 Daniela E. Kirwan
Daniela E. Kirwan Deborah L. W. Chong
Deborah L. W. Chong Jon S. Friedland
Jon S. Friedland
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol. , 19 May 2021
Sec. Microbial Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.631696
This article is part of the Research Topic The Role of Platelet Activation in the Pathophysiology of HIV, TB and Pneumococcal Disease View all 7 articles
In 2019 10 million people developed symptomatic tuberculosis (TB) disease and 1.2 million died. In active TB the inflammatory response causes tissue destruction, which leads to both acute morbidity and mortality. Tissue destruction in TB is driven by host innate immunity and mediated via enzymes, chiefly matrix metalloproteinases (MMPs) which are secreted by leukocytes and stromal cells and degrade the extracellular matrix. Here we review the growing evidence implicating platelets in TB immunopathology. TB patients typically have high platelet counts, which correlate with disease severity, and a hypercoagulable profile. Platelets are present in human TB granulomas and platelet-associated gene transcripts are increased in TB patients versus healthy controls. Platelets most likely drive TB immunopathology through their effect on other immune cells, particularly monocytes, to lead to upregulation of activation markers, increased MMP secretion, and enhanced phagocytosis. Finally, we consider current evidence supporting use of targeted anti-platelet agents in the treatment of TB due to growing interest in developing host-directed therapies to limit tissue damage and improve treatment outcomes. In summary, platelets are implicated in TB disease and contribute to MMP-mediated tissue damage via their cellular interactions with other leukocytes, and are potential targets for novel host-directed therapies.
Tuberculosis (TB) is one of the most important infectious diseases of our time. In 2019 10.0 million people developed symptomatic disease and 1.2 million died, with little change in these figures over the past decade (1). Although many patients are never diagnosed or treated, a significant proportion of deaths occurs in individuals who have received appropriate anti-TB treatment (2). This is largely due to the inflammatory immune response to Mycobacterium tuberculosis (M.tb), the causative agent of TB. This response is highly complex encompassing both the innate and adaptive immune systems, resulting in severe disease manifestations and facilitating onwards transmission (3). In addition, the co-evolution of M.tb and humans has led to bi-directional adaptations that enable the bacteria to persist in a semi-dormant state, and consequently current treatment regimens require a minimum of six months of therapy to ensure eradication of all viable bacilli within the patient (4). Treatment is further complicated by the rise of single-, multi- and extensively drug-resistant disease.
Moreover, even when successfully treated, patients can suffer long-term impaired lung function and reduced quality of life (5–9). This is long established (10–15) and, given that TB primarily affects adults within the economically active age group, has significant public health and economic implications. Pulmonary rehabilitation leads to improvements in pulmonary function following TB (16), so it is important to identify patients with post-TB sequelae so that they can be appropriately managed pending the development of therapies that can reverse or prevent progression of sequelae including lung fibrosis.
Despite these clear indications that heightened immune activity in TB impacts its morbidity and mortality, current treatment regimens focus solely on bacterial killing. Consequently, there is a drive to develop interventions targeting inflammatory responses in TB. Designed to be given alongside standard treatment, achieving a more controlled inflammatory response could have a number of benefits including improving drug penetration at the site of disease and limiting the damage sustained by lung tissues, and thereby improve both acute and long-term clinical outcomes. This could have a huge impact particularly in the era of rising antimicrobial drug resistance. However, for this to be successful a detailed understanding of the innate response in TB is needed. Accumulating evidence indicates that platelets play an important role and may be key to unlocking therapies designed to improve prognosis.
Pulmonary TB is characterized by pathophysiology that is disproportionate to the mycobacterial load, and even in paucibacillary disease, patients may present with severe inflammation. The commonly held belief is that following inhalation mycobacteria are phagocytosed by macrophages, the main innate immune effector cells and hosts for bacterial replication. Macrophages become activated and recruit other innate immune cells including neutrophils (17) which are important early responders to TB infection, involved in both pathogen elimination and in inflammation (18). In addition, a secondary cell-mediated immune reaction involving cross-presentation and priming of CD8+ T cells occurs days after initial antigen exposure (19), known as delayed-type hypersensitivity. These processes can lead to the formation of a granuloma whereby lymphocytes and neutrophils surround infected macrophages to isolate the bacteria; if this containment is unsuccessful, bacterial replication and dissemination occur.
A classic feature of pulmonary TB which is often present at diagnosis is cavitation of the lung. Here, the destruction of lung parenchyma leads to the development of large, air-filled spaces that are relatively immunoprivileged sites where M.tb can proliferate exponentially. The presence of cavities is associated with treatment failure (20) and drug resistance (21), and patients with cavitary disease are highly contagious and represent the main drivers of transmission (22). Cavities are formed through the activity of enzymes called matrix metalloproteinases (MMPs) that are the final effectors of the host innate inflammatory response (23, 24). MMPs are a family of zinc-dependent proteases that collectively degrade all components of the extracellular matrix (ECM), and also have immunological functions including modulating cytokine and chemokine activity, activating defensins, and cleaving proteinase-activated receptors (25, 26). They are tightly regulated by mechanisms such as gene expression, compartmentalization, secretion as proenzymes, enzymatic inactivation, and by specific tissue inhibitors of metalloproteinases (TIMPs). MMPs are responsible for the turnover of normal tissues; if produced in excess they cause tissue damage. In pulmonary TB MMP-1, interstitial collagenase, is the main effector of lung degradation and cavity formation (24, 27). MMP concentrations in plasma and bronchoalveolar lavage fluid (BALF) have consistently been shown to be elevated in patients with acute TB, and correlate with clinical and radiological disease severity (27–30). Genetic polymorphisms that result in greater MMP-1 secretion in pulmonary TB have been identified as a risk factor for developing fibrosis following TB (31).
Persistent inflammation in TB can lead to fibrosis, and following TB treatment many patients suffer long-term lung damage, with reported rates of impairment of up to 68% (8, 32–34). Affected patients can have restrictive, obstructive, or mixed patterns on spirometric testing (34) and this can manifest as various chronic lung diseases including pulmonary fibrosis, emphysema, and bronchiectasis (35). Consequently, TB survivors have shortened life expectancies, with one model predicting an average loss of 3.6 years of life (36), and this is associated with a reduced perceived mental health-related quality of life (HRQoL) even after microbiological cure (37). The extent of radiological disease severity at diagnosis is the most important determinant of residual pulmonary function post treatment (33, 38, 39), however the mechanisms that link initial bacterial infection to the development of fibrosis are poorly understood.
The initial detection of M.tb and subsequent rapid innate immune response may be initiated and accelerated by platelets. Numerous observational studies report thrombocytosis in patients with TB (40–45) and, in some, platelet count correlated with levels of acute phase reactants such as C-reactive Protein (CRP) and with disease severity (45–47). Sahin et al. identified thrombocytosis in 44/100 pulmonary TB patients, and they had higher plateletcrit and platelet distribution width (PDW) than healthy controls. Thrombocytosis was associated with clinically and radiologically advanced disease, and platelet count correlated with CRP and erythrocyte sedimentation rate (ESR) values in pulmonary TB patients but not in patients with community acquired pneumonia (48). In a South Korean study, platelet counts were higher in TB patients than in controls, and in TB patients, the mean platelet volume (MPV) correlated with CRP levels (49). Other studies, however, have associated thrombocytopenia with tuberculosis. For example, in 128 Indian patients, thrombocytopenia was observed in 37.5% (50).
Platelet-associated gene transcripts are upregulated in TB patients (51). As platelet RNA is formed in megakaryocytes prior to platelet formation, the megakaryocyte itself may be influenced by disease states and platelets may be pre-programmed to respond specifically to TB. This has been shown in other diseases such as cancer, where the platelet transcriptome can be used to accurately diagnose various cancers and may predict outcomes (52).
Changes in platelet structure and function have been described in TB patients. Patients with acute TB have increased numbers of alpha granules (53) which contain pro-inflammatory mediators such as tumor necrosis factor alpha (TNFα) and interleukin-1 beta (IL-1β), known to be elevated in TB patients (54). In contrast, patients with chronic TB exhibited elongated platelets and increased numbers of alpha and dense granules. The latter contain a range of mediators including adenosine diphosphate (ADP), adenosine triphosphate (ATP), serotonin, and ionized calcium which is involved in the coagulation cascade. These differences may reflect distinct functional roles at different disease stages, with a predominantly pro-inflammatory phenotype in acute TB and a mixed inflammatory-thrombotic state in chronic disease.
