 Rafael de Freitas e Silva
Rafael de Freitas e Silva Esther von Stebut
Esther von Stebut- 1Department of Natural Sciences, University of Pernambuco, Garanhuns, Brazil
- 2Department of Dermatology, Medical Faculty, University of Cologne, Cologne, Germany
Leishmaniasis are Neglected Tropical Diseases affecting millions of people every year in at least 98 countries and is one of the major unsolved world health issues. Leishmania is a parasitic protozoa which are transmitted by infected sandflies and in the host they mainly infect macrophages. Immunity elicited against those parasites is complex and immune checkpoints play a key role regulating its function. T cell receptors and their respective ligands, such as PD-1, CTLA-4, CD200, CD40, OX40, HVEM, LIGHT, 2B4 and TIM-3 have been characterized for their role in regulating adaptive immunity against different pathogens. However, the exact role those receptors perform during Leishmania infections remains to be better determined. This article addresses the key role immune checkpoints play during Leishmania infections, the limiting factors and translational implications.
Introduction
Global warming is progressively changing the distribution of pathogenic microbes, and consequently humans are more exposed to new or re-emerging infections, especially those transmitted by vectors and reservoirs (1–3). Leishmaniasis affects millions of people living in endemic regions and kills thousands of them every year, and its distribution is changing due to the occurrence of sandflies in new areas (4–7). Those diseases are listed by the World Health Organization (WHO) as important Neglected Tropical Diseases (NTDs) affecting mainly poor people living with less than one dollar per day (8).
Leishmaniasis are complex parasitic protozoan infections, usually transmitted through the bite of infected hematophagous invertebrates, capable of surviving and proliferating in human tissues and blood (9–11). So far, the best option to control the diseases is through vector and host reservoir control, and by treating infected individuals (12, 13). Nevertheless, the drugs are often inaccessible due to high cost and toxicity and there is currently no safe and effective vaccine to be applied in humans (14–16). One of the reasons for the lack of therapeutic approaches is the complex immunity against these parasites and, therefore, many gaps in understanding this puzzle (17). In this regard, this review aims to address some key pieces which were initially observed in cancer and chronical viral infections.
Development of immunity is accompanied by an upregulation of receptors (and their ligands) with regulatory function, also known as immune checkpoints (18–20). Some of them are potentially stimulatory and others induce an inhibitory effect. For this, it was demonstrated that the upregulation of co-inhibitory receptors and their ligands on antigen presenting cells (APCs) and activated T cells are capable to suppress the immunity by delivering negative signals, rendering T cells suppressed in their function (21–24). Nonetheless, antibody blocking can restore T cell capacity to destroy cancer or viral infected cells. Among those, anti-Programmed Death (PD)-1 and anti-Programmed Death Ligand (PD-L)1 are currently used to treat melanoma and other types of cancer (25, 26). Those treatments paved the way for studies with similar interventions for chronic infections, including leishmaniasis.
This review wants to address the following question: what is the role of immune checkpoints in leishmaniasis? Some investigations are ongoing; however, given the limited understanding of their function, dissecting it would significantly amplify our knowledge regarding the function of the human immune system, how it reacts to the intracellular parasite Leishmania and other similar infections. More importantly, it would widen treatment perspectives for the affected individuals.
An Overview on Anti-Leishmania Immunity
Immunity against Leishmania is complex and depends on many factors, such as genetic diversity, parasite species and isolates (27–29). Leishmania spp. are inoculated into the skin as metacyclic promastigotes (30) and once the parasites are in close contact with the body, immunity is triggered (Figure 1). The complement system has an important, although limited function in this task, since glycoproteins, such as GP63 (also known as Leishmanolysin), from the surface of the parasites are capable to bind complement factor C3b and inactivate it (C3bi), blocking the capacity to lyse the parasites and enhancing its recognition by complement receptor-3 (CR3) on macrophages (31–33). As soon as phagocytic cells reach the entry site, they engulf free parasites and factors such as chemokine (C-C motif) ligand 3 (CCL3) are secreted by neutrophils, which in turn attract dendritic cells (DCs) (34–36). C-C-chemokine receptor type 2 (CCR2)-driven monocytes secrete reactive oxygen species (ROS) to kill free parasites and these cells migrate to draining lymph nodes and differentiate to monocyte-derived DCs (9, 37–39). DCs displaying Leishmania antigens coordinate the secretion of interleukin (IL)-12 which instructs the differentiation of T helper type (Th)1 cells to produce and secrete IFN-γ (40–42). IFN-γ levels collectively produced by CD4+ Th1 and other activated cells types, such as CD8+ T cells and natural killer (NK) cells, is, so far, known as the best correlate of protection in leishmaniasis (43, 44). Protection takes place by production of nitric oxide (NO) by the inducible NO synthase (iNOS) in macrophages in order to kill the amastigotes (45–48).
