
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol., 25 March 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.624360
This article is part of the Research TopicPathogens, Pathobionts and AutoimmunityView all 13 articles
 Hongxia Zhang1†
Hongxia Zhang1† Man Liu1†
Man Liu1† Weilong Zhong1Yanping Zheng1
Weilong Zhong1Yanping Zheng1 Yanni Li1Liping Guo1Yujie Zhang2Ying Ran1
Yanni Li1Liping Guo1Yujie Zhang2Ying Ran1 Jingwen Zhao1
Jingwen Zhao1 Lu Zhou1,3*
Lu Zhou1,3* Bangmao Wang1*
Bangmao Wang1*The gut–liver axis has been increasingly recognized as a major autoimmunity modulator. However, the implications of intestinal barrier in the pathogenesis of autoimmune hepatitis (AIH) remain elusive. Here, we investigated the functional role of gut barrier and intestinal microbiota for hepatic innate immune response in AIH patients and murine models. In this study, we found that AIH patients displayed increased intestinal permeability and pronounced RIP3 activation of liver macrophages. In mice models, intestinal barrier dysfunction increased intestinal bacterial translocation, thus amplifying the hepatic RIP3-mediated innate immune response. Furthermore, GSK872 dampened RIP3 activation and ameliorated the activation and accumulation of liver macrophages in vitro and in vivo experiments. Strikingly, broad-spectrum antibiotic ablation significantly alleviated RIP3 activation and liver injury, highlighting the causal role of intestinal microbiota for disease progression. Our results provided a potentially novel mechanism of immune tolerance breakage in the liver via the gut-liver axis. In addition, we also explored the therapeutic and research potentials of regulating the intestinal microbiota for the therapy of AIH.
Autoimmune hepatitis (AIH) is a chronic immune-mediated inflammatory liver disease. Although genetic and environmental factors are involved in the pathogenesis of AIH, the underlying mechanisms remain unclear (1). In recent years, great importance has been attached to the role of intestinal barrier in the pathogenesis of diverse immune-mediated diseases (2–4). In particular, the liver is continuously exposed to gut-derived antigens through the portal vein, which influence its innate and adaptive immune responses (5, 6). It’s known that intestinal barrier disruption can trigger bacteria and bacterial products translocation, which consecutively activate immune cells to release various proinflammatory cytokines and chemokines in the liver (7, 8). Clinically, primary sclerosing cholangitis is a remarkable example of chronic biliary inflammation highly associated with inflammatory bowel disease, indicating that the gut-liver axis plays an important role on the pathogenesis (9, 10).
Macrophages represent a key cellular component of the liver essential for maintaining tissue homeostasis and ensuring rapid responses to hepatic injury (11). Researches have reported the key role for liver macrophages in AIH. H Grønbaek et al. studied 121 AIH patients in a cross-sectional design and demonstrated macrophage activation paralleling disease activity, severity and treatment response, suggesting a role for macrophage activation in AIH (12). Besides, Assis David N reported a distinct genetic and immunopathogenic basis for AIH at the macrophage migration inhibitory factor locus, which indicated that macrophages play a role in pathogenesis and as biomarkers of AIH (13, 14). Liver macrophages consist of ontogenically distinct populations termed as Kupffer cells and monocyte-derived macrophages (15). As macrophages accumulate gut-derived products such as lipopolysaccharide (LPS) and undergone activation, necrosis of macrophages and uncontrolled release of inflammatory cytokine and chemokine results in inflammation and fibrosis of liver tissues (16, 17). Thus, macrophage cell death has been considered to be a major contributor of immune-mediated liver injury (18). Receptor interacting protein kinase 3 (RIP3) has been increasingly recognized as a central player in necroptosis and RIP3 kinase activity supports the recruitment of mixed lineage kinase domain-like (MLKL) to trigger membrane leakage with the consequent release of inflammatory cytokines and chemokines (19–21). Recently, we reported that probiotics application in experimental autoimmune hepatitis (EAH) mice could improve the intestinal barrier and downregulate the RIP3 signaling of liver macrophages (22). Hence, we hypothesized that the activation of RIP3 signaling pathway may be a potential mechanism of gut-liver axis in AIH pathogenesis and thus can be a novel treatment target.
In this study, we demonstrated that intestinal barrier damage and RIP3-mediated activation of liver macrophages existed in AIH patients. As reveled by the tandem model of dextran sulfate sodium (DSS) - concanavalin A (Con A), the disruption of intestinal barrier prior to hepatitis aggravated the activation and accumulation of liver macrophages. This finding highlighted RIP3 as an important interface that mediated liver inflammation. Furthermore, the RIP3-mediated activation of liver macrophage in EAH mice was canceled by gut sterilization, suggesting that immune responses in the liver are potentially regulated by gut microbiota.
All experimental procedures were performed according to the guidelines of the Institutional Animal Care and Use Committee at Tianjin Medical University and followed the International Association of Veterinary Editors guidelines for the Care and Use of Laboratory Animal. The animal use protocol listed below has been reviewed and approved by the Animal Ethical and Welfare Committee of Tianjin Medical University, Approval No. IRB2020-WZ-119.
Sixty-eight patients with AIH including thirty-nine without cirrhosis (AIH-n) and twenty-nine with cirrhosis (AIH-c) and fifteen controls were included. The patients were recruited from the Gastroenterology Department at Tianjin Medical University General Hospital. AIH was diagnosed with the following criteria: (1) patients conformed with 1999 revised International Autoimmune Hepatitis Group score≥10 and/or (2) 2008 IAIHG simplified AIH score≥6 and/or (3)histological features indicative of AIH (23, 24). Patients data were collected prior to corticosteroid therapy. The control subjects (CTRL) were selected from the Health Management Center of Tianjin Medical University General Hospital and matched the patients with AIH in terms of age and gender. Inclusion criteria for the CTRL group were as follows: (1) normal ranges of liver function test, (2) an absence of hepatitis B/C virus antigen, (3) normal abdominal ultrasound tests, and (4) an absence of autoimmune diseases and family history. Blood was collected from the individuals. Feces were collected from six AIH-n patients. Intestinal mucosal biopsy specimens were collected from fourteen patients (six AIH-n patients and eight AIH-c patients) and six controls. Liver biopsy specimens were collected from six AIH-n patients and four patients with hepatic cyst.
Twenty-four female SPF C57BL/6 mice (6 weeks of age) were purchased from Beijing Animal Study Centre and reared under specific pathogen-free conditions in Animal Centre of the Tianjin Medical University. The mice were randomly divided into four groups (n=6 per group) including the CTRL group, DSS group, Con A group and DSS-Con A group. 1%DSS (MP Biomedicals) was dissolved in sterile distilled water ad libitum for 7 days to induce disruption of intestinal barrier integrity. Con A (15 mg/kg, Solarbio) was i.v. administered into the tail vein of mice 12 hours before liver resection (Supplementary Figure 1A). A RIP3 kinase inhibitor GSK872 (Merck) was diluted in 1 mg/mL dimethyl sulfoxide (DMSO). Another 12 C57BL/6 mice were randomized divided into two groups including DSS-Con A group and GSK872-pretreated group (n=6 per group) provided with 1% DSS water for 7 days and intraperitoneally treated with either GSK872 (1 mg/kg) or an equal volume of DMSO 1 h prior to Con A administration.
Another 26 female C57BL/6 mice were randomly divided into three groups (n=6 per group) including the CTRL group, EAH group and antibiotic mixture (Abx) group. The rest mice (n=8) were used to extract hepatic antigen S100 as previous description (25). All mice except the CTRL group were injected intraperitoneally with 0.5ml S100 emulsified in an equal volume of complete Freund’s adjuvant (CFA, sigma, USA) on day 7 and day 14 to induce EAH. The Abx group was pretreated with antibiotic mixture (0.5 g/L vancomycin, 1 g/L ampicillin, 1 g/L metronidazole, and 1 g/L neomycin; Sigma–Aldrich) for 2 weeks prior to S100 administration to deplete endogenous commensal microbiota. On day 28, all the animals were sacrificed under anesthesia (Supplementary Figure 2A).
Fecal samples from patients with AIH were mixed at equal weight. One gram of the mixed feces was diluted in 5 mL sterile PBS solution, then initial filtered, concentrated, homogenized, step by step filtered, centrifuged. The supernatant was collected and centrifuged at 5,000 × g for 10 min at 4°C, then the supernatant of feces from AIH patients (AIH-s) was collected and filter-sterilized through 0.22 μm filters (26).
Human Caco-2 cells (BNCC 338148) were cultured in Modified Eagle’s Medium (MEM) (Gibco) supplemented with 20% fetal bovine serum and a penicillin-streptomycin solution. The cells were incubated in a humidified incubator containing 5% CO2 at 37°C and were seeded in a 24-well plate at a density of 1×105 cells per well. In the stimulation experiment (AIH-s group), the cells were pre-treated with 10% AIH-s for 24h, and the control groups were treated 10% inactive AIH-s or PBS for 24h. Mouse macrophage cell line RAW264.7 was plated in Dulbecco modified eagle medium (Gibco) supplemented with 10% FBS, 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Gibco). The cells were cultured under same conditions as above. RAW264.7 cells were seeded in a 12-well plate at a density of 1×105 cells per well. In LPS experiments (LPS group), the cells were treated with LPS (3 mg/mL, Solarbio Biotech) for 24 h. In GSK872 experiments (LPS-GSK872 group), the cells were treated with LPS (3 μg/mL) and GSK872 (3 µM) for 24 h.
Intestinal permeability was determined through fluorescein isothiocyanate (FITC)-dextran assay. FITC-D (4kDa, Sigma-Aldrich) was dissolved in normal saline infusion (50 mg/mL) and administered to mice through gavage at 6 mg/10 g body weight. Whole blood was collected 4h after FITC-D administration by using heparinized microhematocrit capillary tubes via eye bleed. Plasma was extracted from the blood through centrifugation at 4°C for 10 min at 3,000 rpm. Fluorescence intensity was analyzed using a plate reader. FITC-D concentration of each mouse was detected based on the FITC-D standard curve.
