 Yesim Yilmaz Demirdag
Yesim Yilmaz Demirdag Sudhir Gupta
Sudhir Gupta- Division of Basic and Clinical Immunology, Department of Medicine, University of California, Irvine, Irvine, CA, United States
Bacterial respiratory tract infections are the hallmark of primary antibody deficiencies (PADs). Because they are also among the most common infections in healthy individuals, PADs are usually overlooked in these patients. Careful evaluation of the history, including frequency, chronicity, and presence of other infections, would help suspect PADs. This review will focus on infections in relatively common PADs, discussing diagnostic challenges, and some management strategies to prevent infections.
Introduction
Primary immunodeficiencies (PIDs), also known as inborn errors of immunity (IEI), are genetic disorders classically characterized by increased susceptibility to infections. According to the recent International Union of Immunologic Societies (IUIS) report, primary antibody deficiencies coalesce under the 3rd category: Predominantly antibody deficiencies (PADs) (Table 1) (1). These immune deficiencies occur due to defects in B cell development or function. They are the most common types of IEIs worldwide (2–4). Patients have low levels of one or more immunoglobulin isotypes and/or inadequate production of pathogen-specific antibodies, and they develop infectious and noninfectious manifestations (Table 2) (5, 6, 23, 37, 45).
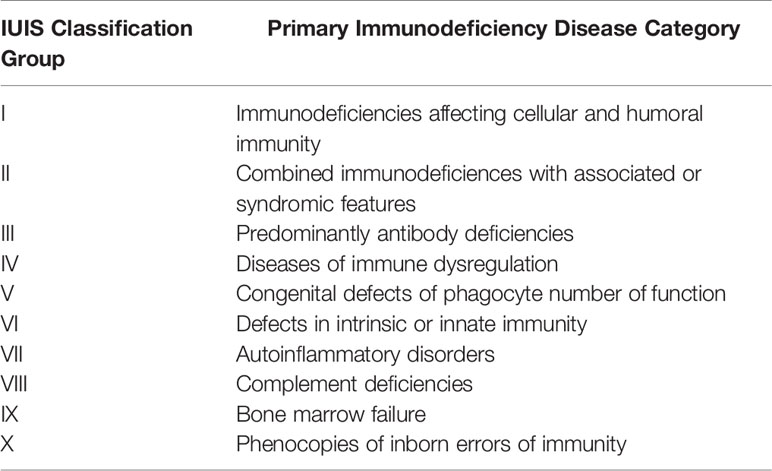
Table 1 Classification of inborn errors of immunity according to the 2019 IUIS report.
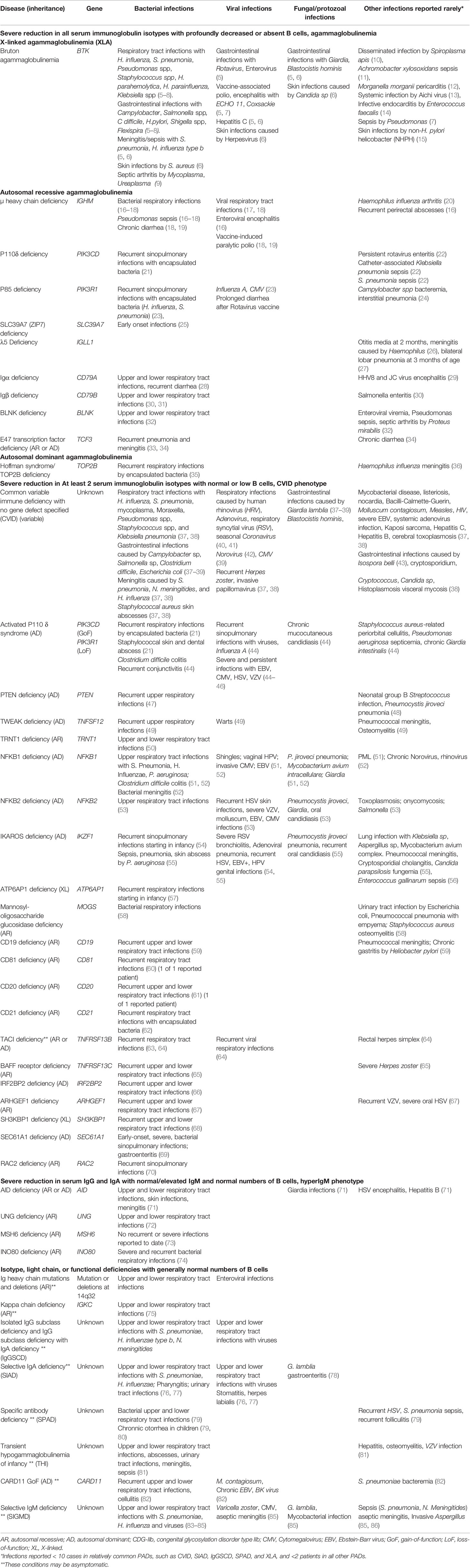
Table 2 Common and rarely reported infections in predominantly antibody deficiencies.
Early diagnosis and timely initiation of immunoglobulin replacement therapy (IgRT) may prevent infections, and therefore, alter the clinical course of PADs. Although sinopulmonary infections with encapsulated bacteria are the most common infections, PADs may also present with infections caused by other usual or unusual microorganisms affecting various organs. A thorough understanding of the types of infections that these patients are susceptible will be invaluable to suspect PADs and for early diagnosis. Here, we will summarize best known and relatively common PADs while focusing on their infectious manifestations. We will use primary antibody deficiencies and predominantly antibody deficiencies interchangeably.
Specific PADs are grouped into four subcategories based on their immunologic findings.
Severe Reduction in All Serum Immunoglobulin Isotypes With Profoundly Decreased or Absent B Cells: Agammaglobulinemia
These conditions are associated with circulating B cells below 2% of total lymphocytes, and undetectable or very low levels of all isotypes of serum immunoglobulins. About 85%–90% of patients in this group have X-linked agammaglobulinemia (XLA) due to mutations in the Bruton tyrosine kinase gene (BTK) (87), and 5% of patients have immunoglobulin μ heavy chain deficiency.
X-Linked Agammaglobulinemia
Classically, affected boys begin to suffer from infections at 4 to 6 months of age. Late-onset and milder cases have also been reported (88, 89). The estimated incidence ranges from 1:200,000 to 1:100,000 live births (90). Neutropenia is seen in 11% to 22% of patients, which resolves once the appropriate IgRT is initiated (5). Normal IgA and IgM levels have been reported in a few patients (5, 7).
Sinopulmonary infections are the most common infections in XLA, but gastrointestinal infections, and more invasive infections may also occur (Table 2). For exampleanalyses of large cohorts revealed that up to 8% of patients may present with meningitis caused by Streptococcus pneumoniae, Neisseria meningitides, and Haemophilus influenzae, or encephalitis caused by ECHO virus, Coxackie virus, and poliovirus (5–8). Enteroviruses also cause dermatomyositis-like presentation (5, 91). Enteroviral infections have been particularly challenging for patients with XLA because they are usually chronic, systemic, resistant to therapy, and may be fatal.
In a US registry consisted of 201 patients with XLA, Giardia lamblia was the most common etiology of gastrointestinal infections, and Pseudomonas spp was the most common etiology of sepsis, accounting for 12 of 46 and 6 of 29 patients, respectively (5).
Recently, refractory cellulitis, pyoderma gangrenosum, and bacteremia due to non-Helicobacter pylori Helicobacter (NHPH) such as Helicobacter bilis (Flexispira) and Helicobacter cinaedi have been reported in XLA (15, 92). In addition, case reports describing septic arthritis due to Mycoplasma or Ureoplasma (9), a disseminated infection caused by Spiroplasma apis (a honey bee pathogen) (10), sepsis due to Achromobacter xylosoxidans mimicking juvenile idiopathic arthritis (11), Morganella morganii pericarditis (12), and infective endocarditis due to Enterococcus faecalis (14) have been published. Aichi virus, a common contaminant in ponds, sewages, and shellfish, causing self-limiting gastroenteritis in immunocompetent persons, may cause chronic and severe infection including fever, bloody diarrhea, chronic hepatitis, and splenomegaly in XLA (13).
Autosomal Recessive and Autosomal Dominant Agammaglobulinemia
Immunoglobulin μ heavy chain (IGHM) deficiency is associated with more severe clinical manifestation compared to XLA (16, 17, 19). Like in XLA, neutropenia may be seen in 30% of these patients (16, 19). In addition to bacterial respiratory tract infections, these patients are at increased risk for pseudomonas sepsis, arthritis, skin abscesses, chronic diarrhea, and enteroviral central nervous system (CNS) infections (16–19). In a study on 19 patients with IGHM, 7 patients had significant enteroviral infections, and 4 had pseudomonas sepsis before the IgRT was started (19).
Other PADs in this group are extremely rare and include defects in phosphatidylinositol 3–kinase δ (PI3Kδ) signaling pathway, which include PIK3CD and PIK3R1 deficiencies, encoding catalytic and regulatory subunits of PI3Kδ, respectively. Biallelic loss of function (LoF) mutations in these genes cause early-onset bacterial sinopulmonary infections, viral infections, oral thrush, esophageal candida infection, Campylobacter bacteremia as well as transient neutropenia and thrombocytopenia (Table 2) (21, 23, 24, 93).
