
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 01 April 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.635131
This article is part of the Research TopicPre-Conference Research Topic: 16th International Symposium on SchistosomiasisView all 35 articles
 Silvain Pinaud1,2†‡
Silvain Pinaud1,2†‡ Guillaume Tetreau1,2‡
Guillaume Tetreau1,2‡ Pierre Poteaux1,2
Pierre Poteaux1,2 Richard Galinier1,2
Richard Galinier1,2 Cristian Chaparro1,2
Cristian Chaparro1,2 Damien Lassalle1,2Anaïs Portet1,2†Elodie Simphor1,2
Damien Lassalle1,2Anaïs Portet1,2†Elodie Simphor1,2 Benjamin Gourbal1,2
Benjamin Gourbal1,2 David Duval1,2*
David Duval1,2*Aerolysins initially characterized as virulence factors in bacteria are increasingly found in massive genome and transcriptome sequencing data from metazoans. Horizontal gene transfer has been demonstrated as the main way of aerolysin-related toxins acquisition in metazoans. However, only few studies have focused on their potential biological functions in such organisms. Herein, we present an extensive characterization of a multigene family encoding aerolysins - named biomphalysin - in Biomphalaria glabrata snail, the intermediate host of the trematode Schistosoma mansoni. Our results highlight that duplication and domestication of an acquired bacterial toxin gene in the snail genome result in the acquisition of a novel and diversified toxin family. Twenty-three biomphalysin genes were identified. All are expressed and exhibited a tissue-specific expression pattern. An in silico structural analysis was performed to highlight the central role played by two distinct domains i) a large lobe involved in the lytic function of these snail toxins which constrained their evolution and ii) a small lobe which is structurally variable between biomphalysin toxins and that matched to various functional domains involved in moiety recognition of targets cells. A functional approach suggests that the repertoire of biomphalysins that bind to pathogens, depends on the type of pathogen encountered. These results underline a neo-and sub-functionalization of the biomphalysin toxins, which have the potential to increase the range of effectors in the snail’s immune arsenal.
A diverse array of toxins widely distributed among organisms has emerged as a key factor involved in virulence or defense factors of host/pathogen interactions (1–6). Accordingly, toxins can be found in a wide variety of organisms from bacteria to metazoans and exhibit a highly specific role on a broad range of cellular pathways, thereby triggering a wide variety of physiological effects such as neurotoxicity or cytolysis/necrosis (7–11). Selective pressures operating in host-pathogen interactions have generated a large repertoire of specific toxins that harbor a distinct selectivity (12–16). Among the most common toxins present in all kingdoms, Pore-Forming Toxins (PFT) are a large class of biological weapons used by prokaryotes as virulence factors and by eukaryotes in defense responses or predation functions (17, 18). Overall, these toxins are secreted as soluble protoxins and undergo a conformational change to form oligomeric pores in membranes subsequent to the interaction with specific receptors located at the membrane surface of targeted cells (19–21). In some cases, proteolytic cleavages are required in order to form the active toxin (22–25). Many studies have highlighted the extensive range of residues recognized by PFTs, which include: i) lipids, ii) sugars or iii) membrane proteins (20, 26–29). However, only a few targets have been identified so far for PFTs. Proteins present on immune cell membranes like CCR and CXR (chemokine receptors) or the C5a receptor, the LPS-induced TNF-α factor (LITAF), the CD59 or HAVCR1 (hepatitis A virus cellular receptor) targeted by leukocidins (30, 31), intermedilysin (32), ε-toxin (33) or hemolysin BL (34) respectively, are well-established.
Among PFTs, several members of the aerolysin family, which derive their name from the bacterium genus Aeromonas, have been identified in many metazoan genomes as inherited by horizontal gene transfer (HGT) (18, 35). In plants, production of cytolytic enterolobin in seeds seems to provide protection from herbivore grazing and/or insect attack (36). In Cnidaria, hydralysin and nylysin-1b are thought to play a role in host defense against predators and for killing prey (35, 37). In vertebrates, Dln1/Aep1 which shows a high affinity for mannose glycans is proposed to be an immune defense molecule for the zebrafish Danio rerio (38, 39) and βγ-CAT protects the frog Bombina maxima against the pathogenic bacterium, Comamonas sp. (40). Interestingly, this aerolysin-like toxin harbors other non-immune functions such as that involved in tissue repair (41), highlighting the diversified functions that these toxins may have. Another aerolysin-related protein, named biomphalysin, characterized in the Schistosomiasis vector snail Biomphalaria glabrata was proposed to have been acquired by HGT (42). Phylogenetic incongruence between aerolysin-related gene trees and species phylogeny revealed that intronless biomphalysin gene clustered with genes from a cnidarian N. vectensis and Gram-negative bacteria (42, 43). When produced recombinantly, this toxin was shown to bind to the membranes of the metazoan parasite Schistosoma mansoni and to participate in its elimination through cytolysis (42). In addition, a genome-wide study revealed that the biomphalysin locus was strongly correlated with resistance to S. mansoni (44) and a transcriptomic analysis indicated that re-exposure to S. mansoni triggered an increased representation of biomphalysin transcripts and protein release in the hemolymph of B. glabrata snails (45).
In this study, we report the expansion of this Biomphalysin family. Interestingly, data from the released genome of B. glabrata provide evidence for the presence of multiple loci coding for biomphalysins in the snail (45, 46). We have focused on the comprehensive characterization of the expansion of this β-PFT biomphalysin family by combining genomic, transcriptomic, structural and functional analyses in B. glabrata. Together, our results provide evidence that these toxin genes were diversified and used as potential weapons in the snail’s immune defense arsenal.
The 23 biomphalysin protein sequences were retrieved from the genome of B. glabrata. To rule out eventual errors in genome assembly, PCRs were performed on snail genomic DNA using specific primer pairs of the predicted coding DNA sequence of each biomphalysin gene (Table S2). The protein sequence (GenBank ACC Number: P09167) corresponding to the crystallized aerolysin from Aeromonas hydrophila was used as outgroup. Sequences were aligned using Clustal Omega from the Seaview software version 5.0.4 (47, 48). The alignment was then subjected to Gblocks program (49) with the less stringent parameters to trim the non-aligned amino acids. Then we used IQ-TREE v1.6.12 (50), an online phylogenetic tool (http://www.iqtree.org/) to determine the best model for a maximum likelihood analysis using the Bayesian information criterion (BIC). The phylogenetic tree was further generated using the Maximum Likelihood method from PhyML v3.0 software (51), with the following parameters (model: WAG/Amino acid equilibrium frequencies: empirical/Proportion of Invariable site: estimated/Gamma distribution parameter: estimated/Number of substitution rate categories: 4/Tree topology search: NNIs/Starting tree: BioNJ). Approximate Likelihood-Ratio Test (aLRT) was carried out to assess the robustness of the branches on the trees inferred from the maximum likelihood method. Values are shown on each branch of the trees generated. Tree is only presented in topology format (Figure 1).
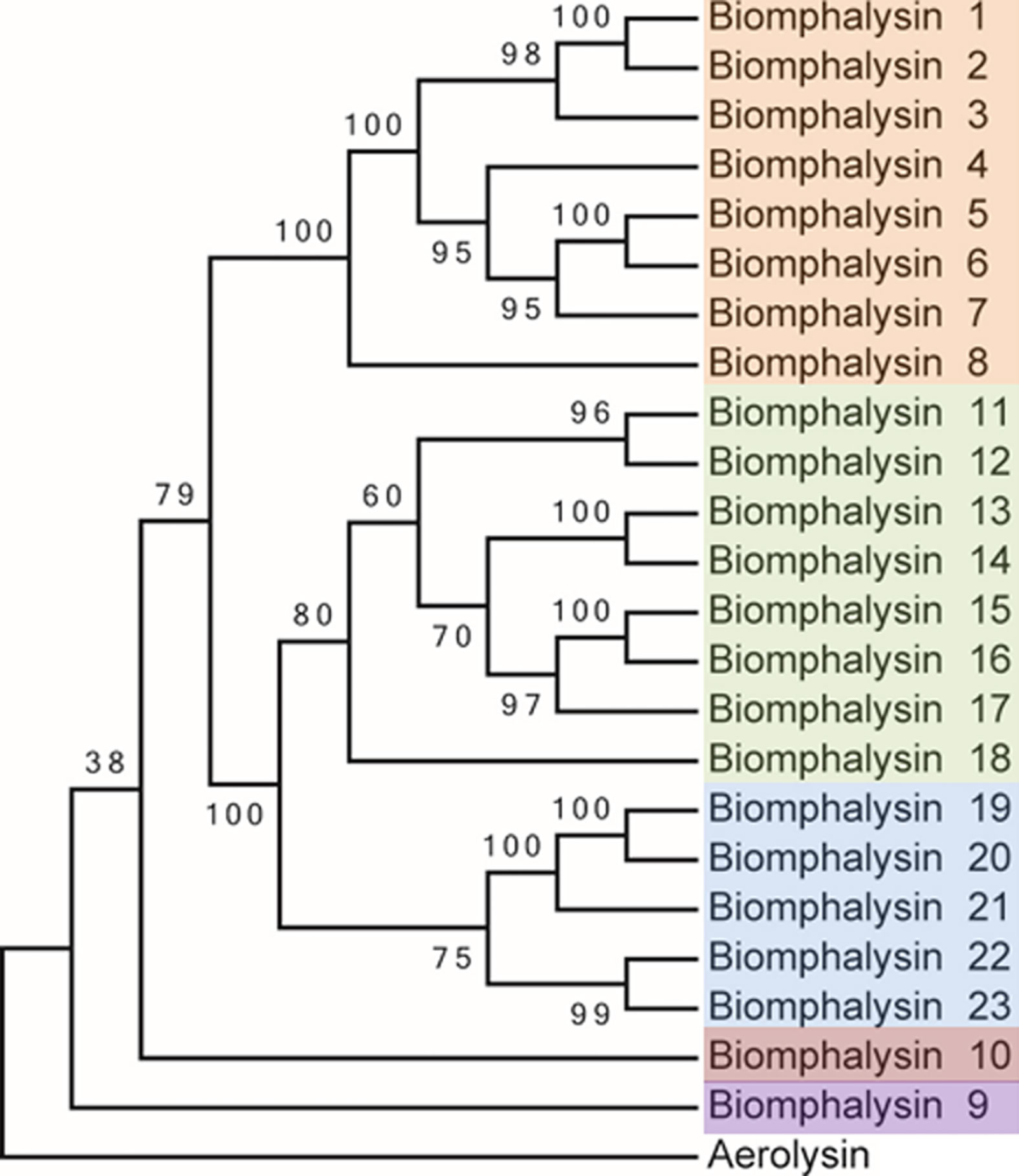
Figure 1 Phylogenetic analysis of the protein sequences from the 23 biomphalysins from Biomphalaria glabrata. The three clusters identified in this analysis are highlighted in orange, green and blue for the clusters I, II and III that contain biomphalysins 1 to 8, 11 to 18 and 19 to 23, respectively. Biomphalysin 9 in purple and 10 in red do not group within biomphalysin clusters. The aerolysin sequence from Aeromonas hydrophila (GenBank P09167) was used as an outgroup. aLRT was carried out on the tree inferred from the Maximum Likelihood method. Ratio test values multiplied by 100 are shown at each branch of the trees. The accession number of each gene and protein used is listed in Table S1.
A second phylogenetic analysis was conducted including the 23 biomphalysin protein sequences together with 52 proteins presenting sequence similarities from other organisms. The aerolysin and MTX2 domains (IPR005830 and IPR004991) were identified in each of the biomphalysin protein sequences by searching against the Conserved Domains Database (http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi) (52). These sequences were used as a query for a BLAST search against the NCBI database to identify similar sequences in other species. A total of 85 sequences were used for the phylogenetic analysis performed as described previously. These included 53 sequences from Gastropoda, 22 from Anthozoa and 10 from bacteria (Figure 2). The accession numbers and corresponding names of each sequence used in phylogenetic analysis are indicated in Table S9 for all phylogenetic trees performed.
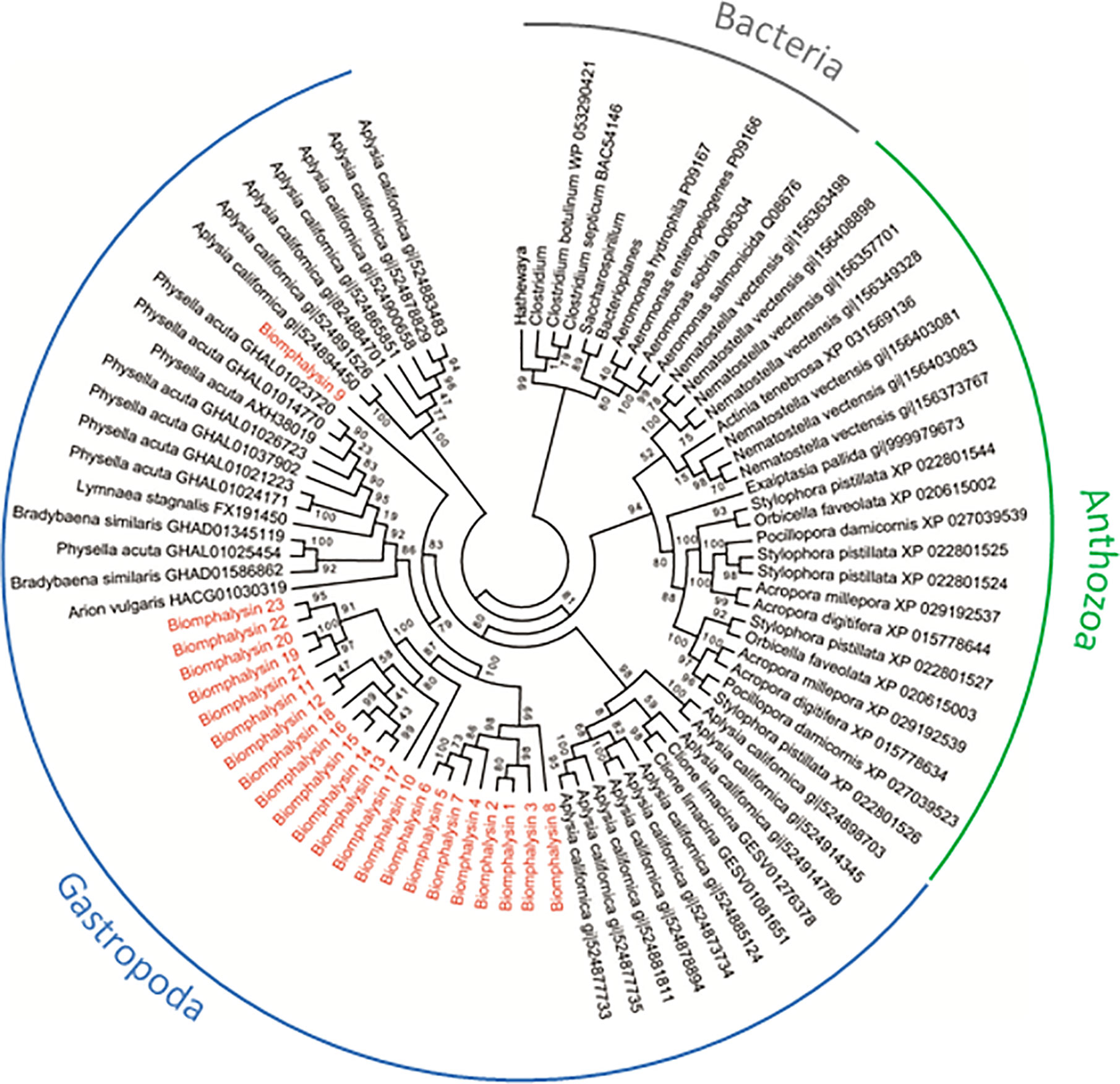
Figure 2 Phylogenetic tree of biomphalysins with other proteins bearing ETX/MTX2 domain. Phylogenetic analysis was conducted from 87 ETX/MTX2 protein domain sequences from biomphalysins and related sequences from other organisms. Protein sequences were aligned using Clustal Omega. Multiple alignment was trimmed using Gblocks, and tree was constructed by Maximum likelihood analysis using PhyML. Values on the branches indicate the robustness of the branches of the tree inferred by ML method. Biomphalysins (red) clustered together with other Gastropoda sequences (blue). Sequences from Anthozoa (green) and Bacteria (gray) formed two other distinct clusters. The accession number of each gene and protein used is listed in Table S9.
