 Jeremy Bechelli
Jeremy Bechelli Claire S. Rumfield1,3
Claire S. Rumfield1,3 David H. Walker
David H. Walker Steven Widen
Steven Widen Kamil Khanipov
Kamil Khanipov Rong Fang
Rong Fang- 1Department of Pathology, University of Texas Medical Branch at Galveston, Galveston, TX, United States
- 2Department of Biological Sciences, Sam Houston State University, Huntsville, TX, United States
- 3Laboratory of Tumor Immunology and Biology, National Cancer Institute, National Institutes of Health, Bethesda, MD, United States
- 4Center for Biodefense and Emerging Infectious Diseases, University of Texas Medical Branch, Galveston, TX, United States
- 5Department of Biochemistry & Molecular Biology, University of Texas Medical Branch at Galveston, Galveston, TX, United States
- 6Department of Pharmacology and Toxicology, University of Texas Medical Branch at Galveston, Galveston, TX, United States
We recently reported that the in vitro and in vivo survivals of Rickettsia australis are Atg5-dependent, in association with an inhibited level of anti-rickettsial cytokine, IL-1β. In the present study, we sought to investigate how R. australis interacts with host innate immunity via an Atg5-dependent autophagic response. We found that the serum levels of IFN-γ and G-CSF in R. australis-infected Atg5flox/floxLyz-Cre mice were significantly less compared to Atg5flox/flox mice, accompanied by significantly lower rickettsial loads in tissues with inflammatory cellular infiltrations including neutrophils. R. australis infection differentially regulated a significant number of genes in bone marrow-derived macrophages (BMMs) in an Atg5-depdent fashion as determined by RNA sequencing and Ingenuity Pathway Analysis, including genes in the molecular networks of IL-1 family cytokines and PI3K-Akt-mTOR. The secretion levels of inflammatory cytokines, such as IL-1α, IL-18, TNF-α, and IL-6, by R. australis-infected Atg5flox/floxLyz-Cre BMMs were significantly greater compared to infected Atg5flox/flox BMMs. Interestingly, R. australis significantly increased the levels of phosphorylated mTOR and P70S6K at a time when the autophagic response is induced. Rapamycin treatment nearly abolished the phosphorylated mTOR and P70S6K but did not promote significant autophagic flux during R. australis infection. These results highlight that R. australis modulates an Atg5-dependent autophagic response, which is not sensitive to regulation by mTORC1 signaling in macrophages. Overall, we demonstrate that R. australis counteracts host innate immunity including IL-1β-dependent inflammatory response to support the bacterial survival via an mTORC1-resistant autophagic response in macrophages.
Introduction
Rickettsiae are Gram-negative, obligately intracellular bacteria transmitted to humans by arthropod vectors. Highly virulent rickettsial species, such as Rickettsia rickettsii, can cause life-threatening disease with fatality as high as 40% (1). Clinically, rickettsial illnesses often present with fever, headache, and petechial rash. Although microvascular endothelial cells are the primary target of rickettsial infection (2), rickettsiae effectively invade macrophages and other types of cells, such as dendritic cells and hepatocytes. We, and others, have recently demonstrated that Rickettsia invades and survives in human macrophages, while macrophages play an important role in the pathogenesis of rickettsioses (3–9). However, it remains poorly understood how virulent rickettsiae utilize macrophages to develop a systemic infection in mammalian hosts.
Autophagy is an intracellular, bulk degradation process in which a portion of a cytoplasmic component of the cell is engulfed in double-membrane structures known as autophagosomes and is subsequently degraded upon fusion with lysosomes (10, 11). Atg5 (autophagy-related gene 5) is essential for autophagic vesicle formation as part of the ATG12-ATG5-ATG16 complex (12). A number of studies have employed Atg5-conditional knockout mice to investigate the interactions of autophagy with pathogenic microbes (13–15). Atg5flox/flox Lyz-Cre mice were generated by breeding Atg5 flox/flox mice to mice expressing the Cre recombinase from the endogenous lysozyme M locus (14), leading to an autophagy deficit mainly in macrophages resulting from deletion of the ATG5 gene. The mechanistic (formerly “mammalian”) target of rapamycin (mTOR) is an atypical serine-threonine protein kinase that plays a critical role in maintaining a balance between cellular anabolism and catabolism (16). The mTOR affects several aspects of cellular functions, including metabolism, aging, growth, apoptosis, and autophagy (17). It forms two complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2), with distinct composition and function (18). Toll-like receptor activation, cytokines, and low concentrations of amino acids will activate mTORC1 signaling leading to the phosphorylation of p70S6 kinase (p70S6K), mTOR and S6 kinase, which are established markers of mTORC1 activation (19–22). The mTORC1 is a master regulator of autophagy, since inhibition of mTORC1 was required to initiate the autophagy process (16, 23). Increasing evidence shows that mTORC1 has been implicated in the regulation of fusion of autophagosome to lysosome and the termination of autophagic flux (16).
Although autophagy has been considered a cornerstone of intracellular surveillance and host defense, intracellular bacteria and viruses are known to subvert or modify autophagy to facilitate their infection course (24–26). Recently, we have demonstrated that R. australis accumulates and co-localizes with LC3 (+) autophagosomes in macrophages, but not accompanied by a significantly reduced level of SQSTM1/p62 (4). In addition, pharmacological inhibition of mTOR signaling promotes the survival of R. australis in macrophages (4). Thus, R. australis induces a modified autophagic response, instead of autophagic flux. Using Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice, our recent studies clearly demonstrated that: 1) R. australis supports its infection in macrophages by inhibiting anti-rickettsial effect mediated by IL-1β via Atg5-dependent autophagic response; 2) Atg5-dependent autophagic response in macrophages facilitates the systemic infection of R. australis in association with suppressed serum levels of IL-1β (4). Notably, it is unknown how R. australis subverts the elements of the host innate immune system to support rickettsial infection via a modified autophagic response. Thus, we determined whether inflammatory and anti-inflammatory cytokines other than IL-1β were regulated by Atg5-dependent autophagic response via utilizing Atg5 flox/flox and Atg5 flox/flox Lyz-Cre mice. In addition, we investigated the interactions of R. australis with mTOR and its effect on the autophagic response in macrophages. Our studies demonstrate that R. australis induces a modified autophagic response while activating mTORC1 signaling in macrophages. Our findings suggest that R. australis subverts host innate immunity to support rickettsial infection in association with Atg5-dependent regulation of G-CSF and IFN-γ, in addition to IL-1β.
Materials and Methods
Rickettsiae and Mice
Rickettsia australis (Cutlack strain) was cultivated in Vero cells and purified by either Renografin density gradient centrifugation or using a Renografin “cushion” as previously described (27–29) for use in in vitro infections. The concentration of rickettsiae propagated in cell culture was determined by plaque assay after purification as described previously (28). Rickettsial stocks were stored at -80°C until use. All the experiments described in this study were conducted in a certified biosafety level 3 (BSL3) laboratory at UTMB. Wild type (WT) B6 mice were purchased from The Jackson Laboratory (catalog number 000664). Atg5 flox/flox Lyz-Cre (autophagy deficient), and Atg5 flox/flox (control) mice were kindly provided by Dr. Noboru Mizushima at the University of Tokyo and Dr. Herbert Virgin IV at Washington University School of Medicine in St. Louis (14, 15, 30). For in vivo experiments, mice were maintained and handled in a certified animal biosafety level-3 (ABSL3) facility at UTMB and inoculated intravenously (i.v.) through the tail vein with R. australis at the doses indicated. R. australis used in animal studies was grown in embryonated chicken egg yolk sac culture as described previously (31). Animals were monitored daily for signs of illness and sacrificed at indicated times. In vivo experiments were performed according to the Guide for the Care and Use of Laboratory Animals guidelines and approved by the Institutional Animal Care and Use Committee at UTMB.
Generation of Bone Marrow-Derived Macrophages
Primary bone marrow-derived macrophages (BMMs) were generated from 6-8 week old female WT B6 mice, Atg5 flox/flox mice and Atg5 flox/flox Lyz-Cre mice as previously described (4, 14, 15, 30, 32). Briefly, femurs and tibias were dissected, bone marrow was flushed using sterile medium, and cells were cultivated in low-endotoxin DMEM containing 10% (v/v) fetal bovine serum (FBS; Hyclone, Utah, SV30160) supplemented with either 20% supernatant from L929 cell culture or recombinant M-CSF (PeproTech, NJ, 315-02) at 37 °C in 5% CO2. Cells were harvested on day 6 and characterized by flow cytometric analysis by staining with anti-F4/80 and CD11b antibodies. Cultures were used when approximately 90% of these cells stained positive for F4/80 and CD11b. BMMs were plated at a density of 1 × 106 cells/well in 24-well plates in RPMI 1640 containing 10% FBS, and experiments were initiated within 24 hrs.
Macrophage In Vitro Infections
Primary mouse BMMs were infected with R. australis at a multiplicity of infection (MOI) of 5. In order to synchronize the internalization of bacteria, rickettsiae were centrifuged onto the cells at 560 × g for 5 min and incubated at 37°C in 5% CO2. Cells were collected and washed for further experiments at indicated times for each experiment, and uninfected cells served as the negative control.
Immunoblotting of Molecules Involved in Autophagy and mTOR Signaling
To assess the conversion of LC3-I to the lipidated LC3-II form, cells were lysed with RIPA lysis buffer (EMD Millipore, MA, 20-188) containing protease and phosphatase inhibitors (Roche, IN). Cell lysates were centrifuged to obtain soluble proteins, separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes and probed with polyclonal antibodies against LC3B (Cell Signaling Technology, MA, 4108). Bands were visualized using appropriate secondary antibodies and enhanced chemiluminescence (ECL) detection reagents (Thermo Scientific, Pierce, IL, 32106). Beta-actin was used to determine equal loading of the gel and was detected with mouse monoclonal antibody (mAb, Sigma, MO, A1978). Blotting against SQSTMI was performed using antibodies directed against SQSTM1 (Cell Signaling Technology, MA, 5114). Antibodies directed against phospho-mTOR (Ser2448) (D9C2) XP® rabbit mAb and phospho-p70 S6 Kinase (Thr389) (1A5) mouse mAb (Cell Signaling Technology, MA) were used for the analysis of the mTOR signaling pathway according to the manufacturer’s instructions. Densitometric analysis was quantitatively measured using Image J (33).
Immunofluorescence Microscopy
For immunofluorescence detection of LC3 puncta in R. australis-infected BMMs, cells were first seeded on glass coverslips in 12-well plates one day before infection as described previously (4). At 1 h post-infection (p.i.), cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 20 min, permeabilized with 0.5% Triton-X in PBS for 20 min and blocked with 3% BSA in PBS for 30 min. Samples were incubated with rabbit monoclonal antibody against LC3B (Cell Signaling Technology, #3868) followed by appropriate secondary antibody. Nuclei were stained with DAPI in ProLong® Gold Antifade Mountant (Life Technology, NY, P-36931). Coverslips were sealed with nail polish, and visualized by confocal microscopy with a 20 × lens (Olympus Fluoview 1000) using FV10-ASW software (Olympus, PA).
The levels of LC3 in cells were quantified as previously described (34). In brief, using Image J, an outline was drawn around each cell and circularity, area, mean fluorescence measured along with several adjacent background readings. The total corrected cellular fluorescence (TCCF) = integrated density – (area of selected cell × mean fluorescence of background readings) was calculated. This TCCF was then equalized against the mean TCCF of neighboring interphase cells in the same field of view, with results presented as fold increase over interphase levels. Box plots and statistical analysis (2-sided unpaired Student t tests) were performed using GraphPad Prism 5.
