
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 20 April 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.631714
This article is part of the Research TopicPhagocytes in Immunity: Linking Material Internalization to Immune Responses and Therapeutic StrategiesView all 17 articles
 Charles Yin1,2
Charles Yin1,2 Bryan Heit1,2,3*
Bryan Heit1,2,3*The rapid and efficient phagocytic clearance of apoptotic cells, termed efferocytosis, is a critical mechanism in the maintenance of tissue homeostasis. Removal of apoptotic cells through efferocytosis prevents secondary necrosis and the resultant inflammation caused by the release of intracellular contents. The importance of efferocytosis in homeostasis is underscored by the large number of inflammatory and autoimmune disorders, including atherosclerosis and systemic lupus erythematosus, that are characterized by defective apoptotic cell clearance. Although mechanistically similar to the phagocytic clearance of pathogens, efferocytosis differs from phagocytosis in that it is immunologically silent and induces a tissue repair response. Efferocytes face unique challenges resulting from the internalization of apoptotic cells, including degradation of the apoptotic cell, dealing with the extra metabolic load imposed by the processing of apoptotic cell contents, and the coordination of an anti-inflammatory, pro-tissue repair response. This review will discuss recent advances in our understanding of the cellular response to apoptotic cell uptake, including trafficking of apoptotic cell cargo and antigen presentation, signaling and transcriptional events initiated by efferocytosis, the coordination of an anti-inflammatory response and tissue repair, unique cellular metabolic responses and the role of efferocytosis in host defense. A better understanding of how efferocytic cells respond to apoptotic cell uptake will be critical in unraveling the complex connections between apoptotic cell removal and inflammation resolution and maintenance of tissue homeostasis.
Efferocytosis is the process of rapid and efficient clearance of apoptotic cells by both professional and non-professional phagocytic cells (1, 2). From an evolutionary perspective, efferocytosis is an ancient mechanism that allowed early multicellular organisms to regulate their growth through the disposal of dying cells during development (3). In complex multicellular organisms, efferocytosis is critical in growth and development, for the resolution of inflammation, and for maintaining tissue homeostasis (4–6). Mechanistically, efferocytosis closely resembles phagocytosis—the internalization and clearance of pathogens and other foreign particulates (7). Indeed, though efferocytosis utilizes a distinct and well-characterized set of cell surface receptors (e.g. TAM family receptors, Tim4, αV integrins) and soluble opsonins (e.g. Gas6, MFGE8, CD93) that bind to ligands found on the plasma membrane of apoptotic cells (e.g. phosphatidylserine), much of the processes downstream of apoptotic cell internalization such as intracellular trafficking of apoptotic cell cargo and cellular responses to internalized of apoptotic cell contents are either thought to be wholly analogous to phagocytosis or to be poorly understood (1, 7–10). Strikingly, efferocytes such as macrophages can distinguish between normal apoptotic cells and those infected with an intracellular pathogen, despite the fact that much of the contents of an infected apoptotic cell (e.g. lipids, nucleic acids, proteins) are identical to that of a non-infected cell, allowing the efferocyte to mount an appropriate immunological response to pathogens within efferocytosed cells (3). This demonstrates that efferocytosis is a distinct process from phagocytosis, that and efferocytes are fine-tuned to be able to distinguish between apoptotic versus pathogenic cargo.
The major efferocytic cell—or efferocyte—within the body is the macrophage (11). These immune cells are responsible for clearance of apoptotic cells and debris across many tissues (12, 13). There is emerging evidence that efferocytic macrophages form a distinct subset of tissue-resident macrophages that differ in both function and pattern of gene expression compared to other tissue-resident macrophage populations (14, 15). Indeed, A-Gonzalez et al. (14) found that tissue-resident murine efferocytic macrophages from across a range of different tissues share a common transcriptional profile, which is characterized by downregulation of proinflammatory cytokines such as IL1β and expression of the mannose receptor CD206 (14). Interestingly, although this population upregulates several anti-inflammatory genes, it’s gene expression profile does not co-cluster with alternatively activated (M2) macrophages (14). This suggests that efferocytic macrophages cannot simply to be thought of as “anti-inflammatory” macrophages, and instead occupy a distinct space in the macrophage transcriptional landscape.
The purpose of this review will be to discuss the distinct cellular responses elicited upon uptake of apoptotic cells by an efferocyte, with a focus on macrophages as the major efferocytic cell population within the body. In particular, we will review differences in trafficking of apoptotic cell cargo and presentation of antigens following internalization, alterations in cell signaling and transcriptional regulation, as well as explore how changes in cargo trafficking and gene expression contribute to the anti-inflammatory phenotype that characterize efferocytes. Further, we will explore recent advances in our understanding of how efferocytes deal with the metabolic stress of internalizing apoptotic cells, how efferocytes respond upon uptake of infected apoptotic cells, and the role of efferocytosis in host defense.
Following recognition, apoptotic cells are engulfed by the efferocyte into a plasma membrane-derived vacuole termed an efferosome (8). Similar to phagosomes that contain internalized pathogens, efferosomes undergo a highly regulated series of sequential fusions with early endosomes, late endosomes, and finally lysosomes (Figure 1) (16–18). These fusion events are regulated by proteins including Rab GTPases and SNAREs, with the fusion events delivering the hydrolytic enzymes which degrade the apoptotic cell within the efferosome (19–23). This process is termed efferosome maturation and is analogous to the maturation processes observed following phagocytosis and endocytosis (8, 24).
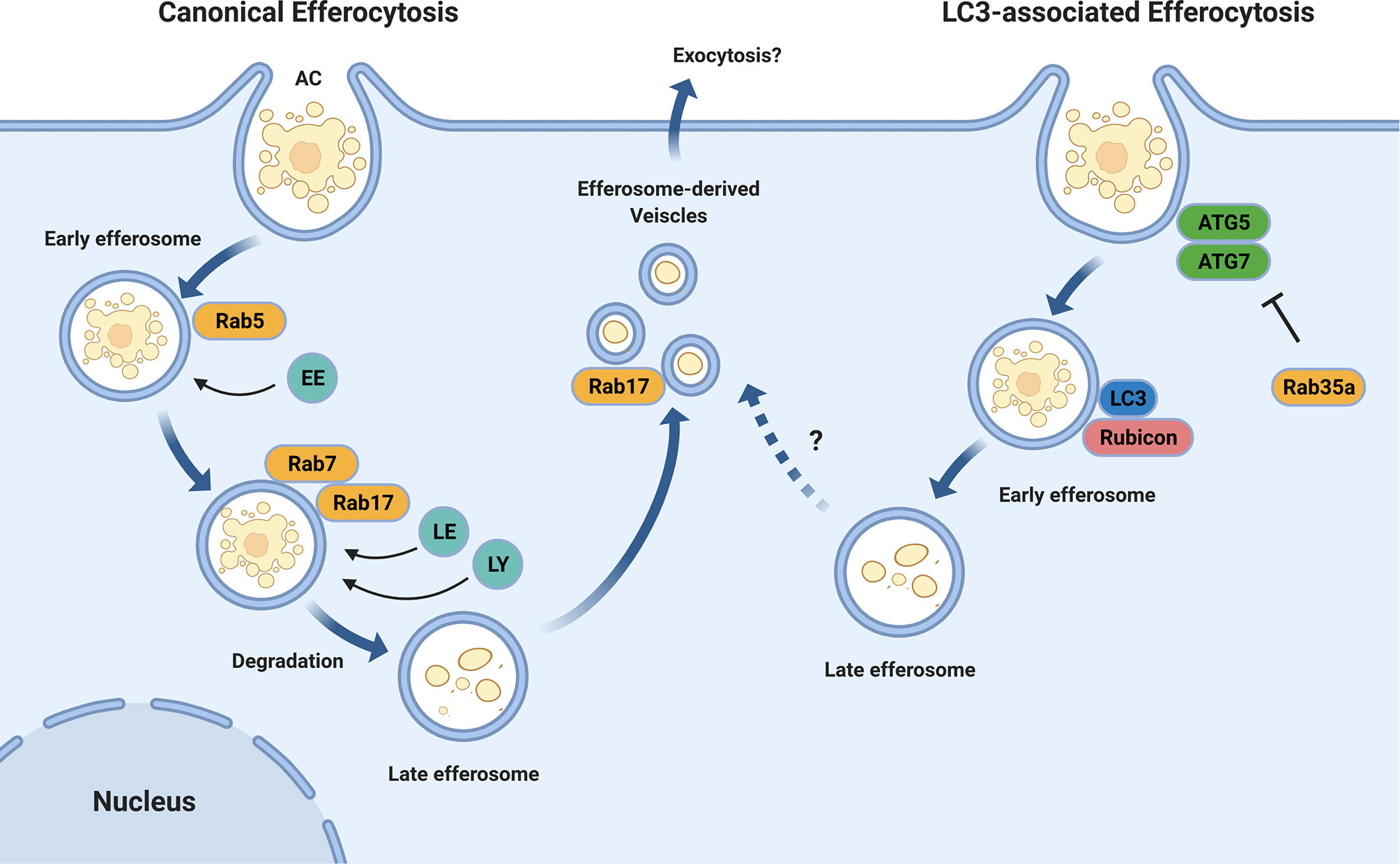
Figure 1 Efferosome Maturation Pathways. Efferocytosis can occur through the canonical endo-lysosomal maturation pathway (left) in which the GTPases Rab5 and Rab7 mediate the sequential fusion of early endosomes (EE), late endosomes (LE), and lysosomes (LY) with the maturing efferosome. Unlike phagocytosis, this efferosome maturation pathway also involves Rab17 which directs the degraded contents from the efferosome to the recycling endosome from where they may be exocytosed, thereby avoiding the delivery of these materials to antigen loading compartments. In addition to the canonical pathway, some efferosomes may mature through an LC3-mediated, autophagy-like pathway (right). In this pathway, the efferosome recruits the protein LC3 which then mediates a rapid degradation of the efferosome in a fashion which suppresses antigen presentation. Similar to LC3-associated phagocytosis, the recruitment of the autophagy-related proteins ATG5 and ATG7, as well as Rubicon to the nascent efferosome appear to be important for efferocytosis through this pathway. The activity of ATG5 and ATG7 are inhibited by Rab35a, which is activated downstream of TLR signaling. Figure produced using BioRender.
