 Ahmed Sheriff
Ahmed Sheriff Stefan Kayser
Stefan Kayser Patrizia Brunner3
Patrizia Brunner3 Birgit Vogt
Birgit Vogt
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 10 February 2021
Sec. Molecular Innate Immunity
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.630430
This article is part of the Research TopicDiagnostic and Therapeutic Applications of Pentraxin and Pentraxin-Associated ProteinsView all 20 articles
C-reactive protein (CRP) is the best-known acute phase protein. In humans, almost every type of inflammation is accompanied by an increase of CRP concentration. Until recently, the only known physiological function of CRP was the marking of cells to initiate their phagocytosis. This triggers the classical complement pathway up to C4, which helps to eliminate pathogens and dead cells. However, vital cells with reduced energy supply are also marked, which is useful in the case of a classical external wound because an important substrate for pathogens is disposed of, but is counterproductive at internal wounds (e.g., heart attack or stroke). This mechanism negatively affects clinical outcomes since it is established that CRP levels correlate with the prognosis of these indications. Here, we summarize what we can learn from a clinical study in which CRP was adsorbed from the bloodstream by CRP-apheresis. Recently, it was shown that CRP can have a direct effect on blood pressure in rabbits. This is interesting in regard to patients with high inflammation, as they often become tachycardic and need catecholamines. These two physiological effects of CRP apparently also occur in COVID-19. Parts of the lung become ischemic due to intra-alveolar edema and hemorrhage and in parallel CRP increases dramatically, hence it is assumed that CRP is also involved in this ischemic condition. It is meanwhile considered that most of the damage in COVID-19 is caused by the immune system. The high amounts of CRP could have an additional influence on blood pressure in severe COVID-19.
Inflammation in humans is deeply evolutionary rooted. A quick and intense inflammatory response is required for the efficient eradication of injury and was highly beneficial in times where external wounds or life-threatening infections where the main—if not only—cause of damage to the body (1). Inflammation as a weapon against environmental risks and triggers is unfortunately a two-edged sword, because it is dangerous when turned against the own body. In modern times, an elevated inflammatory function is thought to be associated with higher risk to develop atherosclerosis, diabetes and other age-related diseases, which are not caused by pathogens (2). But an enhanced immune system can not only facilitate cardiovascular disease, it also exacerbates acute incidents, which are “sterile.” In order to heal internal wounds, the body needs the inflammatory reaction to eliminate dead cells. At the same time activating proliferation and repair mechanisms as well as restoring tissue homeostasis is essential. This immune response is, however, since centuries specialized on the thorough eradication of cells around a wound to minimize the risk of infection. Even if these cells are only energy deprived but still viable. In the setting of an internal wound, as e.g., myocardial infarction or stroke, this effect is often destructive and threatening as it only aggravates deterioration and involves severe collateral damage (3, 4).
Likewise, an excessive immune reaction not justified by its external trigger induces more negative than positive effects on the body (5). During a systemic inflammatory response syndrome or sepsis the inflammation is so enormous and disproportionate that it causes widespread tissue injury and might finally result in multiple organ failure (6). Here, the whole body is often affected by inflammation showing hard to control hemodynamic instability.
Although a multitude of proteins are involved in inflammation, most of them do not actively participate in the elimination of pathogens or human cells (1). One of the acute-phase mediators directly involved in these pro-inflammatory processes is C-reactive protein (CRP) which was discovered by Tillett and Francis (7). CRP is well-established as one of the most reliable markers of inflammation, rising dramatically during any type of inflammation.
There are several very good and extensive reviews published, summarizing the role of CRP as unspecific inflammatory marker and its history from discovery to world-wide used lab marker (8–11). Even though CRP has been investigated in numerous clinical studies and its association and correlation with the progress of certain diseases is evidently clear (12–19), evidence that it is a mediator of the respective disease in humans was missing while strong evidence exists for different animal species (16, 20–22). In addition, CRP can dissociate into monomers physiologically, although it is still under debate if it thereby exerts different molecular functions than the pentameric form (23). The transition of pentameric CRP to monomeric CRP was described in specific inflammatory microenvironments (24, 25). Pro-inflammatory isoforms of pentameric and monomeric CRP were reported (26). The circulating CRP is pentameric and the actual source of all further processes. Despite this extensive in-depth knowledge, the widespread opinion is still that in humans CRP is only an unspecific biomarker.
We want to focus on the controversy/debate that CRP also in humans is not only a marker but an active pro-inflammatory protein, which contributes causally to the severity of tissue damage and the outcome of various diseases (27).
CRP is secreted by the liver into the blood circulation where it efficiently detects and opsonizes bacteria upon their infiltration (28, 29). By marking these pathogens, it initiates their phagocytosis via activation of complement. This mechanism is mainly caused by the binding of CRP to the phosphorylcholine groups in the membrane of bacteria. These groups, however, are also present in all human cells, albeit not accessible on healthy cells. Cells that are apoptotic, necrotic, energy-depleted or simply exposed to inflammatory environments, often being acidic and hypoxic, undergo conformational and biochemical changes in their membrane (30). One of these being the partial hydrolyzation of phosphatidylcholine (PC) to lyso-phosphatdiylcholine (LPC) by another acute-phase-protein, namely the secretory phospholipase A2 type IIa (sPLA2 IIa) (31–34). This makes the phosphorylcholine group accessible to the binding of CRP. Hence, CRP irreversibly marks dying, dead, damaged or hypoxic/ischemic cells. Subsequently, the classical complement pathway is activated and the CRP-marked cells are disposed by phagocytosis (35–40). See Figure 1 for the hypothesized pathomechanism of CRP after an acute phase response caused by inflamed or hypoxic/ischemic tissue.
