 Claus Moser1†
Claus Moser1† Peter Østrup Jensen1,2†
Peter Østrup Jensen1,2† Kim Thomsen1
Kim Thomsen1 Mette Kolpen1
Mette Kolpen1 Morten Rybtke2
Morten Rybtke2 Anne Sofie Lauland1Hannah Trøstrup3
Anne Sofie Lauland1Hannah Trøstrup3 Tim Tolker-Nielsen2*
Tim Tolker-Nielsen2*- 1Department of Clinical Microbiology, Rigshospitalet, Copenhagen University Hospital, Copenhagen, Denmark
- 2Costerton Biofilm Center, Department of Immunology and Microbiology, Faculty of Health and Medical Sciences, University of Copenhagen, Copenhagen, Denmark
- 3Department of Plastic Surgery and Breast Surgery, Zealand University Hospital, Roskilde, Denmark
Pseudomonas aeruginosa is a key pathogen of chronic infections in the lungs of cystic fibrosis patients and in patients suffering from chronic wounds of diverse etiology. In these infections the bacteria congregate in biofilms and cannot be eradicated by standard antibiotic treatment or host immune responses. The persistent biofilms induce a hyper inflammatory state that results in collateral damage of the adjacent host tissue. The host fails to eradicate the biofilm infection, resulting in hindered remodeling and healing. In the present review we describe our current understanding of innate and adaptive immune responses elicited by P. aeruginosa biofilms in cystic fibrosis lung infections and chronic wounds. This includes the mechanisms that are involved in the activation of the immune responses, as well as the effector functions, the antimicrobial components and the associated tissue destruction. The mechanisms by which the biofilms evade immune responses, and potential treatment targets of the immune response are also discussed.
Introduction
Biofilms consist of microbes located in densely packed slow growing microcolonies embedded in a self-produced protective biopolymer matrix. In this life-mode, the microbes attain the highest levels of resistance to our present assortment of antibiotics and the immune system (1, 2). Accordingly, biofilms are a common cause of persistent infections (3), and biofilm-based infections are a major socio-economic burden implicating hospitalization, patient suffering, reduced life quality, increased mortality risk and lost employment. It is estimated that about 60%–70% of hospital acquired infections are caused by microbial biofilms (4). The immune response to biofilms results in collateral damage of adjacent tissues and therefore is an important aspect of biofilm infection pathology (5).
The vast majority of studies of the immune response against bacteria have focused on infections caused by bacteria in the planktonic state. Accordingly, considerably less is known about the immune response to bacteria growing in biofilm-based infections. However, recent in vivo and in vitro studies have begun to reveal the nature of both the innate and adaptive immune responses to biofilms (5, 6).
Planktonic bacteria are recognized by the innate immune systems pathogen recognition receptors (PRRs) through interaction with pathogen-associated molecular patterns (PAMPs), such as the flagellum and lipopolysaccharide (LPS) recognized via Toll-like receptor 5 and 4, respectively (7). Basically, biofilm growing bacteria activate the immune system through the same pathways as planktonic growing bacteria (5, 6). However, when residing in a biofilm the bacteria are embedded in extracellular polymeric substances and the classical PAMPs are less exposed to the immune system. In addition, PAMPs can be down-regulated in biofilm growing bacteria, as has been shown for flagella in P. aeruginosa (8, 9). Thus, in the case of biofilm infections the extracellular matrix components of the biofilms play an important role for the immune response (5, 6, 10).
The inflammatory state induced by biofilm unusually involves activation of both the innate and the adaptative immune response due to the chronic nature of biofilm-associated infections. Neither immune response is capable of eradicating biofilm, but they instead lead to extensive secondary damage.
The present review is focused on interactions between P. aeruginosa biofilms and the immune system (Figure 1). P. aeruginosa is involved in several persistent biofilm infections, including cystic fibrosis (CF) lung infections, chronic wound infections, urinary tract infections with or without catheters, and tracheal tube related ventilator-associated pneumonia (11–13). These infections are difficult or impossible to eradicate with antibiotics alone due to the special physiological state of bacteria in biofilms (2). The immune response has detrimental effects, as it causes destruction of the lungs of CF patients and maintains the inflammatory state of chronic wounds (11, 14). Knowledge about the mechanisms involved in activation, regulation, and evasion of the immune responses, as well as the nature of the antimicrobial components produced by the immune cells, and the associated tissue destruction has increased in recent years and will be discussed in the present review. Organ-system specific immune responses can differ substantially due to significant differences in tissue anatomy and physiology and is discussed when appropriate. Measurement of adaptive immune response during chronic persistent infections has proven an important clinical tool and will be described. Even though the role of the adaptive immune response has long been well recognized as being crucial during healing of wounds and in particular in inflammatory skin disease, the study of the role of the adaptive immune response in chronic wounds with P. aeruginosa biofilm infection has only just recently taken off (15, 16). Therefore, we have not included a detailed description of P. aeruginosa biofilm in chronic wound infections in the section of adaptive immune response. The understanding of all these components of host responses during biofilm infections may eventually form a basis for development of new and effective treatments against biofilm-based infections.
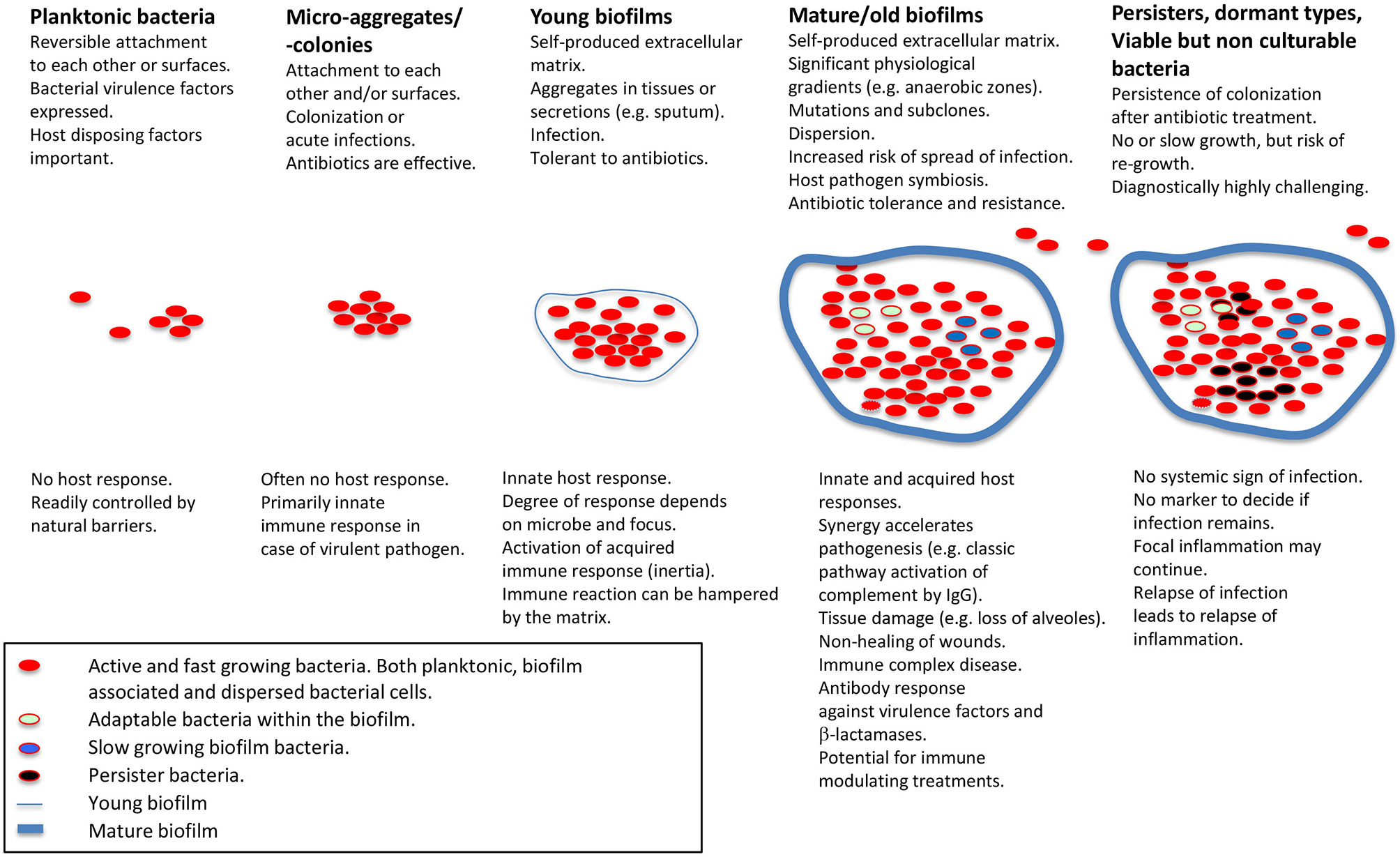
Figure 1 Schematic presentation of biofilm stages and host response. Applies for non-foreign body-related biofilm infections, which is the main focus of the present review. Modified from Moser et al. (5) with permission from John Wiley & Sons, Inc.
Biofilm Formation of P. aeruginosa During Chronic Infection
Biofilm formation by P. aeruginosa occur along with the production of several extracellular matrix components such as type IV pili (17–19), Cup fimbria (20), exopolysaccharides (21–23), CdrA adhesin (24), extracellular DNA (25), LecA/LecB lectins (26, 27) and Fap amyloids (28). The selection during chronic infection of P. aeruginosa variants that over-produce some of these biofilm matrix components is strong evidence for the involvement of biofilms in chronic infections (9, 29–32). Moreover, the presence of biofilms in CF lungs and chronic wounds has been demonstrated by microscopy (33, 34). P. aeruginosa can synthesize three different exopolysaccharides designated Pel, Psl, and alginate, although some strains only produce a subset of these exopolymers (21–23, 35). Overproduction of alginate enables mucoid P. aeruginosa strains to form persistent infections in the lungs of cystic fibrosis (CF) patients (29). Moreover, P. aeruginosa rugose small colony variants that overproduce Psl and Pel exopolysaccharide show enhanced persistence in CF lungs (9, 30, 31), and in chronic wounds (32). Evidence has been presented that Psl protects P. aeruginosa from host defenses in the initial phase of infection of the CF lung (36). Thus, it is possible that an extracellular biofilm matrix dominated by Psl is important in the initial stage of chronic lung infection before the bacteria mutate to produce a biofilm matrix dominated by alginate.
The host immune response plays an important role in the course of biofilm infections, and substantially affects the environment faced by the bacteria. The initial response to the presence of pathogens is an accumulation of activated neutrophils that may reduce the local O2 concentration due to O2 consumption accelerated by the respiratory burst and the production of reactive O2 species (ROS) and nitric oxide (NO) (37–39). Thus, O2 consumption by the neutrophils may result in O2 depletion in infected parts of the body (40). The restricted O2 availability accelerates stratified growth in P. aeruginosa biofilms, resulting in low metabolic activity in the center of biofilm as a consequence of nutrient depletion. However, micro-oxic conditions are sufficient to support growth of P. aeruginosa due to a highly flexible respiratory apparatus (41, 42) Moreover, bacteria may obtain energy under the anaerobic conditions prevalent in biofilm infections via anaerobic respiration or fermentation (43). Anaerobic respiration can occur by denitrification, where nitrogen oxides are utilized as alternative terminal electron acceptors (44, 45). The source of these N-oxides is suggested to originate from the rapid reaction of NO and O2 produced by activated neutrophils (44) resulting in the formation of peroxynitrite (ONOO-) (46), which may dismutate to nitrate and nitrite (47). The concentration of and in CF sputum (43, 48–50) may support P. aeruginosa growth at rates similar to those found in CF pulmonary biofilm (45). These findings suggest that the growth rate of P. aeruginosa during chronic CF lung infection is determined primarily by the number of surrounding neutrophils (51) which deplete O2 and produce and which can be used by the bacteria for anaerobic respiration. As biofilm formation, neutrophil accumulation and O2 depletion are common factors in multiple chronic infections, this interaction between host cells and pathogen is likely to occur also in other infections (44).
Innate Immune Responses During P. aeruginosa Biofilm Infections
Innate immunity fights infections from the moment of first contact and is composed of germline-encoded, non-clonal cellular and humoral mechanisms. These mechanisms enable nonspecific defense against pathogens without former interactions with infectious microbial invaders (52). The main components of the innate immune response engaged in response to P. aeruginosa biofilm include neutrophils, macrophages, dendritic cells, NK cells, and the complement system.
The most solid demonstration of a role of innate immune responses to bacterial biofilm has been obtained by introducing human neutrophils and macrophages to P. aeruginosa biofilms devoid of planktonic bacteria (53–56). The observed response comprises neutrophil accumulation, respiratory burst, penetration, phagocytosis, production of cytokines and eradication of the biofilm bacteria. In addition, P. aeruginosa cultures with increased bacterial aggregation induced stronger respiratory burst by neutrophils and cytokine release by macrophages (57).
