 Deepti Suri1*
Deepti Suri1* Amit Rawat1
Amit Rawat1 Ankur Kumar Jindal1
Ankur Kumar Jindal1 Pandiarajan Vignesh1
Pandiarajan Vignesh1 Anju Gupta1
Anju Gupta1 Rakesh Kumar Pilania1
Rakesh Kumar Pilania1 Vibhu Joshi1
Vibhu Joshi1 Kanika Arora1
Kanika Arora1 Rajni Kumrah1
Rajni Kumrah1 Gummadi Anjani1
Gummadi Anjani1 Amita Aggarwal2
Amita Aggarwal2 Shubha Phadke2
Shubha Phadke2 Fouzia N. Aboobacker3
Fouzia N. Aboobacker3 Biju George3
Biju George3 Eunice Sindhuvi Edison3
Eunice Sindhuvi Edison3 Mukesh Desai4
Mukesh Desai4 Prasad Taur4
Prasad Taur4 Vijaya Gowri4
Vijaya Gowri4 Ambreen Abdulwahab Pandrowala4
Ambreen Abdulwahab Pandrowala4 Sagar Bhattad5Swati Kanakia6
Sagar Bhattad5Swati Kanakia6 Marco Gottorno7
Marco Gottorno7 Isabella Ceccherini7
Isabella Ceccherini7 Adriana Almeida de Jesus8Raphaela Goldbach-Mansky8
Adriana Almeida de Jesus8Raphaela Goldbach-Mansky8 Michael S. Hershfield9
Michael S. Hershfield9 Surjit Singh1
Surjit Singh1- 1Post Graduate Institute of Medical Education and Research (PGIMER), Chandigarh, India
- 2Sanjay Gandhi Post Graduate Institute of Medical Sciences (SGPGI), Lucknow, India
- 3Christian Medical College and Hospital, Vellore, India
- 4Bai Jerbai Wadia Hospital for Children, Mumbai, India
- 5Aster Cauvery Medical Institute Hospital, Bengaluru, India
- 6Lilavati Hospital and Research Center, Mumbai, India
- 7Center for Autoinflammatory Diseases and Immunodeficiency, Istituto di Ricovero e Cura a Carattere Scientifico Instituto Giannina Gaslini, Genoa, Italy
- 8Translational Autoinflammatory Diseases Section, Laboratory of Clinical Immunology and Microbiology, National Institute of Allergy and Infectious Diseases, National Institutes of Health, Bethesda, MD, United States
- 9Duke University Medical Centre, Durham, NC, United States
Background: Systemic autoinflammatory diseases (SAID) are rare inherited disorders involving genes regulating innate immune signaling and are characterized by periodic or chronic multi-systemic inflammation.
Objective: To describe spectrum of clinical, immunological, molecular features, and outcomes of patients with SAID in India.
Methods: Request to share data was sent to multiple centers in India that are involved in care and management of patients with Inborn Errors of Immunity. Six centers provided requisite data that were compiled and analyzed.
Results: Data on 107 patients with SAID were collated—of these, 29 patients were excluded due to unavailability of complete information. Twelve patients (15%) had type 1 interferonopathies, 21 (26%) had diseases affecting inflammasomes, 30 patients (41%) had non-inflammasome related conditions and 1five patients (19%) had Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis (PFAPA). Type1 interferonopathies identified in the cohort included patients with Deficiency of Adenosine Deaminase 2 (DADA2) (six patients; five families); STING-associated vasculopathy infantile-onset (SAVI) (three patients, one family); Spondyloenchondro-dysplasia with Immune Dysregulation (SPENCD) (two patients). Diseases affecting inflammasomes include Mevalonate Kinase Deficiency (eight patients); Cryopyrin-Associated Periodic Syndromes (CAPS) (seven patients); NLR Family, Pyrin domain-containing 12 (NLRP12) (two patients); Familial Mediterranean fever (FMF) (two patients); Autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (APLAID) (two patients). TNF receptor-associated periodic syndrome (TRAPS) (three patients); A20 haploinsufficiency (four patients); Deficiency of Interleukin 1 Receptor Antagonist (DIRA) (two patients) were categorized as non-inflammasome related conditions. There were significant delays in diagnosis Corticosteroids and other immunosuppressive agents were used for treatment as anti-IL-1 drugs and other biological agents were and still are not available in India. Eight (16.3%) patients had so far succumbed to their illness.
Conclusions: This is the first nationwide cohort of patients with SAID from India. Clinical manifestations were diverse. Overlapping of clinical features with other relatively common rheumatological disorders often resulted in delays in diagnosis. More nationwide efforts are needed to enhance awareness of SAID among health care professionals and there is an urgent need to make targeted immunotherapies universally available.
Introduction
Systemic autoinflammatory diseases (SAID) are complex inherited disorders caused by defects in several genes regulating innate immune signaling and are characterized by periodic or chronic multisystem sterile inflammation (1–3).
The term “autoinflammatory disorders” was coined in 1999 by Daniel Kastner's group when they proposed a new group of immunological diseases (4). The paper described genetic background of familial Hibernian fever, and rechristened it as “TNF receptor-associated periodic syndrome (TRAPS).” It also linked it with previously described mutations in Pyrin (MEFV)gene that causes familial Mediterranean fever (FMF) (4–6). In 2010, Kastner et al. defined autoinflammatory diseases as “clinical disorders marked by abnormally increased inflammation, mediated predominantly by cells and molecules of the innate immune system with a significant host predisposition” (1, 7). Euro fever registry and Pediatric Rheumatology International Trials Organization (PRINTO) have also proposed classification criteria for different hereditary recurrent fever syndromes (8).
SAIDs can be monogenic and polygenic or multifactorial (9, 10). Monogenic SAID (e.g., TRAPS, FMF) follow Mendelian inheritance and result from pathogenic variants in a single gene. On the other hand, disorders such as systemic juvenile idiopathic arthritis, Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis (PFAPA) syndrome and Adult-Onset Still Disease have polygenic or multifactorial etiology. The 2019 International Union of Immunological Societies (IUIS) Expert Committee classified monogenic SAID into 3 major groups: Type 1 interferonopathies, defects affecting the inflammasome and non-inflammasome-related conditions (11).
Over the last 2 decades due to an increasing awareness and availability of high throughput genetic sequencing techniques, there has been an exponential increase in discovery of genes responsible for SAID (2, 12, 13). Further, molecular insights of these disorders have provided the basis for new therapeutic interventions leading to improved outcomes and long-term survivals. There is paucity of data on SAID from India with published literature comprising of only anecdotal case reports (14–21). In this manuscript we describe clinical features, molecular profile, treatment and outcome in patients with monogenic SAID from six centers in our country. This paper reports nationwide cohort on SAID.
Patients and Methods
Centers supported by the Foundation for Primary Immunodeficiency Diseases (FPID), USA, and other institutions involved in care of patients with Inborn Errors of Immunity (IEI)across India were contacted to share details of patients with SAID on a template designed by lead author (DS). Data including demographics, prominent clinical manifestations, laboratory investigations, molecular results, treatment regimens, and long-term outcomes were collated on predesigned Microsoft Excel sheet. Findings of radiology and histopathology were also recorded. Participating centers included Postgraduate Institute of Medical Education and Research (PGIMER), Chandigarh, North India (52 patients); Sanjay Gandhi Postgraduate Institute of Medical Sciences (SGPGIMS), Lucknow, North India (25 patients); Christian Medical College, Vellore (12 patients), Bai Jerbai Wadia Hospital for Children (BJWHC), Mumbai, West India (10 patients); and ASTER CMI Hospitals, Bengaluru, South India (seven patients), Lilavati Hospital and Research Center,Mumbai, West India (one patient).
