 Bettina Tosetti1,2
Bettina Tosetti1,2 Beate Ward1Daniela Grumme1,2
Beate Ward1Daniela Grumme1,2 Marc Herb1,2
Marc Herb1,2 Michael Schramm1,2
Michael Schramm1,2 Olaf Utermöhlen1,3Lukas C. Heukamp4
Olaf Utermöhlen1,3Lukas C. Heukamp4 Martin Krönke1,2,3,5*
Martin Krönke1,2,3,5* Oleg Krut6*
Oleg Krut6*- 1Institute for Medical Microbiology, Immunology and Hygiene, University of Cologne, Cologne, Germany
- 2Cologne Cluster of Excellence in Cellular Stress Responses in Aging-Associated Diseases, University of Cologne, Cologne, Germany
- 3Center for Molecular Medicine Cologne, University of Cologne, Cologne, Germany
- 4Institute for Hematopathology Hamburg, Hamburg, Germany
- 5German Center for Infection Research, Bonn-Cologne, Germany
- 6Paul-Ehrlich-Institut, Langen, Germany
Although the crucial role of professional phagocytes for the clearance of S. aureus infections is well-established, several studies indicate an adverse role of leukocytes in the dissemination of S. aureus during infection. Since only little is known about macrophages in this context, we analyzed the role of macrophages, and in particular reactive oxygen species deficiency, for the seeding of S. aureus metastases. Infection of bone marrow-derived macrophages (BMDM) with S. aureus revealed that NADPH oxidase 2 (NOX2-) deficient, but not NOX1- or NOX4-deficient, BMDM failed to clear intracellular S. aureus. Despite of larger intracellular bacterial burden, NOX2-deficient BMDM showed significantly improved survival. Intravenous injection of mice with in vitro-infected BMDMs carrying intracellular viable S. aureus led to higher bacterial loads in kidney and liver of mice compared to injection with plain S. aureus. An even higher frequency of liver abscesses was observed in mice infected with S. aureus-loaded nox2−/− BMDM. Thus, the improved intracellular survival of S. aureus and improved viability of NOX2-deficient BMDM is associated with an aggravated metastatic dissemination of S. aureus infection. A combination of vancomycin and the intracellularly active antibiotic rifampicin led to complete elimination of S. aureus from liver within 48 h, which was not achieved with vancomycin treatment alone, underscoring the impact of intracellular S. aureus on the course of disease. The results of our study indicate that intracellular S. aureus carried by macrophages are sufficient to establish a systemic infection. This suggests the inclusion of intracellularly active antibiotics in the therapeutic regimen of invasive S. aureus infections, especially in patients with NADPH oxidase deficiencies such as chronic granulomatous disease.
Introduction
Staphylococcus aureus bacteremia remains worldwide the leading cause of both community acquired and nosocomial life-threatening systemic infections. S. aureus strains colonizing the anterior nares were found to be a frequent cause of bacteremia (1–3). Hence, breaching of the skin barrier represents one possible first step in the onset of invasive and systemic S. aureus infections. Despite the availability of effective antibiotics and the achievements of intensive medical care, invasive and systemic S. aureus infections remain a life-threatening medical challenge. However, the pathophysiologic mechanisms enabling the dissemination of S. aureus to inner organs remain elusive. Professional phagocytes, especially neutrophils, are crucial for the clearance of the bacteria, as several naturally occurring genetic disorders impairing neutrophil function are associated with an increased incidence and severity of S. aureus infections (4). Similarly, macrophages play a crucial role in the control of S. aureus infection (5, 6).
S. aureus infections belong to the signature diseases of the inherited chronic granulomatous disease (CGD), which can be caused by several mutations in genes encoding the phagocyte NADPH oxidase NOX2 or its subunits p22phox, p40phox, p47phox and p67phox (7). The absence or malfunction of NOX2 in neutrophils of CGD patients results in a defective oxidative burst and impaired killing of phagocytosed microbes (8, 9). In addition, mutations of CYBC1/EROS, a protein that is essential for Gp91-p22Phox heterodimer expression, have been implicated in decreased NOX2 function and CGD (10). CGD is characterized by recurrent infections with a narrow spectrum of fungi and catalase-positive bacteria including S. aureus (11). The importance of professional phagocytes for the immune response against S. aureus is also underlined by the broad range of virulence factors enabling S. aureus to escape efficient recognition by the host immune system or destruction by the oxidative burst of professional phagocytes (12). In addition, S. aureus can survive intracellularly within neutrophils in vitro and in vivo (13–15). Indeed, bloodstream neutrophils may act as Trojan horse for S. aureus enabling dissemination of surviving bacteria (16).
Unlike neutrophils, the role of monocytes and macrophages in systemic infections with S. aureus is less clear to date. Compared to neutrophils, these types of professional phagocytes lack myeloperoxidase, produce a diminished oxidative burst and exhibit an overall lesser bactericidal activity (17–23). Given their longevity and immediate presence at the site of infection, we hypothesized that macrophages might also be important for the dissemination of S. aureus in vivo.
In general, tissue-resident macrophages belong to the first line of defense against invading microbes (24–26). After detection, macrophages engulf the microbes by phagocytosis and inactivate, kill and degrade them in phagolysosomes (27, 28). To this end, macrophages employ an array of directly antimicrobial mechanisms, for example, the generation of reactive oxygen species (ROS) (29–31) and reactive nitrogen species (RNS) (32, 33) and the delivery of microbicidal lysosomal acid hydrolases into maturing phagosomes (34, 35).
The interaction of S. aureus with macrophages is multifaceted (6, 36, 37). On the one hand, macrophages use a wide array of antimicrobial mechanisms to kill S. aureus (6) and, in the absence of macrophages, bacterial burden and mortality following S. aureus infection are markedly increased (5, 38–40). On the other hand, S. aureus has evolved multiple strategies to survive within, manipulate and escape from macrophages (6). While the majority of phagocytosed S. aureus are killed by macrophages such as Kupffer cells that filter the bloodstream, a small proportion of bacteria survives (12, 41). Surviving S. aureus can escape from macrophages and result in formation of micro-abscesses in the liver and, potentially, lead to dissemination throughout the host. Whether this dissemination is caused by S. aureus that killed and escaped from macrophages or by migrating macrophages carrying intracellular S. aureus, is not known.
Here, we analyzed the general capacity of S. aureus-infected murine BMDM to enhance the systemic dissemination of S. aureus metastases. Specifically, we assessed the individual role of NOX1,−2 and-4 for ROS production and the survival of both, intracellular S. aureus and infected host cells. The intracellular survival of S. aureus in neutrophils and macrophages continuously embraces the hypothesis that intracellularly active antibiotics improve invasive S. aureus infections (42). Although rifampicin has been shown to be especially active against intracellular S. aureus (43) a recent clinical study testing adjunctive rifampicin for S. aureus bacteremia revealed no clear benefit over standard antibiotic therapy (44). However, patients suffering CGD were not included in this study. Because NOX2-deficiency aggravates the infectious challenge caused by increased loads of intracellular S. aureus in BMDM, we tested adjunctive rifampicin treatment to reveal a potential benefit of targeting intracellular S. aureus in NOX2-related disorders like CGD.
Materials and Methods
Mice
6–7 weeks old female C57BL/6J mice for in vivo experiments were obtained from Charles River Laboratories (Sulzfeld, Germany). Gp91 phox (nox2−/−) (45) and NOX4 knockout (nox4−/−) mice (46) were kindly provided by Ralf Brandes (Goethe University Frankfurt). NOX1 knockout mice (47) were kindly provided by Karl-Heinz Krause (University of Geneva) and nmf333 mice harboring the Y121H (p22Y121H) point mutation in the gene encoding p22phox (48), were obtained from J. Woo (Stanford University Medical Center, Stanford).
All mice were backcrossed at least 10 times to the C57BL/6J background. Mice were kept under specific pathogen-free conditions at the animal facilities of the Medical Center of the University of Cologne. All animal experiments have been carried out with local ethical committee approval (AZ 84-02.04.2014.A013) and adhering to the guidelines of German jurisdiction and the guidelines for the welfare and use of animals in research. All efforts were made to minimize suffering of the animals.
Bacterial Strains
Staphylococcus aureus strain MW2, a community acquired MRSA strain also known as USA 400, was obtained from the Network on Antimicrobial Resistance in Staphylococcus aureus (www.narsa.net). S. aureus MW2 constitutively expressing GFP under control of the spa-derived constitutive promoter was constructed by transformation of MW2 with plasmid pCN-F7-GFP (49). Wildtype MW2 was used for ROS measurements, all other experiments were conducted with MW2-GFP. MW2-GFP was selected on 5 μg/ml erythromycin on agar plates and during overnight (ON) cultures. For experiments S. aureus were inoculated 1:100 from ON culture into fresh Luria- Bertani (LB) broth and grown at 37°C to an OD600 of 0.3. Bacteria were harvested, washed, and the concentration was adjusted to 1 × 109 CFU/ml in PBS.
