 Baoying Cheng1†
Baoying Cheng1† Xin Li1†
Xin Li1† Kai Dai1†
Kai Dai1† Shengshun Duan1
Shengshun Duan1 Zhouyi Rong1
Zhouyi Rong1 Yingmin Chen1
Yingmin Chen1 Liangcheng Lü1
Liangcheng Lü1 Zhaoji Liu1
Zhaoji Liu1 Xiaohua Huang2
Xiaohua Huang2 Huaxi Xu1
Huaxi Xu1 Yun-Wu Zhang1*
Yun-Wu Zhang1* Honghua Zheng1,2*
Honghua Zheng1,2*- 1Fujian Provincial Key Laboratory of Neurodegenerative Disease and Aging Research, Institute of Neuroscience, School of Medicine, Xiamen University, Xiamen, China
- 2Basic Medical Sciences, School of Medicine, Xiamen University, Xiamen, China
Triggering receptor expressed on myeloid cells-2 (TREM2) and colony-stimulating factor 1 receptor (CSF1R) are crucial molecules for microgliopathy, which is characterized by microglia dysfunction and has recently been proposed as the neuropathological hallmark of neurological disorders. TREM2 and CSF1R are receptors expressed primarily in microglia in the brain and modulate microglial activation and survival. They are thought to be in close physical proximity. However, whether there is a direct interaction between these receptors remains elusive. Moreover, the physiological role and mechanism of the interaction of TREM2 and CSF1R remain to be determined. Here, we found that TREM2 interacted with CSF1R based on a co-immunoprecipitation assay. Additionally, we found that CSF1R knockdown significantly reduced the survival of primary microglia and increased the Trem2 mRNA level. In contrast, CSF1R expression was increased in Trem2-deficient microglia. Interestingly, administration of CSF1, the ligand of CSF1R, partially restored the survival of Trem2-deficient microglia in vitro and in vivo. Furthermore, CSF1 ameliorated Aβ plaques deposition in Trem2-/-; 5XFAD mouse brain. These findings provide solid evidence that TREM2 and CSF1R have intrinsic abilities to form complexes and mutually modulate their expression. These findings also indicate the potential role of CSF1 in therapeutic intervention in TREM2 variant-bearing patients with a high risk of Alzheimer’s disease (AD).
Introduction
Triggering receptor expressed on myeloid cells-2 (TREM2), an immunomodulatory receptor dominantly expressed in myeloid lineage cells such as dendritic cells, osteoclasts and microglia, is essential in activation and survival of myeloid cells. The genetic variants of TREM2 result in an increased risk of several neurodegenerative disorders, including Alzheimer’s disease (AD) (1–5). TREM2 consists of an extracellular V-type Ig domain followed by a short stalk (ectodomain = 19–172 aa), leading to a single transmembrane helix that interacts with DNAX-activation protein 12 (DAP12, also known as TYROBP) containing the immunoreceptor tyrosine-based activation motif (ITAM) to mediate downstream signaling (6, 7). We have previously demonstrated that TREM2 deficiency reduced the viability and proliferation of primary microglia, and induced cell cycle arrest at the G1/S checkpoint (8), indicating an important role of TREM2 in microglia survival.
Colony stimulating factor 1 receptor (CSF1R) is also predominantly expressed in monocyte, macrophage and bone marrow cell precursors, fetal trophoblast and chorioma cells, as well as microglia in the central nervous system (CNS) (9–11). Studies have revealed that people with CSF1R variants bear an adult-onset neurodegenerative disorder characterized by dementia and motor impairments, named adult-onset leukoencephalopathy with axonal spheroids and pigmented glia (ALSP) (12, 13). Thus, TREM2 and CSF1R are considered crucial molecules for “microgliopathy”, which is characterized by reactive gliosis and microglia dysfunction and has been recently proposed as the neuropathological hallmark of neurological disorders (5).
The activation of CSF1R by its ligand colony-stimulating factor 1 (CSF1), also known as macrophage CSF (M-CSF), regulates the survival, proliferation and chemotaxis of macrophages and osteoclasts (14). TREM2 is involved in CSF1-mediated survival and proliferation in macrophages and osteoclast precursors and CSF1 signals through CSF1R to activate β-catenin in a TREM2-dependent manner in those cells (15, 16). In Trem2-/- mice, the β-catenin deficit in osteoclast precursors reduces proliferation and prematurely accelerates osteoclast maturation (15). Another study showed that CSF1 stimulation resulted in DAP12 phosphorylation by Src kinases activated by CSF1R in osteoclasts (17). These results suggest that TREM2 and CSF1R are in close physical proximity (6). However, whether there is a direct interaction and regulation between these receptors remains unknown. Moreover, given the crucial role of TREM2 and CSF1R in microglia survival, the precise mechanisms by which these receptors regulate microglia survival and function still await full exploration.
In the current study, we examined the interaction of CSF1R and TREM2 using transient transfection of these two proteins in HEK293T cells. We demonstrated that TREM2 interacted with CSF1R. Moreover, CSF1R was significantly increased in Trem2-/- microglia whereas downregulation of CSF1R impaired the survival of microglia and increased the Trem2 mRNA level. Interestingly, activating CSF1R by CSF1 resulted in significant activation of the Akt signaling pathway, and partially restored the attenuated cell survival in Trem2-/- microglia and in Trem2-/- mouse brain. Furthermore, administration of CSF1 significantly ameliorates Aβ plaques accumulation in Trem2-/-; 5XFAD mouse brain. Thus, our results indicate that TREM2 interacts with CSF1R but is not necessary for CSF1R-mediated microglia survival. This study also provides a viable target for therapeutic intervention by enhancing CSF1R function in TREM2-loss-of-function patients.
Materials and Methods
Reagents
Phospho-CSF1R (Tyr723) (49C10) Rabbit mAb (RRID: AB_2085229), β-catenin (6B3) Rabbit mAb (RRID: AB_331149), anti-Phospho-Akt (Ser473) (RRID: AB_329825) and anti-total-Akt antibodies (RRID: AB_329827) were all bought from Cell Signaling Technology. Anti-GAPDH (RRID: AB_2630358), anti-CSF1R (RRID: AB_2085251), anti-Aβ (MOAB2), anti-β-actin antibodies (RRID: AB_306371) and Alexa Fluor 647 Donkey Anti-Rat IgG H&L (RRID: AB_2813835), as well as the BrdU Cell proliferation ELISA Kit, were all from Abcam. Rat Anti-mouse CD68 monoclonal antibody (RRID: AB_322219) was from Bio-Rad. Anti-HA-tag (RRID: AB_11042321) antibody and anti-Myc-tag antibody (RRID: AB_11182162) were from Proteintech. Rabbit anti-Iba1 (RRID: AB_2687911) was from Wako. Alexa Fluor 488 Goat anti-Rabbit (RRID: AB_10374301), Alexa Fluor 594 Goat anti-Rabbit (RRID: AB_10374440), Alexa Fluor 647 Goat anti-Rabbit antibodies (RRIDAB_10371940) and TRIzol reagent were all from Thermo Fisher Scientific. Granulocyte-macrophage colony stimulating factor (GM-CSF) and CSF1 were from R&D Systems. Basic Glial Cells Nucleofector Kit was purchased from LONZA. ReverTra Ace qPCR RT Master Mix was from TOYOBO. FastStart Universal SYBR Green Master mix was from Roche. CellTiter 96R Aqueous One Solution (MTS assay) and DeadEnd™ Fluorometric TUNNEL System were from Promega.
Culture and Treatment of Primary Microglia
Primary microglial cells were prepared as described previously (8). Briefly, mixed glial cells from newborn (postnatal 1 or 2 d) Trem2-/- or WT C57BL/6 pups were cultured in Dulbecco modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100 U/ml penicillin/streptomycin in poly-L-lysine (Sigma-Aldrich)-coated cell culture flasks (Thermo). The media was replaced the next day with fresh DMEM plus 10% FBS and 25 ng/ml GM-CSF. Microglia cells were harvested by shaking at a speed of 200 rpm for 20 min after 10 days in culture. The isolated WT microglial cells were subjected to CSF1R knockdown by electroporation. Isolated microglia from Trem2-/- or WT littermates were plated for MTS assay, BrdU incorporation assay, or terminal deoxynucleotidyl transferase-mediated-deoxyuridine triphosphate nick-end labeling (TUNEL) staining. For rescue experiments, Trem2-/- or WT microglia were treated with 50 ng/mL CSF1 for 24 hours and used for qRT-PCR, Western blotting, MTS assay, BrdU incorporation assay, or TUNEL staining.
Animals and Brain Stereotaxic Injection
Short-Term Injection of CSF1 in Trem2-/- Mouse Brain
TREM2 knockout (KO) mice (Trem2-/-; C57BL/6) and wild type (WT) C57BL/6 littermates were obtained from the UC Davis Knockout Mouse Project (KOMP) repository as described previously (18) and were group housed (12 hr/12 hr light/dark) in the Laboratory Animal Center of Xiamen University. Brain stereotaxic injection was performed as described previously (19). Briefly, two-month-old Trem2-/- and age matched WT mice were anesthetized and placed in a stereotaxic frame. A skin incision was made, and holes were drilled at x (1.0 mm from bregma) and y (−0.5 mm from bregma). Sterile PBS (3.0 μL) with or without 30 ng CSF1 was delivered into the right cerebral ventricle at 0.15 µL/min at z-depths of 2.3 mm. The syringe was left in place for 10 min after each injection before being withdrawn slowly. Following injection daily for three days, mice were anesthetized, and the brains were dissected. For biochemical analysis, hippocampus and cortex from the left brain were dissected out, snap-frozen in liquid nitrogen, and stored at −80 °C for later extraction of protein and RNA. For histologic analysis, the right hemisphere was fixed with 4% PFA overnight at 4 °C and transferred to 30% sucrose for 48 hours before being embedded for cryostat sectioning.
Long-Term Injection of CSF1 in Trem2-/-; 5XFAD Mouse Brain
Long-term injection of CSF1 was carried out according to our previous study (20). Briefly, male Trem2-/-; 5XFAD mice (3 to 3.5 months of age) were anaesthetized and stereotaxically injected with 3 μl of 10 ng/μL CSF1 into the right lateral ventricle per week (0.5 mm lateral to the midline, 1.0 mm posterior to the bregma, at a depth of 2.3 mm). Mice were anaesthetized and perfused 2.5 months later. Vehicles were used as Artificial cerebrospinal fluid (ACSF) - treated controls in these experiments. Brains were obtained and post-fixed overnight at 4°C in 4% PFA in PBS. Coronal sections from mouse brains were subjected to Iba1 (1:400), CD68(1:200) and MOAB2 (1:400) immunofluorescence staining and were assessed by independent observer blind to animal treatment groups.