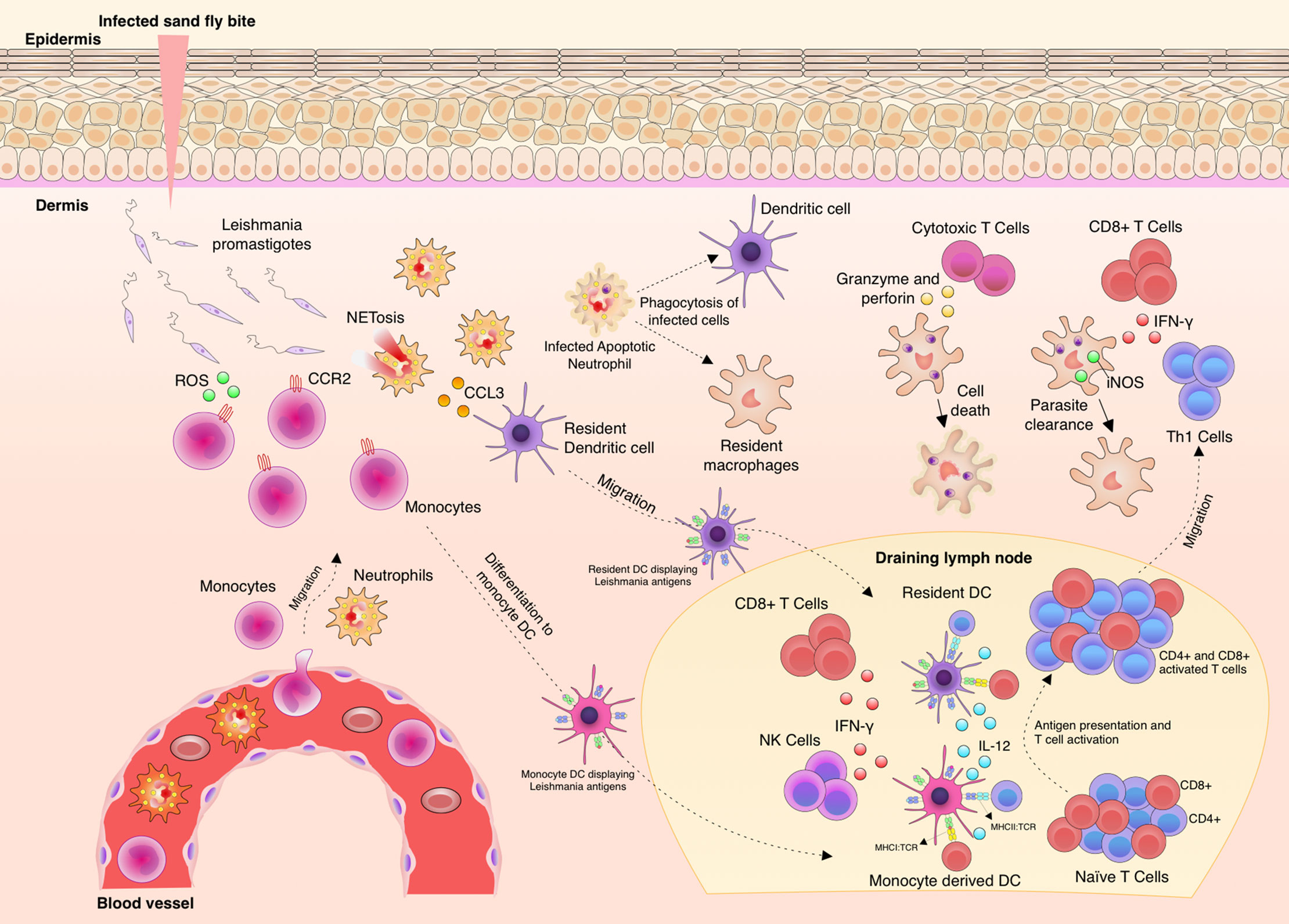
Figure 1 Aspects of immunity against Leishmania parasites. Upon Leishmania entry into the dermis, different phagocytic cells infiltrate to the site, such as neutrophils and monocytes. The parasites are phagocyted by these infiltrating cells and also by resident macrophages and tissue dendritic cells (DC). Neutrophils produce increasing levels of chemokine (C-C motif) ligand 3 (CCL3) to attract dendritic cells to the site. C-C chemokine receptor type 2 (CCR2)+ monocytes produce and release reactive oxygen species (ROS) to kill free parasites. Then, adaptive immunity is elicited through the migration of monocytes and tissue DCs carrying Leishmania antigens to the draining lymph node. These cells present parasite antigens and produce Interleukin (IL)-12 and thus induce CD4+Th1 cell differentiation, Th1 cells migrate to the infection site and finally produce and secrete Interferon (IFN)-γ. Activation of infected macrophage by the action of IFN-γ leads to the production of nitric oxide (NO) by iNOS and thus Leishmania killing. IFN-γ is also locally produced by natural killer (NK) and CD8+ T cells. IL-10 production and parasite persistence are necessary to maintain memory cells.
