 Nick P. Goplen
Nick P. Goplen In Su Cheon
In Su Cheon Jie Sun
Jie Sun
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 29 March 2021
Sec. Immunological Memory
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.636118
This article is part of the Research TopicResident Memory T Cells: Guardians of the Balance of Local Immunity and PathologyView all 11 articles
Following respiratory viral infections or local immunizations, lung resident-memory T cells (TRM) of the CD8 lineage provide protection against the same pathogen or related pathogens with cross-reactive T cell epitopes. Yet, it is now clear that, if homeostatic controls are lost following viral pneumonia, CD8 TRM cells can mediate pulmonary pathology. We recently showed that the aging process can result in loss of homeostatic controls on CD8 TRM cells in the respiratory tract. This may be germane to treatment modalities in both influenza and coronavirus disease 2019 (COVID-19) patients, particularly, the portion that present with symptoms linked to long-lasting lung dysfunction. Here, we review the developmental cues and functionalities of CD8 TRM cells in viral pneumonia models with a particular focus on their capacity to mediate heterogeneous responses of immunity and pathology depending on immune status.
“Infectious diseases are no respecters of wealth, power, or personal merit. Pandemic infectious disease is one situation where we cannot accept Margaret Thatcher's view [there is no such thing as society]. With a fast spreading respiratory virus, for example, everyone is ultimately in the same boat” (Peter C. Doherty concluding remarks in Pandemics, 2013). Respiratory viruses that infect the lower airways such as influenza virus and severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) can cause severe acute lung injury (ALI) and are serious public health challenges. A year after the initial outbreak, SARS-CoV2 infection has resulted in more than 95 million cases and 2 million deaths globally (https://coronavirus.jhu.edu). Conventional T cells, particularly CD8 cytotoxic T cells, play important roles in the control of respiratory viral infection (1, 2). Additionally, CD8 T cells can form a long-lived immunological memory that protects from reinfection of the same or related viruses (3). Among the different subsets of memory CD8 T cells, tissue-resident memory T cells (TRM) that reside within the respiratory tract provide superior immunity against viral re-infections (4). Therefore, vaccines that can elicit robust CD8 TRM cells are highly promising for the prevention/amelioration of future pandemics. Conversely, recent studies have suggested that exaggerated CD8 TRM cell presence and/or uncontrolled CD8 TRM cell function could lead to chronic pathogenic sequelae in the lungs (5, 6). Here, we will review recent literature on pulmonary CD8 TRM cell development and maintenance and discuss their roles in immune protection as opposed to how they may provoke pulmonary pathologies when not tightly regulated. We primarily use influenza virus infection studies as the model for this review.
Pulmonary CD8 TRM cells poised for rapid responsiveness, contribute substantially to immune protection of the host against previously encountered viral pathogens (4). As in other organs, pulmonary TRM cell function appears to be dependent on in situ proliferation and the production of IFN-γ which activates the vasculature enabling recruitment of innate and adaptive responses (4, 7–10). Compared to T effector (TEM), T central (TCM), and T peripheral (TPM) memory cells that collectively circulate through blood, lymph, peripheral and secondary lymphoid organs, TRM cells are transcriptionally and functionally distinct (11–16). The lung is one of few sites where CD8 TRM cells are relatively short-lived and not permanently lodged in tissues compared to the limited number of organs investigated (17–19). Their loss over time has been attributed to migration from the parenchyma to the airways where they encounter a hostile environment eventually leading to their apoptosis (19). Additionally, pulmonary TRM cells can re-enter the circulation and migrate to the draining lymph nodes where they re-establish residency, contributing to their loss from lung tissue (18). Of note, lung TRM cell loss can be mitigated by local prime-boost strategies and/or repeated antigen exposure (20). Given the potential for their short life-span and their importance in clearing subsequent respiratory viral infections, it is critical to understand the environmental and immune-status cues that regulate TRM cell differentiation, maintenance, and function in the lung in order to exploit their benefits through immunotherapies such as vaccines.
Counterparts to TRM cells discovered in mice exist in all organs investigated in humans (11, 21). The lung faces constant microbial exposure, yet histology snapshots suggest the distal airways are remarkably sterile environments in the absence of acute infection. Accordingly, in situ estimates suggest human lung explants contain as many as 10 billion memory T cells (22). There is a diverse antigen-specific CD4 and CD8 T cell presence in most lungs including up to 10% of T cells that respond to influenza virus challenge with proliferation (22). Like CD8 T cells, CD4 T cells in the human lung appear transcriptionally primed for response (23, 24). While the resident CD4:CD8 memory T cell ratios vary by compartment (airway vs. parenchyma), 20–50% of pulmonary CD8 T cells expected to be critical for anti-viral memory responses, display a recently activated phenotype indicated by HLA-DR antigen on their surface (22, 25, 26), suggesting active vigilance.
Tracking of donor lung T cells following pulmonary transplantation, indicates TRM cells are found sparsely in the blood at any given time, similar to what is observed in mouse studies (6, 26, 27). Further, donor and recipient airway TRM cell transcriptional profiles overlap indicating a shared signature imparted by the lung microenvironment despite disparate HLA matches (26). As in mouse studies, a substantial fraction of human lung CD8 TRM cells express multiple inhibitory receptors, suggesting a strong stimulus may be needed for their re-activation (24). Relative to peripheral blood memory T cells, human CD69+ pulmonary CD8 TRM cells almost universally express CD29, CD49a, CXCR6, and PSGL-1 with heterogenous expression of CD103 and CD101. Despite this heterogeneity, strong stimulation through the T Cell Receptor (TCR) results in proliferation of the majority of human TRM cells with their progeny exhibiting enhanced polyfunctional capacity relative to their parents (28). This suggests TRM cells act as sentinels in human lung mucosa and are important for maintaining sterility of alveolar spaces.
Recent barcode lineage-tracing and single-cell transcriptome analyses found that a subset of T cell clones possesses a heightened capacity to form TRM cells, as enriched expression of TRM-fate-associated genes is already apparent in circulating effector T cell clones (13). Consistently, following initial trafficking to the lung, TRM-like phenotypes are observed as early as 2 weeks following influenza infection and these phenotypes, but not numbers, are stable in the airways, lung parenchyma, and trachea for up to 3 months (17, 29). Pulmonary TRM cells have been defined inconsistently throughout the literature, as warranting caution when comparing studies.
While pulmonary CD8 TRM cell definition(s), differentiation, maintenance, and functions have largely been established from monoclonal T cell receptor (TCR) transgenic models, polyclonal experiments give a more heterogeneous and physiological relevant picture of TRM cells coexisting within the same tissue, but have not been widely reviewed. Markers (e.g., CD69, CD103, CD49a, CXCR6, and PD-1) typically used to identify pulmonary CD8 TRM cells in mice are heterogeneously co-expressed within TRM populations (5, 6, 27, 29–32). For example, E-cadherin in the lung is expressed in the cell-cell junctions between bronchiole epithelium (33). Although E-cadherin-binding CD103 is intrinsically important for cytotoxic capacity (34) and is expressed on nearly 100% of TRM in the skin, CD103 is heterogeneously expressed in lung TRM cells, inhibits TRM cell motility, and is not required for heterosubtypic protection against influenza. Conversely, although the collagen IV-binding integrin CD49a is a less common marker used for the identification than CD103, it is required for the heterosubtypic immunity against influenza infection (28, 29).
