 Lisanne M. A. Janssen
Lisanne M. A. Janssen Michiel van der Flier
Michiel van der Flier Esther de Vries
Esther de Vries- 1Department of Tranzo, Tilburg University, Tilburg, Netherlands
- 2Department of Pediatrics, Amalia Children’s Hospital, Nijmegen, Netherlands
- 3Department of Pediatric Infectious Diseases and Immunology, Wilhelmina Children’s Hospital, University Medical Center Utrecht, Utrecht, Netherlands
- 4Laboratory of Medical Microbiology and Immunology, Elisabeth-Tweesteden Hospital, Tilburg, Netherlands
Background: Diagnostic delay in common variable immunodeficiency disorders (CVID) is considerable. There is no generally accepted symptom-recognition framework for its early detection.
Objective: To systematically review all existing data on the clinical presentation of CVID.
Methods: PubMed, EMBASE and Cochrane were searched for cohort studies, published January/1999-December/2019, detailing the clinical manifestations before, at and after the CVID-diagnosis.
Results: In 51 studies (n=8521 patients) 134 presenting and 270 total clinical manifestations were identified. Recurrent upper and/or lower respiratory infections were present at diagnosis in 75%. Many patients had suffered severe bacterial infections (osteomyelitis 4%, meningitis 6%, septicemia 8%, mastoiditis 8%). Bronchiectasis (28%), lymphadenopathy (27%), splenomegaly (13%), inflammatory bowel disease (11%), autoimmune cytopenia (10%) and idiopathic thrombocytopenia (6%) were also frequently reported. A bimodal sex distribution was found, with male predominance in children (62%) and female predominance in adults (58%). 25% of CVID-patients developed other manifestations besides infections in childhood, this percentage was much higher in adults (62%). Immune-dysregulation features, such as granulomatous-lymphocytic interstitial lung disease and inflammatory bowel disease, were more prominent in adults.
Conclusions: The shift from male predominance in childhood to female predominance in adults suggests differences in genetic and environmental etiology in CVID and has consequences for pathophysiologic studies. We confirm the high frequency of respiratory infections at presentation, but also show a high incidence of severe bacterial infections such as sepsis and meningitis, and immune dysregulation features including lymphoproliferative, gastrointestinal and autoimmune manifestations. Early detection of CVID may be improved by screening for antibody deficiency in patients with these manifestations.
Introduction
Common variable immunodeficiency disorders (CVID) is a collection of heterogeneous clinical manifestations linked by low serum levels of immunoglobulins and primary failure of specific antibody production (1–3). The rates of serious comorbidities and resulting mortality of patients with CVID drastically exceed the respective rates in the general population, imposing a high disease burden to the individual patient (4, 5). Although CVID is the most common symptomatic primary immunodeficiency (PID), it is still a rare disease with a greatly varying observed prevalence between countries, ranging in “industrialized countries” from 6.9/100,000 in Finland to 0.6/100,000 in Spain (6–14) and even lower observed prevalence rates (<0.5/100,000) in “developing” countries (15). Therefore, CVID has a low prevalence in primary care and general hospital settings, where non-immunologists have little knowledge of this disease. Also, respiratory infections and non-infectious complications of CVID such as lymphoproliferation, granulomatous disease and autoimmunity are much more prevalent without concomitant CVID. This makes it challenging to front-line clinicians to recognize CVID in these cases. Because of the variability of presenting clinical manifestations, patients visit various physicians of different specialties in search of a diagnosis, which increases the risk of missing the overarching clinical pattern and thereby overlooking the underlying hypogammaglobulinemia (16).
Timely diagnosis and optimal management are likely to result in improved clinical and quality-of-life outcomes for patients with CVID, higher participation in society (school, work) and lower health care costs (4, 17–19). Reducing diagnostic delay is therefore crucial; current approaches mainly comprise improving education and awareness of clinicians in both primary and secondary care. Already a long time ago, the Jeffrey Modell Foundation (JMF) developed ten (mainly pediatric) (20) and the European Society for Immunodeficiencies (ESID) six (adult) ‘Warnings Signs’ to indicate PIDs (21). Unfortunately, these signs have turned out to have a low sensitivity for timely PID diagnosis (22, 23).
In order to improve our insight in the early presentation of CVID and to assist physicians in its timely detection, we aimed to systematically identify and collate existing published cohort studies on the presenting clinical manifestations at and before diagnosis. In addition, we included the overall clinical manifestations during disease follow-up in our systematic review; this was done separately for children and adults to evaluate age-related differences and similarities in pediatric and adult onset CVID.
Methods
Search Strategy
We searched EMBASE, Cochrane and PubMed from January 1999 to December 2019 (inclusive) using a combination of subject headings and free text incorporating the terms ‘common variable immunodeficiency’, ‘late onset hypogammaglobulinemia’, and ‘diagnosis’, and limited to English language and humans. Reference lists of included studies were also searched for potentially relevant studies (snowball method). The complete search strategy is detailed in the supplementary appendix eSearch. The protocol of this systematic review has been registered on PROSPERO with registration number CRD42019121384.
Study Selection
We considered all primary research studies for selection, either retrospective or prospective, of any study design (e.g., case series, cohort), describing the clinical manifestations for a minimum of 10 patients with CVID. Two researchers (LJ and EV) independently screened titles and abstracts of all papers, excluding clearly irrelevant studies. Hereafter, they independently reviewed the full text of remaining papers to assess eligibility. If multiple updates of a cohort were published, the most recent study with the largest dataset describing the total clinical picture of their CVID cohort was included, in order to avoid duplicates of patients in our review. The large European multicenter study by Gathmann et al. (24) was excluded for analysis to avoid overlapping data, because this study collated data from multiple centers that already published a substantial amount of their data as single-center cohorts in more detail. Three European multicenter studies (25–27) partially overlapped in their included centers; in this case the largest multicenter study by Chapel et al. describing the overall clinical picture of CVID was included (26) (for details about the handling of overlapping data, see Supplementary Table 1). Studies that selected cases based on the presence of only certain clinical features of CVID (e.g., only granulomatous, pulmonary, gastrointestinal or autoimmune manifestations) were excluded to avoid giving disproportionate weight to those features in the data synthesis, unless the total number of CVID patients from which these cases were selected was also reported. When the same center/registry published an article about their total cohort and another article in which children and/or adults were separately described, these children- and adult-specific overlapping data were only included in the subgroup analysis for children vs adults. Any uncertainties regarding study selection were discussed between LJ, MF, and EV.
Quality Assessment
After assembling a shortlist of studies eligible for potential inclusion, LJ assessed the risk of bias in these studies to ensure that only those studies with an acceptable risk of bias were included. This quality assessment was checked by EV. Because there is no validated quality checklist for assessing retrospective cohort studies, we constructed a checklist based on relevant items from the MOOSE (meta-analysis of observational studies in epidemiology) reporting guideline for observational studies (28), the STROBE (strengthening the reporting of observational studies in epidemiology) reporting guideline for cohort studies (29) and CASP (critical appraisal skills program) guidelines for case-control and cohort studies (checklist in Supplementary Table 2) (30). Quality was assessed as ‘acceptable’ or ‘unacceptable’ in three domains: definition of CVID, selection of cases, and methods for extracting data on included cases. ‘Acceptable’ for case definition required cases to be defined according to the diagnostic criteria of ESID/Pan-American Group for Immunodeficiency (PAGID) (3), the ESID Registry working definitions for clinical diagnosis of PID (www.esid.org), the International Union of Immunological Societies (IUIS) criteria (31), the World Health Organization (WHO) scientific group (32) or the international consensus document (ICON) (33) (Supplementary Table 3), or - if no reference was made to which diagnostic criteria were used - description of the inclusion criteria corresponding to the above described diagnostic criteria. Although we only included articles about pediatric CVID that reported to have only included established CVID patients, we cannot completely rule out that a few of these patients actually had transient hypogammaglobulinemia of infancy. ‘Acceptable’ for case selection required that at least two of the participants’ baseline characteristics were clearly documented and that the characteristics of cases were sufficiently consistent with the current knowledge regarding CVID (i.e., the age and sex distribution of cases matched the known epidemiology of CVID). ‘Acceptable’ for data extraction required the use of a standardized data collection format and/or the objective measurement of signs (e.g., CT confirmation of bronchiectasis, biopsy confirmation of granulomas). Disagreements between the two reviewers (LJ and EV) were discussed with a third reviewer (MF) until agreement was achieved. Only studies considered by the two reviewers to be acceptable for case definition and to pass in at least one other domain were included, and data related to the ‘unacceptable’ domain were not included in this review.
Data Extraction
Data were extracted from included studies by LJ using a standardized Microsoft Excel spread sheet and were checked by EV and MF. We extracted study characteristics including year of publication, country, recruitment periods, number and type of centers, study design, number of patients, age and sex. Clinical manifestations were recorded, and numbers of patients with each manifestation were noted. This was done separately for the clinical manifestations at presentation (at or before diagnosis) and overall (at any timepoint). When this distinction was not described, the manifestations were collected as ‘overall’ in the standardized format. When a clinical manifestation was not discussed in a study, we made no assumption about whether or not that manifestation had occurred in that population but recorded this item as ‘missing data’ for the respective study in the standardized format. We deliberately chose to use the exact wordings of the included studies, to avoid interpretation bias. Clinical manifestations were never counted twice. For example, where one study separately described sinusitis and otitis, another study only mentioned ‘upper respiratory tract infections’. In addition, we recorded whether in a cohort, children, adults or both were described. A pediatric cohort was defined as age during follow-up <18 years old, which was comparable to the cut-off value for children’s age provided by the original studies. We did not contact the authors of included papers to collect additional information.