Severe Reduction in at Least 2 Serum Immunoglobulin Isotypes With Normal or Low B Cells: CVID Phenotype
Common Variable Immunodeficiency (CVID)
As the most common symptomatic IEI, CVID affects 1:50,000 to 1:10,000 people (4, 37, 94, 95). Patients are usually diagnosed between the 3rd and the 5th decades. According to the European Society for Immunodeficiencies Database (ESID), 33.7% of patients had the onset before 10 years, and, in a large US cohort, 28% of patients had the diagnosis before 21 years (37, 94). The most common clinical manifestations include infections and infection-related complications such as bronchiectasis (37, 38, 94). Otitis media, failure to thrive, and developmental delay were reported more in pediatric-onset CVID compared to adult-onset (96). Non-infectious manifestations, such as autoimmunity, may precede infections and, they are associated with increased mortality (38).
Because of the significant clinical and immunological heterogeneity, the definition of CVID has undergone several revisions. According to the most recent consensus, the diagnosis should be based on laboratory findings, which include low IgG (< 2SD for age-adjusted levels), low IgA, and/or low IgM, and suboptimal response to T-cell-independent or T-cell-dependent vaccines. Some patients may also have low CD4+ T cells. Other conditions such as medications, protein loss, and various disorders that may cause low immunoglobulins should be excluded (97).
The most common causes of pneumonia in CVID are S. pneumonia and H. influenza, followed by Mycoplasma pneumonia, Pseudomonas spp, Staphylococcus spp, and Klebsiella pneumonia (37, 38) (Table 2). The frequency and severity of respiratory infections decrease with IgRT. However, despite IgRT, a prospective study showed that respiratory infections may still occur more commonly in patients with PADs and, common circulating upper respiratory viruses may be detected more often in patients with PADs, including CVID, compared to healthy controls. The most common respiratory virus isolated in this study was human rhinovirus (HRV), followed by adenovirus, respiratory syncytial virus, and seasonal coronavirus (non-pandemic). Using a multiplex PCR approach, this study also showed that HRV and H. influenzae might frequently co-occur in patients with PADs (40). Furthermore, prolonged shedding of HRV after an acute infection has been shown by another group in patients with CVID and XLA compared to healthy controls (40 days versus 10 days) (41). Pneumocystis jiroveci pneumonia was reported in six patients with CVID, three were on systemic steroid treatment and, one was on systemic steroids and 6-mercaptopurine (37).
Large cohorts and observational studies reported gastrointestinal manifestations in 47% to 67% of patients with CVID. Gastrointestinal infections were seen up to 24% of patients with gastrointestinal symptoms (38, 39, 98). In a study on 50 patients with CVID and gastrointestinal symptoms, 14, six, and five patients developed infections by G. lamblia, Clostridium jejuni, and Salmonella, respectively (98). G. lamblia was also the most common gastrointestinal infection in other cohorts (38, 38). Undetectable serum IgA levels increase the risk of gastrointestinal infections (38). Although Helicobacter pylori is not a typical infection in CVID, perhaps due to recurrent antibiotics treatment, a recent study reported H. pylori infection in at least 26% of patients with gastrointestinal symptoms. Some of these patients had intestinal metaplasia, which persisted for at least 3 years after the treatment of H. pylori (98). Chronic diarrhea due to Giardia lamblia and Isospora belli triggered the CVID diagnosis in a 62-year-old woman (43).
Studies on large cohorts also revealed that Norovirus and Cytomegalovirus (CMV) were among the most common viral causes of gastrointestinal infections in CVID (38, 39, 98). Others reported that Norovirus can be very severe, causing malabsorption and enteropathy requiring parenteral nutrition (42, 99). Although most patients with PADs, efficiently clear oral polio vaccine (OPV)-related virus (100), viral shedding up to 28 years after OPV was reported in one patient (101).
Adenovirus, CMV, and EBV infections were the cause of death in eight of 13 patients in a cohort of 25 patients with CVID who received hematopoietic stem cell transplantation (HSCT) because of lymphoma or treatment-refractory immune dysregulation (102).
Recurrent shingles is the most common viral skin infections in CVID, and the risk factors include steroid therapy, chemotherapy, and older age (38, 103).
Activated Phosphoinositide 3-Kinase Delta Syndrome (APDS)
Activated phosphoinositide 3-kinase delta syndrome (APDS) is caused by autosomal dominant (AD) gain-of-function (GoF) mutations in PIK3CD (APDS1), AD loss-of-function (LoF) mutations in PIK3R1 (APDS2) or PTEN (phosphatase and tensin homolog deleted on chromosome 10). More than 200 patients have been reported with APDS (21). Patients develop recurrent respiratory infections, usually caused by S. pneumoniae and H. influenza during childhood, followed by lymphoproliferation, progressive lymphopenia, autoinflammatory disease, early onset bronchiectasis, and lymphoma. Short stature and neurodevelopmental delay were reported in some cohorts (45, 104). According to these reports, about 20% of patients developed conjunctivitis, which progressed to preorbital cellulitis in some. Persistent Epstein-Barr virus (EBV) or CMV viremia, EBV lymphoproliferative disease, and CMV lymphadenitis are characteristics of APDS. Chronic EBV viremia was seen in all patients in a series of 9 with APDS (105). Other infections are summarized in Table 2 (45, 104, 106, 107).
Disseminated CMV infection and chronic diarrhea due to vaccine-induced rotavirus were reported in one patient with APDS2 who had tolerated oral polio and Bacillus Calmette–Guérin (BCG) vaccines (23).
Other Monogenic Defects Associated With CVID Phenotype
Genetic defects may be responsible in up to 30% of patients with CVID who had at least one of the following criteria: younger age of onset, autoimmune/inflammatory conditions, low B cells, and/or family history of hypogammaglobulinemia (51). For example, mutations in the gene encoding TACI (transmembrane activator and calcium-modulator and cyclophilin ligand interactor) are among the most common genetic defects attributed to CVID phenotype. The inheritance pattern may be autosomal dominant or autosomal recessive with the characteristics including, like in many other primary immunodeficiencies, incomplete penetrance and variable expressivity. Mutations in TACI were found in 4 of 19 unrelated patients with CVID phenotype and one of 16 patients with selective IgA deficiency (SIAD) in one study (63). Another group reported TACI mutations in 26 of 176 patients with CVID phenotype (64). Interestingly, none of those patients had a family history of immune deficiency, and none of their 10 familial cases of CVID had TACI mutations. Clinically, patients with TACI mutations present with recurrent sinopulmonary infections as well as autoimmunity, and to date, studies suggest that clinical phenotypes of TACI mutations are influenced by additional genetic and environmental changes (108).
According to a recent large US CVID cohort, the second most common monogenic mutations following TACI mutations were autosomal dominant variants in nuclear factor κ B subunit 1 (NFKB1), accounting 14 of 235 patients (109). In addition to infections, the majority of these patients also had one or more of the following manifestations: lymphocytic/granulomatous infiltrate, enteropathy, and autoimmunity. In another cohort, infections in five patients with NFKB1 deficiency were summarized (51). In addition to sinopulmonary infections, these patients suffered from shingles, P. jiroveci pneumonia, Clostridium difficile colitis, Mycobacterium avium intracellulare (MAI), and progressive multifocal leukoencephalopathy (PML). The latter was the cause of death in 1 of 2 patients.
Heterozygous AD mutations in IKZF1, encoding the zinc-finger transcription factor IKAROS were recently reported in patients with CVID phenotype (54, 55, 110). These patients have progressive loss of serum immunoglobulins and B cells. While S. pneumoniae was a common pathogen, increased susceptibility to viral or fungal infections was not observed in these patients.
Other monogenic conditions associated with CVID phenotype have been reported in a limited number of patients. Although their clinical presentations are similar and consist of recurrent infections starting early in life, there are some deviations. For example, infections in BAFF receptor deficiency may start as late as 70 years of age (65). In addition, some defects are associated with specific clinical manifestations, such as hamartoma and macrocephaly in PTEN deficiency, sideroblastic anemia and progressive developmental delay in TRNT1 deficiency, and severe neurologic disease and dysmorphic facial features in mannosyl-oligosaccharide glucosidase deficiency (58, 111, 112).
Severe Reduction in Serum IgG and IgA With Normal/Elevated IgM and Normal Numbers of B Cells: HyperIgM Phenotype
These conditions result from defects in class-switch recombination (CSR). Activation-induced deaminase (AID) plays a significant role in CSR and somatic hypermutation (SHM). Together, SHM and CSR are crucial for a pathogen-specific, high-affinity antibody response. Uracil-DNA glycosylase (UNG) also plays a significant role in CSR and SHM (113, 114). Consequently, defects in AID or UNG genes result in recurrent and severe infections, autoimmunity (AID deficiency), and lymphoma (UNG deficiency) (Table 2) (19, 71, 115).
In this group, MSH6 deficiency may not be associated with susceptibility to recurrent infections (73).
Isotype, Light Chain, or Functional Deficiencies With Generally Normal Numbers of B Cells
Selective IgA Deficiency
Selective IgA deficiency (SIAD) is defined as undetectable serum IgA levels, and normal IgG and IgM levels after age 4 (116). The prevalence ranges from 1:18.500 in Eastern Asian population to 1:500 in Caucasians (116–118). Some chromosomal disorders, including chromosome 18 abnormalities, trisomy 21, and 22q11.2 microdeletion are associated with SIAD (119–123). In addition, familial clustering may occur (76, 124–126). SIAD may be transient in children, or sometimes, it may evolve to CVID (76, 127, 128).