Pharmacological Inhibition of mTORC1 Signaling
Rapamycin, the pharmacologic gold standard for inhibiting mTOR, which acts by associating with FK-506 binding protein 12, was used to selectively inhibit mTORC1 (35). To inhibit mTORC1 signaling, cells were treated with 50 ng/mL of rapamycin (Sigma-Aldrich, St. Louis MO) for 4 hours prior to infection with rickettsiae. The cell number was counted, morphology observed by microscopy, and cell viability determined with the trypan blue dye exclusion method (36). The inhibitory effect of rapamycin on mTOR was examined by immunoblotting with antibodies against phospho-mTOR (Ser2448) (D9C2) XP® rabbit mAb and phospho-p70 S6 Kinase (Thr389) (1A5) mouse mAb (Cell Signaling Technology, MA).
Measurement of Cytokines by ELISA
Supernatants from Rickettsia-infected cells and uninfected controls were filter-sterilized and stored as aliquots at - 80°C. The concentration of IL-18 present in the supernatant of R. australis-infected BMMs was measured by ELISA using the Mouse IL-18 ELISA Kit (MBL International Corporation), following the manufacturer’s instructions. The detection limit for the ELISA cytokine concentrations was 25 pg/mL for IL-18. Samples were assayed in duplicate and are presented as the average of two independent experiments. Absorbance values were obtained using a VersaMax ELISA microplate reader (Molecular Devices, Sunnyvale, CA), and the concentrations were calculated from values obtained within the linear range of the standard curve.
Bio-plex Assay for Cytokine Analysis
Cell culture supernatants and mouse sera were processed according to the manufacturer’s instructions and then analyzed using a Bio-plex 200 system (Bio-Rad, Hercules, CA). Briefly, the samples were filter sterilized and subsequently centrifuged for 10 minutes at 450 × g at 4°C to remove debris. The resulting supernatants were collected and aliquoted into 96-well plates and processed for analysis on the Bio-plex system. The cytokines were coupled to cytokine-specific multi-plex beads (Bio-Rad) in the Bio-Plex mouse cytokine immunoassay following the manufacturer’s instructions. The pre-designed assay kit measured the concentrations of cytokines/chemokines including interleukin (IL)-1α, IL-6, IL-10, IFN-γ, granulocyte colony-stimulating factor (G-CSF), and tumor necrosis factor (TNF)-α.
Next-Generation Sequencing (NGS) and Pathway Analysis
RNA-seq analysis (next-generation sequencing) was performed on R. australis-infected BMMs of Atg5 flox/flox mice and Atg5 flox/flox Lyz-Cre mice at 24 h p.i. as described previously (37). Briefly, 1 μg of total RNA from uninfected and infected BMMs was poly A+ selected and fragmented using divalent cations and heat (94°C, 8 min). Illumina TruSeq v2 sample preparation kits (Illumina Inc., San Diego, CA) were used for the RNA-Seq library construction. NGS was performed at the NGS core facility, Sealy Center for Molecular Medicine, the University of Texas Medical Branch (UTMB). Sample libraries were sequenced by the Illumina HiSeq 1500 using a 2 × 50 base paired-end run protocol, with TruSeq v3 sequencing-by-synthesis chemistry. Reads were aligned to the mouse GRCm38 reference genome using the STAR alignment program, version 2.5.3a, with the recommended ENCODE options. GFOLD V1.1.4 was used to calculate the fold change differences between treatments. GFOLD generalizes the fold change by considering the posterior distribution of log fold change to overcome the problem of no replicate samples. Heatmap and hierarchical cluster analysis were generated to demonstrate the expression patterns of the top 100 genes differentially expressed. The dataset was filtered for a Log2 (fold change) ≥ 2.5 or ≤ -2.5 and then uploaded to Ingenuity Pathway Analysis (IPA) software (Ingenuity Systems, Redwood City, CA) for further analysis of the Atg5-dependent regulation of transcriptome profile of mouse macrophages during R. australis infection.
Histopathological Analyses
Formalin-fixed, hematoxylin and eosin (H&E)-stained tissue sections from infected and uninfected Atg5 flox/flox and Atg5 flox/flox Lyz-Cre mice were evaluated by a pathologist via both low-magnification and high-magnification microscopy. Images were taken using an Olympus BX41 photomicroscope (Olympus America, Inc., Center Valley, PA) or using a Revolution Microscope and an iPad Pro® tablet (Echo Laboratory, San Diego, CA). The histopathology slides were read by a board-certified Pathologist who has extensive experience with pathological analysis of rickettsial infections.
Statistical Analysis
The one-way analysis of variance (ANOVA) with Bonferroni’s procedure was used for comparisons of multiple experimental groups, and a Student t-test or Welch’s t-test was used for two-group comparisons depending on whether the variance between two groups was significantly different. The statistical analyses were performed using GraphPad Prism software version 5.01. p values of 0.05 or less were the threshold for statistical significance.
Results
R. australis Subverts Host Innate Immunity in Association With an Atg5-Dependent Autophagic Response
We have previously reported that a type 1 immune cytokine profile is closely associated with the host protective immunity against rickettsial diseases (38, 39). MyD88-dependent cytokines, including IFN-γ, IL-6, IL-12, and IL-1β, are the early signatures of a protective host innate immune response against R. australis (38). Inhibiting systemic production of IL-1β by Atg5-dependent autophagic response in macrophages contributes to the enhanced R. australis infection in vivo, as evidenced by our previous published studies employing Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice (4). The important question in this study is whether other components in host innate immunity, in addition to IL-1β, were subverted by R. australis to support rickettsial infection in vivo in an Atg5-dependent mechanism. To answer this question, we determined the association of the Atg5-dependent autophagic response with four pro-inflammatory cytokines, IL-1α, TNF-α,IFN-γ and G-CSF, and one anti-inflammatory cytokine, IL-10, in host innate immunity against R. australis. First, no significant difference in cytokine levels was identified in uninfected Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice (Figure 1). On day 4 p.i., a time point in host innate immunity at which serum levels of IL-1β and R. australis load in tissues were determined in our previous studies, the systemic production levels of IL-1α, TNF-α, and IL-10 in R. australis-infected Atg5 flox/flox Lyz-Cre mice were not significantly different from those in infected Atg5 flox/flox mice (Figure 1). Surprisingly, serum levels of IFN-γ and G-CSF in Atg5 flox/flox Lyz-Cre animals were significantly reduced in association with a previously documented lower R. australis load in tissues (4), compared to Atg5 flox/flox animals (Figure 1). Therefore, our results suggest that R. australis modulates Atg5-dependent regulation of G-CSF and IFN-γ, in addition to IL-1β, in host innate immunity to support rickettsial infection in vivo.
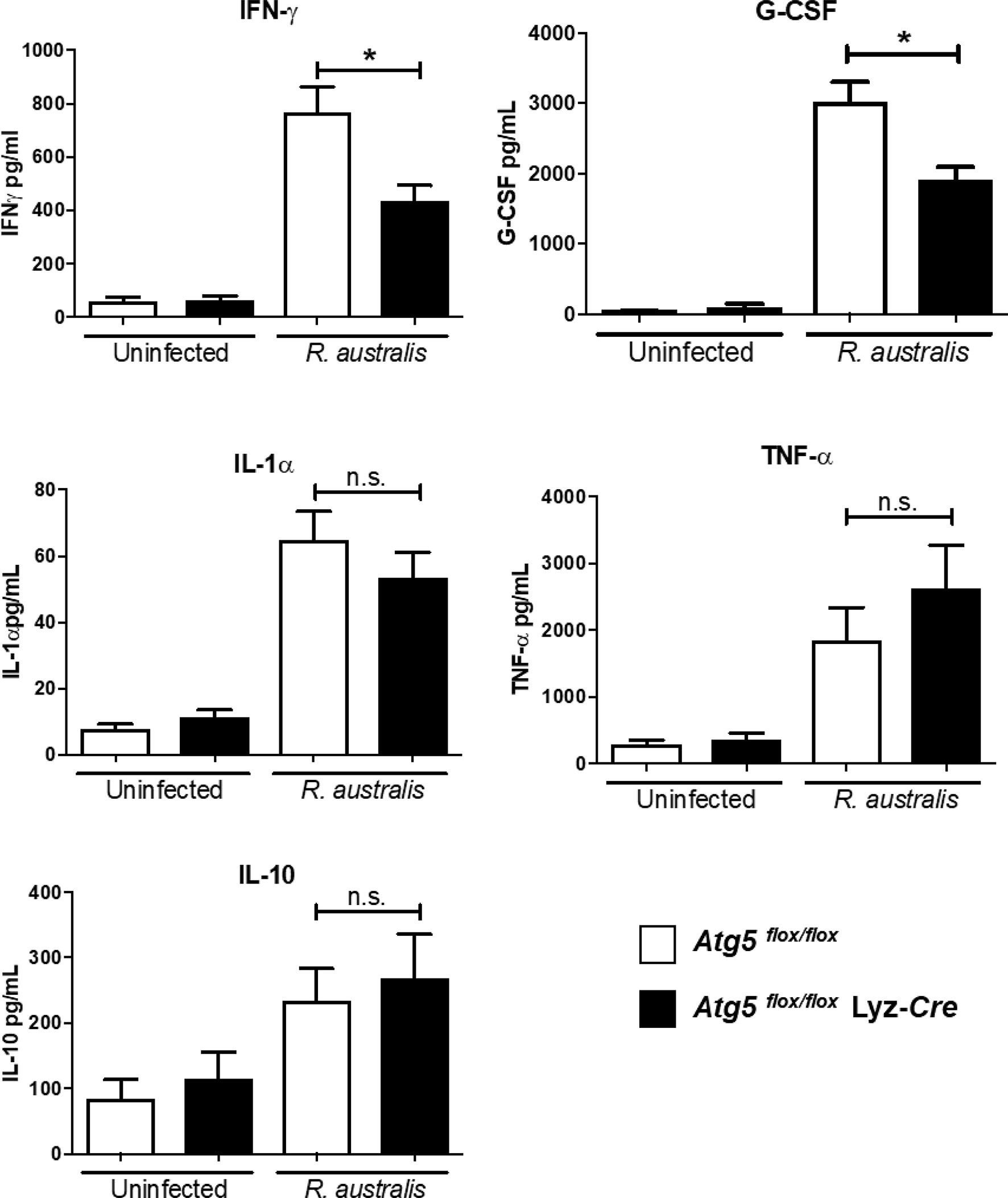
Figure 1 (previous Figure 6). R. australis subverts host innate immunity against rickettsioses via Atg5-dependent autophagic response. Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice were infected with R. australis i.v. at a dose of 3 × 105 PFU per mouse. On day 4 p.i., mice were euthanized, and serum was collected. Systemic production levels of cytokines/chemokines including IFN-γ, G-CSF, TNF-α, IL-1α, and IL-10 in mouse serum were determined by Bioplex assay. Results are means ± SE of data from three independent experiments containing 4-6 mice per group. *p<0.05; n.s., not statistically significant.
Inflammatory Cellular Accumulation Upon Infection With R. australis in Tissue of Atg5 flox/flox Mice Is Quantitatively and Qualitatively Different Compared to Atg5 flox/flox Lyz-Cre Mice
Histologic analysis of the H&E-stained liver and lung sections of infected animals showed inflammatory cell infiltrations and lesions in both R. australis-infected Atg5 flox/flox and infected Atg5 flox/flox Lyz-Cre mice on day 4 p.i. compared to uninfected controls (Figure 2). Livers of infected Atg5 flox/flox Lyz-Cre mice showed numerous perivascular foci of inflammatory infiltration either around the central vein and portal triad or in the lobules (Figure 2A). Compared to Atg5 flox/flox mice, deficiency in Atg5 in macrophages resulted in thrombosis and infarction in the liver (Figure 2B). In the liver of infected Atg5 flox/flox Lyz-Cre mice, inflammatory foci were randomly distributed throughout the tissue. These cellular infiltrations consisted of mainly macrophages, but also lymphocytes and neutrophils (Figures 2C, D). Furthermore, R. australis-infected Atg5 flox/flox Lyz-Cre mice showed interstitial pneumonia in the lungs (Figures 2E, F).
