
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 10 March 2021
Sec. Vaccines and Molecular Therapeutics
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.624310
This article is part of the Research Topic Fighting an Elusive Enemy: Staphylococcus aureus and its Antibiotic Resistance, Immune-Evasion and Toxic Mechanisms View all 20 articles
 Hatice Karauzum1†
Hatice Karauzum1† Arundhathi Venkatasubramaniam1†
Arundhathi Venkatasubramaniam1† Rajan P. Adhikari1Tom Kort1
Rajan P. Adhikari1Tom Kort1 Frederick W. Holtsberg1Ipsita Mukherjee1Mark Mednikov1
Frederick W. Holtsberg1Ipsita Mukherjee1Mark Mednikov1 Roger Ortines1
Roger Ortines1 Nhu T. Q. Nguyen2Thien M. N. Doan2Binh An Diep2
Nhu T. Q. Nguyen2Thien M. N. Doan2Binh An Diep2 Jean C. Lee3
Jean C. Lee3 M. Javad Aman1*
M. Javad Aman1*Staphylococcus aureus causes a wide range of diseases from skin infections to life threatening invasive diseases such as bacteremia, endocarditis, pneumonia, surgical site infections, and osteomyelitis. Skin infections such as furuncles, carbuncles, folliculitis, erysipelas, and cellulitis constitute a large majority of infections caused by S. aureus (SA). These infections cause significant morbidity, healthcare costs, and represent a breeding ground for antimicrobial resistance. Furthermore, skin infection with SA is a major risk factor for invasive disease. Here we describe the pre-clinical efficacy of a multicomponent toxoid vaccine (IBT-V02) for prevention of S. aureus acute skin infections and recurrence. IBT-V02 targets six SA toxins including the pore-forming toxins alpha hemolysin (Hla), Panton-Valentine leukocidin (PVL), leukocidin AB (LukAB), and the superantigens toxic shock syndrome toxin-1 and staphylococcal enterotoxins A and B. Immunization of mice and rabbits with IBT-V02 generated antibodies with strong neutralizing activity against toxins included in the vaccine, as well as cross-neutralizing activity against multiple related toxins, and protected against skin infections by several clinically relevant SA strains of USA100, USA300, and USA1000 clones. Efficacy of the vaccine was also shown in non-naïve mice pre-exposed to S. aureus. Furthermore, vaccination with IBT-V02 not only protected mice from a primary infection but also demonstrated lasting efficacy against a secondary infection, while prior challenge with the bacteria alone was unable to protect against recurrence. Serum transfer studies in a primary infection model showed that antibodies are primarily responsible for the protective response.
Staphylococcus aureus (SA) is an opportunistic pathogen responsible for a wide range of clinical infections, ranging from superficial skin lesions, deep-seated abscesses, and osteomyelitis to life threatening sepsis, endocarditis and pneumonia (1, 2). SA has a propensity for acquiring resistance to antibiotics, with epidemics of penicillin-, methicillin- and vancomycin-resistant strains occurring since 1940 (3). Methicillin-resistant SA (MRSA) represents ~50% of SA infections in the US (4). Therefore, development of an effective vaccine against various SA diseases is of utmost importance. An effective vaccine can not only have a major impact on public health, but it will also significantly reduce the burden of antimicrobial resistance. Prior efforts for development of vaccines and immunotherapeutics have largely focused on the surface proteins or polysaccharides (ClfA, SdrG, IsdB, MntC, CP5, and CP8) (5, 6). Unfortunately, to-date these vaccines have failed to meet their clinical endpoints in human efficacy trials. Opsonic antibodies represent a reliable correlate of protection for several other vaccines such as pneumococcal, meningococcal and H. influenzae vaccines (7). Remarkably, all the SA vaccines advanced into clinic were strong inducers of antibodies that promoted opsonophagocytic activity (8–10), putting in question whether the paradigm of opsonic antibodies as correlate of immunity can be applied to SA. In the case of one of these vaccine candidates, the single component vaccine consisting of the iron-regulated protein IsdB (V710, Merck), the trial was stopped due to safety concerns after ~8,000 patients undergoing cardiothoracic surgery were recruited. Among those patients who developed a SA infection, a higher number of multi-organ failures and death occurred in the vaccine arm compared to placebo (11). A follow up study in a subset of these patients suggested that a combination of immunization with IsdB, SA infection, and the immunological status of the host (reflected in low or undetectable serum IL-2 and IL-17 at the time of vaccination) contributed to the catastrophic outcome (12). Furthermore, both studies in mice (13) and rabbits (14) showed a vaccination induced disease enhancement when whole cell vaccine or crude surface antigens were used for immunization, raising the potential for induction of a deleterious immune response by cell associated antigens.
Over the past two decades great advances have been made in our understanding of the immunological effects of the large number of toxins produced by SA and their role in pathogenesis (15–17). These toxins, in particular the cytolytic and superantigenic toxins, mediate a wide range of immune subversive and tissue destructive functions to support infection (18, 19). The cytolytic toxins comprise the single subunit alpha hemolysin (α-toxin, Hla) and a family of bicomponent toxins, including the closely related Panton-Valentine leukocidin (PVL), leukocidin ED (LukED), gamma hemolysins (HlgAB and HlgCB), as well as phylogenetically more distant leukocidin AB (LukAB; also referred to as LukGH) (18, 20). These cytolysins are involved in disruption of skin and mucosal barriers (Hla, PVL), killing of key innate immune cells at the first line of defense against SA (PVL, LukAB, LukED, HlgA/CB), platelet aggregation (Hla), and lysis of human red blood cells to extract iron for bacterial growth (HlgAB) (20). Superantigens (SAgs), comprising the staphylococcal enterotoxins (SEs such as SEA, SEB, SEC, SED, SEK) and toxic shock syndrome toxin-1 (TSST-1), can cause massive polyclonal activation of T cells leading to toxic shock, induce intoxication and dysregulation of a variety of specialized T cell subsets, and induce anergy and lymphocyte apoptosis (21, 22). Collectively, these toxins create a smoke screen rendering the host unable to mount an effective immune response (23). We hypothesized that vaccination with key staphylococcal toxins can protect against SA infections by protecting tissues and the immune system, thus preventing the complications of disease via clinical protection even in the absence of sterile immunity. To this end, we have developed a multicomponent vaccine IBT-V02, comprised of toxoids for Hla (HlaH35LH48L), subunits of PVL (LukSmut9, LukFmut1), LukAB (LukABmut50) and a fusion of three SAgs (TSST-1, SEB, and SEA; TBA225) and reported the attenuation and efficacy of individual components or partial combinations in bacteremia, pneumonia, and toxic shock models in mice and rabbits (24–27).
Skin and soft tissue infections (SSTI) caused by SA are of special significance worldwide due to their prevalence, the possibility of other disease complications and the associated costs (28, 29). Skin infections typically include furuncles, carbuncles, impetigo, cellulitis, and skin abscesses (30), and methicillin-resistant SA (MRSA) isolates have been found to play a major role in these infections, particularly clones such as USA100, USA300, ST-80, and USA1000 (31–34). Toxins secreted by these and other strains of SA serve as key virulence factors in the pathogenesis of SA skin infections, specifically Hla (35), PVL (36, 37) and SAgs (38, 39). Up to 50% of patients with a primary skin infection can experience one or more repeated bouts of recurrent infection over 1 year, even after successful initial treatment by incision and drainage and antibiotics (40–42). Healthy adults, patients with underlying chronic diseases like diabetes, cancer, vascular disease, eczema, lung disease, infection with human immunodeficiency virus, as well as specific populations (military personnel, athletes, injection drug users, prisoners) are at risk of primary and recurrent skin infections caused by SA (30, 43). Hence there is an urgent need for a vaccine providing protection against SA acute skin infections (SA-ASI) and its recurrence.
Here we report that IBT-V02 can induce a strong neutralizing antibody response in mice and rabbits and provide strong protection against primary and secondary SA-ASI. Importantly, we demonstrate that the vaccine protects in both naïve and immunologically pre-exposed mice, a finding that is relevant to human vaccine development since a large portion of the population is or has been colonized with SA. We further show that the vaccine can be administered during the acute phase of primary infection to protect against a subsequent encounter with SA.
IBT-V02 consists of rationally designed toxoid formulations representative of pore-forming and superantigenic toxins: Hla, PVL, LukAB, SEA, SEB, as well as TSST-1. The Hla toxoid harbors two mutations in the N-terminal domain (H35L and H48L) that renders the protein unable to heptamerize and is thus fully non-toxic (27, 44). We have previously described mutagenesis of the S and F subunits of our PVL toxoid LukSmut9 and LukFmut1 (24). LukSmut9 toxoid contains three mutations (T28F/K97A/S209A) and LukFmut1 toxoid harbors a single mutation (K102A). Each of these toxoids are fully attenuated in combination with the wild type or mutant PVL counterpart or non-canonical partners (24). LukABmut50 is a dimeric toxoid form of LukAB with mutations introduced in LukA (D39A) and LukB (R23E) that render the protein unable to form a pore in the plasma membrane of target cells (25). TBA225 is a fusion protein of the three superantigen toxoids for TSST-1, SEB and SEA (26). Each of the toxoids are mutated in the MHC Class II binding sites and therefore unable to crosslink T cells and antigen presenting cells, resulting in a loss of superantigenicity. The individual mutants are: TSST-1L30R/D27A/I46A, SEBL45R/Y89A/Y94A, and SEAL48R/D70R/Y92A/H225A. The three mutants are fused together via a flexible linker 3x(GGGGS) in the order of TSST-1, SEB, and SEA (26).
The individual toxoids were expressed in E. coli and purified by multistep column chromatography. The LukAB subunits were expressed from a single plasmid and copurified as a heterodimer. The individual components were blended at equal weight ratios. Figure 1 shows the SDS-PAGE (A) and Western blot (B) analysis of the blended vaccine components and their respective molecular weights. Additionally, using SEC-HPLC, the high level of purity of the individual components could be demonstrated (Figure 1C).