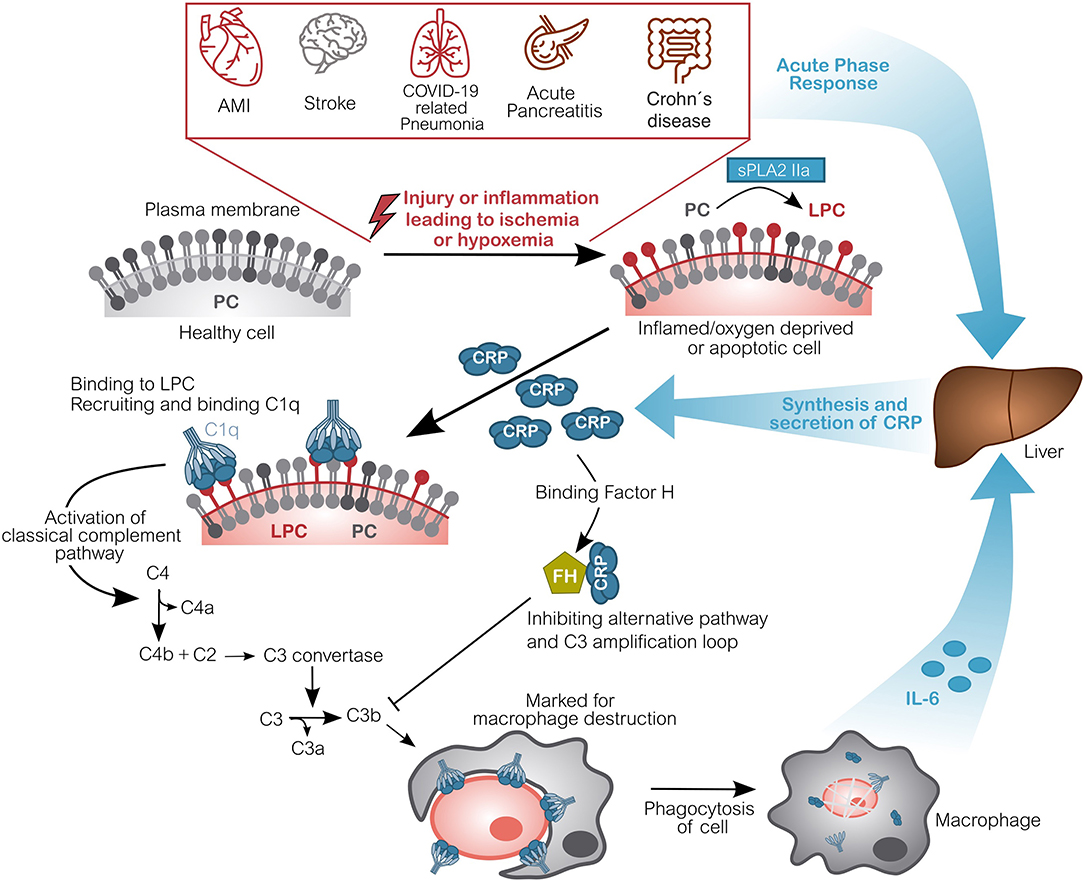
Figure 1. Molecular pathomechanism of CRP-mediated damage in ischemic or hypoxemic tissue. Inflammation or acute oxygen-deprivation happens for example in acute myocardial infarction (AMI), stroke, during COVID-19 related pneumonia, acute pancreatitis as well as during an acute Crohn's disease relapse. This leads to energy-depleted, hypoxic or even ischemic tissue. Cells within this tissue display a modified outer cell membrane: Phosphatidylcholine (PC) is converted into lyso-phosphatidylcholine (LPC) by phospholipase (sPLA2 IIa). Due to the lack of energy, this alteration cannot be reversed. CRP subsequently binds to LPC on anaerobic cells and recruits complement factors (C1q-C4), activating the classical complement pathway. These opsonized cells will be disposed by phagocytes, which in turn induce CRP synthesis by secretion of IL-6. CRP also binds Factor H, which inhibits the alternative complement pathway and actually protects healthy host cells from disposal (16, 22, 79). Although CRP is drawn pentameric, it should be noted that after binding to ischemic cells, the recruitment of C1q and particularly FH is potentially carried out by dissociated, monomeric CRP. Figure adapted from (41). AMI, Acute Myocardial Infarction; CRP, C-reactive protein; C1q, Complement component 1q; C2-C4(a/b), Complement component 2-4(a/b); FH, Factor H; IL-6, Interleukin 6; LPC, Lysophosphatidylcholine; PC, Phosphatidylcholine; sPLA2 IIa secretory phospholipase A2 type IIa.
In the setting of an internal wound this generates a vicious cycle: The primary inflammation triggered by e.g., ischemia (e.g., acute myocardial infarction or stroke) activates a switch to anaerobic metabolism and a striking synthesis and secretion of CRP mediated by IL-6. CRP is circulated to the wound, where it mediates the disposal of dead and dying cells. High CRP concentrations cause more cells to be marked, including still viable cells, which could have regenerated their membrane after restoration of the oxygen flow and switching back to aerobic metabolism. The phagocytosis of these cells in turn produces IL-6, inducing the synthesis of additional CRP, subsequently amplifying the immune response. Thereby, CRP causally contributes to the tissue damage and scarring after an incident [Figure 1, adapted from (41)] (22, 42–44). It should be noted that CRP is synthesized and secreted as a pentamer by the liver, however, it can dissociate into monomers within the microenvironment of the inflamed/ischemic tissue and might exert the drawn functions as monomer (Figure 1).
Although not every step of this molecular mechanism has been proven and shown in detail, there have been convincing proof-of-concept studies in animals substantiating this hypothesis. A large body of data obtained either in rats, porcine models or in vitro in the infarcted myocardium of humans has demonstrated that CRP plays an active role in exacerbating ischemia and reperfusion-induced damage (16, 22, 45–50).
Recent studies exceedingly revealed that CRP modulates signaling cascades besides the classical complement pathway (51–54). This shows that CRP has direct physiological effects on not only inflammation but also the function of e.g., endothelial cells, be it their metabolism, differentiation or migration (55–57). In the context of endothelial cells, it is also discussed if CRP might have protective effects in atherosclerotic lesions. It was shown that, although CRP induces complement activation, it protects the bound cells from the formation of final complement components (58, 59), mainly by recruiting complement factor H (60). The role of CRP in atherosclerosis is however still under debate and cannot be compared to acute ischemic incidents regarding the circulating concentration of CRP.
One of these recent studies showed a direct, quick and extreme effect of CRP on blood pressure in rabbits (61). Human CRP was intravenously injected in vivo to reach a level of 50 mg/L. It dramatically reduced the arterial blood pressure within minutes, while the heart rate remained the same and did not counteract as expected and necessary to maintain the oxygen supply of the organism. The effect persisted for more than 17 min.
While it has been shown that blood pressure and heart rate as well as adrenergic receptor (AR) signaling can affect CRP concentrations (62–64), a direct influence of CRP on hemodynamic variables has hardly been investigated so far. Other in vivo studies administering CRP were performed in rats and humans (65–67). However, the recombinant CRP used in the studies never achieved concentrations of 50 mg/l and blood pressure was measured not directly but only hours after injection (65, 67). Hence, the acute effects might have been overlooked.
The direct and acute effect of high CRP concentrations on blood pressure gives a first hint, why critically ill patients, suffering from e.g., sepsis or acute pancreatitis, can develop hardly controllable hemodynamic variables with preceding elevated CRP levels (68).
