 Syamal K. Datta
Syamal K. Datta
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 14 April 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.629807
This article is part of the Research TopicGenerating and Sustaining Stable Autoantigen-specific CD4 and CD8 Regulatory T Cells in LupusView all 11 articles
Autoantigen-directed tolerance can be induced by certain nucleosomal histone peptide epitope/s in nanomolar dosage leading to sustained remission of disease in mice with spontaneous SLE. By contrast, lupus is accelerated by administration of intact (whole) histones, or whole nucleosomes in microparticles from apoptotic cells, or by post-translationally acetylated histone-peptides. Low-dose therapy with the histone-peptide epitopes simultaneously induces TGFβ and inhibits IL-6 production by DC in vivo, especially pDC, which then induce CD4+CD25+ Treg and CD8+ Treg cells that suppress pathogenic autoimmune response. Both types of induced Treg cells are FoxP3+ and act by producing TGFβ at close cell-to-cell range. No anaphylactic adverse reactions, or generalized immunosuppression have been detected in mice injected with the peptides, because the epitopes are derived from evolutionarily conserved histones in the chromatin; and the peptides are expressed in the thymus during ontogeny, and their native sequences have not been altered. The peptide-induced Treg cells can block severe lupus on adoptive transfer reducing inflammatory cell reaction and infiltration in the kidney. In Humans, similar potent Treg cells are generated by the histone peptide epitopes in vitro in lupus patients’ PBMC, inhibiting anti-dsDNA autoantibody and interferon production. Furthermore, the same types of Treg cells are generated in lupus patients who are in very long-term remission (2-8 years) after undergoing autologous hematopoietic stem cell transplantation. These Treg cells are not found in lupus patients treated conventionally into clinical remission (SLEDAI of 0); and consequently they still harbor pathogenic autoimmune cells, causing subclinical damage. Although antigen-specific therapy with pinpoint accuracy is suitable for straight-forward organ-specific autoimmune diseases, Systemic Lupus is much more complex. The histone peptide epitopes have unique tolerogenic properties for inhibiting Innate immune cells (DC), T cells and B cell populations that are both antigen-specifically and cross-reactively involved in the pathogenic autoimmune response in lupus. The histone peptide tolerance is a natural and non-toxic therapy suitable for treating early lupus, and also maintaining lupus patients after toxic drug therapy. The experimental steps, challenges and possible solutions for successful therapy with these peptide epitopes are discussed in this highly focused review on Systemic Lupus.
This chronicle with historical perspective focuses first on the early steps of pathogenic autoantibody production in lupus, especially on the role of Th cells, and then how they can be regulated. In human lupus, it is now established that the main genetic risk loci for lupus susceptibility in GWAS are located in MHC class II and IRF5 regions, which respectively determine autoantigen presentation and associated activating cytokines production required to recruit autoreactive T helper (Th) cells (1–8). Genetic studies in families with rheumatic autoimmune diseases also support the initiating role of T cells in lupus (9), in addition to complement genes in the MHC locus (10). Importantly, augmented Th cell activity which is prevalent in lupus (11–13), can overcome the need for TLR abnormalities contributing to lupus (14). B cells and other professional APCs are activated to present autoantigens as the disease progresses. However, normally macrophages, such as, tingible body MΦ and DCs remain tolerogenic when handling dying (apoptotic) cells that can provide the autoantigens for lupus if mishandled, as described below (15–18). The professional APC become effectively activated in vivo to present these apoptotic autoantigens after the apoptotic cell derived DNA and/or RNA containing autoantigens are presented in IgG immune complexes (IC) that are bound by the APC to dually stimulate their TLR and FcγR (19, 20). Hence, Th cell mediated class-switched IgG autoantibodies specific for the DNA or RNA containing autoantigens have to be made first for IC formation activating the APC. Moreover, B cells become efficient antigen presenter to lupus Th cells that have been primed first by other APC, or if the B cells have developed high affinity receptors after undergoing somatic mutation and expansion with TFH cell help in germinal centers (19, 21). However, high level expression of X-linked TLR7, due to incomplete X-chromosome inactivation (22), can contribute to lupus development early on, by independently activating DC and other APC, which in turn causes widespread T-cell activation (23, 24). To accomplish the above effect, striking studies have recently shown that IRF5 is first activated by TLR7 using the adaptor TASL, which interacts with SLC15A4, an amino acid transporter in endolysosome, to recruit IRF5 (25). The X-inked gene CXorf21-a encoding TASL and the gene for SLC15A4 were known to be associated with lupus susceptibility, as discussed in ref (26). Of course intrinsic defects in B cells and APC are critically important for lupus pathogenesis. With disease progression, other pathogenic players in T cell, B cell and unconventional APC populations evolve and are recruited to participate in amplifying the autoimmune inflammatory response, especially in extra-follicular sites, to cite a few (27–32), and reviewed elsewhere [Tsokos, 2020 #2492] (33, 34). Those pathogenic contributors might be kept in check by establishing regulatory mechanisms at the earliest steps of the disease, which is the focus of this review on Lupus, and this topic.
Step by step experiments and ensuing hypothesis based on their results at each stage led to cellular and molecular characterization of the pathogenic Th cells of lupus and how the Th cells become capable in helping pathogenic autoantibody production.
First of all, certain distinctive properties of pathogenic anti-DNA autoantibodies were crucial for isolating and characterizing the Th cells that specifically help them. The pathogenic anti-dsDNA autoantibodies that are deposited in kidneys with lupus nephritis have distinct features, as they are complement-fixing IgG in isotypes, with cationic charge, and clonally restricted by isoelectric focusing, and are able to cause glomerulonephritis in vivo (35–41). Moreover, their antigen combining V regions share recurrent idiotype and fine-specificity patterns for autoantigens (39, 42). Sequence analysis of the pathogenic autoantibodies confirmed their clonal expansion, as they shared VH region CDR3 sequences containing numerous cationic residues generated by somatic mutation (43–45), a signature of Th cell drive. Contemporary studies had shown that immune complexes with cationic charge preferentially bind to anionic residues in glomerular basement membrane proteoglycans and collagen (46–48). It was shown later that glomerular binding of these “anti-DNA” antibodies could also be mediated via histones in nucleosomes bound in situ (49–52).
As described above, pathogenic anti-dsDNA antibodies in lupus are class-switched (35, 36) and clonally expanded (43, 44) suggesting a T helper cell dependent response, but it was mysterious up to 1980s and early 1990s how the Th cells actually helped Pathogenic anti-dsDNA autoantibody-producing B cells, because conventional Th cells do not recognize DNA.
In the first step, it was established that special autoimmune T helper (Th) cell subsets expanded the select population of pathogenic anti-dsDNA autoantibody producing B cells in mice with lupus (41, 53). The production of these pathogenic autoantibodies is also driven by select Th cells that are detectable in patients with active lupus nephritis (54–56).
In the next step, to define their antigenic specificity, the autoimmune Th cells were cloned from lupus prone mice, and also from patients with lupus nephritis (~ 800 clones). Prior to these studies, isolation of the pathogenic T helper (Th) cells of lupus was not done, because their antigenic specificities were unknown. However, using their special functional property of inducing the pathogenic variety of anti-dsDNA autoantibodies as a selection marker, the lupus nephritis-inducing Th cells were isolated. Only 12-15% of activated T cells in lupus patients and mouse models, could induce the production of pathogenic IgG anti-DNA autoantibodies (53–57). When administered into young pre-clinical stage lupus-prone mice, pathogenic autoantibody-inducing Th clones could rapidly induce immune-deposit glomerulonephritis (57, 58). Sequences of antigen-binding CDR loops of the TCRs of these pathogenic Th clones of lupus show recurrent motifs of anionic residues, indicating their selection by autoantigens with cationic residues (56, 57, 59). Indeed, a majority of such pathogenic Th clones produced IL-2 and IFN-γ in response to nucleosomes that contain histone peptides bearing cationic determinants, and nucleosome-specific T cells are detectable in pre-clinical stage lupus-prone mice before pathogenic autoantibodies are detectable in their serum (56, 60, 61). In addition, immunization of pre-clinical lupus mice, but not normal mice, with whole nucleosome particles induces accelerated lupus nephritis indicating the need for pre-existing autoimmune T and B cells in a lupus-prone background (60).
Thus the relevant autoantigens for pathogenic anti-dsDNA autoantibody inducing Th cells of lupus were discovered by an unbiased experimental approach, using pathogenic autoantibody inducing Th cells as sensors to detect the relevant autoantigen epitopes. This property provided the lupus Th cells the ability of “linked recognition” (62) for interaction with pathogenic anti-DNA autoantibody producing B cells (Figure 1). In this way, for the first time a true autoantigen for spontaneous SLE; namely endogenous nucleosomes from host’s apoptotic cells, and not some speculative component in microbes (63, 64), was found to be the real text book-like hapten- carrier link between the pathogenic Th and B cell in lupus for cognate interaction (57, 60),—and from that critical experimental step further studies led to identification of the histone peptide epitopes in nucleosomes recognized by those Th cells, and showed how to harness those particular epitopes for regulatory T cell induction for lupus therapy, as described below. All this was possible in 1980s and early 1990s by cloning the select population of pathogenic anti-dsDNA autoantibody-producing B cells, and then the special autoimmune T helper cells that drive such B cells in lupus. To emphasize again, despite the obstacle that the antigenic specificities of lupus T cells were then unknown, using the experimental steps described above, the pathogenic anti-dsDNA autoantibody-inducing T helper cells were cloned to define the structure and specificity of their receptor genes in human and murine lupus. To achieve the ultimate goal of understanding the cause, and designing a cure for spontaneous autoimmune diseases like lupus, it was essential at that time to identify the major autoantigen/s that drives the pathogenic autoimmune response. In lupus, DNA is a target antigen for autoantibodies but paradoxically immunization of mice with DNA does not cause lupus. The studies with the pathogenic autoantibody-inducing T helper clones in early 1990s led to the initial identification of one of the major immunogens that drives the pathogenic T helper cells of lupus (57, 60).
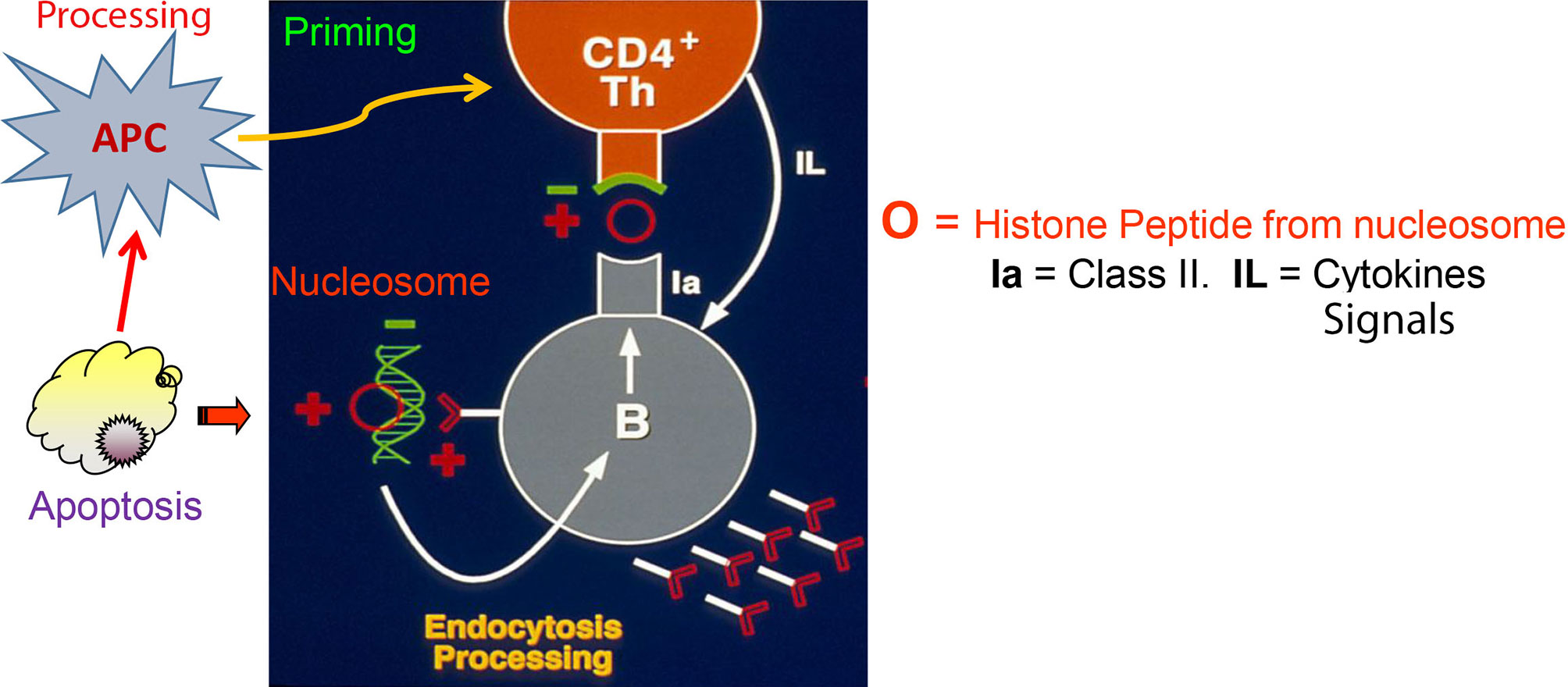
Figure 1 Autoimmune T and B cell interaction in lupus based on Nucleosome-derived autoantigens (based on work done from early 1980s through early1990s; references in the Text). Figure shows that Th cells that induce the production of pathogenic anti-DNA autoantibodies possess anionic residues in CDR3 of their TCRs (green). The lupus Th cells recognize peptides with reciprocal cationic charge (red), such as peptides from histones in nucleosomes presented by the pathogenic anti-DNA autoantibody producing B cells bearing BCRs with cationic residues generated by somatic mutations in CDR3 of their receptors (red). The pathogenic BCRs bind to anionic residues in DNA (green) that are complexed with cationic histones in nucleosomes, which are then endocytosed and processed for presentation to the interacting Th cells. Nucleosomes accumulate due to defective clearance of apoptotic cells in lupus, and are processed by activated APC to prime the pathogenic Th cells; a lupus-specific event initiated early in life. This figure is extensively modified from a figure in J Exp Med (60).
Many studies soon followed that demonstrated or suggested mechanisms that could initiate or amplify the response of pathogenic Th cells to nucleosomal peptides in lupus. Briefly, in lupus, products from apoptotic cells accumulate and become immunogenic because, scavenging molecules in phagocytic cells, such as Marco and other scavenger receptors are functionally deficient in lupus, and so are complement components such as C1q, which facilitate phagocytosis of apoptotic cells without causing an immune response (65–68). Nucleosomes, HMGB1, DNA or RNA from apoptotic cell components not being disposed of properly, act as endogenous TLR ligands, stimulating cells of the innate and adaptive immune system (19, 23, 60, 69–74). For example, HMGB1 chromosomal protein from apoptotic cells that have not been removed properly, forms inflammatory complexes with other accumulating debris like DNA or nucleosomes particles, stimulating immune cells via TLR 2, TLR 4, and RAGE on the cell-surface, or TLR9 in the endosome/lysosome (74–76). Similarly, accumulating extra-cellular nucleosomes in micro particles containing DNA, or ribonucleoproteins containing RNA can stimulate cells of the innate immune system respectively by TLR9 or by TLR 7/8 (29, 31, 77, 78), thus augmenting autoantigen presentation to pathogenic Th cells by those activated APC. The case for TLR 9 is actually more complex, because in early stages, TLR9 actually protects against lupus (79), possibly by promoting tolerance in APC, B cells and helping Treg generation (80, 81). The other possible autoantigen-derived epitope with cationic charge, which could be recognized in a lupus B cell-linked fashion by the pathogenic autoantibody inducing Th cells possessing reciprocally charged anionic residues in their CDR3 region, would be derived from CDR3 region peptides of somatically mutated anti-DNA autoantibodies, as suggested (57, 60); and this possibility was independently demonstrated to be true by several laboratories (82–85). This issue is dealt in other contributions to this research topic.
