and that host-derived inflammatory mediators and growth factors are direct risk factors for carcinogenesis (182).
Colorectal cancer (CRC) is the fourth leading cause of cancer-related deaths worldwide (183). About 10% of CRC cases are hereditary, and the rest are sporadic. Risk factors include age, genetics, diet, and environmental factors (184, 185). Unhealthy behaviors such as physical inactivity, smoking, consumption of red and processed meat, and alcohol consumption increase risk of CRC (186–189). Some diseases, including obesity and type II diabetes, are also associated with increased risk of CRC development (190). Chronic inflammation is one of the major risks of CRC. Patients with inflammatory bowel diseases, including ulcerative colitis and Crohn's disease, are at risk for development of colitis-associated CRC (191, 192). The susceptibility of animal models of CRC, such as APCMin/+ mice (which carry a germline mutation in Apc gene) and azoxymethane-treated mice, is enhanced when dysbiosis is induced by the inflammatory agent dextran sodium sulfate (193, 194).
Further, the intestinal microbiota has emerged as an important factor in CRC initiation and progression. The current view is that CRC initiation is triggered by local mucosal colonization by pathogenic bacteria. Healthy human colon is protected by a mucosal barrier that separates the microbiome from direct contact with the colonic epithelium of the host (195). Reduction of the mucosal barrier increases the contact between the microbiota and colonic epithelial cells and thus constitutes a significant primary step in inciting modifications in the biology of cells and inflammation. Specific changes within the intestinal microbial community are observed in CRC patients, such as increased abundance of Fusobacteria, Alistipes, Porphyromonadaceae, Coriobacteridae, Staphylococcaceae, Akkermansia, and Methanobacteriales, while representation of Bifidobacterium, Lactobacillus, Ruminococcus, Faecalibacterium, Roseburia, and Treponema is decreased (196).
Recently, bacterial biofilms were observed in direct contact with CRC tumors upon mucosal degradation. Biofilms exist in the healthy gut mostly within the lumen and away from the epithelium. The fact that the biofilms were in direct contact with the CRC tumors, especially those located in the right colon of humans (determined as proximal host colon to the hepatic flexure) suggests that bacteria or bacterial products are connected to the initiation cell transformation events (197).
Although it was initially thought that biofilms were only on the tumors, patients with biofilm-positive tumors also had biofilms on their tumor-free mucosa. These findings suggest that in patients with biofilm-positive tumors, the luminal environment may provide an ideal landscape for biofilm development. However, the factors that facilitate biofilm development in the colon are difficult to identify as our knowledge of regulatory signals for bacterial biofilm formation is limited. Additionally, presence of bacterial biofilms on tumors is associated with reduced colonic epithelial cell E-cadherin, enhanced epithelial cell IL-6 and Stat3 activation as well as crypt epithelial cell proliferation (197). These observations have generated significant interest over the last few years in how bacterial biofilms facilitate CRC development.
To confirm that the human biofilms are indeed carcinogenic, three murine models of carcinogenesis were evaluated by Tomkovich et al.: germ-free ApcMinΔ850/+ (129SvEv) mice, (b) germ-free ApcMinΔ850/+ Il10−/− (129SvEv) mice, and conventional ApcMinΔ716/+ (C57BL/6) mice. In all three models, inocula prepared from biofilm-covered human mucosa induced colon tumors primarily in the distal colon at 12 weeks after inoculation. In contrast, inocula prepared from biofilm-negative mucosa did not (198). This study suggested that the biofilm itself might contribute to CRC pathology. Facultative anaerobic pathogens and pathobiont strains thrive in an inflammatory environment due to their ability to utilize inflammation-derived molecules such as nitrites and oxides as electron acceptors (199). In addition, microbial metabolism is altered under dysbiotic conditions, conferring new microbial phenotypes such as enhanced cellular adherence and invasion, mucus utilization, and production of metabolites and toxins (200). Microbiome profiling revealed that biofilms of distinct commensal bacteria, including Bacteriodes fragilis, Fusobacterium spp., and E. coli, were enriched in the CRC tumors, and these bacteria were able to promote CRC tumor development in genetically predisposed animals (184, 201–203).
Studies in murine models also showed that enterotoxigenic B. fragilis (ETBF) and colibactin-producing E. coli (CPEC) secrete carcinogenic toxins that are associated with the propagation of tumors (204–208). Furthermore, CPEC and ETBF were detected in patients with familial adenomatous polyposis, a premalignant disease that can develop into CRC (209).
Fusobacterium nucleatum accelerates tumorigenesis by inducing oncogenic and inflammatory responses in the gut through the production of the adhesin molecule FadA (210). However, whether FadA has toxigenic properties similar to those of colibactin or B. fragilis toxin is currently unknown. Finally, another enteric bacterium, Campylobacter jejuni, produces a genotoxin called cytolethal descending toxin (CDT) that induces colorectal cancer and changes in microbial composition and transcriptomic responses. Germ-free ApcMin/+ mice colonized with human clinical isolates of C. jejuni developed significantly more and larger tumors than the uncolonized mice or mice colonized with the cdtB mutant (211). Overall, these studies indicate a role for biofilm and toxin production by certain bacterial species as a driver for CRC pathogenesis.
It is plausible that toxins contribute to the onset of cancer by acting as direct environmental stressors. Toxins may also cause host cell DNA alterations. In germ-free mice that receive fresh feces from CRC patients, colon epithelia is renewed, more precancerous lesions are observed, and there is increased tissue and blood DNA methylation in intestinal tissues (212); this does not occur in mice given feces from healthy controls.
Recent studies showed a striking association between specific host microbes and aberrant DNA methylation in CRC. Addition or removal of acetyl and methyl residues at specific histone regions led to a corresponding gain or loss of DNA methylation at CpG dinucleotides, which led to an altered epigenomic state. CRC tumors can be grouped by CpG island methylator phenotype (CIMP); high CIMP, low CIMP or no CIMP (213). In CRC tumors where Fusobacterium species were substantially enriched, the tumors had a unique genetic and epigenetic profiles. The epigenetic changes were associated with high CIMP events and somatic mutations (213–215). However, additional studies are needed to determine whether bacterial toxins or whether certain bacterial species can directly promote oncolytic events by causing changes in DNA methylation. Additionally, dietary and digestive factors that are metabolized by microbiota can cause changes in the metabolic landscape and can alter the immune cell function (216). For instance, some short-chain fatty acids such as butyrate have anti-inflammatory properties and protect the host against colitis by increasing the level of colonic regulatory T cells and change the metabolism of epithelium (217–219). Therefore, dysbiosis of microbiota and localized enrichment of bacterial species may also directly influence the immune metabolism and function acting as a promoter or suppressor of tumor oncogenesis.
These data suggest that restricting the biofilm-mediated colonization by toxin-producing bacteria and reversing the dysbiosis would be a first step to reduce the tumorigenesis. Three strategies are feasible: (i) inhibition of biofilm formation, (ii) inhibition of toxin production or activity, or (iii) targeted inhibition of tumor-associated bacterial growth (Figure 3). Consistent with the idea that inhibition of toxin production is a viable strategy, toxin-negative mutants of CPEC and ETBF do not elicit tumor formation in the azoxymethane/dextran sulfate sodium model of CRC (220, 221). Suppression of tumorigenesis has also been achieved by using a small-molecule inhibitor that directly targets the toxin colibactin (222). In support of utility of targeted inhibition of the toxin-producing bacteria, when the growth of CPEC and other Enterobacteriaceae are inhibited with the oral administration of sodium tungstate, a metabolic inhibitor that targets molybdoenzymes, development of malignancies was blocked (Figure 3) (223).