Statistical Analysis
We used MetaXL (version 5.3, EpiGear International, Queensland, Australia) to calculate proportions and standard errors (SEs) of proportions for each clinical manifestation in each included study (34). To combine the results of multiple cohorts, we calculated pooled proportions of each clinical manifestation using the metan command. Anticipating high heterogeneity between included studies, we performed random effects meta-analysis using the DerSimonian and Laird method and standard methods to calculate I2 as an estimate of heterogeneity. In addition, we conducted two subgroup analyses using the same techniques: 1) children vs adults, and 2) clinical manifestations at presentation vs overall clinical manifestations during the disease course. A subgroup analysis based on age was conducted because the few available studies on differences between pediatric-onset and adult-onset CVID have yielded limited and conflicting data (35–37). In order to improve our understanding of the early presentation of CVID, which can assist clinicians in timely detection of this condition, we focused our analysis on clinical manifestations at or prior to diagnosis.
Results
Search Results
After removal of duplicates, we identified 1604 papers. We excluded 1453 after screening titles and abstracts, and a further 96 after full-text assessment (Figure 1), based on the inclusion criteria (see Method). Reference lists of included studies yielded 21 additional eligible studies. There was full consensus between the authors regarding study inclusion.
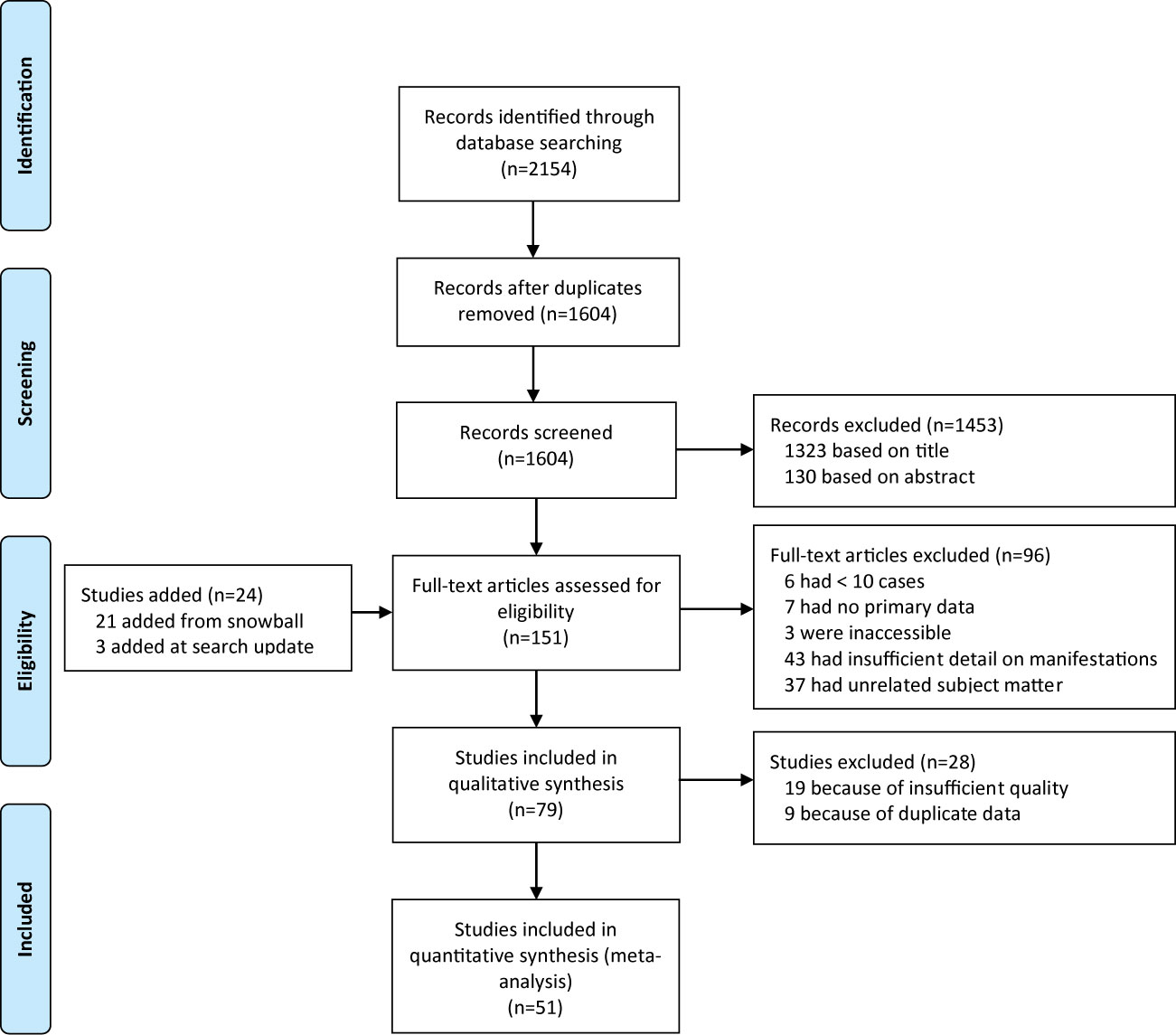
Figure 1 Flow chart showing the study selection process.
Characteristics of Included Studies
The 51 included studies described clinical manifestations in a total of 8521 patients (Table 1) (5, 6, 19, 26, 35–81). 50 studies were conducted in one or more centers in one country only, in 18 different countries in total; 1 study included multiple centers from different countries (26). Most were cohort studies (5 prospective, 42 retrospective) and three compared cases with controls. All 51 studies extracted data from written/typed hospital records. The majority of studies (n=39) identified cases from hospital records alone; others also used regional, national, or continental registries of primary immunodeficiencies (n=12). Three studies also obtained data from a patient and/or parent-completed questionnaire (41, 45, 55). Fifteen studies reported clinical manifestations of their total CVID cohort, but reported in more detail on patients based on the presence of only certain clinical features of CVID: asthma and allergic diseases (39, 81), autoimmune manifestations (63, 65, 66), gastric cancer (49), gastrointestinal manifestations (41, 76), granulomatous manifestations (64, 78), or pulmonary manifestations (43, 45, 46, 69, 75).
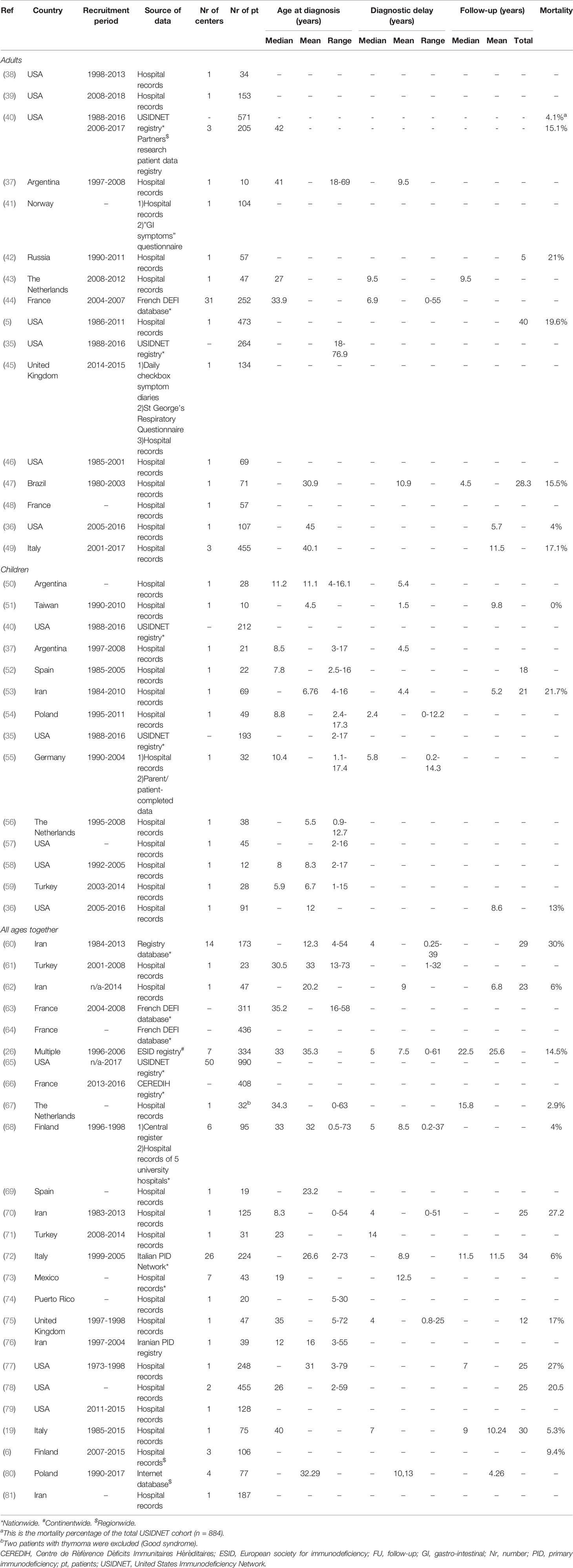
Table 1 Characteristics of included studies.