The role of Th1 cells is well verified in the two main mouse models of L. major infection: the susceptible mouse strain BALB/c shows a weak Th1 and strong Th2 immunity that results from the contribution of distinct factors such as an IL-4-mediated down regulation of the IL-12Rβ on Th2 cells or increased production of IL-12(p40)2 homodimers that antagonize the effect of the IL-12 active form on IL-12R (41, 42, 49); on the other hand, Leishmania-resistant C57BL/6 mice are good controllers of the infection with a strong Th1 response (50–52).
Another important component are Th17 cells and its main cytokine IL-17 that contributes activating iNOS and inducing production of GM-CSF, IL-1β, IL-6, IL-8, TNF-α, and chemokines (53). Th17 cells have been implicated in the recruitment of neutrophils to the lesions caused by Leishmania, and disease progression was prevented in IL-17A-deficient BALB/c mice (54, 55). Nevertheless, disease progression was not affected in IL-17-deficient C57BL/6 mice (56).
The role of CD8+ T cells is still controversial and has been associated with more inflammation, increased Granzyme B, TNF-α and IL-1β production, inflammasome activation, tissue damage and disease severity (57–59). On the other hand, it has been shown that CD8+ T cell-derived IFN-γ is a key factor for disease control (60, 61).
Additionally, regulatory T cells (Treg) are necessary to control the activation of the immune response and excessive inflammation by diverse mechanisms, such as inhibitory receptors and enzymes, and production of IL-10 (62). Moreover, evidence indicates that they are necessary to control the inflammation process and contribute to parasite persistence in resistant mice (63–65). In this regard, it was shown that Langerhans cells (LCs), during Leishmania infection, induced expansion of Treg. Moreover, Treg-derived IL-10, retinoic acid independent, contributes parasite persistence and selective depletion of Treg induces larger lesions (66, 67).
In humans, the immunity against Leishmania is more complex, and often many findings achieved in the mouse models cannot be directed translated to humans (17, 68, 69). The key players during the immunity in CL and VL are similar, however, the tissue milieu is distinct and that influences the course of immunity and final outcome.
Main Clinical Manifestations
Even so, in many cases immunity is unable to properly control parasite growth and they end up replicating as amastigotes in macrophage phagolysosomes (70, 71). From the point of inoculation, some species can have a dermis tropism, causing localized or disseminated skin lesions, or mucocutaneous lesions. L. braziliensis, L. major, L. panamensis, L. mexicana, and L. guyanensis are species associated with these clinical forms. Other Leishmania species have a tropism for the mononuclear phagocyte system from spleen, liver, and bone marrow, and can cause visceral leishmaniasis (VL), which is the deadliest form of leishmaniasis if left untreated (6, 72, 73). Thus different species of the parasite are involved with distinct clinical forms (11, 74).
This broad clinical spectrum adds another layer of complexity to understand immunity against Leishmania (28, 75, 76). The activation of the immune system has been used to detect antibodies and/or cellular immunity for diagnostic purposes, and, so far, it is not fully understood how it works to contain the infection and how can we boost it (77–81). Overall, immunity against Leishmania parasites is very complex and involves many cellular and molecular players that act against the different species of parasites, which can cause infection, and also the intrinsic factors, that influence it (Figure 1).
What are the Immune Checkpoints in Leishmaniasis?
Different stimulatory and inhibitory immune checkpoints pathways are activated or deactivated during an immune response. Those interactions are cell to cell (trans), usually between a T cell and a macrophage, DC, monocyte and neutrophil. However, it has been shown that some interactions are on the same cell (cis) with important consequences for the T cells (82). Altogether, different checkpoints receptors have been discovered with distinct functions that are relevant to consider and to investigate in the context of intracellular infections such as Leishmania (Figure 2).