After seeing such a dramatic drop in blood pressure the influence of CRP on AR signal transduction was investigated in vitro (61). ARs signal via calcium (Ca2+) as second messenger was measured in real-time. CRP triggered calcium signaling in a dose-dependent manner in two different human cell lines, expressing either α- or β-adrenergic receptors. Further, CRP induced an additional calcium increase that came on top of AR agonists phenlyephrine or isoprenaline. This indicates a molecular mechanism that is independent of adrenoceptor activation.
Effects of CRP on endothelial cells have mostly been attributed to its effect on endothelial nitric oxide synthase (eNOS), although studies have to be interpreted with care, as contaminating products in commercial CRP solutions, such as sodium azide, were possibly often causally involved (69–71). eNOS is activated by an increase in intracellular calcium, leading to vasodilatation (72, 73), which could explain the drop of blood pressure in vivo. The mechanism underlying CRP's induction of calcium influx is still undiscovered and should be investigated in future studies.
Therefore, it was demonstrated that one molecule of the inflammation cascade has an influence on blood pressure. The direct influence of other inflammatory mediators needs to be investigated.
Many reports following the hypothesis that CRP has pathological effects suffer from a mean to shut down its activity. Knock-out mice do not represent appropriate models, because CRP in mice fulfills different functions than in humans and does not act as an acute-phase protein (74). Other animal models like e.g., transgenic rabbits expressing human CRP have been used but here most of the investigations focused on atherosclerosis and not on acute incidents (75).
In humans the use of CRP-lowering drugs has so far not been successful, since CRP as acute-phase protein increases drastically within hours and its circulating levels need to be lowered quickly in an acute setting. This cannot be achieved by targeting its synthesis or using approaches, which take several days to affect CRP levels (16, 21, 76).
Selective immuno-adsorption of CRP from the serum avoids these problems and has been shown to efficiently reduce CRP concentrations by ~60% within hours (77–79). The elimination of pathogenic substances from the blood by means of extracorporeal apheresis is an established therapy in the clinical routine of numerous diseases.
CRP apheresis aims to remove CRP from the blood plasma after an infarction to reduce acute tissue damage and ischemic reperfusion damage (41). It has most extensively been utilized after acute myocardial infarction (AMI).
Patients recovering from a heart attack often suffer from a reduced quality of life and a very high risk of subsequent serious complications (e.g., heart failure, arrhythmias, second heart attack, death), which imposes an enormous burden on the healthcare system. It was observed that this risk correlates significantly with the size of myocardial injury and scarring (80, 81).
It has long been known that inflammation, mainly mediated by the innate immune system, expands myocardial injury. However, anti-inflammatory strategies to mitigate myocardial necrosis have so far failed, perhaps because these processes are also necessary for the healing and repair of the heart (3, 4, 82, 83). Whereas, baseline CRP values are recognized as a determinant of the incidence of cardiovascular disease (12, 14), serum CRP concentration after AMI correlates with the clinical outcome (19, 42, 44, 84–87). It is textbook knowledge that high CRP peaks in the first 72 h after AMI correlate with larger infarct size and higher mortality and the incidence of additional adverse cardiovascular events (42–44). This has been documented for four decades and is consistent with the described pathological function of CRP to eliminate cells in the area at risk (27, 30, 88, 89). This region comprises cells that might recover after revascularization and reperfusion, but are eventually destroyed by immune-mediated mechanisms, as explained above and shown in detail in numerous experimental approaches specifically focusing on AMI (20, 46, 48, 85, 90, 91).
It has therefore been suggested earlier to target CRP in AMI, but the therapeutic approaches were not clinically relevant or sufficiently rapid (16, 92–94). This changed when it was shown that specific extracorporeal removal of CRP by CRP apheresis resulted in a significant reduction of the infarct area and stabilization of the left ventricular ejection fraction (LVEF) in a preclinical trial in pigs (22, 50). An interesting observation is that the scar morphology of the animals after CRP apheresis was completely different from controls, supporting the hypothesis that CRP is directly involved in tissue destruction and scarring after the incident (22). Consequently, CRP apheresis has been used in one clinical trial and two case reports in patients with ST-elevation myocardial infarction (STEMI) (77, 79, 95).
In the CAMI-1 trial it was investigated whether specific depletion of CRP can reduce the size of myocardial infarction in humans. Eighty-three patients were included and the treatment was safe and well-tolerated (79). The extent of increase of CRP concentration during the first 32 h after STEMI significantly correlated with the infarct size in control patients. Patients with similar initial CRP increase who subsequently underwent CRP apheresis, had smaller infarct sizes and better LVEF and wall motion (strains) when compared to control patients. Surprisingly, some patients treated with CRP apheresis had not even minor infarct scars and a normal LVEF (96).
It is possible that the supply bottleneck in ischemic tissue does not immediately lead to tissue necrosis, but rather to a conversion of the energy metabolism into anaerobic glycolysis, which leads to a significant lack of energy of the individual cardiomyocytes (97). Afterwards, the cardiomyocytes go into stunning until the metabolism switches back to the aerobic one which eliminates the energy deficiency. This suggests that the cardiomyocytes only survive if they are not marked by CRP and thus disposed of by phagocytes.
This effect can possibly be applied to other ischemia-mediated injuries as for example stroke. Here, similar mechanisms to AMI take place and inflammation plays a crucial role during the occlusion but also subsequent therapeutic reperfusion of the tissue (98, 99). The inflammatory response after stroke has been discovered to be a key prognostic factor for patients (100, 101). High CRP concentrations during the first 48 h after the incident significantly predict immediate and long-term mortality as well as the overall prognosis (13, 101, 101–103). This is supported by pre-clinical evidence in a stroke model in rats and subsequent application of high amounts of human CRP (49). This strongly supports the hypothesis that CRP plays a similar pathological role as in AMI.
SARS-CoV-2 can lead to COVID-19 and induces pulmonary fibrosis and cardiac complications in a minor percentage of infected individuals, among other organ deterioration (104). A major therapeutic approach focuses on the treatment of acute respiratory distress syndrome, as it is the leading cause of mortality, followed by cardiac and septic complications. In the severe course of the disease a massive increase in the CRP concentration accompanied by an initial cytokine storm is followed by pulmonary fibrosis (105, 106). Intra-alveolar edema and hemorrhage is a common observation in the lungs of COVID-19 patients which leads to ischemic alveolar tissue. It may be assumed that CRP itself triggers tissue damage by binding to these ischemic cells and is thus also causally involved in the enlargement of the destroyed tissue and contributes to irreversible tissue destruction (22, 79). Both IL-6 and CRP increase dramatically in the course of clinical manifestation of COVID-19 (107) and rising CRP levels were shown to significantly predict the respiratory decline in patients (105). CRP levels also correlate with CT findings of COVID-19 patients (108). These findings further support the hypothesis that a significant increase in CRP is a signal of lung deterioration and disease progression.
