 Chioma Enweasor1
Chioma Enweasor1 Cameron H. Flayer
Cameron H. Flayer Angela Haczku
Angela Haczku- 1UC Davis Lung Center, University of California, Davis, CA, United States
- 2Center for Immunology and Inflammatory Diseases, Division of Rheumatology, Allergy and Immunology, Massachusetts General Hospital, Harvard Medical School, Boston, MA, United States
Despite recent advances in using biologicals that target Th2 pathways, glucocorticoids form the mainstay of asthma treatment. Asthma morbidity and mortality remain high due to the wide variability of treatment responsiveness and complex clinical phenotypes driven by distinct underlying mechanisms. Emerging evidence suggests that inhalation of the toxic air pollutant, ozone, worsens asthma by impairing glucocorticoid responsiveness. This review discusses the role of oxidative stress in glucocorticoid resistance in asthma. The underlying mechanisms point to a central role of oxidative stress pathways. The primary data source for this review consisted of peer-reviewed publications on the impact of ozone on airway inflammation and glucocorticoid responsiveness indexed in PubMed. Our main search strategy focused on cross-referencing “asthma and glucocorticoid resistance” against “ozone, oxidative stress, alarmins, innate lymphoid, NK and γδ T cells, dendritic cells and alveolar type II epithelial cells, glucocorticoid receptor and transcription factors”. Recent work was placed in the context from articles in the last 10 years and older seminal research papers and comprehensive reviews. We excluded papers that did not focus on respiratory injury in the setting of oxidative stress. The pathways discussed here have however wide clinical implications to pathologies associated with inflammation and oxidative stress and in which glucocorticoid treatment is essential.
Introduction: Asthma Phenotypes, Glucocorticoid Resistance, and Oxidative Stress
Asthma is a highly heterogenous disease that can be classified into subsets by a number of different categories. Establishment of the appropriate subsets determines treatment approaches (1, 2). According to severity, asthma has mild, moderate, and severe forms (3–5). Asthma severity worsens during exacerbations associated with oxidative stress, the most common causes of which are viral respiratory infections and indoor/outdoor air pollution, including exposure to O3 (6–11). Severe asthma is often more difficult to treat than the moderate or mild form of the disease (12, 13).
According to the predominant inflammatory cell type in the airways, asthma can be classified as eosinophilic, neutrophilic, mixed, or paucigranulocytic (3–5). Airway epithelial damage leads to oxidative stress, release of pro-inflammatory mediators and influx of both eosinophils and neutrophils (10). Neutrophils are the predominant inflammatory cells in severe asthma exacerbations (2, 5, 14). These cells are poorly controlled by glucocorticoids (15). Whether a causative allergen can be identified, asthma is also categorized as either allergic or non-allergic (16). Allergic (atopic) asthma is characterized by increased levels of IgE, eosinophilia, exhaled nitric oxide (NO), and Th2-type cytokines (16). Such “Th2 high” asthma can generally be treated with glucocorticoids and biologicals targeting the Th2 cytokine pathways (17). Approximately half of asthmatics however suffer from “Th2 low” asthma in which these pathogenic features cannot be identified. Thus, although Th2 low asthma patients are often resistant to corticosteroids, they cannot benefit from biologic treatment targeting the Th2 pathway either (18, 19). Especially in Th2 low asthma, corticosteroid resistance (the inability to increase FEV1 by 15% after a 7-day course of oral corticosteroids at 20 mg/day) (20) remains a significant clinical problem that continues to increase asthma morbidity and mortality (21–23).
The underlying molecular pathways of glucocorticoid resistant asthma are complex and generally associated with impaired expression and function of the glucocorticoid receptor (GR). GR-α, the classical glucocorticoid receptor isoform (24–27) has a dominant-negative inhibitor, GR‐β, that does not bind corticosteroids. Overexpression of GR‐β is due to abnormal activation of proinflammatory signaling pathways with emerging evidence for a contribution of oxidative stress (8, 10, 28–31). Oxidative stress is defined as an imbalance between reactive oxygen species and the capability of the biological system to detoxify the reactive intermediates or to repair the damage caused by oxidative free radicals (32, 33).
The common causes of oxidative stress potentially linked to glucocorticoid resistance in asthma are summarized in Table 1. Amongst the environmental causes our review is focused on inhalational exposure to the toxic air pollutant, ozone (O3) as it was found to be a significant contributor to respiratory illness. Specifically, O3 induces airway hyperreactivity in mouse models of asthma (6, 86, 87, 89, 91, 98–107), in Th2 low asthma in rhesus macaques (94, 108) and in healthy human subjects and patients with asthma and COPD (6, 7, 59, 109–116). Ground-level (tropospheric) O3 is generated by the action of sunlight’s UV rays from precursors (mostly air pollutants containing hydrocarbons, volatile organic compounds [VOC] and nitrogen oxides emitted during fossil fuel combustion). In cities with high O3 levels people had an over 30% increased risk of dying from lung disease (117) and children playing outdoor sports had a three times greater chance of developing asthma (118, 119).

Table 1 Common causes of oxidative stress linked to glucocorticoid resistance in asthma.
Against O3-induced inflammatory injury, the lung mounts immuno-protective mechanisms such as production of the epithelial-cell derived collectin, surfactant protein D (SP-D) (101). Constitutive expression of this molecule in airway epithelial cells is promoted by glucocorticoid dependent transcription (120–122). O3-induced oxidative stress not only destroys the biologically active tertiary molecular structure of SP-D (91, 123–125) but it also diminishes glucocorticoid responsiveness and SP-D expression in airway epithelial cells in vivo and in vitro (122). Below we discuss the potential significance of O3-induced oxidative stress in glucocorticoid responsiveness in asthma.
O3-induced airway inflammation and glucocorticoid resistance
O3 exposure results in accumulation of reactive oxygen species (ROS) most likely through lipid peroxidation processes of the pulmonary surfactant phospholipids (60) and cell membranes (126–128). ROS in turn rapidly activate the release of alarmins IL-1β, IL-6, IL-23, IL-33, TNF-α, and TSLP (Figure 1A) leading to a cascade of proinflammatory changes in structural and immune cells in the respiratory mucosal tissue (106, 116, 122, 129, 131–136). Activation of the RORγt proinflammatory signaling pathway leads to mRNA transcription of the IL-17A and IL-22 genes (Figure 1B) (131, 137–146). The IL-1 family of cytokines together with IL-17A and IL-22 induce influx and activation of neutrophils (129, 130). IL-33 has also been implicated in O3 -induced airway inflammation (106, 129, 132–136). IL-33 transcription as well as release is upregulated by O3 in the lung in a time dependent manner (106, 134). In the absence of IL-33 or the IL-33 receptor (ST2) acute O3-exposure induced epithelial cell injury with protein leak and myeloid cell recruitment and inflammation were enhanced (134). While E-cadherin and zonula occludens 1 and reactive oxygen species expression in neutrophils and airway hyperreactivity were diminished in knockout mice. The enhancement of neutrophil influx was abolished by administration of recombinant IL-33 suggesting a protective role of IL-33 in O3-induced epithelial barrier injury in mice.
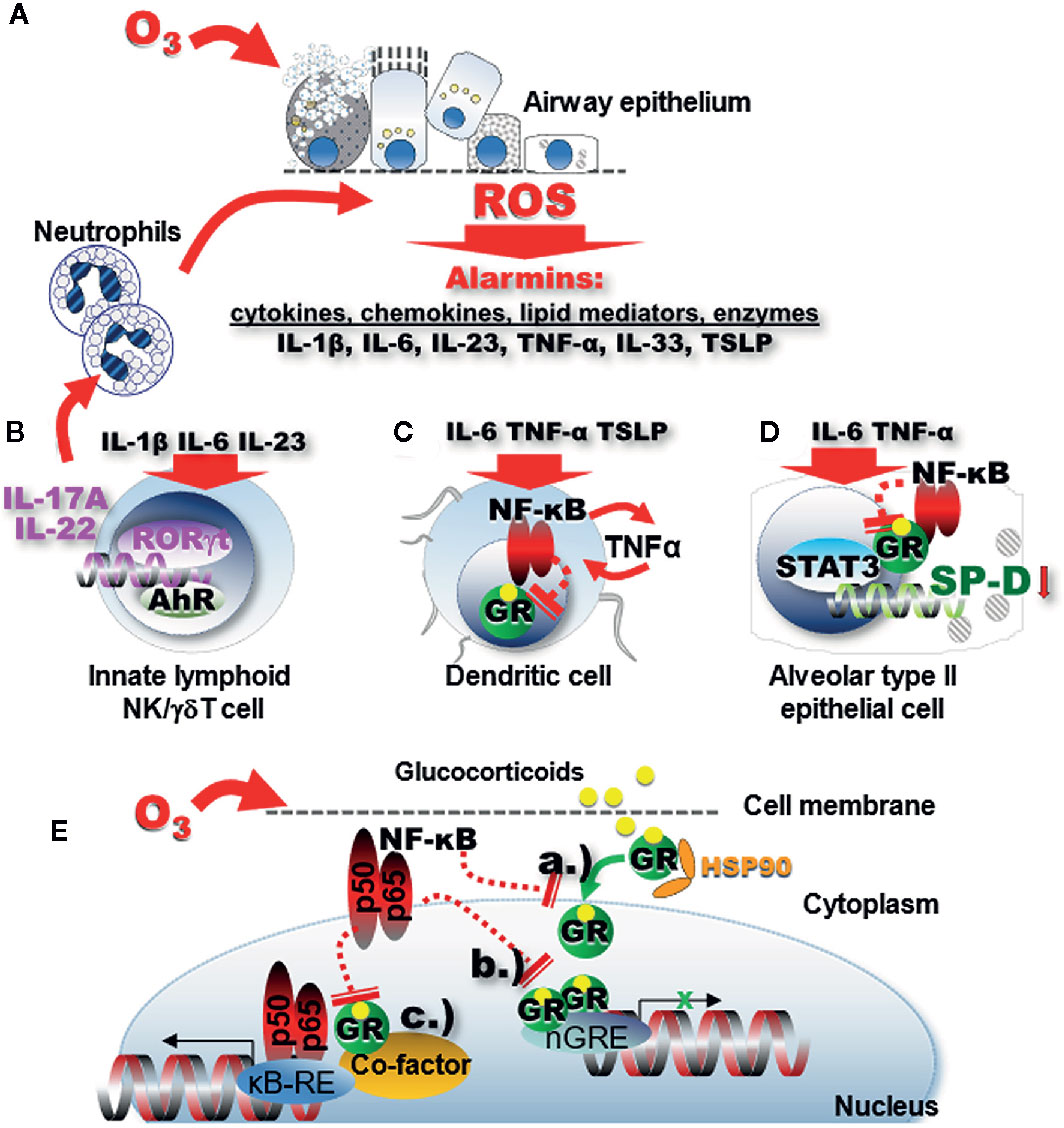
Figure 1 Oxidative stress leads to impaired GR function through proinflammatory signaling. (A) Ozone inhalation generates ROS inducing release of alarmins through lipid peroxidation and proinflammatory activation of immune cells in the respiratory mucosal tissue. (B) IL-1β, IL-6, and IL-23 activate the RORγt proinflammatory signaling pathway that leads to mRNA activation of the IL-17A and IL-22 genes. The IL-1 family of cytokines and the related IL-33 together with IL-17A and IL-22 induce influx and activation of neutrophils (129, 130). In turn, activated neutrophils in the airway mucosal tissue will release more ROS. (C) Proinflammatory signaling activates NF-kB that in turn inhibits expression and function of the GR. Diminished GR function further activates NF-kB forming a vicious proinflammatory cycle. (D) The GR uses non-canonical transactivation of the surfactant protein D gene (sftpd) through STAT3. GR function impairment in alveolar type II epithelial cells leads to inhibition of the immunoprotective SP-D. (E) NF-kB inhibits expression of the GR and interferes with GR function through a.) inhibition of GR nuclear translocation b.) steric hindrance of nGRE binding, and c.) interference with transcription factor “tethering”.