The efferosome maturation process bears many similarities to phagosome maturation, including the recruitment of the Rab GTPases Rab5 and Rab7 (23, 25, 26). Rab5 is recruited to efferosomes as the apoptotic cell is internalized, and remains bound to the efferosome for several minutes following the release of the efferosome from the plasma membrane (19, 27). Here, Rab5 mediates the fusion of the efferosome with early endosomes, beginning the degradative process which will ultimately disassemble the apoptotic cell (19, 20). Rab5 is exchanged for Rab7 several minutes after efferosome formation, with Rab7 mediating the fusion of late endosomes and lysosomes to the efferosome – thus generating a highly hydrolytic environment capable of the complete degradation of the apoptotic cell (18, 27). Recent work by our group and others have demonstrate important differences between the regulation of efferosome maturation versus phagosome maturation (27, 28). Efferosome acidification is a central process that facilitates the degradation of apoptotic cargo through activation of lysosomal proteases (29, 30).
Efferosomes have also been shown to employ LC3-associated phagocytosis (LAP, Figure 1), a noncanonical form of autophagy that involves recruitment of autophagy mediators including the class III phosphatidylinositol-3-kinase (PI3KCIII) complex ATG5 and ATG7 to the surface of nascent efferosomes (31, 32). These elements then direct the rapid maturation of the efferosome and processing of the apoptotic cargo in a manner that suppresses antigen presentation and serves to polarize macrophages towards an anti-inflammatory phenotype (33). While the exact signals which allow for LAP to be employed for efferosome maturation remain unknown, work from the Medzhitov group has demonstrated that phagosome-derived Toll-like receptor (TLR) signaling is required to direct materials into the classical phagocytic (e.g. non-LAP) pathway where they then undergo antigen presentation (34, 35). This indicates that the detection of pathogen products via TLR’s serves not only to induce the expression of genes involved in inflammation and antigen presentation, but also induces immediate differences in the trafficking of cargo bearing TLR ligands compared to those lacking these ligands (36). Rab39a may serve to inhibit LAP following phagocytosis, as this GTPase inhibits autophagy following TLR signaling, and is required for the delivery of MHC I to phagosomes for antigen cross-presentation (37, 38). However, there are no published studies of the role of Rab39a in efferocytosis, and therefore its role in efferocytosis-associated LAP remains unclear. Interestingly, LAP and the formation of LC3-associated efferosomes is dependent on the Beclin1-interacting protein Rubicon (39). Rubicon is a negative regulator of canonical autophagy and downregulation of this protein results in an increase in the number of autophagosomes (39, 40). Indeed, deletion of Rubicon in a mouse model of autoimmune disease significantly increases susceptibility to the development of systemic lupus erythematosus-like features in these animals, potentially due to altered processing of apoptotic cells (41).
Differences in acidification and trafficking of efferosomes, as compared to phagosomes, also plays a role in ensuring the immunologically silent degradation of apoptotic cells (34, 42, 43). There is conflicting evidence in the literature on the kinetics of efferosome maturation as compared to phagosome maturation (35, 42). Erwig et al. reported that in murine macrophages, early maturation and acidification of efferosomes containing apoptotic neutrophils proceeded at a faster rate than phagosomes containing IgG-opsonized neutrophils (42). Inhibition of the small GTPase RhoA using a small molecule inhibitor was sufficient to negate these differences (42). In contrast, Blander and Medzhitov have shown that efferosome maturation proceeded at a slower rate than phagosomes (35). Of note, in the case of Blander and Medzhitov, the phagocytic target employed was inactivated Escherichia coli and the authors argue that it was activation of TLR2 and TLR4 signaling that drove accelerated phagosome maturation (35). In contrast, the IgG-coated neutrophils used by Erwig and colleagues would not have stimulated TLRs in the same fashion (42).
Our group has recently demonstrated that efferosome localization appears to play a role in distinguishing the fate of apoptotic cargo (28). Canonically, phagosomes undergo dynein-mediated trafficking towards the cell centre as they mature, where lysosomes are concentrated due to a similar dynein-mediated trafficking pathway (16, 44–46). Thus, by moving to the cell centre, phagosomes can efficiently undergo fusion with lysosomes to acquire the hydrolytic enzymes that degrade phagosome cargos (29, 47). In contrast, we have shown that while efferosomes also undergo an initial migration towards the cell centre where they fuse with lysosomes, they subsequently fragment into smaller efferosome-derived vesicles (EDVs) which migrate away from the cell centre and towards the periphery (28). At the periphery, EDVs undergo fusion with the recycling endosome compartment, presumably to facilitate exocytosis of degraded apoptotic cargo or resorption of nutrients (28). This process is driven by the small GTPase Rab17 (Figure 1), which is required for both the fragmentation of efferosomes into EDVs and for the movement of the EDVs to the cell periphery (27, 28). Macrophages that overexpress a dominant-negative mutant of Rab17 accumulate efferosomes at the cell center (28). Furthermore, the presence of Rab17 on efferosomes also prevents the delivery of MHC class II, circumventing autoantigen presentation from degraded apoptotic cargo (27). Expression of a dominant-negative Rab17 impairs this pathway, leading to MHC II accumulation in mature efferosomes (27).
The presence of three processes that work simultaneously to limit antigen presentation of efferosome-derived antigens – LAP, accelerated maturation, and Rab17-mediated redirection of cargo out of the maturing efferosome – indicates that limiting autoantigen presentation is a fundamental response of phagocytes following efferocytosis. Moreover, efferocytes engage in non-trafficking-based mechanisms to limit autoimmune responses to efferocytosed materials. As described later in this review, efferocytosis is often accompanied by the upregulation of cytokines such as IL-10 which suppress the activity of mature T cells and promotes the formation of Treg cells from naive T cells (48). Consequentially, T cell responses are inhibited following efferocytosis. For example, Rodriguez-Fernandez et al. demonstrated that in human DCs, efferocytosis of PtdSer-containing liposomes biased the stimulation of autologous T cells from a proliferative to a tolerogenic profile, likely through altered cytokine expression by the DCs (49). Consistent with these mechanisms acting to limit autoreactivity, emerging evidence indicate that defects in the suppression of antigen presentation following efferocytosis is a driver of autoimmune disease (50, 51). In mouse models of systemic lupus erythematosus, dysregulated expression of specific pro-efferocytic receptors such as Tim4, C1q or CLM-1 result in either deficient apoptotic cell clearance or inappropriate antigen presentation that then promotes the development of autoimmune disease in these mice (52). In humans, mutations in efferocytic receptors, especially in MERTK and its opsonins, are associated with a similar increase in the risk of autoimmune disorders including multiple sclerosis and rheumatoid arthritis, highlighting the importance of efficient efferocytosis in limiting autoimmunity (53–57).
Interestingly, some professional antigen presenting cells have mechanisms that allow efferosome-derived antigens to be cross-presented on MHC I. A recent study by Canton et al. demonstrated that type 1 conventional dendritic cells use the receptor DNGR-1 to recognize actin-myosin complexes exposed to the efferosome lumen during the early stage of efferosome maturation (58). Recognition of actin-myosin complexes leads to an alternative maturation pathway where the efferosome does not acquire its normal degradative capacity, and instead, Syk-induced NADPH oxidase activity damages the efferosomal membrane, releasing the efferosome’s cargo into the cytosol. Once in the cytosol, the efferocytosed materials are processed and presented via the canonical MHC I presentation pathway [reviewed in (59)]. Interestingly, the restriction of this process to the early stages of efferosome maturation suggests that this process may only occur in response to engulfed cells that have pre-exposed actin-myosin complexes – e.g. cells which have lost membrane integrity as they progress through late stage apoptosis, or cells which have died a lytic form of cell death such as necroptosis or necrosis (9). Alternatively, this pathway may enable the routine “screening” of apoptotic cell-derived antigens via MHC I, which because it relies on T cells previously activated to the same antigen presented on MHC II by professional antigen presenting cells, lacks the autoimmune potential of MHC II presentation (60).
Differences between the cellular response of phagocytes to efferocytosis of apoptotic cells versus phagocytosis of pathogens require that there be efferocytosis-specific signal transduction events and transcriptional regulation (1, 2). We are just beginning to develop an understanding of the key transcriptional factors that control the cellular events that occur following efferocytosis. Two key families of transcriptional factors that drive this response are members of the liver X receptor (LXR) and peroxisome proliferator-activated receptor (PPAR) families of nuclear receptors (61–63). These transcription factor families bind to the same DNA motifs, and often act as heterodimers, meaning that their functions are often overlapping and redundant (64). Both receptor families bind to many of the same ligands, notably lipid-derived metabolites, with their activation leading to the preferential formation of heterodimers that then bind to direct 5’ – RGKTCA – 3’ repeats (65). Once bound, these LXRs and PPARs coordinate with other transcription factors to either activate or repress transcription (64, 66, 67).