Complement deposits were found by pathologists in the lungs of deceased COVID-19 patients. Among them was especially C1Q. Since C1Q also inhibits antiviral CD8+ effector T-cell responses, a higher frequency of CD8+PD1+ T-cells was found, possibly indicating T-cell exhaustion (109). Despite the depletion of the T cells, massive destruction in the lungs is found along with the extreme levels of CRP in the aggravated COVID-19 patients. C1Q is known to bind CRP after CRP binds the lysophosphatidylcholine of ischemic cells (22).
In addition, cardiac involvement was observed by MRI analysis in 78% of patients, and persistent myocardial inflammation was observed in 60% of patients with recent COVID-19 disease, regardless of pre-existing conditions, the severity and overall course of the acute disease and the time from initial diagnosis (110). Myocardial inflammation was suggested as the underlying mechanism (111, 112).
CRP apheresis provides a therapeutic approach to rapidly decrease the high CRP levels in COVID-19 patients before lung deterioration can progress (113). Until now, the therapeutic option of reducing the extremely high amount of CRP has been used once in the early phase of COVID-19 and in end-stage patients with one case being reported (113, 114). A clinical study in this indication would be beneficial and is currently planned.
The understanding of CRP has undergone two basic transitions. First, CRP has been established as a general biomarker of inflammation and infection in clinical practice. Then, its role as a stable and highly useful prognostic factor for cardiovascular and cerebral disease in healthy individuals has been widely acknowledged and utilized (17, 115). However, the characterization of CRP as not only a biomarker but also a mediator or even trigger of destruction of tissue in humans is still widely ignored (27, 46, 49).
CRP as an archaic protein of the innate immune system physiologically disposes cells and responds to almost every change in tissue homeostasis. From the perspective of the body's energy balance, one has to ask oneself why CRP is produced in large quantities by the liver in situations where it seems wiser to keep a proper energy reserve. Certainly not to provide us with a meaningful biomarker. It is more logical to recognize that it is provided in potentially septic wounds during enemy defense, to eliminate further cells, so that they do not serve the enemy for propagation. This function has unfortunately a negative effect on typically aseptic inner wounds. Further, the demonstration of a direct effect of CRP on blood pressure shows us that the molecular functions of CRP are still not comprehensively described and that the role of this protein is largely underestimated in critically ill patients.
Accepting CRP as an active inflammatory protein offers the promising possibility to therapeutically target CRP whenever the inflammatory reaction is too extensive or not beneficial. Ongoing and future clinical trials will illuminate whether this therapeutic approach will continue to prove its value.
AS and PB conceptualized and wrote the first draft. SK and BV made substantial corrections.
AS was CEO and shareholder of Pentracor GmbH. SK was employed by Pentracor GmbH. BV was shareholder and employee of Pentracor GmbH. PB was employed by iAdsorb GmbH.
1. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol. (2017) 18:826–31. doi: 10.1038/ni.3790
2. Ridker PM. On evolutionary biology, inflammation, infection, and the causes of atherosclerosis. Circulation. (2002) 105:2–4. doi: 10.1161/circ.105.1.2
3. Anzai T. Inflammatory mechanisms of cardiovascular remodeling. Circul J. (2018) 82:629–35. doi: 10.1253/circj.CJ-18-0063
4. Ong S-B, Hernández-Reséndiz S, Crespo-Avilan GE, Mukhametshina RT, Kwek X-Y, Cabrera-Fuentes HA, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Therap. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
5. Neher MD, Weckbach S, Flierl MA, Huber-Lang MS, Stahel PF. Molecular mechanisms of inflammation and tissue injury after major trauma–is complement the “bad guy”? J Biomed Sci. (2011) 18:90–90. doi: 10.1186/1423-0127-18-90
6. Day JRS, Taylor KM. The systemic inflammatory response syndrome and cardiopulmonary bypass. Int J Surg. (2005) 3:129–40. doi: 10.1016/j.ijsu.2005.04.002
7. Tillett WS, Francis T. Serological reactions in pneumonia with a non-protein somatic fraction of pneumococcus. J Exp Med. (1930) 52:561–71.
8. Szalai AJ, Agrawal A, Greenhough TJ, Volanakis JE. C-reactive protein: structural biology and host defense function. Clin Chem Lab Med. (1999) 37:265–70. doi: 10.1515/CCLM.1999.046
9. Volanakis JE. Human C-reactive protein: expression, structure, and function. Mol Immunol. (2001) 38:189–97. doi: 10.1016/s0161-5890(01)00042-6
10. Verma S, Szmitko PE, Ridker PM. C-reactive protein comes of age. Nat Clin Pract Cardiovasc Med. (2005) 2:29–36. doi: 10.1038/ncpcardio0074
11. Casas JP, Shah T, Hingorani AD, Danesh J, Pepys MB. C-reactive protein and coronary heart disease: a critical review. J Intern Med. (2008) 264:295–314. doi: 10.1111/j.1365-2796.2008.02015.x
12. Koenig W, Sund M, Frohlich M, Fischer HG, Lowel H, Doring A, et al. C-Reactive protein, a sensitive marker of inflammation, predicts future risk of coronary heart disease in initially healthy middle-aged men: results from the MONICA (Monitoring Trends and Determinants in Cardiovascular Disease) Augsburg Cohort Study, 1984 to 1992. Circulation. (1999) 99:237–42.