The blood was centrifuged at 3000 rpm for 10 min, and the plasma was then stored at −80°C. LPS, D-lactic acid (DLA), and diamine oxidase (DAO) plasma concentrations were quantified with ELISA kits (SenBeiJia Biotech) in accordance with the manufacturer’s instructions. Zonulin plasma concentrations were quantified using ELISA kits (Elabscience). Plasma alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were tested by using the automated chemistry analyzer from the clinical laboratory of the Tianjin Medical University General Hospital.
The liver and intestinal tissues of patients and mice were collected and fixed in 4% paraformaldehyde. The paraffin-embedded liver and intestinal tissues were sectioned at approximately 5 µm and stained with hematoxylin and eosin (HE) following the standard HE protocol. Pathological changes in the liver and intestinal tissues were evaluated by two independent and experienced pathologists. Intestinal tissue sections from patients were stained with primary anti- zonula occludens-1 (ZO-1) antibody (ab96587, Abcam, Cambridge, MA, USA) or anti-Occludin antibody (ab216327, Abcam, Cambridge, MA, USA) at 4°C overnight and incubated with second antibody for 30 min at 37°C. The staining index was calculated by multiplying percentage positive cells rating by intensity rating in every field for quantitative analysis.
Caco-2 cell monolayers were fixed in cold methanol for 5 min at -20°C. Monolayers were then washed and blocked with bovine serum albumin (BSA) for 30 min at room temperature. Cells were then incubated with either anti-ZO-1 (1:50, Abcam, USA) or anti-Occludin (1:150, Abcam, USA) overnight at 4°C. Cells were then incubated with the secondary antibody for 1 h at room temperature. Cell nuclei were stained with DAPI. Double immunofluorescence analyses for liver macrophages were performed with 4 mm-thick frozen sections. Slides were fixed with acetone, blocked with 5% bovine serum albumin, and incubated with primary antibodies against CD68 (ab955, Abcam, Cambridge, MA, USA) and MAC387 (ab92507, Abcam, Cambridge, MA, USA) at 4°C overnight. The slices were restored to room temperature the next day, incubated with the corresponding secondary antibodies for 1h at 37°C, underwent DAPI reaction, sealed, and observed under a fluorescence microscope.
Total RNA was extracted from liver tissues with TRIzol (Thermo Scientific Inc.), followed by cDNA reverse transcription using the FastKing RT kit (TIANGEN). Real-time-PCR was performed using SYBR® Select Master Mix (Thermo Scientific Inc.). Oligonucleotide primers for target genes are listed in Table 1 and Table 2. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was employed as an endogenous control. The relative mRNA expression levels of the target gene were evaluated by calculating the fold-changes normalized to the GAPDH for each sample using 2−ΔΔCt methods. All cDNA samples were analyzed in triplicate.

Table 1 The Oligonucleotide primers used in Realtime-PCR analysis.

Table 2 The Oligonucleotide primers used in Realtime-PCR analysis.
The liver and intestinal tissues were dissolved in RIPA, PMSF, and protease inhibitors. After homogenization, the protein concentrations were determined using bicinchoninic acid protein assay (Thermo Scientific Inc.). Proteins were separated using SDS-polyacrylamide gel electrophoresis system and then blotted onto a polyvinylidene fluoride membrane (Invitrogen, USA). Primary anti- RIP3(ab62344, Abcam, Cambridge, MA, USA), anti-MLKL (ab196436, Abcam, Cambridge, MA, USA), anti-TNF-α (ab183218, Abcam, Cambridge, MA, USA), anti-IL-6 (ab229381, Abcam, Cambridge, MA, USA), anti-ZO-1 (Abcam, USA), anti-Occludin (Abcam, USA), and anti-GAPDH (CST) antibody were then applied, and anti-GAPDH antibody was employed as the loading control. After incubation with horseradish peroxidase-conjugated secondary antibodies, the chemiluminescent signal was detected. Band intensity was determined by image processor program (Image J).
MAbs specific for CD45, CD11b, and F4/80 were obtained from BD Biosciences. Single-cell suspensions of lymphocyte were harvested from mouse liver. The cells were suspended in buffer, incubated with the above antibody for 30 min, and examined on a FACSCalibur flow cytometer (Becton Dickinson, USA). RAW264.7 cells seeded in a 12-well plate at a density of 1×105 cells per well were stained with Annexin V/PI (BD Pharmingen, San Diego, CA, USA) to detect the apoptosis state in accordance with the manufacturer specifications. Data were analyzed using FlowJo 7.6 software.
Transient genetic silencing of RIP3 was performed by reverse transfection of RAW264.7 cells with 20–30 Nm Silencer® Select siRNAs (Life Technologies, Inc.) using Lipofectamine® RNAiMax reagent (Life Technologies, Inc.), and Opti-MEM® medium (Life Technologies, Inc.). Negative nontarget siRNA was used as control. Knockdown efficiency was confirmed by Western blot analysis and qRT-PCR.
16S rRNA gene sequencing procedure was performed by GENEWIZ, lnc. (Suzhou, China). Total fecal bacteria DNA extractions were acquired by QIAamp ® Fast DNA Stool Mini Kit (QIAamp, Germany). The microbial 16S V3-V4 region was amplified with indexes and adaptors-linked universal primers (341F: ACTCCTACGGGAGGCAGCA G,806R: GGACTACHVGGGTWTCTAAT). PCR was performed using KAPA HiFi Hotstart PCR kit high fidelity enzyme in triplicate. Amplicon libraries were quantified by Qubit 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, US) and then sequenced on Illumina HiSeq platform (Illumina, San Diego, US) for paired end reads of 250 bp. After the singletons were discarded and the chimeras were removed, the tags were clustered into operational taxonomic units (OTUs) using USEARCH (v7.0.1090) at 97% similarity. A representative sequence of each OTU was subjected to taxonomy-based analysis using the RDP database. Heatmap was created using R. Cluster analysis. Alpha and beta diversities were analyzed using QIIME. The relative abundance of bacteria was expressed as the percentage.
Data were presented as mean ± SD. Comparisons among different groups were performed by unpaired one-way ANOVA or Student’s t-test using SPSS 22.0. p<0.05 was considered statistically significant.
The main clinical and demographic features of enrolled patients and controls are shown in Supplementary Table 1. Plasma LPS, DLA, and DAO tests revealed an increased intestinal permeability in AIH-n group compared with that in the CTRL group (Figure 1A). To further assess the integrity of the intestinal barrier in these patients, we detected the structural proteins including ZO-1 and Occludin in the ileocecal junction through IHC staining (Figure 1B). The staining index of ZO-1 and Occludin in AIH-n group was significantly decreased compared with that in the CTRL group. Zonulin is the only physiological modulator of intercellular tight junctions involved in the trafficking of macromolecules and therefore in tolerance/immune response balance (27). Hence, the plasma zonulin levels were evaluated and was found to be significantly increased in the two AIH groups compared with that in the CTRL group (Figure 1C), indicating that breakage of the intestinal barrier integrity is an early event in the pathogenesis of AIH.
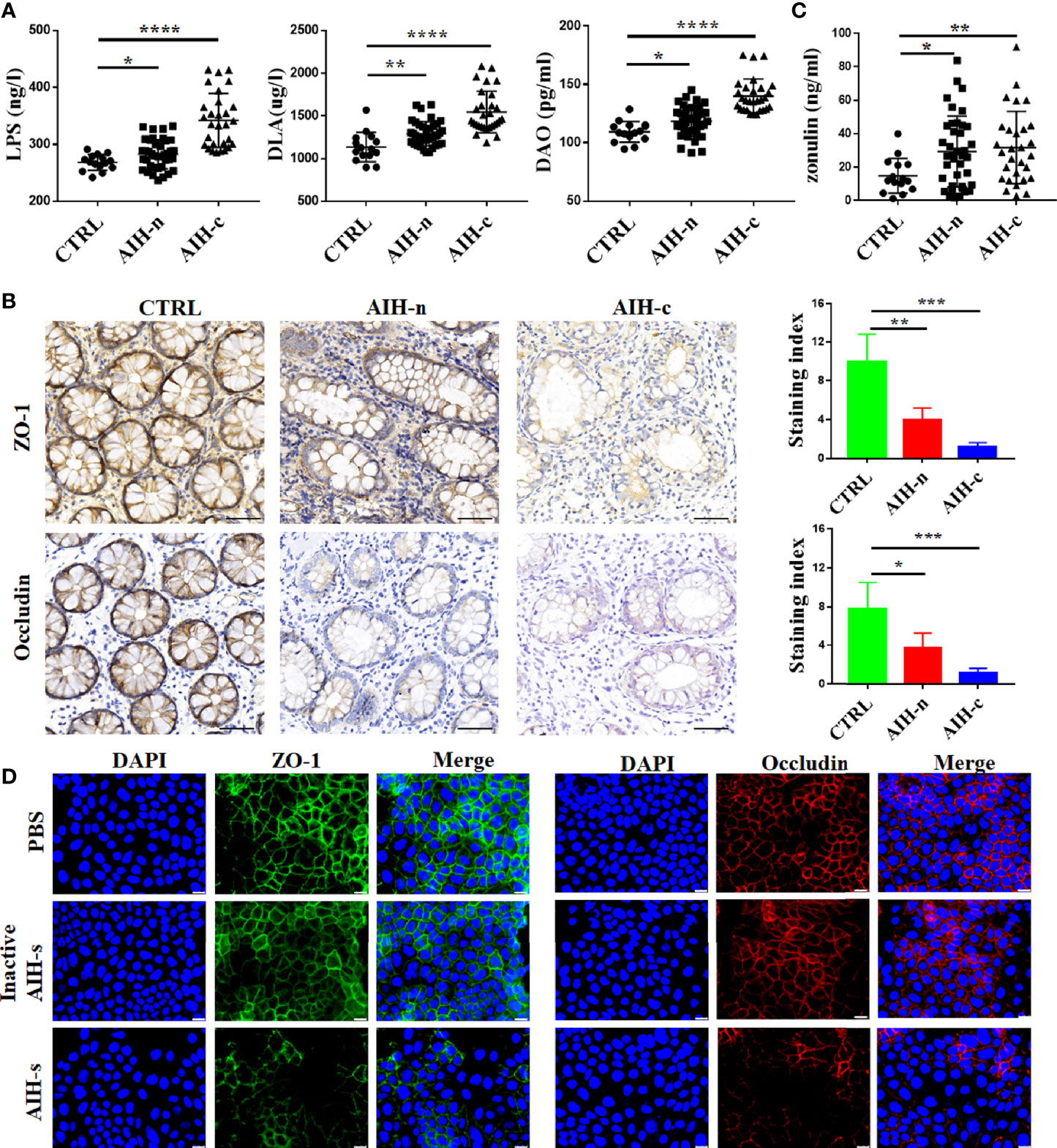
Figure 1 Increased intestinal permeability and loss of epithelial barrier integrity in AIH patients. (A) The plasma concentrations of LPS, DLA and DAO in CTRL group (n=15), AIH-n group (n=39) and AIH-c group (n=29). (B) Expression of ZO-1 and Occludin in the colon were assessed by immunostaining. (C) The plasma concentration of zonulin in the three groups. (D) Representative immunostaining of ZO-1 and Occludin in Caco-2 cells. Scale bars: 50μm. The data were presented as means ± SD (Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Previous data have demonstrated that treatment-naïve AIH patients had compositional and functional alterations of gut microbiome (28). To investigate the effects of gut dysbiosis on intestinal barrier function, we used a vitro model in which Caco-2 epithelial cell monolayers treated with the supernatant of feces from AIH patients (AIH-s). As shown in Figure 1D, the expressions of ZO-1 and Occludin were dramatically decreased in the AIH-s group compared to the control groups (inactive AIH-s group and PBS group).