Whole genome and transcriptome screening by use of the Interproscan software were performed with the Interproscan database to find IPR005830 and IPR004991 signature characteristic to aerolysin and aerolysin-like toxins. The genome of B. glabrata (preliminary assembly v4.3) and the RNA-seq data from naïve and infected snails were used as data sources (45, 53).
The presence of a signal peptide was determined by a primary structure analysis performed using SignalP 3.0. Protein domain searches were performed using SMART (http://smart.embl-heidelberg.de/) and Motif Scan (http://hits.isb-sib.ch/cgi-bin/PFSCAN) software. α-Helical and β-sheet regions were identified by secondary structure prediction using Jpred3 and HHpred servers (http://toolkit.tuebingen.mpg.de/hhpred). Sequences were searched for putative transmembrane domains (TMDs) using the PRED-TMBB server.
3D Prediction of biomphalysin and their isolated small lobe structures were performed using the I-Tasser V5.1 (https://zhanglab.ccmb.med.umich.edu/I-TASSER/) and TM align https://zhanglab.ccmb.med.umich.edu/TM-align/) servers (54, 55). The 3D structure was obtained by multiple threading using the I-Tasser server, which combines two protein structure prediction methods: threading and ab initio prediction (56). Structural similarities between bacterial aerolysin and biomphalysin proteins were determined by calculating a TM-score (Table S5). A TM-score greater than 0.5 reveals significant alignment, whereas a TM-score less than 0.17 indicates a random similarity.
Multiple structural alignment were performed using the ConSurfWeb from the predicted structure of different biomphalysins. The conservation scores are projected onto the protein sequence of biomphalysin 1 (Figure 5).
Up to 20Kb region surrounding each biomphalysin (when size and position on the scaffold allowed) was screened for the presence of TEs through a similarity search using BLAST with the Biomphalaria specific database of repeat elements as well as using RepeatMasker. We performed a visual inspection for conserved sequences in these 10Kb regions surrounding the biomphalysin genes by producing a dotplot using gepard v. 1.4 (57).
Transcription factor motifs were searched using the 2016 version of the JASPAR (58) database of transcription factors (Table S8) and the Motif Occurrence Detection Suite MOODS v. 1.9.2 (59) by using the provided script (moods_dna.py) (Table S7) and the data was parsed, filtered and summarized by in house developed scripts. The search for conserved domains was carried out by using MEME (60) and searching for motifs between 12 and 50 bases long. Shorter motifs did not produce significant results except for TATA boxes and a poly-C stretch.
High quality reads were recovered from previously published RNA-seq data (45) and they were aligned on nucleic sequences of the 23 biomphalysins. Briefly, two pools of 20 uninfected snails originating from Brazil (BgBRE) were sampled and RNA was extracted from their whole body using Trizol® Reagent (Sigma Aldrich) prior to Illumina sequencing. Best quality reads (Phred >29) from both libraries (uninfected 1 & uninfected 2) were aligned as previously described (45) on each of the 23 biomphalysins using Bowtie2 (v2.2.4) and samtools (v0.1.18), which were run on a local Galaxy Project server. Raw counts were normalized with upper-quartile division per libraries (61).
Seven organs (mantel, head-foot, hemocytes, albumen gland, stomach, hepatopancreas and ovotestis) from 5 individual B. glabrata snails (BgBRE2) (± 8 mm) were dissected and immediately transferred in liquid nitrogen for mRNA total extraction using Norgen Biotek (Ontario, Canada) RNA extraction kit following the manufacturer’s protocol. DNAse treatment (ThermoFischer, USA) was carried on to eliminate potential traces of contaminant genomic DNA. Total RNA (500 ng) was reverse transcribed (RT) using Maxima H Minus First Strand cDNA synthesis kit (ThermoFischer, USA) following the manufacturer’s protocol. Equimolar random hexamer primer and oligo (dT)18 primer were used. Absence of genomic DNA and quality of cDNA synthesis was verified by using myoglobin primers (forward: 5’-GAT GTT CGC CAA TGT TCC C-3’ and reverse: 5’-AGC GAT CAA GTT TCC CCA G-3’) targeting intron/exon junctions. All primer couples targeting biomphalysin genes were designed with perlprimer software (Table S2). Specificity of amplification was validated by Sanger sequencing of amplicons. For tissue-specific expression, PCR amplifications were performed on the 7 organs in biological quintuplicate on cDNAs (diluted 5-fold with nuclease-free water). PCR reactions were performed with GoTaq® G2 Hot Start Polymerase kit (Promega, Madison, USA) and performed on a thermocycler (Eppendorf, Hamburg, Germany). PCR protocol was as follows: 4 min at 95°C for initial polymerase activation then 35 cycles constituted of 20s denaturation at 95°C, 30s annealing at 48°C and 30s extension at 72°C with 5 min at 72°C of final extension. PCR products were separated by electrophoresis on 1.5% agarose gels and sequenced.
Snail (BgBRE2) exposed to Gram-positive, Gram-negative bacteria or yeast were conducted according to previously described procedures (62). Briefly, snails were bathed with 108/mL of each microorganisms for 1 h. Then, snails were washed extensively. For S. mansoni infection, each snail was exposed for 6 h to 10 miracidia from Guadeloupe (SmGH2) in 5 mL of pond water. In this snail/parasite interaction, SmGH2 is totally incompatible with BgBRE2. For each immune challenge, 12 replicates of a pool of three snails were performed at 6, 12, 24 and 48 h after exposition. Non-exposed snails (8 replicates of a pool of three snails) were used as control for the evaluation of the basal expression level of tested biomphalysins.
RNA extractions were performed according to the Trizol™ Reagent procedure (ThermoFisher Scientific, Paris, France). DNase treatment and reverse transcription of RNA (4µg) into cDNA were performed with Maxima H minus first strand cDNA synthesis kit (ThermoFisher Scientific, Waltham, MA, USA) using random hexamer according to the manufacturer’s instructions. Real-time RT-PCR analyses were performed using the LightCycler 480 System (Roche) performed in the same way as described in (63). Only primer couples (Table S2) with amplification efficiency of 2 were retained. The cycling program is as follows: denaturation step at 95°C for 2 min, 40 cycles of amplification (denaturation at 95°C for 10 s, annealing at 60°C for 15 s, and elongation at 72°C for 22 s). QPCR was ended by a melting curve step from 65 to 97°C with a heating rate of 0.11°C/s and continuous fluorescence measurement. For each reaction, the cycle threshold (Ct) was determined using the second derivative method of the LightCycler 480 Software release 1.5 (Roche). A mean value of Ct was calculated. The relative expression of biomphalysins was normalized to the housekeeping gene S19 ribosomal protein gene used as reference gene. Then, normalized expression of biomphalysins was compared with non-exposed snails (used as control) with the ΔΔCt method. The normality of the dataset was tested using Shapiro Wilk test, and significant differences were analyzed by pairwise Mann Whitney U test. Differences were considered significant and robust when p<0.005.
Codon usage were compared between sequences from the 23 biomphalysins, 13 aerolysins/Pertussis Toxin for bacteria and 25 randomly selected B. glabrata predicted transcript (1600-1800 pb) (Table S10). The size of B. glabrata transcripts was chosen to be in the range of biomphalysin genes (1600-1800 pb) to avoid biases due to differences in gene length. Nucleotide sequences were compared using codon usage tools of CAIcal server (http://genomes.urv.es/CAIcal/) (64). Mean codon usage was compared for each triplet between biomphalysin transcripts, Biomphalaria transcripts and bacteria transcripts by PCA (Principal Component Analysis) using ClustVis web tool (http://biit.cs.ut.ee/clustvis/) (65). Local transcriptome of B. glabrata (45), the 23 nucleic sequences of biomphalysins and 13 aerolysin-like sequences were used to calculate the GC content using geecee tools (v5.0.0) on local Galaxy server.
Interactome experiments were conducted to determine which snail plasma proteins are capable of binding to the surface of pathogens. Experimental procedure was previously described (66). Briefly, hemolymph was collected from the head–foot region of unexposed snails. Hemolymph was centrifugated to keep only the free-cell compartment which was incubated during 20 min at 26°C with living organisms such as E. coli, M. luteus, S. cerevisiae, S. mansoni and E. caproni. Then, organisms were washed twice with CBSS (Chernin’s balanced salt solution) and resuspended in a lysate buffer (urea, 7 M; thiourea, 2 M; Tris, 30 mM; CHAPS, 4%; pH 8.5) to extract proteins from organisms but also snail plasma proteins associated with their membrane. Extracted proteins were subjected to 2D electrophoresis and silver staining. Extracted proteins from living organisms exposed to CBSS were used as control. Five independent experiments were performed. Then, a comparative analysis between 2D gels from non-exposed and exposed organisms to snail cell-free hemolymph was performed. Only additional spots present in all the three replicates of “organisms exposed to cell-free hemolymph” samples and absent from all the profiles of pathogens alone were collected and submitted to mass spectrometry analysis for identification. To identify biomphalysin proteins present in these additional spots, the obtained peptides by mass spectrometry analysis (nano LCMS/MS, nano-LC1200 system coupled to a Q-TOF 6550 mass spectrometer) were matched to all translated transcripts encoding the different biomphalysin proteins.
To identify all of the gene encoding analogues of aerolysin toxins in B. glabrata, we conducted an exhaustive sequence search of the genome and transcriptomes of B. glabrata. Using the gene sequence from the previously identified biomphalysin 1 (42), only a single partial sequence of 395 bases (Locus_25218_Transcript_19/20) was identified from a local BLAST search of RNA-seq data from uninfected snails (53). However, a tBLASTn search using the same query and database allowed 10 additional hits to be found, with potential homologies to biomphalysin 1 with an E-value score ranging from 1e-79 to 5e-38. As most of these hits harbored an aerolysin or epsilon toxin signature in their sequence (InterPro accession: IPR005830 and IPR004991, respectively), InterProScan and sequence similarity searches were conducted to investigate the presence of additional aerolysin-like genes in the genome of B. glabrata (preliminary assembly v4.3) (46) and the RNA-seq data from S. mansoni infected snails (45). A total of 23 unique loci were identified on 20 different genomic scaffolds (Table S1), including the biomphalysin gene named biomphalysin 1 as well as others, named from biomphalysin 2 to 23. Interestingly, all of the biomphalysin genes identified were devoid of introns, which represents an evolutionary conserved characteristic of this gene family in B. glabrata.
Several biomphalysins were found in the same scaffold. Biomphalysins 1 & 2, 11 & 22 and 20 & 21 were located in tandem in large scaffolds, scaffold 10 (1 Mb), 2594 (72 kb) and 2201 (88 kb), respectively. This suggests that these gene pairs could result from duplication events. Interestingly, although present on different scaffolds, biomphalysins 5 and 6 exhibited a high sequence homology of the two gene parts located in the 5’ and 3’ regions of the genes, as evidenced by comparative sequence dot blot analyses performed with Gepard (Figure S1). A similar observation was made for biomphalysins 11 and 22 in the 5’ part of the sequence. No other highly conserved regions were detected by Gepard analysis including signature of transposable elements in the 20 Kb vicinity of biomphalysin genes, which was also investigated by using RepeatMasker software and by performing BLAST searches against the repeat database (Dfam and Repbase).
To rule out potential errors in genome sequencing and assembly, each biomphalysin gene was amplified and sequenced by Sanger sequencing. PCRs were performed on snail genomic DNA using specific primer pairs to amplify the predicted coding DNA sequence of each biomphalysin gene (Table S2). The putative translated regions were 1725 bp long on average (1698 -1758 bp). The newly identified biomphalysins exhibited a 46.8 to 94.7% and 33.1 to 95.1% identity as compared to biomphalysin 1 based on the nucleic and deduced amino acid sequences, respectively (Tables S3 and S4). Moreover, protein sequence similarities to aerolysin toxin (Accession number: AAA21938) ranged from 23.6 to 30.2% (Table S4). The lowest identity was found between biomphalysins 9 and 23 (28.7%), while the highest was observed for biomphalysins 1 and 2 (95.1%). Their level of pairwise similarity was also very dissimilar, ranging from 43.1% between biomphalysins 9 and 23 to 97.7% between biomphalysins 1 and 2 (Table S4).
The deduced amino-acid sequences of the 23 biomphalysins were compared by performing a phylogenetic analysis. At least three clusters of biomphalysins could be observed and they were supported by high bootstrap values (Figure 1). The first cluster included biomphalysins 1 to 8, the second biomphalysins 11 to 18 and the third included biomphalysins 19 to 23. Biomphalysins 9 and 10 were isolated from the others and biomphalysin 9 was the closest to the aerolysin sequence of Aeromonas hydrophila used as an outgroup (Figure 1).
Biomphalysin proteins are composed of an average of 575 residues (from 565 to 585 amino acids) and all harbored a predicted N-terminal signal peptide for secretion composed of 17-23 amino acids that adopted an alpha-helical conformation (Figure S2). A structural feature shared by all aerolysin-like toxins is the presence of many β-sheet elements that represent approximately 40% of the total sequence. Most of them are present in the common core, also called aerolysin domain, consisting of five β-strands with an insertion loop corresponding to the pore-forming transmembrane domain (TMD) (Figure S2). This TMD is found in all biomphalysins and is shared by members of the β-PFTs as it plays a crucial role in the formation of amphipathic barrels by oligomerization for membrane insertion (67, 68). This predicted membrane spanning domain flanked by two polar residue regions is found in all biomphalysin sequences (Figure S3A). Moreover, an alternating pattern of hydrophobic and hydrophilic residues located in this beta-hairpin loop is conserved in all biomphalysins, highlighting their ability to perforate the lipid layer of targeted organisms (Figures S3B, C) (69). Interestingly, the amino acids involved in oligomerization and cytolytic activity were highly conserved in all biomphalysin genes (Figure S2). By analogy with site-directed mutagenesis performed on aerolysin proteins, amino acids Asp235 and Cys255 of biomphalysin 1 could fulfill a crucial role in the oligomeric assembly into a ring-like structure (70, 71). While Tryp466 and Tryp468 mutations may modify the ability of the aerolysin toxin to oligomerize (72) Tryp420 and His428 could be involved in binding to membrane target moieties (72, 73). The C terminal part of the protein tends to be divergent between some biomphalysins and could be associated to a differential lytic activation (74).
To determine whether such an expansion and/or diversification of the aerolysin-like genes occurred in other invertebrates, we also searched in several Sequence Databases of NCBI (Nucleotide, Protein, EST). Next, a phylogenetic analysis was performed using sequences of the aerolysin domain of aerolysin-like proteins from two bacteria genera Firmicutes and Gammaproteobacteria and different invertebrates, including the Gastropoda B. glabrata, Aplysia californica, Physella acuta, Bradybaena similaris, Lymnaea stagnalis and Arion vulgaris, and the Anthozoa Nematostella vectensis, Exaiptasia pallida, Pocillopora damicornis, Stylophora pistillata, Acropora millepora, Acropora digitifera, Orbicella faveolata and Actina tenebrosa (Figure 2). Bacterial toxins all clustered together, with two different sub-clusters corresponding to the two bacterial genera used, and they were clearly separated from invertebrate aerolysin-like sequences (Figure 2).