Activated neutrophils in the airway mucosal tissue will release more ROS. Release of alarmins and influx of inflammatory cells into the airways are the pathological hallmark of severe asthma exacerbations (2, 5, 10, 14). In the healthy lung, the primary inflammatory cells recruited to the airways following O3 inhalation are the neutrophilic granulocytes (147, 148). These cells appear in the airways within minutes and accumulate in significant numbers as early as 1–2 h after exposure (89, 106, 149). In healthy human subjects exposed to O3 under experimental conditions, a significant airway neutrophilia was associated with a decrease in lung function (7, 147, 150) indicating the pathological significance of these cells. Interestingly, when O3 exposure is combined with allergic sensitization in mouse models, asthmatic non-human primates (rhesus macaques) and in allergic human subjects, a marked influx of both eosinophilic and neutrophilic granulocytes is observed (10, 94, 101, 106). While neutrophilia in healthy volunteers could be attenuated by fluticasone propionate (147), studies on mice (107), dogs (151) rhesus macaques (152), and asthma patients (153, 154) showed controversial results on the effectiveness of glucocorticoids in inhibiting O3-induced exacerbation of asthmatic airway inflammation. Because asthmatic patients respond to O3 with an enhanced airway neutrophilic influx compared with non-asthmatic controls (155), the observation that neutrophils are poorly responsive to glucocorticoids (15) raises a serious concern related to asthma treatment. Indeed, recent studies demonstrated that O3 impaired the effects of glucocorticoid treatment in a mouse model of allergen-induced asthma in vivo as well as in human cell lines and primary epithelial cells in vitro (58, 122, 156). What are the underlying molecular mechanisms of O3-induced glucocorticoid resistant neutrophilic airway inflammation in asthma?
Role of Aryl Hydrocarbon Receptor (AhR) Signaling, IL-17A, and IL-22 in Glucocorticoid Resistant Asthma
The AhR is an intracellular, small molecule ligand-activated transcription factor that regulates gene expression of inflammation-related genes for myeloid and structural cells. AhR is a sensor of xenobiotic chemicals (such as aromatic hydrocarbons) or endogenous indole derivatives [such as kynureine (157)]. AhR mediates environmental signals and is involved in cell differentiation, cell adhesion, mucus and cytokine production (158–160). Upon ligand binding, the AhR complex translocates into the nucleus and heterodimerizes with AhR Nuclear Translocator (ARNT) to induce gene transcription. AhR is an important activator of the genes encoding cytochrome P450 and the cytokines IL-17A and IL-22. The effects of AhR on cell differentiation (including Th17 or Treg polarization) depend on the nature of the ligand and the local cytokine milieu (161, 162).
There are a number of potential mechanisms through which AhR may contribute to glucocorticoid resistance either as a promoter or as an inhibitor. First, glucocorticoid responsiveness of airway neutrophilia is regulated by the circadian clock molecule BMAL1 (Brain and Muscle ARNT-Like 1 or aryl hydrocarbon receptor nuclear translocator-like protein 1 [ARNTL]) (163, 164). BMAL1 function is strongly affected by environmental stressors (165) that can be mediated by AhR: Following agonist-induced activation, AhR enters the nucleus, where it can form a heterodimer with BMAL1 impairing its normal transcriptional activities (166) and promoting glucocorticoid resistance. Second, AhR interferes with the action of NF-κB, a pro-inflammatory transcription factor and antagonist of glucocorticoid action (see discussion below). For example, NF-κB induces AhR expression, but AhR then regulates NF-κB signaling (159) thereby enabling the glucocorticoid action. Third, by interacting with the function of other transcription factors, AhR promotes IL-22 (RORγt), IL-10, and IL-21 (cMaf) as well as aiolos and its own expression (through STAT3). Through aiolos, AhR inhibits expression of IL-2 (159), an inducer of glucocorticoid resistance (167). Thus, on the one hand AhR promotes Th17 cell differentiation, on the other, it induces Th17 cell plasticity into IL-10 producing protective Tr1 cells. While both IL-17A and IL-22 can elicit airway neutrophilia, IL-22 can also play a protective role when produced during epithelial or tissue damage. Recently, chronic ozone exposure induced lung inflammation, airway hyperresponsiveness and tissue remodeling was reported to be associated with increased tryptophan and lipoxin A4 (activators of AhR), and recruitment of IL-17A and IL-22-expressing cells. T cell-specific AhR deletion enhanced lung inflammation indicating that O3 exposure activates AhR, to control airway inflammation by reduction of IL-22 expression (168).
IL-17A has been identified as a central player in the pathogenesis of severe asthma exacerbations (169). In human severe asthma patients high levels of IL-17A were found in induced sputum and bronchial biopsies (170). IL-6, the cytokine most prominently induced by O3 in the lung (89, 171), and IL-23 (131) directly activate ROR-γt leading to IL-17A expression upon O3 inhalation (Figure 1B). IL-17A signaling controls neutrophilic airway inflammation (172) mainly through stimulating the release of IL-8 and other pro-neutrophilic factors in the airways (131, 137–146) (Figure 1B). The importance of this cytokine in O3 exposure-induced exacerbation of allergic airway inflammation was supported in a mouse model in which significant inhibition of IL-17A gene expression by the combined targeting of p38 MAPK activation and oxidative stress was critical in synergistically attenuating airway hyperresponsiveness, eosinophilic and neutrophilic inflammation (107).
IL-17A was also implicated in glucocorticoid resistant asthma (169, 173). For instance, Th17 cells, the main cellular source of this cytokine, were refractory to inhibition with glucocorticoids in asthma, especially, when IL-17A and IL-22 were co-expressed in these cells (169). Increased counts of dual-positive Th2/Th17 cells detected in the BAL fluid of severe asthma patients, were resistant to dexamethasone-induced cell death (169). Glucocorticoid resistance of IL-17 producing cells may be elicited by an elevated expression level of the mitogen-activated protein-extracellular signal-regulated kinase 1 (MEK1) as the MEK-ERK1/2 signaling pathway was shown to interfere with glucocorticoids (174). In a mouse model of airway inflammation, co-administration of dexamethasone with an anti-IL-17A monoclonal antibody significantly inhibited pro-neutrophilic cytokines and the p38 MAPK, NF-kB signaling pathway and reversed O3-induced glucocorticoid insensitivity (144).
While Th17 cells were identified as the main producers of IL-17A, O3-induced asthma exacerbation in mice did not show T cell activation or migration of T cells into the lung prior to the O3- prompted neutrophil influx (106). These results implied that Th17 cells don’t participate in IL-17A release in the early phases of the O3-response. Mathews et al. proposed that the source of IL-17A in response to acute O3 exposure is the γδ T cell (140). In addition, innate lymphoid cells were shown to be essential and sufficient to elicit development of O3-induced neutrophilia (106) and the ensuing airway hyperresponsiveness in mice. These studies suggest the importance of innate immune players in O3-induced IL-17A pathways. Interestingly, when compared with Th2 cells ILC2s were found to be relatively steroid resistant in severe asthmatics (51, 156), although they were responsive to steroids in eosinophilic respiratory conditions (175). Increased IL-17A expression was associated with a reduction in GR-α but induced expression of GR‐β in asthmatic airway epithelial cells indicating that the steroid insensitivity in severe asthmatics may be a result of a reciprocal regulation of GR-α and GR-β by IL-17 cytokines. Thus, in addition to Th cells, both IL-17A and IL-22 can be produced by ILC3, γδ T and NK cells, after stimulation with IL-1β, TGF-β, IL-6, or IL-23 and the transcription factor RORγt (168). Figure 1B illustrates that IL-17A-mediated neutrophilia in response to oxidative stress feeds back to a vicious cycle by releasing additional ROS into the lung tissue. Further, neutrophils have high constitutive GR‐β expression that may help them resist apoptosis in response to corticosteroid treatment (25). Taken together, oxidative stress-induced IL-17A contributes to glucocorticoid resistance due to an increased activation of phosphokinase signaling pathways, reduction of GR-α, increase of GR-β in IL-17A producing innate immune and T cells thereby promoting neutrophilia.
Role of the Glucocorticoid Receptor (GR) in O3-Induced Glucocorticoid Resistance
Glucocorticoids have significant anti-inflammatory, immunosuppressive and immunomodulatory effects and remain the mainstay of asthma treatment (176). A subset of patients however is refractory to glucocorticoids (12, 177, 178), making their asthma difficult to control (179). Glucocorticoid insensitivity in rare cases, can be a primary genetic trait, but more commonly, it is acquired during inflammatory exacerbations (176). Constitutive GR expression is essential for an adequate glucocorticoid action. Corticosteroid insensitivity can be mediated by decreased function and expression of the GR. Expression of the GR gene (NR3C1) is regulated by complex transcriptional and post translational processes that are modified by airway inflammation (169, 180, 181).
How does the GR work? Glucocorticoids go through the cell membrane and bind to the GR that rearranges the stable GR-heat-shock protein (HSP)90 complex into an activated glucocorticoid-GR complex that translocates to the nucleus (Figure 1E). When two of these complexes form homodimers, they bind to specific glucocorticoid response elements (GRE) in the DNA sequence. GRE are located in the promoter regions of glucocorticoid-responsive genes (176). After the recruitment of co-activators or co-repressors, the GR modulates the rate of gene transcription by transactivation or transrepression. Transactivation is triggered by GRE which acts in “trans”, i.e., intermolecularly (this may be considered the opposite of “cis”-acting i.e., intramolecular). On the other hand, transrepression (i.e., inhibition) is the activity of a second transcription factor through protein-protein interaction [reviewed by (182, 183)] (Figure 1E). The repressed molecule is usually a transcription factor whose function is to up-regulate gene transcription. Transrepression was first observed in the action of the GR to inhibit the transcriptional promoting activity of the proinflammatory transcription factors AP-1 and NF-κB. Transactivation and transrepression are both important in mediating the anti-inflammatory effects of glucocorticoids. Transactivation GRE up-regulates anti-inflammatory genes such as the NF-κB inhibitor IκBα, the AP-1 inhibitor glucocorticoid-inducible leucine zipper (GILZ) and IL-10. In a mechanism called “tethering” the GR can also interact with other transcription factors (NF-κB, AP-1, signal transducers, and activators of transcription [STAT] or CAAT Enhancer Binding Protein (C/EBP)], and modulate activation of target genes in a monomeric form (184–186). The activated monomeric GR binds to HDAC (histone deacetylase) and interferes with the activation of the κB responsive element (κB-RE) by p65 and p50 heterodimer subunits of NF-κB. Although the main function of HDACs is to modify histones and chromatin structure, HDAC isoforms can have different regulatory functions in the cytoplasm and nucleus. For instance, HDAC1 is considered to be a transcriptional co-activator (187). On the other hand, impairment of HDAC2 function is implicated in corticosteroid resistance of asthmatic and COPD patients (58, 97). Oxidative stress can lead to the reduction of HDAC2 via activation of phosphoinositide 3 (PI3K). PI3K induces nitric oxide levels in the asthmatic airways that further hinders the functional ability of HDAC2, as reported in asthmatic smokers (178). Moreover, treatment with theophylline, a medication that restores HDAC2 activity, glucocorticoid sensitivity is also restored (178).
GR expression levels are regulated by transcriptional and post translational mechanisms such as kinase-dependent phosphorylation as well as by homologous ligand down-regulation (by GR agonists) that can be significantly modified by increased NF-κB expression during O3-induced oxidative stress (180). Phosphorylation-dephosphorylation is also important in the function of the transcription regulator enzyme, RNA polymerase II. The GR inhibits transcription activation through dephosphorylating RNA polymerase II (188).
Enhanced expression of NF-κB in the nuclear fraction of immune cells paralleled with an impairment of GR nuclear translocation, DNA binding and a decrease in the expression of GR (70). Mutual transrepression has been demonstrated between the GR and NF-κB as well as AP-1. In the highly inflamed airways during oxidative-stress related asthma exacerbation excessive NF-κB and AP-1 activation could be responsible for impaired GR function (27, 176, 189–191). NF-kB not only hinders GR nuclear translocation and directly interferes with GRE-mediated gene transactivation but it can also indirectly “tether” to the GR transcription complex. Importantly, while GR expression is ubiquitous, it is differentially regulated in individual cell types (192). For example cell type-specific increases in NF-κB, in airway epithelial and dendritic cells (Figures 1C, D), upon O3 inhalation, may significantly inhibit GR expression and modulate allergic airway inflammation [reviewed in (72)].