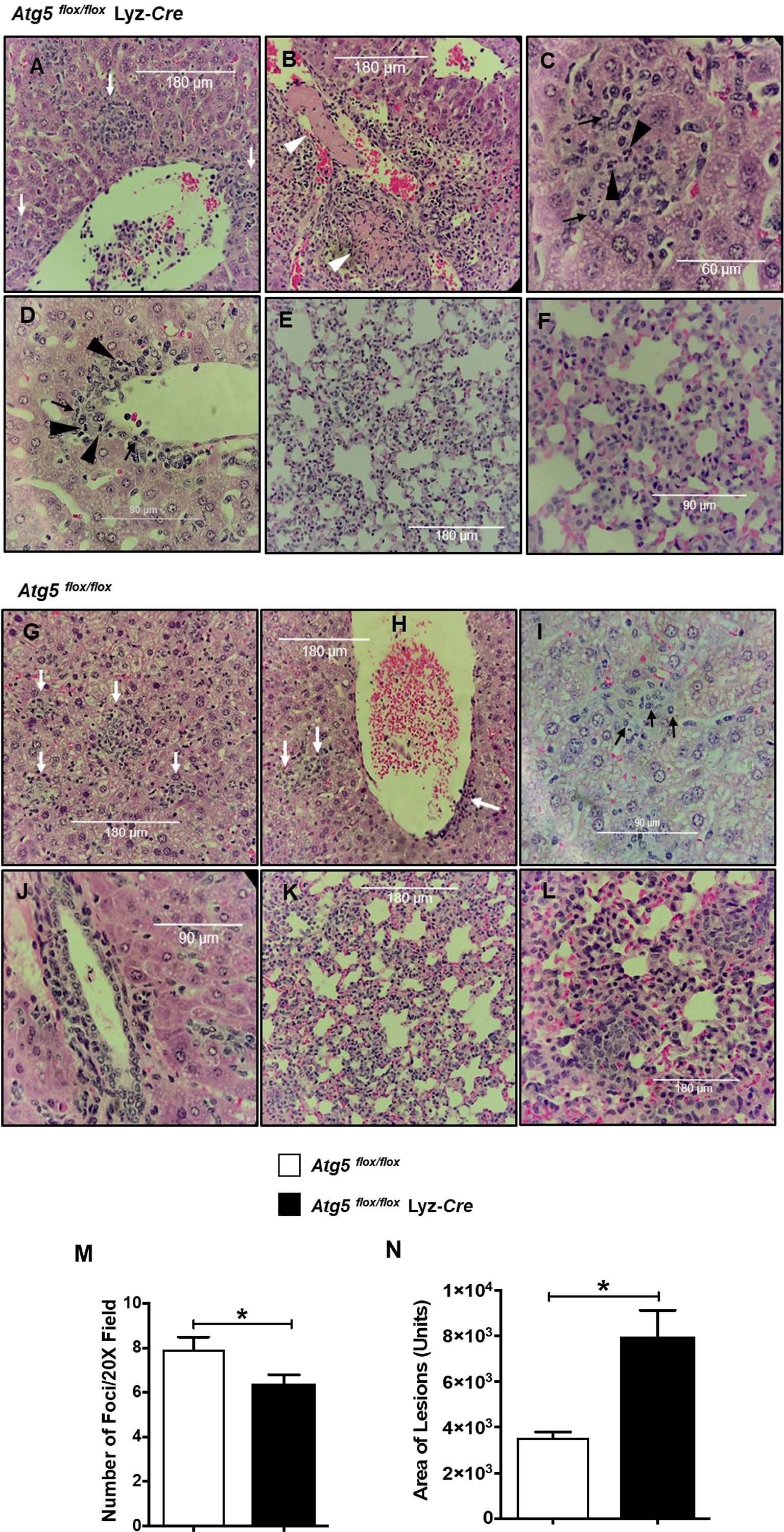
Figure 2 (previous Figure 7). Inflammatory cellular accumulation upon infection with R. australis in tissues of Atg5 flox/flox mice and Atg5 flox/flox Lyz-Cre mice. Mice were infected i.v. with R. australis (3 × 105 PFU per mouse). On day 4 p.i., mice were sacrificed, and tissues were isolated and analyzed. Histological analysis of livers and lungs from infected Atg5 flox/flox Lyz-Cre mice (A–F) and Atg5 flox/flox mice (G–L). Foci of inflammatory infiltration are indicated by white arrows. Thrombus or necrotic cells related to thrombosis is shown as white arrowheads (B). Polymorphonuclear neutrophils (PMNs) (black arrowheads) and macrophages (black arrows) are shown in livers. Furthermore, the size (M) and frequency (N) of inflammatory lesions in livers were analyzed using ImageJ (magnification, ×20). Images were taken using an Olympus BX41 photomicroscope (Olympus America, Inc., Center Valley, PA) or using a Revolution Microscope and an iPad Pro® tablet (Echo Laboratory, San Diego, CA). *p<0.05.
Evaluation of histopathological changes in livers of infected Atg5flox/flox mice showed cellular infiltration in widely distributed foci consisting of macrophages, but fewer or no neutrophils compared to Atg5 flox/flox Lyz-Cre mice (Figures 2G–J). The lungs of R. australis-infected Atg5 flox/flox mice also showed interstitial pneumonia (Figures 2K, L). Interestingly, the frequency of pathological lesions was significantly reduced in livers of infected Atg5 flox/flox Lyz-Cre mice compared to Atg5 flox/flox mice (Figure 2M). In contrast, the inflammatory lesions in the livers of infected Atg5 flox/flox Lyz-Cre mice were significantly greater in size compared to infected Atg5 flox/flox mice (Figure 2N). Considering the greater R. australis load in tissues of Atg5 flox/flox mice vs Atg5 flox/flox Lyz-Cre mice, these results suggest that R. australis subverts host innate immunity via Atg5 to support their infection in association with histopathological changes featured by increased frequency and reduced size of cellular infiltrations consisting of few or no neutrophils.
Atg5-Dependent Gene Regulation by R. australis Infection in Macrophages
No prior studies have examined the Atg5-dependent regulation of host gene expression by Rickettsia in macrophages. We performed four comparisons in this study, including infected Atg5 (+) vs uninfected Atg5 (+), infected Atg5 (-) vs infected Atg5 (+), infected Atg5 (-) vs infected Atg5 (+), and uninfected Atg5 (-) vs uninfected Atg5 (+). As shown in Figure 3, Atg5 (-) refers to Atg5 flox/flox Lyz-Cre while Atg5 (+) refers to Atg5 flox/flox. First, a list of the top 100 differentially expressed genes in each comparison was used to generate a heat map (Figure 3A). Approximately half of these 100 genes were upregulated in infected macrophages in all four comparisons, notably pro-inflammatory cytokines (IL-1 family of cytokines and TNF-alpha) and chemokines (CCL5 and CXCL10), that may be of particular interest for the purpose of our studies. Moreover, 34 genes were differentially expressed in each comparison, suggesting that the expression levels of these genes are closely associated with Atg5-dependent autophagy and/or R. australis infection (Figure 3A). We generated a Venn diagram showing the overlap of significantly modulated genes among four comparisons InteractiVenn (Figure 3B). No gene was shared among the four comparisons. The majority of modulated genes were unique to either infected Atg5 (-) vs uninfected Atg5 (-), infected Atg5 (+) vs uninfected Atg5 (+), or uninfected Atg5 (-) vs uninfected Atg5 (+).
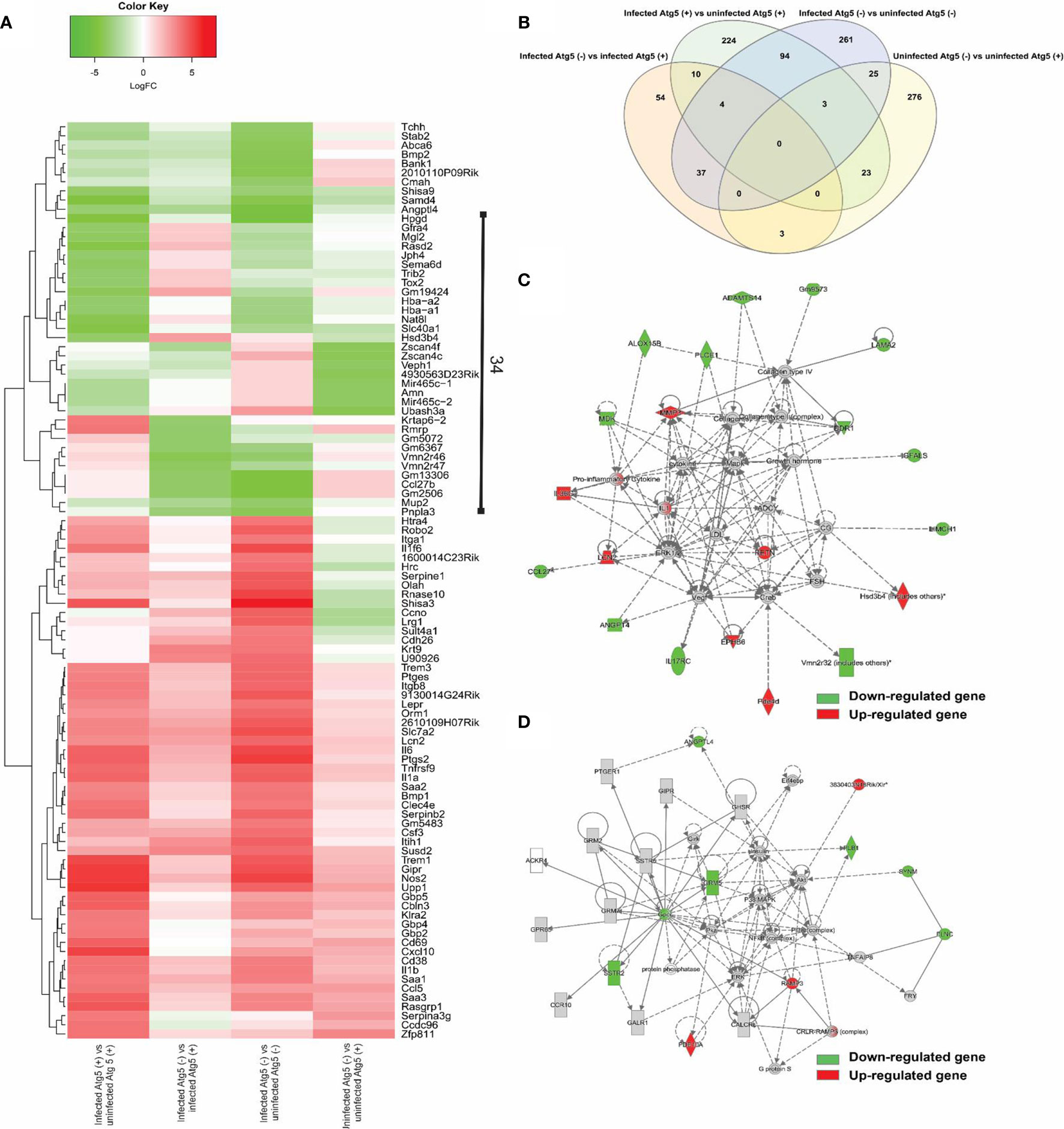
Figure 3 (previous Figure 3). Comparative transcriptional analysis of R. australis-infected Atg5 flox/flox Lyz-Cre and Atg5 flox/flox BMMs. BMMs were isolated from Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice, and then infected with R. australis at an MOI of 5. At 24 h p.i., cells were collected and total RNA was extracted. RNA-seq analysis was performed as described in Materials and Methods. Atg5 (+), Atg5 flox/flox; Atg5 (-), Atg5 flox/flox Lyz-Cre. (A), Heatmap and hierarchical clustering of the top 100 genes differentially regulated by Atg5 during R. australis infection in mouse macrophages. The expression levels of genes are indicated by the color bar above the heatmap. Red color indicates the increased expression whereas green color indicates the decreased expression in four comparisons. (B), Venn diagram showing overlap of significantly modulated genes for each of the four comparisons. (C, D), IPA molecular networks analysis of differentially expressed genes in IL-1 family cytokines signaling and PI3K-Akt-mTOR signaling in R. australis-infected Atg5 flox/flox Lyz-Cre BMMs vs R. australis-infected Atg5 flox/flox BMMs. Red, up-regulated; green, down-regulated.