Increased platelet activity can be detected by quantifying markers released from platelet granules upon activation. Elevated plasma concentrations of platelet factor 4 (PF4; CXCL4), a component of alpha granules specific to platelets, are found in patients with pulmonary TB and correlate with radiological disease severity (55). In a cohort of Peruvian patients with newly diagnosed, drug-sensitive smear-positive pulmonary TB, plasma levels of PF4, platelet-derived growth factor (PDGF)-BB, C-C motif chemokine ligand 5 (CCL5; RANTES), MMP-9, soluble CD40 ligand (sCD40L), and Pentraxin-3 (PTX-3; TNF-stimulated gene (TSG)-14) were elevated at baseline compared to age- and sex-matched controls. Fifty percent of these patients were followed up during their anti-TB treatment, and the plasma concentrations of all of these markers increased at Day 14 and then decreased, returning to normal by Day 60 (56). Levels of platelet-derived mediators such as PF4 have been shown to correlate with disease progression and severity in other chronic inflammatory conditions including inflammatory bowel disease (57), atherosclerosis (58), and rheumatoid arthritis (59); therefore, platelet-derived mediators may also drive inflammatory processes in TB, and could be important in the resolution of inflammation and/or development of fibrosis.
Platelets are present at the site of TB disease either as a result of extravasation of platelets that localize to the lesion, or secondary to platelet biogenesis within the lung itself. Megakaryocytes have been identified within the lungs and may have the capacity to scale up platelet generation in response to specific stimuli (60). This would support a role of platelets as first responders in M.tb infection, placing them at close proximity to any mycobacterial intruders. Lung tissue from M.tb-infected Balb/C mice has demonstrated the presence of the platelet marker CD41 which is not detected in uninfected lung. CD41 expression mostly occurred in association with anucleate cells consistent with the morphology of platelets, predominantly within the alveoli, but also in nucleated cells, indicating platelet phagocytosis and/or adherence to leukocytes (56). Similarly, platelet-specific marker CD42b was observed within epithelioid cells and multinuclear giant cells in human TB lung granulomas (61). Microthrombi occur around TB cavities and have been proposed to prevent dissemination (62). Platelet aggregations and platelet-neutrophil adhesions have been observed within pulmonary lesions and mycobacteria have been visualized within platelets, located mainly alongside the mitochondria (53). Such leukocyte-platelet interactions may be critical in regulating the immune response in TB. PDGF-BB, P-selectin, and RANTES concentrations were elevated in BALF from TB patients compared to patients with non-TB respiratory disease. The formation of platelet-leukocyte aggregates also correlates with secreted levels of platelet-derived mediators in other inflammatory diseases such as atherosclerosis (63). To further support the notion that platelet activity may be involved in immune responses in TB, concentrations of P-selectin, a platelet activation marker, positively correlate with levels of well-characterised markers of disease severity including IL-1β, MMP-1, -3, -7, -8 and -9 in TB patients (56).
Platelets can sense pathogens directly via expressed Toll-like receptors (TLRs) such as TLR2 and TLR4 (64, 65) to lead to activation and release of reactive oxygen species (66) and pro-inflammatory cytokines (67). Activated platelets also interact directly with leukocytes to facilitate cellular recruitment towards the site of infection (68). These adhesive interactions can form platelet-monocyte aggregates (PMA) and platelet-neutrophil aggregates (PNA) which lead to cell activation and enhance immune function such as cytokine or MMP production. Platelets can stimulate neutrophils to release neutrophil extracellular traps (NETs) (69), which are increased in the plasma of TB patients (70) and have been associated with severe TB-associated lung damage and subsequent sequelae (71). These adhesive interactions are mediated by the two main families of cell adhesion molecules, selectins and integrins.
Selectins are expressed on most leukocytes and consist of three family members, L-, P- and E-selectin (CD62L, CD62P and CD62E respectively). These single transmembrane glycoproteins initiate the leukocyte adhesion cascade to aid leukocyte tethering and rolling along inflamed endothelium prior to transmigration (72). Platelets only express P-selectin, which is translocated from alpha granules to the surface membrane in response to agonists such as thrombin, and can be measured in soluble form as a marker of platelet activation; soluble P-selectin levels are higher in TB patients compared to healthy controls (56, 73). P-selectin ligation can activate platelets leading to aggregation, enhanced adhesion, and activation of other platelet expressed integrins such as GPIIb/IIIa (74). Moreover, P-selectin on platelets can interact with P-selectin glycoprotein ligand-1 (PSGL1) expressed on leukocytes to form PMA (75). This interaction has a central role in the pathology of chronic inflammatory diseases including atherosclerosis (76) and heart disease (77). Increased circulating PMA are found in TB patients compared to healthy controls (78), and treatment of M.tb-infected whole blood with anti-P-selectin antibody decreases PMA (78).
Integrins are composed of two subunits, α and β, to form heterodimers that facilitate platelet binding to inflamed endothelium or leukocytes. Platelets express three families of integrins, β1 (CD29), β2 (CD18), and β3 (CD61). β1 integrins recognize sub-endothelial ECM proteins such as collagen (79) or endothelial ligands like VCAM-1 (CD106) (80), whilst β2 integrins bind to intracellular cell adhesion molecule-1 or -2 (ICAM-1/CD54 or ICAM-2/CD102), found on the endothelium, epithelium, or platelets (81, 82). Platelets express two β3 integrins, αvβ3 and αIIbβ3 (GPIIb/IIIa or CD41/CD61). αIIbβ3 is the most dominant integrin expressed on platelets (83) and can recognize arginine-glycine-aspartic acid (RGD)-containing ligands such as fibrinogen and fibrin (84). Ligation of integrins on platelets initiates “inside-out” or “outside-in” signaling pathways to result in functional outcomes such as cell activation, adhesion, or degranulation (85).
Other important adhesive interactions demonstrated between platelets and leukocytes include the binding of surface expressed glycoprotein-Ibα (GPIbα, CD42a), part of a receptor complex that binds von Willibrand factor (vWF), to αMβ2 (CD11b/CD18, Mac-1) on monocytes (86). In addition, platelets express ICAM-2 (87), the ligand for αMβ2 and αLβ2 (CD11a/CD18, LFA-1) expressed on monocytes. In atherosclerosis platelet-derived CD40L (CD154) can interact with CD40 (TNF receptor) expressed on leukocytes to form platelet-leukocyte aggregates (88). Whilst these cellular interactions have been observed in vitro or in animal models, the specific interactions important in PMA formation in TB patients remain unclear and require further investigation.
In TB, cellular networks involving resident and influxing leukocytes, stromal cells, and other cells enable amplification of initial responses, leading to secretion of MMPs and other pro-inflammatory mediators (89). There is evidence that cross-talk between platelets and leukocytes, particularly monocytes, may be key to driving TB immunopathology. Platelets have been shown to significantly upregulate the MMP secretion from M.tb-infected monocytes (56) and are important in the development of granulomas and macrophage differentiation in TB (61). M.tb-infected monocytes co-cultured with platelets may phagocytose them and differentiate into larger, multinucleated giant cells with enhanced phagocytosis and shared traits with multinucleated giant cells observed in TB granulomas. Moreover, on extended culture, macrophages differentiated with both platelets and M.tb upregulate genes involved in leukocyte chemotaxis, including CXCL5 and PPBP/CXCL7, and ECM receptor interactions compared to those cultured with M.tb alone. Although the effect of platelets on inflammatory responses in TB depends upon the stage, site, and severity of TB infection, they appear to predominantly steer monocyte differentiation towards an anti-inflammatory phenotype: the presence of M.tb does not affect cytokine secretion from platelets themselves, but has been shown to raise IL-10 and reduce TNFα secretion by platelet-transformed macrophages (61). Similarly, M.tb-infected peripheral blood mononuclear cells (PBMCs) incubated with platelets led to decreased secretion of pro-inflammatory cytokines TNFα, IL-1β, IL-6 and IFNγ but increased IL-10 compared to M.tb-infected PBMCs incubated without platelets (78).
What remain unexplored are the mechanisms by which platelets induce leukocyte activation during M.tb infection to cause reported cellular responses. Data from in vitro studies utilizing uninfected cells show that following engagement of platelet expressed P-selectin and PSGL1 on leukocytes, signal transduction pathways are initiated to enhance tyrosine phosphorylation and activation of mitogen-activated protein kinase (MAPK) (90). MAPK are phosphorylation-dependent signal-transducing enzymes involved in immune responses and cellular regulation (91). The p38 and extracellular signal-related kinase (ERK)/MAPK pathways are key in regulating MMP and cytokine secretion during M.tb infection (92–96). Therefore, we hypothesize that P-selectin-PSGL1 binding between platelets and leukocytes in M.tb infection could lead to MMP and pro-inflammatory cytokine gene and protein production via MAPK signaling, although this requires confirmation. In contrast, phosphatidyl inositol 3-kinase (PI3K) signaling negatively regulates MMP-1 secretion from M.tb-infected macrophages (97). PSGL1 activation can induce signaling via PI3K (98) and activation of mTOR and Rho-associated kinases (ROCKs) in macrophages to facilitate cell motility and phagocytosis (99). Whether platelets interacting with macrophages during M.tb infection also utilize PSGL1-PI3K/mTOR signaling is unknown.