Likewise, early sampling of mouse lungs challenged with P. aeruginosa biofilms has shown that the innate immune response involves intense accumulation of activated neutrophils in the airways (54, 56, 58–60). Early accumulation of neutrophils at the site of P. aeruginosa biofilm infection is also evident from experimentally infected chronic wounds in mice (14).
Innate Immune Response in CF Patients With Chronic P. aeruginosa Lung Infection
The innate immune response has gained particular attention in patients with CF and chronic P. aeruginosa lung infection, due to the association between accumulation of neutrophils in endobronchial secretions and reduced functionality of the lungs (61). The recruited endobronchial neutrophils display inflammatory activity as indicated by continuing respiratory burst (37, 62) and generation of nitric oxide (44). Accordingly, destruction of the lung tissue has been correlated with oxidative and proteolytic lesions of endobronchial neutrophil activity (63, 64). Chronic lung infections in CF patients are associated with defective apical ion transport due to mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) (65). Infected CF lungs are dominated by P. aeruginosa growing as endobronchial biofilms surrounded by numerous neutrophils (33) and scarce planktonic bacteria, which are subject to phagocytosis by neutrophils (33, 37). The neutrophil response in infected endobronchial secretions in CF resembles the response in experimental in vitro and in vivo biofilms, where high numbers of neutrophils accumulate close to the biofilm (33) and depletion of molecular oxygen (O2) is accelerated (37). This is caused by the reduction of O2 to superoxide during the neutrophils’ active respiratory burst (66). Thus, the response of neutrophils to biofilms during chronic lung infection in CF may contribute considerably to the O2-depletion in infected CF lungs (40). Furthermore, as active neutrophils primarily rely on ATP generated by anaerobic glycolysis (67), the high intake of glucose by neutrophils in CF lungs (68) as well as the enhanced level of L-lactate in sputum from CF patients with chronic P. aeruginosa lung infection (69), is in agreement with a high activity of neutrophils during biofilm infection in CF lungs. The neutrophil response to planktonic P. aeruginosa likewise includes stimulation of the respiratory burst (37), suggesting that neutrophil activation may also include a response to planktonic P. aeruginosa in infected CF lungs. Moreover, activation of neutrophils in infected CF airways may be triggered by alginate (70), LPS or immune complexes (71). The intensity of the neutrophil response may be enhanced by priming with LPS (72) and soluble factors of the innate immune response, such as platelet-activating factor, TNF-α, IL-8 and leukotriene B4 (73–77). Additionally, the migration through inflamed tissue may lead to stimulation of neutrophils due to multiple engagements of integrins and inflammatory cytokines (78). The presence of infectious agents is actually not needed to stimulate the respiratory burst, as seen in response to injury of the intestine in mice (79). The apparent lack of significantly disturbed capacity of neutrophils in CF patients (76) suggests that the reaction of neutrophils to P. aeruginosa biofilms seen in CF patients may also apply to infectious P. aeruginosa biofilms in non-CF patients. Accordingly, biopsies from chronic wounds have revealed biofilm surrounded by high numbers of neutrophils (60, 80–82). Similarly, neutrophils accumulate in high numbers at infectious biofilm in prosthetic knees (83, 84), and the accumulation of neutrophils was intensified and prolonged by P. aeruginosa biofilms in experimental chronic wounds and peritoneal infection (14). Thus, the induction of the biofilm life style observed during interaction between P. aeruginosa and neutrophils in vitro (85–87) may be highly relevant for the formation of biofilm in vivo.
The capability of the innate immune system to recognize invading microorganisms is aided by PRRs that recognize and bind to conserved microbial PAMPs leading to stimulation of the host response. Numerous varieties of PRRs, and their matching ligands are known, but PRRs reacting with PAMPs specifically expressed in microbial biofilm have not been described. PRRs may exist as intra- and extra-cellular membrane-bound receptors, cytoplasmic receptors, or soluble receptors. Since their discovery Toll-like receptors (TLRs) have advanced to become a very well-known family of PRRs. One group of TLRs is expressed on the surface of host cells where they mainly recognize microbial membrane components including lipoproteins, proteins and lipids, while other TLRs are intracellular and recognize microbial nucleic acids (88).
In the airways of chronically infected CF patients, TLR5 was the only MyD88-dependent TLR that was increased on neutrophils (89). This increased expression is possibly facilitated by G-CSF, IL-8 and TNF-α, and by the interaction of bacterial lipoprotein with TLR2 and TLR1 (88). TLR5 is a flagellin receptor (90) and its augmented expression on neutrophils in CF lungs is challenging to explain since flagella are lacking in mucoid biofilms P. aeruginosa isolated from CF lungs (91). The absence of flagella in nonmucoid biofilms, however, intensifies the bactericidal activity of neutrophils in vitro due to release of bactericidal amounts of lactoferrin (92), which may prevent biofilm formation (93, 94). Even though the significance of TLR5 expression for the outcome of biofilm infections is unclear, it may reinforce phagocytosis of planktonic, flagellin-intact P. aeruginosa subpopulations in the CF lungs (94). In support of this, neutrophils only ingested planktonic bacteria in infected airways of CF patients (33, 37), and P. aeruginosa with dysfunctional flagella survived for longer time during lung infection in mice (95). The capability of planktonic P. aeruginosa to provoke a stronger TLR-mediated response than biofilm P. aeruginosa has also been observed for the expression of IL-8 by epithelial cell lines (96). Bacterial eDNA, which is a matrix constituent of biofilms (25, 97), may stimulate neutrophils without involving TLR9 resulting in increased IL-8 production and intracellular signaling (98, 99). Alginate is an abundant component of the matrix in biofilm formed by mucoid P. aeruginosa, and is regarded as the strongest virulence factor in chronic lung infection in CF patients (100). Alginate may increase the respiratory burst of neutrophils (101), and monocytes may respond to alginate by initiating the production of cytokines (102). The activation of monocytes by alginate generated by P. aeruginosa may be mediated by TLR2 and TLR4 (103), while the PRRs involved in the activation of neutrophils remain elusive. The matrix of P. aeruginosa biofilms may contain other polysaccharide components, such as Psl and Pel, which may stimulate an innate response to biofilm (104). Recent evidence suggests that the specific exopolysaccharide composition of P. aeruginosa biofilms is a determinant of the neutrophil response (10). A biofilm with a matrix composed primarily of Psl and alginate polysaccharides was found to be particularly efficient in activating neutrophils (10). It remains, however, to be determined if the innate response against exopolysaccharide expression in biofilm is distinctly stronger than the innate response against exopolysaccharide expression in planktonic cells. In that case, we suggest considering exopolysaccharide as a subgroup of PAMPs termed “biofilm associated molecular patterns” (BAMPs) (Figure 2).
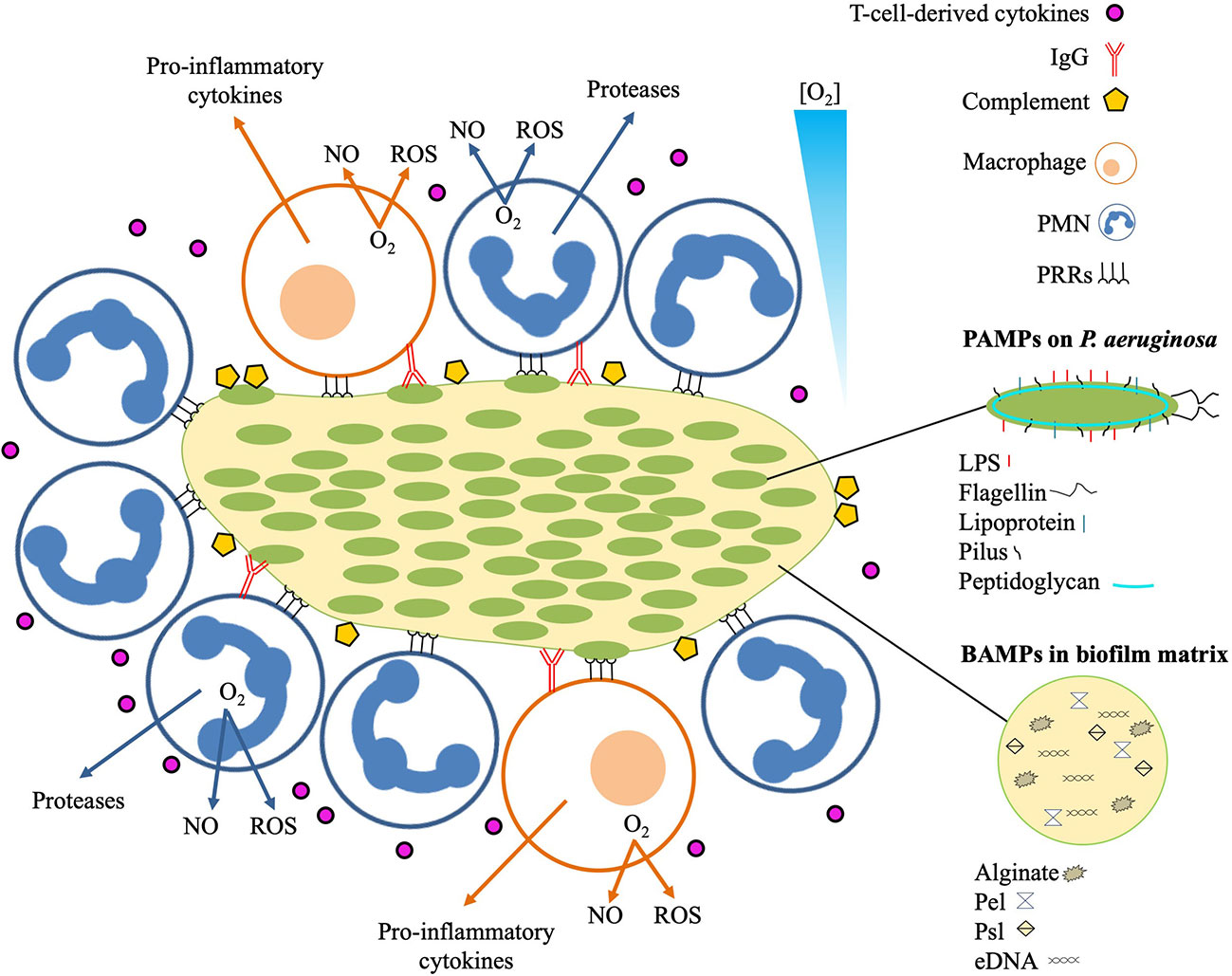
Figure 2 Local immune response to infectious P. aeruginosa biofilm. The innate immune response recognizes pathogen associated molecular patterns (PAMPs) expressed on P. aeruginosa, and biofilm-associated molecular patterns (BAMPs) present in the biofilm matrix. Detection of BAMPs and PAMPs by PMNs and macrophages is mediated by pattern recognition receptors (PRRs). Binding of BAMPs and PAMPs to PRRs stimulates the PMNs and macrophages resulting in consumption of O2 for liberation of tissue-toxic reactive oxygen species (ROS) and nitric oxide (NO). Additional responses by the PMNs include secretion of proteases that may cause proteolytic tissue lesions while the macrophage may further enhance the inflammation by emitting pro-inflammatory cytokines such as TNF-α, Il-1, IL-6, IL-8, and IL-12. The effector cells of the adaptive immune response mainly reside distantly such as the T-cells and the B-cells in the secondary lymphoid organs and the plasma cells in the bone marrow. Activated T-cells may release cytokines that further reinforces the inflammation by stimulating the accumulation and activation of PMNs and production of IgG. The contribution of the increased accumulation of activated PMNs to the local inflammation is further accelerated by binding of antigens to IgG, leading to immune complex mediated stimulation of the PMNs and activation of the classical complement pathway.
Although the soluble and the membrane-bound receptors of the complement system are among the most studied PRRs, a pivotal role of the complement system for the outcome of biofilm infections remains to be firmly established. Infectious biofilm may establish in spite of complement activation even in patients with intact complement systems. In this respect, P. aeruginosa may secrete elastase and alkaline protease that inactivate the complement system (105). Further protection may be provided by alginate with O acetylation which prevents complement opsonization of mucoid P. aeruginosa biofilms (106). The involvement of the complement system in CF lung infections has been demonstrated by the frequent isolation of activated complement (C3c) in the sputum from chronically infected CF patients (107). Furthermore, the matrix polysaccharide, Psl, protects mucoid bacteria from opsonization and killing by complement components in human serum (108). However, whether complement activation requires biofilm formation is unlikely since planktonic bacteria induce stronger activation of the complement system (109). However, P. aeruginosa isolated from CF sputum may escape activated complement system (110).