Definition of Said
Several definitions have been proposed for SAID (4, 8, 11, 22). For the purpose of this study we have used European Society for Immunodeficiencies (ESID) working group definition for the categorization of SAID. ESID has defined “unclassified autoinflammatory diseases” to be characterized by recurrent fever (temperature >38°C) having occurred on at least six occasions with exclusion of other known infective/inflammatory autoimmune disorders and documented evidence of increased inflammatory markers [erythrocyte sedimentation rate (ESR), C-reactive protein (CRP)], age of onset under 40 years and predominantly but not exclusively with systemic symptoms (23). In the present study all patients who fulfilled ESID working group definition and had molecular confirmation of monogenic SAID were included. Patients with polygenic SAID (e.g., systemic juvenile idiopathic arthritis, chronic non-infectious osteitis) and infantile inflammatory bowel disease were excluded.
All patients were further classified into three subtypes according to 2019 IUIS classification for SAID (11). Coatamer complex one protein alpha subunit (COPA) syndrome was classified as Type 1 interferonopathy (24, 25). Patients without molecular confirmation of diagnosis and/or could not be classified in accordance with IUIS classification were also excluded. Some patients included in this series have been reported earlier and these have been duly cited (14, 17, 19, 26, 27).
Molecular Investigations
Molecular analysis of patients for PGIMER, Chandigarh was performed at Pediatric Allergy Immunology Laboratory at PGIMER, Chandigarh or in collaboration with international centers, namely Center for Autoinflammatory Diseases and Immunodeficiency, Genoa, Italy (1three patients) and National Institutes of Health (NIH), USA (three patients). Measurement of plasma adenosine deaminase 2 (ADA2) activity in extracts of dried plasma spots was performed in the laboratory of Dr. Michael Hershfield at Duke University School of Medicine, Durham NC, USA (28).
Laboratory Investigation at PGIMER, Chandigarh
Molecular analysis of Nucleotide binding oligomerization domain 2 (NOD 2) gene in patients suspected to have Blau syndrome (11/14 patients) and Adenosine deaminase 2(ADA2) gene for Adenosine Deaminase 2 (ADA2) deficiency was performed in-house in Pediatric Immunology Laboratory, Advanced Pediatric Center by Sanger sequencing. Exon-4 of NOD2 gene was amplified using specified oligonucleotide primers and results were analyzed using Codon Code Aligner software (Codon Code Corporation, Massachusetts, USA). Screening of hotspot region (exon 2) of ADA2 gene was also performed in patients clinically suspected to have Deficiency of Adenosine Deaminase 2 (DADA2).
Molecular analysis in most patients at other centers was carried out at commercial laboratories that use targeted gene panel by Next Generation Sequencing (NGS) techniques. Sanger sequencing was used to confirm the variants obtained by NGS.
Results
Data on 107 patients with SAID were collated from various centers in India. Of these, 19 patients had to be excluded as molecular confirmation was not available. Ten patients with variants of unknown significance (VUS) in genes associated with SAIDs were also excluded if found inconsistent with clinical profiles or non-pathogenic based on predictive analysis tools. Remaining 78 patients (Figure 1) included Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis (PFAPA) (15 patients (19%) from PGIMER); type 1 interferonopathies (1two patients, 15%); diseases affecting inflammasomes (21 patients, 26%); and non-inflammasome related conditions (30 patients, 38%%). Clinical details of patients with PFAPA (1five patients) and Blau syndrome (14 patients) are not being presented in the current manuscript (Suri et al., manuscript in submission).
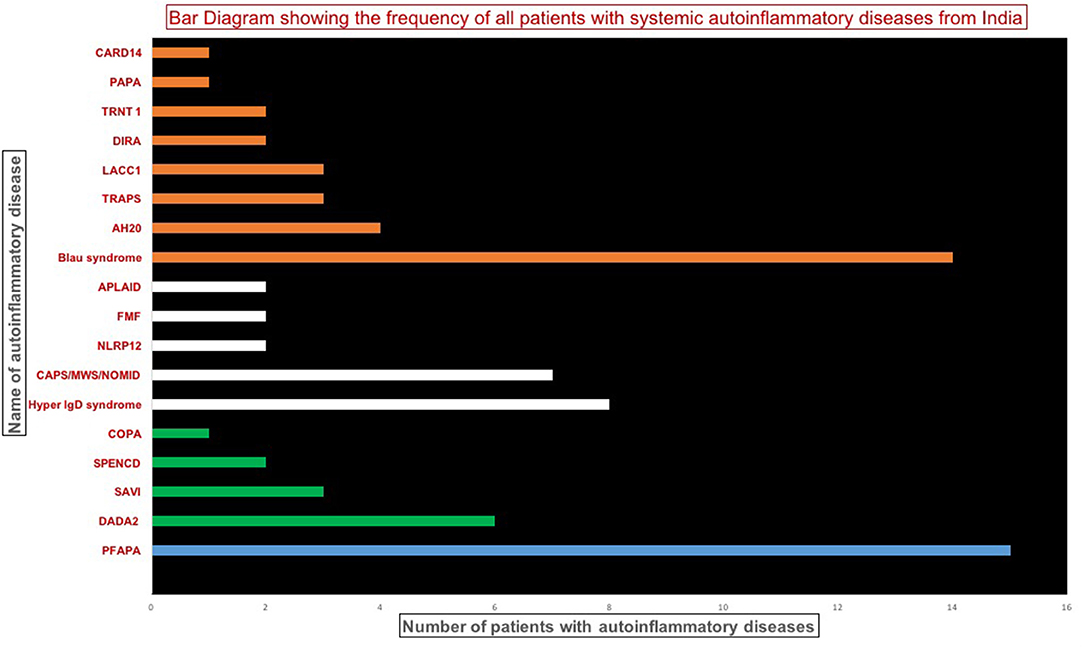
Figure 1. Bar diagram showing the frequency of patients with various systemic autoinflammatory disease. DADA2, Deficiency of adenosine deaminase 2; SAVI, STING-associated vasculopathy infantile-onset; SPENCD, Spondyloenchondro-dysplasia with Immune Dysregulation; CAPS, Cryopyrin-Associated Periodic Syndromes; NLRP12, NLR Family, Pyrin domain-containing 12; FMF, Familial Mediterranean fever; APLAID, Autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation; TRAPS, TNF receptor-associated periodic syndrome; HA20, A20 haploinsufficiency; LACC1, Laccase Domain Containing 1; DIRA, Deficiency of Interleukin 1 Receptor Antagonist; TRNT1, TRNA nucleotidyl transferase; PAPA, Pyogenic Arthritis; Pyoderma gangrenosum and Acne; COPA, Coatamer complex 1 protein alpha subunit (COPA) syndrome; CARD14, Caspase Recruitment Domain Family Member 14.
Clinical Profile Of Patients With Type 1 Interferonopathies
Type 1 interferonopathies identified in the cohort included patients with DADA2 (six patients; five families); STING-associated vasculopathy infantile-onset (SAVI) (three patients from one family); Spondylo enchondro dysplasia with Immune Dysregulation (SPENCD) (two patients) and Coatamer complex one protein alpha subunit (COPA syndrome) (one patient) (Table 1).