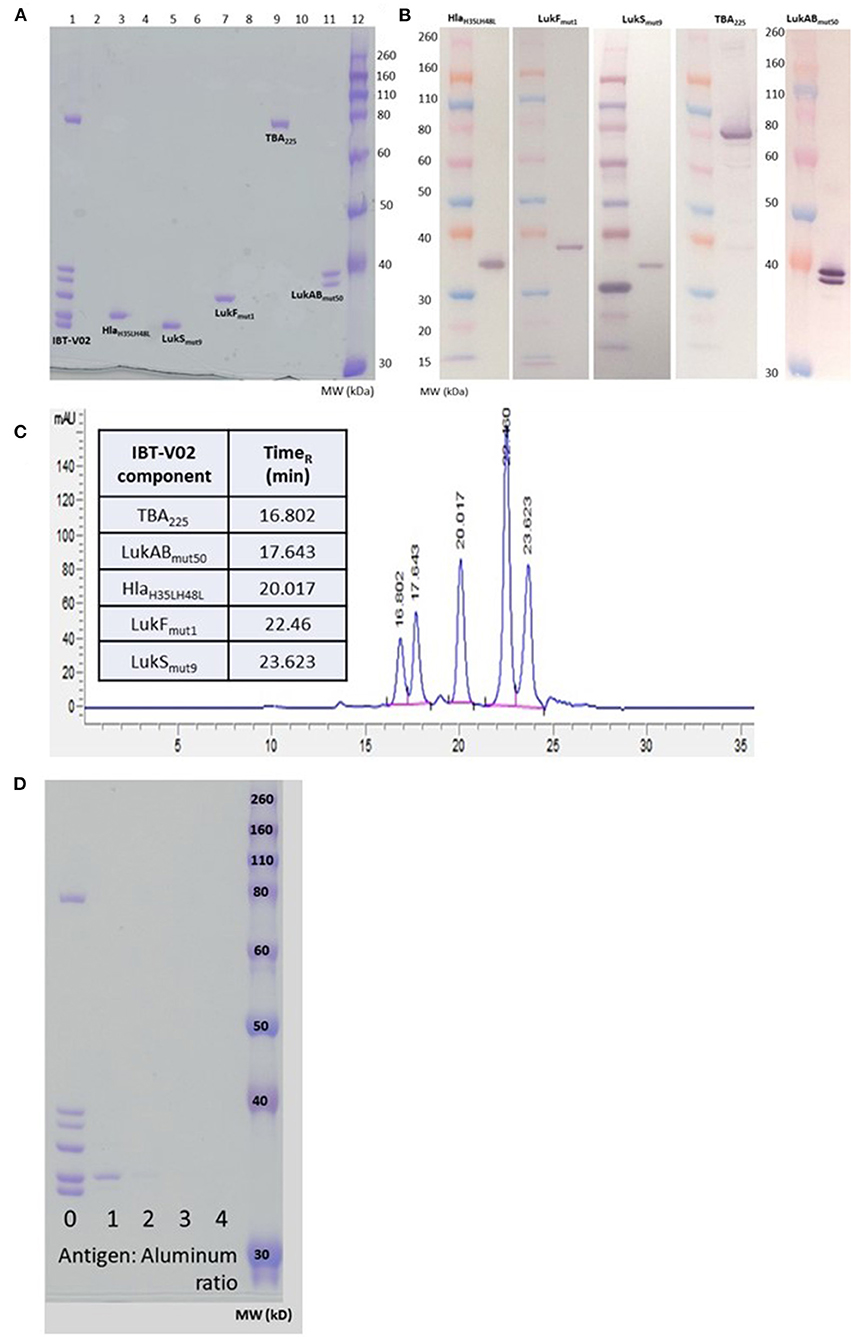
Figure 1. Biochemical characterization of IBT-V02 components. (A) SDS PAGE of blended (Lane 1) and individual components (Lanes 3, 5, 7, 9, 11). (B) Western Blot analysis and (C) SEC-HPLC of individual components. (D) SDS PAGE to determine protein adsorption of blended components to Al(OH)3.
We had previously demonstrated the immunogenicity of the IBT-V02 components in combination with Alhydrogel® (24–26). To optimize the formulation for the multicomponent vaccine we incubated the blended vaccine components with Alhydrogel® at ratios of 1, 2, 3, and 4 (protein:Alhydrogel®) for 60 min at room temperature (RT) followed by centrifugation. To detect any unadsorbed protein in the samples, the supernatants were run on SDS-PAGE and proteins visualized by Coomassie Blue staining. As shown in Figure 1D, a minimum of three-fold excess was needed for complete adsorption by Alhydrogel®.
Immunogenicity was evaluated in groups of 5 BALB/c mice vaccinated on days 0, 14, and 28 with either IBT-V02 (50 μg/mouse) or individual components (10 μg/mouse) formulated with Alhydrogel® at a ratio of 1:5. Sera were collected on day 42. Individual serum samples were used to determine total antigen specific IgG titers by a multiplex assay developed in house. Pooled sera from each group were also used to determine the neutralization titers against target antigens Hla, PVL, LukAB, SEA, SEB, TSST-1. Antibody titers were determined as the effective dilution of the sera that produced 50% maximal response (ED50). As shown in Figure 2A, IgG titers to Hla, LukS, or LukF generated by immunization with IBT-V02 were not significantly different from the titers induced by immunization with the individual toxoids, suggesting that blending the 5 toxoid components did not have a negative impact on the response to these antigens. Whereas, IBT-V02 induced robust serum antibody titers to LukAB (ED50 titers ranging from 37,608 to 84,051), SEA (ED50 titers from 2,070 to 10,352), SEB (ED50 titers from 30,898 to 104,426), and TSST-1 (ED50 titers from 28,681 to 86,701), these titers were significantly lower compared to titers generated to the individual toxoids. Nonetheless, the neutralizing antibody titers of serum samples were comparable between IBT-V02 and individual components (Figure 2B). Of the two PVL components, LukSmut9 appeared to make a larger contribution to PVL neutralization than LukFmut1 (Figure 2B). Overall, the data showed that IBT-V02 induces strong binding and neutralizing antibody titers in mice.
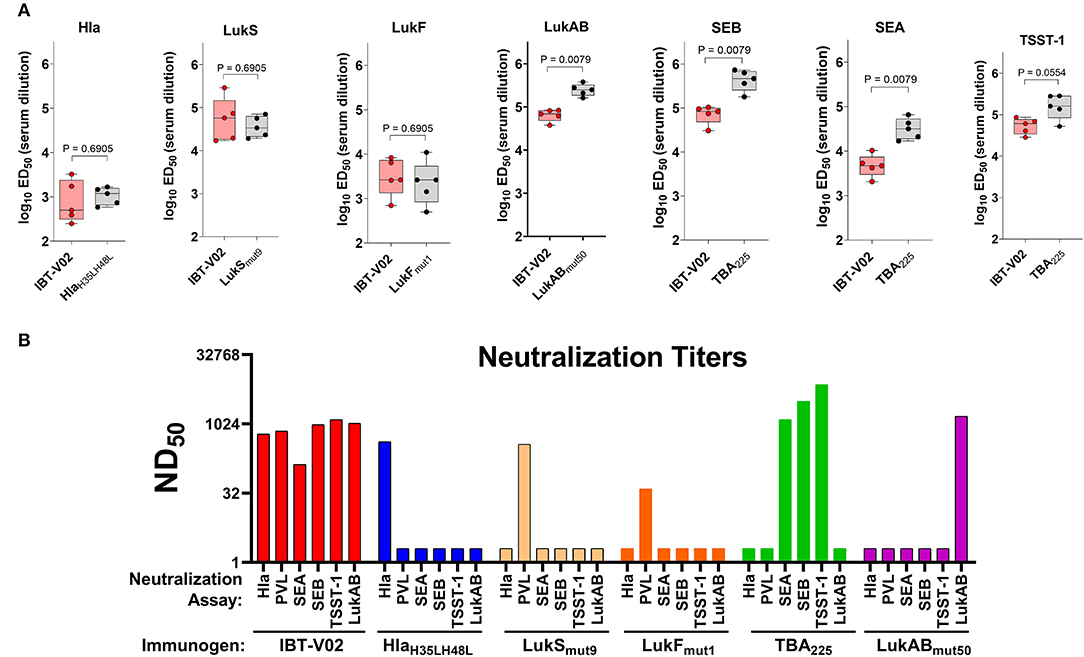
Figure 2. Immunogenicity of IBT-V02 in BALB/c mice, n = 5/group. (A) Serum IgG binding titers and (B) Serum toxin neutralizing titers of mice immunized with blended 5-component toxoid vaccine (IBT-V02) or with each individual toxoid tested against wildtype target antigens Hla, LukS, LukF, LukAB, SEA, SEB, and TSST-1.
To test the efficacy of IBT-V02 in an acute skin infection model, groups of 10 mice were vaccinated with 75, 50, or 25 μg of the vaccine candidate, or 75 μg of BSA as control with the same schedule described above. Mice were challenged with 1 × 107 CFU of USA300 (LAC) on day 42 by subcutaneous (SC) infection and were monitored for 14 days for lesion size, weight change, and any sign of disease. All mice vaccinated with 50 or 75 μg of IBT-V02 showed significantly smaller lesions than control animals at all time points (Figure 3A). The efficacy was also reflected in reduced weight loss (Figure 3B) and lower lesion severity scores (Figure 3C).