Liver macrophages, which consist of resident macrophages (Kupffer cells) and monocytes-derived macrophages, maintain liver immune homeostasis. Therefore, macrophage heterogeneity and activation status in the liver tissues of AIH patients were studied. Double immunofluorescence analyses for CD68 and MAC387 revealed that the number of resident and infiltrating macrophages significantly increased in the liver tissues of AIH-n group (Figure 2A). Furthermore, intracellular staining of inflammatory cytokines such as TNF-α and IL-6 co-stained with CD68 showed that the liver macrophages in AIH patients are significantly activated and a majority of the inflammatory signature derives from liver macrophages (Figures 2B, C). Besides, the mRNA expression of related inflammatory cytokines and chemokines in liver tissues was investigated (Figure 2D). TNF-α, IL-6, and IL-1β expression was significantly increased in the AIH-n group. CCL2 expression was also increased, and that of CCR2 was not significantly different.
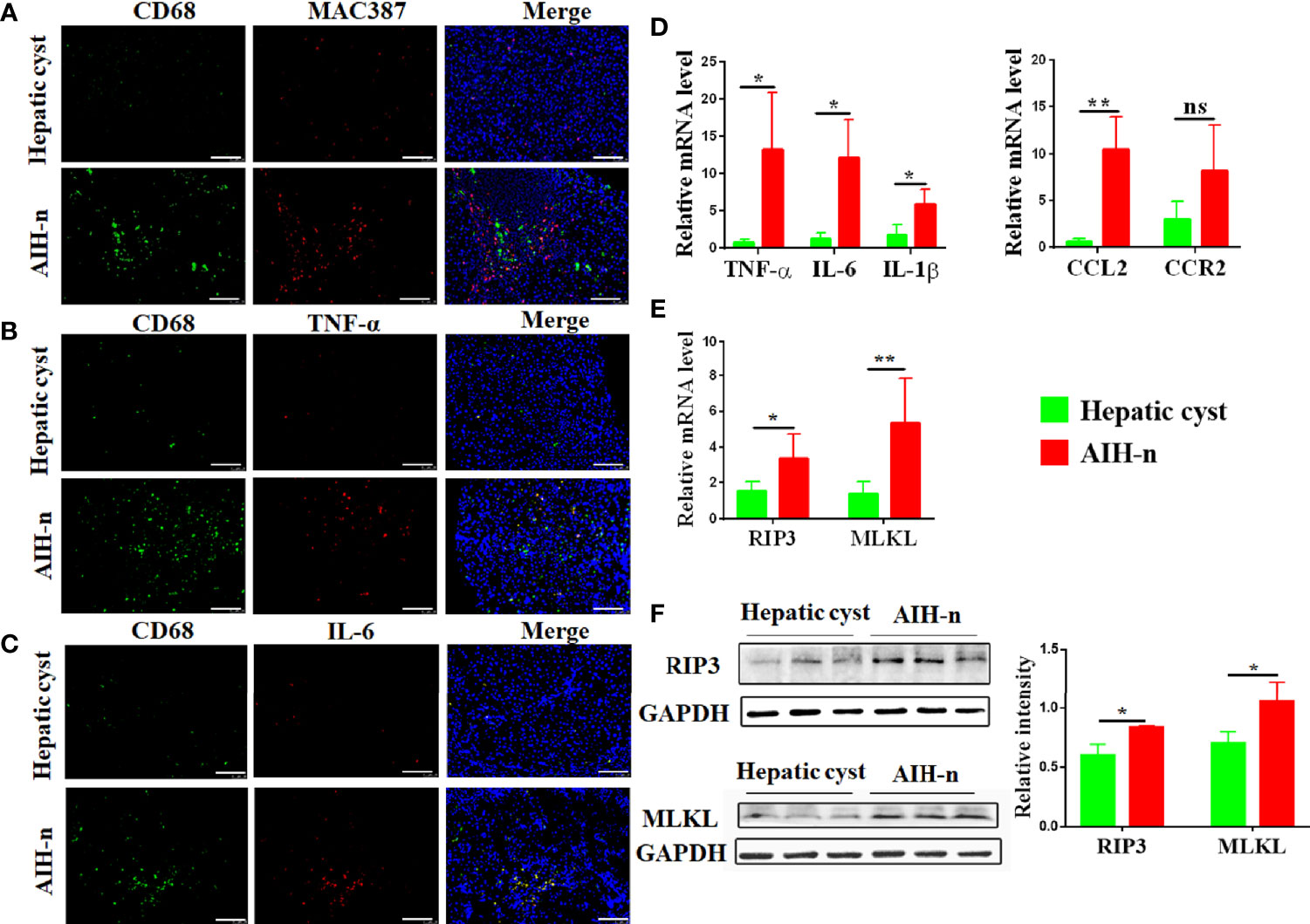
Figure 2 Activation and infiltrating of macrophages in the liver tissue of AIH patients. (A–C) Double-immunofluorescence staining for CD68 and MAC387 (A) CD68 and TNF-α (B) CD68 and IL-6 (C) in liver tissues of patients with AIH and hepatic cyst. (D) RT-qPCR analysis of TNF-α, IL-6, IL-1β, CCL2 and CCR2 on tissue homogenates from the liver of the two groups. (E) RT-qPCR analysis of RIP3 and MLKL in the liver of the two groups. (F) Protein levels of RIP3 and MLKL in the liver of the two groups were detected and the relative intensity was quantified. Scale bars: 100μm. The data were presented as means ± SD (Student’s t-test, *p < 0.05, **p < 0.01, ns: p < 0.05).
RIP3 has been increasingly recognized as a key inflammatory signal adapter that mediates programmed necroptosis and the consequent release of inflammatory cytokines and chemokines (29, 30). Therefore, the activation of RIP3 and MLKL (the direct downstream effector of RIP3) in the liver tissues of AIH-n group was further explored. As shown in Figure 2E, the AIH-n group had significantly higher mRNA expression of RIP3 and MLKL than the hepatic cyst group. Concordantly, the protein expression of RIP3 and MLKL was also increased in the AIH-n group (Figure 2F).
Clinical data indicated an increase in the intestinal permeability and alterations of liver immune homeostasis in AIH-n patients. Hence, we hypothesized that the enteropathy and breakage of the intestinal barrier integrity are not epiphenomena but could play a pathogenic role in AIH by regulating liver inflammation. For hypothesis testing, the tandem model of DSS-Con A was employed. As shown in Supplementary Figure 1B, the body weight of mice in each group did not differ significantly (p > 0.05). In addition, the liver index and spleen index were significantly increased in the Con A group compared with those in the CTRL group, but no difference was observed between the Con A and DSS-Con A groups (Supplementary Figure 1C, D). Furthermore, the gut barrier integrity of the tandem model was assessed. The results showed that the DSS-Con A group had significantly higher plasma concentration of FITC-dextran compared with the Con A group (Figure 3A). The mRNA and protein expression levels of ZO-1 and Occludin were significantly decreased in the gut mucosa of DSS-Con A group compared with those of Con A group (Figures 3B, C).