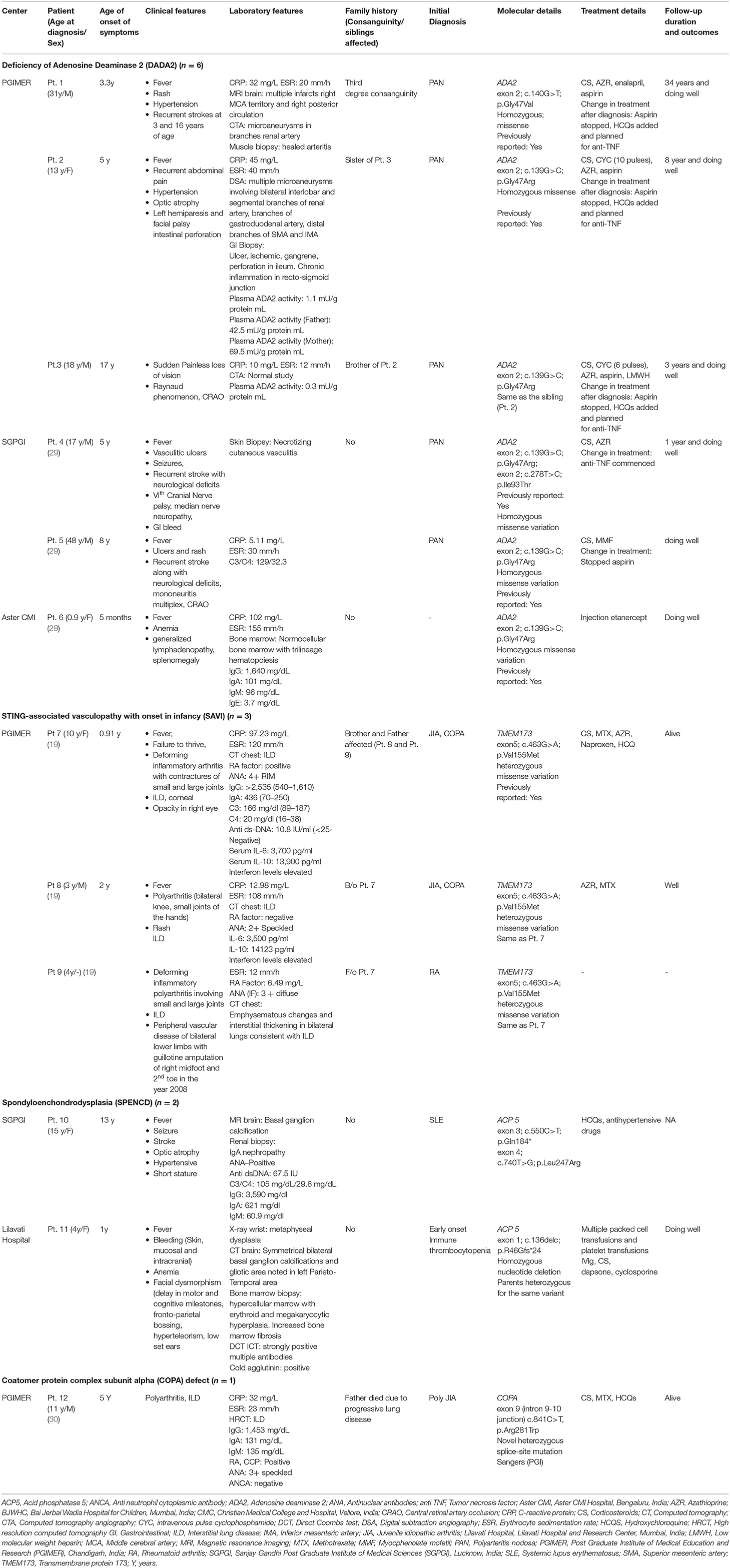
Table 1. Clinical manifestations, molecular profile, treatment, and outcome of patients with type I interferonopathies (n = 12).
DADA2
Age of onset of symptoms in patients with DADA2 ranged from 5 months to 17 years while age at diagnosis ranged from 9 months to 48 years. All patients with DADA 2 were diagnosed and managed as polyartritis nodosa (PAN). Family history was contributory in three patients (patient no. 2, 3, and 5). Predominant clinical features included fever (4/5), recurrent stroke (3/5), vasculitic rash (3/5), and retinal changes (2/5). Patient one had presented with hypertensive stroke at 3.3 years of aging 1992 and had second episode at the age of 16 years in 2002. The diagnosis of DADA2 was established in 2018 after three decades of follow-up.
Patient no 3 was diagnosed to have central retinal artery occlusion. Inflammatory markers were persistently normal. His sister (patient two) was under treatment and follow up for PAN. She had presented with recurrent abdominal pain with perforation peritonitis and catheter angiography had revealed microaneurysms in mesenteric arteries and renal arteries (Figure 2). In view of family historyDAD2 was suspected and mutation in ADA2 gene was detected. Establishment of diagnosis lead to stoppage of aspirin and commencement of anti-TNF agents.
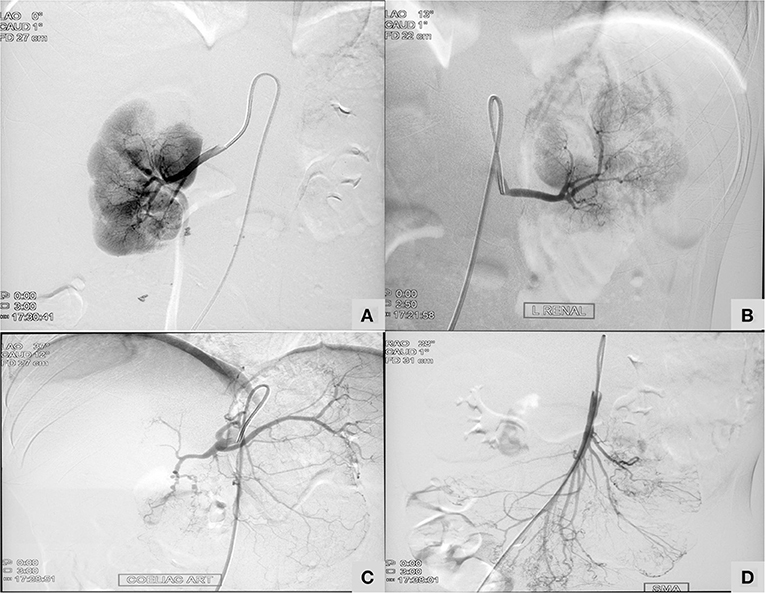
Figure 2. A 13-year-old girl with Deficiency of Adenosine Deaminase 2 (Table 1; Patient two) showing micronaeurysm on intervation catheter angiography in bilateral renal arteries (A,B), celiac artery (C), and superior mesenteric artery (D).
SAVI
A 10-year-old girl (patient no. 7), previously reported (19) had presented with fever, polyarthritis, and interstitial lung disease (ILD). Initial diagnosis of juvenile idiopathic arthritis with ILD was considered. Younger sibling (patient no. eight) and father (patient no. nine) of index patient also had similar symptoms. Father gave history of gangrene of both lower limbs with amputation of right midfoot and left 2nd toe. Exome sequencing revealed pathogenic variant in Transmembrane protein 173 (TMEM173) gene confirming the diagnosis of SAVI.
SPENCD
A 13-year-old girl (patient no 10) had persistent pyrexia, decreased vision with bilateral optic atrophy, hypertensive stroke, seizures, and proteinuria. Investigations showed hypergammaglobulinemia and positive antinuclear antibodies (ANA) with elevated anti-double stranded DNA (dsDNA) but normal complements. Initial diagnosis of systemic lupus erythematosus was proffered. Renal biopsy revealed IgA nephropathy. Magnetic Resonance Imaging (MRI) brain showed basal ganglia calcifications. Exome sequencing revealed pathogenic variant in Acid phosphatase 5 (ACP 5)gene which was confirmed on Sanger sequencing.