Furthermore, CD103 is expressed at a substantially lower frequency on the TRM cells that form the bulk of the protective response vs. influenza nucleoprotein (Db-NP366−374) in C57BL/6 mice compared to another immune-dominant epitope from viral polymerase peptide (Db-PA224−233) (5). Nonetheless, parabiosis studies indicate both of these phenotypically different populations exhibit similar degrees of tissue residency 2 months following infection (6). Though the significance is unclear, this immunodominant population (responding to Db-NP366−374) in a secondary response that mostly lacks CD103 expression, abundantly expresses classic exhaustion markers (PD-1, TIM-3, LAG-3, and TIGIT) relative to Db-PA224−233 and Kb-OVASIINFEKL –specific TRM and memory CD8 T cells in the circulation (5, 6). These insights from various studies highlight the marked epitope-specific CD8 TRM cell heterogeneity within the pool of polyclonal TRM cells directed against the same pathogen. Indeed, data from organ donors indicates a diverse TCR repertoire against influenza virus, suggesting that heterogeneity is quintessential in the local pulmonary response (28).
It is becoming clearer that local immune interactions influence CD8 TRM cell numbers without affecting the circulating memory pool. Alveolar macrophages (AMs) are a self-renewing population of airway-resident cells seeded early in embryonic development (35). AMs maintain lung homeostasis and respond to inflammatory cues. Absence or dysfunction of AMs in severe influenza infection leads to exacerbated pulmonary pathology and enhanced mortality (36, 37). In studies where we were investigating the effects of PPAR-γ in the macrophage compartment on influenza severity, intrinsic absence increased the density of pulmonary TRM cells and long-term stromal disrepair indicated by persistent inflammation and collagen deposition (38, 39). We subsequently found that depletion of AMs prior to influenza infection, but not during the CD8 T cell contraction phase, enhanced TRM cell density without affecting the circulatory memory compartment (Figure 1) (38). This suggests AMs have an early influence on the lung microenvironment that governs in situ TRM cell differentiation. It is not currently clear what subtype of CD169+ AMs are responsible for limiting the TRM cell compartment nor by what means. Conversely, bone-marrow derived monocytes trafficking to the site of infection enhance the early antigen-presentation required for TRM cell differentiation in the lung (40). Yet, inflammatory macrophages in the gut mediate heterogeneous TRM cell differentiation by contributing to the pro-inflammatory milieu (41).
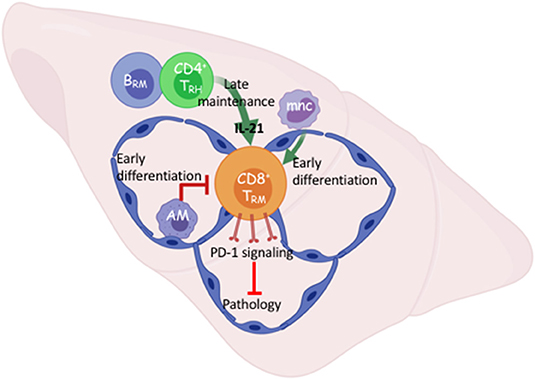
Figure 1. Early and late cellular networks involved in Trm cell differentiation and maintenance. After viral respiratory pneumonia, early pulmonary CD8 TRM cell differentiation is driven by re-encounter with antigen presented via interstitial classic monocytes (mnc) and opposed by alveolar macrophages that maintain lung homeostasis. B cell-dependent tissue-resident CD4 helper T cells (TRH) support TRM cell maintenance through IL-21 dependent survival. TRM intrinsic PD-1 signaling prevents pathology in the absence of infectious virus. Created with BioRender.
In contrast to the limiting of the TRM cell compartment by innate resident macrophages, we and others have recently shown that a population of CD4 tissue-resident helper T (TRH) cells aid the persistence of pulmonary CD8 TRM cells following influenza infection (42, 43). This novel population of TRH cells simultaneously exhibits T follicular helper (TFH)-like properties that enhance the local B cell response and tissue-resident memory T cell features. CD4 TRH cells are the major cellular sources of IL-21 in the tissue, and blockade of IL-21 signaling at the memory stage diminished CD8 TRM cell survival specifically in the Db-NP366−374 population.
While the influenza response in the lung is not an active chronic infection, viral RNA remnants may cause persistent pathology (44). In persistent viral infection in the brain, provision of IL-21 by T follicular-like tissue-resident CD4 T cells likely promotes ATP production in local CD8 T cells through enhancing electron transport chain efficiency (45). Our data suggests this could be a means by which local CD8 T cells differentiate and persist in response to IL-21. Nonetheless, a local interaction between CD8 and CD4 T cells is required for optimal TRM cell responses following both acute and persistent viral infections (Figure 1). Importantly, this cellular network was responsible for local secondary protection against heterologous infection mediated by the influenza-specific CD8 TRM cells. Interestingly, TRH cell development requires the presence of B cells (43); thus there exists a local interplay among adaptive immune cells for the maintenance of pulmonary lymphocyte memory following viral pneumonia. Understanding how the local cellular networks modulate immune protection may aid the development of mucosal vaccines. Additionally, understanding the molecular cues governing their persistence will likely be important to elicit proper TRM cell responses through immunotherapies.
Unlike the majority of inflamed organs investigated, where it merely enhances TRM cell differentiation, local antigen signals are required for the establishment of pulmonary CD8 TRM cell (17, 46). As briefly mentioned above, TRM cells with TCRs of different specificities against influenza epitopes, exhibit different phenotypes and have distinct requirements for their maintenance (5). At the transcriptional level, polyclonal CD8 TRM cells also vary in their programs between TRM cells of different specificities (5, 6). The TCR is likely playing an active role in these differences. Just as the quality of TCR signals can determine CD8 T cell fate in the circulation, lower affinity TCR signals enhance the potential to differentiate into pulmonary TRM cells (47–49).
Furthermore, the duration and amount of antigenic signals seem important for establishing the diversity of the TRM cell pool against a given respiratory pathogen. For instance, the differential persistence of influenza NP vs. PA antigen at the memory phase clearly dictates the distinct phenotypes of the TRM cells against the two antigens (5). Influenza virion contains many more NP molecules than PA molecules and NP proteins and/or NP366−374 peptide-MHC-I complex are present for a longer period and potentially in a much higher amount than PA proteins or PA peptide-MHC-I complex at the memory phase (50). In agreement, influenza NP-specific (Db-NP366−374), but not PA-specific (Db-PA224−233), TRM cells receive chronic TCR signaling at the memory phase, leading to the development of an “exhausted-like” phenotype (characterized by the high expression of co-inhibitory molecules including PD-1 and Tim-3) in Db-NP366−374 TRM cells (5). Interestingly, like the persistence of true exhausted CD8 T cells during chronic viral infection, the persistence of “exhausted-like” Db-NP366−374 TRM cells is also dependent on the continuous presence of pMHC-I and co-stimulatory signaling as the induced depletion of MHC-I or the late blockade of CD28 diminished Db-NP366−374 TRM cell magnitude (5). How these antigenic signals in the lung work in concert with the main cytokine (TGF-β) responsible for TRM cell differentiation across a breadth of tissues is unclear.
TGF-β is an integrin-activated cytokine with widely varying effects on white blood cells from the hematopoietic stem cell (HSC) stage through to terminal differentiation (51). TGF-β mediates the fine line between immune-tolerance and appropriate activation of both the innate and adaptive immune systems (52–58). As with most of its cell-type dependent functions, effects of TGF-β on CD8 T cells can be stimulatory or inhibitory, depending on the state of differentiation (57, 59). TGF-β can raise the threshold of TCR-induced activation on naïve CD8 T cells, whereas it can induce either TCM-like or TRM-like differentiation in recently activated CD8 T cells (57, 60–62). TGF-β mediates TRM cell differentiation by imparting a partially shared transcriptional footprint across a breadth of organs, however, it is the tissues themselves that govern the uniqueness of the footprint such as what metabolites TRM cells use to persist (61, 63, 64). Similar to most peripheral sites, TGF-β is essential for differentiation of pulmonary TRM cells of numerous antigen specificities (5, 41, 65). Interestingly, low affinity TCR-pMHC interactions leave CD8 T cells more susceptible to TGF-βR signaling which could explain their proclivity toward TRM cell differentiation (47, 49). For respiratory viral infections, the effects of TGF-β signaling on TRM cell generation is Smad4-independent, which may suggest non-canonical TGF-β R signaling pathways are vital for pulmonary TRM cell differentiation (65, 66). Thus, it is likely the context and tissue dependent circumstances of T cell activation may govern how TGF-β contributes to TRM cell heterogeneity.