Glucocorticoid resistance linked to oxidative stress through defective nuclear translocation and GRE binding (Figure 1E) (reviewed by Spiers et al. (28, 193, 194). That nuclear translocation of the GR is susceptible to highly pro-oxidative environments was shown by a cultured, fluorescently labeled chimeric GR. Okamoto and colleagues (193) demonstrated that nuclear translocation of GR following acute dexamethasone treatment was impaired in the presence of hydrogen peroxide. This effect was reduced by administration of exogenous antioxidants or by replacing serine for a redox-sensitive cysteine residue. The dissociation of heat shock proteins from the cytosolic GR is also impaired in a pro-oxidative environment, indicating that there may be multiple pathways involved in the cellular response to glucocorticoids (193, 194). Thus, a balanced oxidative state is critical for normal function of the GR.
GR function is also reduced when the molecule is phosphorylated. For example, the proinflammatory signaling molecule, p38MAPK can phosphorylate the GR that blocks nuclear translocation and the ability to bind to DNA leading to decreased ability of the GR to regulate transcription of anti-inflammatory genes (178). Similarly, activation of the MEK-ERK1/2 pathway was shown to antagonize the inhibitory action of glucocorticoids in Th17 cells (174).
Additional mechanisms involve increased expression of GRβ, an isomer of GRα that suppresses the ability of GRα to bind to GRE and induce anti-inflammatory genes. Increase in GRβ is caused by a rise in pro-inflammatory cytokines or through super-antigen such as staphylococcus enterotoxin-induced activation of T lymphocytes (176). Reduced GR expression was reported in asthmatic and COPD patients with insensitivity to corticosteroid treatment (189–191). GR expression can be reduced by homologous ligand down-regulation (upon administration of GR agonists) or other pathways such as transrepression of the GRα isoform by NF-κB in the inflamed tissue (27). It is unclear whether low levels of GR mRNA are due to suppression of promoter activation, decreased mRNA stability, or both (27) during oxidative stress. Importantly, the expression and function of the human GR is distinct from other species. For example, it is unclear whether transrepression of the GRα by NF-κB plays a role in corticosteroid resistance in mice as existence of the dominant negative GRβ isoform [responsible for glucocorticoid resistance (195)] could not be demonstrated in these rodents. Ligand-induced GR down regulation is seen in various tissues and cell types except in T cells (27) suggesting that innate immune and tissue cells may be more susceptible for glucocorticoid resistance. Further studies are still needed to identify the cell types ultimately responsible for mediating the effects of corticosteroid insensitivity in the lung.
Airway Epithelial Cell Function Is Constitutively Regulated by Endogenous Glucocorticoids
Alveolar type II epithelial cells are the major source of pulmonary surfactant, as well as the immunoprotective lung collectins, surfactant protein (SP)-A and SP-D. SP-D, a glucocorticoid-dependent airway epithelial cell product is critical in the maintenance of pulmonary immune homeostasis (196–203). Individual susceptibility to the effects of O3 exposure suggests that inflammatory responsiveness is genetically regulated (204). This is supported by strain dependence of the inflammatory response to O3 observed in mice (205–207). A failure of protective immune mechanisms likely plays an important role in shaping the O3 effects in the lung. A differential ability of Balb/c and C57BL/6 mice to respond to allergen (208) or O3 (89), was inversely proportionate to the amount of SP-D in the lung of these mouse strains (89, 209). Further, when compared to wild-type C57BL/6 mice, the naturally low SP-D producer Balb/c or the SP-D knockout (C67BL/6) animals displayed increased susceptibility to and a prolonged recovery period from airway inflammation after allergen or O3 exposure (89, 210–212).
In addition, O3-induced exacerbation of Th2-type airway inflammation in allergen challenged mice was associated with the appearance of abnormal, lower order oligomeric molecular formations of SP-D. Interestingly, in asthmatic rhesus macaques, O3 induced de-oligomerization of SP-D was restored by treatment with a flaxseed derivative anti-oxidant (94). Thus, oxidative damage can cause conformational change in the SP-D molecule resulting in a potential loss of its immunoprotective function (91, 213). Glucocorticoids were shown to be necessary for expression of SP-D in epithelial cells (120, 121, 214, 215). Interestingly however, there is no glucocorticoid response elements in the promoter region of the SP-D gene (sftpd). This DNA region however contains an evolutionarily conserved STAT3/6 response element in a prominent proximal location. IL-4/IL-13 (activators of STAT6) as well as IL-6 (activator of STAT3) directly upregulated SP-D synthesis in airway epithelial cells in vitro and in mice in vivo (89, 210). Between SP-D and the STAT3/6-activating IL-6 (89) as well as Th2 cytokines IL-4/IL-13 (216), respectively, negative regulatory feedback mechanisms were identified. In these, inflammatory transcriptional signaling by STAT3/6 would upregulate SP-D synthesis and release. In turn, increased amounts of this protein in the airways would suppress further inflammation through inhibition of proinflammatory cytokine transcription. Lastly, there are indications that STAT3 can be directly phosphorylated by H2O2 (the molecular product of O3 when mixed in water) in airway epithelial cells in vitro (217). O3 and glucocorticoid treatment had antagonistic effects on SP-D expression and function in the lung, with O3 inhibiting glucocorticoid-induced sftpd transcription in vivo in mice and in vitro, in human airway epithelial cell cultures. These results indicated that glucocorticoids sustain vital functions in airway epithelium such as SP-D production, aimed at promoting immune homeostasis. This function is directly perturbed by O3-induced oxidative stress.
Antioxidant Approach for Asthma Treatment
As we discussed, there is a marked role for oxidative stress in asthma, especially in severe exacerbations associated with glucocorticoid resistance. Although this fact has been well established, and according to a WHO estimate, more than 80% of the Earth’s inhabitants used Traditional Medicine/Complementary and Alternative Medicine (TCAM) for their primary healthcare needs (218), a large variety of nutritional, pharmacological, and environmental antioxidant clinical approaches to asthma treatment have been controversial and generally disappointing (33).
Emerging evidence from experimental models shows that successful targeting of oxidative stress in asthma is dependent on activation of NRF2 (Nuclear factor-erythroid 2 related factor 2). NRF2 is an ubiquitous master transcription factor that works through antioxidant response elements (AREs) to induce antioxidant enzyme and cytoprotective protein mRNA expression. Under baseline, “unstressed” conditions, the Kelch-like ECH-associated protein 1 (Keap1) inhibits cellular NRF2 in the cytoplasm and promotes its proteasomal degradation. NRF2 is activated by diverse stimuli such as oxidants, pro-oxidants, antioxidants, and chemopreventive agents (219). NRF2 induces cellular rescue pathways against oxidative injury, abnormal inflammatory and immune responses, apoptosis, and carcinogenesis (219). In a mouse model of asthma Sussan and colleagues used cell-specific activation of NRF2 in club cells of the airway epithelium and found a significantly reduced allergen-induced airway hyperresponsiveness, inflammation, mucus, Th2 cytokine secretion, oxidative stress, and airway leakiness and increased airway levels of tight junction proteins zonula occludens-1 and E-cadherin on the epithelial cell surface. Pharmacological activation of NRF2 during allergen challenge reduced allergic inflammation and airway hyperresponsiveness (220). Administration of the ROS inhibitors, N-acetyl cysteine or apocynin in a mouse model, had no effect on acute injury and lung inflammation but GR-1 antibody depletion of neutrophils significantly reduced ROS production in neutrophils, epithelial cells, interstitial macrophages, and eosinophils (134). In the same study, administration of IL-33 attenuated, while absence of IL-33/ST2 signaling enhanced O3-induced airway inflammation and oxidative stress, and diminished zonula occludens-1 and E-cadherin expression highlighting the complex role this cytokine plays during lung injury (134).
In a different study, activation of NRF2 decreased the viability of the wild-type but not of the NRF2-deficient ILC2s resembling the pro-apoptotic effect of glucocorticoids albeit without the involvement of caspase 3-dependent apoptosis or necroptosis. In mice NRF2 activation decreased the number of pulmonary ILC2s and eosinophils suggesting NRF2 activation as a potential alternative strategy for steroid-resistant allergic inflammation (29). Lack of NRF2 in the lung exacerbates oxidative insults including supplemental respiratory therapy (e.g., hyperoxia, mechanical ventilation), cigarette smoke, allergen, virus, bacterial endotoxin and other inflammatory agents (e.g., carrageenin), environmental pollution (e.g., particles, O3), and bleomycin (219, 221). Bioinformatic studies elucidated functional AREs and NRF2-directed genes that are critical components of signaling mechanisms in pulmonary protection by NRF2. Association of loss of function with promoter polymorphisms in NRF2 or somatic and epigenetic mutations in KEAP1 and NRF2 has been found in cohorts of patients with acute lung injury/acute respiratory distress syndrome or lung cancer (219).
The role of non-enzymatic antioxidants was studied in a multiple linear regression analysis that revealed significant associations of vitamin C, vitamin E, beta-cryptoxanthin, lutein/zeaxanthin, beta-carotene, and retinol with FEV1% in a large population study (93). Since removal of ROS and RNS from the cells by antioxidants could impair the action of NRF2, one might speculate that antioxidant vitamin administration with simultaneous NRF2 activation could be beneficial in oxidative stress-induced asthma exacerbation, which is a highly proinflammatory condition. To this effect, a dietary flaxseed compound (LGM2605) a synthetic form of the lignan secoisolariciresinol digluconate (SDG) was identified as both an antioxidant and an activator of NRF2. SDG demonstrated strong protective actions against different sources of oxidative damage (222, 223) supporting the potential for antioxidant approaches for asthma treatment. A cohort of asthmatic macaques from the California National Primate Research Center was identified to naturally develop airway hyperresponsiveness (224). These animals display no overt airway inflammation or Th2 cell activation and their peripheral blood mononuclear cells are unresponsive to glucocorticoids (224). Thus, these animals represent “Th2 low” glucocorticoid resistant asthmatic patients and are therefore uniquely poised for investigation of novel alternative or adjuvant approaches to glucocorticoid treatment. A 7-days treatment with LGM2605 of these macaques that received a single exposure to O3 or air (as control) not only prevented the O3-induced exacerbation of airway hyperresponsiveness but also significantly improved baseline lung function (94). These studies highlight the significance of oxidative stress in the effect of O3 on airway hyperresponsiveness and support the idea that anti-oxidant treatment may be beneficial in glucocorticoid resistant, Th2 low asthma.
Conclusions
Severe glucocorticoid resistant asthma continues to increase morbidity and mortality despite the advent of new powerful biological treatments that target proinflammatory cytokines. Scientific and clinical evidence is emerging that alternative and adjuvant therapeutic approaches could significantly contribute to reducing and/or controlling severe asthmatic symptoms. Harnessing antioxidant mechanisms may have a special importance in this effort as oxidative stress has been clearly demonstrated to worsen steroid resistance in severe asthma. The pathways we discussed here are however widely applicable to clinical conditions associated with inflammation and oxidative stress and in which glucocorticoid treatment is essential. One recent example of this is the wide variability of effectiveness observed by dexamethasone treatment of severe COVID-19 patients (225–227). Our assessment of the literature raised a number of interesting questions that require future clarifications. For example, what is the importance of the different cell types in mediating glucocorticoid resistance in asthma? Does the nature of oxidative stress depend on its etiology? What role do AhR-related mechanisms play and how does transcriptional regulation of the circadian clock figure into glucocorticoid responsiveness? Is it possible to increase expression and the protective function of molecules like SP-D? What are the effects of simultaneous molecular targeting of oxidative stress, inflammation, and NRF2 pathways? How feasible it is to translate experimental data to human studies and ultimately to clinical application? Greater understanding of how oxidative stress affects asthma and steroid resistance may lead to novel therapies that could improve the lives of millions.
Author Contributions
CF drafted the ozone-related sections. CE drafted the glucocorticoid resistance-related sections. AH revised the draft, edited, and finalized the paper. All authors contributed to the article and approved the submitted version.