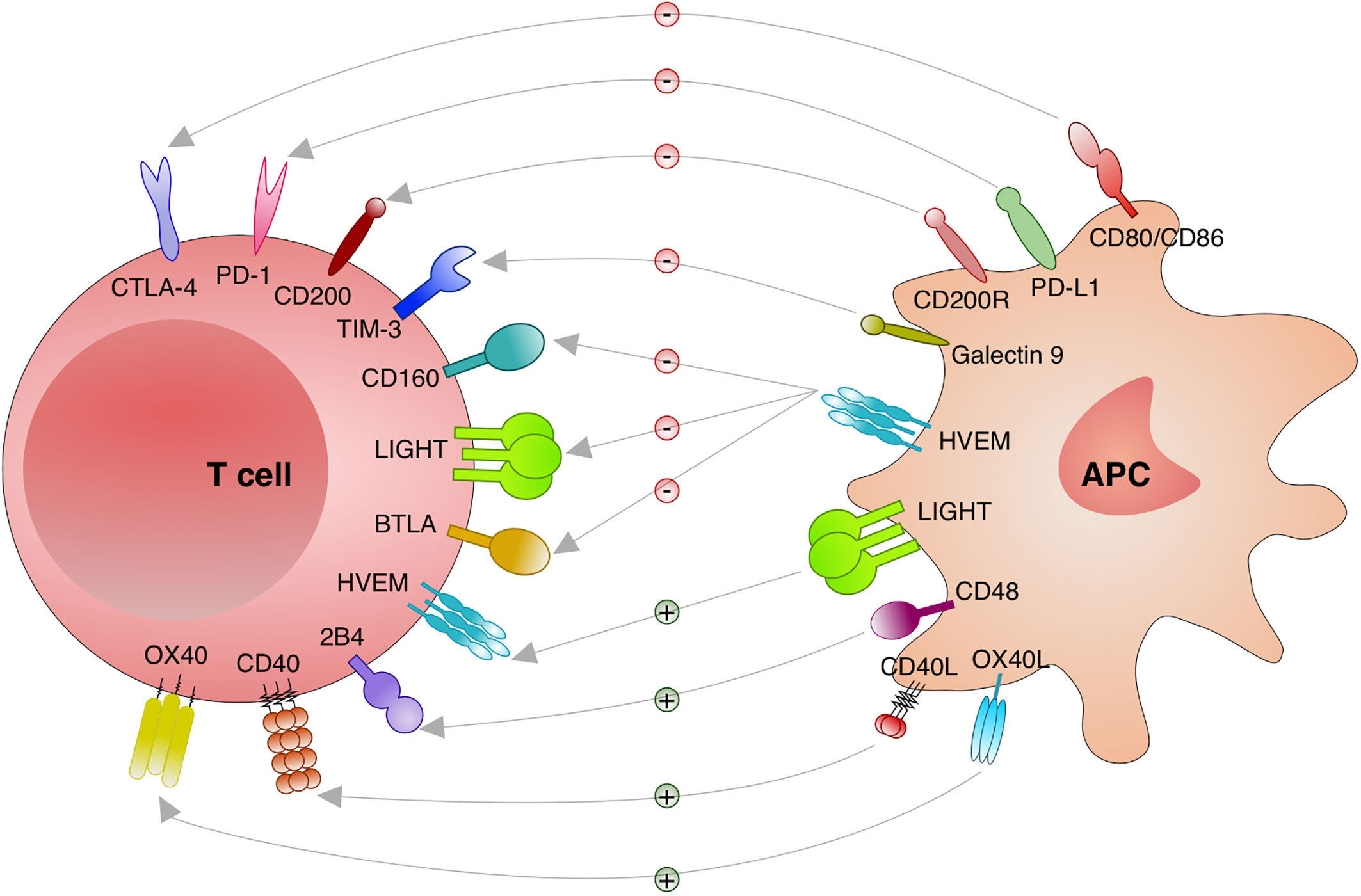
Figure 2 Immune checkpoints investigated in leishmaniasis. Distinct immune checkpoints have been investigated for the function during the immunity against Leishmania. Cytotoxic T lymphocyte attenuator 4 (CTLA-4; CD152) binds to CD80/CD86 and blocks its stimulatory effect on T cells. Programmed cell death protein 1 (PD-1; CD279) binds to its ligands PD-L1 or PD-L2 and inhibit T cells. CD200 (OX-2) and CD200R engagement results in an important inhibitory signal to T cells. T cell immunoglobulin and mucin domain-containing protein (TIM)-3 and Galectin 9 engagement deliver an inhibitory signal to T cells. Binding of CD160, lymphotoxin LT-like (LIGHT) and B and T lymphocyte attenuator (BTLA; CD272) to herpesvirus entry mediator (HVEM) induces inhibitory signals in T cells. When HVEM from T cells binds to LIGHT on antigen presenting cells (APC) stimulatory effects on T cells are induced. Engagement of CD40 and CD40 Ligand (L) delivers a stimulatory effect to T cells. 2B4 and CD48 binding results in stimulatory effects to T cells. Binding of OX40 to OX40L results in stimulatory effects to T cells.