LXRs are well-characterized regulators of cholesterol, glucose and fatty acid metabolism (68). The two members of the LXR family, LXRα and LXRβ, are both activated following efferocytic apoptotic cell uptake, and in turn increase the cell’s efferocytic capacity via two distinct mechanisms (69). The first mechanism – described in-detail later in this review – is the upregulation of the metabolic pathways required to process the large quantities of lipids, sterols and proteins present in an efferocytosed apoptotic cell. The second mechanism is the upregulation of efferocytic receptors and signaling molecules. Stimulation of LXRs in vivo with apoptotic thymocytes has been shown to upregulate MERTK, a key efferocytic receptor involved in apoptotic cell recognition and uptake (69, 70). This enhances the efferocytic capacity of the efferocyte, and increases MERTK-mediated anti-inflammatory activity via increased activation of SOCS3, a suppressor of cytokine-induced JAK/STAT signaling (71). Conversely, peritoneal macrophages isolated from LXR double-knockout mice have been shown to have diminished capacity to engage in efferocytosis, without any impairment in the phagocytosis of E. coli (69). Indeed, activation of LXRα/β appears to be required to shift macrophages away from a pro-inflammatory state following efferocytosis, with exposure of LXR double-knockout macrophages to apoptotic thymocytes resulting in increased expression of several pro-inflammatory mediators including IL1β, MCP-1 and the scavenger receptor MARCO (69).
Given the functional overlap between LXRs and PPARs, it is of no surprise that the observed role of the PPAR family in efferocytosis closely parallels the role of LXRs. As with LXRs, PPARs have previously been implicated in macrophage polarization and in enhancing lipids metabolism and synthesis of lipid-derived molecules such as eicosanoids and arachidonic acid (72). Similar to the LXR family of transcription factors, activation of certain members of the PPAR family, including PPARγ and PPARδ, appear to directly enhance efferocytic activity in macrophages (63, 66, 73). Majai et al. demonstrated that downregulation of PPARγ activity using a small-molecule inhibitor resulted in a diminished capacity of human monocyte-derived macrophages to efferocytosed apoptotic neutrophils (66). This resulted from the downregulation of several key efferocytic receptors including CD36, AXL, TG2 and PTX3 (66). Using PPARγ-specific agonists, Zizzo & Cohen demonstrated that PPARγ activation leads directly to upregulation of MERTK and its opsonin Gas6 in macrophages, as well as to polarization of macrophages to a pro-efferocytic M2c phenotype (74). Furthermore, efferocytosis of apoptotic cells by macrophages has been shown to directly suppress key inflammatory pathways, including activation of PKCα, a kinase involved in many cellular functions including inflammatory cytokine transcription and the generation of bactericidal free radicals (64). Indeed, activation of PPARγ in response to efferocytosis of apoptotic cells in murine macrophages has been shown to attenuate reactive oxygen species formation in response to proinflammatory mediators (64). Similarly, efferocytosis induces the expression of SOCS1 and SOCS3, which in turn inhibit Jak/STAT signaling through inflammatory cytokine receptors, thereby reducing the responsiveness of efferocytic macrophages to inflammatory stimuli (75). Finally, the uptake of apoptotic thymocytes by murine bone marrow-derived macrophages has been shown by Mukundan and colleagues to upregulate PPARδ and stimulate PPARδ-dependent expression of C1qb, a member of the complement cascade that has been identified as an opsonin involved in the efferocytic clearance of apoptotic macrophages (63).
Defects in efferocytosis have been implicated in the pathogenesis of several inflammatory and autoimmune disorders, including atherosclerosis (76, 77). Our group recently discovered that atherosclerotic macrophages upregulated the hematopoietic transcription factor GATA2 in response to modified lipoproteins (78). Upregulation of GATA2 led to the downregulation of multiple proteins required for efficient efferocytosis, including downregulation of the efferocytic receptor αX integrin, multiple signaling molecules required for these receptors function including multiple Src-family kinases, impaired efferosome-lysosome fusion via decreased expression of Rab7, and impairment in multiple degradative pathways needed for the degradation of apoptotic cargos including lysosomal acidification (10, 78). Interestingly, mutations in the GATA2 gene has been linked to increased risk of cardiovascular disease in human cohort studies (79). It remains to be seen whether there are other transcription factors that act to impair efferocytosis during autoimmune or inflammatory diseases.
A key feature of efferocytosis is the limitation of inflammation and the resolution of inflammatory responses (5, 9). We have previously discussed how efferosome maturation acts to prevent antigen presentation on MHC II, and how efferocytosis activates transcriptional programs that restrain inflammation (27, 78). It is well established that efferocytosis induces the production of anti-inflammatory mediators (80, 81). Meagher et al. showed as early as 1992 that the uptake of apoptotic neutrophils by macrophages does not lead to release of the pro-inflammatory mediator thromboxane A2, in contrast with phagocytosis of bacterial pathogens (80). Only a few years later Fadok and colleagues demonstrated that efferocytosis in macrophages resulted in suppression of a host of proinflammatory molecules including IL1β, IL8, IL10, GM-CSF and TNFα (81). Furthermore, these investigators determined that efferocytosis upregulated anti-inflammatory mediators including TGFβ and prostaglandin E2 (81).
More recent studies have demonstrated that efferocytic macrophages carry anti-inflammatory functions and gene expression signatures. A landmark study in 2017 showed that pro-efferocytic macrophages across various tissues carried a distinct gene expression signature that differentiated them from other tissue-resident macrophages (14). In particular, this pro-efferocytic signature is characterized by downregulation of the inflammatory cytokine IL1β (14). Campana et al. further demonstrated that in a sterile liver inflammation model, efferocytosis of apoptotic hepatocytes induced a M2-like phenotype and activation of the STAT3-IL6-IL10 pathway (82). Finally, in an acute coronary ligature model, Howangyin and colleagues demonstrated that mouse macrophages lacking the efferocytic receptor MERTK and its opsonin MFGE8 had decreased production of the vascular tissue repair factor VEGF-A and increased tissue damage in a model of myocardial infarct (83).
Beyond simply downregulating the production of pro-inflammatory factors, there is growing evidence that efferocytosis also directly induces the resolution of inflammation (61, 84). Specialized pro-resolving mediators (SPMs) are a class of signaling molecules including resolvins and lipoxins that are derived from free fatty acids that play a key role in limiting inflammation in physiological settings (85). The work of Cai et al. demonstrated that mice lacking the efferocytic receptor MERTK have decreased levels of LXA4 and RvD1 when challenged with zymosan in a model of inducible peritonitis (86). These authors further demonstrated that activation of MERTK using a cross-linking antibody resulted in decreased levels of the enzyme 5-lipooxygenase in the macrophage nucleus, which has previously been shown to result in increased SPM production (86). Interestingly, SPM signaling enhances the efferocytic capacity of macrophages and reduces their sensitivity to efferocytosis-induced cell stress, suggesting that SPM production may be a self-reinforcing stimuli which acts in an autocrine or paracrine manner to enhance the efferocytic capacity within a tissue when apoptotic cells are present (87, 88).
The uptake and degradation of apoptotic cells places a unique metabolic demand on efferocytes (89). These cells must not only quickly degrade the apoptotic cell, but must also ensure that components of the degraded apoptotic cell - especially excess lipids and cholesterol - are redistributed and not allowed to accumulate within the efferocyte (90, 91). A failure to prevent the accumulation of metabolites such as cholesterol and lipids is a source of significant cellular stress that promotes inflammation and can lead to the death of the efferocyte (92, 93). Evidence indicates that efferocytes such as macrophages have unique means of dealing with this additional metabolic load (94, 95). Lipid catabolism is enhanced via a distinctive metabolome characterized by an increase in the generation of ATP from the β-oxidation, accompanied by a concordant enhancement of the mitochondrial electron transport chain, fatty acid oxidation, and oxidative phosphorylation (Figure 2) (94). These adaptations allow efferocytes to rapidly process excess lipids obtained from internalized apoptotic cells.