13. Arenillas JF, Alvarez-Sabin J, Molina CA, Chacon P, Montaner J, Rovira A, et al. C-reactive protein predicts further ischemic events in first-ever transient ischemic attack or stroke patients with intracranial large-artery occlusive disease. Stroke. (2003) 34:2463–8. doi: 10.1161/01.STR.0000089920.93927.A7
14. Danesh J, Wheeler JG, Hirschfield GM, Eda S, Eiriksdottir G, Rumley A, et al. C-reactive protein and other circulating markers of inflammation in the prediction of coronary heart disease. N Engl J Med. (2004) 350:1387–97. doi: 10.1056/NEJMoa032804
15. Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. (2005) 352:20–8. doi: 10.1056/NEJMoa042378
16. Pepys MB, Hirschfield GM, Tennent GA, Gallimore JR, Kahan MC, Bellotti V, et al. Targeting C-reactive protein for the treatment of cardiovascular disease. Nature. (2006) 440:1217–21. doi: 10.1038/nature04672
17. Peters SA, Visseren FL, Grobbee DE. Biomarkers. Screening for C-reactive protein in CVD prediction. Nat Rev Cardiol. (2013) 10:12–4. doi: 10.1038/nrcardio.2012.164
18. Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, et al. Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet. (2018) 391:319–28. doi: 10.1016/S0140-6736(17)32814-3
19. Mani P, Puri R, Schwartz GG, Nissen SE, Shao M, Kastelein JJP, et al. Association of initial and serial c-reactive protein levels with adverse cardiovascular events and death after acute coronary syndrome: a secondary analysis of the VISTA-16 trial. JAMA Cardiol. (2019) 4:314–20 doi: 10.1001/jamacardio.2019.0179
20. Barrett TD, Hennan JK, Marks RM, Lucchesi BR. C-reactive-protein-associated increase in myocardial infarct size after ischemia/reperfusion. J Pharmacol Exp Ther. (2002) 303:1007–13. doi: 10.1124/jpet.102.040600
21. Szalai AJ, McCrory MA, Xing D, Hage FG, Miller A, Oparil S, et al. Inhibiting C-reactive protein for the treatment of cardiovascular disease: promising evidence from rodent models. Mediators Inflamm. (2014) 2014:353614. doi: 10.1155/2014/353614
22. Sheriff A, Schindler R, Vogt B, Abdel-Aty H, Unger JK, Bock C, et al. Selective apheresis of C-reactive protein: a new therapeutic option in myocardial infarction? J Clin Apher. (2015) 30:15–21. doi: 10.1002/jca.21344
23. Boncler M, Wu Y, Watala C. The multiple faces of c-reactive protein-physiological and pathophysiological implications in cardiovascular disease. Molecules. (2019) 24:2062. doi: 10.3390/molecules24112062
24. Braig D, Nero TL, Koch H-G, Kaiser B, Wang X, Thiele JR, et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat Commun. (2017) 8:14188. doi: 10.1038/ncomms14188
25. McFadyen JD, Kiefer J, Braig D, Loseff-Silver J, Potempa LA, Eisenhardt SU, et al. Dissociation of c-reactive protein localizes and amplifies inflammation: evidence for a direct biological role of c-reactive protein and its conformational changes. Front Immunol. (2018) 9:1351. doi: 10.3389/fimmu.2018.01351
26. Thiele JR, Zeller J, Bannasch H, Stark GB, Peter K, Eisenhardt SU. Targeting C-reactive protein in inflammatory disease by preventing conformational changes. Med Inflamm. (2015) 2015:372432. doi: 10.1155/2015/372432
27. Kunze R. C-reactive protein: from biomarker to trigger of cell death? Ther Apher Dial. (2019) 23:494–6. doi: 10.1111/1744-9987.12802
28. Szalai AJ, Briles DE, Volanakis JE. Role of complement in C-reactive-protein-mediated protection of mice from Streptococcus pneumoniae. Infect Immunity. (1996) 64:0–4853.
29. Mold C, Rodic-Polic B, Du Clos TW. Protection from < em>Streptococcus pneumoniae </em> infection by c-reactive protein and natural antibody requires complement but not Fcγ receptors. J Immunol. (2002) 168:6375–81. doi: 10.4049/jimmunol.168.12.6375
30. Sparkes BL, Woods K, Roth M, Welti R, Fleming SD. (2009) Phospholipase A2 alters membrane lipid composition during ischemia/reperfusion (39.55). The Journal of Immunology 182(1 Supplement), 39.55-39.55
31. Nijmeijer R, Lagrand WK, Baidoshvili A, Lubbers YT, Hermens WT, Meijer CJ, et al. Secretory type II phospholipase A2 binds to ischemic myocardium during myocardial infarction in humans. Cardiovasc Res. (2002) 53:138−46. doi: 10.1016/s0008-6363(01)00480-1
32. Nijmeijer R, Willemsen M, Meijer CJ, Visser CA, Verheijen RH, Gottlieb RA, et al. Type II secretory phospholipase A2 binds to ischemic flip-flopped cardiomyocytes and subsequently induces cell death. Am J Physiol Heart Circ Physiol. (2003) 285:H2218–24. doi: 10.1152/ajpheart.00887.2002
33. Murakami M, Taketomi Y, Sato H, Yamamoto K. Secreted phospholipase A2 revisited. J Biochem. (2011) 150:233–55. doi: 10.1093/jb/mvr088
34. Yagami T, Yamamoto Y, Koma H. The role of secretory phospholipase A(2) in the central nervous system and neurological diseases. Mol Neurobiol. (2014) 49:863–76. doi: 10.1007/s12035-013-8565-9
35. Kushner I, Kaplan MH. Studies of acute phase protein: I. An immunohistochemical method for the localization of cx-reactive protein in rabbits association with necrosis in local inflammatory lesions. J Exp Med. (1961) 114:961–74. doi: 10.1084/jem.114.6.961
36. Kushner I, Rakita L, Kaplan MH. Studies of acute-phase protein. II Localization of Cx-reactive protein in heart in induced myocardial infarction in rabbits. J Clin Invest. (1963) 42:286–92. doi: 10.1172/jci104715
37. Narkates AJ, Volanakis JE. C-reactive protein binding specificities: artificial and natural phospholipid bilayers. Ann N Y Acad Sci. (1982) 389:172–82. doi: 10.1111/j.1749-6632.1982.tb22135.x
38. Vogt B, Fuhrnrohr B, Muller R, Sheriff A. CRP and the disposal of dying cells: consequences for systemic lupus erythematosus and rheumatoid arthritis. Autoimmunity. (2007) 40:295–8. doi: 10.1080/08916930701358925
39. Goda T, Miyahara Y. Calcium-independent binding of human C-reactive protein to lysophosphatidylcholine in supported planar phospholipid monolayers. Acta Biomaterialia. (2017) 48:206–14. doi: 10.1016/j.actbio.2016.10.043
40. Sproston NR, Ashworth JJ. Role of C-reactive protein at sites of inflammation and infection. Front Immunol. (2018) 9:754–4. doi: 10.3389/fimmu.2018.00754
41. Kayser S, Brunner P, Althaus K, Dorst J, Sheriff A. Selective apheresis of c-reactive protein for treatment of indications with elevated CRP concentrations. J Clin Med. (2020)9:2947. doi: 10.3390/jcm9092947
42. Dimitrijević O, Stojcevski BD, Ignjatović S, Singh NM. Serial measurements of C-reactive protein after acute myocardial infarction in predicting one-year outcome. Int Heart J. (2006) 47:833–42. doi: 10.1536/ihj.47.833
43. Reindl M, Reinstadler SJ, Feistritzer HJ, Klug G, Tiller C, Mair J, et al. Relation of inflammatory markers with myocardial and microvascular injury in patients with reperfused ST-elevation myocardial infarction. Eur Heart J Acute Cardiovasc Care. (2017) 6:640–9. doi: 10.1177/2048872616661691
44. Stumpf C, Sheriff A, Zimmermann S, Schaefauer L, Schlundt C, Raaz D, et al. C-reactive protein levels predict systolic heart failure and outcome in patients with first ST-elevation myocardial infarction treated with coronary angioplasty. Arch Med Sci. (2017) 13:1086–93. doi: 10.5114/aoms.2017.69327
45. Hack CE, Wolbink GJ, Schalkwijk C, Speijer H, Hermens WT, van den Bosch H. A role for secretory phospholipase A2 and C-reactive protein in the removal of injured cells. Immunol Today. (1997) 18:111–5.