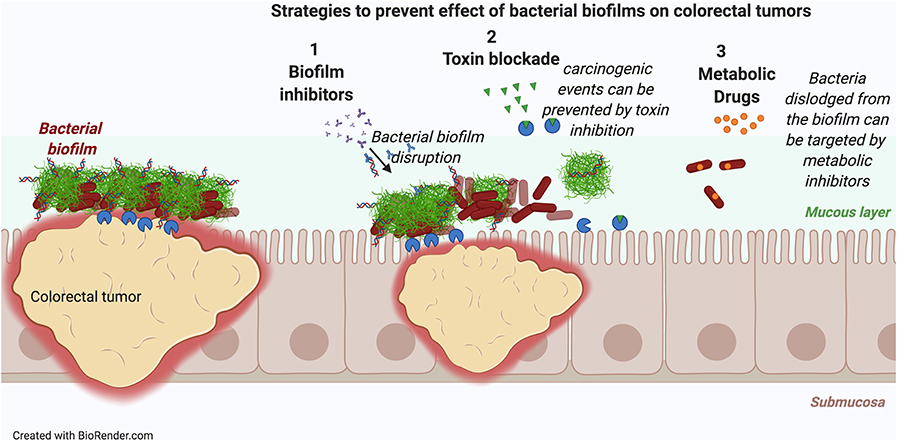
Figure 3. Novel strategies to intercept colorectal cancer. Three novel strategies either alone or in combination could reduce carcinogenesis triggered in biofilm associated colorectal tumors: (i) Inhibition of biofilm formation to dissociate the biofilms from the tumors, (ii) inhibition of toxin production by biofilm forming bacteria, (iii) inhibition of biofilm-associated bacterial growth by metabolic inhibitors.
Conclusions and Outlook
In the past decade, it has become increasingly apparent that the gut microbiota and infections with bacterial pathogens profoundly impact complex human diseases. In this review, we highlighted novel findings that indicate that biofilm-forming bacteria that produce the amyloid curli in the gastrointestinal tract are linked to autoimmune diseases, neurodegenerative diseases, and CRC. In healthy individuals, bacterial biofilms occupy the gut, and the extracellular matrix material educates the immune system to reinforce the epithelial barrier, preventing the leakage of bacterial ligands including pathogenic amyloids. However, the recent studies suggest that biofilms that harbor amyloid proteins like curli can initiate or accelerate pathogenic processes in a number of human diseases. The emerging picture suggests that in individuals that are genetically susceptible to chronic diseases, bacterial amyloids have pathogenic effects. For instance, in individuals who carry genetic risk factors such as HLA-B27, infections with invasive Salmonella, a pathogen that expresses curli, triggers autoimmunity and joint inflammation. Furthermore, enteric infections such as those with uropathogenic Escherichia coli trigger disease flares in SLE patients. It is important to note that the translocation of curli or curli-expressing bacteria into tissues is critical for the generation of autoimmune responses; this indicates that leakage of other amyloids or amyloid-producing bacteria from the gut or invasive infections may trigger similar responses. Consistent with this idea, infections with curli-producing enteric bacteria and other invasive bacteria that express amyloids, such as B. burgdorferi, S. aureus, Pseudomonas spp, and M. tuberculosis, trigger autoimmune reactions primarily affecting the joints (e.g., arthritis, septic arthritis).
Amyloid proteins are pathogenic in neurodegenerative diseases, and it was anticipated that bacterial amyloids like curli would influence aggregation of human amyloids like Aβ and αSyn. As predicted, recent work has shown that the colonization of the gut microbiota with curli-expressing E. coli increases αSyn pathology in mice that are predisposed to develop PD. However, whether curli can directly seed αSyn or whether it indirectly causes neuroinflammation and subsequent neurodegeneration is not known. Given these findings, treatments that reduce the pathological amyloid content in the microbiome or that reverse bacterial amyloid-induced neuroinflammation have potential as treatments for AD and PD.
One of the groundbreaking findings of the past several years was the demonstration that bacterial biofilms are associated with colonic tumors in humans and in animal models of CRC. Carcinogenic toxins produced by bacteria have been identified, but our current understanding of the direct effects of biofilms on the initiation and progression of CRC is limited. Given that we know how toxic amyloid intermediates form during initial stages of biofilm establishment, further work will be required to illuminate how curli or other amyloids contained in biofilms contribute to the onset of CRC by acting as a direct environmental stressor.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
ÇT was supported by NIH grants AI153325, AI151893, and AI148770.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Deo PN, Deshmukh R. Oral microbiome: unveiling the fundamentals. J Oral Maxillofac Pathol. (2019) 23:122–8. doi: 10.4103/jomfp.JOMFP_152_19
2. Cani PD. Human gut microbiome: hopes, threats and promises. Gut. (2018) 67:1716–25. doi: 10.1136/gutjnl-2018-316723
3. Shanahan F, van Sinderen D, O'Toole PW, Stanton C. Feeding the microbiota: transducer of nutrient signals for the host. Gut. (2017) 66:1709–17. doi: 10.1136/gutjnl-2017-313872
4. Berer K, Gerdes LA, Cekanaviciute E, Jia X, Xiao L, Xia Z, et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci USA. (2017) 114:10719–24. doi: 10.1073/pnas.1711233114
5. Hill-Burns EM, Debelius JW, Morton JT, Wissemann WT, Lewis MR, Wallen ZD, et al. Parkinson's disease and Parkinson's disease medications have distinct signatures of the gut microbiome. Mov Disord. (2017) 32:739–49. doi: 10.1002/mds.26942
6. Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. (2012) 490:55–60. doi: 10.1038/nature11450
7. Tilg H, Adolph TE, Gerner RR, Moschen AR. The intestinal microbiota in colorectal cancer. Cancer Cell. (2018) 33:954–64. doi: 10.1016/j.ccell.2018.03.004
8. Dobell C, Leeuwenhoek Av. Antony van Leeuwenhoek and his “Little animals”; being some account of the father of protozoology and bacteriology and his multifarious discoveries in these disciplines. New York, NY: Harcourt, Brace and company (1932). doi: 10.5962/bhl.title.13354
9. NIH HMP Working Group, Peterson J, Garges S, Giovanni M, McInnes P, Wang L, et al. The NIH human microbiome project. Genome Res. (2009) 19:2317–23. doi: 10.1101/gr.096651.109
10. Ursell LK, Metcalf JL, Parfrey LW, Knight R. Defining the human microbiome. Nutr Rev. (2012) 70(Suppl.1):38. doi: 10.1111/j.1753-4887.2012.00493.x
11. Davenport ER, Sanders JG, Song SJ, Amato KR, Clark AG, Knight R. The human microbiome in evolution. BMC Biol. (2017) 15:127–7. doi: 10.1186/s12915-017-0454-7
12. Senghor B, Sokhna C, Ruimy R, Lagier J. Gut microbiota diversity according to dietary habits and geographical provenance. Human Microbiome J. (2018) 7–8:1–9. doi: 10.1016/j.humic.2018.01.001
13. Thursby E, Juge N. Introduction to the human gut microbiota. Biochem J. (2017) 474:1823–36. doi: 10.1042/BCJ20160510
14. Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, et al. Metagenomic analysis of the human distal gut microbiome. Science. (2006) 312:1355–9. doi: 10.1126/science.1124234
