 Meifang Liu
Meifang Liu Shujuan Liang
Shujuan Liang Cai Zhang
Cai Zhang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 March 2021
Sec. NK and Innate Lymphoid Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.624687
This article is part of the Research Topic The Role of Natural Killer Cells in Autoimmune Diseases View all 7 articles
Autoimmune diseases generally result from the loss of self-tolerance (i.e., failure of the immune system to distinguish self from non-self), and are characterized by autoantibody production and hyperactivation of T cells, which leads to damage of specific or multiple organs. Thus, autoimmune diseases can be classified as organ-specific or systemic. Genetic and environmental factors contribute to the development of autoimmunity. Recent studies have demonstrated the contribution of innate immunity to the onset of autoimmune diseases. Natural killer (NK) cells, which are key components of the innate immune system, have been implicated in the development of multiple autoimmune diseases such as systemic lupus erythematosus, type I diabetes mellitus, and autoimmune liver disease. However, NK cells have both protective and pathogenic roles in autoimmunity depending on the NK cell subset, microenvironment, and disease type or stage. In this work, we review the current knowledge of the varied roles of NK cell subsets in systemic and organic-specific autoimmune diseases and their clinical potential as therapeutic targets.
Autoimmune diseases generally result from the loss of self-tolerance (i.e., failure of the immune system to distinguish self from non-self), which leads to the production of autoantibodies and self-reactive lymphocytes that cause tissue damage (1, 2). Although most autoimmune diseases are relatively uncommon, they are associated with significant morbidity and mortality. Nearly 100 distinct autoimmune diseases have been identified to date; these are organ-specific (e.g., primary biliary cirrhosis [PBC]) or are characterized by systemic immune dysfunction involving multiple organs [e.g., systemic lupus erythematosus (SLE)] (3, 4). Despite significant advances in the diagnosis and treatment of autoimmune diseases, many details of their pathogenesis and etiology have yet to be elucidated.
Autoimmune diseases principally develop as a result of abnormal activation of T and B cells. However, there is increasing evidence that natural killer (NK) cells—which link innate and adaptive immunity—play an important role in their development, for example in SLE, type 1 diabetes mellitus (T1DM), and autoimmune liver disease (ALD) (5–7). NK cells are cytotoxic lymphocytes that are critical for the defense against infections and tumors (8). NK cell activation is governed by the integration of activating and inhibitory signals from cell surface receptors (9, 10); NK cells detect cells that are under stress as a result of infection or malignancy and rapidly respond by secreting cytotoxic granules or death receptor ligands. In addition to their direct cytotoxicity, NK cells exert immunoregulatory functions in innate and adaptive immune responses by producing various cytokines and chemokines such as interferon (IFN)-γ, tumor necrosis factor (TNF)-α, granulocyte-macrophage colony-stimulating factor (GM-CSF), and chemokine (C-C motif) ligand (CCL)5 (11–13). They also exhibit immunologic memory, which challenges the conventional distinction between innate and adaptive immunity (14–18). NK cells shape the adaptive immune response through secreted cytokines and chemokines or crosstalk with other immune cells such as T and B cells and dendritic cells (DCs). Thus, NK cell hyperactivation or dysfunction is associated with the pathogenesis of various inflammatory and autoimmune diseases. However, NK cells have both protective and pathogenic functions in these diseases (19, 20) depending on the NK cell subset, microenvironment, and disease type and developmental stage. In this review, we discuss recent research on the diversity of NK cells and their roles in autoimmune diseases.
NK cells are a heterogeneous population of innate lymphocytes comprising subsets with distinct phenotypes or cytokine secretion patterns (Figure 1) (20, 21). In humans, conventional (c)NK cells are divided into 2 major subsets based on the relative surface expression levels of cluster of differentiation (CD)56 and CD16 (FcγRIII). CD56brightCD16− NK cells (also termed CD56bright NK cells) are mostly present in secondary lymphoid tissues and produce cytokines such as IFN-γ, TNF-α, GM-CSF, interleukin (IL)-10, and IL-13 upon stimulation, thereby serving an immunoregulatory function in the maintenance of immune homeostasis (12, 13). Although CD56bright NK cells have low cytotoxicity, this effect is enhanced under inflammatory conditions (12, 22). CD56dimCD16+ NK cells (also termed CD56dim NK cells) exist predominantly in peripheral blood and express high levels of CD16; as they mature and become cytotoxic, they also express the terminal differentiation marker CD57 (23–25).
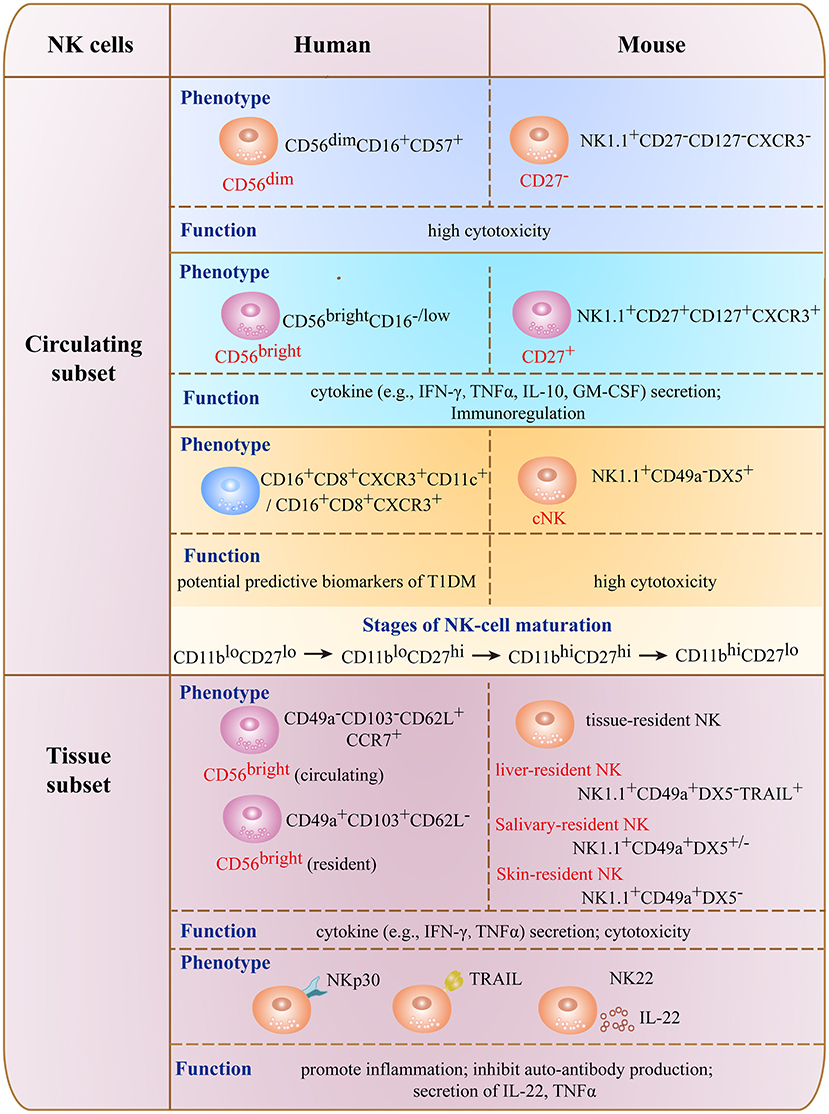
Figure 1. Phenotype and functions of major NK cell subsets in mouse and human.
Circulating CD56bright NK cells in peripheral blood are thought to be the precursors of CD56dim NK cells (26). In murine NK cells lacking CD56 expression, CD27, CD127, and C-X-C motif chemokine receptor (CXCR)3 are important markers that distinguish NK cell subsets (27–29). CD27−, CD127−, and CXCR3− NK cells have potent cytotoxicity resembling that of CD56dim NK cells in humans, whereas NK cells positive for these markers are responsible for cytokine secretion and have low cytolytic capacity like the human CD56bright subset.
There are 4 stages in the maturation of human and murine NK cells—i.e., CD11bloCD27lo → CD11bloCD27hi → CD11bhiCD27hi → CD11bhiCD27lo (30). Murine NK cells can also be divided into cNK and tissue-resident (tr)NK cells based on their production of IFN-γ and cytotoxicity (31–33). cNK cells are widely distributed and are CD49a−DX5+, whereas trNK cells have variable distribution in the uterus, skin, kidneys, salivary glands, and adipose tissue; the 2 cell types have common as well as unique features (31, 34–37).
Some factors in the tissue microenvironment (e.g., cytokines and ligands for NK cell receptors) contribute to differences in the composition of NK cell subsets and diversity of trNK cells. For example, liver-derived transforming growth factor (TGF)-β regulates and maintains the CD56bright phenotype of human liver-resident NK cells by suppressing T-bet expression (38); and salivary gland-derived TGF-β directs the differentiation of salivary gland innate lymphoid cells including salivary-resident NK cells by inhibiting eomesodermin (Eomes) expression (34). Glomerulus-specific expression of NK group 2 member D (NKG2D) ligands was shown to be correlated with increased infiltration, maturation, and activation of NK cells in kidney during the development of lupus nephritis (39). NK cells can also be classified as NK1, NK2, or NK22 cells based on their cytokine secretion profiles (40, 41). With the development of detection methods such as mass cytometry, 2 NK cell subsets (CD16+CD8+CXCR3+ and CD16+CD8+CXCR3+CD11c+) were recently identified in patients at high risk for developing T1DM that can potentially serve as predictive biomarkers for the disease (42).
SLE is a progressive autoimmune disease with variable clinical manifestations affecting several organs including skin, lungs, blood, heart, and nervous system (43). It is characterized by the presence of nuclear autoantibodies along with abnormally activated T cells and hyperactive B cells, which form immune complexes that lead to inflammation (4, 44–46).
While adaptive immune mechanisms leading to organ damage in SLE have been extensively studied, the contribution of innate immune cells—especially NK cells—remains unclear. Several studies have provided evidence that NK cells are involved in SLE pathogenesis. For instance, the number of NK cells was markedly reduced in lpr mice, an SLE model; adoptive transfer of NK cells delayed the onset of autoimmunity, indicating a protective role for NK cells in SLE (47). It was also suggested that NK cells could delay SLE onset by suppressing autoantibody secretion by B cells. Recent studies have investigated the direct cytotoxic action and cytokine profiles of NK cells in the pathogenesis of SLE. For example, the increased cytotoxicity and proinflammatory phenotype of NK cells was shown to be correlated with downregulation of CD3ξ expression in patients with SLE (48); the authors also demonstrated that caspase-3 activity was higher in NK cells from patients with SLE than those from healthy controls and that this contributed to the downregulation of CD3ξ expression in the cells, which enhanced their proinflammatory phenotype.
NK cell subsets with different phenotypes, distributions, functions, and development have distinct roles in SLE. The proportion of NK cells (especially mature CD56dim NK cells) was lower but that of CD56bright NK cells was higher in the peripheral blood of SLE patients compared to healthy controls (49–51). It is worth noting that CD56dim NK cells in active SLE patients showed increased IFN-γ production and an activated phenotype that included high expression of activating receptors (e.g., NKp44, NKp46, and CD69) and low expression of CD158a/h/g (51). The reduced number of circulating CD56dim NK cells in SLE can be attributed to the migration of highly cytotoxic (CD56dim) NK cells from peripheral blood to kidneys in SLE patients and the consequent damage to local tissue. Many factors such as increased expression of NKG2D ligands [e.g., ribonucleic acid export [RAE]-1 and mouse UL16-binding protein-like transcript (MULT)-1], CD226 ligands (e.g., CD112 and CD115), proinflammatory cytokines (e.g., TNF-α), and chemoattractant chemokines [e.g., C-X3-C motif chemokine ligand (CX3CL)1] in kidney tissue potentially contribute to the migration of CD56dim NK cells (39, 52–54). Similar NK cell migration was also observed in an animal SLE model, in which circulating DX5+ NK cells (mostly CD226+) infiltrated into the kidneys of MRL/lpr mice with an enhanced phenotype, which may be responsible for the kidney injury observed in SLE (52). Additionally, plasmocytoid (p)DC-secreted IFN-α mediated the activation-induced cell death of circulating NK cells in patients with active SLE, thereby contributing to the loss of circulating NK cells (52). A recent study showed that CD56bright NK cells may contribute to SLE development. Serum IL-15 level was elevated in SLE patients, particularly those with active disease (55, 56); and an increased number of peripheral blood Ki67+CD56bright NK cells was strongly correlated with elevated serum IL-15, clinical severity, and active nephritis in SLE patients (57). The high serum IL-15 level in SLE may be attributable to type I IFN-mediated DC activation; moreover, IL-15 induces the expression of Ki67 in NK cells, which stimulates NK cell proliferation and contributes to pathogenesis of SLE. However, the precise mechanisms by which CD56bright NK cells promote SLE or mediate tissue injury remain unclear. It is thought that inflamed tissues recruit NK cells and alter their effector function by transforming their phenotype from one of low toxicity to that of cytotoxic CD56bright NK cells via mechanisms that are as yet unknown (22). Thus, multiple factors in the local environment contribute to kidney infiltration by NK cells (especially CD56dim NK cells) and tissue injury.