A 4-year-old girl (patient no 11) had presented with bleeding manifestations (skin, mucosal and intracranial bleed) since infancy (Figure 3). She had steroid refractory anemia and thrombocytopenia with no autoantibodies and hypocellular bone marrow. She was later noted to have short stature and metaphyseal dysplasia along with bilateral basal ganglia calcification. Targeted gene panel revealed homozygous nucleotide deletion in exon 1 of ACP5 gene.
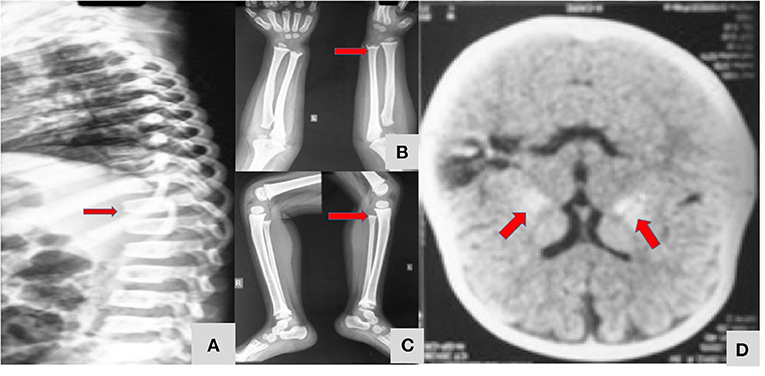
Figure 3. A 4 year-old-girl with Spondyloenchondrodysplasia (Table 1; Patient 11) showing fish mouth vertebra (A), metaphyseal dysplasia of long bones of upper and lower limbs (B,C), and bilateral symmetrical basal ganglia calcification (D).
COPA Syndrome
The index patient (patient no 12), previously reported (30) was diagnosed to have COPA syndrome when they had presented with rheumatoid factor positive deforming polyarthritis and interstitial lung disease. His father also had arthritis and had succumbed to progressive lung disease.
Clinical Profile of Patients With Defects Affecting the Inflammasomes
Twenty-one patients were classified to have inflammasomopathies (n = 21, 26%). These included Mevalonate Kinase Deficiency (HyperIgD syndrome) (eight patients); Cryopyrin-Associated Periodic Syndromes (CAPS) (seven patients); NLR Family, Pyrin domain-containing 12 (NLRP12) (two patients); FMF (two patients); Autoinflammation and PLCG2-associated antibody deficiency and immune dysregulation (APLAID) (two patients) (Table 2).
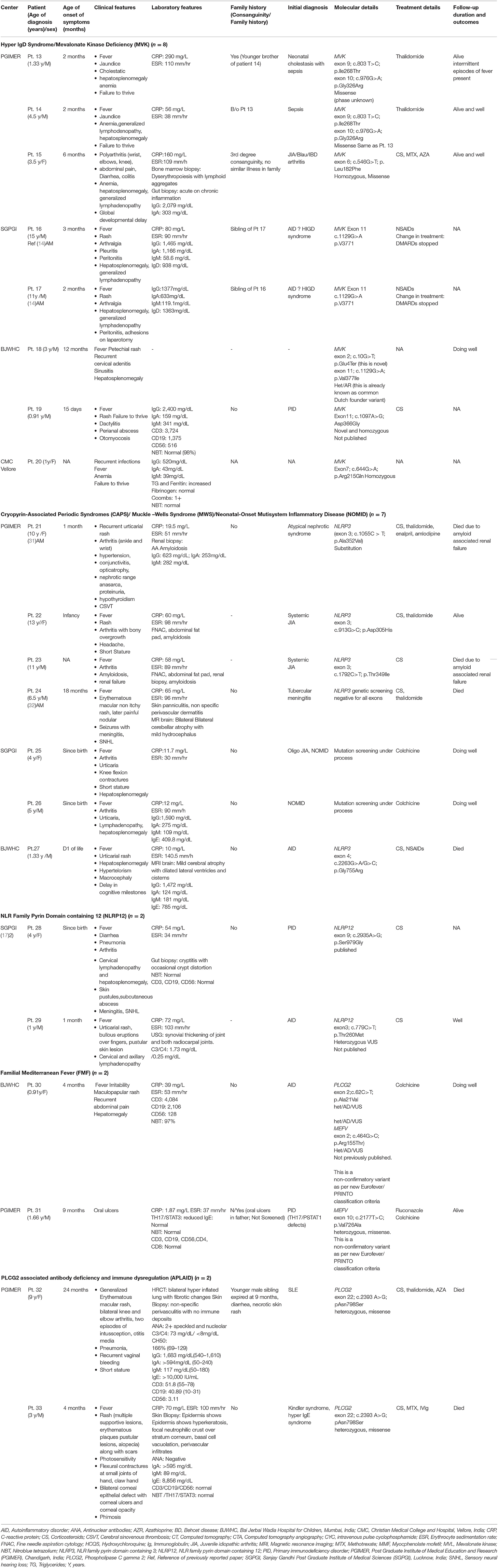
Table 2. Clinical manifestations, molecular profile, treatment, and outcomes of patients with defect affecting the inflammasome (n = 21).
Hyper IgD Syndrome
Patients with Hyper Ig D syndrome (Patient no. 13–20) had onset of symptoms during infancy (15 days−1 year) with predominant clinical features being fever (7/8), rash (4/8), lymphadenopathy (4/8), hepatosplenomegaly (5/8), and anemia (4/8). Initial diagnosis of neonatal sepsis was considered in two patients (patient no. 13–14). Three patients were found to have the V377I Dutch founder variant and 2 had c.1129G>A variant which is fairly common in South India particularly Kerala (15).
Cryopyrin-Associated Periodic Syndromes CAPS
In this cohort we report seven patients with CAPS caused by mutations in NLR family pyrin domain containing 3 (NLRP3) gene. All patients had been symptomatic since early infancy but there were significant delays in diagnosis. Age at diagnosis ranged from 15 months to 13 years. Most of these patients were initially diagnosed as JIA. Seven patients had classical phenotype of NOMID with infantile onset of fevers, urticarial rash, arthritis, and progressive deformities with bony overgrowths (Figure 4). Sensory neural hearing loss and headache was found in only1 patient. Of the seven patients, pathogenic variants in NLRP3 gene were identified in four patients (patient no. 21–23, 27) while no mutation could be identified in patient no 24 on exome sequencing. Molecular studies of two patients (patient 25, 26) are awaited. Three patients (patient no 21,22, and 23) had developed amyloidosis when by the time diagnosis of CAPS was established and two. patients (patient no 21 and 23) succumbed to their illness. Drugs used for treatment included corticosteroids, thalidomide and colchicine as anti-interleukin 1 (anti-IL1) therapy was not easily accessible. 4/7 patients with CAPS died while 3 were alive at the time of this report. Patient no. 22 has been on thalidomide for 12 years which has resulted in normalization of inflammatory parameters but she continues to have significant growth retardation, deformities, and intermittent headaches.
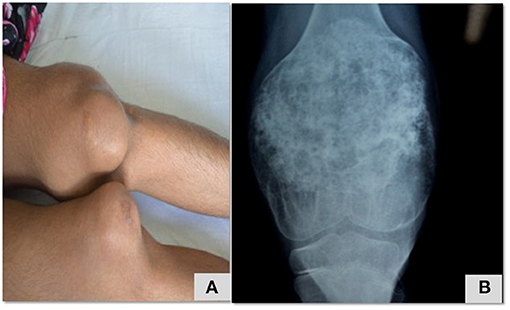
Figure 4. Swelling of knee joint with enlarged, deformed femora and patellae due to overgrowth arthropathy (A), heterogeneously calcified tumor-like protrusions originating from the growth plate (B) in a child with NOMID (Table 2; patient 22).