15. Sender R, Fuchs S, Milo R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. (2016) 14:e1002533. doi: 10.1371/journal.pbio.1002533
16. Palmer C, Bik EM, DiGiulio DB, Relman DA, Brown PO. Development of the human infant intestinal microbiota. PLoS Biol. (2007) 5:e177. doi: 10.1371/journal.pbio.0050177
17. Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. (2001) 292:1115–8. doi: 10.1126/science.1058709
18. Lundin A, Bok CM, Aronsson L, Bjorkholm B, Gustafsson JA, Pott S, et al. Gut flora, Toll-like receptors and nuclear receptors: a tripartite communication that tunes innate immunity in large intestine. Cell Microbiol. (2008) 10:1093–103. doi: 10.1111/j.1462-5822.2007.01108.x
19. Wen L, Duffy A. Factors influencing the gut microbiota, inflammation, and type 2 diabetes. J Nutr. (2017) 147:1468S–75S. doi: 10.3945/jn.116.240754
20. Wilkins LJ, Monga M, Miller AW. Defining dysbiosis for a cluster of chronic diseases. Sci Rep. (2019) 9:12918-y. doi: 10.1038/s41598-019-49452-y
21. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, et al. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. (2014) 384:766–81. doi: 10.1016/S0140-6736(14)60460-8
22. Ng SC, Shi HY, Hamidi N, Underwood FE, Tang W, Benchimol EI, et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet. (2018) 390:2769–78. doi: 10.1016/S0140-6736(17)32448-0
23. Schnabel RB, Yin X, Gona P, Larson MG, Beiser AS, McManus DD, et al. 50 year trends in atrial fibrillation prevalence, incidence, risk factors, and mortality in the Framingham Heart Study: a cohort study. Lancet. (2015) 386:154–62. doi: 10.1016/S0140-6736(14)61774-8
24. Loftus PA, Wise SK. Epidemiology and economic burden of asthma. Int Forum Allergy Rhinol. (2015) 5(Suppl.1):7. doi: 10.1002/alr.21547
25. Scales CD Jr, Smith AC, Hanley JM, Saigal CS, Urologic diseases in America project. Prevalence of kidney stones in the United States. Eur Urol. (2012) 62:160–5. doi: 10.1016/j.eururo.2012.03.052
26. Menke A, Casagrande S, Geiss L, Cowie CC. Prevalence of and trends in diabetes among adults in the United States, 1988–2012. JAMA. (2015) 314:1021–9. doi: 10.1001/jama.2015.10029
27. Costerton JW. Introduction to biofilm. Int J Antimicrob Agents. (1999) 11:217–9. doi: 10.1016/S0924-8579(99)00018-7
28. Henrici AT. Studies of freshwater bacteria: I. A direct microscopic technique. J Bacteriol. (1933) 25:277–87. doi: 10.1128/JB.25.3.277-287.1933
29. Flemming HC, Wingender J. The biofilm matrix. Nat Rev Microbiol. (2010) 8:623–33. doi: 10.1038/nrmicro2415
30. Zogaj X, Bokranz W, Nimtz M, Romling U. Production of cellulose and curli fimbriae by members of the family Enterobacteriaceae isolated from the human gastrointestinal tract. Infect Immun. (2003) 71:4151–8. doi: 10.1128/IAI.71.7.4151-4158.2003
31. Sakellaris H, Hannink NK, Rajakumar K, Bulach D, Hunt M, Sasakawa C, et al. Curli loci of Shigella spp. Infect Immun. (2000) 68:3780–3. doi: 10.1128/IAI.68.6.3780-3783.2000
32. Sharma D, Misba L, Khan AU. Antibiotics versus biofilm: an emerging battleground in microbial communities. Antimicrob Resist Infect Control. (2019) 8:76–3. doi: 10.1186/s13756-019-0533-3
33. Barzegari A, Kheyrolahzadeh K, Hosseiniyan Khatibi SM, Sharifi S, Memar MY, Zununi Vahed S. The battle of probiotics and their derivatives against biofilms. Infect Drug Resist. (2020) 13:659–72. doi: 10.2147/IDR.S232982
34. Römling U. Characterization of the rdar morphotype, a multicellular behaviour in Enterobacteriaceae. Cell Mol Life Sci. (2005) 62:1234–46. doi: 10.1007/s00018-005-4557-x
35. Da Re S, Ghigo JM. A CsgD-independent pathway for cellulose production and biofilm formation in Escherichia coli. J Bacteriol. (2006) 188:3073–87. doi: 10.1128/JB.188.8.3073-3087.2006
36. Uhlich GA, Cooke PH, Solomon EB. Analyses of the red-dry-rough phenotype of an Escherichia coli O157:H7 strain and its role in biofilm formation and resistance to antibacterial agents. Appl Environ Microbiol. (2006) 72:2564–72. doi: 10.1128/AEM.72.4.2564-2572.2006
37. Zhao J, Sun Y. Regulation of c-di-GMP metabolism and biofilm formation in Yersinia pestis. Sheng Wu Gong Cheng Xue Bao. (2017) 33:1513–24. doi: 10.13345/j.cjb.170073
38. Tytgat HLP, Nobrega FL, van der Oost J, de Vos WM. Bowel biofilms: tipping points between a healthy and compromised Gut? Trends Microbiol. (2019) 27:17–25. doi: 10.1016/j.tim.2018.08.009
39. Costerton JW, Lewandowski Z, Caldwell DE, Korber DR, Lappin-Scott HM. Microbial biofilms. Annu Rev Microbiol. (1995) 49:711–45. doi: 10.1146/annurev.mi.49.100195.003431
40. Costerton JW, Geesey GG, Cheng KJ. How bacteria stick. Sci Am. (1978) 238:86–95. doi: 10.1038/scientificamerican0178-86
41. Gallo PM, Rapsinski GJ, Wilson RP, Oppong GO, Sriram U, Goulian M, et al. Amyloid-DNA composites of bacterial biofilms stimulate autoimmunity. Immunity. (2015) 42:1171–84. doi: 10.1016/j.immuni.2015.06.002
42. Lappin-Scott H, Burton S, Stoodley P. Revealing a world of biofilms–the pioneering research of Bill Costerton. Nat Rev Microbiol. (2014) 12:781–7. doi: 10.1038/nrmicro3343
43. Chorell E, Andersson E, Evans ML, Jain N, Götheson A, Åden J, et al. Bacterial chaperones CsgE and CsgC differentially modulate human α-synuclein amyloid formation via transient contacts. PLoS ONE. (2015) 10:e0140194. doi: 10.1371/journal.pone.0140194
44. Olsén A, Jonsson A, Normark S. Fibronectin binding mediated by a novel class of surface organelles on Escherichia coli. Nature. (1989) 338:625–55. doi: 10.1038/338652a0
45. McCrate OA, Zhou X, Reichhardt C, Cegelski L. Sum of the parts: composition and architecture of the bacterial extracellular matrix. J Mol Biol. (2013) 425:22. doi: 10.1016/j.jmb.2013.06.022
46. Hung C, Zhou Y, Pinkner JS, Dodson KW, Crowley JR, Heuser J, et al. Escherichia coli biofilms have an organized and complex extracellular matrix structure. mBio. (2013) 4:13. doi: 10.1128/mBio.00645-13
47. Maurer JJ, Brown TP, Steffens WL, Thayer SG. The occurrence of ambient temperature-regulated adhesins, curli, and the temperature-sensitive hemagglutinin tsh among avian Escherichia coli. Avian Dis. (1998) 42:106–18. doi: 10.2307/1592582
48. Olsen A, Arnqvist A, Hammar M, Normark S. Environmental regulation of curli production in Escherichia coli. Infect Agents Dis. (1993) 2:272–4.