Most knowledge regarding the role of NK cells in SLE is derived from studies on peripheral blood; less is known about the functions of local NK cells in target tissues or trNK cells in SLE development. Recent studies using mouse SLE models have demonstrated a correlation between the number kidney-infiltrating NK cells and an active disease state (39, 52). The microenvironment of lupus nephritis promotes the maturation and maintenance of resident NK cells, as evidenced by a significantly larger CD11bhiCD27lo NK cell fraction in the kidneys of SLE model mice (39). Notably, the kidney immune cell profile of SLE patients was recently established by single-cell RNA sequencing (58, 59); 21 subsets of leukocyte were identified including 2 clusters of trNK cells (CD56brightCD16− and CD56dimCD16+) with both pro- and anti-inflammatory activities (58). Further studies are needed to determine the contribution of specific NK subsets to the pathogenesis of SLE.
SS is a chronic, multisystem disorder characterized by lymphocyte infiltration of target glands (e.g., lachrymal and salivary) and sicca symptoms (60, 61). SS can be divided into primary (p)SS or secondary SS depending on whether it occurs alone or in conjunction with other systemic autoimmune diseases such as SLE or rheumatoid arthritis (RA). The hallmarks of pSS are progressive focal infiltration of immune cells (mainly T and B cells), hypergammaglobulinemia, and the presence of autoantibodies, underscoring the importance of adaptive immunity in SS pathophysiology (62, 63). However, recent findings have suggested that innate immunity—especially NK cells—plays a role in SS pathogenesis (64–66). The relative and total numbers of circulating NK cells and CD56dim NK cells were lower while the number of CD56bright NK cells was higher in pSS patients compared to healthy control subjects (67, 68). The ratio of CD56bright to CD56dim NK cells was correlated with IgG level and showed diagnostic utility for pSS with good sensitivity and specificity (67). The lower numbers of circulating NK cells and CD56dim NK cells in pSS patients may lead to their infiltration into glands and focal immune injury.
NK cells in salivary glands are predominantly tissue-resident; only a very small fraction is derived from peripheral blood (69). Salivary gland NK cells promote pSS progression by inducing IFN-γ production (70, 71). IL-33 expression is upregulated in the salivary glands of pSS patients and acts synergistically with IL-12 and IL-23 to stimulate IFN-γ secretion by NK cells, which contributes to pSS pathogenesis (72, 73). The overexpression of IL-12 in SS was also shown to participate in the differentiation of helper T (Th)1 cells and IFN-γ production (74, 75); and activation of IFN signaling and the recruitment of pDCs to the salivary glands of pSS patients may promote NK cell activation and IFN-γ production, thereby aggravating disease pathogenesis (71, 76, 77).
Different NK cell subsets in the salivary glands may play distinct roles in the pathogenesis of pSS. For example, NKp30+ NK cells are thought to accumulate in the minor salivary glands of pSS patients. The interaction of NKp30 with its ligand B7–H6 present on DCs and salivary gland epithelial cells induces IFN-γ secretion by NK cells, which enhances focal inflammation and cellular damage (78). NK22 cells—a subset of NK cells that produce IL-22—have also been detected in the salivary glands of pSS patients (79). Elevated IL-22 in the salivary glands acting synergistically with IL-17 and IL-23 plays a proinflammatory role in pSS (79, 80). On the other hand, NK22 cells secrete B cell-activating factor, which may contribute to B cell-mediated immunity in the development of pSS (81). Thus, NK cell subsets in the salivary gland have a pathogenic role in pSS. However, it was recently reported that a TNF-related apoptosis-inducing ligand (TRAIL)+ NK cells exerted a protective function in chronic infection-associated pSS by specifically eliminating activated CD4+ T cells in salivary glands (82). Taken together, most of the current data suggests a disease-promoting role for NK cells, while some NK cell subsets may have a protective function against pSS development.
SSc, also known as scleroderma, is an immune-associated multisystem rheumatic disease characterized by vasculopathy, immune activation, and tissue fibrosis of skin and internal organs caused by abnormal production and deposition of collagen (83, 84). Although SSc is uncommon, affecting about 1 in 10,000 people worldwide, it has high morbidity and mortality (85). SSc can be divided into limited and diffuse cutaneous forms (lcSSc and dcSSc, respectively) (86).
The etiology and progression of SSc depend on multiple factors including immune activation (87, 88). Toll-like receptor (TLR)-mediated DC activation may be responsible for the variable immune responses observed in patients with different SSc phenotypes. Type I IFN-induced TLR expression by DCs may also contribute to the pathogenesis of SSc (89–91). However, the role of NK cells in SSc is not fully understood. Changes in the percentages, phenotype, and functions of peripheral blood NK cells have been observed in patients with SSc (92, 93); one study suggested that these changes were dependent on the SSc subtype, with increased NK cell numbers in dcSSc but not lcSSc (93). The activating receptor killer cell immunoglobulin-like receptor (KIR), two Ig domains and short cytoplasmic tail (KIR2DS)2 but not the corresponding inhibitory receptor was shown to be expressed on NK cells from SSc patients (94). Additionally, a linear increase in activated CD56bright NK cells with SSc progression from early to definite SSc was demonstrated following TLR1/2 stimulation, highlighting the contribution of NK cells to SSc onset (95). NK cells secrete cytokines such as TNF-α, IL-6, and macrophage inflammatory protein (MIP)-1α and crosstalk with other immune cell types including DCs in response to TLR stimulation, thereby aggravating inflammation (96). Although the number of circulating NK cells is preserved in SSc, the cells show an unusual phenotype with decreased expression of chemokine receptors [CX3C chemokine receptor (CX3CR)1 and CXCR4], NKG2D, and CD69 (92). The reduced percentage of circulating CX3CR1+ NK cells in SSc patients may be due to their recruitment to target tissues in response to upregulation of CX3CL1 and NKG2D ligands in the inflamed endothelium. Moreover, NK cells from SSc patients induce endothelial cell activation, possibly exacerbating endothelial injury and contributing to SSc pathogenesis (92). A recent cytometry by time-of-flight study quantifying the proportions and phenotypes of circulating immune cells in patients with systemic autoimmune disease (including SSc) identified 12 cell populations that were altered compared to healthy controls, including a notable decrease in the size of the CD56hi NK cell fraction (97). Thus, significant changes in immune cell populations occur in early-stage SSc, although how this contributes to disease onset remains to be determined. Most of the available evidence suggests a pathogenic role for NK cells in SSc that is related to disease stage and subtype (i.e., early or late stage of lcSSc and dcSSc).
RA, one of the most prevalent chronic inflammatory diseases, is characterized by persistent synovitis, production of autoantibodies (especially against rheumatoid factor and citrullinated peptide), and cartilage and bone destruction, which lead to systemic complications including pulmonary, cardiovascular, skeletal, and psychological disorders (98, 99). The pathogenesis of RA is heterogeneous and complex and involves genetic and environmental factors, although the detailed mechanisms are not fully understood.
Genome-wide analyses have suggested that immune regulatory mechanisms underlie RA (100). RA results from the loss of immune self-tolerance, autoantigen presentation, and aberrant inflammatory cytokine production caused by abnormal activation of innate and adaptive immune systems (101). Although adaptive immunity (e.g., autoantibody production mediated by CD4+ T cells and B cells) predominates, innate immune cells (e.g., DCs, macrophages, mast cells, and NK cells) have also been implicated in RA pathogenesis (98, 102, 103).
Single-nucleotide polymorphisms in NK cell-related genes—especially NKG2D and MHC class I polypeptide-related sequence A (MICA)—were shown to be associated with RA susceptibility and severity (104–107). Early studies reported conflicting findings regarding the roles of NK cells in RA, which may be attributable to the fact that peripheral but not local synovial NK cells were examined; moreover, the different NK subsets were not distinguished, and the data were derived from different cohorts (108–110). The total numbers and percentages of peripheral NK cells were shown to be abnormally elevated in patients with active RA; this along with disease activity, autoantibody levels, and Th17/regulatory T cell (Treg) imbalance was correlated with increased serum IL-2 level (111). Higher percentages of peripheral NK cells were also found to be associated with elevated serum IL-15 level in RA patients compared to healthy controls. Circulating NK cells from RA patients showed decreased expression of the activating receptor NKp46 and higher expression of the inhibitory receptors CD158b and CD158e, which was associated with an impaired response to IL-15 (112). Decreased numbers of total peripheral NK cells and CD56dim NK cells, but not the CD56bright population, was observed in seropositive (i.e., positive for anticyclic citrullinated peptide antibodies and/or rheumatoid factor) patients with early RA (113). A decreased number of CD56dim NK cells was associated with CD16-triggered NK cell apoptosis, and CD16 also triggered IFN-γ and TNF-α production by CD56dim NK cells, thereby contributing to systemic inflammation (113).
Many studies have reported the accumulation of NK cells in the inflamed joints of RA patients, particularly in the active stage of the disease (114–116). The synovial CD56bright NK cell population in inflamed joints expands and rapidly secretes IFN-γ in response to monocyte/macrophage-secreted IL-12, IL-15, and IL-18; IFN-γ activates macrophages and promotes TNF-α production (114, 115). The interaction between NK cells and monocytes/macrophages may constitute a positive feedback loop that leads to uncontrolled, persistent inflammation in RA. In addition to IFN-γ and TNF-α, GM-CSF plays an important role in RA (117, 118). Recently, synovial NK cells were shown to potentiate inflammatory arthritis through secretion of GM-CSF in mice (119). Synovial NK cells produce GM-CSF in an IL-18–dependent manner, which promotes neutrophil infiltration into inflamed joints and persistent arthritis. The suppressor of cytokine signaling (SOCS) family member cytokine-inducible SH2-containing protein (CIS) was identified as a direct negative regulator of GM-CSF signaling, suggesting that it can be a therapeutic target (119). Synovial fluid NK cells express high levels of the activating receptors NKG2D, DNAX accessory molecule (DNAM)-1, NKp44, and NKp46 and inhibitory receptor CD94/NKG2A, while synovial fibroblasts express multiple ligands for NK cell receptors. These activating receptors mediate the cytotoxic effects of NK cells on synovial fibroblasts, thus promoting local joint inflammation in RA (120). Synovial fluid NK cells also express high levels of C-C chemokine receptor (CCR)6, which promotes NK cell migration to the inflamed joint (121). An NK22 subset was identified in the synovial fluid of RA patients that plays an important role in the pathogenesis of RA by secreting IL-22 and TNF-α, which enhanced the proliferation of fibroblast-like synoviocytes (121, 122).
Most of the existing evidence indicates that synovial NK cells exert pathogenic effects in RA by directly promoting cell or tissue injury either by inducing cytotoxicity (e.g., in synovial fibroblasts) expressing activating ligands for NK cell receptors or by secreting inflammatory cytokines (e.g., IFN-γ, TNF-α, GM-CSF, and IL-22) that induce the activation of macrophages and neutrophils, thereby indirectly aggravating inflammation and tissue damage. The contribution of distinct NK subsets and their crosstalk with other cells in the inflammatory microenvironment at different stages of RA development await further study.