Risk of Bias of Included Studies
Most included studies defined cases using the diagnostic criteria of PAGID and ESID (27 studies); other used criteria were: the ESID Registry working diagnosis criteria (8 studies), International consensus document (3 studies), IUIS criteria (2 studies), and WHO classification (4 studies). Seven studies did not report which criteria were used but did describe a CVID diagnosis that corresponded to the above approved classifications. One study reported to use both the diagnostic criteria of PAGID/ESID and the WHO classification (51). Lack of routine B and T cell immunophenotyping in most studies prohibited an accurate assessment of potential late-onset combined immunodeficiency (LOCID). 37 studies (73%) included all consecutive cases within the study period, with a further 11 studies (22%) describing why a proportion of potentially eligible cases were excluded. In the remaining 3 studies (6%), the proportion of consecutively included cases was unclear.
A weakness of the included studies was lack of clarity at which point in the diagnostic and follow-up pathway clinical features were recorded. Twenty studies explicitly stated when clinical manifestations occurred [at or before diagnosis (n=4), and both at/or before diagnosis and during follow-up (n=16)]. The remaining 31 studies were unclear as to when the reported clinical manifestations occurred during the disease course.
Pooled Frequencies of Clinical Characteristics From Meta-Analysis
Pooled frequencies of demographic information are shown in Table 2. In pediatric CVID patients, males were in the majority (62%, 95% CI 54-69), while females predominated in the adult CVID patients (58%, 95% CI 53-64). The high pooled proportion of consanguinity in the pediatric and total cohort should be interpreted with caution (31% and 20% respectively; only one study reported this for adults). This proportion varied substantially per country. In an Argentinian cohort none had a history of consanguinity (37), while the rate of consanguinity was very high in an Iranian cohort (72%) (53).
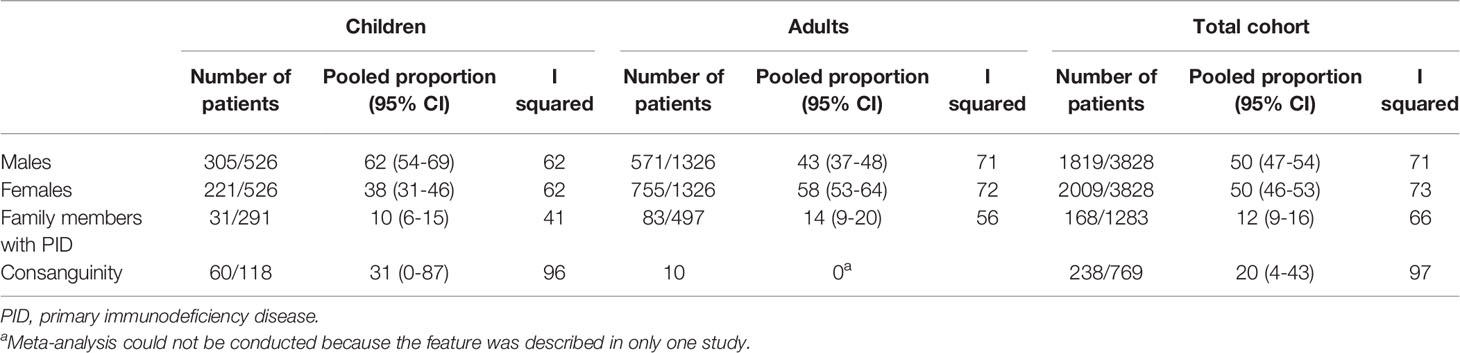
Table 2 Demographic parameters in adult-, pediatric-, and total cohorts.
In total, 147 out of a potential of 270 meta-analyses were conducted. For the remaining 123 clinical manifestations, meta-analysis was not possible since the features were each reported in only one study. The high heterogeneity (I2) statistics in the meta-analyses (mostly >80%) indicated that the degree of heterogeneity between studies was greater than that expected by chance alone and confirmed the appropriateness of random-effects meta-analysis to generate pooled proportions.
There were 49 specific clinical manifestations for which it was possible to calculate pooled proportions for the subgroup at presentation, i.e. ‘at or before diagnosis’; these are shown in Figure 2 in comparison with overall, i.e. ‘at, before or after’ diagnosis. The most frequent clinical manifestations at presentation (reported in ≥39% of patients) are shown above the grey dotted horizontal lines. A history of upper and/or lower respiratory infections was present at diagnosis in three-quarters of patients (upper respiratory tract infections in 73%, lower respiratory tract infections in 73%, sinusitis in 59%, pneumonia in 57%, bronchitis in 57% and otitis in 39%) and severe bacterial infections in 8% (septicemia), 8% (mastoiditis), 6% (meningitis), and 4% (osteomyelitis).
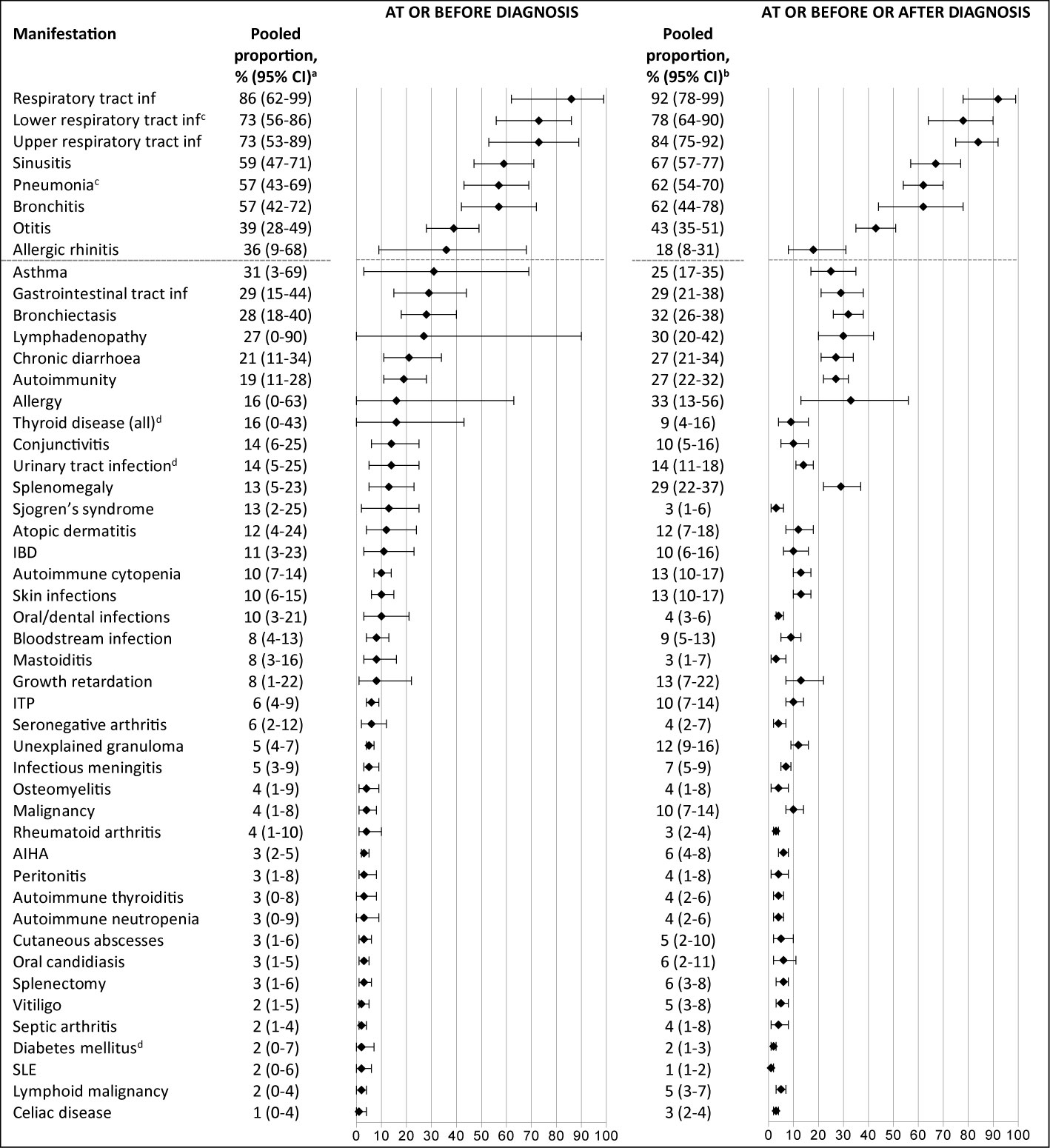
Figure 2 Frequency of reported clinical manifestations at presentation vs overall clinical manifestations during the disease course.The most frequent clinical manifestations at presentation (reported in ≥39% of patients) are shown above the grey dotted horizontal lines. aNumber of patients ranged from 44 to 1137; number of studies ranged from 2-15. bNumber of patients ranged from 51 to 4061; number of studies ranged from 2-31. cPneumonia and lower respiratory tract infections were not combined into one category, as they were often mentioned as two separate categories in the included studies. dThe prevalence of this clinical manifestation is similar or lower to lifetime prevalence estimates in general population. IBD, inflammatory bowel disease; inf, infections; ITP, idiopathic thrombocytopenic purpura; AIHA, autoimmune hemolytic anemia; SLE, systemic lupus erythematosus.
Bronchiectasis was already present in almost one third of the patients at or before the CVID diagnosis was made (28%, 95% CI 18-40). Non-infectious manifestations that were frequently present at diagnosis were: lymphadenopathy (27%), splenomegaly (13%), inflammatory bowel disease (11%), and autoimmune hematological manifestations (autoimmune cytopenia (10%) and idiopathic thrombocytopenia (6%)). The pooled prevalences at presentation of urinary tract infection (14%, 95% CI 5-25), thyroid disease (16%, 95% CI 0-43), and diabetes mellitus (2%, 95% CI 0-7) correspond to the estimated lifetime prevalence estimates in the general population of 30% (82), 12% (83), and 0.9% (type 1) (84), respectively. An overview of all reported clinical manifestations at and before diagnosis with – when known – lifetime prevalence estimates from the general population is included in Supplementary Table 4.