Funding
T32ES007059 (CF) and T32 HL007013 (CF); P30ES023513 Pilot Grant (AH), R21AI116121 (AH), R41AI132012, and 2R42AI132012 (AH); T32HL116275 (CF).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Lefaudeux D, De Meulder B, Loza MJ, Peffer N, Rowe A, Baribaud F, et al. U-BIOPRED clinical adult asthma clusters linked to a subset of sputum omics. J Allergy Clin Immunol (2017) 139(6):1797–807. doi: 10.1016/j.jaci.2016.08.048
2. Barnes PJ. Targeting cytokines to treat asthma and chronic obstructive pulmonary disease. Nat Rev Immunol (2018) 18(7):454–66. doi: 10.1038/s41577-018-0006-6
3. Chung KF, Wenzel SE, Brozek JL, Bush A, Castro M, Sterk PJ, et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur Respir J (2014) 43(2):343–73. doi: 10.1183/09031936.00202013
4. Chung KF. Managing severe asthma in adults: lessons from the ERS/ATS guidelines. Curr Opin Pulm Med (2015) 21(1):8–15. doi: 10.1097/MCP.0000000000000116
5. Chung KF. Asthma phenotyping: a necessity for improved therapeutic precision and new targeted therapies. J Intern Med (2016) 279(2):192–204. doi: 10.1111/joim.12382
6. Peden DB. The role of oxidative stress and innate immunity in O(3) and endotoxin-induced human allergic airway disease. Immunol Rev (2011) 242(1):91–105. doi: 10.1111/j.1600-065X.2011.01035.x
7. Alexis NE, Carlsten C. Interplay of air pollution and asthma immunopathogenesis: a focused review of diesel exhaust and ozone. Int Immunopharmacol (2014) 23(1):347–55. doi: 10.1016/j.intimp.2014.08.009
8. Jesenak M, Zelieskova M, Babusikova E. Oxidative Stress and Bronchial Asthma in Children-Causes or Consequences? Front Pediatr (2017) 5:162. doi: 10.3389/fped.2017.00162
9. Chau-Etchepare F, Hoerger JL, Kuhn BT, Zeki AA, Haczku A, Louie S, et al. Viruses and non-allergen environmental triggers in asthma. J Invest Med (2019) 67(7):1029–41. doi: 10.1136/jim-2019-001000
10. Bontinck A, Maes T, Joos G. Asthma and air pollution: recent insights in pathogenesis and clinical implications. Curr Opin Pulm Med (2020) 26(1):10–9. doi: 10.1097/MCP.0000000000000644
11. Feddema JJ, Claassen E. Prevalence of viral respiratory infections amongst asthmatics: Results of a meta-regression analysis. Respir Med (2020) 173:106020. doi: 10.1016/j.rmed.2020.106020
12. Barnes PJ. Therapeutic approaches to asthma-chronic obstructive pulmonary disease overlap syndromes. J Allergy Clin Immunol (2015) 136(3):531–45. doi: 10.1016/j.jaci.2015.05.052
13. Nabe T. Steroid-Resistant Asthma and Neutrophils. Biol Pharm Bull (2020) 43(1):31–5. doi: 10.1248/bpb.b19-00095
14. Busse WW. A role for neutrophils in asthma exacerbations. Nat Med (2017) 23(6):658–59. doi: 10.1038/nm.4351
15. Bruijnzeel PL, Uddin M, Koenderman L. Targeting neutrophilic inflammation in severe neutrophilic asthma: can we target the disease-relevant neutrophil phenotype? J Leukoc Biol (2015) 98(4):549–56. doi: 10.1189/jlb.3VMR1214-600RR
16. Pakkasela J, Ilmarinen P, Honkamaki J, Tuomisto LE, Andersen H, Piirila P, et al. Age-specific incidence of allergic and non-allergic asthma. BMC Pulm Med (2020) 20(1):9. doi: 10.1186/s12890-019-1040-2
17. Peters MC, Wenzel SE. Intersection of biology and therapeutics: type 2 targeted therapeutics for adult asthma. Lancet (2020) 395(10221):371–83. doi: 10.1016/S0140-6736(19)33005-3
18. Fahy JV. Type 2 inflammation in asthma–present in most, absent in many. Nat Rev Immunol (2015) 15(1):57–65. doi: 10.1038/nri3786
19. Fajt ML, Wenzel SE. Asthma phenotypes and the use of biologic medications in asthma and allergic disease: the next steps toward personalized care. J Allergy Clin Immunol (2015) 135(2):299–310; quiz 11. doi: 10.1016/j.jaci.2014.12.1871
20. Nimmagadda SR, Spahn JD, Leung DY, Szefler SJ. Steroid-resistant asthma: evaluation and management. Ann Allergy Asthma Immunol (1996) 77(5):345–55; quiz 55-6. doi: 10.1016/S1081-1206(10)63332-7
21. Liang TZ, Chao JH. Inhaled Corticosteroids. StatPearls. Treasure Island (FL): StatPearls Publishing (2020).
22. Tripple JW, Ameredes BT, Calhoun WJ. Outpatient Management of Chronic Asthma in 2020. JAMA J Am Med Assoc (2020) 323(6):561–62. doi: 10.1001/jama.2019.19986
23. Vervloet M, van Dijk L, Spreeuwenberg P, Price D, Chisholm A, Van Ganse E, et al. The Relationship Between Real-World Inhaled Corticosteroid Adherence and Asthma Outcomes: A Multilevel Approach. J Allergy Clin Immunol Pract (2020) 8(2):626–34. doi: 10.1016/j.jaip.2019.09.003
24. Oakley RH, Jewell CM, Yudt MR, Bofetiado DM, Cidlowski JA. The dominant negative activity of the human glucocorticoid receptor beta isoform. Specificity and mechanisms of action. J Biol Chem (1999) 274(39):27857–66. doi: 10.1074/jbc.274.39.27857
25. Strickland I, Kisich K, Hauk PJ, Vottero A, Chrousos GP, Klemm DJ, et al. High constitutive glucocorticoid receptor beta in human neutrophils enables them to reduce their spontaneous rate of cell death in response to corticosteroids. J Exp Med (2001) 193(5):585–93. doi: 10.1084/jem.193.5.585
26. Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. Proc Natl Acad Sci U S A (2001) 98(12):6865–70. doi: 10.1073/pnas.121455098
27. Schaaf MJ, Cidlowski JA. Molecular mechanisms of glucocorticoid action and resistance. J Steroid Biochem Mol Biol (2002) 83(1-5):37–48. doi: 10.1016/S0960-0760(02)00263-7
28. Spiers JG, Chen HJ, Sernia C, Lavidis NA. Activation of the hypothalamic-pituitary-adrenal stress axis induces cellular oxidative stress. Front Neurosci (2014) 8:456. doi: 10.3389/fnins.2014.00456
29. Nagashima R, Kosai H, Masuo M, Izumiyama K, Noshikawaji T, Morimoto M, et al. Nrf2 Suppresses Allergic Lung Inflammation by Attenuating the Type 2 Innate Lymphoid Cell Response. J Immunol (2019) 202(5):1331–39. doi: 10.4049/jimmunol.1801180
30. Decrue F, Gorlanova O, Usemann J, Frey U. Lung functional development and asthma trajectories. Semin Immunopathol (2020) 42(1):17–27. doi: 10.1007/s00281-020-00784-2
31. Fuertes E, van der Plaat DA, Minelli C. Antioxidant genes and susceptibility to air pollution for respiratory and cardiovascular health. Free Radic Biol Med (2020) 151:88–98. doi: 10.1016/j.freeradbiomed.2020.01.181
32. Betteridge DJ. What is oxidative stress? Metabolism (2000) 49(2 Suppl 1):3–8. doi: 10.1016/S0026-0495(00)80077-3
33. Sahiner UM, Birben E, Erzurum S, Sackesen C, Kalayci O. Oxidative stress in asthma: Part of the puzzle. Pediatr Allergy Immunol (2018) 29(8):789–800. doi: 10.1111/pai.12965
34. Castanhinha S, Sherburn R, Walker S, Gupta A, Bossley CJ, Buckley J, et al. Pediatric severe asthma with fungal sensitization is mediated by steroid-resistant IL-33. J Allergy Clin Immunol (2015) 136(2):312–22.e7. doi: 10.1016/j.jaci.2015.01.016
35. Erzurum SC. New Insights in Oxidant Biology in Asthma. Ann Am Thorac Soc (2016) 13 Suppl 1:S35–9. doi: 10.1513/AnnalsATS.201506-385MG
36. Van Cauwenberge P, Van Zele T, Bachert C. Chronic rhinonsinusitis and nasal polyposis: the etiopathogenesis revealed? Verh K Acad Geneeskd Belg (2008) 70(5-6):305–22.
37. Liang Z, Zhang Q, Thomas CM, Chana KK, Gibeon D, Barnes PJ, et al. Impaired macrophage phagocytosis of bacteria in severe asthma. Respir Res (2014) 15:72. doi: 10.1186/1465-9921-15-72
38. Schroeder BO, Ehmann D, Precht JC, Castillo PA, Kuchler R, Berger J, et al. Paneth cell alpha-defensin 6 (HD-6) is an antimicrobial peptide. Mucosal Immunol (2015) 8(3):661–71. doi: 10.1038/mi.2014.100
39. Harrison OJ, Linehan JL, Shih HY, Bouladoux N, Han SJ, Smelkinson M, et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science (2019) 363(6422):eaat6280. doi: 10.1126/science.aat6280
40. Mileva M, Bakalova R, Tancheva L, Galabov S. Effect of immobilization, cold and cold-restraint stress on liver monooxygenase activity and lipid peroxidation of influenza virus-infected mice. Arch Toxicol (2002) 76(2):96–103. doi: 10.1007/s00204-001-0306-6
41. Mileva M, Marazova K, Galabov AS. Gastric mucosal lesions in influenza virus infected mice: role of gastric lipid peroxidation. Methods Findings Exp Clin Pharmacol (2003) 25(7):521–4. doi: 10.1358/mf.2003.25.7.778090
42. Garcia GL, Valenzuela A, Manzoni T, Vaughan AE, Lopez CB. Distinct Chronic Post-Viral Lung Diseases upon Infection with Influenza or Parainfluenza Viruses Differentially Impact Superinfection Outcome. Am J Pathol (2020) 190(3):543–53. doi: 10.1016/j.ajpath.2019.11.003
43. Mattos MS, Ferrero MR, Kraemer L, Lopes GAO, Reis DC, Cassali GD, et al. CXCR1 and CXCR2 Inhibition by Ladarixin Improves Neutrophil-Dependent Airway Inflammation in Mice. Front Immunol (2020) 11:566953. doi: 10.3389/fimmu.2020.566953
44. Castro SM, Guerrero-Plata A, Suarez-Real G, Adegboyega PA, Colasurdo GN, Khan AM, et al. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am J Respir Crit Care Med (2006) 174(12):1361–9. doi: 10.1164/rccm.200603-319OC
45. Mochizuki H, Todokoro M, Arakawa H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation (2009) 32(4):252–64. doi: 10.1007/s10753-009-9128-0
46. Mata M, Martinez I, Melero JA, Tenor H, Cortijo J. Roflumilast inhibits respiratory syncytial virus infection in human differentiated bronchial epithelial cells. PLoS One (2013) 8(7):e69670. doi: 10.1371/journal.pone.0069670
47. Yoshida S, Noguchi A, Kikuchi W, Fukaya H, Igarashi K, Takahashi T. Elevation of Serum Acid Sphingomyelinase Activity in Children with Acute Respiratory Syncytial Virus Bronchiolitis. Tohoku J Exp Med (2017) 243(4):275–81. doi: 10.1620/tjem.243.275
48. Koziol-White CJ, Cooper PR, Lee W, Gern JE, Haczku A, Panettieri RA. Rhinovirus 16 (RV16) Induces Airway Hyperresponsiveness (AHR) and Differential Mediator Release in Human lung slices ex vivo. J Allergy Clin Immunol (2011) 127(2):Ab55–5. doi: 10.1016/j.jaci.2010.12.228
49. Korfhagen TR, Kitzmiller J, Chen G, Sridharan A, Haitchi HM, Hegde RS, et al. SAM-pointed domain ETS factor mediates epithelial cell-intrinsic innate immune signaling during airway mucous metaplasia. Proc Natl Acad Sci U S A (2012) 109(41):16630–5. doi: 10.1073/pnas.1208092109
50. Han M, Hong JY, Jaipalli S, Rajput C, Lei J, Hinde JL, et al. IFN-gamma Blocks Development of an Asthma Phenotype in Rhinovirus-Infected Baby Mice by Inhibiting Type 2 Innate Lymphoid Cells. Am J Respir Cell Mol Biol (2017) 56(2):242–51. doi: 10.1165/rcmb.2016-0056OC
51. Liu S, Verma M, Michalec L, Liu W, Sripada A, Rollins D, et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J Allergy Clin Immunol (2018) 141(1):257–68.e6. doi: 10.1016/j.jaci.2017.03.032