Isolation and Culture of Bone Marrow-Derived Macrophages
For in vitro differentiation of bone marrow cells into bone marrow-derived macrophages (BMDM), bone marrow was prepared from the tibias and femurs of mice from the C57/BL6 J Background since this is the background of used KO mouse lines. The erythrocytes were lysed with Tris-buffered ammonium chloride (8.3% NH4Cl, 0.1 M Tris). Bone marrow cells were cultured in VLE RPMI 1640 medium (Biochrom), supplemented with 10% FCS, penicillin (100 U/ml) and streptomycin (100 μg/ml), 2 mM HEPES, 200 nM sodium pyruvate and 10 ng/ml recombinant M-CSF (Peprotech) to differentiate bone marrow cells into BMDM. BMDM were used for experiments at day 8 of in vitro differentiation unless specified otherwise. More than 90% of these cells were F4/80+/Cd11+ BMDM as determined by flow cytometry. Antibiotics were removed 16 h prior to infection of BMDM (50).
Measurement of ROS Production by BMDM
For ROS measurements BMDM were seeded in a density of 1 × 105 cells /well in sterile white 96 well LumiNunc plates (Thermo Scientific) in antibiotic free medium 16 h before measurement. After washing cells twice with cold HBSS containing magnesium sulfate (200 mg/L) and calciumchloride (185.4 mg/L) (Sigma), either viable non-opsonized or opsonized S. aureus MW2 [5% normal mouse serum (NMS)] were added to respective wells using MOI50 or MOI10 as indicated. Cells treated with 5% normal mouse serum (NMS; Innovative research) in HBSS served as non-infected control. Upon addition of bacteria or mouse serum, infection was synchronized by centrifugation at 840 g at 4°C in a swing bucket rotor for 5 min. Subsequently supernatant was aspirated and fresh HBSS was added to the cells. 2x Luminol/HRP mix in HBSS was added to each well. Finally, 10 μM PMA (Sigma) as positive control or 100 μg/ml Pam2CSK4 (Invivogen) as control for TLR2 dependent ROS production in HBSS was added immediately before measurement. ROS production was measured in a plate reader preheated to 37°C in 60 s intervals over 60 min (Tristar, Berthold Instruments). For evaluation first cell-free values were subtracted from each sample, subsequently values of non-infected or untreated samples were subtracted. Plotted was the corrected RLU per minute in mean and SD of triplicates.
Assessment of Opsonophagocytic Clearing of S. aureus Using Flow Cytometry
To assess uptake and clearing of bacteria in BMDM, cells were harvested and adjusted to 5 × 106 cells/ml. Assays were performed in triplicates using a total of 2.5 × 106 cells/ml. BMDM were infected with S. aureus MW2 GFP (MOI15) opsonized with 5% NMS in suspension. After addition of bacteria, samples were mixed and infection was synchronized by centrifugation at 840 g at 4°C for 2 min, subsequently incubated for 5 min at 37°C rotating end over end, and thereafter the pellets were resuspended. This scheme was performed thrice. Remaining extracellular S. aureus were removed with Lysostaphin (Sigma; 2.5 mg/ml) in a final concentration of 0.2 mg/ml on ice for 20 min, followed by washing with PBS twice. The pellets were resuspended in 1 ml of culture medium without antibiotics and 50 μl of samples were analyzed directly (t0) and at additional time points (30 min, 1 h, 6 h and 24 h p. i.) for FACS analysis. Bacterial clearing was determined by calculating the amount of GFP positive macrophages (percentage within M1; see supp. Supplementary Figure 1) of respective time points in percent of t0. Samples were incubated rotating end over end at 37°C. After the 6 h time point Gentamycin (Sigma; 50 μg/ml) was added to the samples, to enable assessment of viability 24 h post infection. BMDM viability was assessed by trypan blue exclusion using the Countess (Life Technologies) and was calculated as percent of viable non-infected BMDM.
Infection of Mice
BMDM on day 8 of differentiation were infected with S. aureus-GFP using MOI30, which results in close to 100% loading of BMDM (Supplementary Figure 1B). After Lysostaphin treatment, BMDM were washed and 2.5 × 106 cells in 300 μl were administered intravenously into the tail vein of mice. 5 h post infection the amount of S. aureus present in infected cells was determined after lysis of BMDM in H2O at pH11 and subsequent plating in 10x serial dilutions on agar plates. Control mice were injected with 4 × 106 plain S. aureus corresponding to the CFUs retrieved from S. aureus infected BMDM.
For determination of the bacterial load, mice were euthanized and sacrificed by cervical dislocation 24 h, 72 h and on day 7 post injection. Organs were homogenized in 0.1% Triton X-100 in gentleMACS M tubes using the predefined program for protein isolation using the gentleMACS (Miltenyi Biotec) and plated in 10-fold serial dilutions on Mueller-Hinton agar plates using the Eddy Jet spiral plater (IUL Instruments) in mode log50. After overnight incubation of the plates at 37°C, the CFUs were counted using the Countermat flash plate reader (IUL Instruments).
Statistical Analysis
In vitro results and log transformed CFU values of in vivo experiments were analyzed by unpaired two-sided students t-test or 1-way ANOVA with Bonferroni post-test between selected pairs (viability data in Figures 2E–G). Contingency table analysis was performed using fisher' exact test. Statistical analysis and plotting of data was performed using GraphPad Prism 5.01.
Results
S. aureus-Induced ROS Production Depends on Functional NOX2 and Is Independent of TLR2
Macrophages do not always succeed in S. aureus killing, thereby providing a niche for persistence, which fosters continued infection (36). It has been speculated for a long time that professional phagocytes might serve as Trojan horses for S. aureus and lead to the frequently observed dissemination from the primary focus of infection (15). Whereas the role of neutrophils has been evaluated in greater detail, macrophages, although being a source of bacterial persistence, have been far less studied. A prerequisite for bacterial persistence is a perfect symbiosis of intracellular S. aureus and host cells, which can only be achieved when the bacteriocidal activities of macrophages are kept at bay. One of the most important bacteriocidal mechanisms of macrophages, the oxidative burst is generated by the NOX family of NADPH oxidases. Next to NOX2, at least two additional NOX isoforms, namely NOX1 and NOX4, are expressed by macrophages and might be involved in ROS generation. Indeed, wildtype BMDM strongly responded with ROS production to opsonized S. aureus (Figure 1A, left). The kinetics of ROS production was comparable to that of BMDM stimulated pharmacologically with 10 μM PMA used as positive control (Figure 1A, right). Treatment with the NOX inhibitor DPI led to a significant inhibition of ROS production (Figure 1A). We next scrutinized the relative contribution of NOX-isoenzymes 1, 2 and 4 to the oxidative burst of macrophages in response to S. aureus. To this end, BMDM with genetically defined deficiencies for individual NOX isoforms were employed, including nox1−/−, nox2−/− and nox4−/− as well as p22Y121H, a non-functional mutant of the NADPH oxidase subunit p22phox that is common to isoforms NOX1,−2,−3, and−4. The oxidative burst induced by S. aureus in BMDM from nox1−/− and nox4−/− mice was MOI-dependent and comparable to that of wildtype BMDM (Figure 1B). By contrast, macrophages isolated from nox2−/− or p22Y121H mice showed completely abolished production of S. aureus-induced ROS. These findings demonstrate that mainly the NADPH oxidase NOX2 is responsible for ROS production by BMDM upon S. aureus infection and raised the question about the control of NOX2 activation.