In Figure 3, all clinical manifestations that were present in ≥10% of patients are shown (as presented, whether or not likely related to CVID); the CVID-associated manifestations are also shown when present in <10% when they were considered important to incorporate by the authors (based on obvious relation with CVID in the current literature or consensus in the field). We grouped these manifestations into eleven distinct clinical categories according to the body system affected and the clinical phenotypes described by Chapel et al. (26). An overview of all reported clinical manifestations is included in Supplementary Table 5. Many CVID patients developed non-infectious manifestations during follow-up: bronchiectasis in 32%, lymphadenopathy in 30%, splenomegaly in 29%, polyclonal lymphocytic infiltration in 29%, and autoimmune manifestations in 27%. In addition, a substantial number of patients developed malignancies (10%) and atopic diseases during the entire disease course [asthma (25%), allergic rhinitis (18%)].
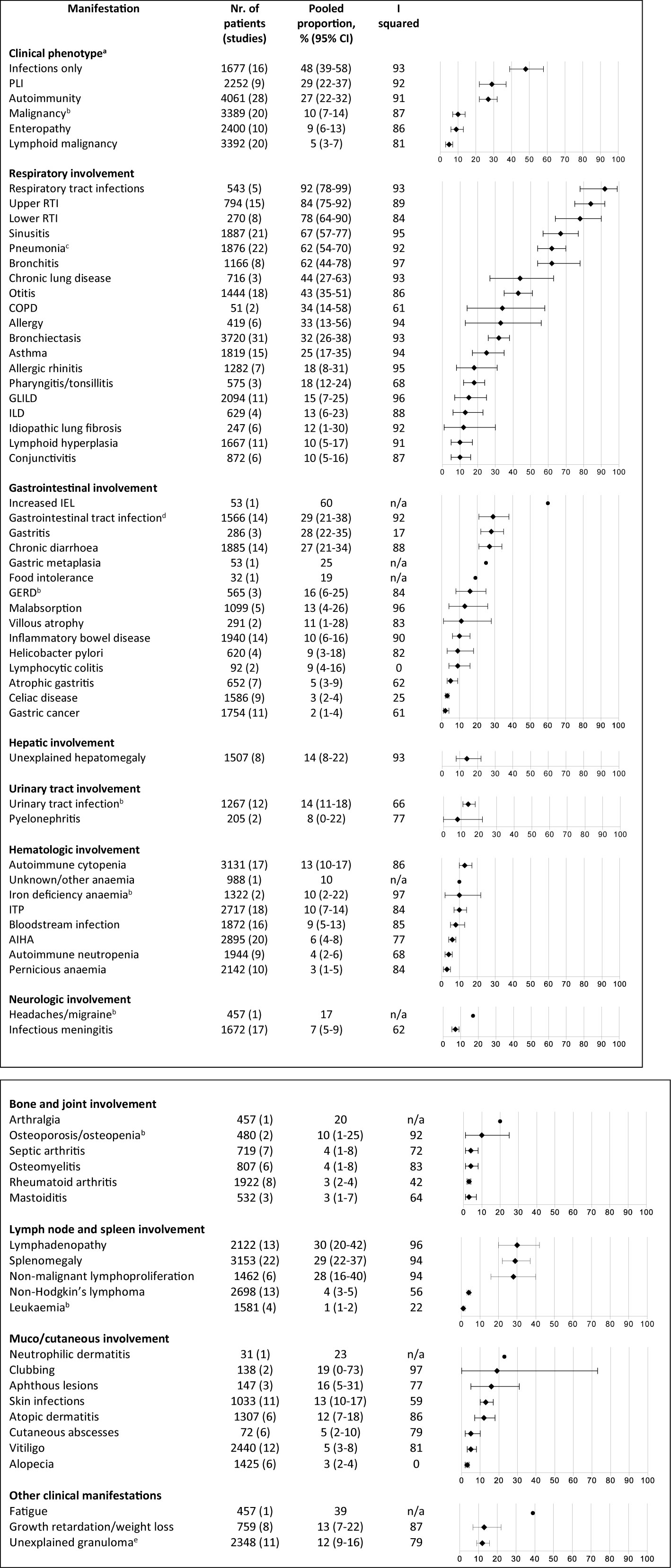
Figure 3 Frequency of reported clinical manifestations of patients with common variable immunodeficiency. All clinical manifestations that were present in ≥10% of patients are shown; the CVID-associated manifestations are also shown when present in <10% when they were considered important to incorporate by the authors (based on current literature and consensus in the field). aDerived from Chapel et al. (26). bThe prevalence of this clinical manifestation is similar or lower to lifetime prevalence estimates in general population. cStreptococcus pneumoniae 15%, 95% CI 8-23; Hemophilus influenzae 19%, 95% CI 8-33; Moraxella catarrhalis 7%, 95% CI 0-19; Staphylococcus aureus 7%, 95% CI 3-12; Mycobacterial infection 1%, 95% CI 0-2; Pneumocystis jiroveci 1%, 95% CI 0-2%; Pseudomonas 6%, 95% CI 2-10; Aspergillus 3%, 95% CI 1-5; Mycoplasma 2%, 95% CI 0-4. dGiardia intestinalis 13%, 95% CI 7-21; Candida species 10%, 95% CI 4-19; Salmonella species 6%, 95% CI 2-12; Campylobacter species 4%, 95% CI 1-8.eIntestinal granulomatosis 1%, 95% CI 0-4; liver granuloma 3%, 95% CI 1-6; granuloma in lymph node 2%, 95% CI 0-5; granuloma in spleen 1%, 95% CI 0-2; skin granuloma 1%, 95% CI 0-2. AIHA, autoimmune hemolytic anemia; COPD, chronic obstructive pulmonary disease; CVID, common variable immunodeficiency disorders; GERD, gastro-esophageal reflux disease; GLILD, granulomatous and lymphocytic interstitial lung disease; IEL, increased intraepithelial lymphocytes; ILD, interstitial lung disease; ITP, idiopathic thrombocytopenic purpura; PLI, polyclonal lymphocytic infiltration; RTI, respiratory tract infection.
Three quarters of the children (76%, 95% CI 57-91) developed no other complications besides infections during the reported follow-up periods (See Figure 4), while this percentage was much lower in the adults (38%, 95% CI 27-49). Certain infectious features of CVID, such as otitis, were more common in children (55%, 95% CI 45-64) than in adults (32%, 95% CI 27-37), whereas certain immune dysregulation features, such as granulomatous-lymphocytic interstitial lung disease, chronic diarrhea and inflammatory bowel disease were more prominent in adults (33%, 95% CI 13-57; 34%, 95% CI 22-48; 18%, 95% CI 6-33; respectively) than in children (6%, 95% CI 4-9; 17%, 95% CI 11-24; 3%, 95% CI 1-6, respectively). Bronchiectasis were more common in adults (36%, 95% CI 27-46) than in children (16%, 95% CI 9-25). An overview of all reported clinical manifestations in children and adults is included in Supplementary Table 6.

Figure 4 Frequency of reported clinical manifestations in children vs adults. IBD, inflammatory bowel disease; inf, infections; GLILD, granulomatous and lymphocytic interstitial lung disease; ITP, idiopathic thrombocytopenic purpura; AIHA, autoimmune hemolytic anemia.
Discussion
To our knowledge, our study is the first systematic review and meta-analysis of pooled clinical manifestations in patients with CVID. Our findings can help clinicians to recognize CVID, and to estimate how common a clinical manifestation is in pediatric and adult CVID. We identified 134 different presenting clinical manifestations in patients diagnosed with CVID (the limited number of data impeded splitting up between children and adults). In addition, we identified 270 different clinical manifestations occurring during the entire course of the disease (147 in children and 170 in adults). Most frequent presenting manifestations were recurrent upper respiratory tract infections (73%), lower respiratory tract infections (73%), sinusitis (59%), pneumonia (57%), bronchitis (57%) and otitis (39%), concurrent with the first two ESID and first three JMF warning signs for PID (20, 21). However, these manifestations are also frequent in the general population and may lack discriminating value, unless their unusually recurrent and persistent nature is recognized (85). Other alerts to potential PID that are in line with the JMF warning signs that may be more discriminatory include severe bacterial infections (osteomyelitis in 4%, meningitis in 6%, septicemia in 8%, mastoiditis in 8%), which are clearly more frequent than lifetime prevalence in the general population. Recent studies already demonstrated a high incidence of antibody deficiency in patients with pneumococcal meningitis (86–88), confirming our finding of a high frequency of infectious meningitis in CVID, but we also show a high incidence of bloodstream infections, mastoiditis and to a somewhat lesser extent osteomyelitis in CVID patients. This suggests that the incidence of CVID may also be increased in patients with bloodstream infections, mastoiditis and osteomyelitis without other clear predispositions and suggest screening for CVID could be useful in these patients. This finding warrants further exploration.