For invertebrate sequences, the phylogenetic analysis segregates aerolysin-like proteins from Gastropoda and Anthozoa. Interestingly, the phylogenetic closeness of species did induce a higher phylogenetic similarity in the sequences of aerolysin-like proteins. Main branches present relatively good support values (ranging from 79 to 100%) (Figure 2). Aerolysin-like proteins from the Gastropoda A. californica were more similar to those from C. Limacina as they both belonged to Opistobranchs class. At the opposite, the five others gastropod species which belong to Panpulmonata clade present higher phylogenetic similarities of their aerolysin-like proteins. On the same way in Anthozoa class, aerolysin-like sequences from hard corals species (Scleractinia) and from soft coral ones (Actinaria) formed two different clusters. Aerolysin-like proteins cluster according to the taxonomic classification of the organism from which they have been identified. This suggests that the multiple toxins of each species could result from a species-specific diversification. However in B. glabrata, biomphalysin 9 was not clustered with the 22 other biomphalysins and instead, positioned at a basal node of all the other aerolysin-related proteins from panpulmonata species, suggesting a markedly different evolutionary process between the biomphalysin genes. Also, aerolysin-like proteins from A. californica are separated into two different clusters for which one is more basal than aerolysin-like proteins from all other invertebrate species used in this study.
GC content and the codon usage are genus-specific signatures and common features used to detect recent horizontal transfer events by comparing the sequences of the gene supposedly acquired by HGT (here, biomphalysins) (42) and gene sequences from the recipient species (here, other genes from B. glabrata). The GC content of biomphalysin genes (41.0%) was similar to B. glabrata transcripts but was significantly lower than the GC% of bacterial toxin nucleic sequences (55.8%, Fisher’s exact test, p-value = 0.0232) (Figure 3A). Concerning the codon composition, most of the variance (78.1%) was explained by the first axis of the principal component analysis (PCA), which opposed bacterial sequences to snail ones, while the second axis which explained 21.9% of variance, separated the biomphalysins from other genes of B. glabrata (Figure 3B). GC% and codon usage of biomphalysin and bacterial toxin genes have distinct signatures and their analysis indicates that biomphalysins are more closely related to endogenous Biomphalaria transcripts than to bacterial toxin genes, which suggests an old domestication of biomphalysin genes by the snail.
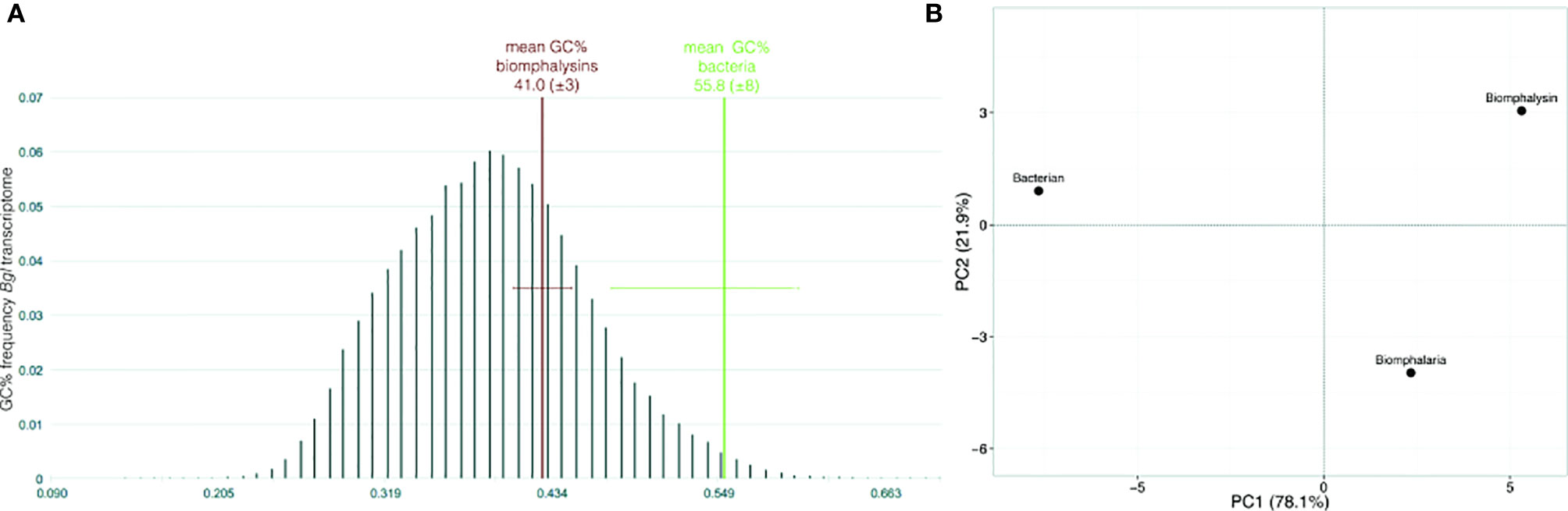
Figure 3 Nucleotide support for domestication. (A) Box-plot representation of GC content and (B) principal component analysis on codon usage comparing nucleotide sequences of biomphalysins (N = 23) with other Biomphalaria glabrata sequences (± 1700 bp; N = 25) and bacterial toxin sequences (N = 13). Distribution of GC% was indicated for Biomphalaria glabrata transcripts while only the mean and standard deviation were indicated for biomphalysin and bacterial toxin sequences.
Despite the apparent divergence and the low sequence homology between the 23 biomphalysins, their structures exhibit high levels of similarity (Figure 4). We previously reported that the anti-parasitic biomphalysin 1 displayed the same structural features as the bacterial aerolysin toxin (42). A structural modeling for each biomphalysin protein was performed using the best alignment template against the Protein Data Bank (PDB) database (http://rcsb.org/) using the I-Tasser server (54, 75). Structural similarities are indicated by a TM score ranging from 0 (totally dissimilar proteins) to 1 (exactly similar structure). All biomphalysins exhibited a structure close to the aerolysin with a TM score ranging from 0.64 to 0.80 (Table S5). The analysis allowed for the prediction of two distinct lobes like for the aerolysin toxin from Aeromonas hydrophila: a small lobe named Domain I and a large lobe constituted of three structural domains (Domain II, III and IV) (Figures S2, 4). Despite differences in the primary structure, highly similar conformations were observed between the different members, with an average pairwise TM score of 0.77 (Table S5). An analysis of the conservation of residue positions among the biomphalysin family was performed with the Consurf software (Figure 5) (76). Conservation scores mapped onto the 3D structure of biomphalysin 1 revealed that most conserved residues are located in Domain II, which is involved in oligomerization (70) and moiety binding in aerolysin (77, 78), and in Domain III, which is necessary for the stabilization of the channel (79) and for toxin insertion into lipid bilayers via the transmembrane α-hairpin loop (67). Conserved residues are strategically located to maintain the highly organized typical topological arrangement of a β-PFT. Indeed, the most conserved residues shape the two α-helices and the different β-strands from Domains II and III (Figures S2, 5). The structure of this large lobe appears to be conserved despite a divergent sequence with an average TM score of 0.93 ± 0.02 (Table S6). Interestingly, many variable amino acids were found in loop regions and particularly in the loop from TMD (Figure 5). Nevertheless, the stretches of alternating hydrophobic and hydrophilic residues, which are key elements for the transmembrane feature, are also conserved (Figure S3B).
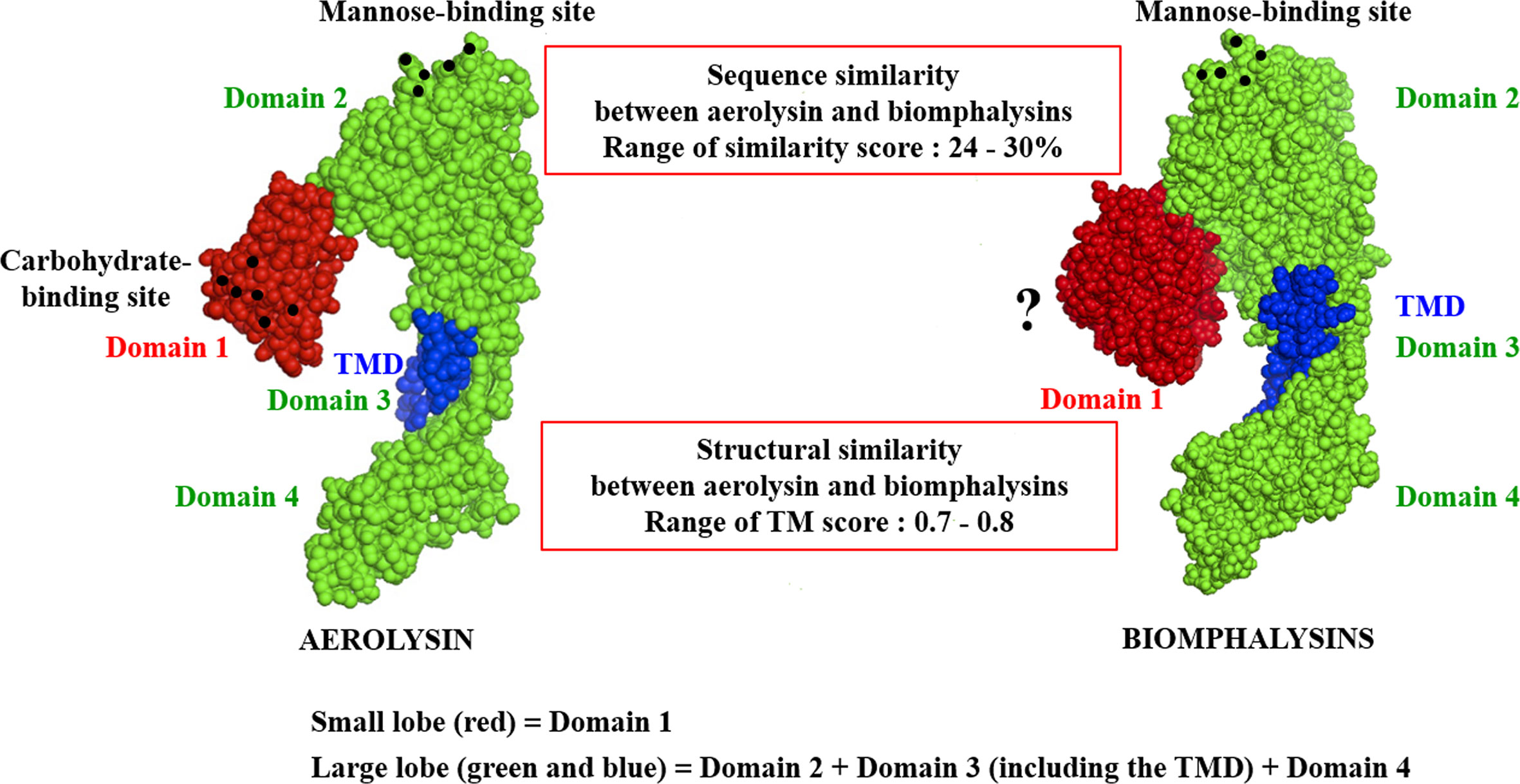
Figure 4 Structural schematic representation of biomphalysins. Images of 3D structure of aerolysin (PDB accession code: 1PRE) and predicted biomphalysin 1 were generated using PyMOL in sphere style. The small domain of both proteins is colored in red, the large domain in green and the TMD in blue. Amino acid residues involved in glycan recognition are symbolized by black spheres. “?” means that no motif has been detected using motif prediction software such as the SMART and MotifScan programs.
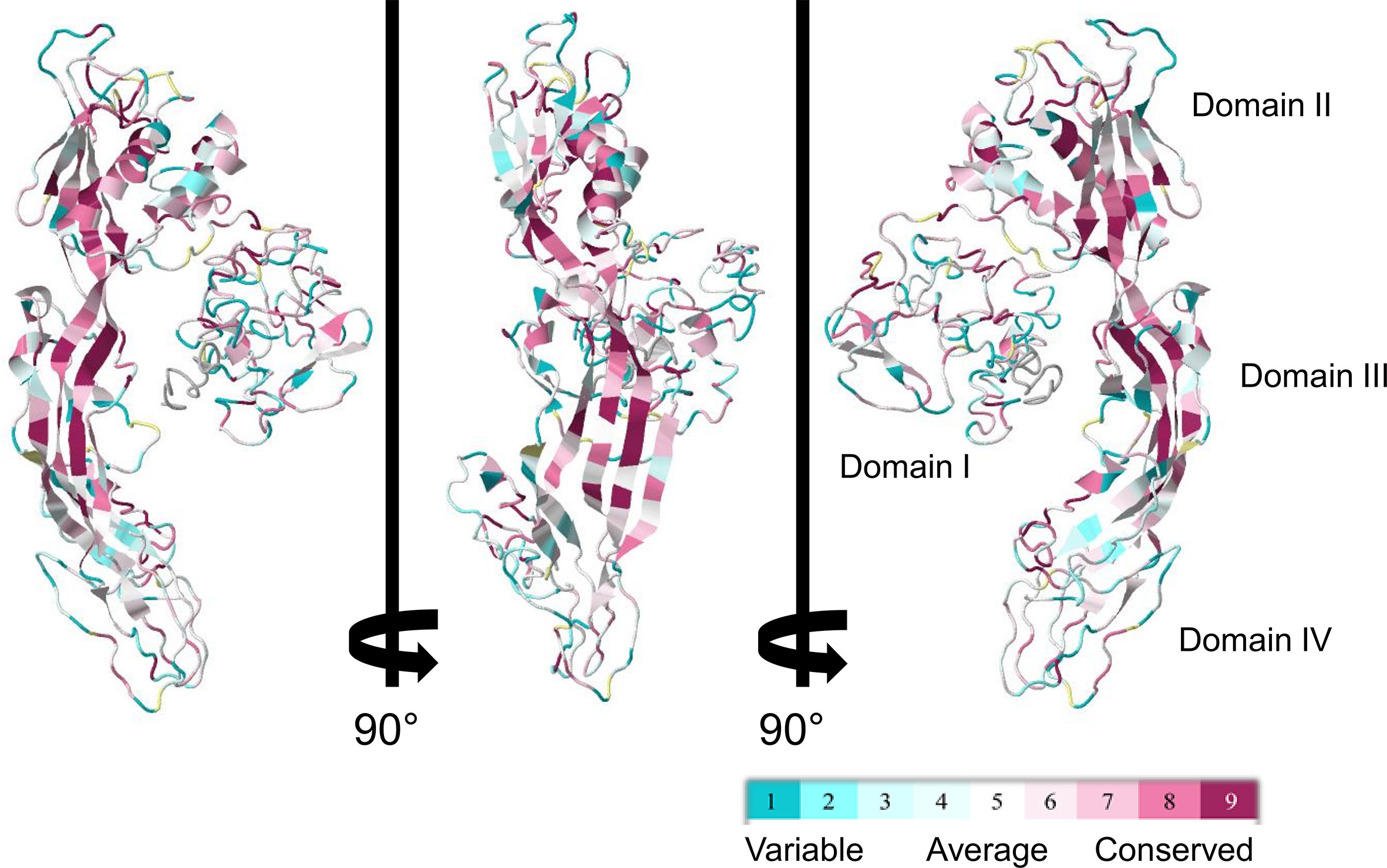
Figure 5 ConSurf sequence conservation score of biomphalysin proteins. ConSurf image was generated using alignment of all 23 biomphalysin proteins and by plotting alignment scores on the predicted structure of biomphalysin 1. Variable residues are shown in cyan while the most conserved ones are in dark red. Images are rotated 90° around the y axis.