As mentioned previously, a subset of influenza-specific TRM cells display an exhausted-like phenotype including high expression of PD-1. When PD-L1-PD-1 signaling in influenza infected mice is blocked at the memory stage, the magnitude of the Db-NP366−374, but not Db-PA224−233, TRM cell responses was augmented (5). Furthermore, late PD-L1 blockade increases effector cytokine, particularly TNF, production by Db-NP366−374 TRM cells, indicating targeting the checkpoint molecule PD-1 “rejuvenates” the exhausted-like TRM cells following influenza infection. Consequently, TRM cell-mediated protective immunity was enhanced upon secondary heterologous viral challenge (5). Unexpectedly, pulmonary inflammation and fibrosis were drastically exacerbated following PD-L1 blockade in a CD8 T cell-dependent manner. It is possible that enhanced production of effector molecules from an increased number of CD8 TRM, mediates diffuse alveolar damage in the absence of molecular regulation such as PD-1 signaling (67–69) (Figure 1). Failure to acutely repair this CD8-dependent airway damage, could result in exacerbated collagen deposition or impaired degradation suggesting macrophage and/or fibroblast involvement (5, 6). These results suggest that there is a fine balance on TRM cell-mediated protective immunity and lung pathology following viral pneumonia. These data also indicate that the gradual TRM cell loss in the respiratory tract is perhaps a host-protective mechanism to avoid potential collateral damage to a vital organ. There are also examples of CD8 TRM cells causing pathology in the skin and intestine when homeostatic controls are lost and diseases like vitiligo, psoriasis, or celiac may emerge following destruction of melanocytes, epidermal or mucosal barrier tissues, respectively (70–73). Collectively, these data indicate that one's immune-status is an important regulator of the potential harm to local tissue brought on by unruly TRM cell activation.
Many hurdles exist with regards to provoking efficacious adaptive immune responses in those of advanced age (>70 years)—the demographic that may benefit most from vaccines for emerging pathogens. To understand how immune responses in aged and young hosts proceed differently, we need to understand how the innate and adaptive systems differ globally during the natural aging process. Low-grade systemic inflammation under homeostatic conditions is a hallmark signature of aging, but to what degree it impairs protective immune responses is unclear. This so-called “inflamm-aging” may in-part, be mediated by enhanced myelopoiesis during aging, another hallmark of aging (74). Interestingly plasma cell accumulation in the bone marrow has been shown to drive the myeloid bias with age. Plasma cells remodel bone marrow stroma that govern hematopoiesis, via provision of tumor necrosis factor (TNF), a principle “inflamm-aging” cytokine (75). The skewing of hematopoietic output leads to an age-related decline of naive lymphocytes in the circulation (74–76). Aside from decreased B cell numbers, there is a wide range of age-related functional changes in peripheral B cells that could affect antibody responses to vaccines in the elderly (77–79). Bone marrow is not the only primary lymphoid tissue that suffers age-related output predicaments that might influence vaccine efficacy in the elderly.
Thymic involution starts in the earliest years of life and drops output of naive T cells ~10-fold past the age of 40 (80). This impacts the circulatory T cell compartment as there are fewer recent thymic emigrants seeding secondary lymphoid tissue. For unknown reasons, this affects the diversity of the naïve CD8 compartment more than the CD4 T cell compartment (81). Thus, with age, CD8 memory T cells are enriched and TCR repertoires are likely narrowed across tissues (80–85). Notably, if memory CD8 T cells are formed early in life, they likely provide life-long diverse secondary responses (86, 87).
However, the ability to generate new memory is dependent on naive CD8 T cells, which in our later years (mouse and human), skew to a more differentiated state with the majority exhibiting immuno-senescence, characterized by high signaling thresholds for activation and proliferation (88–91). Moreover, once lymphocytes exit their developmental sites and emigrate to secondary lymph tissue, they encounter age-related stromal deterioration influencing their organization within lymph nodes (92). The above confounders likely affect naïve lymphocyte generation, maintenance, activation and in sum, negatively impact formation of protective immunity toward pathogens and vaccines (85, 93).
One of the first clinical observations in the current pandemic was that mortality and severe morbidity in COVID-19 disproportionately affects those of advanced age (94). This is also true of most severe influenza seasons (95). Severe influenza-like illness are associated with delayed, but prolonged innate and adaptive responses during the effector phase (96). We have recently examined pulmonary CD8 TRM cell responses in young (2 months) and aged (20–22 months) C57BL/6 mice following influenza infection. Aging is associated with the decreased potential of circulating memory T cell generation (97). In sharp contrast, lungs from aged mice have 40-fold more CD8 TRM cells compared to those of young lungs (6). Transfer of CD8 T cells from young mice into the aged hosts results in increased accumulation of memory T cells derived from young mice in the aged lungs following influenza infection. This indicates that the aged environment provokes exaggerated accumulation of TRM cells (6). We found higher levels of Tgfb1 transcript in the aged lungs and the accumulation of TRM cells in aged hosts was largely TGF-β dependent (Figure 2). Relatedly, Chikungunya virus infection in aged mice leads to heightened and dysregulated TGF-β production that exacerbates pathology (98).
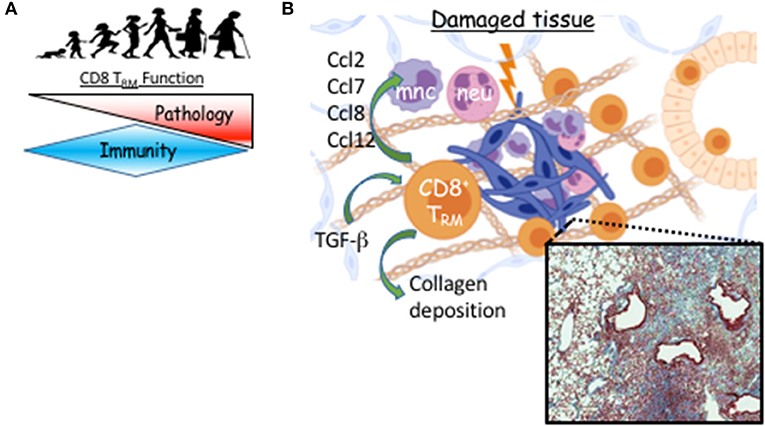
Figure 2. TRM cell-mediated long-term sequelae post viral pneumonia during aging. (A) TRM function switches from immune protection to pathology as we age. (B) Following viral pneumonia, CD8 TRM cells accumulate in aged lungs where their differentiation is TGF-b–dependent. Instead of providing increased immune protection, they provoke pathology, likely through direct or indirect recruitment of myeloid cells that contribute to unresolved inflammation and prevention of collagen degradation. Micrograph is Masson's trichrome stained lung from aged mouse 60 days post-H1N1 infection. Blue is digitally enhanced collagen deposition which is dependent on CD8 TRM cells (6). Created with BioRender.
Of note, alveolar macrophage numbers and function dwindle with age (99). Given the suppressive roles of alveolar macrophages in TRM cell generation (38), it could be possible that diminished alveolar macrophage function may aid the exaggerated development of TRM cells during aging. Notably, many factors change in the aged lung that have not been investigated in the context of TRM accumulation. DAVID analysis of the aged lung transcriptome indicates decreased cell cycle with increased extracellular matrix and cell adhesion gene programs (100). Human Lung Cell Atlas (HLCA) data indicates these changes are accompanied by increases in fibroblasts and neuroendocrine populations and a drop in Type II pneumocytes (100, 101). Additionally, the stroma may be more apt to prompt inflammation in lungs of aged individuals (102). Nevertheless, the data indicate that the aged environment enhances TRM cell accumulation after a single de novo response, suggesting that the aged lung is fertile ground for TRM cell differentiation. In contrast, there is a reduced generation of lung TRM cells following influenza infection in infant mice, largely due to T cell-intrinsic defects (103).