Inhibitory Checkpoints
Programmed Cell Death 1 (PD-1) and Programmed Death-Ligands 1 (PD-L1) and 2 (PD-L2)
The programmed cell death protein 1 (PD-1; CD279) and its ligands PD-L1 (B7-H1) and PD-L2 (B7-DC), which are members of the B7/CD28 family are important axis which were investigated in leishmaniasis (83–85). Engagement of PD-1 to PD-L1 or PD-L2 induces phosphorylation of tyrosine residues in PD-1 intracellular domain which recruits tyrosine phosphatases such as Src homology 2 domain-containing phosphatase (SHP)-2. Thereafter, distinct kinases necessary for cell survival and proliferation are dephosphorylated (86–89). The resulting effect of this signaling cascade is T cell inhibition and exhaustion (90, 91). It has been shown in different models that PD-1/PD-L1 are important to maintain immune homeostasis by inducing peripheral tolerance and protecting tissues from autoimmune attack (24). The role of these molecules during acquisition of immunity against many infectious agents is not well established and PD-1/PD-L1 may suppress immunity allowing chronic infection (92–94). Infection by L. mexicana can cause a localized cutaneous lesion (LCL) or a diffuse form (DCL) and it has been observed that patients with DCL have low numbers of CD8+ T cells in their lesions when compared to LCL (83). Moreover, CD8+ T cells isolated from the blood of DCL exhibited less capacity to proliferate under antigen specific stimulation, less IFN-γ production, and enhanced expression of PD-1 (83). This cell phenotype observed has been typically associated with CD8+ T cell exhaustion (25). However, particularly in this study, other exhaustion markers have not been extensively investigated and this could give a better idea on the level of CD8+ T cell exhaustion undergone by those infected patients. During human CL, higher frequencies of CD4+PD-1+ T cells were observed in active CL disease patients compared to post-treated patients and uninfected individuals (95). Interestingly, it was observed a continuously increasing frequency of PD-1+ T cells together with the development of CD4+ T cells subpopulations (95). Lower frequencies of PD-1+ among T naïve (CD45RO-CCR7+) and higher frequencies among T terminally differentiated (CD45RO-CCR7-) (95).
According to the most recent investigations, a distinct pattern of co-inhibitory receptors might also be associated with each species of Leishmania involved, such as the case for L. major infection, which causes CL. The main role of these checkpoints could be to limit T cell activation during the initial phase of the immunity, during antigen presentation by APCs (96). Specific molecules, e.g. on parasites, might be involved in inducing the expression of checkpoint receptors by different cell subsets upon infection, such as the case of lipophosphoglycan (LPG). This molecule is expressed on the surface of Leishmania parasites, and when mice were vaccinated with different LPG concentrations, CD8+ T cells upregulated PD-1 on its surface, but not CD4+ T cells (97). Additionally, it was recently shown in BALB/c footpad infection with L. amazonensis that either anti-PD-1 or anti-PD-L1 treatment reduced the parasite burden and this effect was potentially mediated by CD8+IFN-γ+ T cells (98). Among the different clinical forms of leishmaniasis, there is evidence supporting the role of PD-L1 as one regulator of anti-VL immunity (85, 99, 100). However, there is very few evidence supporting the same role for PD-L1 during CL. Since both CL and VL take place in different organs in the body, PD-L1 may act distinctly in those sites and its role is not fully understood yet.
It has been shown in L. donovani infections that blocking of PD-L1 results in an increased survival of CD8+ T cells and partially restores their function, a notion supported by resulting lower parasites numbers upon treatment (101). Other authors have reported the same effect in that infection by L. donovani induced PD-1 expression on CD8+ T cells and its ligand on splenic DCs (102, 103). Treatment with antibody to block the PD-1/PD-L1 interaction was able to partially restore the function of CD8+ T cells, which may indicate that other checkpoint receptors were involved in CD8+ T cell exhaustion (102).