ALD is a chronic liver disorder caused by the loss of tolerance to self-antigens in the liver. There are 3 types of ALD—namely, autoimmune hepatitis (AIH), primary sclerosing cholangitis (PSC), and PBC (123)—that have similar pathogenesis but different patterns of liver injury. AIH is characterized by infiltration of inflammatory cells around the portal tracts, which causes interface hepatitis. In PSC, the large hepatic bile ducts are targeted, leading to biliary tree obliteration, biliary cirrhosis, and portal hypertension. Damage to the small bile ducts in PBC results in portal tract destruction and biliary cirrhosis (123).
Adaptive immune responses are responsible for the progressive destruction of liver parenchyma in ALD. However, innate immune cells (especially NK cells) also contribute to ALD physiopathology (7). NK cells are enriched in the liver, accounting for up to 30–50% of total liver lymphocytes (124, 125). A direct role for NK cells in hepatocellular damage has been reported in AIH (126), and recent data from both animal models and clinical studies have provided further evidence for the contributions of different functional NK cell subsets in AIH. In a mouse model of polyinosinic:polycytidylic acid-induced hepatitis, which mimics human AIH histopathology, NK cells accumulated and were activated in the liver and caused hepatocyte injury, suggesting a pathogenic role for NK cells in AIH (127). In a concanavalin A-induced AIH model, IL-17C enhanced IL-2 expression in intrahepatic CD4+ T cells, which promoted NK cell activation and increased the number of liver NK cells and their activation; this was mostly due to a change in the CD3−NK1.1+NKP46+ NK cell fraction post treatment. There was also a significant upregulation of the surface markers CD25 and CD69 in liver NK cells, indicating that the change in the functional subset of NK cells in this model was induced by the cytokine environment (128). Additionally, murine CD49a+DX5− lrNK cells were shown to play a pathogenic role during viral infection by inhibiting virus-specific T cell immunity in a programmed cell death protein (PD)-1/programmed death ligand (PD-L)1–dependent manner (129). Conversely, CD49a−DX5+ cNK cells—but not CD49a+DX5− lrNK cells—in mice exerted a protective effect during acute infection with the hepatitis-B virus by promoting the antiviral activity of CD8+ T cells via IFN-γ secretion (130). Thus, distinct hepatic NK cell subsets (especially circulating and liver-resident populations) have different functions during AIH development. A clinical study found enhanced liver infiltration of NK cells in acute AIH patients, with higher numbers of CD56bright but not CD56dim NK cells before treatment that were markedly reduced after corticosteroid therapy. Infiltrated NK cells also expressed a high level CD161—a Th17 plasticity-associated marker—as well as the activation molecules perforin and granzyme B, and were also more resistant to Treg-mediated suppression, suggesting that inadequate regulation by exhausted Forkhead box (FOX)P3+ Tregs was responsible for the functional bias of NK cells and their pathogenic effects in AIH (131). A study combining clinical samples and experimental AIH demonstrated that a decrease in the size of the CD56dim NK cell fraction in peripheral blood was negatively correlated with disease progression in AIH patients. In an animal model, hepatic accumulation of CXCR3− cNK cells—equivalent to CD56dim NK cells in humans—was accompanied by a reduction in the numbers of these cells in the periphery (blood, spleen, and bone marrow); this peripheral cNK cell type redistribution was associated with AIH progression (132). The same authors also observed that although the number of CD49a+ lrNK cells did not increase significantly, they showed an activation phenotype identical to cNK cells and contributed to AIH injury. These data imply that targeting NK cell activation and migration is a potential therapeutic strategy for AIH.
In patients with PBC, the number of cytotoxic NK cells was shown to be increased in peripheral blood and liver. These NK cells kill biliary epithelial cells in a TLR4-, TLR3-, or TRAIL-dependent manner (133–135). Given their expression of TRAIL, lrNK cells may be primarily responsible for PBC pathogenesis. Infiltration of NK cells into the liver was dependent on the C-X-C chemokine receptor (CXCR)1/C-X-C motif ligand (CXCL)8 axis and involved CD56dim cells, which exerted cytotoxic effects against autologous biliary epithelial cells (136). In patients with PSC, the number of NK cells was increased in peripheral blood but not the liver (137, 138). The expression of ligands of inhibitory KIRs was significantly reduced in PSC patients (139), while that of ligands for activating NK cell receptors was upregulated (140, 141). These data strongly suggest that NK cells are activated and cytotoxic in PSC. A recent study reported higher proportions of CCR7+ NK cells in the peripheral blood and liver of PSC patients based on increased expression of the CCR7 ligand CCL21, suggesting that CCR7+ NK cell infiltration contributes to PSC pathogenesis (142). Data from the same study also showed a significantly higher number of CXCR3+ NK cells in peripheral blood but not in the liver of PSC patients. In contrast to the earlier view that NK cells play a pathogenic role, it was recently demonstrated that lrNK cells have an immunosuppressive function in the pathogenesis of PSC. In a dominant-negative TGF-β receptor (TGFβR)II mouse model that mimics key phenotypic features of human PBC, the progression of PSC was negatively correlated with lrNK cell counts. It was further demonstrated that DX5−CD11chi lrNK cells colocalized with and inhibited the proliferation of CD4+ T cells (143). These findings strongly suggest that liver-infiltrating NK cells participate in ALD development, although the detailed mechanisms require further investigation.
MS is a chronic disease of the central nervous system (CNS) (144) that results from immune-mediated inflammation, demyelination, and subsequent axonal damage (145); it is characterized by progressive motor disability and cognitive deficits, and affects younger adults (146). The incidence of MS is increasing worldwide. Despite decades of research, the etiology of MS is still unknown. Increasing evidence points to the critical role of autoimmunity in MS development and progression (144, 147), with many studies focusing on the contribution of T and B cells (144, 147, 148). In fact, therapies targeting T and B cells have demonstrated clinical success in the treatment of MS; these are reviewed in detail elsewhere (149–151).
MS was previously considered as an organ-specific T cell-mediated autoimmune disease (152, 153). However, therapies targeting B cells have also been effective (154). Data from both MS patients and experimental autoimmune encephalomyelitis (EAE), an animal model of MS, indicate that NK cells are associated with disease activity and therapeutic response in MS (155, 156), although their precise role is controversial On one hand, blockade of the interaction between the NK cell inhibitory receptor NKG2A and its ligand Qa-1 with an antibody that is equivalent to human leukocyte antigen (HLA)-E alleviated CNS inflammation in EAE by promoting the cytotoxic action of NK cells on T cells and microglia (157, 158). Enrichment of CNS-resident NK cells by treatment with an IL-2/IL-2 antibody complex blocked MS progression by suppressing the myelin-reactive Th17 response (142). The same study also demonstrated that NK cells created a Th17-polarizing cytokine environment by increasing the levels of IL-1β, TGF-β, TNF, and proinflammatory cytokines including MCP-1 and MIP-1α; IL-2 complex treatment reversed this effect, highlighting its therapeutic potential for MS (142). MS disease severity was negatively correlated with the accumulation of NK cells in the CNS, which involved CX3CR1/CX3CL1 signaling (159). The number of peripheral CD3−CD56+ NK cells was higher IFN-β-treated RR-MS patients compared to untreated patients although NKG2D+CD3−CD56+ NK cell count and endogenous IL-22 level in CD3−CD56+ NK cells were lower in untreated RR-MS and CIS patients than in treated RR-MS patients, indicating that NK cells have a therapeutic role in MS while IFN-β treatment may direct them toward a proinflammatory phenotype (40). On the other hand, NK cells were shown to exacerbate EAE by promoting the expansion of M1 macrophages and encephalitogenic T cells into the CNS via secretion of IFN-γ (143). Through a mechanism involving KIR two Ig domains and long cytoplasmic tail (KIR2DL)4-HLA-G–mediated conjugation of human NK cells and oligodendrocytes (OLs), activated NK cells were polarized to express IFN-γ and exert cytotoxic effects against OLs, suggesting a mechanism by which NK cells promote MS pathogenesis (160). Data from MS patients and an EAE model also support a pathogenic role for NK cells in MS development. In EAE, contact with NK cells induced the release of IL-15 by neural stem cells (NSCs), which promoted NK cell proliferation and survival, thus contributing to CNS damage. Especially during the later stages of EAE, reduced surface expression of Qa-1 on NSCs resulted in their killing by NK cells; depleting NK cells during this phase alleviated disease severity (161). Most of these studies focused on total NK cells, whether different NK cell subsets play distinct roles in MS pathology remains an open question.
The major NK cell populations in the cerebrospinal fluid (CSF) and CNS are CD56bright NK cells (162, 163). Along with a significant increase in total CD3−CD56+ NK cells, the regulatory CD56bright NK cell fraction was also increased and persisted following alemtuzumab treatment (164). In relapsing–remitting (RR)-MS–the most common disease subtype—and clinically isolated syndrome, dysfunction of CD3−CD16+CD56dim (CD56dim) and CD3−CD16−CD56bright (CD56bright) NK cell subsets was shown to be associated with disease progression; specifically, the size of the latter cell population was significantly increased in IFN-β-treated RR-MS patients compared to untreated patients and healthy subjects (165). Another study demonstrated that a lower NK/CD4+ T cell ratio in IFN-β-treated early RR-MS patients had prognostic value for disease activity compared to the CD56dim subset, while CD56bright NK cell count was negatively correlated with CD4+ T cell and more specifically, CD4+IL-17A+ T cell numbers (166). These data indicate that CD56bright NK cells have an immune regulatory function in MS that involves suppressing activated T cells and T cell subsets such as Th17 cells; moreover, CD56dim NK cells appear to play a role in reducing disease activity (166). Interestingly, recent data from trials of various MS drugs such as IFN-β, fingolimod, and daclizumab indicate that the size of NK cell populations—specifically CD56bright cells—is increased upon treatment (167, 168). A mass cytometry-based investigation of circulating CD56bright cells in patients with RR-MS treated with daclizumab beta revealed an upregulation of CD56 in total NK cells along with multiple phenotypic changes in the CD56bright cell population (169). One study examining the roles and biological features of CD27high and CD27low/− NK cells during the pre-disease onset stage in EAE found that the numbers of CD27low/− NK cells in the spleen, lymph nodes, and bone marrow were increased whereas the number of CD27high cells was decreased; this was accompanied by enhanced cytotoxicity of CD27low/− NK cells and reduced IFN-γ production of CD27high NK cells. Adoptive transfer of CD27low/− NK cells aggravated EAE; and CD27high but not CD27low/− cells inhibited CD4+ T cell proliferation and Th17 cell differentiation. Moreover, these 2 subsets exhibited distinct roles in inducing the maturation of antigen-presenting bone marrow-derived macrophages and DCs. Thus, CD27+ NK cell subsets have different functions at the early stage of MS (170). Regulating CD56+ NK cell subsets is a potential therapeutic strategy for preventing MS onset.
NK cells contribute to MS pathogenesis by causing direct damage to CNS components (e.g., OLs) or exerting regulatory effects on autologous T cells (Th17 cells) by modulating the expression of activating and inhibitory receptors or secreting cytokines (IFNγ, IL-15, etc). Whether the functions of NK cells in MS are beneficial or detrimental is an open question, as most studies were conducted on peripheral NK cells rather than local tissue-resident or accumulated NK cells and have yielded conflicting findings. Novel experimental approaches are needed to evaluate the complex dynamics of NK cell subsets during MS development and their potential as therapeutic targets.
T1DM is a typical organ-specific disease characterized by immune-mediated destruction of insulin-producing β cells in pancreatic islets (171), resulting in the loss of control of blood glucose levels even with insulin replacement therapy; this can in turn lead to ketoacidosis, severe hypoglycemia, and secondary complications (172). The innate and adaptive immune responses are involved in the destruction of pancreatic β cells (173–176), and recent studies have focused on the possible roles of NK cells in the initiation and progression of T1DM (177–180).