Th1, TFH, TPH, Gamma Delta Th, CD8 Th, CD4-CD8- Th cells and more, participate in contributing to the pathogenic response in lupus. However, is it mainly Th1 → TFH cells initiating/sustaining pathogenic autoantibody production; whereas the others evolve as amplifiers at extrafollicular inflammatory sites? First of all as mentioned above, only particular subclasses of IgG anti-DNA antibodies are more closely associated with a pathogenic potential in lupus patients and mice, and these pathogenic IgG antibodies belong to Th1-induced isotype classes. In lupus patients, Th1-induced anti-DNA IgG1antibodies are always elevated before the occurrence of renal relapse, and IgG1 plus IgG2 anti-DNA antibodies are found in patient’s renal eluates, whereas in lupus prone mice, murine Th1-induced IgG2a, IgG2b, and IgG3 anti-DNA are more frequently eluted from kidneys with active nephritis (39, 42, 86–88). In contrast to TFH cells which conventionally produce IL-21; Th1 cytokine IFNγ not only mediates class switch for the nephritogenic isotypes, but Th1 derived IFNγ signal is also critical for autoantibody production by germinal center B cells (89, 90). Furthermore, many non-autoantigen specific, bystander TFH cells expand as a secondary event with progression of disease, which could amplify (but not initiate) anti-DNA autoantibody production (91). Indeed, Ig class-switch recombination (CSR), during T and B cell cognate interaction, which is Th1 IFNγ cytokine dependent in lupus, may occur before the TFH IL-21 driven expansion of autoantibody producing B cells in germinal center, which comes later (92). Moreover, Th1-biased GC TFH cells have been reported (93), and another group reported that the differentiation and function of a Th1-derived TFH1-like cell population is driven by IL-12 signaling, which is important for differentiation of Th1 cells in the first place (94–96).
Therefore, Th1 → TFH1 evolution/transition is a possibility in pathogenic anti-DNA autoantibody production in lupus.
And then there are the potent TPH cells with TFH like phenotype but are CXCR5-; they help lupus B cells also by producing IL-21; and IL-10–producing CCR6+T cells populate lymph nodes of SLE patients. These Th cells probably evolve after receiving cytokine and other signals from activated B cells and other APC at extra-follicular sites, as the disease progresses (27, 97).
In addition, helper activity of CD8+ and CD4-/CD8- αβ and γδ TCR+ Th cells, in pathogenic autoantibody production in human SLE has been reported (54, 55). The subset of T cells in humans that are CD4-/CD8- and αβ TCR+ with pathogenic anti-DNA autoantibody-inducing ability in SLE is interesting, because such Th cells were considered to be unique to MRL-lpr mice with lupus. However, similar pathogenic autoantibody-inducing T cells with double negative phenotype that express “forbidden”, autoreactive T cell receptors were described in non-lpr lupus prone mice (41, 53, 98) and then in human lupus (54, 55). Although these double negative T cells might be secondary events in lupus compared to the CD4+ Th cells, they make an important contribution to pathogenesis of the disease. The CD4-/CD8- and αβ TCR+ T cells also have important role in target organ inflammation (99, 100).
In the next step, the critical peptide autoepitopes recognized by lupus nephritis-inducing Th cells were localized initially to be in the core histones of nucleosomes, at amino acid (aa) positions: 10-33 of H-2B, 16-39 and 71-94 of H4, and 85-102 of H3 (61). Altogether 154 overlapping 15-mer peptides spanning the entire length of all four core histones were tested to find the buried epitopes in nucleosomal histones that were recognized specifically autoimmune Th cells that cause lupus in mouse models. In addition, another dominant epitope was identified in position 22-42 of H1’, by mass spectrometry analysis of naturally processed peptides eluted from class II molecules of lupus B cell (APC) lines fed with chromatin (101). Remarkably, all these epitopes are located in regions of histones that contact with DNA in the nucleosome, and they are also targeted by autoantibodies from lupus B cells (B-cell epitopes), indicating that the epitopes could be protected from degradation during autoantigen processing and thus preferentially presented to the Th cells (61, 101–103). Surprisingly, the nucleosomal peptides have the features of “universal epitopes” (104), for instance, the peptide epitopes are promiscuously recognized by pathogenic Th cells derived from lupus-prone SNF1 mice (MHC I-Ad/q) even when presented by APC bearing I-A molecules of all other mouse haplotypes, and human HLA-DR as well! Due to reciprocal charge interaction, the lupus TCRs probably contact the nucleosomal peptide-complexed with MHC promiscuously to sustain TCR signaling (105, 106). The promiscuity of lupus TCRs influences their selection in the thymus of lupus-prone mice and ability to generate Treg cells for tolerance spreading in the periphery, as described below (80, 107–109).
Nested Epitopes for CD8 T Cells. The tolerogenic nucleosomal peptide autoepitopes bind to MHC class II as described above, but CD8+ Treg cells were also induced by injection of the epitopes. Indeed algorithms showed, MHC class I-binding motifs were nested in their sequences, as described (108, 110). The rationale being that the relatively long chain peptides epitopes would be processed further by APC for cross-presentation to CD8 T cells (111). For an example, H471–94 nucleosomal epitope has the nested CD8 sequence shown in bold letters TYTEHAKRKTVTAMDVVYALKRQG, and similarly individually distinct nested CD8 epitopes were detected in each of the longer peptide epitopes from the nucleosomes with CD4 Treg inducing ability, as detailed (108).
(Experimental Steps of More Recent Publications in Mouse Models and Then in Human Lupus Are Described in Brief Below):
The major autoepitopes for lupus nephritis-inducing Th cells were localized to H1’22-42, H385-102, H416-39 and H471-94, as described above. These peptide epitopes stimulate both autoimmune Th cells and B cells. In lupus-prone mice, tolerance therapy at High doses (300μg I.V.) of the peptide epitopes halted the progression of established lupus nephritis. However, high-dose may not be suitable in humans. Therefore, low-dose tolerance therapy was developed with 300 fold lower doses by injecting lupus-prone mice with 1 µg nucleosomal histone peptide autoepitopes S.C. every 2 wk (108). This sub-nanomolar peptide therapy lowered autoantibody levels, blocked nephritis progression and markedly diminished inflammatory cell infiltration in kidneys, thus restoring normal life span. H471-94 was the most effective autoepitope in this study. Low-dose tolerance therapy induced regulatory cell subsets of CD8+ suppressor Treg, and CD4+CD25+ Treg cells, which contained autoantigen-specific and cross-reactive autoantigen-directed Treg cells. The Treg cells suppressed IFN-γ production by pathogenic lupus Th cells in response to nucleosomal epitopes at up to 1:100 ratio of Treg or Treg : Th cells, and diminished autoantibody production in vitro by up to 90-100% by inhibiting nucleosome-stimulated T cell help to nuclear autoantigen-specific B cells. The induced CD4+CD25+ Treg and CD8+ Treg cells produced, and required TGF-β1 for immunosuppression; moreover, they effectively suppressed lupus autoimmunity upon adoptive transfer in vivo. For their suppressor function, the CD4+CD25+ Treg cells were partially cell contact-dependent, but CD8+ Treg cells were contact-independent. Thus, this work demonstrated that low-dose tolerance with the conserved histone autoepitopes durably ameliorates the regulatory defect in SLE by inducing TGF-β producing Treg cells, and without causing adverse side effects such as, generalized immunosuppression or allergic/anaphylactic reactions (Figures 2 and 3).
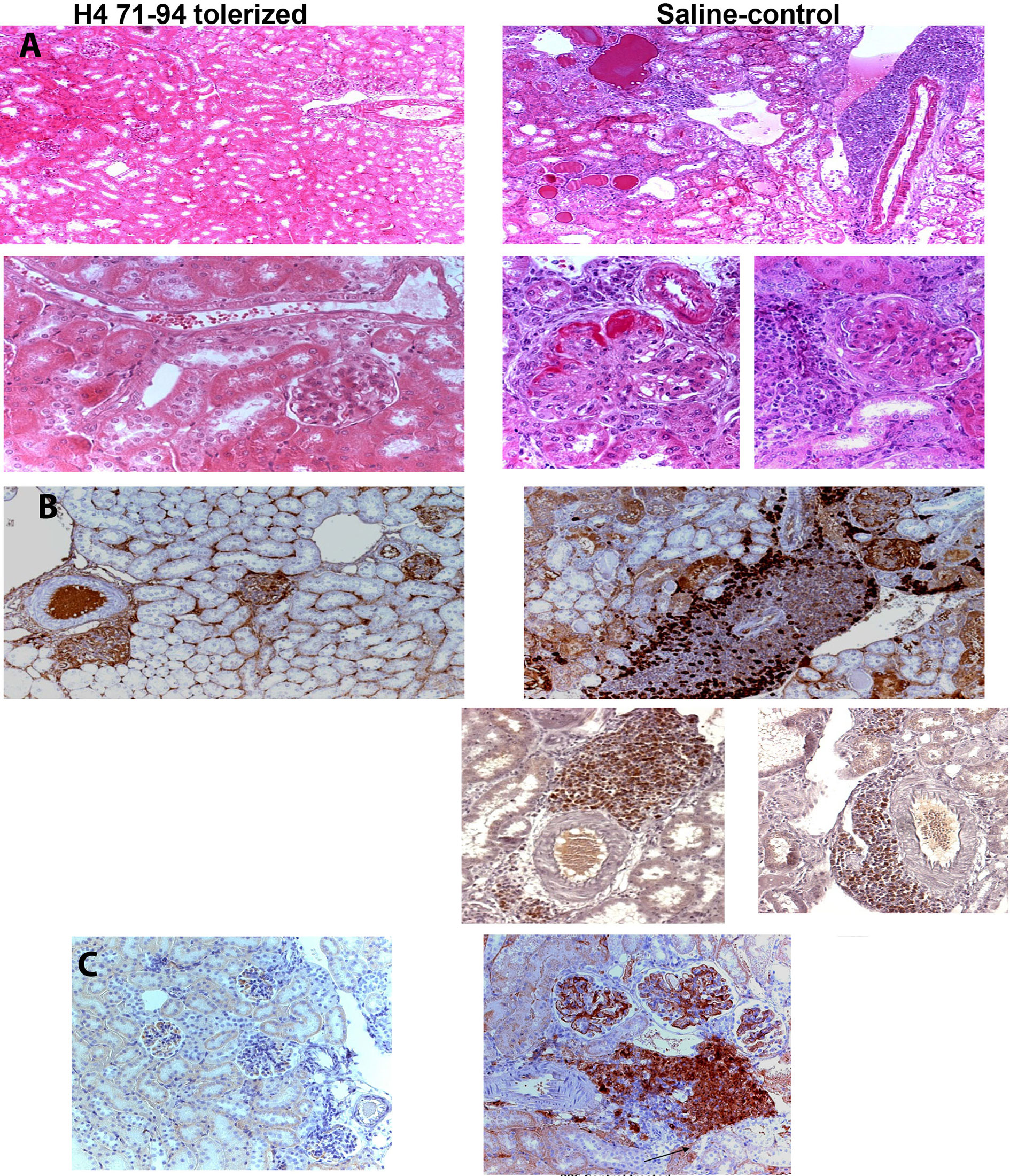
Figure 2 (A) Renal histology from lupus prone mice tolerized by histone peptide epitope (left) or age-matched saline-injected control mice (right). H&E staining; ×100 magnification shown in Upper panels. The saline control shows marked interstitial infiltrate of mononuclear cells with perivascular distribution, hyalinized and sclerotic glomeruli and tubules engorged with casts. Lower panels (original magnification, ×400) show in further detail the differences between mice that underwent peptide-epitope therapy (left) and control mice (right). Kidneys from the former group of mice show marked thickening of basement membranes and advanced sclerosis and crescent formation in glomeruli, and perivascular, interstitial infiltrates of mononuclear cells. (B) Immunohistochemistry (original magnification was ×200). Brown color shows positive staining for IgG deposits in glomeruli of lupus-prone mice, in both peptide-treated (left upper panel) and control groups (right upper panel). However, marked cellular infiltrates around blood vessels containing CD4+ T cells (on the right, in upper panel), CD8+ T cells (on left in lower panel), and CD138+ plasma cells (on right side in lower panel) were found only in kidneys of control mice, although both groups had IgG immune complex deposits. (C) Immunohistochemistry showing glomerular, and interstitial-perivascular infiltration of Th17 cells in control (PBS)-injected control lupus mice (Right side). This inflammatory cell infiltration was prevented in age-matched control mice by low-dose tolerance therapy with nucleosomal histone peptide epitope (Left Panel). Figure partially derived from J Immunol (80, 108), (Originally published in The Journal of Immunology. Kang H-K, Michaels MA, Berner BR, Datta SK. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol (2005) 174:3247-55. And Kang H-K, Liu M, Datta SK. Low-Dose Peptide Tolerance Therapy of Lupus Generates Plasmacytoid Dendritic Cells That Cause Expansion of Autoantigen-Specific Regulatory T Cells and Contraction of Inflammatory Th17 Cells J Immunol (2007) 178:7849-58. Copyright © [2005 and 2007] The American Association of Immunologists, Inc.).