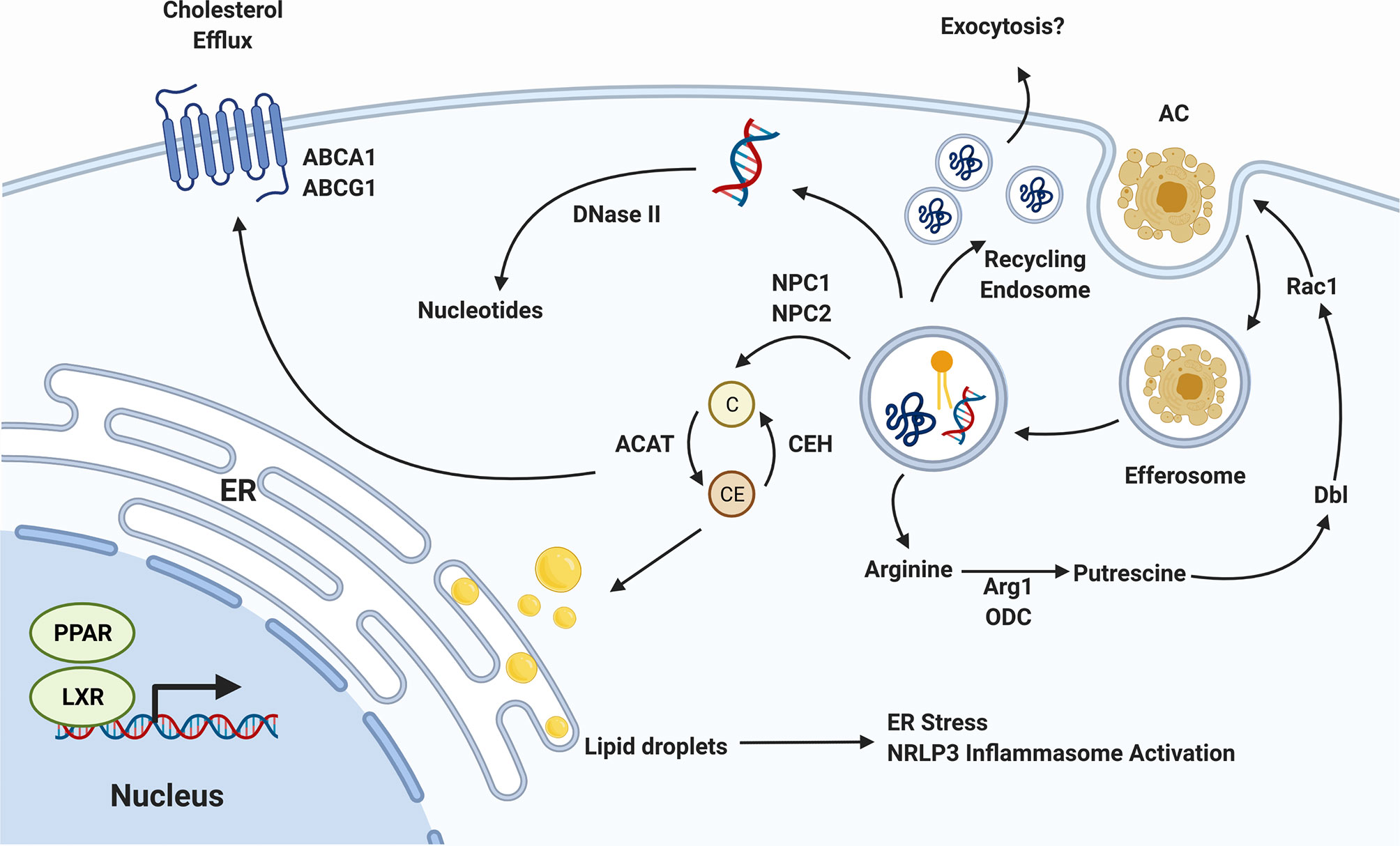
Figure 2 Efferocyte Metabolism. The biomolecules released as efferocytosed apoptotic cells are degraded must be processed by the efferocyte, incurring a significant metabolic load. Cholesterol (C) is exported from the efferosome to cytosolic carriers which, in the presence of cholesterol transporters such as pABCA1 and ABCG1, can export this cholesterol to circulating high density lipoprotein. In the absence of sufficient export, cholesterol is esterified into cholesterol esters (CE) which can accumulate in the endoplasmic reticulum (ER). DNA is degraded in the efferosome by DNase II, and proteins by a range of cathepsin and other proteases, with the resulting nucleotides and amino acids transported into the cytosol where they are recycled. The amino acid arginine is converted in the cytosol to the putrescine, which activates Dbl to enhance Rac1 activity, thereby promoting the efferocytosis of additional apoptotic cells. Lastly, the activation or PPAR and LXR nuclear receptors by lipid-derived metabolites induces a pro-efferocytic metabolic profile via upregulation of cholesterol export machinery and upregulation of lipid β-oxidation. Figure prepared in BioRender.
Efferocytes have multiple molecular mechanisms in place to deal with the metabolic stress induced by cholesterol accumulation, most of which converge on increasing the rate of cholesterol export from the cell (96, 97). Following uptake of an apoptotic cell, cholesterol is exported from the efferosome by NPC1 and NPC2 to cytosolic cholesterol carriers (98). These carriers transport cholesterol throughout the cell, but in the absence of cholesterol export, these carriers ultimately deliver cholesterol to the ER (98, 99). Here, cholesterol accumulates within the ER membrane, eventually forming lipid droplets (100). Unaddressed, these droplets can accumulate to the point where they induce the ER’s unfolded protein response, leading to apoptosis of the efferocyte (92). Efferocytes such as macrophages increase the expression of genes involved in cholesterol export to avoid this fate, notably the cholesterol efflux pumps ABCA1 and ABCG1, which export cytosolic cholesterol to lipid-poor apolipoproteins and HDL (Figure 2) (101, 102). Macrophages have multiple pathways by which these cholesterol efflux pumps can be induced. This includes the induction of ABCA1 transcription by LXR following apoptotic cell uptake (103). In parallel, signaling through the efferocytic receptor BAI1 induces a signaling through the BAI1/ELMO/Rac1 pathway that leads to upregulation of ABCA1 in an LXR-independent manner (104). Both the LXR-dependent and -independent pathways enable macrophages to export excess cholesterol absorbed during efferocytosis, thus maintaining cholesterol homeostasis within the cell and avoiding death of the efferocyte (103, 104). The consequences of impaired cholesterol efflux can be dire. The increased ER stress caused by lipid droplet formation not only leads to death via the unfolded protein response but is also inflammatory due to activation of the NLRP3 inflammasome (105). In addition to causing cell death, the accumulation of cholesterol can directly impair efferocytosis. In one study, Viaud et al. inhibited lysosomal acid lipase, an enzyme required for hydrolysis of cholesterol esters within lysosomes into free cholesterol prior to their export via NPC1/2 to cytosolic carriers (106). This resulted in accumulation of cholesterol esters within the lysosome interfered with Rac1 activation, blocking the engulfment of additional apoptotic cells (106).
In addition to excess lipids and cholesterol, efferocytes must also deal with excess amino acids, short peptides, and apoptotic cell DNA (107). While amino acids and peptides are exported from efferosomes by lysosomal transporters, and via trafficking to the recycling endosome, apoptotic cell DNA is degraded by DNase II in professional efferocytes such as macrophages (108). This is a critical step in maintaining the immunologically silent nature of efferocytosis, with deletion of DNase II from macrophages resulting in the upregulation of pro-inflammatory mediators such as TNFα, likely via activation of TLR9 by partially digested DNA fragments containing unmethylated CpG motifs (108, 109).
Another important alteration to cellular metabolism following efferocytosis are those allowing for additional rounds of efferocytosis (110). Professional efferocytes such as macrophages must often clear multiple apoptotic cells in succession, and impaired clearance of multiple apoptotic cells is regarded as a marker of defective efferocytosis (110, 111). Several components of cellular metabolism are altered in order to facilitate continuous efferocytosis. Wang et al. showed that efferocytic uptake of apoptotic cells induced Drp1-mediated mitochondrial fission along with mitochondrial calcium ion release (110). When this fission process was inhibited macrophages lost their ability to successively engulf apoptotic cells. These macrophages exhibited defective sealing of the efferosome and decreased continuous efferocytic capacity (110). Interestingly, this process is accompanied by a loss of mitochondrial membrane potential driven by the uncoupling protein Ucp2, increased glucose uptake via SLC2A1, and a shift to glycolysis over oxidative phsophroylation (2, 111, 112). In parallel, these cells upregulate the lactate transporter SLC16A1, enabling the rapid export of the end-product of glycolysis (112). This shift in cellular energetics may be required to sustain rapid, successive apoptotic cell uptake and degradation, although how the decoupling of oxidative phosphorylation observed in these studies occurs in cells seemingly also requiring increased oxidative phosphorylation for the β-oxidation of fatty acids remains unresolved (94, 95, 112). Broadly speaking, mitochondrial fission and fusion are important processes that serve to regulate mitochondrial DNA segregation, mitochondrial reactive oxygen species levels and calcium homeostasis (113). These processes have also been shown to be coupled to particular metabolic states in macrophages (113, 114). For example, classically activated, pro-inflammatory macrophages require massive upregulation of glycolysis within the cell (114). Nair et al. demonstrated that blockade of mitochondrial fission with Mdivi-1, a mitochondrial division inhibitor, led to reversal of metabolic reprogramming towards glycolysis in macrophages treated with LPS (115). Therefore, alteration of mitochondrial fusion and fission following efferocytosis may represent alignment with the unique metabolic state adopted by efferocytes following apoptotic cell internalization. Finally, recent work has shown that apoptotic cell-derived arginine and ornithine are converted by macrophages into to putrescine through the activity of the enzymes arginase 1 and ornithine decarboxylase (116). Putrescine subsequently increases Rac1 activity through upregulation of the GTP exchange factor Dbl, enhancing the ability of the efferocyte to engulf additional apoptotic cells (Figure 2) (116). During efferocytosis, macrophages further process putrescine into other polyamines such as spermidine and spermine, but these don’t appear to have the same efferocytosis-enhancing effect as putrescine (116). However, it should be noted that some polyamines, in particular spermidine, confer protection from atherosclerosis by promoting enhanced cholesterol efflux and appear to have cardioprotective effects in animal models of heart failure (117, 118).
An often-underappreciated role of efferocytosis is its role in host defense (3, 119). Efferocytosis plays an important role in control of intracellular pathogens, most notably, control of Mycobacterium tuberculosis (120). In its natural life cycle, M. tuberculosis is internalized by macrophages, where it persists within the phagosome by halting phagosome maturation prior to acidification (121). But while M. tuberculosis can proliferate within these phagosomes, the infected macrophages eventually undergo apoptosis and are cleared through efferocytosis by other, healthy macrophages (3, 121). Because the bacterium is trapped within the apoptotic cell, it is unable to inhibit efferosome maturation as efficiently as it inhibits phagosome maturation (122). Consequentially, the clearance of infected macrophages by efferocytosis is an important mechanism for controlling M. tuberculosis through killing within fully matured efferosomes (3, 120). Importantly, the efferocytic degradation of M. tuberculosis-infected apoptotic cells results in antigen presentation on MHC II – unlike what is observed with uninfected apoptotic cells. While the exact mechanism which allows for the normally non-immunogenic efferocytic pathway to result in antigen presentation remains unclear, it is mediated at least in part by annexin 1, which is required for cross-presentation of M. tuberculosis antigens on MHC I to CD8+ T cells (123).