Co-Immunoprecipitation Assay
HEK293T cells were co-transfected with HA-tagged CSF1R and Myc-tagged TREM2. Twenty-four hours later, cells were harvested and lysed in 1% TNEN (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 2 mM EDTA, 1% NP-40) lysis buffer supplemented with protease inhibitors. For all experiments, total protein concentration was determined by bicinchoninic acid (BCA) assay (Boster). Lysates were immunoprecipitated using anti-Myc or HA antibodies in the presence of Protein G Dynabeads (Thermo Fisher), followed by immunoblot analysis.
CSF1R Knockdown by siRNA
Electroporation in primary microglia was performed as described by us previously (8). Briefly, knockdown of CSF1R with Csf1r-specific siRNA (GenePharma) was achieved by electroporation using an Amaxa Nucleofector and a glial specific Nucleofector kit (LONZA) according to the manufacturer’s instructions. Each electroporation reaction contained 106 cells and 200 nM siRNA. Transfected cells were plated and used for qRT-PCR, Western blotting, MTS assay.
TUNEL Staining
Cells were fixed in 4% polyformaldehyde (Sigma-Aldrich) for 15 min, and then subjected to TUNEL staining according to the manufacturer’s instructions (Dead End Fluorometric TUNNEL System, promega, G3250). DAPI was used to stain cell nuclei. Seven different fields per HPF were selected randomly to quantify the number of TUNEL-positive cells. TUNEL-positive cell nuclei were counted manually and compared with the total microglia number counted manually as DAPI and given as a percentage. The quantification data were from at least three independent experiments.
Reverse Transcription and Quantitative Real-Time PCR
Total RNA was isolated from mouse brain or primary microglia using TRIzol reagent (Thermo Fisher Scientific). Reverse transcription was performed using ReverTra Ace qPCR RT Master Mix (TOYOBO) and quantitative PCR (qPCR) was performed using the FastStart Universal SYBR Green Master mix (Roche). The set of β-actin primers was used as an internal control for each specific gene amplification. The qPCR was performed on a LightCycler 480 (Roche). The real-time value for each sample was averaged and compared using the cycle threshold (CT) method, where the amount of target RNA (△CT) was normalized to the endogenous β-actin reference (CT) and then normalized against control levels. The primer sequences were as follows: β-actin-forward: 5’-TCTTGGGTATGGAATCCTGTGGCA-3’; β-actin-reverse: 5’-TCTCCTTCTGCATCCTGTCAGCAA-3’; Csf1r-forward: 5’-GGTTGTAGAGCCGGGTGAAA-3’; Csf1r-reverse: 5’-AAGAGTGGGCCGGATCTTTG-3’; Trem2-forward: 5’-TGCTGGCAAAGGAAAGGTG-3’; and Trem2-reverse: 5’-GTTGAGGGCTTGGGACAGG-3’.
Western Blotting
Samples were homogenized and incubated in RIPA Lysis and Extraction Buffer (Boster), supplemented with Protease and Phosphatase Inhibitor Cocktail (Thermo). Protein concentrations were determined using the BCA Protein Assay Kit (Boster) according to the manufacturer’s instructions. Equal amounts of total proteins were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to PVDF membranes (Millipore, IPVH00010). After blocking, the membranes were blotted using a primary antibody (1:1,000) and detected with horseradish peroxidase-conjugated secondary antibody (1:10,000). Proteins were visualized using ECL Western blotting detection reagents (Yeasen). Immunoreactive bands were quantified using ImageJ software (8).
MTS Assay
Cell viability was analyzed using the MTS assay. Briefly, microglia were cultured in 96-well plates at a concentration of 2 × 104/well and treated with or without 50 ng/ml CSF1. After 24 hours, 20 μl of MTS One Solution Reagent was pipetted into each well of the 96-well assay plate containing the cells in 100 μl culture medium. Then the plate was incubated at 37°C for 2 hours in a humidified, 5% CO2 atmosphere before recording the absorbance at 490 nm using an automated ELISA plate reader (Varioskan Flash, Thermo).
BrdU Incorporation Assay
The BrdU incorporation assay was performed using the BrdU cell proliferation ELISA kit (Abcam) according to the manufacturer’s protocol. Briefly, microglial cells were seeded at a density of 104 cells/well and BrdU was added to the cells for 24 hours for incorporation. Cells were then fixed, permeabilized, and denatured to enable the detection of incorporated BrdU through anti-BrdU antibody. After the incubation of horseradish peroxidase-conjugated secondary antibody, BrdU-positive ELISA signals were quantified using a spectrophotometer (Varioan Flash; Thermo Fisher Scientific) at a dual wavelength of 450/550 nm and normalized against total cell numbers.
Immunofluorescence Staining
The coronal brain slices were fixed with 4% PFA in PBS for 30 min at room temperature (RT). Before staining, the slices were washed thoroughly with PBS. The slices were then permeabilized with 0.1% Triton X-100 for 15 min at RT. The nonspecific binding sites were blocked with 5% normal goat serum in PBS for 30 min at RT and the slices were incubated with the mouse anti-Iba1 primary antibody (1:400; Wako Chemicals) and/or MOAB2 (1:400) and/or CD68 (1:200) overnight at 4°C. The slices were then washed three times with PBS and incubated with the Alexa Fluor 488-conjugated goat anti-mouse secondary antibody (1:500; Thermo) for 2 hours at RT. The slices were washed three times with PBS and sealed with an anti-fade reagent (Life Technologies, P36935). The images were captured with an Olympus FV10-ASW confocal microscope. Five different fields per high-power field (HPF) in hippocampi and cortices of at least five sections per mouse from three to five mice were randomly selected to quantify the number of microglia double stained with Iba1 and/or MOAB2 with Olympus FV10-ASW 4.0 Viewer software.
Statistics
Data were presented as mean ± SEM. Two groups were compared using two-tailed Student’s t-tests. For more than two groups, one- or two-way ANOVA was used, as appropriate, followed by Dunnett’s or Tukey’s post hoc adjustment. All analyses were performed with GraphPad Prism 7 software. p < 0.05 was considered statistically significant.
Results
TREM2 Interacts With CSF1R
It has been proposed that TREM2 and CSF1R are in close physical proximity (6). We thus explored their potential interactions by immunoprecipitation assays. We first generated full-length TREM2 or CSF1R plasmid and found that full-length TREM2 interacted with full-length CSF1R (Figures 1A, B). We then generated various TREM2 or CSF1R truncated constructs that respectively carry an additional Myc tag or HA tag at the carboxyl terminus (Figure 1A) to find the specific fragment of their interaction. We found that full-length TREM2 did not interact with the CSF1R amino-terminal fragment (NTF, 1 aa - 514 aa) (Figure 1C) but interacted with the CSF1R carboxyl-terminal fragment (CTF, 514 aa - 972 aa) (Figure 1C), which possesses the transmembrane domain. Reciprocally, full-length CSF1R also interacted with TREM2-CTF (164 aa - 230 aa) that contains the transmembrane domain (Figure 1D), and TREM2-NTF (1 aa -171 aa) that has no transmembrane domain (Figure 1D), indicating that the transmembrane of TREM2 and CSF1R are colocalized and interact with each other. Taken together, these results indicate that these two receptors can interact with one another.
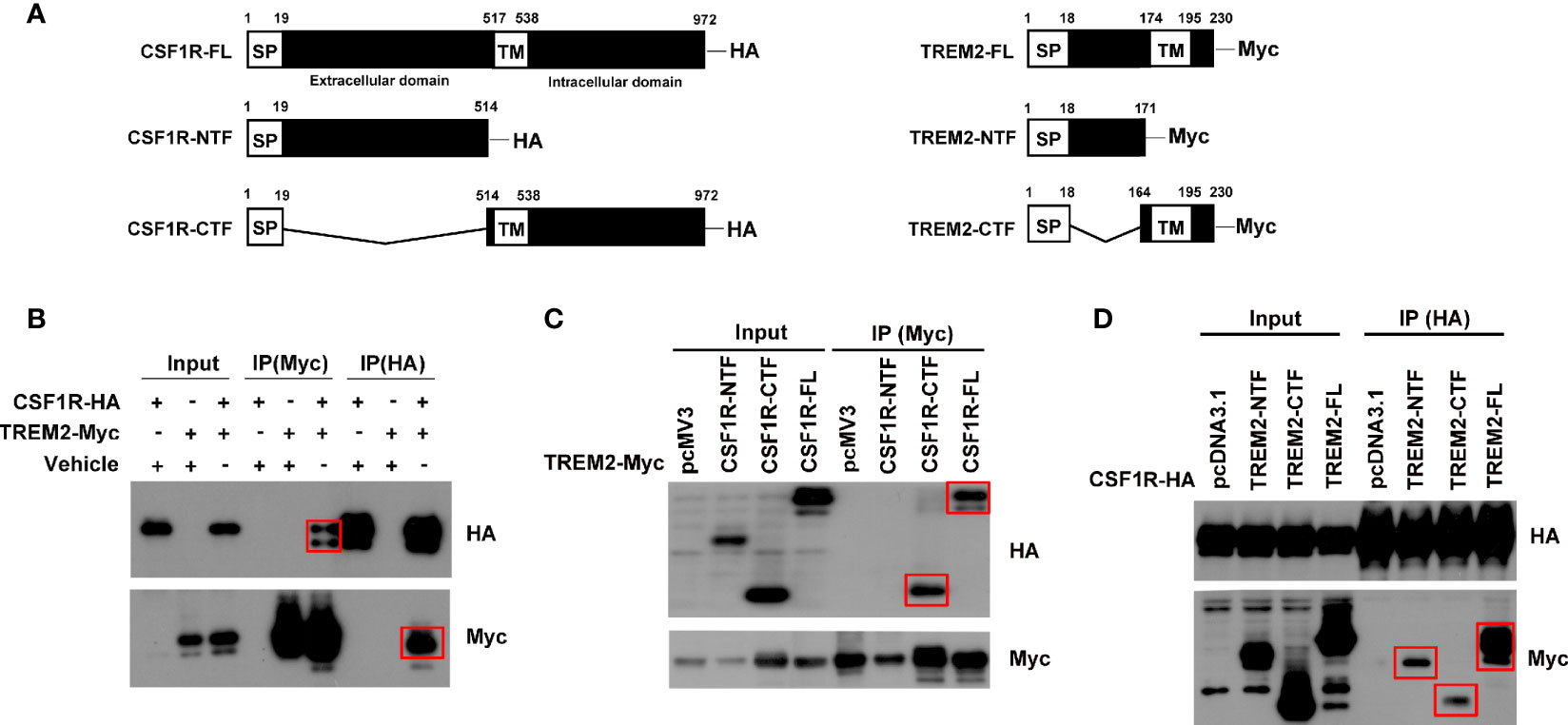
Figure 1 TREM2 interacts with CSF1R. HEK293T cells were transfected with different combinations of indicated plasmids, and Western blotting was performed using HA and Myc-tag antibodies. (A) Schematic diagram of fragments of CSF1R and TREM2. (B) Co-IP of HA-tagged full-length CSF1R with Myc-tagged full-length TREM2. (C) Co-IP of HA-tagged CSF1R fragments with Myc-tagged full-length TREM2. (D) Co-IP of HA-tagged full-length CSF1R with Myc-tagged TREM2 fragments. Red rectangles indicate positive bands.