Although, the majority of patients are asymptomatic, recurrent bacterial, and viral respiratory infections, autoimmunity (e.g., celiac disease), and allergies are seen more commonly in SIAD than in general population. In addition, individuals with SIAD more often undergo tonsillectomy and adenoidectomy, and they develop pharyngitis, stomatitis, herpes labialis, and urinary tract infections more often than the general population (76, 77, 129–131).
While the recommendation is to avoid a definite diagnosis of SIAD before age 4, one study showed that children with undetectable IgA levels suffered from infections more often than children with detectable but low IgA levels, and atopy was more common in both groups than in children with normal IgA levels (121).
In some patients, SIAD and specific antibody deficiency, or SIAD and IgG subclass deficiency may co-occur. Bronchiectasis and recurrent infections are more commonly reported in these patients than in patients with isolated SIAD (123).
Like in many other PADs, G. lamblia has been an increasingly reported gastrointestinal pathogen in SIAD (78, 131).
In patients older than 12 years, one study reported that, although the prevalence of H. pylori-associated dyspepsia was not higher in SIAD, those patients who had H. pylori infection experienced more esophagogastroduodenoscopy (EGD)-proven gastritis, duodenal ulcers, and nodular lymphoid hyperplasia (132). Other studies showed that H. pylori infection was one of the most common gastrointestinal infections in children (< 17 years) with SIAD (133, 134). In addition, H. pylori infection may be associated with more severe periodontitis in SIAD (129).
Interestingly, patients with SIAD are not susceptible to rotavirus disease, and in fact, they may develop higher levels of rotavirus-specific IgG1 than healthy individuals (135).
Specific Antibody Deficiency
Specific antibody deficiency (SPAD or SAD) is defined as an inadequate antibody response to polysaccharide antigens in patients who have normal serum immunoglobulin levels and IgG subclasses. It may be the most common PIDs in children (3), and it may be transient (79, 136). In addition to bacterial respiratory tract infections, very rarely, invasive infections and even bronchiectasis may develop in undiagnosed patients (Table 2) (136, 137).
IgG Subclass Deficiency
IgG subclass deficiency (IgGSCD) is diagnosed when one or more IgG subclass levels are 2SDs below the age-adjusted range in patients with normal total IgG levels. Patients present with recurrent bacterial and viral respiratory infections and atopy (3, 138–141). IgGSCD has also been reported more commonly in patients with chronic obstructive pulmonary disease (COPD) exacerbation (141).
In children, the most common IgGSCD is IgG2 deficiency followed by IgG3 deficiency, and maybe transient (142, 143). In adults, IgG3 deficiency may be the most common IgGSCD (144). IgG3 deficiency was also the most common immune deficiency in children and adults with refractory rhinosinusitis (145).
IgGSCD and SPAD may co-occur (140). In children, this combination may be more common in boys, may be associated with bronchiectasis, and may progress to CVID or other immunodeficiencies (143).
Selective IgM Deficiency
Selective IgM deficiency (SIGMD), defined as low serum IgM levels (< 2SD), and normal IgA and IgG levels, can be seen in children and adults (83, 146–148). Its prevalence was reported as 0.37% in healthy blood donors (147). About 45% of symptomatic patients may also have SPAD or IgGSCD (83, 84). Respiratory infections, and rarely, bronchiectasis, sepsis, and meningitis may be seen in up to 80% of symptomatic patients (Table 2) (84–86, 149).
In addition, atopic diseases and autoimmunity are common, especially in adults with SIGMD (85, 148).
COVID-19 and PADs
Coronavirus disease -19 (COVID-19) caused by severe acute respiratory distress syndrome coronavirus 2 (SARS-CoV-2), has been one of the major pandemics in human history. Like other immunocompromised states, PADs are presumed to be a risk factor for severe SARS-CoV-2 infection. Though data are limited, a large study including PADs (N:53) who developed COVID-19 reported that 13 of 40 patients with PAD who also had associated co-morbidities died whereas all 13 patients who had no COVID-19 associated co-morbidities survived (150).
An effective and timely production of pathogen-specific neutralizing antibodies should prevent viral replication and progression to severe disease. Therefore, one would expect worse clinical outcome in patients with PADs. In addition, SARS-CoV-2-specific T cells were recently demonstrated to be important in controlling and resolution of COVID-19 (151). Recently, two young adults with XLA (one had underlying bronchiectasis) developed COVID-19 pneumonia, but did not require intensive care. Another three patients with XLA had prolonged COVID-19 with increased proinflammatory responses recovered after treatment with convalescent serum (152, 153). Another group reported that two patients with agammaglobulinemia due to absent B cells had milder infection than five patients with CVID who had detectable B cells (154). These observations may be explained by presence of normal T cells in the cases of XLA, and defective T cells in some patients with CVID, and emphasize the importance of T cells in COVID-19. Detailed examination of clinical and immunologic response to SARS-CoV-2 in patients with PADs will provide indispensable knowledge on the pathogenesis of COVID-19.
Challenges in Diagnosing Infections in PADs
Although infections are the most common manifestations of PADs, identifying the pathogen is not always easy because of high chance of false-negative serologic testing due to defects in pathogen-specific antibody production. In addition, false-positive serology may be seen in patients who are on IgRT. Therefore, direct identification of the microorganism by blood or tissue cultures, immunofluorescense staining, pcr testing, or amplicon sequencing are necessary for accurate diagnosis. Recently, next-generation sequencing (NGS) and matrix-assisted laser desorption/ionization time-of-flight mass-spectrometry (MALDI-TOF MS) successfully identified the pathogens in patients with PADs (13, 15, 155).
Relationship Between IgG Levels and Infections
Life-long IgRT, administered either intravenously (IVIG) or subcutaneously (SCIG), is the standard treatment of the majority of PADs (7, 139, 140). In general, IgRT does not prevent or treat non-infectious manifestations.
Serum IgG trough levels, defined as IgG levels just before the next dose, strongly correlate with the dose and interval (8, 156). However, some patients with bronchiectasis or chronic lung disease require twice as much IgRT dose to achieve the same IgG trough levels (103, 157). Bacterial infections and bronchiectasis are more commonly seen in patients with low IgG levels (64, 103, 158, 159). IgRT may not significantly reduce viral respiratory infections (40).
A meta-analysis showed a progressively reduced number of infections with increasing IgG trough levels from 660 to 960 mg/dL. Beyond this level, the reduction in infection rate was not statistically significant (156). In another study, a positive correlation between total IgG trough levels and specific anti- pneumococcus and anti- H. influenza IgG levels were observed, and no serious lung infections developed in patients with IgG trough levels ≥700 mg/dL (160). In XLA, IgG trough levels > 800 mg/dl may prevent the onset of bronchiectasis, chronic sinusitis, and enteroviral infections (8).
On the other hand, IgG trough levels that prevent bacterial infections varied from 5 g/L to 17 gr/L in different patients, suggesting that the dose should be individualized based on clinical symptoms (103, 161). However, adjusting the IgRT dose based on frequency of infections alone may not be sufficient to prevent chronic lung damage (8, 157, 162). In addition, very low serum IgA and IgM levels have been associated with more severe radiographic lung disease and higher chance of bacterial colonization (159, 163). These findings suggest that higher trough IgG levels should be aimed in selected patients, such as, patients with chronic lung damage, history of viral CNS infections, and very low IgA or IgM levels. For example, two of three patients with enteroviral meningoencephalitis recovered from infections with higher dose of IVIG achieving trough levels between 3,100 and 6,300 mg/dL (8). High dose IgRT, however, had no beneficial effect in severe Norovirus infection in patients with CVID (99).
A recent systematic review and meta-analysis showed that weekly SCIG therapy resulted in higher IgG trough levels, and a linear decrease in incidence of infections with every 100 mg/dl increase in IgG trough levels. This was not observed in patients who received IVIG (164). Another recent study found that IVIG rather than SCIG therapy was associated with higher risk of bronchiectasis (165). This observation require confirmation by larger, controlled studies.
Other Therapies to Prevent Infections
Prophylactic antibiotics have been used to prevent infections in PADs (166). However, their negative effect on airway and gastrointestinal microbiota continues to be a major concern (159).
Administration of conjugated pneumococcal vaccine may achieve protective antibody levels and prevent pneumococcal infections in SIAD and IgSCD.
Although hematopoietic stem cell transplantation is not recommended in PADs, mostly due to the risks of GVHD, associated mortality, or no benefit, reduced intensity conditioning showed promising results in XLA (167, 168).