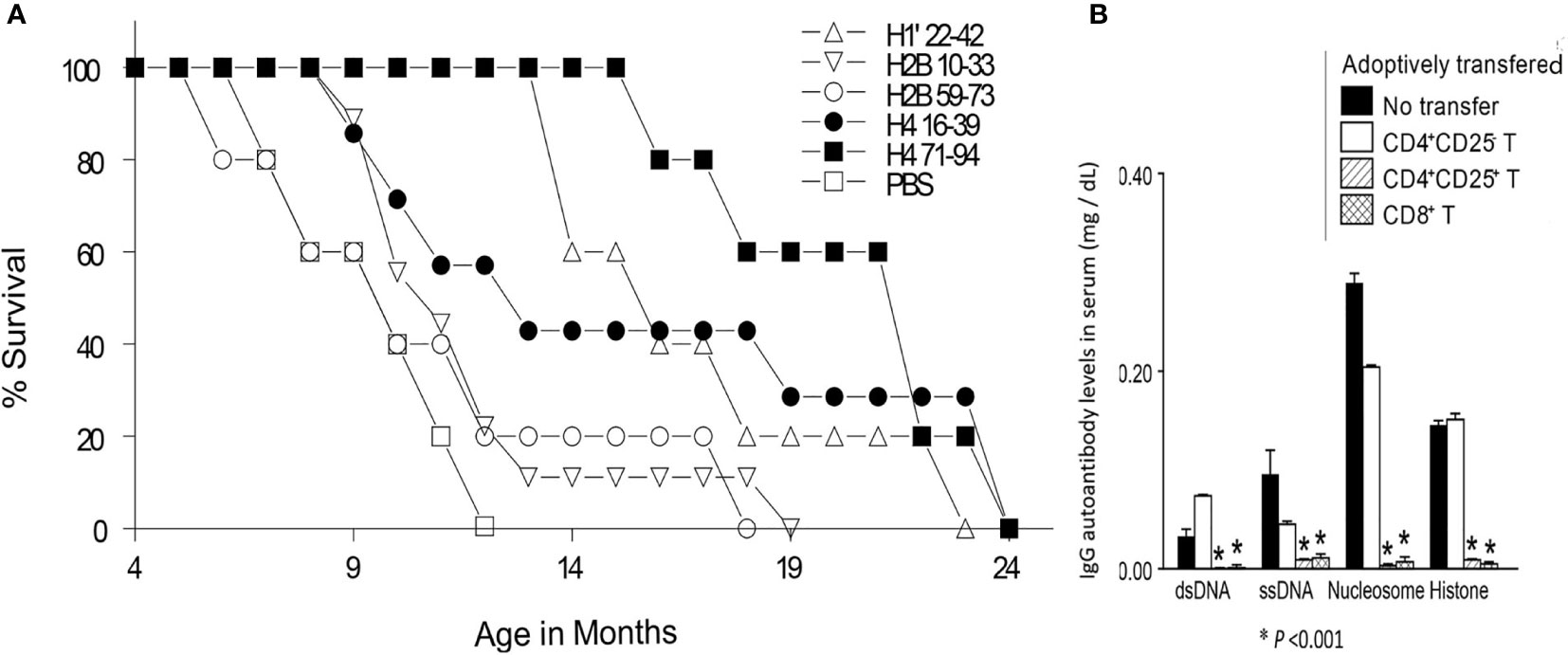
Figure 3 (A) Percent survival, of age-matched, lupus-diseased mice, injected subcutaneously with different nucleosomal histone peptides in low-doses, or with saline (PBS) every two week. (B) In a rigorous test for potency of suppression, Treg cells from H471-94 peptide tolerized mice suppressed lupus acceleration upon adoptive transfer in vivo. Pre-clinical lupus-prone mice were immunized (not tolerized) by another histone peptide at 100 fold higher doses with adjuvant CFA, leading them to produce augmented levels of autoantibodies and develop severe nephritis rapidly, and this accelerated disease was suppressed by adoptive transfer of CD4+CD25+ Treg and CD8+Treg cells, but not CD4+CD25- effector T cells from H471–94-treated tolerized mice. Y-axis values are for IgG autoantibody levels in serum of recipients (mg/dL). *P <0.001. Parts of this Figure are from J Immunol (80, 108). Experimental details are in those references. (Originally published in The Journal of Immunology. Kang H-K, Michaels MA, Berner BR, Datta SK. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol (2005) 174:3247-55. And Kang H-K, Liu M, Datta SK. Low-Dose Peptide Tolerance Therapy of Lupus Generates Plasmacytoid Dendritic Cells That Cause Expansion of Autoantigen-Specific Regulatory T Cells and Contraction of Inflammatory Th17 Cells J Immunol (2007) 178:7849-58. Copyright © [2005 and 2007] The American Association of Immunologists, Inc.)
As noted above, low-dose tolerance of mice with lupus using just a single nucleosomal peptide epitope (H471-94) could halt the progression of lupus nephritis by generating potent Treg cells that suppressed autoimmune T and B cells specific for a broad spectrum of nuclear autoantigens, and markedly inhibited inflammatory cell reaction and infiltration in kidneys. Next step was to determine how this therapy with only 0.36 nM of peptide injected subcutaneously (S.C.) every 2 weeks, induced in vivo TGFβ-producing CD8+Treg, and CD4+25+ Treg cell subsets containing regulatory cells that were autoantigen-specific, as established by the following approach (80). In order to track which APC had captured the histone peptide after tolerance therapy; DC, macrophages and B cells were isolated from local lymph nodes and spleens of lupus-prone mice injected with low-dose H471-94 peptide S.C., and then those APC were tested for their ability to stimulate cognate H471-94-specific T cell hybridomas in culture. The T cell hybridomas are highly sensitive and specific sensors detecting cognate peptide-MHC II on APCs presenting attomole concentration of the histone peptide. Only DC and B cells from spleen of histone peptide-injected mice stimulated the T hybridomas. Thus, during tolerance therapy, the subcutaneously injected H471-94 peptide, which is highly soluble and charged, was rapidly absorbed systemically and captured by APC in the spleen. However, splenic DC, but not B cells or macrophages was responsible for the tolerogenic effect of the peptide therapy. Adoptive transfer of plasmacytoid DC or whole DC, but not B cells from the H471-94 peptide treated mice suppressed responses of autoimmune T cells to nucleosome peptides up to 80% by Treg cells induced in the un-manipulated lupus mouse recipients, and blocked development of nephritis and autoantibody production in a lupus acceleration assay. The DC from the H471-94 peptide injected mice expressed a tolerogenic phenotype upon capturing the S.C. injected H471-94 peptide, expressing relatively low levels of CD80, CD86, CD40 and MHC class II. Compared to controls, the peptide epitope treated animal’s DC, especially plasmacytoid DC (pDC) produced increased amounts of TGFβ but decreased amount of IL-6 on stimulation by nucleosomes and other TLR-ligands, surprisingly the TLR-9 pathway was important for this tolerogenic effect (80). Moreover, these H471-94 peptide-tolerized pDC ameliorated lupus autoimmune disease by simultaneously inducing/expanding contained autoantigen-specific and cross-reactive autoantigen-directed Treg, and suppressing effector Th1 and Th17 cells that infiltrate the kidneys causing lupus nephritis. As an aside, these studies initially showed that inflammatory Th17 infiltrate the kidneys of mice with lupus nephritis (80), which was then demonstrated also in human lupus nephritis (99, 112, 113).
Altogether, these studies early on showed the pathogenic importance of tubulo-interstitial region infiltration in lupus nephritis kidneys by various inflammatory cells in addition to monocyte/macrophages; such as extrafollicular germinal center like accumulation of CD4 and CD8 T cells, and B cells and plasma cells to set up residence in organized perivascular foci, as well as Th17 cells; and importantly, this infiltration was inhibited by the histone peptide epitope tolerance therapy resulting in its beneficial effect (80, 108) (Figure 2). As discussed below, similar to these therapeutic results, the role of locally active Treg cells migrating into the kidney and suppressing lupus nephritis has been recently demonstrated in other systems (114–116). Interestingly, Figure 2B, shows that IgG immune complex deposits were equally present in the kidneys in both peptide-treated and control lupus-prone mice, but interstitial infiltrates of interacting T and B cells and APC were prominent only in the control mice with severe nephritis. This observation is consistent with the demonstration that lupus B cells can contribute to nephritis even without autoantibody production, but just by autoantigen presentation and providing cytokine and other membrane signals to pathogenic Th cells (117); and that Belimumab has beneficial effect in patients with active lupus nephritis (118), which is surprising, but we now know that mature memory B cells also express BAFFR like immature transitional B cells (119). All these intricate pathogenic interactions were prevented by tolerance therapy with the histone peptide epitopes (Figure 2).
It is noteworthy that the immune response against nuclear autoantigens is inter-connected by cross-reactive recognition at the B cell level (38, 39, 42), and importantly at the Th cell level (61, 101, 102, 105, 109). Thus the same lupus Th clone can help either a B cell specific for nucleosomes, or a B cell specific for dsDNA, or for ssDNA, or histone, or HMG, because each B cell can take up and process the whole chromatin particle by recognizing its own specific epitope in the chromatin, and then present the Th clone’s relevant histone peptide epitope derived from chromatin processing, and that results in linked intermolecular help (56, 60, 120). Therefore, suppressing the Th cells of lupus could block spreading of response to multiple epitopes in chromatin (Figure 4). This hypothesis of “Tolerance Spreading” was experimentally supported as described above showing that progression of established lupus nephritis in the lupus-prone mouse models can be delayed, diminishing proteinuria and prolonging life by administering the nucleosomal peptide epitopes singly in high dose IV or low dose SC in tolerogenic regimens (80, 108, 109).
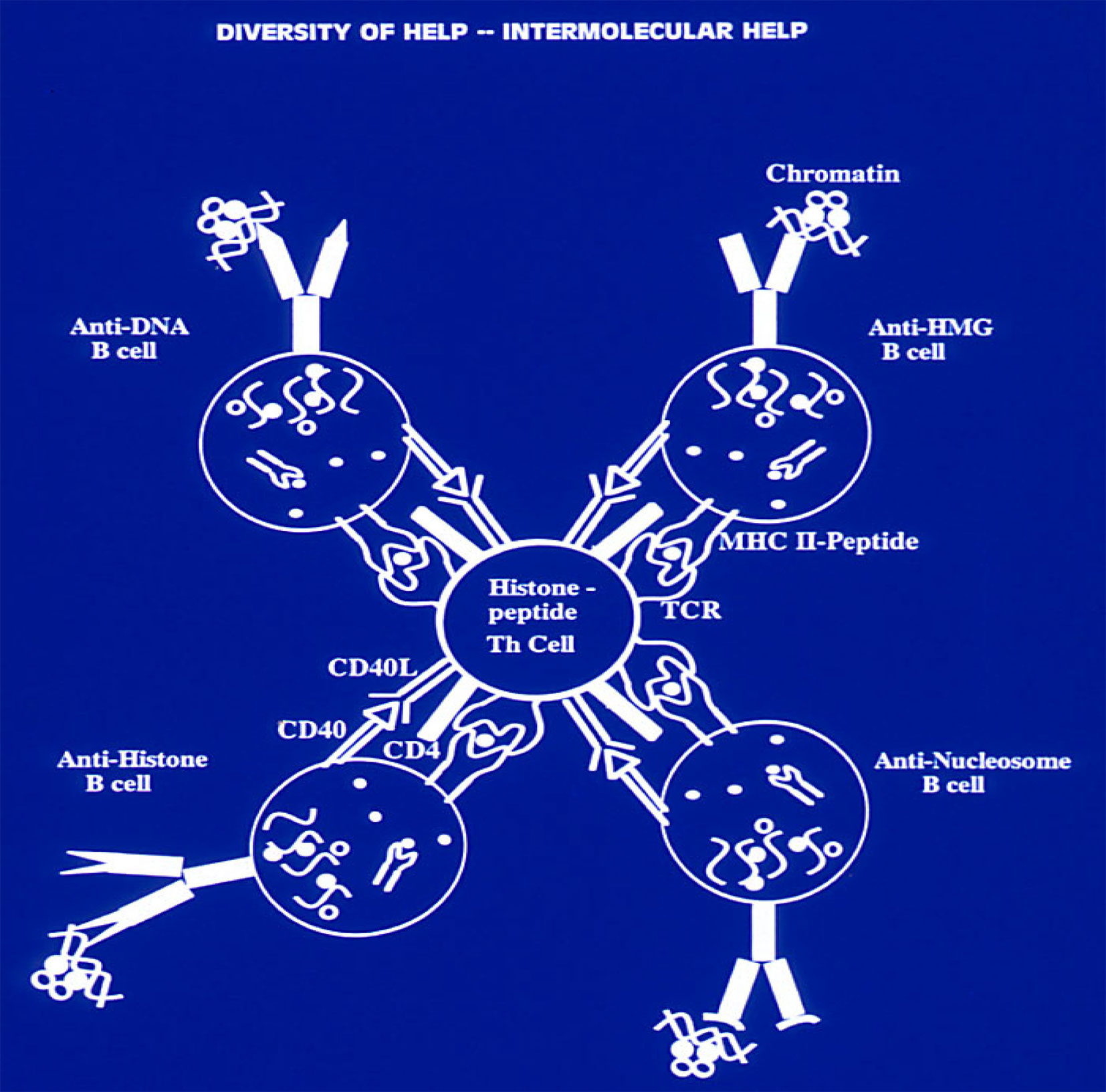
Figure 4 Production of a variety of anti-nuclear autoantibodies by inter-molecular T-cell help in SLE. A lupus Th cell with specificity for an individual nucleosomal histone peptide can help either a B cell specific for nucleosomes, or a B cell specific for dsDNA, or for ssDNA, or histone, or HMG, because each B cell can take up and process the whole chromatin particle by recognizing its own specific epitope in the chromatin, and then present to the Th clone its relevant histone peptide epitope derived from chromatin processing, which results in “inter-molecular help” This principle of linked “inter-molecular help” for a variety of B-cell epitopes in the complex chromatin particle would also apply to other Th cells of lupus which induce other pathogenic autoantibodies; and forms the basis for “Tolerance-Spreading” as described in the text. Modified from Ref (109, 120). (Originally published in The Journal of Immunology. Kaliyaperumal A, Michaels MA, Datta SK. Antigen-specific therapy of murine lupus nephritis using nucleosomal peptides: Tolerance spreading impairs pathogenic function of autoimmune T and B cells. J Immunol (1999) 162:5775-83. Copyright © [1999] The American Association of Immunologists, Inc.).
Indeed, the production of a variety of pathogenic autoantibodies to nuclear autoantigens was inhibited by tolerance therapy with any one of the epitopes. Due to promiscuous recognition described above, multiple autoimmune T cells with different TCRs can respond to the same peptide from a nucleosomal histone, and on the other hand, an individual autoimmune T cell can recognize multiple nucleosome-derived peptides that are distinct in sequence (61, 105). Therefore, when injected in a soluble tolerance-inducing form, in the absence of adjuvants, even one peptide epitope can tolerize autoimmune Th cells of diverse specificity for chromatin-derived autoantigens and conversely suppressing Th cells with specificty for one nuclear autoantigenic epitope deprives help for multiple autoimmune B cells of lupus. Thus, tolerance induced by any one of the dominant peptide epitopes can suppress autoimmune response to other nucleosome-derived pathogenic epitopes (“Tolerance Spreading”). Indeed, such “cross-reactive” suppression directed at the broad spectrum of pathogenic autoimmune response is more desirable in lupus therapy rather than pinpoint precision for antigen-specificity, which is the goal of some studies using modern techniques (121). Furthermore, the peptide epitopes are very effective in tolerance induction because, they are simultaneously recognized by autoimmune T and B cells, and they may inhibit autoimmune B cells and DC directly in lupus (80, 101, 108, 109).
Despite “Tolerance Spreading to other lupus autoepitopes”, the histone peptide therapy resulted in autoantigen-specific regulation because, pathogenic autoimmune responses in lupus-prone subjects were preferentially and selectively downregulated, without any suppression of immune responses to foreign antigens, as detailed in these References (80, 108, 109). Moreover, ability of the treated animals to survive environmental microbes/pathogens appeared to be intact and robust as compared to untreated controls and non-autoimmune “normal” mice housed in the same “dirty” mouse facilities (80, 108, 109). Finally, experiments showed that the Treg cells generated by the peptide epitope therapy suppressed T cell response and T cell helper activity specifically directed to the peptide autoepitopes, but not for a foreign antigen, such as Hen Egg Lysozyme (80, 108).