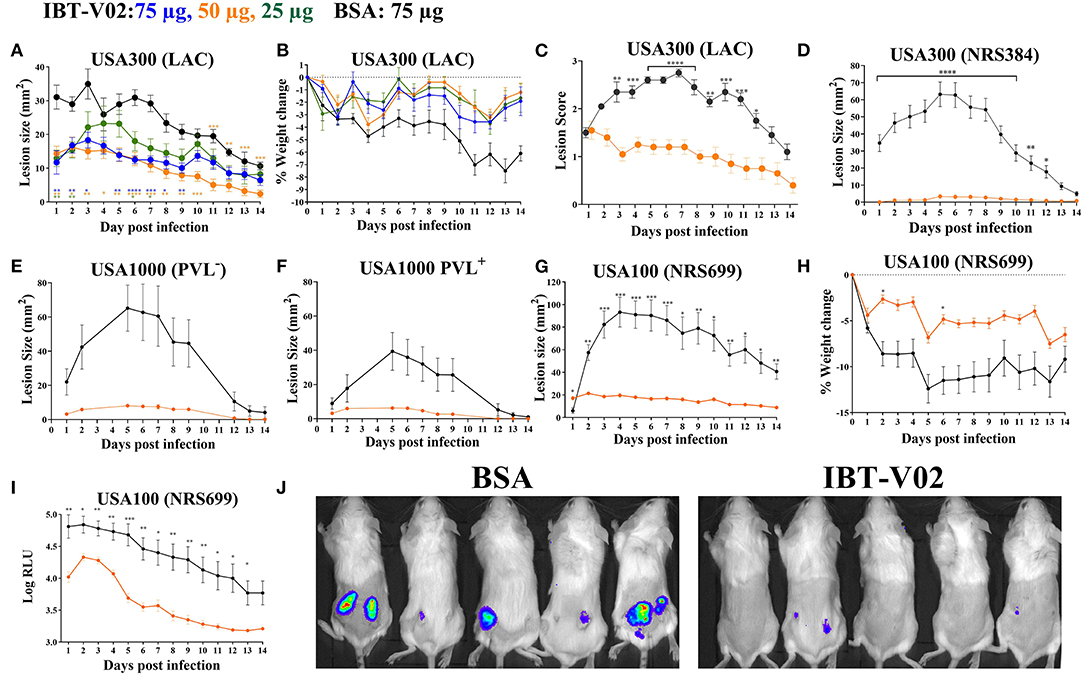
Figure 3. Efficacy of IBT-V02 in mouse skin infection. (A–C) Efficacy of IBT-V02 at 75, 50, and 25 μg doses against skin infection with USA300 (LAC), n = 5/group. (A) Lesion size, (B) weight loss and (C) lesion scores measured for 14 days. (D–J) Efficacy of IBT-V02 vs. BSA at 50 μg/mouse. Lesion size after infection with (D) USA300 (NRS384), n = 10/group, (E) USA1000 PVL−, n = 5/group or (F) USA1000 PVL+, n = 5/group. (G–J) Efficacy of IBT-V02 at 50 μg/mouse against luminescent USA100 (NRS699), n = 10/group. (G) Lesion size, (H) weight loss, (I) relative light units (RLU) in skin as a readout of bacterial burden, and (J) bioluminescent imaging of USA100-infected mice on day 7 post infection. Data were analyzed using GraphPad PRISM v8.4.3. Statistical analysis for lesion size performed using 2-way ANOVA, Sidak's multiple comparisons test.
The efficacy of IBT-V02 (50 μg/mouse) was further tested using another USA300 isolate (NRS384), a USA100 clone (NRS699) transduced with lux operon, as well as two USA1000 isolates. IBT-V02 showed remarkable efficacy against all strains as measured by lesion size, weight loss, or bacterial burden measured as luminescence intensity (Figures 3D–J).
To evaluate a potential protective role of vaccine elicited CD4 T cells, mice were immunized with either IBT-V02 or BSA, and CD4 T cells were depleted 72 h prior to and 48 h post-infection with 1 × 107 CFU of USA300 (NRS384) using an anti-CD4 monoclonal antibody (mAb clone GK1.5). Mice treated with isotype control antibodies were used as controls. As shown in Figure 4A, mice treated with anti-CD4 mAb showed lesions similar in size to mice treated with isotype control antibodies, indicating that the protective efficacy of IBT-V02 in mice was not driven by vaccine generated CD4 T cells. Interestingly, depletion of CD4 T cells in mice immunized with BSA resulted in larger lesions when compared to isotype treated BSA control. However, areas under the curve for mouse lesions showed no significant differences between these two groups (Figure 4A). Sera collected 2 weeks after the last immunization were characterized for toxin binding IgG titers (Figure 4B) as well as toxin neutralizing antibodies (Figure 4C). All mice demonstrated robust IgG titers toward all target antigens, with the exception of one mouse that showed low titers toward SEA (Figure 4B). Individual serum samples tested against Hla and serum pools tested against remaining target antigens all showed robust toxin neutralizing antibody titers (Figure 4C). Next, we evaluated the protective role of vaccine-generated serum antibodies by passive serum transfer studies. Immune sera were generated in ICR (CD1) mice through repeated immunization (4x 2-weeks apart). Sera were collected, pooled, and characterized for toxin-specific IgG binding titers (Supplementary Figure 1A) and toxin neutralizing titers (Supplementary Figure 1B). Serum pools demonstrated high toxin binding as well as neutralizing antibody titers against all target antigens (Supplementary Figures 1A,B). Naïve BALB/c mice were then treated with 500 μl of neat IBT-V02 immune sera intraperitoneally (IP) 4 h prior to intradermal infection with 1 × 107 CFU of USA300 (NRS384). Sera collected from naïve mice were administered to control animals, and the treatment was repeated 4 days post-infection. Lesions were monitored and measured for 14 days. As shown in Figure 4D, mice treated with immune sera developed significantly smaller lesions when compared to mice treated with naïve sera. The potency of the vaccine generated serum antibodies was highlighted even more when immune serum was diluted in PBS prior to treatment and still showed protective efficacy at 1:2 and 1:5 dilutions (Figure 4E).
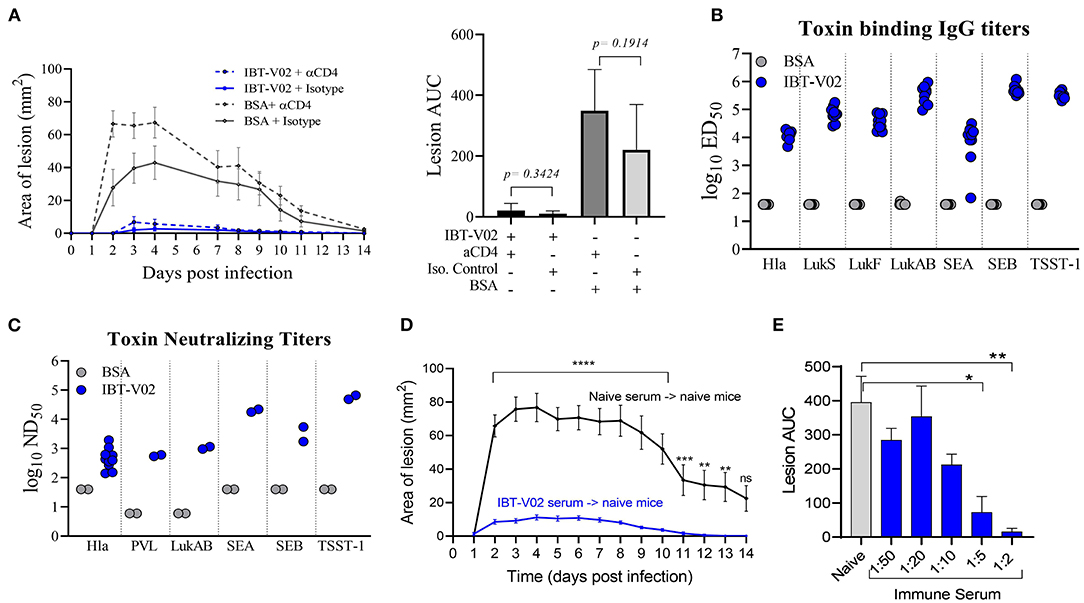
Figure 4. Protection against USA300 (NRS384) is mediated by vaccine-generated serum antibodies. (A) Lesion size and area under the curve after depletion of CD4 T cells in BSA-immunized and IBT-V02 immunized mice during infection, n = 5/group. (B) Toxin binding IgG titers tested on individual serum samples. (C) Toxin neutralizing titers tested on individual serum samples against Hla, and titers tested on serum pools/group against PVL, LukAB, SEA, SEB, and TSST-1 prior to CD4 T cells depletion. (D) Lesion size after passive transfer of naïve or immune sera previously generated in CD1 mice into BALB/c mice, n = 10/group. Statistical analysis performed using two-way ANOVA, Sidak's multiple comparisons test. (E) Area under the curve of infection-induced lesions after passive transfer of naïve sera or different dilutions of immune sera. Statistical analysis was performed by t-test using GraphPad PRISM v8.4.3. Siginificance symbols: *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001; ****P ≤ 0.0001.
The immunogenicity as well as the efficacy of IBT-V02 was further evaluated in a rabbit model of acute skin infection. Two groups of sixteen New Zealand White rabbits (NZWR) were immunized ID three times 2 weeks apart with 100 μg of IBT-V02. Rabbits were infected ID with 9.94 × 109 CFU of MRSA USA300 (NRS384), and the lesions were monitored for 7 days post-infection and pictures were taken daily. High total IgG titers (ED50) were achieved against TSST-1, SEB, SEA, Hla, LukS-PV, LukF-PV, and LukAB (Figure 5A). Immunization of rabbits also induced highly significant neutralizing titers toward the target antigens of IBT-V02 as well as cross neutralizing activity toward closely related bi-component pore forming toxins HlgAB Figure 5B) and HlgCB, consistent with previous findings (27). Lesions of rabbits immunized with IBT-V02 were significantly smaller (Figure 5C) and had lower bacterial burden (Figure 5D) on day 7 post-infection compared to controls. Figure 5E shows representative images of lesions on days 1, 4, and 7 post-infection.