The intense buildup of neutrophils associated to P. aeruginosa biofilm infections in CF, chronic wounds and implanted devices, would be anticipated to eliminate the biofilm. However, specific defects may weaken the immune defense. Thus, as a consequence of the basic defect in CFTR, both neutrophils and macrophages in the CF lungs exhibit blunted phagocytic capacity that could contribute to poor bacterial clearance and altered efferocytosis (111, 112). Moreover, the failing bactericidal activity of the summoned neutrophils may rely on rhamnolipids produced by P. aeruginosa (56). Synthesis of rhamnolipid depends on quorum sensing (QS) (60) indicating the ability of P. aeruginosa biofilm to contain bacterial densities necessary to achieve the quora required to activate QS-dependent rhamnolipid production (56, 59, 60) in chronic wounds (81) and lungs of infected CF patients (33). Rhamnolipids protects the biofilm against approaching functional neutrophils by inducing cellular necrosis (60). Intriguingly, the molecule OdDHL may attract neutrophils (113) and may thus attract and lure the neutrophils to the site of infection where they are killed by rhamnolipids. The QS-regulated attenuation of the host response may facilitate the initial establishment of biofilm infection (6). However, succeeding lung infection in CF patients involves extensive genetic adaptions with frequent mutations, e.g. in the QS regulator gene lasR (114). Dysfunctional QS of the lasR mutants may result in defective proteolytic neutralization of chemotactic cytokines allowing the pro-inflammatory cytokines to attract increased numbers of neutrophils to the lungs leading to intensified pulmonary inflammation (115). The size of bacterial aggregates may also contribute to the protection of bacteria against the immune response offered from biofilm formation. In fact, when the size of aggregated P. aeruginosa with deficient QS exceeded diameters of 5 µm, phagocytosis by human neutrophils was inhibited (116).
P. aeruginosa in biofilms can produce additional virulence factors, such as pyocyanin, that may cause cellular damage and immune modulations in cystic fibrosis lungs (117). Pyocyanin has been associated to broader functions, such as impairment of ciliary beat frequency and mucin hypersecretion, which in turn create a positive loop for biofilm formation and dysregulated immune responses in the CF lung (118).
It may be expected that the infectious biofilm in CF lungs would succumb due to the potent antibiofilm activity of antimicrobial peptides produced by neutrophils and lung epithelial cells (119). However, the low pH in CF lungs may impair the antimicrobial activity of antimicrobial peptides (120, 121). In addition, the defective distribution of salts in CF lung may have crucial effect on the optimal functionality of some antimicrobial peptides (122). Other environmental conditions in CF lungs may contribute significantly to the reduced activity of antimicrobial peptides. These conditions include proteolytic degradation of antimicrobial peptides by bacterial proteases (123) and by host proteases (124) and inhibition of antimicrobial peptides by binding to complexes of LPS, F-actin, mucins, and host derived DNA (125).
Innate Immune Response to P. aeruginosa Infection in Chronic Wounds
Whereas the majority of our knowledge on immune responses to P. aeruginosa biofilms comes from studies of CF lung infections, studies of chronic wound infections has recently shed additional light on the topic. The prevalence of recalcitrant wounds is expanding epidemically alongside with obesity and lifestyle diseases. The host response to bacterial intruders in chronic wounds is hallmarked by a persistent inflammatory phase. This phase comprises continuous oxidative damage, senescence of fibroblasts and skewing of constructive growth factors required for tissue resolution. The pathoetiology also includes low mitogenic-activity, high protease combined with low inhibitor-activity, microbiota changes, the etiology behind the original insult and the specific invading pathogen. Accumulating evidence emphasizes the paramount impact of infectious bacterial biofilm on the host response in the wound and the implication for recovery.
Unfortunately, it is challenging to achieve appropriate numbers of participants for conducting randomized studies on intervention in patients with recalcitrant wounds due to different chronicity definitions and patient heterogeneity. In addition, it is not feasible to extrapolate the results from chronic wounds of one etiology to another since many patients may suffer from several diseases (126).
The impact of infection with P. aeruginosa on wound chronicity is well described in clinical settings and experimental models (80, 81, 127). The presence of biofilm is now commonly recognized as a leading cause of chronic infections with persisting pathology despite antibacterial therapy and continuous induction of the host response (128). Certain components of P. aeruginosa biofilms, such as rhamnolipids, are likely playing important roles for persistence of infection as it causes cellular necrosis and killing of neutrophils (56, 59, 60). Other studies support the capability of P. aeruginosa to attenuate bactericidal components of the host defense (53, 129)
The endogenous antimicrobial peptides (AMPs) are phylogenetically ancient and constitute a crucial part of the skin’s innate defense to infection (130). AMPs may be made by keratinocytes and infiltrating granulocytes and macrophages in response to infection, wound healing, trauma, or chronic inflammation. In addition, AMPs possess regenerative properties (131). AMPs are amphipathic molecules (132), which enables interaction with phospholipids of microbial membranes leading to pore formation and bactericidal cell lysis (133). The endogenous antimicrobial host defense protein S100A8/A9 belongs to the alarmin group and displays various activities. S100A8/A9 is expressed in actively healing wounds in human and murines (134, 135), but S100A8/A9 is absent in chronic, colonized venous leg ulcers in humans (136, 137) possibly resulting from the distorted local host response. This is suspected to cause deterioration of wound healing.
Relevant animal models are valuable tools for obtaining knowledge on the interplay between host and pathogen. Accordingly, animal models have enabled detailed descriptions of disposing factors, infectious agents and host response to infection. There are obvious limitations when comparing murine to humane wound healing and regeneration. Mice heal with predominantly contraction in a looser attached skin with higher hair density and thinner dermis versus the humane granulation healing. There are also significant differences in the immune response, with more neutrophils in the humane circulating blood versus a higher number of lymphocytes in mouse blood in addition to substantial differences with regards to the antimicrobial peptides. Despite this, mice represent a generally accepted experimental animal of choice.
To study the interaction between biofilm and the host response, we have established a chronic wound model which enables examinations of P. aeruginosa biofilm-infected wound closure in two mouse strains. One strain is relatively resistant to P. aeruginosa infection and consists of C3H/HeN mice. The other strain is made up of BALB/c mice which are susceptible to the infection (14, 138). The C3H/HeN mice have Th1-dominated response towards the infectious agents Leishmania major and Candida species. On the contrary, the response of the BALB/c mice against these agents is Th2-dominated. The direction of the Th response has essential effects on mortality rates and clearance of infection (138). A dichotomized early response in the mouse model of chronic wounds has been indicated by the attenuated local IL-1β inflammatory response to P. aeruginosa biofilm during the first 5 days of infection in C3H/HeN mice as compared to the BALB/c mice (14). Furthermore, our group recently demonstrated that P. aeruginosa biofilm may decrease the intensity of local neutrophil response in several murine wounds which may compromise the control of infection. The connection between the slow healing and the genotype in BALB/c mice has been confirmed by another group (198), which makes this strain of mice an excellent choice of animal model for wound healing. In this context, comparing the spontaneous healing of P. aeruginosa biofilm infected wounds in C3H/HeN and BALB/c mice with the S100A8/A9 expression, could be highly valuable for further evaluation of the significance of S100A8/A9.
Adaptive Immune Responses During P. aeruginosa Biofilm Infections
The adaptive immune system discriminates the host proteins and other potential antigens from foreign molecules, to ensure that the lymphocytic and humoral antibody mediated effector functions do not result in excessive damage to the infected organism. However, the adaptive immune reaction is extensively superior in the specific response, as compared to the innate responses. Furthermore, recognition of the identical or similar pathogen upon reinfection by the adaptive immune system advances rapid clonal expansion of up to a 1000-fold antigen specific effector and central memory cells at subsequent exposures. The developed memory is the premise for immunity to subsequent infections. Compared to innate responses, which cannot discriminate between primary and secondary responses, the secondary responses of the adaptive immune system is substantially faster, more potent and with enhanced affinity as compared to primary exposure (139, 140). Activation of the adaptive immune system often results in clearance of the infection by planktonic bacteria, due to the combined activity of the innate and adaptive immune systems augmenting both the immune reactions. However, in the case of chronic biofilm infections the pathogens are not eliminated. Instead, the synergy of the innate and adaptive immune mechanisms, the latter with inertia at first encounter, is a central component of biofilm pathogenesis (5, 141–143).
Activation of the adaptive host responses is facilitated through dendritic cells (DC) required for sufficient activation at the first pathogen encounter and macrophages (Mφ) (144). Immature DCs in the peripheral tissue are effective in antigen uptake and are especially abundant at pathogen exposed regions, as the mucosal surfaces and in the secondary lymphoid tissue (145, 146). DCs mature following antigen uptake, and from inflammatory cytokine impact, into mature DCs dedicated in antigen processing and presentation (145, 146). Therefore, the DCs are essential in linking the innate and adaptive immune systems, and have the exclusive capacity to prime naïve T-cells into subsequent Th1, Th2, or Th17 cells and responses (145–147). Due to the limited presence of DCs in tissues, isolation is highly challenging, especially in human studies. Our own studies using a chronic P. aeruginosa lung infection model revealed commitment of pulmonary DCs during the infection (148). Pulmonary DCs was demonstrated as early as 2 days of initiation onset (148). Interestingly, an increased number of DCs in the regional lymph node was not detected until day 7 (148). The fraction of activated pulmonary DCs increased during the 10-day observation period, when demonstrated by CD80 and CD86 expression (148). In contrast, the percentage of activated DCs in the lymph node decreased at day 10 (148). The cytokine release of the DCs from the lung and lymph node were in general paralleled. Interestingly however, the initial release of the pro-inflammatory cytokines IL-6 and IL-12 reached a maximum at days 2–3, followed by an increased IL-10 production at day 7 (148). This observation, likely represents an essential controlling role of the DCs in induction of the adaptive immune system effector functions, impacted by the adjacent innate responses (148). This is supported by observations from another study, where P. aeruginosa QS signal molecules diminished the murine DC IL-12 production, while the IL-10 release remained. In addition, antigen specific T-cell proliferation was down regulated by QS exposed DCs. These results indicates that DCs are inhibited in T-cell stimulation by the P. aeruginosa QS signals, and by this mechanism contribute to the P. aeruginosa biofilm pathology (6, 149).
From previous observations of GM-CSF and G-CSF on DCs, we hypothesized that the increased G-CSF would impact the DC response in chronically pulmonary P. aeruginosa infected CF patients, besides recruiting PMNs from the bone marrow (150). Indeed, the GM-CSF/G-CSF ratio and the IFN-γ response correlated, and interestingly also correlated to a better lung function. In contrast, IL-3 and IFN-γ responses correlated inversely (150–156). DCs seem to impact host responses in biofilm infections and represent a potential therapeutic target.
As mentioned above, the innate and adaptive immune effector elements function in collaboration. As a consequence of the persistent biofilm infection, the adjacent tissue is impacted by the injurious oxidative radicals and enzymes originating from the inflammatory cells. Besides the pathogen related virulence factors, elastases, proteases, and other exoenzymes resulting from the inflammation expedites degradation of crucial surface molecules of the immune cell, further adding to impaired anti-biofilm mechanisms of the host responses (107, 157–160). The ineffective host response is considered the key basis of the biofilm related pathology, since antibodies against several bacterial virulence factors, such as elastase, lipopolysaccharide, and flagella have been reported, which presumable should improve biofilm outcome (161–163). However, these virulence factors are considered to be involved in pathogenesis, predominantly during the initial phases and to support development from microbial colonization to infection per se. Although, the bacterial virulence factors are less involved in the direct chronic biofilm pathology, the antibody mediated precipitation of virulence factors and other microbe antigens results in formation of immune complexes deposited in the tissues. Since, this leads to activation of the complement system and PMN opsonization, tissue damage is the consequence (100).
A special situation of the adaptive immune response and chronic P. aeruginosa infection of airways is the induction of a mucosal antibody response represented by specific secretory IgA (sIgA). The IgG responses can be regarded as an element of the systemic immune response, and primarily get access to mucosal surfaces through inflamed epithelium. In contrast, sIgA is the primary antibody of mucosal surfaces, and it is produced in double the amount of IgG, and is secreted to the mucosal surfaces as dimeric sIgA bound to the secretory component (164). At the surfaces, sIgA functions through immune exclusion by binding to the pathogen and its PAMPs without activation of complement and opsonization. In CF sIgA has been found in sinuses and correlating to chronic sinusitis, whereas IgG dominates in the lower airways, where it correlates to inflammation of the respiratory airways (165). sIgA was also found to correlate to an early detection of P. aeruginosa of the lower airways of CF patients (165).
T-Cell Response and Clinical Outcome in CF Patients With Chronic P. aeruginosa Lung Infection
The biofilm infection and host response interplay has been best characterized for CF patients with pulmonary chronic P. aeruginosa biofilm infections (6). Early intensive antibiotic therapy, maintenance antibiotic treatment strategy between exacerbations, and planned elective intravenous antibiotic courses has become standard of care in CF (11). However, the natural course of the pulmonary chronic P. aeruginosa infection revealed a dichotomized outcome. A poor outcome, and a pronounced or rapid escalation in antibody response, was reported for most CF patients (166). However, for a small group of CF patients the humoral response was modest and these patients had a beneficial outcome (166). In addition, the intensified antibiotic treatment strategy in CF, resulting in significantly superior outcomes correlates to less pronounced antibody responses in CF (167).