Considering that structural information revealed the presence of variable loops mostly located in Domain II, which could contribute to the specificity of interaction with its target, protein-ligand binding sites were predicted based on biomphalysin three-dimensional structures. In addition, a mannose-6-phosphate binding domain was identified in Domain II of all biomphalysins (Figure S4). This carbohydrate binding pocket is located in the vicinity of the two α-helices, a well-characterized site in aerolysin toxin involved in the binding of the mannose residue from glycosyl phosphatidylinositol (GPI)-anchored receptors (78). It contains several key residues for its binding activity, such as arginine, glycine and tryptophan (Figures S2, S4) (68, 80, 81).
The small lobe (Domain I) of the aerolysin toxin presents a binding site similar to the carbohydrate-binding domain of C-type lectins, also called Aerolysin Pertussis Toxin (APT) domain (IPR005138) (26). For all biomphalysin proteins, no lectin motif was found in the amino acid sequence. As most residues were variables in the small lobe of biomphalysin proteins, a similarity structural search was performed using the I-TASSER software against proteins whose tridimensional structures were resolved by crystallography (54). TM scores for the isolated small lobe ranged from 0.45 to 0.87 and were therefore much lower than those of the large lobe (0.89 to 0.97) (Table S6). No significant predictions were obtained for four of the small lobes as their similarity score was below the threshold of 0.5, considered as the limit of prediction significance (54). The analysis of the small lobes of the nineteen other biomphalysins gave relevant structural alignments to five different crystallized proteins or domains of crystallized proteins (Table 1).

Table 1 Best predicted hit for the structure of the small lobes of each biomphalysin.
Despite an overall similar architecture, the small lobe of the different biomphalysins was predicted to fold into different states (Figure 6). For example, the small lobes of the biomphalysin 3 and 20 were respectively similar to a capsid protein and a kinase domain of the PDK1 (phosphoinositide-dependent protein kinase 1), that shaped a protein substrate docking site (82, 83). Eleven of the small lobes matched with a high TM score (higher than 0.6) to members of the complement system, especially complement factor B, which is part of the innate immune system that enhances the immune response against pathogens through their interacting features (84). Interestingly, the best hit of structural prediction for four of the remaining biomphalysins corresponded to a crystal of a protein complex composed of the complement fragment C3b, Factors B and D. In this complex, the small lobes of biomphalysins 6, 12, 15 and 22 are structured in the same way as the domain Ba of Factor B, characterized to interact with the terminal end of C3b, mainly with the MG7 (MacroGlobulin) and the CUB (complement C1r/C1s, Uegf, Bmp1) domains (85). This indicates that the structural diversification of the small lobe might have led to a functional diversification of biomphalysins. This small lobe presents the highest variability of folding units that are independent from the large lobe, leading to a potential broad spectrum of targeted organisms. This unit module could be considered as a diversified moiety-binding domain associated with the aerolysin domain that is involved in the lytic function.
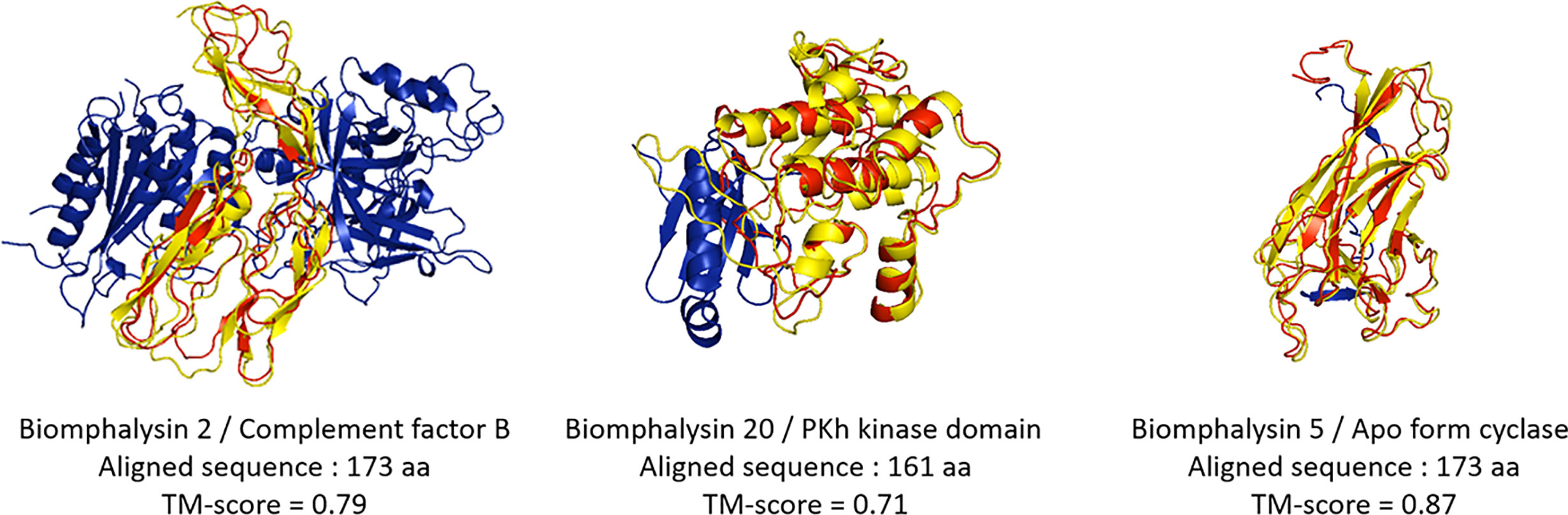
Figure 6 Structural prediction of the biomphalysin small lobes. Three-dimensional structure prediction of small lobe of biomphalysin proteins 2, 5 and 20 was performed using I-Tasser server. A C-score was determined for each prediction (see Table S7). The small lobe of biomphalysin proteins 2, 5 and 20 were investigated against RCSB Protein Data Bank (PDB). A TM-score was calculated between the small lobes and the best hit found by I-Tasser server. A score greater than 0.5 reveals significant alignment. 3D superposition of each small lobe of biomphalysin 2, 5 and 20 and the respective best hit were performed by TM align and visualized with PyMOL. The best structural alignment was reported between the predicted isolated structure of the small lobe in red and the significant template hit identified in yellow.
High quality reads from a previous RNA-seq experiment that included two pools of 20 uninfected snails were used (45). They were aligned with high-stringency to the 23 biomphalysin genes using Bowtie2, counted and normalized by upper-quartile for each library. Every biomphalysin was detected in uninfected snails with different levels of expression (Figure 7A). Biomphalysins 1, 2, 4, 20 and 21 were the most represented whereas biomphalysins 5, 14 and 16 were the least expressed. Transcription of all biomphalysins highlights that none of them are pseudogenes.
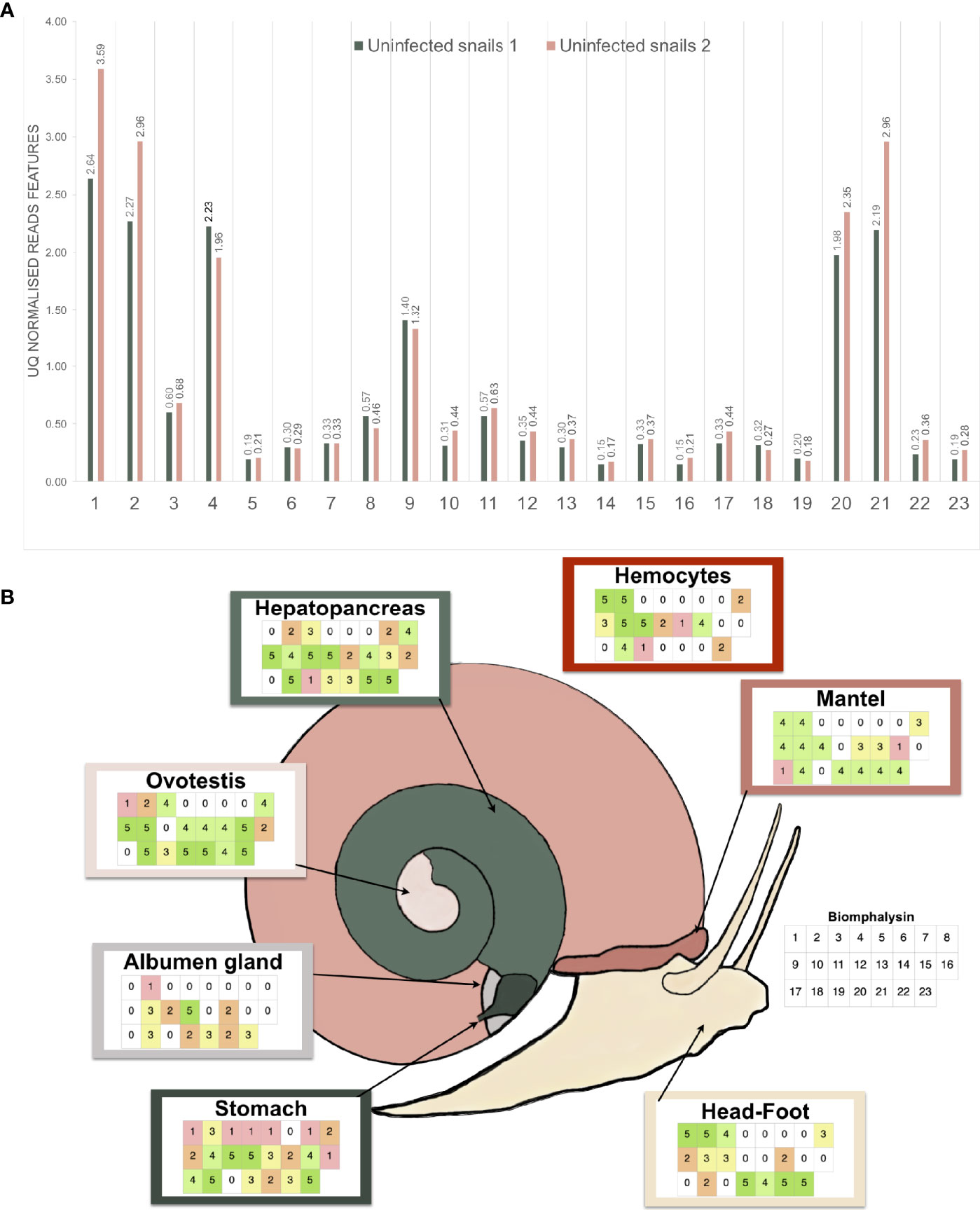
Figure 7 Expression analysis of the biomphalysin family. (A) Constitutive expression of biomphalysins members in whole snails rnaseq dataset (Pinaud et al., 2016). Transcripts abundance for all 23 biomphalysins retrieved from RNAseq experiment performed on two pools of 20 uninfected snails. Libraries from uninfected snails 1 and 2 are highlighted in light-green and pink, respectively. Counts were normalized by upper-quartile (UQ) division for each library. (B) Patterns of expression of biomphalysins in different snail tissues. Each table represents the presence of the 23 biomphalysin transcripts in each tissue by RT-PCR, from biomphalysin 1 in the upper left case to biomphalysin 23 in the lower right case of each table. A code color was used and corresponds to the number of snails for which the tested biomphalysin was detected in a given tissue. As example, an uncolored case indicates that the biomphalysin studied was not expressed in any snail.
To further explore the patterns of expression of the biomphalysin genes, we analyzed their transcript representation by RT-PCR in seven different tissues (stomach, albumen gland, ovotestis, hepatopancreas, hemocytes, mantel, head-foot). This revealed a mosaic pattern of expression of these genes in naïve snails at tissue and individual levels (Figure 7B). While some biomphalysins, such as biomphalysins 2, 10, 14, 18 and 23, were ubiquitously expressed in all tested tissues, others were specifically expressed in some tissues, such as biomphalysin 7, that was restricted to the hepatopancreas and stomach or the biomphalysin 17, which was only located in the mantel and stomach (Figure 7B). Moreover, a high variation in the number of biomphalysins expressed could be observed between the tissues. The highest number of biomphalysins expressed was observed in the stomach, which contained transcripts for 22 out of the 23 biomphalysins, while albumen gland and hemocytes solely displayed 11 and 12 biomphalysins expressed, respectively. Such high variability in tissue expression suggests that the different biomphalysins might have different functions. Many transcriptional factor binding sites were identified in the promoter regions of the biomphalysins but none were found conserved for all the 23 biomphalysins (Tables S7, S8). The diversity of the cis regulatory elements found in the promoter regions suggests that each biomphalysin gene could be regulated spatially and temporally by specific transcription factors, supporting the hypothesis of a functional divergence in this family (Table S8). Consequently, we suggest that each biomphalysin gene could be associated with a functional diversification and sub-functionalization of the snail’s innate immune system to mount an efficient response to different pathogens or to regulate host–commensal bacteria communities or may even be involved in non-immune functions, such as cellular turn-over or nutrition.
To explore the role of biomphalysins in immunity against different pathogens, snails were exposed to either bacteria, yeasts or Schistosoma parasites to investigate the effects of immune challenges on the expression of different members of the biomphalysin family. We focused on the most expressed biomphalysins in uninfected snails (Figure 7A) and measured their relative expression ratio by RT-qPCR in response to an exposure of potential pathogens (Figure 8). Expressions of biomphalysins 2, 4, 20 and 21 are significantly up-regulated at 6h post-microbial exposure. Among the biomphalysins monitored, microorganism-induced expression of biomphalysin 4 is the highest from 3 to 4 fold (p< 0.005). When compared to the control, after 6 hours of exposure, their expression tends to decrease with a significant reduction at 48h post-exposure with E. coli and S. cerevisiae challenges for biomphalysins 2, 20 and 21 (from 2.5 to 7.8 fold). Interestingly, the expression of biomphalysin 4 and 21 are induced significantly 6h after S. mansoni exposure, 2.3- and 4.2-fold respectively. Their level remains elevated compared to the control for up to 48h, with a significance for biomphalysin 4 overexpression (3.3- to 2.6- fold at 24 and 48h, respectively). Unlike to biomphalysin 1 expression which is not modulated by Schistosoma exposure (42), biomphalysins 4 and 21 may play a role in the response to parasite infection by forming a homo- or heteromultimeric pore complex to sustain a lytic response. Taken together, our findings suggest that biomphalysins repertoire may be crucial to mount an efficient and specific immune response against various pathogens.