Author Contributions
YY prepared the first draft of the manuscript. YY and SG contributed to editing and reviewed and authorized the final version. All authors contributed to the article and approved the submitted version.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human Inborn Errors of Immunity: 2019 Update of the IUIS Phenotypical Classification. J Clin Immunol (2020) 40(1):66–81. doi: 10.1007/s10875-020-00758-x
2. Gathmann B, Grimbacher B, Beauté J, Dudoit Y, Mahlaoui N, Fischer A, et al. The European internet-based patient and research database for primary immunodeficiencies: Results 2006-2008. Clin Exp Immunol (2009) 157 Suppl 1(Suppl 1):3–11. doi: 10.1111/j.1365-2249.2009.03954.x
3. Javier FC, Moore CM, Sorensen RU. Distribution of primary immunodeficiency diseases diagnosed in a pediatric tertiary hospital. Ann Allergy Asthma Immunol (2000) 84(1):25–30. doi: 10.1016/S1081-1206(10)62736-6
4. Kobrynski L, Powell RW, Bowen S. Prevalence and Morbidity of Primary Immunodeficiency Diseases, United States 2001–2007. J Clin Immunol (2014) 34(8):954–61. doi: 10.1007/s10875-014-0102-8
5. Winkelstein JA, Marino MC, Lederman HM, Jones SM, Sullivan K, Burks AW, et al. X-linked agammaglobulinemia: Report on a United States registry of 201 patients. Medicine (Baltimore) (2006) 85(4):193–202. doi: 10.1097/01.md.0000229482.27398.ad
6. Lougaris V, Soresina A, Baronio M, Montin D, Martino S, Signa S, et al. Long-term follow-up of 168 patients with X-linked agammaglobulinemia reveals increased morbidity and mortality. J Allergy Clin Immunol (2020) 146(2):429–37. doi: 10.1016/j.jaci.2020.03.001
7. Plebani A, Soresina A, Rondelli R, Amato GM, Azzari C, Cardinale F, et al. Clinical, immunological, and molecular analysis in a large cohort of patients with X-linked agammaglobulinemia: An Italian Multicenter Study. Clin Immunol (2002) 104(3):221–30. doi: 10.1006/clim.2002.5241
8. Quartier P, Debré M, De Blic J, De Sauverzac R, Sayegh N, Jabado N, et al. Early and prolonged intravenous immunoglobulin replacement therapy in childhood agammaglobulinemia: A retrospective survey of 31 patients. J Pediatr (1999) 134(5):589–96. doi: 10.1016/S0022-3476(99)70246-5
9. Jhaveri VV, Lasalvia MT. Invasive Ureaplasma Infection in Patients Receiving Rituximab and Other Humoral Immunodeficiencies-A Case Report and Review of the Literature. Open Forum Infect Dis (2019) 6(10):ofz399. doi: 10.1093/ofid/ofz399
10. Etienne N, Bret L, Le Brun C, Lecuyer H, Moraly J, Lanternier F, et al. Disseminated spiroplasma apis infection in patient with agammaglobulinemia, France. Emerg Infect Dis (2018) 24(12):2382–6. doi: 10.3201/eid2412.180567
11. Janarthanan M, Gollapalli S, Sankaranarayanan S. Achromobacter xylosoxidans Sepsis Unveiling X-linked Agammaglobulinemia Masquerading as Systemic-onset Juvenile Idiopathic Arthritis. Indian Pediatr (2019) 56(5):423–5. doi: 10.1007/s13312-019-1541-3
12. Cho YK, Kook H, Woo YJ, Choi YY, Ma JS, Hwang TJ. Morganella morganii pericarditis in a child with X-linked agammaglobulinemia. Pediatr Int (2010) 52(3):489–91. doi: 10.1111/j.1442-200X.2010.03036.x
13. Bucciol G, Moens L, Payne K, Wollants E, Mekahli D, Levtchenko E, et al. Chronic Aichi Virus Infection in a Patient with X-Linked Agammaglobulinemia. J Clin Immunol (2018) 38(7):748–52. doi: 10.1007/s10875-018-0570-3
14. Shakoor A, El-Isa A, Kinsella E, Halas R, Leonov A. Uncommon Infections in Children Suggest Underlying Immunodeficiency: A Case of Infective Endocarditis in a 3-Year-Old Male. Case Rep Infect Dis (2018) 1–3. doi: 10.1155/2018/9380763
15. Inoue K, Sasaki S, Yasumi T, Imai K, Kusunoki T, Morio T, et al. Helicobacter cinaedi-Associated Refractory Cellulitis in Patients with X-Linked Agammaglobulinemia. J Clin Immunol (2020) 40(8):1132–7. doi: 10.1007/s10875-020-00830-6
16. Yel L, Minegishi Y, Coustan-Smith E, Buckley RH, Trübel H, Pachman LM, et al. Mutations in the mu heavy chain gene in patients with agammaglobulinemia. N Engl J Med (1996) 335(20):1486–93. doi: 10.1056/NEJM199611143352003
17. Silva P, Justicia A, Regueiro A, Farinã S, Couselo JM, Loidi L. Autosomal recessive agammaglobulinemia due to defect in μ heavy chain caused by a novel mutation in the IGHM gene. Genes Immun (2017) 18(3):197–9. doi: 10.1038/gene.2017.14
18. Lopez Granados E, Porpiglia AS, Hogan MB, Matamoros N, Krasovec S, Pignata C, et al. Clinical and molecular analysis of patients with defects in μ heavy chain gene. J Clin Invest (2002) 112(1):136–42. doi: 10.1172/JCI0215658
19. Yazdani R, Abolhassani H, Kiaee F, Habibi S, Azizi G, Tavakol M, et al. Comparison of Common Monogenic Defects in a Large Predominantly Antibody Deficiency Cohort. J Allergy Clin Immunol Pract (2019) 7(3):864–78.e9. doi: 10.1016/j.jaip.2018.09.004
20. Meffre E, LeDeist F, De Saint-Basile G, Deville A, Fougereau M, Fischer A, et al. A human non-XLA immunodeficiency disease characterized by blockage of B cell development at an early ProB cell stage. J Clin Invest (1996) 98(7):1519–26. doi: 10.1016/S0165-2478(97)85883-X
21. Nunes-Santos CJ, Uzel G, Rosenzweig SD. PI3K pathway defects leading to immunodeficiency and immune dysregulation. J Allergy Clin Immunol (2019) 143(5):1676–87. doi: 10.1016/j.jaci.2019.03.017
22. Sogkas G, Fedchenko M, Dhingra A, Jablonka A, Schmidt RE, Atschekzei F. Primary immunodeficiency disorder caused by phosphoinositide 3–kinase δ deficiency. J Allergy Clin Immunol (2018) 9:543. doi: 10.1016/j.jaci.2018.06.039
23. Tang P, Upton JEM, Barton-Forbes MA, Salvadori MI, Clynick MP, Price AK, et al. Autosomal Recessive Agammaglobulinemia Due to a Homozygous Mutation in PIK3R1. J Clin Immunol (2018) 38(1):88–95. doi: 10.1007/s10875-017-0462-y
24. Conley ME, Dobbs AK, Quintana AM, Bosompem A, Wang YD, Coustan-Smith E, et al. Agammaglobulinemia and absent B lineage cells in a patient lacking the p85α subunit of PI3K. J Exp Med (2012) 209(3):463–70. doi: 10.1084/jem.20112533
25. Anzilotti C, Swan DJ, Boisson B, Deobagkar-Lele M, Oliveira C, Chabosseau P, et al. An essential role for the Zn 2+ transporter ZIP7 in B cell development. Nat Immunol (2019) 20(3):350–61. doi: 10.1038/s41590-018-0295-8
26. Minegishi Y, Coustan-Smith E, Wang YH, Cooper MD, Campana D, Conley ME. Mutations in the human λ5/14.1 Gene result in B cell deficiency and agammaglobulinemia. J Exp Med (1998) 187(1):71–7. doi: 10.1084/jem.187.1.71
27. Gemayel KT, Litman GW, Sriaroon P. Autosomal recessive agammaglobulinemia associated with an IGLL1 gene missense mutation. Ann Allergy Asthma Immunol (2016) 117(4):439–41. doi: 10.1016/j.anai.2016.07.038
28. Minegishi Y, Coustan-Smith E, Rapalus L, Ersoy F, Campana D, Conley ME. Mutations in Igα (CD79a) result in a complete block in B-cell development. J Clin Invest (1999) 104(8):1115–21. doi: 10.1172/JCI7696
29. Khalili A, Plebani A, Vitali M, Abolhassani H, Lougaris V, Mirminachi B, et al. Autosomal recessive agammaglobulinemia: A novel non-sense mutation in CD79a. J Clin Immunol (2014) 34(2):138–41. doi: 10.1007/s10875-014-9989-3
30. Ferrari S, Lougaris V, Caraffi S, Zuntini R, Yang J, Soresina A, et al. Mutations of the Igβ gene cause agammaglobulinemia in man. J Exp Med (2007) 204(9):2047–51. doi: 10.1084/jem.20070264
31. Dobbs AK, Yang T, Farmer D, Kager L, Parolini O, Conley ME. Cutting Edge: A Hypomorphic Mutation in Igβ (CD79b) in a Patient with Immunodeficiency and a Leaky Defect in B Cell Development. J Immunol (2007) 179(4):2055–9. doi: 10.4049/jimmunol.179.4.2055
32. NaserEddin A, Shamriz O, Keller B, Alzyoud RM, Unger S, Fisch P, et al. Enteroviral Infection in a Patient with BLNK Adaptor Protein Deficiency. J Clin Immunol (2015) 35(4):356–60. doi: 10.1007/s10875-015-0164-2