Taken together, these studies indicate that platelets have a key role in directing cellular outcomes in response to M.tb through interactions with cell adhesion molecules and by maneuvering intracellular signaling pathways as summarized by Figure 1. A better understanding of these cellular interactions may reveal which signaling mechanisms could be targeted to decrease TB immunopathology.
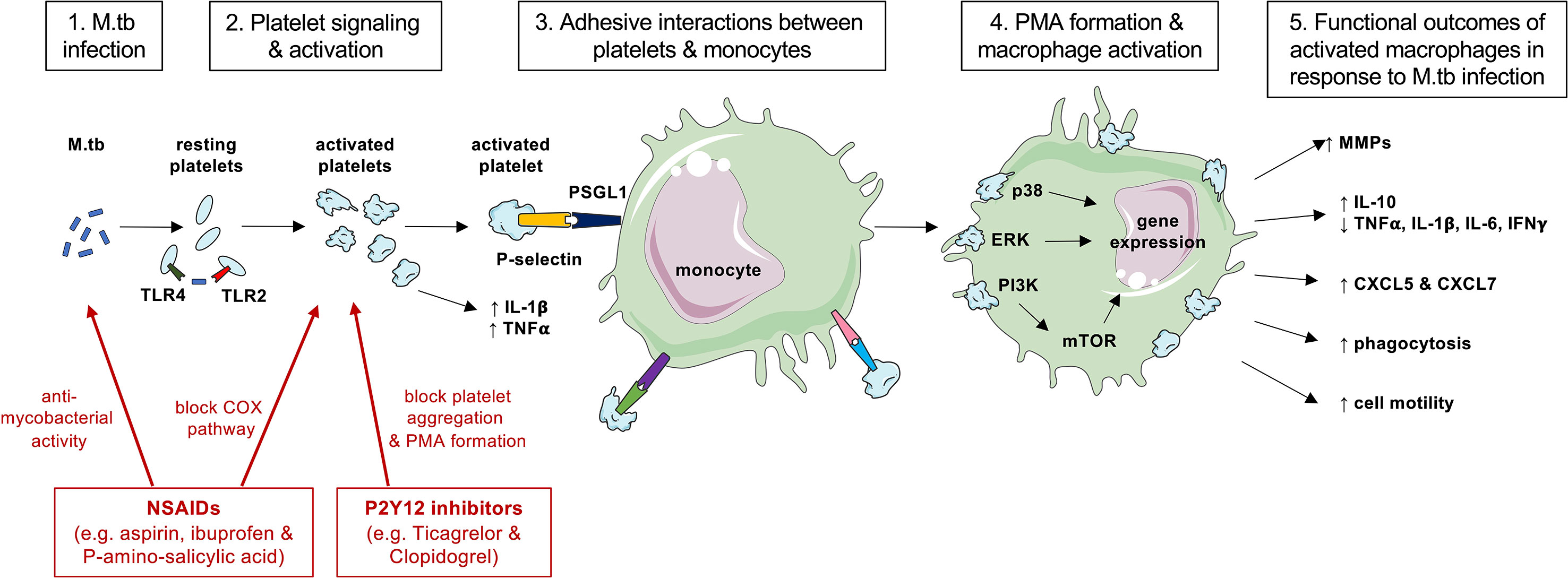
Figure 1 A summary of cellular interactions between platelets and monocyte during M.tb infection and actions of anti-platelet therapies. Platelets may be able to directly sense M.tb infection (step 1) via TLR2 and TLR4 to become activated and degranulate to release pro-inflammatory cytokines (step 2). Activated platelets upregulate surface P-selectin expression and bind to PSGL1 on monocytes (step 3) leading to PMA formation (step 4). Ligation of PSGL1 can initiate signal transduction via MAPK and induce gene expression (step 4) to result in cellular functional outcomes such as MMP and cytokine production or phagocytosis (step 5). Anti-platelet agents such as non-steroidal anti-inflammatory drugs (NSAIDs) and P2Y12 inhibitors (shown in red) may exhibit direct anti-mycobacterial activity on M.tb or block the actions of platelets to decrease PMA formation and MMP expression that cause collateral host tissue damage. This figure was created with images adapted from Servier Medical Art by Servier. Original images are licensed under a Creative Commons Attribution 3.0 Unported License.
Imbalances in eicosanoids have been associated with the development of TB (100, 101). Eicosanoids are lipid mediators derived from the activity of the enzymes cyclooxygenase (COX)-1 and -2 on arachidonic acid, which enter either the cyclooxygenase or the lipoxygenase pathway. The cyclooxygenase pathway leads to prostaglandins D, E and F, prostacyclin, and thromboxane A2 (TXA A2) production. Prostaglandins are powerful vasodilators, inhibit platelet aggregation, and act as signaling molecules linking the innate immune system to acute inflammatory pathways (102), whereas thromboxanes are vasoconstrictors that trigger platelet aggregation (103). The lipoxygenase pathway causes the production of lipoxins, which reduce pro-inflammatory cytokine production and neutrophil recruitment (104). Non-steroidal anti-inflammatory drugs (NSAIDs) inhibit COX-1 and -2, and include aspirin (Acetylsalicylic acid, ASA), the most commonly prescribed anti-platelet agent. Aspirin irreversibly inhibits both COX enzymes and displays powerful anti-thrombotic effects at low doses as well as anti-inflammatory activity at higher doses.
Repurposing anti-platelet and anti-inflammatory NSAIDs for use in TB is attractive because they are already approved for use and have well-established safety profiles. Data indicate they may be effective at enhancing TB control (105); animal and human studies are presented in Table 1. NSAIDs have some direct anti-mycobacterial activity (104, 109, 113, 116, 124, 125), directly interfering with M.tb metabolism via downregulation of genes involved in energy production (126). However, their main effect in TB is likely to be due to their action on the host immune system, particularly by modulating inflammatory pathways that are active in TB disease (104, 107, 111, 116, 117). Administration of ibuprofen to TB-infected C3HeB/FeJ mice led to fewer and smaller lung lesions, decreased neutrophil infiltration, reduced bacillary load, and enhanced survival (115). The effects of NSAID treatment may be either protective, by enabling effective clearance of pathogens, or harmful by causing tissue destruction, particularly where immune activity is disproportionate to the magnitude of the infective insult (118). This will depend upon multiple factors including timing, route of administration, and dose, all of which must be considered when proposing NSAIDs as a potential adjunctive treatment.
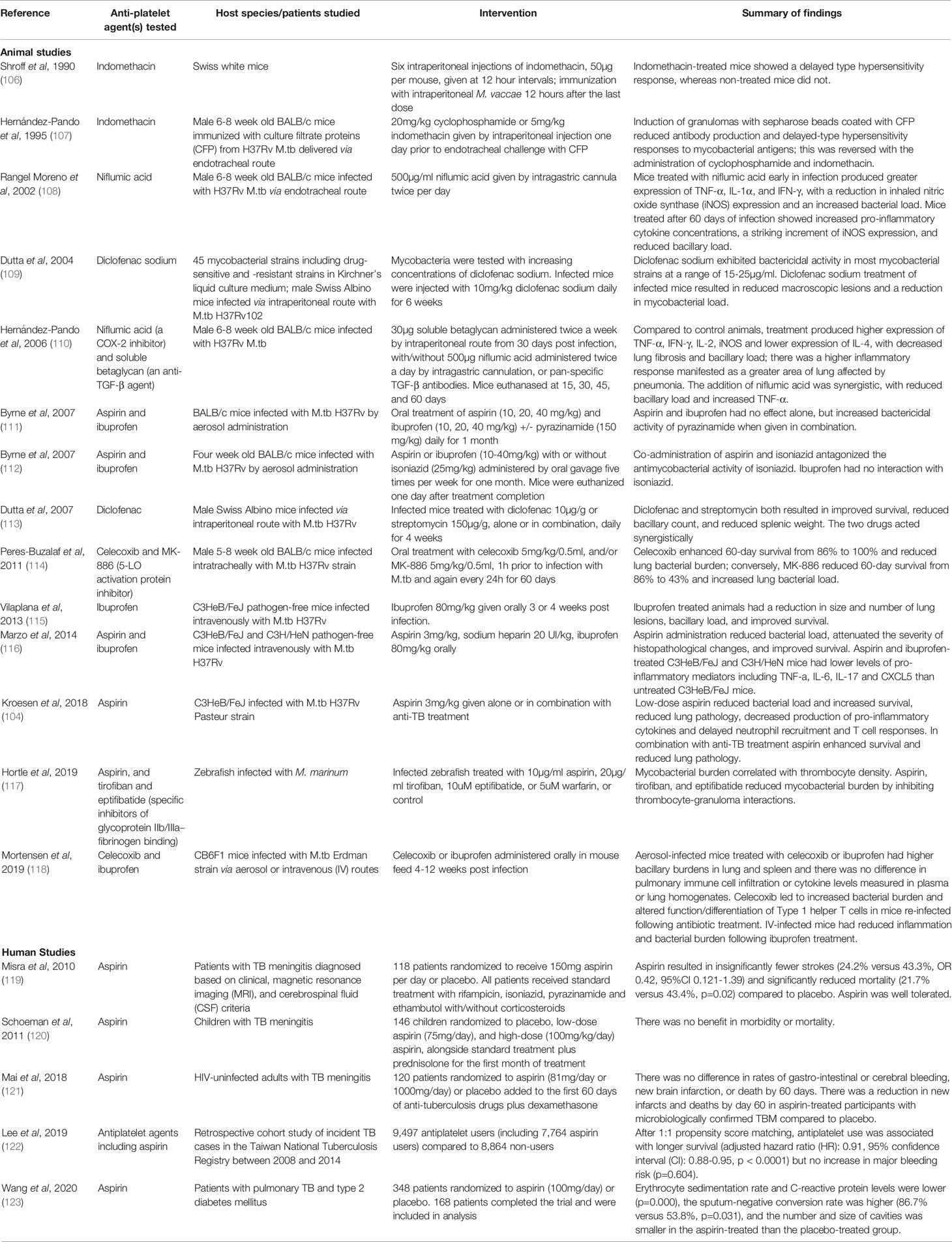
Table 1 Animal and human studies investigating the effects of anti-platelet agents in tuberculosis.