As described above, the nucleosomal histone peptide epitopes when administered in nanomolar doses (1µg) subcutaneously (S.C.) every 2 weeks or even every month to lupus-prone mice, are effective in delaying or even preventing nephritis. This dose is lower by almost 1000 fold compared to some other peptides being tried as therapeutic agents. Furthermore, a major histone peptide epitope, administered singly, can suppress lupus disease also via nasal tolerance (122, 123). The peptide epitopes are a constituent of nucleosome, a highly conserved, ubiquitous self-antigen produced during ongoing apoptosis in generative lymphoid organs and recognized by developing cells of the immune system bearing appropriate receptors. Therefore, anaphylactic reactions were not observed with these self-peptides when administered in close to 1000 lupus-prone mice for various studies. The histone peptide epitopes, administered S.C. in a very low doses, generate Treg cells that suppress by producing minute amounts of TGFβ that act in close range cell to cell interaction, rather than causing Th2 deviation with consequent allergic reactions seen in the case of therapy of other autoimmune diseases, such as, EAE/MS and diabetes in NOD mice using other peptides. The histone peptide epitopes induce stable Treg that are autoepitope-specific and cross-reactive autoantigen-directed Treg cells by simultaneously decreasing IL-6 and increasing TGF-β production by DCs, which consequently caused Smad-3 phosphorylation in the peptide epitope-specific auto-immune CD4+ Th cells, and the peptide tolerance therapy is effective even at an age when they manifest clinically active disease. A single dominant epitope such as H471-94, is capable of inhibiting the diverse autoimmune process in lupus, because, potent and durable regulatory T cells (Treg) are generated by low-dose tolerance therapy mediating “tolerance spreading”. Both sets of regulatory T cells act via TGFβ in close range, and suppress autoimmune Th and B cells and other autoantigen presenting cells.
Although the phenomenon of low-dose tolerance was well known (124), most work since then have dealt with tolerance induction to foreign antigens in non-autoimmune subjects. However, the histone peptide induced low-dose tolerance was achieved in subjects with spontaneous SLE, whose immune system is already primed for autoimmune response against ubiquitous nucleosomal self-epitopes. The Autoantigen-specific and cross-reactive autoantigen-directed Treg cells generated by the peptide epitope therapy was effective even in the presence of complex lupus abnormalities, such as hyperactivity of lupus T and B cells and DC; particularly IFN-α producing pDC.
Unlike the peptide epitopes in low doses, whole (intact) histones worsen lupus (60) probably by binding to DNA or reciprocally charged molecules in vivo to make complex nucleosome like particle structures. Moreover, processing by APC of intact histones generates altered epitopes by post-translational modifications, such as acetylation or citrulination (125). Therefore, intact whole (complete) histones should not be used for tolerance induction.
Another group induced Treg cells by continuous infusion of a model laboratory antigen using hemagglutinin (HA)-specific TCR−Tg mouse system, and they also targeted the peptide to a surface receptor DEC-205 on DC, and administered considerable quantities of TGFβ in vivo (126). However, those approaches for therapy of diabetes were found to be deleterious (127). Moreover, in vivo administration of TGF−β as a drug in the presence of high IL−6 levels in lupus could induce pathogenic Th17 cells and TFH cells, instead of generating Treg cells (128, 129).
Remarkably, pathogenic anti-DNA autoantibody inducing Th cells in human lupus recognized the same immunodominant histone peptide autoepitopes identified in murine lupus (61, 102), and those T cells in lupus patient’s PBMC respond by producing IFNγ. To reiterate, IFNγ-dependent IgG autoantibody subclasses cause lupus nephritis by fixing C’ and binding to inflammatory Fcγ receptors in pathogenic cells (39, 130). Furthermore, the peptide autoepitopes for Th cells of human lupus have the property of promiscuous HLA-DR binding, and as in lupus-prone mice, they are located in the native nucleosome at sites that contact with DNA, and they reside in histone regions that are also targeted by lupus B-cells (autoantibodies), thus being protected during antigen processing. Therefore, these immunodominant epitopes could probably be used as “universal” tolerogens in lupus patients despite their diversity of HLA alleles. The pathogenic role of nucleosome epitope-specific Th cells in human lupus have been confirmed by other laboratories (131, 132). Similar principles apply to other autoantigens in lupus, such as, Sm, RNP (133), but this review is focused on anti-DNA response whose pathogenic role in human lupus nephritis is well characterized. Remarkably, very recent approaches using latest technology to identify immunodominant epitopes for influenza hemagglutinin-specific memory T cells (134), showed results that are similar in outcome to the histone peptide approach performed two decades ago to identify the recurrent epitopes for pathogenic anti-DNA inducing memory T cells of lupus (61, 102).
Unexpectedly, prolonged remission achieved by patients with refractory lupus after autologous hematopoietic stem cell transplantation (HSCT) have a different mechanistic basis than “clinical remission” in conventional drug-treated patients, who do not achieve true immunologic remission, although they have a Systemic Lupus Disease Activity Index (SLEDAI) of 0–2 (80% of the drug induced remission patients were at zero level). In patients with stem cell transplant induced remission, CD4+CD25highFoxP3+ Treg, and CD8+FoxP3+ Treg cells are generated, accompanied by almost total suppression of pathogenic T cells that respond to the histone peptide autoepitopes (135).
Detailed experiments in the above ref (135)., demonstrated that the post-transplant CD8 Treg cells suppressive activity was nucleosomal histone peptide-specific, as well as nonspecific, but directed to cross-reactive autoreactive and activated T cells. Both types of Treg cell’s suppressive activity was mainly TGF-β-dependent, but independent of cell-cell contact. The post-transplant patients’ CD8 Treg cells were stably FoxP3+ and they expressed markedly increased levels of CTLA-4, CD103, PD-1, PD-L1 and LAP, when compared to CD8 T cells from the same patients before undergoing transplantation. By contrast, the pre-transplant lupus patient’s CD8 T cells have cell-contact dependent helper activity for autoantibody production. The CD8 Treg found only in post-transplant patients are considerably more potent in suppressive activity compared to the CD4+CD25high Treg cells that appear during clinical “remission” in lupus patients treated by conventional drugs, in whom autoimmune response of CD4 T cells to nucleosome-derived autoepitopes persists even during “clinical remission” (SLEDAI of zero). Therefore, autologous HSCT leads to the generation of a newly differentiated population of LAPhighCD103high CD8TGF-β Treg cells that maintain the lupus patients in “true immunological remission”, unlike patients with conventional drug therapy. Remarkably, very similar, highly potent CD8 Treg cells are also generated by low-dose nucleosomal peptide tolerance therapy that can prevent or treat lupus disease in mouse models of spontaneous SLE, as described above.
As stated, autoantibodies in lupus that belong to IFN-γ (Th1) dependent IgG subclasses fix complement and bind to activating FcγR on inflammatory cells to mediate pathogenicity. A CD4 T cell population in untreated lupus patients PBMC produces IFN-γ in response to histone peptide autoepitopes, and this autoimmune IFN-γ production response was almost completely suppressed in fresh PBMC from lupus patients in remission post-transplant. Removal of CD8 T cells (total) from the PBMC of post-transplant patients in remission, restored the IFN-γ response of CD4 T cells to nucleosomes and histone epitopes, much more strongly than removal of CD4+CD25high cell subset enriched for Treg. Therefore, the latter subset probably cannot restore immunologic remission in conventionally treated lupus patients although they are increased in such patients after “clinical remission” (SLEDAI of 0–2).
The Post-transplant CD8 T cells suppressed by secreting mainly TGF-β and they expressed high levels of TGF-β latency-associated peptide (LAP), but they produced IL-10 to a much lesser extent; which is desirable because IL-10, by causing expansion of autoimmune B cells, is deleterious in lupus (136).
The return of potent CD8TGF−β Treg cells after HSCT in refractory lupus patients, or after nucleosomal peptide epitope tolerance therapy in lupus-prone mice is an important biomarker for a state of true Immunologic Remission. These CD8+CD103+FoxP3+ TGFβ producing Treg are highly effective in controlling lupus, as shown in autologous stem cell transplant patients in remission above; and after corticosteroid pulse therapy induced remission in patients with lupus nephritis (Tsai YG et al. Plos One 2014, 9:e81344); as well as in murine models of lupus (108, 137, 138), and graft-versus-host lupus (138–140). And the above category of CD8+FoxP3+ TGFβ producing Treg that are highly effective in controlling lupus disease, are quite different from another variety of CD8+ Treg that are FoxP3-negative, cytotoxic and contact-dependent, and with varying surface phenotypes found in organ-specific autoimmune diseases (141–143). Those FoxP3-Ly49+(CD158e+ in humans)CD122hiHelios+CXCR5+ CD8 Treg cells were decreased as a percentage of total CD8 T cell population in lupus (144, 145), but such changes in proportion could be due to many reasons that cause shifts in various CD8 T cell subsets in lupus (146). Therefore, no cause and effect relationship of the latter CD8 Treg with spontaneous lupus disease in humans has been established yet. Anyway, target organ pathology in lupus nephritis, is inhibited by the TGFβ producing CD4+FoxP3+ Treg and CD8+FoxP3+ Treg cells induced by histone peptide epitope tolerance therapy (80, 108), or by targeted nanoparticle therapy (140) induced CD8+ Treg, which are quite different from the cytotoxic CD8 Treg (144, 145). Indeed, CD8+CD103+FoxP3+ TGFβ producing Treg cells, which maintain lupus patients in long term immunological remission after autologous bone marrow transplantation (135), or that induced in lupus patients’ PBMC by the histone peptide epitopes in vitro (147), have their highly effective suppressor counterparts in several models of autoimmune diseases including lupus (148–151). The role of locally active tissue-resident TGFβ producing Treg cells migrating into the kidney and its lymph nodes to suppress lupus nephritis pathogenesis, like those induced by histone peptide epitope therapy, has been recently demonstrated in other Treg inducing systems (114–116, 152).
As low-dose tolerance induced by the histone peptide epitopes effectively inhibited lupus disease in mouse models, the effect of the epitopes on lupus patients’ PBMC cultures was tested in vitro. As discussed above, the major Peptide Autoepitopes for nucleosome-specific T Cells of human lupus were identical in sequence to the peptide autoepitopes for pathogenic T cells of lupus-prone mice (61, 102), and they shared similar properties of promiscuous MHC class II binding and being B cell autoantibody epitopes as well. Thus the peptide epitopes could be effective tolerogens for inhibiting both autoimmune T and B cell populations in lupus patients with diverse HLA alleles (61, 102, 105, 147).
Indeed, in PBMC cultures from inactive lupus patients and healthy subjects, addition of the histone peptide epitopes induced CD4+CD25highFoxP3+ or CD4+CD45RA+FoxP3low Treg cells, as well as CD8+CD25+FoxP3+ Treg cells with stable FoxP3 expression and suppressive activity (147). In the case of PBMC from patients with active lupus, dexamethasone or hydroxychloroquine were additionally needed for Treg-induction by the peptide epitopes in cultures. The peptide-induced regulatory T cells in lupus PBMC depended on TGFβ/ALK-5/pSmad 2/3 signaling, and TGF-β precursor LAP was expressed by those Treg cells, indicating that TGFβ production was responsible for their suppressive activity, and a positive feedback mechanism as well. The peptide epitope-induced Treg cells also inhibited type I IFN related gene expression in lupus PBMC. Expression of major members of Type I IFN genes themselves, as well as type I IFN induced genes (ISG) were markedly reduced by histone peptide epitopes in TLR9-stimulated PBMC of lupus patients. As stated above, pDCs in lupus are the main producers of Type I IFN upon stimulation by nuclear autoantigens complexed with anti-nuclear autoantibodies (7, 153, 154), and in lupus-prone mice, histone peptide epitopes act on pDC rendering them to become tolerogenic (80). Secondly, expression of 13 ISG genes, which have been reported to be upregulated in patients with active lupus (153, 155, 156) were also inhibited by the peptide epitopes. Moreover, the histone peptide Th cell epitopes, which were also shared by autoantibody producing B cell epitopes in lupus, could inhibit production of pathogenic autoantibodies by PBMC from active lupus patients as potently as an anti-IL6 antibody. Experimental details are in reference (147). Importantly, a mixture of the peptide epitopes (cocktail) was more effective in uniformly suppressing pathogenic activities in Human lupus PBMC cultures, as compared to single epitopes, because patients are heterogeneous in contrast to inbred lupus-prone mice. For example, suppression by a histone peptide cocktail #1 (C1), which is a mixture of H122-42, H3115-135 and H416-39 at a concentration of 1.5 μM for each peptide or histone peptide epitope cocktail #2 (C2), which is a mixture of H122-42, H3115-135 and H416-39 at a concentration of 4 μM of each peptide were very efficient in suppressing pathogenic autoantibody production and type I IFN related gene expression in lupus PBMC (147). Thus, low-dose histone peptide epitopes could durably inhibit pathogenic autoimmune response in human lupus by diverse pathways.
Generalized immunosuppression can control manifestations of active lupus, but despite their toxicity the drugs fail to achieve true immunological remission. Such drug therapies should be followed by autoantigen specific suppression of pathogenic autoimmune cells in lupus to prevent flares and continuing organ damage. In contrast to lupus patients, normal subjects have regulatory mechanisms including regulatory T cells that prevent abnormal pathogenic response to nuclear autoantigens from cells undergoing apoptosis routinely in the body (135, 147, 157).
The tolerogenic histone peptide epitopes have the potential for prophylactically repairing the functional deficiency of regulatory T cells in lupus (135, 147, 157, 158). The above studies in mouse models in vivo, and with lupus patient’s cells in vitro, showed that the peptide autoepitopes have the ability to bring about durable regulatory mechanisms; probably because of desirable properties mentioned above, and summarized here. The histone peptide epitopes are derived from nucleosomes of apoptotic cells produced daily in the body, which are cleared silently in the normal host without causing any immune response (17). Indeed large scale apoptosis occurs daily in generative organs, such as bone marrow and thymus and the products are used to “educate” the cells of developing immune system. The epitopes with their native sequences intact are called “unaltered peptide ligands (UPL)”; they are derived from nucleosomes of apoptotic cells that are naturally processed and displayed to developing lymphocytes during ontogeny (101, 107, 159, 160), and therefore, unlike artificially altered peptide ligands (APL), or post-translationally acetylated or citrullinated histone peptides, the unaltered histone peptide epitopes described here are not associated with anaphylactic/allergic reactions or worsening of lupus (80, 108, 109). In fact Treg cells are generated in the thymus, even in lupus-prone mice, in a natural response to the native unaltered histone peptide epitopes (107),. Only 1 µg (0.34 nanomolar) of the histone peptide epitope/s administered biweekly is effective in low-dose tolerance therapy of lupus-prone mice; that dosage would be around 0.2 to 2 mg range in humans with lupus. The histone peptides are rapidly absorbed systemically after S.C. injection, because they possess numerous charged residues making them highly soluble. As soon as they reach the lymphoid organs the peptides render APCs, especially pDC tolerogenic by inducing TGFβ and inhibiting IL-6, and consequently the peptide epitope presenting DC generate long-lasting Treg containing autoantigen-specific and cross-reactive autoantigen-directed Treg and Treg cells that suppress lupus (80, 108). Because the peptide epitopes operate by being taken up extremely rapidly by DC in vivo rendering them tolerogenic, short half life due to decay of the epitopes is not a problem. Moreover, the histone peptide therapy induced stable autoantigen-specific and cross-reactive autoantigen-directed regulatory or suppressive T cells generated in vivo are effective in suppressing disease upon transfer into lupus-prone mice (80, 108). Both MHC class II, and nested MHC class I binding determinants are present in the peptide epitope sequences so that they can genarate both CD4 Treg and CD8 Treg cells (80, 108). The epitopes are recognized by autoimmune T cells irrespective of the HLA type of lupus patients (102, 105, 135, 147, 159), similar to “universal epitopes” (104). Tolerance therapy with the histone peptide epitopes is effective even in mice with established lupus disease (80, 108, 109). The peptides can generate Treg in lupus patient’s PBMC even in the presence of other conventional maintenance medicines such as hydroxychloroquine or corticosteroids (147). The peptide autoepitopes from histones induce “linked tolerance” to other nuclear antigen autoepitopes recognized by pathogenic T and B cell of lupus (cross-reactive, “tolerance spreading”), but not to foreign antigens or other organ-derived autoantigens. In addition to generation of Treg cells, the peptides also exert tolerogenic effect directly on pathogenic lupus B cells and DC (80, 101, 147); suppressing autoantibody production irrespective of the degree of Treg induction (135, 147). Regulatory mechanisms against abnormal autoimmune response to nuclear autoantigens in asymptomatic subjects could be enforced by the relatively innocuous tolerance therapy with histone peptides (147), which suggests that apparently healthy relatives or ANA positive subjects at risk for developing lupus as predicted by GWAS bio-markers, could be protected prophylactically with these peptide epitopes.