33. Ben-Ali M, Yang J, Chan KW, Ben-Mustapha I, Mekki N, Benabdesselem C, et al. Homozygous transcription factor 3 gene (TCF3) mutation is associated with severe hypogammaglobulinemia and B-cell acute lymphoblastic leukemia. J Allergy Clin Immunol (2017) 40(4):1191–4.e4. doi: 10.26226/morressier.57bc1755d462b80290b4d590
34. Qureshi S, Sheikh MDA, Qamar FN. Autosomal Recessive Agammaglobulinemia - first case with a novel TCF3 mutation from Pakistan. Clin Immunol (2019) 198:100–101. doi: 10.1016/j.clim.2018.07.016
35. Hoffman HM, Bastian JF, Bird LM. Humoral immunodeficiency with facial dysmorphology and limb anomalies: A new syndrome. Clin Dysmorphol (2001) 10(1):1–8. doi: 10.1097/00019605-200101000-00001
36. Hügle B, Hoffman H, Bird LM, Gebauer C, Suchowerskyj P, Sack U, et al. Hoffman syndrome: New patients, new insights. Am J Med Genet Part A (2011) 155A(1):149–53. doi: 10.1002/ajmg.a.33678
37. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012). doi: 10.1182/blood-2011-09-377945
38. Oksenhendler E, Gérard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis (2008) 119(7):1650–7. doi: 10.1086/587669
39. Khan R, Habbal M, Scaffidi MA, Bukhari AA, Rumman A, Al Ghamdi S, et al. Gastrointestinal Disease in Patients with Common Variable Immunodeficiency: A Retrospective Observational Study. J Can Assoc Gastroenterol (2020) 3(4):162–8. doi: 10.1093/jcag/gwz004
40. Ponsford MJ, Price C, Farewell D, Greene G, Moore C, Perry M, et al. Increased Respiratory Viral Detection and Symptom Burden Among Patients with Primary Antibody Deficiency: Results from the BIPAD Study. J Allergy Clin Immunol Pract (2020) 9(2):735–44.e6. doi: 10.1016/j.jaip.2020.08.016
41. Peltola V, Waris M, Kainulainen L, Kero J, Ruuskanen O. Virus shedding after human rhinovirus infection in children, adults and patients with hypogammaglobulinaemia. Clin Microbiol Infect (2013) 19(7):E322–7. doi: 10.1111/1469-0691.12193
42. Woodward J, Gkrania-Klotsas E, Kumararatne D. Chronic norovirus infection and common variable immunodeficiency. Clin Exp Immunol (2017) 188(3):363–70. doi: 10.1111/cei.12884
43. Silva GBE, Fernandes KP, Segundo GRS. Common variable immunodeficiency and isosporiasis: first report case. Rev Soc Bras Med Trop (2012) 45(6):768–9. doi: 10.1590/S0037-86822012000600023
44. Elkaim E, Neven B, Bruneau J, Mitsui-Sekinaka K, Stanislas A, Heurtier L, et al. Clinical and immunologic phenotype associated with activated phosphoinositide 3-kinase δ syndrome 2: A cohort study. J Allergy Clin Immunol (2016) 138(1):210–8.e9. doi: 10.1016/j.jaci.2016.03.022
45. Coulter TI, Chandra A, Bacon CM, Babar J, Curtis J, Screaton N, et al. Clinical spectrum and features of activated phosphoinositide 3-kinase δ syndrome: A large patient cohort study. J Allergy Clin Immunol (2017) 139(2):597–606.e4. doi: 10.1016/j.jaci.2016.06.021
46. Angulo I, Vadas O, Garçon F, Banham-Hall E, Plagnol V, Leahy TR, et al. Phosphoinositide 3-kinase δ gene mutation predisposes to respiratory infection and airway damage. Science (80-) (2013) 342(6160):866–71. doi: 10.1126/science.1243292
47. Browning MJ, Chandra A, Carbonaro V, Okkenhaug K, Barwell J. Cowden’s syndrome with immunodeficiency. J Med Genet (2015) 52(12):856–9. doi: 10.1136/jmedgenet-2015-103266
48. Tsujita Y, Mitsui-Sekinaka K, Imai K, Yeh TW, Mitsuiki N, Asano T, et al. Phosphatase and tensin homolog (PTEN) mutation can cause activated phosphatidylinositol 3-kinase δ syndrome–like immunodeficiency. J Allergy Clin Immunol (2016) 138(6):1672–80.e10. doi: 10.1016/j.jaci.2016.03.055
49. Wang HY, Ma CA, Zhao Y, Fan X, Zhou Q, Edmonds P, et al. Antibody deficiency associated with an inherited autosomal dominant mutation in TWEAK. Proc Natl Acad Sci U S A (2013) 110(13):5127–32. doi: 10.1073/pnas.1221211110
50. Wiseman DH, May A, Jolles S, Connor P, Powell C, Heeney MM, et al. A novel syndrome of congenital sideroblastic anemia, B-cell immunodeficiency, periodic fevers, and developmental delay (SIFD). Blood (2013) 122(1):112–23. doi: 10.1182/blood-2012-08-439083
51. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova JL, et al. Genetic diagnosis using whole exomesequencing in common variable immunodeficiency. Front Immunol (2016) 7:220. doi: 10.3389/fimmu.2016.00220
52. Tuijnenburg P, Lango Allen H, Burns SO, Greene D, Jansen MH, Staples E, et al. Loss-of-function nuclear factor κB subunit 1 (NFKB1) variants are the most common monogenic cause of common variable immunodeficiency in Europeans. J Allergy Clin Immunol (2018) 142(4):1285–96. doi: 10.1016/j.jaci.2018.01.039
53. Klemann C, Camacho-Ordonez N, Yang L, Eskandarian Z, Rojas-Restrepo JL, Frede N, et al. Clinical and immunological phenotype of patients with primary immunodeficiency due to damaging mutations in NFKB2. Front Immunol (2019) 10:297. doi: 10.3389/fimmu.2019.00297
54. Eskandarian Z, Fliegauf M, Bulashevska A, Proietti M, Hague R, Smulski CR, et al. Assessing the functional relevance of variants in the Ikaros family zinc finger protein 1 (IKZF1) in a cohort of patients with primary immunodeficiency. Front Immunol (2019) 10:568. doi: 10.3389/fimmu.2019.01490
55. Boutboul D, Kuehn HS, Van De Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest (2018) 128(7):3071–87. doi: 10.1172/JCI98164
56. Chen QY, Wang XC, Wang WJ, Zhou QH, Liu DR, Wang Y. B-cell deficiency: A de novo IKZF1 patient and review of the literature. J Investig Allergol Clin Immunol (2018) 28(1):53–6. doi: 10.18176/jiaci.0207
57. Jansen EJR, Timal S, Ryan M, Ashikov A, Van Scherpenzeel M, Graham LA, et al. ATP6AP1 deficiency causes an immunodeficiency with hepatopathy, cognitive impairment and abnormal protein glycosylation. Nat Commun (2016) 7:11600. doi: 10.1038/ncomms11600
58. Sadat MA, Moir S, Chun T-W, Lusso P, Kaplan G, Wolfe L, et al. Glycosylation, Hypogammaglobulinemia, and Resistance to Viral Infections. N Engl J Med (2014) 370(17):1615–25. doi: 10.1056/NEJMoa1302846
59. van Zelm MC, Reisli I, van der Burg M, Castaño D, van Noesel CJM, van Tol MJD, et al. An Antibody-Deficiency Syndrome Due to Mutations in the CD19 Gene. N Engl J Med (2006). doi: 10.1056/NEJMoa051568
60. Van Zelm MC, Smet J, Adams B, Mascart F, Schandené L, Janssen F, et al. CD81 gene defect in humans disrupts CD19 complex formation and leads to antibody deficiency. J Clin Invest (2010) 120(4):1265–74. doi: 10.1172/JCI39748
61. Kuijpers TW, Bende RJ, Baars PA, Grummels A, Derks IAM, Dolman KM, et al. CD20 deficiency in humans results in impaired T cell-independent antibody responses. J Clin Invest (2010) 120(1):214–22. doi: 10.1172/JCI40231
62. Rosain J, Miot C, Lambert N, Rousselet MC, Pellier I, Picard C. CD21 deficiency in 2 siblings with recurrent respiratory infections and hypogammaglobulinemia. J Allergy Clin Immunol Pract (2017). doi: 10.1016/j.jaip.2017.04.011
63. Castigli E, Wilson SA, Garibyan L, Rachid R, Bonilla F, Schneider L, et al. TACI is mutant in common variable immunodeficiency and IgA deficiency. Nat Genet (2005) 5(6):1765–7.e3. doi: 10.1038/ng1601
64. Zhang L, Radigan L, Salzer U, Behrens TW, Grimbacher B, Diaz G, et al. Transmembrane activator and calcium-modulating cyclophilin ligand interactor mutations in common variable immunodeficiency: Clinical and immunologic outcomes in heterozygotes. J Allergy Clin Immunol (2007) 20(5):1178–85. doi: 10.1016/j.jaci.2007.10.001
65. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Böhm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci U S A (2009) 106(33):13945–50. doi: 10.1073/pnas.0903543106
66. Keller MD, Pandey R, Li D, Glessner J, Tian L, Henrickson SE, et al. Mutation in IRF2BP2 is responsible for a familial form of common variable immunodeficiency disorder. J Allergy Clin Immunol (2016) 138(2):544–50.e4. doi: 10.1016/j.jaci.2016.01.018