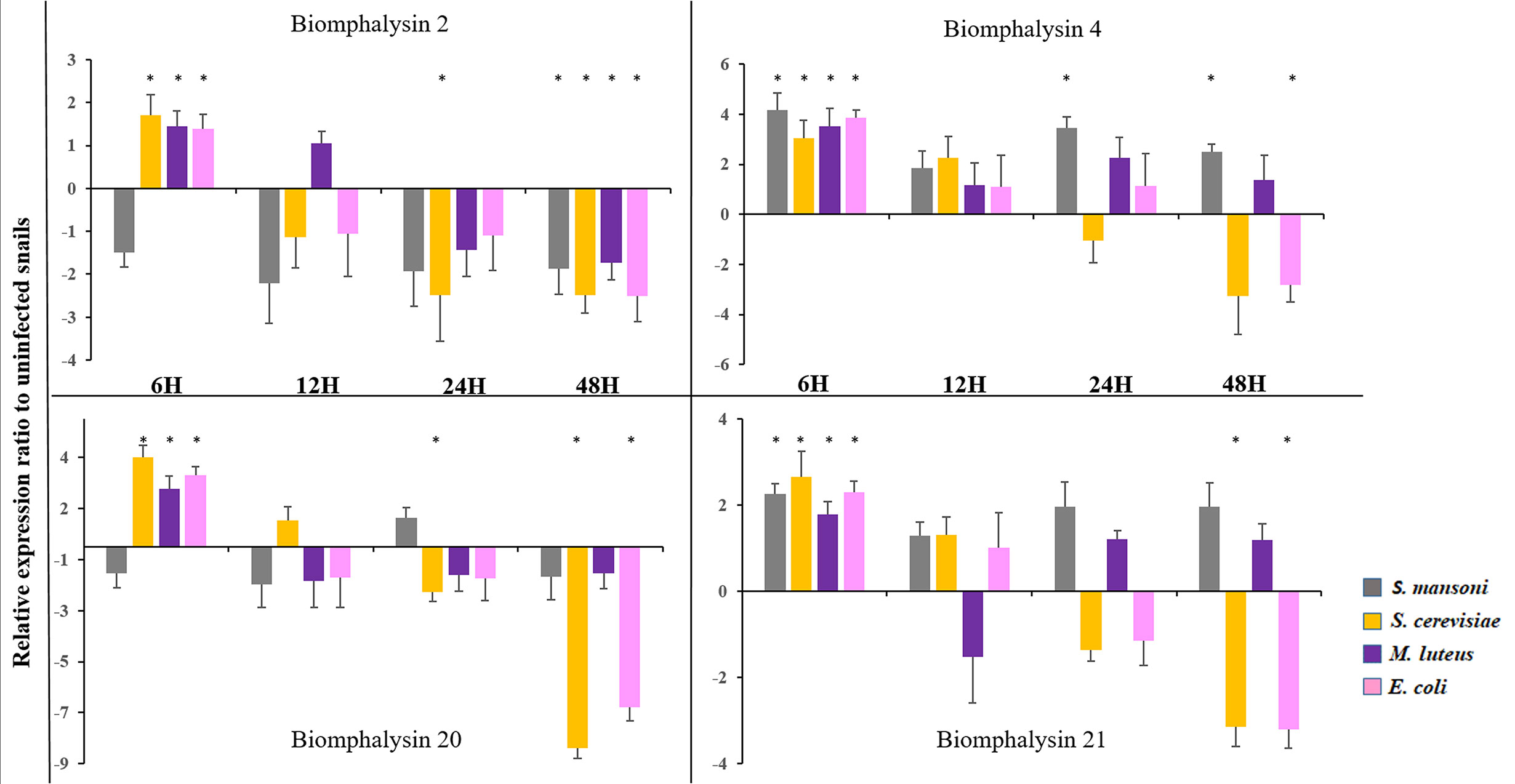
Figure 8 Expression of Biomphalysin 2, 4, 20 and 21 Expression of biomphalysins was monitored 6, 12, 24 and 48 h after immune challenge with Schistosoma mansoni (gray), Saccharomyces cerevisiae (orange), Micrococcus luteus (purple) and Escherichia coli (pink). Expression normalization was done using the S19 housekeeping gene for each experimental point. Data are represented as mean 2- ΔΔCt in challenged snails relative to unexposed snails. Values of 2- ΔΔCt less than 1 were transformed for a better visualization as follow: -1/(2- ΔΔCt). * indicates a significant over- or under-expression in challenged condition compared to control (Mann-Whitney U test, p < 0.005).
As changes in gene expression do not always correlate with changes at the protein level, investigations of the interaction of biomphalysins with pathogen proteins during infection are required. A recent interactome approach was performed by confronting soluble proteins from the snail cell-free hemolymph with outer membrane proteins of living pathogens (66). This approach allowed for the identification of biomphalysin as a pathogen-interacting protein (66). Biomphalysins interact in vivo with gram-negative (Escherichia coli) and gram-positive bacteria (Micrococcus luteus), yeast (Saccharomyces cerevisiae) and two metazoan parasites, Echinostoma caproni and S. mansoni) (66). Using peptide MS/MS spectra recovered from the 2D-gels spots identified as biomphalysin, we sought to identify the biomphalysins that were associated with the different pathogen tested, by mapping them onto the deduced amino acid sequences of the 23 biomphalysins (Table 2). Some peptides were identified in 4 out of 5 interactions, and others were only present in a single interaction. Next, we calculated the number of biomphalysins potentially involved in the interaction with each pathogen tested. E. coli and E. caproni were recognized by at least one biomphalysin, and a minimum of two biomphalysins were required to sense the presence of M. luteus, S. cerevisiae and S. mansoni (Table 2). This is consistent with the fact that three different spots were identified in the gels for these three pathogens; thus, in contrast with what we initially thought, this suggests that two different biomphalysin members - and not different isoforms of the same biomphalysin - are involved in binding to different pathogens (66).
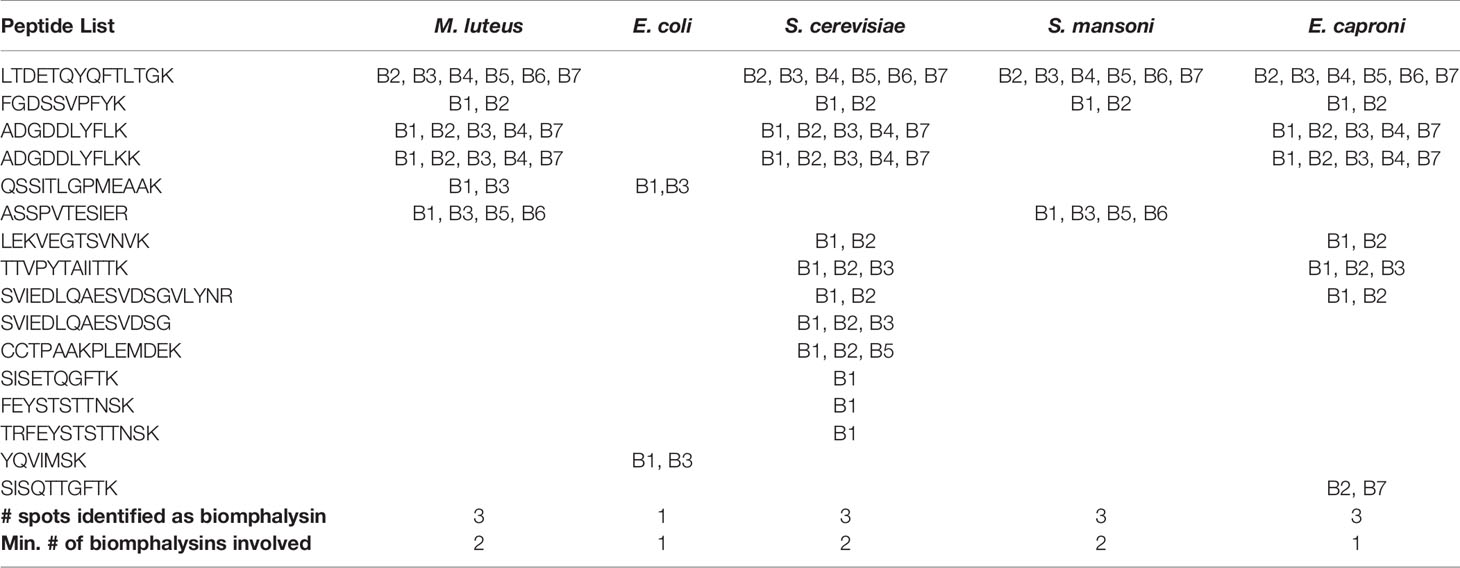
Table 2 Biomphalysin identification revealed by an interactome approach between pathogens and cell-free hemolymph.
In natural environments, Biomphalaria glabrata snails face complex communities of pathogens. It has been extensively demonstrated that B. glabrata are confronted with metazoan parasites (nematodes or trematodes) and play the role of the intermediate vector in the human Schistosoma life cycle (86–89). Albeit rare, crustacean infection has been experimentally demonstrated too (90). Despite being pulmonated, Biomphalaria snails are dependent on wetland ecosystems and must also be confronted with fungal (91) or microbial infections (92). Finally, it has also been demonstrated that B. glabrata may be associated with complex endosymbiotic bacterial microbiota (93–97) or numerous endosymbiotic viruses (98). Since Biomphalaria is one of the main intermediate hosts of Schistosoma, significant efforts have been made to decipher the interaction between the host’s defense mechanisms and the parasite’s infective strategies. Genome sequencing and annotation have made it possible to predict which genes are potentially responsible for the encoding factors involved in snail immune defense (46, 99). Based on comparative approaches between susceptible and resistant snail strains, several candidate genes were proposed as those involved in parasite recognition or elimination (100–104). In addition, small RNA-mediated gene silencing approaches have made substantial advances in functional characterization of key genes involved in host/parasite interaction (45, 105–110). Combined with biochemical approaches such as immunoprecipitation and binding assays, the trimeric protein complex composed of FREPs (Fibrinogen-Related Proteins), TEP (ThioEster-containing Protein) and Biomphalysin appears to be crucial in the recognition and elimination of the parasites (111–113), but these different proteins are not the only pieces in the complex molecular interaction between B. glabrata and S. mansoni (103). Depending on the parasite and snail strains, several other genetic factors have been demonstrated to influence the probability of success in infection, known as compatibility polymorphism (44, 114–117) characterized by a reciprocal arms race between host immune weapons and parasite countermeasures. Concerning the snail’s immune response against other pathogens and intruders, functional data remain scattered despite several transcriptomic approaches conducted to decipher the signaling pathways and to identify key genes (43, 62, 118–120). However, the exposure of snail recombinant proteins or cell-free plasma against some pathogens revealed that some FREPs, biomphalysins, BgTEP1 and LBP/BPI are able to coat the microbial surface suggesting a major role in opsonization and/or in targeted lysis (66, 121–123). Faced with a wide variety of pathogens, the diversification of immune molecules appears to be the evolutionary path considered in invertebrates to mount a specific and effective defense response (99, 124–126). In Biomphalaria, some large repertoires of genes that encode immune receptors such as FREPs, Toll-like receptor (TLR) or TEPs have been reported with more than 30, 50 or 11 genes respectively (46, 63, 99, 127), while the number of effector genes identified may appear more restricted (128).
In this work, we report the characterization of 23 aerolysin-like genes in the snail B. glabrata after the initial discovery of biomphalysin 1 (42). The diversification of this family to 23 members likely occurred through multiple gene duplication events from at least one horizontally transferred ancestral gene. However, the phylogenetic analysis performed on aerolysin-related proteins revealed that biomphalysin sequences are not all distributed in a single monophyletic cluster. Since all biomphalysins except for biomphalysin 9 are distributed in a clade that includes other gastropods, we hypothesize that at least two HGT events potentially occurred without excluding the possibility of a different evolutionary history from a single horizontally acquired ancestral gene, which contributed to a high sequence diversity within the biomphalysin family (Figure 2). Considering the phylogenetic incongruence between biomphalysin 1 and aerolysin-related proteins and that biomphalysin genes are intron-less (42), it was proposed that biomphalysins were acquired from bacteria. This HGT of bacterial toxins was previously reported between species belonging to the different kingdoms (18, 35). The higher similarity in GC content and codon usage between the biomphalysins and other genes from B. glabrata compared to bacterium relatives, further support an ancient transfer followed by host domestication. HGT-derived genes gradually adapt the compositional traits of their host genome (GC content and codon usage) so that they are suitable for the host transcriptional and translational machineries, which lead to the functional integration into the host biological pathways (129, 130). Diversification of aerolysin-like families by gene duplication seems to be a shared feature among all invertebrates including toxins. This species-specific diversity of aerolysin-related toxins observed among different invertebrates strongly highlights that these acquired genes must confer an adaptive advantage for each recipient organism. In cnidaria, hydralysins are suspected to play a major role in protecting against predators or for killing prey (35, 37). In Gastropoda such as Aplysia, Physella or Lymnaea, the presence of toxin-related proteins could be used to escape predators as a defensive mechanism in Physella acuta and Lymnaea stagnalis (131–133). The success for aerolysin-related gene retention by various metazoan organisms could be linked to the absence of a similar preexisting metabolic network in the recipient organisms (134–136). Thus, the acquisition of new functions can easily be integrated into the recipient’s metabolome to provide an immediate benefit (137, 138) since they can be described as self-contained units (35). Aerolysin-related proteins are composed of two structurally independent lobes, with one involved in substrate interaction and the other in lytic activity (Figure 4). Notably, biomphalysin 1 has been shown to directly interact with the surface of Schistosoma sporocysts and its lytic activity was promoted by additional plasmatic factors (42). In the snail, all biomphalysins are expressed and all exhibited a tissue-specific pattern of expression. Some tissues such as the stomach expressed almost all biomphalysin genes while others, like the albumen gland, only expressed half of these genes (Figure 7). Together, this highlights that biomphalysin transcription and/or translation is successfully regulated by the recipient and suggests that each biomphalysin might have suffered a different selection pressure leading to different biological functions in the snail, including the sensing or elimination of pathogenic invaders.
The structure of the large lobe is highly conserved in all biomphalysins, which supports the conservation of their lytic activity (Figure 5). Conversely, structural predictions revealed high divergence between the (isolated) small lobes of biomphalysins and bacterial aerolysin (Table S5). While the small lobe of bacterial aerolysin is a carbohydrate-binding site (APT domain) (26), those of biomphalysins matched to different domains involved in moiety recognition or protein interaction. Interestingly, for about half of biomphalysins, the small lobe is structurally close to the Factor B (Figure 6), a key component of the activate complement of the alternative pathway in vertebrates (139). The interaction between Factor B and the cleaved complement component C3b to form and induce the stability of the C3 convertase is mediated by the Ba domain composed by three N-terminal CCP (Complement Control Protein) modules for which some small lobes of biomphalysins have close structural similarities (85, 140, 141). The CCP module, also known as the sushi domain, is harbored by many proteins involved in cell adhesion, in blood coagulation or in innate immunity (142). For example, human ApoH and Decay-Accelerating Factor (DAF), two CCP-containing proteins, have been shown to interact with pathogens such plasmodium and E. coli, respectively (143, 144). Together, this suggests that ligand-binding specificity of biomphalysins could be related to the structural nature of their small lobes. The interactome results showed that the repertoire of biomphalysins that bind to pathogens depends on the nature of the pathogen considered (66). A recent remarkable work based on recombinant proteins revealed the association between FREPs and BgTEP1 (113), thus confirming the previous results obtained by Mone and collaborators (111). In addition, biomphalysins can interact directly with the surface of S. mansoni without plasmatic cofactors (42) and have been shown to be associated with FREPs and BgTEP1 (113). This may suggest a double recognition of pathogens by at least FREP3 and biomphalysin (s) to induce an efficient lytic response since each protein component considered separately is unable to kill parasites (42, 113). Thus, we speculate that BgTEP1 and 2 might serve as the missing pieces of the puzzle between biomphalysin and FREP3, that are required to induce the lytic-pore formation. The biomphalysin-dependent recognition pathway can be achieved by single or multiple toxins, since tryptic peptides collected and analyzed by mass spectrometry identified in the present or previous study (113) could match to biomphalysins 1, 2, 3, 4 and 7. Since interactions between biomphalysins, C3-like proteins and FREPs are increasingly reported (111–113), their functional association could represent the proto-complement present in protostome invertebrates, and particularly in the Lophotrochozoa phylum. This proto-complement complex defined in a phylogenetic approach based on transcriptomic data from multiple invertebrates contains at least a C3 like-TEP, a serine protease C2/factor B and complement receptors (145, 146), to induce an activated complement cascade on the pathogen surface leading to the non-self opsonization and elimination. Some molecules of this proto-complement have been characterized from several mollusk species like Complement factor C2/B from Bivalvia such as Ruditapes decussates (147) or Sinonovacula constricta (148), C3-like factors from mussels (149) or clams (150), but few studies led to the characterization of the whole system (145, 151). While a diversity of C3-like proteins within the TEP superfamily has been reported (63, 99) as well as a large repertoire of carbohydrate-binding proteins such as FREPs (46, 152) and VIgL domain-containing proteins in B. glabrata (53, 112), no complement factor C2/B-like protein has yet been identified. As the small lobe from several biomphalysins is structurally close to the CCP domain of Factor B, we speculated that biomphalysins could be involved in the activation of the proto-complement system by binding with TEPs.