The focus of the present study is on the subversion of the host response in macrophages by R. australis observed in an Atg5-competent vs. Atg5-compromised mice. The networks generated by IPA analysis illustrate the interrelationships between genes and the temporal changes in gene modulation. Two molecular networks, in which IL-1 family cytokines (Figure 3C) and PI3K-Akt-mTOR (Figure 3D) were central hub molecules, were identified by IPA. The first network (Figure 3C) consists of 22 genes differentially expressed in infected Atg5 (-) and infected Atg5 (+) BMMs. In IL-1 family cytokine signaling, nine and thirteen genes were up- and down-regulated, respectively, by R. australis infection in Atg5-deficient macrophages compared to Atg5-competent macrophages. Thus, Atg5 specifically down-regulated nine genes during R. australis infection, including IL-1, IL-36G, lipocalin-2 (LCN2), resistin (RETN), NADPH-dependent 3-keto-steroid reductase (Hsd3b4), matrix metallopeptidase 3 (MMP3), and ephrin type-B receptor 6 (EPHB6), some of which have been shown to contribute to host innate immunity against infections (40). Therefore, Atg5 benefits R. australis infection in macrophages, at least in part in association with inhibiting the members of IL-1 family cytokines in the host innate immune system. In Figure 3D, the hub molecules are PI3K, Akt, insulin, and eukaryotic initiation factor 4E-binding protein 1 (Eif4ebp) (41). When mTORC1 is active, it phosphorylates (activates) p70S6 kinase (S6K) and the eIF4e binding protein (42). Thus, the PI3K-Akt-mTOR signaling pathway was regulated by R. australis infection. Compared to infected Atg5 flox/flox BMMs, 4 genes including RAMP3 and PDE10A were upregulated while 7 genes including ANGPTL4 and Gpcr were downregulated in infected Atg5 flox/flox Lyz-Cre BMMs by R. australis infection (Figure 3D). These results suggest that the enhanced R. australis infection in Atg5-competent macrophages is associated with the alterations of the key cellular signaling pathway, PI3K-Akt- mTOR.
R. australis Suppresses the Production of Inflammatory Cytokines by Infected Macrophages via a Modified Atg5-Dependent Autophagic Response
Our recently published studies have shown that R. australis induces a modified Atg5-dependent autophagic response to benefit its replication through inhibiting the secretion of rickettsicidal IL-1β, although IL-1β-independent mechanisms also contribute to this process (4). To explore IL-1β-independent factors involved in supporting R. australis infection in macrophages, we measured the secretion levels of five cytokines in R. australis-infected Atg5 flox/flox Lyz-Cre BMMs and Atg5 flox/flox BMMs at 24 h p.i. Uninfected BMMs of Atg5-conditional knockout mice did not produce any significant levels of the examined cytokines (Figure 4). The deficiency of Atg5-dependent autophagy did not change the levels of either IFN-γ or G-CSF secreted by infected macrophages (Figure 4). Interestingly, R. australis-infected Atg5 flox/flox Lyz-Cre BMMs produced significantly greater levels of IL-6, IL-1α, and TNF-α compared to infected Atg5 flox/flox BMMs (Figure 4). The enhanced production of IL-6, IL-1α and TNF-α was associated with reduced R. australis infection in Atg5-deficient macrophages (4). These results suggest that R. australis exploits the Atg5-dependent autophagic response to inhibit the production of IL-6, IL-1α, and TNF-α in macrophages to support their infection.
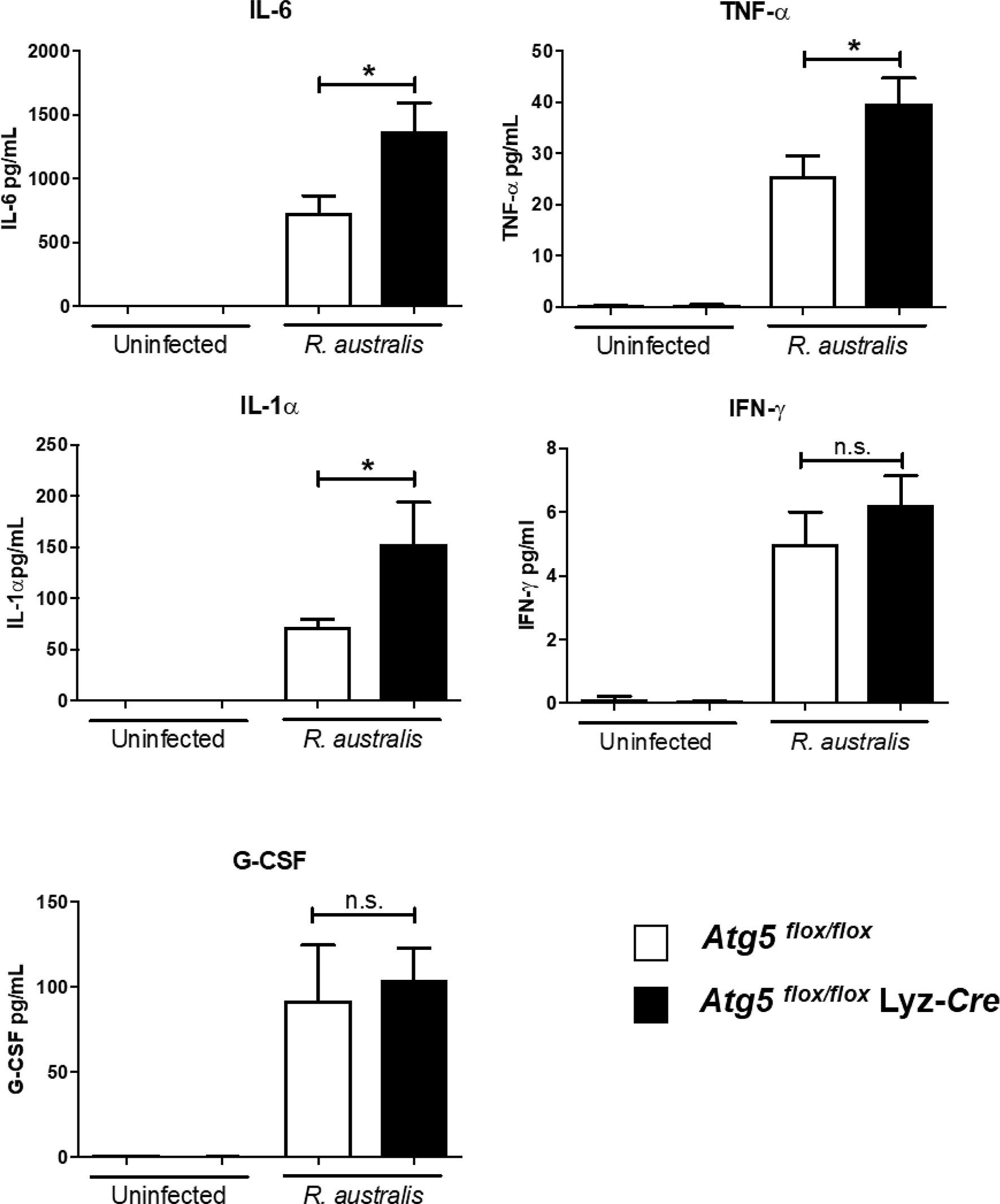
Figure 4 (previous Figure 4). R. australis counteracts the production of pro-inflammatory cytokines by infected macrophages via Atg5-dependent autophagy. BMMs were isolated from Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice, and they were infected with R. australis at an MOI of 5. At 24 h p.i., supernatant was harvested. Production levels of cytokines including IL-6, TNF-α, IL-1α, IFN-γ and G-CSF in the supernatant were assessed by Bioplex assay. Results are means ± SE of data from three independent experiments. *p<0.05; n.s., not statistically significant.
IL-18 is an important member of the IL-1 family of cytokines (43–45). To determine the effect of autophagy on production of IL-18 by R. australis-infected macrophages, we employed BMMs from both Atg5- and Atg16l1-conditional knockout mice. R. australis infection resulted in significantly greater production levels of IL-18 in Atg5 flox/flox Lyz-Cre BMMs compared to Atg5 flox/flox BMMs (Figure 5A). Furthermore, the secretion levels of IL-18 by R. australis-infected Atg16l1 flox/flox BMMs were significantly less than those by infected Atg16l1 flox/flox Lyz-Cre BMMs (Figure 5B). We had demonstrated that deletion of Atg16l1 significantly reduces the concentrations of R. australis (4). Thus, the reduced IL-18 production by Atg16l1 flox/flox BMMs was associated with a greater rickettsial load in these BMMs compared to Atg16l1 flox/flox Lyz-Cre BMMs. These results clearly revealed that R. australis suppressed the production of IL-18 in an autophagy-dependent manner, consistent with the results of Atg5-dependent regulation of IL-1 family cytokines at the transcriptional level by this bacterium, as shown in Figure 3C.