52. Ortega H, Nickle D, Carter L. Rhinovirus and asthma: Challenges and opportunities. Rev Med Virol (2020) 20:e2193. doi: 10.1002/rmv.2193
53. Bakadia BM, Boni BOO, Ahmed AAQ, Yang G. The impact of oxidative stress damage induced by the environmental stressors on COVID-19. Life Sci (2020) 26:118653. doi: 10.1016/j.lfs.2020.118653
54. Laforge M, Elbim C, Frere C, Hemadi M, Massaad C, Nuss P, et al. Tissue damage from neutrophil-induced oxidative stress in COVID-19. Nat Rev Immunol (2020) 20(9):515–16. doi: 10.1038/s41577-020-0407-1
55. Mrityunjaya M, Pavithra V, Neelam R, Janhavi P, Halami PM, Ravindra PV. Immune-Boosting, Antioxidant and Anti-inflammatory Food Supplements Targeting Pathogenesis of COVID-19. Front Immunol (2020) 11:570122. doi: 10.3389/fimmu.2020.570122
56. Thomas T, Stefanoni D, Reisz JA, Nemkov T, Bertolone L, Francis RO, et al. COVID-19 infection alters kynurenine and fatty acid metabolism, correlating with IL-6 levels and renal status. JCI Insight (2020) 5(14):e140327. doi: 10.1172/jci.insight.140327
57. Holguin F. Oxidative stress in airway diseases. Ann Am Thorac Soc (2013) 10 Suppl:S150–7. doi: 10.1513/AnnalsATS.201305-116AW
58. Milara J, Navarro A, Almudever P, Lluch J, Morcillo EJ, Cortijo J. Oxidative stress-induced glucocorticoid resistance is prevented by dual PDE3/PDE4 inhibition in human alveolar macrophages. Clin Exp Allergy (2011) 41(4):535–46. doi: 10.1111/j.1365-2222.2011.03715.x
59. Reid CE, Considine EM, Watson GL, Telesca D, Pfister GG, Jerrett M. Associations between respiratory health and ozone and fine particulate matter during a wildfire event. Environ Int (2019) 129:291–98. doi: 10.1016/j.envint.2019.04.033
60. da Costa Loureiro L, da Costa Loureiro L, Gabriel-Junior EA, Zambuzi FA, Fontanari C, Sales-Campos H, et al. Pulmonary surfactant phosphatidylcholines induce immunological adaptation of alveolar macrophages. Mol Immunol (2020) 122:163–72. doi: 10.1016/j.molimm.2020.04.010
61. Reid CE, Brauer M, Johnston FH, Jerrett M, Balmes JR, Elliott CT. Critical Review of Health Impacts of Wildfire Smoke Exposure. Environ Health Perspect (2016) 124(9):1334–43. doi: 10.1289/ehp.1409277
62. Pratt JR, Gan RW, Ford B, Brey S, Pierce JR, Fischer EV, et al. A national burden assessment of estimated pediatric asthma emergency department visits that may be attributed to elevated ozone levels associated with the presence of smoke. Environ Monit Assess (2019) 191(Suppl 2):269. doi: 10.1007/s10661-019-7420-5
63. Reid CE, Maestas MM. Wildfire smoke exposure under climate change: impact on respiratory health of affected communities. Curr Opin Pulm Med (2019) 25(2):179–87. doi: 10.1097/MCP.0000000000000552
64. Li XN, Ma LY, Ji H, Qin YH, Jin SS, Xu LX. Resveratrol protects against oxidative stress by activating the Keap-1/Nrf2 antioxidant defense system in obese-asthmatic rats. Exp Ther Med (2018) 16(6):4339–48. doi: 10.3892/etm.2018.6747
65. Lynch SV, Boushey HA. The microbiome and development of allergic disease. Curr Opin Allergy Clin Immunol (2016) 16(2):165–71. doi: 10.1097/ACI.0000000000000255
66. Martinez Y, Mas D, Betancur C, Gebeyew K, Adebowale T, Hussain T, et al. Role of the Phytochemical Compounds like Modulators in Gut Microbiota and Oxidative Stress. Curr Pharm Des (2020) 26(22):2642–56. doi: 10.2174/1381612826666200515132218
67. Wright RJ, Cohen RT, Cohen S. The impact of stress on the development and expression of atopy. Curr Opin Allergy Clin Immunol (2005) 5(1):23–9. doi: 10.1097/00130832-200502000-00006
68. Chida Y, Sudo N, Sonoda J, Hiramoto T, Kubo C. Early-life psychological stress exacerbates adult mouse asthma via the hypothalamus-pituitary-adrenal axis. Am J Respir Crit Care Med (2007) 175(4):316–22. doi: 10.1164/rccm.200607-898OC
69. Haczku A, Tong E, Krytska K, Kierstein S, Panettieri RA. Combination of stress and asthma inhibits the glucocorticoid receptor (GR) and CAAT Enhancer binding protein (C/EBP)-mediated production of the immunoprotective surfactant protein D (SP-D) in the lung. Am J Respir Crit Care Med (2008) 177:A64.
70. Bailey MT, Kierstein S, Sharma S, Spaits M, Kinsey SG, Tliba O, et al. Social stress enhances allergen-induced airway inflammation in mice and inhibits corticosteroid responsiveness of cytokine production. J Immunol (2009) 182(12):7888–96. doi: 10.4049/jimmunol.0800891
71. Miller GE, Gaudin A, Zysk E, Chen E. Parental support and cytokine activity in childhood asthma: the role of glucocorticoid sensitivity. J Allergy Clin Immunol (2009) 123(4):824–30. doi: 10.1016/j.jaci.2008.12.019
72. Haczku A, Panettieri RA Jr. Social stress and asthma: the role of corticosteroid insensitivity. J Allergy Clin Immunol (2010) 125(3):550–8. doi: 10.1016/j.jaci.2009.11.005
73. Barnes P, FitzGerald G, Brown M, Dollery C. Nocturnal asthma and changes in circulating epinephrine, histamine, and cortisol. N Engl J Med (1980) Jul 31303(5):263–7. doi: 10.1056/NEJM198007313030506
74. Skloot GS. Nocturnal asthma: mechanisms and management. Mt Sinai J Med (2002) May69(3):140–7. doi: 10.1136/bmj.288.6428.1397
75. Sutherland ER. Nocturnal asthma. J Allergy Clin Immunol (2005) 116(6):1179–86; quiz 87. doi: 10.1016/j.jaci.2005.09.028
76. Buttgereit F, Gibofsky A. Delayed-release prednisone - a new approach to an old therapy. Expert Opin Pharmacother (2013) 14(8):1097–106. doi: 10.1517/14656566.2013.782001
77. Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci (2013) 34(9):518–30. doi: 10.1016/j.tips.2013.07.003
78. Sundar IK, Yao H, Sellix MT, Rahman I. Circadian molecular clock in lung pathophysiology. Am J Physiol Lung Cell Mol Physiol (2015) 309(10):L1056–75. doi: 10.1152/ajplung.00152.2015
79. Truong KK, Lam MT, Grandner MA, Sassoon CS, Malhotra A. Timing Matters: Circadian Rhythm in Sepsis, Obstructive Lung Disease, Obstructive Sleep Apnea, and Cancer. Ann Am Thorac Soc (2016) 13(7):1144–54. doi: 10.1513/AnnalsATS.201602-125FR
80. Zaslona Z, Case S, Early JO, Lalor SJ, McLoughlin RM, Curtis AM, et al. The circadian protein BMAL1 in myeloid cells is a negative regulator of allergic asthma. Am J Physiol Lung Cell Mol Physiol (2017) 312(6):L855–L60. doi: 10.1152/ajplung.00072.2017
81. Sundar IK, Sellix MT, Rahman I. Redox regulation of circadian molecular clock in chronic airway diseases. Free Radic Biol Med (2018) 119:121–28. doi: 10.1016/j.freeradbiomed.2017.10.383
82. Wilkinson ST, Tome ME, Briehl MM. Mitochondrial adaptations to oxidative stress confer resistance to apoptosis in lymphoma cells. Int J Mol Sci (2012) 13(8):10212–28. doi: 10.3390/ijms130810212
83. Estrela JM, Salvador R, Marchio P, Valles SL, Lopez-Blanch R, Rivera P, et al. Glucocorticoid receptor antagonism overcomes resistance to BRAF inhibition in BRAF(V600E)-mutated metastatic melanoma. Am J Cancer Res (2019) 9(12):2580–98. doi: 10.1016/S0959-8049(20)31182-5
84. Janssen-Heininger Y, Reynaert NL, van der Vliet A, Anathy V. Endoplasmic reticulum stress and glutathione therapeutics in chronic lung diseases. Redox Biol (2020) 33:101516. doi: 10.1016/j.redox.2020.101516
85. Ederle C, Charles AL, Khayath N, Poirot A, Meyer A, Clere-Jehl R, et al. Mitochondrial Function in Peripheral Blood Mononuclear Cells (PBMC) Is Enhanced, Together with Increased Reactive Oxygen Species, in Severe Asthmatic Patients in Exacerbation. J Clin Med (2019) 8(10):1613–26. doi: 10.3390/jcm8101613
86. Li F, Xu M, Wang M, Wang L, Wang H, Zhang H, et al. Roles of mitochondrial ROS and NLRP3 inflammasome in multiple ozone-induced lung inflammation and emphysema. Respir Res (2018) 19(1):230. doi: 10.1186/s12931-018-0931-8
87. Xu M, Wang L, Wang M, Wang H, Zhang H, Chen Y, et al. and NLRP3 inflammasome in acute ozone-induced murine model of airway inflammation and bronchial hyperresponsiveness. Free Radical Res (2019) 53(7):780–90. doi: 10.1080/10715762.2019.1630735
88. Hansen S, Holmskov U. Lung surfactant protein D (SP-D) and the molecular diverted descendants: conglutinin, CL-43 and CL-46. Immunobiology (2002) 205(4-5):498–517. doi: 10.1078/0171-2985-00150
89. Kierstein S, Poulain FR, Cao Y, Grous M, Mathias R, Kierstein G, et al. Susceptibility to ozone-induced airway inflammation is associated with decreased levels of surfactant protein D. Respir Res (2006) 7(1):85. doi: 10.1186/1465-9921-7-85
90. Starosta V, Griese M. Oxidative damage to surfactant protein D in pulmonary diseases. Free Radical Res (2006) 40(4):419–25. doi: 10.1080/10715760600571248
91. Yousefi S, Sharma SK, Stojkov D, Germic N, Aeschlimann S, Ge MQ, et al. Oxidative damage of SP-D abolishes control of eosinophil extracellular DNA trap formation. J Leukoc Biol (2018) 104(1):205–14. doi: 10.1002/JLB.3AB1117-455R
92. Vandewalle J, Libert C. Glucocorticoids in Sepsis: To Be or Not to Be. Front Immunol (2020) 11:1318. doi: 10.3389/fimmu.2020.01318
93. Schunemann HJ, Grant BJ, Freudenheim JL, Muti P, Browne RW, Drake JA, et al. The relation of serum levels of antioxidant vitamins C and E, retinol and carotenoids with pulmonary function in the general population. Am J Respir Crit Care Med (2001) 163(5):1246–55. doi: 10.1164/ajrccm.163.5.2007135
94. Flayer CH, Larson ED, Joseph A, Kao S, Qu W, Van Haren A, et al. Ozone-induced enhancement of airway hyperreactivity in rhesus macaques: Effects of antioxidant treatment. J Allergy Clin Immunol (2020) 145(1):312–23. doi: 10.1016/j.jaci.2019.08.034
95. Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J (2005) 25(3):552–63. doi: 10.1183/09031936.05.00117504
96. Dozor AJ. The role of oxidative stress in the pathogenesis and treatment of asthma. Ann N Y Acad Sci (2010) 1203:133–7. doi: 10.1111/j.1749-6632.2010.05562.x
97. Barnes PJ. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol (2013) 131(3):636–45. doi: 10.1016/j.jaci.2012.12.1564
98. Michalec L, Choudhury BK, Postlethwait E, Wild JS, Alam R, Lett-Brown M, et al. CCL7 and CXCL10 orchestrate oxidative stress-induced neutrophilic lung inflammation. J Immunol (2002) 168(2):846–52. doi: 10.4049/jimmunol.168.2.846
99. Cho HY, Kleeberger SR. Genetic mechanisms of susceptibility to oxidative lung injury in mice. Free Radic Biol Med (2007) 42(4):433–45. doi: 10.1016/j.freeradbiomed.2006.11.021
100. Kierstein S, Cao Y, Krytska K, Sharma S, Keslacy S, Amrani Y, et al. Exacerbation of Allergen-Induced Airway Hyperresponsiveness after Ozone Exposure Is Associated with Prolonged Survival of Eosinophils and Inhibition of Fas and FasL. Am J Respir Crit Care Med (2007) 175:A463.