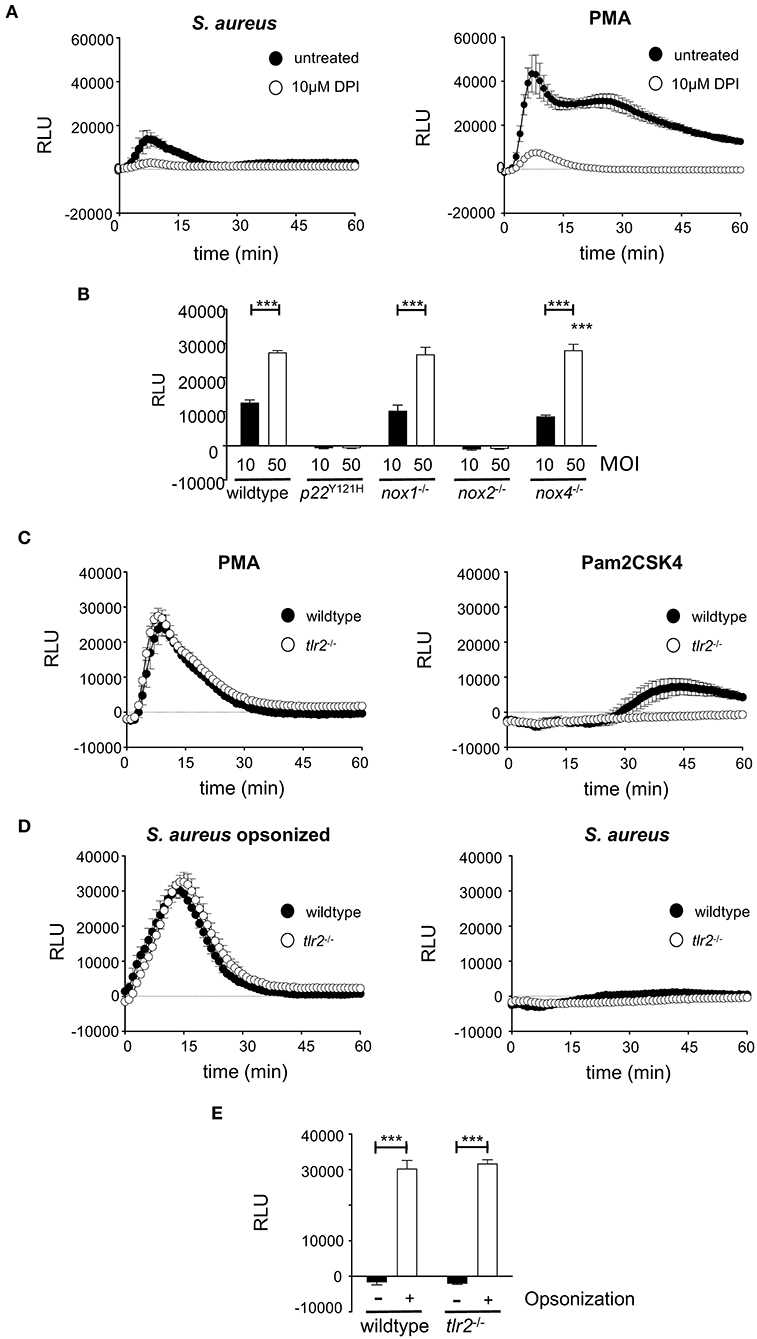
Figure 1. Opsonophagocytosis of S. aureus leads to NOX2 dependent ROS production in macrophages. For ROS measurement, 1 × 105 wildtype or knockout BMDM were seeded in a white 96-well plate without antibiotics 16 h prior to measurement. ROS production was measured using Luminol in a Luminometer preheated to 37°C. (A) ROS production in wildtype BMDM incubated with opsonized S. aureus (MOI50; left) or 10μM PMA (right) in the absence (closed circles) or presence of 10 μM DPI. (B) Peak value of ROS production at 13 min after infection with opsonized S. aureus MOI10 (closed bars) or MOI50 (open bars) in wildtype and NADPH-oxidase deficient BMDMs. Shown are mean and SD of triplicates corrected for non-infected control of a representative experiment performed at least three times. Statistical significance was analyzed by unpaired two-sided Student's t-test using GraphPad Prism 5.01 between indicated conditions (***=p < 0.001). (C) ROS production in wildtype (closed circles) or tlr2−/− BMDM (open circles) stimulated with 10 μM PMA (left), or 100 ng/ml Pam2CSK4 (right). (D) ROS production in wildtype (closed circles) or tlr2−/− BMDM (open circles) upon infection with S. aureus MW2 opsonized with 5% mouse serum (MOI50; left) or non opsonized S. aureus (MOI50; right). (E) Peak value of ROS production at 13 min after infection with non-opsonized (closed bars) or opsonized S. aureus (open bars) in wildtype and tlr2−/− BMDM. Shown are means and SD of triplicates of a representative experiment performed at least three times. Statistical significance was analyzed by unpaired two-sided Student's t-test using GraphPad Prism 5.01 between indicated conditions (***=p < 0.001). NOX, NADPH Oxidas; ROS, reactive oxygen species; PMA, phorbol 12-myristate 13-acetate; DPI, diphenyleniodonium; MOI, multiplicity of infection; BMDMs, bone marrow derived macrophages; SD, standard deviation; TLR2, Toll-like receptor 2; NMS, normal mouse serum; p.i., post infection; CFU, colony forming unit; i.v., intravenous; s.c., subcutaneous.
NOX2 is activated by opsonophagocytic receptors like FCγR and macrophage-1 antigen (Mac-1) (36, 51). Indeed, opsonized S. aureus led to strong ROS production (Figures 1C,D). Infection with non-opsonized S. aureus did not stimulate any measurable production of ROS at this time after infection (Figure 1D). TLR2-dependent activation of JNK results in inhibition of ROS production upon S. aureus infection and increased bacterial survival (52). Pam2CSK4, a synthetic TLR2 ligand, did not induce TLR2-dependent ROS production within the first 30 min (Figure 1C). Furthermore, opsonized S. aureus induced comparable production of ROS in either TLR2-deficient or -proficient BMDM (Figures 1C–E). Thus, the induction of NOX2-mediated ROS production by phagocytic receptors seems to overrule any possibly antagonistic action of TLR2.
Elimination of Phagocytosed S. aureus and Survival of Infected BMDM Depend on NOX2 Activity
To investigate the impact of the reduced oxidative burst in NOX2-deficient BMDM on the survival of both, intracellular S. aureus and host cell, we determined the kinetics of relative numbers of S. aureus-positive BMDM and the viability of host cells. ROS inhibition in BMDM by treatment with DPI significantly impaired the clearance of intracellular S. aureus over the first 24 h (Figure 2A). At 24 h post infection about 25% of DPI-treated BMDM were positive for GFP-expressing S. aureus, whereas only 10% of untreated BMDM carried S. aureus. As expected, NOX2-deficient BMDM, like DPI-treated BMDM, showed a markedly impaired clearance of intracellular S. aureus (Figure 2B). In contrast, nox1−/− and nox4−/− BMDM cleared S. aureus like wildtype BMDM (Figures 2C,D), which corresponds to their normal ability to elicit an oxidative burst. Thus, ROS production by NOX2 in BMDM is a prerequisite for the ability to clear phagocytosed S. aureus.