One of the most reliable alerts to potential CVID was CVID in the family (12% of the total reviewed population). Both the six ESID and ten JMF warning signs make no mention of other presenting manifestations than frequent and/or severe infections, such as bronchiectasis (28%), lymphadenopathy (27%), splenomegaly (13%), chronic diarrhea (21%), inflammatory bowel disease (11%), or autoimmune hematological manifestations (autoimmune cytopenia (10%) and idiopathic thrombocytopenia (6%)). Our results suggest that also lymphoproliferative, gastrointestinal and autoimmune manifestations should be included in warning signs for predicting PID. This is important, because we still fail to detect the disease early enough. Increasing awareness of this varied and complex presentation of CVID can lead to earlier detection and initiation of treatment.
Our findings show a male predominance in children with CVID (62%), but a female predominance in adults (58%). This is also observed in atopic disease and we previously described this in patients with unclassified primary antibody deficiency (89), but it has not previously been recognized in CVID. This sex shift may indicate that etiology differs in different age groups. Early childhood male predominance suggests X-linked heredity is present in some boys diagnosed with the disease; adult female predominance suggests sex hormone effects, environmental exposure, and epigenetic influences may play a role (90). This implicates that future studies that attempt to define mechanisms that underpin CVID should be stratified according to sex.
There were clear differences in clinical manifestations occurring during the disease course between children and adults with CVID. Overall prevalence of bronchiectasis was 36% in adults vs 16% in children. Persistence of an ‘infection-only’ phenotype was much more prevalent in pediatric than in adult CVID (76 vs 38%). During childhood, three quarters of patients developed no other complications besides infections, while this percentage was much lower during follow up in adults (38%). Immune-dysregulation features, such as granulomatous-lymphocytic interstitial lung disease (15 vs 6%), chronic diarrhea (34 vs 17%), and inflammatory bowel disease (18 vs 3%) were more prominent in adults compared to children. A possible explanation could be longer ongoing inflammation and longer follow-up in the adults (43). One is an adult for many more years than one is a child, thus there are many more physician visits in the adult years and more opportunities for CVID complications to be observed. The different signs and symptoms observed in CVID between pediatric and adult age, with more non-infectious disease complications in adults, suggest that different monitoring strategies for children and adults during follow-up may be warranted.
Most common non-infectious manifestations included bronchiectasis (32%), lymphadenopathy (30%), splenomegaly (29%), polyclonal lymphocytic infiltration (29%), and autoimmune manifestations (27%). While only a quarter of CVID patients had features of immune dysregulation at presentation, this increased to about half of the patients throughout the course of the disease. This suggests that these manifestations more often occur later in the disease course. It is crucial that CVID patients are monitored for the development of these complications, because some of these are difficult to treat and associated with increased mortality (4,14). The coincidence of immunodeficiency and immune dysregulation can be explained by several mechanisms. Immunodeficiency may result in insufficient clearance of microbial antigens, and the resulting persistent antigenic exposure could then trigger granulomatous disease and autoimmunity (91). Both complications have been linked to hyperplastic germinal centers enriched with polyclonal/self-reactive B-cell clones (92), and immature B cell development (25) in CVID. In addition, low numbers of regulatory T-cells (91, 93), and an increasing number of genetic defects (94) have been associated with immune dysregulation in CVID. Additional factors, such as commensal microbial dysbiosis and epigenetic modifications remain to be better elucidated (95).
Interestingly, we found high pooled prevalences of atopic diseases both at presentation of CVID and during the entire disease course (asthma 31 vs 25%, allergic rhinitis 36 vs 18%). The pooled prevalences of asthma and allergic rhinitis are higher in CVID compared to the estimated lifetime prevalences in the general population of 13.6% (96) and 6.6% (97), respectively. This should be interpreted with caution because of the considerable heterogeneity between the studies (I2 >80). Also, not all patients underwent cutaneous or in vitro testing or spirometry to support these diagnoses, nor was it reported how often the asthma was atopic in nature. It is possible that symptoms derived from the deficient immune system were interpreted as atopic disease, on the other hand, atopic disorders could actually be more prevalent in CVID. Overlap between the symptoms of atopic diseases and immunodeficiency may lead to delayed diagnosis, so it is important to consider CVID in patients with atopic diagnoses who are insufficiently responsive to standard treatment and who also have infections. To further elucidate the association between atopic diseases and CVID, a prospective multi-center study in a large unselected CVID cohort would be needed.
A substantial number of patients developed malignancies during the disease course (10%, 95% CI 7-14). This pooled prevalence is comparable with the result of a previous focused meta-analysis of malignancy prevalence in CVID (8.6%, 95% CI 7.1-10) (98). Also, in alignment with previous reports the most common malignancies were lymphoid malignancies (5%, 95% CI 3-7) and gastric cancers (2%, 95% CI 1-4) (49, 72, 98–100). The lack of data on controls impedes comparison of our results to the normative population, but the prevalences are higher compared to the lifetime prevalence estimates in the United States population (lymphoid malignancies 2.3%, gastric cancers 0.8%) (101). The prevalence of cases with lung-, colorectal-, uterine-, liver-, and pancreatic cancer and leukemia were similar to what one might expect in the general population according to the lifetime risk statistics based on the United States population (Supplementary Table 4) (101).
Strengths and Limitations
This analysis collates data from >8000 patients (850 children, 2998 adults, 4673 not specified/both) in 51 studies from 18 different countries. The included studies were conducted in Europe, North- and South America, and Asia; there were no studies from Australia or Africa. Our review adhered to rigorous methods, including a systematic search strategy, and explicit inclusion criteria (102). The findings therefore present the most comprehensive and internationally relevant presenting manifestations for clinicians worldwide.
The study has some limitations and potential sources of bias. The main limitations reflect deficits in the design and reporting of the included studies. Accuracy of our systematic analysis depends on the quality of the published and supplementary data that we included. All studies provided data on cases only, and not on controls. Therefore, we were unable to compare the frequency of clinical manifestations in CVID patients to the frequency in the general population. Publication bias could have led to overrepresentation of more complex cases of CVID, and therefore higher incidences of non-infectious complications. We did not include unpublished data. Heterogeneity between included studies was high. Most included studies provided little motivation for the selection of the clinical manifestations studied, thus it is difficult to account with certainty for the variation in number and choice of the selected clinical manifestations. The variation in reported clinical phenotypes and complications between cohorts may stem from differences in study populations (for instance, due to access to health care, rate at which patients are properly diagnosed, degree of consanguinity, or population genetic differences), use of different methods to diagnose findings, underreporting of histological diagnoses because biopsies are not performed, and the use of different definitions for CVID. Full consensus regarding the definition of CVID does not yet exist (103). Also, in a few series, a small proportion of pediatric and adult patients had opportunistic infections and/or a low CD4 T-cell count, and those patients should actually be classified as a combined immunodeficiency. This phenotype has been re-named in adults as late onset combined immunodeficiency (LOCID) by the IUIS. In addition, given the rapid progress in next-generation sequencing, in non-consanguineous populations, a causative mutation may currently be identified in ~25% of CVID patients (104). Ideally, patients with monogenic diseases and LOCID should have been excluded from our analysis, but it was not possible to identify them exactly in the described cohorts (105). Transient hypogammaglobulinemia of infancy may have been misdiagnosed as CVID in some of the children included in the different series. As we only included children diagnosed at age >4 years it is unlikely this accounts for a large percentage of included children.
Implications for Future Research
Our study identified two key limitations in the current evidence base on CVID presentation. First, we found relatively few studies that explicitly reported data on clinical signs and symptoms at or before diagnosis of the disease. We lack data on the frequency and time of onset of symptoms from the first symptoms at home to the final diagnosis. Second, only few studies compared pediatric with adult CVID. Further large, multicenter, prospective cohort studies, separately describing children and adults, would address these gaps.
Conclusions
In conclusion, this meta-analysis confirms the high frequency of upper and/or lower respiratory tract infections in CVID at presentation, but also shows a remarkably high incidence of severe bacterial infections (osteomyelitis in 4%, meningitis in 6%, septicemia in 8%, mastoiditis in 8%) compared to lifetime prevalence in the general population. This suggests that the incidence of CVID may also be high in patients with severe bacterial infections without other clear predispositions and suggests screening for CVID might be useful in these patients. These findings warrant further exploration. In addition, CVID patients commonly present with other manifestations than frequent or severe infections – which are not included in ESID and JMF warning signs for identifying patients with primary immunodeficiencies. Not only the infectious, but also the immune dysregulation features (shown in Figure 2), should alert to the possibility of CVID, regardless whether they occur with or without recurrent infections. The bimodal sex distribution in patients with CVID implicates that future studies that attempt to define mechanisms that underpin CVID should be stratified according to sex.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Author Contributions
LJ, MF, and EV devised the study. LJ, MF, and EV acquired the data. LJ carried out data analysis and drafted the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of Interest
MF received research support to study innovative antibody preparations from CSL Behring outside the submitted work. EV received an unrestricted research grant for the unPAD study outside the submitted work from Shire/Takeda.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.620709/full#supplementary-material
Abbreviations
CASP, critical appraisal skills program; CVID, common variable immunodeficiency disorders; ESID, European Society of Immunodeficiency; ICON, international consensus document; LOCID, late onset combined immunodeficiency; MOOSE, meta-analysis of observational studies in epidemiology; PID, primary immunodeficiency; SE, standard error; STROBE, strengthening the reporting of observational studies in epidemiology.