Our data suggest that memory T cells can robustly accumulate in mucosal tissue during aging following a single round of viral challenge. Yet, aged individuals still have impaired protective responses following vaccines or respiratory viral infections which has been attributed to memory CD8 T cell function (104). To resolve the discrepancy, we performed single cell (sc) RNA-seq on young or aged TRM cells against the major influenza protective epitope Db-NP366−374. Our results found that TRM cells isolated from aged lungs lack a subpopulation characterized by high expression of molecules involved in TCR signaling and effector function (6). Consequently, we found that aged mice exhibit impaired TRM cell-mediated protective immunity against heterologous viral rechallenge compared to those of young mice. Thus, aging facilitates the accumulation of dysfunctional TRM cells in the respiratory tract, which explains the phenomena that aged individuals have increased susceptibility of influenza-associated severe diseases despite the robust presence of influenza-specific TRM cells in the respiratory tract. Given the current spread of SARS-CoV2 infection among the elderly population, it would be important to determine whether SARS-CoV2-specific TRM cells exhibit similar functional impairment during aging as the TRM cell-mediated protection would be a key determinant of respiratory immunity during secondary exposure to the virus.
If these newly formed TRM cells are not providing protection, what is their role in the tissue during aging? To address the question, we depleted either circulating, or circulating plus resident CD8 T cells and examined the long-term effects on organ-level transcription and histopathology (6). Depletion of the resident CD8 T cells that were not providing protection against subsequent influenza infection, led to resolution of pulmonary inflammation in aged hosts while concomitantly decreasing the inflammatory environment at the transcriptional level, particularly, chemokines involved in recruiting monocytes and neutrophils (Figure 2) (6). Further, long-term age-related infection-induced exacerbation of collagen deposition was mitigated in the absence of parenchymal CD8 T cells (Figure 2). Establishment of pulmonary TRM in IAV infection models depends on local presentation of antigen, likely via monocyte-derived macrophages and/or dendritic cells, which we find sustained in the aged lung parenchyma (40, 46, 105). Infiltrating monocyte-derived macrophages have been shown to exacerbate collagen-deposition following influenza infection (106). Collectively, this could indicate the aged environment provokes accumulation of pulmonary TRM cells that support ongoing inflammation of the organ contributing to its poor repair following respiratory viral pneumonia.
As discussed above, SARS-CoV2 infection disproportionately affects aged individuals. Of particular relevance is the observation of severe COVID-19 patients presenting both with CD8 T cell lymphopenia in the blood, but large number of TRM-like CD8 T cells in the airways (107). Notably, emerging evidence has suggested that a large proportion of COVID-19 patients exhibit pulmonary and extrapulmonary symptoms 6 months after recovery from the acute morbidity (108). Particularly, it is predicted that a large number of severe COVID-19 patients will develop persistent lung damage and fibrosis as observed in patients infected with SARS-CoV and MERS (109–113). Notably, TGF-β activating integrin is upregulated in fibrotic lung lesions in COVID-19 patients 2 months post-infection, which could support fibrosis and TRM cell maintenance (114). It would be critically significant to examine whether malfunctional CD8 TRM cells contribute to the long-term fibrotic sequelae of SARS-CoV2 infection.
While viral-specific pathogenic CD8 T cells have not been found in human tissue to-date, plausible candidates may now be on the radar. Age-associated granzyme K-expressing CD8 T cells are enriched in the T effector memory compartment in human blood (81). Age-associated CD8 T cell counterparts in mice were identified by expression of the effector molecule granzyme K, the checkpoint molecule PD-1, integrin CD49d, and the transcription factor TOX and are enriched in blood and across tissues (spleen, peritoneum, lungs, liver, and white adipose tissue) with age. The aged environment conferred this phenotype to young CD8 T cells in adoptive transfer models. While the TCR repertoires of age-associated CD8 T cells were clonally narrowed within each host across tissues, between hosts, their TCR sequences were diverse, suggesting either microbial-specific or stochastic differentiation. It is important to note that these age-associated CD8 T cells are transcriptionally distinct from senescent virtual memory CD8 T cells also enriched with age (88). It's unclear how granzyme K+ age-associated CD8 T cells behave in an immune response. While their phenotype (PD-1Hi TOX+) is typically associated with CD8 T cell exhaustion, recombinant granzyme K augmented cytokine and chemokine production from senescent fibroblasts in vitro (81). Activation of local age-associated CD8 T cells may thus provoke inflammation and potentially influence tissue remodeling and senescence associated secretion phenotypes.
Examples of age-related increases in lung tissue disrepair abound and are found commonly in idiopathic pulmonary fibrosis (IPF) (115, 116). IPF is an interstitial pneumonic disease that results in alveoli involved in gas exchange being progressively replaced by scar tissue with a 20% 5 year survivability (117). No treatment can reverse the process once started. As its namesake would suggest, IPF has no known single cause and it is unclear how the tissue becomes damaged and fails to repair. It is notable that IPF shares some features of viral pneumonia sequelae including COVID-19, most prominent of which is collagen accumulation which can lead to fibrosis (118). We described an increased number of CD8 T cells in the parenchyma surrounding lesions in IPF patients (5). It is plausible that these patients lost the battle for homeostatic control of local memory T cells that can mediate bystander inflammation. Of note, respiratory T cells have a role in dysfunctional wound repair resulting in fibrosis in acute lung injury models (119). Further, one of the frontline treatments (Nintedanib) that slows development of IPF by presumably targeting the kinase activities of PDGF, FGF, and VEGF receptors, inhibits src family tyrosine kinases, including the crucial T cell activating kinase Lck, with similar IC50 values (120, 121). This could implicate dampened T cell activity as a partial mechanism slowing fibrotic progression in the lung. Thus, while lung damage and repair models can happen in lymphocyte-scarce environments, certain T cell subsets exacerbate fibrosis and the jury may need to be recalled as to whether local T cells play a role in IPF pathogenesis and potentially viral pneumonic sequelae in humans.
Although pulmonary resident memory CD8 T cells have shown outstanding immune-protective capacity, this does not seem to be the case in aged hosts following respiratory viral infections. In contrast, resident CD8 T cells mediate pathology during the disease course leading to non-resolution of lung inflammation in aged hosts. Unexpectedly, aged hosts accumulate local TRM cells despite a poor response in the circulation (6). This suggests efforts should be retooled to restore their protective immunity (122) and mitigate their pathogenic capacity rather than recruit more to the mucosa. These opposing features of TRM cells in young and aged hosts may identify a balance between immune protection and pathology and shed light on their teleological existence in a vital organ. While recent work has highlighted the cellular and molecular networks that mediate pulmonary TRM density in young healthy hosts, we are just beginning to understand the potential they have to mediate damage when homeostatic controls are lost, e.g. through the aging process. Understanding the mechanisms modulating the balance of TRM cell-mediated immunity vs. pathogenicity will be important to selectively harness the beneficial function of TRM cells and simultaneously mitigate their pathogenic potential.