Thus the peptide epitope therapy might be most suitable for maintaining lupus patients in true immunological remission after clinical remission has been induced by more toxic immunosuppressive agents. To summarize, unlike pinpoint antigen-specific therapy suitable for straight-forward organ-specific autoimmune diseases, the histone peptide epitopes directly or indirectly (through Treg cells they induce) suppress Innate immune cells (DC), T cells and B cells involved in the pathogenic autoimmune response in the complex systemic autoimmune disease, Lupus.
The histone peptide epitopes could also be used to develop sensitive diagnostic and/or prognostic tools (peptide-MHC tetramers) or assays (intracellular cytokine response) for tracking pathogenic Th cells that may appear prior to manifestation of the disease and elevation of autoantibodies. Indeed, understanding mechanism/s for generation of unusual and potent CD8+ Treg cells by the peptide therapy will be of therapeutic value in a broad spectrum of immune mediated diseases, and Immunologic Monitoring with the peptide epitopes may serve as biomarkers for true immunologic remission (supplementing conventional measures of clinical remission, such as, SLEDAI SLAM, BILAG).
Early phase clinical trials have shown promising outcome with autoantigen peptide therapy for inducing antigen-specific tolerance in several autoimmune diseases, such as Multiple Sclerosis and Type 1 Diabetes (161–167). These results are encouraging for clinical trials with histone peptides for lupus in the near future, but several distinct features of this lupus therapy need to be addressed to reach that goal. Unlike pinpoint antigen-specific therapy suitable for straight-forward organ-specific autoimmune diseases, the histone peptide epitopes have unique tolerogenic properties with broad autoreactivity-specific inhibitory effect. By rendering Innate immune cells (DC) tolerogenic, the histone peptides induce Treg cells that suppress T and B cell populations which are both antigen-specifically and cross-reactively involved in the pathogenic autoimmune response in the complex systemic autoimmune disease, Lupus:
Multiple laboratories have shown that histone peptide epitope/s or other peptide epitopes administered without IL-2 injection, can induce generation of effective Treg in vivo, which inhibit disease in various lupus-prone mice (80, 82–85, 108, 122, 168). Although lupus T cells are deficient in IL-2 production (169, 170), that situation is relative not absolute, as lupus patients do not succumb to recurrent infections found in IL-2 knockout Immunodeficiency. A possibility in the case of these peptide epitopes is that in low doses, they could transiently activate autoreactive T cells, which could then provide small amounts of IL-2 for generating the regulatory T cells. Indeed this proposed mechanism (171), has actually been demonstrated by a similar situation occurring in the thymus where IL-2 is produced by a small population of self-reactive CD4 single positive (CD4SP) thymocytes, which then stimulates Treg precursor cells to differentiate (172). Those regulatory T cells induced in vivo may then be sustained also by other signals such as from ICOS, or TNFR2 (Tseng WY et al. Proc Natnl Acad Sci USA 2019, 116:21666-21672) (173). Still in view of the benefits of low dose IL-2 therapy in all autoimmune diseases, whether deficient in IL-2 or not (129, 169, 170, 174–177); adjunct therapy with low-dose IL-2 will be beneficial in the peptide-epitope therapy of lupus, as stated in the theme of this Topic. Moreover, Low dose IL-2 and corticosteroids in maintenance dose, actually were necessary for the peptide epitopes’ induction of Treg cells in ACTIVE lupus patients’ PBMC in vitro (147). Indeed, corticosteroids themselves induce Treg cells by various mechanisms to some extent (Tsai YG et al. Plos One 2014, 9:e81344) (178, 179), and thus could potentiate the autoantigen-specific and cross-reactive autoantigen-directed Treg response by the peptide epitopes (147).
How can durable immunoregulatory mechanisms be established in the inflammatory environment of lupus? In lupus patients, tolerance therapy with histone peptide epitope would be optimal after inflammatory burden is reduced by drugs. Nevertheless, in animal models, the peptides alone are effective in ameliorating established lupus nephritis (80, 108, 109). The regulatory T cells are more stable in inflammatory environment because they were induced by the peptide epitopes in vivo, in contrast to Treg cells induced/expanded in vitro. Furthermore, dexamethasone or hydroxychloroquine in maintenance doses actually supported Treg-induction by the peptides in lupus patients’ PBMC cultures, indicating that drugs that counter the increased activity of IRF5 and TLR pathways in lupus APC would be of added benefit (147). The histone peptide epitopes also can directly regulate autoimmune B cells and DC in lupus, in addition to generating Treg cells (80, 101, 109); and indeed the peptides could suppress autoantibody production to baseline levels in lupus patient’s PBMC even before significantly increasing Treg cell numbers in culture (147). The select histone peptide epitopes, which are tolerogenic, can directly reduce IL-6 and increase TGFβ production by DC (80), a situation which renders the DC not only be able to induce Treg, but also become susceptible to suppression by Treg (180). This property of inducing TGFβ production and simultaneously decreasing IL-6 production by DC, especially pDC, in turn induces TGF-β signal (Smad-3 phosphorylation) in target auto-immune CD4+ T cells converting them to stable Treg cells; a property highly beneficial for lupus therapy (80, 147), also because TFH cell differentiation is inhibited in germinal centers under such conditions (128).
Since apoptotic cells have immunosuppressive properties (17, 181); the unaltered histone peptide epitopes derived from apoptotic platform may have intrinsic tolerogenic property (80, 147). In very low doses, without immunostimulatory adjuvants, the histone peptide epitopes could possibly activate latent TGFβ, by inducing expression of the integrin αvβ8 in resting pDC, as shown in other systems (182).
Treg cells require continued antigen-specific stimulation from DC to maintain lineage stability and high affinity regulatory cell function (183, 184). In lupus-prone mice, regulatory T cells induced by the peptide epitopes are detectable up to six weeks after S.C. injection. Subcutaneous injection route works for the highly soluble and charged histone peptide epitopes which are very rapidly absorbed systemically (80). In humans many protein drugs, and mAb biologics, insulin, IVIg etc., are administered S.C. without causing local/systemic inflammatory response.
However, despite the promising beneficial effects in animal models of established lupus disease, there is always the possibility of adverse autoreactive response to the peptide epitopes in patients with lupus, although they might be selected at the earliest, pre-clinical stage of disease. Another issue is that peptide epitope cocktails in low doses were more effective than a single peptide epitope in suppressing lupus manifestations in human lupus PBMC, but cocktails may be more immunogenic when injected in the skin (147).
Therefore, peptide delivery should be considered in fail-safe tolerogenic vehicles, such as Nanoparticles (NP), which are described in detail by experts in this field in other articles as part of this research topic. Just as a brief synopsis, the peptide epitopes may be delivered within the nanoparticles, or administered around the same time, but separately from tolerogenic nanoparticles (185). There are many issues in choosing the right nanoparticles for such therapy, specifically for lupus; for instance, liposome derived NP can activate complement, and rapamycin containing NP may interfere with initial Treg generation (186, 187), although rapamycin is effective in maintaining Treg, once they are induced (188). It is noteworthy that injected nanoparticles might be nonspecifically immunosuppressive, like silica particles, by overloading the immune system’s APCs, which phagocytose and engorge themselves with any foreign particles (189, 190). Therefore, targeted nanoparticles designed to be directed against potentially autoreactive T cells are much more promising (140, 191), as addressed by articles from experts in this research topic.
Finally, emerging studies on epigenetic or metabolic mechanisms for Treg cell stability (192–194), and correcting other abnormalities in lupus T cells, such as, metabolic (12, 13), could be potentiated by utilizing the benefits of peptide epitope therapy, in the near future.
SD wrote and edited this review, and is accountable for the content of the work.
This is a review of published work. Details of funding are in those publications cited in this review, and the work by the author’s group at that time had been supported partially by funding from National Institutes of Health (NIAID for 43 years, and NIAMS for 30 years), and Alliance for Lupus Research (TIL grant for 6 years).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The studies reviewed (and cited) in this paper are from collaborative work with many talented scientists, who were mainly at Northwestern University Feinberg School of Medicine at the time the studies were done, especially: Arunan Kaliyaperumal, Hee-Kap Kang, Li Zhang, Chandra Mohan, Sharlene Adams, Sumi Rajagopalan (Shivakumar), Rosalind Ramsey Goldman, and Richard Burt.
1. Alarcón-Riquelme ME, Ziegler JT, Molineros J, Howard TD, Moreno-Estrada A, Sánchez-Rodríguez E, et al. Genome-Wide Association Study in an Amerindian Ancestry Population Reveals Novel Systemic Lupus Erythematosus Risk Loci and the Role of European Admixture. Arthritis Rheumatol (Hoboken NJ) (2016) 68(4):932–43. doi: 10.1002/art.39504
2. Barcellos LF, May SL, Ramsay PP, Quach HL, Lane JA, Nititham J, et al. High-density SNP screening of the major histocompatibility complex in systemic lupus erythematosus demonstrates strong evidence for independent susceptibility regions. PloS Genet (2009) 5(10):e1000696. doi: 10.1371/journal.pgen.1000696
3. Bentham J, Morris DL, Graham DSC, Pinder CL, Tombleson P, Behrens TW, et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat Genet (2015) 47(12):1457–64. doi: 10.1038/ng.3434
4. Martin JE, Assassi S, Diaz-Gallo LM, Broen JC, Simeon CP, Castellvi I, et al. A systemic sclerosis and systemic lupus erythematosus pan-meta-GWAS reveals new shared susceptibility loci. Hum Mol Genet (2013) 22(19):4021–9. doi: 10.1093/hmg/ddt248
5. Krausgruber T, Blazek K, Smallie T, Alzabin S, Lockstone H, Sahgal N, et al. IRF5 promotes inflammatory macrophage polarization and TH1-TH17 responses. Nat Immunol (2011) 12(3):231–8. doi: 10.1038/ni.1990
6. Stone RC, Feng D, Deng J, Singh S, Yang L, Fitzgerald-Bocarsly P, et al. Interferon regulatory factor 5 activation in monocytes of systemic lupus erythematosus patients is triggered by circulating autoantigens independent of type I interferons. Arthritis Rheumatol (2012) 64(3):788–98. doi: 10.1002/art.33395
7. Niewold TB, Kelly JA, Flesch MH, Espinoza LR, Harley JB, Crow MK. Association of the IRF5 risk haplotype with high serum interferon-alpha activity in systemic lupus erythematosus patients. Arthritis Rheumatol (2008) 58(8):2481–7. doi: 10.1002/art.23613
8. Yan J, Hedl M, Abraham C. Myeloid Cell-Intrinsic IRF5 Promotes T Cell Responses through Multiple Distinct Checkpoints In Vivo, and IRF5 Immune-Mediated Disease Risk Variants Modulate These Myeloid Cell Functions. J Immunol (Baltimore Md 1950) (2020) 205(4):1024–38. doi: 10.4049/jimmunol.1900743
9. Wang Y, Chen S, Chen J, Xie X, Gao S, Zhang C, et al. Germline genetic patterns underlying familial rheumatoid arthritis, systemic lupus erythematosus and primary Sjögren’s syndrome highlight T cell-initiated autoimmunity. Ann Rheumatic Dis (2020) 79(2):268–75. doi: 10.1136/annrheumdis-2019-215533
10. Kamitaki N, Sekar A, Handsaker RE, de Rivera H, Tooley K, Morris DL, et al. Complement genes contribute sex-biased vulnerability in diverse disorders. Nature (2020) 582(7813):577–81. doi: 10.1038/s41586-020-2277-x
11. Desai-Mehta A, Lu L, Ramsey-Goldman R, Datta SK. Hyperexpression of CD40 ligand by B and T cells in human lupus and its role in pathogenic autoantibody production. J Clin Invest (1996) 97:2063–73. doi: 10.1172/JCI118643
12. Yin Y, Choi SC, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T cell metabolism reverses lupus. Sci Trans Med (2015) 7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835
13. Li W, Elshikha AS, Cornaby C, Teng X, Abboud G, Brown J, et al. T cells expressing the lupus susceptibility allele Pbx1d enhance autoimmunity and atherosclerosis in dyslipidemic mice. JCI Insight (2020) 5(11):e138274. doi: 10.1172/jci.insight.138274
14. Giles JR, Neves AT, Marshak-Rothstein A, Shlomchik MJ. Autoreactive helper T cells alleviate the need for intrinsic TLR signaling in autoreactive B cell activation. JCI Insight (2017) 2(4):e90870. doi: 10.1172/jci.insight.90870
15. Steinman RM, Turley S, Mellman I, Inaba K. The induction of tolerance by dendritic cells that have captured apoptotic cells. J Exp Med (2000) 191(3):411–6. doi: 10.1084/jem.191.3.411
16. Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol (2003) 21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040
17. Voll RE, Herrmann M, Roth EA, Stach C, Kalden JR, Girkontaite I. Immunosuppressive effects of apoptotic cells. Nature (1997) 390:350–1. doi: 10.1038/37022
18. Skoberne M, Beignon AS, Larsson M, Bhardwaj N. Apoptotic cells at the crossroads of tolerance and immunity. Curr Topics Microbiol Immunol (2005) 289:259–92. doi: 10.1007/3-540-27320-4_12
19. Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, Marshak-Rothstein A. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature (2002) 416:595–8. doi: 10.1038/416603a
20. Means TK, Latz E, Hayashi F, Murali MR, Golenbock DT, Luster AD. Human lupus autoantibody-DNA complexes activate DCs through cooperation of CD32 and TLR9. J Clin Invest (2005) 115(2):407–17. doi: 10.1172/JCI23025
21. Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity (2008) 28(6):799–809. doi: 10.1016/j.immuni.2008.03.019
22. Souyris M, Cenac C, Azar P, Daviaud D, Canivet A, Grunenwald S, et al. TLR7 escapes X chromosome inactivation in immune cells. Sci Immunol (2018) 3(19):eaap8855. doi: 10.1126/sciimmunol.aap8855
23. Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, et al. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natnl Acad Sci USA (2006) 103(26):9970–5. doi: 10.1073/pnas.0603912103
24. Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, et al. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity (2007) 27(5):801–10. doi: 10.1016/j.immuni.2007.09.009
25. Heinz LX, Lee J, Kapoor U, Kartnig F, Sedlyarov V, Papakostas K, et al. TASL is the SLC15A4-associated adaptor for IRF5 activation by TLR7-9. Nature (2020) 581(7808):316–22. doi: 10.1038/s41586-020-2282-0
26. Pisetsky DS. The role of TASL in the pathogenesis of SLE: X marks the spot. Ann Rheumatic Dis (2021) 80(1):6–7. doi: 10.1136/annrheumdis-2020-218643
27. Bocharnikov AV, Keegan J, Wacleche VS, Cao Y, Fonseka CY, Wang G, et al. PD-1hiCXCR5- T peripheral helper cells promote B cell responses in lupus via MAF and IL-21. JCI Insight (2019) 4(20):e130062. doi: 10.1172/jci.insight.130062
28. Phalke S, Marrack P. Age (autoimmunity) associated B cells (ABCs) and their relatives. Curr Opin Immunol (2018) 55:75–80. doi: 10.1016/j.coi.2018.09.007
29. Kang HK, Chiang MY, Ecklund D, Zhang L, Ramsey-Goldman R, Datta SK. Megakaryocyte progenitors are the main APCs inducing Th17 response to lupus autoantigens and foreign antigens. J Immunol (Baltimore Md 1950) (2012) 188(12):5970–80. doi: 10.4049/jimmunol.1200452
30. Finkielsztein A, Schlinker AC, Zhang L, Miller WM, Datta SK. Human megakaryocyte progenitors derived from hematopoietic stem cells of normal individuals are MHC class II-expressing professional APC that enhance Th17 and Th1/Th17 responses. Immunol Lett (2015) 163(1):84–95. doi: 10.1016/j.imlet.2014.11.013
31. Chen CI, Zhang L, Datta SK. Hematopoietic stem and multipotent progenitor cells produce IL-17, IL-21 and other cytokines in response to TLR signals associated with late apoptotic products and augment memory Th17 and Tc17 cells in the bone marrow of normal and lupus mice. Clin Immunol (Orlando Fla) (2016) 162:9–26. doi: 10.1016/j.clim.2015.10.007
32. Zufferey A, Speck ER, Machlus KR, Aslam R, Guo L, McVey MJ, et al. Mature murine megakaryocytes present antigen-MHC class I molecules to T cells and transfer them to platelets. Blood Adv (2017) 1(20):1773–85. doi: 10.1182/bloodadvances.2017007021
33. Rahman A, Isenberg DA. Systemic lupus erythematosus. N Engl J Med (2008) 358(9):929–39. doi: 10.1056/NEJMra071297
34. Liu Z, Davidson A. Taming lupus-a new understanding of pathogenesis is leading to clinical advances. Nat Med (2012) 18(6):871–82. doi: 10.1038/nm.2752
35. Rothfield NF, Stollar BD. The relation of immunoglobulin class, pattern of anti-nuclear antibody, and complement-fixing antibodies to DNA in sera from patients with systemic lupus erythematosus. J Clin Invest (1967) 46:1785–94. doi: 10.1172/JCI105669
36. Koffler D, Carr RI, Agnello V, Thoburn R, Kunkel HG. Antibodies to polynucleotides in human sera; antigenic specificity and relation to disease. J Exp Med (1971) 134:294–312. doi: 10.1084/jem.134.1.294
37. Ebling F, Hahn BH. Restricted subpopulations of DNA antibodies in kidneys of mice with systemic lupus: comparison of antibodies in serum and renal eluates. Arthritis Rheumatol (1980) 23:392–403. doi: 10.1002/art.1780230402
38. Gavalchin J, Nicklas J, Eastcott JW, Madaio MP, Stollar BD, Schwartz RS, et al. Lupus prone (SWR x NZB)F1 mice produce potentially nephritogenic autoantibodies inherited from the normal SWR parent. J Immunol (1985) 134:885–94.