49. Simm R, Morr M, Kader A, Nimtz M, Romling U. GGDEF and EAL domains inversely regulate cyclic di-GMP levels and transition from sessility to motility. Mol Microbiol. (2004) 53:1123–34. doi: 10.1111/j.1365-2958.2004.04206.x
50. Gerstel U, Romling U. Oxygen tension and nutrient starvation are major signals that regulate agfD promoter activity and expression of the multicellular morphotype in Salmonella typhimurium. Environ Microbiol. (2001) 3:638–48. doi: 10.1046/j.1462-2920.2001.00235.x
51. Hufnagel DA, Tükel C, Chapman MR. Disease to dirt: the biology of microbial amyloids. PLoS Pathog. (2013) 9:e1003740. doi: 10.1371/journal.ppat.1003740
52. Chapman MR, Robinson LS, Pinkner JS, Roth R, Heuser J, Hammar M, et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. (2002) 295:851–5. doi: 10.1126/science.1067484
53. Wang X, Smith DR, Jones JW, Chapman MR. In vitro polymerization of a functional Escherichia coli amyloid protein. J Biol Chem. (2007) 282:3713–9. doi: 10.1074/jbc.M609228200
54. Larsen P, Nielsen JL, Otzen D, Nielsen PH. Amyloid-like adhesins produced by floc-forming and filamentous bacteria in activated sludge. Appl Environ Microbiol. (2008) 74:1517–26. doi: 10.1128/AEM.02274-07
55. Maury CP. The emerging concept of functional amyloid. J Intern Med. (2009) 265:329–34. doi: 10.1111/j.1365-2796.2008.02068.x
56. Otzen D. Functional amyloid: turning swords into plowshares. Prion. (2010) 4:256–64. doi: 10.4161/pri.4.4.13676
57. Cheng N, He R, Tian J, Ye PP, Ye RD. Cutting edge: TLR2 is a functional receptor for acute-phase serum amyloid A. J Immunol. (2008) 181:22–6. doi: 10.4049/jimmunol.181.1.22
58. Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol. (2012) 188:1098–107. doi: 10.4049/jimmunol.1101121
59. Knowles TP, Vendruscolo M, Dobson CM. The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol. (2014) 15:384–96. doi: 10.1038/nrm3810
60. Tukel C, Raffatellu M, Humphries AD, Wilson RP, Andrews-Polymenis HL, Gull T, et al. CsgA is a pathogen-associated molecular pattern of Salmonella enterica serotype Typhimurium that is recognized by Toll-like receptor 2. Mol Microbiol. (2005) 58:289–304. doi: 10.1111/j.1365-2958.2005.04825.x
61. Tukel C, Wilson RP, Nishimori JH, Pezeshki M, Chromy BA, Baumler AJ. Responses to amyloids of microbial and host origin are mediated through toll-like receptor 2. Cell Host Microbe. (2009) 6:45–53. doi: 10.1016/j.chom.2009.05.020
62. Tükel C, Nishimori JH, Wilson RP, Winter MG, Keestra AM, van Putten, Jos PM, et al. Toll-like receptors 1 and 2 cooperatively mediate immune responses to curli, a common amyloid from enterobacterial biofilms. Cell Microbiol. (2010) 12:1495–505. doi: 10.1111/j.1462-5822.2010.01485.x
63. Winter SE, Lopez CA, Bäumler AJ. The dynamics of gut-associated microbial communities during inflammation. EMBO Rep. (2013) 14:319–27. doi: 10.1038/embor.2013.27
64. Bian Z, Brauner A, Li Y, Normark S. Expression of and cytokine activation by Escherichia coli curli fibers in human sepsis. J Infect Dis. (2000) 181:602–12. doi: 10.1086/315233
65. Humphries A, Deridder S, Baumler AJ. Salmonella enterica serotype Typhimurium fimbrial proteins serve as antigens during infection of mice. Infect Immun. (2005) 73:5329–38. doi: 10.1128/IAI.73.9.5329-5338.2005
66. Miller AL, Pasternak JA, Medeiros NJ, Nicastro LK, Tursi SA, Hansen EG, et al. In vivo synthesis of bacterial amyloid curli contributes to joint inflammation during S. Typhimurium infection. PLoS Pathog. (2020) 16:e1008591. doi: 10.1371/journal.ppat.1008591
67. Nishimori JH, Newman TN, Oppong GO, Rapsinski GJ, Yen JH, Biesecker SG, et al. Microbial amyloids induce interleukin 17A (IL-17A) and IL-22 responses via Toll-like receptor 2 activation in the intestinal mucosa. Infect Immun. (2012) 80:4398–408. doi: 10.1128/IAI.00911-12
68. Oppong GO, Rapsinski GJ, Newman TN, Nishimori JH, Biesecker SG, Tükel Ç. Epithelial cells augment barrier function via activation of the Toll-like receptor 2/phosphatidylinositol 3-kinase pathway upon recognition of Salmonella enterica serovar Typhimurium curli fibrils in the gut. Infect Immun. (2013) 81:478–86. doi: 10.1128/IAI.00453-12
69. Oppong GO, Rapsinski GJ, Tursi SA, Biesecker SG, Klein-Szanto AJ, Goulian M, et al. Biofilm-associated bacterial amyloids dampen inflammation in the gut: oral treatment with curli fibres reduces the severity of hapten-induced colitis in mice. NPJ Biofilms Microbiomes. (2015) 1:10.1038/npjbiofilms.2015.19. doi: 10.1038/npjbiofilms.2015.19
70. Shah C, Baral R, Bartaula B, Shrestha LB. Virulence factors of uropathogenic Escherichia coli (UPEC) and correlation with antimicrobial resistance. BMC Microbiol. (2019) 19:204–3. doi: 10.1186/s12866-019-1587-3
71. Subashchandrabose S, Mobley HLT. Virulence and fitness determinants of uropathogenic Escherichia coli. Microbiol Spectr. (2015) 3:10.1128/microbiolspec.UTI-2012. doi: 10.1128/microbiolspec.UTI-0015-2012
72. Hadjifrangiskou M, Gu AP, Pinkner JS, Kostakioti M, Zhang EW, Greene SE, et al. Transposon mutagenesis identifies uropathogenic Escherichia coli biofilm factors. J Bacteriol. (2012) 194:6195–205. doi: 10.1128/JB.01012-12
73. Karigoudar RM, Karigoudar MH, Wavare SM, Mangalgi SS. Detection of biofilm among uropathogenic Escherichia coli and its correlation with antibiotic resistance pattern. J Lab Physicians. (2019) 11:17–22. doi: 10.4103/JLP.JLP_98_18
74. Kai-Larsen Y, Luthje P, Chromek M, Peters V, Wang X, Holm A, et al. Uropathogenic Escherichia coli modulates immune responses and its curli fimbriae interact with the antimicrobial peptide LL-37. PLoS Pathog. (2010) 6:e1001010. doi: 10.1371/journal.ppat.1001010
75. Subashchandrabose S, Hazen TH, Brumbaugh AR, Himpsl SD, Smith SN, Ernst RD, et al. Host-specific induction of Escherichia coli fitness genes during human urinary tract infection. Proc Natl Acad Sci USA. (2014) 111:18327–32. doi: 10.1073/pnas.1415959112
76. Nicastro L, Tükel Ç. Bacterial amyloids: the link between bacterial infections and autoimmunity. Trends Microbiol. (2019) 27:954–63. doi: 10.1016/j.tim.2019.07.002
77. Bolon B. Cellular and molecular mechanisms of autoimmune disease. Toxicol Pathol. (2012) 40:216–29. doi: 10.1177/0192623311428481
78. Ring GH, Lakkis FG. Breakdown of self-tolerance and the pathogenesis of autoimmunity. Semin Nephrol. (1999) 19:25–33. doi: 10.1055/s-2008-1040823
79. Carter JD, Hudson AP. Reactive arthritis: clinical aspects and medical management. Rheum Dis Clin North Am. (2009) 35:21–44. doi: 10.1016/j.rdc.2009.03.010
80. Cunningham MW, McCormack JM, Fenderson PG, Ho MK, Beachey EH, Dale JB. Human and murine antibodies cross-reactive with streptococcal M protein and myosin recognize the sequence GLN-LYS-SER-LYS-GLN in M protein. J Immunol. (1989) 143:2677–83.