NG and JS wrote and IC was responsible for editing the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by NIH RO1s (Grant Numbers: AG047156, AI112844, AG069264, AI147394, and AI154598).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Topham DJ, Tripp RA, Doherty PC. CD8+ T cells clear influenza virus by perforin or Fas-dependent processes. J Immunol. (1997) 159:5197–200. Available online at: https://www.jimmunol.org/content/159/11/5197.long
2. Guo H, Santiago F, Lambert K, Takimoto T, Topham DJ. T cell-mediated protection against lethal 2009 pandemic H1N1 influenza virus infection in a mouse model. J Virol. (2011) 85:448–55. doi: 10.1128/JVI.01812-10
3. Guo H, Topham DJ. Multiple distinct forms of CD8+ T cell cross-reactivity and specificities revealed after 2009 H1N1 influenza A virus infection in mice. PLoS ONE. (2012) 7:e46166. doi: 10.1371/journal.pone.0046166
4. McMaster SR, Wilson JJ, Wang H, Kohlmeier JE. Airway-resident memory CD8 T cells provide antigen-specific protection against respiratory virus challenge through rapid IFN-gamma production. J Immunol. (2015) 195:203–9. doi: 10.4049/jimmunol.1402975
5. Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, et al. PD-1(hi) CD8(+) resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol. (2019) 4:eaaw1217. doi: 10.1126/sciimmunol.aaw1217
6. Goplen NP, Wu Y, Son YM, Li C, Wang ZI, Cheon S, et al. Tissue-resident CD8(+) T cells drive age-associated chronic lung sequelae after viral pneumonia. Sci Immunol. (2020) 5:eabc4557. doi: 10.1126/sciimmunol.abc4557
7. Beura LK, Mitchell JS, Thompson EA, Schenkel JM, Mohammed J, Wijeyesinghe S, et al. Intravital mucosal imaging of CD8(+) resident memory T cells shows tissue-autonomous recall responses that amplify secondary memory. Nat Immunol. (2018) 19:173–82. doi: 10.1038/s41590-017-0029-3
8. Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, Masopust D. T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science. (2014) 346:98–101. doi: 10.1126/science.1254536
9. Decman V, Laidlaw BJ, Dimenna LJ, Abdulla S, Mozdzanowska K, Erikson J, et al. Cell-intrinsic defects in the proliferative response of antiviral memory CD8 T cells in aged mice upon secondary infection. J Immunol. (2010) 184:5151–9. doi: 10.4049/jimmunol.0902063
10. Park SL, Zaid A, Hor JL, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol. (2018) 19:183–91. doi: 10.1038/s41590-017-0027-5
11. Kumar BV, Ma W, Miron M, Garnot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep. (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
12. Gerlach C, Moseman EA, Loughhead SM, Alvarez D, Zwijnenburg AJ, Waanders L, et al. The chemokine receptor CX3CR1 defines three antigen-experienced CD8 T cell subsets with distinct roles in immune surveillance and homeostasis. Immunity. (2016) 45:1270–84. doi: 10.1016/j.immuni.2016.10.018
13. Kok L, Dijkgraaf FE, Urbanus J, Besser K, Vredevoogd DW, Cardoso RF, et al. A committed tissue-resident memory T cell precursor within the circulating CD8+ effector T cell pool. J Exp Med. (2020) 217:e20191711. doi: 10.1084/jem.20191711
14. Milner JJ, Toma C, Yu B, Zhang K, Omilusik K, Phan AT, et al. Runx3 programs CD8(+) T cell residency in non-lymphoid tissues and tumours. Nature. (2017) 552:253–7. doi: 10.1038/nature24993
15. Behr FM, Chuwonpad A, Stark R, van Gisbergen KP. Armed and ready: transcriptional regulation of tissue-resident memory CD8 T cells. Front Immunol. (2018) 9:1770. doi: 10.3389/fimmu.2018.01770
16. Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, et al. Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science. (2016) 352:459–63. doi: 10.1126/science.aad2035
17. Wu T, Hu Y, Lee YT, Bouchard KR, Benechet A, Khanna K, et al. Lung-resident memory CD8 T cells (TRM) are indispensable for optimal cross-protection against pulmonary virus infection. J Leukoc Biol. (2014) 95:215–24. doi: 10.1189/jlb.0313180
18. Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity. (2018) 48:327–38.e5. doi: 10.1016/j.immuni.2018.01.015
19. Hayward SL, Scharer CD, Cartwright EK, Takamura S, Li ZT, Boss JM, et al. Environmental cues regulate epigenetic reprogramming of airway-resident memory CD8(+) T cells. Nat Immunol. (2020) 21:309–20. doi: 10.1038/s41590-019-0584-x
20. Van Braeckel-Budimir N, Varga SM, Badovinac VP, Harty JT. Repeated antigen exposure extends the durability of influenza-specific lung-resident memory CD8(+) T cells and heterosubtypic immunity. Cell Rep. (2018) 24:3374–82.e3. doi: 10.1016/j.celrep.2018.08.073
21. Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, et al. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell. (2014) 159:814–28. doi: 10.1016/j.cell.2014.10.026
22. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS, et al. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, antigen specificity. PLoS ONE. (2011) 6:e16245. doi: 10.1371/journal.pone.0016245
23. Oja AE, Piet B, Helbig C, Stark R, van der Zwan D, Blaauwgeers H, et al. Trigger-happy resident memory CD4(+) T cells inhabit the human lungs. Mucos Immunol. (2018) 11:654–67. doi: 10.1038/mi.2017.94
24. Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, et al. Programs for the persistence, vigilance and control of human CD8(+) lung-resident memory T cells. Nat Immunol. (2016) 17:1467–78. doi: 10.1038/ni.3589
25. Wang Z, Zhu L, Nguyen THO, Wan Y, Sant S, Quinones-Parra SM, et al. Clonally diverse CD38(+)HLA-DR(+)CD8(+) T cells persist during fatal H7N9 disease. Nat Commun. (2018) 9:824. doi: 10.1038/s41467-018-03243-7
26. Snyder ME, Finlayson MO, Connors JT, Dogra P, Senda T, Bush E, et al. Generation and persistence of human tissue-resident memory T cells in lung transplantation. Sci Immunol. (2019) 4:eaav5581. doi: 10.1126/sciimmunol.aav5581
27. Takamura S, Yagi H, Hakata Y, Motozono C, McMaster SR, Masumoto T, et al. Specific niches for lung-resident memory CD8+ T cells at the site of tissue regeneration enable CD69-independent maintenance. J Exp Med. (2016) 213:3057–73. doi: 10.1084/jem.20160938
28. Pizzolla A, Nguyen TH, Sant S, Jaffar J, Loudovaris T, Mannering SI, et al. Influenza-specific lung-resident memory T cells are proliferative and polyfunctional and maintain diverse TCR profiles. J Clin Investig. (2018) 128:721–33. doi: 10.1172/JCI96957
29. Reilly EC, Emo Lambert K, Buckley PM, Reilly NS, Smith I, Chaves FA, et al. TRM integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc Natl Acad Sci USA. (2020) 117:12306–14. doi: 10.1073/pnas.1915681117
30. Takamura S, Kato S, Motozono C, Shimaoka T, Ueha S, Matsuo K, et al. Interstitial-resident memory CD8(+) T cells sustain frontline epithelial memory in the lung. J Exp Med. (2019) 216:2736–47. doi: 10.1084/jem.20190557