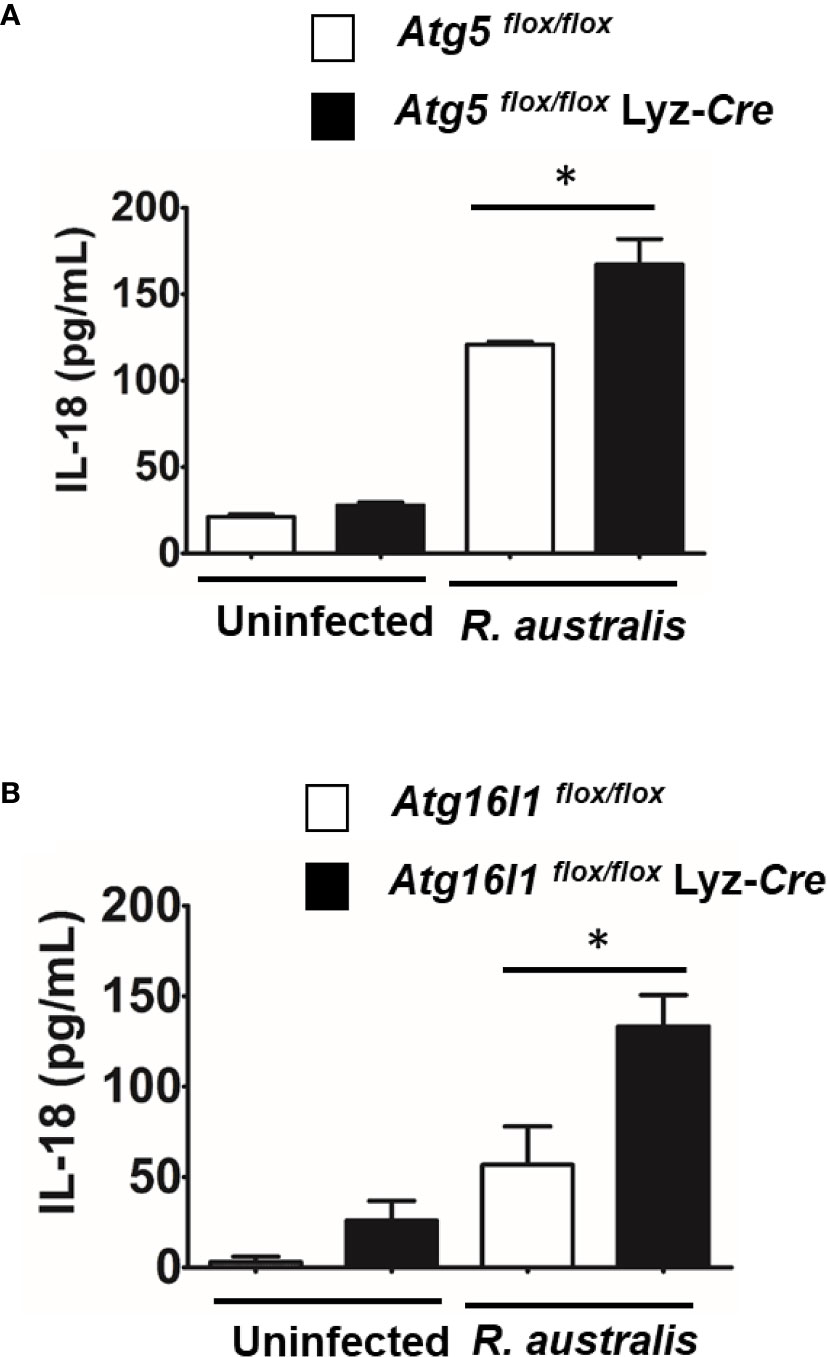
Figure 5 (previous Figure 5). R. australis suppressed the production of IL-18 by infected macrophages via Atg5-dependent autophagy. BMMs were isolated from Atg5 flox/flox Lyz-Cre and Atg5 flox/flox mice, and Atg16l1 flox/flox Lyz-Cre and Atg16l1 flox/flox mice. Macrophages were infected with R. australis at an MOI of 5. At 24 h p.i., supernatant was harvested. Concentrations of IL-18 in the supernatant of infected Atg5 flox/flox Lyz-Cre BMMs and Atg5 flox/flox BMMs (A), and infected Atg16l1 flox/flox Lyz-Cre BMMs and Atg16l1 flox/flox BMMs (B) were assessed by ELISA. Results are means ± SE of data from two independent experiments. *p < 0.05.
R. australis Induces Activation of mTORC1 Signaling
As described above (Figure 3D), R. australis differentially regulated the genes in the PI3K-Akt-mTOR signaling pathway in macrophages via an Atg5-dependent mechanism. Since inhibition of mTORC1 by rapamycin treatment promotes R. australis infection (4), mTORC1 activation is likely important to regulate the cellular environment during rickettsial infection. This was the impetus to study whether R. australis infection has an impact on mTORC1 signaling and if mTORC1 signaling regulates autophagy induction. To address these questions, we infected B6 BMMs with R. australis and determined the phosphorylation of mTOR and P70S6 kinase as a read-out for mTORC1 activity. Surprisingly, R. australis infection induced direct phosphorylation of mTOR on serine 2448 and phosphorylation of P70S6 kinase (threonine 389) in B6 BMMs compared to uninfected controls at as early as 1 h p.i. when autophagic response is induced by these bacteria (Figure 6A) (4). Densitometry analysis from three independent experiments showed that the expression levels of phosphorylated mTOR and P70S6 in R. australis-infected macrophages were significantly greater than in uninfected macrophages (Figures 6B, C). These results suggest that infection with R. australis leads to both direct and indirect activation of mTORC1 in infected macrophages (4). To confirm that the increase in phosphorylated mTOR and P70S6 kinase represents the activation of mTORC1 signaling, B6 BMM macrophages were pre-treated with rapamycin prior to infection. Blockage of mTORC1 signaling by treatment with rapamycin for less than 6 h nearly abolished the increase in both phosphorylated mTOR and P70S6 (Figure 6). Thus, these results demonstrated that R. australis infection activated mTORC1 signaling in mouse macrophages, which can be effectively eliminated by treatment with rapamycin.
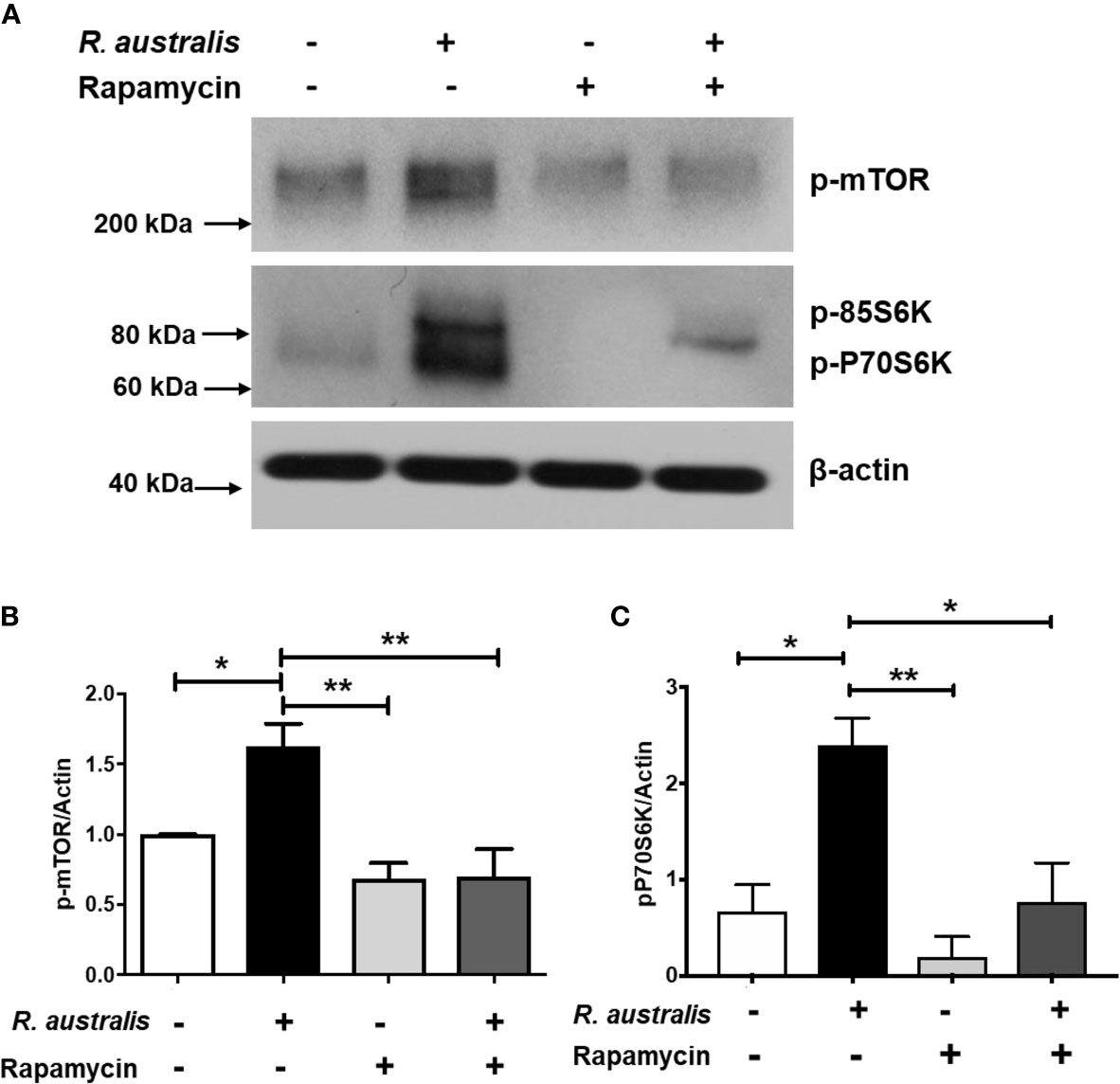
Figure 6 (previous Figure 1). R. australis activates mTORC1 in mouse macrophages. BMMs of WT B6 mice were isolated and infected with R. australis at an MOI of 5. To inhibit mTORC1 signaling, cells were treated with 50 ng/mL of rapamycin for 4 hours prior to infection with R. australis. At 1 h p.i., cells were collected, and cell lysates were immunoblotted with antibodies directed against phosphorylated mTOR, phosphorylated p70S6K, and β-actin (A). The activation of phosphorylated mTOR (B) and phosphorylated p70S6K (C) was analyzed by densitometry using β-actin as a normalization control with three independent replicates. *p < 0.05; **p < 0.01.
R. australis Induces Autophagic Response While Activating mTORC1 Signaling
Our previous studies have reported that R. australis induces ATG5 (+) LC3 (+) autophagosomes with less likely degradative autophagy in macrophages at 1 h p.i (4). As shown in Figure 6, R. australis activated mTORC1 at the same time when Atg5-dependent autophagic response was induced in the same type of cells. This is interesting to us because mTOR-dependent autophagy would have decreased phosphorylation of mTOR. To this end, we stimulated B6 BMMs with rapamycin in the context of R. australis infection and analyzed autophagy induction by examining the levels of LC3-II, SQSTM1 and the conversion of soluble LC3-I to lipid bound LC3-II. At 1 h p.i., consistent with our previous report, R. australis alone induced significantly increased levels of LC3-II compared to uninfected controls without a significantly reduced level of SQSTM1/p62, indicating a modified autophagic response (Figures 7A, B). In contrast, rapamycin treatment slightly increased levels of LC3-II/LC3-I and slightly reduced levels of SQSTM1/p62 in R. australis-infected macrophages at 1 h p.i. (Figures 7A, B). However, these changes were not statistically significant (Figure 7B). At 3 h p.i., we did not find any significant change in expression levels of LC3-II, LC3-II/LC3-I conversion, or SQSTM1/p62 in R. australis-infected macrophages treated with rapamycin compared to controls (Figures 7A, B). Since R. australis is known to modify autophagy into an Atg5-dependent autophagic response (4), we decided to determine the quantity of LC3 (+) autophagosomes by immunofluorescence confocal microscopy. Indeed, at 1 h p.i., LC3 puncta staining was significantly increased in R. australis-infected BMMs treated with rapamycin compared to macrophages infected with rickettsiae alone (Figures 7C, D). However, R. australis-infected macrophages with rapamycin treatment did not show significantly increased LC3-II fluorescence intensities compared to rapamycin-treated uninfected cells. In other words, we did not observe synergistic or additive effects by R. australis infection in rapamycin-treated macrophages at 1 h p.i. (Figures 7C, D). Thus, inhibition of mTORC1 most likely enhanced the accumulation of autophagosomes in R. australis-infected macrophages. These data suggest that R. australis induces a modified mTORC1-independent autophagic response in macrophages.