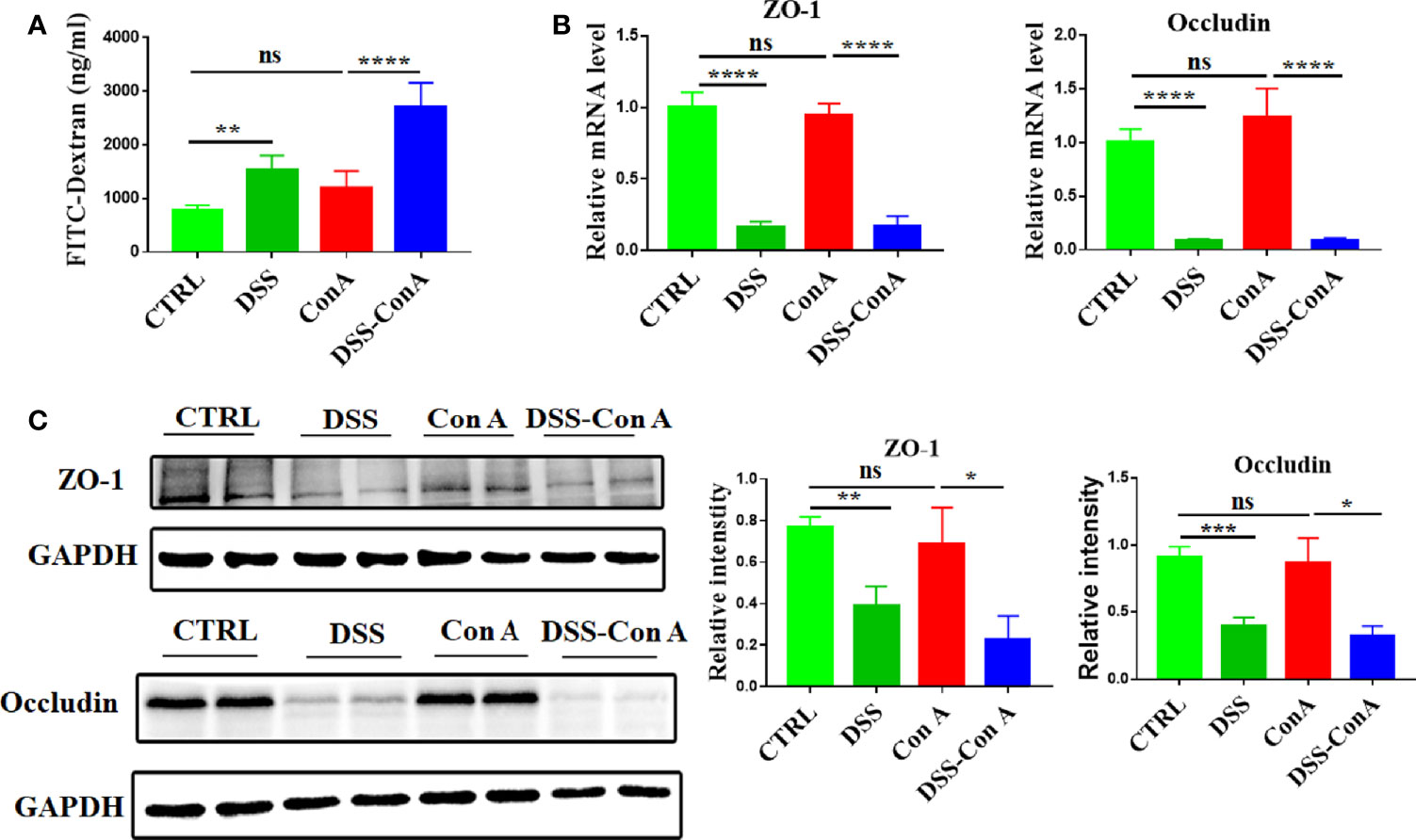
Figure 3 Breakage of intestinal barrier integrity by 1%DSS. (A) FITC-dextran in vivo permeability assay in the four groups. (B) RT-qPCR analysis of ZO-1 and Occludin on tissue homogenates from the colon of the four groups. (C) Protein levels of ZO-1 and Occludin in the colon of the four groups were detected and the relative intensity was quantified. (n=6). The data were presented as means ± SD (Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: p < 0.05).
Next, the inflammation and immunity of liver tissues were evaluated, and our data showed that the breakage of the intestinal barrier aggravated the Con A-mediated liver inflammation in the portal area (Figure 4A). The plasma transaminase levels of DSS-Con A group significantly increased compared with those of Con A group (Figure 4B). Moreover, the mice in the DSS-Con A group had a severe inflammatory cytokine milieu with higher mRNA expression of TNF-α, IL-6, and IL-1β in the liver tissues compared with that in Con A group, and the CCL2 expression was also significantly increased in DSS-Con A group (Supplementary Figure 3A, B). In addition, immunofluorescence staining of inflammatory cytokines in F4/80+ cells indicated that the liver macrophages in DSS-Con A group had a more severe inflammatory signature compared with that in Con A group (Figure 4C). The CD45+ F4/80+ CD11b+ population of liver mononuclear cells were analyzed via flow cytometry to examine the state of the resident and infiltrating macrophages in the liver following the intestinal barrier breakage. The number of CD45+ F4/80hi CD11blo liver resident Kupffer cells was significantly decreased, whereas that of CD45+ F4/80lo CD11bhi infiltrating macrophages significantly increased in Con A group compared with those of CTRL group. However, no difference was found between Con A and DSS-Con A groups. The ratio of infiltrating macrophages to Kupffer cells significantly increased in DSS-Con A group compared with that in Con A group (Figure 4D). These results suggested that the intestinal barrier breakage contributes to Con A-mediated liver injury by promoting the activation and infiltration of liver macrophages.
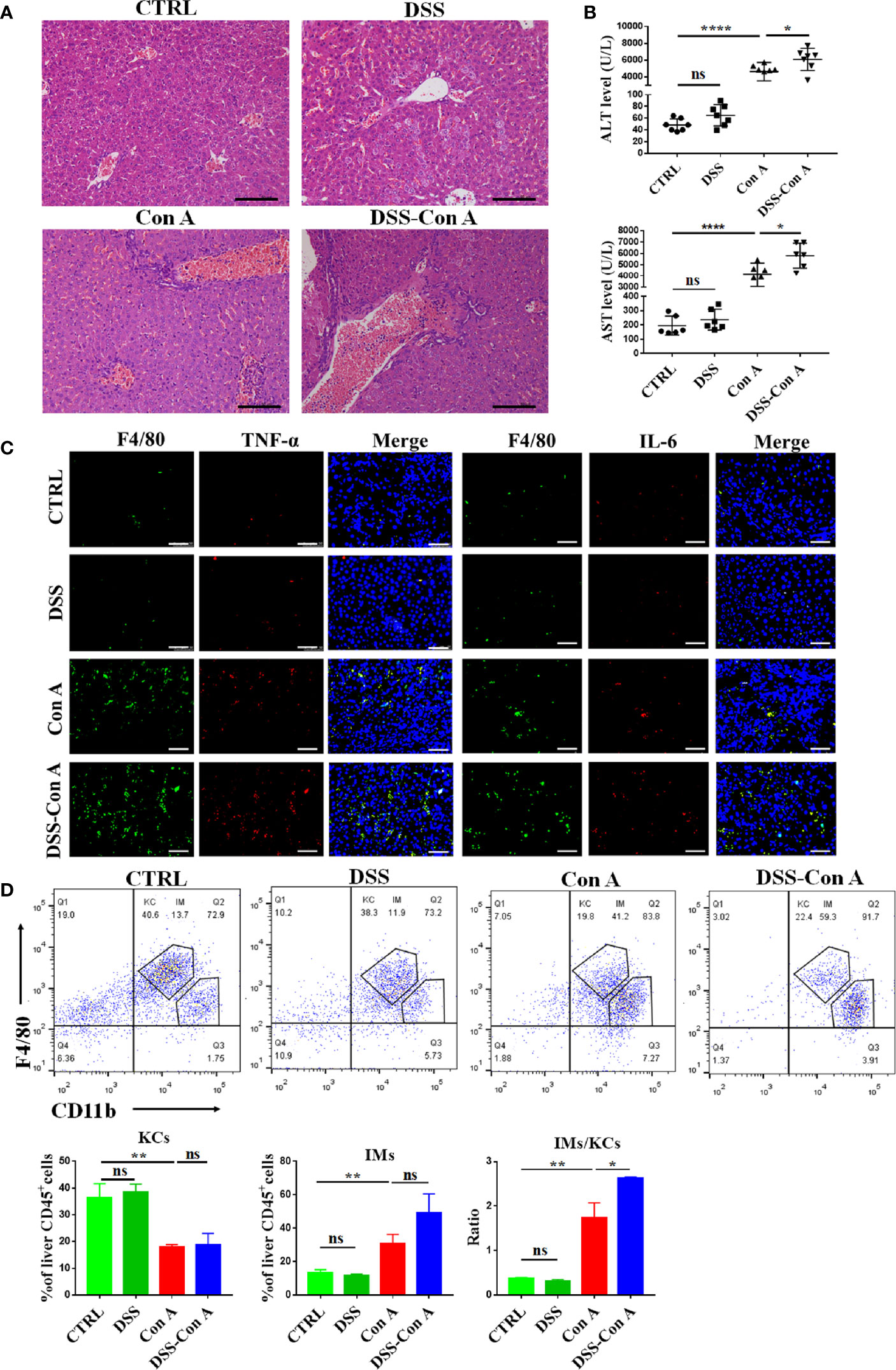
Figure 4 Breakage of the intestinal barrier aggravated liver injury and infiltration of liver macrophages. (A) HE staining of liver tissues in four groups. (B) The plasma concentrations of ALT and AST in four groups. (C) Double-immunofluorescence staining for F4/80 and TNF-α, F4/80 and IL-6 of liver tissues in four groups. (D) Representative flow cytometry plots and percentage of CD45+F4/80hiCD11blo Kupffer cells and CD45+F4/80loCD11bhi infiltrating macrophages in mononuclear cells from liver of the four groups. (n=6). Scale bars: 50μm. The data were presented as means ± SD (Student’s t-test, *p < 0.05, **p < 0.01, ****p < 0.0001, ns: p < 0.05).
The mechanism underlying the augmented activation and accumulation of liver macrophage was studied under this tandem model. The protein expression of RIP3 and MLKL in the liver tissue was significantly upregulated in DSS-Con A group compared with that in Con A group (Figure 5A). The relative mRNA expression of RIP3 and MLKL also increased (Supplementary Figure 3C). As shown in Figure 5B, p-RIP3-positive cells and p-MLKL-positive cells were markedly increased in the liver tissues from DSS-Con A group, particularly in macrophages. Further, the mice were treated with GSK872 to investigate whether the inhibition of RIP3 signaling pathway can ameliorate the activation and infiltration of liver macrophages in the DSS-Con A group. The liver of GSK872-pretreated group showed markedly diminished RIP3 and MLKL expression (Figure 5C) and significantly inhibited mRNA expression levels of inflammatory cytokines and chemokines including TNF-α, IL-6, IL-1β, and CCL2 (Figures 5D, E). GSK872 also inhibited the infiltration of CD45+ F4/80lo CD11bhi macrophages, leading to a significantly decreased ratio of infiltrating macrophages to Kupffer cells (Figure 5F). Accordingly, the GSK872-pretreated group had significantly reduced inflammation in the portal area of liver tissues and decreased plasma transaminases levels compared with the DSS-Con A group (Figures 5G, H). Besides, the annexin V/PI apoptosis assay for further assessment of cell death showed that breakage of the intestinal barrier significantly induced the late apoptosis of liver macrophages and GSK872 markedly decreased the early and late apoptosis rates (Supplementary Figure 4). All together, these results indicated that the pronounced activation and infiltration of liver macrophages in the DSS-Con A group are regulated by RIP3 signaling pathway.