In conclusion, it appears that biomphalysin acquisition from a proto-aerolysin and expansion underline a neo- and sub-functionalization to supplement the snail immune system’s arsenal. The diverse biomphalysins appear to be key players in the immune repertoires as they harbor two essential functions: one in pathogen recognition and the second in pathogen elimination through their cytolytic activity. Therefore, a systematic molecular investigation on each biomphalysin member is expected to further unravel their specific functions in response to different pathogens.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was reviewed and approved by DRAAF Languedoc-Roussillon (Direction Régionale de l’Alimentation, de l’Agriculture et de la Forêt), Montpellier, France (Authorization #007083).
DD designed the research. SP, GT, RG, and BG substantially participated in conception and improvement of research. GT and RG performed the phylogenetic analysis. DD performed structural predictions. CC performed global analysis on promoter and search on duplication events on whole genome. SP and PP performed gene expression analysis. SP, AP, and GT performed interaction experiments. GT, SP, and DD led the manuscript writing. DL, PP, and ES annotate biomphalysin genes. All authors contributed to the article and approved the submitted version.
This work was funded by ANR AeroSNAIL (number ANR-19-CE11-0016-01). DL received a PhD allocation (2017–2020) granted by the Fondation pour la Recherche Médicale (FRM). ES is supported by a PhD funding from the Region Occitanie (BioResist project) and the University of Perpignan Via Domitia graduate school ED305. This study is set within the framework of the “Laboratoires d’Excellences (LABEX)” TULIP (ANR-10-LABX-41). The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Nathalie Arancibia and Anne Rognon for their technical assistance and Marie Buysse for technical help in molecular biology as Master student. Also, DD thanks Laurent Maveyraud and Jérôme Duval for their helpful comments.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.635131/full#supplementary-material
Supplementary Figure 1 | Dot plot of the different biomphalysins and their surrounding genomic regions. The first row and column represent all the biomphalysins ordered from 1 to 23, followed by the (up to 20 kbp) genomic regions surrounding and including the biomphalysin. Blue circle, conserved structure between biomphalysins 5 and 6. Green circle, similarity region near biomphalysins 11 and 22.
Supplementary Figure 2 | Sequence alignment and predicted topology of biomphalysin proteins. Multiple alignment of the biomphalysin proteins was performed using CLUSTALO. Conserved amino acid residues identified in 75% of all sequences are highlighted with a black box. Amino acids containing similar chemical properties are shown in a gray box. Key amino acids whose roles are described as important for oligomerization and lytic activity are indicated in white on a red background. The positions of secondary structure elements were predicted using the server PROMALS3D. α-helices and β-strands are indicated by “h” and “e”, respectively. The putative peptide signal indicated in brown were predicted on SignalP 4.1 server (153). The Transmembrane Domain boxed with a blue dashed line was predicted using the PRED –TMBB server. The signal peptide is in brown, the small lobe in red and the large lobe containing the aerolysin motif (IPR005830) in green. Structural domains according to the predicted structure of biomphalysin 1 are colored coded with domain 1 in red, domain 2 in green, domain 3 in blue and domain 4 in purple. The predicted residues involved in mannose binding are shown in a yellow box.
Supplementary Figure 3 | TMD characterization in biomphalysin proteins. (A) Multiple alignment of the region surrounding the transmembrane domain of all biomphalysin proteins was performed using the HHpred server. The Transmembrane Domain boxed in blue was predicted using the PRED –TMBB server and confronted to the TMD of aerolysin protein (AFP82959) from Aeromonas hydrophyla. Hydrophylics (Ser and Thr) and Hydrophobics (Val, Ile, Leu and Ala) are shown in red and blue respectively. (B) A hydropathy plot of the predicted TMD from all biomphalysin proteins was done by the method of Kyte and DooLittle (1982). An alternation of hydrophilic and hydrophobic residues was observed for all predicted TMD. For example, TMD sequence of biomphalysin 2 is given in this plot. (C). Structure of the TMD of biomphalysin 2 was predicted by the I Tasser server from the complete sequence of the protein. Close view on TMD was generated by PyMOLWin. Beta sheets represented by arrows forms a beta hairpin similar to the loop region in domain III of aerolysin toxin. All biomphalysins contains a transmembrane beta hairpin required to drive membrane insertion.
Supplementary Figure 4 | Carbohydrate binding site prediction on biomphalysin proteins. (A) Surface representation of biomphalysin16 protein interacting with alpha-D mannose-6-phosphate (PDB: M6P). M6P are shown as spherical structures with gray carbon, phosphate and oxygen atoms. Ribbon representation of biomphalysin 16 was superimposed on a transparent surface map. (B) Close-up of the binding site. Alpha-D mannose-6-phosphate bound to the predicted pocket is located in the domain II of biomphalysin protein. The predicted residues involved in mannose binding are shown.
β-PFT, Beta-Pore Forming Toxin; CDS, Coding DNA Sequence; GPI, Glycosyl Phosphatidyl-Inositol; HGT, Horizontal Gene Transfer; M6P, Mannose-6-Phosphate; PCA, Principal Component Analysis; PDB, Protein Data Bank; TMD, Trans-Membrane Domain.
1. Beceiro A, Tomás M, Bou G. Antimicrobial resistance and virulence: a successful or deleterious association in the bacterial world? Clin Microbiol Rev (2013) 26:185–230. doi: 10.1128/CMR.00059-12
2. Dal Peraro M, van der Goot FG. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol (2016) 14:77–92. doi: 10.1038/nrmicro.2015.3
3. Parisi MG, Parrinello D, Stabili L, Cammarata M. Cnidarian Immunity and the Repertoire of Defense Mechanisms in Anthozoans. Biol (Basel) (2020) 9:283. doi: 10.3390/biology9090283
4. Zhang Y. Why do we study animal toxins? Dongwuxue Yanjiu (2015) 36:183–222. doi: 10.13918/j.issn.2095-8137.2015.4.183
5. Schmitt CK, Meysick KC, O’Brien AD. Bacterial toxins: friends or foes? Emerg Infect Dis (1999) 5:224–34. doi: 10.3201/eid0502.990206
6. Podobnik M, Anderluh G. Pore-forming toxins in Cnidaria. Semin Cell Dev Biol (2017) 72:133–41. doi: 10.1016/j.semcdb.2017.07.026
7. Los FCO, Randis TM, Aroian RV, Ratner AJ. Role of Pore-Forming Toxins in Bacterial Infectious Diseases. Microbiol Mol Biol Rev: MMBR (2013) 77:173–207. doi: 10.1128/MMBR.00052-12
8. Rudkin JK, McLoughlin RM, Preston A, Massey RC. Bacterial toxins: Offensive, defensive, or something else altogether? PloS Pathog (2017) 13:e1006452. doi: 10.1371/journal.ppat.1006452
9. Sachkova MY, Landau M, Surm JM, Macrander J, Singer SA, Reitzel AM, et al. Toxin-like neuropeptides in the sea anemone Nematostella unravel recruitment from the nervous system to venom. Proc Natl Acad Sci USA (2020) 44:27481–92. doi: 10.1101/2020.05.28.121442
10. Sachkova MY, Singer SA, Macrander J, Reitzel AM, Peigneur S, Tytgat J, et al. The Birth and Death of Toxins with Distinct Functions: A Case Study in the Sea Anemone Nematostella. Mol Biol Evol (2019) 36:2001–12. doi: 10.1093/molbev/msz132
11. Otto M. Staphylococcus aureus toxins. Curr Opin Microbiol (2014) 17:32–7. doi: 10.1016/j.mib.2013.11.004
12. Bäumler A, Fang FC. Host specificity of bacterial pathogens. Cold Spring Harb Perspect Med (2013) 3:a010041. doi: 10.1101/cshperspect.a010041
13. Bliven KA, Maurelli AT. Evolution of Bacterial Pathogens Within the Human Host. Microbiol Spectr (2016) 1:10. doi: 10.1128/microbiolspec.VMBF-0017-2015
14. Masri L, Branca A, Sheppard AE, Papkou A, Laehnemann D, Guenther PS, et al. Host-Pathogen Coevolution: The Selective Advantage of Bacillus thuringiensis Virulence and Its Cry Toxin Genes. PloS Biol (2015) 13:e1002169. doi: 10.1371/journal.pbio.1002169
15. Endo H, Tanaka S, Imamura K, Adegawa S, Kikuta S, Sato R. Cry toxin specificities of insect ABCC transporters closely related to lepidopteran ABCC2 transporters. Peptides (2017) 98:86–92. doi: 10.1016/j.peptides.2017.04.003
16. Oliveira D, Borges A, Simões M. Staphylococcus aureus Toxins and Their Molecular Activity in Infectious Diseases. Toxins (Basel) (2018) 10:252. doi: 10.3390/toxins10060252
17. Knapp O, Stiles B, Popoff MR. The Aerolysin Like Toxin Family of cytolitic, Pore Forming Toxins. Open Toxinol J (2010) 3:53–68. doi: 10.2174/1875414701003010053
18. Szczesny P, Iacovache I, Muszewska A, Ginalski K, van der Goot FG, Grynberg M. Extending the Aerolysin Family: From Bacteria to Vertebrates. PloS One (2011) 6:e20349. doi: 10.1371/journal.pone.0020349
19. Iacovache I, van der Goot FG, Pernot L. Pore formation: An ancient yet complex form of attack. Biochim Biophys Acta (BBA) Biomembranes (2008) 1778:1611–23. doi: 10.1016/j.bbamem.2008.01.026
20. Cirauqui N, Abriata LA, van der Goot FG, Dal Peraro M. Structural, physicochemical and dynamic features conserved within the aerolysin pore-forming toxin family. Sci Rep (2017) 7:13932. doi: 10.1038/s41598-017-13714-4
21. Iacovache I, De Carlo S, Cirauqui N, Dal Peraro M, van der Goot FG, Zuber B. Cryo-EM structure of aerolysin variants reveals a novel protein fold and the pore-formation process. Nat Commun (2016) 7:12062. doi: 10.1038/ncomms12062
22. Howard SP, Buckley JT. Activation of the hole-forming toxin aerolysin by extracellular processing. J Bacteriol (1985) 163:336–40. doi: 10.1128/JB.163.1.336-340.1985
23. Nonaka S, Salim E, Kamiya K, Hori A, Nainu F, Asri RM, et al. Molecular and Functional Analysis of Pore-Forming Toxin Monalysin From Entomopathogenic Bacterium Pseudomonas entomophila. Front Immunol (2020) 11:520. doi: 10.3389/fimmu.2020.00520
24. Opota O, Vallet-Gély I, Vincentelli R, Kellenberger C, Iacovache I, Gonzalez MR, et al. Monalysin, a novel ß-pore-forming toxin from the Drosophila pathogen Pseudomonas entomophila, contributes to host intestinal damage and lethality. PloS Pathog (2011) 7:e1002259. doi: 10.1371/journal.ppat.1002259
25. Abrami L, Fivaz M, Decroly E, Seidah NG, Jean F, Thomas G, et al. The pore-forming toxin proaerolysin is activated by furin. J Biol Chem (1998) 273:32656–61. doi: 10.1074/jbc.273.49.32656
26. Rossjohn J, Buckley JT, Hazes B, Murzin AG, Read RJ, Parker MW. Aerolysin and pertussis toxin share a common receptor-binding domain. EMBO J (1997) 16:3426–34. doi: 10.1093/emboj/16.12.3426
27. Abrami L, Fivaz M, van Der Goot FG. Adventures of a pore-forming toxin at the target cell surface. Trends Microbiol (2000) 8:168–72. doi: 10.1016/S0966-842X(00)01722-4
28. Soto C, Del Valle A, Valiente PA, Ros U, Lanio ME, Hernández AM, et al. Differential binding and activity of the pore-forming toxin sticholysin II in model membranes containing diverse ceramide-derived lipids. Biochimie (2017) 138:20–31. doi: 10.1016/j.biochi.2017.04.003
29. De S, Bubnys A, Alonzo F,3, Hyun J, Lary JW, Cole JL, et al. The Relationship between Glycan Binding and Direct Membrane Interactions in Vibrio cholerae Cytolysin, a Channel-forming Toxin. J Biol Chem (2015) 290:28402–15. doi: 10.1074/jbc.M115.675967
30. Tam K, Schultz M, Reyes-Robles T, Vanwalscappel B, Horton J, Alonzo F,3, et al. Staphylococcus aureus Leukocidin LukED and HIV-1 gp120 Target Different Sequence Determinants on CCR5. MBio (2016) 7:e02024–16. doi: 10.1128/mBio.02024-16
31. Haapasalo K, Wollman AJM, de Haas CJC, van Kessel KPM, van Strijp JAG, Leake MC. Staphylococcus aureus toxin LukSF dissociates from its membrane receptor target to enable renewed ligand sequestration. FASEB J (2019) 33:3807–24. doi: 10.1096/fj.201801910R
32. Johnson S, Brooks NJ, Smith RA, Lea SM, Bubeck D. Structural basis for recognition of the pore-forming toxin intermedilysin by human complement receptor CD59. Cell Rep (2013) 3:1369–77. doi: 10.1016/j.celrep.2013.04.029
33. Ivie SE, McClain MS. Identification of amino acids important for binding of Clostridium perfringens epsilon toxin to host cells and to HAVCR1. Biochemistry (2012) 51:7588–95. doi: 10.1021/bi300690a
34. Liu J, Zuo Z, Sastalla I, Liu C, Jang JY, Sekine Y, et al. Sequential CRISPR-Based Screens Identify LITAF and CDIP1 as the Bacillus cereus Hemolysin BL Toxin Host Receptors. Cell Host Microbe (2020) 28:402–10. doi: 10.1016/j.chom.2020.05.012
35. Moran Y, Fredman D, Szczesny P, Grynberg M, Technau U. Recurrent horizontal transfer of bacterial toxin genes to eukaryotes. Mol Biol Evol (2012) 29:2223–30. doi: 10.1093/molbev/mss089
36. Castro-Faria-Neto HC, Martins MA, Bozza PT, Perez SA, Correa-Da-Silva AC, Lima MC, et al. Pro-inflammatory activity of enterolobin: a haemolytic protein purified from seeds of the Brazilian tree Enterolobium contortisiliquum. Toxicon (1991) 29:1143–50. doi: 10.1016/0041-0101(91)90211-9
37. Sher D, Fishman Y, Zhang M, Lebendiker M, Gaathon A, Mancheno JM, et al. Hydralysins, a new category of beta-pore-forming toxins in cnidaria. J Biol Chem (2005) 280:22847–55. doi: 10.1074/jbc.M503242200