81. Mehlin C, Headley CM, Klebanoff SJ. An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J Exp Med. (1999) 189:907–18. doi: 10.1084/jem.189.6.907
82. Lasa I, Penadés JR. Bap: a family of surface proteins involved in biofilm formation. Res Microbiol. (2006) 157:99–107. doi: 10.1016/j.resmic.2005.11.003
83. Besingi RN, Wenderska IB, Senadheera DB, Cvitkovitch DG, Long JR, Wen ZT, et al. Functional amyloids in Streptococcus mutans, their use as targets of biofilm inhibition and initial characterization of SMU_63c. Microbiology. (2017) 163:488–501. doi: 10.1099/mic.0.000443
84. Dueholm MS, Petersen SV, Sønderkær M, Larsen P, Christiansen G, Hein KL, et al. Functional amyloid in Pseudomonas. Mol Microbiol. (2010) 77:1009–20. doi: 10.1111/j.1365-2958.2010.07269.x
85. Ohnishi S, Koide A, Koide S. The roles of turn formation and cross-strand interactions in fibrillization of peptides derived from the OspA single-layer beta-sheet. Protein Sci. (2001) 10:2083–92. doi: 10.1110/ps.15901
86. Alteri CJ, Xicohténcatl-Cortes J, Hess S, Caballero-Olín G, Girón JA, Friedman RL. Mycobacterium tuberculosis produces pili during human infection. Proc Natl Acad Sci USA. (2007) 104:5145–50. doi: 10.1073/pnas.0602304104
87. Schwartz K, Syed AK, Stephenson RE, Rickard AH, Boles BR. Functional amyloids composed of phenol soluble modulins stabilize Staphylococcus aureus biofilms. PLoS Pathog. (2012) 8:e1002744. doi: 10.1371/journal.ppat.1002744
88. Colmegna I, Cuchacovich R, Espinoza LR. HLA-B27-associated reactive arthritis: pathogenetic and clinical considerations. Clin Microbiol Rev. (2004) 17:348–69. doi: 10.1128/CMR.17.2.348-369.2004
89. Nikkari S, Rantakokko K, Ekman P, Mottonen T, Leirisalo-Repo M, Virtala M, et al. Salmonella-triggered reactive arthritis: use of polymerase chain reaction, immunocytochemical staining, and gas chromatography-mass spectrometry in the detection of bacterial components from synovial fluid. Arthritis Rheum. (1999) 42:84–9. doi: 10.1002/1529-0131(199901)42:1<84::AID-ANR11>3.0.CO;2-C
90. Nikkari S, Merilahti-Palo R, Saario R, Soderstrom KO, Granfors K, Skurnik M, et al. Yersinia-triggered reactive arthritis. Use of polymerase chain reaction and immunocytochemical staining in the detection of bacterial components from synovial specimens. Arthritis Rheum. (1992) 35:682–7. doi: 10.1002/art.1780350613
91. Sibilia J, Limbach FX. Reactive arthritis or chronic infectious arthritis? Ann Rheum Dis. (2002) 61:580–7. doi: 10.1136/ard.61.7.580
92. Manasson J, Shen N, Garcia Ferrer HR, Ubeda C, Iraheta I, Heguy A, et al. Gut microbiota perturbations in reactive arthritis and postinfectious spondyloarthritis. Arthritis Rheumatol. (2018) 70:242–54. doi: 10.1002/art.40359
93. Bian Z, Yan ZQ, Hansson GK, Thoren P, Normark S. Activation of inducible nitric oxide synthase/nitric oxide by curli fibers leads to a fall in blood pressure during systemic Escherichia coli infection in mice. J Infect Dis. (2001) 183:612–9. doi: 10.1086/318528
94. Tursi SA, Lee EY, Medeiros NJ, Lee MH, Nicastro LK, Buttaro B, et al. Bacterial amyloid curli acts as a carrier for DNA to elicit an autoimmune response via TLR2 and TLR9. PLoS Pathog. (2017) 13:e1006315. doi: 10.1371/journal.ppat.1006315
95. Horta-Baas G, Romero-Figueroa MDS, Montiel-Jarquin AJ, Pizano-Zarate ML, Garcia-Mena J, Ramirez-Duran N. Intestinal dysbiosis and rheumatoid arthritis: a link between Gut microbiota and the pathogenesis of rheumatoid arthritis. J Immunol Res. (2017) 2017:4835189. doi: 10.1155/2017/4835189
96. Dueholm MS, Albertsen M, Otzen D, Nielsen PH. Curli functional amyloid systems are phylogenetically widespread and display large diversity in operon and protein structure. PLoS ONE. (2012) 7:e51274. doi: 10.1371/journal.pone.0051274
97. Larsen P, Nielsen JL, Dueholm MS, Wetzel R, Otzen D, Nielsen PH. Amyloid adhesins are abundant in natural biofilms. Environ Microbiol. (2007) 9:3077–90. doi: 10.1111/j.1462-2920.2007.01418.x
98. Doaty S, Agrawal H, Bauer E, Furst DE. Infection and lupus: which causes which? Curr Rheumatol Rep. (2016) 18:13–4. doi: 10.1007/s11926-016-0561-4
99. Danza A, Ruiz-Irastorza G. Infection risk in systemic lupus erythematosus patients: susceptibility factors and preventive strategies. Lupus. (2013) 22:1286–94. doi: 10.1177/0961203313493032
100. Gladman DD, Hussain F, Ibañez D, Urowitz MB. The nature and outcome of infection in systemic lupus erythematosus. Lupus. (2002) 11:234–9. doi: 10.1191/0961203302lu170oa
101. Petri M. Infection in systemic lupus erythematosus. Rheum Dis Clin North Am. (1998) 24:423–56. doi: 10.1016/S0889-857X(05)70016-8
102. Pachucki RJ, Corradetti C, Kohler L, Ghadiali J, Gallo PM, Nicastro L, et al. Persistent bacteriuria and antibodies recognizing curli/eDNA complexes from Escherichia coli are linked to flares in systemic lupus erythematosus. Arthritis Rheumatol. (2020) 72:1872–81. doi: 10.1002/art.41400
103. Goedert M. Neurodegeneration. Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Aβ, tau, and α-synuclein. Science. (2015) 349:1255555. doi: 10.1126/science.1255555
104. Calsolaro V, Edison P. Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimer's Dement. (2016) 12:719–32. doi: 10.1016/j.jalz.2016.02.010
105. Refolo V, Stefanova N. Neuroinflammation and glial phenotypic changes in alpha-synucleinopathies. Front Cell Neurosci. (2019) 13:263. doi: 10.3389/fncel.2019.00263
106. Collins SM, Surette M, Bercik P. The interplay between the intestinal microbiota and the brain. Nat Rev. (2012) 10:735–42. doi: 10.1038/nrmicro2876
107. Powell N, Walker MM, Talley NJ. The mucosal immune system: master regulator of bidirectional gut-brain communications. Nat Rev Gastroenterol Hepatol. (2017) 14:143–59. doi: 10.1038/nrgastro.2016.191
108. Dinan TG, Cryan JF. The microbiome-gut-brain axis in health and disease. Gastroenterol Clin North America. (2017) 46:77–89. doi: 10.1016/j.gtc.2016.09.007
109. Erny D, de Angelis H, Lena A, Jaitin D, Wieghofer P, Staszewski O, et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci. (2015) 18:965–77. doi: 10.1038/nn.4030
110. Gheorghe CE, Martin JA, Manriquez FV, Dinan TG, Cryan JF, Clarke G. Focus on the essentials: tryptophan metabolism and the microbiome-gut-brain axis. Curr Opin Pharmacol. (2019) 48:137–45. doi: 10.1016/j.coph.2019.08.004
111. Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci. (2018) 12:49. doi: 10.3389/fnins.2018.00049
112. Holmqvist S, Chutna O, Bousset L, Aldrin-Kirk P, Li W, Björklund T, et al. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. (2014) 128:805–20. doi: 10.1007/s00401-014-1343-6
113. Bravo JA, Forsythe P, Chew MV, Escaravage E, Savignac HM, Dinan TG, et al. Ingestion of Lactobacillus strain regulates emotional behavior and central GABA receptor expression in a mouse via the vagus nerve. PNAS. (2011) 108:16050–5. doi: 10.1073/pnas.1102999108
114. Pretorius E, Bester J, Kell DB. A bacterial component to Alzheimer's-type dementia seen via a systems biology approach that links iron dysregulation and inflammagen shedding to disease. J Alzheimers Dis. (2016) 53:1237–56. doi: 10.3233/JAD-160318
115. Montacute R, Foley K, Forman R, Else KJ, Cruickshank SM, Allan SM. Enhanced susceptibility of triple transgenic Alzheimer's disease (3xTg-AD) mice to acute infection. J Neuroinflam. (2017) 14:5. doi: 10.1186/s12974-017-0826-5