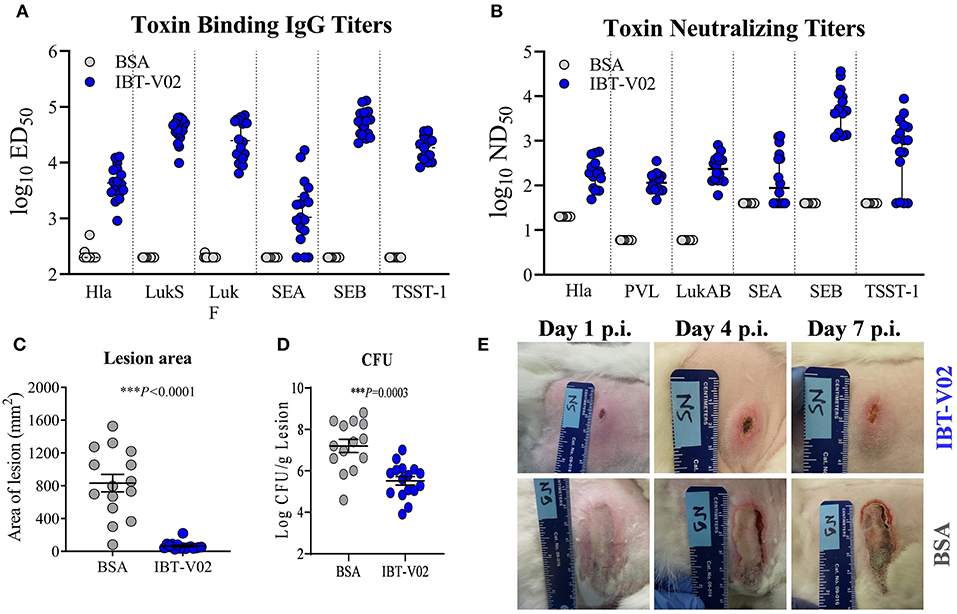
Figure 5. Efficacy of IBT-V02 in a rabbit dermonecrosis model with USA300 (NRS384). (A) Serum IgG binding titers and (B) Serum toxin neutralizing titers of rabbits immunized with BSA or IBT-V02 tested against target antigens Hla, PVL (LukS & LukF), LukAB, SEA, SEB, and TSST-1. (C) Lesion area and (D) bacterial burden on day 7 post infection (E) Lesion images of BSA or IBT-V02 immunized rabbits on days 1, 4, and 7 post infection. Data analyzed using GraphPad PRISM v8.4.3. Statistical analysis performed with Mann-Whitney test. Shown are mean values with SEM.
Pre-clinical evaluation of vaccine candidates is commonly performed in naïve animals, and this has been the case for SA vaccine candidate vaccines evaluated to-date. However, contrary to mice, most humans are not naïve for SA since a large part of the population is persistently or intermittently colonized by SA and demonstrate pre-existing memory against a wide range of staphylococcal antigens (45–48). Therefore, we evaluated whether the efficacy of IBT-V02 in mice was affected by cutaneous pre-exposure to SA. As shown in Figure 6A, groups of 10 mice were intradermally injected with (pre-exposed to) 1 × 105 CFU of SA USAS300 (NRS384) or left untreated as a control on study day 0. The animals were then vaccinated with IBT-V02 on days 28 and 42. Animals were rested for an additional 4 weeks and then challenged ID with either 1 × 107 or 1 × 108 CFU. Mice were monitored for another 2 weeks for lesion size and general health. Mice pre-exposed to SA exhibited the same course of lesion development and healing as the naïve animals (Figure 6B), indicating that cutaneous pre-exposure to SA neither provides protection against subsequent infection nor has an impact on vaccine efficacy. Similarly, IBT-V02 protective efficacy was comparable between naïve and pre-exposed animals.
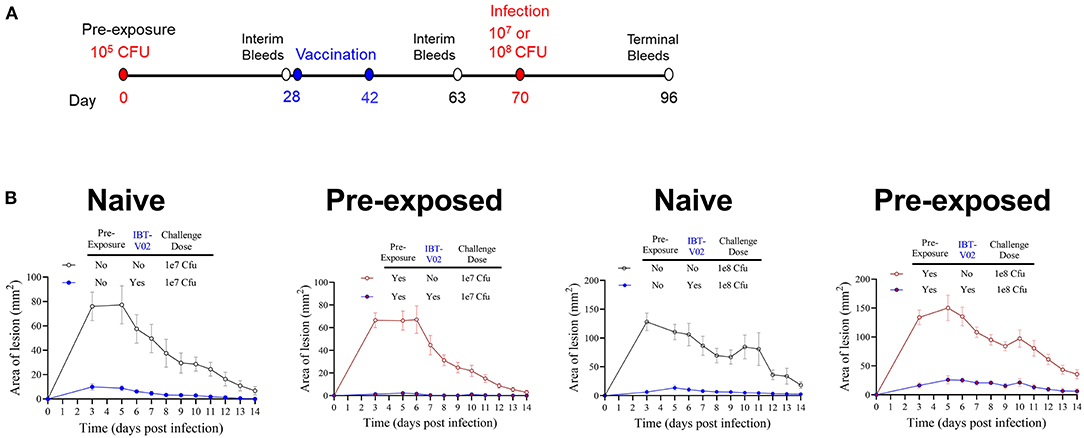
Figure 6. Efficacy of IBT-V02 in pre-exposed mice. (A) Infection and vaccination schedule. (B) Lesion areas of mice with or without intradermal pre-exposure prior to infection with 107 or 108 CFU of USA300 (NRS384), n = 5/group, shown are mean values with SEM.
Recurrence of SA skin infections is frequently seen and an important public health problem. A vaccine that could protect against recurrence is therefore of high value. The ideal time to vaccinate individuals against recurrence may be during the active infection. Here we asked the question whether immunization started in the acute phase of a primary infection can protect against secondary infection with a higher challenge dose. Two groups of 10 mice were infected intradermally with 1 × 107 CFU of SA USA300 (NRS384) on study day 0. One group received IBT-V02 (IM) on study days 2, 16, and 30 while the other group served as infection only control. Both groups were infected with 1 × 108 CFU on day 49 and monitored for another 14 days. Animals were bled on days 0, 44, and 71 for serological analysis. The study design is shown in Figure 7A.
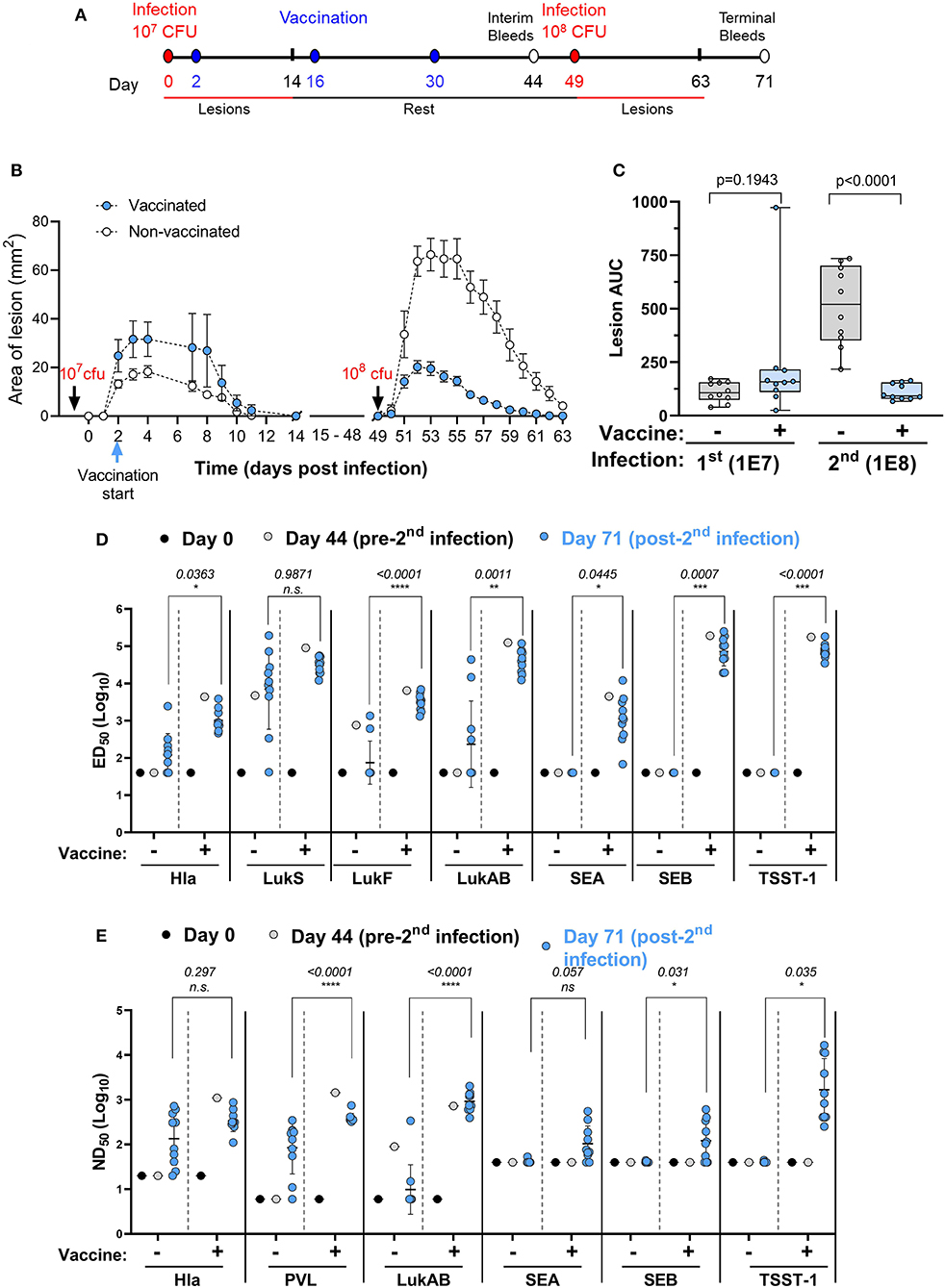
Figure 7. Efficacy of IBT-V02 during active infection. (A) Infection and vaccination schedule. (B) Lesion areas of vaccinated and non-vaccinated mice after primary (107 CFU) and secondary infection (108 CFU) with USA300. (C) Area under curve of infection-induced lesions of individual mice. (D) Serum IgG binding titers with lower limits of detection (LLOD) at 40, and (E) Serum toxin neutralizing titers with LLOD of 20 (Hla), 6 (PVL & LukAB), and 40 (SEA, SEB, TSST-1) of vaccinated and non-vaccinated mice on serum pools collected on days 0 and 44 and on individual serum samples collected on day 71 tested against target antigens Hla, PVL (LukS & LukF), LukAB, SEA, SEB, and TSST-1, n = 10 mice/group. Data were analyzed using GraphPad PRISM v8.4.3. Statistical analysis was performed using t-test. Shown are log-transformed mean values with SD.
As shown in Figure 7B, vaccination during acute infection did not affect the lesion size as further supported by the AUC analysis (Figure 7C). After the second infection with a high dose of USA300, the control animals exhibited much larger lesions than the primary lesions (Figures 7B,C), further confirming the findings in our previous experiment (Figure 6). However, animals vaccinated during the acute phase exhibited highly significant reduction in lesion size compared to non-vaccinated mice (Figures 7B,C).