By investigating specific cytokine release from re-stimulated peripheral blood mononuclear cells (PBMCs), and later on cytokine measurements from unspecific stimulated T cells, a Th1/Th2 cytokine dichotomy in chronically infected CF patients was revealed (168, 169). Chronically infected CF patients had a Th2 dominated cytokine response with increased IL-4 (and IL-5, IL-10) production and diminished IFN-γ production. In addition, a similar Th1/Th2 cytokine dichotomy was later demonstrated in bronchoalveolar lavage fluid from subgroups of CF patients (170, 171). Interestingly, IFN-γ release from PBMCs correlated to an improved lung function, suggesting a potential beneficial effect of IFN-γ (168). Inbred mouse strains with chronic P. aeruginosa lung infection showed a pronounced pulmonary IFN-γ level in the relatively resistant C3H/HeN mouse (138, 172). Reinfection of the susceptible BALB/c mice resulted in a pulmonary Th1 response similar to the C3H/HeN mice and resembled the course of a primary infection in the C3H/HeN mice (173).
The explanation for the improved outcome of a Th1 dominated response in CF patients with chronic P. aeruginosa lung infection is incomplete, especially since the Th1 dominated response would be more appropriate towards intracellular pathogens. However, phagocytosis of apoptotic PMNs by alveolar macrophages before the PMNs progress into necrosis and thereby increase inflammation, is believed to be involved (174). Reduction of IL-8, the most important PMN chemoattractant is another likely mechanism (175, 176). A diminished Th2 response would presumably result in a reduced antibody response, due to reduced B and plasma-cell stimulation, and subsequently decreased immune complex formation and tissue damage.
Additional T cell subsets have been described, including the Th17 subset, characterized by production of IL-17 and sometimes IL-22 (177). Th17 cells are induced by TGF-β (178) and may be of interest in CF, since IL-17 induces the PMN mobiliser G-CSF and chemoattractant IL-8 (179, 180). In this way, Th17 may add to pulmonary pathology of chronic P. aeruginosa lung infections (179, 180). In sputum from stable CF patients and in chronically infected CF patients, IL-17 and IL-23, was increased as compared to CF patients without chronic P. aeruginosa lung infections (179). Interestingly, such difference was not observed in CF patients infected with Staphylococcus aureus (179). A substantially decreased fraction of peripheral Th17 cells in CF patients has been reported, and interpreted as augmented homing of the cells to the lungs, increasing the pulmonary inflammation (181). Determinations of cytokines related to Th subsets were conducted in children with CF, and demonstrated increase of both IL-17A and the Th2 related cytokines IL-5 and IL-13 in children with symptoms (180). In contrast, such relationship was not observed for Th1 related cytokines, indicating a correlation between Th2 and Th17 subsets in CF (180). Such a Th2-Th17 axis could dispose for P. aeruginosa lung infections, but this has not been clarified yet (171, 180, 182). Interestingly, T cell suppressive neutrophil myeloid-derived suppressor cells (MDSCs) has recently been reported in CF (183, 184). The presence of neutrophil MDSCs in peripheral blood correlated to improved lung function in CF in contrast to what would be expected (183). Down regulation of the harmful and dominating Th2 and Th17 response axis, could be the mechanism behind this observation.
The role of regulatory T cells (Treg), Th22, and additional T cell subsets has only been sparsely studied in biofilm infections. However, decreased levels and reduced functions of these immune cells in CF patients have been suggested and may result in augmented IL-17 and IL-8 production (182, 185).
Novel Potential Treatment Options Towards P. aeruginosa Biofilm Infections
The administration of preformed antibodies or immunoglobulins to treat various infectious diseases is known as passive immunization therapy. Passive immunotherapy using avian IgY immunoglobulins (yolk) targeting P. aeruginosa represents an alternative to conventional antibiotic therapeutics. IgY is the predominant serum antibody in chickens and is the avian homologue of mammalian IgG (186). It accumulates in the egg yolk from the blood and provides the offspring with humoral immunity. Hyperimmunization of chickens with specific antigens, provides high yields of specific IgY antibodies in the egg yolk (187). In vitro studies with IgY targeting P. aeruginosa showed firm binding to flagella and interference with the adhesion of bacteria to epithelial cells (188). Potentially, such effect could prevent bacteria from colonizing the respiratory tract. Additionally, our group has also observed promising effects of anti-P. aeruginosa IgY. In in vitro studies, respiratory PMN burst and bacterial killing of P. aeruginosa were shown to be significantly increased in the presence of anti-P. aeruginosa IgY (189). Anti-P. aeruginosa IgY seems to affect aggregation of bacteria resulting in immobilization and increased surface hydrophobicity, enhancing non-Fc receptor mediated phagocytosis (190). The observed in vitro effects of anti-P. aeruginosa IgY, were in accordance with in vivo observations in an acute murine pneumonia model, where we demonstrated a 2-log reduction in pulmonary bacteria, which was paralleled by decreased inflammation in the airways of anti-P. aeruginosa IgY treated mice as compared to mice receiving non-specific IgY (191).
Potentially, anti-bacterial immunotherapies by means of pathogen specific IgY augments PMN mediated phagocytic effects and reduce the level of airway colonization in CF and may even potentiate the action of anti-pseudomonal antibiotics (192). Moreover, a clinical study examining the effects of oral prophylactic immunotherapy with anti-P. aeruginosa IgY in non-chronically infected CF patients has shown promising results (193).
Recombinant S100A8/A9 also show promising therapeutic properties. Our group found that immune modulation of P. aeruginosa-biofilm infected wounds on BALB/c mice by 4-days local application of recombinant S100A8/A9, combined with systemically administered ciprofloxacin, significantly reduced the bacterial load of the wounds (194). Since in vitro synergistic effect between S100A8/A9 and ciprofloxacin was not observed, the effect is highly dependent on host cells (194). Human studies and animal experiments indicate impairment of the S100A8/A9 response and that the level of S100A8/A9 is inappropriate in non-healing wounds. We are currently investigating this area to improve the understanding of the pathophysiological multifaceted role of S100A8/A9 in biofilm-infected wounds.
In adjunctive therapies of non-healing wounds with an inappropriate anti-biofilm host response, autologous fibrin rich patches containing thrombocytes and leucocytes are a promising treatment strategy (195). A three layered 3C patch, is produced by centrifugation of the patient’s whole blood in a specially developed device (195). The 3C patch is subsequently applied to the chronic wound (196). In an open study on chronic wounds of various backgrounds, an accelerated healing with 3C patches was revealed in the majority of the patients (197). The effect is most likely caused by production of healing growth factors and cytokines, e.g. PDGF-bb, from thrombocytes (195). In support of these observations, a substantial PMN activity was observed inside 3C patches in terms of respiratory burst, PMN phagocytosis activity and anti-biofilm action (196).
Conclusions and Perspectives
Knowledge of the immune responses and bacterial defense mechanisms under conditions of biofilm infections is important as it constitutes an important part of the pathology of biofilm infections. As documented in the present review, our knowledge of immune responses to biofilm infections has increased considerably in recent years and is likely to provide important treatment tools against biofilm infections in the future. We may eventually be able to damping harmful immune system activities, or to activate parts of the immune system that can eradicate biofilm infections without causing detrimental collateral damage. In addition, antibiotic augmenting effects of the immune system could be identified. Alternatively, we may be able to manipulate the bacteria and down-regulate or eliminate the components of biofilms that are responsible for the recalcitrance towards immune system activities.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by a grant (DFF-7016-00039) to TT-N from the Danish Council for Independent Research. CM is supported by Novo Nordisk Fonden (Borregaard Clinical Scientist Fellowship in translational research; grant no. NNF17OC0025074).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Hall CW, Mah T-F.Molecular mechanisms of biofilm-based antibiotic resistance and tolerance in pathogenic bacteria. FEMS Microbiol Rev (2017) 41:276–301. doi: 10.1093/femsre/fux010 doi: 10.1093/femsre/fux010
2. Ciofu O, Tolker-Nielsen T.Tolerance and Resistance of Pseudomonas aeruginosa Biofilms to Antimicrobial Agents—How P. aeruginosa Can Escape Antibiotics. Front Microbiol (2019) 10:913. doi: 10.3389/fmicb.2019.00913 doi: 10.3389/fmicb.2019.00913
3. Costerton JW, Stewart PS, Greenberg EP.Bacterial biofilms: a common cause of persistent infections. Sci (New York NY) (1999) 284:1318–22. doi: 10.1126/science.284.5418.1318 doi: 10.1126/science.284.5418.1318
4. Bryers JD.Medical biofilms. Biotechnol bioengineering (2008) 100:1–18. doi: 10.1002/bit.21838 doi: 10.1002/bit.21838
5. Moser C, Pedersen HT, Lerche CJ, Kolpen M, Line L, Thomsen K, et al.Biofilms and host response - helpful or harmful. APMIS: Acta pathologica microbiologica immunologica Scandinavica (2017) 125:320–38. doi: 10.1111/apm.12674 doi: 10.1111/apm.12674
6. Jensen PØ, Givskov M, Bjarnsholt T, Moser C.The immune system vs. Pseudomonas aeruginosa biofilms. FEMS Immunol Med Microbiol (2010) 59:292–305. doi: 10.1111/j.1574-695X.2010.00706.x doi: 10.1111/j.1574-695X.2010.00706.x
7. Raoust E, Balloy V, Garcia-Verdugo I, Touqui L, Ramphal R, Chignard M.Pseudomonas aeruginosa LPS or flagellin are sufficient to activate TLR-dependent signaling in murine alveolar macrophages and airway epithelial cells. PloS One (2009) 4:e7259. doi: 10.1371/journal.pone.0007259 doi: 10.1371/journal.pone.0007259
8. Tart AH, Wolfgang MC, Wozniak DJ.The alternative sigma factor AlgT represses Pseudomonas aeruginosa flagellum biosynthesis by inhibiting expression of fleQ. J Bacteriol (2005) 187:7955–62. doi: 10.1128/JB.187.23.7955-7962.2005 doi: 10.1128/JB.187.23.7955-7962.2005
9. Starkey M, Hickman JH, Ma L, Zhang N, De Long S, Hinz A, et al.Pseudomonas aeruginosa rugose small-colony variants have adaptations that likely promote persistence in the cystic fibrosis lung. J Bacteriol (2009) 191:3492–503. doi: 10.1128/JB.00119-09 doi: 10.1128/JB.00119-09