The ability of macrophages to recognize intracellular pathogens within infected apoptotic cells is a relatively new finding, and the mechanisms that underlie this process remain incompletely defined. It is thought that engagement of TLRs within the maturing efferosome is required, with TLR4 known to be required for the recognition of infected apoptotic cells in other models (124). The detection of pathogens within apoptotic cells is not restricted to bacteria. Efferocytosis of apoptotic cells infected by the herpes simplex virus appears to trigger recognition within the efferosome and subsequent preparation and cross-presentation of viral antigens to CD8+ T cells, where it plays an important role in the control of the virus in a mouse models of infection (125).
It is also unclear whether differences exist in how efferocytes handle the processing of excess cholesterol, lipids and nucleic acids derived from apoptotic cell uptake should that cell be infected with an intracellular pathogen. Indeed, the lipidomic response to pathogen phagocytosis appears to be the opposite of that following efferocytosis. Lipid synthesis is increased following pathogen phagocytosis, including synthesis of ceramides on the phagosome itself (126), and an accompanying upregulation of lipogenesis via TLR-mediated activation of the transcription factors sterol regulatory element binding transcription factor 1 and 2 (SREBP1/2) (127–130). To our knowledge however, no study to date has examined whether a similar phenomenon occurs in maturing efferosomes or whether there is any difference in how efferocytes handle excess lipids and other metabolites following uptake of an infected apoptotic cell.
Efferocytosis is an essential homeostatic mechanism which clears apoptotic cells and debris before the dying cell progresses to necrosis and induces an inflammatory response (1, 13). Although mechanistically similar to phagocytosis, efferocytosis is mediated by a distinct set of receptors, engages a unique maturation pathway, and ultimately results in the efficient degradation of internalized apoptotic cells while avoiding antigen presentation and inflammation (16). To engage in efferocytosis, macrophages take on a unique gene expression and metabolic profile to ensure they are equipped with the necessary metabolic capacity to process the contents of multiple dying cells (14, 94). In this review, we explored several cellular responses to apoptotic cell uptake observed in efferocytes, especially in professional efferocytic cells such as macrophages.
The process of efferosome maturation is similar to that of phagosome maturation, with processes ultimately resulting in cargo degradation (8). However, while phagosomes acquire antigen-presentation machinery – resulting in the presentation of phagosome-derived antigens on both MHC I and II – efferosomes avoid this process and instead dispose of apoptotic cargo in an immunologically silent fashion (1, 28). Similarly, while phagocytosis results in activation of several signaling cascades that lead to generation of a pro-inflammatory response, efferocytosis engages a different set of pathways which upregulate anti-inflammatory and tissue remodeling mediators. This is accomplished through the activation of distinct transcription factors in cells undergoing phagocytosis versus efferocytosis. Indeed, efferocytic macrophages carry a common gene expression signature associated with these functions that distinguish them from pro-inflammatory, tissue patrolling, and other tissue-resident macrophages (14). Efferocytosis of apoptotic cells also appears to induce a unique set of metabolic adaptations designed to permit the efferocyte to effectively deal with the increase burden of lipids, cholesterol and other apoptotic cell-derived macromolecules, while simultaneously priming the cell to engage in additional rounds of efferocytosis (94, 110). Finally, efferocytosis has a role in host defense against intracellular pathogens, including both bacteria and viruses (119, 120, 125).
Although there have been significant advances in our understanding of efferocytosis over the past few decades, there remain significant gaps in our understanding – especially regarding the role of efferocytosis in pathogen clearance, and in our understanding of the metabolic reprogramming of efferocytes. In particular, we current lack a detailed mechanistic understanding of how efferocytes are able to distinguish between infected versus non-infected apoptotic cells. It is further unclear how efferocytes respond metabolically to infected apoptotic cells. Furthermore, it remains unclear whether differences in immunological outcomes following pathogen versus apoptotic cell uptake are solely the result of differences in the receptors used to recognize each type of cargo, or if the processing of cargo within the phagosome or efferosome also plays a significant role. Finally, we are only beginning to unravel the complexities of immunometabolic responses to apoptotic cell uptake and further work is needed to fully define how efferocytes are able to cope efficiently with the massive intake of lipids, proteins and nucleic acids. With a growing body of evidence that defects in efferocytosis are involved in inflammatory and autoimmune disease, a clearer understanding of how professional efferocytes such as macrophages respond to apoptotic cell uptake will be crucial in furthering our understanding of the pathogenesis of these disorders and identifying potential therapeutic options.
CY and BH contributed equally to the authoring of this manuscript. All authors contributed to the article and approved the submitted version.
This study was funded by a Heart and Stroke Foundation of Canada Grant-In-Aid and an Ontario Ministry of Research and Innovation Early Research Award to BH. CY was funded by a Vanier PhD Scholarship and Canadian Institutes for Health Research (CIHR) MD/PhD studentship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
LAP, LC3-associated phagocytosis; PI3KCIII, class III phosphatidylinositol-3-kinase; TLR, Toll-like receptor; DCs, dendritic cells; PtdSer, phosphatidylserine; LXR, liver X receptor; PPAR, peroxisome proliferator-activated receptor; SPM, pro-resolving mediator.
1. Henson PM. Cell Removal: Efferocytosis. Annu Rev Cell Dev Biol (2017) 33:1–18. doi: 10.1146/annurev-cellbio-111315-125315
2. Morioka S, Maueröder C, Ravichandran KS. Living on the Edge: Efferocytosis at the Interface of Homeostasis and Pathology. Immunity (2019) 50(5):1149–62. doi: 10.1016/j.immuni.2019.04.018
3. Martin CJ, Peters KN, Behar SM. Macrophages Clean Up: Efferocytosis and Microbial Control. Curr Opin Microbiol (2014) 17:17–23. doi: 10.1016/j.mib.2013.10.007.Macrophages
4. Brill A, Torchinsky A, Carp H, Toder V. The role of apoptosis in normal and abnormal embryonic development. J Assist Reprod Genet (1999) 16:512–9. doi: 10.1023/A:1020541019347
5. Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis (2017) 22(10):1189–204. doi: 10.1007/s10495-017-1413-z
6. Tabas I. 2016 Russell Ross Memorial Lecture in Vascular Biology. Arterioscler Thromb Vasc Biol (2016) 37(2):183–9. doi: 10.1161/ATVBAHA.116.308036
7. Freeman SA, Grinstein S. Phagocytosis: Receptors, signal integration, and the cytoskeleton. Immunol Rev (2014) 262:193–215. doi: 10.1111/imr.12212
8. Kinchen JM, Ravichandran KS. Phagosome maturation: going through the acid test. Nat Rev Mol Cell Biol (2008) 9:781–95. doi: 10.1038/nrm2515
9. Nagata S. Apoptosis and Clearance of Apoptotic Cells. Annu Rev Immunol (2018) 361829:1–18. doi: 10.1146/annurev-immunol
10. Blackburn JWD, Lau DHC, Liu EY, Ellins J, Vrieze AM, Pawlak EN, et al. Soluble CD93 is an apoptotic cell opsonin recognized by α x β 2. Eur J Immunol (2019) 49(4):1–23. doi: 10.1002/eji.201847801
11. Thorp E, Subramanian M, Tabas I. The role of macrophages and dendritic cells in the clearance of apoptotic cells in advanced atherosclerosis. Eur J Immunol (2011) 41:2515–8. doi: 10.1002/eji.201141719
12. Gregory CD, Pound JD. Cell death in the neighbourhood: Direct microenvironmental effects of apoptosis in normal and neoplastic tissues. J Pathol (2011) 223:177–94. doi: 10.1002/path.2792
13. Gordon S, Plüddemann A. Macrophage Clearance of Apoptotic Cells: A Critical Assessment. Front Immunol (2018) 9:127. doi: 10.3389/fimmu.2018.00127
14. A-Gonzalez N, Quintana JA, García-Silva S, Mazariegos M, González de la Aleja A, Nicolás-Ávila JA, et al. Phagocytosis imprints heterogeneity in tissue-resident macrophages. J Exp Med (2017) 214:1281–96. doi: 10.1084/jem.20161375
15. Cochain C, Vafadarnejad E, Arampatzi P, Pelisek J, Winkels H, Ley K, et al. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ Res (2018) 122:1661–74. doi: 10.1161/CIRCRESAHA.117.312509
16. Kinchen JM, Doukoumetzidis K, Almendinger J, Stergiou L, Tosello-Trampont A, Sifri CD, et al. A pathway for phagosome maturation during engulfment of apoptotic cells. Nat Cell Biol (2008) 10:556–66. doi: 10.1038/ncb1718
17. Roberts RL, Barbieri MA, Ullrich J, Stahl PD. Dynamics of rab5 activation in endocytosis and phagocytosis. J Leukoc Biol (2000) 68:627–32.