Increased Expression of CSF1R in Trem2-/- Microglia
Protein interactions mediate many types of cellular processes. To further explore the potential mutual effects of CSF1R and TREM2 on their expression, we determined the effect of TREM2 on CSF1R expression in primary microglia. The mRNA and protein levels of CSF1R in primary microglia from Trem2-/- or WT mice were detected by qPCR and Western blotting assay, respectively (Figures 2A–D). As expected, Csf1r mRNA was significantly increased in Trem2-/- microglia when compared to that in WT cells (Figure 2B). Similarly, CSF1R protein level was also enhanced in Trem2-/- microglia compared with WT cells (Figures 2C, D). Conversely, to further explore the effects of CSF1R on TREM2 expression, two independent siRNAs (Si1 and Si2) were used to knock down the expression of Csf1r in microglia and the knockdown efficacy was verified by qRT-PCR (Figure 2E). Compared to non-targeting (NT) control, Trem2 mRNA transfected with Csf1r siRNAs was significantly increased (Figure 2F). Collectively, these data demonstrated that CSF1R and TREM2 mutually modulate their expression and the expression of CSF1R is increased in Trem2 KO microglia.
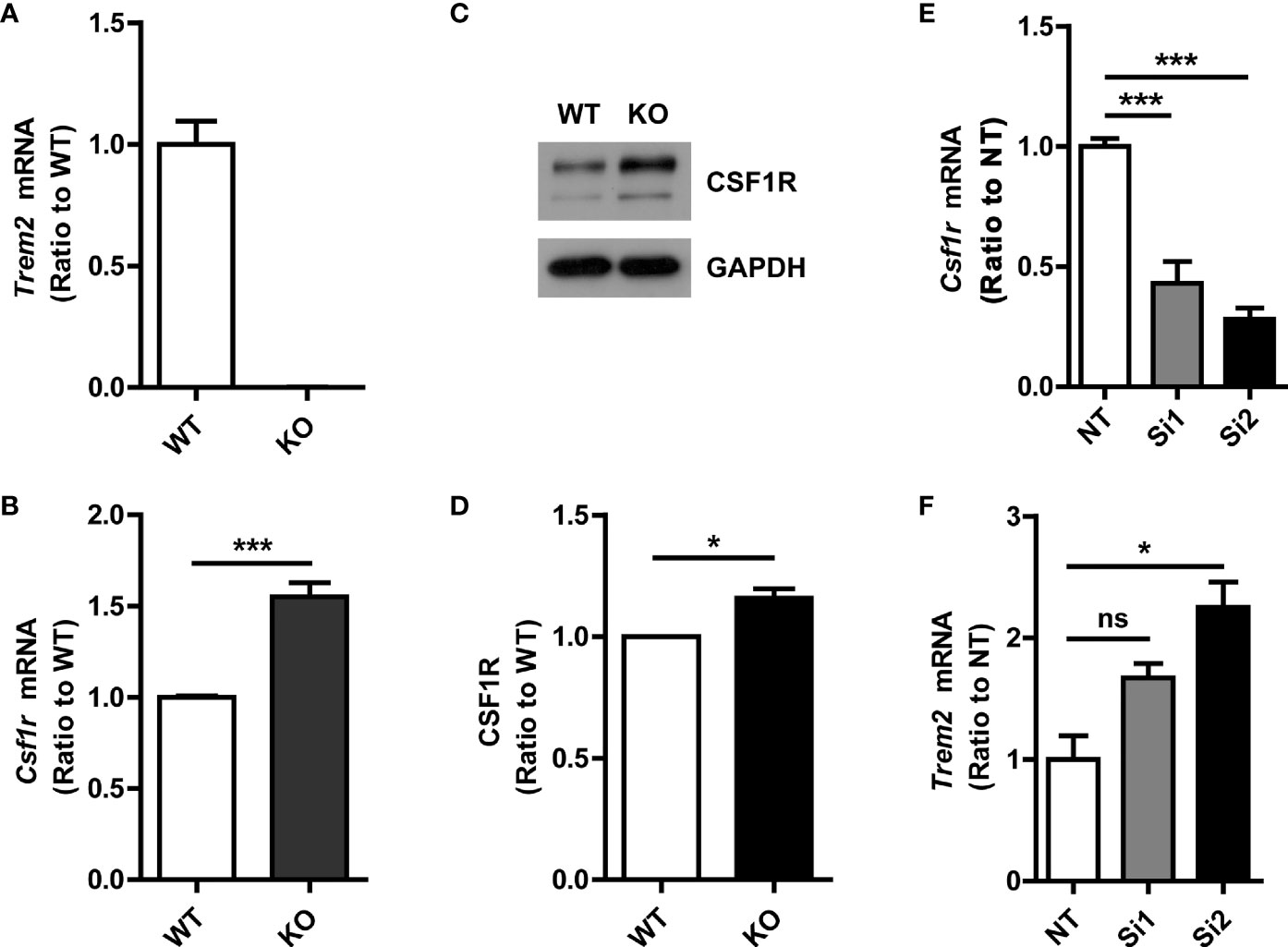
Figure 2 Increased CSF1R expression in Trem2-/- (knockout, KO) microglia. (A) Trem2 KO in mouse primary microglia was confirmed by qRT-PCR. (B) Csf1r mRNA increased in Trem2 KO primary microglia. (C) Representative images of Western blotting. (D) CSF1R was increased in Trem2 KO primary microglia. Protein levels of CSF1R were quantified by densitometry and are presented as ratios to GAPDH. (E) Csf1r was knocked down in primary microglia by two independent siRNAs and the knockdown efficiency was assessed by qRT-PCR. (F) Trem2 mRNA was increased in Csf1r knockdown primary microglia. One-way ANOVA with Dunnett’s multiple comparison test. Data are plotted as mean ± SEM (n=3). *p<0.05; ***p<0.001; ns, not significant.
CSF1R Knockdown Significantly Decreases Microglial Survival
Colony-stimulating factor 1 (CSF1) regulates the survival, proliferation, and differentiation of mononuclear phagocytic cells and is the primary regulator of mononuclear phagocyte production in vivo (21). Moreover, genetic loss of the CSF1R blocks the normal population of resident microglia in the brain that originates from the yolk sac during early development. Microglia are >99% depleted at embryonic day 16 and day 1 post-partum brain in mice homozygous for a null mutation in the Csflr gene or in mice for selective CSF1R inhibitors extensive treatment (22, 23). To this end, we tested the effects of transitory CSF1R depletion on microglia survival. CSF1R was significantly knocked-down by two independent siRNAs, which was verified by qPCR (Figure 3A) and Western blotting assay (Figures 3B, C). Through genetic manipulation, we found that cell viability was suppressed significantly in Csf1r knockdown microglia (Figure 3D) as examined by MTS assay. To further quantify the apoptotic and necrotic status of Csf1r knockdown microglia by TUNEL staining, we assessed the number of TUNEL-positive cells (green) that co-localized with DAPI compared with total DAPI-positive cells (blue) in those cells (Figures 3E, F). The number of TUNEL-positive cells that co-localized with DAPI was significantly increased in Csf1r knockdown microglia compared with the NT control microglia (Figure 3E). These results suggest that CSF1R is critical for microglia survival.