10. Rybtke M, Jensen PØ, Nielsen CH, Tolker-Nielsen T.The extracellular polysaccharide matrix of Pseudomonas aeruginosa biofilms is a determinant of polymorphonuclear leukocyte responses. Infect Immun (2020) 89:e00631–20. doi: 10.1128/IAI.00631-20 doi: 10.1128/IAI.00631-20
11. Høiby N, Bjarnsholt T, Moser C, Bassi GL, Coenye T, Donelli G, et al.ESCMID ∗ guideline for the diagnosis and treatment of biofilm infections 2014. Clin Microbiol Infection (2015) 21:S1–S25. doi: 10.1016/j.cmi.2014.10.024 doi: 10.1016/j.cmi.2014.10.024
12. Tolker-Nielsen T.Biofilm Development. Microbiol Spectr (2015) 3:MB–0001-2014. doi: 10.1128/microbiolspec.MB-0001-2014 doi: 10.1128/microbiolspec.MB-0001-2014
13. Tolker-Nielsen T.Pseudomonas aeruginosa biofilm infections: from molecular biofilm biology to new treatment possibilities. APMIS Supplementum (2014) 138:1–51. doi: 10.1111/apm.12335 doi: 10.1111/apm.12335
14. Trøstrup H, Thomsen K, Christophersen LJ, Hougen HP, Bjarnsholt T, Jensen PØ, et al.Pseudomonas aeruginosa biofilm aggravates skin inflammatory response in BALB/c mice in a novel chronic wound model. Wound Repair Regeneration: Off Publ Wound Healing Soc [and] Eur Tissue Repair Soc (2013) 21:292–9. doi: 10.1111/wrr.12016 doi: 10.1111/wrr.12016
15. Connor LM, Tang SC, Cognard E, Ochiai S, Hilligan KL, Old SI, et al. Th2 responses are primed by skin dendritic cells with distinct transcriptional profiles. J Exp Med (2017) 214:125–42. doi: 10.1084/jem.20160470
16. Sweere JM, Ishak H, Sunkari V, Bach MS, Manasherob R, Yadava K, et al. The Immune Response to Chronic Pseudomonas aeruginosa Wound Infection in Immunocompetent Mice. Adv Wound Care (New Rochelle) (2020) 9:35–47. doi: 10.1089/wound.2019.1039
17. Deziel E, Comeau Y, Villemur R. Initiation of biofilm formation by Pseudomonas aeruginosa 57RP correlates with emergence of hyperpiliated and highly adherent phenotypic variants deficient in swimming, swarming, and twitching motilities. J Bacteriol (2001) 183:1195–204. doi: 10.1128/JB.183.4.1195-1204.2001
18. Chiang P, Burrows LL. Biofilm formation by hyperpiliated mutants of Pseudomonas aeruginosa. J Bacteriol (2003) 185:2374–8. doi: 10.1128/JB.185.7.2374-2378.2003
19. O’Toole GA, Kolter R. Flagellar and twitching motility are necessary for Pseudomonas aeruginosa biofilm development. Mol Microbiol (1998) 30:295–304. doi: 10.1046/j.1365-2958.1998.01062.x
20. Vallet I, Olson JW, Lory S, Lazdunski A, Filloux A. The chaperone/usher pathways of Pseudomonas aeruginosa: identification of fimbrial gene clusters (cup) and their involvement in biofilm formation. Proc Natl Acad Sci U.S.A. (2001) 98:6911–6. doi: 10.1073/pnas.111551898
21. Friedman L, Kolter R. Two genetic loci produce distinct carbohydrate-rich structural components of the Pseudomonas aeruginosa biofilm matrix. J Bacteriol (2004) 186:4457–65. doi: 10.1128/JB.186.14.4457-4465.2004
22. Jackson KD, Starkey M, Kremer S, Parsek MR, Wozniak DJ. Identification of psl, a locus encoding a potential exopolysaccharide that is essential for Pseudomonas aeruginosa PAO1 biofilm formation. J Bacteriol (2004) 186:4466–75. doi: 10.1128/JB.186.14.4466-4475.2004
23. Høiby N, Döring G, Schiøtz PO. Pathogenic mechanisms of chronic Pseudomonas aeruginosa infections in cystic fibrosis patients. Antibiotics chemotherapy (1987) 39:60–76. doi: 10.1159/000414334
24. Borlee BR, Goldman AD, Murakami K, Samudrala R, Wozniak DJ, Parsek MR. Pseudomonas aeruginosa uses a cyclic-di-GMP-regulated adhesin to reinforce the biofilm extracellular matrix. Mol Microbiol (2010) 75:827–42. doi: 10.1111/j.1365-2958.2009.06991.x
25. Whitchurch CB, Tolker-Nielsen T, Ragas PC, Mattick JS. Extracellular DNA required for bacterial biofilm formation. Science (2002) 295:1487. doi: 10.1126/science.295.5559.1487
26. Tielker D, Hacker S, Loris R, Strathmann M, Wingender J, Wilhelm S, et al. Pseudomonas aeruginosa lectin LecB is located in the outer membrane and is involved in biofilm formation. Microbiology (2005) 151:1313–23. doi: 10.1099/mic.0.27701-0
27. Diggle SP, Stacey RE, Dodd C, Camara M, Williams P, Winzer K. The galactophilic lectin, LecA, contributes to biofilm development in Pseudomonas aeruginosa. Environ Microbiol (2006) 8:1095–104. doi: 10.1111/j.1462-2920.2006.001001.x
28. Dueholm MS, Søndergaard MT, Nilsson M, Christiansen G, Stensballe A, Overgaard MT, et al. Expression of Fap amyloids in Pseudomonas aeruginosa, P. fluorescens, and P. putida results in aggregation and increased biofilm formation. Microbiologyopen (2013) 2:365–82. doi: 10.1002/mbo3.81
30. Malone JG, Jaeger T, Spangler C, Ritz D, Spang A, Arrieumerlou C, et al. YfiBNR mediates cyclic di-GMP dependent small colony variant formation and persistence in Pseudomonas aeruginosa. PloS Pathog (2010) 6:e1000804. doi: 10.1371/journal.ppat.1000804
31. Evans TJ. Small colony variants of Pseudomonas aeruginosa in chronic bacterial infection of the lung in cystic fibrosis. Future Microbiol (2015) 10:231–9. doi: 10.2217/fmb.14.107
32. Gloag ES, Marshall CW, Snyder D, Lewin GR, Harris JS, Santos-Lopez A, et al. Pseudomonas aeruginosa Interstrain Dynamics and Selection of Hyperbiofilm Mutants during a Chronic Infection. mBio (2019) 10(4):e01698-19. doi: 10.1128/mBio.01698-19
33. Bjarnsholt T, Jensen PØ, Fiandaca MJ, Pedersen J, Hansen CR, Andersen CB, et al. Pseudomonas aeruginosa biofilms in the respiratory tract of cystic fibrosis patients. Pediatr pulmonology (2009) 44:547–58. doi: 10.1002/ppul.21011
34. Fazli M, Bjarnsholt T, Kirketerp-Møller K, Jørgensen B, Andersen AS, Krogfelt KA, et al. Nonrandom distribution of Pseudomonas aeruginosa and Staphylococcus aureus in chronic wounds. J Clin Microbiol (2009) 47:4084–9. doi: 10.1128/JCM.01395-09
35. Harmsen M, Yang L, Pamp SJ, Tolker-Nielsen T. An update on Pseudomonas aeruginosa biofilm formation, tolerance, and dispersal. FEMS Immunol Med Microbiol (2010) 59:253–68. doi: 10.1111/j.1574-695X.2010.00690.x
36. Mishra M, Byrd MS, Sergeant S, Azad AK, Parsek MR, McPhail L, et al. Pseudomonas aeruginosa Psl polysaccharide reduces neutrophil phagocytosis and the oxidative response by limiting complement-mediated opsonization. Cell Microbiol (2012) 14:95–106. doi: 10.1111/j.1462-5822.2011.01704.x
37. Kolpen M, Hansen CR, Bjarnsholt T, Moser C, Christensen LD, van Gennip M, et al. Polymorphonuclear leucocytes consume oxygen in sputum from chronic Pseudomonas aeruginosa pneumonia in cystic fibrosis. Thorax (2010) 65:57–62. doi: 10.1136/thx.2009.114512
38. Kolpen M, Bjarnsholt T, Moser C, Hansen CR, Rickelt LF, Kühl M, et al. Nitric oxide production by polymorphonuclear leucocytes in infected cystic fibrosis sputum consumes oxygen. Clin Exp Immunol (2014) 177:310–9. doi: 10.1111/cei.12318
39. Balridge CW, Gerard RW. The extra respiration of phagocytosis. Am J Physiol (1933) 103:235–6. doi: 10.1152/ajplegacy.1932.103.1.235
40. Worlitzsch D, Tarran R, Ulrich M, Schwab U, Cekici A, Meyer KC, et al. Effects of reduced mucus oxygen concentration in airway Pseudomonas infections of cystic fibrosis patients. J Clin Invest (2002) 109:317–25. doi: 10.1172/JCI0213870
41. Alvarez-Ortega C, Harwood CS. Responses of Pseudomonas aeruginosa to low oxygen indicate that growth in the cystic fibrosis lung is by aerobic respiration. Mol Microbiol (2007) 65:153–65. doi: 10.1111/j.1365-2958.2007.05772.x
42. Borrero-de Acuna JM, Rohde M, Wissing J, Jansch L, Schobert M, Molinari G, et al. Protein Network of the Pseudomonas aeruginosa Denitrification Apparatus. J bacteriology (2016) 198:1401–13. doi: 10.1128/JB.00055-16
43. Zumft WG. Cell biology and molecular basis of denitrification. Microbiol Mol Biol Rev MMBR (1997) 61:533–616. doi: 10.1128/.61.4.533-616.1997
44. Kolpen M, Kühl M, Bjarnsholt T, Moser C, Hansen CR, Liengaard L, et al. Nitrous oxide production in sputum from cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. PloS One (2014) 9:e84353. doi: 10.1371/journal.pone.0084353
45. Line L, Alhede M, Kolpen M, Kühl M, Ciofu O, Bjarnsholt T, et al. Physiological levels of nitrate support anoxic growth by denitrification of Pseudomonas aeruginosa at growth rates reported in cystic fibrosis lungs and sputum. Front Microbiol (2014) 5:554. doi: 10.3389/fmicb.2014.00554
46. Huie RE, Padmaja S. The reaction of no with superoxide. Free Radical Res Commun (1993) 18:195–9. doi: 10.3109/10715769309145868
47. Kissner R, Koppenol WH. Product distribution of peroxynitrite decay as a function of pH, temperature, and concentration. J Am Chem Soc (2002) 124:234–9. doi: 10.1021/ja010497s
48. Jones KL, Hegab AH, Hillman BC, Simpson KL, Jinkins PA, Grisham MB, et al. Elevation of nitrotyrosine and nitrate concentrations in cystic fibrosis sputum. Pediatr pulmonology (2000) 30:79–85. doi: 10.1002/1099-0496(200008)30:2<79::aid-ppul1>3.0.co;2-1
49. Grasemann H, Ioannidis I, Tomkiewicz RP, de Groot H, Rubin BK, Ratjen F. Nitric oxide metabolites in cystic fibrosis lung disease. Arch Dis childhood (1998) 78:49–53. doi: 10.1136/adc.78.1.49
50. Palmer KL, Brown SA, Whiteley M. Membrane-bound nitrate reductase is required for anaerobic growth in cystic fibrosis sputum. J bacteriology (2007) 189:4449–55. doi: 10.1128/JB.00162-07
51. Kragh KN, Alhede M, Jensen PØ, Moser C, Scheike T, Jacobsen CS, et al. Polymorphonuclear leukocytes restrict growth of Pseudomonas aeruginosa in the lungs of cystic fibrosis patients. Infection Immun (2014) 82:4477–86. doi: 10.1128/IAI.01969-14
52. Kimbrell DA, Beutler B. The evolution and genetics of innate immunity. Nat Rev Genet (2001) 2:256–67. doi: 10.1038/35066006
53. Jesaitis AJ, Franklin MJ, Berglund D, Sasaki M, Lord CI, Bleazard JB, et al. Compromised host defense on Pseudomonas aeruginosa biofilms: characterization of neutrophil and biofilm interactions. J Immunol (2003) 171:4329–39. doi: 10.4049/jimmunol.171.8.4329
54. Bjarnsholt T, Jensen PØ, Burmølle M, Hentzer M, Haagensen JA, Hougen HP, et al. Pseudomonas aeruginosa tolerance to tobramycin, hydrogen peroxide and polymorphonuclear leukocytes is quorum-sensing dependent. Microbiology (2005) 151:373–83. doi: 10.1099/mic.0.27463-0
55. Leid JG, Willson CJ, Shirtliff ME, Hassett DJ, Parsek MR, Jeffers AK. The exopolysaccharide alginate protects Pseudomonas aeruginosa biofilm bacteria from IFN-gamma-mediated macrophage killing. J Immunol (2005) 175:7512–8. doi: 10.4049/jimmunol.175.11.7512
56. Jensen PØ, Bjarnsholt T, Phipps R, Rasmussen TB, Calum H, Christoffersen L, et al. Rapid necrotic killing of polymorphonuclear leukocytes is caused by quorum-sensing-controlled production of rhamnolipid by Pseudomonas aeruginosa. Microbiology (2007) 153:1329–38. doi: 10.1099/mic.0.2006/003863-0
57. Pestrak MJ, Chaney SB, Eggleston HC, Dellos-Nolan S, Dixit S, Mathew-Steiner SS, et al. Pseudomonas aeruginosa rugose small-colony variants evade host clearance, are hyper-inflammatory, and persist in multiple host environments. PloS Pathog (2018) 14:e1006842. doi: 10.1371/journal.ppat.1006842
58. Jensen PØ, Moser C, Kobayashi O, Hougen HP, Kharazmi A, Høiby N. Faster activation of polymorphonuclear neutrophils in resistant mice during early innate response to Pseudomonas aeruginosa lung infection. Clin Exp Immunol (2004) 137:478–85. doi: 10.1111/j.1365-2249.2004.02554.x