67. Bouafia A, Lofek S, Bruneau J, Chentout L, Lamrini H, Trinquand A, et al. Loss of ARHGEF1 causes a human primary antibody deficiency. J Clin Invest (2019) 138(2):544–50.e4. doi: 10.1172/JCI120572
68. Keller B, Shoukier M, Schulz K, Bhatt A, Heine I, Strohmeier V, et al. Germline deletion of CIN85 in humans with X chromosome-linked antibody deficiency. J Exp Med (2018) 215(5):1327–36. doi: 10.1084/jem.20170534
69. Schubert D, Klein MC, Hassdenteufel S, Caballero-Oteyza A, Yang L, Proietti M, et al. Plasma cell deficiency in human subjects with heterozygous mutations in Sec61 translocon alpha 1 subunit (SEC61A1). J Allergy Clin Immunol (2018) 141(4):1427–38. doi: 10.1016/j.jaci.2017.06.042
70. Alkhairy OK, Rezaei N, Graham RR, Abolhassani H, Borte S, Hultenby K, et al. RAC2 loss-of-function mutation in 2 siblings with characteristics of common variable immunodeficiency. J Allergy Clin Immunol (2015) 135(5):1380–4.e1-5. doi: 10.1016/j.jaci.2014.10.039
71. Quartier P, Bustamante J, Sanal O, Plebani A, Debré M, Deville A, et al. Clinical, immunologic and genetic analysis of 29 patients with autosomal recessive hyper-IgM syndrome due to Activation-Induced Cytidine Deaminase deficiency. Clin Immunol (2004) 11(1):22–9. doi: 10.1016/j.clim.2003.10.007
72. Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat Immunol (2003) 4(10):1023–8. doi: 10.1038/ni974
73. Gardès P, Forveille M, Alyanakian M-A, Aucouturier P, Ilencikova D, Leroux D, et al. Human MSH6 Deficiency Is Associated with Impaired Antibody Maturation. J Immunol (2012) 188(4):2023–9. doi: 10.4049/jimmunol.1102984
74. Kracker S, Di Virgilio M, Schwartzentruber J, Cuenin C, Forveille M, Deau MC, et al. An inherited immunoglobulin class-switch recombination deficiency associated with a defect in the INO80 chromatin remodeling complex. J Allergy Clin Immunol (2015) 135(4):998–1007.e6. doi: 10.1016/j.jaci.2014.08.030
75. Sala P, Colatutto A, Fabbro D, Mariuzzi L, Marzinotto S, Toffoletto B, et al. Immunoglobulin K light chain deficiency: A rare, but probably underestimated, humoral immune defect. Eur J Med Genet (2016) 59(4):219–22. doi: 10.1016/j.ejmg.2016.02.003
76. Lougaris V, Sorlini A, Monfredini C, Ingrasciotta G, Caravaggio A, Lorenzini T, et al. Clinical and Laboratory Features of 184 Italian Pediatric Patients Affected with Selective IgA Deficiency (SIgAD): a Longitudinal Single-Center Study. J Clin Immunol (2019) 39(5):470–5. doi: 10.1007/s10875-019-00647-y
77. Jorgensen GH, Gardulf A, Sigurdsson MI, Sigurdardottir ST, Thorsteinsdottir I, Gudmundsson S, et al. Clinical symptoms in adults with selective IgA deficiency: A case-control study. J Clin Immunol (2013) 33(4):742–7. doi: 10.1007/s10875-012-9858-x
78. Eren M, Saltik-Temizel IN, Yüce A, Çaǧlar M, Koçak N. Duodenal appearance of giardiasis in a child with selective immunoglobulin A deficiency. Pediatr Int (2007) 49(3):409–11. doi: 10.1111/j.1442-200X.2007.02357.x
79. Ruuskanen O, Nurkka A, Helminen M, Viljanen MK, Käyhty H, Kainulainen L. Specific antibody deficiency in children with recurrent respiratory infections: A controlled study with follow-up. Clin Exp Immunol (2013) 172(2):238–44. doi: 10.1111/cei.12053
80. Boyle RJ, Le C, Balloch A, Tang MLK. The clinical syndrome of specific antibody deficiency in children. Clin Exp Immunol (2006) 146(3):486–92. doi: 10.1016/j.jaci.2005.12.435
81. Moschese V, Graziani S, Avanzini MA, Carsetti R, Marconi M, La Rocca M, et al. A prospective study on children with initial diagnosis of transient hypogammaglobulinemia of infancy: Results from the Italian Primary Immunodeficiency Network. Int J Immunopathol Pharmacol (2008) 21(2):343–52. doi: 10.1177/039463200802100211
82. Snow AL, Xiao W, Stinson JR, Lu W, Benjamin CD, Zheng L, et al. Congenital B cell lymphocytosis explained by novel germline CARD11 mutations. J Exp Med (2012) 209(12):2247–61. doi: 10.1084/jem.20120831
83. Goldstein MF, Goldstein AL, Dunsky EH, Dvorin DJ, Belecanech GA, Shamir K. Selective IgM immunodeficiency: Retrospective analysis of 36 adult patients with review of the literature. Ann Allergy Asthma Immunol (2006) 97(6):717–30. doi: 10.1016/S1081-1206(10)60962-3
84. Yel L, Ramanuja S, Gupta S. Clinical and immunological features in igm deficiency. Int Arch Allergy Immunol (2009). doi: 10.1159/000222682
85. Lee Lucuab-Fegurgur D, Gupta S. Comprehensive clinical and immunological features of 62 adult patients with selective primary IgM deficiency. Am J Clin Exp Immunol (2019) 8(6):55–67. doi: 10.1016/j.anai.2018.09.027
86. Hong R, Gupta S. Selective immunoglobulin M deficiency in an adult with Streptococcus pneumoniae sepsis and invasive aspergillosis. J Investig Allergol Clin Immunol (2008) 18(3):214–8.
87. Conley ME, Broides A, Hernandez-Trujillo V, Howard V, Kanegane H, Miyawaki T, et al. Genetic analysis of patients with defects in early B-cell development. Immunol Rev (2005) 203:216–34. doi: 10.1111/j.0105-2896.2005.00233.x
88. Usui K, Sasahara Y, Tazawa R, Hagiwara K, Tsukada S, Miyawaki T, et al. Recurrent pneumonia with mild hypogammaglobulinemia diagnosed as X-linked agammaglobulinemia in adults. Respir Res (2001) 2(3):188–92. doi: 10.1186/rr56
89. Martignani C, Massaro G, Bruno AG, Biffi M, Ziacchi M, Diemberger I. Acute primary purulent pericarditis in an adult patient with unknown X-linked agammaglobulinemia. Immunobiology (2020) 225(1):151861. doi: 10.1016/j.imbio.2019.10.010
90. El-Sayed ZA, Abramova I, Aldave JC, Al-Herz W, Bezrodnik L, Boukari R, et al. X-linked agammaglobulinemia (XLA):Phenotype, diagnosis, and therapeutic challenges around the world. World Allergy Organ J (2019) 12(3):100018. doi: 10.1016/j.waojou.2019.100018
91. Halliday E, Winkelstein J, Webster ADB. fEnteroviral infections in primary immunodeficiency (PID): A survey of morbidity and mortality. J Infect (2003) 46(1):1–8. doi: 10.1053/jinf.2002.1066
92. Turvey SE, Leo SH, Boos A, Deans GD, Prendiville J, Crawford RI, et al. Successful approach to treatment of helicobacter bilis infection in x-linked agammaglobulinemia. J Clin Immunol (2012) 32(6):1404–8. doi: 10.1007/s10875-012-9750-8
93. Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. PI3Kδ and primary immunodeficiencies. Nat Rev Immunol (2016) 16(11):702–14. doi: 10.1038/nri.2016.93
94. Gathmann B, Mahlaoui N, Gérard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 134(1):116–26. doi: 10.1016/j.jaci.2013.12.1077
95. Slade CA, Bosco JJ, Giang TB, Kruse E, Stirling RG, Cameron PU, et al. Delayed diagnosis and complications of predominantly antibody deficiencies in a cohort of Australian adults. Front Immunol (2018) 9:694. doi: 10.3389/fimmu.2018.00694
96. Sanchez LA, Maggadottir SM, Pantell MS, Lugar P, Rundles CC, Sullivan KE, et al. Two Sides of the Same Coin: Pediatric-Onset and Adult-Onset Common Variable Immune Deficiency. J Clin Immunol (2017) 37(6):592–602. doi: 10.1007/s10875-017-0415-5
97. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract (2016) 4(1):38–59. doi: 10.1016/j.jaip.2015.07.025
98. Malamut G, Verkarre V, Suarez F, Viallard JF, Lascaux AS, Cosnes J, et al. The enteropathy associated with common variable immunodeficiency: The delineated frontiers with celiac disease. Am J Gastroenterol (2010) 105(10):2262–75. doi: 10.1038/ajg.2010.214
99. Brown LAK, Ruis C, Clark I, Roy S, Brown JR, Albuquerque AS, et al. A comprehensive characterization of chronic norovirus infection in immunodeficient hosts. J Allergy Clin Immunol (2019) 144(5):1450–3. doi: 10.1016/j.jaci.2019.07.036
100. Halsey NA, Pinto J, Espinosa-Rosales F, Faure-Fontenla MA, Da Silva E, Khan AJ, et al. Search for poliovirus carriers among people with primary immune deficiency diseases in the United States, Mexico, Brazil, and the United Kingdom. Bull World Health Organ (2004) 82(1):3–8.