Data from TB patients suggest that anti-platelet agents can be beneficial. One of the first drugs in use for TB, P-amino-salicylic acid (PAS), is an aspirin homologue that was deemed effective despite an unknown mechanism of action when it was first given to a patient in 1944. We showed that PAS modulates anti-inflammatory immune activity, and suppresses MMP-1 secretion through a PGE2-dependent mechanism without affecting M.tb replication (93). PAS therefore represents the first example of a host-directed therapy used to treat TB. More recently, a Taiwanese population-based study of patients with drug-sensitive TB found that use of antiplatelet drugs was associated with significantly improved overall survival and a lower 12-month mortality rate, as well as lower rates of smear positivity and fewer cavities. Benefits were greater with aspirin compared to non-aspirin anti-platelet agents such as clopidogrel (122). In a randomized controlled trial, giving 100mg aspirin to diabetic patients with pulmonary TB resulted in decreased secretion of inflammatory mediators including ESR and CRP, a higher sputum-negative conversion rate (87% versus 54%), and fewer and smaller cavities following treatment, compared to placebo-treated control patients (123). In TB meningitis, both inflammation and thrombosis are associated with higher mortality and complications including stroke in survivors; randomized controlled trials of aspirin have shown benefits in morbidity and mortality (119, 121). Further studies are currently underway to evaluate NSAIDS including meloxicam and ibuprofen as adjuncts to standard TB treatment regimens (127–129).
Drugs blocking other platelet signaling pathways are potentially important in the management of TB. ADP is the endogenous ligand for P2Y12 and P2Y13 receptors, which are implicated in platelet aggregation. ADP induces MCP-1 expression to enhance macrophage migration, and P2Y13 expression is increased in TB patients (130). Ticagrelor and clopidogrel, both P2Y12 inhibitors, given to healthy volunteers significantly reduced inflammatory and prothrombotic mechanisms including PMA formation and pro-inflammatory cytokine release following endotoxin challenge (131). In a double-blind placebo-controlled study a single dose of oral ticagrelor reduced PMA formation in healthy volunteers, and this was associated with an increase in pro-inflammatory cytokines in blood exposed to the TLR2 ligand Pam3CSK4 and a decrease in blood exposed to TLR4 ligand LPS, suggesting that platelets may differentially modulate cytokine responses depending upon the receptors involved (132). The role of such compounds and of inhibitors of platelet signaling pathways, proposed in Figure 1, as potential therapy targets in TB, are not yet known but merit detailed investigation and this represents a major avenue of future research.
In this review, we have presented substantial evidence that platelets are a key component of the innate immune response to tuberculosis. Both observational and experimental data show that the administration of anti-platelet agents in patients with TB may be effective at limiting disease manifestations and improving long-term outcomes in patients who are successfully treated. These findings are highly promising, however much remains to be understood about the exact mechanisms of platelet engagement with mycobacteria and other cells in the orchestration of the complex immune response to M.tb. Research is needed to ascertain the effect of therapeutic blockade of specific targets within the platelet on downstream signaling and leukocyte activity. In particular, the timing of anti-platelet administration will be of utmost importance to ensure optimal use, enabling interruption of harmful tissue destructive processes whilst preserving, or even enhancing, the immune system’s anti-infective properties.
All authors contributed to the article and approved the submitted version.
DK is supported by MRC Clinical Research Training Fellowship (MR/P019978/2).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
2. Reynolds M, Glaser L, Phin N, Muzyamba MC, Mirza A, Russel RH, et al. Tuberculosis in England: 2019 Report. London: UK Public Health England (2019).
3. Kaufmann SH, Dorhoi A. Inflammation in Tuberculosis: Interactions, Imbalances and Interventions. Curr Opin Immunol (2013) 25(4):441–9. doi: 10.1016/j.coi.2013.05.005
4. Ehrt S, Schnappinger D, Rhee KY. Metabolic Principles of Persistence and Pathogenicity in Mycobacterium Tuberculosis. Nat Rev Microbiol (2018) 16(8):496–507. doi: 10.1038/s41579-018-0013-4
5. Dhar R, Singh S, Talwar D, Mohan M, Tripathi SK, Swarnakar R, et al. Bronchiectasis in India: Results From the European Multicentre Bronchiectasis Audit and Research Collaboration (EMBARC) and Respiratory Research Network of India Registry. Lancet Global Health (2019) 7(9):e1269–e79. doi: 10.1016/S2214-109X(19)30327-4
6. Maguire GP, Anstey NM, Ardian M, Waramori G, Tjitra E, Kenangalem E, et al. Pulmonary Tuberculosis, Impaired Lung Function, Disability and Quality of Life in a High-Burden Setting. Int J Tuberc Lung Dis (2009) 13(12):1500–6.
7. Byrne AL, Marais BJ, Mitnick CD, Lecca L, Marks GB. Tuberculosis and Chronic Respiratory Disease: A Systematic Review. Int J Infect Dis (2015) 32:138–46. doi: 10.1016/j.ijid.2014.12.016
8. Amaral AF, Coton S, Kato B, Tan WC, Studnicka M, Janson C, et al. Tuberculosis Associates With Both Airflow Obstruction and Low Lung Function: BOLD Results. Eur Respir J (2015) 46(4):1104–12. doi: 10.1183/13993003.02325-2014
9. Miller TL, McNabb SJ, Hilsenrath P, Pasipanodya J, Weis SE. Personal and Societal Health Quality Lost to Tuberculosis. PloS One (2009) 4(4):e5080. doi: 10.1371/journal.pone.0005080
10. Snider GL, Doctor L, Demas TA, Shaw AR. Obstructive Airway Disease in Patients With Treated Pulmonary Tuberculosis. Am Rev Respir Dis (1971) 103(5):625–40. doi: 10.1164/arrd.1971.103.5.625
11. Birath G, Caro J, Malmberg R, Simonsson BG. Airways Obstruction in Pulmonary Tuberculosis. Scand J Respir Dis (1966) 47(1):27–36.