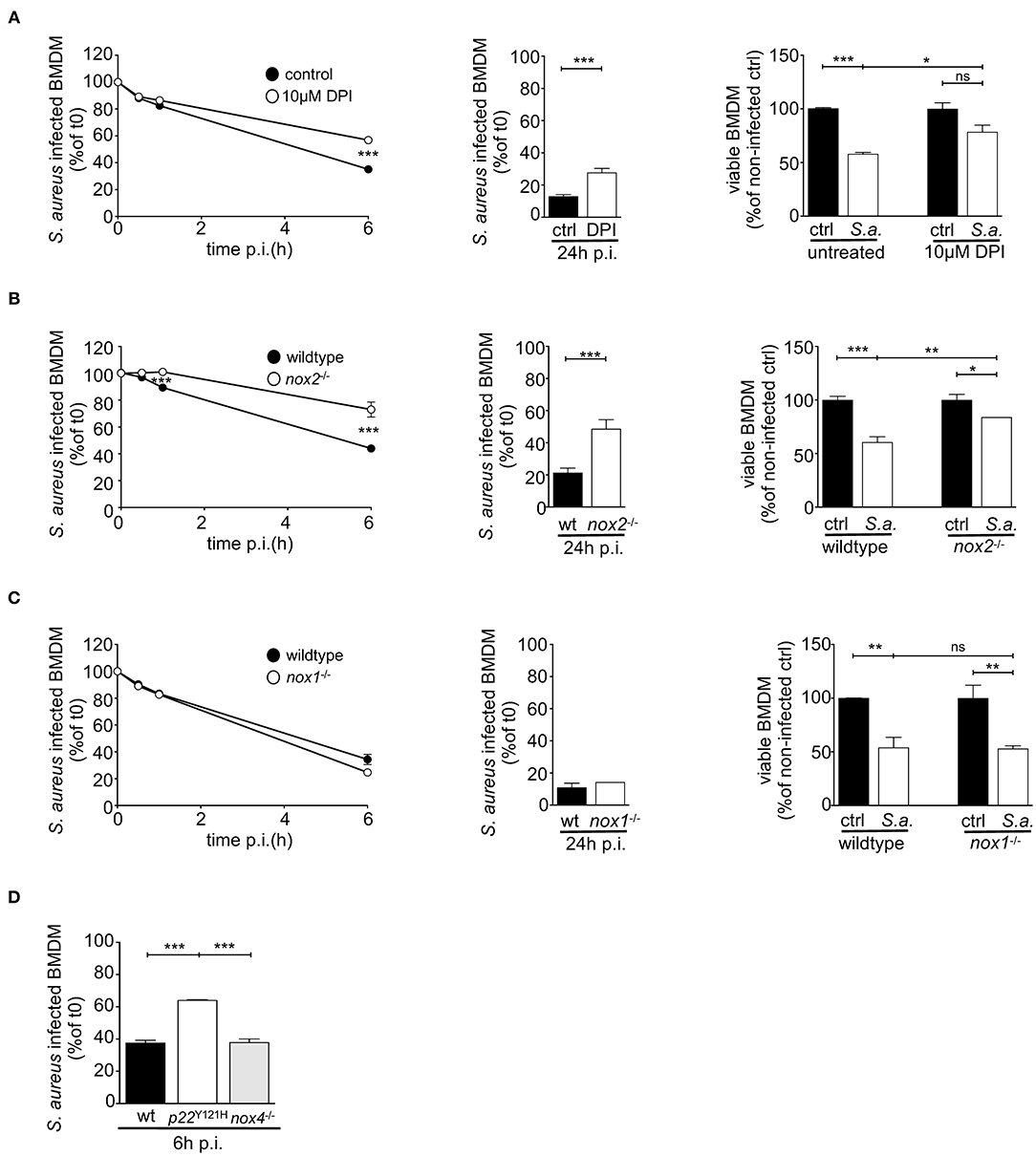
Figure 2. S. aureus clearing and survival of infected BMDM depend on functional NOX2. BMDM were infected with GFP-expressing S. aureus MW2 (MOI15) opsonized with 5% NMS in suspension and assessed 24 h p.i. by flow cytometry for clearing of S. aureus and for BMDM viability (right panel). (A) BMDM pre-treated with 10 μM DPI (open circles) or left untreated (closed circles), (B) wildtype (closed circles) and nox2−/− BMDM (open circles), (C) wildtype (closed circles) and nox1−/− BMDM (open circles). Statistical significance was analyzed by unpaired two-sided Student's t-test using GraphPad Prism 5.01 between indicated conditions (** = p < 0.01; ***= p < 0.001). The viabilities of BMDMs at 24 h p.i. were assessed by trypan blue exclusion and depicted as bar charts (A–C, right). Represented is the percentage of viable cells referring to corresponding non-infected cells for each genotype in mean and SD of triplicates. Statistical significance was analyzed by 1-way ANOVA with Bonferroni post-test between selected pairs as implemented in GraphPad Prism 5.01 between indicated conditions (*= p < 0.05; ** = p < 0.01; ***= p < 0.001). (D) Clearing kinetics of wildtype, p22phoxY121H and nox4−/− at 6h post infection. Shown are means and SD of triplicates as percent of corresponding values at t0. Shown are representative experiments performed three times. NOX, NADPH Oxidas; ROS, reactive oxygen species; PMA, phorbol 12-myristate 13-acetate; DPI, diphenyleniodonium; MOI, multiplicity of infection; BMDMs, bone marrow derived macrophages; SD, standard deviation; TLR2, Toll-like receptor 2; NMS, normal mouse serum; p.i., post infection; CFU, colony forming unit; i.v., intravenous; s.c., subcutaneous.
The question arose whether the reduced relative numbers of S. aureus-positive BMDM result from successful ROS-dependent killing of intracellular S. aureus or rather are secondary to killing of the BMDM by S. aureus. Therefore, we investigated the viability of BMDM. DPI-treated as well as nox2−/− BMDM had a substantial advantage in survival upon infection, despite their higher bacterial burden (Figures 2A–C; right). In fact, the viability of non-infected wildtype BMDM was less than 60% indicating that S. aureus infection led to cell death in 40% of infected BMDM and suggesting that S. aureus may have escaped into the culture supernatant and subsequently killed by gentamycin present in the culture medium. This scenario is consistent with a continuous cycle of phagocytosis, intracellular S. aureus replication, host cell death, bacterial release and re-uptake by macrophages (which may only occur in the absence of antibiotics) as recently proposed by Jubrail and colleagues (37, 53). In contrast, S. aureus induced cell death in only 15% of NOX2-deficient or DPI-treated BMDM (Figures 2A,B; right). Together, these data suggest that the oxidative burst not only reduces the number of BMDM carrying phagocytosed S. aureus but also contributes to death of infected macrophages. Furthermore, a defective oxidative burst likely renders BMDM an intracellular niche for S. aureus survival and replication.
NOX2 Deficiency Enhances Dissemination of S. aureus by Macrophages
The survival of S. aureus inside BMDM and in particular the prolonged survival of nox2−/− macrophages observed in our experiments in vitro raised the question about possible functional consequences in vivo, such as enhanced pathogenicity and dissemination of S. aureus in mice. Therefore, we next investigated the course of infection in mice challenged either with S. aureus internalized by macrophages or with the corresponding dose of plain S. aureus injected as a bacterial suspension. Wildtype or nox2−/− BMDM were infected in vitro with S. aureus and subsequently administered i.v. into wildtype C57BL/6J mice. The dose of plain S. aureus administered as a bacterial suspension corresponded to the number of live S. aureus recovered from infected nox2−/− BMDM (4 × 106 CFU). Mice challenged with BMDM loaded with intracellular S. aureus showed larger CFU counts in kidneys and liver, and less prominently in spleen, on day 7 after infection than mice challenged with plain S. aureus (Figures 3A–G). Notably, when nox2−/− BMDM were used as vehicles, they produced by trend the highest bacterial load in kidneys and liver. Intravenous injection of plain S. aureus also induced bacterial dissemination in mice. Animals infected by plain bacteria more efficiently cleared S. aureus from kidney and liver than animals injected with BMDM-associated S. aureus (Figures 3B,C,G–I).
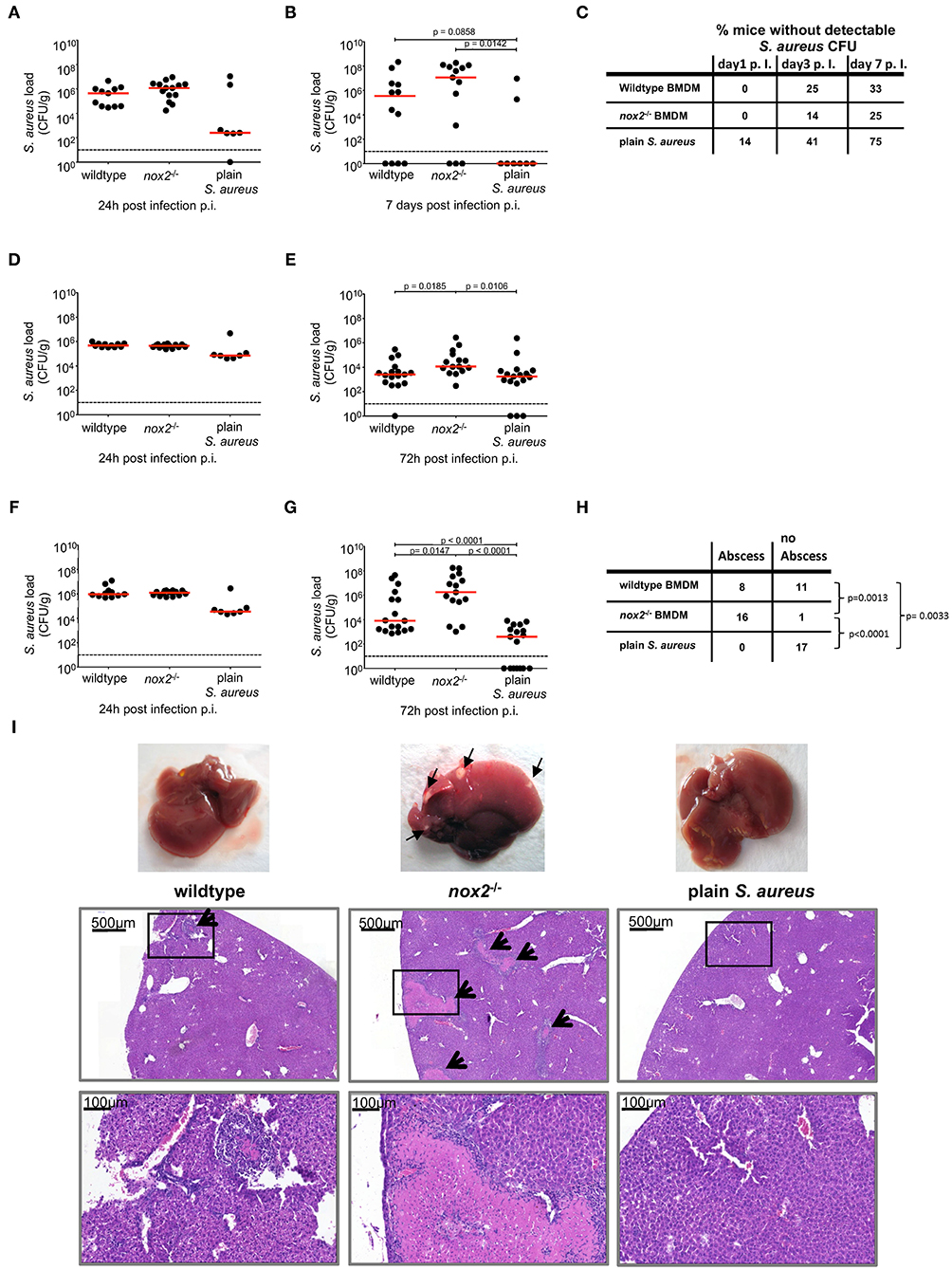
Figure 3. Bacterial load in kidney, spleen and liver from mice upon injection of BMDM infected with S. aureus. BMDM of wildtype or nox2−/− mice were infected with S. aureus MW2 GFP (MOI30) on day 8 post isolation from bone marrow. Upon infection 2.5 × 106 BMDM were injected in PBS intravenously into naive C57Bl6J mice. Mice injected with 4 × 106 plain S. aureus MW2 GFP, corresponding to the CFU recovered from infected BMDM served a control. Represented are individual values for each organ in CFU/g from two independent experiments. Red line indicates median, dashed line represents limit of detection. Student's t-test was performed on log-transformed values. CFU numbers were analyzed. in kidneys (A) 24 h (wt: n=11; nox2−/−: n=14; plain S. aureus: n=7) and (B) 7 days (wildtype: n=12; nox2−/−: n=13; plain S. aureus: n=8). (C) Percentage of mice without detectable CFU in kidneys for indicated timepoints. CFU numbers in spleen (D) 24 h and (E) 72 h (wildtype: n=17; nox2−/−: n=15; plain S. aureus: n=17) p.i. CFU numbers in liver (F) 24 h and (G) 72 h p.i. (H) Contingency table for presence or absence of visible abscesses in liver 72 h post infection analyzed by Fisher‘s exact test. (I) Representative pictures from livers 72 h post infection with micrographs from respective Hematoxylin-Eosin staining on formalin fixed tissue. Black arrows indicate abscesses. Scale bar represents 500 μm in upper and 100 μm in lower micrographs, respectively. Rectangular zone in upper pictures indicates position of bottom pictures. NOX, NADPH Oxidas; ROS, reactive oxygen species; PMA, phorbol 12-myristate 13-acetate; DPI, diphenyleniodonium; MOI, multiplicity of infection; BMDMs, bone marrow derived macrophages; SD, standard deviation; TLR2, Toll-like receptor 2; NMS, normal mouse serum; p.i., post infection; CFU, colony forming unit; i.v., intravenous; s.c., subcutaneous.