NLRP12
Variants in NLRP12 gene were identified t in two patients (patient no 28, 29). Both children were symptomatic since early infancy. Patient no. 28 had presented with recurrent episodes of fever, and infections (skin and subcutaneous abscess, diarrhea, meningitis, pneumonia), arthritis, sensorineural hearing loss and hepatosplenomegaly while patient no. 29 in addition had urticarial rash, pustular skin lesions, and lymphadenopathy. Heterozygous mutation in exon 3 in NLR family pyrin domain containing12 (NLRP12) gene was identified. Corticosteroids were used for treatment in patient 28 and is currently well.
FMF
Clinical profile of patients (Patient no 30, 31) with variants in MEFV gene is summarized in Table 2. Patient no. 30 had presented with periodic fever, rash, and abdominal pain. Targeted panel revealed variants of unknown significance in MEFV gene and Phospholipase C Gamma 2 (PLCG 2) gene. The patient is doing well on colchicine.
Nine months old boy (patient 31) had presented with recurrent oral ulcers. In view of family history of oral ulceration exome sequencing was performed. Heerozygyous issensense mutation in MEFV gene was identified. Symptomatic improvement has been noted after initiation of colchicine.
APLAID
Clinical profiles of patients no. 32, 33 with PLCG2 variants are summarized in Table 2. Patient no. 32 had erythematous macular rash (Figure 5), large joint arthritis, episodes of intussusception along with recurrent sinopulmonary infections. A de-novo heterozygous missense mutation in exon 22 of PLCG2 gene that resulted in substitution of serine by asparagine at codon 798 (pAsn798Ser), was validated using Sanger sequencing. The Asn798Ser variant has a minor allele frequency of 0.08, 0.07, and 0.16% in the 1,000 genomes, ExAC and internal databases, respectively. The in-silico predictions of the variant were found damaging by PolyPhen-2 (HumDiv), damaging by Sorting Intolerant from Tolerant (SIFT), likelihood ratio test (LRT) and Mutation Taster 2.
Figure 5. Maculopapular erythematous rash over lower limb (A,B) that was initially diagnosed as Henoch Scholein purpura in a patient with PLCG2 (Table 2; patient no 32).
Patients no. 33 had scaring photosensitive rash and a provisional diagnosis of Kindler syndrome was made (Mahajan et al. manuscript in submission). He was also detected to have same mutations in PLCG2 gene as patient no. 31. Both these patients were unrelated and belonged to different ethnic backgrounds. They had multiple relapses and both succumbed to their illness.
Clinical Profile of Patients With Non-Inflammasome Related Conditions
Thirty patients (38%) in our cohort were grouped under non-inflammasome related conditions that included TNF receptor-associated periodic syndrome (TRAPS) (three patients); Deficiency of the Interleukin 1 Receptor Antagonist(DIRA) (two patients); Pyogenic sterile Arthritis, Pyoderma Gangrenosum, Acne syndrome (PAPA) (one patient); A20 haploinsufficiency (four patient); CCA-adding transfer RNA nucleotidyl transferase (TRNT1) (two patients); Caspase recruitment domain-containing protein 14 (CARD 14) (one patients); and Laccase Domain Containing 1(LACC1) (three patients from one family) (Table 3). Patients with Blau syndrome (14 patients are not being presented in this paper (Suri et al., manuscript in submission). Patient with LACC1 has also been reported previously (27) (Table 3).
Table 3. Clinical manifestations, molecular profile, treatment and outcomes of patients with non-inflammasome-related conditions (n = 16).
TRAPS
Patient no. 34 had recurrent episodes of fever lasting for 2–3 weeks every 3–4 months with rash since 18 months of age. These f episodes were associated with pain abdomen, myalgias, arthritis, periorbital edema (Figure 6), and subcutaneous swellings. She had received multiple courses of antimicrobials in view of marked polymorphonuclear leukocytosis. Her father was also symptomatic and used to have fever and intermittent subcutaneous swelling and rash. Father was diagnosed to have acute rheumatic fever in childhood. In view of periodic fever, with systemic manifestations and family history suggestive of autosomal dominant disorder, diagnosis of TRAPS was proffered and confirmed on exome sequencing. She was initially managed with corticosteroids followed by injection etanercept. She remains well at follow up.

Figure 6. Unilateral periorbitaledema without conjunctivitis noted in child with TRAPS during flares (Table 3; Patient no 34).
A 45 years old female (patient no. 35) who had been under follow-up of Dermatology services was diagnosed to have pustular psoriasis since early adolescence. She would have intermittent flares with rash, fever and arthritis. I Her disease was refractory to methotrexate, cyclosporin and corticosteroids. In view of recurrent episodes of fevers, markedly elevated inflammatory parameters, sterile neutrophilic infiltrates on skin biopsy, a possibility of autoinflammatory disease was considered. Whole exome sequencing revealed variant in TNF Receptor Superfamily Member 1A(TNFRSF1A) mutation. She could not be initiated on biological agents due to financial constraints and succumbed to her illness 2 years after diagnosis was established.
A 10-year-old boy (patient no 36) had presented with high grade fever without focus. He reported having febrile episodes lasting for 3–4 weeks with variable afebrile periods since early childhood. These episodes were associated with rash over the trunk and limbs, myalgia and limp, abdominal pain, vomiting, and periorbital swelling. The inflammatory parameters were elevated and targeted panel for autoinflammatory diseases confirmed the diagnosis of TRAPS. The patient demonstrated partial response to etanercept which was changed to tocilizumab to which he responded well.
DIRA
Patient no 37 as has been previously reported (26), was the first Indian patient with large deletion in Interleukin 1 Receptor Antagonist (IL1RN) gene. She is doing well on Anakinra at 6 years of follow-up supported by National Institutes of Health, USA.
Patient no 38 had presented at day 7 of life with paucity of movement of both upper limbs. Inflammatory parameters were increased with sterile blood cultures. X-rays showed bilateral humerus, rib and clavicular involvement. He was treated with oral prednisolone 2 mg/Kg with slow tapper over 4 months. He responded dramatically and bone lesions healed. He developed pustules at follow up. Deletion in IL1RN gene as in patient no 37 was not detected on Western blot analysis. Results of whole exome sequencing are awaited. He is currently doing well and off corticosteroids. However, ESR remains elevated.
PAPA Syndrome
A 4-years old girl (patient no 39) had been unwell for 2.5 years when she presented with periodic fevers associated with painful oral ulcers, abdominal pain with hematochezia and colitis. Over the years, she developed multiple pyoderma gangrenosum lesion over extremities, angle of mouth and gluteal region that caused complete destruction of left cheek and lower lip. The lesion were difficult to heal and resulted in fistulae formation. She was initially suspected to have inflammatory bowel disease and oral prednisolone and azathioprine were initiated. Injection infliximab (3 doses) were also commenced. There was partial response in skin lesions and colitis initially. However, lesions reoccurred, and she succumbed to her illness.
A20 Haploinsufficiency
Patient 40 was 2 years old when she had presented with recurrent oral ulcers and genital ulcers (Figure 7). She had colitis, refectory ulcers requiring repeated hospitalization. Markers of inflammation were elevated, and Human Leucocyte Antigen 51 (HLA B 51) allele was detected. Considering a possibility of Bechet's disease, she was commenced on corticosteroid and azathioprine. At 5 years, she was readmitted with persistent headache, blurring of vision and relapse of oro-genital ulcers. She had papilledema and magnetic resonance imaging (MRI) of brain revealed type 2 Arnold Chiari malformation. When younger brother also developed recurrent oral ulcers, genetic studies were performed in the index patent and targeted panel revealed a novel variant in Tumor necrosis factor alpha-induced protein 3 (TNFAIP3) gene (Table 3). She is doing well on follow up. Mother is carrier for the same variant while the younger sibling does not carry this variant. Patient 41 had also presented with early onset inflammatory bowel disease and oral ulcers and a novel variant in TNFAIP3) gene was found.