12. Lancaster JF, Tomashefski JF. Tuberculosis–a Cause of Emphysema. Am Rev Respir Dis (1963) 87:435–7. doi: 10.1164/arrd.1963.87.3P1.435
13. Simpson DG, Uschner M, Mc CJ. Respiratory Function in Pulmonary Tuberculosis. Am Rev Respir Dis (1963) 87:1–16. doi: 10.1164/arrd.1963.87.1.1
14. Garvin A, Lundsgaard C, Van Slyke DD. Studies of Lung Volume: Ii. Tuberc Men J Exp Med (1918) 27(1):87–94. doi: 10.1084/jem.27.1.87
15. Garvin A, Lundsgaard C, Van Slyke DD. Studies of Lung Volume: Iii. Tuberc Women J Exp Med (1918) 27(1):129–42. doi: 10.1084/jem.27.1.129
16. Ando M, Mori A, Esaki H, Shiraki T, Uemura H, Okazawa M, et al. The Effect of Pulmonary Rehabilitation in Patients With Post-Tuberculosis Lung Disorder. Chest (2003) 123(6):1988–95. doi: 10.1378/chest.123.6.1988
17. Ferrero E, Biswas P, Vettoretto K, Ferrarini M, Uguccioni M, Piali L, et al. Macrophages Exposed to Mycobacterium Tuberculosis Release Chemokines Able to Recruit Selected Leucocyte Subpopulations: Focus on Gammadelta Cells. Immunology (2003) 108(3):365–74. doi: 10.1046/j.1365-2567.2003.01600.x
18. Hilda JN, Das S, Tripathy SP, Hanna LE. Role of Neutrophils in Tuberculosis: A Bird’s Eye View. Innate Immun (2020) 26(4):240–7. doi: 10.1177/1753425919881176
19. Hinchey J, Lee S, Jeon BY, Basaraba RJ, Venkataswamy MM, Chen B, et al. Enhanced Priming of Adaptive Immunity by a Proapoptotic Mutant of Mycobacterium Tuberculosis. J Clin Invest (2007) 117(8):2279–88. doi: 10.1172/JCI31947
20. Chatterjee A, D’Souza D, Vira T, Bamne A, Ambe GT, Nicol MP, et al. Strains of Mycobacterium Tuberculosis From Western Maharashtra, India, Exhibit a High Degree of Diversity and Strain-Specific Associations With Drug Resistance, Cavitary Disease, and Treatment Failure. J Clin Microbiol (2010) 48(10):3593–9. doi: 10.1128/JCM.00430-10
21. Kempker RR, Rabin AS, Nikolaishvili K, Kalandadze I, Gogishvili S, Blumberg HM, et al. Additional Drug Resistance in Mycobacterium Tuberculosis Isolates From Resected Cavities Among Patients With Multidrug-Resistant or Extensively Drug-Resistant Pulmonary Tuberculosis. Clin Infect Dis (2012) 54(6):e51–4. doi: 10.1093/cid/cir904
22. Dye C, Williams BG. The Population Dynamics and Control of Tuberculosis. Science (2010) 328(5980):856–61. doi: 10.1126/science.1185449
23. Price NM, Farrar J, Tran TT, Nguyen TH, Tran TH, Friedland JS. Identification of a Matrix-Degrading Phenotype in Human Tuberculosis In Vitro and In Vivo. J Immunol (2001) 166:4223–30. doi: 10.4049/jimmunol.166.6.4223
24. Ong CW, Elkington PT, Friedland JS. Tuberculosis, Pulmonary Cavitation, and Matrix Metalloproteinases. Am J Respir Crit Care Med (2014) 190(1):9–18. doi: 10.1164/rccm.201311-2106PP
25. Parks WC, Wilson CL, López-Boado YS. Matrix Metalloproteinases as Modulators of Inflammation and Innate Immunity. Nat Rev Immunol (2004) 4(8):617–29. doi: 10.1038/nri1418
26. Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a Matrix Metalloprotease-1 Receptor That Promotes Invasion and Tumorigenesis of Breast Cancer Cells. Cell (2005) 120(3):303–13. doi: 10.1016/j.cell.2004.12.018
27. Ugarte-Gil CA, Elkington P, Gilman RH, Coronel J, Tezera LB, Bernabe-Ortiz A, et al. Induced Sputum MMP-1, -3 & -8 Concentrations During Treatment of Tuberculosis. PloS One (2013) 8(4):e61333. doi: 10.1371/journal.pone.0061333
28. Singh S, Kubler A, Singh UK, Singh A, Gardiner H, Prasad R, et al. Antimycobacterial Drugs Modulate Immunopathogenic Matrix Metalloproteinases in a Cellular Model of Pulmonary Tuberculosis. Antimicrob Agents Chemother (2014) 58(8):4657–65. doi: 10.1128/AAC.02141-13
29. Elkington P, Shiomi T, Breen R, Nuttall RK, Ugarte-Gil CA, Walker NF, et al. MMP-1 Drives Immunopathology in Human Tuberculosis and Transgenic Mice. J Clin Invest (2011) 121(5):1827–33. doi: 10.1172/JCI45666
30. Walker NF, Clark SO, Oni T, Andreu N, Tezera L, Singh S, et al. Doxycycline and HIV Infection Suppress Tuberculosis-Induced Matrix Metalloproteinases. Am J Respir Crit Care Med (2012) 185(9):989–97. doi: 10.1164/rccm.201110-1769OC
31. Wang CH, Lin HC, Lin SM, Huang CD, Liu CY, Huang KH, et al. MMP-1(-1607G) Polymorphism as a Risk Factor for Fibrosis After Pulmonary Tuberculosis in Taiwan. Int J Tuberc Lung Dis (2010) 14(5):627–34.
32. Menezes AM, Hallal PC, Perez-Padilla R, Jardim JR, Muiño A, Lopez MV, et al. Tuberculosis and Airflow Obstruction: Evidence From the PLATINO Study in Latin America. Eur Respir J (2007) 30(6):1180–5. doi: 10.1183/09031936.00083507
33. Ross J, Ehrlich RI, Hnizdo E, White N, Churchyard GJ. Excess Lung Function Decline in Gold Miners Following Pulmonary Tuberculosis. Thorax (2010) 65(11):1010–5. doi: 10.1136/thx.2009.129999
34. Pasipanodya JG, Miller TL, Vecino M, Munguia G, Garmon R, Bae S, et al. Pulmonary Impairment After Tuberculosis. Chest (2007) 131(6):1817–24. doi: 10.1378/chest.06-2949
35. Hnizdo E, Singh T, Churchyard G. Chronic Pulmonary Function Impairment Caused by Initial and Recurrent Pulmonary Tuberculosis Following Treatment. Thorax (2000) 55(1):32–8. doi: 10.1136/thorax.55.1.32
36. Hoger S, Lykens K, Beavers SF, Katz D, Miller TL. Longevity Loss Among Cured Tuberculosis Patients and the Potential Value of Prevention. Int J Tuberc Lung Dis (2014) 18(11):1347–52. doi: 10.5588/ijtld.14.0242
37. Daniels KJ, Irusen E, Pharaoh H, Hanekom S. Post-Tuberculosis Health-Related Quality of Life, Lung Function and Exercise Capacity in a Cured Pulmonary Tuberculosis Population in the Breede Valley District, South Africa. S Afr J Physiother (2019) 75(1):1319. doi: 10.4102/sajp.v75i1.1319
38. Plit ML, Anderson R, Van Rensburg CE, Page-Shipp L, Blott JA, Fresen JL, et al. Influence of Antimicrobial Chemotherapy on Spirometric Parameters and Pro-Inflammatory Indices in Severe Pulmonary Tuberculosis. Eur Respir J (1998) 12(2):351–6. doi: 10.1183/09031936.98.12020351
39. Willcox PA, Ferguson AD. Chronic Obstructive Airways Disease Following Treated Pulmonary Tuberculosis. Respir Med (1989) 83(3):195–8. doi: 10.1016/S0954-6111(89)80031-9
40. Robson SC, White NW, Aronson I, Woollgar R, Goodman H, Jacobs P. Acute-Phase Response and the Hypercoagulable State in Pulmonary Tuberculosis. Br J Haematol (1996) 93(4):943–9. doi: 10.1046/j.1365-2141.1996.d01-1722.x
41. Feng Y, Yin H, Mai G, Mao L, Yue J, Xiao H, et al. Elevated Serum Levels of CCL17 Correlate With Increased Peripheral Blood Platelet Count in Patients With Active Tuberculosis in China. Clin Vaccine Immunol (2011) 18(4):629–32. doi: 10.1128/CVI.00493-10
42. Turken O, Kunter E, Sezer M, Solmazgul E, Cerrahoglu K, Bozkanat E, et al. Hemostatic Changes in Active Pulmonary Tuberculosis. Int J Tuberc Lung Dis (2002) 6(10):927–32. doi: 10.5588/09640569513039
43. Bozóky G, Ruby E, Góhér I, Tóth J, Mohos A. [Hematologic Abnormalities in Pulmonary Tuberculosiss]. Orv Hetil (1997) 138(17):1053–6.
44. Morris CD, Bird AR, Nell H. The Haematological and Biochemical Changes in Severe Pulmonary Tuberculosis. Q J Med (1989) 73(272):1151–9.