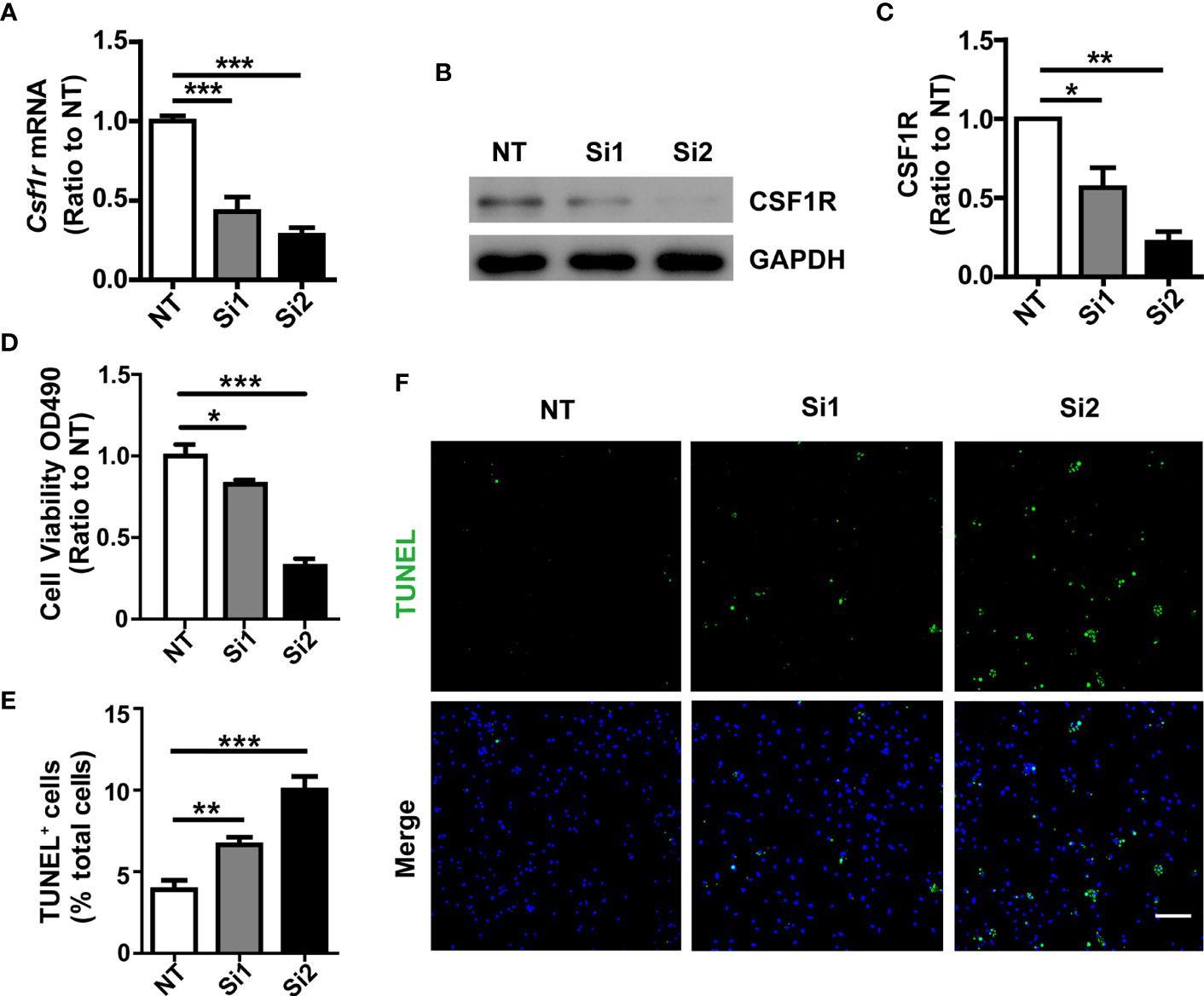
Figure 3 Decreased cell survival in Csf1r knockdown microglia. (A) Csf1r was knocked down in primary microglia by two independent siRNAs and the knockdown efficiency was assessed by qRT-PCR. (B, C) Csf1r knockdown efficiency was assessed by Western blotting. (D) Knockdown of Csf1r suppressed microglial viability as examined by MTS assay. (E, F) The apoptosis and necrosis of Csf1r knockdown microglia were assessed by TUNEL staining. TUNEL-positive cells (green) that co-localized with DAPI were quantified as a percentage of total DAPI-positive cells (blue). The number of TUNEL-positive cells that co-localized with DAPI increased in Csf1r knockdown microglia compared with nontarget control (NT). Scale bar, 50 µm. Two-way ANOVA with Tukey’s multiple comparison test. Data are plotted as mean ± SEM (n=3). *p<0.05; **p<0.01; ***p<0.001.
CSF1 Activates Akt Signaling in Trem2-/- Microglia
Lack of TREM2 impairs proliferation and β-catenin activation in osteoclast precursors (OcP) in response to CSF1, the ligand of CSF1R (15). Moreover, TREM2 deficiency reduced the viability and proliferation of microglia, and decreased the stability of β-catenin (8). Given that the known ligands for CSF1R are CSF1 and IL34 (24), we then detected these two ligands levels in Trem2-/- microglia and found that Csf1 mRNA was not changed whereas IL-34 mRNA was increased in Trem2-/- microglia when compared to those in wild-type (WT) microglia (Supplementary Figures 1A, B), suggesting that these two ligands of CSF1R may play different role in Trem2-/- mouse brain. To elucidate whether TREM2 is necessary for CSF1/CSF1R signaling in microglia, Trem2-/- or WT microglia were cultured in DMEM containing 50 ng/ml CSF1 for 5 min, 15 min, 30 min, and 60 min, respectively. Consistent with previous results in DAP12-deficient macrophages and Trem2-deficient OcP (15), the expression of β-catenin in total cell lysates was reduced in the absence of TREM2 and was not restored in Trem2-/- microglia in response to transient CSF1 treatment (Supplementary Figures 2A, B). We then analyzed the phosphorylation of CSF1R and Akt in those cells. The phosphorylation of CSF1R and Akt were elevated both in Trem2-/- and in WT microglia in response to CSF1 (Figures 4A–C), which was also consistent with previous studies that TREM2 was not required for CSF1R-dependent phosphorylation of Akt in OcP cells (15). The increase was highest at 5 min of CSF1 treatment and was more apparent in Trem2-/- microglia than in WT microglia (Figures 4A–C). These results suggest that TREM2 is not necessary for CSF1-mediated CSF1R and Akt phosphorylation in Trem2-deficient microglia.
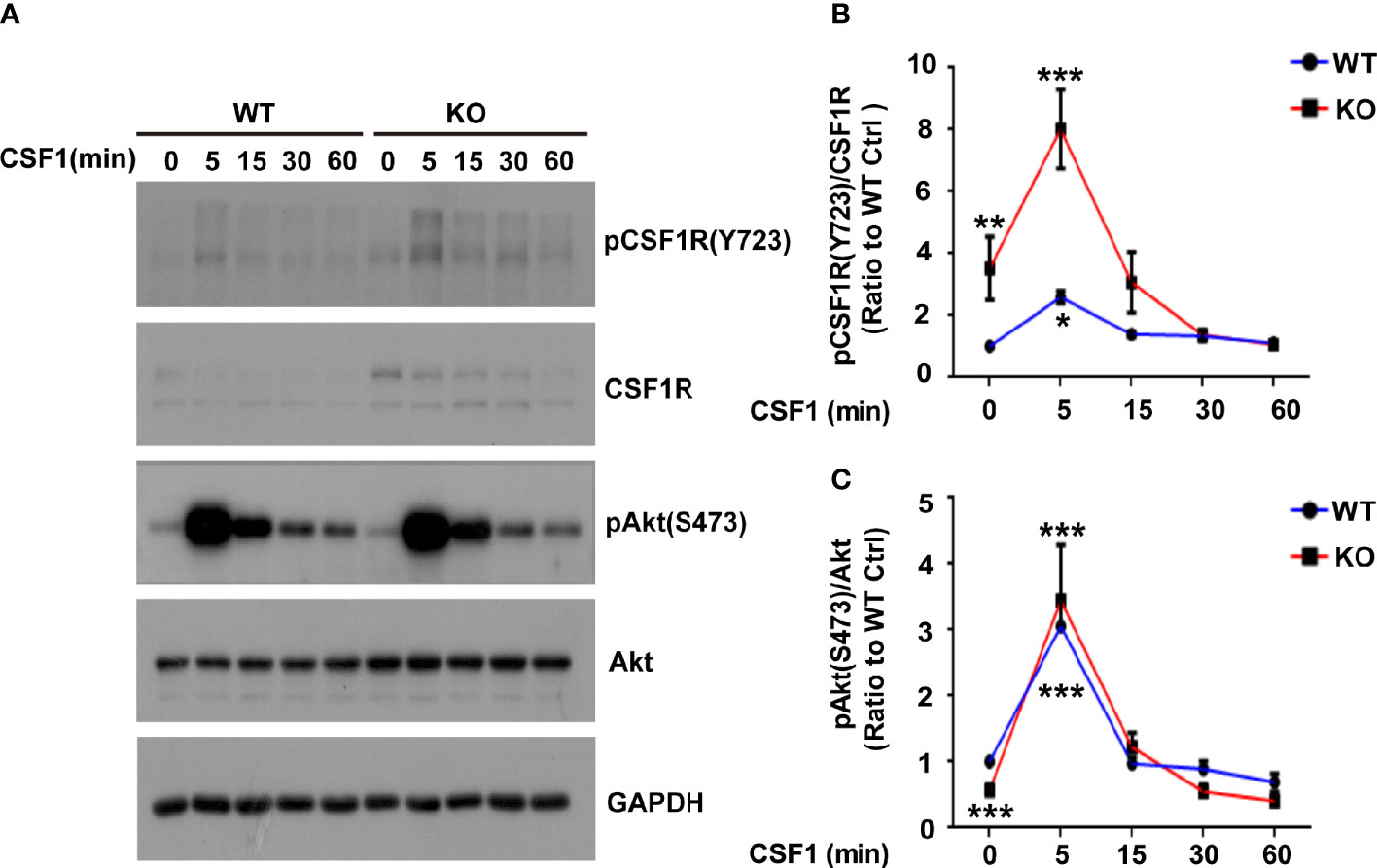
Figure 4 The Akt signal is activated in response to CSF1 in WT and Trem2 KO primary microglia. (A) Primary microglia from WT or Trem2 KO mice were treated with CSF1 (50 ng/ml) for 5, 15, 30 or 60 mins and cell lysates were analyzed by Western blotting. Representative images of Western blotting for the total and phosphorylation of CSF1R and Akt are shown. (B) Protein levels of phosphorylated CSF1R at Tyr723 site (pCSF1RTyr723) were quantified by densitometry and are presented as ratios to GAPDH and total CSF1R. The pCSF1RTyr723 was increased to the highest level at 5-min CSF1 treatment and gradually returned to the normal level. (C) Protein levels of phosphorylated Akt (Ser473) were quantified by densitometry and are presented as ratios to GAPDH and total Akt. The pAktSer473 was increased to the highest level in response to 5-min treatment of CSF1. Two-way ANOVA with Tukey’s multiple comparison test. Data are plotted as mean ± SEM (n=3). Ratio to WT microglia without CSF1 treatment, *p<0.05; **p<0.01; ***p<0.001.
CSF1 Administration Restores Cellular Survival in Trem2-/- Microglia In Vivo and In Vitro
Given that CSF1R was increased in Trem2-/- microglia and that Akt, an important molecule responsible for maintaining cell survival, was significantly activated in Trem2-/- microglia in response to CSF1, it is conceivable that CSF1 may have remedial action in microglia survival and microgliosis in Trem2-/- microglia and/or in Trem2-/- mouse brain. Accordingly, to further evaluate the impact of CSF1 on Trem2-deficient microglia survival, Trem2-/- or WT microglia were treated with 50 ng/ml CSF1 for 24 hours. As expected, CSF1 promoted the cell viability in Trem2-/- microglia as by the MTS assays (Figure 5A), although it did not completely restore the effect to the levels in WT cells. Moreover, the proliferation of microglia in Trem2-/- microglia was significantly increased in response to CSF1 (Figure 5B). Additionally, apoptosis was significantly suppressed in Trem2-/- microglia upon CSF1 treatment (Figures 5C, D). To evaluate the role of CSF1 in regulating Trem2-/- microglial survival in vivo, Trem2-/- or WT mice were intralateroventricularly microinjected with 30 ng CSF1 daily for three days. We found that CSF1 treatment resulted in a significant increase in the number of microglia in Trem2-/- and WT mouse brain though this response in Trem2-deficient mice was not the same as in WT mice (Figures 6A–C). These results showed that CSF1 treatment partially restored the number of microglia in the Trem2-/- mouse brain, indicating that activation of the CSF1R signaling pathway partially restores the survival and/or promotes the proliferation of Trem2-/- microglia both in vitro and in vivo.