We further measured the total IgG and neutralizing titers against the toxins represented by IBT-V02 on serum pools collected from mice on days 0 and 44 and on individual serum samples collected on day 71. The first infection in non-vaccinated mice induced very low levels of total IgG against Hla and LukAB and moderate levels of antibodies against LukS-PV and LukF-PV (Figure 7D, day 0 vs. day 44 in the no vaccine groups). However, the second infection in the no vaccine group did boost the antibodies against pore forming toxin antigens Hla, LukS-PV, and LukAB, but not LukF-PV (Figure 7D, day 44 vs. day 71 in the no vaccine groups). Vaccination induced high IgG titers against all antigens and the antibody levels were not significantly changed after the second challenge (Figure 7D, see the vaccine groups, day 44 vs. day 71). In all cases, except for LukS, the day 71 titers (post-second challenge) were significantly higher in the vaccinated than in non-vaccinated mice (Figure 7D). The primary infection failed to induce any appreciable neutralizing titers against Hla and PVL, but the titers were moderately increased after the second infection, whereas the modest LukAB neutralizing activity seen in the serum pool remained low after the second infection (Figure 7E, the no vaccine groups). Vaccination during infection induced high neutralizing activity against all three cytolysins which was retained upon high dose reinfection (Figure 7D). Whereas, vaccinated groups demonstrated robust IgG binding titers (Figure 7D) and neutralizing titers (Figure 7E) against SEA, SEB, and TSST-1, serum antibodies generated against these SAgs were not detected in the no vaccine group (Figures 7D,E), which was not surprising as USA300 (NRS384) does not harbor any of these SAgs.
S. aureus (SA) is the most common cause of purulent acute skin infections in humans, leading to a high burden of disease in both healthy and immunocompromised individuals with an estimated 8.7 million ambulatory visits alone in the US (30, 49, 50). SA also complicates burn and surgical wound sites and lesions of atopic dermatitis (51, 52). A major problem for patients with acute skin infection is the high rate of recurrence that can be as high as 70% in 1 year (41, 53–55). Recurrence is hardly reduced by successful treatment of the primary lesion using incision and drainage and/or antibiotics (56). SA produces a wide range of cytolytic and superantigenic toxins with a wide range of pathogenic and immune subversive activities severely affecting the course of disease including skin infections (20, 21, 35). Here we present an entirely toxoid multi-component SA vaccine (IBT-V02) targeting Hla, PVL, LukAB, and three superantigens (SEA, SEB, and TSST-1) and demonstrate its immunogenicity and protective efficacy in mouse and rabbit models of dermonecrosis. We demonstrate efficacy of the vaccine in both naïve mice and mice pre-exposed to SA as well as when administered during acute infection.
The role of cytolytic toxins in the pathogenesis of SA skin infections is well-documented through human epidemiological studies and animal models (57). Prominent among these toxins is Hla, which is the main driver of the skin dermonecrotic lesions caused by SA in mouse and rabbit models (35, 58). Hla is highly hemolytic for rabbit red blood cells but not for human erythrocytes (59), and its main pathogenic activity in humans is likely due to its ability to damage the skin and mucosal barriers by targeting epithelial cells (60–63), endothelium (64, 65), and keratinocytes (66), as well as pro-inflammatory properties through inflammasome activation (35). Hla also contributes to biofilm formation by SA wound isolates (67). Furthermore, Hla causes platelet aggregation that can contribute to micro-thrombi leading to organ failure (68). Hla monoclonal antibodies or vaccines are protective against dermonecrosis in mice (69, 70). In humans, antibodies against Hla have been shown to correlate with a better clinical outcome in patients with SSTI (71). PVL, a prominent toxin produced by the epidemic clone USA300 and a growing number of strains around the world (72), is implicated in the epidemiology of SSTIs. PVL was linked to community-acquired MRSA (CA-MRSA) outbreaks including SA skin infection in the 1900s (2). In a meta-analysis, the presence of the pvl gene was associated with abscesses and furuncles (73). Whereas, wild type mice do not respond to PVL, in non-obese diabetic (NOD)/severe combined immune deficiency (SCID)/IL2rγnull (NSG) mice reconstituted with human umbilical cord blood cells and administered fresh human neutrophils, a 1–2 log lower USA300 inoculum is needed to induce consistent skin lesions compared to an isogenic PVL-negative strain (37). Consistently, the PVL-negative SA strain induced smaller lesions compared to the parental strain (37). Both PVL and LukAB cause dose dependent skin inflammation in rabbits (74). Staphylococcal superantigens (SAgs) have been implicated in colonization of skin with SA and the etiology of atopic dermatitis (AD), a chronic disease that pre-disposes patients to recurrent SA skin infections. The majority of SA isolates from atopic eczema produce SAgs (75), and SAgs are likely a major factor in the Th2 type inflammatory response in AD patients (34). Consistent with this, SAgs facilitate epithelial presentation of allergens to Th2 cells (76). Adhesion molecules that bind to SA, such as fibronectin and laminin, are also upregulated as a result of the Th2 cytokine IL-4 released in the inflammatory environment of skin (77). IL-4 and IL-13 have also been shown to suppress IFNγ- or TNFα-induced expression of antimicrobial peptides in keratinocytes (77). Thus, neutralization of superantigens in the skin is expected to change the immunological environment to unfavorable conditions for colonization and infection. Thus, an effective vaccine for SA-ASI should target these cytolytic toxins as well as major superantigens.
The 5-component vaccine IBT-V02 presented here includes toxoids of Hla, both PVL subunits, LukAB as well as a fusion of toxoids for TSST-1, SEB, SEA formulated in Alhydrogel (Al(OH)3). All individual components of the vaccine were characterized and published previously and shown to be highly attenuated and to induce broad neutralizing antibody responses (24–27). Here we show that the blended vaccine formulated with Al(OH)3 is highly immunogenic in mice and rabbits, inducing a strong total IgG and neutralizing antibody response. We tested the vaccine for efficacy against dermonecrosis caused by SA-ASI with several clinically relevant strains of CA-MRSA (ST-8), USA300 and USA1000 (ST-59), and USA100 (ST-5) clones. IBT-V02 showed protective efficacy against all the strains tested with highly significant reduction in lesion size, lesion score, weight loss, and bacterial burden. The protective efficacy in this model was primarily mediated by serum antibodies as depletion of CD4 cells at the time of infection in vaccinated or control animals had no significant impact on the vaccine efficacy, whereas adoptive hyperimmune serum transfer to naïve animals was highly protective.
About two thirds of the human population is persistently or intermittently colonized with SA and, as a result, most people have immunological memory toward staphylococcal antigens. Yet most vaccine candidates have been tested in immunologically naïve animals. To evaluate whether pre-exposure has an impact on the efficacy of IBT-V02, we exposed naïve mice to a low dose of USA300 by the cutaneous route, similar to the most common route of human exposure, before vaccination with IBT-V02 and challenge. The results of this study demonstrated that (i) pre-exposure to SA does not elicit a protective immune response, and (ii) vaccine efficacy is identical between naïve and pre-exposed animals.
Recurrent SSTI is a major public health problem, a burden on the families of patients, a significant driver of healthcare costs, and a driver of antibiotic use, thus contributing to antibiotic resistance (40). Some patients can experience repeated bouts of recurrent infections over months and even years after primary infection. Given that ~0.4% of patients with SA skin infections develop systemic infections (56), recurrence further increases the risk of invasive disease. While patients with comorbidities are generally considered more prone to recurrence, a major portion of recurrent skin infections are in patients with no major comorbidities (56). To address the issue of recurrent infections, we developed a re-infection model in mice using two subsequent intradermal infections with increasing challenge doses. Our data show that not only an asymptomatic exposure, but even a full-blown, symptomatic primary infection, does not protect against SA re-infection.
Our data further show that IBT-V02 can be administered during the acute phase of the first infection to protect against a subsequent reinfection. Mice that were vaccinated during the first infection showed significantly less severe lesions upon a second challenge with a ten-fold higher dose. This is important because patient compliance with requests to return for vaccination against a possible future recurrence could present a roadblock to prevention, especially when the primary infection is not severe or life threatening. Offering the vaccine at the time of infection for diseases with the risk of recurrence, such as SA SSTI or bacteremia, C. difficile infections, or urinary tract infections, could significantly increase vaccine coverage and reduce the burden of disease.
In summary, we have presented here the first fully toxoid multi-component vaccine against acute and recurrent SA skin infections. Importantly, we have demonstrated efficacy of the toxoid vaccine against a secondary skin infection. Future studies are needed to further characterize the immune response that is critical for protection against secondary infection including the role of the immune response against superantigens in more SAg-sensitive animal models as well as in future clinical trials. Our findings along with a body of evidence on the role of anti-toxin antibodies in improvement of clinical outcome of SA disease [reviewed extensively in (57)] strongly support testing of this vaccine for prevention and mitigation of recurrent SA-ASI in human clinical trials.
IBT-V02, composed of 5 components representing 7 toxoids, was characterized by SDS, WB, and SE-HPLC. For WB, antigen-specific primary and secondary antibodies were used. For SEC-HPLC, 10–40 μg of each IBT-V02 component was injected in an Agilent Technologies 1260 Infinity Series instrument using an AdvanceBio SEC 300 Å 7.8 × 300 mm LC column with a mobile phase of 50 mM sodium phosphate buffer + 150 mM NaCl, pH 7.0, running at a flow rate of 0.5 mL/min. The chromatogram generated by the Agilent OpenLabs software plotted absorbance at 280 nm as a function of retention time. All analyses of the peaks were performed by the auto-integrate function in the OpenLabs software.
IBT-V02 was incubated with Alhydrogel (Brenntag Bio) at various antigen: aluminum ratios for 1 h at room temperature. After incubation, the antigen-adjuvant mixture was centrifuged, and the supernatant was run on an SDS-PAGE gel. Adsorption of antigens to Alhydrogel was indicated by a thin or negligible band of protein visible on the gel as compared to a control without Alhydrogel adsorption, depicted by the non-adsorbed complete antigen band.