101. Dunn G, Klapsa D, Wilton T, Stone L, Minor PD, Martin J. Twenty-Eight Years of Poliovirus Replication in an Immunodeficient Individual: Impact on the Global Polio Eradication Initiative. PloS Pathog (2015) 11(8):e1005114. doi: 10.1371/journal.ppat.1005114
102. Wehr C, Gennery AR, Lindemans C, Schulz A, Hoenig M, Marks R, et al. Multicenter experience in hematopoietic stem cell transplantation for serious complications of common variable immunodeficiency. J Allergy Clin Immunol (2015) 135(4):988–97.e6. doi: 10.1016/j.jaci.2014.11.029
103. Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: Relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol (2010) 125(6):1354–60.e4. doi: 10.1016/j.jaci.2010.02.040
104. Maccari ME, Abolhassani H, Aghamohammadi A, Aiuti A, Aleinikova O, Bangs C, et al. Disease evolution and response to rapamycin in activated phosphoinositide 3-kinase δ syndrome: The European society for immunodeficiencies-activated phosphoinositide 3-kinase δ syndrome registry. Front Immunol (2018) 9:543. doi: 10.3389/fimmu.2018.00543
105. Lucas CL, Kuehn HS, Zhao F, Niemela JE, Deenick EK, Palendira U, et al. Dominant-activating germline mutations in the gene encoding the PI(3)K catalytic subunit p110δ result in T cell senescence and human immunodeficiency. Nat Immunol (2014) 15(1):88–97. doi: 10.1038/ni.2771
106. Hartman HN, Niemela J, Hintermeyer MK, Garofalo M, Stoddard J, Verbsky JW, et al. Gain of Function Mutations of PIK3CD as a Cause of Primary Sclerosing Cholangitis. J Clin Immunol (2015) 35(1):11–4. doi: 10.1007/s10875-014-0109-1
107. Dominguez-Pinilla N, Allende LM, Rosain J, Gallego M del C, Chaves F, Deswarte C, et al. Disseminated abscesses due to Mycoplasma faucium in a patient with activated PI3Kδ syndrome type 2. J Allergy Clin Immunol Pract (2018) 6(5):1796–8.e2. doi: 10.1016/j.jaip.2018.02.014
108. Rachid R, Castigli E, Geha RS, Bonilla FA. TACI mutation in common variable immunodeficiency and IgA deficiency. Curr Allergy Asthma Rep (2006) 6(5):357–62. doi: 10.1007/s11882-996-0004-9
109. Abolhassani H, Hammarström L, Cunningham-Rundles C. Current genetic landscape in common variable immune deficiency. Blood (2020) 135(9):656–67. doi: 10.1016/B978-0-12-816768-7.00018-1
110. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B Cells in Patients with Heterozygous Mutations in IKAROS. N Engl J Med (2016) 374(11):1032–43. doi: 10.1056/NEJMoa1512234
111. Driessen GJ, IJspeert H, Wentink M, Yntema HG, van Hagen PM, van Strien A, et al. Increased PI3K/Akt activity and deregulated humoral immune response in human PTEN deficiency. J Allergy Clin Immunol (2016) 138(6):1744–7.e5. doi: 10.1016/j.jaci.2016.07.010
112. Frans G, Moens L, Schaballie H, Wuyts G, Liston A, Poesen K, et al. Homozygous N-terminal missense mutation in TRNT1 leads to progressive B-cell immunodeficiency in adulthood. J Allergy Clin Immunol (2017) 139(1):360–3.e6. doi: 10.1016/j.jaci.2016.06.050
113. Kavli B, Andersen S, Otterlei M, Liabakk NB, Imai K, Fischer A, et al. B cells from hyper-IgM patients carrying UNG mutations lack ability to remove uracil from ssDNA and have elevated genomic uracil. J Exp Med (2005) 201(12):2011–21. doi: 10.1084/jem.20050042
114. Methot SP, Di Noia JM. Molecular Mechanisms of Somatic Hypermutation and Class Switch Recombination. Adv Immunol (2017) 133:37–87. doi: 10.1016/bs.ai.2016.11.002
115. Kracker S, Gardes P, Mazerolles F, Durandy A. Immunoglobulin class switch recombination deficiencies. Clin Immunol (2010) 135(2):193–203. doi: 10.1007/978-1-4419-6448-9_15
116. Yel L. Selective IgA Deficiency. J Clin Immunol (2010) 30(1):10–6. doi: 10.1007/s10875-009-9357-x
117. Yazdani R, Azizi G, Abolhassani H, Aghamohammadi A. Selective IgA Deficiency: Epidemiology, Pathogenesis, Clinical Phenotype, Diagnosis, Prognosis and Management. Scand J Immunol (2017) 85(1):3–12. doi: 10.1111/sji.12499
118. Janzi M, Kull I, Sjöberg R, Wan J, Melén E, Bayat N, et al. Selective IgA deficiency in early life: Association to infections and allergic diseases during childhood. Clin Immunol (2009) 133(1):78–85. doi: 10.1016/j.clim.2009.05.014
119. Latiff AHA, Kerr MA. The clinical significance of immunoglobulin A deficiency. Ann Clin Biochem (2007) 44(Pt 2):131–9. doi: 10.1258/000456307780117993
120. Lewkonia RM, Lin CC, Haslam RHA. Selective IgA deficiency with 18q+ and 18q- karyotypic anomalies. J Med Genet (1980) 17(6):453–6. doi: 10.1136/jmg.17.6.453
121. Burgio GR, Duse M, Monafo V, Ascione A, Nespoli L. Selective IgA deficiency: Clinical and immunological evaluation of 50 pediatric patients. Eur J Pediatr (1980) 133(2):101–6. doi: 10.1007/BF00441577
122. McGoey RR, Gedalia A, Marble M. Monosomy 18p and immunologic dysfunction: Review of the literature and a new case report with thyroiditis, IgA deficiency, and systemic lupus erythematosus. Clin Dysmorphol (2011) 20(2):127–30. doi: 10.1097/MCD.0b013e3283414db7
123. Aghamohammadi A, Cheraghi T, Gharagozlou M, Movahedi M, Rezaei N, Yeganeh M, et al. IgA deficiency: Correlation between clinical and immunological phenotypes. J Clin Immunol (2009) 29(1):130–6. doi: 10.1007/s10875-008-9229-9
124. Soler-Palacín P, Cobos-Carrascosa E, Martín-Nalda A, Caracseghi F, Hernández M, Figueras-Nadal C. Is familial screening useful in selective immunoglobulin A deficiency? An Pediatr (2016) 84(2):70–8. doi: 10.1016/j.anpede.2015.10.010
125. Rezaei N, Abolhassani H, Kasraian A, Mohammadinejad P, Sadeghi B, Aghamohammadi A. Family study of pediatric patients with primary antibody deficiencies. Iran J Allergy Asthma Immunol (2013) 12(4):377–82.
126. Karaca NE, Severcan EU, Bilgin BG, Azarsiz E, Akarcan S, Gunaydın NC, et al. Familial inheritance and screening of first-degree relatives in common variable immunodeficiency and immunoglobulin A deficiency patients. Int J Immunopathol Pharmacol (2018) 32:2058738418779458. doi: 10.1177/2058738418779458
127. Aghamohammadi A, Mohammadi J, Parvaneh N, Rezaei N, Moin M, Espanol T, et al. Progression of selective IgA deficiency to common variable immunodeficiency. Int Arch Allergy Immunol (2008) 147(2):87–92. doi: 10.1159/000135694
128. Espanol T, Catala M, Hernandez M, Caragol I, Bertran JM. Development of a common variable immunodeficiency in IgA-deficient patients. Clin Immunol Immunopathol (1996) 80(3 Pt 1):333–5. doi: 10.1006/clin.1996.0132
129. Jorgensen GH, Arnlaugsson S, Theodors A, Ludviksson BR. Immunoglobulin a deficiency and oral health status: A case-control study. J Clin Periodontol (2010) 37(1):1–8. doi: 10.1111/j.1600-051X.2009.01494.x
130. Ludvigsson JF, Neovius M, Hammarström L. Risk of Infections Among 2100 Individuals with IgA Deficiency: a Nationwide Cohort Study. J Clin Immunol (2016) 36(2):134–40. doi: 10.1007/s10875-015-0230-9
131. Moschese V, Chini L, Graziani S, Sgrulletti M, Gallo V, Di Matteo G, et al. Follow-up and outcome of symptomatic partial or absolute IgA deficiency in children. Eur J Pediatr (2019) 178(1):51–60. doi: 10.1007/s00431-018-3248-1
132. Magen E, Waitman DA, Goldstein N, Schlesinger M, Dickstein Y, Kahan NR. Helicobacter pylori infection in patients with selective immunoglobulin a deficiency. Clin Exp Immunol (2016) 184(3):332–7. doi: 10.1111/cei.12765
133. Aytekin C, Tuygun N, Gokce S, Dogu F, Ikinciogullari A. Selective IgA deficiency: Clinical and laboratory features of 118 children in Turkey. J Clin Immunol (2012) 32(5):961–6. doi: 10.1007/s10875-012-9702-3
134. Langford TD, Housley MP, Boes M, Chen J, Kagnoff MF, Gillin FD, et al. Central importance of immunoglobulin A in host defense against Giardia spp. Infect Immun (2002) 70(1):11–8. doi: 10.1128/IAI.70.1.11-18.2002
135. Istrate C, Hinkula J, Hammarström L, Svensson L. Individuals with selective IgA deficiency resolve rotavirus disease and develop higher antibody titers (IgG, IgG1) than IgA competent individuals. J Med Virol (2008) 80(3):531–5. doi: 10.1002/jmv.21101
136. Wolpert J, Knutsen AP. Natural history of selective antibody deficiency to bacterial polysaccharide antigens in children. Pediatr Asthma. Allergy Immunol (1998) 12(3):183–91. doi: 10.1089/pai.1998.12.183
137. Vendrell M, De Gracia J, Rodrigo MJ, Cruz MJ, Alvarez A, Garcia M, et al. Antibody production deficiency with normal IgG levels in bronchiectasis of unknown etiology. Chest (2005) 127(1):197–204. doi: 10.1378/chest.127.1.197
138. Ozkan H, Atlihan F, Genel F, Targan S, Gunvar T. IgA and/or IgG subclass deficiency in children with recurrent respiratory infections and its relationship with chronic pulmonary damage. J Investig Allergol Clin Immunol (2005) 15(1):69–74.