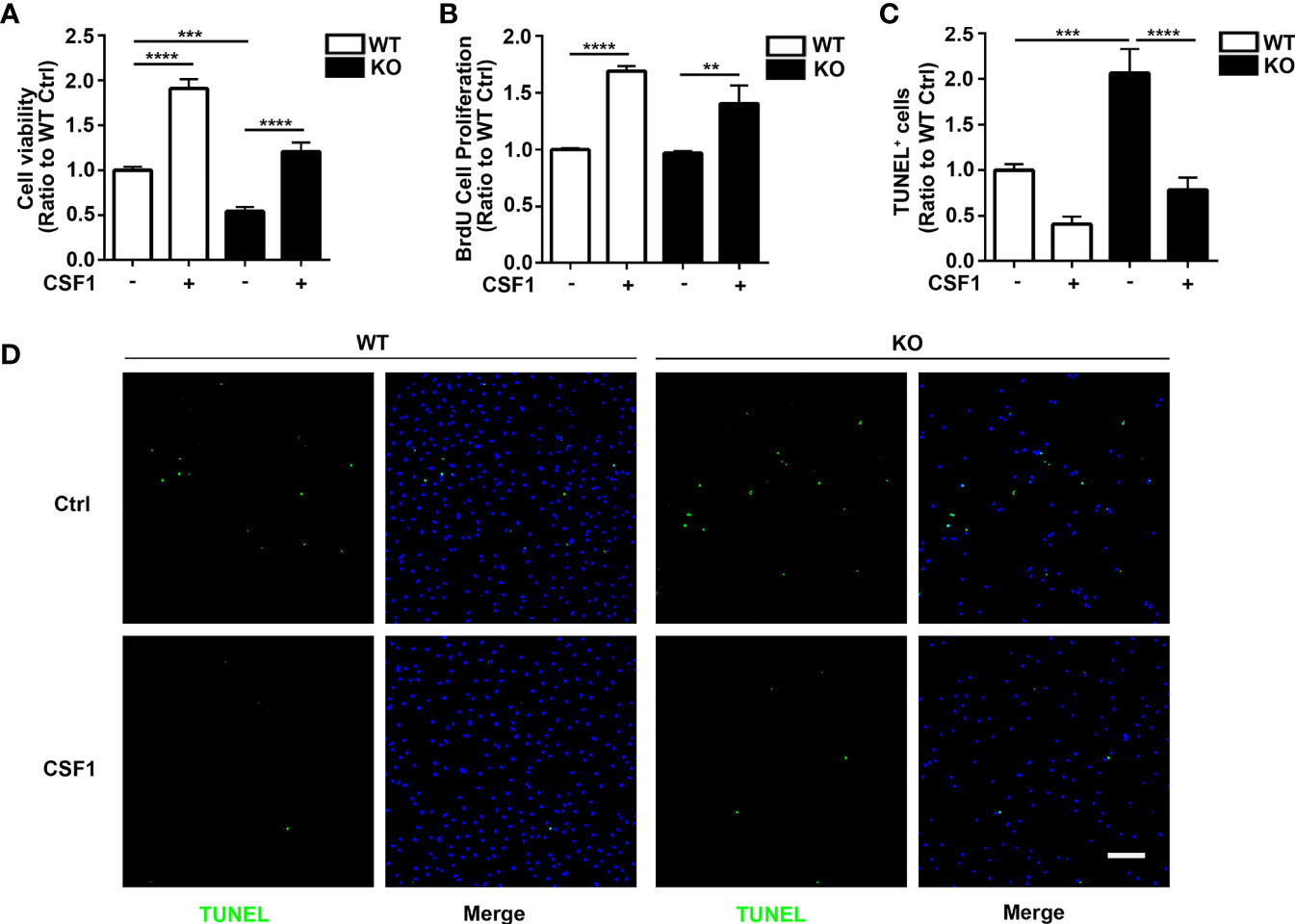
Figure 5 CSF1 restores cell growth and viability in Trem2 KO microglia. (A) Cell viability of Trem2 KO microglia was enhanced in response to CSF1 treatment using MTS assays. (B) CSF1 promotes Trem2 KO microglia proliferation using BrdU incorporation assay. (C) Apoptosis and necrosis of Trem2 KO microglia were assessed by TUNEL staining. TUNEL-positive, apoptotic cells (green) were quantified as a percentage of total DAPI-positive cells (blue). CSF1 inhibited the apoptosis and necrosis of Trem2 KO microglia. (D) Representative images of TUNEL staining that co-localized with DAPI. Scale bar, 50 µm. Two-way ANOVA with Tukey’s multiple comparison test. Data are plotted as mean ± SEM (n=3). **p<0.01; ***p<0.001; ****p<0.0001.
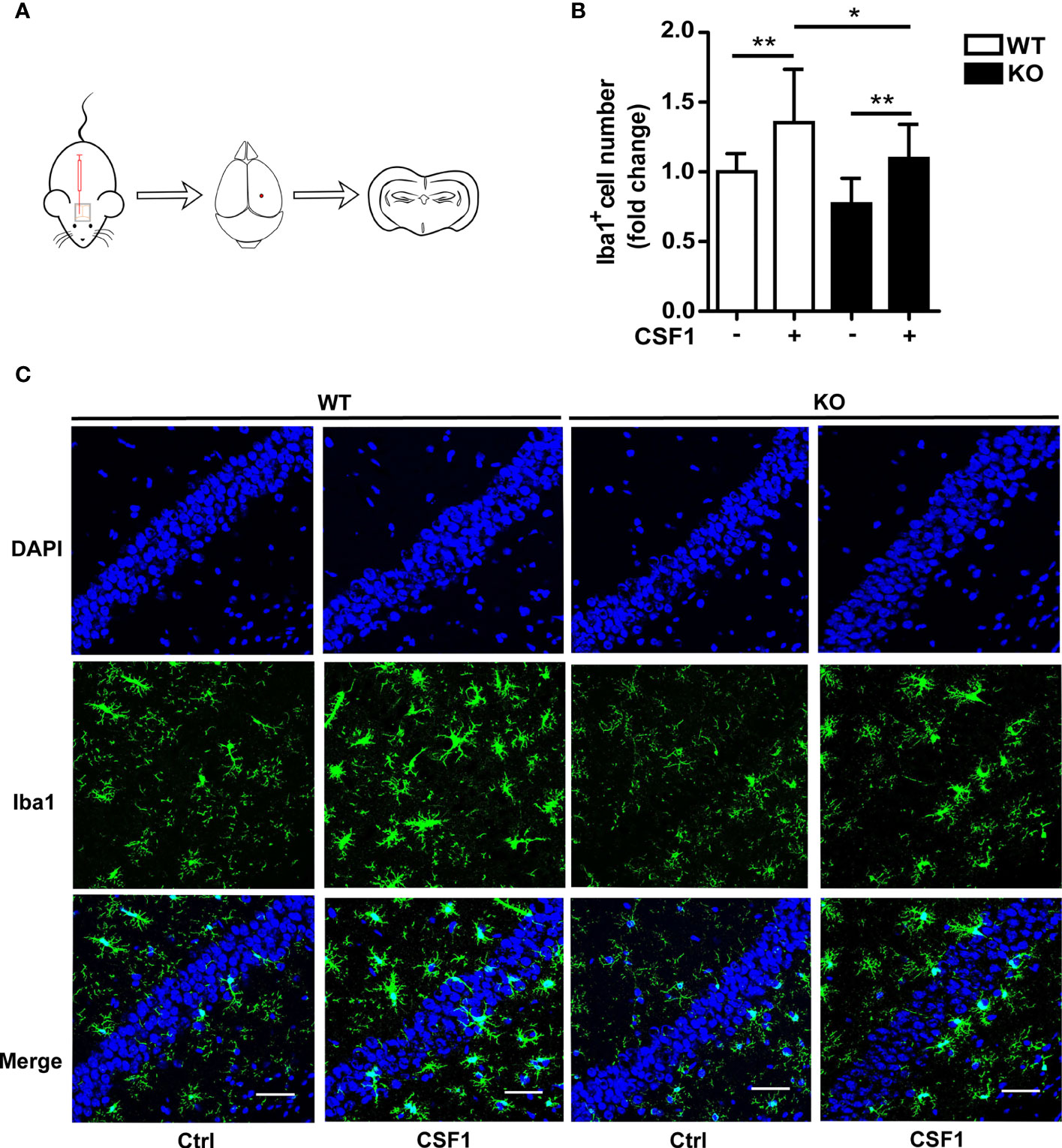
Figure 6 CSF1 administration contributes to microglial survival in Trem2 KO mouse brain. (A) Schematic map of CSF1 intracerebroventricular injection in mouse brain. Two-month-old Trem2 KO and WT littermates were intralateroventricularly microinjected with 30 ng CSF1 daily for three days. Hippocampi and cortex from the left hemispheres of brains were dissected for protein and RNA extraction, and the right hemispheres of brains were embedded for cryostat sectioning and immunofluorescence. (B) Coronal sections from Trem2 KO or WT mouse brain were stained with Iba-1 (green) for microglia. Number of microglia double stained with DAPI (blue) and Iba1 (green) was quantified per HPF in Trem2 KO or WT mice. Microglia number response to CSF1 was assessed by quantifying the cell number ratio of CSF1 group to control group in WT or Trem2 KO brain. (C) Representative images of microglial immunofluorescent staining are shown. Two-way ANOVA with Tukey’s multiple comparison test. Data are plotted as mean ± SEM (n=3). *p<0.05; **p<0.01. Scale bar, 50 µm.