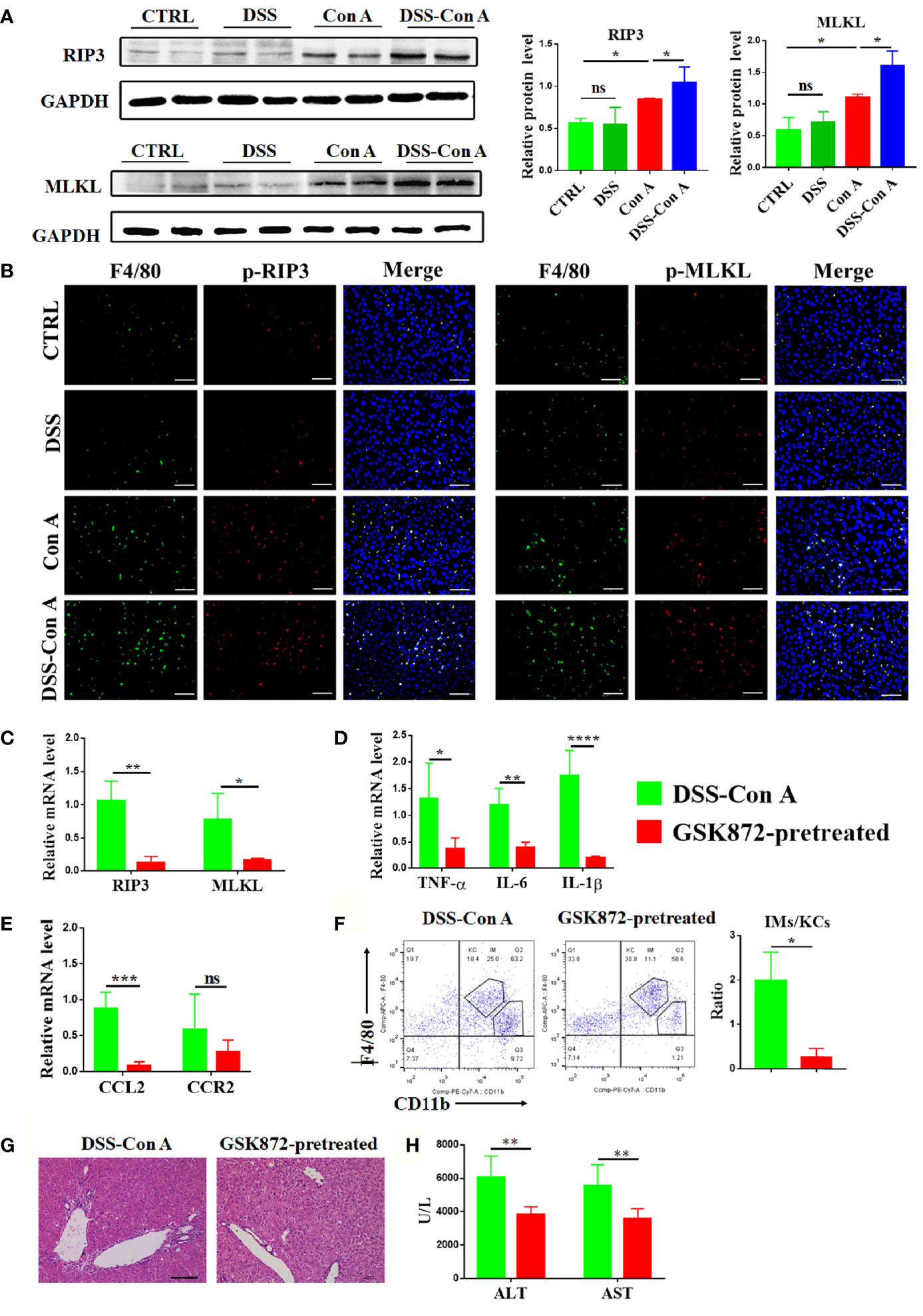
Figure 5 Breakage of intestinal barrier aggravated the activation of RIP3 signaling pathway of the liver tissue. (A) Protein levels of RIP3 and MLKL in the liver of the four groups were detected and the relative intensity was quantified. (B) Representative double-immunofluorescence staining for F4/80 and p-RIP3, F4/80 and p-MLKL of liver tissues in four groups. (C) RT-qPCR analysis of RIP3 and MLKL on liver tissue of DSS-ConA group and GSK872-pretreated group. (D) RT-qPCR analysis of TNF-α, IL-6, and IL-1β on liver tissue of the two groups. (E) RT-qPCR analysis of CCL2 and CCR2 on liver tissue of the two groups. (F) Representative flow cytometry plots and percentage of CD45+F4/80hi CD11blo Kupffer cells and CD45+ F4/80lo CD11bhi infiltrating macrophages in mononuclear cells from liver of the two groups. (G) HE staining of the liver tissue from the two groups. (H) The plasma concentrations of ALT and AST of the two groups. (n=6). Scale bars: 50μm. The data were presented as means ± SD (Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: p < 0.05).
The expression levels of macrophage-related cytokines and chemokines were analyzed by activating or inhibiting the RIP3 signaling pathway in vitro to further explore the effect of RIP3 signaling pathway on liver macrophages. The results showed that the relative mRNA expression of RIP3 and MLKL was upregulated after LPS stimulation in RAW264.7 cells (Supplementary Figure 5A). The protein level of RIP3 and MLKL also significantly increased in the LPS group (Figure 6A). Upon the activation of RIP3 signaling pathway by LPS, the relative mRNA expression levels of cytokines and chemokines including TNF-α, IL-6, IL-1β, CCL2, and CCR2 as well as the protein expression of key cytokines such as TNF-α and IL-6 were also significantly increased (Supplementary Figure 5B, C). By contrast, the relative mRNA expression of cytokines and chemokines were down-regulated when the RIP3 signaling pathway was inhibited with GSK872 (Figures 6B–D). The protein level of TNF-α and IL-6 in the LPS-GSK872 group also decreased but the difference was not statistically significant (Supplementary Figure 5C). Besides, the annexin V/PI apoptosis assay showed that LPS significantly induced the early and late apoptosis of RAW264.7 cells and GSK872 markedly decreased the late apoptosis rate (Supplementary Figure 6). Furthermore, RIP3 was efficiently knocked down by RIP3 siRNA when compared with CTRL siRNA (Figures 6E, F). Accordingly, the protein level of key cytokines such as TNF-α and IL-6 also significantly decreased in the RIP3 siRNA group (Figure 6F) and the relative mRNA expression of macrophage-related cytokines and chemokines including TNF-α, IL-6, IL-1β, CCL2, and CCR2 also significantly decreased in the RIP3 siRNA group (Figures 6G, H). All these results emphasized that the RIP3 signaling pathway can be activated by intestinal LPS to regulate the activation and accumulation of macrophages.
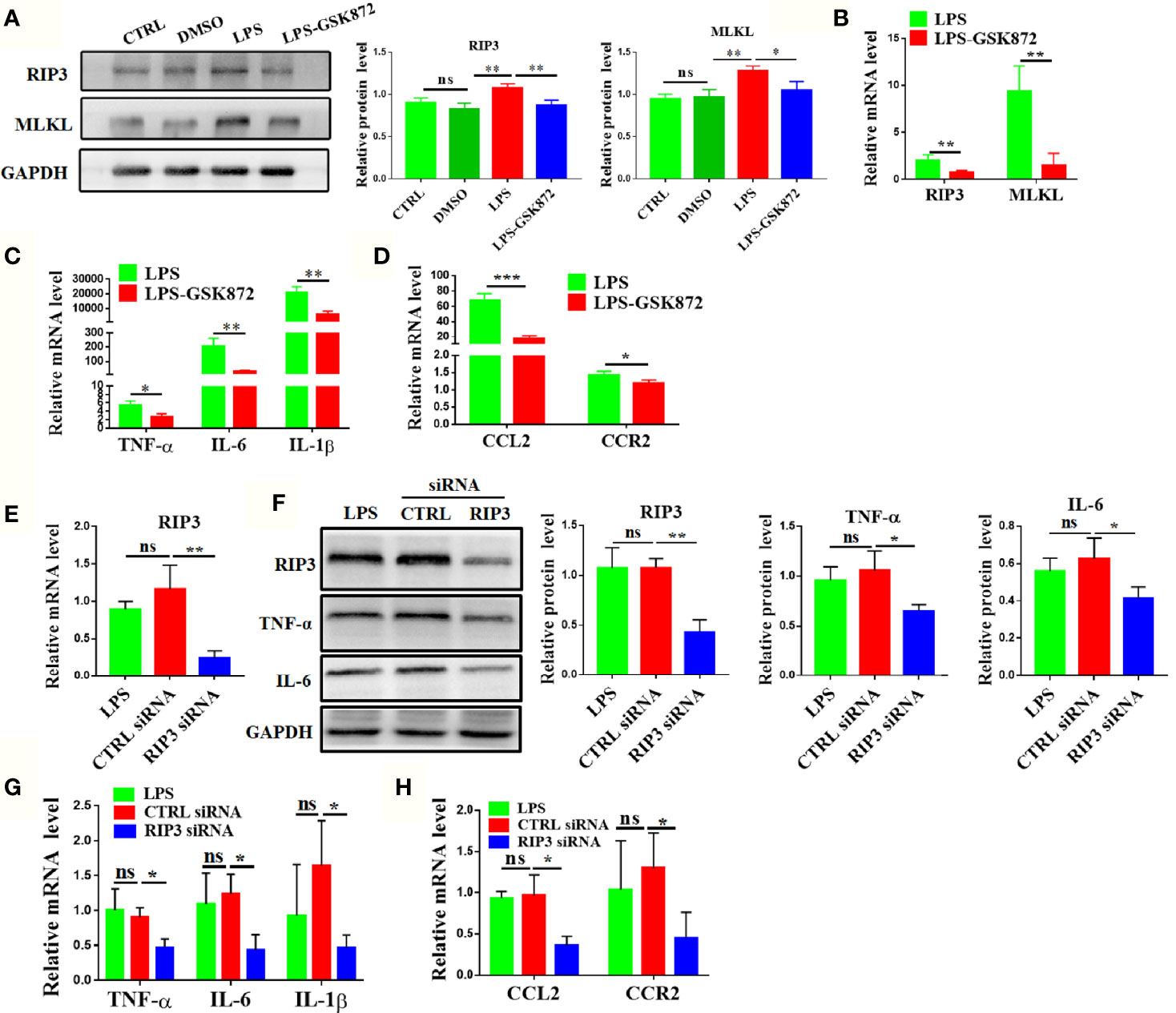
Figure 6 RIP3 signaling pathway regulates the expression of macrophage-related cytokines and chemokines in RAW264.7 cell lines. (A, B) Relative expression of RIP3 and MLKL was inhibited by GSK872. (C, D) Relative expression of TNF-α, IL-6, and IL-1β (C), CCL2 and CCR2(D) was down-regulated by GSK872. (E, F) Relative expression of RIP3 was inhibited by RIP3 siRNA and key cytokines such as TNF-α and IL-6 were detected and the relative intensity was quantified. (G, H) Relative expression of TNF-α, IL-6, and IL-1β (G), CCL2 and CCR2(H) was down-regulated by RIP3 siRNA. The data were presented as means ± SD of three independent experiments (Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ns: p > 0.05).