116. Claesson MJ, Jeffery IB, Conde S, Power SE, O'Connor EM, Cusack S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature. (2012) 488:178–84. doi: 10.1038/nature11319
117. Franceschi C, Bonafè M, Valensin S, Olivieri F, Luca MD, Ottaviani E, et al. Inflamm-aging: an evolutionary perspective on immunosenescence. Ann N Y Acad Sci. (2000) 908:244–54. doi: 10.1111/j.1749-6632.2000.tb06651.x
118. Goodall EF, Wang C, Simpson JE, Baker DJ, Drew DR, Heath PR, et al. Age-associated changes in the blood-brain barrier: comparative studies in human and mouse. Neuropathol Appl Neurobiol. (2018) 44:328–40. doi: 10.1111/nan.12408
119. Braniste V, Al-Asmakh M, Kowal C, Anuar F, Abbaspour A, Tóth M, et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci Transl Med. (2014) 6:263ra158. doi: 10.1126/scitranslmed.3009759
120. Sochocka M, Donskow-Łysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer's disease-a critical review. Mol Neurobiol. (2019) 56:1841–51. doi: 10.1007/s12035-018-1188-4
121. Perov S, Lidor O, Salinas N, Golan N, Tayeb-Fligelman E, Deshmukh M, et al. Structural insights into Curli CsgA cross-β fibril architecture inspire repurposing of anti-amyloid compounds as anti-biofilm agents. PLoS Pathog. (2019) 15:1007978. doi: 10.1371/journal.ppat.1007978
122. Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal. (2015) 8:2005965. doi: 10.1126/scisignal.2005965
123. Friedland RP. Mechanisms of molecular mimicry involving the microbiota in neurodegeneration. J Alzheimers Dis. (2015) 45:349–62. doi: 10.3233/JAD-142841
124. Rapsinski GJ, Wynosky-Dolfi MA, Oppong GO, Tursi SA, Wilson RP, Brodsky IE, et al. Toll-like receptor 2 and NLRP3 cooperate to recognize a functional bacterial amyloid, curli. Infect Immun. (2015) 83:693–701. doi: 10.1128/IAI.02370-14
125. Laflamme N, Lacroix S, Rivest S. An essential role of interleukin-1β in mediating NF-κB activity and COX-2 transcription in cells of the blood–brain barrier in response to a systemic and localized inflammation but not during endotoxemia. J Neurosci. (1999) 19:10923–30. doi: 10.1523/JNEUROSCI.19-24-10923.1999
126. Terrando N, Eriksson LI, Ryu JK, Yang T, Monaco C, Feldmann M, et al. Resolving postoperative neuroinflammation and cognitive decline. Ann Neurol. (2011) 70:986–95. doi: 10.1002/ana.22664
127. Chen SG, Stribinskis V, Rane MJ, Demuth DR, Gozal E, Roberts AM, et al. Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged fischer 344 rats and Caenorhabditis elegans. Sci Rep. (2016) 6:srep34477. doi: 10.1038/srep34477
128. Miraglia F, Colla E. Microbiome, Parkinson's disease and molecular mimicry. Cells. (2019) 8:30222. doi: 10.3390/cells8030222
129. Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer's disease-associated β-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. (2018) 99:56–63.e3. doi: 10.1016/j.neuron.2018.06.030
130. Kumar DKV, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer's disease. Sci Transl Med. (2016) 8:340ra72. doi: 10.1126/scitranslmed.aaf1059
131. Park S, Moon JC, Shin SY, Son H, Jung YJ, Kim N, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. (2016) 478:924–8. doi: 10.1016/j.bbrc.2016.08.052
132. Kobayashi N, Masuda J, Kudoh J, Shimizu N, Yoshida T. Binding sites on tau proteins as components for antimicrobial peptides. Biocontrol Sci. (2008) 13:49–56. doi: 10.4265/bio.13.49
133. Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer's disease. Alzheimer's Dement. (2018) 14:1602–14. doi: 10.1016/j.jalz.2018.06.3040
134. Braak H, Ghebremedhin E, Rüb U, Bratzke H, Del Tredici K. Stages in the development of Parkinson's disease-related pathology. Cell Tissue Res. (2004) 318:121–34. doi: 10.1007/s00441-004-0956-9
135. Pellegrini C, Antonioli L, Colucci R, Ballabeni V, Barocelli E, Bernardini N, et al. Gastric motor dysfunctions in Parkinson's disease: current pre-clinical evidence. Parkinsonism Relat Disord. (2015) 21:1407–14. doi: 10.1016/j.parkreldis.2015.10.011
136. Park H, Lee JY, Shin CM, Kim J, Kim TJ, Kim JW. Characterization of gastrointestinal disorders in patients with parkinsonian syndromes. Parkinsonism Relat Disord. (2015) 21:455–60. doi: 10.1016/j.parkreldis.2015.02.005
137. Sampson TR, Debelius JW, Thron T, Janssen S, Shastri GG, Ilhan ZE, et al. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson's disease. Cell. (2016) 167:1469–80.e12. doi: 10.1016/j.cell.2016.11.018
138. Sampson TR, Challis C, Jain N, Moiseyenko A, Ladinsky MS, Shastri GG, et al. A gut bacterial amyloid promotes α-synuclein aggregation and motor impairment in mice. Elife. (2020) 9:e53111. doi: 10.7554/eLife.53111
139. Christensen LFB, Jensen KF, Nielsen J, Vad BS, Christiansen G, Otzen DE. Reducing the amyloidogenicity of functional amyloid protein FapC increases its ability to inhibit α-synuclein fibrillation. ACS Omega. (2019) 4:4029–39. doi: 10.1021/acsomega.8b03590
140. Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. (2013) 154:103–17. doi: 10.1016/j.cell.2013.05.057
141. Vasconcelos B, Stancu I, Buist A, Bird M, Wang P, Vanoosthuyse A, et al. Heterotypic seeding of Tau fibrillization by pre-aggregated Abeta provides potent seeds for prion-like seeding and propagation of Tau-pathology in vivo. Acta Neuropathol. (2016) 131:549–69. doi: 10.1007/s00401-015-1525-x
142. Zhou Y, Smith D, Leong BJ, Brännström K, Almqvist F, Chapman MR. Promiscuous cross-seeding between bacterial amyloids promotes interspecies biofilms. J Biol Chem. (2012) 287:35092–103. doi: 10.1074/jbc.M112.383737
143. Lundmark K, Westermark GT, Olsén A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: cross-seeding as a disease mechanism. Proc Natl Acad Sci USA. (2005) 102:6098–102. doi: 10.1073/pnas.0501814102
144. Friedland RP, McMillan JD, Kurlawala Z. What are the molecular mechanisms by which functional bacterial amyloids influence amyloid beta deposition and neuroinflammation in neurodegenerative disorders? Int J Mol Sci. (2020) 21:51652. doi: 10.3390/ijms21051652
145. Voth S, Gwin M, Francis CM, Balczon R, Frank DW, Pittet J, et al. Virulent Pseudomonas aeruginosa infection converts antimicrobial amyloids into cytotoxic prions. FASEB J. (2020) 34:9156–79. doi: 10.1096/fj.202000051RRR
146. Kim S, Kwon S, Kam T, Panicker N, Karuppagounder SS, Lee S, et al. Transneuronal propagation of pathologic α-synuclein from the gut to the brain models Parkinson's disease. Neuron. (2019) 103:627–41.e7. doi: 10.1016/j.neuron.2019.05.035
147. Challis C, Hori A, Sampson TR, Yoo BB, Challis RC, Hamilton AM, et al. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci. (2020) 23:327–36. doi: 10.1038/s41593-020-0589-7
148. Van Den Berge N, Ferreira N, Gram H, Mikkelsen TW, Alstrup AKO, Casadei N, et al. Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol. (2019) 138:535–50. doi: 10.1007/s00401-019-02040-w
149. Uemura N, Yagi H, Uemura MT, Hatanaka Y, Yamakado H, Takahashi R. Inoculation of α-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener. (2018) 13:5. doi: 10.1186/s13024-018-0257-5
150. Uemura N, Yagi H, Uemura MT, Yamakado H, Takahashi R. Limited spread of pathology within the brainstem of α-synuclein BAC transgenic mice inoculated with preformed fibrils into the gastrointestinal tract. Neurosci Lett. (2020) 716:134651. doi: 10.1016/j.neulet.2019.134651