Regarding CD4+ T cells, Esch et al., 2013 have observed that CD4+ T cells from L. infantum-infected dogs have a significant increase in PD-1 expression which was associated to some extent with CD4+ T cell exhaustion. By blocking PD-L1 in vitro, an increase in the production of ROS in monocytes derived phagocytes was found (85). In murine models it has been described that infection with L. donovani restricted the expansion of specific CD8+ T cells. Moreover, these cells were also more prone to undergo apoptosis and upregulated PD-1, and this dysfunctional phenotype was partially recovered by blocking of PD-L1. Upon PD-L1 inhibition, the authors also detected lower numbers of parasites which might reflect a better infection outcome (102). Furthermore, it has been shown that PD-L1 inhibition restored the function of CD4+ and CD8+ T cells in the bone marrow of L. donovani-infected mice. Upregulation of PD-L1 by macrophages and of PD-1 by CD4+, CD8+ T and Tregs IL-10+Foxp3+ upon infection was also observed. Phenotypically, blocking of PD-L1 led to decreased parasite burdens and inhibited autophagy, which is regarded as one of the mechanism used by Leishmania to obtain nutrients in the host cell (99). Other studies have also previously described an upregulation of PD-1 and PD-L1 on T cells and macrophages, respectively, upon infection with L. infantum or L. donovani (85, 102). Nevertheless, it was also demonstrated Pdl1-/- mice infection with L. major induced lower frequencies of a population of CD4+Ly6Chi effector T cells and higher frequencies of Foxp3+ Tregs compared with WT mice (95).
Different species of mammals can act as reservoirs for the parasite, i.e. dogs are an important reservoir for human infection. In the case of dogs VL, it has been shown that there is an impaired CD4+ T cell function, IFN-γ production, combined with T cell exhaustion through PD-1 expression (104). On the other hand, one work has shed light upon the role of PD-L1 on regulatory B cells during dog VL, since the specific blocking of PD-L1 on B cells restored Th1 responses (84). The PD-1/PD-L1 axis might exert its function during infection with Leishmania by reducing the phagocytic capacity of macrophages. However, it is not yet clear which other cell subsets may also be involved since they can also upregulate PD-L1 upon Leishmania infection, e.g. regulatory B cells and DCs. The exact role of PD-L1 expression on these cell subsets is not yet clear, and they might contribute to disease progression and T cell exhaustion. In the case of the PD-1 and PD-L1 pathway it has also been shown for other protozoan infections, blocking the pathway may rescue the function of exhausted cells, such as CD8+ T cells and B cells, and this could lead to parasite control and less mortality (105).
PD-L2 is another ligand that binds PD-1 with a two to six fold higher affinity than PD-L1 (106), and it is expressed by macrophages, DCs, bone marrow-derived mast cells, B cells, and intestinal stromal cells (107–111). Expression of PD-L2 is induced by IL-4, IFN-γ and granulocyte-monocyte colony stimulating factor (GM-CSF) (112, 113). More importantly, comparing the roles of PD-L1 and PD-L2 during murine infection with L. mexicana, it was observed that Pdl2-/- mice displayed larger lesions and higher parasite burden when compared to Pdl1-/- and WT mice. The most important difference were increased levels of L. mexicana IgM and IgG2a that the authors associated with poor disease resolution (114). Those results emphasize a potential role of PD-L2 regulating T-independent (IgM) and -dependent (IgG2a) B cell responses.
In summary, the PD-1 and PD-L1/PD-L2 axis likely regulates immunity against Leishmania parasites, and specific blocking of PD-1/PD-L1 potentially boosts T cell responses which are necessary for parasite elimination. Nevertheless, it is not fully clear how this axis works to control parasite proliferation or killing and the role of the different cells that express PD-1/PD-L1.
Cytotoxic T Lymphocyte Attenuator 4 (CTLA-4; CD152)
CTLA-4 was discovered many years ago and it is one of the most classical co-inhibitory receptors constitutionally expressed in intracellular vesicles in Foxp3+ regulatory T cells (Tregs) and conventional T and B cells during immune activation (115). The first works addressing its function reported that mice lacking CTLA-4 undergo massive lymphocyte proliferation and organ failure (116, 117). CTLA-4 acts by: trans-endocytosis of CD80 and CD86 on APCs to inside the T cells, depleting these ligands (118); inducing IL-10 or TGF-β which down modulate co-stimulatory ligands on APCs (119); finally, it is also suggested that binding of CTLA-4 to CD80/CD86 in DCs induces indoleamine 2,3-dioxygenase (IDO) which degrades tryptophan (120). Lack of tryptophan is also an important mechanism of cell cycle arrest and T cell suppression (121). In 2011, the first anti-CTLA-4 antibody was approved for treatment of melanoma, a type of skin cancer (122, 123).