59. van Gennip M, Moser C, Christensen LD, Bjarnsholt T, Calum H, Jensen PØ, et al. Augmented effect of early antibiotic treatment in mice with experimental lung infections due to sequentially adapted mucoid strains of Pseudomonas aeruginosa. J antimicrobial chemotherapy (2009) 64:1241–50. doi: 10.1093/jac/dkp352
60. Alhede M, Bjarnsholt T, Jensen PØ, Phipps RK, Moser C, Christophersen L, et al. Pseudomonas aeruginosa recognizes and responds aggressively to the presence of polymorphonuclear leukocytes. Microbiology (2009) 155:3500–8. doi: 10.1099/mic.0.031443-0
61. Mayer-Hamblett N, Aitken ML, Accurso FJ, Kronmal RA, Konstan MW, Burns JL, et al. Association between pulmonary function and sputum biomarkers in cystic fibrosis. Am J respiratory Crit Care Med (2007) 175:822–8. doi: 10.1164/rccm.200609-1354OC
62. Houston N, Stewart N, Smith DS, Bell SC, Champion AC, Reid DW. Sputum neutrophils in cystic fibrosis patients display a reduced respiratory burst. J cystic fibrosis Off J Eur Cystic Fibrosis Soc (2013) 12:352–62. doi: 10.1016/j.jcf.2012.11.004
63. Sagel SD, Chmiel JF, Konstan MW. Sputum biomarkers of inflammation in cystic fibrosis lung disease. Proc Am Thoracic Soc (2007) 4:406–17. doi: 10.1513/pats.200703-044BR
64. Starosta V, Rietschel E, Paul K, Baumann U, Griese M. Oxidative changes of bronchoalveolar proteins in cystic fibrosis. Chest (2006) 129:431–7. doi: 10.1378/chest.129.2.431
65. Knowles MR, Boucher RC. Mucus clearance as a primary innate defense mechanism for mammalian airways. J Clin Invest (2002) 109:571–7. doi: 10.1172/JCI0215217
66. Babior BM, Kipnes RS, Curnutte JT. Biological defense mechanisms. The production by leukocytes of superoxide, a potential bactericidal agent. J Clin Invest (1973) 52:741–4. doi: 10.1172/JCI107236
67. Borregaard N, Herlin T. Energy metabolism of human neutrophils during phagocytosis. J Clin Invest (1982) 70:550–7. doi: 10.1172/JCI110647
68. Chen DL, Ferkol TW, Mintun MA, Pittman JE, Rosenbluth DB, Schuster DP. Quantifying pulmonary inflammation in cystic fibrosis with positron emission tomography. Am J respiratory Crit Care Med (2006) 173:1363–9. doi: 10.1164/rccm.200506-934OC
69. Bensel T, Stotz M, Borneff-Lipp M, Wollschlager B, Wienke A, Taccetti G, et al. Lactate in cystic fibrosis sputum. J cystic fibrosis Off J Eur Cystic Fibrosis Soc (2011) 10:37–44. doi: 10.1016/j.jcf.2010.09.004
70. Pedersen SS. Lung infection with alginate-producing, mucoid Pseudomonas aeruginosa in cystic fibrosis. APMIS Suppl (1992) 28:1–79. doi: 10.1136/thx.47.1.6
71. Kronborg G, Fomsgaard A, Jensen ET, Kharazmi A, Høiby N. Induction of oxidative burst response in human neutrophils by immune complexes made in vitro of lipopolysaccharide and hyperimmune serum from chronically infected patients. APMIS Acta pathologica microbiologica immunologica Scandinavica (1993) 101:887–94. doi: 10.1111/j.1699-0463.1993.tb00197.x
72. Kharazmi A, Rechnitzer C, Schiøtz PO, Jensen T, Baek L, Høiby N. Priming of neutrophils for enhanced oxidative burst by sputum from cystic fibrosis patients with Pseudomonas aeruginosa infection. Eur J Clin Invest (1987) 17:256–61. doi: 10.1111/j.1365-2362.1987.tb01245.x
73. Downey DG, Bell SC, Elborn JS. Neutrophils in cystic fibrosis. Thorax (2009) 64:81–8. doi: 10.1136/thx.2007.082388
74. Kharazmi A, Nielsen H, Bendtzen K. Modulation of human neutrophil and monocyte chemotaxis and superoxide responses by recombinant TNF-alpha and GM-CSF. Immunobiology (1988) 177:363–70. doi: 10.1016/S0171-2985(88)80004-4
75. Yuo A, Kitagawa S, Kasahara T, Matsushima K, Saito M, Takaku F. Stimulation and priming of human neutrophils by interleukin-8: cooperation with tumor necrosis factor and colony-stimulating factors. Blood (1991) 78:2708–14. doi: 10.1182/blood.V78.10.2708.2708
76. Jones AM, Martin L, Bright-Thomas RJ, Dodd ME, McDowell A, Moffitt KL, et al. Inflammatory markers in cystic fibrosis patients with transmissible Pseudomonas aeruginosa. Eur respiratory J (2003) 22:503–6. doi: 10.1183/09031936.03.00004503
77. Jensen PØ, Moser C, Kharazmi A, Presler T, Koch C, Høiby N. Increased serum concentration of G-CSF in cystic fibrosis patients with chronic Pseudomonas aeruginosa pneumonia. J cystic fibrosis Off J Eur Cystic Fibrosis Soc (2006) 5:145–51. doi: 10.1016/j.jcf.2005.12.004
78. Nathan C, Srimal S, Farber C, Sanchez E, Kabbash L, Asch A, et al. Cytokine-induced respiratory burst of human neutrophils: dependence on extracellular matrix proteins and CD11/CD18 integrins. J Cell Biol (1989) 109:1341–9. doi: 10.1083/jcb.109.3.1341
79. Campbell EL, Bruyninckx WJ, Kelly CJ, Glover LE, McNamee EN, Bowers BE, et al. Transmigrating neutrophils shape the mucosal microenvironment through localized oxygen depletion to influence resolution of inflammation. Immunity (2014) 40:66–77. doi: 10.1016/j.immuni.2013.11.020
80. Bjarnsholt T, Kirketerp-Møller K, Jensen PØ, Madsen KG, Phipps R, Krogfelt K, et al. Why chronic wounds will not heal: a novel hypothesis. Wound Repair regeneration Off Publ Wound Healing Soc [and] Eur Tissue Repair Soc (2008) 16:2–10. doi: 10.1111/j.1524-475X.2007.00283.x
81. Kirketerp-Møller K, Jensen PØ, Fazli M, Madsen KG, Pedersen J, Moser C, et al. Distribution, organization, and ecology of bacteria in chronic wounds. J Clin Microbiol (2008) 46:2717–22. doi: 10.1128/JCM.00501-08
82. Fazli M, Bjarnsholt T, Kirketerp-Møller K, Jørgensen A, Andersen CB, Givskov M, et al. Quantitative analysis of the cellular inflammatory response against biofilm bacteria in chronic wounds. Wound Repair regeneration Off Publ Wound Healing Soc [and] Eur Tissue Repair Soc (2011) 19:387–91. doi: 10.1111/j.1524-475X.2011.00681.x
83. Trampuz A, Hanssen AD, Osmon DR, Mandrekar J, Steckelberg JM, Patel R. Synovial fluid leukocyte count and differential for the diagnosis of prosthetic knee infection. Am J Med (2004) 117:556–62. doi: 10.1016/j.amjmed.2004.06.022
84. Trampuz A, Zimmerli W. Prosthetic joint infections: update in diagnosis and treatment. Swiss Med Wkly (2005) 135:243–51.
85. Mathee K, Ciofu O, Sternberg C, Lindum PW, Campbell JI, Jensen P, et al. Mucoid conversion of Pseudomonas aeruginosa by hydrogen peroxide: a mechanism for virulence activation in the cystic fibrosis lung. Microbiology (1999) 145( Pt 6):1349–57. doi: 10.1099/13500872-145-6-1349
86. Walker TS, Tomlin KL, Worthen GS, Poch KR, Lieber JG, Saavedra MT, et al. Enhanced Pseudomonas aeruginosa biofilm development mediated by human neutrophils. Infection Immun (2005) 73:3693–701. doi: 10.1128/IAI.73.6.3693-3701.2005
87. Parks QM, Young RL, Poch KR, Malcolm KC, Vasil ML, Nick JA. Neutrophil enhancement of Pseudomonas aeruginosa biofilm development: human F-actin and DNA as targets for therapy. J Med Microbiol (2009) 58:492–502. doi: 10.1099/jmm.0.005728-0
88. Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol (2010) 11:373–84. doi: 10.1038/ni.1863
89. Koller B, Kappler M, Latzin P, Gaggar A, Schreiner M, Takyar S, et al. TLR expression on neutrophils at the pulmonary site of infection: TLR1/TLR2-mediated up-regulation of TLR5 expression in cystic fibrosis lung disease. J Immunol (2008) 181:2753–63. doi: 10.4049/jimmunol.181.4.2753
90. Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, et al. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature (2001) 410:1099–103. doi: 10.1038/35074106
91. Garrett ES, Perlegas D, Wozniak DJ. Negative control of flagellum synthesis in Pseudomonas aeruginosa is modulated by the alternative sigma factor AlgT (AlgU). J bacteriology (1999) 181:7401–4. doi: 10.1128/JB.181.23.7401-7404.1999
92. Leid JG, Kerr M, Selgado C, Johnson C, Moreno G, Smith A, et al. Flagellum-mediated biofilm defense mechanisms of Pseudomonas aeruginosa against host-derived lactoferrin. Infection Immun (2009) 77:4559–66. doi: 10.1128/IAI.00075-09
93. Singh PK, Parsek MR, Greenberg EP, Welsh MJA. component of innate immunity prevents bacterial biofilm development. Nature (2002) 417:552–5. doi: 10.1038/417552a
94. Byrd MS, Pang B, Mishra M, Swords WE, Wozniak DJ. The Pseudomonas aeruginosa exopolysaccharide Psl facilitates surface adherence and NF-kappaB activation in A549 cells. mBio (2010) 1(3):e00140-10. doi: 10.1128/mBio.00140-10
95. Balloy V, Verma A, Kuravi S, Si-Tahar M, Chignard M, Ramphal R. The role of flagellin versus motility in acute lung disease caused by Pseudomonas aeruginosa. J Infect Dis (2007) 196:289–96. doi: 10.1086/518610
96. Roussel L, LaFayette S, Nguyen D, Baglole CJ, Rousseau S. Differential Contribution of the Aryl-Hydrocarbon Receptor and Toll-Like Receptor Pathways to IL-8 Expression in Normal and Cystic Fibrosis Airway Epithelial Cells Exposed to Pseudomonas aeruginosa. Front Cell Dev Biol (2016) 4:148. doi: 10.3389/fcell.2016.00148
97. Allesen-Holm M, Barken KB, Yang L, Klausen M, Webb JS, Kjelleberg S, et al. A characterization of DNA release in Pseudomonas aeruginosa cultures and biofilms. Mol Microbiol (2006) 59:1114–28. doi: 10.1111/j.1365-2958.2005.05008.x
98. Alvarez ME, Fuxman Bass JI, Geffner JR, Fernandez Calotti PX, Costas M, Coso OA, et al. Neutrophil signaling pathways activated by bacterial DNA stimulation. J Immunol (2006) 177:4037–46. doi: 10.4049/jimmunol.177.6.4037
99. Fuxman Bass JI, Gabelloni ML, Alvarez ME, Vermeulen ME, Russo DM, Zorreguieta A, et al. Characterization of bacterial DNA binding to human neutrophil surface. Lab investigation; J Tech Methods Pathol (2008) 88:926–37. doi: 10.1038/labinvest.2008.59
100. Koch C, Høiby N. Pathogenesis of cystic fibrosis. Lancet (1993) 341:1065–9. doi: 10.1016/0140-6736(93)92422-P
101. Pedersen SS, Kharazmi A, Espersen F, Høiby N. Pseudomonas aeruginosa alginate in cystic fibrosis sputum and the inflammatory response. Infection Immun (1990) 58:3363–8. doi: 10.1128/IAI.58.10.3363-3368.1990
102. Otterlei M, Sundan A, Skjak-Braek G, Ryan L, Smidsrod O, Espevik T. Similar mechanisms of action of defined polysaccharides and lipopolysaccharides: characterization of binding and tumor necrosis factor alpha induction. Infection Immun (1993) 61:1917–25. doi: 10.1128/IAI.61.5.1917-1925.1993
103. Flo TH, Ryan L, Latz E, Takeuchi O, Monks BG, Lien E, et al. Involvement of toll-like receptor (TLR) 2 and TLR4 in cell activation by mannuronic acid polymers. J Biol Chem (2002) 277:35489–95. doi: 10.1074/jbc.M201366200
104. Ryder C, Byrd M, Wozniak DJ. Role of polysaccharides in Pseudomonas aeruginosa biofilm development. Curr Opin Microbiol (2007) 10:644–8. doi: 10.1016/j.mib.2007.09.010
105. Kharazmi A. Mechanisms involved in the evasion of the host defence by Pseudomonas aeruginosa. Immunol Lett (1991) 30:201–5. doi: 10.1016/0165-2478(91)90026-7
106. Pier GB, Coleman F, Grout M, Franklin M, Ohman DE. Role of alginate O acetylation in resistance of mucoid Pseudomonas aeruginosa to opsonic phagocytosis. Infection Immun (2001) 69:1895–901. doi: 10.1128/IAI.69.3.1895-1901.2001
107. Schiøtz PO, Sørensen H, Høiby M. Activated complement in the sputum from patients with cystic fibrosis. Acta Pathol Microbiol Scand C (1979) 87C:1–5.