101. Kierstein S, Krytska K, Sharma S, Amrani Y, Salmon M, Panettieri RA Jr., et al. Ozone inhalation induces exacerbation of eosinophilic airway inflammation and hyperresponsiveness in allergen-sensitized mice. Allergy (2008) 63(4):438–46. doi: 10.1111/j.1398-9995.2007.01587.x
102. Yang IA, Fong KM, Zimmerman PV, Holgate ST, Holloway JW. Genetic susceptibility to the respiratory effects of air pollution. Thorax (2008) 63(6):555–63. doi: 10.1136/thx.2007.079426
103. Yang IA, Fong KM, Zimmerman PV, Holgate ST, Holloway JW. Genetic susceptibility to the respiratory effects of air pollution. Postgrad Med J (2009) 85(1006):428–36. doi: 10.1136/thx.2007.079426
104. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al. Oxidative stress-induced antibodies to carbonyl-modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med (2011) 184(7):796–802. doi: 10.1164/rccm.201010-1605OC
105. Wiegman CH, Li F, Clarke CJ, Jazrawi E, Kirkham P, Barnes PJ, et al. A comprehensive analysis of oxidative stress in the ozone-induced lung inflammation mouse model. Clin Sci (Lond) (2014) 126(6):425–40. doi: 10.1042/CS20130039
106. Yang Q, Ge MQ, Kokalari B, Redai IG, Wang X, Kemeny DM, et al. Group 2 innate lymphoid cells mediate ozone-induced airway inflammation and hyperresponsiveness in mice. J Allergy Clin Immunol (2016) 137(2):571–8. doi: 10.1016/j.jaci.2015.06.037
107. Bao A, Yang H, Ji J, Chen Y, Bao W, Li F, et al. Involvements of p38 MAPK and oxidative stress in the ozone-induced enhancement of AHR and pulmonary inflammation in an allergic asthma model. Respir Res (2017) 18(1):216. doi: 10.1186/s12931-017-0697-4
108. Murphy SR, Schelegle ES, Edwards PC, Miller LA, Hyde DM, Van Winkle LS. Postnatal exposure history and airways: oxidant stress responses in airway explants. Am J Respir Cell Mol Biol (2012) 47(6):815–23. doi: 10.1165/rcmb.2012-0110OC
109. Moore BD, Hyde D, Miller L, Wong E, Frelinger J, Schelegle ES. Allergen and ozone exacerbate serotonin-induced increases in airway smooth muscle contraction in a model of childhood asthma. Respiration (2012) 83(6):529–42. doi: 10.1159/000336835
110. Moore BD, Hyde DM, Miller LA, Wong EM, Schelegle ES. Persistence of serotonergic enhancement of airway response in a model of childhood asthma. Am J Respir Cell Mol Biol (2014) 51(1):77–85. doi: 10.1165/rcmb.2013-0387OC
111. Ciencewicki JM, Verhein KC, Gerrish K, McCaw ZR, Li J, Bushel PR, et al. Effects of mannose-binding lectin on pulmonary gene expression and innate immune inflammatory response to ozone. Am J Physiol Lung Cell Mol Physiol (2016) 311(2):L280–91. doi: 10.1152/ajplung.00205.2015
112. Duan L, Li J, Ma P, Yang X, Xu S. Vitamin E antagonizes ozone-induced asthma exacerbation in Balb/c mice through the Nrf2 pathway. Food Chem Toxicol (2017) 107(Pt A):47–56. doi: 10.1016/j.fct.2017.06.025
113. Li R, Jiang N, Liu Q, Huang J, Guo X, Liu F, et al. Impact of Air Pollutants on Outpatient Visits for Acute Respiratory Outcomes. Int J Environ Res Public Health (2017) 14(1):47–58. doi: 10.3390/ijerph14010047
114. Li Z, Xu X, Thompson LA, Gross HE, Shenkman EA, DeWalt DA, et al. Longitudinal Effect of Ambient Air Pollution and Pollen Exposure on Asthma Control: The Patient-Reported Outcomes Measurement Information System (PROMIS) Pediatric Asthma Study. Acad Pediatr (2019) 19(6):615–23. doi: 10.1016/j.acap.2019.03.010
115. Marques Mejias MA, Tomas Perez M, Hernandez I, Lopez I, Quirce S. Asthma Exacerbations in the Pediatric Emergency Department at a Tertiary Hospital: Association With Environmental Factors. J Invest Allergol Clin Immunol (2019) 29(5):365–70. doi: 10.18176/jiaci.0364
116. Wiegman CH, Li F, Ryffel B, Togbe D, Chung KF. Oxidative Stress in Ozone-Induced Chronic Lung Inflammation and Emphysema: A Facet of Chronic Obstructive Pulmonary Disease. Front Immunol (2020) 11:1957. doi: 10.3389/fimmu.2020.01957
117. Jerrett M, Burnett RT, Pope CA,3, Ito K, Thurston G, Krewski D, et al. Long-term ozone exposure and mortality. N Engl J Med (2009) 360(11):1085–95. doi: 10.1056/NEJMoa0803894
118. McConnell R, Berhane K, Gilliland F, London SJ, Islam T, Gauderman WJ, et al. Asthma in exercising children exposed to ozone: a cohort study. Lancet (2002) 359(9304):386–91. doi: 10.1016/S0140-6736(02)07597-9
119. Peden DB. The “envirome” and what the practitioner needs to know about it. Ann Allergy Asthma Immunol (2019) 123(6):542–49. doi: 10.1016/j.anai.2019.09.014
120. Deterding RR, Shimizu H, Fisher JH, Shannon JM. Regulation of surfactant protein D expression by glucocorticoids in vitro and in vivo. Am J Respir Cell Mol Biol (1994) 10(1):30–7. doi: 10.1165/ajrcmb.10.1.8292379
121. Cao Y, Tao JQ, Bates SR, Beers MF, Haczku A. IL-4 induces production of the lung collectin surfactant protein-D. J Allergy Clin Immunol (2004) 113(3):439–44. doi: 10.1016/j.jaci.2003.11.031
122. Flayer CH, Ge MQ, Hwang JW, Kokalari B, Redai IG, Jiang Z, et al. Ozone Inhalation Attenuated the Effects of Budesonide on Aspergillus fumigatus-Induced Airway Inflammation and Hyperreactivity in Mice. Front Immunol (2019) 10:2173. doi: 10.3389/fimmu.2019.02173
123. Zhang L, Ikegami M, Crouch EC, Korfhagen TR, Whitsett JA. Activity of pulmonary surfactant protein-D (SP-D) in vivo is dependent on oligomeric structure. J Biol Chem (2001) 276(22):19214–9. doi: 10.1074/jbc.M010191200
124. Yousefi S, Sharma SK, Flayer CH, Stojkov D, Ge MQ, Germic N, et al. Oxidative Damage Abolishes the Ability of SP-D to Block Eosinophil Extracellular DNA Trap Release During Exacerbation of Allergic Airway Inflammation. Am J Respir Crit Care Med (2018) 197:205–14. doi: 10.1002/JLB.3AB1117-455R
125. Yousefi S, Qu WX, Flayer CH, Stojkov D, Germic N, Aeschlimann S, et al. Surfactant Protein D (SP-D) Inhibits Neutrophil Extracellular DNA Trap Formation: Effects of S-nitrosylation. J Allergy Clin Immunol (2019) 143(2):Ab192–Ab92. doi: 10.1016/j.jaci.2018.12.588
126. Kelly FJ, Mudway I, Krishna MT, Holgate ST. The free radical basis of air pollution: focus on ozone. Respir Med (1995) 89(10):647–56. doi: 10.1016/0954-6111(95)90131-0
127. Girotti AW. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J Lipid Res (1998) 39(8):1529–42. doi: 10.1016/S0022-2275(20)32182-9
128. Bromberg PA. Mechanisms of the acute effects of inhaled ozone in humans. Biochim Biophys Acta (2016) 1860(12):2771–81. doi: 10.1016/j.bbagen.2016.07.015
129. Mumby S, Chung KF, Adcock IM. Transcriptional Effects of Ozone and Impact on Airway Inflammation. Front Immunol (2019) 10:1610. doi: 10.3389/fimmu.2019.01610
130. Sokolowska M, Quesniaux VFJ, Akdis CA, Chung KF, Ryffel B, Togbe D. Acute Respiratory Barrier Disruption by Ozone Exposure in Mice. Front Immunol (2019) 10:2169. doi: 10.3389/fimmu.2019.02169
131. Brand JD, Mathews JA, Kasahara DI, Wurmbrand AP, Shore SA. Regulation of IL-17A expression in mice following subacute ozone exposure. J Immunotoxicol (2016) 13(3):428–38. doi: 10.3109/1547691X.2015.1120829
132. Mathews JA, Krishnamoorthy N, Kasahara DI, Cho Y, Wurmbrand AP, Ribeiro L, et al. IL-33 Drives Augmented Responses to Ozone in Obese Mice. Environ Health Perspect (2017) 125(2):246–53. doi: 10.1289/EHP272
133. Shore SA. Mechanistic Basis for Obesity-related Increases in Ozone-induced Airway Hyperresponsiveness in Mice. Ann Am Thorac Soc (2017) 14(Supplement_5):S357–S62. doi: 10.1513/AnnalsATS.201702-140AW
134. Michaudel C, Mackowiak C, Maillet I, Fauconnier L, Akdis CA, Sokolowska M, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J Allergy Clin Immunol (2018) 142(3):942–58. doi: 10.1016/j.jaci.2017.11.044
135. Kasahara DI, Wilkinson JE, Cho Y, Cardoso AP, Huttenhower C, Shore SA. The interleukin-33 receptor contributes to pulmonary responses to ozone in male mice: role of the microbiome. Respir Res (2019) 20(1):197. doi: 10.1186/s12931-019-1168-x
136. Kasahara DI, Shore SA. IL-33, diet-induced obesity, and pulmonary responses to ozone. Respir Res (2020) 21(1):98. doi: 10.1186/s12931-020-01361-9
137. Pichavant M, Goya S, Meyer EH, Johnston RA, Kim HY, Matangkasombut P, et al. Ozone exposure in a mouse model induces airway hyperreactivity that requires the presence of natural killer T cells and IL-17. J Exp Med (2008) 205(2):385–93. doi: 10.1084/jem.20071507
138. Kasahara DI, Kim HY, Williams AS, Verbout NG, Tran J, Si H, et al. Pulmonary inflammation induced by subacute ozone is augmented in adiponectin-deficient mice: role of IL-17A. J Immunol (2012) 188(9):4558–67. doi: 10.4049/jimmunol.1102363
139. Kasahara DI, Kim HY, Mathews JA, Verbout NG, Williams AS, Wurmbrand AP, et al. Pivotal role of IL-6 in the hyperinflammatory responses to subacute ozone in adiponectin-deficient mice. Am J Physiol Lung Cell Mol Physiol (2014) 306(6):L508–20. doi: 10.1152/ajplung.00235.2013
140. Mathews JA, Williams AS, Brand JD, Wurmbrand AP, Chen L, Ninin FM, et al. gammadelta T cells are required for pulmonary IL-17A expression after ozone exposure in mice: role of TNFalpha. PLoS One (2014) 9(5):e97707. doi: 10.1371/journal.pone.0097707
141. Mathews JA, Kasahara DI, Ribeiro L, Wurmbrand AP, Ninin FM, Shore SA. gammadelta T Cells Are Required for M2 Macrophage Polarization and Resolution of Ozone-Induced Pulmonary Inflammation in Mice. PLoS One (2015) 10(7):e0131236. doi: 10.1371/journal.pone.0131236
142. Williams AS, Mathews JA, Kasahara DI, Wurmbrand AP, Chen L, Shore SA. Innate and ozone-induced airway hyperresponsiveness in obese mice: role of TNF-alpha. Am J Physiol Lung Cell Mol Physiol (2015) 308(11):L1168–77. doi: 10.1152/ajplung.00393.2014
143. Che L, Jin Y, Zhang C, Lai T, Zhou H, Xia L, et al. Ozone-induced IL-17A and neutrophilic airway inflammation is orchestrated by the caspase-1-IL-1 cascade. Sci Rep (2016) 6:18680. doi: 10.1038/srep18680