139. Meyts I, Bossuyt X, Proesmans M, De B. Isolated IgG3 deficiency in children: To treat or not to treat? Case presentation and review of the literature. Pediatr Allergy Immunol (2006) 17(7):544–50. doi: 10.1111/j.1399-3038.2006.00454.x
140. Abrahamian F, Agrawal S, Gupta S. Immunological and clinical profile of adult patients with selective immunoglobulin subclass deficiency: Response to intravenous immunoglobulin therapy. Clin Exp Immunol (2010) 159(3):344–50. doi: 10.1111/j.1365-2249.2009.04062.x
141. Leitao Filho FS, Ra SW, Mattman A, Schellenberg RS, Criner GJ, Woodruff PG, et al. Serum IgG subclass levels and risk of exacerbations and hospitalizations in patients with COPD. Respir Res (2018) 19(1):30. doi: 10.1186/s12931-018-0733-z
142. Kutukculer N, Karaca NE, Demircioglu O, Aksu G. Increases in serum immunoglobulins to age-related normal levels in children with IgA and/or IgG subclass deficiency. Pediatr Allergy Immunol (2007) 18(2):167–73. doi: 10.1111/j.1399-3038.2006.00491.x
143. Schatorjé EJH, de Jong E, van Hout RWNM, García Vivas Y, de Vries E. The Challenge of Immunoglobulin-G Subclass Deficiency and Specific Polysaccharide Antibody Deficiency – a Dutch Pediatric Cohort Study. J Clin Immunol (2016) 36(2):141–8. doi: 10.1007/s10875-016-0236-y
144. Khokar A, Gupta S. Clinical and Immunological Features of 78 Adult Patients with Primary Selective IgG Subclass Deficiencies. Arch Immunol Ther Exp (Warsz) (2019) 67(5):325–34. doi: 10.1007/s00005-019-00556-3
145. Vanlerberghe L, Joniau S, Jorissen M. The prevalence of humoral immunodeficiency in refractory rhinosinusitis: A retrospective analysis. B-ENT (2006) 2(4):161–6.
146. Louis AG, Gupta S. Primary selective IgM deficiency: An ignored immunodeficiency. Clin Rev Allergy Immunol (2014) 46(2):104–11. doi: 10.1007/s12016-013-8375-x
147. Entezari N, Adab Z, Zeydi M, Saghafi S, Jamali M, Kardar GA, et al. The prevalence of Selective Immunoglobulin M Deficiency (SIgMD) in Iranian volunteer blood donors. Hum Immunol (2016) 77(1):7–11. doi: 10.1016/j.humimm.2015.09.051
148. Gupta S, Gupta A. Selective IgM deficiency-An underestimated primary immunodeficiency. Front Immunol (2017) 8:1056. doi: 10.3389/fimmu.2017.01056
149. Hobbs JR, Milner RDG, Watt PJ. Gamma-M Deficiency Predisposing to Meningococcal Septicaemia. Br Med J (1967) 4(5579):583–6. doi: 10.1136/bmj.4.5579.583
150. Meyts I, Bucciol G, Quinti I, Neven B, Fischer A, Seoane E, et al. Coronavirus Disease 2019 in patients with inborn errors of immunity: an international study. J Allergy Clin Immunol (2020) 147(2):520–31. doi: 10.1016/j.jaci.2020.09.010
151. Rydyznski Moderbacher C, Ramirez SI, Dan JM, Grifoni A, Hastie KM, Weiskopf D, et al. Antigen-Specific Adaptive Immunity to SARS-CoV-2 in Acute COVID-19 and Associations with Age and Disease Severity. Cell (2020) 183(4):996–1012.e19. doi: 10.1016/j.cell.2020.09.038
152. Jin H, Reed JC, Liu STH, Ho HEN, Lopes JP, Ramsey NB, et al. Three patients with X-linked agammaglobulinemia hospitalized for COVID-19 improved with convalescent plasma. J Allergy Clin Immunol Pract (2020) 8(10):3594–6.e3. doi: 10.1016/j.jaip.2020.08.059
153. Soresina A, Moratto D, Chiarini M, Paolillo C, Baresi G, Focà E, et al. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr Allergy Immunol (2020) 31(5):565–9. doi: 10.1111/pai.13263
154. Quinti I, Lougaris V, Milito C, Cinetto F, Pecoraro A, Mezzaroma I, et al. A possible role for B cells in COVID-19? Lesson from patients with agammaglobulinemia. J Allergy Clin Immunol (2020) 146(1):211–3.e4. doi: 10.1016/j.jaci.2020.04.013
155. Lecuit M, Eloit M. The diagnosis of infectious diseases by whole genome next generation sequencing: A new era is opening. Front Cell Infect Microbiol (2014) 4:25. doi: 10.3389/fcimb.2014.00025
156. Lee JL, Mohamed Shah N, Makmor-Bakry M, Islahudin FH, Alias H, Noh LM, et al. A Systematic Review and Meta-regression Analysis on the Impact of Increasing IgG Trough Level on Infection Rates in Primary Immunodeficiency Patients on Intravenous IgG Therapy. J Clin Immunol (2020) 40(5):682–98. doi: 10.1007/s10875-020-00788-5
157. De Gracia J, Vendrell M, Álvarez A, Pallisa E, Rodrigo MJ, De La Rosa D, et al. Immunoglobulin therapy to control lung damage in patients with common variable immunodeficiency. Int Immunopharmacol (2004) 4(6):745–53. doi: 10.1016/j.intimp.2004.02.011
158. Quinti I, Soresina A, Guerra A, Rondelli R, Spadaro G, Agostini C, et al. Effectiveness of immunoglobulin replacement therapy on clinical outcome in patients with primary antibody deficiencies: Results from a multicenter prospective cohort study. J Clin Immunol (2011) 31(3):315–22. doi: 10.1007/s10875-011-9511-0
159. Pulvirenti F, Camilli R, Giufrè M, Milito C, Pimentel de Araujo F, Mancini F, et al. Risk factors for Haemophilus influenzae and pneumococcal respiratory tract colonization in CVID. J Allergy Clin Immunol (2018) 142(6):1999–2002.e3. doi: 10.1016/j.jaci.2018.08.014
160. Chua I, Lagos M, Charalambous BM, Workman S, Chee R, Grimbacher B. Pathogen-specific IgG antibody levels in immunodeficient patients receiving immunoglobulin replacement do not provide additional benefit to therapeutic management over total serum IgG. J Allergy Clin Immunol (2011) 127(6):1410–1. doi: 10.1016/j.jaci.2011.01.035
161. Bonagura VR, Marchlewski R, Cox A, Rosenthal DW. Biologic IgG level in primary immunodeficiency disease: The IgG level that protects against recurrent infection. J Allergy Clin Immunol (2008) 122(1):210–2. doi: 10.1016/j.jaci.2008.04.044
162. Janssen WJM, Mohamed Hoesein F, Van de Ven AAJM, Maarschalk J, van Royen F, de Jong PA, et al. IgG trough levels and progression of pulmonary disease in pediatric and adult common variable immunodeficiency disorder patients. J Allergy Clin Immunol (2017) 140(1):303–6.e4. doi: 10.1016/j.jaci.2016.11.050
163. Berbers RM, Mohamed Hoesein FAA, Ellerbroek PM, van Montfrans JM, Dalm VASH, van Hagen PM, et al. Low IgA Associated With Oropharyngeal Microbiota Changes and Lung Disease in Primary Antibody Deficiency. Front Immunol (2020) 11:1245. doi: 10.3389/fimmu.2020.01245
164. Shrestha P, Karmacharya P, Wang Z, Donato A, Joshi AY. Impact of IVIG vs. SCIG on IgG trough level and infection incidence in primary immunodeficiency diseases: A systematic review and meta-analysis of clinical studies. World Allergy Organ J (2019) 12(10):100068. doi: 10.1016/j.waojou.2019.100068
165. Stubbs A, Bangs C, Shillitoe B, Edgar JD, Burns SO, Thomas M, et al. Bronchiectasis and deteriorating lung function in agammaglobulinaemia despite immunoglobulin replacement therapy. Clin Exp Immunol (2018) 191(2):212–9. doi: 10.1111/cei.13068
166. Milito C, Pulvirenti F, Cinetto F, Lougaris V, Soresina A, Pecoraro A, et al. Double-blind, placebo-controlled, randomized trial on low-dose azithromycin prophylaxis in patients with primary antibody deficiencies. J Allergy Clin Immunol (2019) 144(2):584–93.e7. doi: 10.1016/j.jaci.2019.01.051
167. Howard V, Myers LA, Williams DA, Wheeler G, Turner EV, Cunningham JM, et al. Stem cell transplants for patients with X-linked agammaglobulinemia. Clin Immunol (2003) 107(2):98–102. doi: 10.1016/S1521-6616(03)00045-7
Keywords: primary immunodeficiencies, infections, common variable immunodeficiency, immunoglobulin therapy, selective IgA deficiency, specific antibody deficiency, IgG subclass deficiency, selective IgM deficiency
Citation: Demirdag YY and Gupta S (2021) Update on Infections in Primary Antibody Deficiencies. Front. Immunol. 12:634181. doi: 10.3389/fimmu.2021.634181
Received: 27 November 2020; Accepted: 07 January 2021;
Published: 11 February 2021.
Edited by:
Andrew R. Gennery, Newcastle University, United KingdomReviewed by:
Hassan Abolhassani, Karolinska Institute, SwedenIsabella Quinti, Sapienza University of Rome, Italy
Copyright © 2021 Demirdag and Gupta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yesim Yilmaz Demirdag, RHJ5ZXNpbXlpbG1hekBnbWFpbC5jb20=