38. Chen LL, Xie J, Cao DD, Jia N, Li YJ, Sun H, et al. The pore-forming protein Aep1 is an innate immune molecule that prevents zebrafish from bacterial infection. Dev Comp Immunol (2018) 82:49–54. doi: 10.1016/j.dci.2018.01.003
39. Jia N, Liu N, Cheng W, Jiang YL, Sun H, Chen LL, et al. Structural basis for receptor recognition and pore formation of a zebrafish aerolysin-like protein. EMBO Rep (2016) 17:235–48. doi: 10.15252/embr.201540851
40. Xiang Y, Yan C, Guo X, Zhou K, Li S, Gao Q, et al. Host-derived, pore-forming toxin-like protein and trefoil factor complex protects the host against microbial infection. Proc Natl Acad Sci USA (2014) 111:6702–7. doi: 10.1073/pnas.1321317111
41. Gao ZH, Deng CJ, Xie YY, Guo XL, Wang QQ, Liu LZ, et al. Pore-forming toxin-like protein complex expressed by frog promotes tissue repair. FASEB J (2019) 33:782–95. doi: 10.1096/fj.201800087R
42. Galinier R, Portela J, Mone Y, Allienne JF, Henri H, Delbecq S, et al. Biomphalysin, a new beta pore-forming toxin involved in Biomphalaria glabrata immune defense against Schistosoma mansoni. PloS Pathog (2013) 9:e1003216. doi: 10.1371/journal.ppat.1003216
43. Lassalle D, Tetreau G, Pinaud S, Galinier R, Crickmore N, Gourbal B, et al. Glabralysins, Potential New beta-Pore-Forming Toxin Family Members from the Schistosomiasis Vector Snail Biomphalaria glabrata. Genes (Basel) (2020) 11:65. doi: 10.3390/genes11010065
44. Tennessen JA, Bonner KM, Bollmann SR, Johnstun JA, Yeh JY, Marine M, et al. Genome-Wide Scan and Test of Candidate Genes in the Snail Biomphalaria glabrata Reveal New Locus Influencing Resistance to Schistosoma mansoni. PloS Negl Trop Dis (2015) 9:e0004077. doi: 10.1371/journal.pntd.0004077
45. Pinaud S, Portela J, Duval D, Nowacki FC, Olive M-A, Allienne J-F, et al. A Shift from Cellular to Humoral Responses Contributes to Innate Immune Memory in the Vector Snail Biomphalaria glabrata. PloS Pathog (2016) 12:e1005361. doi: 10.1371/journal.ppat.1005361
46. Adema CM, Hillier LW, Jones CS, Loker ES, Knight M, Minx P, et al. Whole genome analysis of a schistosomiasis-transmitting freshwater snail. Nat Commun (2017) 8:15451. doi: 10.1038/ncomms16153
47. Gouy M, Guindon S, Gascuel O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol (2010) 27(2):221–4. doi: 10.1093/molbev/msp259
48. Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci (1996) 12:543–8. doi: 10.1093/bioinformatics/12.6.543
49. Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol (2000) 17:540–52. doi: 10.1093/oxfordjournals.molbev.a026334
50. Trifinopoulos J, Nguyen LT, von Haeseler A, Minh BQ. W-IQ-TREE: a fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res (2016) 44:W232–235. doi: 10.1093/nar/gkw256
51. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol (2010) 59:307–21. doi: 10.1093/sysbio/syq010
52. Marchler-Bauer A, Lu S, Anderson JB, Chitsaz F, Derbyshire MK, DeWeese-Scott C, et al. CDD: a Conserved Domain Database for the functional annotation of proteins. Nucleic Acids Res (2011) 39:D225–9. doi: 10.1093/nar/gkq1189
53. Dheilly NM, Duval D, Mouahid G, Emans R, Allienne JF, Galinier R, et al. A family of variable immunoglobulin and lectin domain containing molecules in the snail Biomphalaria glabrata. Dev Comp Immunol (2015) 48:234–43. doi: 10.1016/j.dci.2014.10.009
54. Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf (2008) 9:40. doi: 10.1186/1471-2105-9-40
55. Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res (2005) 33:2302–9. doi: 10.1093/nar/gki524
56. Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc (2010) 5:725–38. doi: 10.1038/nprot.2010.5
57. Krumsiek J, Arnold R, Rattei T. Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics (2007) 23:1026–8. doi: 10.1093/bioinformatics/btm039
58. Mathelier A, Fornes O, Arenillas DJ, Chen CY, Denay G, Lee J, et al. JASPAR 2016: a major expansion and update of the open-access database of transcription factor binding profiles. Nucleic Acids Res (2016) 44:D110–115. doi: 10.1093/nar/gkv1176
59. Korhonen J, Martinmaki P, Pizzi C, Rastas P, Ukkonen E. MOODS: fast search for position weight matrix matches in DNA sequences. Bioinformatics (2009) 25:3181–2. doi: 10.1093/bioinformatics/btp554
60. Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, et al. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res (2009) 37:W202–208. doi: 10.1093/nar/gkp335
61. Dillies MA, Rau A, Aubert J, Hennequet-Antier C, Jeanmougin M, Servant N, et al. A comprehensive evaluation of normalization methods for Illumina high-throughput RNA sequencing data analysis. Brief Bioinform (2013) 14:671–83. doi: 10.1093/bib/bbs046
62. Deleury E, Dubreuil G, Elangovan N, Wajnberg E, Reichhart J-M, Gourbal B, et al. Specific versus Non-Specific Immune Responses in an Invertebrate Species Evidenced by a Comparative de novo Sequencing Study. PloS One (2012) 7:e32512. doi: 10.1371/journal.pone.0032512
63. Duval D, Pichon R, Lassalle D, Laffitte M, Gourbal B, Galinier R. A New Assessment of Thioester-Containing Proteins Diversity of the Freshwater Snail Biomphalaria glabrata. Genes (Basel) (2020) 11:69. doi: 10.3390/genes11010069
64. Puigbo P, Bravo IG, Garcia-Vallve S. E-CAI: a novel server to estimate an expected value of Codon Adaptation Index (eCAI). BMC Bioinf (2008) 9:65. doi: 10.1186/1471-2105-9-65
65. Metsalu T, Vilo J. ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res (2015) 43:W566–570. doi: 10.1093/nar/gkv468
66. Tetreau G, Pinaud S, Portet A, Galinier R, Gourbal B, Duval D. Specific pathogen recognition by multiple innate immune sensors in an invertebrate. Front Immunol (2017) 8:1249. doi: 10.3389/fimmu.2017.01249
67. Iacovache I, Paumard P, Scheib H, Lesieur C, Sakai N, Matile S, et al. A rivet model for channel formation by aerolysin-like pore-forming toxins. EMBO J (2006) 25:457–66. doi: 10.1038/sj.emboj.7600959
68. Degiacomi MT, Iacovache I, Pernot L, Chami M, Kudryashev M, Stahlberg H, et al. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat Chem Biol (2013) 9:623–9. doi: 10.1038/nchembio.1312
69. Rai AK, Chattopadhyay K. Revisiting the membrane interaction mechanism of a membrane-damaging beta-barrel pore-forming toxin Vibrio cholerae cytolysin. Mol Microbiol (2015) 97:1051–62. doi: 10.1111/mmi.13084
70. Buckley JT, Wilmsen HU, Lesieur C, Schulze A, Pattus F, Parker MW, et al. Protonation of Histidine- 132 Promotes Oligomerization of the Channel-Forming Toxin Aerolysin. Biochemistry (1995) 50:16450–5. doi: 10.1021/bi00050a028
71. Rossjohn J, Feil SC, McKinstry WJ, Tsernoglou D, Van Der Goot G, Buckley JT, et al. Aerolysin - A paradigm for membrane insertion of beta-sheet protein toxins? J Struct Biol (1998) 121:92–100. doi: 10.1006/jsbi.1997.3947
72. Van der Goot FG, Ausio J, Wong KR, Pattus F, Buckley JT. Dimerization stabilizes the pore-forming toxin aerolysin in solution. J Biol Chem (1993) 268:18272–9. doi: 10.1016/S0021-9258(17)46840-2
73. Green MJ, Buckley JT. Site-Directed Mutagenesis of the Hole-Forming Toxin Aerolysin: Studies on the Roles of Histidines in Receptor Binding and Oligomerization of the Monomer. Biochemistry (1990) 29:2177–80. doi: 10.1021/bi00460a031
74. Zhang D, Pridgeon JW, Klesius PH. Expression and activity of recombinant proaerolysin derived from Aeromonas hydrophila cultured from diseased channel catfish. Vet Microbiol (2013) 165:478–82. doi: 10.1016/j.vetmic.2013.04.023
75. Rose PW, Prlic A, Altunkaya A, Bi C, Bradley AR, Christie CH, et al. The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res (2017) 45:D271–81. doi: 10.1093/nar/gkw1000
76. Ashkenazy H, Abadi S, Martz E, Chay O, Mayrose I, Pupko T, et al. ConSurf 2016: an improved methodology to estimate and visualize evolutionary conservation in macromolecules. Nucleic Acids Res (2016) 44:W344–350. doi: 10.1093/nar/gkw408
77. MacKenzie CR, Hirama T, Buckley JT. Analysis of Receptor Binding by the Channel-forming Toxin Aerolysin Using Surface Plasmon Resonance. J Biol Chem (1999) 274:22604–9. doi: 10.1074/jbc.274.32.22604
78. Abrami L, Velluz MC, Hong Y, Oshishi K, Mehlert A, Ferguson M, et al. The glycan core of GPI-anchored proteins modulates aerolysin binding but is not sufficient: the polypeptide moiety is required for the toxin receptor interaction. FEBS Lett (2002) 512:249–54. doi: 10.1016/S0014-5793(02)02274-3
79. Rossjohn J, Raja SM, Nelson KL, Feil SC, van der Goot FG, Parker MW, et al. Movement of a Loop in Domain 3 of Aerolysin Is Required for Channel Formation. Biochemistry (1998) 37:741–6. doi: 10.1021/bi9721039
80. Zelent B, Odili S, Buettger C, Shiota C, Grimsby J, Taub R, et al. Sugar binding to recombinant wild-type and mutant glucokinase monitored by kinetic measurement and tryptophan fluorescence. Biochem J (2008) 413:269–80. doi: 10.1042/BJ20071718
81. Antonik PM, Volkov AN, Broder UN, Re DL, van Nuland NA, Crowley PB. Anomer-Specific Recognition and Dynamics in a Fucose-Binding Lectin. Biochemistry (2016) 55:1195–203. doi: 10.1021/acs.biochem.5b01212
82. Xu X, Chen Y, Fu Q, Ni D, Zhang J, Li X, et al. The chemical diversity and structure-based discovery of allosteric modulators for the PIF-pocket of protein kinase PDK1. J Enzyme Inhib Med Chem (2019) 34:361–74. doi: 10.1080/14756366.2018.1553167
83. Najafov A, Shpiro N, Alessi DR. Akt is efficiently activated by PIF-pocket- and PtdIns(3,4,5)P3-dependent mechanisms leading to resistance to PDK1 inhibitors. Biochem J (2012) 448:285–95. doi: 10.1042/BJ20121287
84. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement System Part II: Role in Immunity. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257
85. Forneris F, Ricklin D, Wu J, Tzekou A, Wallace RS, Lambris JD, et al. Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science (2010) 330:1816–20. doi: 10.1126/science.1195821
86. Guerino LR, Carvalho JF, Magalhães LA, Zanotti-Magalhães EM. Susceptibility of Biomphalaria glabrata submitted to concomitant infection with Angiostrongylus costaricensis and Schistosoma mansoni. Braz J Biol (2017) 77:451–8. doi: 10.1590/1519-6984.15215
87. Fernández MV, Hamann MI. Larval digenean communities in Biomphalaria species in two contrasting wetlands from South America. Dis Aquat Organ (2018) 130:159–64. doi: 10.3354/dao03256
88. dos Santos Bonfim TC, Maldonado A Jr, Tunholi VM, Tunholi-Alves VM, Faro MJ, Mota EM, et al. Biochemical and histopathological alterations in Biomphalaria glabrata due to co-infection by Angiostrongylus cantonensis and Echinostoma paraensei. J Invertebr Pathol (2014) 115:80–5. doi: 10.1016/j.jip.2013.10.005
89. Zanardi VS, Barbosa LM, Simões FM, Thiengo SC, Blanton RE, Ribeiro Junior G, et al. Prevalence of Infection of Biomphalaria glabrata by Schistosoma mansoni and the risk of urban Schistosomiasis mansoni in Salvador, Bahia, Brazil. Rev Soc Bras Med Trop (2019) 52:e20190171. doi: 10.1590/0037-8682-0171-2019
90. Sullivan JT, Yeung JT. Tissue invasion of laboratory-reared Biomphalaria glabrata by a harpacticoid copepod. J Invertebr Pathol (2011) 107:159–60. doi: 10.1016/j.jip.2011.03.002
91. Duarte GF, Rodrigues J, Fernandes ÉK, Humber RA, Luz C. New insights into the amphibious life of Biomphalaria glabrata and susceptibility of its egg masses to fungal infection. J Invertebr Pathol (2015) 125:31–6. doi: 10.1016/j.jip.2014.12.013
92. Duval D, Galinier R, Mouahid G, Toulza E, Allienne JF, Portela J, et al. A novel bacterial pathogen of Biomphalaria glabrata: a potential weapon for schistosomiasis control? PloS Negl Trop Dis (2015) 9:e0003489. doi: 10.1371/journal.pntd.0003489
93. Allan ERO, Tennessen JA, Sharpton TJ, Blouin MS. Allelic Variation in a Single Genomic Region Alters the Microbiome of the Snail Biomphalaria glabrata. J Hered (2018) 109:604–9. doi: 10.1093/jhered/esy014
94. Silva TM, Melo ES, Lopes AC, Veras DL, Duarte CR, Alves LC, et al. Characterization of the bacterial microbiota of Biomphalaria glabrata (Say, 1818) (Mollusca: Gastropoda) from Brazil. Lett Appl Microbiol (2013) 57:19–25. doi: 10.1111/lam.12068
95. Huot C, Clerissi C, Gourbal B, Galinier R, Duval D, Toulza E. Schistosomiasis Vector Snails and Their Microbiota Display a Phylosymbiosis Pattern. Front Microbiol (2019) 10:3092. doi: 10.3389/fmicb.2019.03092
96. Chevalier FD, Diaz R, McDew-White M, Anderson TJC, Le Clec’h W. The hemolymph of Biomphalaria snail vectors of schistosomiasis supports a diverse microbiome. Environ Microbiol (2020) 12:5450–66. doi: 10.1101/2020.04.22.056168
97. Osório JB, de Mattos Pereira L, Giongo A, Marconatto L, Potriquet J, Candido RRF, et al. Mollusk microbiota shift during Angiostrongylus cantonensis infection in the freshwater snail Biomphalaria glabrata and the terrestrial slug Phillocaulis soleiformis. Parasitol Res (2020) 119:2495–503. doi: 10.1007/s00436-020-06743-y
98. Galinier R, Tetreau G, Portet A, Pinaud S, Duval D, Gourbal B. First characterization of viruses from freshwater snails of the genus Biomphalaria, the intermediate host of the parasite Schistosoma mansoni. Acta Trop (2017) 167:196–203. doi: 10.1016/j.actatropica.2016.12.021
99. Castillo MG, Humphries JE, Mourão MM, Marquez J, Gonzalez A, Montelongo CE. Biomphalaria glabrata immunity: Post-genome advances. Dev Comp Immunol (2020) 104:103557. doi: 10.1016/j.dci.2019.103557
100. Coustau C, Gourbal B, Duval D, Yoshino TP, Adema CM, Mitta G. Advances in gastropod immunity from the study of the interaction between the snail Biomphalaria glabrata and its parasites: A review of research progress over the last decade. Fish Shellfish Immunol (2015) 1:5–16. doi: 10.1016/j.fsi.2015.01.036