CSF1 Administration Ameliorates Aβ Plaques Accumulation in Trem2-/-; 5XFAD Mouse Brain
Due to the significant increase in Trem2-/- microglial survival in response to CSF1, we next want to know whether CSF1 may play remedial role in Aβ plaques accumulation in Trem2-/-; 5XFAD mouse brain. Thus, Trem2-/-; 5XFAD mice were treated with 30 ng CSF1 or ACSF weekly for 2.5 months (Figure 7A). Interestingly, MOAB2 staining of plaques revealed that CSF1 treatment resulted in a significant reduction in Aβ plaques accumulation in Trem2-/-; 5XFAD mouse brain (Figures 7B, C). As expected, CSF1 promoted microglial activation in Trem2-/-; 5XFAD mice as assessed by Iba1 immunofluorescence assays (Figures 7B, D). Meanwhile, there was a dramatic enhancement in CD68+ Iba1+ microglial cells associated with plaques in Trem2-/-; 5XFAD mouse brain in response to CSF1 treatment, indicating that CSF1 remarkably activated microglia into a phagocytic state (Figures 7B, E). These results showed that CSF1 treatment ameliorates Aβ plaques accumulation in the Trem2-/-; 5XFAD mouse brain, indicating that activation of the CSF1R signaling pathway maybe a therapeutic strategy in AD treatment.
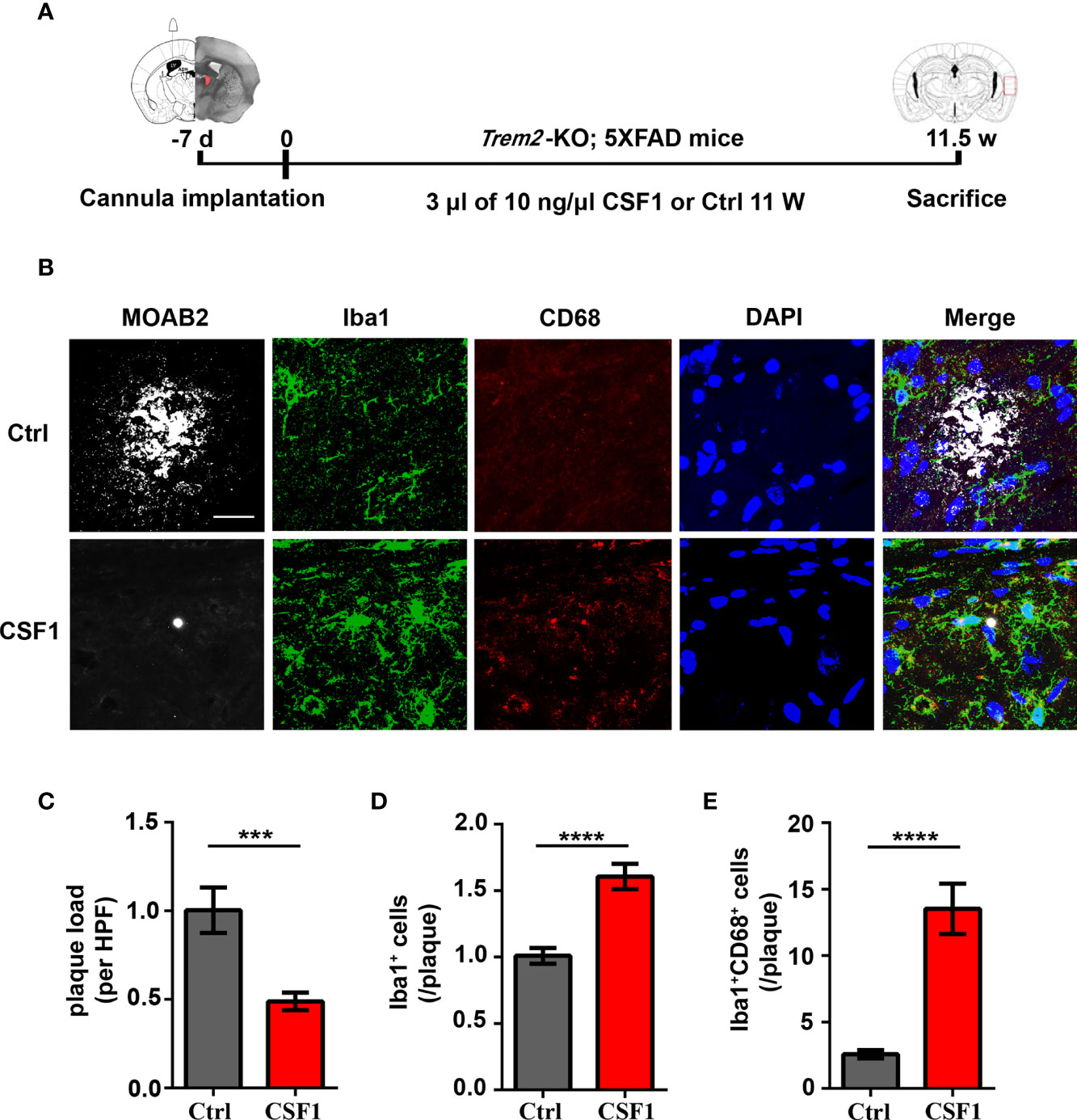
Figure 7 CSF1 administration ameliorates Aβ plaques accumulation in Trem2-/-; 5XFAD mouse brain. (A) Schematic timeline of CSF1 administration in Trem2-/-; 5XFAD mouse brain. Trem2-/-;5XFAD mice were treated with 30 ng CSF1 or artificial cerebrospinal fluid-treated controls weekly for 2.5 months. The right hemispheres of the brains were embedded for cryostat sectioning and immunofluorescence. (B) Coronal sections of the brains were stained with Iba-1 (green) for microglia, CD68 (red) for phagocytic marker and MOAB2 (gray) for Aβ plaques. Representative images of microglial immunofluorescent staining are shown. (C) Immunofluorescence density of Aβ plaques stained with MOAB2 (gray) was quantified per HPF. Aβ plaques immunofluorescence density response to CSF1 was assessed by quantifying the plaques ratio of CSF1 group to control group in Trem2-/-;5XFAD mouse brain. (D) Number of microglia double stained with DAPI (blue) and Iba1 (green) was quantified per HPF. Microglia number response to CSF1 was assessed by quantifying the cell number ratio of CSF1 group to control group in Trem2-/-;5XFAD mouse brain. (E) Number of microglia double stained with CD68 (red) and Iba1 (green) associated with plaques was quantified per HPF. Phagocytic microglia number response to CSF1 was assessed by quantifying the co-staining cell number ratio of CSF1 group to those of control group in Trem2-/-;5XFAD mouse brain. Student t test. Data are plotted as mean ± SEM (n=3). ***p<0.001; ****p<0.0001. Scale bar, 50 µm.
Discussion
Herein, we demonstrate the physical association of CSF1R and TREM2 and that TREM2 is not necessary for CSF1/CSF1R signaling mediated microglial survival (Figure 8). We further observed that forced knockdown of CSF1R decreased cell survival, which was consistent with previous studies myeloid lineage cells (22, 23). Interestingly, CSF1R was significantly enhanced in Trem2-/- microglia and activating CSF1R by CSF1 resulted in significant restoration of the attenuated survival in Trem2-/- microglia and in Trem2-/- mouse brain due to activation of the Akt signaling pathway (Figure 8).
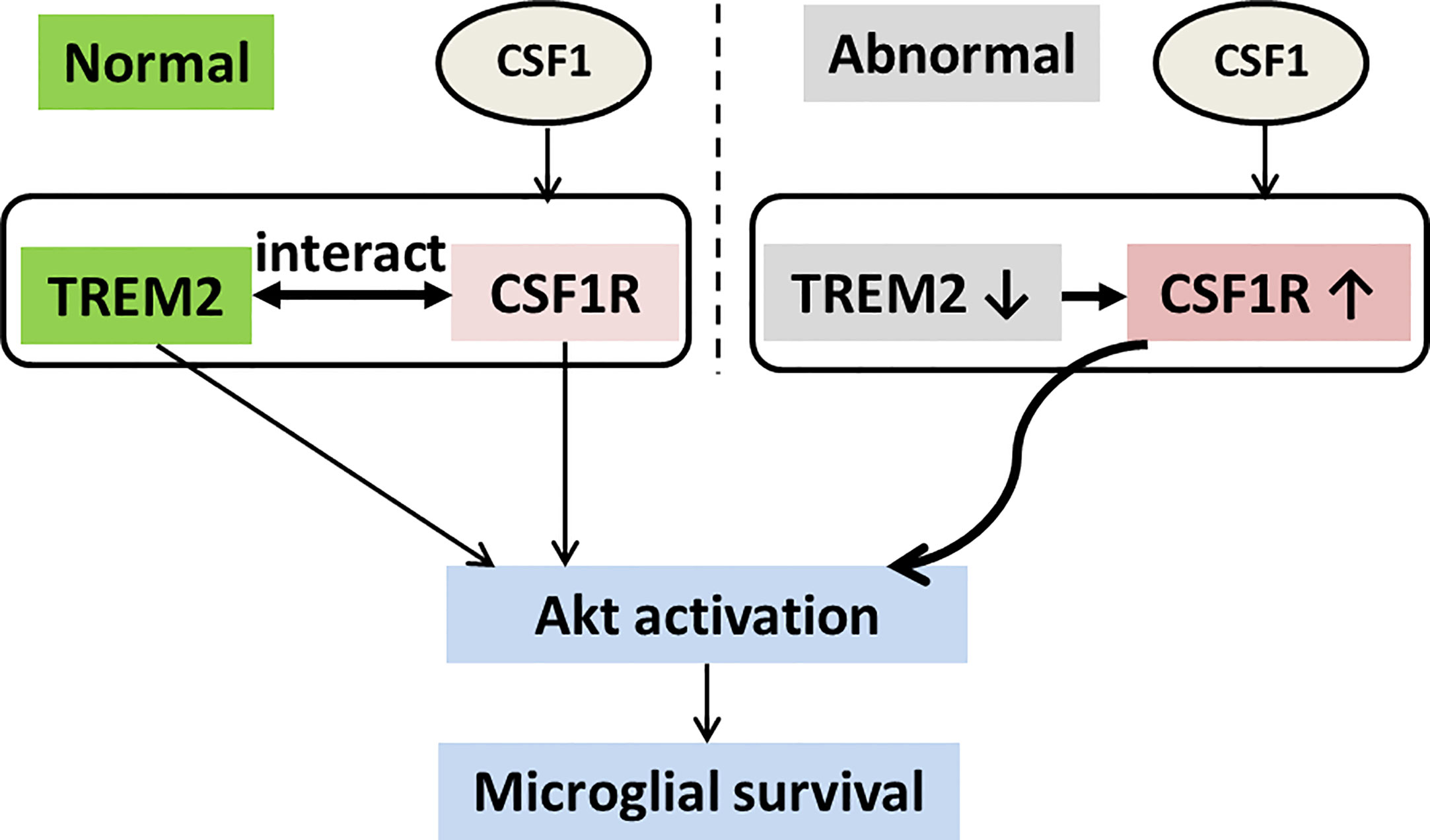
Figure 8 Schematic model of TREM2 and CSF1R interaction in microglial survival. TREM2 and CSF1R interact with each other, both of which promote microglial survival through the Akt pathway. CSF1R expression is increased and has elevated response to CSF1 for Akt activation in Trem2 deficient microglia.