31. Wein AN, McMaster SR, Takamura S, Dunbar PR, Cartwright EK, Hayward SL, et al. CXCR6 regulates localization of tissue-resident memory CD8 T cells to the airways. J Exp Med. (2019) 216:2748–62. doi: 10.1084/jem.20181308
32. Walsh DA, Borges da Silva H, Beura LK, Peng C, Hamilton SE, Masopust D, et al. The functional requirement for CD69 in establishment of resident memory CD8(+) T cells varies with tissue location. J Immunol. (2019) 203:946–55. doi: 10.4049/jimmunol.1900052
33. El-Hashash AH, Turcatel G, Varma S, Berika M, Al Alam D, Warburton D. Eya1 protein phosphatase regulates tight junction formation in lung distal epithelium. J Cell Sci. (2012) 125:4036–48. doi: 10.1242/jcs.102848
34. Le Floc'h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. Alpha E beta 7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med. (2007) 204:559–70. doi: 10.1084/jem.20061524
35. Hashimoto D, Chow A, Noizat C, Teo P, Beasely MB, Leboeuf M, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. (2013) 38:792–804. doi: 10.1016/j.immuni.2013.04.004
36. Hussell T, Bell TJ. Alveolar macrophages: plasticity in a tissue-specific context. Nat Rev Immunol. (2014) 14:81–93. doi: 10.1038/nri3600
37. Purnama C, Ng SL, Tetlak P, Setiagani YA, Kandasamy M, Baalasubramanian S, et al. Transient ablation of alveolar macrophages leads to massive pathology of influenza infection without affecting cellular adaptive immunity. Eur J Immunol. (2014) 44:2003–12. doi: 10.1002/eji.201344359
38. Goplen NP, Huang S, Zhu B, Cheon IS, Son YM, Wang Z, et al. Tissue-resident macrophages limit pulmonary CD8 resident memory T cell establishment. Front Immunol. (2019) 10:2332. doi: 10.3389/fimmu.2019.02332
39. Huang S, Goplen NP, Zhu B, Cheon IS, Son YM, Wang Z, et al. Macrophage PPAR-gamma suppresses long-term lung fibrotic sequelae following acute influenza infection. PLoS ONE. (2019) 14:e0223430. doi: 10.1371/journal.pone.0223430
40. Dunbar PR, Cartwright EK, Wein AN, Tsukamoto T, Tiger Li ZR, Kumar N, et al. Pulmonary monocytes interact with effector T cells in the lung tissue to drive TRM differentiation following viral infection. Mucos Immunol. (2020) 13:161–71. doi: 10.1038/s41385-019-0224-7
41. Bergsbaken T, Bevan MJ. Proinflammatory microenvironments within the intestine regulate the differentiation of tissue-resident CD8(+) T cells responding to infection. Nat Immunol. (2015) 16:406–14. doi: 10.1038/ni.3108
42. Son YM, Cheon IS, Wu Y, Li C, Wang Z, Gao X, et al. Tissue-resident CD4(+) T helper cells assist the development of protective respiratory B and CD8(+) T cell memory responses. Sci Immunol. (2021) 6:eabb6852. doi: 10.1126/sciimmunol.abb6852
43. Swarnalekha N, Schreiner D, Litzler LC, Iftikhar S, Kirchmeier D, Kunzil M, et al. T resident helper cells promote humoral responses in the lung. Sci Immunol. (2021) 6:eabb6808. doi: 10.1126/sciimmunol.abb6808
44. Keeler SP, Agapov EV, Hinojosa ME, Letvin AN, Wu K, Holtzman MJ. Influenza A virus infection causes chronic lung disease linked to sites of active viral RNA remnants. J Immunol. (2018) 201:2354–68. doi: 10.4049/jimmunol.1800671
45. Ren HM, Kolawole EM, Ren M, Jin G, Netherby-Winslow CS, Wade Q, et al. IL-21 from high-affinity CD4 T cells drives differentiation of brain-resident CD8 T cells during persistent viral infection. Sci Immunol. (2020) 5:eabb5590. doi: 10.1126/sciimmunol.abb5590
46. McMaster SR, Wein AN, Dunbar PR, Hayward SL, Cartwright EK, Denning TL, et al. Pulmonary antigen encounter regulates the establishment of tissue-resident CD8 memory T cells in the lung airways and parenchyma. Mucos Immunol. (2018) 11:1071–8. doi: 10.1038/s41385-018-0003-x
47. Knudson KM, Goplen NP, Cunningham CA, Daniels MA, Teixeiro E. Low-affinity T cells are programmed to maintain normal primary responses but are impaired in their recall to low-affinity ligands. Cell Rep. (2013) 4:554–65. doi: 10.1016/j.celrep.2013.07.008
48. Zehn D, Lee SY, Bevan MJ. Complete but curtailed T-cell response to very low-affinity antigen. Nature. (2009) 458:211–4. doi: 10.1038/nature07657
49. Fiege JK, Stone IA, Fay EJ, Markman MW, Wijeyesinghe S, Macchietto M, et al. The impact of TCR signal strength on resident memory T cell formation during influenza virus infection. J Immunol. (2019) 203:936–45. doi: 10.4049/jimmunol.1900093
50. Ballesteros-Tato A, Leon B, Lee BO, Lund FE, Randall TD. Epitope-specific regulation of memory programming by differential duration of antigen presentation to influenza-specific CD8(+) T cells. Immunity. (2014) 41:127–40. doi: 10.1016/j.immuni.2014.06.007
51. Sitnicka E, Ruscetti FW, Priestley GV, Wolf NS, Bartelmez SH. Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood. (1996) 88:82–8. doi: 10.1182/blood.V88.1.82.bloodjournal88182
52. Christ M, McCartney-Francis NL, Kulkarni AB, Ward JM, Mizel DE, Mackall CL, et al. Immune dysregulation in TGF-beta 1-deficient mice. J Immunol. (1994) 153:1936–46. Available online at: https://www.jimmunol.org/content/153/5/1936.long
53. Gorham JD, Guler ML, Fenoglio D, Gubler U, Murphy KM. Low dose TGF-beta attenuates IL-12 responsiveness in murine Th cells. J Immunol. (1998) 161:1664–70. Available online at: https://www.jimmunol.org/content/161/4/1664
54. Fahlen L, Read S, Gorelik L, Hurst SD, Coffman RL, Flavell RA, et al. T cells that cannot respond to TGF-beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med. (2005) 201:737–46. doi: 10.1084/jem.20040685
55. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med. (2005) 201:1061–7. doi: 10.1084/jem.20042276
56. Laouar Y, Sutterwala FS, Gorelik L, Flavell RA. Transforming growth factor-beta controls T helper type 1 cell development through regulation of natural killer cell interferon-gamma. Nat Immunol. (2005) 6:600–7. doi: 10.1038/ni1197
57. Ma C, Zhang N. Transforming growth factor-beta signaling is constantly shaping memory T-cell population. Proc Natl Acad Sci USA. (2015) 112:11013–7. doi: 10.1073/pnas.1510119112
58. Arumugam V, Bluemn T, Wesley E, Schmidt AM, Kambayashi T, Malarkannan S, et al. TCR signaling intensity controls CD8+ T cell responsiveness to TGF-beta. J Leukoc Biol. (2015) 98:703–12. doi: 10.1189/jlb.2HIMA1214-578R
59. Zhang N, Bevan MJ. TGF-beta signaling to T cells inhibits autoimmunity during lymphopenia-driven proliferation. Nat Immunol. (2012) 13:667–3. doi: 10.1038/ni.2319
60. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol. (2013) 14:1285–93. doi: 10.1038/ni.2745
61. Nath AP, Braun A, Ritchie RC, Carbone FR, Mackay LK, Gebhardt T, et al. Comparative analysis reveals a role for TGF-beta in shaping the residency-related transcriptional signature in tissue-resident memory CD8+ T cells. PLoS ONE. (2019) 14:e0210495. doi: 10.1371/journal.pone.0210495