144. Fei X, Zhang PY, Zhang X, Zhang GQ, Bao WP, Zhang YY, et al. IL-17A Monoclonal Antibody Partly Reverses the Glucocorticoids Insensitivity in Mice Exposed to Ozonec. Inflammation (2017) 40(3):788–97. doi: 10.1007/s10753-017-0523-7
145. Mathews JA, Krishnamoorthy N, Kasahara DI, Hutchinson J, Cho Y, Brand JD, et al. Augmented Responses to Ozone in Obese Mice Require IL-17A and Gastrin-Releasing Peptide. Am J Respir Cell Mol Biol (2018) 58(3):341–51. doi: 10.1165/rcmb.2017-0071OC
146. Zhang JH, Yang X, Chen YP, Zhang JF, Li CQ. Nrf2 Activator RTA-408 Protects Against Ozone-Induced Acute Asthma Exacerbation by Suppressing ROS and gammadeltaT17 Cells. Inflammation (2019) 42(5):1843–56. doi: 10.1007/s10753-019-01046-6
147. Alexis NE, Lay JC, Haczku A, Gong H, Linn W, Hazucha MJ, et al. Fluticasone propionate protects against ozone-induced airway inflammation and modified immune cell activation markers in healthy volunteers. Environ Health Perspect (2008) 116(6):799–805. doi: 10.1289/ehp.10981
148. Alexis NE, Lay JC, Hazucha M, Harris B, Hernandez ML, Bromberg PA, et al. Low-level ozone exposure induces airways inflammation and modifies cell surface phenotypes in healthy humans. Inhal Toxicol (2010) 22(7):593–600. doi: 10.3109/08958371003596587
149. Hyde DM, Hubbard WC, Wong V, Wu R, Pinkerton K, Plopper CG. Ozone-induced acute tracheobronchial epithelial injury: relationship to granulocyte emigration in the lung. Am J Respir Cell Mol Biol (1992) 6(5):481–97. doi: 10.1165/ajrcmb/6.5.481
150. Kim CS, Alexis NE, Rappold AG, Kehrl H, Hazucha MJ, Lay JC, et al. Lung function and inflammatory responses in healthy young adults exposed to 0.06 ppm ozone for 6.6 hours. Am J Respir Crit Care Med (2011) 183(9):1215–21. doi: 10.1164/rccm.201011-1813OC
151. Stevens WH, Adelroth E, Wattie J, Woolley MJ, Ellis R, Dahlback M, et al. Effect of inhaled budesonide on ozone-induced airway hyperresponsiveness and bronchoalveolar lavage cells in dogs. J Appl Physiol (1985) (1994) 77(6):2578–83. doi: 10.1152/jappl.1994.77.6.2578
152. Joad JP, Kott KS, Bric JM, Schelegle ES, Gershwin LJ, Plopper CG, et al. The effects of inhaled corticosteroids on intrinsic responsiveness and histology of airways from infant monkeys exposed to house dust mite allergen and ozone. Toxicol Appl Pharmacol (2008) 226(2):153–60. doi: 10.1016/j.taap.2007.09.005
153. Vagaggini B, Taccola M, Cianchetti S, Carnevali S, Bartoli ML, Bacci E, et al. Ozone exposure increases eosinophilic airway response induced by previous allergen challenge. Am J Respir Crit Care Med (2002) 166(8):1073–7. doi: 10.1164/rccm.2201013
154. Vagaggini B, Cianchetti S, Bartoli M, Ricci M, Bacci E, Dente FL, et al. Prednisone blunts airway neutrophilic inflammatory response due to ozone exposure in asthmatic subjects. Respiration (2007) 74(1):61–8. doi: 10.1159/000096078
155. Hernandez ML, Lay JC, Harris B, Esther CR Jr., Brickey WJ, Bromberg PA, et al. Atopic asthmatic subjects but not atopic subjects without asthma have enhanced inflammatory response to ozone. J Allergy Clin Immunol (2010) 126(3):537–44 e1. doi: 10.1016/j.jaci.2010.06.043
156. Jia Y, Fang X, Zhu X, Bai C, Zhu L, Jin M, et al. IL-13(+) Type 2 Innate Lymphoid Cells Correlate with Asthma Control Status and Treatment Response. Am J Respir Cell Mol Biol (2016) 55(5):675–83. doi: 10.1165/rcmb.2016-0099OC
157. Takenaka MC, Gabriely G, Rothhammer V, Mascanfroni ID, Wheeler MA, Chao CC, et al. Control of tumor-associated macrophages and T cells in glioblastoma via AHR and CD39. Nat Neurosci (2019) 22(5):729–40. doi: 10.1038/s41593-019-0370-y
158. Beamer CA, Shepherd DM. Role of the aryl hydrocarbon receptor (AhR) in lung inflammation. Semin Immunopathol (2013) 35(6):693–704. doi: 10.1007/s00281-013-0391-7
159. Gutierrez-Vazquez C, Quintana FJ. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity (2018) 48(1):19–33. doi: 10.1016/j.immuni.2017.12.012
160. Michaudel C, Maillet I, Fauconnier L, Quesniaux V, Chung KF, Wiegman C, et al. Interleukin-1alpha Mediates Ozone-Induced Myeloid Differentiation Factor-88-Dependent Epithelial Tissue Injury and Inflammation. Front Immunol (2018) 9:916. doi: 10.3389/fimmu.2018.00916
161. Quintana FJ. The aryl hydrocarbon receptor: a molecular pathway for the environmental control of the immune response. Immunology (2013) 138(3):183–9. doi: 10.1111/imm.12046
162. Quintana FJ, Sherr DH. Aryl hydrocarbon receptor control of adaptive immunity. Pharmacol Rev (2013) 65(4):1148–61. doi: 10.1124/pr.113.007823
163. Gibbs J, Ince L, Matthews L, Mei J, Bell T, Yang N, et al. An epithelial circadian clock controls pulmonary inflammation and glucocorticoid action. Nat Med (2014) 20(8):919–26. doi: 10.1038/nm.3599
164. Ince LM, Zhang Z, Beesley S, Vonslow RM, Saer BR, Matthews LC, et al. Circadian variation in pulmonary inflammatory responses is independent of rhythmic glucocorticoid signaling in airway epithelial cells. FASEB J (2019) 33(1):126–39. doi: 10.1096/fj.201800026RR
165. Sundar IK, Yao H, Sellix MT, Rahman I. Circadian clock-coupled lung cellular and molecular functions in chronic airway diseases. Am J Respir Cell Mol Biol (2015) 53(3):285–90. doi: 10.1165/rcmb.2014-0476TR
166. Jaeger C, Tischkau SA. Role of Aryl Hydrocarbon Receptor in Circadian Clock Disruption and Metabolic Dysfunction. Environ Health Insights (2016) 10:133–41. doi: 10.4137/EHI.S38343
167. Leung DY, Szefler SJ. New insights into steroid resistant asthma. Pediatr Allergy Immunol (1998) 9(1):3–12. doi: 10.1111/j.1399-3038.1998.tb00293.x
168. Michaudel C, Bataille F, Maillet I, Fauconnier L, Colas C, Sokol H, et al. Ozone-Induced Aryl Hydrocarbon Receptor Activation Controls Lung Inflammation via Interleukin-22 Modulation. Front Immunol (2020) 11:144. doi: 10.3389/fimmu.2020.00144
169. Ramakrishnan RK, Al Heialy S, Hamid Q. Role of IL-17 in asthma pathogenesis and its implications for the clinic. Expert Rev Respir Medi (2019) 13(11):1057–68. doi: 10.1080/17476348.2019.1666002
170. Chesne J, Braza F, Mahay G, Brouard S, Aronica M, Magnan A. IL-17 in severe asthma. Where do we stand? Am J Respir Crit Care Med (2014) 190(10):1094–101. doi: 10.1164/rccm.201405-0859PP
171. Johnston RA, Schwartzman IN, Flynt L, Shore SA. Role of interleukin-6 in murine airway responses to ozone. Am J Physiol Lung Cell Mol Physiol (2005) 288(2):L390–7. doi: 10.1152/ajplung.00007.2004
172. Linden A, Dahlen B. Interleukin-17 cytokine signalling in patients with asthma. Eur Respir J (2014) 44(5):1319–31. doi: 10.1183/09031936.00002314
173. Banuelos J, Cao Y, Shin SC, Lu NZ. Immunopathology alters Th17 cell glucocorticoid sensitivity. Allergy (2017) 72(3):331–41. doi: 10.1111/all.13051
174. Tsitoura DC, Rothman PB. Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4+ T cells. J Clin Invest (2004) 113(4):619–27. doi: 10.1172/JCI200418975
175. Walford HH, Lund SJ, Baum RE, White AA, Bergeron CM, Husseman J, et al. Increased ILC2s in the eosinophilic nasal polyp endotype are associated with corticosteroid responsiveness. Clin Immunol (2014) 155(1):126–35. doi: 10.1016/j.clim.2014.09.007
176. Barnes PJ. Glucocorticosteroids. Handb Exp Pharmacol (2017) 237:93–115. doi: 10.1007/164_2016_62
177. Haczku A, Alexander A, Brown P, Assoufi B, Li B, Kay AB, et al. The effect of dexamethasone, cyclosporine, and rapamycin on T-lymphocyte proliferation in vitro: comparison of cells from patients with glucocorticoid-sensitive and glucocorticoid-resistant chronic asthma. J Allergy Clin Immunol (1994) 93(2):510–9. doi: 10.1016/0091-6749(94)90361-1
178. Trevor JL, Deshane JS. Refractory asthma: mechanisms, targets, and therapy. Allergy (2014) 69(7):817–27. doi: 10.1111/all.12412
179. Wang W, Li JJ, Foster PS, Hansbro PM, Yang M. Potential therapeutic targets for steroid-resistant asthma. Curr Drug Targets (2010) 11(8):957–70. doi: 10.2174/138945010791591412
180. Pujols L, Mullol J, Picado C. Alpha and beta glucocorticoid receptors: relevance in airway diseases. Curr Allergy Asthma Rep (2007) 7(2):93–9. doi: 10.1007/s11882-007-0005-3
181. Mukherjee M, Svenningsen S, Nair P. Glucocortiosteroid subsensitivity and asthma severity. Curr Opin Pulm Med (2017) 23(1):78–88. doi: 10.1097/MCP.0000000000000337
182. Baschant U, Lane NE, Tuckermann J. The multiple facets of glucocorticoid action in rheumatoid arthritis. Nat Rev Rheumatol (2012) 8(11):645–55. doi: 10.1038/nrrheum.2012.166
183. Ingawale DK, Mandlik SK. New insights into the novel anti-inflammatory mode of action of glucocorticoids. Immunopharmacol Immunotoxicol (2020) 42(2):59–73. doi: 10.1080/08923973.2020.1728765
184. Reichardt HM, Kaestner KH, Tuckermann J, Kretz O, Wessely O, Bock R, et al. DNA binding of the glucocorticoid receptor is not essential for survival. Cell (1998) 93(4):531–41. doi: 10.1016/S0092-8674(00)81183-6
185. Reichardt HM, Tuckermann JP, Gottlicher M, Vujic M, Weih F, Angel P, et al. Repression of inflammatory responses in the absence of DNA binding by the glucocorticoid receptor. EMBO J (2001) 20(24):7168–73. doi: 10.1093/emboj/20.24.7168
186. Gross KL, Lu NZ, Cidlowski JA. Molecular mechanisms regulating glucocorticoid sensitivity and resistance. Mol Cell Endocrinol (2009) 300(1-2):7–16. doi: 10.1016/j.mce.2008.10.001
187. Zupkovitz G, Tischler J, Posch M, Sadzak I, Ramsauer K, Egger G, et al. Negative and positive regulation of gene expression by mouse histone deacetylase 1. Mol Cell Biol (2006) 26(21):7913–28. doi: 10.1128/MCB.01220-06
188. Ito K, Chung KF, Adcock IM. Update on glucocorticoid action and resistance. J Allergy Clin Immunol (2006) 117(3):522–43. doi: 10.1016/j.jaci.2006.01.032