SA strains USA300 (LAC), USA300 (NRS384), were obtained from the BEI resources. Strain USA300 (SF8300) and USA1000 (ST59, PVL+) (78) was provided by Dr. Binh Diep at UCSF. Strain USA1000 (PVL-) was obtained from a repository collected from patients with SA sepsis (79). Bioluminescent NRS699 strain (NRS699 lux) was generated in house: Strain SAP140, an RN4220 strain with pRP1195 plasmid (Temperature sensitive plasmid with Lux ABCDE cassette) was received as a gift from Dr. Roger Plaut, FDA (80). The SAP140 phage lysate was made at 30°C using 80α phage. The transduction was carried out at 30°C to move the temperature sensitive plasmid into recipient NRS699 strain (Gift from Dr. Jean C. Lee) using a standard transduction protocol (80). Integration of plasmid into the recipient chromosome was carried out by shifting the growth temperature to 43°C in the each transductant strain (80). Three clones were screened under IVIS camera to confirm the bioluminescent phenotype.
For mouse skin infection with USA300 (LAC) and USA100 (NRS699) bacteria were incubated at 37°C, shaking at 200 rpm in tryptic soy broth (TSB) to mid-exponential phase. The culture was centrifuged and washed twice with PBS prior to injection. Inoculum was verified by CFU/ml determinations. For mouse skin infection with USA300 (NRS384) and USA1000, Brain-Heart Infusion (BHI) broth was inoculated with a swab of SA from a freshly grown blood agar plate, and culture was grown for 18 h at 37°C, shaking at 230 rpm. The culture was centrifuged at 3,000 rpm for 10 min, and the pellet was washed twice in PBS before suspensions in PBS. To ensure single cell suspension, bacterial preparations were passed through a syringe with a 27-gauge needle before freezing aliquots at −80°C until further use. For the rabbit ABSSSI model, USA300 (NRS384) was prepared as previously described (81).
Female Balb/C mice were purchased from Charles River. The starting age of mice for each experiment was 6 weeks. Mice were maintained under pathogen-free conditions and fed laboratory chow and water ad libitum. Mice were immunized intramuscularly on each side of the tail base (50 μl each side) three times 2 weeks apart with a total of 50 μg of IBT-V02 (10 μg each antigen) in 250 μg Alhydrogel. For serological analyses, the mice were bled via the retro-orbital (RO) or tail vein route prior to and 10–14 days after the final immunization. Two weeks after the last immunization, the backs of the mice were shaved and 50 μl of S. aureus suspension was administered via intradermal (ID) or subcutaneous (SC) injection. The animals were monitored daily for weight loss. Lesions were measured daily for 14 days post-infection using calipers. The areas of the lesions were calculated using the formula Area (A) = Length (L)/2 x Width (W)/2 x π. Statistical significance was determined using two-way ANOVA with Sidak's multiple comparisons analysis (GraphPad PRISM v8.4.3). As described in the text, some experiments required two immunizations after pre-exposure or during acute infection. For passive serum transfer studies, female CD1 mice, 8 weeks of age were purchased from ENVIGO and were immunized four times 2 weeks apart with 50 μg of IBT-V02 formulated in 250 μg Alhydrogel. Terminal bleeds were performed 2 weeks after the final immunization, and pooled sera from these animals were stored at −80°C until further use. Pooled sera from naïve CD1 mice purchased from ENVIGO served as controls. Mice were maintained under pathogen-free conditions and fed laboratory chow and water ad libitum. All mouse work was conducted in accordance with protocols that were approved by institutional animal care and use committees (IACUC) of Integrated BioTherapeutics, where mouse studies were performed.
Animal experiments performed at the Brigham and Women's Hospital were conducted in accordance with the recommendations and guidelines in the Public Health Service Policy on Humane Care and Use of Laboratory Animals, and animal use protocols were approved by the Partners Healthcare Institutional Animal Care and Use Committee. Mice that were challenged subcutaneously with luminescent USA100 (NRS 699) were monitored daily for weight loss and lesion size. Mice were imaged with an IVIS Lumina 3 system, and bioluminescence was quantified with Living Image 4.7 software.
The rabbit ABSSSI model was reviewed and approved by the University of California San Francisco Institutional Animal Care and Use Committee. Experiments were conducted in a facility certified by the Association for Assessment and Accreditation of Laboratory Animal Care International. Female and male New Zealand White (NZW) rabbits were purchased from Western Oregon Rabbit Co. The starting age range of rabbits for each experiment was 8–11 weeks. Rabbits were immunized intradermally on the right and left dorsal lumbar skin three times 2 weeks apart with a total of 100 μg of IBT-V02 (20 μg each antigen) in 500 μg Alhydrogel. Animals were bled prior to immunization to obtain naïve sera and 2 weeks after the last immunization to obtain immune sera. The vaccinated rabbits were challenged in the ABSSSI model as previously described (81).
A multiplex assay to detect serum IgG titers to SA antigens has been previously developed at IBT using the Luminex xMAP® technology. Briefly, IBT-V02 target antigens Hla, LukS-PV, LukF-PV, LukAB, SEA, SEB, and TSST-1 were coupled to carboxylated MagPlex microsphere beads with distinct spectral regions via a carbodiimide reaction. Antigen-coupled beads were incubated with serum samples at a starting dilution of 1:40 in a two-fold 8-point dilution series at room temperature (RT) for 2 h. Samples were washed and incubated with a PE-conjugated goat anti-mouse IgG antibody (Biolegend, San Diego, CA.) for 1 hour at RT. The samples were washed and acquired using a Luminex200. Data were analyzed using a 4-parameter (4PL) curve fit in XLFit (Microsoft). IgG titers were expressed as the effective dilution at the point of the 4PL curve where 50% (ED50) of antigen was detected by toxin-specific antibodies present in the serum sample.
Hla TNAs were performed as previously described (82). In brief, 4% rabbit red blood cells (RRBCs) were co-cultured with wild-type Hla ± serially diluted serum samples. Cells were centrifuged after 30 min and absorbance determined at OD416nm. PVL and LukAB TNAs were performed with human promyelocytic leukemia (HL-60) cells as previously described (25). In brief, differentiated HL-60 cells were incubated with either PVL or LukAB ± serially diluted serum samples for 3 h, and CellTiter Glo was added to the culture to measure cell viability. SAg TNAs were performed with PBMCS from healthy volunteers. Cells were co-cultured with SEA, SEB, or TSST-1 in the presence or absence of serially diluted serum samples for 48 h, supernatants were collected, and IFNγ was measured in the supernatants as a readout of superantigenicity as we previously described (26). Data were analyzed using a 4-parameter (4PL) curve fit in XLFit (Microsoft). Toxin neutralizing activity was defined as the effective dilution of sera at the point of the 4PL curve at which 50% of toxin activity was neutralized (ND50).
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Integrated Biotherapeutics IACUC.
HK, AV, JL, BD, and MA designed experiments. IM, MM, RO, NN, AV, RA, TK, and JL carried out experiments and analyzed data. JL, BD, MA, FH, and HK interpreted data. HK, AV, and MA wrote manuscript. MA acquired funding. All authors contributed to the article and approved the submitted version.
Research reported in this paper was supported by the Cooperative Agreement Number IDSEP160030 from ASPR/BARDA and by awards from Wellcome Trust, Germany's Federal Ministry of Education and Research, the UK Global Antimicrobial Resistance Innovation Fund (GAMRIF) and the Bill & Melinda Gates Foundation, as administrated by CARB-X. The content is solely the responsibility of the authors and does not necessarily represent the official views of the Department of Health and Human Services Office of the Assistant Secretary for Preparedness and Response, other funders, or CARB-X. The work was further supported by NIAID Grant R01-AI111205.
MA and HK have stocks and RA and FH have stock options in Integrated Biotherapeutics Inc. JL serves on the Scientific Advisory Board of Integrated BioTherapeutics.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Excellent technical assistance was provided by William J. Eagen. We thank Dr. Roger Plaut, for strain SAP140, an RN4220 strain with pRP1195 plasmid (temperature sensitive plasmid with Lux ABCDE cassette).