References
1. Picard C, Bobby Gaspar H, Al-Herz W, Bousfiha A, Casanova J-L, Chatila T, et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J Clin Immunol (2018) 38(1):96–128. doi: 10.1007/s10875-017-0464-9
2. Bonilla FA, Barlan I, Chapel H, Costa-Carvalho BT, Cunningham-Rundles C, de la Morena MT, et al. International Consensus Document (ICON): Common Variable Immunodeficiency Disorders. J Allergy Clin Immunol Pract (2016) 4(1):38–59. doi: 10.1016/j.jaip.2015.07.025
3. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan-American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol (1999) 93(3):190–7. doi: 10.1006/clim.1999.4799
4. Odnoletkova I, Kindle G, Quinti I, Grimbacher B, Knerr V, Gathmann B, et al. The burden of common variable immunodeficiency disorders: a retrospective analysis of the European Society for Immunodeficiency (ESID) registry data. Orphanet J Rare Dis (2018) 13(1):201. doi: 10.1186/s13023-018-0941-0
5. Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood (2012) 119(7):1650–7. doi: 10.1182/blood-2011-09-377945
6. Selenius JS, Martelius T, Pikkarainen S, Siitonen S, Mattila E, Pietikainen R, et al. Unexpectedly High Prevalence of Common Variable Immunodeficiency in Finland. Front Immunol (2017) 8:1190. doi: 10.3389/fimmu.2017.01190
7. CEREDIH: The French PID study group. The French national registry of primary immunodeficiency diseases. Clin Immunol (2010) 135(2):264–72. doi: 10.1016/j.clim.2010.02.021
8. Matamoros Florí N, Mila Llambi J, Español Boren T, Raga Borja S, Fontan Casariego G. Primary immunodeficiency syndrome in Spain: first report of the National Registry in Children and Adults. J Clin Immunol (1997) 17(4):333–9. doi: 10.1023/A:1027382916924
9. Stray-Pedersen A, Abrahamsen TG, Frøland SS. Primary immunodeficiency diseases in Norway. J Clin Immunol (2000) 20(6):477–85. doi: 10.1023/A:1026416017763
10. Kilic SS, Ozel M, Hafizoglu D, Karaca NE, Aksu G, Kutukculer N. The prevalences [correction] and patient characteristics of primary immunodeficiency diseases in Turkey–two centers study. J Clin Immunol (2013) 33(1):74–83. doi: 10.1007/s10875-012-9763-3
11. Edgar JDM, Buckland M, Guzman D, Conlon NP, Knerr V, Bangs C, et al. The United Kingdom Primary Immune Deficiency (UKPID) Registry: report of the first 4 years’ activity 2008-2012. Clin Exp Immunol (2014) 175(1):68–78. doi: 10.1111/cei.12172
12. Marschall K, Hoernes M, Bitzenhofer-Grüber M, Jandus P, Duppenthaler A, Wuillemin WA, et al. The Swiss National Registry for Primary Immunodeficiencies: report on the first 6 years’ activity from 2008 to 2014. Clin Exp Immunol (2015) 182(1):45–50. doi: 10.1111/cei.12661
13. Ludviksson BR, Sigurdardottir ST, Johannsson JH, Haraldsson A, Hardarson TO. Epidemiology of Primary Immunodeficiency in Iceland. J Clin Immunol (2015) 35(1):75–9. doi: 10.1007/s10875-014-0107-3
14. Westh L, Mogensen TH, Dalgaard LS, Bernth Jensen JM, Katzenstein T, Hansen A-BE, et al. Identification and Characterization of a Nationwide Danish Adult Common Variable Immunodeficiency Cohort. Scand J Immunol (2017) 85(6):450–61. doi: 10.1111/sji.12551
15. Weifenbach N, Schneckenburger AAC, Lötters S. Global Distribution of Common Variable Immunodeficiency (CVID) in the Light of the UNDP Human Development Index (HDI): A Preliminary Perspective of a Rare Disease. J Immunol Res (2020) 2020:8416124. doi: 10.1155/2020/8416124
16. Ilkjaer FV, Rasmussen LD, Martin-Iguacel R, Westh L, Katzenstein TL, Hansen A-BE, et al. How to Identify Common Variable Immunodeficiency Patients Earlier: General Practice Patterns. J Clin Immunol (2019) 39(7):641–52. doi: 10.1007/s10875-019-00666-9
17. Quinti I, Di Pietro C, Martini H, Pesce AM, Lombardi F, Baumghartner M, et al. Health related quality of life in common variable immunodeficiency. Yonsei Med J (2012) 53(3):603–10. doi: 10.3349/ymj.2012.53.3.603
18. Rider NL, Kutac C, Hajjar J, Scalchunes C, Seeborg FO, Boyle M, et al. Health-Related Quality of Life in Adult Patients with Common Variable Immunodeficiency Disorders and Impact of Treatment. J Clin Immunol (2017) 37(5):461–75. doi: 10.1007/s10875-017-0404-8
19. Graziano V, Pecoraro A, Mormile I, Quaremba G, Genovese A, Buccelli C, et al. Delay in diagnosis affects the clinical outcome in a cohort of cvid patients with marked reduction of iga serum levels. Clin Immunol (2017) 180:1–4. doi: 10.1016/j.clim.2017.03.011
21. https://esid.org/Working-Parties/Clinical-Working-Party/Resources/6-Warning-Signs-for-PID-in-Adultshttps://esid.org/Working-Parties/Clinical-Working-Party/Resources/6-Warning-Signs-for-PID-in-Adults.
22. Arkwright PD, Gennery AR. Ten warning signs of primary immunodeficiency: a new paradigm is needed for the 21st century. Ann N Y Acad Sci (2011) 1238:7–14. doi: 10.1111/j.1749-6632.2011.06206.x
23. Arslan S, Ucar R, Caliskaner AZ, Reisli I, Guner SN, Sayar EH, et al. How effective are the 6 European Society of Immunodeficiency warning signs for primary immunodeficiency disease? Ann Allergy Asthma Immunol (2016) 116(2):151–5.e1. doi: 10.1016/j.anai.2015.12.001
24. Gathmann B, Mahlaoui N, Gerard L, Oksenhendler E, Warnatz K, Schulze I, et al. Clinical picture and treatment of 2212 patients with common variable immunodeficiency. J Allergy Clin Immunol (2014) 134(1):116–26. doi: 10.1016/j.jaci.2013.12.1077
25. Wehr C, Kivioja T, Schmitt C, Ferry B, Witte T, Eren E, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. (2008) 111(1):77–85. doi: 10.1182/blood-2007-06-091744
26. Chapel H, Lucas M, Lee M, Bjorkander J, Webster D, Grimbacher B, et al. Common variable immunodeficiency disorders: division into distinct clinical phenotypes. Blood. (2008) 112(2):277–86. doi: 10.1182/blood-2007-11-124545
27. Packwood K, Drewe E, Staples E, Webster D, Witte T, Litzman J, et al. NOD2 polymorphisms in clinical phenotypes of common variable immunodeficiency disorders. Clin Exp Immunol (2010) 161(3):536–41. doi: 10.1111/j.1365-2249.2010.04216.x
28. Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D, et al. Meta-analysis of observational studies in epidemiology: a proposal for reporting. Meta-analysis Of Observational Studies in Epidemiology (MOOSE) group. JAMA (2000) 283(15):2008–12. doi: 10.1001/jama.283.15.2008
29. von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP. Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. BMJ. (2007) 335(7624):806–8. doi: 10.1136/bmj.39335.541782.AD
30. Critical Appraisal Skills Programme. CASP (case control and cohort questions) Checklist. (2018). Available at: www.casp-uk.net (Accessed 2021-03-05).
31. Primary immunodeficiency diseases. Report of an IUIS Scientific Committee. International Union of Immunological Societies. Clin Exp Immunol (1999) 118 Suppl:1–28. doi: 10.1046/j.1365-2249.1999.00109.x
32. Primary immunodeficiency diseases. Report of a WHO scientific group. Clin Exp Immunol (1997) 109 Suppl:1–28.
33. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank MM, Hsu JT, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol (2015) 136(5):1178–86. doi: 10.1016/j.jaci.2015.04.049
34. MetaXL User Guide. Sunrise Beach, Queensland, Australia: EpiGear International Pty Ltd (2011-2016) Available at: www.epigear.com.