151. Arotcarena M, Dovero S, Prigent A, Bourdenx M, Camus S, Porras G, et al. Bidirectional gut-to-brain and brain-to-gut propagation of synucleinopathy in non-human primates. Brain. (2020) 143:1462–75. doi: 10.1093/brain/awaa096
152. Honarpisheh P, Reynolds CR, Blasco Conesa MP, Moruno Manchon JF, Putluri N, Bhattacharjee MB, et al. Dysregulated gut homeostasis observed prior to the accumulation of the brain amyloid-β in Tg2576 mice. Int J Mol Sci. (2020) 21:51711. doi: 10.3390/ijms21051711
153. Pellegrini C, Daniele S, Antonioli L, Benvenuti L, D'Antongiovanni V, Piccarducci R, et al. Prodromal intestinal events in Alzheimer's disease (AD): colonic dysmotility and inflammation are associated with enteric AD-related protein deposition. Int J Mol Sci. (2020) 21:103523. doi: 10.3390/ijms21103523
154. Semar S, Klotz M, Letiembre M, Van Ginneken C, Braun A, Jost V, et al. Changes of the enteric nervous system in amyloid-β protein precursor transgenic mice correlate with disease progression. J Alzheimers Dis. (2013) 36:7–20. doi: 10.3233/JAD-120511
155. Han X, Tang S, Dong L, Song L, Dong Y, Wang Y, et al. Loss of nitrergic and cholinergic neurons in the enteric nervous system of APP/PS1 transgenic mouse model. Neurosci Lett. (2017) 642:59–65. doi: 10.1016/j.neulet.2017.01.061
156. Puig KL, Lutz BM, Urquhart SA, Rebel AA, Zhou X, Manocha GD, et al. Overexpression of mutant amyloid-β protein precursor and presenilin 1 modulates enteric nervous system. J Alzheimers Dis. (2015) 44:1263–78. doi: 10.3233/JAD-142259
157. Van Ginneken C, Schäfer K, Van Dam D, Huygelen V, De Deyn PP. Morphological changes in the enteric nervous system of aging and APP23 transgenic mice. Brain Res. (2011) 1378:43–53. doi: 10.1016/j.brainres.2011.01.030
158. Minter MR, Zhang C, Leone V, Ringus DL, Zhang X, Oyler-Castrillo P, et al. Antibiotic-induced perturbations in gut microbial diversity influences neuro-inflammation and amyloidosis in a murine model of Alzheimer's disease. Sci Rep. (2016) 6:30028. doi: 10.1038/srep30028
159. Minter MR, Hinterleitner R, Meisel M, Zhang C, Leone V, Zhang X, et al. Antibiotic-induced perturbations in microbial diversity during post-natal development alters amyloid pathology in an aged APP SWE /PS1 ΔE9 murine model of Alzheimer's disease. Sci Rep. (2017) 7:11047. doi: 10.1038/s41598-017-11047-w
160. Harach T, Marungruang N, Duthilleul N, Cheatham V, Mc Coy KD, Frisoni G, et al. Reduction of Abeta amyloid pathology in APPPS1 transgenic mice in the absence of gut microbiota. Sci Rep. (2017) 7:41802. doi: 10.1038/srep41802
161. Li B, He Y, Ma J, Huang P, Du J, Cao L, et al. Mild cognitive impairment has similar alterations as Alzheimer's disease in gut microbiota. Alzheimer's Dement. (2019) 15:1357–66. doi: 10.1016/j.jalz.2019.07.002
162. Li Z, Zhu H, Guo Y, Du X, Qin C. Gut microbiota regulate cognitive deficits and amyloid deposition in a model of Alzheimer's disease. J Neurochem. (2020) 155:448–61. doi: 10.1111/jnc.15031
163. Kim M, Kim Y, Choi H, Kim W, Park S, Lee D, et al. Transfer of a healthy microbiota reduces amyloid and tau pathology in an Alzheimer's disease animal model. Gut. (2020) 69:283–94. doi: 10.1136/gutjnl-2018-317431
164. Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Saecker A, et al. NLRP3 is activated in Alzheimer's disease and contributes to pathology in APP/PS1 mice. Nature. (2013) 493:674–8. doi: 10.1038/nature11729
165. Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte R, Tejera D, et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer's disease. Nature. (2017) 552:355–61. doi: 10.1038/nature25158
166. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature. (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
167. Tejera D, Mercan D, Sanchez-Caro JM, Hanan M, Greenberg D, Soreq H, et al. Systemic inflammation impairs microglial Aβ clearance through NLRP3 inflammasome. EMBO J. (2019) 38:101064. doi: 10.15252/embj.2018101064
168. Shen H, Guan Q, Zhang X, Yuan C, Tan Z, Zhai L, et al. New mechanism of neuroinflammation in Alzheimer's disease: the activation of NLRP3 inflammasome mediated by gut microbiota. Prog Neuropsychopharmacol Biol Psychiatry. (2020) 100:109884. doi: 10.1016/j.pnpbp.2020.109884
169. Brown GC. The endotoxin hypothesis of neurodegeneration. J Neuroinflammation. (2019) 16:7. doi: 10.1186/s12974-019-1564-7
170. Zhan X, Stamova B, Jin L, DeCarli C, Phinney B, Sharp FR. Gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology. (2016) 87:2324–32. doi: 10.1212/WNL.0000000000003391
171. Kim C, Lv G, Lee JS, Jung BC, Masuda-Suzukake M, Hong C, et al. Exposure to bacterial endotoxin generates a distinct strain of α-synuclein fibril. Sci Rep. (2016) 6:30891. doi: 10.1038/srep30891
172. Branton WG, Ellestad KK, Maingat F, Wheatley BM, Rud E, Warren RL, et al. Brain microbial populations in HIV/AIDS: α-proteobacteria predominate independent of host immune status. PLoS ONE. (2013) 8:54673. doi: 10.1371/journal.pone.0054673
173. Alonso R, Pisa D, Fernández-Fernández AM, Carrasco L. Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer's disease. Front Aging Neurosci. (2018) 10:159. doi: 10.3389/fnagi.2018.00159
174. Pisa D, Alonso R, Fernández-Fernández AM, Rábano A, Carrasco L. Polymicrobial infections in brain tissue from Alzheimer's disease patients. Sci Rep. (2017) 7:5903. doi: 10.1038/s41598-017-05903-y
175. Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer's disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. (2019) 5:aau3333. doi: 10.1126/sciadv.aau3333
176. Mente K. Letter re: gram-negative bacterial molecules associate with Alzheimer disease pathology. Neurology. (2017) 88:2338. doi: 10.1212/WNL.0000000000004046
177. Angelucci F, Cechova K, Amlerova J, Hort J. Antibiotics, gut microbiota, and Alzheimer's disease. J Neuroinflammation. (2019) 16:4. doi: 10.1186/s12974-019-1494-4
178. Panza F, Lozupone M, Solfrizzi V, Watling M, Imbimbo BP. Time to test antibacterial therapy in Alzheimer's disease. Brain. (2019) 142:2905–29. doi: 10.1093/brain/awz244
179. Kowalski K, Mulak A. Brain-gut-microbiota axis in Alzheimer's disease. J Neurogastroenterol Motil. (2019) 25:48–60. doi: 10.5056/jnm18087
180. de Martel C, Ferlay J, Franceschi S, Vignat J, Bray F, Forman D, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol. (2012) 13:607–15. doi: 10.1016/S1470-2045(12)70137-7
181. zur Hausen H. The search for infectious causes of human cancers: where and why. Virology. (2009) 392:1–10. doi: 10.1016/j.virol.2009.06.001
182. Wroblewski LE, Peek RM, Wilson KT. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev. (2010) 23:713–39. doi: 10.1128/CMR.00011-10
183. Arnold M, Sierra MS, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global patterns and trends in colorectal cancer incidence and mortality. Gut. (2017) 66:683–91. doi: 10.1136/gutjnl-2015-310912
184. Sears C, Garrett W. Microbes, microbiota, and colon cancer. Cell Host Microbe. (2014) 15:317–28. doi: 10.1016/j.chom.2014.02.007
185. Brennan CA, Garrett WS. Gut microbiota, inflammation, and colorectal cancer. Annu Rev Microbiol. (2016) 70:395–411. doi: 10.1146/annurev-micro-102215-095513
186. Golshiri P, Rasooli S, Emami M, Najimi A. Effects of physical activity on risk of colorectal cancer: a case–control study. Int J Prev Med. (2016) 7:175991. doi: 10.4103/2008-7802.175991