39. Gavalchin J, Datta SK. The NZB x SWR model of lupus nephritis. II. Autoantibodies deposited in renal lesions show a restricted idiotypic diversity. J Immunol (1987) 138:138–48.
40. Vlahakos DV, Foster MH, Adams S, Katz M, Ucci AA, Barrett KJ, et al. Anti-DNA antibodies form immune deposits at distinct glomerular and vascular sites. Kidney Internatnl (1992) 41:1690–700. doi: 10.1038/ki.1992.242
41. Datta SK, Patel H, Berry D. Induction of a cationic shift in IgG anti-DNA autoantibodies. Role of T helper cells with classical and novel phenotypes in three murine models of lupus nephritis. J Exp Med (1987) 165:1252–68. doi: 10.1084/jem.165.5.1252
42. Gavalchin J, Seder RA, Datta SK. The NZB x SWR model of lupus nephritis. I. cross-reactive idiotypes of monoclonal anti-DNA antibodies in relation to antigenic specificity, charge and allotype. Identification of interconnected idiotype families inherited from the normal SWR and the autoimmune NZB parents. J Immunol (1987) 138:128–37.
43. O’Keefe TL, Bandyopadhyay S, Datta SK, Imanishi-Kari T. Variable region sequences of an idiotypically connected family of pathogenic anti-DNA autoantibodies. J Immunol (1990) 144:4275–83.
44. Radic MZ, Weigert M. Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol (1994) 12:487–520. doi: 10.1146/annurev.iy.12.040194.002415
45. Shlomchik MJ, Mascelli M, Shan H, Radic MZ, Pisetsky D, Marshak-Rothstein A, et al. Anti-DNA antibodies from autoimmune mice arise by clonal expansion and somatic mutation. J Exp Med (1990) 171:265–92. doi: 10.1084/jem.171.1.265
46. Rennke HG, Cotran RS, Venkatachalam MA. Role of molecular charge in glomerular permeability. Tracer studies with cationized ferritin. J Cell Biol (1975) 67:638–46. doi: 10.1083/jcb.67.3.638
47. Kanwar YS, Farquhar MG. Anionic sites in glomerular basement membrane. In vivo and in vitro localization to the laminae rarae by cationic probes. J Cell Biol (1976) 81:137–53. doi: 10.1083/jcb.81.1.137
48. Kohro-Kawata J, Wang P, Kawata Y, Matsuzaki M, Nakamura K. Highly cationic anti-DNA antibodies in patients with lupus nephritis analyzed by two-dimensional electrophoresis and immunoblotting. Electrophoresis (1998) 19(8-9):1511–5. doi: 10.1002/elps.1150190849
50. van der Vlag J, Berden JH. Lupus nephritis: role of antinucleosome autoantibodies. Semin Nephrol (2011) 31(4):376–89. doi: 10.1016/j.semnephrol.2011.06.009
51. Olin AI, Mörgelin M, Truedsson L, Sturfelt G, Bengtsson AA. Pathogenic mechanisms in lupus nephritis: Nucleosomes bind aberrant laminin β1 with high affinity and colocalize in the electron-dense deposits. Arthritis Rheumatol (Hoboken NJ) (2014) 66(2):397–406. doi: 10.1002/art.38250
52. Kalaaji M, Mortensen E, Jørgensen L, Olsen R, Rekvig OP. Nephritogenic lupus antibodies recognize glomerular basement membrane-associated chromatin fragments released from apoptotic intraglomerular cells. Am J Pathol (2006) 168(6):1779–92. doi: 10.2353/ajpath.2006.051329
53. Sainis K, Datta SK. CD4+ T cell lines with selective patterns of autoreactivity as well as CD4-/CD8- T helper cell lines augment the production of idiotypes shared by pathogenic anti-DNA autoantibodies in the NZB x SWR model of lupus nephritis. J Immunol (1988) 140:2215–24.
54. Shivakumar S, Tsokos GC, Datta SK. T cell receptor alpha/beta expressing double negative (CD4-/CD8-) and CD4+ T helper cells in humans augment the production of pathogenic anti-DNA autoantibodies associated with lupus nephritis. J Immunol (1989) 143:103–12.
55. Rajagopalan S, Zordan T, Tsokos GC, Datta SK. Pathogenic anti-DNA autoantibody inducing T helper cell lines from patients with active lupus nephritis: Isolation of CD4-/CD8- T helper cell lines that express the γδ T-cell receptor. Proc Natl Acad Sci USA (1990) 87:7020–4. doi: 10.1073/pnas.87.18.7020
56. Desai-Mehta A, Mao C, Rajagopalan S, Robinson T, Datta SK. Structure and specificity of T-cell receptors expressed by pathogenic anti-DNA autoantibody-inducing T cells in human lupus. J Clin Invest (1995) 95:531–41. doi: 10.1172/JCI117695
57. Adams S, Leblanc P, Datta SK. Junctional region sequences of T-cell receptor β-chain genes expressed by pathogenic anti-DNA autoantibody-inducing helper T cells from lupus mice: Possible selection by cationic autoantigens. Proc Natl Acad Sci United States America (1991) 88(24):11271–5. doi: 10.1073/pnas.88.24.11271
58. Mohan C, Shi Y, Laman JD, Datta SK. Interaction between CD40 and its ligand gp39 in the development of murine lupus nephritis. J Immunol (1995) 154:1470–80.
59. Mao C, Osman GE, Adams S, Datta SK. T cell receptor alpha-chain repertoire of pathogenic autoantibody-inducing T cells in lupus mice. J Immunol (1994) 152:1462–70.
60. Mohan C, Adams S, Stanik V, Datta SK. Nucleosome: A major immunogen for pathogenic autoantibody-inducing T cells of lupus. J Exp Med (1993) 177(5):1367–81. doi: 10.1084/jem.177.5.1367
61. Kaliyaperumal A, Mohan C, Wu W, Datta SK. Nucleosomal peptide epitopes for nephritis-inducing T helper cells of murine lupus. J Exp Med (1996) 183:2459–69. doi: 10.1084/jem.183.6.2459
62. Mitchison NA. T-cell-B-cell cooperation. Nat Rev Immunol (2004) 4(4):308–12. doi: 10.1038/nri1334
63. Datta SK, Schwartz RS. Genetics of expression of xenotropic virus and autoimmunity in NZB mice. Nature (1976) 263:412–5. doi: 10.1038/263412b0
64. Datta SK, McConahey PJ, Manny N, Theofilopoulos AN, Dixon FJ, Schwartz RS. Genetic studies of autoimmunity and retrovirus expression in crosses of NZB mice. II. The viral envelope glycoprotein gp 70. J Exp Med (1978) 147:872–81. doi: 10.1084/jem.147.3.872
65. Cook HT, Botto M. Mechanisms of Disease: the complement system and the pathogenesis of systemic lupus erythematosus. Nat Clin Pract Rheumatol (2006) 2(6):330–7. doi: 10.1038/ncprheum0191
66. Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther (2011) 13(1):202. doi: 10.1186/ar3206
67. Rogers NJ, Lees MJ, Gabriel L, Maniati E, Rose SJ, Potter PK, et al. A defect in Marco expression contributes to systemic lupus erythematosus development via failure to clear apoptotic cells. J Immunol (Baltimore Md 1950) (2009) 182(4):1982–90. doi: 10.4049/jimmunol.0801320
68. Munoz LE, Lauber K, Schiller M, Manfredi AA, Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol (2010) 6(5):280–9. doi: 10.1038/nrrheum.2010.46
69. Casciola-Rosen LA, Anhalt G, Rosen A. Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J Exp Med (1994) 179:1317–30. doi: 10.1084/jem.179.4.1317
70. Mevorach D, Mascarenhas JO, Gershov D, Elkon KB. Complement-dependent clearance of apoptotic cells by human macrophages. J Exp Med (1998) 188:2313–20. doi: 10.1084/jem.188.12.2313
71. Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature (2000) 407(6805):784–8. doi: 10.1038/35037722
72. Poon IK, Lucas CD, Rossi AG, Ravichandran KS. Apoptotic cell clearance: basic biology and therapeutic potential. Nat Rev Immunol (2014) 14(3):166–80. doi: 10.1038/nri3607
73. Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Sci (N Y NY) (2006) 312(5780):1669–72. doi: 10.1126/science.1124978
74. Lartigue A, Colliou N, Calbo S, Francois A, Jacquot S, Arnoult C, et al. Critical role of TLR2 and TLR4 in autoantibody production and glomerulonephritis in lpr mutation-induced mouse lupus. J Immunol (Baltimore Md 1950) (2009) 183(10):6207–16. doi: 10.4049/jimmunol.0803219
75. Magna M, Pisetsky DS. The role of HMGB1 in the pathogenesis of inflammatory and autoimmune diseases. Mol Med (2014) 20:138–46. doi: 10.2119/molmed.2013.00164
77. Berland R, Fernandez L, Kari E, Han JH, Lomakin I, Akira S, et al. Toll-like receptor 7-dependent loss of B cell tolerance in pathogenic autoantibody knockin mice. Immunity (2006) 25(3):429–40. doi: 10.1016/j.immuni.2006.07.014
78. Mobarrez F, Svenungsson E, Pisetsky DS. Microparticles as autoantigens in systemic lupus erythematosus. Eur J Clin Invest (2018) 48(12):e13010. doi: 10.1111/eci.13010
79. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity (2006) 25(3):417–28. doi: 10.1016/j.immuni.2006.07.013
80. Kang H-K, Liu M, Datta SK. Low-Dose Peptide Tolerance Therapy of Lupus Generates Plasmacytoid Dendritic Cells That Cause Expansion of Autoantigen-Specific Regulatory T Cells and Contraction of Inflammatory Th17 Cells. J Immunol (Baltimore Md 1950) (2007) 178(12):7849–58. doi: 10.4049/jimmunol.178.12.7849
81. Tilstra JS, John S, Gordon RA, Leibler C, Kashgarian M, Bastacky S, et al. B cell-intrinsic TLR9 expression is protective in murine lupus. J Clin Invest (2020) 130(6):3172–87. doi: 10.1172/jci132328
82. Singh RR, Ebling FM, Sercarz EE, Hahn BH. Immune tolerance to autoantibody-derived peptides delays development of autoimmunity in murine lupus. J Clin Invest (1995) 96:2990–6. doi: 10.1172/JCI118371
83. Sharabi A, Zinger H, Zborowsky M, Sthoeger ZM, Mozes E. A peptide based on the complementarity-determining region 1 of an autoantibody ameliorates lupus by up-regulating CD4+CD25+ cells and TGF-beta. Proc Natl Acad Sci U S A (2006) 103(23):8810–5. doi: 10.1073/pnas.0603201103
84. Aas-Hanssen K, Thompson KM, Bogen B, Munthe LA. Systemic Lupus Erythematosus: Molecular Mimicry between Anti-dsDNA CDR3 Idiotype, Microbial and Self Peptides-As Antigens for Th Cells. Front Immunol (2015) 6:382:382. doi: 10.3389/fimmu.2015.00382
85. Nyland JF, Stoll ML, Jiang F, Feng F, Gavalchin J. Mechanisms involved in the p62-73 idiopeptide-modulated delay of lupus nephritis in SNF(1) mice. Lupus (2012) 21(14):1552–64. doi: 10.1177/0961203312461964
86. Ghiggeri GM, D’Alessandro M, Bartolomeo D, Degl’Innocenti ML, Magnasco A, Lugani F, et al. An Update on Antibodies to Necleosome Components as Biomarkers of Sistemic Lupus Erythematosus and of Lupus Flares. Int J Mol Sci (2019) 20(22):5799. doi: 10.3390/ijms20225799
87. Ravirajan CT, Rowse L, MacGowan JR, Isenberg DA. An analysis of clinical disease activity and nephritis-associated serum autoantibody profiles in patients with systemic lupus erythematosus: a cross-sectional study. Rheumatol (Oxford England) (2001) 40(12):1405–12. doi: 10.1093/rheumatology/40.12.1405
88. Takahashi S, Fossati L, Iwamoto M, Merino R, Motta R, Kobayakawa T, et al. Imbalance towards Th1 predominance is associated with acceleration of lupus-like autoimmune syndrome in MRL mice. J Clin Invest (1996) 97(7):1597–604. doi: 10.1172/jci118584