18. Harrison RE, Bucci C, Vieira OV, Schroer TA, Grinstein S. Phagosomes fuse with late endosomes and/or lysosomes by extension of membrane protrusions along microtubules: role of Rab7 and RILP. Mol Cell Biol (2003) 23:6494–506. doi: 10.1128/MCB.23.18.6494
19. Mills IG, Jones AT, Clague MJ. Involvement of the endosomal autoantigen EEA1 in homotypic fusion of early endosomes. Curr Biol (1998) 8:881–4. doi: 10.1016/S0960-9822(07)00351-X
20. Gorvel JP, Chavrier P, Zerial M, Gruenberg J. Rab5 Controls Early Endosome Fusion in Vitro. Cell (1991) 64:915–25. doi: 10.1016/0092-8674(91)90316-Q
21. Simonsen A, Gaullier JM, D’Arrigo A, Stenmark H. The Rab5 effector EEA1 interacts directly with syntaxin-6. J Biol Chem (1999) 274:28857–60. doi: 10.1074/jbc.274.41.28857
22. Chen YA, Scheller RH. SNARE-mediated membrane fusion. Nat Rev Mol Cell Biol (2001) 2:98–106. doi: 10.1038/35052017
23. Taefehshokr N, Yin C, Heit B. Rab GTPases in the differential processing of phagocytosed pathogens versus efferocytosed apoptotic cells. Histol Histopathol (2020) 18252. doi: 10.14670/HH-18-252
24. Flannagan RS, Jaumouillé V, Grinstein S. The Cell Biology of Phagocytosis. Annu Rev Pathol (2011) 7:61–98. doi: 10.1146/annurev-pathol-011811-132445
25. Poteryaev D, Datta S, Ackema K, Zerial M, Spang A. Identification of the switch in early-to-late endosome transition. Cell (2010) 141:497–508. doi: 10.1016/j.cell.2010.03.011
26. Cui Y, Zhao Q, Gao C, Ding Y, Zeng Y, Ueda T, et al. Activation of the Rab7 GTPase by the MON1-CCZ1 Complex Is Essential for PVC-to-Vacuole Trafficking and Plant Growth in Arabidopsis. Plant Cell (2014) 26:2080–97. doi: 10.1105/tpc.114.123141
27. Yin C, Kim Y, Argintaru D, Heit B. Rab17 mediates differential antigen sorting following efferocytosis and phagocytosis. Cell Death Dis (2016) 7:e2529. doi: 10.1038/cddis.2016.431
28. Yin C, Argintaru D, Heit B. Rab17 mediates intermixing of phagocytosed apoptotic cells with recycling endosomes. Small GTPases (2017) 1–9. doi: 10.1080/21541248.2017.1308852
29. Yin J, Huang Y, Guo P, Hu S, Yoshina S, Xuan N, et al. GOP-1 promotes apoptotic cell degradation by activating the small GTPase Rab2 in C. elegans. J Cell Biol (2017) 216:1775–94. doi: 10.1083/jcb.201610001
30. Ward MG, Li G, Hao M. Apoptotic beta-cells induce macrophage reprogramming under diabetic conditions. J Biol Chem (2018) 293(42):16160–73. doi: 10.1074/jbc.RA118.004565
31. Li B, Castano AP, Hudson TE, Nowlin BT, Lin S-L, Bonventre JV, et al. The melanoma-associated transmembrane glycoprotein Gpnmb controls trafficking of cellular debris for degradation and is essential for tissue repair. FASEB J (2010) 24:4767–81. doi: 10.1096/fj.10.154757
32. Heckmann BL, Green DR. LC3-associated phagocytosis at a glance. J Cell Sci (2019) 132. doi: 10.1242/jcs.231472
33. Wan J, Weiss E, Ben Mkaddem S, Mabire M, Choinier P-M, Picq O, et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci Transl Med (2020) 12:eaaw8523. doi: 10.1126/scitranslmed.aaw8523
34. Blander JM, Medzhitov R. Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature (2006) 440:808–12. doi: 10.1038/nature04596
35. Blander JM, Medzhitov R. Regulation of Phagosome Maturation by Signals from Toll-Like Receptors. Science (2011) 1014:1014–8. doi: 10.1126/science.1096158
36. Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol (2006) 7:1029–35. doi: 10.1038/ni1006-1029
37. Seto S, Sugaya K, Tsujimura K, Nagata T, Horii T, Koide Y. Rab39a interacts with phosphatidylinositol 3-kinase and negatively regulates autophagy induced by lipopolysaccharide stimulation in macrophages. PloS One (2013) 8:1–13. doi: 10.1371/journal.pone.0083324
38. Cruz FM, Colbert JD, Rock KL. The GTP ase Rab39a promotes phagosome maturation into MHC -I antigen-presenting compartments. EMBO J (2020) 39:1–21. doi: 10.15252/embj.2019102020
39. Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol (2015) 17:893–906. doi: 10.1038/ncb3192
40. Bhargava HK, Tabata K, Byck JM, Hamasaki M, Farrell DP, Anishchenko I, et al. Structural basis for autophagy inhibition by the human Rubicon-Rab7 complex. Proc Natl Acad Sci USA (2020) 117:17003–10. doi: 10.1073/pnas.2008030117
41. Wong SW, Sil P, Martinez J. Rubicon: LC3-associated phagocytosis and beyond. FEBS J (2018) 285:1379–88. doi: 10.1111/febs.14354
42. Erwig L-P, McPhilips KA, Wynes MW, Ivetic A, Ridley AJ, Henson PM. Differential regulation of phagosome maturation in macrophages and dendritic cells mediated by Rho GTPases and ezrin–radixin–moesin (ERM) proteins. Proc Natl Acad Sci USA (2006) 103:12825–30. doi: 10.1155/2015/359153
43. Canton J, Khezri R, Glogauer M, Grinstein S. Contrasting phagosome pH regulation and maturation in human M1 and M2 macrophages. Mol Biol Cell (2014) 25:3330–41. doi: 10.1091/mbc.E14-05-0967
44. De Luca M, Cogli L, Progida C, Nisi V, Pascolutti R, Sigismund S, et al. RILP regulates vacuolar ATPase through interaction with the V1G1 subunit. J Cell Sci (2015) 128:2565–5. doi: 10.1242/jcs.175323
45. Trivedi PC, Bartlett JJ, Pulinilkunnil T. Lysosomal Biology and Function: Modern View of Cellular Debris Bin. Cells (2020) 9:1–35. doi: 10.3390/cells9051131
46. Rai A, Pathak D, Thakur S, Singh S, Dubey AK, Mallik R. Dynein Clusters into Lipid Microdomains on Phagosomes to Drive Rapid Transport toward Lysosomes. Cell (2016) 164:722–34. doi: 10.1016/j.cell.2015.12.054
47. Fairn GD, Grinstein S. How nascent phagosomes mature to become phagolysosomes. Trends Immunol (2012) 33(8):1–9. doi: 10.1016/j.it.2012.03.003
48. Heo YJ, Bin JY, Oh HJ, Park MK, Heo YM, Cho M, et al. IL-10 suppresses Th17 cells and promotes regulatory T cells in the CD4+ T cell population of rheumatoid arthritis patients. Immunol Lett (2010) 127:150–6. doi: 10.1016/j.imlet.2009.10.006
49. Rodriguez-Fernandez S, Pujol-Autonell I, Brianso F, Perna-Barrull D, Cano-Sarabia M, Garcia-Jimeno S, et al. Phosphatidylserine-liposomes promote tolerogenic features on dendritic cells in human type 1 diabetes by apoptotic mimicry. Front Immunol (2018) 9:253. doi: 10.3389/fimmu.2018.00253
50. Yurdagul A, Doran AC, Cai B, Fredman G, Tabas IA. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front Cardiovasc Med (2017) 4:1–10. doi: 10.3389/fcvm.2017.00086
51. Abdolmaleki F, Farahani N, Hayat SMG, Pirro M, Bianconi V, Barreto GE, et al. The role of efferocytosis in autoimmune diseases. Front Immunol (2018) 9:1645. doi: 10.3389/fimmu.2018.01645
52. Kimani SG, Geng K, Kasikara C, Kumar S, Sriram G, Wu Y, et al. Contribution of Defective PS Recognition and Efferocytosis to Chronic Inflammation and Autoimmunity. Front Immunol (2014) 5:566. doi: 10.3389/fimmu.2014.00566
53. Zizzo G, Guerrieri J, Dittman LM, Merrill JT, Cohen PL. Circulating levels of soluble MER in lupus reflect M2c activation of monocytes/macrophages, autoantibody specificities and disease activity. Arthritis Res Ther (2013) 15:R212–R227. doi: 10.1186/ar4407
54. Waterborg CEJ, Beermann S, Broeren MGA, Bennink MB, Alves-filho JC. Protective role of the Mer Tyrosine Kinase via efferocytosis in rheumatoid arthritis Models. Front Immunol (2018) 9:742. doi: 10.3389/fimmu.2018.00742
55. Ma GZM, Stankovich J, Kilpatrick TJ, Binder MD, Field J, Bahlo M, et al. Polymorphisms in the receptor tyrosine kinase MERTK gene are associated with Multiple Sclerosis susceptibility. PloS One (2011) 6:2–7. doi: 10.1371/journal.pone.0016964
56. Zaza R, Ibayyan L, El-Khateeb M, Bahou YG, Khreisat E, Al-Khateeb W, et al. Association of genetic polymorphisms of MERTK with multiple sclerosis among Jordanians. BioMed Res (2017) 28:399–404.