Disruption of the intestinal barrier lead to bacterial translocation, which consecutively activates immune cells to release various proinflammatory cytokines and chemokines (31). To test whether the gut commensal bacteria are required for RIP3 activation, EAH mouse model displaying dysbiosis in fecal microbiomes was used. Figures 7A, B shows that as measured by observed index and fisher index, the EAH group had significantly decreased alpha-diversity compared with the CTRL group. Principal component analysis based on weighted UniFrac distances revealed a different structure between the two groups (Figure 7C). The gut microbiota of all the samples in the two groups were dominated by three major phyla: Bacteroidetes, Firmicutes, and Proteobacteria (Figure 7D). Compared with those of the CTRL group, higher abundance of Bacteroidetes and lower abundance of Firmicutes and Proteobacteria were found in the EAH group. This phenomenon resulted in a decreased Firmicutes/Bacteroidetes (F/B) ratio (0.19) in the EAH group compared with that in the CTRL group (1.32). Genus-level analysis revealed that the mice in the EAH group had increased relative abundance of potential pathogenic bacteria, such as Bacteroides and Prevotellaceae_UCG-001, and a relatively low abundance of Lactobacillus and Ruminiclostridium (Figure 7E). Gut microbiota was also compared between the two groups through linear discriminant analysis effect size (LEfSe) to identify the specific microbiota linked to EAH. Prevotellaceae, which is associated with autoimmune diseases such as rheumatoid arthritis (32), were highly abundant in the EAH group (Figure 7F).
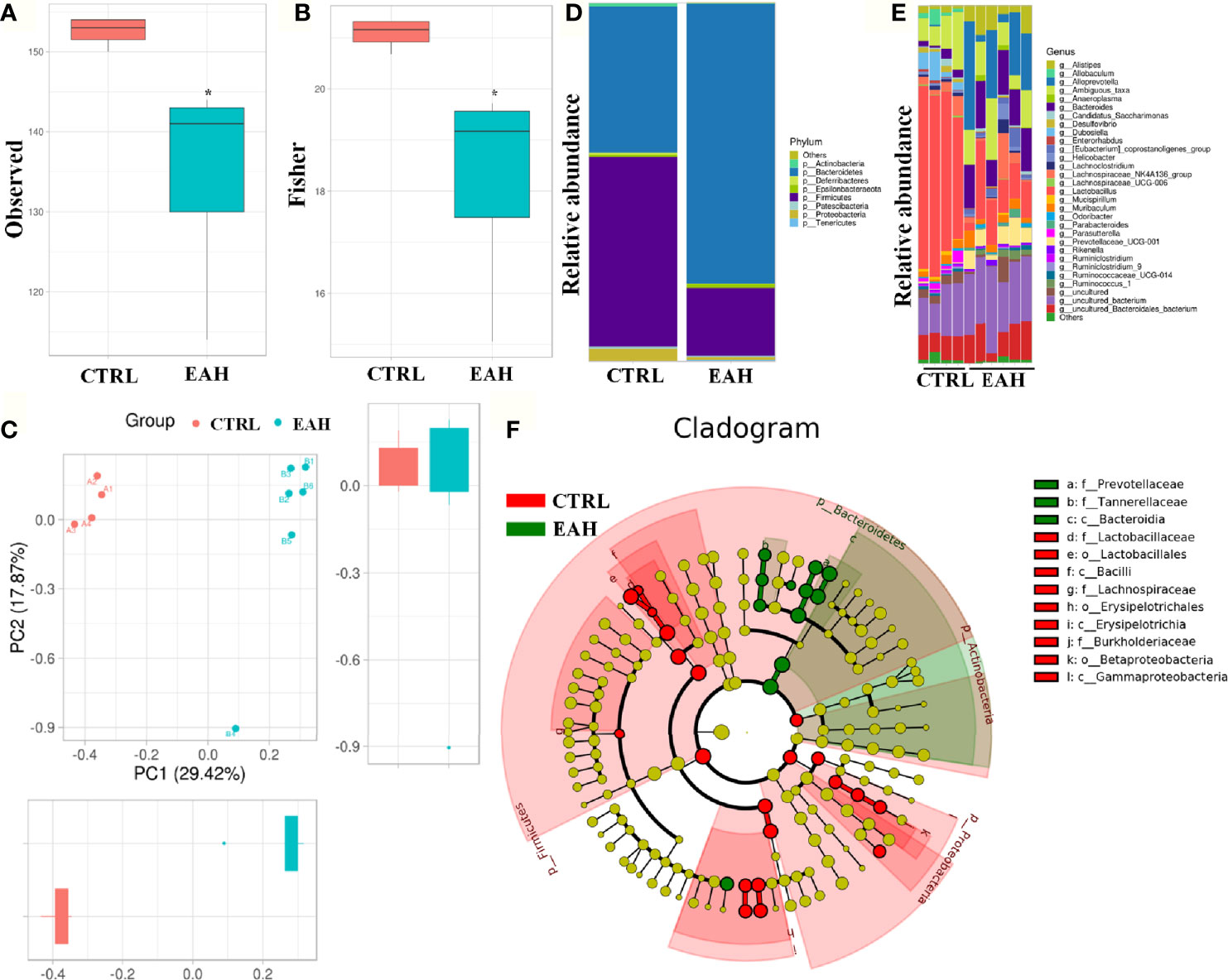
Figure 7 Alteration of the gut microbiota composition in EAH mice. (A, B) Observed (A) and Fisher (B) diversity indexes of the gut microbiota in CTRL group (n=4) and EAH group (n=6). (C) ANOSIM based on weighted UniFrac distances. (D) Bar charts of the gut microbiota composition at the phylum level in CTRL group and EAH group. (E) Bar charts of the gut microbiota composition at the genus level in each mouse. (F) Cladogram generated from the LEfSe. (*p < 0.05).
To analyze the role of gut commensal microbiota in liver immunity, the mice were treated with broad-spectrum antibiotic mixture prior to EAH induction. As shown in Supplementary Figure 2B, the mice in the EAH group had lower body weight than those in the CTRL group after 4 weeks, whereas no significant weight loss was found in the Abx group. In addition, the liver index and spleen index were significantly increased in the EAH group compared with those in the CTRL group, whereas no significant increase was observed in the Abx group (Supplementary Figure 2C, D). OTU comparison among the three groups revealed lower abundance of OTUs in the Abx group compared with those in the CTRL and EAH groups (28 OTUs vs. 176 OTUs vs. 181 OTUs). Among the 28 OTUs in the Abx group, 25 were shared by the three groups (Figures 8A, B). Analysis of Chao1 index and Shannon index indicated that the community richness and diversity also significantly decreased in the Abx group relative to those in the other two groups (Figure 8C). Remarkably, none of the mice in Abx group had developed hepatitis, and the transaminase level of the mice in the Abx group nearly returned to normal (Figures 8D, E). The intestinal permeability of these mice was also evaluated, and the results showed that the plasma FITC-D and LPS levels of Abx-treated mice were significantly decreased compared with those of the EAH mice (Figure 8F). The RIP3 signaling pathway was significantly inhibited with lower expression of inflammation cytokines and chemokines in liver tissues of the Abx group compared with those of the EAH group (Figures 8G–I). Besides, we evaluated the changes of liver macrophages by immunofluorescence and the results revealed that the mice in EAH group had increased number of macrophages in the liver tissue and the macrophages was significantly activated than that in the CTRL group, however, the broad-spectrum antibiotic mixture significantly alleviated the accumulation and activation of liver macrophages as shown in Supplementary Figure 7.

Figure 8 RIP3-mediated activation and infiltration of liver macrophages requires gut commensal microbiota. (A, B) Bacterial OUT heatmap (A) and Venn diagrams (B) in the CTRL group (n=4), EAH group (n=6) and Abx group (n=6). (C) The community richness and diversity of the gut microbiota were evaluated with Chao1 and Shannon indexes. (D) HE staining of the liver tissue from three groups. (E) The plasma concentrations of ALT and AST of three groups. (F)The plasma concentrations of FITC-D and LPS of three groups. (G–I) RT-qPCR analysis of RIP3 and MLKL (G) TNF-α, IL-6, and IL-1β (H) CCL2 and CCR2 (I) in three groups. Scale bars: 100μm. The data were presented as means ± SD (Student’s t-test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, ns: p > 0.05).
It’s known that alterations in the gut microbiota have been related with most autoimmune diseases, but in most cases, it remains unclear whether these changes are a cause or effect of the disease or merely a reflection of epidemiological differences between groups. The gut–liver axis has clinical importance as a potential therapeutic target in a wide range of chronic liver diseases (33–35). Recent evidence suggests that the intestinal environment specifically, modifications of the microbiome profile, regulate the pathogenesis of AIH by inducing intestinal inflammation and increasing gut permeability (36). In this present study, we try to explore the causality between the leaky gut/dysbiosis and AIH, demonstrating that loss of gut barrier integrity breaks liver immune homeostasis and augments liver injury.