Macroscopical examination of explanted livers revealed that all livers from mice injected with infected nox2−/− BMDM contained one or more subcapsular abscesses of at least 1 mm in diameter (Figure 3I). Therefore, we assessed the frequency of subcapsular liver abscesses at 72 h post infection in the three experimental groups. The incidence of liver abscesses was significantly increased in the group that received nox2−/− BMDM carrying S. aureus as compared to the group that received wildtype BMDM carrying S. aureus (Figure 3H). No subcapsular abscesses were detected in livers of mice that received plain S. aureus. Histological analysis of HE-stained sections revealed relatively small abscesses (about 1.5 × 104 μm2) for mice that received wildtype BMDM carrying S. aureus (Figure 3I). By contrast, liver sections of mice that received nox2−/− BMDM carrying S. aureus revealed large abscesses with extensive necrosis and excessive inflammatory infiltrates. In one field of view, in total eight abscesses could be detected, which had an average area of about 3 × 105 μm2. As expected, in the livers from mice receiving plain S. aureus, no histopathological signs of infection were detected, consistent with the relatively low CFU count at 72 h post infection. These data suggest that intracellular S. aureus internalized by macrophages induce a disseminated and long-lasting infection in mice, especially when compared to plain bacteria. Moreover, the aggravation of the infection caused by nox2−/− BMDM-associated S. aureus underscores the pathogenic potential of a weakened but surviving host cell to provide a niche for S. aureus survival and replication.
Antibiotic Targeting of Intracellular S. aureus Ameliorates the Course of Infection
S. aureus infections are treated with antibiotics like ß-lactams or vancomycin that are extracellularly active, sometimes in combination with intracellularly active antibiotics like clindamycin or rifampin. Our results suggest that BMDM carrying viable S. aureus either die and release S. aureus into the extracellular milieu, where they are exposed to antibiotics. BMDM carrying viable S. aureus are yet also able to survive and initiate and maintain an infection with enhanced severity in vivo. We therefore hypothesized that a combined treatment with an extracellularly active antibiotic together with an intracellularly active antibiotic should be superior to treatment with an extracellularly active antibiotic alone. To test this hypothesis, mice were challenged with S. aureus-containing wildtype BMDM (Figure 4A), S. aureus-containing nox2−/− BMDM (Figure 4B) or with plain S. aureus (Figure 4C). After 1 h of infection, mice subcutaneously received the extracellularly active antibiotic vancomycin alone, or in combination with the intracellularly active rifampicin. Rifampicin was chosen because of its excellent ability to eradicate intracellular S. aureus in host cells (43). Injection of antibiotics was repeated at 6, 24 and 30 h post infection to account for the limited half-life of vancomycin. At 48 h p.i., livers were explanted and analyzed for bacterial loads. First, we observed that wildtype and nox2−/− BMDM carrying S. aureus produced 2–3 logs greater numbers of S. aureus (CFU/g) than plain S. aureus (Figure 4, compare A, B, and C), which is probably the result of almost instant exposure of extracellular S. aureus to the bacteriocidal antibiotic vancomycin and to the innate host defense. In the two BMDM groups, vancomycin alone reduced the bacterial load in livers in a statistically significant manner (Figures 4A,B) suggesting that S. aureus must escape BMDM over time and are subsequently eliminated by exposure to vancomycin. When mice were infected with plain S. aureus, vancomycin reduced the bacterial burden to a lesser, although still significant, extent leaving a large residual population of viable S. aureus particularly in the liver (Figure 4C). This finding suggests that a large proportion of plain S. aureus are protected from vancomycin in vivo, probably by internalization into professional phagocytes. Indeed, the combined treatment of mice with vancomycin and rifampicin resulted in complete elimination of S. aureus (Figures 4A–C), which underscores a potential benefit of intracellularly active antibiotics in the treatment of S. aureus infections.
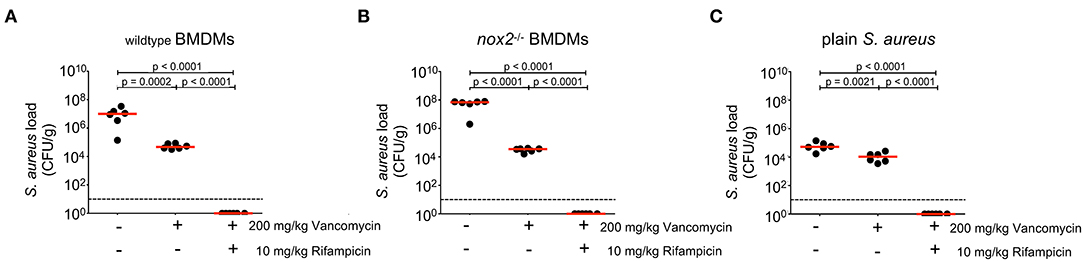
Figure 4. Effects of antibiotic treatment on bacterial load in organs 24 h upon injection of BMDM infected with S. aureus. BMDM of (A) wildtype or (B) nox2−/− mice were infected with S. aureus MW2 GFP (MOI30). 2.5 × 106 infected BMDMs were injected in PBS i.v. into naive C57Bl6J mice (n=6 per genotype and treatment). (C) Mice injected with 4 × 106 plain S. aureus MW2 GFP, corresponding to the CFU recovered from infected BMDM (n=6 per treatment). 200 mg/kg vancomycin was injected s.c. either alone or in combination with 10 mg/kg Rifampicin at 1 h, 6 h, 24 h, and 30 h after i.v. infection. At 48 h p.i. livers of infected mice were homogenized and plated on Mueller-Hinton plates. Shown are individual values for each liver in CFU/g. Red line indicates median, dashed line represents limit of detection. Student's t-test was performed on log-transformed values. NOX, NADPH Oxidas; ROS, reactive oxygen species; PMA, phorbol 12-myristate 13-acetate; DPI, diphenyleniodonium; MOI, multiplicity of infection; BMDMs, bone marrow derived macrophages; SD, standard deviation; TLR2, Toll-like receptor 2; NMS, normal mouse serum; p.i., post infection; CFU, colony forming unit; i.v., intravenous; s.c., subcutaneous.