Evidence for the contribution of NK cells to the pathogenesis of T1DM has come from experimental models [e.g., BioBreeding diabetes-prone rat and non-obese diabetic (NOD) mouse] and clinical studies. NK cells are among the first immune cells to migrate to pancreatic islets (181, 182) and are involved in all stages of T1DM (183–185). The destructive effect of NK cells on pancreatic β cells was demonstrated by the decreased inflammation of pancreatic islets and T1DM remission induced by NK cell depletion (181, 186). Pancreatic NK cells of NOD mice with an inflammatory phenotype were found to express high levels of CD69 and CD25 and a low level of CD62 ligand (CD62L), suggesting their involvement in the development of T1DM (187). NK cells were also shown to contribute to the elimination of pancreatic β cells in an NKp46-dependent manner, leading to T1DM development (182). In NOD mice expressing an impaired NKG2D receptor in peripheral NK cells, exposure of pancreatic islet cells to the NKG2D ligand RAE-1 resulted in the downregulation of NKG2D and reduced NK cell cytotoxicity and IFN-γ secretion (188). NK cells in pancreatic islets were found to indirectly contribute to the destruction of pancreatic β cells by facilitating T cell activation (6, 189, 190). However, other studies have suggested that NK cells have a protective role in T1DM. For example, it was reported that NK cells limit the destruction of pancreatic β cells and contribute to T1DM remission by decreasing the numbers of autoreactive cytotoxic T cells (191); and a later study showed that increasing the proportion of NKG2D+ NK cells and inducing IFN-γ secretion in this population which enhancing the protective effect of complete Freund's adjuvant (CFA) in NOD mice (192). The mechanism underlying the modulation of NK cell function in the CFA treatment model is not clear, because the reason for the increased proportion of NK cells in the periphery and whether this influences the infiltration or functional status of pancreatic NK cells are unknown. NK1.1+/c-Kit+ NK cells were identified in the spleen, lymph, and islets of NOD mice that exerted direct cytolytic activity against activated insulin-specific CD8+ T cells in a PD-1/PD-L1–dependent manner (193). Adoptive transfer of these NK cells delayed diabetes development, suggesting an immunoregulatory and protective role for NK cells in T1DM (179, 193, 194).
Clinical data on the functions of NK cells in T1DM patients are scarce and ambiguous (179, 180, 195). Nonetheless, NK cells are known to play an important role in the immunopathogenesis of T1DM (196). A reduced number and decreased cytolytic activity of NK cells along with downregulation of activating receptors (e.g., NKG2D, NKp46, NKp30) or increased expression of inhibitory receptors (e.g., KIR2DL3) in peripheral blood mononuclear cells have been reported in T1DM patients (182, 195, 197–200), highlighting an association between NK cells and T1DM disease stage or risk (42, 201). A decrease in cytolytic activity of NK cells toward group B coxsackievirus (CV-B)–infected pancreatic β cells contributed to the persistence of CV-B and triggered autoimmunity in T1DM patients (197, 202). However, other studies found no correlation between T1DM clinical status and abnormalities in the number or function of NK cells (203, 204). One report showed that NK cells were rarely detected even in heavily inflamed pancreatic islets of T1DM patients, suggesting that they are not required for the death of pancreatic β cells (205). A bias in functional NK cell subsets has been observed that may be correlated with the development and progression of T1DM; although the number of CD3−CD56+ NK cells is reduced in patients with longstanding T1DM, there were no differences in the ratio of CD56high/CD16− and CD56low/CD16+ NK cells between patients with longstanding or recent-onset T1DM and controls (195). Newly diagnosed young T1DM patients had fewer NK cells and effector CD56dimCD16+ and CD56dimCD16− NK cell subsets compared to controls; moreover, T1DM patients with diabetic ketoacidosis (DKA) had fewer CD56brightCD16− regulatory NK cells compared to patients without DKA (200). Hence, the relative proportions of NK cell subsets may be altered depending on the degree of metabolic impairment. Two novel NK cell subsets (CD16+CD8+ and CD16+CD8−) were identified in children at high risk of T1DM using dimensional reduction and computational unsupervised clustering approaches; and further analyses revealed that CD16+CD8+ (as CD16+CD8+CXCR3+) and CD16+CD8+CXCR3+CD11c+ NK cells were more abundant in high-risk individuals. However, whether CD11c expression reflects the activation status of NK cells in autoimmunity is unclear. The inconsistent results obtained in different studies may be attributable to the use of NK cell samples from peripheral blood, the lack of human pancreas specimens and functional markers of specific NK subsets, and the limited knowledge of the translocation of NK cells from peripheral to local organs during T1DM development.
The existing evidence demonstrates that NK cells are involved in the development of autoimmune diseases. The inflammatory microenvironment facilitates the migration of circulating NK cells into inflamed tissues and shapes the effector function of infiltrating and trNK cells, which involves upregulation of activating receptors and downregulation of inhibitory receptors, cell activation, and enhanced cytolytic activity and cytokine production (e.g., IFN-γ, TNF-α, and GM-CSF) leading to direct cell or tissue injury. NK cells can also bind autoantibodies through CD16 (FcγRIIIa) to mediate antibody-dependent cellular cytotoxicity. The cytokines secreted by NK cells recruit and activate other immune cells such as macrophages, neutrophils, and autoreactive T and B cells, thereby indirectly enhancing inflammation and tissue damage. However, some NK cells subsets may have a protective role in autoimmune diseases that involves direct killing of immature DCs and autoreactive T and B cells, or secretion of IL-10 to block the activation and effector functions of autoreactive T and B cells. The protective and pathogenic mechanisms of NK cells in autoimmune diseases are summarized in Figure 2.
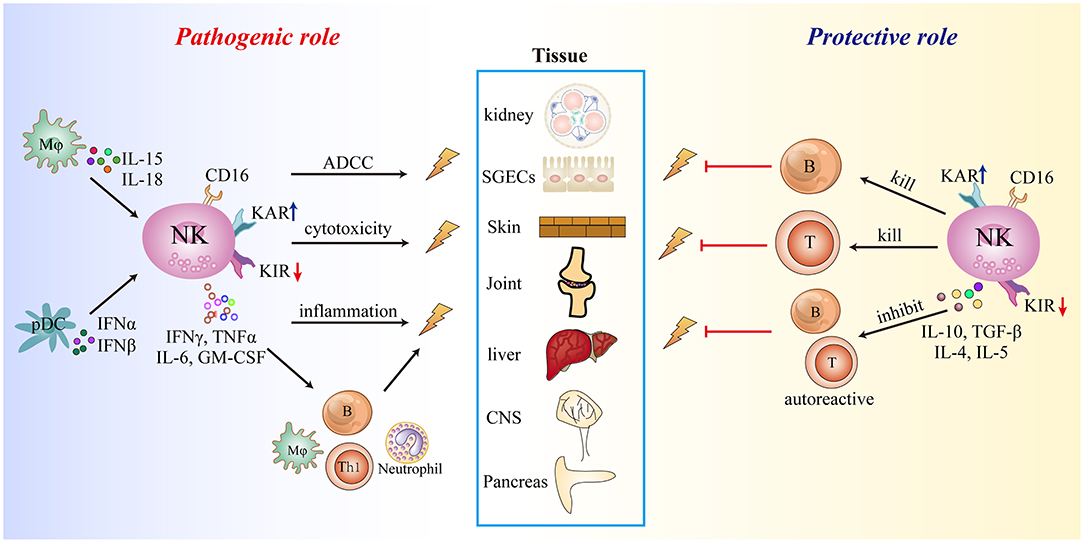
Figure 2. Summary of the role of NK cells in autoimmune diseases. KAR, killer cell-activating receptor; KIR, killer cell immunoglobulin-like inhibitory receptor.
There are many open questions regarding the precise functions and mechanisms of NK cells in autoimmune diseases. Based on current data, it is difficult to reconcile the contradictory roles (protective or pathogenic) of NK cells even in a single disease. Some early studies focused on bulk peripheral NK cells rather than specific NK cell subsets, especially local tissue-infiltrating or trNK cells. Single-cell RNA sequencing, mass spectrometry, and bioinformatics approaches have aided investigations of NK cell heterogeneity across different tissues (including diseased vs. healthy tissue) and the identification of distinct NK cell subsets involved in pro- and anti-inflammatory responses. The type of NK cell that accumulates in a tissue may determine the ultimate outcome of NK cell-mediated immune responses. Moreover, the tissue-specific microenvironment (e.g., cytokines, chemokines, ligands of NK cell receptors, and apoptosis-related molecules) and crosstalk between NK cells and other immune or stromal cells may shape the properties and functions of particular NK cell subsets. Notably, NK cells may have both beneficial or detrimental effects in the same autoimmune disease at different stages (e.g., onset, progression, relapse, or remission). Thus, future investigations should focus on tissue-specific NK cells and their interactions with other cells at the site of inflammation during different stages of disease development. Elucidating the precise roles of NK cells in disease initiation, progression, and resolution will provide insight into the pathogenic mechanisms of autoimmune diseases and that can guide the design of new therapeutic interventions.
ML, SL, and CZ were involved in the search and analysis of the literature, design and writing of the manuscript, and revision of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81873883, 82000525, 91842305, and 81771686), and the Medical and health science and Technology Development Plan of Shandong Province (2018WS065).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Herzog S, Jumaa H. Self-recognition and clonal selection: autoreactivity drives the generation of B cells. Curr Opin Immunol. (2012) 24:166–72. doi: 10.1016/j.coi.2012.02.004
2. Lee S, Ko Y, Kim TJ. Homeostasis and regulation of autoreactive B cells. Cell Mol Immunol. (2020) 17:561–9. doi: 10.1038/s41423-020-0445-4
3. Wang L, Wang FS, Gershwin ME. Human autoimmune diseases: a comprehensive update. J Intern Med. (2015) 278:369–95. doi: 10.1111/joim.12395
4. Durcan L, O'Dwyer T, Petri M. Management strategies and future directions for systemic lupus erythematosus in adults. Lancet. (2019) 393:2332–43. doi: 10.1016/S0140-6736(19)30237-5
5. Segerberg F, Lundtoft C, Reid S, Hjorton K, Leonard D, Nordmark G, et al. Autoantibodies to killer cell immunoglobulin-like receptors in patients with systemic lupus erythematosus induce natural killer cell hyporesponsiveness. Front Immunol. (2019) 10:2164. doi: 10.3389/fimmu.2019.02164
6. Berhani O, Glasner A, Kahlon S, Duev-Cohen A, Yamin R, Horwitz E, et al. Human anti-NKp46 antibody for studies of NKp46-dependent NK cell function and its applications for type 1 diabetes and cancer research. Eur J Immunol. (2019) 49:228–41. doi: 10.1002/eji.201847611
7. Hudspeth K, Pontarini E, Tentorio P, Cimino M, Donadon M, Torzilli G, et al. The role of natural killer cells in autoimmune liver disease: a comprehensive review. J Autoimmun. (2013) 46:55–65. doi: 10.1016/j.jaut.2013.07.003
8. O'Brien KL, Finlay DK. Immunometabolism and natural killer cell responses. Nat Rev Immunol. (2019) 19:282–90. doi: 10.1038/s41577-019-0139-2
9. Sivori S, Vacca P, Del Zotto G, Munari E, Mingari MC, Moretta L. Human NK cells: surface receptors, inhibitory checkpoints, and translational applications. Cell Mol Immunol. (2019) 16:430–41. doi: 10.1038/s41423-019-0206-4
10. Chen S, Dong Z. NK cell recognition of hematopoietic cells by SLAM-SAP families. Cell Mol Immunol. (2019) 16:452–9. doi: 10.1038/s41423-019-0222-4