The gut barrier is a fundamental gatekeeper to prevent translocation of bacterial components and the intestinal epithelial barrier (IEB) are crucial to prevent the passage of commensal bacteria and pathogens, from the lumen into the systemic circulation (37). The IEB is a single layer of epithelial cells held together by a complex junctional system composed of tight junctions, adherent junctions, and desmosomes (38). Recent studies indicated that leaky gut and increased intestinal permeability contributed to disease initiation and progression (39–41). Alterations of gut barrier integrity are found in patients affected by extraintestinal autoimmune diseases, but a direct causal link between enteropathy and triggering of autoimmunity is yet to be established. It was reported that, in mice models of T1D, loss of gut barrier integrity can lead to activation of islet-reactive T cells within the intestinal mucosa (42). Remarkably, Vieira et al. found that translocation of a gut pathobiont, Enterococcus gallinarum, to the liver and other systemic tissues triggers autoimmune responses in a genetic background predisposing to autoimmunity (43). In the present study, we demonstrated that the expression of tight junction proteins and the intestinal permeability is altered in AIH patients. Those alterations were detected at an early stage in AIH progression that is concomitant with the activation of the liver macrophages. Dysbiosis and reduction of tight junction proteins could ultimately lead to intestinal bacterial translocation with increased serum LPS levels and broken liver immune homeostasis that we observed in AIH patients and EAH mice.
The presence of loss of gut barrier integrity in patients and mice models of autoimmune diseases, such as T1D and systemic lupus erythematosus, has been known for long time but a causal link between the intestinal alterations and induction autoimmunity was never established (44). Our data demonstrate that loss of gut barrier integrity and modifications of the structural proteins prior to hepatitis, break liver immune tolerance, thus augment liver injury. Different triggering events have been reported such as viral infections or any factor that perturbs the liver environment leading to inflammation, tissue damage, and the release of sequestered liver antigen resulting in the stimulation of autoreactive immune cells (34). Here, we show that breakage of the gut barrier is one of those events that unleashes liver autoimmunity and provokes liver injury. The liver is particularly enriched in macrophages, which protect against infection, control host–microbiota mutualism, and maintain liver homeostasis (15, 45). Our data showed that intestinal barrier disruption increased activation and accumulation of liver macrophages thus aggravated liver injury. In the present study, we analyzed subsets of liver immune cells via flow cytometry and found the ratio of CD45+ F4/80hi CD11blo infiltrating macrophages to CD45+ F4/80lo CD11bhi Kupffer cells significantly increased in DSS-Con A group.
RIP3 is an essential part of the cellular machinery that executes “programmed” or “regulated” necrosis. Bacterial products such as LPS can activate the RIP3 signaling pathway (46, 47). Upon activation, the necrosome complex phosphorylates MLKL, leading to fatal permeabilization of the plasma membrane that exerts pro-inflammatory functions (21). In agreement with the induced gene expression of necrosome-related genes, Western blot analyses of liver specimen revealed the elevated levels of RIP3 protein in DSS-Con A mice and EAH mice. Whether RIP3 regulates the activation and infiltration of liver macrophage in DSS-Con A mice remains unclear. Thus, these mice were treated with GSK872 to dampen the RIP3 activation and thus significantly ameliorated the activation and accumulations of liver macrophages. The same results were observed in vitro. Our data are in accordance with a recent report showing that the RIP3 expression is up-regulated in liver tissues and macrophages of humans and mice with liver fibrosis; in addition, the absence of RIP3 in macrophages could alleviate inflammation and macrophage or neutrophil accumulation in mice after carbon tetrachloride or bile duct ligation treatment (48). It’s worth noting that the link between the RIP3 pathway and AIH is not definitively clear without gene knock-out (KO). However, it’s reported that RIP3 regulates stem cells generation through modulating cell cycle progression genes and RIP3 KO displayed lower expression of cell cycle genes and a slower proliferation rate compared to wild type (49). Besides, Patrick-Simon Welz et al. demonstrated that genetic deficiency in RIP3 prevented the development of spontaneous pathology in both the small intestine and colon of mice, which may prevent us from studying the role of the intestinal barrier in AIH (50). For the above reasons, we used GSK872, a RIP3 specific inhibitor in vivo and in vitro experiments to illustrate the role of RIP3 pathway in AIH. Our data suggested that the loss of intestinal barrier augmented liver macrophage activation and infiltration and the RIP3 signaling pathway might be the underlying molecular mechanism of intestinal barrier disruption on AIH pathogenesis.
The gut microbiota has a strong impact in AIH pathogenesis as demonstrated both in humans and mice models, but it is still unclear how commensal bacteria modulate liver autoimmunity. In fact, while in other autoimmune diseases such as rheumatoid arthritis, autoimmune encephalomyelitis and multiple sclerosis, the gut microbiota plays a clear triggering role (51, 52). In AIH, key epitopes that might trigger the disease might be sought among environmental agents especially within the intestinal microbiome (1, 53). Our data showed that gut inflammation by itself in EAH mice depleted of endogenous microbiota is not capable to activate liver autoimmunity, which suggests that the commensal gut microbiota is required for the activation of innate immune response. The intestinal microbiota maintains gut barrier integrity, shapes the mucosal immune system and balances host defense with microbial metabolites, components, and attachment to host cells (54). While the role of gut microbiota in liver immunity is still controversial. In some cases, it plays a beneficial effect, for example, in SPF and gnotobiotic mice, gut microbiota and commensal D-lactate programs Kupffer cells to capture and kill circulating pathogens (55). On the contrary, the gut microbiota contributes to a mouse model of spontaneous bile duct inflammation and GF mice develop a milder biliary affection, thus suggesting that commensal strains are important to trigger liver autoimmunity (56).
Our study showed that the intestinal barrier and gut microbiota in patients with AIH and murine models were destroyed. Diminished intestinal barrier function contributed to the activation and accumulation of liver macrophages via RIP3 signaling pathway. This phenomenon further aggravated the immune response in the inflamed liver. Our results revealed the novel mechanism of immune tolerance breakage in the liver via the gut–liver axis. In addition, therapeutic and research potentials of regulating the intestinal microbiota for AIH therapy were explored.
We have uploaded the 16S rRNA data into the SRA database successfully. The SRA records will be accessible with the following link: https://www.ncbi.nlm.nih.gov/sra/PRJNA678135.
The studies involving human participants were reviewed and approved by the Animal Ethical and Welfare Committee of Tianjin Medical University. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Animal Ethical and Welfare Committee of Tianjin Medical University.
LZ and BW designed the study. HZ, ML, WZ and YZ performed the experiments. YL, LG, YZ, YR, WC and JZ analyzed the results. HZ and LZ wrote the paper. All authors contributed to the article and approved the submitted version.
This study was supported by the grants (81860109 and 81470834) from the National Natural Science Foundation of China.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the National Natural Science Foundation of China for their financial support. The funders had no role in study design, data collection, data analysis, interpretation, writing and revision of the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.624360/full#supplementary-material
1. Mack CL, Adams D, Assis DN, Kerkar N, Manns MP, Mayo MJ, et al. Diagnosis and management of autoimmune hepatitis in adults and children: 2019 practice guidance and guidelines from the American Association for the study of liver diseases. Hepatology (2020) 72:671–722. doi: 10.1002/hep.31065
2. Freedman SN, Shahi SK, Mangalam AK. The “Gut Feeling”: Breaking Down the Role of Gut Microbiome in Multiple Sclerosis. Neurotherapeutics (2018) 15:109–25. doi: 10.1007/s13311-017-0588-x
3. Vatanen T, Franzosa EA, Schwager R, Tripathi S, Arthur TD, Vehik K, et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature (2018) 562:589–94. doi: 10.1038/s41586-018-0620-2
4. Nakamoto N, Sasaki N, Aoki R, Miyamoto K, Suda W, Teratani T, et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat Microbiol (2019) 4:492–503. doi: 10.1038/s41564-018-0333-1
5. Crispe IN. The liver as a lymphoid organ. Annu Rev Immunol (2009) 27:147–63. doi: 10.1146/annurev.immunol.021908.132629
6. Thomson AW, Knolle PA. Antigen-presenting cell function in the tolerogenic liver environment. Nat Rev Immunol (2010) 10:753–66. doi: 10.1038/nri2858
7. Albillos A, de Gottardi A, Rescigno M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J Hepatol (2020) 72:558–77. doi: 10.1016/j.jhep.2019.10.003
8. Camilleri M. Leaky gut: mechanisms, measurement and clinical implications in humans. Gut (2019) 68:1516–26. doi: 10.1136/gutjnl-2019-318427
9. Lazaridis KN, LaRusso NF. Primary Sclerosing Cholangitis. N Engl J Med (2016) 375:1161–70. doi: 10.1056/NEJMra1506330
10. Eksteen B. The Gut-Liver Axis in Primary Sclerosing Cholangitis. Clin Liver Dis (2016) 20:1–14. doi: 10.1016/j.cld.2015.08.012