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.624310/full#supplementary-material
1. Kobayashi SD, Malachowa N, DeLeo FR. Pathogenesis of Staphylococcus aureus abscesses. Am J Pathol. (2015) 185:1518–27. doi: 10.1016/j.ajpath.2014.11.030
2. Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. (2015) 28:603–61. doi: 10.1128/CMR.00134-14
3. Chambers HF, Deleo FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol. (2009) 7:629–41. doi: 10.1038/nrmicro2200
4. Klevens RM, Morrison MA, Nadle J, Petit S, Gershman K, Ray S, et al. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. (2007) 298:1763–71. doi: 10.1001/jama.298.15.1763
5. Bagnoli F, Bertholet S, Grandi G. Inferring reasons for the failure of Staphylococcus aureus vaccines in clinical trials. Front Cell Infect Microbiol. (2012) 2:1–4. doi: 10.3389/fcimb.2012.00016
6. Fowler VG Jr, Proctor RA. Where does a Staphylococcus aureus vaccine stand? Clin Microbiol Infect. (2014) 20(Suppl. 5):66–75. doi: 10.1111/1469-0691.12570
7. Plotkin SA. Correlates of protection induced by vaccination. Clin Vaccine Immunol. (2010) 17:1055–65. doi: 10.1128/CVI.00131-10
8. Fattom A, Schneerson R, Watson DC, Karakawa WW, Fitzgerald D, Pastan I, et al. Laboratory and clinical evaluation of conjugate vaccines composed of Staphylococcus aureus type 5 and type 8 capsular polysaccharides bound to Pseudomonas aeruginosa recombinant exoprotein A. Infect Immun. (1993) 61:1023–32. doi: 10.1128/IAI.61.3.1023-1032.1993
9. Inoue M, Yonemura T, Baber J, Shoji Y, Aizawa M, Cooper D, et al. Safety, tolerability, and immunogenicity of a novel 4-antigen Staphylococcus aureus vaccine (SA4Ag) in healthy Japanese adults. Hum Vaccin Immunother. (2018) 14:2682–91. doi: 10.1080/21645515.2018.1496764
10. Stranger-Jones YK, Bae T, Schneewind O. Vaccine assembly from surface proteins of Staphylococcus aureus. Proc Natl Acad Sci U S A. (2006) 103:16942–7. doi: 10.1073/pnas.0606863103
11. Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: a randomized trial. JAMA. (2013) 309:1368–78. doi: 10.1001/jama.2013.3010
12. McNeely TB, Shah NA, Fridman A, Joshi A, Hartzel JS, Keshari RS, et al. Mortality among recipients of the Merck V710 Staphylococcus aureus vaccine after postoperative S. aureus infections: an analysis of possible contributing host factors. Hum Vaccin Immunother. (2014) 10:3513–6. doi: 10.4161/hv.34407
13. Karauzum H, Haudenschild CC, Moore IN, Mahmoudieh M, Barber DL, Datta S K. Lethal CD4 T cell responses induced by vaccination against Staphylococcus aureus bacteremia. J Infect Dis. (2017) 215:1231–9. doi: 10.1093/infdis/jix096
14. Spaulding AR, Salgado-Pabon W, Merriman JA, Stach CS, Ji Y, Gillman AN, et al. Vaccination against Staphylococcus aureus pneumonia. J Infect Dis. (2014) 209:1955–62. doi: 10.1093/infdis/jit823
15. Aman MJ. Superantigens of a superbug: major culprits of Staphylococcus aureus disease? Virulence. (2017) 8:607–10. doi: 10.1080/21505594.2016.1255399
16. Miller LS, Cho JS Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol. (2011) 11:505–18. doi: 10.1038/nri3010
17. Vandenesch F, Naimi T, Enright MC, Lina G, Nimmo GR, Heffernan H, et al. Community-acquired methicillin-resistant Staphylococcus aureus carrying Panton-Valentine leukocidin genes: worldwide emergence. Emerg Infect Dis. (2003) 9:978–84. doi: 10.3201/eid0908.030089
18. Aman MJ, Adhikari RP. Staphylococcal bicomponent pore-forming toxins: targets for prophylaxis and immunotherapy. Toxins (Basel). (2014) 6:950–72. doi: 10.3390/toxins6030950
19. Vandenesch F, Lina G, Henry T. Staphylococcus aureus hemolysins, bi-component leukocidins, and cytolytic peptides: a redundant arsenal of membrane-damaging virulence factors? Front Cell Infect Microbiol. (2012) 2:1–15. doi: 10.3389/fcimb.2012.00012
20. Spaan AN, van Strijp JAG, Torres VJ. Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol. (2017) 15:435–47. doi: 10.1038/nrmicro.2017.27
21. Kotzin BL, Leung DY, Kappler J, Marrack P. Superantigens and their potential role in human disease. Adv Immunol. (1993) 54:99–166. doi: 10.1016/S0065-2776(08)60534-9
22. Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. (1990) 248:705–11. doi: 10.1126/science.2185544
23. Broker BM, Mrochen D, Peton V. The T Cell response to Staphylococcus aureus. Pathogens. (2016) 5:1–23. doi: 10.3390/pathogens5010031
24. Karauzum H, Adhikari RP, Sarwar J, Devi VS, Abaandou L, Haudenschild C, et al. structurally designed attenuated subunit vaccines for S. aureus LukS-PV and LukF-PV confer protection in a mouse bacteremia model. PLoS ONE. (2013) 8:e65384. doi: 10.1371/journal.pone.0065384
25. Kailasan S, Kort T, Mukherjee I, Liao GC, Kanipakala T, Williston N, et al. Rational design of toxoid vaccine candidates for Staphylococcus aureus leukocidin AB (LukAB). Toxins (Basel). (2019) 11:339. doi: 10.3390/toxins11060339
26. Venkatasubramaniam A, Adhikari RP, Kort T, Liao GC, Conley S, Abaandou L, et al. TBA225, a fusion toxoid vaccine for protection and broad neutralization of staphylococcal superantigens. Sci Rep. (2019) 9:3279. doi: 10.1038/s41598-019-39890-z
27. Tran VG, Venkatasubramaniam A, Adhikari RP, Krishnan S, Wang X, Le VTM, et al. Efficacy of active immunization with attenuated alpha-hemolysin and panton-valentine leukocidin in a rabbit model of Staphylococcus aureus necrotizing pneumonia. J Infect Dis. (2019) 221:267–75. doi: 10.1093/infdis/jiz437
28. Kaye KS, Petty LA, Shorr AF, Zilberberg MD. Current epidemiology, etiology, and burden of acute skin infections in the United States. Clin Infect Dis. (2019) 68:S193–9. doi: 10.1093/cid/ciz002
29. Suaya JA, Eisenberg DF, Fang C, Miller LG. Skin and soft tissue infections and associated complications among commercially insured patients aged 0-64 years with and without diabetes in the U.S. PLoS ONE. (2013) 8:e60057. doi: 10.1371/journal.pone.0060057
30. McCaig LF, McDonald LC, Mandal S, Jernigan DB. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg Infect Dis. (2006) 12:1715–23. doi: 10.3201/eid1211.060190
31. King MD, Humphrey BJ, Wang YF, Kourbatova EV, Ray SM, Blumberg HM. Emergence of community-acquired methicillin-resistant Staphylococcus aureus USA 300 clone as the predominant cause of skin and soft-tissue infections. Ann Intern Med. (2006) 144:309–17. doi: 10.7326/0003-4819-144-5-200603070-00005
32. Mairi A, Touati A, Lavigne JP Methicillin-resistant Staphylococcus aureus ST80 clone: a systematic review. Toxins (Basel). (2020) 12:119. doi: 10.3390/toxins12020119
33. Singh J, Johnson RC, Schlett CD, Elassal EM, Crawford KB, Mor D, et al. Multi-body-site microbiome and culture profiling of military trainees suffering from skin and soft tissue infections at fort Benning, Georgia. mSphere. (2016) 1:e00232–16. doi: 10.1128/mSphere.00232-16
34. Nair N, Kourbatova E, Poole K, Huckabee CM, Murray P, Huskins WC, et al. Molecular epidemiology of methicillin-resistant Staphylococcus aureus (MRSA) among patients admitted to adult intensive care units: the STAR*ICU trial. Infect Control Hosp Epidemiol. (2011) 32:1057–63. doi: 10.1086/662178
35. Berube BJ, Bubeck Wardenburg J. Staphylococcus aureus alpha-toxin: nearly a century of intrigue. Toxins (Basel). (2013) 5:1140–66. doi: 10.3390/toxins5061140
36. Baba-Moussa L, Sina H, Scheftel JM, Moreau B, Sainte-Marie D, Kotchoni SO, et al. Staphylococcal Panton-Valentine leucocidin as a major virulence factor associated to furuncles. PLoS ONE. (2011) 6:e25716. doi: 10.1371/journal.pone.0025716
37. Tseng CW, Biancotti JC, Berg BL, Gate D, Kolar SL, Muller S, et al. Increased susceptibility of humanized NSG mice to panton-valentine leukocidin and Staphylococcus aureus skin infection. PLoS Pathog. (2015) 11:e1005292. doi: 10.1371/journal.ppat.1005292
38. Kim CK, Karau MJ, Greenwood-Quaintance KE, Tilahun AY, Krogman A, David CS, et al. Superantigen-producing Staphylococcus aureus elicits systemic immune activation in a murine wound colonization model. Toxins (Basel). (2015) 7:5308–19. doi: 10.3390/toxins7124886
39. Varshney AK, Wang X, Scharff MD, Macintyre J, Zollner RS, Kovalenko OV, et al. Staphylococcal enterotoxin B-specific monoclonal antibody 20B1 successfully treats diverse Staphylococcus aureus infections. J Infect Dis. (2013) 208:2058–66. doi: 10.1093/infdis/jit421
40. Creech CB, Al-Zubeidi DN, Fritz SA. Prevention of recurrent staphylococcal skin infections. Infect Dis Clin North Am. (2015) 29:429–64. doi: 10.1016/j.idc.2015.05.007
41. Fritz SA, Camins BC, Eisenstein KA, Fritz JM, Epplin EK, Burnham CA, et al. Effectiveness of measures to eradicate Staphylococcus aureus carriage in patients with community-associated skin and soft-tissue infections: a randomized trial. Infect Control Hosp Epidemiol. (2011) 32:872–80. doi: 10.1086/661285
42. Montgomery CP, David MZ, Daum RS. Host factors that contribute to recurrent staphylococcal skin infection. Curr Opin Infect Dis. (2015) 28:253–8. doi: 10.1097/QCO.0000000000000156
43. Johnson JK, Khoie T, Shurland S, Kreisel K, Stine OC, Roghmann MC. Skin and soft tissue infections caused by methicillin-resistant Staphylococcus aureus USA300 clone. Emerg Infect Dis. (2007) 13:1195–200. doi: 10.3201/eid1308.061575
44. Menzies BE Kernodle DS Site-directed mutagenesis of the alpha-toxin gene of Staphylococcus aureus: role of histidines in toxin activity in vitro and in a murine model. Infect Immun. (1994) 62:1843–7. doi: 10.1128/IAI.62.5.1843-1847.1994