The effects of CSF1 are mediated by the CSF1R tyrosine kinase, and the subsequent phosphorylation of downstream molecules (25). Triggering this phosphorylation cascade increases gene transcription and protein translation, leading to the survival and proliferation of target cells (14). TREM2 is involved in CSF1-mediated survival and proliferation in macrophages and osteoclast precursors. CSF1/CSF1R signals through calmodulin-dependent kinases downstream of DAP12 activates β-catenin in a TREM2-dependent manner (15, 16). Moreover, another study showed that CSF1/CSF1R activated Src kinases, resulting in DAP12 phosphorylation (17). This mechanism suggests that these receptors are in close physical proximity and the functional interaction may potentially influence each other; TREM2 has been proposed as the most obvious candidate receptor responsible for the crosstalk with CSF1R (26). Our study revealed that TREM2 and CSF1R interacted with each other through transmembrane regions. However, the interaction of CSF1R and TREM2 in physiological conditions needs to be further confirmed in future work using practicable TREM2 antibodies. Previously published work revealed an essential role of DAP12 for TREM2 signaling and for CSF1-induced proliferation and survival of macrophages (17), which suggests that the association of DAP12 with a cognate receptor is required (16). As such, whether DAP12 affects the interaction of CSF1R and TREM2 or whether DAP12 is a component of the interaction complex remains elusive. Additionally, given that DAP10 can also interact with DAP12–TREM2 complex and DAP10 was required for the effective activation of phosphoinositide 3-kinase (PI3K) growth factor receptor-bound protein 2 (Grb2), and therefore protein kinase B (Akt) and ERK signaling (27), it may be interesting to investigate whether DAP10 also interacts with the CSF1R-TREM2 complex.
Moreover, it has been proposed that the cytoplasmic domain of CSF1R can translocate to the nucleus (28), but a detailed mechanism for whether the interaction between CSF1R and TREM2 is necessary to target cytoplasmic CSF1R proteins translocated to the nucleus remain elusive. Additionally, recent studies reported that a soluble form of TREM2 (sTREM2) derived from proteolytic cleavage of the cell surface receptor promotes microglial survival in a PI3K/Akt-dependent manner (20), meaning it is desirable to investigate in future whether CSF1R is the receptor that mediates the effects of sTREM2 on microglial survival.
We also found that CSF1R and TREM2 mutually regulate their expression. It is intriguing to note that TREM2 deficiency in microglia led to enhanced CSF1R protein and mRNA level. As expected, knockdown of Csf1r in microglia significantly increased Trem2 mRNA level. These results suggest that CSF1R and TREM2 are complementary in their expression though detail mechanisms need to be clarified. Since both CSF1R and TREM2 are essential in microglial survival, the reactive enhancement of CSF1R in Trem2 KO microglia may be a salvage measure for microglial survival. Thus, CSF1R and TREM2 have overlapping functions in microglial survival. Emerging evidence suggests that the Akt signaling pathway plays a critical role in cellular survival (29). We have previously reported that Akt was decreased in Trem2 KO microglia (8), which indicates TREM2 also acts through Akt signaling to mediate microglial survival. Interestingly, the activation of Akt in response to CSF1 in Trem2-deficient microglia indicates that CSF1 may be a potential therapeutic molecule for TREM2-loss-of-function neurodegenerative disease. Additionally, Akt inhibitor administration in those experiments can further support our results. Nevertheless, whether the crosstalk of TREM2 with CSF1R signaling and the activation of Akt are involved in the migration of microglia remain to be determined. Furthermore, the similar effects of Akt activation in response to CSF1 in wildtype or Trem2 KO microglia indicate that TREM2 is not necessary for CSF1/CSF1R/Akt signaling and that there is an unknown signaling pathway of TREM2 in the regulation of microglial survival, which needs to be clarified in future. It is still unclear from our findings whether CSF1R promotes the stabilization of β-catenin and perhaps the subsequent activation of other survival signaling such as Wnt/β-catenin or PI3K/Akt, although previous studies demonstrated that TREM2 signaling via its associated adaptor DAP12 synergizes with CSF1R signaling to promote survival of macrophages (15, 16) as well as the survival of primary microglia (8).
Interestingly, the expression of two ligands for CSF1R in Trem2-/- microglia was different. The increased IL34 mRNA and unaltered Csf1 mRNA level in those cells imply their different roles in Trem2-/- mouse brain. These results also indicate that the increase of IL34 in Trem2-/- microglia cannot restore microglial viability although the increase needs to be verified by ELISA assay. CSF1 supplements are practical to restore microglial viability in Trem2-/- mice. Thus one of the most intriguing avenues of this research is that CSF1 can restore cell survival in TREM2 deficient microglia. Since systemic administration of human recombinant CSF1 ameliorates memory deficits in a transgenic mouse model of AD (30, 31), it would not be surprising if future work shows that administration of CSF1 influences this signaling chain in AD therapy.
Our studies also provide direct evidence for the critical role of CSF1R in microglial survival though the molecular mechanisms of how CSF1R deficiency affects the survival of microglia remain elusive. Signaling by CSF1 through CSF1R induced the stabilization and nuclear translocation of beta-catenin, which activated genes involved in the cell cycle. Thus, CSF1R deficiency may result in cell cycle arrest and accelerated apoptosis of microglia. Detailed studies need to be conducted, however, to demonstrate those effects. Csf1r-/- mice of most strain backgrounds die perinatally and the surviving mice exhibit multiple developmental deficits with an almost total lack of microglia. In contrast, Trem2-/- mice are grossly normal in growth without apparent microglia reduction in young or adult brains but with fewer and abnormal microglia in aged brains (32). Moreover, Csf1r haploinsufficiency in humans leads to ALSP, an adult-onset progressive dementia predominantly affecting the cerebral white matter. However, the loss of functions in TREM2 in humans resulted in a severe form of dementia with bone cystic lesions known as Nasu-Hakola disease (NHD), a lethal form of progressive, early onset dementia (33).
The phenotype of the Csf1r-/- mouse is more severe than that of the Trem2-/- mouse, indicating that CSF1R is more important than TREM2 in mouse growth. Additionally, microglia are reduced by more than 94% in 3-week-old Csf1r-null brains (23, 34) and are almost eliminated from the brain upon CSF1R inhibitor treatment (35, 36). Collectively, CSF1R functions directly in microglia survival and the pro-survival effects of TREM2 cannot replace that of CSF1R but only have a synergistic effect with it. These two proteins are required for survival of microglia. While they share most functions, a key difference is that CSF1R modulates microglia survival more efficiently than TREM2. Accordingly, further studies are needed to extend the TREM2 and CSF1R signaling pathway and to identify nuances that are crucial to fully understand their function.
In summary, we show here for the first time that TREM2 interacts with CSF1R and they mutually affect their expression. Importantly, activating CSF1R signaling by CSF1 protects against the phenotypes in Trem2-deficient microglia. Nevertheless, there is a potential risk of CSF1R activation in this therapeutic strategy as several studies have shown that CSF1R activation resulted in tumorigenesis (37–39). Thus, the brain-targeting strategies of CSF1R activation need to be seriously considered. Given that augmented β-amyloid (Aβ) accumulates due to a dysfunctional response of microglia in Trem2-/-; 5XFAD mouse brain, which fail to cluster around Aβ plaques and become apoptotic (40–42), we found CSF1 can ameoliate the augmented Aβ accumulation in Trem2-deficient 5XFAD mouse brain by activating microglia into a phagocytic state. Interestingly, studies suggested that CSF1R inhibitors also reduce plaque burden in AD mouse models (35, 43–45). In those studies, microglia influence plaque morphologies during peak pathological progression. The combined data demonstrate a critical role of CSF1R in modulating microglia survival in AD progress. Further studies of behavior tests are required to understand whether CSF1R can be used as a therapeutic target against these diseases.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by Animal Ethics Committee of Xiamen University.