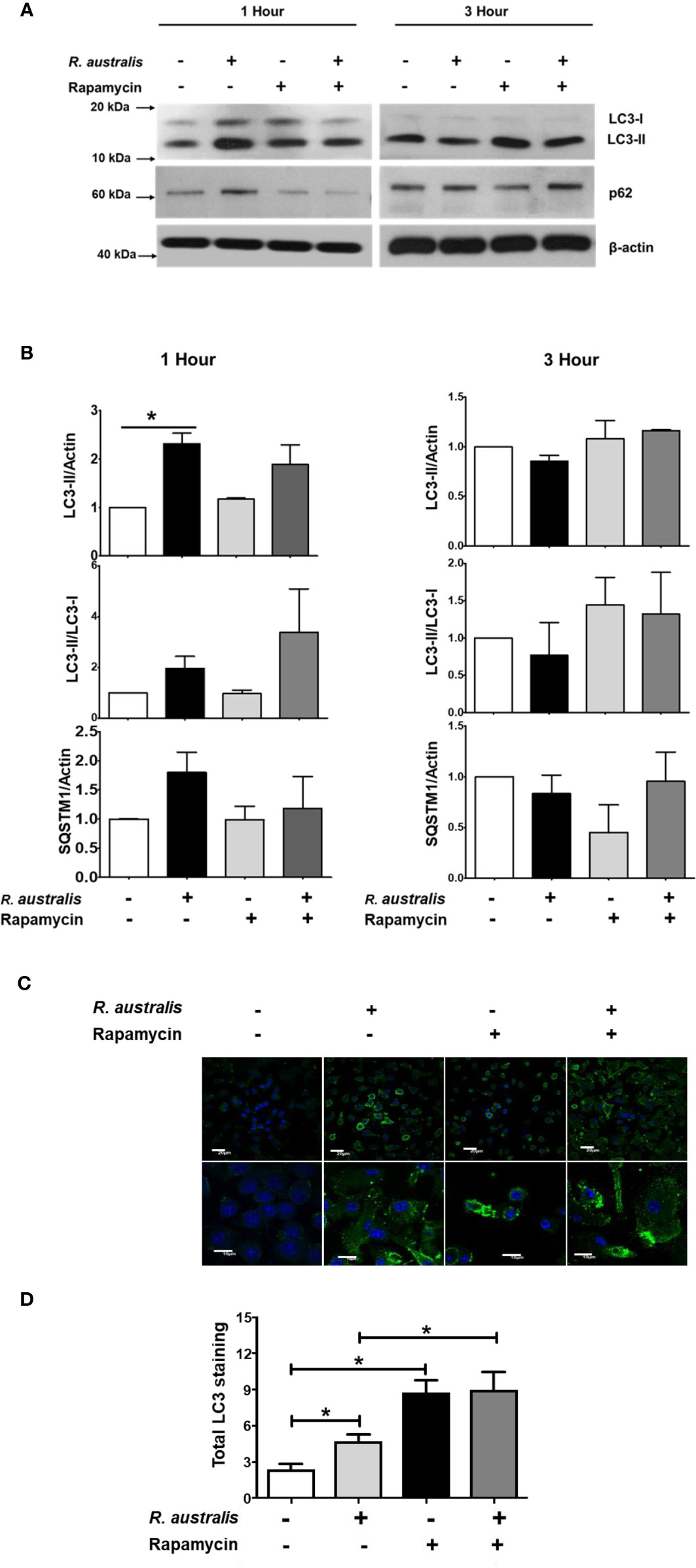
Figure 7 (previous Figure 2). Interactions of mTORC1 and autophagy with R. australis in macrophages. BMMs of WT B6 mice were isolated and infected with R. australis at an MOI of 5. To inhibit mTORC1 signaling, cells were treated with 50 ng/mL of rapamycin for 4 hours prior to infection with R. australis. Cells were collected at 1 h and 3 h p.i., and cell lysates were immunoblotted with antibodies directed against LC3-II, p62 and β-actin (A). (B), The ratios of LC3-II/Actin, LC3-II/LC3-I, and SQSTM1/Actin in uninfected and infected BMMs with or without rapamycin treatment at 1 h and 3 h p.i. were analyzed by densitometry. (C), Representative confocal immunofluorescence microscopic images of uninfected and infected BMMs with or without rapamycin treatment at 1 h p.i. at a magnification of 20x. Green, LC3 puncta; blue, nuclei (DAPI). Bar = 20 µm in the upper and 10 µm in the bottom row, respectively. (D), Total LC3 staining was quantified using Image J software. Microscopy data represent two to three independent experiments. Data shown are mean ± SE. Group comparison was not labeled if not statistically significant. *p < 0.05.
Discussion
Our recent report on interactions of autophagy with R. australis paved the way for the present study (4). Autophagy is a fundamental cellular homeostasis program that degrades surplus cellular contents in cytoplasm to provide sources of energy (46). Increasing evidence has demonstrated various mechanisms by which host autophagy interacts with invading microbial pathogens. However, the interplay of autophagy with the host inflammatory response to infectious agents in vivo and the subsequent pathological changes in tissues are not well understood. Studies revealing such roles of autophagy are likely to provide insightful information toward understanding the pathogenesis of these infectious diseases and the potential development of novel therapeutics targeting autophagy. In this regard, our studies demonstrated several novel findings. R. australis, a member in the transitional group of the Rickettsia genus, was revealed to activate mTORC1 signaling in primary mouse macrophages. Moreover, mTORC1 control of autophagy was dysregulated during R. australis infection at least at 1 h p.i. (Figure 8). Our studies also demonstrated that deficiency of Atg5 enhanced the host control of R. australis in vivo accompanied by numerous infiltrations of inflammatory cells in tissues, including neutrophils. R. australis infection significantly regulated mammalian host genes and subverted host innate inflammatory responses to support the infection by modifying Atg5-dependent autophagy.
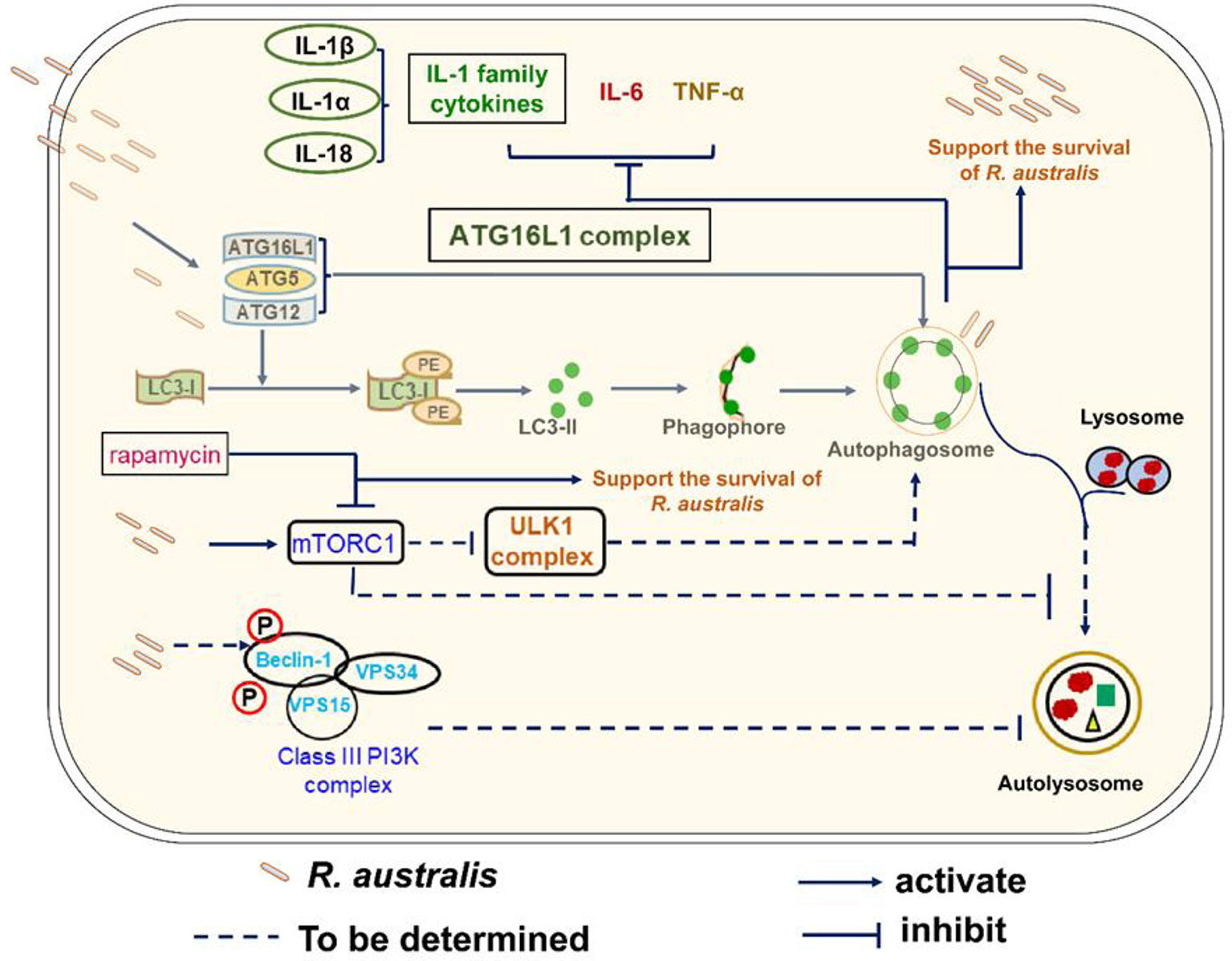
Figure 8 Schematic diagram of interactions of Atg5-dependent autophagy and mTORC1 with R. australis in macrophages. This schematic diagram depicts the molecular mechanisms involved in the modulation of autophagy and activation of mTORC1 by R. australis in order to benefit bacterial infection. R. australis activates the ATG5-ATG12-ATG16L1 complex leading to accumulation of LC3 (+) autophagosomes. Atg5 (+) LC3 (+) autophagosomes induced by R. australis in macrophages inhibit production levels of IL-1 family cytokines including IL-1β, IL-1α and IL-18 as well as other inflammatory cytokines such as IL-6 and TNF-α, the effects of which favor rickettsial infection. R. australis activates mTORC1 signaling at 1 h p.i. in macrophages when the Atg5-dependent autophagic response is induced. Thus, R. australis-induced autophagic response is resistant to regulation by mTORC1 signaling. It would be interesting to explore whether R. australis-induced autophagosomes can fuse with lysosome and mature into autolysosomes, and how ULK and class III PI3K complex are involved in this process in the future. Solid lines represent pathways demonstrated in our current and previous studies, while dashed lines refer to the mechanisms hypothesized but not tested.