45. Unsal E, Aksaray S, Köksal D, Sipit T. Potential Role of Interleukin 6 in Reactive Thrombocytosis and Acute Phase Response in Pulmonary Tuberculosis. Postgrad Med J (2005) 81(959):604–7. doi: 10.1136/pgmj.2004.030544
46. Tozkoparan E, Deniz O, Ucar E, Bilgic H, Ekiz K. Changes in Platelet Count and Indices in Pulmonary Tuberculosis. Clin Chem Lab Med (2007) 45(8):1009–13. doi: 10.1515/CCLM.2007.194
47. Baynes RD, Bothwell TH, Flax H, McDonald TP, Atkinson P, Chetty N, et al. Reactive Thrombocytosis in Pulmonary Tuberculosis. J Clin Pathol (1987) 40(6):676–9. doi: 10.1136/jcp.40.6.676
48. Sahin F, Yazar E, Yildiz P. Prominent Features of Platelet Count, Plateletcrit, Mean Platelet Volume and Platelet Distribution Width in Pulmonary Tuberculosis. Multidiscip Respir Med (2012) 7(1):38. doi: 10.1186/2049-6958-7-38
49. Lee MY, Kim YJ, Lee HJ, Cho SY, Park TS. Mean Platelet Volume in Mycobacterium Tuberculosis Infection. BioMed Res Int (2016) 2016:7508763. doi: 10.1155/2016/7508763
50. Kutiyal AS. A Study of Haematological and Haemostasis Parameters and Hypercoagulable State in Tuberculosis Patients in Northern India and the Outcome With Anti Tubercular Therapy. J Clin Diagn Res (2017) 11(2):OC09–13. doi: 10.7860/JCDR/2017/24022.9249
51. Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, et al. An Interferon-Inducible Neutrophil-Driven Blood Transcriptional Signature in Human Tuberculosis. Nature (2010) 466(7309):973–7. doi: 10.1038/nature09247
52. Best MG, Sol N, Kooi I, Tannous J, Westerman BA, Rustenburg F, et al. RNA-Seq of Tumor-Educated Platelets Enables Blood-Based Pan-Cancer, Multiclass, and Molecular Pathway Cancer Diagnostics. Cancer Cell (2015) 28(5):666–76. doi: 10.1016/j.ccell.2015.09.018
53. Khechinashvili GN, Khvitiya NG. Protective Characteristics of Platelets in Tuberculosis. Bull Exp Biol Med (2004) 138(5):513–4. doi: 10.1007/s10517-005-0084-x
54. Law K, Weiden M, Harkin T, Tchou-Wong K, Chi C, Rom WN. Increased Release of Interleukin-1 Beta, Interleukin-6, and Tumor Necrosis Factor-Alpha by Bronchoalveolar Cells Lavaged From Involved Sites in Pulmonary Tuberculosis. Am J Respir Crit Care Med (1996) 153(2):799–804. doi: 10.1164/ajrccm.153.2.8564135
55. Buyukasik Y, Soylu B, Soylu AR, Ozcebe OI, Canbakan S, Haznedaroglu IC, et al. In Vivo Platelet and T-Lymphocyte Activities During Pulmonary Tuberculosis. Eur Respir J (1998) 12(6):1375–9. doi: 10.1183/09031936.98.12061375
56. Fox KA, Kirwan DE, Whittington AM, Krishnan N, Robertson BD, Gilman RH, et al. Platelets Regulate Pulmonary Inflammation and Tissue Destruction in Tuberculosis. Am J Respir Crit Care Med (2018) 198(2):245–55. doi: 10.1164/rccm.201710-2102OC
57. Ye L, Zhang Y-P, Yu N, Jia Y-X, Wan S-J, Wang F-Y. Serum Platelet Factor 4 is a Reliable Activity Parameter in Adult Patients With Inflammatory Bowel Disease: A Pilot Study. Medicine (2017) 96(11):e6323–e. doi: 10.1097/MD.0000000000006323
58. Domschke G, Gleissner CA. CXCL4-Induced Macrophages in Human Atherosclerosis. Cytokine (2019) 122:154141. doi: 10.1016/j.cyto.2017.08.021
59. Yeo L, Adlard N, Biehl M, Juarez M, Smallie T, Snow M, et al. Expression of Chemokines CXCL4 and CXCL7 by Synovial Macrophages Defines an Early Stage of Rheumatoid Arthritis. Ann Rheum Dis (2016) 75(4):763–71. doi: 10.1136/annrheumdis-2014-206921
60. Lefrançais E, Ortiz-Muñoz G, Caudrillier A, Mallavia B, Liu F, Sayah DM, et al. The Lung is a Site of Platelet Biogenesis and a Reservoir for Haematopoietic Progenitors. Nature (2017) 544(7648):105–9. doi: 10.1038/nature21706
61. Feng Y, Dorhoi A, Mollenkopf HJ, Yin H, Dong Z, Mao L, et al. Platelets Direct Monocyte Differentiation Into Epithelioid-Like Multinucleated Giant Foam Cells With Suppressive Capacity Upon Mycobacterial Stimulation. J Infect Dis (2014) 210(11):1700–10. doi: 10.1093/infdis/jiu355
62. Kuhn C, Askin F. Lung and Mediastinum. In: Kissane J, editor. Anderson’s Pathology, 8th ed. St. Louis: CV Mosby (1985). p. 838–54.
63. Gremmel T, Ay C, Riedl J, Kopp CW, Eichelberger B, Koppensteiner R, et al. Platelet-Specific Markers are Associated With Monocyte-Platelet Aggregate Formation and Thrombin Generation Potential in Advanced Atherosclerosis. Thromb Haemost (2016) 115(3):615–21. doi: 10.1160/th15-07-0598
64. Cognasse F, Hamzeh H, Chavarin P, Acquart S, Genin C, Garraud O. Evidence of Toll-Like Receptor Molecules on Human Platelets. Immunol Cell Biol (2005) 83(2):196–8. doi: 10.1111/j.1440-1711.2005.01314.x
65. Andonegui G, Kerfoot SM, McNagny K, Ebbert KV, Patel KD, Kubes P. Platelets Express Functional Toll-Like Receptor-4. Blood (2005) 106(7):2417–23. doi: 10.1182/blood-2005-03-0916
66. Blair P, Rex S, Vitseva O, Beaulieu L, Tanriverdi K, Chakrabarti S, et al. Stimulation of Toll-Like Receptor 2 in Human Platelets Induces a Thromboinflammatory Response Through Activation of Phosphoinositide 3-Kinase. Circ Res (2009) 104(3):346–54. doi: 10.1161/CIRCRESAHA.108.185785
67. Aslam R, Speck ER, Kim M, Crow AR, Bang KW, Nestel FP, et al. Platelet Toll-Like Receptor Expression Modulates Lipopolysaccharide-Induced Thrombocytopenia and Tumor Necrosis Factor-Alpha Production In Vivo. Blood (2006) 107(2):637–41. doi: 10.1182/blood-2005-06-2202
68. Rossaint J, Margraf A, Zarbock A. Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front Immunol (2018) 9:2712. doi: 10.3389/fimmu.2018.02712
69. Zucoloto AZ, Jenne CN. Platelet-Neutrophil Interplay: Insights Into Neutrophil Extracellular Trap (NET)-Driven Coagulation in Infection. Front Cardiovasc Med (2019) 6:85. doi: 10.3389/fcvm.2019.00085
70. van der Meer AJ, Zeerleder S, Blok DC, Kager LM, Lede IO, Rahman W, et al. Neutrophil Extracellular Traps in Patients With Pulmonary Tuberculosis. Respir Res (2017) 18(1):181. doi: 10.1186/s12931-017-0663-1
71. Muefong CN, Sutherland JS. Neutrophils in Tuberculosis-Associated Inflammation and Lung Pathology. Front Immunol (2020) 11:962. doi: 10.3389/fimmu.2020.00962
72. Nourshargh S, Alon R. Leukocyte Migration Into Inflamed Tissues. Immunity (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
73. Eini P, Shirvani M, Hajilooi M, Esna-Ashari F. Comparison of L-Selectin Blood Level and Gene Polymorphism in Tuberculosis Patients With Healthy Individuals. J Clin Lab Anal (2018) 32(6):e22409. doi: 10.1002/jcla.22409
74. Théorêt JF, Yacoub D, Hachem A, Gillis MA, Merhi Y. P-Selectin Ligation Induces Platelet Activation and Enhances Microaggregate and Thrombus Formation. Thromb Res (2011) 128(3):243–50. doi: 10.1016/j.thromres.2011.04.018
75. Martins P, van Gils JM, Mol A, Hordijk PL, Zwaginga JJ. Platelet Binding to Monocytes Increases the Adhesive Properties of Monocytes by Up-Regulating the Expression and Functionality of β(1) and β(2) Integrins. J Leukoc Biol (2006) 79(3):499–507. doi: 10.1189/jlb.0605318
76. Linden MD, Jackson DE. Platelets: Pleiotropic Roles in Atherogenesis and Atherothrombosis. Int J Biochem Cell Biol (2010) 42(11):1762–6. doi: 10.1016/j.biocel.2010.07.012