62. Mackay LK, Wynne-Jones E, Freestone D, Pellicci DG, Mielke LA, Newman DM, et al. T-box transcription factors combine with the cytokines TGF-beta and IL-15 to control tissue-resident memory t cell fate. Immunity. (2015) 43:1101–11. doi: 10.1016/j.immuni.2015.11.008
63. Mohammed J, Beura LK, Bobr A, Astry B, Chicoine B, Kashem SW, et al. Stromal cells control the epithelial residence of DCs and memory T cells by regulated activation of TGF-beta. Nat Immunol. (2016) 17:414–21. doi: 10.1038/ni.3396
64. Frizzell H, Fonseca R, Chriso SN, Evard M, Cruz-Gomez S, Zanluqui NG, et al. Organ-specific isoform selection of fatty acid-binding proteins in tissue-resident lymphocytes. Sci Immunol. (2020) 5:eaay9283. doi: 10.1126/sciimmunol.aay9283
65. Hu Y, Lee YT, Kaech SM, Garvy B, Cauley LS. Smad4 promotes differentiation of effector and circulating memory CD8 T cells but is dispensable for tissue-resident memory CD8 T cells. J Immunol. (2015) 194:2407–14. doi: 10.4049/jimmunol.1402369
66. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
67. Hashimoto S, Kobayashi A, Kooguchi K, Kitamura Y, Onodera H, Nakajima H, et al., Upregulation of two death pathways of perforin/granzyme and FasL/Fas in septic acute respiratory distress syndrome. Am J Respir Crit Care Med. (2000) 161:237–43. doi: 10.1164/ajrccm.161.1.9810007
68. Panoskaltsis-Mortari A, Ingbar DH, Jung P, Haddad IY, Bitterman PB, Wangensteen OD, et al. KGF pretreatment decreases B7 and granzyme B expression and hastens repair in lungs of mice after allogeneic BMT. Am J Physiol Lung Cell Mol Physiol. (2000) 278:L988–99. doi: 10.1152/ajplung.2000.278.5.L988
69. Mintern JD, Guillonneau C, Carbone FR, Doherty PC, Turner SJ. Cutting edge: tissue-resident memory CTL down-regulate cytolytic molecule expression following virus clearance. J Immunol. (2007) 179:7220–4. doi: 10.4049/jimmunol.179.11.7220
70. Riding RL, Harris JE. The role of memory CD8(+) T cells in vitiligo. J Immunol. (2019) 203:11–9. doi: 10.4049/jimmunol.1900027
71. Meresse B, Chen Z, Ciszewski C, Tretiakova M, Bhagat G, Krausz TN, et al. Coordinated induction by IL15 of a TCR-independent NKG2D signaling pathway converts CTL into lymphokine-activated killer cells in celiac disease. Immunity. (2004) 21:357–66. doi: 10.1016/j.immuni.2004.06.020
72. Cheuk S, Schlums H, Gallais Serezal I, Martine E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8(+) T cells poised for cytotoxic function in human skin. Immunity. (2017) 46:287–300. doi: 10.1016/j.immuni.2017.01.009
73. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature. (2017) 543:252–6. doi: 10.1038/nature21379
74. Ho YH, del Toro R, Rivera-Torres J, Rak J, Korn C, García-García A, et al. Remodeling of bone marrow hematopoietic stem cell niches promotes myeloid cell expansion during premature or physiological aging. Cell Stem Cell. (2019) 25:407–18 e406. doi: 10.1016/j.stem.2019.06.007
75. Pioli PD, Casero D, Montecino-Rodriguez E, Morrison SL, Dorshkind K. Plasma cells are obligate effectors of enhanced myelopoiesis in aging bone marrow. Immunity. (2019) 51:351–66.e6. doi: 10.1016/j.immuni.2019.06.006
76. Kirschner K, Chandra T, Kiselev V, Flores-Santa Cruz D, Macaulay IC, Park HJ, et al. Proliferation drives aging-related functional decline in a subpopulation of the hematopoietic stem cell compartment. Cell Rep. (2017) 19:1503–11. doi: 10.1016/j.celrep.2017.04.074
77. Rubtsov AV, Rubtsova K, Fischer A, Meehan RT, Gillis JZ, Kappler JW, et al. Toll-like receptor 7 (TLR7)-driven accumulation of a novel CD11c(+) B-cell population is important for the development of autoimmunity. Blood. (2011) 118:1305–15. doi: 10.1182/blood-2011-01-331462
78. Henry C, Zheng NY, Huang M, Cavanov A, Rojas KT, Kaur K, et al. Influenza virus vaccination elicits poorly adapted b cell responses in elderly individuals. Cell Host Microbe. (2019) 25:357–66.e6. doi: 10.1016/j.chom.2019.01.002
79. Riley RL, Khomtchouk K, Blomberg BB. Age-associated B cells (ABC) inhibit B lymphopoiesis and alter antibody repertoires in old age. Cell Immunol. (2017) 321:61–7. doi: 10.1016/j.cellimm.2017.04.008
80. Thome JJ, Grinshpun B, Kumar BV, Kubota M, Ohmura Y, Lerner H, et al. Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci Immunol. (2016) 1:eaah6506. doi: 10.1126/sciimmunol.aah6506
81. Mogilenko DA, Shpynov O, Andhey PS, Arthur L, Swain A, Esaulova E, et al. Comprehensive profiling of an aging immune system reveals clonal GZMK(+) CD8(+) T cells as conserved Hallmark of inflammaging. Immunity. (2020) 54:99–115.e12. doi: 10.1016/j.immuni.2020.11.005
82. Sant S, Grzelak L, Wang Z, Pizzolla A, Koutsakos M, Crowe J, et al. Single-cell approach to influenza-specific CD8(+) T cell receptor repertoires across different age groups, tissues, and following influenza virus infection. Front Immunol. (2018) 9:1453. doi: 10.3389/fimmu.2018.01453
83. Ahmed M, Lanzer KG, Yager EJ, Adams PS, Johnson LL, Blackman MA. Clonal expansions and loss of receptor diversity in the naive CD8 T cell repertoire of aged mice. J Immunol. (2009) 182:784–92. doi: 10.4049/jimmunol.182.2.784
84. Qi Q, Liu Y, Cheng Y, Glanville J, Zhang D, Lee JY, et al. Diversity and clonal selection in the human T-cell repertoire. Proc Natl Acad Sci USA. (2014) 111:13139–44. doi: 10.1073/pnas.1409155111
85. Messaoudi I, Lemaoult J, Guevara-Patino JA, Metzner BM, Nikolich-Zugich J. Age-related CD8 T cell clonal expansions constrict CD8 T cell repertoire and have the potential to impair immune defense. J Exp Med. (2004) 200:1347–58. doi: 10.1084/jem.20040437
86. Valkenburg SA, Venturi V, Dang TH, Bird NL, Doherty PC, Turner SJ, et al. Early priming minimizes the age-related immune compromise of CD8(+) T cell diversity and function. PLoS Pathog. (2012) 8:e1002544. doi: 10.1371/journal.ppat.1002544
87. Hammarlund E, Lewis MW, Hansen SG, Strelow LI, Nelson JA, Sexton GJ, et al. Duration of antiviral immunity after smallpox vaccination. Nat Med. (2003) 9:1131–7. doi: 10.1038/nm917
88. Hussain T, Quinn KM. Similar but different: virtual memory CD8 T cells as a memory-like cell population. Immunol Cell Biol. (2019) 97:675–84. doi: 10.1111/imcb.12277
89. Quinn KM, Fox A, Harland KL, Russ BE, Li J, Nguyen THO, et al. Age-related decline in primary CD8(+) T cell responses is associated with the development of senescence in virtual memory CD8(+) T cells. Cell Rep. (2018) 23:3512–24. doi: 10.1016/j.celrep.2018.05.057
90. Moskowitz DM, Zhang DW, Hu B, Le Saux S, Yanes RE, Ye Z, et al. Epigenomics of human CD8 T cell differentiation and aging. Sci Immunol. (2017) 2:eaag0192. doi: 10.1126/sciimmunol.aag0192
91. White JT, Cross EW, Kedl RM. Antigen-inexperienced memory CD8(+) T cells: where they come from and why we need them. Nat Rev Immunol. (2017) 17:391–400. doi: 10.1038/nri.2017.34