11. Schuster IS, Coudert JD, Andoniou CE, Degli-Esposti MA. “Natural regulators”: NK cells as modulators of T cell immunity. Front Immunol. (2016) 7:235. doi: 10.3389/fimmu.2016.00235
12. Tian Z, Gershwin ME, Zhang C. Regulatory NK cells in autoimmune disease. J Autoimmun. (2012) 39:206–15. doi: 10.1016/j.jaut.2012.05.006
13. Fu B, Tian Z, Wei H. Subsets of human natural killer cells and their regulatory effects. Immunology. (2014) 141:483–9. doi: 10.1111/imm.12224
14. Peng H, Jiang X, Chen Y, Sojka DK, Wei H, Gao X, et al. Liver-resident NK cells confer adaptive immunity in skin-contact inflammation. J Clin Invest. (2013) 123:1444–56. doi: 10.1172/JCI66381
15. Hammer Q, Romagnani C. About training and memory: NK-cell adaptation to viral infections. Adv Immunol. (2017) 133:171–207. doi: 10.1016/bs.ai.2016.10.001
16. O'Sullivan TE, Sun JC, Lanier LL. Natural killer cell memory. Immunity. (2015) 43:634–45. doi: 10.1016/j.immuni.2015.09.013
17. Chen Y, Tian Z, Peng H. Immunological memory: ILC1s come into view. Cell Mol Immunol. (2019) 16:895–6. doi: 10.1038/s41423-019-0311-4
18. Wang X, Peng H, Tian Z. Innate lymphoid cell memory. Cell Mol Immunol. (2019) 16:423–9. doi: 10.1038/s41423-019-0212-6
19. Jiao G, Wang B. NK cell subtypes as regulators of autoimmune liver disease. Gastroenterol Res Pract. (2016) 2016:6903496. doi: 10.1155/2016/6903496
20. Zhang C, Tian Z. NK cell subsets in autoimmune diseases. J Autoimmun. (2017) 83:22–30. doi: 10.1016/j.jaut.2017.02.005
21. Liu M, Zhang C. The role of innate lymphoid cells in immune-mediated liver diseases. Front Immunol. (2017) 8:695. doi: 10.3389/fimmu.2017.00695
22. Pesce S, Moretta L, Moretta A, Marcenaro E. Human NK cell subsets redistribution in pathological conditions: a role for CCR7 receptor. Front Immunol. (2016) 7:414. doi: 10.3389/fimmu.2016.00414
23. Freud AG, Mundy-Bosse BL, Yu J, Caligiuri MA. The broad spectrum of human natural killer cell diversity. Immunity. (2017) 47:820–33. doi: 10.1016/j.immuni.2017.10.008
24. Poli A, Michel T, Theresine M, Andres E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. (2009) 126:458–65. doi: 10.1111/j.1365-2567.2008.03027.x
25. Lopez-Verges S, Milush JM, Pandey S, York VA, Arakawa-Hoyt J, Pircher H, et al. CD57 defines a functionally distinct population of mature NK cells in the human CD56dimCD16+ NK-cell subset. Blood. (2010) 116:3865–74. doi: 10.1182/blood-2010-04-282301
26. Cichocki F, Grzywacz B, Miller JS. Human NK cell development: one road or many? Front Immunol. (2019) 10:2078. doi: 10.3389/fimmu.2019.02078
27. Vossen MT, Matmati M, Hertoghs KM, Baars PA, Gent MR, Leclercq G, et al. CD27 defines phenotypically and functionally different human NK cell subsets. J Immunol. (2008) 180:3739–45. doi: 10.4049/jimmunol.180.6.3739
28. Allan DSJ, Cerdeira AS, Ranjan A, Kirkham CL, Aguilar OA, Tanaka M, et al. Transcriptome analysis reveals similarities between human blood CD3(-) CD56(bright) cells and mouse CD127(+) innate lymphoid cells. Sci Rep. (2017) 7:3501. doi: 10.1038/s41598-017-03256-0
29. Marquardt N, Wilk E, Pokoyski C, Schmidt RE, Jacobs R. Murine CXCR3+CD27bright NK cells resemble the human CD56bright NK-cell population. Eur J Immunol. (2010) 40:1428–39. doi: 10.1002/eji.200940056
30. Fu B, Wang F, Sun R, Ling B, Tian Z, Wei H. CD11b and CD27 reflect distinct population and functional specialization in human natural killer cells. Immunology. (2011) 133:350–9. doi: 10.1111/j.1365-2567.2011.03446.x
31. Zhou J, Tian Z, Peng H. Tissue-resident NK cells and other innate lymphoid cells. Adv Immunol. (2020) 145:37–53. doi: 10.1016/bs.ai.2019.11.002
32. Sun H, Sun C, Xiao W, Sun R. Tissue-resident lymphocytes: from adaptive to innate immunity. Cell Mol Immunol. (2019) 16:205–15. doi: 10.1038/s41423-018-0192-y
33. Wang Y, Zhang C. the roles of liver-resident lymphocytes in liver diseases. Front Immunol. (2019) 10:1582. doi: 10.3389/fimmu.2019.01582
34. Cortez VS, Cervantes-Barragan L, Robinette ML, Bando JK, Wang Y, Geiger TL, et al. Transforming growth factor-beta signaling guides the differentiation of innate lymphoid cells in salivary glands. Immunity. (2016) 44:1127–39. doi: 10.1016/j.immuni.2016.03.007
35. O'Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, et al. Adipose-resident group 1 innate lymphoid cells promote obesity-associated insulin resistance. Immunity. (2016) 45:428–41. doi: 10.1016/j.immuni.2016.06.016
36. Sojka DK, Plougastel-Douglas B, Yang L, Pak-Wittel MA, Artyomov MN, Ivanova Y, et al. Tissue-resident natural killer (NK) cells are cell lineages distinct from thymic and conventional splenic NK cells. Elife. (2014) 3:e01659. doi: 10.7554/eLife.01659
37. Victorino F, Sojka DK, Brodsky KS, McNamee EN, Masterson JC, Homann D, et al. Tissue-Resident NK cells mediate ischemic kidney injury and are not depleted by anti-asialo-GM1 antibody. J Immunol. (2015) 195:4973–85. doi: 10.4049/jimmunol.1500651
38. Harmon C, Jameson G, Almuaili D, Houlihan DD, Hoti E, Geoghegan J, et al. Liver-derived TGF-beta maintains the eomes(hi)Tbet(lo) phenotype of liver resident natural killer cells. Front Immunol. (2019) 10:1502. doi: 10.3389/fimmu.2019.01502
39. Spada R, Rojas JM, Perez-Yague S, Mulens V, Cannata-Ortiz P, Bragado R, et al. NKG2D ligand overexpression in lupus nephritis correlates with increased NK cell activity and differentiation in kidneys but not in the periphery. J Leukoc Biol. (2015) 97:583–98. doi: 10.1189/jlb.4A0714-326R
40. Tahrali I, Kucuksezer UC, Akdeniz N, Altintas A, Uygunoglu U, Aktas-Cetin E, et al. CD3(-)CD56(+) NK cells display an inflammatory profile in RR-MS patients. Immunol Lett. (2019) 216:63–9. doi: 10.1016/j.imlet.2019.10.006
41. Tang Q, Ahn YO, Southern P, Blazar BR, Miller JS, Verneris MR. Development of IL-22-producing NK lineage cells from umbilical cord blood hematopoietic stem cells in the absence of secondary lymphoid tissue. Blood. (2011) 117:4052–5. doi: 10.1182/blood-2010-09-303081
42. Barcenilla H, Akerman L, Pihl M, Ludvigsson J, Casas R. Mass cytometry identifies distinct subsets of regulatory T cells and natural killer cells associated with high risk for type 1 diabetes. Front Immunol. (2019) 10:982. doi: 10.3389/fimmu.2019.00982
43. Agmon-Levin N, Theodor E, Segal RM, Shoenfeld Y. Vitamin D in systemic and organ-specific autoimmune diseases. Clin Rev Allergy Immunol. (2013) 45:256–66. doi: 10.1007/s12016-012-8342-y
44. Nusbaum JS, Mirza I, Shum J, Freilich RW, Cohen RE, Pillinger MH, et al. Sex differences in systemic lupus erythematosus: epidemiology, clinical considerations, and disease pathogenesis. Mayo Clin Proc. (2020) 95:384–94. doi: 10.1016/j.mayocp.2019.09.012
45. Salem D, Subang R, Kuwana M, Levine JS, Rauch J. T cells from induced and spontaneous models of SLE recognize a common T cell epitope on beta2-glycoprotein I. Cell Mol Immunol. (2019) 16:685–93. doi: 10.1038/s41423-018-0013-3
46. Wu BX, Zhao LD, Zhang X. CXCR4 and CXCR5 orchestrate dynamic germinal center reactions and may contribute to the pathogenesis of systemic lupus erythematosus. Cell Mol Immunol. (2019) 16:724–6. doi: 10.1038/s41423-019-0244-y
47. Takeda K, Dennert G. The development of autoimmunity in C57BL/6 lpr mice correlates with the disappearance of natural killer type 1-positive cells: evidence for their suppressive action on bone marrow stem cell proliferation, B cell immunoglobulin secretion, and autoimmune symptoms. J Exp Med. (1993) 177:155–64. doi: 10.1084/jem.177.1.155
48. Suarez-Fueyo A, Bradley SJ, Katsuyama T, Solomon S, Katsuyama E, Kyttaris VC, et al. Downregulation of CD3zeta in NK cells from systemic lupus erythematosus patients confers a proinflammatory phenotype. J Immunol. (2018) 200:3077–86. doi: 10.4049/jimmunol.1700588
49. Henriques A, Teixeira L, Ines L, Carvalheiro T, Goncalves A, Martinho A, et al. NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol. (2013) 32:805–13. doi: 10.1007/s10067-013-2176-8
50. Spada R, Rojas JM, Barber DF. Recent findings on the role of natural killer cells in the pathogenesis of systemic lupus erythematosus. J Leukoc Biol. (2015) 98:479–87. doi: 10.1189/jlb.4RU0315-081RR
51. Liu M, Liu J, Zhang X, Xiao Y, Jiang G, Huang X. Activation status of CD56(dim) natural killer cells is associated with disease activity of patients with systemic lupus erythematosus. Clin Rheumatol. (2020) doi: 10.1007/s10067-020-05306-x
52. Huang Z, Fu B, Zheng SG, Li X, Sun R, Tian Z, et al. Involvement of CD226+ NK cells in immunopathogenesis of systemic lupus erythematosus. J Immunol. (2011) 186:3421–31. doi: 10.4049/jimmunol.1000569
53. Inoue A, Hasegawa H, Kohno M, Ito MR, Terada M, Imai T, et al. Antagonist of fractalkine (CX3CL1) delays the initiation and ameliorates the progression of lupus nephritis in MRL/lpr mice. Arthritis Rheum. (2005) 52:1522–33. doi: 10.1002/art.21007
54. Nakatani K, Yoshimoto S, Iwano M, Asai O, Samejima K, Sakan H, et al. Fractalkine expression and CD16+ monocyte accumulation in glomerular lesions: association with their severity and diversity in lupus models. Am J Physiol Renal Physiol. (2010) 299:F207–16. doi: 10.1152/ajprenal.00482.2009
55. Lin SJ, Kuo ML, Hsiao HS, Lee PT, Chen JY, Huang JL. Activating and inhibitory receptors on natural killer cells in patients with systemic lupus erythematosis-regulation with interleukin-15. PLoS ONE. (2017) 12:e0186223. doi: 10.1371/journal.pone.0186223
56. Lin SJ, Kuo ML, Hsiao HS, Lee PT, Lee WI, Chen JY, et al. Cytotoxic function and cytokine production of natural killer cells and natural killer T-like cells in systemic lupus erythematosis regulation with interleukin-15. Mediators Inflamm. (2019) 2019:4236562. doi: 10.1155/2019/4236562
57. Hudspeth K, Wang S, Wang J, Rahman S, Smith MA, Casey KA, et al. Natural killer cell expression of Ki67 is associated with elevated serum IL-15, disease activity and nephritis in systemic lupus erythematosus. Clin Exp Immunol. (2019) 196:226–36. doi: 10.1111/cei.13263
58. Arazi A, Rao DA, Berthier CC, Davidson A, Liu Y, Hoover PJ, et al. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol. (2019) 20:902–14. doi: 10.1038/s41590-019-0398-x
59. Nehar-Belaid D, Hong S, Marches R, Chen G, Bolisetty M, Baisch J, et al. Mapping systemic lupus erythematosus heterogeneity at the single-cell level. Nat Immunol. (2020) 21:1094–106. doi: 10.1038/s41590-020-0743-0
60. Cafaro G, Croia C, Argyropoulou OD, Leone MC, Orlandi M, Finamore F, et al. One year in review 2019: Sjogren's syndrome. Clin Exp Rheumatol. (2019) 37 Suppl 118:3–15.