189. Adcock IM, Caramori G. Cross-talk between pro-inflammatory transcription factors and glucocorticoids. Immunol Cell Biol (2001) 79(4):376–84. doi: 10.1046/j.1440-1711.2001.01025.x
190. Adcock IM, Barnes PJ. Molecular mechanisms of corticosteroid resistance. Chest (2008) 134(2):394–401. doi: 10.1378/chest.08-0440
191. Adcock IM, Ford PA, Bhavsar P, Ahmad T, Chung KF. Steroid resistance in asthma: mechanisms and treatment options. Curr Allergy Asthma Rep (2008) 8(2):171–8. doi: 10.1007/s11882-008-0028-4
192. Kalinyak JE, Dorin RI, Hoffman AR, Perlman AJ. Tissue-specific regulation of glucocorticoid receptor mRNA by dexamethasone. J Biol Chem (1987) 262(22):10441–4. doi: 10.1016/S0021-9258(18)60980-9
193. Okamoto K, Tanaka H, Ogawa H, Makino Y, Eguchi H, Hayashi S, et al. Redox-dependent regulation of nuclear import of the glucocorticoid receptor. J Biol Chem (1999) 274(15):10363–71. doi: 10.1074/jbc.274.15.10363
194. Zhu HJ, Dai YF, Wang O, Li M, Lu L, Zhao WG, et al. Generalized glucocorticoid resistance accompanied with an adrenocortical adenoma and caused by a novel point mutation of human glucocorticoid receptor gene. Chin Med J (Engl) (2011) 124(4):551–5. doi: 10.3760/cma.j.issn.0366-6999.2011.04.013
195. Geng CD, Pedersen KB, Nunez BS, Vedeckis WV. Human glucocorticoid receptor alpha transcript splice variants with exon 2 deletions: evidence for tissue- and cell type-specific functions. Biochemistry (2005) 44(20):7395–405. doi: 10.1021/bi047485e
196. Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol (2005) 5(1):58–68. doi: 10.1038/nri1528
197. Kishore U, Greenhough TJ, Waters P, Shrive AK, Ghai R, Kamran MF, et al. and SP-D: structure, function and receptors. Mol Immunol (2006) 43(9):1293–315. doi: 10.1016/j.molimm.2005.08.004
198. Pastva AM, Wright JR, Williams KL. Immunomodulatory roles of surfactant proteins A and D: implications in lung disease. Proc Am Thorac Soc (2007) 4(3):252–7. doi: 10.1513/pats.200701-018AW
199. Haczku A. Protective role of the lung collectins surfactant protein A and surfactant protein D in airway inflammation. J Allergy Clin Immunol (2008) 122(5):861–79; quiz 80-1. doi: 10.1016/j.jaci.2008.10.014
200. Forbes LR, Haczku A. SP-D and regulation of the pulmonary innate immune system in allergic airway changes. Clin Exp Allergy (2010) 40(4):547–62. doi: 10.1111/j.1365-2222.2010.03483.x
201. Orgeig S, Hiemstra PS, Veldhuizen EJ, Casals C, Clark HW, Haczku A, et al. Recent advances in alveolar biology: evolution and function of alveolar proteins. Respir Physiol Neurobiol (2010) 173 Suppl:S43–54. doi: 10.1016/j.resp.2010.04.023
202. Hillaire ML, Haagsman HP, Osterhaus AD, Rimmelzwaan GF, van Eijk M. Pulmonary surfactant protein D in first-line innate defence against influenza A virus infections. J Innate Immunity (2013) 5(3):197–208. doi: 10.1159/000346374
203. Briana DD, Malamitsi-Puchner A. Perinatal biomarkers implying ‘Developmental Origins of Health and Disease’ consequences in intrauterine growth restriction. Acta Paediatr (2020) 109(7):1317–22. doi: 10.1111/apa.15022
204. Horvath SM, Gliner JA, Folinsbee LJ. Adaptation to ozone: duration of effect. Am Rev Respir Dis (1981) 123(5):496–9. doi: 10.1164/arrd.1981.123.5.496
205. Backus-Hazzard GS, Howden R, Kleeberger SR. Genetic susceptibility to ozone-induced lung inflammation in animal models of asthma. Curr Opin Allergy Clin Immunol (2004) 4(5):349–53. doi: 10.1097/00130832-200410000-00004
206. Lu FL, Johnston RA, Flynt L, Theman TA, Terry RD, Schwartzman IN, et al. Increased pulmonary responses to acute ozone exposure in obese db/db mice. Am J Physiol Lung Cell Mol Physiol (2006) 290(5):L856–65. doi: 10.1152/ajplung.00386.2005
207. Shore SA. Obesity and asthma: lessons from animal models. J Appl Physiol (1985) (2007) 102(2):516–28. doi: 10.1152/japplphysiol.00847.2006
208. Takeda K, Haczku A, Lee JJ, Irvin CG, Gelfand EW. Strain dependence of airway hyperresponsiveness reflects differences in eosinophil localization in the lung. Am J Physiol Lung Cell Mol Physiol (2001) 281(2):L394–402. doi: 10.1152/ajplung.2001.281.2.L394
209. Atochina EN, Beers MF, Tomer Y, Scanlon ST, Russo SJ, Panettieri RA Jr., et al. Attenuated allergic airway hyperresponsiveness in C57BL/6 mice is associated with enhanced surfactant protein (SP)-D production following allergic sensitization. Respir Res (2003) 4(1):15. doi: 10.1186/1465-9921-4-15
210. Haczku A. Role and regulation of lung collectins in allergic airway sensitization. Pharmacol Ther (2006) 110(1):14–34. doi: 10.1016/j.pharmthera.2005.08.008
211. Groves AM, Gow AJ, Massa CB, Hall L, Laskin JD, Laskin DL. Age-related increases in ozone-induced injury and altered pulmonary mechanics in mice with progressive lung inflammation. Am J Physiol Lung Cell Mol Physiol (2013) 305(8):L555–68. doi: 10.1152/ajplung.00027.2013
212. Ge MQ, Kokalari B, Flayer CH, Killingbeck SS, Redai IG, MacFarlane AWT, et al. Cutting Edge: Role of NK Cells and Surfactant Protein D in Dendritic Cell Lymph Node Homing: Effects of Ozone Exposure. J Immunol (2016) 196(2):553–7. doi: 10.4049/jimmunol.1403042
213. Forbes LR, Ducka B, Fehrenbach M, Hortobagyi L, Zhu X, Kierstein S, et al. (SP-D) Binds to Signal Regulatory Protein Alpha (SIRP-a) on Alveolar Macrophages and Dendritic Cells and Promotes Allergen-Induced Lymph-Node Directed Migration. J Allergy Clin Immunol (2010) 125(2):AB126. doi: 10.1016/j.jaci.2009.12.498
214. Mariencheck W, Crouch E. Modulation of surfactant protein D expression by glucocorticoids in fetal rat lung. Am J Respir Cell Mol Biol (1994) 10(4):419–29. doi: 10.1165/ajrcmb.10.4.8136157
215. Rust K, Bingle L, Mariencheck W, Persson A, Crouch EC. Characterization of the human surfactant protein D promoter: transcriptional regulation of SP-D gene expression by glucocorticoids. Am J Respir Cell Mol Biol (1996) 14(2):121–30. doi: 10.1165/ajrcmb.14.2.8630261
216. Haczku A, Cao Y, Vass G, Kierstein S, Nath P, Atochina-Vasserman EN, et al. IL-4 and IL-13 form a negative feedback circuit with surfactant protein-D in the allergic airway response. J Immunol (2006) 176(6):3557–65. doi: 10.4049/jimmunol.176.6.3557
217. Edenborough KM, Gilbertson BP, Brown LE. A mouse model for the study of contact-dependent transmission of influenza A virus and the factors that govern transmissibility. J Virol (2012) 86(23):12544–51. doi: 10.1128/JVI.00859-12
218. Arteaga-Badillo DA, Portillo-Reyes J, Vargas-Mendoza N, Morales-Gonzalez JA, Izquierdo-Vega JA, Sanchez-Gutierrez M, et al. Asthma: New Integrative Treatment Strategies for the Next Decades. Med (Kaunas) (2020) 56(9):438–58. doi: 10.3390/medicina56090438
219. Cho HY, Kleeberger SR. Nrf2 protects against airway disorders. Toxicol Appl Pharmacol (2010) 244(1):43–56. doi: 10.1016/j.taap.2009.07.024
220. Sussan TE, Gajghate S, Chatterjee S, Mandke P, McCormick S, Sudini K, et al. Nrf2 reduces allergic asthma in mice through enhanced airway epithelial cytoprotective function. Am J Physiol Lung Cell Mol Physiol (2015) 309(1):L27–36. doi: 10.1152/ajplung.00398.2014
221. Kikuchi N, Ishii Y, Morishima Y, Yageta Y, Haraguchi N, Itoh K, et al. Nrf2 protects against pulmonary fibrosis by regulating the lung oxidant level and Th1/Th2 balance. Respir Res (2010) 11:31. doi: 10.1186/1465-9921-11-31
222. Kinniry P, Amrani Y, Vachani A, Solomides CC, Arguiri E, Workman A, et al. Dietary flaxseed supplementation ameliorates inflammation and oxidative tissue damage in experimental models of acute lung injury in mice. J Nutr (2006) 136(6):1545–51. doi: 10.1093/jn/136.6.1545
223. Pietrofesa RA, Velalopoulou A, Albelda SM, Christofidou-Solomidou M. Asbestos Induces Oxidative Stress and Activation of Nrf2 Signaling in Murine Macrophages: Chemopreventive Role of the Synthetic Lignan Secoisolariciresinol Diglucoside (LGM2605). Int J Mol Sci (2016) 17(3):322. doi: 10.3390/ijms17030322
224. Capitanio JP, Miller LA, Schelegle ES, Mendoza SP, Mason WA, Hyde DM. Behavioral inhibition is associated with airway hyperresponsiveness but not atopy in a monkey model of asthma. Psychosom Med (2011) 73(4):288–94. doi: 10.1097/PSY.0b013e3182155c83
225. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in Hospitalized Patients with Covid-19 - Preliminary Report. N Engl J Med (2020). doi: 10.1056/NEJMoa2021436
226. Group WHOREAfC-TW, Sterne JAC, Murthy S, Diaz JV, Slutsky AS, Villar J, et al. Association Between Administration of Systemic Corticosteroids and Mortality Among Critically Ill Patients With COVID-19: A Meta-analysis. JAMA J Am Med Assoc (2020) 324(13):1330–41. doi: 10.1001/jama.2020.17023
227. Tomazini BM, Maia IS, Cavalcanti AB, Berwanger O, Rosa RG, Veiga VC, et al. Effect of Dexamethasone on Days Alive and Ventilator-Free in Patients With Moderate or Severe Acute Respiratory Distress Syndrome and COVID-19: The CoDEX Randomized Clinical Trial. JAMA (2020) 324(13):1307–16. doi: 10.1001/jama.2020.17021
Keywords: asthma, oxidative stress, air pollution, ozone, glucocorticoid resistance, IL-17A, neutrophils
Citation: Enweasor C, Flayer CH and Haczku A (2021) Ozone-Induced Oxidative Stress, Neutrophilic Airway Inflammation, and Glucocorticoid Resistance in Asthma. Front. Immunol. 12:631092. doi: 10.3389/fimmu.2021.631092
Received: 19 November 2020; Accepted: 18 January 2021;
Published: 26 February 2021.
Edited by:
Bernahrd Ryffel, Centre National de la Recherche Scientifique (CNRS), FranceReviewed by:
Chloé Michaudel, INRA Centre Jouy-en-Josas, FranceGarry M. Walsh, University of Aberdeen, United Kingdom
Copyright © 2021 Enweasor, Flayer and Haczku. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Haczku, aGFjemt1QHVjZGF2aXMuZWR1