92. Masters R, Hall A, Bartley JM, Keilich SR, Lorenzo EC, Jellison ER, et al. Assessment of lymph node stromal cells as an underlying factor in age-related immune impairment. J Gerontol A Biol Sci Med Sci. (2019) 74:1734–43. doi: 10.1093/gerona/glz029
93. Yager EJ, Ahmed M, Lanzer K, Randall TD, Woodland DL, Blackman MA. Age-associated decline in T cell repertoire diversity leads to holes in the repertoire and impaired immunity to influenza virus. J Exp Med. (2008) 205:711–23. doi: 10.1084/jem.20071140
94. Zhang X, Tan Y, Ling Y, Lu G, Liu F, Yi Z, et al. Viral and host factors related to the clinical outcome of COVID-19. Nature. (2020) 583:437–40. doi: 10.1038/s41586-020-2355-0
95. Acosta E, Hallman SA, Dillon LY, Ouellette N, Bourbeau R, Herring DA, et al. Determinants of influenza mortality trends: age-period-cohort analysis of influenza mortality in the United States, 1959-2016. Demography. (2019) 56:1723–46. doi: 10.1007/s13524-019-00809-y
96. Wong SS, Oshansky CM, Guo XJ, Ralston J, Wood T, Seeds R, et al. Severe influenza is characterized by prolonged immune activation: results from the SHIVERS cohort study. J Infect Dis. (2018) 217:245–56. doi: 10.1093/infdis/jix571
97. Po JL, Gardner EM, Anaraki F, Katsikis PD, Murasko DM. Age-associated decrease in virus-specific CD8+ T lymphocytes during primary influenza infection. Mech Ageing Dev. (2002) 123:1167–81. doi: 10.1016/S0047-6374(02)00010-6
98. Uhrlaub JL, Pulko V, DeFilippis VR, Broeckel R, Streblow DN, Coleman GD, et al. Dysregulated TGF-beta production underlies the age-related vulnerability to chikungunya virus. PLoS Pathog. (2016) 12:e1005891. doi: 10.1371/journal.ppat.1005891
99. Evren E, Ringqvist E, Willinger T. Origin and ontogeny of lung macrophages: from mice to humans. Immunology. (2020) 160:126–38. doi: 10.1111/imm.13154
100. Chow RD, Majety M, Chen S. The aging transcriptome and cellular landscape of the human lung in relation to SARS-CoV-2. Nat Commun. (2021) 12:4. doi: 10.1038/s41467-020-20323-9
101. Travaglini KJ, Nabhan AN, Penland L, Sinha R, Gillich A, Sit RV, et al. A molecular cell atlas of the human lung from single-cell RNA sequencing. Nature. (2020) 587:619–25. doi: 10.1038/s41586-020-2922-4
102. Parikh P, Wicher S, Khandalavala K, Pabelick CM, Britt RD Jr, Prakash YS. Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol. (2019) 316:L826–42. doi: 10.1152/ajplung.00424.2018
103. Zens KD, Chen JK, Guyer RS, Wu FL, Cvetkovski F, Miron M, et al. Reduced generation of lung tissue-resident memory T cells during infancy. J Exp Med. (2017) 214:2915–32. doi: 10.1084/jem.20170521
104. Zhou X, McElhaney JE. Age-related changes in memory and effector T cells responding to influenza A/H3N2 and pandemic A/H1N1 strains in humans. Vaccine. (2011) 29:2169–77. doi: 10.1016/j.vaccine.2010.12.029
105. Anderson KG, Sung H, Skon CN, Lefançois L, Deisinger A, Vezys V, et al. Cutting edge: intravascular staining redefines lung CD8 T cell responses. J Immunol. (2012) 189:2702–6. doi: 10.4049/jimmunol.1201682
106. Misharin AV, Morales-Nebreda L, Reyfman PA, Cuda CM, Walter JM, McQuattie-Pimentel AC, et al. Monocyte-derived alveolar macrophages drive lung fibrosis and persist in the lung over the life span. J Exp Med. (2017) 214:2387–404. doi: 10.1084/jem.20162152
107. Liao M, liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
108. Huang C, Huang L, Wang Y, Li X, Ren L, Gu X, et al. 6-month consequences of COVID-19 in patients discharged from hospital: a cohort study. Lancet. (2021) 397:220–32. doi: 10.1016/S0140-6736(20)32656-8
109. Hsu HH, Tzao C, Wu CP, Chang WC, Tsai CL, Tung HJ, et al. Correlation of high-resolution CT, symptoms, and pulmonary function in patients during recovery from severe acute respiratory syndrome. Chest. (2004) 126:149–58. doi: 10.1378/chest.126.1.149
110. Fang Y, Zhou J, Ding X, Ling G, Yu S. Pulmonary fibrosis in critical ill patients recovered from COVID-19 pneumonia: preliminary experience. Am J Emerg Med. (2020) 38:2134–8. doi: 10.1016/j.ajem.2020.05.120
111. Xu YH, Dong JH, An WM, Lv XY, Yin XP, Zhang JZ, et al. Clinical and computed tomographic imaging features of novel coronavirus pneumonia caused by SARS-CoV-2. J Infect. (2020) 80:394–400. doi: 10.1016/j.jinf.2020.02.017
112. George PM, Wells AU, Jenkins RG. Pulmonary fibrosis and COVID-19: the potential role for antifibrotic therapy. Lancet Respir Med. (2020) 8:807–15. doi: 10.1016/S2213-2600(20)30225-3
113. Jenkins G. Demystifying pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. (2020) 319:L554–9. doi: 10.1152/ajplung.00365.2020
114. Foster CC, Davis RA, Hausner SH, Sutcliffe JL. Alphalphavbeta6 targeted molecular PET/CT imaging of lung post SARS-CoV-2 infection. J Nucl Med, (2020) 61:1717–9. doi: 10.2967/jnumed.120.255364
115. Sueblinvong V, Neujahr DC, Todd Mills S, Roser-Page S, Ritzenthaler JD, Guidot D, et al. Predisposition for disrepair in the aged lung. Am J Med Sci. (2012) 344:41–51. doi: 10.1097/MAJ.0b013e318234c132
116. Thannickal VJ, Murthy M, Balch WE, Chandel NS, Meiners S, Eickelberg O, et al. Blue journal conference. Aging and susceptibility to lung disease. Am J Respir Critic Care Med. (2015) 191:261–9. doi: 10.1164/rccm.201410-1876PP
117. Bonella F, Stowasser S, Wollin L. Idiopathic pulmonary fibrosis: current treatment options and critical appraisal of nintedanib. Drug Des Devel Ther. (2015) 9:6407–19. doi: 10.2147/DDDT.S76648
118. Huang Y, Tan C, Wu J, Chen M, Wang Z, Luo L, et al. Impact of coronavirus disease 2019 on pulmonary function in early convalescence phase. Respir Res. (2020) 21:163. doi: 10.1186/s12931-020-01429-6
119. Yang D, Chen X, Wang J, Lou Q, Lou Y, Li L, et al. Dysregulated lung commensal bacteria drive interleukin-17B production to promote pulmonary fibrosis through their outer membrane vesicles. Immunity. (2019) 50:692–706.e7. doi: 10.1016/j.immuni.2019.02.001
120. Rudd CE. CD4, CD8 and the TCR-CD3 complex: a novel class of protein-tyrosine kinase receptor. Immunol Today. (1990) 11:400–6. doi: 10.1016/0167-5699(90)90159-7
121. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1434–45. doi: 10.1183/09031936.00174914
Keywords: viral pneumonia, influenza, resident memory, pathology, homeostasis, age
Citation: Goplen NP, Cheon IS and Sun J (2021) Age-Related Dynamics of Lung-Resident Memory CD8+ T Cells in the Age of COVID-19. Front. Immunol. 12:636118. doi: 10.3389/fimmu.2021.636118
Received: 30 November 2020; Accepted: 26 January 2021;
Published: 29 March 2021.
Edited by:
Vandana Kalia, University of Washington, United StatesReviewed by:
Linda M. Wakim, The University of Melbourne, AustraliaCopyright © 2021 Goplen, Cheon and Sun. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nick P. Goplen, Z29wbGVuLm5pY2hvbGFzQG1heW8uZWR1; Jie Sun, c3VuLmppZUBtYXlvLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.