89. Jackson SW, Jacobs HM, Arkatkar T, Dam EM, Scharping NE, Kolhatkar NS, et al. B cell IFN-γ receptor signaling promotes autoimmune germinal centers via cell-intrinsic induction of BCL-6. J Exp Med (2016) 213(5):733–50. doi: 10.1084/jem.20151724
90. Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, et al. IFN-γ receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med (2016) 213(5):715–32. doi: 10.1084/jem.20151722
91. Ritvo PG, Saadawi A, Barennes P, Quiniou V, Chaara W, El Soufi K, et al. High-resolution repertoire analysis reveals a major bystander activation of Tfh and Tfr cells. Proc Natl Acad Sci U S A (2018) 115(38):9604–9. doi: 10.1073/pnas.1808594115
92. Roco JA, Mesin L, Binder SC, Nefzger C, Gonzalez-Figueroa P, Canete PF, et al. Class-Switch Recombination Occurs Infrequently in Germinal Centers. Immunity (2019) 51(2):337–50.e7. doi: 10.1016/j.immuni.2019.07.001
93. Velu V, Mylvaganam GH, Gangadhara S, Hong JJ, Iyer SS, Gumber S, et al. Induction of Th1-Biased T Follicular Helper (Tfh) Cells in Lymphoid Tissues during Chronic Simian Immunodeficiency Virus Infection Defines Functionally Distinct Germinal Center Tfh Cells. J Immunol (Baltimore Md 1950) (2016) 197(5):1832–42. doi: 10.4049/jimmunol.1600143
94. Powell MD, Read KA, Sreekumar BK, Jones DM, Oestreich KJ. IL-12 signaling drives the differentiation and function of a T(H)1-derived T(FH1)-like cell population. Sci Rep (2019) 9(1):13991. doi: 10.1038/s41598-019-50614-1
95. Ma X, Nakayamada S, Kubo S, Sakata K, Yamagata K, Miyazaki Y, et al. Expansion of T follicular helper-T helper 1 like cells through epigenetic regulation by signal transducer and activator of transcription factors. Ann Rheumatic Dis (2018) 77(9):1354–61. doi: 10.1136/annrheumdis-2017-212652
96. Morita R, Schmitt N, Bentebibel SE, Ranganathan R, Bourdery L, Zurawski G, et al. Human blood CXCR5(+)CD4(+) T cells are counterparts of T follicular cells and contain specific subsets that differentially support antibody secretion. Immunity (2011) 34(1):108–21. doi: 10.1016/j.immuni.2010.12.012
97. Facciotti F, Larghi P, Bosotti R, Vasco C, Gagliani N, Cordiglieri C, et al. Evidence for a pathogenic role of extrafollicular, IL-10-producing CCR6(+)B helper T cells in systemic lupus erythematosus. Proc Natl Acad Sci U S A (2020) 117(13):7305–16. doi: 10.1073/pnas.1917834117
98. Adams S, Zordan T, Sainis K, Datta SK. T cell receptor Vβ genes expressed by IgG anti-DNA autoantibody inducing T cells in lupus nephritis: Forbidden receptors and double negative T cells. Eur J Immunol (1990) 20:1435–43. doi: 10.1002/eji.1830200705
99. Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol (2008) 181(12):8761–6. doi: 10.4049/jimmunol.181.12.8761
100. Holoshitz J, Koning F, Coligan JE, De Bruyn J, Strober S. Isolation of CD4- CD8- mycobacteria-reactive T lymphocyte clones from rheumatoid arthritis synovial fluid. Nature (1989) 339(6221):226–9. doi: 10.1038/339226a0
101. Kaliyaperumal A, Michaels MA, Datta SK. Naturally processed chromatin peptides reveal a major autoepitope that primes pathogenic T and B cells of lupus. J Immunol (2002) 168:2530–7. doi: 10.4049/jimmunol.168.5.2530
102. Lu L, Kaliyaperumal A, Boumpas DT, Datta SK. Major peptide autoepitopes for nucleosome-specific T cells of human lupus. J Clin Invest (1999) 104:345–55. doi: 10.1172/JCI6801
103. Stemmer C, Richalet-Secordel P, Van Bruggen MCJ, Kramers C, Berden JHM, Muller S. Dual reactivity of several monoclonal anti-nucleosome antibodies for double stranded DNA and a short segment of histone H3. J Biol Chem (1996) 271:21257–61. doi: 10.1074/jbc.271.35.21257
104. Panina-Bordignon P, Tan A, Termijtelen A, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes: promiscous binding to human MHC class II and promiscous recognition by T cells. Eur J Immunol (1989) 19:2237–42. doi: 10.1002/eji.1830191209
105. Shi Y, Kaliyaperumal A, Lu L, Southwood S, Sette A, Michaels MA, et al. Promiscuous presentation and recognition of nucleosomal autoepitopes in lupus: Role of autoimmune T cell receptor alpha chain. J Exp Med (1998) 187:367–78. doi: 10.1084/jem.187.3.367
106. Wilson DB, Wilson DH, Schroder K, Pinilla C, Blondelle S, Houghten RA, et al. Specificity and degeneracy of T cells. Mol Immunol (2004) 40(14-15):1047–55. doi: 10.1016/j.molimm.2003.11.022
107. Michaels MA, Kang HK, Kaliyaperumal A, Satyaraj E, Shi Y, Datta SK. A defect in deletion of nucleosome-specific autoimmune T cells in lupus-prone thymus: role of thymic dendritic cells. J Immunol (2005) 175(9):5857–65. doi: 10.4049/jimmunol.175.9.5857
108. Kang H-K, Michaels MA, Berner BR, Datta SK. Very low-dose tolerance with nucleosomal peptides controls lupus and induces potent regulatory T cell subsets. J Immunol (2005) 174:3247–55. doi: 10.4049/jimmunol.174.6.3247
109. Kaliyaperumal A, Michaels MA, Datta SK. Antigen-specific therapy of murine lupus nephritis using nucleosomal peptides: Tolerance spreading impairs pathogenic function of autoimmune T and B cells. J Immunol (1999) 162:5775–83.
110. Kuttler C, Nussbaum AK, Dick TP, Rammensee HG, Schild H, Hadeler KP. An algorithm for the prediction of proteasomal cleavages (erratum in J. Mol. Biol. Aug. 4, 301:229). J Mol Biol (2000) 298:417–29. doi: 10.1006/jmbi.2000.3683
111. Rosalia RA, Quakkelaar ED, Redeker A, Khan S, Camps M, Drijfhout JW, et al. Dendritic cells process synthetic long peptides better than whole protein, improving antigen presentation and T-cell activation. Eur J Immunol (2013) 43(10):2554–65. doi: 10.1002/eji.201343324
112. Ghali JR, Holdsworth SR, Kitching AR. Targeting IL-17 and IL-23 in Immune Mediated Renal Disease. Curr Med Chem (2015) 22(38):4341–65. doi: 10.2174/0929867322666151030163022
113. Kitching AR, Holdsworth SR. The emergence of TH17 cells as effectors of renal injury. J Am Soc Nephrol JASN (2011) 22(2):235–8. doi: 10.1681/asn.2010050536
114. Stremska ME, Dai C, Venkatadri R, Wang H, Sabapathy V, Kumar G, et al. IL233, an IL-2-IL-33 hybrid cytokine induces prolonged remission of mouse lupus nephritis by targeting Treg cells as a single therapeutic agent. J Autoimmun (2019) 102:133–41. doi: 10.1016/j.jaut.2019.05.005
115. Rose A, von Spee-Mayer C, Kloke L, Wu K, Kühl A, Enghard P, et al. IL-2 Therapy Diminishes Renal Inflammation and the Activity of Kidney-Infiltrating CD4+ T Cells in Murine Lupus Nephritis. Cells (2019) 8(10):1234. doi: 10.3390/cells8101234
116. Yan JJ, Lee JG, Jang JY, Koo TY, Ahn C, Yang J. IL-2/anti-IL-2 complexes ameliorate lupus nephritis by expansion of CD4(+)CD25(+)Foxp3(+) regulatory T cells. Kidney Int (2017) 91(3):603–15. doi: 10.1016/j.kint.2016.09.022
117. Chan OT, Madaio MP, Shlomchik MJ. The central and multiple roles of B cells in lupus pathogenesis. Immunol Rev (1999) 169:107–21. doi: 10.1111/j.1600-065x.1999.tb01310.x
118. Furie R, Rovin BH, Houssiau F, Malvar A, Teng YKO, Contreras G, et al. Two-Year, Randomized, Controlled Trial of Belimumab in Lupus Nephritis. New Engl J Med (2020) 383(12):1117–28. doi: 10.1056/NEJMoa2001180
119. Müller-Winkler J, Mitter R, Rappe JCF, Vanes L, Schweighoffer E, Mohammadi H, et al. Critical requirement for BCR, BAFF, and BAFFR in memory B cell survival. J Exp Med (2021) 218(2):e20191393. doi: 10.1084/jem.20191393
120. Datta SK, Kaliyaperumal A. Nucleosome-driven autoimmune response in lupus - Pathogenic T helper cell epitopes and costimulatory signals. In "B Lymphocytes and Autoimmunity" (Editors, N. Chiorazzi, R. G. Lahita, K. Pavelka, M. Ferrarini). Ann N Y Acad Sci (1997) 815:155–70. doi: 10.1111/j.1749-6632.1997.tb52057.x
121. Salinas VH, Stüve O. Systems Approaches to Unravel T Cell Function and Therapeutic Potential in Autoimmune Disease. J Immunol (Baltimore Md 1950) (2021) 206(4):669–75. doi: 10.4049/jimmunol.2000954
122. Wu HY, Ward FJ, Staines NA. Histone Peptide-induced nasal tolerance: suppression of murine lupus. J Immunol (2002) 169:1126–34. doi: 10.4049/jimmunol.169.2.1126
123. Wu HY, Monsonego A, Weiner HL. The mechanism of nasal tolerance in lupus prone mice is T-cell anergy induced by immature B cells that lack B7 expression. J Autoimmun (2006) 26(2):116–26. doi: 10.1016/j.jaut.2005.11.005
124. Mitchison NA. Induction of immunological paralysis in two zones of dosage. Proc R Soc London B (1964) 161:275–92. doi: 10.1098/rspb.1964.0093
125. Dieker J, Berden JH, Bakker M, Briand JP, Muller S, Voll R, et al. Autoantibodies against Modified Histone Peptides in SLE Patients Are Associated with Disease Activity and Lupus Nephritis. PloS One (2016) 11(10):e0165373. doi: 10.1371/journal.pone.0165373
126. Daniel C, Weigmann B, Bronson R, von Boehmer H. Prevention of type 1 diabetes in mice by tolerogenic vaccination with a strong agonist insulin mimetope. J Exp Med (2011) 208(7):1501–10. doi: 10.1084/jem.20110574
127. Bergman ML, Lopes-Carvalho T, Martins AC, Grieco FA, Eizirik DL, Demengeot J. Tolerogenic insulin peptide therapy precipitates type 1 diabetes. J Exp Med (2017) 214(7):2153–6. doi: 10.1084/jem.20160471
128. Papillion A, Powell MD, Chisolm DA, Bachus H, Fuller MJ, Weinmann AS, et al. Inhibition of IL-2 responsiveness by IL-6 is required for the generation of GC-T(FH) cells. Sci Immunol (2019) 4(39):eaaw7636. doi: 10.1126/sciimmunol.aaw7636
129. Ballesteros-Tato A, Papillion A. Mechanisms of action of low-dose IL-2 restoration therapies in SLE. Curr Opin Immunol (2019) 61:39–45. doi: 10.1016/j.coi.2019.07.003
130. Balomenos D, Rumold R, Theofilopoulos AN. Interferon-gamma is required for lupus-like disease and lymphoaccumulation in MRL-lpr mice. J Clin Invest (1998) 101:364–71. doi: 10.1172/JCI750
131. Voll RE, Roth EA, Girkontaite I, Fehr H, Herrman M, Lorenz HM, et al. Histone-specific Th0 and Th1 clones derived from systemic lupus erythematosus patients induce double-stranded DNA antibody production. Arthritis Rheumatol (1997) 40:2162–71. doi: 10.1002/art.1780401210
132. Bruns A, Blass S, Hausdorf G, Burmester GR, Hiepe F. Nucleosomes are major T and B cell autoantigens in systemic lupus erythematosus. Arthritis Rheumatol (2000) 43:2307–15. doi: 10.1002/1529-0131(200010)43:10<2307::AID-ANR19>3.0.CO;2-J
133. Hoffman RW, Takeda Y, Sharp GC, Lee DR, Hill DL, Kaneoka H, et al. Human T cell clones reactive against U-snRNP autoantigens from connective tissue disease patients and healthy individuals. J Immunol (1993) 151:6460–9.