Discussion
Although macrophages play a crucial role in the control of S. aureus infection (5), macrophages do not always succeed in S. aureus killing, thereby providing a reservoir for bacterial persistence (36). It has been speculated for a long time that neutrophils might serve as Trojan horses for S. aureus and drive dissemination from the primary focus of infection (15). However, whereas the role of neutrophils has been evaluated in greater detail, macrophages, although being a source of bacterial persistence, have been far less studied. We here show that the NADPH oxidase NOX2 plays a crucial role in determining the survival of both intracellular S. aureus and infected bone marrow-derived macrophages. Furthermore, NOX2-deficient BMDM carrying viable intracellular S. aureus were important drivers of systemic dissemination of the pathogen and aggravation of infection. Eventually, in vivo experiments reveal that rifampicin in adjunction of vancomycin completely eradicates S. aureus from liver, kidneys and spleen suggesting a therapeutical potential, targeting S. aureus-carrying macrophages in systemic S. aureus infections, especially in NOX2 deficiency related disorders.
Based on the early observations of Rogers and colleagues and clinical data from the UK, neutrophils were proposed as Trojan horses for the metastasis of S. aureus infection (12, 15, 54, 55). Gresham and co-workers were able to show that S. aureus-infected murine neutrophils obtained from the peritoneal cavity at 24 h post infection readily establish infection when transferred into healthy recipient mice (13). However, neutrophils are short-lived cells and die rapidly when unable to control S. aureus infection (14, 56). Several studies analyzed intracellular survival of S. aureus in macrophages (36, 37, 57–60). Since macrophages, amongst others, eliminate apoptotic neutrophils and were shown to play an important role in clearance of S. aureus upon local infection of the lungs (61), these long-lived cells are also suited as Trojan horse.
Kubica et al. observed that some S. aureus survive for several days in human monocyte-derived macrophages before eventually lysing the host cell and subsequently undergo extracellular replication (60). More recently, Jubrail et al. have shown that, although intracellular antimicrobial mechanisms initially kill S. aureus rapidly after ingestion, they become progressively exhausted despite ongoing phagocytosis leading to host cell lysis and re-uptake by macrophages, which ultimately results in an intracellular pool of persisting S. aureus (37). In line with their observations, our present study provides in vitro evidence that S. aureus is able to survive within BMDM despite the presence of functional NOX2. As expected, NOX2-deficient macrophages were even more permissive, showing a markedly improved survival upon infection in vitro.
ROS represent only one among many other mechanisms to destroy phagocytosed pathogens, which include RNS, proteolytic enzymes, antimicrobial peptides, as well as acidification of the phagolysosome, nutrient restriction, autophagy or extracellular traps. However, mutations in lysosomal enzymes (e.g., myeloperoxidase) processing superoxide radicals into highly active antimicrobial compounds like hypochlorite are less associated with S. aureus infections than NADPH oxidase deficiency (62). Indeed, the outstanding role of NOX2 for the containment of S. aureus infections is reflected by CGD, a genetic immune deficiency affecting the function of the phagocyte NADPH oxidase NOX2, where S. aureus infection is one of the signature complications (8). Although ROS are major, directly acting defense factors against bacteria, other indirect ROS-dependent events severing intracellular processing of S. aureus have been postulated, such as the antagonism of v-ATPase-mediated proton influx inhibiting phagosomal acidification and maturation (37, 63). In addition, the link between Nox2-derived ROS production and the induction of a highly bactericidal form of phagocytosis called LC3-associated phagocytosis was recently discovered and is crucial for degradation of many pathogens (31, 40, 64–67). These reports are in line with our observation that nox2−/− BMDM are more permissive for intracellular S. aureus than wildtype BMDM resulting in better survival of both S. aureus and host cells, which in turn leads to greater metastatic dissemination of S. aureus.
This study has some limitations. Although bone marrow cells were differentiated in vitro using M-CSF, which drives differentiation into the macrophage lineage, it is not clear whether these in vitro-generated BMDM reflect natural macrophages in all their functional facets such as in vivo trafficking, self-renewal potential in specific tissues, polarization into pro- or anti-inflammatory phenotypes and anti-bacterial defense. Furthermore, only few S. aureus strains were investigated in a single strain of mice (C57BL/6J), which does not allow for generalizations. However, this study is consistent with and extends previous excellent reports from many other laboratories, who analyzed the interaction of macrophages and S. aureus in great detail (36, 37), for review (6, 53). In this study, we recapitulated these observations with BMDMs of different genotypes and found no reason to believe that BMDM were fundamentally different to in vivo differentiated macrophages in this context.
Evidence from previous work and the present study is accumulating, indicating that wildtype macrophages, and especially NOX2-deficient macrophages, provide a protective niche for S. aureus enabling the dissemination and aggravation of infection. As to clinical practice, an efficient antibiotic treatment strategy should address intracellular S. aureus, which is especially important in CGD, where macrophages lack NOX2 function (8). Indeed, the ISDA Clinical Practice Guidelines recommend adjunctive rifampicin in specific instances at the B-III level, meaning there is moderate evidence to support a recommendation for or against use, where the evidence comes from opinions of respected authorities, based on clinical experience or descriptive studies, or reports of expert committees (68). A more recent clinical study testing adjunctive rifampicin for S. aureus bacteremia revealed no clear overall benefit over standard antibiotic therapy (44). Although subgroup analyses suggested some benefit in those groups with methicillin-sensitive S. aureus infection treated with flucloxacillin as the only backbone antibiotic (p=0.01), the authors concluded that, with 20 subgroups analyzed, one statistically significant association might have occurred by chance (44). Whereas the clinical use of rifampicin remains controversial, rifampicin is intracellularly effective even within the acidic vacuoles of neutrophils (69) as well as in non-phagocytic cells (43). We show here that a combination therapy of vancomycin and rifampicin in mice infected intravenously with either intracellular S. aureus inside wildtype BMDM or nox2−/− BMDM or with plain bacteria resulted in complete clearance of S. aureus from the liver. In contrast, mice receiving vancomycin alone only reduced the CFU number and the remaining bacterial loads still ranged between 1 × 104 and 1 × 105 CFU/g. Thus, it still might be a worthwhile clinical study to test adjunctive rifampicin treatment of invasive S. aureus infections, especially in patients with CGD.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Ethics commitee University of Cologne (AZ 84-02.04.2014.A013).
Author Contributions
BT, MK, and OK planned and supervised the project. OK, BT, MS, BW, and DG performed experiments. LH performed histopathology. BT, OK, MS, and OU wrote manuscript. MK, MS, MH, BT, and OK revised the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was partially supported by Deutsche Forschungsgemeinschaft (DFG SFB 670 Cell autonomous immunity).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank R. Brandes, K.-H. Krause, and J. Woo for providing us with knock-out mouse lines.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.633629/full#supplementary-material
Supplementary Figure 1. Flow cytometry analysis of S. aureus clearing in BMDMs. BMDMs were infected with GFP-expressing S. aureus MW2 opsonized with 5% NMS and assessed for clearing by flow cytometry. (A) representative dot-plot to demonstrate gating strategy of BMDM population. 5000 events within gate R1 were measured and the percentage of GFP positive BMDMs within this gate was assessed by histogram plot analysis of counts versus channel FL1-H (GFP fluorescence). (B) representative histograms of wildtype (left) or NOX2-/- BMDMs infected with MOI 1.5, MOI15 and MOI30 to define conditions for optimal loading and clearing assays. M1, this marker represents the gate for GFP positive BMDMs. The grey filled plot represents non infected BMDMs. Fluorescence of infected BMDM samples was color-coded corresponding to specific time points p.i.: t0, red line; t6h p.i., dark blue line; and t24h p.i., light blue line. The corresponding fluorescence values are presented in plots. T0 reflects the time point after infection followed by immediate lysis of extracellular S. aureus by lysostaphin. (C) Representative histograms of BMDMs infected with an MOI15 of GFP-expressing S. aureus MW2. These plots correspond to results as shown in Figures 2A–D, respectively. Note different scale of Y-axis for the top panel.