57. Binder MD, Fox AD, Merlo D, Johnson LJ, Giuffrida L, Calvert SE, et al. Common and Low Frequency Variants in MERTK Are Independently Associated with Multiple Sclerosis Susceptibility with Discordant Association Dependent upon HLA-DRB1*15:01 Status. PloS Genet (2016) 12:1–25. doi: 10.1371/journal.pgen.1005853
58. Canton J, Blees H, Henry CM, Buck MD, Schulz O, Rogers NC, et al. The receptor DNGR-1 signals for phagosomal rupture to promote cross-presentation of dead-cell-associated antigens. Nat Immunol (2021) 22:140–53. doi: 10.1038/s41590-020-00824-x
59. Rock KL, Reits E, Neefjes J. Present Yourself! By MHC Class I and MHC Class II Molecules. Trends Immunol (2016) 37:724–37. doi: 10.1016/j.it.2016.08.010
60. Zhang N, Bevan MJ. CD8(+) T cells: foot soldiers of the immune system. Immunity (2011) 35:161–8. doi: 10.1016/j.immuni.2011.07.010
61. Korns D, Frasch SC, Fernandez-Boyanapalli R, Henson PM, Bratton DL. Modulation of macrophage efferocytosis in inflammation. Front Immunol (2011) 2:57. doi: 10.3389/fimmu.2011.00057
62. Rébé C, Raveneau M, Chevriaux A, Lakomy D, Sberna A-L, Costa A, et al. Induction of transglutaminase 2 by a liver X receptor/retinoic acid receptor alpha pathway increases the clearance of apoptotic cells by human macrophages. Circ Res (2009) 105:393–401. doi: 10.1161/CIRCRESAHA.109.201855
63. Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-gonzalez RR, et al. PPAR- δ senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat Med (2009) 15:1266–73. doi: 10.1038/nm.2048
64. Johann AM, von Knethen A, Lindemann D, Brüne B. Recognition of apoptotic cells by macrophages activates the peroxisome proliferator-activated receptor-γ and attenuates the oxidative burst. Cell Death Differ (2006) 13:1533–40. doi: 10.1038/sj.cdd.4401832
65. Penvose A, Keenan JL, Bray D, Ramlall V, Siggers T. Comprehensive study of nuclear receptor DNA binding provides a revised framework for understanding receptor specificity. Nat Commun (2019) 10:1–15. doi: 10.1038/s41467-019-10264-3
66. Majai G, Sarang Z, Csomós K, Zahuczky G, Fésüs L. PPARγ-dependent regulation of human macrophages in phagocytosis of apoptotic cells. Eur J Immunol (2007) 37:1343–54. doi: 10.1002/eji.200636398
67. Wang LH, Yang XY, Zhang X, Huang J, Hou J, Li J, et al. Transcriptional Inactivation of STAT3 by PPARγ Suppresses IL-6-Responsive Multiple Myeloma Cells. Immunity (2004) 20:205–18. doi: 10.1016/S1074-7613(04)00030-5
68. Wang B, Tontonoz P. Liver X receptors in lipid signalling and membrane homeostasis. Nat Rev Endocrinol (2018) 14:452–63. doi: 10.1038/s41574-018-0037-x
69. A-Gonzalez N, Bensinger SJ, Hong C, Beceiro S, Bradley MN, Zelcer N, et al. Apoptotic cells promote their own clearance and immune tolerance through activation of the nuclear receptor LXR. Immunity (2009) 31:245–58. doi: 10.1016/j.immuni.2009.06.018
70. Pastore M, Grimaudo S, Pipitone RM, Lori G, Raggi C, Petta S, et al. Role of myeloid-epithelial-reproductive tyrosine kinase and macrophage polarization in the progression of atherosclerotic lesions associated with nonalcoholic fatty liver disease. Front Pharmacol (2019) 10:604. doi: 10.3389/fphar.2019.00604
71. Zhang B, Fang L, Wu HM, Ding PS, Xu K, Liu RY. Mer receptor tyrosine kinase negatively regulates lipoteichoic acid-induced inflammatory response via PI3K/Akt and SOCS3. Mol Immunol (2016) 76:98–107. doi: 10.1016/j.molimm.2016.06.016
72. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med (2002) 53:409–35. doi: 10.1146/annurev.med.53.082901.104018
73. Croasdell A, Duffney PF, Kim N, Lacy SH, Sime PJ, Phipps RP. PPARy and the Innate Immune System Mediate the Resolution of Inflammation. PPAR Res (2015) 2015:1–20. doi: 10.1155/2015/549691
74. Zizzo G, Cohen PL. The PPAR-γ antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: A key role for PPAR-γ in human macrophage polarization. J Inflamm (United Kingdom) (2015) 12:1–16. doi: 10.1186/s12950-015-0081-4
75. Gordon P, Okai B, Hoare JI, Erwig LP, Wilson HM. SOCS3 is a modulator of human macrophage phagocytosis. J Leukoc Biol (2016) 100:771–80. doi: 10.1189/jlb.3a1215-554rr
76. Thorp E, Tabas I. Mechanisms and consequences of efferocytosis in advanced atherosclerosis. J Leukoc Biol (2009) 86:1089–95. doi: 10.1189/jlb.0209115
77. Linton MF, Babaev VR, Huang J, Linton EF, Tao H, Yancey PG. Macrophage Apoptosis and Efferocytosis in the Pathogenesis of Atherosclerosis. Circ J (2016) 80:2259–68. doi: 10.1253/circj.CJ-16-0924
78. Yin C, Vrieze AM, Rosoga M, Akingbasote J, Pawlak EN, Jacob RA, et al. Efferocytic Defects in Early Atherosclerosis Are Driven by GATA2 Overexpression in Macrophages. Front Immunol (2020) 11:594136. doi: 10.3389/fimmu.2020.594136
79. Muiya NP, Wakil S, Al-Najai M, Tahir AI, Baz B, Andres E, et al. A study of the role of GATA2 gene polymorphism in coronary artery disease risk traits. Gene (2014) 544:152–8. doi: 10.1016/j.gene.2014.04.064
80. Meagher LC, Savill JS, Baker A, Fuller RW, Haslett C. Phagocytosis of apoptotic neutrophils does not induce macrophage release of thromboxane B2. J Leukoc Biol (1992) 52:269–73. doi: 10.1002/jlb.52.3.269
81. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-β, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi: 10.1172/JCI1112
82. Campana L, Starkey Lewis PJ, Pellicoro A, Aucott RL, Man J, O’Duibhir E, et al. The STAT3–IL-10–IL-6 Pathway Is a Novel Regulator of Macrophage Efferocytosis and Phenotypic Conversion in Sterile Liver Injury. J Immunol (2018) 200:1169–87. doi: 10.4049/jimmunol.1701247
83. Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, et al. Myeloid-Epithelial-Reproductive Receptor Tyrosine Kinase and Milk Fat Globule Epidermal Growth Factor 8 Coordinately Improve Remodeling after Myocardial Infarction via Local Delivery of Vascular Endothelial Growth Factor. Circulation (2016) 133:826–39. doi: 10.1161/CIRCULATIONAHA.115.020857
84. Tabas I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol (2009) 10:36–46. doi: 10.1038/nri2675
85. Lehmann C, Homann J, Ball AK, Blöcher R, Kleinschmidt TK, Basavarajappa D, et al. Lipoxin and resolvin biosynthesis is dependent on 5-lipoxygenase activating protein. FASEB J (2015) 29:5029–43. doi: 10.1096/fj.15-275487
86. Cai B, Thorp EB, Doran AC, Subramanian M, Sansbury BE, Lin C-S, et al. MerTK cleavage limits proresolving mediator biosynthesis and exacerbates tissue inflammation. Proc Natl Acad Sci (2016) 113:6526–31. doi: 10.1073/pnas.1524292113
87. Lee HN, Surh YJ. Resolvin D1-mediated NOX2 inactivation rescues macrophages undertaking efferocytosis from oxidative stress-induced apoptosis. Biochem Pharmacol (2013) 86:759–69. doi: 10.1016/j.bcp.2013.07.002
88. Rymut N, Heinz J, Sadhu S, Hosseini Z, Riley CO, Marinello M, et al. Resolvin D1 promotes efferocytosis in aging by limiting senescent cell-induced MerTK cleavage. FASEB J (2020) 34:597–609. doi: 10.1096/fj.201902126R
89. Han CZ, Ravichandran KS. Metabolic connections during apoptotic cell engulfment. Cell (2011) 147:1442–5. doi: 10.1016/j.cell.2011.12.006
90. Westover E. Cholesterol in Health and Disease. J Clin Invest (2002) 110:583–90. doi: 10.1172/JCI200216381.Imagine
91. Chistiakov DA, Melnichenko AA, Myasoedova VA, Grechko AV, Orekhov AN. Mechanisms of foam cell formation in atherosclerosis. J Mol Med (2017) 95:1153–65. doi: 10.1007/s00109-017-1575-8
92. Li Y, Schwabe RF, DeVries-Seimon T, Yao PM, Gerbod-Giannone MC, Tall AR, et al. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-α and interleukin-6: Model of NF-κB- and map kinase-dependent inflammation in advanced atherosclerosis. J Biol Chem (2005) 280:21763–72. doi: 10.1074/jbc.M501759200
93. Lara-Guzmán OJ, Gil-Izquierdo Á, Medina S, Osorio E, Álvarez-Quintero R, Zuluaga N, et al. Oxidized LDL triggers changes in oxidative stress and inflammatory biomarkers in human macrophages. Redox Biol (2018) 15:1–11. doi: 10.1016/j.redox.2017.11.017