61. Mariette X, Criswell LA. Primary Sjogren's syndrome. N Engl J Med. (2018) 378:931–9. doi: 10.1056/NEJMcp1702514
62. Ambrosi A, Wahren-Herlenius M. Update on the immunobiology of Sjogren's syndrome. Curr Opin Rheumatol. (2015) 27:468–75. doi: 10.1097/BOR.0000000000000195
63. Voulgarelis M, Tzioufas AG. Pathogenetic mechanisms in the initiation and perpetuation of Sjogren's syndrome. Nat Rev Rheumatol. (2010) 6:529–37. doi: 10.1038/nrrheum.2010.118
64. Srivastava A, Makarenkova HP. Innate immunity and biological therapies for the treatment of Sjogren's syndrome. Int J Mol Sci. (2020) 21:9172. doi: 10.3390/ijms21239172
65. Kiripolsky J, McCabe LG, Kramer JM. Innate immunity in Sjogren's syndrome. Clin Immunol. (2017) 182:4–13. doi: 10.1016/j.clim.2017.04.003
66. Kramer JM. Early events in Sjogren's syndrome pathogenesis: the importance of innate immunity in disease initiation. Cytokine. (2014) 67:92–101. doi: 10.1016/j.cyto.2014.02.009
67. Ming B, Wu T, Cai S, Hu P, Tang J, Zheng F, et al. The increased ratio of blood CD56(bright) NK to CD56(dim) NK is a distinguishing feature of primary Sjogren's syndrome. J Immunol Res. (2020) 2020:7523914. doi: 10.1155/2020/7523914
68. Davies R, Hammenfors D, Bergum B, Jakobsen K, Solheim M, Vogelsang P, et al. Patients with primary Sjogren's syndrome have alterations in absolute quantities of specific peripheral leucocyte populations. Scand J Immunol. (2017) 86:491–502. doi: 10.1111/sji.12622
69. Peng H, Tian Z. Diversity of tissue-resident NK cells. Semin Immunol. (2017) 31:3–10. doi: 10.1016/j.smim.2017.07.006
70. Hall JC, Casciola-Rosen L, Berger AE, Kapsogeorgou EK, Cheadle C, Tzioufas AG, et al. Precise probes of type II interferon activity define the origin of interferon signatures in target tissues in rheumatic diseases. Proc Natl Acad Sci USA. (2012) 109:17609–14. doi: 10.1073/pnas.1209724109
71. Gottenberg JE, Cagnard N, Lucchesi C, Letourneur F, Mistou S, Lazure T, et al. Activation of IFN pathways and plasmacytoid dendritic cell recruitment in target organs of primary Sjogren's syndrome. Proc Natl Acad Sci USA. (2006) 103:2770–5. doi: 10.1073/pnas.0510837103
72. Awada A, Nicaise C, Ena S, Schandene L, Rasschaert J, Popescu I, et al. Potential involvement of the IL-33-ST2 axis in the pathogenesis of primary Sjogren's syndrome. Ann Rheum Dis. (2014) 73:1259–63. doi: 10.1136/annrheumdis-2012-203187
73. Jung SM, Lee J, Baek SY, Lee JH, Lee J, Park KS, et al. The Interleukin 33/ST2 axis in patients with primary Sjogren syndrome: expression in serum and salivary glands, and the clinical association. J Rheumatol. (2015) 42:264–71. doi: 10.3899/jrheum.140234
74. Nocturne G, Mariette X. Advances in understanding the pathogenesis of primary Sjogren's syndrome. Nat Rev Rheumatol. (2013) 9:544–56. doi: 10.1038/nrrheum.2013.110
75. Nanke Y, Kobashigawa T, Yago T, Kawamoto M, Yamanaka H, Kotake S. Detection of IFN-gamma+IL-17+ cells in salivary glands of patients with Sjogren's syndrome and Mikulicz's disease: potential role of Th17*Th1 in the pathogenesis of autoimmune diseases. Nihon Rinsho Meneki Gakkai Kaishi. (2016) 39:473–7. doi: 10.2177/jsci.39.473
76. Mavragani CP, Crow MK. Activation of the type I interferon pathway in primary Sjogren's syndrome. J Autoimmun. (2010) 35:225–31. doi: 10.1016/j.jaut.2010.06.012
77. Ogawa Y, Shimizu E, Tsubota K. Interferons and dry eye in Sjogren's syndrome. Int J Mol Sci. (2018) 19:3548. doi: 10.3390/ijms19113548
78. Rusakiewicz S, Nocturne G, Lazure T, Semeraro M, Flament C, Caillat-Zucman S, et al. NCR3/NKp30 contributes to pathogenesis in primary Sjogren's syndrome. Sci Transl Med. (2013) 5:195ra96. doi: 10.1126/scitranslmed.3005727
79. Ciccia F, Guggino G, Rizzo A, Ferrante A, Raimondo S, Giardina A, et al. Potential involvement of IL-22 and IL-22-producing cells in the inflamed salivary glands of patients with Sjogren's syndrome. Ann Rheum Dis. (2012) 71:295–301. doi: 10.1136/ard.2011.154013
80. Sisto M, Lorusso L, Tamma R, Ingravallo G, Ribatti D, Lisi S. Interleukin-17 and−22 synergy linking inflammation and EMT-dependent fibrosis in Sjogren's syndrome. Clin Exp Immunol. (2019) 198:261–72. doi: 10.1111/cei.13337
81. Cella M, Otero K, Colonna M. Expansion of human NK-22 cells with IL-7, IL-2, and IL-1beta reveals intrinsic functional plasticity. Proc Natl Acad Sci USA. (2010) 107:10961–6. doi: 10.1073/pnas.1005641107
82. Schuster IS, Wikstrom ME, Brizard G, Coudert JD, Estcourt MJ, Manzur M, et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity. (2014) 41:646–56. doi: 10.1016/j.immuni.2014.09.013
83. Denton CP, Khanna D. Systemic sclerosis. Lancet. (2017) 390:1685–99. doi: 10.1016/S0140-6736(17)30933-9
84. Ramos PS, Silver RM, Feghali-Bostwick CA. Genetics of systemic sclerosis: recent advances. Curr Opin Rheumatol. (2015) 27:521–9. doi: 10.1097/BOR.0000000000000214
85. Orlandi M, Barsotti S, Lepri G, Codullo V, Di Battista M, Guiducci S, et al. One year in review 2018: systemic sclerosis. Clin Exp Rheumatol. (2018) 36 (Suppl. 113):3–23.
86. Lopez-Cacho JM, Gallardo S, Posada M, Aguerri M, Calzada D, Mayayo T, et al. Association of immunological cell profiles with specific clinical phenotypes of scleroderma disease. Biomed Res Int. (2014) 2014:148293. doi: 10.1155/2014/148293
87. van Bon L, Cossu M, Radstake TR. An update on an immune system that goes awry in systemic sclerosis. Curr Opin Rheumatol. (2011) 23:505–10. doi: 10.1097/BOR.0b013e32834b0dac
88. Fullard N, O'Reilly S. Role of innate immune system in systemic sclerosis. Semin Immunopathol. (2015) 37:511–7. doi: 10.1007/s00281-015-0503-7
89. van Bon L, Popa C, Huijbens R, Vonk M, York M, Simms R, et al. Distinct evolution of TLR-mediated dendritic cell cytokine secretion in patients with limited and diffuse cutaneous systemic sclerosis. Ann Rheum Dis. (2010) 69:1539–47. doi: 10.1136/ard.2009.128207
90. Farina GA, York MR, Di Marzio M, Collins CA, Meller S, Homey B, et al. Poly(I:C) drives type I IFN- and TGFbeta-mediated inflammation and dermal fibrosis simulating altered gene expression in systemic sclerosis. J Invest Dermatol. (2010) 130:2583–93. doi: 10.1038/jid.2010.200
91. Agarwal SK, Wu M, Livingston CK, Parks DH, Mayes MD, Arnett FC, et al. Toll-like receptor 3 upregulation by type I interferon in healthy and scleroderma dermal fibroblasts. Arthritis Res Ther. (2011) 13:R3. doi: 10.1186/ar3221
92. Benyamine A, Magalon J, Sabatier F, Lyonnet L, Robert S, Dumoulin C, et al. Natural killer cells exhibit a peculiar phenotypic profile in systemic sclerosis and are potent inducers of endothelial microparticles release. Front Immunol. (2018) 9:1665. doi: 10.3389/fimmu.2018.01665
93. Horikawa M, Hasegawa M, Komura K, Hayakawa I, Yanaba K, Matsushita T, et al. Abnormal natural killer cell function in systemic sclerosis: altered cytokine production and defective killing activity. J Invest Dermatol. (2005) 125:731–7. doi: 10.1111/j.0022-202X.2005.23767.x
94. Momot T, Koch S, Hunzelmann N, Krieg T, Ulbricht K, Schmidt RE, et al. Association of killer cell immunoglobulin-like receptors with scleroderma. Arthritis Rheum. (2004) 50:1561–5. doi: 10.1002/art.20216
95. Cossu M, van Bon L, Nierkens S, Bellocchi C, Santaniello A, Dolstra H, et al. The magnitude of cytokine production by stimulated CD56(+) cells is associated with early stages of systemic sclerosis. Clin Immunol. (2016) 173:76–80. doi: 10.1016/j.clim.2016.09.004
96. Barranco C. Systemic sclerosis: the future is CD56-bright. Nat Rev Rheumatol. (2016) 12:624. doi: 10.1038/nrrheum.2016.168
97. van der Kroef M, van den Hoogen LL, Mertens JS, Blokland SLM, Haskett S, Devaprasad A, et al. Cytometry by time of flight identifies distinct signatures in patients with systemic sclerosis, systemic lupus erythematosus and Sjogrens syndrome. Eur J Immunol. (2020) 50:119–29. doi: 10.1002/eji.201948129
98. McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Engl J Med. (2011) 365:2205–19. doi: 10.1056/NEJMra1004965
99. Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. (2016) 388:2023–38. doi: 10.1016/S0140-6736(16)30173-8
100. Wellcome Trust Case Control C. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. (2007) 447:661–78. doi: 10.1038/nature05911
101. Calabresi E, Petrelli F, Bonifacio AF, Puxeddu I, Alunno A. One year in review 2018: pathogenesis of rheumatoid arthritis. Clin Exp Rheumatol. (2018) 36:175–84.