101. Pila EA, Li H, Hambrook JR, Wu X, Hanington PC. Schistosomiasis from a Snail’s Perspective: Advances in Snail Immunity. Trends Parasitol (2017) 33:845–57. doi: 10.1016/j.pt.2017.07.006
102. Portet A, Pinaud S, Tetreau G, Galinier R, Cosseau C, Duval D, et al. Integrated multi-omic analyses in Biomphalaria-Schistosoma dialogue reveal the immunobiological significance of FREP-SmPoMuc interaction. Dev Comp Immunol (2017) 75:16–27. doi: 10.1016/j.dci.2017.02.025
103. Mitta G, Gourbal B, Grunau C, Knight M, Bridger JM, Théron A. The Compatibility Between Biomphalaria glabrata Snails and Schistosoma mansoni: An Increasingly Complex Puzzle. Adv Parasitol (2017) 97:111–45. doi: 10.1016/bs.apar.2016.08.006
104. Knight M, Ittiprasert W, Arican-Goktas HD, Bridger JM. Epigenetic modulation, stress and plasticity in susceptibility of the snail host, Biomphalaria glabrata, to Schistosoma mansoni infection. Int J Parasitol (2016) 46:389–94. doi: 10.1016/j.ijpara.2016.03.003
105. Hanington PC, Forys MA, Dragoo JW, Zhang SM, Adema CM, Loker ES. Role for a somatically diversified lectin in resistance of an invertebrate to parasite infection. Proc Natl Acad Sci USA (2010) 107:21087–92. doi: 10.1073/pnas.1011242107
106. Hanington PC, Forys MA, Loker ES. A somatically diversified defense factor, FREP3, is a determinant of snail resistance to schistosome infection. PloS Negl Trop Dis (2012) 6:e1591. doi: 10.1371/journal.pntd.0001591
107. Baeza Garcia A, Pierce RJ, Gourbal B, Werkmeister E, Colinet D, Reichhart JM, et al. Involvement of the cytokine MIF in the snail host immune response to the parasite Schistosoma mansoni. PloS Pathog (2010) 6:e1001115. doi: 10.1371/journal.ppat.1001115
108. Pila EA, Gordy MA, Phillips VK, Kabore AL, Rudko SP, Hanington PC. Endogenous growth factor stimulation of hemocyte proliferation induces resistance to Schistosoma mansoni challenge in the snail host. Proc Natl Acad Sci USA (2016) 113:5305–10. doi: 10.1073/pnas.1521239113
109. Pila EA, Tarrabain M, Kabore AL, Hanington PC. A Novel Toll-Like Receptor (TLR) Influences Compatibility between the Gastropod Biomphalaria glabrata, and the Digenean Trematode Schistosoma mansoni. PloS Pathog (2016) 12:e1005513. doi: 10.1371/journal.ppat.1005513
110. Allan ER, Tennessen JA, Bollmann SR, Hanington PC, Bayne CJ, Blouin MS. Schistosome infectivity in the snail, Biomphalaria glabrata, is partially dependent on the expression of Grctm6, a Guadeloupe Resistance Complex protein. PloS Negl Trop Dis (2017) 11:e0005362. doi: 10.1371/journal.pntd.0005362
111. Mone Y, Gourbal B, Duval D, Du Pasquier L, Kieffer-Jaquinod S, Mitta G. A Large Repertoire of Parasite Epitopes Matched by a Large Repertoire of Host Immune Receptors in an Invertebrate Host/Parasite Model. PloS Neglected Trop Dis (2010) 4:e813. doi: 10.1371/journal.pntd.0000813
112. Wu XJ, Dinguirard N, Sabat G, Lui HD, Gonzalez L, Gehring M, et al. Proteomic analysis of Biomphalaria glabrata plasma proteins with binding affinity to those expressed by early developing larval Schistosoma mansoni. PloS Pathog (2017) 13:e1006081. doi: 10.1371/journal.ppat.1006081
113. Li H, Hambrook JR, Pila EA, Gharamah AA, Fang J, Wu X, et al. Coordination of humoral immune factors dictates compatibility between Schistosoma mansoni and Biomphalaria glabrata. Elife (2020) 9:e51708. doi: 10.7554/eLife.51708
114. Tennessen JA, Bollmann SR, Peremyslova E, Kronmiller BA, Sergi C, Hamali B, et al. Clusters of polymorphic transmembrane genes control resistance to schistosomes in snail vectors. Elife (2020) 9:e59395. doi: 10.7554/eLife.59395
115. Tennessen JA, Théron A, Marine M, Yeh JY, Rognon A, Blouin MS. Hyperdiverse gene cluster in snail host conveys resistance to human schistosome parasites. PloS Genet (2015) 11:e1005067. doi: 10.1371/journal.pgen.1005067
116. Ittiprasert W, Knight M. Reversing the resistance phenotype of the Biomphalaria glabrata snail host Schistosoma mansoni infection by temperature modulation. PloS Pathog (2012) 8:e1002677. doi: 10.1371/journal.ppat.1002677
117. Mone Y, Ribou A-C, Cosseau C, Duval D, Theron A, Mitta G, et al. An example of molecular co-evolution: Reactive oxygen species (ROS) and ROS scavenger levels in Schistosoma mansoni/Biomphalaria glabrata interactions. Int J For Parasitol (2011) 41:721–30. doi: 10.1016/j.ijpara.2011.01.007
118. Zhang SM, Loker ES, Sullivan JT. Pathogen-associated molecular patterns activate expression of genes involved in cell proliferation, immunity and detoxification in the amebocyte-producing organ of the snail Biomphalaria glabrata. Dev Comp Immunol (2016) 56:25–36. doi: 10.1016/j.dci.2015.11.008
119. Sullivan JT, Belloir JA. Activation of an innate immune response in the schistosome-transmitting snail Biomphalaria glabrata by specific bacterial PAMPs. Dev Comp Immunol (2014) 42:256–60. doi: 10.1016/j.dci.2013.09.016
120. Hanelt B, Lun CM, Adema CM. Comparative ORESTES-sampling of transcriptomes of immune-challenged Biomphalaria glabrata snails. J Invertebr Pathol (2008) 99:192–203. doi: 10.1016/j.jip.2008.06.002
121. Zhang SM, Zeng Y, Loker ES. Characterization of immune genes from the schistosome host snail Biomphalaria glabrata that encode peptidoglycan recognition proteins and gram-negative bacteria binding protein. Immunogenetics (2007) 59:883–98. doi: 10.1007/s00251-007-0245-3
122. Portet A, Galinier R, Pinaud S, Portela J, Nowacki F, Gourbal B, et al. BgTEP: An Antiprotease Involved in Innate Immune Sensing in Biomphalaria glabrata. Front Immunol (2018) 9:1206. doi: 10.3389/fimmu.2018.01206
123. Baron OL, van West P, Industri B, Ponchet M, Dubreuil G, Gourbal B, et al. Parental transfer of the antimicrobial protein LBP/BPI protects Biomphalaria glabrata eggs against oomycete infections. PloS Pathog (2013) 9:e1003792. doi: 10.1371/journal.ppat.1003792
124. Pasquier LD. Germline and somatic diversification of immune recognition elements in Metazoa. Immunol Lett (2006) 104:2–17. doi: 10.1016/j.imlet.2005.11.022
125. Lemaitre B, Hoffmann J. The host defense of Drosophila melanogaster. Annu Rev Immunol (2007) 25:697–743. doi: 10.1146/annurev.immunol.25.022106.141615
126. Ghosh J, Buckley KM, Nair SV, Raftos DA, Miller C, Majeske AJ, et al. Sp185/333: a novel family of genes and proteins involved in the purple sea urchin immune response. Dev Comp Immunol (2010) 34:235–45. doi: 10.1016/j.dci.2009.10.008
127. Lu L, Loker ES, Adema CM, Zhang SM, Bu L. Genomic and transcriptional analysis of genes containing fibrinogen and IgSF domains in the schistosome vector Biomphalaria glabrata, with emphasis on the differential responses of snails susceptible or resistant to Schistosoma mansoni. PloS Negl Trop Dis (2020) 14:e0008780. doi: 10.1371/journal.pntd.0008780
128. Baron OL, Deleury E, Reichhart JM, Coustau C. The LBP/BPI multigenic family in invertebrates: Evolutionary history and evidences of specialization in mollusks. Dev Comp Immunol (2016) 57:20–30. doi: 10.1016/j.dci.2015.11.006
129. Husnik F, McCutcheon JP. Functional horizontal gene transfer from bacteria to eukaryotes. Nat Rev Microbiol (2018) 16:67–79. doi: 10.1038/nrmicro.2017.137
130. Lercher MJ, Pál C. Integration of horizontally transferred genes into regulatory interaction networks takes many million years. Mol Biol Evol (2008) 25:559–67. doi: 10.1093/molbev/msm283
131. Melo VMM, Fonseca AM, Vasconcelos IM, Carvalho AFFU. Toxic, antimicrobial and hemagglutinating activities of the purple fluid of the sea hare Aplysia dactylomela Rang, 1828. Braz J Med Biol Res (1998) 31:785–91. doi: 10.1590/S0100-879X1998000600010
132. Schultz JH, Bu L, Adema CM. Comparative immunological study of the snail Physella acuta (Hygrophila, Pulmonata) reveals shared and unique aspects of gastropod immunobiology. Mol Immunol (2018) 101:108–19. doi: 10.1016/j.molimm.2018.05.029
133. Seppälä O, Walser J-C, Cereghetti T, Seppälä K, Salo T, Adema CM. Transcriptome profiling of a multiuse model species Lymnaea stagnalis (Gastropoda) for ecoimmunological research. bioRxiv (2020). doi: 10.1101/2020.09.23.308643
134. Vogan AA, Higgs PG. The advantages and disadvantages of horizontal gene transfer and the emergence of the first species. Biol Direct (2011) 6:1. doi: 10.1186/1745-6150-6-1
135. Keeling PJ, Palmer JD. Horizontal gene transfer in eukaryotic evolution. Nat Rev Genet (2008) 9:605–18. doi: 10.1038/nrg2386
136. Soucy SM, Huang J, Gogarten JP. Horizontal gene transfer: building the web of life. Nat Rev Genet (2015) 16:472–82. doi: 10.1038/nrg3962
137. Chou S, Daugherty MD, Peterson SB, Biboy J, Yang Y, Jutras BL, et al. Transferred interbacterial antagonism genes augment eukaryotic innate immune function. Nature (2015) 518:98–101. doi: 10.1038/nature13965
138. Zhu S, Gao B. Nematode-derived drosomycin-type antifungal peptides provide evidence for plant-to-ecdysozoan horizontal transfer of a disease resistance gene. Nat Commun (2014) 5:3154. doi: 10.1038/ncomms4154
139. Matsumoto M, Fukuda W, Circolo A, Goellner J, Strauss-Schoenberger J, Wang X, et al. Abrogation of the alternative complement pathway by targeted deletion of murine factor B. Proc Natl Acad Sci USA (1997) 94:8720–5. doi: 10.1073/pnas.94.16.8720
140. Xu Y, Narayana SV, Volanakis JE. Structural biology of the alternative pathway convertase. Immunol Rev (2001) 180:123–35. doi: 10.1034/j.1600-065X.2001.1800111.x
141. Bhattacharya AA, Lupher ML Jr, Staunton DE, Liddington RC. Crystal structure of the A domain from complement factor B reveals an integrin-like open conformation. Structure (2004) 12:371–8. doi: 10.1016/j.str.2004.02.012
142. Makou E, Herbert AP, Barlow PN. Creating functional sophistication from simple protein building blocks, exemplified by factor H and the regulators of complement activation. Biochem Soc Trans (2015) 43:812–8. doi: 10.1042/BST20150074
143. Sa e Cunha C, Nyboer B, Heiss K, Sanches-Vaz M, Fontinha D, Wiedtke E, et al. Plasmodium berghei EXP-1 interacts with host Apolipoprotein H during Plasmodium liver-stage development. Proc Natl Acad Sci USA (2017) 114:E1138–47. doi: 10.1073/pnas.1606419114
144. Banadakoppa M, Goluszko P, Liebenthal D, Nowicki BJ, Nowicki S, Yallampalli C. PI3K/Akt pathway restricts epithelial adhesion of Dr + Escherichia coli by down-regulating the expression of decay accelerating factor. Exp Biol Med (2014) 239:581–94. doi: 10.1177/1535370214522183
145. Gorbushin AM. Immune repertoire in the transcriptome of Littorina littorea reveals new trends in lophotrochozoan proto-complement evolution. Dev Comp Immunol (2018) 84:250–63. doi: 10.1016/j.dci.2018.02.018
146. Gorbushin AM. Derivatives of the lectin complement pathway in Lophotrochozoa. Dev Comp Immunol (2019) 94:35–58. doi: 10.1016/j.dci.2019.01.010
147. Prado-Alvarez M, Rotllant J, Gestal C, Novoa B, Figueras A. Characterization of a C3 and a factor B-like in the carpet-shell clam, Ruditapes decussatus. Fish Shellfish Immunol (2009) 26:305–15. doi: 10.1016/j.fsi.2008.11.015
148. Peng M, Li Z, Niu D, Liu X, Dong Z, Li J. Complement factor B/C2 in molluscs regulates agglutination and illuminates evolution of the Bf/C2 family. FASEB J (2019) 33:13323–33. doi: 10.1096/fj.201901142RR
149. Leprêtre M, Almunia C, Armengaud J, Salvador A, Geffard A, Palos-Ladeiro M. The immune system of the freshwater zebra mussel, Dreissena polymorpha, decrypted by proteogenomics of hemocytes and plasma compartments. J Proteomics (2019) 202:103366. doi: 10.1016/j.jprot.2019.04.016
150. Peng M, Niu D, Chen Z, Lan T, Dong Z, Tran TN, et al. Expression of a novel complement C3 gene in the razor clam Sinonovacula constricta and its role in innate immune response and hemolysis. Dev Comp Immunol (2017) 73:184–92. doi: 10.1016/j.dci.2017.03.027
151. Wang L, Zhang H, Zhang D, Lv Z, Liu Z, Wang W, et al. The RNA-seq analysis suggests a potential multi-component complement system in oyster Crassostrea gigas. Dev Comp Immunol (2017) 76:209–19. doi: 10.1016/j.dci.2017.06.009
152. Adema CM, Hertel LA, Miller RD, Loker ES. A family of fibrinogen-related proteins that precipitates parasite-derived molecules is produced by an invertebrate after infection. Proc Natl Acad Sci USA (1997) 94:8691–6. doi: 10.1073/pnas.94.16.8691
Keywords: biomphalaria, biomphalysin, aerolysin, invertebrate immunity, pore-forming toxin (PFT), structure
Citation: Pinaud S, Tetreau G, Poteaux P, Galinier R, Chaparro C, Lassalle D, Portet A, Simphor E, Gourbal B and Duval D (2021) New Insights Into Biomphalysin Gene Family Diversification in the Vector Snail Biomphalaria glabrata. Front. Immunol. 12:635131. doi: 10.3389/fimmu.2021.635131
Received: 29 November 2020; Accepted: 08 March 2021;
Published: 01 April 2021.
Edited by:
Roberta Lima Caldeira, Oswaldo Cruz Foundation (Fiocruz), BrazilReviewed by:
Coenraad Adema, University of New Mexico, United StatesCopyright © 2021 Pinaud, Tetreau, Poteaux, Galinier, Chaparro, Lassalle, Portet, Simphor, Gourbal and Duval. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Duval, ZGF2aWQuZHV2YWxAdW5pdi1wZXJwLmZy
†Present address: Silvain Pinaud, Cambridge Institute, CRUK, Cambridge, United Kingdom Anaïs Portet, Molecular Immunity Unit, University of Cambridge, Cambridge, United Kingdom
Department of Medicine, MRC Laboratory of Molecular Biology, Cambridge, United Kingdom
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.