The mTOR signaling pathway is widely expressed in tissues and cells of mammalian hosts, and functions as an evolutionarily conserved sensor of environmental and endogenous stress (47). The mTOR is a down-stream effector of PI3K-Akt signaling pathway and mediates nutrient-dependent intracellular signaling related to cell growth, proliferation, and differentiation (16, 48). Hepatitis C virus (HCV) activates mTOR via the viral nonstructural protein 5A (NS5A) to enhance cell survival by blocking apoptosis (49). Human cytomegalovirus (HCMV) induces mTOR activation and maintains this activation throughout infection (50). Zullo and Lee reported that several Mycobacterium species activate mTOR signaling and induce autophagy; however, they show that the induction of autophagy in macrophages is mTOR-independent (51). Rapamycin has been reported to mainly inhibit mTORC1, particularly within 24 hour of stimulation (52, 53). In the present study, treatment with rapamycin for 5 hours nearly abolished the increase in phosphorylated mTOR and P70S6 in R. australis-infected macrophages (Figures 6 and 7), suggesting activation of mTORC1 signaling by rickettsiae. Although the results in Figures 6 and 7 provided both direct and indirect evidence that R. australis activates mTORC1 signaling, this conclusion can even be further supported by the unaltered total expression levels of mTOR and P70S6 in these infected macrophages. We are currently performing such experiments in our laboratory. Furthermore, it remains unknown whether R. australis activates mTORC2 signaling or not. It is not surprising to us that R. australis dysregulated the control of autophagy by mTORC1 in macrophages at 1 h p.i. based on two findings previously reported (4, 54). First, R. australis does not induce a canonical autophagy pathway characterized by increased LC3-II and reduced p62 (4). Instead, R. australis manipulates an Atg5-dependent autophagic response in order to facilitate their infection (Figure 8). Secondly, given the recent discovery that R. typhi activates both Class I and III PI3Ks (54), it is interesting to speculate that R. australis activates class III PI3K complex to further trigger the activation of mTORC1 (Figure 8). Increasing evidence indicates that mTORC1 also directly regulates the subsequent steps of the autophagy process, including the nucleation, autophagosome elongation, autophagosome maturation and termination (16). It is, therefore, worthwhile to investigate whether and how mTORC1 is involved in regulating the accumulation of autophagosomes and maturation of autophagosome into autolysosome during R. australis infection. Furthermore, previous studies have demonstrated that inhibition of apoptosis is essential for endothelial cell survival during R. rickettsii infection. R. rickettsii even protects host endothelial cells from staurosporine-induced cell death (55, 56). We also reported that R. australis does not induce cell death in infected mouse BMMs (5). Activation of mTOR is known to promote cell survival (57). Thus, it is an attractive hypothesis that the activation of mTOR, including mTORC1 and/or mTORC2 signaling, in cells infected with rickettsiae may serve as a mechanism for promoting cell survival.
Autophagy has been shown to negatively regulate host inflammatory cytokines, particularly the IL-1 family of cytokines. R. australis inhibits IL-1β secretion in infected BMMs in vitro and in sera by modulating Atg5-dependent autophagy (4). In vitro neutralization of IL-1β by specific antibodies abolishes the enhanced R. australis infection by an Atg5-dependent autophagic response in macrophages (4). R. australis has been reported to induce significant levels of IL-18, another member in IL-1 family cytokines, by B6 BMMs (5). The differences in production levels of IL-18 by BMMs of Atg5- and Atg16l1-conditional knockout mice (Figure 5) from those by B6 BMMs could result from the variabilities in mouse genetic background. Interestingly, IL-1, IL-2, IL-6, TNF-α, IFN-γ, and TGF-β are known to induce autophagy while IL-4, IL-10 and IL-13 inhibit autophagy (58). Our present study demonstrated autophagy-dependent regulation of IL-1α, IL-6, IL-18, and TNF-α in murine macrophages infected with R. australis in vitro (Figures 4 and 5). These inflammatory cytokines upregulated by Atg5-dependent autophagy may contribute to reduced rickettsial loads in tissues of R. australis-infected Atg5-deficient macrophages (4). However, R. australis primarily targets microvascular endothelial cells in vivo while macrophages are the initial target cells at the cutaneous entry site of rickettsiae (3, 59, 60). It is not surprising to see a difference in cytokine profiles in R. australis-infected BMMs and sera of R. australis-infected Atg5-conditional knockout mice. For example, macrophages are not a major producer of IFN- γ, and our in vitro data confirm this conclusion (Figure 4). In contrast, in Rickettsia-infected mice, NK and T cells are both the major cells secreting IFN-γ (28, 31, 38, 39). Although an effective Th1-type cytokine response is critical for controlling rickettsial infection as demonstrated by us and other groups (28, 31, 38, 39, 61–63), the serum of R. australis-infected Atg5 flox/flox Lyz-Cre mice had significantly lower levels of IFN-γ, with lower bacterial loads in tissues, compared to Atg5 flox/flox mice (Figure 6) (4). The lower level of IFN-γ in the serum of Atg5 flox/flox Lyz-Cre mice compared to Atg5 flox/flox animals may result from reduced rickettsial loads in the tissues. Another explanation is that the enhanced systemic production of IFN-γ in Atg5 flox/flox mice may result from robust activation of NK and T cells by autophagy-competent macrophages in host innate immunity. Overall, R. australis ultimately subverted the host innate inflammatory response, which favored its own infection in vivo.
The histopathological data of R. australis-infected Atg5-conditional knockout mice showed that Atg5 flox/flox Lyz-Cre mice had significantly fewer pathological foci with larger size in liver compared to Atg5 flox/flox mice (Figure 2). We hypothesize that the pathological changes in infected Atg5 flox/flox Lyz-Cre mice represent effective host control of pathogenic Rickettsia in infected tissues resulting from recruiting a number of potent innate immune cells. In contrast, the pathological changes in R. australis-infected Atg5 flox/flox mice represent paralysis or subversion of innate host immunity, as manifested by the greater frequency of foci but a limited area of cellular infiltration. Our in vivo pathology results strongly suggest that Atg5-deficiency promotes an inflammatory response consisting of a considerable frequency of neutrophils. IL-1β is a major stimulator of leukocyte recruitment through its ability to up-regulate adhesion to endothelial cells (64, 65). R. australis infection induces significantly greater levels of IL-1β in the serum of infected Atg5 flox/flox Lyz-Cre mice vs Atg5 flox/flox mice (4). The accumulation of neutrophils in the inflammatory foci in liver of infected Atg5 flox/flox Lyz-Cre mice was possibly associated with the greater production levels of IL-1β compared to infected Atg5 flox/flox mice.
This study revealed that R. australis induced Atg5-dependent autophagic response while activating mTORC1 signaling in macrophages. Additionally, our data demonstrated that R. australis modulated Atg5-dependent autophagy to inhibit inflammatory cytokines at both transcriptional and post-transcriptional levels in macrophages, including IL-6, IL-1α, TNF-α, and IL-18. In conclusion, the R. australis-modified autophagic response in macrophages supports the in vivo infection through subversion of host innate immunity against rickettsiae.
Data Availability Statement
The data has been uploaded to the GEO, with accession number GSE171160.
Ethics Statement
The animal study was reviewed and approved by the Institutional Animal Care and Use Committee at UTMB.
Author Contributions
JB and CR performed the experiments. JB and RF wrote the manuscript. JB, CR, and RF designed the experiments and collected data. DW performed the histopathological analysis as a board-certified pathologist, analyzed the data, and revised the manuscript. SW performed the RNA-Seq and data analysis. KK analyzed the RNA-Seq results by IPA analysis and generated Figure 3. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by grant AI133359 to RF from the National Institute of Allergy and Infectious Diseases. JB was supported by the National Institutes of Health T32 Training Grant (AI060549) at the University of Texas Medical Branch.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Seungmin Hwang at the University of Chicago for kindly providing bone marrows of Atg16l1 flox/flox Lyz-Cre and Atg16l1 flox/flox mice.
References
1. Straily A, Drexler N, Cruz-Loustaunau D, Paddock CD, Alvarez-Hernandez G. Notes from the Field: Community-based prevention of Rocky Mountain spotted fever - Sonora, Mexico, 2016. MMWR Morb Mortal Wkly Rep (2016) 65(46):1302–3. doi: 10.15585/mmwr.mm6546a6
2. Sahni SK, Rydkina E. Progress in the functional analysis of rickettsial genes through directed mutagenesis of Rickettsia prowazekii phospholipase D. Future Microbiol (2009) 4(10):1249–53. doi: 10.2217/fmb.09.99
3. Rumfield C, Hyseni I, McBride JW, Walker DH, Fang R. Activation of ASC inflammasome driven by Toll-like receptor 4 contributes to host immunity against rickettsial Infection. Infect Immun (2020) 88(4):e00886–19. doi: 10.1128/IAI.00886-19
4. Bechelli J, Vergara L, Smalley C, Buzhdygan TP, Bender S, Zhang W, et al. Atg5 supports Rickettsia australis infection in macrophages in vitro and in vivo. Infect Immun (2018) 87(1):e00651–18. doi: 10.1128/IAI.00651-18
5. Smalley C, Bechelli J, Rockx-Brouwer D, Saito T, Azar SR, Ismail N, et al. Rickettsia australis activates inflammasome in human and murine macrophages. PloS One (2016) 11(6):e0157231. doi: 10.1371/journal.pone.0157231
6. Curto P, Simões I, Riley SP, Martinez JJ. Differences in intracellular fate of two spotted fever group Rickettsia in macrophage-like cells. Front Cell Infect Microbiol (2016) 6:80. doi: 10.3389/fcimb.2016.00080
7. Curto P, Riley SP, Simões I, Martinez JJ. Macrophages infected by a pathogen and a non-pathogen spotted fever group Rickettsia reveal differential reprogramming signatures early in infection. Front Cell Infect Microbiol (2019) 9:97. doi: 10.3389/fcimb.2019.00097
8. Radulovic S, Price PW, Beier MS, Gaywee J, Macaluso JA, Azad A. Rickettsia-macrophage interactions: host cell responses to Rickettsia akari and Rickettsia typhi. Infect Immun (2002) 70(5):2576–82. doi: 10.1128/iai.70.5.2576-2582.2002
9. Banajee KH, Embers ME, Langohr IM, Doyle LA, Hasenkampf NR, Macaluso KR. Correction: Amblyomma maculatum feeding augments Rickettsia parkeri infection in a rhesus Macaque model: a pilot study. PloS One (2015) 10(8):e0137598. doi: 10.1371/journal.pone.0137598. Erratum for: PLoS One (2015)10(8):e0135175.
10. Mizushima N, Noda T, Yoshimori T, Tanaka Y, Ishii T, George MD, et al. A protein conjugation system essential for autophagy. Nature (1998) 395(6700):395–8. doi: 10.1038/26506
11. Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell (2004) 6(4):463–77. doi: 10.1016/s1534-5807(04)00099-1
12. Li W, Zhang L. Regulation of ATG and autophagy initiation. Adv Exp Med Biol (2019) 1206:41–65. doi: 10.1007/978-981-15-0602-4_2
13. Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, et al. The role of autophagy during the early neonatal starvation period. Nature (2004) 432:1032–6. doi: 10.1038/nature03029
14. Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature (2006) 441:885–89. doi: 10.1038/nature04724
15. Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy (2007) 3:581–5. doi: 10.4161/auto.4782
16. Dossou AS, Basu A. The Emerging Roles of mTORC1 in Macromanaging Autophagy. Cancers (Basel) (2019) 11(10):1422. doi: 10.3390/cancers11101422
17. Weichhart T, Säemann MD. The multiple facets of mTOR in immunity. Trends Immunol (2009) 30(5):218–26. doi: 10.1016/j.it.2009.02.002
18. Jhanwar-Uniyal M, Amin AG, Cooper JB, Das K, Schmidt MH, Murali R. Discrete signaling mechanisms of mTORC1 and mTORC2: Connected yet apart in cellular and molecular aspects. Adv Biol Regul (2017) 64:39–48. doi: 10.1016/j.jbior.2016.12.001
19. Hong S, Mannan AM, Inoki K. Evaluation of the nutrient-sensing mTOR pathway. Methods Mol Biol (2012) 821:29–44. doi: 10.1007/978-1-61779-430-8_3
20. Gelsomino F, Bertolini F, Luppi G, Spallanzani A, Pettorelli E, Reggiani Bonetti L, et al. Cascinu S. A Dose-finding and Biomarker Evaluation Phase Ib Study of Everolimus in Association With 5-Fluorouracil and Pelvic Radiotherapy as Neoadjuvant Treatment of Locally Advanced Rectal Cancer (E-LARC Study). Clin Colorectal Cancer (2017) 16(4):410–15.e1. doi: 10.1016/j.clcc.2017.03.011
21. Zhang F, Hu X, Gu Y, Bian H, Xu Z, Wang Q, et al. URI knockdown induces autophagic flux in gastric cancer cells. Am J Cancer Res (2018) 8(10):2140–9. Available at: www.ajcr.us/ISSN:2156-6976/ajcr0078088.