46. Griselli M, Herbert J, Hutchinson WL, Taylor KM, Sohail M, Krausz T, et al. C-reactive protein and complement are important mediators of tissue damage in acute myocardial infarction. J Exp Med. (1999) 190:1733–40.
47. Gershov D, Kim S, Brot N, Elkon KB. C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med. (2000) 192:1353–64. doi: 10.1084/jem.192.9.1353
48. Nijmeijer R, Lagrand WK, Lubbers YT, Visser CA, Meijer CJ, Niessen HW, et al. C-reactive protein activates complement in infarcted human myocardium. Am J Pathol. (2003) 163:269–75. doi: 10.1016/S0002-9440(10)63650-4
49. Gill R, Kemp JA, Sabin C, Pepys MB. Human C-reactive protein increases cerebral infarct size after middle cerebral artery occlusion in adult rats. J Cereb Blood Flow Metab. (2004) 24:1214–8. doi: 10.1097/01.Wcb.0000136517.61642.99
50. Slagman AC, Bock C, Abdel-Aty H, Vogt B, Gebauer F, Janelt G, et al. Specific removal of C-reactive protein by apheresis in a porcine cardiac infarction model. Blood Purif. (2011) 31:9–17. doi: 10.1159/000320763
51. You YK, Huang XR, Chen HY, Lyu XF, Liu HF, Lan HY. C-reactive protein promotes diabetic kidney disease in db/db mice via the CD32b-Smad3-mTOR signaling pathway. Sci Rep. (2016) 6:26740. doi: 10.1038/srep26740
52. Newling M, Sritharan L, van der Ham AJ, Hoepel W, Fiechter RH, de Boer L, et al. C-reactive protein promotes inflammation through fcγr-induced glycolytic reprogramming of human macrophages. J Immunol. (2019) 203:225–35. doi: 10.4049/jimmunol.1900172
53. Sun W, Wu Y, Gao M, Tian Y, Qi P, Shen Y, et al. C-reactive protein promotes inflammation through TLR4/NF-κB/TGF-β pathway in HL-1 cells. Biosci Rep. (2019) 39:BSR20190888. doi: 10.1042/bsr20190888
54. Zhang L, Shen ZY, Wang K, Li W, Shi JM, Osoro EK, et al. C-reactive protein exacerbates epithelial-mesenchymal transition through Wnt/β-catenin and ERK signaling in streptozocin-induced diabetic nephropathy. Faseb J. (2019) 33:6551–63. doi: 10.1096/fj.201801865RR
55. Schneeweis C, Gräfe M, Bungenstock A, Spencer-Hänsch C, Fleck E, Goetze S. Chronic CRP-exposure inhibits VEGF-induced endothelial cell migration. J Atheroscler Thromb. (2010) 17:203–12. doi: 10.5551/jat.3004
56. Del Giudice M, Gangestad SW. Rethinking IL-6 and CRP: Why they are more than inflammatory biomarkers, and why it matters. Brain Behav Immun. (2018) 70:61–75. doi: 10.1016/j.bbi.2018.02.013
57. Rajab IM, Hart PC, Potempa LA. How C-reactive protein structural isoforms with distinctive bioactivities affect disease progression. Front Immunol. (2020) 11:2126. doi: 10.3389/fimmu.2020.02126
58. Bhakdi S, Torzewski M, Paprotka K, Schmitt S, Barsoom H, Suriyaphol P, et al. Possible protective role for C-reactive protein in atherogenesis: complement activation by modified lipoproteins halts before detrimental terminal sequence. Circulation. (2004) 109:1870–6. doi: 10.1161/01.Cir.0000124228.08972.26
59. Torzewski M, Bhakdi S. Complement and atherosclerosis-united to the point of no return? Clin Biochem. (2013) 46:20–5. doi: 10.1016/j.clinbiochem.2012.09.012
60. Haapasalo K, Meri S. Regulation of the complement system by pentraxins. Front Immunol. (2019) 10:1750. doi: 10.3389/fimmu.2019.01750
61. Bock C, Vogt B, Mattecka S, Yapici G, Brunner P, Fimpel S, et al. C-reactive protein causes blood pressure drop in rabbits and induces intracellular calcium signaling. Front Immunol. (2020) 11:1978. doi: 10.3389/fimmu.2020.01978
62. Abramson JL, Weintraub WS, Vaccarino V. Association between pulse pressure and C-reactive protein among apparently healthy US adults. Hypertension. (2002) 39:197–202. doi: 10.1161/hy0202.104270
63. Anzai T, Yoshikawa T, Takahashi T, Maekawa Y, Okabe T, Asakura Y, et al. Early use of beta-blockers is associated with attenuation of serum C-reactive protein elevation and favorable short-term prognosis after acute myocardial infarction. Cardiology. (2003) 99:47–53. doi: 10.1159/000068449
64. Whelton SP, Narla V, Blaha MJ, Nasir K, Blumenthal RS, Jenny NS, et al. Association between resting heart rate and inflammatory biomarkers (high-sensitivity C-reactive protein, interleukin-6, and fibrinogen) (from the Multi-Ethnic Study of Atherosclerosis). Am J Cardiol. (2014) 113:644–9. doi: 10.1016/j.amjcard.2013.11.009
65. Bisoendial R, Birjmohun R, Keller T, van Leuven S, Levels H, Levi M, et al. In vivo effects of C-reactive protein (CRP)-infusion into humans. Circ Res. (2005) 97:e115–116. doi: 10.1161/01.RES.0000196746.75724.8b
66. Clapp BR, Hirschfield GM, Storry C, Gallimore JR, Stidwill RP, Singer M, et al. Inflammation and endothelial function: direct vascular effects of human C-reactive protein on nitric oxide bioavailability. Circulation. (2005) 111:1530–6. doi: 10.1161/01.CIR.0000159336.31613.31
67. Bisoendial RJ, Kastelein JJ, Peters SL, Levels JH, Birjmohun R, Rotmans JI, et al. Effects of CRP infusion on endothelial function and coagulation in normocholesterolemic and hypercholesterolemic subjects. J Lipid Res. (2007) 48:952–60. doi: 10.1194/jlr.P600014-JLR200