134. Cassotta A, Paparoditis P, Geiger R, Mettu RR, Landry SJ, Donati A, et al. Deciphering and predicting CD4+ T cell immunodominance of influenza virus hemagglutinin. J Exp Med (2020) 217(10):e20200206. doi: 10.1084/jem.20200206
135. Zhang L, Bertucci AM, Ramsey-Goldman R, Burt RK, Datta SK. Regulatory T cell (Treg) subsets return in patients with refractory lupus following stem cell transplantation, and TGF-β producing CD8+ Treg cells are associated with immunological remission of lupus. J Immunol (2009) 183(10):6346–58. doi: 10.4049/jimmunol.0901773
136. Geginat J, Vasco M, Gerosa M, Tas SW, Pagani M, Grassi F, et al. IL-10 producing regulatory and helper T-cells in systemic lupus erythematosus. Semin Immunol (2019) 44:101330. doi: 10.1016/j.smim.2019.101330
137. Singh RP, La Cava A, Hahn BH. pConsensus peptide induces tolerogenic CD8+ T cells in lupus-prone (NZB x NZW)F1 mice by differentially regulating Foxp3 and PD1 molecules. J Immunol (2008) 180(4):2069–80. doi: 10.4049/jimmunol.180.4.2069
138. Zheng SG, Wang JH, Koss MN, Quismorio F Jr., Gray JD, Horwitz DA. CD4+ and CD8+ regulatory T cells generated ex vivo with IL-2 and TGF-beta suppress a stimulatory graft-versus-host disease with a lupus-like syndrome. J Immunol (2004) 172(3):1531–9. doi: 10.4049/jimmunol.172.3.1531
139. Beres AJ, Haribhai D, Chadwick AC, Gonyo PJ, Williams CB, Drobyski WR. CD8+ Foxp3+ regulatory T cells are induced during graft-versus-host disease and mitigate disease severity. J Immunol (Baltimore Md 1950) (2012) 189(1):464–74. doi: 10.4049/jimmunol.1200886
140. Horwitz DA, Bickerton S, Koss M, Fahmy TM, La Cava A. Suppression of Murine Lupus by CD4+ and CD8+ Treg Cells Induced by T Cell-Targeted Nanoparticles Loaded With Interleukin-2 and Transforming Growth Factor β. Arthritis Rheumatol (Hoboken NJ) (2019) 71(4):632–40. doi: 10.1002/art.40773
141. Choi JY, Eskandari SK, Cai S, Sulkaj I, Assaker JP, Allos H, et al. Regulatory CD8 T cells that recognize Qa-1 expressed by CD4 T-helper cells inhibit rejection of heart allografts. Proc Natl Acad Sci U S A (2020) 117(11):6042–6. doi: 10.1073/pnas.1918950117
142. Saligrama N, Zhao F, Sikora MJ, Serratelli WS, Fernandes RA, Louis DM, et al. Opposing T cell responses in experimental autoimmune encephalomyelitis. Nature (2019) 572(7770):481–7. doi: 10.1038/s41586-019-1467-x
143. Tang X, Kumar V. Advances in the Study of CD8+ Regulatory T Cells. Crit Rev Immunol (2019) 39(6):409–21. doi: 10.1615/CritRevImmunol.2020033260
144. Kim HJ, Wang X, Radfar S, Sproule TJ, Roopenian DC, Cantor H. CD8+ T regulatory cells express the Ly49 Class I MHC receptor and are defective in autoimmune prone B6-Yaa mice. Proc Natl Acad Sci U S A (2011) 108(5):2010–5. doi: 10.1073/pnas.1018974108
145. Mishra S, Liao W, Liu Y, Yang M, Ma C, Wu H, et al. TGF-β and Eomes control the homeostasis of CD8+ regulatory T cells. J Exp Med (2021) 218(1):e20200030. doi: 10.1084/jem.20200030
146. Minning S, Xiaofan Y, Anqi X, Bingjie G, Dinglei S, Mingshun Z, et al. Imbalance between CD8(+)CD28(+) and CD8(+)CD28(-) T-cell subsets and its clinical significance in patients with systemic lupus erythematosus. Lupus (2019) 28(10):1214–23. doi: 10.1177/0961203319867130
147. Zhang L, Bertucci AM, Ramsey-Goldman R, Harsha-Strong ER, Burt RK, Datta SK. Major pathogenic steps in human lupus can be effectively suppressed by nucleosomal histone peptide epitope-induced regulatory immunity. Clin Immunol (Orlando Fla) (2013) 149(3):365–78. doi: 10.1016/j.clim.2013.08.008
148. Zhong H, Liu Y, Xu Z, Liang P, Yang H, Zhang X, et al. TGF-β-Induced CD8(+)CD103(+) Regulatory T Cells Show Potent Therapeutic Effect on Chronic Graft-versus-Host Disease Lupus by Suppressing B Cells. Front Immunol (2018) 9:35:35. doi: 10.3389/fimmu.2018.00035
149. Deng W, Xu M, Meng Q, Li Z, Qiu X, Yin S, et al. CD8+CD103+ iTregs inhibit the progression of lupus nephritis by attenuating glomerular endothelial cell injury. Rheumatol (Oxford England) (2019) 58(11):2039–50. doi: 10.1093/rheumatology/kez112
150. Keino H, Masli S, Sasaki S, Streilein JW, Stein-Streilein J. CD8+ T regulatory cells use a novel genetic program that includes CD103 to suppress Th1 immunity in eye-derived tolerance. Invest Ophthalmol Visual Sci (2006) 47(4):1533–42. doi: 10.1167/iovs.04-1454
151. Uss E, Rowshani AT, Hooibrink B, Lardy NM, van Lier RAW, ten Berge IJM. CD103 Is a Marker for Alloantigen-Induced Regulatory CD8+ T Cells. J Immunol (2006) 177(5):2775–83. doi: 10.4049/jimmunol.177.5.2775
152. Tsai YG, Lee CY, Lin TY, Lin CY. CD8+ Treg cells associated with decreasing disease activity after intravenous methylprednisolone pulse therapy in lupus nephritis with heavy proteinuria. PloS One (2014) 9(1):e81344. doi: 10.1371/journal.pone.0081344
153. Chaussabel D, Quinn C, Shen J, Patel P, Glaser C, Baldwin N, et al. A modular analysis framework for blood genomics studies: application to systemic lupus erythematosus. Immunity (2008) 29(1):150–64. doi: 10.1016/j.immuni.2008.05.012
154. Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. Fc gamma RIIa is expressed on natural IFN-alpha-producing cells (plasmacytoid dendritic cells) and is required for the IFN-alpha production induced by apoptotic cells combined with lupus IgG. J Immunol (Baltimore Md 1950) (2003) 171(6):3296–302. doi: 10.4049/jimmunol.171.6.3296
155. Baechler EC, Batliwalla FM, Karypis G, Gaffney PM, Ortmann WA, Espe KJ, et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci USA (2003) 100:2610–5. doi: 10.1073/pnas.0337679100
156. Nikpour M, Dempsey AA, Urowitz MB, Gladman DD, Barnes DA. Association of a gene expression profile from whole blood with disease activity in systemic lupus erythaematosus. Ann Rheumatic Dis (2008) 67(8):1069–75. doi: 10.1136/ard.2007.074765
157. Singh RR, Ebling FM, Albuquerque DA, Saxena V, Kumar V, Giannini EH, et al. Induction of autoantibody production is limited in nonautoimmune mice. J Immunol (2002) 169(1):587–94. doi: 10.4049/jimmunol.169.1.587
158. Kang HK, Chiang MY, Liu M, Ecklund D, Datta SK. The Histone Peptide H4(71-94) Alone Is More Effective than a Cocktail of Peptide Epitopes in Controlling Lupus: Immunoregulatory Mechanisms. J Clin Immunol (2011) 31(3):379–94. doi: 10.1007/s10875-010-9504-4
159. Datta SK. Major peptide autoepitopes for nucleosome-centered T and B cell interaction in human and murine lupus. Ann NY Acad Sci (2003) 987:79–90. doi: 10.1111/j.1749-6632.2003.tb06035.x
160. Wardemann H, Yurasov S, Schaefer A, Young JW, Meffre E, Nussenzweig MC. Predominant autoantibody production by early human B cell precursors. Sci (N Y NY) (2003) 301(5638):1374–7. doi: 10.1126/science.1086907
161. Steinman L, Ho PP, Robinson WH, Utz PJ, Villoslada P. Antigen-specific tolerance to self-antigens in protein replacement therapy, gene therapy and autoimmunity. Curr Opin Immunol (2019) 61:46–53. doi: 10.1016/j.coi.2019.07.011
162. Serra P, Santamaria P. Antigen-specific therapeutic approaches for autoimmunity. Nat Biotechnol (2019) 37(3):238–51. doi: 10.1038/s41587-019-0015-4
163. Chataway J, Martin K, Barrell K, Sharrack B, Stolt P, Wraith DC. Effects of ATX-MS-1467 immunotherapy over 16 weeks in relapsing multiple sclerosis. Neurology (2018) 90(11):e955–62. doi: 10.1212/wnl.0000000000005118
164. Roep BO, Wheeler DCS, Peakman M. Antigen-based immune modulation therapy for type 1 diabetes: the era of precision medicine. Lancet Diabetes Endocrinol (2019) 7(1):65–74. doi: 10.1016/s2213-8587(18)30109-8
165. Walczak A, Siger M, Ciach A, Szczepanik M, Selmaj K. Transdermal application of myelin peptides in multiple sclerosis treatment. JAMA Neurol (2013) 70(9):1105–9. doi: 10.1001/jamaneurol.2013.3022
166. Lutterotti A, Yousef S, Sputtek A, Stürner KH, Stellmann JP, Breiden P, et al. Antigen-specific tolerance by autologous myelin peptide-coupled cells: a phase 1 trial in multiple sclerosis. Sci Trans Med (2013) 5(188):188ra75. doi: 10.1126/scitranslmed.3006168
167. Roep BO, Solvason N, Gottlieb PA, Abreu JRF, Harrison LC, Eisenbarth GS, et al. Plasmid-encoded proinsulin preserves C-peptide while specifically reducing proinsulin-specific CD8+ T cells in type 1 diabetes. Sci Trans Med (2013) 5(191):191ra82. doi: 10.1126/scitranslmed.3006103
168. Suen J-L, Chuang Y-H, Tsai B-Y, Yau PM, Chiang B-L. Treatment of murine lupus using nucleosomal T cell epitopes identified by bone marrow-derived dendritic cells. Arthritis Rheumatol (2004) 50:3250–9. doi: 10.1002/art.20520
169. Linker-Israeli M, Bakke AC, Kitridou RC, Gendler S, Gillis S, Horwitz DA. Defective production of interleukin 1 and interleukin 2 in patients with systemic lupus erythematosus (SLE). J Immunol (Baltimore Md 1950) (1983) 130(6):2651–5.
170. Mizui M, Tsokos GC. Low-Dose IL-2 in the Treatment of Lupus. Curr Rheumatol Rep (2016) 18(11):68. doi: 10.1007/s11926-016-0617-5
171. Zhang L, Datta SK. Prospects for a peptide vaccine for human lupus. Int J Clin Rheumatol (2014) 9(3):229–32. doi: 10.2217/ijr.14.37
172. Hemmers S, Schizas M, Azizi E, Dikiy S, Zhong Y, Feng Y, et al. IL-2 production by self-reactive CD4 thymocytes scales regulatory T cell generation in the thymus. J Exp Med (2019) 216(11):2466–78. doi: 10.1084/jem.20190993
173. Smigiel KS, Richards E, Srivastava S, Thomas KR, Dudda JC, Klonowski KD, et al. CCR7 provides localized access to IL-2 and defines homeostatically distinct regulatory T cell subsets. J Exp Med (2014) 211(1):121–36. doi: 10.1084/jem.20131142
174. Gutierrez-Ramos JC, Andreu JL, Revilla Y, Viñuela E, Martinez C. Recovery from autoimmunity of MRL/lpr mice after infection with an interleukin-2/vaccinia recombinant virus. Nature (1990) 346(6281):271–4. doi: 10.1038/346271a0
175. Humrich JY, Morbach H, Undeutsch R, Enghard P, Rosenberger S, Weigert O, et al. Homeostatic imbalance of regulatory and effector T cells due to IL-2 deprivation amplifies murine lupus. Proc Natl Acad Sci U S A (2010) 107(1):204–9. doi: 10.1073/pnas.0903158107
176. He J, Zhang R, Shao M, Zhao X, Miao M, Chen J, et al. Efficacy and safety of low-dose IL-2 in the treatment of systemic lupus erythematosus: a randomised, double-blind, placebo-controlled trial. Ann Rheumatic Dis (2020) 79(1):141–9. doi: 10.1136/annrheumdis-2019-215396
177. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheumatic Dis (2019) 78(2):209–17. doi: 10.1136/annrheumdis-2018-214229
178. Cari L, De Rosa F, Nocentini G, Riccardi C. Context-Dependent Effect of Glucocorticoids on the Proliferation, Differentiation, and Apoptosis of Regulatory T Cells: A Review of the Empirical Evidence and Clinical Applications. Int J Mol Sci (2019) 20(5):1142. doi: 10.3390/ijms20051142
179. Kim D, Nguyen QT, Lee J, Lee SH, Janocha A, Kim S, et al. Anti-inflammatory Roles of Glucocorticoids Are Mediated by Foxp3(+) Regulatory T Cells via a miR-342-Dependent Mechanism. Immunity (2020) 53(3):581–96. doi: 10.1016/j.immuni.2020.07.002
180. Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Sci (N Y NY) (2003) 299:1033–6. doi: 10.1126/science.1078231
181. Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest (1998) 101:890–8. doi: 10.1172/JCI1112
182. Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, et al. Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature (2007) 449(7160):361–5. doi: 10.1038/nature06110
183. Vahl JC, Drees C, Heger K, Heink S, Fischer JC, Nedjic J, et al. Continuous T cell receptor signals maintain a functional regulatory T cell pool. Immunity (2014) 41(5):722–36. doi: 10.1016/j.immuni.2014.10.012
184. Rubtsov YP, Niec RE, Josefowicz S, Li L, Darce J, Mathis D, et al. Stability of the regulatory T cell lineage in vivo. Sci (N Y NY) (2010) 329(5999):1667–71. doi: 10.1126/science.1191996
185. Kishimoto TK. Development of ImmTOR Tolerogenic Nanoparticles for the Mitigation of Anti-drug Antibodies. Front Immunol (2020) 11:969. doi: 10.3389/fimmu.2020.00969
186. Do MH, Wang X, Zhang X, Chou C, Nixon BG, Capistrano KJ, et al. Nutrient mTORC1 signaling underpins regulatory T cell control of immune tolerance. J Exp Med (2020) 217(1):e20190848. doi: 10.1084/jem.20190848
187. Valle A, Jofra T, Stabilini A, Atkinson M, Roncarolo MG, Battaglia M. Rapamycin prevents and breaks the anti-CD3-induced tolerance in NOD mice. Diabetes (2009) 58(4):875–81. doi: 10.2337/db08-1432
188. Lai ZW, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet (London England) (2018) 391(10126):1186–96. doi: 10.1016/s0140-6736(18)30485-9
189. Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J Controlled Release Off J Controlled Release Soc (2016) 240:332–48. doi: 10.1016/j.jconrel.2016.01.020
190. Miao X, Leng X, Zhang Q. The Current State of Nanoparticle-Induced Macrophage Polarization and Reprogramming Research. Int J Mol Sci. (2017) 18(2):336 doi: 10.3390/ijms18020336
191. Ganugula R, Arora M, Zou D, Agarwal SK, Mohan C, Kumar M. A highly potent lymphatic system-targeting nanoparticle cyclosporine prevents glomerulonephritis in mouse model of lupus. Sci Adv (2020) 6(24):eabb3900. doi: 10.1126/sciadv.abb3900
192. Akamatsu M, Mikami N, Ohkura N, Kawakami R, Kitagawa Y, Sugimoto A, et al. Conversion of antigen-specific effector/memory T cells into Foxp3-expressing T(reg) cells by inhibition of CDK8/19. Sci Immunol (2019) 4(40):eaaw2707. doi: 10.1126/sciimmunol.aaw2707
193. Mikami N, Kawakami R, Sakaguchi S. New Treg cell-based therapies of autoimmune diseases: towards antigen-specific immune suppression. Curr Opin Immunol (2020) 67:36–41. doi: 10.1016/j.coi.2020.07.004
Keywords: autoimmunity, systemic lupus erythematosus, T regulatory cells, T suppressor cells, autoantigen specific tolerance, autoantigen derived peptide epitopes
Citation: Datta SK (2021) Harnessing Tolerogenic Histone Peptide Epitopes From Nucleosomes for Selective Down-Regulation of Pathogenic Autoimmune Response in Lupus (Past, Present, and Future). Front. Immunol. 12:629807. doi: 10.3389/fimmu.2021.629807
Received: 16 November 2020; Accepted: 23 March 2021;
Published: 14 April 2021.
Edited by:
Shohei Hori, The University of Tokyo, JapanCopyright © 2021 Datta. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Syamal K. Datta, c2tkMjU3QG5vcnRod2VzdGVybi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.