However, information about the effect of CTLA-4 blocking as an immunotherapeutic intervention against chronic infections is limited. Interestingly, it was observed that susceptible mice treated with anti-CTLA-4 one day post L. donovani infection displayed reduced parasite numbers and also induced IFN-γ and IL-4 producing cells (124). However, mice lacking CTLA-4 transducing cytoplasmic tail displayed more susceptibility to infection with L. major and increased Th2 responses compared to mice with intact CTLA-4 which were resistant to infection and had stronger Th1 responses (125). Complementary studies have also shown in vivo blocking of CTLA-4 promoted Th2 responses and exacerbated disease in L. major-infected susceptible mice (126, 127). Additionally, in proliferating CD4+ T cells of Ctla4-/- mice increased production of Th2 cytokines was observed (128). Importantly, CTLA-4 engagement results in transforming growth factor (TGF)-β production and this cytokine blocks IFN-γ production inducing parasite survival and proliferation (129, 130). Moreover, blocking of CTLA-4 and neutralization of TGF-β resulted in increased production of IFN-γ and parasite elimination in coculture systems (131, 132). Overall, those remarkable results indicate an important role for CTLA-4 controlling T cell responses during anti-Leishmania imunity with important effects mediated by TGF-β.
In humans, expression of CTLA-4 in CD8+ T cells of patients infected with L. panamensis was described (133). Besides its potential as a potent T cell booster, the role of this receptor during leishmaniasis is still questionable and it needs to be better understood.
CD200 (OX-2)
Another axis that has been lately investigated is the engagement of CD200, which is expressed by APCs, with CD200R on T cells (134–136). Following signaling there is formation of a complex of proteins that inhibits Ras activation and results in inhibition of mitogen kinases such as Phosphoinositide 3-kinase (PI3K) and Extracellular Signal-regulated Kinase (Erk) delivering an important inhibitory signal (137–140). Cortez et al., 2011 have shown a correlation between L. amazonensis and the expression of CD200 as a mechanism to potentiate its virulence in the host. The authors demonstrated that infection of macrophages with L. amazonensis, and not with L. major, induces the expression of CD200 transcripts over time (141). Moreover, the lack of CD200 abrogated parasite proliferation in macrophages and this was rescued by the administration of recombinant Fc-CD200. In vivo, mice which lack CD200 (Cd200-/-) harbored smaller lesions with fewer parasites compared to wild type counterparts. In contrast, the same result was not found in mice infected with L. major, indicating that CD200 has an important role at regulating the amount of parasites and the outcome of the infection specifically for L. amazonensis. Another interesting point showed by this paper is the improvement of iNOS activity in Cd200-/- mice already 1-h post L. amazonensis infection. This suggested that CD200 has a iNOS regulating role during infection. Upon in vitro infection of bone marrow macrophages, Leishmania-induced increased levels of CD200 in bone marrow macrophages and the lack of CD200 (BMM Cd200-/-) inhibited parasite proliferation. Furthermore, it was demonstrated that Leishmania (amazonensis) DNA recognized by TLR9 is necessary to activate TRIFF and MyD88 and consequently activate CD200/CD200R cis interaction (142). The role of CD200 was also investigated in the context of L. donovani infection. The interaction of CD200/CD200R aid activated CD4+CD44+ T cells to produce IL-4, IL-10, TGF-β, and IL-27. Overall this effect abrogated macrophage effector functions and it was restored by the administration of anti-CD200 which induced more IL-2, IL-12 and IFN-γ by T cells, and therefore activated macrophages to produce NO (143). Additionally, immunization with centrin-deleted L. donovani resulted in CD200/CD200R downregulation and consequently suppression of IL-10-producing CD4+ T cells and more Th1 cells compared to WT infection (144).
Collectively, the evidence suggests that Leishmania can also influence the expression of CD200 as escape mechanism, thus the blocking of this axis holds potential to boost T cell responses and control parasite growth. More investigation is required to address distinct the effect of various parasite species, specific cell types involved, and the relevance for the human situation.
Stimulatory Checkpoints
CD40 and CD40-Ligand (CD40L)
CD40 is a member of the TNF receptor superfamily expressed mainly in B cells and its interaction with CD40L (CD154), expressed on CD4+ T cells, stimulates T cells and is crucial to promote germinal center formation and antibody class switching (145–147). It has been shown, in vitro, that infection of peritoneal macrophages from BALB/c mice with L. major induces different isoforms of CD40-induced N-Ras protein, which in turn induces activation of the ERK-1/2 pathway consequently resulting in less production of IL-12 and more of IL-10 (148). Moreover, the inhibition of CD40 induced N-Ras activation and reduced L. major infection. These authors have proposed a model in which the interaction of LPG with TLR2 regulates the different effects mediated by the isoforms of Ras. Thus, this finding highlights one important mechanism by which Leishmania species might directly influence immunity through the modulation of CD40-induced Ras protein. Altogether, more investigation is required to broadly clarify those escape mechanisms used by Leishmania, and to demonstrate its potential to control Leishmania infection in vivo.