68. Balk RA. Systemic inflammatory response syndrome (SIRS): where did it come from and is it still relevant today? Virulence. (2014) 5:20–6. doi: 10.4161/viru.27135
69. Berg CWVD, Taylor KE, Lang D. C-reactive protein-induced in vitro vasorelaxation is an artefact caused by the presence of sodium azide in commercial preparations. Arterioscl Thromb Vasc Biol. (2004) 24:e168–71. doi: 10.1161/01.ATV.0000142807.92781.d9
70. Lafuente N, Azcutia V, Matesanz N, Cercas E, Rodríguez-Mañas L, Sánchez-Ferrer CF, et al. Evidence for sodium azide as an artifact mediating the modulation of inducible nitric oxide synthase by c-reactive protein. J Cardiovasc Pharmacol. (2005) 45:193–6. doi: 10.1097/01.fjc.0000154371.95907.bd
71. Liu C, Wang s, deb a, nath ka, katusic zs, mcconnell jp, et al. proapoptotic, antimigratory, antiproliferative, and antiangiogenic effects of commercial c-reactive protein on various human endothelial cell types in vitro. Circul Res. (2005) 97:135–43. doi: 10.1161/01.RES.0000174612.90094.fd
72. Fleming I, Busse R. Signal transduction of eNOS activation. Cardiovasc Res. (1999) 43:532–41. doi: 10.1016/s0008-6363(99)00094-2
73. Fleming I, Fisslthaler B, Dimmeler S, Kemp BE, Busse R. Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ Res. (2001) 88:E68–75. doi: 10.1161/hh1101.092677
74. Torzewski M, Waqar AB, Fan J. Animal models of C-reactive protein. Mediators Inflamm. (2014) 2014:683598. doi: 10.1155/2014/683598
75. Koike T, Kitajima S, Yu Y, Nishijima K, Zhang J, Ozaki Y, et al. Human C-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. (2009) 120:2088–94. doi: 10.1161/circulationaha.109.872796
76. Jones NR, Pegues MA, McCrory MA, Singleton W, Bethune C, Baker BF, et al. A selective inhibitor of human c-reactive protein translation is efficacious in vitro and in c-reactive protein transgenic mice and humans. Mol Ther Nucleic Acids. (2012) 1:e52. doi: 10.1038/mtna.2012.44
77. Ries W, Sheriff A, Heigl F, Zimmermann O, Garlichs CD, Torzewski J. “First in Man”: Case report of selective c-reactive protein apheresis in a patient with acute st segment elevation myocardial infarction. Case Rep Cardiol. (2018) 2018:4767105. doi: 10.1155/2018/4767105
78. Mattecka S, Brunner P, Hähnel B, Kunze R, Vogt B, Sheriff A. PentraSorb C-reactive protein: characterization of the selective c-reactive protein adsorber resin. Therapeutic Apheresis and Dialysis. (2019) 23:474–81. doi: 10.1111/1744-9987.12796
79. Ries W, Heigl F, Garlichs C, Sheriff A, Torzewski J. Selective C-Reactive Protein-Apheresis in Patients. Ther Apher Dial. (2019) 23:570–4. doi: 10.1111/1744-9987.12804
80. Stone GW, Selker HP, Thiele H, Patel MR, Udelson JE, Ohman EM, et al. Relationship between infarct size and outcomes following primary pci: patient-level analysis from 10 randomized trials. J Am Coll Cardiol. (2016) 67:1674–83. doi: 10.1016/j.jacc.2016.01.069
81. de Waha S, Patel MR, Granger CB, Ohman EM, Maehara A, Eitel I, et al. Relationship between microvascular obstruction and adverse events following primary percutaneous coronary intervention for ST-segment elevation myocardial infarction: an individual patient data pooled analysis from seven randomized trials. Eur Heart J. (2017) 38:3502–10. doi: 10.1093/eurheartj/ehx414
82. Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. (2002) 53:31–47. doi: 10.1016/s0008-6363(01)00434-5
83. Frangogiannis NG. Regulation of the inflammatory response in cardiac repair. Circ Res. (2012) 110:159–73. doi: 10.1161/CIRCRESAHA.111.243162
84. Pietila K, Harmoinen A, Hermens W, Simoons ML, Van de Werf F, Verstraete M. Serum C-reactive protein and infarct size in myocardial infarct patients with a closed versus an open infarct-related coronary artery after thrombolytic therapy. Eur Heart J(1993) 14:915–919. doi: 10.1093/eurheartj/14.7.915
85. Beranek JT. C-reactive protein and complement in myocardial infarction and postinfarction heart failure. Eur Heart J. (1997) 18:1834–6. doi: 10.1093/oxfordjournals.eurheartj.a015180
86. Suleiman M, Khatib R, Agmon Y, Mahamid R, Boulos M, Kapeliovich M, et al. Early inflammation and risk of long-term development of heart failure and mortality in survivors of acute myocardial infarction predictive role of C-reactive protein. J Am Coll Cardiol. (2006) 47:962–8. doi: 10.1016/j.jacc.2005.10.055
87. Liu D, Qi X, Li Q, Jia W, Wei L, Huang A, et al. Increased complements and high-sensitivity C-reactive protein predict heart failure in acute myocardial infarction. Biomed Rep. (2016) 5:761–5. doi: 10.3892/br.2016.793
88. Pepys MB. C-reactive protein fifty years on. Lancet. (1981) 1:653–7. doi: 10.1016/s0140-6736(81)91565-8
89. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med. (1998) 188:2313–20.
90. Lagrand WK, Niessen HW, Wolbink GJ, Jaspars LH, Visser CA, Verheugt FW, et al. C-reactive protein colocalizes with complement in human hearts during acute myocardial infarction. Circulation. (1997) 95:97–103.