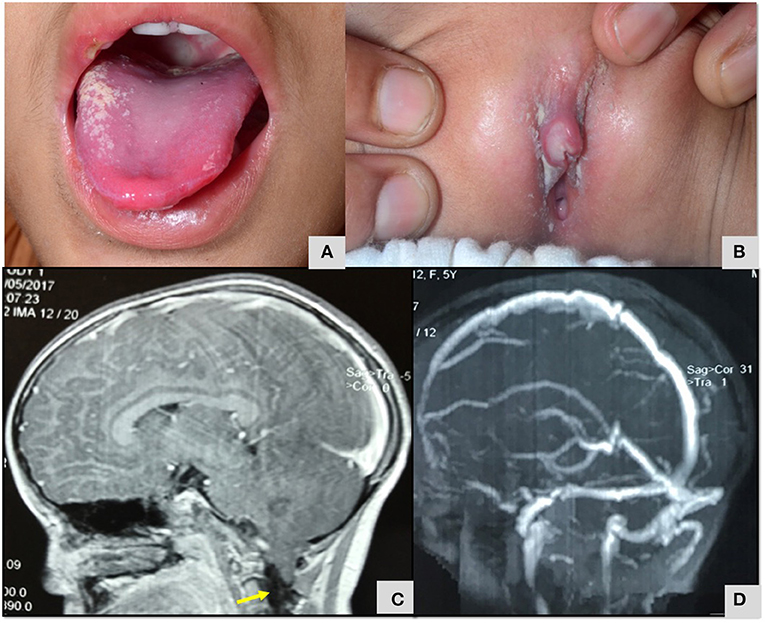
Figure 7. (A,B) Oral cavity erythema and ulceration and genital ulcers in child with A20 haploinsufficiency (Table 3; Patient no 40). (C) MRI Brain T2 weighted sagittal section images demonstrating type 2 Arnold Chiari malformation (Table 3; Patient no 40). (D) MR venography of brain demonstrated normal flow and no evidence of cerebral sinus venous thrombosis (Table 3; Patient no 40).
TRNT1 Deficiency
Patient no. 44, had presented with recurrent fever, and diarrhea. Two elder brothers earlier had died. He was evaluated for primary immune deficiency and was noted to have pan hypogammaglobulinemia. A provisional diagnosis of X-linked agammaglobulinemia was made and intravenous immunoglobulin replacement initiated. Exome sequencing revealed a compound heterozygous mutation in exon 2 of TRNT1 gene. Similarly, patient no. 48 also had recurrent infections, bronchiectasis and hypogammaglobulinemia.
CARD 14: Patient 46had early onset difficult to treat psoriasis and was found to have mutations in CARD 14 gene.
Discussion
SAID were first recognized in 1999 (4). Over the last two decades, knowledge and recognition of SAID has grown at an unprecedented speed and there have been a plethora of publications on this subject (3, 12, 13, 22). Significant improvement in understanding of genetic and pathogenic mechanisms of SAIDs has resulted in remarkable progress in their management. However, the darta from India is limited (14–18, 26, 27, 33–40). There is no national registry for SAID and there is lack of knowledge on nationwide burden of these diseases. This manuscript is the first attempt to collate data from various centers involved in care of patients with SAID and highlight diagnostic difficulties, treatment, and outcomes of such patients in India.
SAID display wide range of clinical manifestations and can affect almost every organ system. They are uncommon and difficult to diagnose clinically. Overlapping clinical features of these disorders with other relatively common rheumatological disorders often lead to delayin diagnosis. At Chandigarh, we diagnosed our first child with NOMID in 2005. This child was being managed as systemic onset JIA for over 10 years. Similarly, patients with Blau syndrome were initially treated as JIA with uveitis, and patients with DADA2 as PAN. Other initial diagnosis included systemic lupus erythematosus, inflammatory bowel disease and Bechet's disease. With improved awareness amongst internists and pediatrician along with availability of affordable diagnostic techniques, these syndromes are now being suspected and diagnosed early.
Diagnosis of most SAID is based on clinical suspicion, family history and demonstration of elevated inflammatory parameters (ESR, CRP, serum amyloid A protein). Distinct interferon signatures and cytokine patterns may be helpful biomarkers to stratify and monitor patients. However, interpretation and standardization of these tests is difficult. Despite expansion of various laboratory investigations, these investigations are not yet available for clinical use in India. Genetic analysis is needed in all patients for confirmation of diagnosis. In the past, most genetic studies were performed in collaboration with international centers. In the last 5 years, molecular diagnostic techniques were established at PGIMER Chandigarh and National Institute of Immunohematology Mumbai, which are Indian Council of Medical Research (ICMR) recognized Centers for Advanced Research (CAR) in Primary Immune Deficiency Diseases. However, diagnostic facilities are still limited. At PGIMER, among SAID, we can perform Sanger sequencing for ADA2 and NOD2 genes. In recent years, NGS based targeted autoinflammatory panels are available in commercial laboratories, albeit expensive. Interpretation of data and functional validation of variants of unknown significance (VUS) detected remains a challenge.
Management of SAID is aimed at suppression of systemic inflammation. Colchicine and glucocorticoids have been traditionally used to treat SAID. However, with improved knowledge and understanding of pathogenic mechanisms of autoinflammation and availability of specific targeted immunotherapies, treatment strategies have been completely revolutionized (41, 42). Anti-IL-1 drugs (anakinra, canakinumab, and rilonacept), have become standard of care for most inflammasomopathies. These agents successfully control inflammation and improve growth and quality of life. Other biologic agents used include anti-TNF drugs (etanercept), anti-IL-6 drugs (tocilizumab), and Janus kinase inhibitors (tofacitinib, baricitinib, and ruxolitinib). Treatment of SAID is extremely challenging in resource constrained settings. Anti-IL-1 drugs are not readily available in India and other developing countries. These drugs have to be imported on a “named-patient-basis” and are exorbitantly expensive. Some biosimilar molecules like anti TNF (adalimumab, infliximab) and anti IL6 (tocilizumab) are available alternative therapies. Though these molecules are cost cheaper in India when compared to Western countries, yet they remain well beyond the scope of average Indian family. Thus, corticosteroids and other conventional immunosuppressive agents still form the main stay of therapy. Patients often require higher doses as the disease progresses and they often develop corticosteroid related side effects. Off late, hematopoietic stem cell transplant (HSCT) is also emerging as a curative option for some SAID (43, 44). However, none of our patients received HSCT.
AID related morbidity and mortality continues to be high. In our cohort (8/49) have died at the time of analysis due to non-availability of treatment and development of complications. Amyloidosis had already developed in four patients at the time of diagnosis and it remains an important cause of death.
There are several limitations of this study. It is a case-based record review report from major primary immunodeficiency diseases centers across the country. Data from various individual rheumatology units could not be collated. Moreover, shared data were not uniform from all centers. Patients with unclassified SAID and patients in whom molecular diagnosis could not be established were excluded from the study.
This is a first comprehensive multicentric report of patients with SAID from India. Varied clinical and molecular spectrum has been reported. Considerable delays in diagnosis were recognized. Application of NGS based targeted panels and whole exome sequencing has helped in identifying known as well as novel gene defects. Establishment of diagnosis in a patient enabled early diagnosis of other family members and provided an opportunity for prenatal diagnosis. Although, ability to diagnose SAID has improved, non-availability of expensive immunotherapies remains a major drawback. In India corticosteroids and conventional immunosuppressive agents continue to remain corner stones for treatment. Lack of availability of targeted immunotherapies for treatment prevents the initiation of effective treatments that can change patients' lives. SAID continue to result in significant morbidity and mortality.