108. Jones CJ, Wozniak DJ. Psl Produced by Mucoid Pseudomonas aeruginosa Contributes to the Establishment of Biofilms and Immune Evasion. mBio (2017) 8(3):e00864-17. doi: 10.1128/mBio.00864-17
109. Jensen ET, Kharazmi A, Garred P, Kronborg G, Fomsgaard A, Mollnes TE, et al. Complement activation by Pseudomonas aeruginosa biofilms. Microb Pathog (1993) 15:377–88. doi: 10.1006/mpat.1993.1087
110. Davies J, Neth O, Alton E, Klein N, Turner M. Differential binding of mannose-binding lectin to respiratory pathogens in cystic fibrosis. Lancet (2000) 355:1885–6. doi: 10.1016/S0140-6736(00)02297-2
111. Di A, Brown ME, Deriy LV, Li C, Szeto FL, Chen Y, et al. CFTR regulates phagosome acidification in macrophages and alters bactericidal activity. Nat Cell Biol (2006) 8:933–44. doi: 10.1038/ncb1456
112. Painter RG, Bonvillain RW, Valentine VG, Lombard GA, LaPlace SG, Nauseef WM, et al. The role of chloride anion and CFTR in killing of Pseudomonas aeruginosa by normal and CF neutrophils. J leukocyte Biol (2008) 83:1345–53. doi: 10.1189/jlb.0907658
113. Zimmermann S, Wagner C, Muller W, Brenner-Weiss G, Hug F, Prior B, et al. Induction of neutrophil chemotaxis by the quorum-sensing molecule N-(3-oxododecanoyl)-L-homoserine lactone. Infection Immun (2006) 74:5687–92. doi: 10.1128/IAI.01940-05
114. Hoffman LR, Kulasekara HD, Emerson J, Houston LS, Burns JL, Ramsey BW, et al. Pseudomonas aeruginosa lasR mutants are associated with cystic fibrosis lung disease progression. J cystic fibrosis Off J Eur Cystic Fibrosis Soc (2009) 8:66–70. doi: 10.1016/j.jcf.2008.09.006
115. LaFayette SL, Houle D, Beaudoin T, Wojewodka G, Radzioch D, Hoffman LR, et al. Cystic fibrosis-adapted Pseudomonas aeruginosa quorum sensing lasR mutants cause hyperinflammatory responses. Sci Adv (2015) 1(6):e1500199. doi: 10.1126/sciadv.1500199
116. Alhede M, Lorenz M, Fritz BG, Jensen PØ, Ring HC, Bay L, et al. Bacterial aggregate size determines phagocytosis efficiency of polymorphonuclear leukocytes. Med Microbiol Immunol (2020) 209:669–80. doi: 10.1007/s00430-020-00691-1
117. Lau GW, Hassett DJ, Ran H, Kong F. The role of pyocyanin in Pseudomonas aeruginosa infection. Trends Mol Med (2004) 10:599–606. doi: 10.1016/j.molmed.2004.10.002
118. Rada B, Leto TL. Pyocyanin effects on respiratory epithelium: relevance in Pseudomonas aeruginosa airway infections. Trends Microbiol (2013) 21:73–81. doi: 10.1016/j.tim.2012.10.004
119. Hiemstra PS, Amatngalim GD, van der Does AM, Taube C. Antimicrobial Peptides and Innate Lung Defenses: Role in Infectious and Noninfectious Lung Diseases and Therapeutic Applications. Chest (2016) 149:545–51. doi: 10.1378/chest.15-1353
120. Pezzulo AA, Tang XX, Hoegger MJ, Abou Alaiwa MH, Ramachandran S, Moninger TO, et al. Reduced airway surface pH impairs bacterial killing in the porcine cystic fibrosis lung. Nature (2012) 487:109–13. doi: 10.1038/nature11130
121. Abou Alaiwa MH, Reznikov LR, Gansemer ND, Sheets KA, Horswill AR, Stoltz DA, et al. pH modulates the activity and synergism of the airway surface liquid antimicrobials beta-defensin-3 and LL-37. Proc Natl Acad Sci United States America (2014) 111:18703–8. doi: 10.1073/pnas.1422091112
122. Goldman MJ, Anderson GM, Stolzenberg ED, Kari UP, Zasloff M, Wilson JM. Human beta-defensin-1 is a salt-sensitive antibiotic in lung that is inactivated in cystic fibrosis. Cell (1997) 88:553–60. doi: 10.1016/s0092-8674(00)81895-4
123. Peschel A. How do bacteria resist human antimicrobial peptides? Trends Microbiol (2002) 10:179–86. doi: 10.1016/s0966-842x(02)02333-8
124. Taggart CC, Greene CM, Smith SG, Levine RL, McCray PB Jr., O’Neill S, et al. Inactivation of human beta-defensins 2 and 3 by elastolytic cathepsins. J Immunol (2003) 171:931–7. doi: 10.4049/jimmunol.171.2.931
125. Bucki R, Byfield FJ, Janmey PA. Release of the antimicrobial peptide LL-37 from DNA/F-actin bundles in cystic fibrosis sputum. Eur respiratory J (2007) 29:624–32. doi: 10.1183/09031936.00080806
126. Trøstrup H, Thomsen K, Calum H, Høiby N, Moser C. Animal models of chronic wound care: the application of biofilms in clinical research. Chronic Wound Care Manage Res (2016) 3:123–32. doi: 10.2147/CWCMR.S84361
127. Høgsberg T, Bjarnsholt T, Thomsen JS, Kirketerp-Møller K. Success rate of split-thickness skin grafting of chronic venous leg ulcers depends on the presence of Pseudomonas aeruginosa: a retrospective study. PloS One (2011) 6:e20492. doi: 10.1371/journal.pone.0020492
128. Høiby N, Ciofu O, Johansen HK, Song Z-J, Moser C, Jensen PØ, et al. The clinical impact of bacterial biofilms. Int J Oral Sci (2011) 3:55–65. doi: 10.4248/IJOS11026
129. Van Der Plas MJA, Bhongir RKV, Kjellström S, Siller H, Kasetty G, Mörgelin M, et al. (2016) Pseudomonas aeruginosa elastase cleaves a C-terminal peptide from human thrombin that inhibits host inflammatory responses. Nat Commun (2016) 7:11567. doi: 10.1038/ncomms11567
130. Zasloff M. Antimicrobial peptides of multicellular organisms. Nature (2002) 415:389–95. doi: 10.1038/415389a
131. Chan JK, Roth J, Oppenheim JJ, Tracey KJ, Vogl T, Feldmann M, et al. Alarmins: awaiting a clinical response. J Clin Invest (2012) 122:2711–9. doi: 10.1172/JCI62423
132. Zhang L-J, Gallo RL. Antimicrobial peptides. Curr biology: CB (2016) 26:R14–19. doi: 10.1016/j.cub.2015.11.017
133. Nakatsuji T, Gallo RL. Antimicrobial peptides: Old Molecules with New Ideas. J Invest Dermatol (2012) 132:887–95. doi: 10.1038/jid.2011.387
134. Thorey IS, Roth J, Regenbogen J, Halle JP, Bittner M, Vogl T, et al. The Ca2+-binding proteins S100A8 and S100A9 are encoded by novel injury-regulated genes. J Biol Chem (2001) 276:35818–25. doi: 10.1074/jbc.M104871200
135. Wu N, Davidson JM. Migration inhibitory factor-related protein (MRP)8 and MRP14 are differentially expressed in free-electron laser and scalpel incisions. Wound Repair Regeneration (2004) 12:327–36. doi: 10.1111/j.1067-1927.2004.012313.x
136. Trøstrup H, Lundquist R, Christensen LH, Jørgensen LN, Karlsmark T, Haab BB, et al. S100A8/A9 deficiency in nonhealing venous leg ulcers uncovered by multiplexed antibody microarray profiling. Br J Dermatol (2011) 165:292–301. doi: 10.1111/j.1365-2133.2011.10384.x
137. Trøstrup H, Holstein P, Christophersen L, Jørgensen B, Karlsmark T, Høiby N, et al. S100A8/A9 is an important host defence mediator in neuropathic foot ulcers in patients with type 2 diabetes mellitus. Arch Dermatol Res (2016) 308:347–55. doi: 10.1007/s00403-016-1646-7
138. Moser C, Johansen HK, Song Z, Hougen HP, Rygaard J, Høiby N. Chronic Pseudomonas aeruginosa lung infection is more severe in Th2 responding BALB/c mice compared to Th1 responding C3H/HeN mice. APMIS Acta pathologica microbiologica immunologica Scandinavica (1997) 105:838–42. doi: 10.1111/j.1699-0463.1997.tb05092.x
139. Janeway CA, Travers P. Immunobiology. 3rd edn. UK/USA: Current Biology Ltd, Churchill Livingstone, Garland Publishing Inc. (1997).
141. Høiby N, Krogh Johansen H, Moser C, Song Z, Ciofu O, Kharazmi A. Pseudomonas aeruginosa and the in vitro and in vivo biofilm mode of growth. Microbes Infection (2001) 3:23–35. doi: 10.1016/S1286-4579(00)01349-6
142. Schaudinn C, Gorur A, Keller D, Sedghizadeh PP, Costerton JW. Periodontitis: an archetypical biofilm disease. J Am Dental Assoc (1939) (2009) 140:978–86. doi: 10.14219/jada.archive.2009.0307
143. Brady RA, Leid JG, Calhoun JH, Costerton JW, Shirtliff ME. Osteomyelitis and the role of biofilms in chronic infection. FEMS Immunol Med Microbiol (2008) 52:13–22. doi: 10.1111/j.1574-695X.2007.00357.x
144. Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature (1998) 392:245–52. doi: 10.1038/32588
145. Kaliński P, Hilkens CM, Wierenga EA, Kapsenberg ML. T-cell priming by type-1 and type-2 polarized dendritic cells: the concept of a third signal. Immunol Today (1999) 20:561–7. doi: 10.1016/S0167-5699(99)01547-9
146. Lai L, Alaverdi N, Maltais L, Morse HC. Mouse cell surface antigens: nomenclature and immunophenotyping. J Immunol (Baltimore Md: 1950) (1998) 160:3861–8.
147. Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu Y-J, et al. Immunobiology of Dendritic Cells. Annu Rev Immunol (2000) 18:767–811. doi: 10.1146/annurev.immunol.18.1.767
148. Damlund DSM, Christophersen L, Jensen PØ, Alhede M, Høiby N, Moser C. Activation of pulmonary and lymph node dendritic cells during chronic Pseudomonas aeruginosa lung infection in mice. APMIS Acta pathologica microbiologica immunologica Scandinavica (2016) 124:500–7. doi: 10.1111/apm.12530
149. Skindersøe ME, Zeuthen LH, Brix S, Fink LN, Lazenby J, Whittall C, et al. Pseudomonas aeruginosa quorum-sensing signal molecules interfere with dendritic cell-induced T-cell proliferation. FEMS Immunol Med Microbiol (2009) 55:335–45. doi: 10.1111/j.1574-695X.2008.00533.x
150. Moser C, Jensen PØ, Pressler T, Frederiksen B, Lanng S, Kharazmi A, et al. Serum concentrations of GM-CSF and G-CSF correlate with the Th1/Th2 cytokine response in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. APMIS: Acta pathologica microbiologica immunologica Scandinavica (2005) 113:400–9. doi: 10.1111/j.1600-0463.2005.apm_142.x
151. Jarrossay D, Napolitani G, Colonna M, Sallusto F, Lanzavecchia A. Specialization and complementarity in microbial molecule recognition by human myeloid and plasmacytoid dendritic cells. Eur J Immunol (2001) 31:3388–93. doi: 10.1002/1521-4141(200111)31:11<3388::AID-IMMU3388>3.0.CO;2-Q
152. Kadowaki N, Ho S, Antonenko S, Malefyt RW, Kastelein RA, Bazan F, et al. Subsets of human dendritic cell precursors express different toll-like receptors and respond to different microbial antigens. J Exp Med (2001) 194:863–9. doi: 10.1084/jem.194.6.863
153. Penna G, Vulcano M, Roncari A, Facchetti F, Sozzani S, Adorini L. Cutting edge: differential chemokine production by myeloid and plasmacytoid dendritic cells. J Immunol (Baltimore Md: 1950) (2002) 169:6673–6. doi: 10.4049/jimmunol.169.12.6673
154. Piccioli D, Tavarini S, Borgogni E, Steri V, Nuti S, Sammicheli C, et al. Functional specialization of human circulating CD16 and CD1c myeloid dendritic-cell subsets. Blood (2007) 109:5371–9. doi: 10.1182/blood-2006-08-038422
155. Demedts IK, Bracke KR, Maes T, Joos GF, Brusselle GG. Different roles for human lung dendritic cell subsets in pulmonary immune defense mechanisms. Am J respiratory Cell Mol Biol (2006) 35:387–93. doi: 10.1165/rcmb.2005-0382OC
156. Ito T, Kanzler H, Duramad O, Cao W, Liu Y-J. Specialization, kinetics, and repertoire of type 1 interferon responses by human plasmacytoid predendritic cells. Blood (2006) 107:2423–31. doi: 10.1182/blood-2005-07-2709
157. Kharazmi A, Döring G, Høiby N, Valerius NH. Interaction of Pseudomonas aeruginosa alkaline protease and elastase with human polymorphonuclear leukocytes in vitro. Infection Immun (1984) 43:161–5. doi: 10.1128/IAI.43.1.161-165.1984
158. Horvat RT, Parmely MJ. Pseudomonas aeruginosa alkaline protease degrades human gamma interferon and inhibits its bioactivity. Infection Immun (1988) 56:2925–32. doi: 10.1128/IAI.56.11.2925-2932.1988
159. Theander TG, Kharazmi A, Pedersen BK, Christensen LD, Tvede N, Poulsen LK, et al. Inhibition of human lymphocyte proliferation and cleavage of interleukin-2 by Pseudomonas aeruginosa proteases. Infection Immun (1988) 56:1673–7. doi: 10.1128/IAI.56.7.1673-1677.1988
160. McCormick LL, Karulin AY, Schreiber JR, Greenspan NS. Bispecific antibodies overcome the opsonin-receptor mismatch of cystic fibrosis in vitro: restoration of neutrophil-mediated phagocytosis and killing of Pseudomonas aeruginosa. J Immunol (Baltimore Md: 1950) (1997) 158:3474–82.