Author Contributions
HZ and Y-WZ conceived the study and contributed new reagents or analytic tools. BC, XL, and KD performed the research. BC, XH, and XL analyzed the data and interpreted the results. HZ and BC wrote the paper. BC and XL prepared the figures. All other authors discussed and revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Natural Science Foundation of China (81771164, 91949129, 81771377, 92049202 and U1705285) and the Open Research Fund of State Key Laboratory of Cellular Stress Biology, Xiamen University (SKLCSB2019KF014). This work was also supported by grant from the Natural Science Foundation of Fujian Province of China (2018D0022 and 2020J01014).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Li Zhong and Xiaofen Chen from Institute of Neuroscience, Xiamen University for their technical help.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.633796/full#supplementary-material
References
1. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med (2013) 368:117–27. doi: 10.1056/NEJMoa1211851
2. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med (2013) 368:107–16. doi: 10.1056/NEJMoa1211103
3. Painter MM, Atagi Y, Liu CC, Rademakers R, Xu H, Fryer JD, et al. TREM2 in CNS homeostasis and neurodegenerative disease. Mol Neurodegener (2015) 10:43. doi: 10.1186/s13024-015-0040-9
4. Ulrich JD, Holtzman DM. TREM2 Function in Alzheimer’s Disease and Neurodegeneration. ACS Chem Neurosci (2016) 7:420–27. doi: 10.1021/acschemneuro.5b00313
5. Zheng H, Cheng B, Li Y, Li X, Chen X, Zhang YW. TREM2 in Alzheimer’s Disease: Microglial Survival and Energy Metabolism. Front Aging Neurosci (2018) 10:395. doi: 10.3389/fnagi.2018.00395
6. Kober DL, Brett TJ. TREM2-Ligand Interactions in Health and Disease. J Mol Biol (2017) 429:1607–29. doi: 10.1016/j.jmb.2017.04.004
7. Klesney-Tait J, Turnbull IR, Colonna M. The TREM receptor family and signal integration. Nat Immunol (2006) 7:1266–1273. doi: 10.1038/ni1411
8. Zheng H, Jia L, Liu CC, Rong Z, Zhong L, Yang L, et al. TREM2 Promotes Microglial Survival by Activating Wnt/beta-Catenin Pathway. J Neurosci (2017) 37:1772–84. doi: 10.1523/JNEUROSCI.2459-16.2017
9. Stanley ER, Guilbert LJ, Tushinski RJ, Bartelmez SH. CSF-1–a mononuclear phagocyte lineage-specific hemopoietic growth factor. J Cell Biochem (1983) 21:151–9. doi: 10.1002/jcb.240210206
10. Tushinski RJ, Oliver IT, Guilbert LJ, Tynan PW, Warner JR, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell (1982) 28:71–81. doi: 10.1016/0092-8674(82)90376-2
11. Raivich G, Haas S, Werner A, Klein MA, Kloss C, Kreutzberg GW. Regulation of MCSF receptors on microglia in the normal and injured mouse central nervous system: a quantitative immunofluorescence study using confocal laser microscopy. J Comp Neurol (1998) 395:342–58. doi: 10.1002/(SICI)1096-9861(19980808)395:3<342::AID-CNE6>3.0.CO;2-2
12. Rademakers R, Baker M, Nicholson AM, Rutherford NJ, Finch N, Soto-Ortolaza A, et al. Mutations in the colony stimulating factor 1 receptor (CSF1R) gene cause hereditary diffuse leukoencephalopathy with spheroids. Nat Genet (2011) 44:200–5. doi: 10.1038/ng.1027
13. Nicholson AM, Baker MC, Finch NA, Rutherford NJ, Wider C, Graff-Radford NR, et al. CSF1R mutations link POLD and HDLS as a single disease entity. Neurology (2013) 80:1033–40. doi: 10.1212/WNL.0b013e31828726a7
14. Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends Cell Biol (2004) 14:628–38. doi: 10.1016/j.tcb.2004.09.016
15. Otero K, Shinohara M, Zhao H, Cella M, Gilfillan S, Colucci A, et al. TREM2 and beta-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol (2012) 188:2612–21. doi: 10.4049/jimmunol.1102836
16. Otero K, Turnbull IR, Poliani PL, Vermi W, Cerutti E, Aoshi T, et al. Macrophage colony-stimulating factor induces the proliferation and survival of macrophages via a pathway involving DAP12 and beta-catenin. Nat Immunol (2009) 10:734–43. doi: 10.1038/ni.1744
17. Zou W, Reeve JL, Liu Y, Teitelbaum SL, Ross FP. DAP12 couples c-Fms activation to the osteoclast cytoskeleton by recruitment of Syk. Mol Cell (2008) 31:422–31. doi: 10.1016/j.molcel.2008.06.023
18. Jay TR, Miller CM, Cheng PJ, Graham LC, Bemiller S, Broihier ML, et al. TREM2 deficiency eliminates TREM2 inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med (2015) 212:287–95. doi: 10.1084/jem.20142322
19. Zhao Y, Tseng IC, Heyser CJ, Rockenstein E, Mante M, Adame A, et al. Appoptosin-Mediated Caspase Cleavage of Tau Contributes to Progressive Supranuclear Palsy Pathogenesis. Neuron (2015) 87:963–75. doi: 10.1016/j.neuron.2015.08.020
20. Zhong L, Chen XF, Wang T, Wang Z, Liao C, Wang Z, et al. Soluble TREM2 induces inflammatory responses and enhances microglial survival. J Exp Med (2017) 214:597–607. doi: 10.1084/jem.20160844
21. Chitu V, Stanley ER. Regulation of Embryonic and Postnatal Development by the CSF-1 Receptor. Curr Top Dev Biol (2017) 123:229–75. doi: 10.1016/bs.ctdb.2016.10.004
22. Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron (2014) 82:380–97. doi: 10.1016/j.neuron.2014.02.040
23. Erblich B, Zhu L, Etgen AM, Dobrenis K, Pollard JW. Absence of colony stimulation factor-1 receptor results in loss of microglia, disrupted brain development and olfactory deficits. PloS One (2011) 6:e26317. doi: 10.1371/journal.pone.0026317
24. Stanley ER, Chitu V. CSF-1 Receptor Signaling in Myeloid Cells. Cold Spring Harb Perspect Biol (2014) 6:a021857. doi: 10.1101/cshperspect.a021857
25. Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y. Biology and action of colony–stimulating factor-1. Mol Reprod Dev (1997) 46:4–10. doi: 10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V
26. McVicar DW, Trinchieri G. CSF-1R, DAP12 and beta-catenin: a menage a trois. Nat Immunol (2009) 10:681–683. doi: 10.1038/ni0709-681
27. Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal (2010) 3:ra38. doi: 10.1126/scisignal.2000500
28. Wilhelmsen K, van der Geer P. Phorbol 12-myristate 13-acetate-induced release of the colony-stimulating factor 1 receptor cytoplasmic domain into the cytosol involves two separate cleavage events. Mol Cell Biol (2004) 24:454–64. doi: 10.1128/MCB.24.1.454-464.2004
29. Song G, Ouyang G, Bao S. The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med (2005) 9:59–71. doi: 10.1111/j.1582-4934.2005.tb00337.x
30. Luo J, Elwood F, Britschgi M, Villeda S, Zhang H, Ding Z, et al. Colony-stimulating factor 1 receptor (CSF1R) signaling in injured neurons facilitates protection and survival. J Exp Med (2013) 210:157–72. doi: 10.1084/jem.20120412
31. Boissonneault V, Filali M, Lessard M, Relton J, Wong G, Rivest S. Powerful beneficial effects of macrophage colony-stimulating factor on beta-amyloid deposition and cognitive impairment in Alzheimer’s disease. Brain (2009) 132:1078–92. doi: 10.1093/brain/awn331
32. Poliani PL, Wang Y, Fontana E, Robinette ML, Yamanishi Y, Gilfillan S, et al. TREM2 sustains microglial expansion during aging and response to demyelination. J Clin Invest (2015) 125:2161–70. doi: 10.1172/JCI77983
33. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R, et al. Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet (2002) 71:656–62. doi: 10.1086/342259
34. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330:841–5. doi: 10.1126/science.1194637
35. Sosna J, Philipp S, Albay R3, Reyes-Ruiz JM, Baglietto-Vargas D, LaFerla FM, et al. Early long-term administration of the CSF1R inhibitor PLX3397 ablates microglia and reduces accumulation of intraneuronal amyloid, neuritic plaque deposition and pre-fibrillar oligomers in 5XFAD mouse model of Alzheimer’s disease. Mol Neurodegener (2018) 13:11. doi: 10.1186/s13024-018-0244-x
36. Dagher NN, Najafi AR, Kayala KM, Elmore MR, White TE, Medeiros R, et al. Colony-stimulating factor 1 receptor inhibition prevents microglial plaque association and improves cognition in 3xTg-AD mice. J Neuroinflamm (2015) 12:139. doi: 10.1186/s12974-015-0366-9
37. Wrobel CN, Debnath J, Lin E, Beausoleil S, Roussel MF, Brugge JS. Autocrine CSF-1R activation promotes Src-dependent disruption of mammary epithelial architecture. J Cell Biol (2004) 165:263–73. doi: 10.1083/jcb.200309102
38. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Rüttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer (2017) 5:53. doi: 10.1186/s40425-017-0257-y
39. Giricz O, Mo Y, Dahlman KB, Cotto-Rios XM, Vardabasso C, Nguyen H, et al. The RUNX1/IL-34/CSF-1R axis is an autocrinally regulated modulator of resistance to BRAF-V600E inhibition in melanoma. JCI Insight (2018) 3:e120422. doi: 10.1172/jci.insight.120422
40. Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, et al. TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell (2015) 160:1061–71. doi: 10.1016/j.cell.2015.01.049
41. Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer’s disease. Mol Neurodegener (2020) 15:40. doi: 10.1186/s13024-020-00391-7
42. Jadhav VS, Lin PBC, Pennington T, Di Prisco GV, Jannu AJ, Xu G, et al. Trem2 Y38C mutation and loss of Trem2 impairs neuronal synapses in adult mice. Mol Neurodegener (2020) 15:62. doi: 10.1186/s13024-020-00409-0
43. Mancuso R, Fryatt G, Cleal M, Obst J, Pipi E, Monzón-Sandoval J, et al. CSF1R inhibitor JNJ-40346527 attenuates microglial proliferation and neurodegeneration in P301S mice. Brain (2019) 142:3243–64. doi: 10.1093/brain/awz241
44. Spangenberg E, Severson PL, Hohsfield LA, Crapser J, Zhang J, Burton EA, et al. Sustained microglial depletion with CSF1R inhibitor impairs parenchymal plaque development in an Alzheimer’s disease model. Nat Commun (2019) 10:3758. doi: 10.1038/s41467-019-11674-z
Keywords: TREM2, CSF1R, interaction, cell survival, microglia
Citation: Cheng B, Li X, Dai K, Duan S, Rong Z, Chen Y, Lü L, Liu Z, Huang X, Xu H, Zhang Y-W and Zheng H (2021) Triggering Receptor Expressed on Myeloid Cells-2 (TREM2) Interacts With Colony-Stimulating Factor 1 Receptor (CSF1R) but Is Not Necessary for CSF1/CSF1R-Mediated Microglial Survival. Front. Immunol. 12:633796. doi: 10.3389/fimmu.2021.633796
Received: 02 February 2021; Accepted: 09 March 2021;
Published: 25 March 2021.
Edited by:
Jorge Correale, Fundación Para la Lucha Contra las Enfermedades Neurológicas de la Infancia (FLENI), ArgentinaReviewed by:
Laura Andrea Pasquini, Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), ArgentinaTaylor Jay, Oregon Health and Science University, United States
Copyright © 2021 Cheng, Li, Dai, Duan, Rong, Chen, Lü, Liu, Huang, Xu, Zhang and Zheng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yun-Wu Zhang, eXVuemhhbmdAeG11LmVkdS5jbg==; Honghua Zheng, aG9uZ2h1YUB4bXUuZWR1LmNu
†These authors have contributed equally to this work