To conclude, more efforts are needed to enhance awareness of autoinflammatory diseases among health care professionals and there is an urgent need to make life saving drugs universally available.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The studies involving human participants were reviewed and approved by Institute Ethics Committee, PGIMER, Chandigarh (Ref No: INT/IEC/2021/SPL-264). Written informed consent from the participants' legal guardian/next of kin was not required to participate in this study in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the minor(s)' legal guardian/next of kin for the publication of any potentially identifiable images or data included in this article.
Disclosure
RG-M and AA are supported by the Intramural Research Program of NIAID, National Institutes of Health, Bethesda, Maryland, USA.
Author Contributions
DS, AR, AJ, PV, AG, and RP: data collection, writing of initial draft, editing of manuscript at all stages of its production, patient management, and review of literature. DS, AR, AJ, PV, AG, RP, VJ, KA, RK, GA, AA, SP, FN, BG, EE, MD, PT, VG, AP, SB, and SK: data collection, management of patients, and review of final manuscript. AR, VJ, KA, RK, SP, FN, BG, and ES: genetic evaluation and data collection. MG, IC, AAdJ, and RG-M: genetic evaluation, review of final manuscript, and critical revision. AR, SB, SK, MG, IC, AAdJ, RG-M, and SS: genetic evaluation and review of the final manuscript. MH: performed ADA2 levels in patients with DADA two patients. DS, AR, and SS: patient management, review of literature, editing and critical revision of manuscript at all stages of its production, and final approval of manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by India Council of Medical Research (ICMR), New Delhi, India, and Department of Health Research, Ministry of Health and Family Welfare, Government of India, New Delhi, India for funding (vide Grant No. GIA/48/2014-DHR). However, the funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Kastner DL, Aksentijevich I, Goldbach-Mansky R. Autoinflammatory disease reloaded: a clinical perspective. Cell. (2010) 140:784–90. doi: 10.1016/j.cell.2010.03.002
2. Marino A, Tirelli F, Giani T, Cimaz R. Periodic fever syndromes and the autoinflammatory diseases (AIDs). J Transl Autoimmun. (2020) 3:100031. doi: 10.1016/j.jtauto.2019.100031
3. Stoffels M, Kastner DL. Old dogs, new tricks: monogenic autoinflammatory disease unleashed. Annu Rev Genomics Hum Genet. (2016) 31:245–72. doi: 10.1146/annurev-genom-090413-025334
4. McDermott MF, Aksentijevich I, Galon J, McDermott EM, Ogunkolade BW, Centola M, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell. (1999) 97:133–44. doi: 10.1016/S0092-8674(00)80721-7
5. Aksentijevich I, Centola M, Deng Z, Sood R, Balow JE, Wood G, et al. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The international FMF Consortium. Cell. (1997) 90:797–807. doi: 10.1016/s0092-8674(00)80539-5
6. Samuels J, Aksentijevich I, Torosyan Y, Centola M, Deng Z, Sood R, et al. Familial Mediterranean fever at the millennium. Clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health. Medicine (Baltimore). (1998) 77:268–97. doi: 10.1097/00005792-199807000-00005
7. Ben-Chetrit E, Gattorno M, Gul A, Kastner DL, Lachmann HJ, Touitou I, et al. Consensus proposal for taxonomy and definition of the autoinflammatory diseases (AIDs): a Delphi study. Ann Rheum Dis. (2018) 77:1558–65. doi: 10.1136/annrheumdis-2017-212515
8. Gattorno M, Hofer M, Federici S, Vanoni F, Bovis F, Aksentijevich I, et al. Classification criteria for autoinflammatory recurrent fevers. Ann Rheum Dis. (2019) 78:1025–32. doi: 10.1136/annrheumdis-2019-215048
9. Navallas M, Inarejos Clemente EJ, Iglesias E, Rebollo-Polo M, Zaki FM, Navarro OM. Autoinflammatory diseases in childhood, part 1: monogenic syndromes. Pediatr Radiol. (2020) 50:415–30. doi: 10.1007/s00247-019-04536-9
10. Navallas M, Inarejos Clemente EJ, Iglesias E, Rebollo-Polo M, Hernández JC, Navarro OM. Autoinflammatory diseases in childhood, part 2: polygenic syndromes. Pediatr Radiol. (2020) 50:431–44. doi: 10.1007/s00247-019-04544-9
11. Tangye SG, Al-Herz W, Bousfiha A, Chatila T, Cunningham-Rundles C, Etzioni A, et al. Human inborn errors of immunity: update on the classification from the international union of immunological societies expert committee. J Clin Immunol. (2020) 40:24–64. doi: 10.1007/s10875-019-00737-x
12. Oda H, Kastner DL. Genomics, biology, and human illness: advances in the monogenic autoinflammatory diseases. Rheum Dis Clin North Am. (2017) 43:327–45. doi: 10.1016/j.rdc.2017.04.011
13. Stone DL, Beck DB, Manthiram K, Park YH, Chae JJ, Remmers E, et al. The systemic autoinflammatory diseases: coming of age with the human genome. J Allergy Clin Immunol. (2020) 146:997–1001. doi: 10.1016/j.jaci.2020.09.014
14. Lawrence A, Hol F, Aggarwal A, Drenth JPH. Hyperimmunoglobulinaemia D syndrome in India: report of two siblings with a novel mutation. Ann Rheum Dis. (2006) 65:1674–6. doi: 10.1136/ard.2006.054494
15. Govindaraj GM, Jain A, Peethambaran G, Bhoyar RC, Vellarikkal SK, Ganapati A, et al. Spectrum of clinical features and genetic variants in mevalonate kinase (MVK) gene of South Indian families suffering from Hyperimmunoglobulin D Syndrome. PLoS ONE. (2020) 15:e0237999. doi: 10.1371/journal.pone.0237999
16. Babu K, Rao AP. Clinical profile in genetically proven blau syndrome: a case series from South India. Ocul Immunol Inflamm. (2020). doi: 10.1080/09273948.2020.1746353. [Epub ahead of print].
17. Gupta L, Ahmed S, Singh B, Prakash S, Phadke S, Aggarwal A. Novel NLRP12 variant presenting with familial cold autoimmunity syndrome phenotype. Ann Rheum Dis. (2019). doi: 10.1136/annrheumdis-2019-216158 [Epub ahead of print].