References
1. Lekstrom-Himes JA, Gallin JI. Immunodeficiency diseases caused by defects in phagocytes. N Engl J Med. (2000) 343:1703–14. doi: 10.1056/NEJM200012073432307
2. Von Eiff C, Becker K, Machka K, Stammer H, Peters G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study Group N Engl J Med. (2001) 344:11–6. doi: 10.1056/NEJM200101043440102
3. Wertheim HF, Vos MC, Ott A, Van Belkum A, Voss A, Kluytmans JA, et al. Risk and outcome of nosocomial Staphylococcus aureus bacteraemia in nasal carriers versus non-carriers. Lancet. (2004) 364:703–5. doi: 10.1016/S0140-6736(04)16897-9
4. Heyworth PG, Cross AR, Curnutte JT. Chronic granulomatous disease. Curr Opin Immunol. (2003) 15:578–84. doi: 10.1016/S0952-7915(03)00109-2
5. Surewaard BG, Deniset JF, Zemp FJ, Amrein M, Otto M, Conly J, et al. Identification and treatment of the Staphylococcus aureus reservoir in vivo. J Exp Med. (2016) 213:1141–51. doi: 10.1084/jem.20160334
6. Pidwill GR, Gibson JF, Cole J, Renshaw SA, Foster SJ. The Role of Macrophages in Staphylococcus aureus Infection. Front Immunol. (2020) 11:620339. doi: 10.3389/fimmu.2020.620339
7. Ben-Ari J, Wolach O, Gavrieli R, Wolach B. Infections associated with chronic granulomatous disease: linking genetics to phenotypic expression. Expert Rev Anti Infect Ther. (2012) 10:881–94. doi: 10.1586/eri.12.77
8. Buvelot H, Posfay-Barbe KM, Linder P, Schrenzel J, Krause KH. Staphylococcus aureus, phagocyte NADPH oxidase and chronic granulomatous disease. FEMS Microbiol Rev. (2017) 41:139–57. doi: 10.1093/femsre/fuw042
9. Yu HH, Yang YH, Chiang BL. Chronic granulomatous disease: a comprehensive review. (2020). Clin Rev Allergy Immunol. doi: 10.1007/s12016-020-08800-x
10. Thomas DC, Charbonnier LM, Schejtman A, Aldhekri H, Coomber EL, Dufficy ER, et al. EROS/CYBC1 mutations: Decreased NADPH oxidase function and chronic granulomatous disease. J Allergy Clin Immunol. (2019) 143:782–5. e781. doi: 10.1016/j.jaci.2018.09.019
11. Foster TJ. Immune evasion by staphylococci. Nat Rev Microbiol. (2005) 3:948–58. doi: 10.1038/nrmicro1289
12. Rogers DE. Studies on bacteriemia. I Mechanisms relating to the persistence of bacteriemia in rabbits following the intravenous injection of staphylococci J Exp Med. (1956) 103:713–42. doi: 10.1084/jem.103.6.713
13. Gresham HD, Lowrance JH, Caver TE, Wilson BS, Cheung AL, Lindberg FP. Survival of Staphylococcus aureus inside neutrophils contributes to infection. J Immunol. (2000) 164:3713–22. doi: 10.4049/jimmunol.164.7.3713
14. Voyich JM, Braughton KR, Sturdevant DE, Whitney AR, Said-Salim B, Porcella SF, et al. Insights into mechanisms used by Staphylococcus aureus to avoid destruction by human neutrophils. J Immunol. (2005) 175:3907–19. doi: 10.4049/jimmunol.175.6.3907
15. Thwaites GE, Gant V. Are bloodstream leukocytes Trojan Horses for the metastasis of Staphylococcus aureus? Nat Rev Microbiol. (2011) 9:215–22. doi: 10.1038/nrmicro2508
16. Houghton AM, Hartzell WO, Robbins CS, Gomis-Ruth FX, Shapiro SD. Macrophage elastase kills bacteria within murine macrophages. Nature. (2009) 460:637–41. doi: 10.1038/nature08181
17. Weiss J, Olsson I. Cellular and subcellular localization of the bactericidal/permeability-increasing protein of neutrophils. Blood. (1987) 69:652–9. doi: 10.1182/blood.V69.2.652.bloodjournal692652
19. Nathan C, Shiloh MU. Reactive oxygen and nitrogen intermediates in the relationship between mammalian hosts and microbial pathogens. Proc Natl Acad Sci U S A. (2000) 97:8841–8. doi: 10.1073/pnas.97.16.8841
20. Lehrer RI, Ganz T. Defensins of vertebrate animals. Curr Opin Immunol. (2002) 14:96–102. doi: 10.1016/S0952-7915(01)00303-X
21. Selsted ME, Ouellette AJ. Mammalian defensins in the antimicrobial immune response. Nat Immunol. (2005) 6:551–7. doi: 10.1038/ni1206
22. Silva MT, Correia-Neves M. Neutrophils and macrophages: the main partners of phagocyte cell systems. Front Immunol. (2012) 3:174. doi: 10.3389/fimmu.2012.00174
23. Prame Kumar K, Nicholls AJ, Wong CHY. Partners in crime: neutrophils and monocytes/macrophages in inflammation and disease. Cell Tissue Res. (2018) 371:551–65. doi: 10.1007/s00441-017-2753-2
24. Davies LC, Jenkins SJ, Allen JE, Taylor PR. Tissue-resident macrophages. Nat Immunol. (2013) 14:986–95. doi: 10.1038/ni.2705
25. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. (2014) 41:21–35. doi: 10.1016/j.immuni.2014.06.013
26. Jung S. Macrophages and monocytes in 2017: macrophages and monocytes: of tortoises and hares. Nat Rev Immunol. (2018) 18:85–6. doi: 10.1038/nri.2017.158
27. Haas A. The phagosome: compartment with a license to kill. Traffic. (2007) 8:311–30. doi: 10.1111/j.1600-0854.2006.00531.x
28. Pauwels AM, Trost M, Beyaert R, Hoffmann E. Patterns, Receptors, and Signals: Regulation of Phagosome Maturation. Trends Immunol. (2017) 38:407–22. doi: 10.1016/j.it.2017.03.006
29. Mastroeni P, Morgan FJ, Mckinley TJ, Shawcroft E, Clare S, Maskell DJ, et al. Enhanced virulence of Salmonella enterica serovar typhimurium after passage through mice. Infect Immun. (2011) 79:636–43. doi: 10.1128/IAI.00954-10
30. Schramm M, Wiegmann K, Schramm S, Gluschko A, Herb M, Utermohlen O, et al. Riboflavin (vitamin B2) deficiency impairs NADPH oxidase 2 (Nox2) priming and defense against Listeria monocytogenes. Eur J Immunol. (2014) 44:728–41. doi: 10.1002/eji.201343940
31. Gluschko A, Herb M, Wiegmann K, Krut O, Neiss WF, Utermohlen O, et al. The beta2 integrin mac-1 induces protective LC3-associated phagocytosis of listeria monocytogenes. Cell Host Microbe. (2018) 23:324–37. e325. doi: 10.1016/j.chom.2018.01.018
32. Alvarez-Dominguez C, Carrasco-Marin E, Lopez-Mato P, Leyva-Cobian F. The contribution of both oxygen and nitrogen intermediates to the intracellular killing mechanisms of C1q-opsonized Listeria monocytogenes by the macrophage-like IC-21 cell line. Immunology. (2000) 101:83–9. doi: 10.1046/j.1365-2567.2000.00083.x
33. Aktan F. iNOS-mediated nitric oxide production and its regulation. Life Sci. (2004) 75:639–53. doi: 10.1016/j.lfs.2003.10.042
34. Weiss G, Schaible UE. Macrophage defense mechanisms against intracellular bacteria. Immunol Rev. (2015) 264:182–203. doi: 10.1111/imr.12266
35. Mitchell G, Chen C, Portnoy D. Strategies Used by Bacteria to Grow in Macrophages. In: Gordon S editors. Myeloid Cells in Health and Disease. Washington, DC: ASM Press (2017) 701–25. doi: 10.1128/microbiolspec.MCHD-0012-2015
36. Flannagan RS, Heit B, Heinrichs DE. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol. (2016) 18:514–35. doi: 10.1111/cmi.12527
37. Jubrail J, Morris P, Bewley MA, Stoneham S, Johnston SA, Foster SJ, et al. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol. (2016) 18:80–96. doi: 10.1111/cmi.12485
38. Prajsnar TK, Cunliffe VT, Foster SJ, Renshaw SA. A novel vertebrate model of Staphylococcus aureus infection reveals phagocyte-dependent resistance of zebrafish to non-host specialized pathogens. Cell Microbiol. (2008) 10:2312–25. doi: 10.1111/j.1462-5822.2008.01213.x