77. Linden MD, Furman MI, Frelinger AL 3rd, Fox ML, Barnard MR, Li Y, et al. Indices of Platelet Activation and the Stability of Coronary Artery Disease. J Thromb Haemost (2007) 5(4):761–5. doi: 10.1111/j.1538-7836.2007.02462.x
78. Kullaya V, van der Ven A, Mpagama S, Mmbaga BT, de Groot P, Kibiki G, et al. Platelet-Monocyte Interaction in Mycobacterium Tuberculosis Infection. Tuberc (Edinburgh Scotland) (2018) 111:86–93. doi: 10.1016/j.tube.2018.05.002
79. White DJ, Puranen S, Johnson MS, Heino J. The Collagen Receptor Subfamily of the Integrins. Int J Biochem Cell Biol (2004) 36(8):1405–10. doi: 10.1016/j.biocel.2003.08.016
80. Elices MJ, Osborn L, Takada Y, Crouse C, Luhowskyj S, Hemler ME, et al. VCAM-1 on Activated Endothelium Interacts With the Leukocyte Integrin VLA-4 at a Site Distinct From the VLA-4/Fibronectin Binding Site. Cell (1990) 60(4):577–84. doi: 10.1016/0092-8674(90)90661-W
81. Smith CW, Rothlein R, Hughes BJ, Mariscalco MM, Rudloff HE, Schmalstieg FC, et al. Recognition of an Endothelial Determinant for CD 18-Dependent Human Neutrophil Adherence and Transendothelial Migration. J Clin Invest (1988) 82(5):1746–56. doi: 10.1172/JCI113788
82. Staunton DE, Dustin ML, Springer TA. Functional Cloning of ICAM-2, a Cell Adhesion Ligand for LFA-1 Homologous to ICAM-1. Nature (1989) 339(6219):61–4. doi: 10.1038/339061a0
83. Jennings LK, Phillips DR. Purification of Glycoproteins Iib and III From Human Platelet Plasma Membranes and Characterization of a Calcium-Dependent Glycoprotein Iib-III Complex. J Biol Chem (1982) 257(17):10458–66. doi: 10.1016/S0021-9258(18)34040-7
84. Litvinov RI, Farrell DH, Weisel JW, Bennett JS. The Platelet Integrin αiibβ3 Differentially Interacts With Fibrin Versus Fibrinogen. J Biol Chem (2016) 291(15):7858–67. doi: 10.1074/jbc.M115.706861
85. Vilahur G, Gutiérrez M, Arzanauskaite M, Mendieta G, Ben-Aicha S, Badimon L. Intracellular Platelet Signalling as a Target for Drug Development. Vascul Pharmacol (2018) 111:22–5. doi: 10.1016/j.vph.2018.08.007
86. Simon DI, Chen Z, Xu H, Li CQ, Dong J, McIntire LV, et al. Platelet Glycoprotein Ibalpha is a Counterreceptor for the Leukocyte Integrin Mac-1 (CD11b/CD18). J Exp Med (2000) 192(2):193–204. doi: 10.1084/jem.192.2.193
87. Diacovo TG, deFougerolles AR, Bainton DF, Springer TA. A Functional Integrin Ligand on the Surface of Platelets: Intercellular Adhesion Molecule-2. J Clin Invest (1994) 94(3):1243–51. doi: 10.1172/JCI117442
88. Gerdes N, Seijkens T, Lievens D, Kuijpers MJ, Winkels H, Projahn D, et al. Platelet CD40 Exacerbates Atherosclerosis by Transcellular Activation of Endothelial Cells and Leukocytes. Arterioscler Thromb Vasc Biol (2016) 36(3):482–90. doi: 10.1161/ATVBAHA.115.307074
89. Sabir N, Hussain T, Mangi MH, Zhao D, Zhou X. Matrix Metalloproteinases: Expression, Regulation and Role in the Immunopathology of Tuberculosis. Cell Prolif (2019) 52(4):e12649. doi: 10.1111/cpr.12649
90. Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. Engagement of P-Selectin Glycoprotein Ligand-1 Enhances Tyrosine Phosphorylation and Activates Mitogen-Activated Protein Kinases in Human Neutrophils. J Biol Chem (1997) 272(45):28750–6. doi: 10.1074/jbc.272.45.28750
91. Johnson GL, Lapadat R. Mitogen-Activated Protein Kinase Pathways Mediated by ERK, JNK, and P38 Protein Kinases. Science (2002) 298(5600):1911–2. doi: 10.1126/science.1072682
92. O’Kane C, Elkington P, Jones M, Caviedes L, Tovar M, Gilman R, et al. STAT3, P38 MAPK, and NF-Kappab Drive Unopposed Monocyte-Dependent Fibroblast MMP-1 Secretion in Tuberculosis. Am J Respir Cell Mol Biol (2010) 43(4):465–74. doi: 10.1165/rcmb.2009-0211OC
93. Rand L, Green J, Saraiva L, Friedland J, Elkington P. Matrix Metalloproteinase-1 is Regulated in Tuberculosis by a P38 MAPK-Dependent, P-Aminosalicylic Acid-Sensitive Signaling Cascade. J Immunol (2009) 182(9):5865–72. doi: 10.4049/jimmunol.0801935
94. Elkington P, Emerson J, Lopez-Pascua L, O’Kane C, Horncastle D, Boyle J, et al. Mycobacterium Tuberculosis Up-Regulates Matrix Metalloproteinase-1 Secretion From Human Airway Epithelial Cells Via a P38 MAPK Switch. J Immunol (2005) 175(8):5333–40. doi: 10.4049/jimmunol.175.8.5333
95. Mercer BA, Kolesnikova N, Sonett J, D’Armiento J. Extracellular Regulated Kinase/Mitogen Activated Protein Kinase is Up-Regulated in Pulmonary Emphysema and Mediates Matrix Metalloproteinase-1 Induction by Cigarette Smoke. J Biol Chem (2004) 279(17):17690–6. doi: 10.1074/jbc.M313842200
96. Reiling N, Blumenthal A, Flad HD, Ernst M, Ehlers S. Mycobacteria-Induced TNF-Alpha and IL-10 Formation by Human Macrophages is Differentially Regulated at the Level of Mitogen-Activated Protein Kinase Activity. J Immunol (2001) 167(6):3339–45. doi: 10.4049/jimmunol.167.6.3339
97. Brace PT, Tezera LB, Bielecka MK, Mellows T, Garay D, Tian S, et al. Mycobacterium Tuberculosis Subverts Negative Regulatory Pathways in Human Macrophages to Drive Immunopathology. PloS Pathog (2017) 13(6):e1006367. doi: 10.1371/journal.ppat.1006367
98. George AL, Bangalore-Prakash P, Rajoria S, Suriano R, Shanmugam A, Mittelman A, et al. Endothelial Progenitor Cell Biology in Disease and Tissue Regeneration. J Hematol Oncol (2011) 4:24. doi: 10.1186/1756-8722-4-24
99. Fox R, Nhan TQ, Law GL, Morris DR, Liles WC, Schwartz SM. PSGL-1 and Mtor Regulate Translation of ROCK-1 and Physiological Functions of Macrophages. EMBO J (2007) 26(2):505–15. doi: 10.1038/sj.emboj.7601522
100. Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, et al. Host-Directed Therapy of Tuberculosis Based on Interleukin-1 and Type I Interferon Crosstalk. Nature (2014) 511(7507):99–103. doi: 10.1038/nature13489
101. Colas RA, Nhat LTH, Thuong NTT, Gómez EA, Ly L, Thanh HH, et al. Proresolving Mediator Profiles in Cerebrospinal Fluid are Linked With Disease Severity and Outcome in Adults With Tuberculous Meningitis. FASEB J (2019) 33(11):13028–39. doi: 10.1096/fj.201901590R
102. Yao C, Narumiya S. Prostaglandin-Cytokine Crosstalk in Chronic Inflammation. Br J Pharmacol (2019) 176(3):337–54. doi: 10.1111/bph.14530
103. Ivanyi J, Zumla A. Nonsteroidal Antiinflammatory Drugs for Adjunctive Tuberculosis Treatment. J Infect Dis (2013) 208(2):185–8. doi: 10.1093/infdis/jit153
104. Kroesen VM, Rodríguez-Martínez P, García E, Rosales Y, Díaz J, Martín-Céspedes M, et al. A Beneficial Effect of Low-Dose Aspirin in a Murine Model of Active Tuberculosis. Front Immunol (2018) 9:798. doi: 10.3389/fimmu.2018.00798
105. Kroesen VM, Groschel MI, Martinson N, Zumla A, Maeurer M, van der Werf TS, et al. Non-Steroidal Anti-Inflammatory Drugs as Host-Directed Therapy for Tuberculosis: A Systematic Review. Front Immunol (2017) 8:772. doi: 10.3389/fimmu.2017.00772
106. Shroff KE, Sainis KB, Sengupta SR, Kamat RS. Role of Antigen-Presenting Cells in Variation in Immunogenicity of Mycobacteria. Clin Exp Immunol (1990) 79(2):285–90. doi: 10.1111/j.1365-2249.1990.tb05192.x
107. Hernández-Pando R, Orozco H, Mancilla R. T-Cell Lung Granulomas Induced by Sepharose-Coupled Mycobacterium Tuberculosis Protein Antigens: Immunosuppressive Phenomena Reversed With Cyclophosphamide and Indomethacin. Immunology (1995) 86(4):506–11.