22. Li SH, Lin WC, Huang TL, Chen CH, Chiu TJ, Fang FM, et al. Significance of mammalian target of rapamycin in patients with locally advanced stage IV head and neck squamous cell carcinoma receiving induction chemotherapy with docetaxel, cisplatin, and fluorouracil. Head Neck (2016) 38 Suppl 1:E844–52. doi: 10.1002/hed.24111
23. Katholnig K, Linke M, Pham H, Hengstschläger M, Weichhart T. Immune responses of macrophages and dendritic cells regulated by mTOR signalling. Biochem Soc Trans (2013) 41(4):927–33. doi: 10.1042/BST20130032
24. Deretic V, Levine B. Autophagy, immunity, and microbial adaptations. Cell Host Microbe (2009) 5(6):527–49. doi: 10.1016/j.chom.2009.05.016
25. Choi Y, Bowman JW, Jung JU. Autophagy during viral infection - a double-edged sword. Nat Rev Microbiol (2018) 16(6):341–54. doi: 10.1038/s41579-018-0003-6
26. Paulus GL, Xavier RJ. Autophagy and checkpoints for intracellular pathogen defense. Curr Opin Gastroenterol (2015) 31(1):14–23. doi: 10.1097/MOG.0000000000000134
27. Hanson BA, Wisseman CL Jr, Waddell A, Silverman DJ. Some characteristics of heavy and light bands of Rickettsia prowazekii on Renografin gradients. Infect Immun (1981) 34(2):596–604. doi: 10.1128/IAI.34.2.596-604.1981
28. Fang R, Ismail N, Shelite T, Walker DH. CD4+ CD25+ Foxp3- T-regulatory cells produce both gamma interferon and interleukin-10 during acute severe murine spotted fever rickettsiosis. Infect Immun (2009) 77(9):3838–49. doi: 10.1128/IAI.00349-09
29. Ammerman NC, Beier-Sexton M, Azad AF. Laboratory maintenance of Rickettsia rickettsii. Curr Protoc Microbiol (2008) 11(1):3A.5.1–5.21. doi: 10.1002/9780471729259.mc03a05s11
30. Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe (2008) 4(5):458–69. doi: 10.1016/j.chom.2008.10.003
31. Fang R, Ismail N, Walker DH. Contribution of NK cells to the innate phase of host protection against an intracellular bacterium targeting systemic endothelium. Am J Pathol (2012) 181(1):185–95. doi: 10.1016/j.ajpath.2012.03.020
32. Zhang X, Goncalves R, Mosser DM. The isolation and characterization of murine macrophages. Curr Protoc Immunol (2008) 83(1):14.1.1–1.14. doi: 10.1002/0471142735.im1401s83
33. Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods (2012) 9(7):671–5. doi: 10.1038/nmeth.2089
34. McCloy RA, Rogers S, Caldon CE, Lorca T, Castro A, Burgess A. Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle (2014) 13(9):1400–12. doi: 10.4161/cc.28401
35. Yu K, Toral-Barza L. Biochemical and pharmacological inhibition of mTOR by rapamycin and an ATP-competitive mTOR inhibitor. Methods Mol Biol (2012) 821:15–28. doi: 10.1007/978-1-61779-430-8_2
36. Altman SA, Randers L, Rao G. Comparison of trypan blue dye exclusion and fluorometric assays for mammalian cell viability determinations. Biotechnol Prog (1993) 9(6):671–4. doi: 10.1021/bp00024a017
37. Thangamani S, Hermance ME, Santos RI, Slovak M, Heinze D, Widen SG, et al. Transcriptional immunoprofiling at the tick-virus-host interface during early stages of tick-borne encephalitis virus transmission. Front Cell Infect Microbiol (2017) 7:494. doi: 10.3389/fcimb.2017.00494
38. Bechelli J, Smalley C, Zhao X, Judy B, Valdes P, Walker DH, et al. MyD88 mediates instructive signaling in dendritic cells and protective inflammatory response during rickettsial infection. Infect Immun (2016) 84(4):883–93. doi: 10.1128/IAI.01361-15
39. Fang R, Ismail N, Soong L, Popov VL, Whitworth T, Bouyer DH, et al. Differential interaction of dendritic cells with Rickettsia conorii: impact on host susceptibility to murine spotted fever rickettsiosis. Infect Immun (2007) 75(6):3112–23. doi: 10.1128/IAI.00007-07
40. Chan YR, Liu JS, Pociask DA, Zheng M, Mietzner TA, Berger T, et al. Lipocalin 2 is required for pulmonary host defense against Klebsiella infection. J Immunol (2009) 182(8):4947–56. doi: 10.4049/jimmunol.0803282
41. Jossé L, Xie J, Proud CG, Smales CM. mTORC1 signalling and eIF4E/4E-BP1 translation initiation factor stoichiometry influence recombinant protein productivity from GS-CHOK1 cells. Biochem J (2016) 473(24):4651–64. doi: 10.1042/BCJ20160845
42. Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol (2008) 6(4):266–75. doi: 10.1038/nrmicro1855
43. Dinarello CA. The IL-1 family of cytokines and receptors in rheumatic diseases. Nat Rev Rheumatol (2019) 15(10):612–32. doi: 10.1038/s41584-019-0277-8
44. Ghose P, Ali AQ, Fang R, Forbes D, Ballard B, Ismail N. The interaction between IL-18 and IL-18 receptor limits the magnitude of protective immunity and enhances pathogenic responses following infection with intracellular bacteria. J Immunol (2011) 187(3):1333–46. doi: 10.4049/jimmunol.1100092
45. Arend WP, Palmer G, Gabay C. IL-1, IL-18, and IL-33 families of cytokines. Immunol Rev (2008) 223:20–38. doi: 10.1111/j.1600-065X.2008.00624.x
46. Chun Y, Kim J. Autophagy: An essential degradation program for cellular homeostasis and life. Cells (2018) 7(12):278. doi: 10.3390/cells7120278
47. Cipponi A, Goode DL, Bedo J, McCabe MJ, Pajic M, Croucher DR, et al. MTOR signaling orchestrates stress-induced mutagenesis, facilitating adaptive evolution in cancer. Science (2020) 368(6495):1127–31. doi: 10.1126/science.aau8768
48. Cao GD, Xu XY, Zhang JW, Chen B, Xiong MM. Phosphorylated Mammalian Target of Rapamycin p-mTOR Is a Favorable Prognostic Factor than mTOR in Gastric Cancer. PloS One (2016) 11(12):e0168085. doi: 10.1371/journal.pone.0168085
49. Peng L, Liang D, Tong W, Li J, Yuan Z. Hepatitis C virus NS5A activates the mammalian target of rapamycin (mTOR) pathway, contributing to cell survival by disrupting the interaction between FK506-binding protein 38 (FKBP38) and mTOR. J Biol Chem (2010) 285(27):20870–81. doi: 10.1074/jbc.M110.112045
50. Kudchodkar SB, Yu Y, Maguire TG, Alwine JC. Human cytomegalovirus infection induces rapamycin-insensitive phosphorylation of downstream effectors of mTOR kinase. J Virol (2004) 78(20):11030–9. doi: 10.1128/JVI.78.20.11030-11039.2004
51. Zullo AJ, Lee S. Mycobacterial induction of autophagy varies by species and occurs independently of mammalian target of rapamycin inhibition. J Biol Chem (2012) 287(16):12668–78. doi: 10.1074/jbc.M111.320135
52. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell (2006) 22(2):159–68. doi: 10.1016/j.molcel.2006.03.029
53. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, et al. Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science (2012) 335(6076):1638–43. doi: 10.1126/science.1215135
54. Voss OH, Gillespie JJ, Lehman SS, Rennoll SA, Beier-Sexton M, Rahman MS, et al. Risk1, a Phosphatidylinositol 3-Kinase Effector, Promotes Rickettsia typhi Intracellular Survival. mBio (2020) 11(3):e00820–20. doi: 10.1128/mBio.00820-20
55. Clifton DR, Goss RA, Sahni SK, van Antwerp D, Baggs RB, Marder VJ, et al. NF-kappa B-dependent inhibition of apoptosis is essential for host cellsurvival during Rickettsia rickettsii infection. Proc Natl Acad Sci USA (1998) 95(8):4646–51. doi: 10.1073/pnas.95.8.4646
56. Bechelli JR, Rydkina E, Colonne PM, Sahni SK. Rickettsia rickettsii infection protects human microvascular endothelial cells against staurosporine-induced apoptosis by a cIAP(2)-independent mechanism. J Infect Dis (2009) 199(9):1389–98. doi: 10.1086/597805
57. Covarrubias AJ, Aksoylar HI, Horng T. Control of macrophage metabolism and activation by mTOR and Akt signaling. Semin Immunol (2015) 27(4):286–96. doi: 10.1016/j.smim.2015.08.001
59. Cragun WC, Bartlett BL, Ellis MW, Hoover AZ, Tyring SK, Mendoza N, et al. The expanding spectrum of eschar-associated rickettsioses in the United States. Arch Dermatol (2010) 146:641–8. doi: 10.1001/archdermatol.2010.48
60. Walker DH, Hudnall SD, Szaniawski WK, Feng HM. Monoclonal antibody-based immunohistochemical diagnosis of rickettsialpox: the macrophage is the principal target. Mod Pathol (1999) 12:529–33.
61. Mansueto P, Vitale G, Cascio A, Seidita A, Pepe I, Carroccio A, et al. New insight into immunity and immunopathology of Rickettsial diseases. Clin Dev Immunol (2012) 2012:967852. doi: 10.1155/2012/967852
62. Li H, Jerrells TR, Spitalny GL, Walker DH. Gamma interferon as a crucial host defense against Rickettsia conorii in vivo. Infect Immun (1987) 55(5):1252–5. doi: 10.1128/IAI.55.5.1252-1255.1987
63. Feng HM, Popov VL, Walker DH. Depletion of gamma interferon and tumor necrosis factor alpha in mice with Rickettsia conorii-infected endothelium: impairment of rickettsicidal nitric oxide production resulting in fatal, overwhelming rickettsial disease. Infect Immun (1994) 62(5):1952–60. doi: 10.1128/IAI.62.5.1952-1960.1994
64. Prince LR, Allen L, Jones EC, Hellewell PG, Dower SK, Whyte MK, et al. The role of interleukin-1beta in direct and toll-like receptor 4-mediated neutrophil activation and survival. Am J Pathol (2004) 165(5):1819–26. doi: 10.1016/s0002-9440(10)63437-2
Keywords: Rickettsia, mTOR signaling, autophagy, macrophages, innate immunity
Citation: Bechelli J, Rumfield CS, Walker DH, Widen S, Khanipov K and Fang R (2021) Subversion of Host Innate Immunity by Rickettsia australis via a Modified Autophagic Response in Macrophages. Front. Immunol. 12:638469. doi: 10.3389/fimmu.2021.638469
Received: 06 December 2020; Accepted: 11 March 2021;
Published: 12 April 2021.
Edited by:
Isabelle Vergne, UMR5089 Institut de Pharmacologie et de Biologie Structurale (IPBS), FranceReviewed by:
Dan-Li Xie, Wenzhou Medical University, ChinaChinnaswamy Jagannath, Weill Cornell Medical College of Cornell University, United States
Oliver Voss, University of Maryland, Baltimore, United States
Copyright © 2021 Bechelli, Rumfield, Walker, Widen, Khanipov and Fang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rong Fang, cm9mYW5nQHV0bWIuZWR1