102. Wu X. Innate lymphocytes in inflammatory arthritis. Front Immunol. (2020) 11:565275. doi: 10.3389/fimmu.2020.565275
103. Fang W, Zhang Y, Chen Z. Innate lymphoid cells in inflammatory arthritis. Arthritis Res Ther. (2020) 22:25. doi: 10.1186/s13075-020-2115-4
104. Mariaselvam CM, Tamouza R, Krishnamoorthy R, Charron D, Misra DP, Jain VK, et al. Association of NKG2D gene variants with susceptibility and severity of rheumatoid arthritis. Clin Exp Immunol. (2017) 187:369–75. doi: 10.1111/cei.12891
105. Iwaszko M, Swierkot J, Kolossa K, Jeka S, Wiland P, Bogunia-Kubik K. Influence of NKG2D genetic variants on response to anti-tnf agents in patients with rheumatoid arthritis. Genes. (2018) 9:64. doi: 10.3390/genes9020064
106. Kirsten H, Petit-Teixeira E, Scholz M, Hasenclever D, Hantmann H, Heider D, et al. Association of MICA with rheumatoid arthritis independent of known HLA-DRB1 risk alleles in a family-based and a case control study. Arthritis Res Ther. (2009) 11:R60. doi: 10.1186/ar2683
107. Elemam NM, Hachim MY, Hannawi S, Maghazachi AA. Differentially expressed genes of natural killer cells can distinguish rheumatoid arthritis patients from healthy controls. Genes. (2020) 11:492. doi: 10.3390/genes11050492
108. Johansson S, Berg L, Hall H, Hoglund P. NK cells: elusive players in autoimmunity. Trends Immunol. (2005) 26:613–8. doi: 10.1016/j.it.2005.08.008
109. Lunemann A, Lunemann JD, Munz C. Regulatory NK-cell functions in inflammation and autoimmunity. Mol Med. (2009) 15:352–8. doi: 10.2119/molmed.2009.00035
110. Deniz G, Erten G, Kucuksezer UC, Kocacik D, Karagiannidis C, Aktas E, et al. Regulatory NK cells suppress antigen-specific T cell responses. J Immunol. (2008) 180:850–7. doi: 10.4049/jimmunol.180.2.850
111. Li B, Guo Q, Wang Y, Su R, Gao C, Zhao J, et al. Increased serum interleukin-2 levels are associated with abnormal peripheral blood natural killer cell levels in patients with active rheumatoid arthritis. Mediators Inflamm. (2020) 2020:6108342. doi: 10.1155/2020/6108342
112. Lin SJ, Hsu CY, Kuo ML, Lee PT, Hsiao HS, Chen JY. Phenotypic and functional characterization of natural killer cells in rheumatoid arthritis-regulation with interleukin-15. Sci Rep. (2020) 10:5858. doi: 10.1038/s41598-020-62654-z
113. Chalan P, Bijzet J, Kroesen BJ, Boots AM, Brouwer E. Altered natural killer cell subsets in seropositive arthralgia and early rheumatoid arthritis are associated with autoantibody status. J Rheumatol. (2016) 43:1008–16. doi: 10.3899/jrheum.150644
114. Dalbeth N, Callan MF. A subset of natural killer cells is greatly expanded within inflamed joints. Arthritis Rheum. (2002) 46:1763–72. doi: 10.1002/art.10410
115. Shen Q, Beucler MJ, Ray SC, Rappleye CA. Macrophage activation by IFN-gamma triggers restriction of phagosomal copper from intracellular pathogens. PLoS Pathog. (2018) 14:e1007444. doi: 10.1371/journal.ppat.1007444
116. Yamin R, Berhani O, Peleg H, Aamar S, Stein N, Gamliel M, et al. High percentages and activity of synovial fluid NK cells present in patients with advanced stage active rheumatoid arthritis. Sci Rep. (2019) 9:1351. doi: 10.1038/s41598-018-37448-z
117. Becher B, Tugues S, Greter M. GM-CSF: from growth factor to central mediator of tissue inflammation. Immunity. (2016) 45:963–73. doi: 10.1016/j.immuni.2016.10.026
118. Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. (2016) 12:37–48. doi: 10.1038/nrrheum.2015.161
119. Louis C, Souza-Fonseca-Guimaraes F, Yang Y, D'Silva D, Kratina T, Dagley L, et al. NK cell-derived GM-CSF potentiates inflammatory arthritis and is negatively regulated by CIS. J Exp Med. (2020) 217:e20191421. doi: 10.1084/jem.20191421
120. Nielsen N, Pascal V, Fasth AE, Sundstrom Y, Galsgaard ED, Ahern D, et al. Balance between activating NKG2D, DNAM-1, NKp44 and NKp46 and inhibitory CD94/NKG2A receptors determine natural killer degranulation towards rheumatoid arthritis synovial fibroblasts. Immunology. (2014) 142:581–93. doi: 10.1111/imm.12271
121. Ren J, Feng Z, Lv Z, Chen X, Li J. Natural killer-22 cells in the synovial fluid of patients with rheumatoid arthritis are an innate source of interleukin 22 and tumor necrosis factor-alpha. J Rheumatol. (2011) 38:2112–8. doi: 10.3899/jrheum.101377
122. Zhu J, Jia E, Zhou Y, Xu J, Feng Z, Wang H, et al. Interleukin-22 secreted by NKp44+ natural killer cells promotes proliferation of fibroblast-like synoviocytes in rheumatoid arthritis. Medicine. (2015) 94:e2137. doi: 10.1097/MD.0000000000002137
123. Doherty DG. Immunity, tolerance and autoimmunity in the liver: a comprehensive review. J Autoimmun. (2016) 66:60–75. doi: 10.1016/j.jaut.2015.08.020
124. Peng H, Tian Z. NK cells in liver homeostasis and viral hepatitis. Sci China Life Sci. (2018) 61:1477–85. doi: 10.1007/s11427-018-9407-2
125. Shi FD, Ljunggren HG, La Cava A, Van Kaer L. Organ-specific features of natural killer cells. Nat Rev Immunol. (2011) 11:658–71. doi: 10.1038/nri3065
126. Kaneda K, Kurioka N, Seki S, Wake K, Yamamoto S. Pit cell-hepatocyte contact in autoimmune hepatitis. Hepatology. (1984) 4:955–8. doi: 10.1002/hep.1840040529
127. Dong Z, Wei H, Sun R, Hu Z, Gao B, Tian Z. Involvement of natural killer cells in PolyI:C-induced liver injury. J Hepatol. (2004) 41:966–73. doi: 10.1016/j.jhep.2004.08.021
128. Huang J, Yuan Q, Zhu H, Yin L, Hong S, Dong Z, et al. IL-17C/IL-17RE augments T cell function in autoimmune hepatitis. J Immunol. (2017) 198:669–80. doi: 10.4049/jimmunol.1600977
129. Zhou J, Peng H, Li K, Qu K, Wang B, Wu Y, et al. Liver-resident NK cells control antiviral activity of hepatic T Cells via the PD-1-PD-L1 axis. Immunity. (2019) 50:403–17.e4. doi: 10.1016/j.immuni.2018.12.024
130. Zheng M, Sun R, Wei H, Tian Z. NK cells help induce anti-hepatitis B virus CD8+ T Cell immunity in mice. J Immunol. (2016) 196:4122–31. doi: 10.4049/jimmunol.1500846
131. Jeffery HC, Braitch MK, Bagnall C, Hodson J, Jeffery LE, Wawman RE, et al. Changes in natural killer cells and exhausted memory regulatory T cells with corticosteroid therapy in acute autoimmune hepatitis. Hepatol Commun. (2018) 2:421–36. doi: 10.1002/hep4.1163
132. Xiao F, Ai G, Yan W, Wan X, Luo X, Ning Q. Intrahepatic recruitment of cytotoxic NK cells contributes to autoimmune hepatitis progression. Cell Immunol. (2018) 327:13–20. doi: 10.1016/j.cellimm.2017.12.008
133. Chuang YH, Lian ZX, Cheng CM, Lan RY, Yang GX, Moritoki Y, et al. Increased levels of chemokine receptor CXCR3 and chemokines IP-10 and MIG in patients with primary biliary cirrhosis and their first degree relatives. J Autoimmun. (2005) 25:126–32. doi: 10.1016/j.jaut.2005.08.009
134. Shimoda S, Harada K, Niiro H, Shirabe K, Taketomi A, Maehara Y, et al. Interaction between Toll-like receptors and natural killer cells in the destruction of bile ducts in primary biliary cirrhosis. Hepatology. (2011) 53:1270–81. doi: 10.1002/hep.24194
135. Liang Y, Yang Z, Li C, Zhu Y, Zhang L, Zhong R. Characterisation of TNF-related apoptosis-inducing ligand in peripheral blood in patients with primary biliary cirrhosis. Clin Exp Med. (2008) 8:1–7. doi: 10.1007/s10238-008-0149-z
136. Maghazachi AA. Role of chemokines in the biology of natural killer cells. Curr Top Microbiol Immunol. (2010) 341:37–58. doi: 10.1007/82_2010_20
137. Panasiuk A, Prokopowicz D, Zak J, Panasiuk B, Wysocka J. Lymphocyte subpopulations in peripheral blood in primary sclerosing cholangitis. Hepatogastroenterology. (2004) 51:1289–91.
138. Hata K, Van Thiel DH, Herberman RB, Whiteside TL. Natural killer activity of human liver-derived lymphocytes in various liver diseases. Hepatology. (1991) 14:495–503. doi: 10.1002/hep.1840140315
139. Karlsen TH, Boberg KM, Olsson M, Sun JY, Senitzer D, Bergquist A, et al. Particular genetic variants of ligands for natural killer cell receptors may contribute to the HLA associated risk of primary sclerosing cholangitis. J Hepatol. (2007) 46:899–906. doi: 10.1016/j.jhep.2007.01.032
140. Wiencke K, Spurkland A, Schrumpf E, Boberg KM. Primary sclerosing cholangitis is associated to an extended B8-DR3 haplotype including particular MICA and MICB alleles. Hepatology. (2001) 34(4 Pt 1):625–30. doi: 10.1053/jhep.2001.27543
141. Pollheimer MJ, Halilbasic E, Fickert P, Trauner M. Pathogenesis of primary sclerosing cholangitis. Best Pract Res Clin Gastroenterol. (2011) 25:727–39. doi: 10.1016/j.bpg.2011.10.009
142. Langeneckert AE, Lunemann S, Martrus G, Salzberger W, Hess LU, Ziegler AE, et al. CCL21-expression and accumulation of CCR7(+) NK cells in livers of patients with primary sclerosing cholangitis. Eur J Immunol. (2019) 49:758–69. doi: 10.1002/eji.201847965
143. Zhao ZB, Lu FT, Ma HD, Wang YH, Yang W, Long J, et al. Liver-resident NK cells suppress autoimmune cholangitis and limit the proliferation of CD4(+) T cells. Cell Mol Immunol. (2020) 17:178–89. doi: 10.1038/s41423-019-0199-z
144. Stys PK, Tsutsui S. Recent advances in understanding multiple sclerosis. F1000Res. (2019) 8:F1000 Faculty Rev-2100. doi: 10.12688/f1000research.20906.1
145. Karussis D. The diagnosis of multiple sclerosis and the various related demyelinating syndromes: a critical review. J Autoimmun. (2014) 48-49:134–42. doi: 10.1016/j.jaut.2014.01.022
146. Filippi M, Bar-Or A, Piehl F, Preziosa P, Solari A, Vukusic S, et al. Multiple sclerosis. Nat Rev Dis Primers. (2018) 4:43. doi: 10.1038/s41572-018-0041-4
147. Cavallo S. Immune-mediated genesis of multiple sclerosis. J Transl Autoimmun. (2020) 3:100039. doi: 10.1016/j.jtauto.2020.100039
148. Arneth B. Contributions of T cells in multiple sclerosis: what do we currently know? J Neurol. (2020). doi: 10.1007/s00415-020-10275-x. [Epub ahead of print].
149. Faissner S, Gold R. Efficacy and safety of the newer multiple sclerosis drugs approved since 2010. CNS Drugs. (2018) 32:269–87. doi: 10.1007/s40263-018-0488-6
150. Piehl F. Current and emerging disease-modulatory therapies and treatment targets for multiple sclerosis. J Intern Med. (2020). doi: 10.1111/joim.13215. [Epub ahead of print].