39. Prajsnar TK, Hamilton R, Garcia-Lara J, Mcvicker G, Williams A, Boots M, et al. A privileged intraphagocyte niche is responsible for disseminated infection of Staphylococcus aureus in a zebrafish model. Cell Microbiol. (2012) 14:1600–19. doi: 10.1111/j.1462-5822.2012.01826.x
40. Prajsnar TK, Serba JJ, Dekker BM, Gibson JF, Masud S, Fleming A, et al. The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy. (2020) 1–15. doi: 10.1080/15548627.2020.1739443
41. Pollitt EJG, Szkuta PT, Burns N, Foster SJ. Staphylococcus aureus infection dynamics. PLoS Pathog. (2018) 14:e1007112. doi: 10.1371/journal.ppat.1007112
42. Norden CW, Budinsky A. Treatment of experimental chronic osteomyelitis due to Staphylococcus aureus with ampicillin/sulbactam. J Infect Dis. (1990) 161:52–3. doi: 10.1093/infdis/161.1.52
43. Krut O, Sommer H, Kronke M. Antibiotic-induced persistence of cytotoxic Staphylococcus aureus in non-phagocytic cells. J Antimicrob Chemother. (2004) 53:167–73. doi: 10.1093/jac/dkh076
44. Thwaites GE, Scarborough M, Szubert A, Nsutebu E, Tilley R, Greig J, et al. Adjunctive rifampicin for Staphylococcus aureus bacteraemia (ARREST): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. (2018) 391:668–78. doi: 10.1016/S0140-6736(17)32456-X
45. Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, et al. Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production. Nat Genet. (1995) 9:202–9. doi: 10.1038/ng0295-202
46. Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, et al. A key role for NOX4 in epithelial cell death during development of lung fibrosis. Antioxid Redox Signal. (2011) 15:607–19. doi: 10.1089/ars.2010.3829
47. Gavazzi G, Banfi B, Deffert C, Fiette L, Schappi M, Herrmann F, et al. Decreased blood pressure in NOX1-deficient mice. FEBS Lett. (2006) 580:497–504. doi: 10.1016/j.febslet.2005.12.049
48. Nakano Y, Longo-Guess CM, Bergstrom DE, Nauseef WM, Jones SM, Banfi B. Mutation of the Cyba gene encoding p22phox causes vestibular and immune defects in mice. J Clin Invest. (2008) 118:1176–85. doi: 10.1172/JCI33835
49. Charpentier E, Anton AI, Barry P, Alfonso B, Fang Y, Novick RP. Novel cassette-based shuttle vector system for gram-positive bacteria. Appl Environ Microbiol. (2004) 70:6076–85. doi: 10.1128/AEM.70.10.6076-6085.2004
50. Herb M, Farid A, Gluschko A, Kronke M, Schramm M. Highly Efficient Transfection of Primary Macrophages with In Vitro Transcribed mRNA. J Vis Exp. (2019) 153. doi: 10.3791/60143
51. Tian W, Li XJ, Stull ND, Ming W, Suh CI, Bissonnette SA, et al. Fc gamma R-stimulated activation of the NADPH oxidase: phosphoinositide-binding protein p40phox regulates NADPH oxidase activity after enzyme assembly on the phagosome. Blood. (2008) 112:3867–77. doi: 10.1182/blood-2007-11-126029
52. Watanabe I, Ichiki M, Shiratsuchi A, Nakanishi Y. TLR2-mediated survival of Staphylococcus aureus in macrophages: a novel bacterial strategy against host innate immunity. J Immunol. (2007) 178:4917–25. doi: 10.4049/jimmunol.178.8.4917
53. Moldovan A, Fraunholz MJ. In or out: Phagosomal escape of Staphylococcus aureus. Cell Microbiol. (2019) 21:e12997. doi: 10.1111/cmi.12997
54. Rogers DE. Staphylococcal infections. Dis Mon. (1958) 8:1–48. doi: 10.1016/S0011-5029(58)80009-7
55. Rogers DE, Melly MA. Further observations on the behavior of staphylococci within human leukocytes. J Exp Med. (1960) 111:533–58. doi: 10.1084/jem.111.4.533
56. Kobayashi SD, Braughton KR, Palazzolo-Ballance AM, Kennedy AD, Sampaio E, Kristosturyan E, et al. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J Innate Immun. (2010) 2:560–75. doi: 10.1159/000317134
57. Baughn R, Bonventre PF. Phagocytosis and intracellular killing of Staphylococcus aureus by normal mouse peritoneal macrophages. Infect Immun. (1975) 12:346–52. doi: 10.1128/IAI.12.2.346-352.1975
58. Elliott GR, Peterson PK, Verbrugh HA, Freiberg MR, Hoidal JR, Quie PG. Influence of subinhibitory concentrations of penicillin, cephalothin, and clindamycin on Staphylococcus aureus growth in human phagocytic cells. Antimicrob Agents Chemother. (1982) 22:781–4. doi: 10.1128/AAC.22.5.781
59. Hebert A, Sayasith K, Senechal S, Dubreuil P, Lagace J. Demonstration of intracellular Staphylococcus aureus in bovine mastitis alveolar cells and macrophages isolated from naturally infected cow milk. FEMS Microbiol Lett. (2000) 193:57–62. doi: 10.1016/S0378-1097(00)00455-9
60. Kubica M, Guzik K, Koziel J, Zarebski M, Richter W, Gajkowska B, et al. A potential new pathway for Staphylococcus aureus dissemination: the silent survival of S. aureus phagocytosed by human monocyte-derived macrophages PLoS ONE. (2008) 3:e1409. doi: 10.1371/journal.pone.0001409
61. Shapiro A, Raman S, Johnson M, Piehl M. Community-acquired MRSA infections in North Carolina children: prevalence, antibiotic sensitivities, and risk factors. N C Med J. (2009) 70:102–7. doi: 10.18043/ncm.70.2.102
62. Lanza F. Clinical manifestation of myeloperoxidase deficiency. J Mol Med (Berl). (1998) 76:676–81. doi: 10.1007/s001090050267
63. Savina A, Jancic C, Hugues S, Guermonprez P, Vargas P, Moura IC, et al. NOX2 controls phagosomal pH to regulate antigen processing during crosspresentation by dendritic cells. Cell. (2006) 126:205–18. doi: 10.1016/j.cell.2006.05.035
64. Neumann Y, Bruns SA, Rohde M, Prajsnar TK, Foster SJ, Schmitz I. Intracellular Staphylococcus aureus eludes selective autophagy by activating a host cell kinase. Autophagy. (2016) 12:2069–84. doi: 10.1080/15548627.2016.1226732
65. Heckmann BL, Green DR. LC3-associated phagocytosis at a glance. J Cell Sci. (2019) 132. doi: 10.1242/jcs.222984
66. Herb M, Gluschko A, Schramm M. LC3-associated phagocytosis - The highway to hell for phagocytosed microbes. Semin Cell Dev Biol. (2020) 101:68–76. doi: 10.1016/j.semcdb.2019.04.016
67. Munoz-Sanchez S, Van Der Vaart M, Meijer AH. Autophagy and Lc3-Associated Phagocytosis in Zebrafish Models of Bacterial Infections. Cells. (2020) 9:2372. doi: 10.3390/cells9112372
68. Liu C, Bayer A, Cosgrove SE, Daum RS, Fridkin SK, Gorwitz RJ, et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis. (2011) 52:e18–55. doi: 10.1093/cid/ciq146
Keywords: chronic granulomatous disease, Staphylococcus aureus, reactive oxygen species, antibiotic treatment, sepsis model, macrophages
Citation: Tosetti B, Ward B, Grumme D, Herb M, Schramm M, Utermöhlen O, Heukamp LC, Krönke M and Krut O (2021) NOX2 Deficiency Permits Sustained Survival of S. aureus in Macrophages and Contributes to Severity of Infection. Front. Immunol. 12:633629. doi: 10.3389/fimmu.2021.633629
Received: 25 November 2020; Accepted: 24 February 2021;
Published: 22 March 2021.
Edited by:
Fabio Bagnoli, GlaxoSmithKline, ItalyReviewed by:
Tiina Kelkka, University of Helsinki, FinlandGalileo Escobedo, General Hospital of Mexico, Mexico
Marie José STASIA, UMR5075 Institut de Biologie Structurale (IBS), France
Copyright © 2021 Tosetti, Ward, Grumme, Herb, Schramm, Utermöhlen, Heukamp, Krönke and Krut. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Martin Krönke, bS5rcm9lbmtlQHVuaS1rb2Vsbi5kZQ==; Oleg Krut, b2xlZy5rcnV0QHBlaS5kZQ==