161. Döring G, Høiby N. Longitudinal study of immune response to Pseudomonas aeruginosa antigens in cystic fibrosis. Infection Immun (1983) 42:197–201. doi: 10.1128/IAI.42.1.197-201.1983
162. Döring G, Goldstein W, Röll A, Schiøtz PO, Høiby N, Botzenhart K. Role of Pseudomonas aeruginosa exoenzymes in lung infections of patients with cystic fibrosis. Infection Immun (1985) 49:557–62. doi: 10.1128/IAI.49.3.557-562.1985
163. Petersen TD, Ciofu O, Pressler T, Giwercman B, Pedersen SS, Høiby N. Quantitative analysis of the IgG and IgG subclass immune responses to chromosomal Pseudomonas aeruginosa beta-lactamase in serum from patients with cystic fibrosis by western blotting and laser scanning densitometry. Thorax (1996) 51:733–8. doi: 10.1136/thx.51.7.733
164. Corthesy B. Multi-faceted functions of secretory IgA at mucosal surfaces. Front Immunol (2013) 4:185. doi: 10.3389/fimmu.2013.00185
165. Mauch RM, Rossi CL, Nolasco da Silva MT, Bianchi Aiello T, Ribeiro JD, Ribeiro AF, et al. Secretory IgA-mediated immune response in saliva and early detection of Pseudomonas aeruginosa in the lower airways of pediatric cystic fibrosis patients. Med Microbiol Immunol (2019) 208:205–13. doi: 10.1007/s00430-019-00578-w
166. Høiby N, Flensborg EW, Beck B, Friis B, Jacobsen SV, Jacobsen L. Pseudomonas aeruginosa infection in cystic fibrosis. Diagnostic and prognostic significance of Pseudomonas aeruginosa precipitins determined by means of crossed immunoelectrophoresis. Scand J Respir Dis (1977) 58:65–79.
167. Johansen HK, Nørregaard L, Gøtzsche PC, Pressler T, Koch C, Høiby N. Antibody response to Pseudomonas aeruginosa in cystic fibrosis patients: a marker of therapeutic success?–A 30-year cohort study of survival in Danish CF patients after onset of chronic P. aeruginosa lung infection. Pediatr pulmonology (2004) 37:427–32. doi: 10.1002/ppul.10457
168. Moser C, Kjaergaard S, Pressler T, Kharazmi A, Koch C, Høiby N. The immune response to chronic Pseudomonas aeruginosa lung infection in cystic fibrosis patients is predominantly of the Th2 type. APMIS Acta pathologica microbiologica immunologica Scandinavica (2000) 108:329–35. doi: 10.1034/j.1600-0463.2000.d01-64.x
169. Moss RB, Hsu YP, Olds L. Cytokine dysregulation in activated cystic fibrosis (CF) peripheral lymphocytes. Clin Exp Immunol (2000) 120:518–25. doi: 10.1046/j.1365-2249.2000.01232.x
170. Brazova J, Sediva A, Pospisilova D, Vavrova V, Pohunek P, Macek M, et al. Differential cytokine profile in children with cystic fibrosis. Clin Immunol (Orlando Fla) (2005) 115:210–5. doi: 10.1016/j.clim.2005.01.013
171. Hartl D, Griese M, Kappler M, Zissel G, Reinhardt D, Rebhan C, et al. Pulmonary T(H)2 response in Pseudomonas aeruginosa-infected patients with cystic fibrosis. J Allergy Clin Immunol (2006) 117:204–11. doi: 10.1016/j.jaci.2005.09.023
172. Moser C, Hougen HP, Song Z, Rygaard J, Kharazmi A, HøIby N. Early immune response in susceptible and resistant mice strains with chronic Pseudomonas aeruginosa lung infection determines the type of T-helper cell response. APMIS Acta pathologica microbiologica immunologica Scandinavica (1999) 107:1093–100. doi: 10.1111/j.1699-0463.1999.tb01514.x
173. Moser C, Jensen PØ, Kobayashi O, Hougen HP, Song Z, Rygaard J, et al. Improved outcome of chronic Pseudomonas aeruginosa lung infection is associated with induction of a Th1-dominated cytokine response. Clin Exp Immunol (2002) 127:206–13. doi: 10.1046/j.1365-2249.2002.01731.x
174. Ware LB, Matthay MA. The acute respiratory distress syndrome. New Engl J Med (2000) 342:1334–49. doi: 10.1056/NEJM200005043421806
175. Cassatella MA, Guasparri I, Ceska M, Bazzoni F, Rossi F. Interferon-gamma inhibits interleukin-8 production by human polymorphonuclear leucocytes. Immunology (1993) 78:177–84.
176. Schnyder-Candrian S, Strieter RM, Kunkel SL, Walz A. Interferon-α and interferon-γ down-regulate the production of interleukin-8 and ENA-78 in human monocytes. J leukocyte Biol (1995) 57:929–35. doi: 10.1002/jlb.57.6.929
177. Stockinger B, Veldhoen M, Martin B. Th17 T cells: linking innate and adaptive immunity. Semin Immunol (2007) 19:353–61. doi: 10.1016/j.smim.2007.10.008
178. Ivanov II, Zhou L, Littman DR. Transcriptional regulation of Th17 cell differentiation. Semin Immunol (2007) 19:409–17. doi: 10.1016/j.smim.2007.10.011
179. Decraene A, Willems-Widyastuti A, Kasran A, De Boeck K, Bullens DM, Dupont LJ. Elevated expression of both mRNA and protein levels of IL-17A in sputum of stable Cystic Fibrosis patients. Respiratory Res (2010) 11:177. doi: 10.1186/1465-9921-11-177
180. Tiringer K, Treis A, Fucik P, Gona M, Gruber S, Renner S, et al. A Th17- and Th2-skewed Cytokine Profile in Cystic Fibrosis Lungs Represents a Potential Risk Factor for Pseudomonas aeruginosa Infection. Am J respiratory Crit Care Med (2013) 187:621–9. doi: 10.1164/rccm.201206-1150OC
181. Bayes HK, Bicknell S, MacGregor G, Evans TJ. T helper cell subsets specific for Pseudomonas aeruginosa in healthy individuals and patients with cystic fibrosis. PloS One (2014) 9:e90263. doi: 10.1371/journal.pone.0090263
182. Bruscia EM, Bonfield TL. Innate and Adaptive Immunity in Cystic Fibrosis. Clinics Chest Med (2016) 37:17–29. doi: 10.1016/j.ccm.2015.11.010
183. Rieber N, Brand A, Hector A, Graepler-Mainka U, Ost M, Schäfer I, et al. Flagellin induces myeloid-derived suppressor cells: implications for Pseudomonas aeruginosa infection in cystic fibrosis lung disease. J Immunol (Baltimore Md: 1950) (2013) 190:1276–84. doi: 10.4049/jimmunol.1202144
184. Öz HH, Zhou B, Voss P, Carevic M, Schroth C, Frey N, et al. Pseudomonas aeruginosa Airway Infection Recruits and Modulates Neutrophilic Myeloid-Derived Suppressor Cells. Front Cell Infect Microbiol (2016) 6:167. doi: 10.3389/fcimb.2016.00167
185. Hector A, Schäfer H, Pöschel S, Fischer A, Fritzsching B, Ralhan A, et al. Regulatory T-Cell Impairment in Cystic Fibrosis Patients with Chronic Pseudomonas Infection. Am J respiratory Crit Care Med (2015) 191:914–23. doi: 10.1164/rccm.201407-1381OC
186. Warr GW, Magor KE, Higgins DA. IgY: clues to the origins of modern antibodies. Immunol Today (1995) 16:392–8. doi: 10.1016/0167-5699(95)80008-5
187. Bhanushali JK, Gilbert JM, McDougald LR. Simple method to purify chicken immunoglobulin G. Poultry Sci (1994) 73:1158–61. doi: 10.3382/ps.0731158
188. Nilsson E, Amini A, Wretlind B, Larsson A. Pseudomonas aeruginosa infections are prevented in cystic fibrosis patients by avian antibodies binding Pseudomonas aeruginosa flagellin. J Chromatogr B Analytical Technol Biomed Life Sci (2007) 856:75–80. doi: 10.1016/j.jchromb.2007.05.029
189. Thomsen K, Christophersen L, Bjarnsholt T, Jensen PØ, Moser C, Høiby N. Anti-Pseudomonas aeruginosa IgY Antibodies Induce Specific Bacterial Aggregation and Internalization in Human Polymorphonuclear Neutrophils. Infection Immun (2015) 83:2686–93. doi: 10.1128/IAI.02970-14
190. Thomsen K, Christophersen L, Jensen PØ, Bjarnsholt T, Moser C, Høiby N. Anti-Pseudomonas aeruginosa IgY antibodies promote bacterial opsonization and augment the phagocytic activity of polymorphonuclear neutrophils. Hum Vaccines Immunotherapeutics (2016) 12:1690–9. doi: 10.1080/21645515.2016.1145848
191. Thomsen K, Christophersen L, Bjarnsholt T, Jensen PØ, Moser C, Høiby N. Anti-Pseudomonas aeruginosa IgY antibodies augment bacterial clearance in a murine pneumonia model. J cystic fibrosis Off J Eur Cystic Fibrosis Soc (2015) 15:171–8. doi: 10.1016/j.jcf.2015.08.002
192. Thomsen K, Christophersen L, Lerche CJ, Holmgaard DB, Calum H, Høiby N, et al. Azithromycin potentiates avian IgY effect against Pseudomonas aeruginosa in a murine pulmonary infection model. Int J antimicrobial Agents (2021) 57:106213. doi: 10.1016/j.ijantimicag.2020.106213
193. Nilsson E, Larsson A, Olesen HV, Wejåker P-E, Kollberg H. Good effect of IgY against Pseudomonas aeruginosa infections in cystic fibrosis patients. Pediatr pulmonology (2008) 43:892–9. doi: 10.1002/ppul.20875
194. Laulund ASB, Trøstrup H, Lerche CJ, Thomsen K, Christophersen L, Calum H, et al. Synergistic effect of immunomodulatory S100A8/A9 and ciprofloxacin against Pseudomonas aeruginosa biofilm in a murine chronic wound model. Pathog Dis (2019) 78(5):ftz027. doi: 10.1093/femspd/ftz027
195. Lundquist R, Holmstrøm K, Clausen C, Jørgensen B, Karlsmark T. Characteristics of an autologous leukocyte and platelet-rich fibrin patch intended for the treatment of recalcitrant wounds. Wound Repair Regeneration: Off Publ Wound Healing Soc [and] Eur Tissue Repair Soc (2013) 21:66–76. doi: 10.1111/j.1524-475X.2012.00870.x
196. Thomsen K, Trøstrup H, Christophersen L, Lundquist R, Høiby N, Moser C. The phagocytic fitness of leucopatches may impact the healing of chronic wounds. Clin Exp Immunol (2016) 184:368–77. doi: 10.1111/cei.12773
197. Game F, Jeffcoate W, Tarnow L, Jacobsen JL, Whitham DJ, Harrison EF, et al. LeucoPatch IItt. LeucoPatch system for the management of hard-to-heal diabetic foot ulcers in the UK, Denmark, and Sweden: an observer-masked, randomised controlled trial. Lancet Diabetes Endocrinol (2018) 6:870–8. doi: 10.2337/db18-638-P
Keywords: biofilm infections, Pseudomonas aeruginosa, innate immune response, adaptive immune response, novel treatment possibilities
Citation: Moser C, Jensen PØ, Thomsen K, Kolpen M, Rybtke M, Lauland AS, Trøstrup H and Tolker-Nielsen T (2021) Immune Responses to Pseudomonas aeruginosa Biofilm Infections. Front. Immunol. 12:625597. doi: 10.3389/fimmu.2021.625597
Received: 03 November 2020; Accepted: 20 January 2021;
Published: 22 February 2021.
Edited by:
Semih Esin, University of Pisa, ItalyReviewed by:
Giovanni Di Bonaventura, University of Studies G. d’Annunzio Chieti and Pescara, ItalyLucia Grassi, Ghent University, Belgium
Copyright © 2021 Moser, Jensen, Thomsen, Kolpen, Rybtke, Lauland, Trøstrup and Tolker-Nielsen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tim Tolker-Nielsen, dHRuQHN1bmQua3UuZGs=
†These authors have contributed equally to this work