35. Sanchez LA, Maggadottir SM, Pantell MS, Lugar P, Rundles CC, Sullivan KE. Two Sides of the Same Coin: Pediatric-Onset and Adult-Onset Common Variable Immune Deficiency. J Clin Immunol (2017) 37(6):592–602. doi: 10.1007/s10875-017-0415-5
36. Baloh C, Reddy A, Henson M, Prince K, Buckley R, Lugar P. 30-Year Review of Pediatric- and Adult-Onset CVID: Clinical Correlates and Prognostic Indicators. J Clin Immunol (2019) 39(7):678–87. doi: 10.1007/s10875-019-00674-9
37. Bezrodnik L, Gaillard MI, Carelli D. Clinical and immunological assessment of 94 patients with primary humoral immunodeficiency: Common variable immunodeficiency, selective IgA deficiency and polysaccharide antibody deficiency syndrome. J Pediatric Infect Dis (2011) 6:159–66. doi: 10.3233/JPI-2011-0320
38. Barton JC, Bertoli LF, Barton JC. Comparisons of CVID and IgGSD: referring physicians, autoimmune conditions, pneumovax reactivity, immunoglobulin levels, blood lymphocyte subsets, and HLA-A and -B typing in 432 adult index patients. J Immunol Res (2014) 2014:542706. doi: 10.1155/2014/542706
39. Bjelac JA, Blanch MB, Fernandez J. Allergic disease in patients with common variable immunodeficiency at a tertiary care referral center. Ann allergy Asthma immunology: Off Publ Am Coll Allergy Asthma Immunol United States (2018) 120:90–2. doi: 10.1016/j.anai.2017.09.075
40. Farmer JR, Ong M-S, Barmettler S, Yonker LM, Fuleihan R, Sullivan KE, et al. Common Variable Immunodeficiency Non-Infectious Disease Endotypes Redefined Using Unbiased Network Clustering in Large Electronic Datasets. Front Immunol (2017) 8:1740. doi: 10.3389/fimmu.2017.01740
41. Jorgensen SF, Reims HM, Frydenlund D, Holm K, Paulsen V, Michelsen AE, et al. A Cross-Sectional Study of the Prevalence of Gastrointestinal Symptoms and Pathology in Patients With Common Variable Immunodeficiency. Am J Gastroenterol (2016) 111(10):1467–75. doi: 10.1038/ajg.2016.329
42. Karaulov AV, Sidorenko IV, Kapustina AS. Major approaches in early diagnostics of common variable immunodeficiency in adults in Moscow. F1000Research (2012) 1:46. doi: 10.12688/f1000research.1-46.v1
43. Maarschalk-Ellerbroek LJ, de Jong PA, van Montfrans JM, Lammers JWJ, Bloem AC, Hoepelman AIM, et al. CT screening for pulmonary pathology in common variable immunodeficiency disorders and the correlation with clinical and immunological parameters. J Clin Immunol (2014) 34(6):642–54. doi: 10.1007/s10875-014-0068-6
44. Oksenhendler E, Gerard L, Fieschi C, Malphettes M, Mouillot G, Jaussaud R, et al. Infections in 252 patients with common variable immunodeficiency. Clin Infect Dis (2008) 46(10):1547–54. doi: 10.1086/587669
45. Sperlich JM, Grimbacher B, Workman S, Haque T, Seneviratne SL, Burns SO, et al. Respiratory Infections and Antibiotic Usage in Common Variable Immunodeficiency. J Allergy Clin Immunol Pract (2018) 6(1):159–168.e3. doi: 10.1016/j.jaip.2017.05.024
46. Bates CA, Ellison MC, Lynch DA, Cool CD, Brown KK, Routes JM. Granulomatous-lymphocytic lung disease shortens survival in common variable immunodeficiency. J Allergy Clin Immunol (2004) 114(2):415–21. doi: 10.1016/j.jaci.2004.05.057
47. Kokron CM, Errante PR, Barros MT, Baracho GV, Camargo MM, Kalil J, et al. Clinical and laboratory aspects of common variable immunodeficiency. Acad Bras Cienc (2004) 76(4):707–26. doi: 10.1590/S0001-37652004000400007
48. Piqueras B, Lavenu-Bombled C, Galicier L, Bergeron-van der Cruyssen F, Mouthon L, Chevret S, et al. Common variable immunodeficiency patient classification based on impaired B cell memory differentiation correlates with clinical aspects. J Clin Immunol (2003) 23(5):385–400. doi: 10.1023/A:1025373601374
49. Pulvirenti F, Pecoraro A, Cinetto F, Milito C, Valente M, Santangeli E, et al. Gastric Cancer Is the Leading Cause of Death in Italian Adult Patients With Common Variable Immunodeficiency. Front Immunol (2018) 9:2546. doi: 10.3389/fimmu.2018.02546
50. Almejun MB, Sajaroff E, Galicchio M, Oleastro M, Bernasconi A, Zelazko M, et al. Immunological characteristics and two novel mutations in TACI in a cohort of 28 pediatric patients with common variable immunodeficiency. J Clin Immunol (2012) 32(1):89–97. doi: 10.1007/s10875-011-9613-8
51. Chan H-Y, Yang Y-H, Yu H-H, Chien Y-H, Chiang L-L, Chiang B-L. Clinical characteristics and outcomes of primary antibody deficiency: a 20-year follow-up study. J Formos Med Assoc (2014) 113(6):340–8. doi: 10.1016/j.jfma.2012.07.005
52. Llobet MP, Soler-Palacin P, Detkova D, Hernandez M, Caragol I, Espanol T. Common variable immunodeficiency: 20-yr experience at a single centre. Pediatr Allergy Immunol (2009) 20(2):113–8. doi: 10.1111/j.1399-3038.2008.00744.x
53. Mohammadinejad P, Aghamohammadi A, Abolhassani H, Sadaghiani MS, Abdollahzade S, Sadeghi B, et al. Pediatric patients with common variable immunodeficiency: long-term follow-up. J Investig Allergol Clin Immunol (2012) 22(3):208–14.
54. Piatosa B, Pac M, Siewiera K, Pietrucha B, Klaudel-Dreszler M, Heropolitanska-Pliszka E, et al. Common variable immune deficiency in children–clinical characteristics varies depending on defect in peripheral B cell maturation. J Clin Immunol (2013) 33(4):731–41. doi: 10.1007/s10875-013-9875-4
55. Urschel S, Kayikci L, Wintergerst U, Notheis G, Jansson A, Belohradsky BH. Common variable immunodeficiency disorders in children: delayed diagnosis despite typical clinical presentation. J Pediatr (2009) 154(6):888–94. doi: 10.1016/j.jpeds.2008.12.020
56. van de Ven AAJM, van de Corput L, van Tilburg CM, Tesselaar K, van Gent R, Sanders EAM, et al. Lymphocyte characteristics in children with common variable immunodeficiency. Clin Immunol (2010) 135(1):63–71. doi: 10.1016/j.clim.2009.11.010
57. Yong PL, Orange JS, Sullivan KE. Pediatric common variable immunodeficiency: immunologic and phenotypic associations with switched memory B cells. Pediatr Allergy Immunol (2010) 21(5):852–8. doi: 10.1111/j.1399-3038.2010.01004.x
58. Ogershok PR, Hogan MB, Welch JE, Corder WT, Wilson NW. Spectrum of illness in pediatric common variable immunodeficiency. Ann Allergy Asthma Immunol (2006) 97(5):653–6. doi: 10.1016/S1081-1206(10)61096-4
59. Erdem SB, Gulez N, Genel F, Karaman S, Nacaroglu HT. Characteristics of the patients followed with the diagnosis of common variable immunodeficiency and the complications. Cent J Immunol (2019) 44(2):119–26. doi: 10.5114/ceji.2019.87060
60. Aghamohammadi A, Abolhassani H, Latif A, Tabassomi F, Shokuhfar T, Torabi Sagvand B, et al. Long-term evaluation of a historical cohort of Iranian common variable immunodeficiency patients. Expert Rev Clin Immunol (2014) 10(10):1405–17. doi: 10.1586/1744666X.2014.958469
61. Ardeniz O, Basoglu OK, Gunsar F, Unsel M, Bayraktaroglu S, Mete N, et al. Clinical and immunological analysis of 23 adult patients with common variable immunodeficiency. J Investig Allergol Clin Immunol (2010) 20(3):222–36.
62. Arshi S, Nabavi M, Bemanian MH, Shakeri R, Taghvaei B, Ghalebaghi B, et al. Phenotyping and follow up of forty-seven Iranian patients with common variable immunodeficiency. Allergol Immunopathol (Madr) (2016) 44(3):226–31. doi: 10.1016/j.aller.2015.04.005
63. Boileau J, Mouillot G, Gerard L, Carmagnat M, Rabian C, Oksenhendler E, et al. Autoimmunity in common variable immunodeficiency: correlation with lymphocyte phenotype in the French DEFI study. J Autoimmun (2011) 36(1):25–32. doi: 10.1016/j.jaut.2010.10.002
64. Boursiquot J-N, Gerard L, Malphettes M, Fieschi C, Galicier L, Boutboul D, et al. Granulomatous disease in CVID: retrospective analysis of clinical characteristics and treatment efficacy in a cohort of 59 patients. J Clin Immunol (2013) 33(1):84–95. doi: 10.1007/s10875-012-9778-9
65. Feuille EJ, Anooshiravani N, Sullivan KE, Fuleihan RL, Cunningham-Rundles C. Autoimmune Cytopenias and Associated Conditions in CVID: a Report From the USIDNET Registry. J Clin Immunol (2018) 38(1):28–34. doi: 10.1007/s10875-017-0456-9
66. Fischer A, Provot J, Jais J-P, Alcais A, Mahlaoui N. Autoimmune and inflammatory manifestations occur frequently in patients with primary immunodeficiencies. J Allergy Clin Immunol (2017) 140(5):1388–93. doi: 10.1016/j.jaci.2016.12.978
67. Van der Hilst JCH, Smits BW, van der Meer JWM. Hypogammaglobulinaemia: cumulative experience in 49 patients in a tertiary care institution. Neth J Med (2002) 60(3):140–7.
68. Kainulainen L, Nikoskelainen J, Ruuskanen O. Diagnostic findings in 95 Finnish patients with common variable immunodeficiency. J Clin Immunol (2001) 21(2):145–9. doi: 10.1023/A:1011012023616
69. Martinez Garcia MA, de Rojas MD, Nauffal Manzur MD, Munoz Pamplona MP, Compte Torrero L, Macian V, et al. Respiratory disorders in common variable immunodeficiency. Respir Med (2001) 95(3):191–5. doi: 10.1053/rmed.2000.1020
70. Mohammadinejad P, Pourhamdi S, Abolhassani H, Mirminachi B, Havaei A, Masoom SN, et al. Primary Antibody Deficiency in a Tertiary Referral Hospital: A 30-Year Experiment. J Investig Allergol Clin Immunol (2015) 25(6):416–25.