108. Rangel Moreno J, Estrada García I, De La Luz García Hernández M, Aguilar Leon D, Marquez R, Hernández Pando R. The Role of Prostaglandin E2 in the Immunopathogenesis of Experimental Pulmonary Tuberculosis. Immunology (2002) 106(2):257–66. doi: 10.1046/j.1365-2567.2002.01403.x
109. Dutta NK, Kumar KA, Mazumdar K, Dastidar SG. In Vitro and In Vivo Antimycobacterial Activity of Antiinflammatory Drug, Diclofenac Sodium. Indian J Exp Biol (2004) 42(9):922–7.
110. Hernández-Pando R, Orozco-Esteves H, Maldonado HA, Aguilar-León D, Vilchis-Landeros MM, Mata-Espinosa DA, et al. A Combination of a Transforming Growth Factor-Beta Antagonist and an Inhibitor of Cyclooxygenase is an Effective Treatment for Murine Pulmonary Tuberculosis. Clin Exp Immunol (2006) 144(2):264–72. doi: 10.1111/j.1365-2249.2006.03049.x
111. Byrne ST, Denkin SM, Zhang Y. Aspirin and Ibuprofen Enhance Pyrazinamide Treatment of Murine Tuberculosis. J Antimicrob Chemother (2007) 59(2):313–6. doi: 10.1093/jac/dkl486
112. Byrne ST, Denkin SM, Zhang Y. Aspirin Antagonism in Isoniazid Treatment of Tuberculosis in Mice. Antimicrob Agents Chemother (2007) 51(2):794–5. doi: 10.1128/AAC.01145-06
113. Dutta NK, Mazumdar K, Dastidar SG, Park JH. Activity of Diclofenac Used Alone and in Combination With Streptomycin Against Mycobacterium Tuberculosis in Mice. Int J Antimicrob Agents (2007) 30(4):336–40. doi: 10.1016/j.ijantimicag.2007.04.016
114. Peres-Buzalaf C, de Paula L, Frantz FG, Soares EM, Medeiros AI, Peters-Golden M, et al. Control of Experimental Pulmonary Tuberculosis Depends More on Immunostimulatory Leukotrienes Than on the Absence of Immunosuppressive Prostaglandins. Prostaglandins Leukotrienes Essent Fatty Acids (2011) 85(2):75–81. doi: 10.1016/j.plefa.2011.04.024
115. Vilaplana C, Marzo E, Tapia G, Diaz J, Garcia V, Cardona PJ. Ibuprofen Therapy Resulted in Significantly Decreased Tissue Bacillary Loads and Increased Survival in a New Murine Experimental Model of Active Tuberculosis. J Infect Dis (2013) 208(2):199–202. doi: 10.1093/infdis/jit152
116. Marzo E, Vilaplana C, Tapia G, Diaz J, Garcia V, Cardona PJ. Damaging Role of Neutrophilic Infiltration in a Mouse Model of Progressive Tuberculosis. Tuberc (Edinburgh Scotland) (2014) 94(1):55–64. doi: 10.1016/j.tube.2013.09.004
117. Hortle E, Johnson KE, Johansen MD, Nguyen T, Shavit JA, Britton WJ, et al. Thrombocyte Inhibition Restores Protective Immunity to Mycobacterial Infection in Zebrafish. J Infect Dis (2019) 220(3):524–34. doi: 10.1101/338111
118. Mortensen R, Clemmensen HS, Woodworth JS, Therkelsen ML, Mustafa T, Tonby K, et al. Cyclooxygenase Inhibitors Impair CD4 T Cell Immunity and Exacerbate Mycobacterium Tuberculosis Infection in Aerosol-Challenged Mice. Commun Biol (2019) 2:288–. doi: 10.1038/s42003-019-0530-3
119. Misra UK, Kalita J, Nair PP. Role of Aspirin in Tuberculous Meningitis: A Randomized Open Label Placebo Controlled Trial. J Neurol Sci (2010) 293(1-2):12–7. doi: 10.1016/j.jns.2010.03.025
120. Schoeman JF, Janse van Rensburg A, Laubscher JA, Springer P. The Role of Aspirin in Childhood Tuberculous Meningitis. J Child Neurol (2011) 26(8):956–62. doi: 10.1177/0883073811398132
121. Mai NT, Dobbs N, Phu NH, Colas RA, Thao LT, Thuong NT, et al. A Randomised Double Blind Placebo Controlled Phase 2 Trial of Adjunctive Aspirin for Tuberculous Meningitis in HIV-Uninfected Adults. eLife (2018) 7:e33478. doi: 10.7554/eLife.33478
122. Lee MR, Lee MC, Chang CH, Liu CJ, Chang LY, Zhang JF, et al. Use of Antiplatelet Agents and Survival of Tuberculosis Patients: A Population-Based Cohort Study. J Clin Med (2019) 8(7):923. doi: 10.3390/jcm8070923
123. Wang W, Du Z, Ni M, Wang Z, Liang M, Sheng H, et al. Aspirin Enhances the Clinical Efficacy of Anti-Tuberculosis Therapy in Pulmonary Tuberculosis in Patients With Type 2 Diabetes Mellitus. Infect Dis (2020) 52(10):721–9. doi: 10.1080/23744235.2020.1778177
124. Maitra A, Bates S, Shaik M, Evangelopoulos D, Abubakar I, McHugh TD, et al. Repurposing Drugs for Treatment of Tuberculosis: A Role for non-Steroidal Anti-Inflammatory Drugs. Br Med Bull (2016) 118(1):138–48. doi: 10.1093/bmb/ldw019
125. Schaller A, Sun Z, Yang Y, Somoskovi A, Zhang Y. Salicylate Reduces Susceptibility of Mycobacterium Tuberculosis to Multiple Antituberculosis Drugs. Antimicrob Agents Chemother (2002) 46(8):2636–9. doi: 10.1128/AAC.46.8.2636-2639.2002
126. Denkin S, Byrne S, Jie C, Zhang Y. Gene Expression Profiling Analysis of Mycobacterium Tuberculosis Genes in Response to Salicylate. Arch Microbiol (2005) 184(3):152–7. doi: 10.1007/s00203-005-0037-9
127. University of Stellenbosch. TB-IRIS NSAID Cox-2 Inhibitor Prevention Trial (Clinicaltrials.Gov Identifier: NCT02060006) (2014). Available at: https://clinicaltrials.gov/ct2/show/NCT02060006.
128. Fundació Institut Germans Trias i Pujol. Potential Efficacy and Safety of Using Adjunctive Ibuprofen for XDR-TB Tuberculosis (NSAIDS-XDRTB) (Clinicaltrials.Gov Identifier NCT02781909) (2016). Available at: https://clinicaltrials.gov/ct2/show/NCT02781909.
129. Anne Margarita Dyrhol Riise Therapeutic Vaccination and Immune Modulation. New Treatment Strategies for the MDR Tuberculosis Pandemic (TBCOX2) (Clinicaltrials.Gov Identifier NCT02503839) (2015). Available at: https://clinicaltrials.gov/ct2/show/NCT02503839.
130. Zhang X, Qin J, Zou J, Lv Z, Tan B, Shi J, et al. Extracellular ADP Facilitates Monocyte Recruitment in Bacterial Infection Via ERK Signaling. Cell Mol Immunol (2018) 15(1):58–73. doi: 10.1038/cmi.2016.56
131. Thomas MR, Outteridge SN, Ajjan RA, Phoenix F, Sangha GK, Faulkner RE, et al. Platelet P2Y12 Inhibitors Reduce Systemic Inflammation and its Prothrombotic Effects in an Experimental Human Model. Arterioscler Thromb Vasc Biol (2015) 35(12):2562–70. doi: 10.1161/ATVBAHA.115.306528
Keywords: tuberculosis, platelets, innate immunity, inflammation, lung fibrosis, anti-platelet drugs
Citation: Kirwan DE, Chong DLW and Friedland JS (2021) Platelet Activation and the Immune Response to Tuberculosis. Front. Immunol. 12:631696. doi: 10.3389/fimmu.2021.631696
Received: 20 November 2020; Accepted: 04 May 2021;
Published: 19 May 2021.
Edited by:
Charles Feldman, University of the Witwatersrand, South AfricaReviewed by:
Carmen Judith Serrano, Mexican Social Security Institute (IMSS), MexicoCopyright © 2021 Kirwan, Chong and Friedland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniela E. Kirwan, ZGtpcndhbkBzZ3VsLmFjLnVr
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.