94. Zhang S, Weinberg S, DeBerge M, Gainullina A, Schipma M, Kinchen JM, et al. Efferocytosis Fuels Requirements of Fatty Acid Oxidation and the Electron Transport Chain to Polarize Macrophages for Tissue Repair. Cell Metab (2019) 29:443–456.e5. doi: 10.1016/j.cmet.2018.12.004
95. Cummings RJ, Barbet G, Bongers G, Hartmann BM, Gettler K, Muniz L, et al. Different tissue phagocytes sample apoptotic cells to direct distinct homeostasis programs. Nature (2016) 539:565–9. doi: 10.1038/nature20138
96. Yvan-Charvet L, Pagler TA, Seimon TA, Thorp E, Welch CL, Witztum JL, et al. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ Res (2010) 106:1861–9. doi: 10.1161/CIRCRESAHA.110.217281
97. Costet P, Lalanne F, Gerbod-Giannone MC, Molina JR, Fu X, Lund EG, et al. Retinoic Acid Receptor-Mediated Induction of ABCA1 in Macrophages. Mol Cell Biol (2003) 23:7756–66. doi: 10.1128/mcb.23.21.7756-7766.2003
98. Subramanian K, Balch WE. NPC1/NPC2 function as a tag team duo to mobilize cholesterol. Proc Natl Acad Sci USA (2008) 105:15223–4. doi: 10.1073/pnas.0808256105
99. Dron JS, Hegele RA. Genetics of Lipid and Lipoprotein Disorders and Traits. Curr Genet Med Rep (2017) 5:115–5. doi: 10.1007/s40142-017-0117-6
100. Tangirala RK, Jerome WG, Jones NL, Small DM, Johnson WJ, Glick JM, et al. Formation of cholesterol monohydrate crystals in macrophage-derived foam cells. J Lipid Res (1994) 35:93–104. doi: 10.1016/S0022-2275(20)40131-2
101. Wang X, Collins HL, Ranalletta M, Fuki IV, Billheimer JT, Rothblat GH, et al. Macrophage ABCA1 and ABCG1, but not SR-BI, promote macrophage reverse cholesterol transport in vivo. J Clin Invest (2007) 117:2216–24. doi: 10.1172/JCI32057
102. Yvan-Charvet L, Wang N, Tall AR. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol (2010) 30:139–43. doi: 10.1161/ATVBAHA.108.179283
103. Tamehiro N, Park MH, Hawxhurst V, Nagpal K, Adams ME, Zannis VI, et al. LXR Agonism Upregulates the Macrophage ABCA1/Syntrophin Protein Complex That Can Bind ApoA-I and Stabilized ABCA1 Protein, but Complex Loss Does Not Inhibit Lipid Efflux. Biochemistry (2015) 54:6931–41. doi: 10.1021/acs.biochem.5b00894
104. Fond AM, Lee CS, Schulman IG, Kiss RS, Ravichandran KS. Apoptotic cells trigger a membrane-initiated pathway to increase ABCA1. J Clin Invest (2015) 125:2748–58. doi: 10.1172/JCI80300
105. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature (2010) 464:1357–61. doi: 10.1038/nature08938
106. Viaud M, Ivanov S, Vujic N, Duta-mare M, Aira L, Barouillet T, et al. Lysosomal cholesterol hydrolysis couples efferocytosis to anti-inflammatory oxysterol production. Circ Res (2019) 122:1369–84. doi: 10.1161/CIRCRESAHA.117.312333.Lysosomal
107. Boada-romero E, Martinez J, Heckmann BL, Green DR, Group I, Park T. Mechanisms and physiology of the clearance of dead cells by efferocytosis. Nat Rev Cell Biol (2020) 21:398–414. doi: 10.1038/s41580-020-0232-1.Mechanisms
108. Kawane K, Ohtani M, Miwa K, Kizawa T, Kanbara Y, Yoshioka Y, et al. Chronic polyarthritis caused by mammalian DNA that escapes from degradation in macrophages. Nature (2006) 443:998–1002. doi: 10.1038/nature05245
109. Saito Y, Hikita H, Nozaki Y, Kai Y, Makino Y, Nakabori T, et al. DNase II activated by the mitochondrial apoptotic pathway regulates RIP1-dependent non-apoptotic hepatocyte death via the TLR9/IFN-β signaling pathway. Cell Death Differ (2019) 26:470–86. doi: 10.1038/s41418-018-0131-6
110. Wang Y, Subramanian M, Yurdagul A, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, et al. Mitochondrial Fission Promotes the Continued Clearance of Apoptotic Cells by Macrophages. Cell (2017) 171:331–345.e22. doi: 10.1016/j.cell.2017.08.041
111. Park D, Han CZ, Elliott MR, Kinchen JM, Trampont PC, Das S, et al. Continued clearance of apoptotic cells critically depends on the phagocyte Ucp2 protein. Nature (2011) 477:220–4. doi: 10.1038/nature10340
112. Morioka S, Perry JSA, Raymond MH, Medina CB, Zhu Y, Zhao L, et al. Efferocytosis induces a novel SLC program to promote glucose uptake and lactate release. Nature (2018) 563(7733):714–8. doi: 10.1038/s41586-018-0735-5
113. Rambold AS, Pearce EL. Mitochondrial Dynamics at the Interface of Immune Cell Metabolism and Function. Trends Immunol (2018) 39:6–18. doi: 10.1016/j.it.2017.08.006
114. Benmoussa K, Garaude J, Acín-Pérez R. How Mitochondrial Metabolism Contributes to Macrophage Phenotype and Functions. J Mol Biol (2018) 430:3906–21. doi: 10.1016/j.jmb.2018.07.003
115. Nair S, Sobotka KS, Joshi P, Gressens P, Fleiss B, Thornton C, et al. Lipopolysaccharide-induced alteration of mitochondrial morphology induces a metabolic shift in microglia modulating the inflammatory response in vitro and in vivo. Glia (2019) 67:1047–61. doi: 10.1002/glia.23587
116. Yurdagul A, Subramanian M, Wang X, Crown SB, Ilkayeva OR, Darville L, et al. Macrophage Metabolism of Apoptotic Cell-Derived Arginine Promotes Continual Efferocytosis and Resolution of Injury. Cell Metab (2020) 31:518–533.e10. doi: 10.1016/j.cmet.2020.01.001
117. Eisenberg T, Abdellatif M, Schroeder S, Primessnig U, Stekovic S, Pendl T, et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med (2016) 22:1428–38. doi: 10.1038/nm.4222
118. Michiels CF, Kurdi A, Timmermans J-P, De Meyer GRY, Martinet W. Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis (2016) 251:319–27. doi: 10.1016/j.atherosclerosis.2016.07.899
119. Zheng DJ, Abou Taka M, Heit B. Role of Apoptotic Cell Clearance in Pneumonia and Inflammatory Lung Disease. Pathogens (Basel Switzerland) (2021) 10. doi: 10.3390/pathogens10020134
120. Martin CJ, Booty MG, Rosebrock TR, Nunes-Alves C, Desjardins DM, Keren I, et al. Efferocytosis is an innate antibacterial mechanism. Cell Host Microbe (2012) 12:289–300. doi: 10.1016/j.chom.2012.06.010
121. Vandal OH, Nathan CF, Ehrt S. Acid resistance in Mycobacterium tuberculosis. J Bacteriol (2009) 191:4714–21. doi: 10.1128/JB.00305-09
122. Behar SM, Martin CJ, Booty MG, Nishimura T, Zhao X, Gan HX, et al. Apoptosis is an innate defense function of macrophages against Mycobacterium tuberculosis. Mucosal Immunol (2011) 4:279–87. doi: 10.1038/mi.2011.3
123. Tzelepis F, Verway M, Daoud J, Gillard J, Hassani-Ardakani K, Dunn J, et al. Annexin1 regulates DC efferocytosis and cross-presentation during Mycobacterium tuberculosis infection. J Clin Invest (2015) 125:752–68. doi: 10.1172/JCI77014
124. Zhang Y, Bliska JB. Role of toll-like receptor signaling in the apoptotic response of macrophages to Yersinia infection. Infect Immun (2003) 71:1513–9. doi: 10.1128/IAI.71.3.1513-1519.2003
125. Subramanian M, Hayes CD, Thome JJ, Thorp E, Matsushima GK, Herz J, et al. An AXL/LRP-1/RANBP9 complex mediates DC efferocytosis and antigen cross-presentation in vivo. J Clin Invest (2014) 124:1296–308. doi: 10.1172/JCI72051
126. Pathak D, Mehendale N, Singh S, Mallik R, Kamat SS. Lipidomics Suggests a New Role for Ceramide Synthase in Phagocytosis. ACS Chem Biol (2018) 13:2280–7. doi: 10.1021/acschembio.8b00438
127. Shimano H, Sato R. SREBP-regulated lipid metabolism: Convergent physiology-divergent pathophysiology. Nat Rev Endocrinol (2017) 13:710–30. doi: 10.1038/nrendo.2017.91
128. Jeon T, Osborne TF. SREBPs: Metabolic integrators in physiology and metabolism. Trends Endocrinol Metab (2012) 23:65–72. doi: 10.1016/j.tem.2011.10.004
129. Im SS, Yousef L, Blaschitz C, Liu JZ, Edwards RA, Young SG, et al. Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab (2011) 13:540–9. doi: 10.1016/j.cmet.2011.04.001
Keywords: efferocytosis, intracellular trafficking, transcriptional regulation, cellular metabolism, inflammation resolution, host defense
Citation: Yin C and Heit B (2021) Cellular Responses to the Efferocytosis of Apoptotic Cells. Front. Immunol. 12:631714. doi: 10.3389/fimmu.2021.631714
Received: 20 November 2020; Accepted: 29 March 2021;
Published: 20 April 2021.
Edited by:
Gregory D. Fairn, St. Michael’s Hospital, CanadaReviewed by:
Luisa Martinez-Pomares, University of Nottingham, United KingdomCopyright © 2021 Yin and Heit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bryan Heit, YmhlaXRAdXdvLmNh
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.