18. Jindal AK, Pilania RK, Suri D, Gupta A, Gattorno M, Ceccherini I, et al. A young female with early onset arthritis, uveitis, hepatic, and renal granulomas: a clinical tryst with Blau syndrome over 20 years and case-based review. Rheumatol Int. (2021) 41:173–81. doi: 10.1007/s00296-019-04316-6
19. Anjani G, Jindal AK, Prithvi A, Kaur A, Rawat A, Sharma M, et al. Deforming polyarthritis in a north indian family-clinical expansion of STING-Associated Vasculopathy with Onset in Infancy (SAVI). J Clin Immunol. (2021) 41:209–11. doi: 10.1007/s10875-020-00872-w
20. Jindal AK, Pilania RK, Rawat A, Singh S. Primary immunodeficiency disorders in india-a situational review. Front Immunol. (2017) 8:714. doi: 10.3389/fimmu.2017.00714
21. Pilania RK, Chaudhary H, Jindal AK, Rawat A, Singh S. Current status and prospects of primary immunodeficiency diseases in Asia. Genes Dis. (2020) 7:3–11. doi: 10.1016/j.gendis.2019.09.004
22. Wekell P, Berg S, Karlsson A, Fasth A. Toward an inclusive, congruent, and precise definition of autoinflammatory diseases. Front Immunol. (2017) 8:497. doi: 10.3389/fimmu.2017.00497
23. Brodszki N, Frazer-Abel A, Grumach AS, Kirschfink M, Litzman J, Perez E, et al. European Society for Immunodeficiencies (ESID) and european reference network on rare primary immunodeficiency, autoinflammatory and autoimmune diseases (ERN RITA) complement guideline: deficiencies, diagnosis, and management. J Clin Immunol. (2020) 40:576–91. doi: 10.1007/s10875-020-00754-1
24. Lepelley A, Martin-Niclós MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. J Exp Med. (2020) 217:e20200600. doi: 10.1084/jem.20200600
25. Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. J Exp Med. (2020) 217:20201045. doi: 10.1084/jem.20201045
26. Mendonca LO, Malle L, Donovan FX, Chandrasekharappa SC, Montealegre Sanchez GA, Garg M, et al. Deficiency of interleukin-1 receptor antagonist (DIRA): report of the first indian patient and a novel deletion affecting IL1RN. J Clin Immunol. (2017) 37:445–51. doi: 10.1007/s10875-017-0399-1
27. Singh A, Suri D, Vignesh P, Anjani G, Jacob P, Girisha KM. LACC1 gene mutation in three sisters with polyarthritis without systemic features. Ann Rheum Dis. (2020) 79:425–6. doi: 10.1136/annrheumdis-2019-216263
28. Ben-Ami T, Revel-Vilk S, Brooks R, Shaag A, Hershfield MS, Kelly SJ, et al. Extending the clinical phenotype of adenosine deaminase 2 deficiency. J Pediatr. (2016) 177:316–20. doi: 10.1016/j.jpeds.2016.06.058
29. Sharma A, Naidu G, Sharma V, Jha S, Dhooria A, Dhir V, et al. Deficiency of adenosine deaminase 2 in adults and children: experience from India. Arthritis Rheumatol. (2021) 73:276–85. doi: 10.1002/art.41500
30. Banday AZ, Kaur A, Jindal AK, Patra PK, Guleria S, Rawat A. Splice-site mutation in COPA gene and familial arthritis - a new frontier. Rheumatology. (2021) 60:e7–e9. doi: 10.1093/rheumatology/keaa354
31. Pandiarajan V, Gupta A, Rowczenio D, Hawkins P, Muralidaran C, Tiewsoh K, et al. Nephrotic syndrome as a presenting feature in a child with NLRP3 mutation. J Clin Rheumatol. (2018). doi: 10.1097/RHU.0000000000000942 [Epub ahead of print].
32. Sankhyan N, Kalra V, Aksentijevich I, Kabra M, Gulati S, Sharma S. Atypical late presentation in neonatal-onset multisystem inflammatory disease (NOMID). J Pediatr Neurol. (2015) 7:301–5. doi: 10.3233/JPN-2009-0297
33. Janarthanan M, Poddar C, Sudharshan S, Seabra L, Crow YJ. Familial Blau syndrome: First molecularly confirmed report from India. Indian J Ophthalmol. (2019) 67:165–7. doi: 10.4103/ijo.IJO_671_18
34. Khubchandani R, Aksentijevich I. Deficiency of adenosine deaminase 2 (DADA2) - a new autoinflammatory disease with multisystem features. Indian Pediatr. (2020) 2020:S097475591600231. doi: 10.1007/s13312-020-2041-1
35. Correa ARE, Gupta N, Bagri N, Vignesh P, Alam S, Yamaguchi S. Mevalonate kinase deficiency as cause of periodic fever in two siblings. Indian Pediatr. (2020) 15:180–1. doi: 10.1007/s13312-020-1742-9
36. Sandhya P, Vellarikkal SK, Nair A, Ravi R, Mathew J, Jayarajan R, et al. Egyptian tale from India: application of whole-exome sequencing in diagnosis of atypical familial Mediterranean fever. Int J Rheum Dis. (2017) 20:1770–5. doi: 10.1111/1756-185X.13042
37. Nair SB, Chavan PP, Athalye AS, Aksentijevich I, Khubchandani RP. Detection of a novel mutation in NLRP3/CIAS1 gene in an Indian child with Neonatal-Onset Multisystem Inflammatory Disease (NOMID). Clin Rheumatol. (2019) 38:403–6. doi: 10.1007/s10067-018-4225-9
38. Raghawan AK, Ramaswamy R, Radha V, Swarup G. HSC70 regulates cold-induced caspase-1 hyperactivation by an autoinflammation-causing mutant of cytoplasmic immune receptor NLRC4. Proc Natl Acad Sci USA. (2019) 22;116:21694–703. doi: 10.1073/pnas.1905261116
39. Gupta A, Tripathy SK, Phulware RH, Arava S, Bagri NK. Cryopyrin-associated periodic fever syndrome in children: a case-based review. Int J Rheum Dis. (2020) 23:262–70. doi: 10.1111/1756-185X.13772
40. Ghosh K, Mishra K, Shah A, Patel P, Shetty S. Novel deleterious sequence change in the NLRP12 gene in a child with the autoinflammatory syndrome, joint hypermobility and cutis Laxa from India. Mediterr J Hematol Infect Dis. (2019) 11:e2019018. doi: 10.4084/mjhid.2019.018
41. Soriano A, Soriano M, Espinosa G, Manna R, Emmi G, Cantarini L, et al. Current therapeutic options for the main monogenic autoinflammatory diseases and PFAPA syndrome: evidence-based approach and proposal of a practical guide. Front Immunol. (2020) 11:865. doi: 10.3389/fimmu.2020.00865
42. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol. (2017) 18:826–31. doi: 10.1038/ni.3790
43. Liu L, Wang W, Wang Y, Hou J, Ying W, Hui X, et al. A Chinese DADA2 patient: report of two novel mutations and successful HSCT. Immunogenetics. (2019) 71:299–305. doi: 10.1007/s00251-018-01101-w
Keywords: systemic autoinflamatory diseases, India, deficiency of adenosine deaminase 2, NOMID/CINCA, hyper IgD syndrome, A20 (TNFAIP3), inflammasome, Type I interferonopathies
Citation: Suri D, Rawat A, Jindal AK, Vignesh P, Gupta A, Pilania RK, Joshi V, Arora K, Kumrah R, Anjani G, Aggarwal A, Phadke S, Aboobacker FN, George B, Edison ES, Desai M, Taur P, Gowri V, Pandrowala AA, Bhattad S, Kanakia S, Gottorno M, Ceccherini I, Almeida de Jesus A, Goldbach-Mansky R, Hershfield MS and Singh S (2021) Spectrum of Systemic Auto-Inflammatory Diseases in India: A Multi-Centric Experience. Front. Immunol. 12:630691. doi: 10.3389/fimmu.2021.630691
Received: 18 November 2020; Accepted: 22 February 2021;
Published: 19 March 2021.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Hal M. Hoffman, University of California, San Diego, United StatesMario Abinun, Newcastle upon Tyne Hospitals NHS Foundation Trust, United Kingdom
Copyright © 2021 Suri, Rawat, Jindal, Vignesh, Gupta, Pilania, Joshi, Arora, Kumrah, Anjani, Aggarwal, Phadke, Aboobacker, George, Edison, Desai, Taur, Gowri, Pandrowala, Bhattad, Kanakia, Gottorno, Ceccherini, Almeida de Jesus, Goldbach-Mansky, Hershfield and Singh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deepti Suri, c3VyaWRlZXB0aUBnbWFpbC5jb20=