11. Sica A, Invernizzi P, Mantovani A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology (2014) 59:2034–42. doi: 10.1002/hep.26754
12. Grønbaek H, Kreutzfeldt M, Kazankov K, Jessen N, Sandahl T, Hamilton-Dutoit S, et al. Single-centre experience of the macrophage activation marker soluble (s)CD163 - associations with disease activity and treatment response in patients with autoimmune hepatitis. Aliment Pharm Ther (2016) 44:1062–70. doi: 10.1111/apt.13801
13. Assis DN, Leng L, Du X, Zhang CK, Grieb G, Merk M, et al. The role of macrophage migration inhibitory factor in autoimmune liver disease. Hepatol (Baltimore Md) (2014) 59:580–91. doi: 10.1002/hep.26664
14. Assis DN, Takahashi H, Leng L, Zeniya M, Boyer JL, Bucala R. A Macrophage Migration Inhibitory Factor Polymorphism Is Associated with Autoimmune Hepatitis Severity in US and Japanese Patients. Digest Dis Sci (2016) 61:3506–12. doi: 10.1007/s10620-016-4322-z
15. Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol (2017) 66:1300–12. doi: 10.1016/j.jhep.2017.02.026
16. Roychowdhury S, McCullough RL, Sanz-Garcia C, Saikia P, Alkhouri N, Matloob A, et al. Receptor interacting protein 3 protects mice from high-fat diet-induced liver injury. Hepatology (2016) 64:1518–33. doi: 10.1002/hep.28676
17. Chenxu G, Minxuan X, Yuting Q, Tingting G, Jing F, Jinxiao L, et al. Loss of RIP3 initiates annihilation of high-fat diet initialized nonalcoholic hepatosteatosis: A mechanism involving Toll-like receptor 4 and oxidative stress. Free Radic Biol Med (2019) 134:23–41. doi: 10.1016/j.freeradbiomed.2018.12.034
18. Shrestha A, Mehdizadeh Gohari I, McClane BA. RIP1, RIP3, and MLKL Contribute to Cell Death Caused by Clostridium perfringens Enterotoxin. mBio (2019) 10:e02985–19. doi: 10.1128/mBio.02985-19
19. Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell (2012) 148:213–27. doi: 10.1016/j.cell.2011.11.031
20. Moriwaki K, Chan FK. RIP3: a molecular switch for necrosis and inflammation. Genes Dev (2013) 27:1640–9. doi: 10.1101/gad.223321.113
21. Wang H, Sun L, Su L, Rizo J, Liu L, Wang LF, et al. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol Cell (2014) 54:133–46. doi: 10.1016/j.molcel.2014.03.003
22. Zhang H, Liu M, Liu X, Zhong W, Li Y, Ran Y, et al. Bifidobacterium animalis ssp. Lactis 420 mitigates autoimmune hepatitis through regulating intestinal barrier and liver immune cells. Front Immunol (2020) 11:569104. doi: 10.3389/fimmu.2020.569104
23. Alvarez F, Berg PA, Bianchi FB, Bianchi L, Burroughs AK, Cancado EL, et al. International Autoimmune Hepatitis Group Report: review of criteria for diagnosis of autoimmune hepatitis. J Hepatol (1999) 31:929–38. doi: 10.1016/s0168-8278(99)80297-9
24. Hennes EM, Zeniya M, Czaja AJ, Pares A, Dalekos GN, Krawitt EL, et al. Simplified criteria for the diagnosis of autoimmune hepatitis. Hepatology (2008) 48:169–76. doi: 10.1002/hep.22322
25. Lohse AW, Manns M, Dienes HP, Meyer ZBK, Cohen IR. Experimental autoimmune hepatitis: disease induction, time course and T-cell reactivity. Hepatology (1990) 11:24–30. doi: 10.1002/hep.1840110106
26. Le Roy T, Llopis M, Lepage P, Bruneau A, Rabot S, Bevilacqua C, et al. Intestinal microbiota determines development of non-alcoholic fatty liver disease in mice. Gut (2013) 62:1787–94. doi: 10.1136/gutjnl-2012-303816
27. Fasano A. Zonulin and its regulation of intestinal barrier function: the biological door to inflammation, autoimmunity, and cancer. Physiol Rev (2011) 91:151–75. doi: 10.1152/physrev.00003.2008
28. Wei Y, Li Y, Yan L, Sun C, Miao Q, Wang Q, et al. Alterations of gut microbiome in autoimmune hepatitis. Gut (2020) 69:569–77. doi: 10.1136/gutjnl-2018-317836
29. Moriwaki K, Chan FK. Necrosis-dependent and independent signaling of the RIP kinases in inflammation. Cytokine Growth Factor Rev (2014) 25:167–74. doi: 10.1016/j.cytogfr.2013.12.013
30. Chan FK, Luz NF, Moriwaki K. Programmed necrosis in the cross talk of cell death and inflammation. Annu Rev Immunol (2015) 33:79–106. doi: 10.1146/annurev-immunol-032414-112248
31. Rizzetto L, Fava F, Tuohy KM, Selmi C. Connecting the immune system, systemic chronic inflammation and the gut microbiome: The role of sex. J Autoimmun (2018) 92:12–34. doi: 10.1016/j.jaut.2018.05.008
32. Alpizar-Rodriguez D, Lesker TR, Gronow A, Gilbert B, Raemy E, Lamacchia C, et al. Prevotella copri in individuals at risk for rheumatoid arthritis. Ann Rheum Dis (2019) 78:590–3. doi: 10.1136/annrheumdis-2018-214514
33. Dean G, Hanauer S, Levitsky J. The Role of the Intestine in the Pathogenesis of Primary Sclerosing Cholangitis: Evidence and Therapeutic Implications. Hepatology (2020) 72:1127–38. doi: 10.1002/hep.31311
34. Dubinkina VB, Tyakht AV, Odintsova VY, Yarygin KS, Kovarsky BA, Pavlenko AV, et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome (2017) 5:41. doi: 10.1186/s40168-017-0359-2
35. Zhu L, Baker SS, Gill C, Liu W, Alkhouri R, Baker RD, et al. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: a connection between endogenous alcohol and NASH. Hepatology (2013) 57:601–9. doi: 10.1002/hep.26093
36. Mieli-Vergani G, Vergani D, Czaja AJ, Manns MP, Krawitt EL, Vierling JM, et al. Autoimmune hepatitis. Nat Rev Dis Primers (2018) 14:18017. doi: 10.1038/nrdp.2018.17
37. Chelakkot C, Ghim J, Ryu SH. Mechanisms regulating intestinal barrier integrity and its pathological implications. Exp Mol Med (2018) 50:103. doi: 10.1038/s12276-018-0126-x
38. Odenwald MA, Turner JR. The intestinal epithelial barrier: a therapeutic target? Nat Rev Gastroenterol Hepatol (2017) 14:9–21. doi: 10.1038/nrgastro.2016.169
39. Costa FR, Francozo MC, de Oliveira GG, Ignacio A, Castoldi A, Zamboni DS, et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J Exp Med (2016) 213:1223–39. doi: 10.1084/jem.20150744
40. Kim JW, Kwok SK, Choe JY, Park SH. Recent Advances in Our Understanding of the Link between the Intestinal Microbiota and Systemic Lupus Erythematosus. Int J Mol Sci (2019) 20:4871. doi: 10.3390/ijms20194871
41. Chiang HI, Li JR, Liu CC, Liu PY, Chen HH, Chen YM, et al. An Association of Gut Microbiota with Different Phenotypes in Chinese Patients with Rheumatoid Arthritis. J Clin Med (2019) 8:1770. doi: 10.3390/jcm8111770
42. Sorini C, Cosorich I, Lo Conte M, De Giorgi L, Facciotti F, Lucianò R, et al. Loss of gut barrier integrity triggers activation of islet-reactive T cells and autoimmune diabetes. Proc Natl Acad Sci U S A (2019) 116:15140–9. doi: 10.1073/pnas.1814558116
43. Manfredo Vieira S, Hiltensperger M, Kumar V, Zegarra-Ruiz D, Dehner C, Khan N, et al. Translocation of a gut pathobiont drives autoimmunity in mice and humans. Science (2018) 359:1156–61. doi: 10.1126/science.aar7201
44. Fasano A. Leaky gut and autoimmune diseases. Clin Rev Allergy Immunol (2012) 42:71–8. doi: 10.1007/s12016-011-8291-x
45. Endo-Umeda K, Nakashima H, Komine-Aizawa S, Umeda N, Seki S, Makishima M. Liver X receptors regulate hepatic F4/80 (+) CD11b(+) Kupffer cells/macrophages and innate immune responses in mice. Sci Rep (2018) 8:9281. doi: 10.1038/s41598-018-27615-7
46. Nikseresht S, Khodagholi F, Nategh M, Dargahi L. RIP1 Inhibition Rescues from LPS-Induced RIP3-Mediated Programmed Cell Death, Distributed Energy Metabolism and Spatial Memory Impairment. J Mol Neurosci (2015) 57:219–30. doi: 10.1007/s12031-015-0609-3
47. Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W, et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PloS One (2016) 11:e155723. doi: 10.1371/journal.pone.0155723
48. Wei S, Zhou H, Wang Q, Zhou S, Li C, Liu R, et al. RIP3 deficiency alleviates liver fibrosis by inhibiting ROCK1-TLR4-NF-kappaB pathway in macrophages. FASEB J (2019) 33:11180–93. doi: 10.1096/fj.201900752R
49. Al-Moujahed A, Tian B, Efstathiou NE, Konstantinou EK, Hoang M, Lin H, et al. Receptor interacting protein kinase 3 (RIP3) regulates iPSCs generation through modulating cell cycle progression genes. Stem Cell Res (2019) 35:101387. doi: 10.1016/j.scr.2019.101387
50. Welz P, Wullaert A, Vlantis K, Kondylis V, Fernández-Majada V, Ermolaeva M, et al. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature (2011) 477:330–4. doi: 10.1038/nature10273
51. Berer K, Mues M, Koutrolos M, Rasbi ZA, Boziki M, Johner C, et al. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature (2011) 479:538–41. doi: 10.1038/nature10554
52. Taurog JD, Richardson JA, Croft JT, Simmons WA, Zhou M, Fernández-Sueiro JL, et al. The germfree state prevents development of gut and joint inflammatory disease in HLA-B27 transgenic rats. J Exp Med (1994) 180:2359–64. doi: 10.1084/jem.180.6.2359
53. Czaja AJ. Factoring the intestinal microbiome into the pathogenesis of autoimmune hepatitis. World J Gastroenterol (2016) 22:9257–78. doi: 10.3748/wjg.v22.i42.9257
54. Kayama H, Okumura R, Takeda K. Interaction Between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annu Rev Immunol (2020) 38:23–48. doi: 10.1146/annurev-immunol-070119-115104
55. McDonald B, Zucoloto AZ, Yu I, Burkhard R, Brown K, Geuking MB, et al. Programing of an Intravascular Immune Firewall by the Gut Microbiota Protects against Pathogen Dissemination during Infection. Cell Host Microbe (2020) 28:660–668.e4. doi: 10.1016/j.chom.2020.07.014
Keywords: autoimmune hepatitis, intestinal barrier, dysbiosis, macrophages, RIP3 signaling pathway, gut-liver axis
Citation: Zhang H, Liu M, Zhong W, Zheng Y, Li Y, Guo L, Zhang Y, Ran Y, Zhao J, Zhou L and Wang B (2021) Leaky Gut Driven by Dysbiosis Augments Activation and Accumulation of Liver Macrophages via RIP3 Signaling Pathway in Autoimmune Hepatitis. Front. Immunol. 12:624360. doi: 10.3389/fimmu.2021.624360
Received: 31 October 2020; Accepted: 02 March 2021;
Published: 25 March 2021.
Edited by:
Gregg Joshua Silverman, New York University, United StatesReviewed by:
Shruti Sharma, Tufts University School of Medicine, United StatesCopyright © 2021 Zhang, Liu, Zhong, Zheng, Li, Guo, Zhang, Ran, Zhao, Zhou and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bangmao Wang, Z2kudG11aEBzb2h1LmNvbQ==; Lu Zhou, bHpob3UwMUB0bXUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.