91. Valtchanova-Matchouganska A, Gondwe M, Nadar A. The role of C-reactive protein in ischemia/reperfusion injury and preconditioning in a rat model of myocardial infarction. Life sciences. (2004) 75:901–10. doi: 10.1016/j.lfs.2003.12.029
92. Heinecke JW. Chemical knockout of C-reactive protein in cardiovascular disease. Nat Chem Biol. (2006) 2:300–1. doi: 10.1038/nchembio0606-300
93. Kitsis RN, Jialal I. Limiting myocardial damage during acute myocardial infarction by inhibiting C-reactive protein. N Engl J Med. (2006) 355:513–5. doi: 10.1056/NEJMcibr063197
94. Krijnen PA, Meischl C, Nijmeijer R, Visser CA, Hack CE, Niessen HW. Inhibition of sPLA2-IIA, C-reactive protein or complement: new therapy for patients with acute myocardial infarction? Cardiovasc Hematol Disord Drug Targets. (2006) 6:113–23. doi: 10.2174/187152906777441830
95. Boljevic D, Nikolic A, Rusovic S, Lakcevic J, Bojic M, Balint B. A promising innovative treatment for st-elevation myocardial infarction: the use of c-reactive protein selective apheresis: case report. Blood Purification. (2020) 49:753–7. doi: 10.1159/000506176
96. Ries W, Heigl F, Garlichs C, Sheriff A, Torzewski J. Die CRP-Apherese: Eine neue Therapiemöglichkeit bei Inflammation. Medicom: Nephro-News (2019).
97. Reinstadler SJ, Stiermaier T, Fuernau G, de Waha S, Desch S, Metzler B, et al. The challenges and impact of microvascular injury in ST-elevation myocardial infarction. Expert Review of Cardiovascular Therapy. (2016) 14:431–43. doi: 10.1586/14779072.2016.1135055
98. Muir KW, Tyrrell P, Sattar N, Warburton E. Inflammation and ischaemic stroke. Curr Opin Neurol. (2007) 20:334–42. doi: 10.1097/WCO.0b013e32813ba151
99. Anrather J, Iadecola C. Inflammation and Stroke: An Overview. Neurotherapeutics. (2016) 13:661–70. doi: 10.1007/s13311-016-0483-x
100. Winbeck K, Poppert H, Etgen T, Conrad B, Sander D. Prognostic relevance of early serial C-reactive protein measurements after first ischemic stroke. Stroke. (2002) 33:2459–64. doi: 10.1161/01.str.0000029828.51413.82
101. Montaner J, Fernandez-Cadenas I, Molina CA, Ribo M, Huertas R, Rosell A, et al. Poststroke C-reactive protein is a powerful prognostic tool among candidates for thrombolysis. Stroke. (2006) 37:1205–10. doi: 10.1161/01.STR.0000217744.89208.4e
102. Muir KW, Weir CJ, Alwan W, Squire IB, Lees KR. C-reactive protein and outcome after ischemic stroke. Stroke. (1999) 30:981–5. doi: 10.1161/01.str.30.5.981
103. Elkind MS, Tai W, Coates K, Paik MC, Sacco RL. High-sensitivity C-reactive protein, lipoprotein-associated phospholipase A2, and outcome after ischemic stroke. Arch Intern Med. (2006) 166:2073–80. doi: 10.1001/archinte.166.19.2073
104. Wadman M, Couzin-Frankel J, Kaiser J, Matacic C. A rampage through the body. Science. (2020) 368:356–60. doi: 10.1126/science.368.6489.356
105. Mueller AA, Tamura T, Crowley CP, DeGrado JR, Haider H, Jezmir JL, et al. Inflammatory biomarker trends predict respiratory decline in COVID-19 patients. Cell Rep Med. (2020) 1:100144. doi: 10.1016/j.xcrm.2020.100144
106. Ruan Q, Yang K, Wang W, Jiang L, Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. (2020) 46:846–8. doi: 10.1007/s00134-020-05991-x
107. Chen G, Wu D, Guo W, Cao Y, Huang D, Wang H, et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J Clin Invest. (2020) 130:2620–9. doi: 10.1172/JCI137244
108. Tan C, Huang Y, Shi F, Tan K, Ma Q, Chen Y, et al. C-reactive protein correlates with computed tomographic findings and predicts severe COVID-19 early. J Med Virol. (2020) 92:856–62. doi: 10.1002/jmv.25871
109. Nienhold R, Ciani Y, Koelzer VH, Tzankov A, Haslbauer JD, Menter T, et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat Commun. (2020) 11:5086. doi: 10.1038/s41467-020-18854-2
110. Puntmann VO, Carerj ML, Wieters I, Fahim M, Arendt C, Hoffmann J, et al. Outcomes of cardiovascular magnetic resonance imaging in patients recently recovered from coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:1265–73. doi: 10.1001/jamacardio.2020.3557
111. Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiology. (2020) 5:819–24. doi: 10.1001/jamacardio.2020.1096
112. Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. (2020) 8:420–2. doi: 10.1016/S2213-2600(20)30076-X
113. Kayser S, Kunze R, Sheriff A. Selective C-reactive protein apheresis for Covid-19 patients suffering from organ damage. Ther Apher Dial. (2020) doi: 10.1111/1744-9987.13532
114. Torzweski J, Heigl F, Zimmermann O, Wagner F, Schumann C, Hettich R, et al. First-in-man: case report of selective c-reactive protein apheresis in a patient with sars-cov-2 infection. Am J Case Rep. (2020) 21:e925020. doi: 10.12659/AJCR.925020
115. Di Napoli M, Schwaninger M, Cappelli R, Ceccarelli E, Di Gianfilippo G, Donati C, et al. Evaluation of C-reactive protein measurement for assessing the risk and prognosis in ischemic stroke: a statement for health care professionals from the CRP Pooling Project members. Stroke. (2005) 36:1316–29. doi: 10.1161/01.STR.0000165929.78756.ed
Keywords: CRP–C-reactive protein, ischemia/reperfusion injury, cardiovascular, COVID-19, inflammation
Citation: Sheriff A, Kayser S, Brunner P and Vogt B (2021) C-Reactive Protein Triggers Cell Death in Ischemic Cells. Front. Immunol. 12:630430. doi: 10.3389/fimmu.2021.630430
Received: 17 November 2020; Accepted: 22 January 2021;
Published: 10 February 2021.
Edited by:
Kenji Daigo, Nippon Medical School, JapanReviewed by:
Michael Torzewski, Robert-Bosch-Hospital, GermanyCopyright © 2021 Sheriff, Kayser, Brunner and Vogt. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed Sheriff, YWhtZWQuc2hlcmlmZkBjaGFyaXRlLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.