71. Musabak UH, Demirel F, Yesillik S, Baysan A, Selcuk A, Kartal O, et al. Adults with common variable immunodeficiency: a single-center experience. Turkish J Med Sci (2017) 47(1):1–12. doi: 10.3906/sag-1503-22
72. Quinti I, Soresina A, Spadaro G, Martino S, Donnanno S, Agostini C, et al. Long-term follow-up and outcome of a large cohort of patients with common variable immunodeficiency. J Clin Immunol (2007) 27(3):308–16. doi: 10.1007/s10875-007-9075-1
73. Ramirez-Vargas N, Arablin-Oropeza SE, Mojica-Martinez D, Yamazaki-Nakashimada MA, de la Luz Garcia-Cruz M, Teran-Juarez LM, et al. Clinical and immunological features of common variable immunodeficiency in Mexican patients. Allergol Immunopathol (Madr) (2014) 42(3):235–40. doi: 10.1016/j.aller.2013.01.007
74. Santaella ML, Font I, Disdier O. Common variable immunodeficiency: experience in Puerto Rico. P R Health Sci J (2005) 24(1):7–10.
75. Thickett KM, Kumararatne DS, Banerjee AK, Dudley R, Stableforth DE. Common variable immune deficiency: respiratory manifestations, pulmonary function and high-resolution CT scan findings. QJM (2002) 95(10):655–62. doi: 10.1093/qjmed/95.10.655
76. Khodadad A, Aghamohammadi A, Parvaneh N, Rezaei N, Mahjoob F, Bashashati M, et al. Gastrointestinal manifestations in patients with common variable immunodeficiency. Dig Dis Sci (2007) 52(11):2977–83. doi: 10.1007/s10620-006-9736-6
77. Cunningham-Rundles C, Bodian C. Common variable immunodeficiency: clinical and immunological features of 248 patients. Clin Immunol (1999) 92(1):34–48. doi: 10.1006/clim.1999.4725
78. Ardeniz O, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Clin Immunol (2009) 133(2):198–207. doi: 10.1016/j.clim.2009.05.001
79. Filion CA, Taylor-Black S, Maglione PJ, Radigan L, Cunningham-Rundles C. Differentiation of Common Variable Immunodeficiency From IgG Deficiency. J Allergy Clin Immunol Pract (2019) 7(4):1277–84. doi: 10.1016/j.jaip.2018.12.004
80. Wiesik-Szewczyk E, Zietkiewicz M, Matyja-Bednarczyk A, Napiorkowska-Baran K, Suchanek H, Jahnz-Rozyk K. The first Polish cohort of adult patients with common variable immunodeficiency from 4 specialized centers: do we provide standards of care? Polish Arch Intern Med (2018) 128(9):563–6. doi: 10.20452/pamw.4315
81. Yazdani R, Heydari A, Azizi G, Abolhassani H, Aghamohammadi A. Asthma and Allergic Diseases in a Selected Group of Patients With Common Variable Immunodeficiency. J Investig Allergol Clin Immunol (2016) 26(3):209–11. doi: 10.18176/jiaci.0062
82. Foxman B. Epidemiology of urinary tract infections: incidence, morbidity, and economic costs. Dis Mon (2003) 49(2):53–70. doi: 10.1067/mda.2003.7
84. Eaton WW, Pedersen MG, Atladóttir HO, Gregory PE, Rose NR, Mortensen PB. The prevalence of 30 ICD-10 autoimmune diseases in Denmark. Immunol Res (2010) 47(1–3):228–31. doi: 10.1007/s12026-009-8153-2
85. de Vries E. Patient-centred screening for primary immunodeficiency, a multi-stage diagnostic protocol designed for non-immunologists: 2011 update. Clin Exp Immunol (2012) 167(1):108–19. doi: 10.1111/j.1365-2249.2011.04461.x
86. Butters C, Phuong LK, Cole T, Gwee A. Prevalence of Immunodeficiency in Children With Invasive Pneumococcal Disease in the Pneumococcal Vaccine Era: A Systematic Review. JAMA Pediatr (2019). doi: 10.1001/jamapediatrics.2019.3203
87. Cowan J, Do TL, Desjardins S, Ramotar K, Corrales-Medina V, Cameron DW. Prevalence of Hypogammaglobulinemia in Adult Invasive Pneumococcal Disease. Clin Infect Dis (2018) 66(4):564–9. doi: 10.1093/cid/cix836
88. Gaschignard J, Levy C, Chrabieh M, Boisson B, Bost-Bru C, Dauger S, et al. Invasive pneumococcal disease in children can reveal a primary immunodeficiency. Clin Infect Dis (2014) 59(2):244–51. doi: 10.1093/cid/ciu274
89. Janssen LMA, Bassett P, Macken T, van Esch J, Pruijt H, Knoops A, et al. Mild Hypogammaglobulinemia Can Be a Serious Condition. Front Immunol (2018) 9:2384. doi: 10.3389/fimmu.2018.02384
90. Ngo ST, Steyn FJ, McCombe PA. Gender differences in autoimmune disease. Front Neuroendocrinol (2014) 35(3):347–69. doi: 10.1016/j.yfrne.2014.04.004
91. Horn J, Manguiat A, Berglund LJ, Knerr V, Tahami F, Grimbacher B, et al. Decrease in phenotypic regulatory T cells in subsets of patients with common variable immunodeficiency. Clin Exp Immunol (2009) 156(3):446–54. doi: 10.1111/j.1365-2249.2009.03913.x
92. Romberg N, Le Coz C, Glauzy S, Schickel J-N, Trofa M, Nolan BE, et al. Patients with common variable immunodeficiency with autoimmune cytopenias exhibit hyperplastic yet inefficient germinal center responses. J Allergy Clin Immunol (2019) 143(1):258–65. doi: 10.1016/j.jaci.2018.06.012
93. Fevang B, Yndestad A, Sandberg WJ, Holm AM, Muller F, Aukrust P, et al. Low numbers of regulatory T cells in common variable immunodeficiency: association with chronic inflammation in vivo. Clin Exp Immunol (2007) 147(3):521–5. doi: 10.1111/j.1365-2249.2006.03314.x
94. Ameratunga R, Lehnert K, Woon S-T, Gillis D, Bryant VL, Slade CA, et al. Review: Diagnosing Common Variable Immunodeficiency Disorder in the Era of Genome Sequencing. Clin Rev Allergy Immunol (2018) 54(2):261–8. doi: 10.1007/s12016-017-8645-0
95. Jorgensen SF, Fevang B, Aukrust P. Autoimmunity and Inflammation in CVID: a Possible Crosstalk between Immune Activation, Gut Microbiota, and Epigenetic Modifications. J Clin Immunol (2019) 39(1):30–6. doi: 10.1007/s10875-018-0574-z
97. Ghouri N, Hippisley-Cox J, Newton J, Sheikh A. Trends in the epidemiology and prescribing of medication for allergic rhinitis in England. J R Soc Med (2008) 101(9):466–72. doi: 10.1258/jrsm.2008.080096
98. Kiaee F, Azizi G, Rafiemanesh H, Zainaldain H, Sadaat Rizvi F, Alizadeh M, et al. Malignancy in common variable immunodeficiency: a systematic review and meta-analysis. Expert Rev Clin Immunol (2019) 15(10):1105–13. doi: 10.1080/1744666X.2019.1658523
99. Vajdic CM, Mao L, van Leeuwen MT, Kirkpatrick P, Grulich AE, Riminton S. Are antibody deficiency disorders associated with a narrower range of cancers than other forms of immunodeficiency? Blood (2010) 116(8):1228–34. doi: 10.1182/blood-2010-03-272351
100. Kralickova P, Milota T, Litzman J, Malkusova I, Jilek D, Petanova J, et al. CVID-Associated Tumors: Czech Nationwide Study Focused on Epidemiology, Immunology, and Genetic Background in a Cohort of Patients With CVID. Front Immunol (2018) 9:3135. doi: 10.3389/fimmu.2018.03135
102. Moher D, Shamseer L, Clarke M, Ghersi D, Liberati A, Petticrew M, et al. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst Rev (2015) 4:1. doi: 10.1186/2046-4053-4-1
103. Ameratunga R, Woon S-T. Perspective: Evolving Concepts in the Diagnosis and Understanding of Common Variable Immunodeficiency Disorders (CVID). Clin Rev Allergy Immunol (2019) 59(1):109–21. doi: 10.1007/s12016-019-08765-6
104. Maffucci P, Filion CA, Boisson B, Itan Y, Shang L, Casanova J-L, et al. Genetic Diagnosis Using Whole Exome Sequencing in Common Variable Immunodeficiency. Front Immunol (2016) 7:220. doi: 10.3389/fimmu.2016.00220
Keywords: humoral immunodeficiency, antibody deficiency, common variable immunodeficiency disorders, non-infectious complications, clinical manifestations
Citation: Janssen LMA, van der Flier M and de Vries E (2021) Lessons Learned From the Clinical Presentation of Common Variable Immunodeficiency Disorders: A Systematic Review and Meta-Analysis. Front. Immunol. 12:620709. doi: 10.3389/fimmu.2021.620709
Received: 23 October 2020; Accepted: 24 February 2021;
Published: 23 March 2021.
Edited by:
Waleed Al-Herz, Kuwait University, KuwaitReviewed by:
Charlotte Cunningham-Rundles, Icahn School of Medicine at Mount Sinai, United StatesHassan Abolhassani, Karolinska Institute, Sweden
Copyright © 2021 Janssen, van der Flier and de Vries. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Esther de Vries, ZS5kZXZyaWVzQHRpbGJ1cmd1bml2ZXJzaXR5LmVkdQ==