188. Biedermann L, Zeitz J, Mwinyi J, Sutter-Minder E, Rehman A, Ott SJ, et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE. (2013) 8:e59260. doi: 10.1371/journal.pone.0059260
189. Rossi M, Jahanzaib Anwar M, Usman A, Keshavarzian A, Bishehsari F. Colorectal cancer and alcohol consumption-populations to molecules. Cancers. (2018) 10:38. doi: 10.3390/cancers10020038
190. Vipperla K, O'Keefe SJ. The microbiota and its metabolites in colonic mucosal health and cancer risk. Nutr Clin Pract. (2012) 27:624–35. doi: 10.1177/0884533612452012
191. Farraye FA, Odze RD, Eaden J, Itzkowitz SH. AGA medical position statement on the diagnosis and management of colorectal neoplasia in inflammatory bowel disease. Gastroenterology. (2010) 138:738–45. doi: 10.1053/j.gastro.2009.12.037
192. Jess T, Rungoe C, Peyrin-Biroulet L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin Gastroenterol Hepatol. (2012) 10:639–45. doi: 10.1016/j.cgh.2012.01.010
193. Tanaka T, Kohno H, Suzuki R, Hata K, Sugie S, Niho N, et al. Dextran sodium sulfate strongly promotes colorectal carcinogenesis in Apc(Min/+) mice: inflammatory stimuli by dextran sodium sulfate results in development of multiple colonic neoplasms. Int J Cancer. (2006) 118:25–34. doi: 10.1002/ijc.21282
194. Neufert C, Becker C, Neurath MF. An inducible mouse model of colon carcinogenesis for the analysis of sporadic and inflammation-driven tumor progression. Nat Protocols. (2007) 2:1998–2004. doi: 10.1038/nprot.2007.279
195. Mirzaei R, Mirzaei H, Alikhani MY, Sholeh M, Arabestani MR, Saidijam M, et al. Bacterial biofilm in colorectal cancer: what is the real mechanism of action? Microb Pathog. (2020) 142:104052. doi: 10.1016/j.micpath.2020.104052
196. Sun J, Kato I. Gut microbiota, inflammation and colorectal cancer. Genes Dis. (2016) 3:130–43. doi: 10.1016/j.gendis.2016.03.004
197. Dejea CM, Wick EC, Hechenbleikner EM, White JR, Mark Welch JL, Rossetti BJ, et al. Microbiota organization is a distinct feature of proximal colorectal cancers. Proc Natl Acad Sci USA. (2014) 111:18321–6. doi: 10.1073/pnas.1406199111
198. Tomkovich S, Dejea CM, Winglee K, Drewes JL, Chung L, Housseau F, et al. Human colon mucosal biofilms from healthy or colon cancer hosts are carcinogenic. J Clin Invest. (2019) 129:1699–712. doi: 10.1172/JCI124196
199. Winter SE, Bäumler AJ. Dysbiosis in the inflamed intestine: chance favors the prepared microbe. Gut Microbes. (2014) 5:71–3. doi: 10.4161/gmic.27129
200. Tsilimigras MCB, Fodor A, Jobin C. Carcinogenesis and therapeutics: the microbiota perspective. Nat Microbiol. (2017) 2:17008. doi: 10.1038/nmicrobiol.2017.8
201. Viljoen KS, Dakshinamurthy A, Goldberg P, Blackburn JM. Quantitative profiling of colorectal cancer-associated bacteria reveals associations between Fusobacterium spp., enterotoxigenic Bacteroides fragilis (ETBF) and clinicopathological features of colorectal cancer. PLoS ONE. (2015) 10:e0119462. doi: 10.1371/journal.pone.0119462
202. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. (2012) 22:292–8. doi: 10.1101/gr.126573.111
203. Chen W, Liu F, Ling Z, Tong X, Xiang C. Human intestinal lumen and mucosa-associated microbiota in patients with colorectal cancer. PLoS ONE. (2012) 7:e39743. doi: 10.1371/journal.pone.0039743
204. Putze J, Hennequin C, Nougayrède J, Zhang W, Homburg S, Karch H, et al. Genetic structure and distribution of the colibactin genomic island among members of the family Enterobacteriaceae. Infect Immun. (2009) 77:4696. doi: 10.1128/IAI.00522-09
205. Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PLoS ONE. (2013) 8:e56964. doi: 10.1371/journal.pone.0056964
206. Raisch J, Buc E, Bonnet M, Sauvanet P, Vazeille E, de Vallée A, et al. Colon cancer-associated B2 Escherichia coli colonize gut mucosa and promote cell proliferation. World J Gastroenterol. (2014) 20:6560–72. doi: 10.3748/wjg.v20.i21.6560
207. Wilson MR, Jiang Y, Villalta PW, Stornetta A, Boudreau PD, Carrá A, et al. The human gut bacterial genotoxin colibactin alkylates DNA. Science. (2019) 363:eaar7785. doi: 10.1126/science.aar7785
208. Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, et al. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. (2014) 63:761–70. doi: 10.1136/gutjnl-2013-304739
209. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science. (2018) 359:592–7. doi: 10.1126/science.aah3648
210. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science. (2017) 358:1443. doi: 10.1126/science.aal5240
211. He Z, Gharaibeh RZ, Newsome RC, Pope JL, Dougherty MW, Tomkovich S, et al. Campylobacter jejuni promotes colorectal tumorigenesis through the action of cytolethal distending toxin. Gut. (2019) 68:289–300. doi: 10.1136/gutjnl-2018-317200
212. Sobhani I, Bergsten E, Couffin S, Amiot A, Nebbad B, Barau C, et al. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. PNAS. (2019) 116:24285–95. doi: 10.1073/pnas.1912129116
213. Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. (2014) 74:1311–8. doi: 10.1158/0008-5472.CAN-13-1865
214. Toyota M, Ohe-Toyota M, Ahuja N, Issa JJ. Distinct genetic profiles in colorectal tumors with or without the CpG island methylator phenotype. PNAS. (2000) 97:710–5. doi: 10.1073/pnas.97.2.710
215. Werner RJ, Kelly AD, Issa JJ. Epigenetics and precision oncology. Cancer J. (2017) 23:281. doi: 10.1097/PPO.0000000000000281
216. Sapienza C, Issa J. Diet, nutrition, and cancer epigenetics. Annu Rev Nutr. (2016) 36:665–81. doi: 10.1146/annurev-nutr-121415-112634
217. Litvak Y, Byndloss MX, Bäumler AJ. Colonocyte metabolism shapes the gut microbiota. Science. (2018) 362:eaat9076. doi: 10.1126/science.aat9076
218. Nagpal R, Wang S, Ahmadi S, Hayes J, Gagliano J, Subashchandrabose S, et al. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci Rep. (2018) 8:12649. doi: 10.1038/s41598-018-30114-4
219. Bultman SJ. Interplay between diet, gut microbiota, epigenetic events, and colorectal cancer. Mol Nutr Food Res. (2017) 61:902. doi: 10.1002/mnfr.201500902
220. Cevallos SA, Lee J, Tiffany CR, Byndloss AJ, Johnston L, Byndloss MX, et al. Increased epithelial oxygenation links colitis to an expansion of tumorigenic bacteria. mBio. (2019) 10:2244. doi: 10.1128/mBio.02244-19
221. Hwang S, Lee CG, Jo M, Park CO, Gwon S, Hwang S, et al. Enterotoxigenic Bacteroides fragilis infection exacerbates tumorigenesis in AOM/DSS mouse model. Int J Med Sci. (2020) 17:145–52. doi: 10.7150/ijms.38371
222. Cougnoux A, Delmas J, Gibold L, Faïs T, Romagnoli C, Robin F, et al. Small-molecule inhibitors prevent the genotoxic and protumoural effects induced by colibactin-producing bacteria. Gut. (2016) 65:278–85. doi: 10.1136/gutjnl-2014-307241
Keywords: curli, biofilm, microbiome, systemic lupus erythematosus, reactive arthritis, Parkinson's disease, Alzheimer's disease, colorectal cancer
Citation: Miller AL, Bessho S, Grando K and Tükel Ç (2021) Microbiome or Infections: Amyloid-Containing Biofilms as a Trigger for Complex Human Diseases. Front. Immunol. 12:638867. doi: 10.3389/fimmu.2021.638867
Received: 07 December 2020; Accepted: 09 February 2021;
Published: 26 February 2021.
Edited by:
Semih Esin, University of Pisa, ItalyReviewed by:
Gislane Lelis Vilela de Oliveira, São Paulo State University, BrazilRicardo Oropeza, National Autonomous University of Mexico, Mexico
Copyright © 2021 Miller, Bessho, Grando and Tükel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Çagla Tükel, Y3R1a2VsQHRlbXBsZS5lZHU=