151. Lee DSW, Rojas OL, Gommerman JL. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat Rev Drug Discov. (2020) 15:1–21. doi: 10.1038/s41573-020-00092-2
152. Dhaeze T, Tremblay L, Lachance C, Peelen E, Zandee S, Grasmuck C, et al. CD70 defines a subset of proinflammatory and CNS-pathogenic TH1/TH17 lymphocytes and is overexpressed in multiple sclerosis. Cell Mol Immunol. (2019) 16:652–65. doi: 10.1038/s41423-018-0198-5
153. Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, et al. Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol. (2008) 172:146–55. doi: 10.2353/ajpath.2008.070690
154. Greenfield AL, Hauser SL. B-cell therapy for multiple sclerosis: entering an era. Ann Neurol. (2018) 83:13–26. doi: 10.1002/ana.25119
155. Van Kaer L, Postoak JL, Wang C, Yang G, Wu L. Innate, innate-like and adaptive lymphocytes in the pathogenesis of MS and EAE. Cell Mol Immunol. (2019) 16:531–9. doi: 10.1038/s41423-019-0221-5
156. Mimpen M, Smolders J, Hupperts R, Damoiseaux J. Natural killer cells in multiple sclerosis: a review. Immunol Lett. (2020) 222:1–11. doi: 10.1016/j.imlet.2020.02.012
157. Lu L, Ikizawa K, Hu D, Werneck MB, Wucherpfennig KW, Cantor H. Regulation of activated CD4+ T cells by NK cells via the Qa-1-NKG2A inhibitory pathway. Immunity. (2007) 26:593–604. doi: 10.1016/j.immuni.2007.03.017
158. Leavenworth JW, Schellack C, Kim HJ, Lu L, Spee P, Cantor H. Analysis of the cellular mechanism underlying inhibition of EAE after treatment with anti-NKG2A F(ab')2. Proc Natl Acad Sci USA. (2010) 107:2562–7. doi: 10.1073/pnas.0914732107
159. Huang D, Shi FD, Jung S, Pien GC, Wang J, Salazar-Mather TP, et al. The neuronal chemokine CX3CL1/fractalkine selectively recruits NK cells that modify experimental autoimmune encephalomyelitis within the central nervous system. FASEB J. (2006) 20:896–905. doi: 10.1096/fj.05-5465com
160. Banerjee PP, Pang L, Soldan SS, Miah SM, Eisenberg A, Maru S, et al. KIR2DL4-HLAG interaction at human NK cell-oligodendrocyte interfaces regulates IFN-gamma-mediated effects. Mol Immunol. (2019) 115:39–55. doi: 10.1016/j.molimm.2018.09.027
161. Liu Q, Sanai N, Jin WN, La Cava A, Van Kaer L, Shi FD. Neural stem cells sustain natural killer cells that dictate recovery from brain inflammation. Nat Neurosci. (2016) 19:243–52. doi: 10.1038/nn.4211
162. Rodriguez-Martin E, Picon C, Costa-Frossard L, Alenda R, Sainz de la Maza S, Roldan E, et al. Natural killer cell subsets in cerebrospinal fluid of patients with multiple sclerosis. Clin Exp Immunol. (2015) 180:243–9. doi: 10.1111/cei.12580
163. Han S, Lin YC, Wu T, Salgado AD, Mexhitaj I, Wuest SC, et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J Immunol. (2014) 192:2551–63. doi: 10.4049/jimmunol.1302884
164. Gilmore W, Lund BT, Li P, Levy AM, Kelland EE, Akbari O, et al. Repopulation of T, B, and NK cells following alemtuzumab treatment in relapsing-remitting multiple sclerosis. J Neuroinflammation. (2020) 17:189. doi: 10.1186/s12974-020-01847-9
165. Laroni A, Armentani E, Kerlero de Rosbo N, Ivaldi F, Marcenaro E, Sivori S, et al. Dysregulation of regulatory CD56(bright) NK cells/T cells interactions in multiple sclerosis. J Autoimmun. (2016) 72:8–18. doi: 10.1016/j.jaut.2016.04.003
166. Mimpen M, Muris AH, Rolf L, Gerlach O, Kuhle J, Hupperts R, et al. Prognostic value of natural killer cell/T cell ratios for disease activity in multiple sclerosis. Eur J Neurol. (2020) 28:901–9. doi: 10.1111/ene.14680
167. Gross CC, Schulte-Mecklenbeck A, Wiendl H, Marcenaro E, Kerlero de Rosbo N, Uccelli A, et al. Regulatory functions of natural killer cells in multiple sclerosis. Front Immunol. (2016) 7:606. doi: 10.3389/fimmu.2016.00606
168. Laroni A, Uccelli A. CD56bright natural killer cells: a possible biomarker of different treatments in multiple sclerosis. J Clin Med. (2020) 9:1450. doi: 10.3390/jcm9051450
169. Ranganath T, Simpson LJ, Ferreira AM, Seiler C, Vendrame E, Zhao N, et al. Characterization of the impact of daclizumab beta on circulating natural killer cells by mass cytometry. Front Immunol. (2020) 11:714. doi: 10.3389/fimmu.2020.00714
170. Gao M, Yang Y, Li D, Ming B, Chen H, Sun Y, et al. CD27 natural killer cell subsets play different roles during the pre-onset stage of experimental autoimmune encephalomyelitis. Innate Immun. (2016) 22:395–404. doi: 10.1177/1753425916658111
171. Bluestone JA, Herold K, Eisenbarth G. Genetics, pathogenesis and clinical interventions in type 1 diabetes. Nature. (2010) 464:1293–300. doi: 10.1038/nature08933
172. Maahs DM, Rewers M. Editorial: mortality and renal disease in type 1 diabetes mellitus–progress made, more to be done. J Clin Endocrinol Metab. (2006) 91:3757–9. doi: 10.1210/jc.2006-1730
173. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest. (2017) 127:2881–91. doi: 10.1172/JCI94549
174. Smith MJ, Simmons KM, Cambier JC. B cells in type 1 diabetes mellitus and diabetic kidney disease. Nat Rev Nephrol. (2017) 13:712–20. doi: 10.1038/nrneph.2017.138
175. Coppieters KT, Roep BO, von Herrath MG. Beta cells under attack: toward a better understanding of type 1 diabetes immunopathology. Semin Immunopathol. (2011) 33:1–7. doi: 10.1007/s00281-010-0236-6
176. Sun L, Xi S, He G, Li Z, Gang X, Sun C, et al. Two to tango: dialogue between adaptive and innate immunity in type 1 diabetes. J Diabetes Res. (2020) 2020:4106518. doi: 10.1155/2020/4106518
177. Rodriguez-Calvo T. Enteroviral infections as a trigger for type 1 diabetes. Curr Diab Rep. (2018) 18:106. doi: 10.1007/s11892-018-1077-2
178. Marca V, Gianchecchi E, Fierabracci A. Type 1 diabetes and its multi-factorial pathogenesis: the putative role of NK cells. Int J Mol Sci. (2018) 19:794. doi: 10.3390/ijms19030794
179. Fraker C, Bayer AL. The expanding role of natural killer cells in type 1 diabetes and immunotherapy. Curr Diab Rep. (2016) 16:109. doi: 10.1007/s11892-016-0806-7
180. Jayasimhan A, Mansour KP, Slattery RM. Advances in our understanding of the pathophysiology of Type 1 diabetes: lessons from the NOD mouse. Clin Sci (Lond). (2014) 126:1–18. doi: 10.1042/CS20120627
181. Poirot L, Benoist C, Mathis D. Natural killer cells distinguish innocuous and destructive forms of pancreatic islet autoimmunity. Proc Natl Acad Sci USA. (2004) 101:8102–7. doi: 10.1073/pnas.0402065101
182. Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, et al. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol. (2010) 11:121–8. doi: 10.1038/ni.1834
183. Perricone R, Perricone C, De Carolis C, Shoenfeld Y. NK cells in autoimmunity: a two-edg'd weapon of the immune system. Autoimmun Rev. (2008) 7:384–90. doi: 10.1016/j.autrev.2008.03.002
184. Shi FD, Van Kaer L. Reciprocal regulation between natural killer cells and autoreactive T cells. Nat Rev Immunol. (2006) 6:751–60. doi: 10.1038/nri1935
185. Flodstrom M, Shi FD, Sarvetnick N, Ljunggren HG. The natural killer cell – friend or foe in autoimmune disease? Scand J Immunol. (2002) 55:432–41. doi: 10.1046/j.1365-3083.2002.01084.x
186. Flodstrom M, Maday A, Balakrishna D, Cleary MM, Yoshimura A, Sarvetnick N. Target cell defense prevents the development of diabetes after viral infection. Nat Immunol. (2002) 3:373–82. doi: 10.1038/ni771
187. Brauner H, Elemans M, Lemos S, Broberger C, Holmberg D, Flodstrom-Tullberg M, et al. Distinct phenotype and function of NK cells in the pancreas of nonobese diabetic mice. J Immunol. (2010) 184:2272–80. doi: 10.4049/jimmunol.0804358
188. Ogasawara K, Hamerman JA, Hsin H, Chikuma S, Bour-Jordan H, Chen T, et al. Impairment of NK cell function by NKG2D modulation in NOD mice. Immunity. (2003) 18:41–51. doi: 10.1016/S1074-7613(02)00505-8
189. Angstetra E, Graham KL, Zhao Y, Irvin AE, Elkerbout L, Santamaria P, et al. An indirect role for NK cells in a CD4(+) T-cell-dependent mouse model of type I diabetes. Immunol Cell Biol. (2012) 90:243–7. doi: 10.1038/icb.2011.16
190. Gur C, Enk J, Kassem SA, Suissa Y, Magenheim J, Stolovich-Rain M, et al. Recognition and killing of human and murine pancreatic beta cells by the NK receptor NKp46. J Immunol. (2011) 187:3096–103. doi: 10.4049/jimmunol.1101269
191. Lee IF, Qin H, Trudeau J, Dutz J, Tan R. Regulation of autoimmune diabetes by complete Freund's adjuvant is mediated by NK cells. J Immunol. (2004) 172:937–42. doi: 10.4049/jimmunol.172.2.937
192. Lee IF, Qin H, Priatel JJ, Tan R. Critical role for IFN-gamma in natural killer cell-mediated protection from diabetes. Eur J Immunol. (2008) 38:82–9. doi: 10.1002/eji.200737189
193. Ehlers M, Papewalis C, Stenzel W, Jacobs B, Meyer KL, Deenen R, et al. Immunoregulatory natural killer cells suppress autoimmunity by down-regulating antigen-specific CD8+ T cells in mice. Endocrinology. (2012) 153:4367–79. doi: 10.1210/en.2012-1247
194. Lang PA, Crome SQ, Xu HC, Lang KS, Chapatte L, Deenick EK, et al. NK cells regulate CD8(+) T cell mediated autoimmunity. Front Cell Infect Microbiol. (2020) 10:36. doi: 10.3389/fcimb.2020.00036
195. Rodacki M, Svoren B, Butty V, Besse W, Laffel L, Benoist C, et al. Altered natural killer cells in type 1 diabetic patients. Diabetes. (2007) 56:177–85. doi: 10.2337/db06-0493
196. Gianchecchi E, Delfino DV, Fierabracci A. NK cells in autoimmune diseases: linking innate and adaptive immune responses. Autoimmun Rev. (2018) 17:142–54. doi: 10.1016/j.autrev.2017.11.018
197. Nekoua MP, Bertin A, Sane F, Alidjinou EK, Lobert D, Trauet J, et al. Pancreatic beta cells persistently infected with coxsackievirus B4 are targets of NK cell-mediated cytolytic activity. Cell Mol Life Sci. (2020) 77:179–94. doi: 10.1007/s00018-019-03168-4
198. Zhang Y, Wang H, Lou X, Qu X, Gao L, Liu X, et al. Decreased percentage of NKG2D+NK cells in patients with incident onset of type 1 diabetes. Clin Exp Pharmacol Physiol. (2017) 44:180–90. doi: 10.1111/1440-1681.12699
199. Qin H, Lee IF, Panagiotopoulos C, Wang X, Chu AD, Utz PJ, et al. Natural killer cells from children with type 1 diabetes have defects in NKG2D-dependent function and signaling. Diabetes. (2011) 60:857–66. doi: 10.2337/db09-1706
200. Oras A, Peet A, Giese T, Tillmann V, Uibo R. A study of 51 subtypes of peripheral blood immune cells in newly diagnosed young type 1 diabetes patients. Clin Exp Immunol. (2019) 198:57–70. doi: 10.1111/cei.13332
201. Fitas AL, Martins C, Borrego LM, Lopes L, Jorns A, Lenzen S, et al. Immune cell and cytokine patterns in children with type 1 diabetes mellitus undergoing a remission phase: A longitudinal study. Pediatr Diabetes. (2018) 19:963–71. doi: 10.1111/pedi.12671
202. Nekoua MP, Dechaumes A, Sane F, Alidjinou EK, Moutairou K, Yessoufou A, et al. Enteroviral pathogenesis of type 1 diabetes: the role of natural killer cells. Microorganisms. (2020) 8:989. doi: 10.3390/microorganisms8070989
203. Dean JW, Peters LD, Fuhrman CA, Seay HR, Posgai AL, Stimpson SE, et al. Innate inflammation drives NK cell activation to impair Treg activity. J Autoimmun. (2020) 108:102417. doi: 10.1016/j.jaut.2020.102417
204. Akesson C, Uvebrant K, Oderup C, Lynch K, Harris RA, Lernmark A, et al. Altered natural killer (NK) cell frequency and phenotype in latent autoimmune diabetes in adults (LADA) prior to insulin deficiency. Clin Exp Immunol. (2010) 161:48–56. doi: 10.1111/j.1365-2249.2010.04114.x
Keywords: local microenvironment, immune tolerance, NK subsets, NK cells, autoimmune diseases
Citation: Liu M, Liang S and Zhang C (2021) NK Cells in Autoimmune Diseases: Protective or Pathogenic? Front. Immunol. 12:624687. doi: 10.3389/fimmu.2021.624687
Received: 31 October 2020; Accepted: 22 February 2021;
Published: 12 March 2021.
Edited by:
Antonio La Cava, University of California, Los Angeles, United StatesReviewed by:
Luc Van Kaer, Vanderbilt University, United StatesCopyright © 2021 Liu, Liang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Cai Zhang, Y2FpemhhbmdzZEBzZHUuZWR1LmNu; Shujuan Liang, bGlhbmdzaGpAd2ZtYy5lZHUuY24=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.