45. de Wit J, Totte JEE, van Buchem FJM, Pasmans S. The prevalence of antibody responses against Staphylococcus aureus antigens in patients with atopic dermatitis: a systematic review and meta-analysis. Br J Dermatol. (2018) 178:1263–71. doi: 10.1111/bjd.16251
46. Kolata JB, Kuhbandner I, Link C, Normann N, Vu CH, Steil L, et al. The fall of a dogma? unexpected high T-Cell memory response to Staphylococcus aureus in humans. J Infect Dis. (2015) 212:830–8. doi: 10.1093/infdis/jiv128
47. Pelzek AJ, Shopsin B, Radke EE, Tam K, Ueberheide BM, Fenyo D, et al. Human memory B Cells targeting Staphylococcus aureus exotoxins are prevalent with skin and soft tissue infection. mBio. (2018) 9:e02125–17. doi: 10.1128/mBio.02125-17
48. Wu Y, Liu X, Akhgar A, Li JJ, Mok H, Sellman BR, et al. Prevalence of IgG and neutralizing antibodies against staphylococcus aureus alpha-toxin in healthy human subjects and diverse patient populations. Infect Immun. (2018) 86:e00671–17. doi: 10.1128/IAI.00671-17
49. Miller LG, Eisenberg DF, Liu H, Chang CL, Wang Y, Luthra R, et al. Incidence of skin and soft tissue infections in ambulatory and inpatient settings, 2005-2010. BMC Infect Dis. (2015) 15:362. doi: 10.1186/s12879-015-1071-0
50. Ray GT, Suaya JA, Baxter R. Incidence, microbiology, and patient characteristics of skin and soft-tissue infections in a U.S. population: a retrospective population-based study. BMC Infect Dis. (2013) 13:252. doi: 10.1186/1471-2334-13-252
51. Lowy FD. Staphylococcus aureus infections. N Engl J Med. (1998) 339:520–32. doi: 10.1056/NEJM199808203390806
52. Ong PY, Leung DY. The infectious aspects of atopic dermatitis. Immunol Allergy Clin North Am. (2010) 30:309–21. doi: 10.1016/j.iac.2010.05.001
53. Williams DJ, Cooper WO, Kaltenbach LA, Dudley JA, Kirschke DL, Jones TF, et al. Comparative effectiveness of antibiotic treatment strategies for pediatric skin and soft-tissue infections. Pediatrics. (2011) 128:e479–87. doi: 10.1542/peds.2010-3681
54. Fritz SA, Hogan PG, Hayek G, Eisenstein KA, Rodriguez M, Epplin EK, et al. Household vs. individual approaches to eradication of community-associated Staphylococcus aureus in children: a randomized trial. Clin Infect Dis. (2012) 54:743–51. doi: 10.1093/cid/cir919
55. Miller LG, Eells SJ, David MZ, Ortiz N, Taylor AR, Kumar N, et al. Staphylococcus aureus skin infection recurrences among household members: an examination of host, behavioral, and pathogen-level predictors. Clin Infect Dis. (2015) 60:753–63. doi: 10.1093/cid/ciu943
56. Talan DA, Mower WR, Krishnadasan A, Abrahamian FM, Lovecchio F, Karras D J, et al. Trimethoprim-sulfamethoxazole vs. placebo for uncomplicated skin abscess. N Engl J Med. (2016) 374:823–32. doi: 10.1056/NEJMoa1507476
57. Miller LS, Fowler VG, Shukla SK, Rose WE, Proctor RA. Development of a vaccine against Staphylococcus aureus invasive infections: evidence based on human immunity, genetics and bacterial evasion mechanisms. FEMS Microbiol Rev. (2020) 44:123–53. doi: 10.1093/femsre/fuz030
58. Inoshima N, Wang Y, Bubeck Wardenburg J. Genetic requirement for ADAM10 in severe Staphylococcus aureus skin infection. J Invest Dermatol. (2012) 132:1513–6. doi: 10.1038/jid.2011.462
59. Venkatasubramaniam A, Kanipakala T, Ganjbaksh N, Mehr R, Mukherjee I, Krishnan S, et al. A critical role for HlgA in Staphylococcus aureus pathogenesis revealed by A switch in the SaeRS two-component regulatory system. Toxins (Basel). (2018) 10:377. doi: 10.3390/toxins10090377
60. Herman A, Croteau G, Sekaly RP, Kappler J, Marrack P. HLA-DR alleles differ in their ability to present staphylococcal enterotoxins to T cells. J Exp Med. (1990) 172:709–17. doi: 10.1084/jem.172.3.709
61. Putra I, Rabiee B, Anwar KN, Gidfar S, Shen X, Babalooee M, et al. Staphylococcus aureus alpha-hemolysin impairs corneal epithelial wound healing and promotes intracellular bacterial invasion. Exp Eye Res. (2019) 181:263–70. doi: 10.1016/j.exer.2019.02.019
62. Ratner AJ, Hippe KR, Aguilar JL, Bender MH, Nelson AL, Weiser JN. Epithelial cells are sensitive detectors of bacterial pore-forming toxins. J Biol Chem. (2006) 281:12994–8. doi: 10.1074/jbc.M511431200
63. Ziesemer S, Eiffler I, Schonberg A, Muller C, Hochgrafe F, Beule AG, et al. Staphylococcus aureus alpha-toxin induces actin filament remodeling in human airway epithelial model cells. Am J Respir Cell Mol Biol. (2018) 58:482–91. doi: 10.1165/rcmb.2016-0207OC
64. Powers ME, Kim HK, Wang Y, Bubeck Wardenburg J. ADAM10 mediates vascular injury induced by Staphylococcus aureus alpha-hemolysin. J Infect Dis. (2012) 206:352–6. doi: 10.1093/infdis/jis192
65. Vann JM, Proctor RA. Cytotoxic effects of ingested Staphylococcus aureus on bovine endothelial cells: role of S. aureus alpha-hemolysin. Microb Pathog. (1988) 4:443–53. doi: 10.1016/0882-4010(88)90029-0
66. Soong G, Chun J, Parker D, Prince A. Staphylococcus aureus activation of caspase 1/calpain signaling mediates invasion through human keratinocytes. J Infect Dis. (2012) 205:1571–9. doi: 10.1093/infdis/jis244
67. Anderson MJ, Schaaf E, Breshears LM, Wallis HW, Johnson JR, Tkaczyk C, et al. Alpha-toxin contributes to biofilm formation among Staphylococcus aureus wound isolates. Toxins (Basel). (2018) 10:157. doi: 10.3390/toxins10040157
68. Surewaard BGJ, Thanabalasuriar A, Zeng Z, Tkaczyk C, Cohen TS, Bardoel BW, et al. alpha-Toxin induces platelet aggregation and liver injury during Staphylococcus aureus sepsis. Cell Host Microbe. (2018) 24:271–84 e3. doi: 10.1016/j.chom.2018.06.017
69. Adhikari RP, Thompson CD, Aman MJ, Lee JC. Protective efficacy of a novel alpha hemolysin subunit vaccine (AT62) against Staphylococcus aureus skin and soft tissue infections. Vaccine. (2016) 34:6402–7. doi: 10.1016/j.vaccine.2016.09.061
70. Kennedy AD, Bubeck Wardenburg J, Gardner DJ, Long D, Whitney AR, Braughton KR, et al. Targeting of alpha-hemolysin by active or passive immunization decreases severity of USA300 skin infection in a mouse model. J Infect Dis. (2010) 202:1050–8. doi: 10.1086/656043
71. Fritz SA, Tiemann KM, Hogan PG, Epplin EK, Rodriguez M, Al-Zubeidi DN, et al. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin Infect Dis. (2013) 56:1554–61. doi: 10.1093/cid/cit123
72. Diep BA, Gill SR, Chang RF, Phan TH, Chen JH, Davidson MG, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. (2006) 367:731–9. doi: 10.1016/S0140-6736(06)68231-7
73. Shallcross LJ, Fragaszy E, Johnson AM, Hayward AC The role of the Panton-Valentine leucocidin toxin in staphylococcal disease: a systematic review and meta-analysis. Lancet Infect Dis. (2012) 13:43–54. doi: 10.1016/S1473-3099(12)70238-4
74. Malachowa N, Kobayashi SD, Braughton KR, Whitney AR, Parnell MJ, Gardner DJ, et al. Staphylococcus aureus leukotoxin GH promotes inflammation. J Infect Dis. (2012) 206:1185–93. doi: 10.1093/infdis/jis495
75. McFadden JP, Noble WC, Camp RD. Superantigenic exotoxin-secreting potential of staphylococci isolated from atopic eczematous skin. Br J Dermatol. (1993) 128:631–2. doi: 10.1111/j.1365-2133.1993.tb00257.x
76. Ardern-Jones MR, Black AP, Bateman EA, Ogg GS. Bacterial superantigen facilitates epithelial presentation of allergen to T helper 2 cells. Proc Natl Acad Sci U S A. (2007) 104:5557–62. doi: 10.1073/pnas.0700733104
77. Cho SH, Strickland I, Tomkinson A, Fehringer AP, Gelfand EW, Leung DY. Preferential binding of Staphylococcus aureus to skin sites of Th2-mediated inflammation in a murine model. J Invest Dermatol. (2001) 116:658–63. doi: 10.1046/j.0022-202x.2001.01331.x
78. Diep BA, Le VT, Badiou C, Le HN, Pinheiro MG, Duong AH, et al. IVIG-mediated protection against necrotizing pneumonia caused by MRSA. Sci Transl Med. (2016) 8:357ra124. doi: 10.1126/scitranslmed.aag1153
79. Adhikari RP, Ajao AO, Aman MJ, Karauzum H, Sarwar J, Lydecker AD, et al. Lower antibody levels to Staphylococcus aureus exotoxins are associated with sepsis in hospitalized adults with invasive S. aureus infections. J Infect Dis. (2012) 206:915–23. doi: 10.1093/infdis/jis462
80. Plaut RD, Mocca CP, Prabhakara R, Merkel TJ, Stibitz S. Stably luminescent Staphylococcus aureus clinical strains for use in bioluminescent imaging. PLoS ONE. (2013) 8:e59232. doi: 10.1371/journal.pone.0059232
81. Le VT, Tkaczyk C, Chau S, Rao RL, Dip EC, Pereira-Franchi EP, et al. Critical role of alpha-toxin and protective effects of its neutralization by a human antibody in acute bacterial skin and skin structure infections. Antimicrob Agents Chemother. (2016) 60:5640–8. doi: 10.1128/AAC.00710-16
Keywords: S. aureus, toxoid vaccine, skin infection, pore-forming toxins, superantigens
Citation: Karauzum H, Venkatasubramaniam A, Adhikari RP, Kort T, Holtsberg FW, Mukherjee I, Mednikov M, Ortines R, Nguyen NTQ, Doan TMN, Diep BA, Lee JC and Aman MJ (2021) IBT-V02: A Multicomponent Toxoid Vaccine Protects Against Primary and Secondary Skin Infections Caused by Staphylococcus aureus. Front. Immunol. 12:624310. doi: 10.3389/fimmu.2021.624310
Received: 31 October 2020; Accepted: 08 February 2021;
Published: 10 March 2021.
Edited by:
Fabio Bagnoli, GlaxoSmithKline, ItalyReviewed by:
Pietro Speziale, University of Pavia, ItalyCopyright © 2021 Karauzum, Venkatasubramaniam, Adhikari, Kort, Holtsberg, Mukherjee, Mednikov, Ortines, Nguyen, Doan, Diep, Lee and Aman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: M. Javad Aman, amFtYW5AaW50ZWdyYXRlZGJpb3RoZXJhcGV1dGljcy5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.