OX40 (CD134)
OX40 is predominantly expressed by activated T cells and when it binds to its ligand OX40L (CD252), expressed by DCs, macrophages and B cells, induces the expression of survival proteins Bcl-2 and Bcl-XL that blocks apoptosis and allows cell cycle progression (149–151). There are evidences indicating OX40/OX40L axis promoting Th2 responses and other refusing this effect (152–155). Zubairi et al. (2004) has shown that treatment of L. donovani-infected C57BL/6 mice with OX40-Fc alone or in combination with anti-CTLA-4 diminishes parasite burdens through and IFN-γ and IL-12 dependent manner (155). In contrast, a distinct outcome was observed after infecting Ox40l-/- C57BL/6 mice with L. donovani (156). Ox40l-/- C57BL/6 infected mice eliminated parasites in chronic phase and displayed stronger IFN-γ responses compared to WT mice (156). Differently, OX40L-deficient BALB/c mice were resistant to L. major infection and exhibited reduced production of Th2 cytokines (154). Likewise, transgenic BALB/c mice overexpressing OX40L displayed pronounced Th2 responses accompanied by a high parasite burden (154). Interestingly, Ox40l-/- BALB/c mice infected with L. mexicana or L. major developed increased lesions compared to Ox40l+/+ counterparts (157). Nevertheless, a Th2 cytokine response bias was verified only for L. major infection (157). This pathway has a strong potential to regulate Leishmania immunity and it is yet to clarify the exact mechanisms involved during responses against different species.
Herpes Virus Entry Mediator (HVEM)
The herpesvirus entry mediator (HVEM) is an important member of the tumor necrosis factor (TNF) superfamily with distinct ligands: lymphotoxin LT-like (LIGHT) and LTα3 that are members of the TNF superfamily; and, B and T lymphocyte attenuator (BTLA; CD272) and CD160 that are members of the Immunoglobulin (Ig) superfamily (158–162). HVEM has cysteine-rich domains 1 and 2 (CRD1, CRD2), where BTLA and CD160 bind to CRD1 and LIGHT binds to CRD2 (163–165). Upon binding, cross linking of HVEM induces a cascade resulting in NF-κB activation (166–168). In another way, HVEM can also act as ligand and pass signals to cells expressing BTLA (169). HVEM has a similar effect (as LIGHT) and controls IL-12 secretion (170). Although there is some evidence on the role of LIGHT/HVEM signaling during Leishmania infection (171), the role of one of its main ligands, BTLA, during the infection is not clear yet.
Lymphotoxin Exhibits Inducible Expression and Competes With Herpes Simplex Virus Glycoprotein d for Herpes Virus Entry Mediator, a Receptor Expressed by T Cells (LIGHT) or Tumor Necrosis Factor SuperFamily member 14 (TNFSF14)
LIGHT protein is recognized by HVEM and when HVEM is expressed on T cells and interacts with LIGHT from APCs, this results in a T cell stimulatory effect (172). LIGHT can also be expressed by activated T cells, activated NK cells and immature DCs (173, 174). In addition to binding to HVEM, LIGHT can also recognize and bind another receptor known as Lymphotoxin-β Receptor (LTβR) (175). This receptor is found in non-myeloid cells such as epithelial and stromal cells, and myeloid cells such as immature DCs, but not on lymphocytes (176). The axis LIGHT/LTβR is more related with development of the immune system and maintenance of immune responses (177–179). However, interaction of LIGHT with either receptors has been associated with potentiation of inflammatory responses (172). Some data has been presented supporting the role of LIGHT, which is a ligand for HVEM, and an important molecule for the secretion of IL-12p40 by DCs and macrophages (180). LIGHT has an important function in CD8α+ DCs during the primary response against L. major, but no effect in a secondary response in the presence of CD40 (180). Moreover, LIGHT/HVEM has a critical role controlling parasite burden during the course of L. donovani infection through the production of T cell-derived IFN-γ and TNF-α (171). However, LIGHT/LTβR suppress anti-Leishmania immunity in the liver of L. donovani infected mice (171). Table 1 summarizes the main role of inhibitory and stimulatory checkpoints in Leishmania infection.
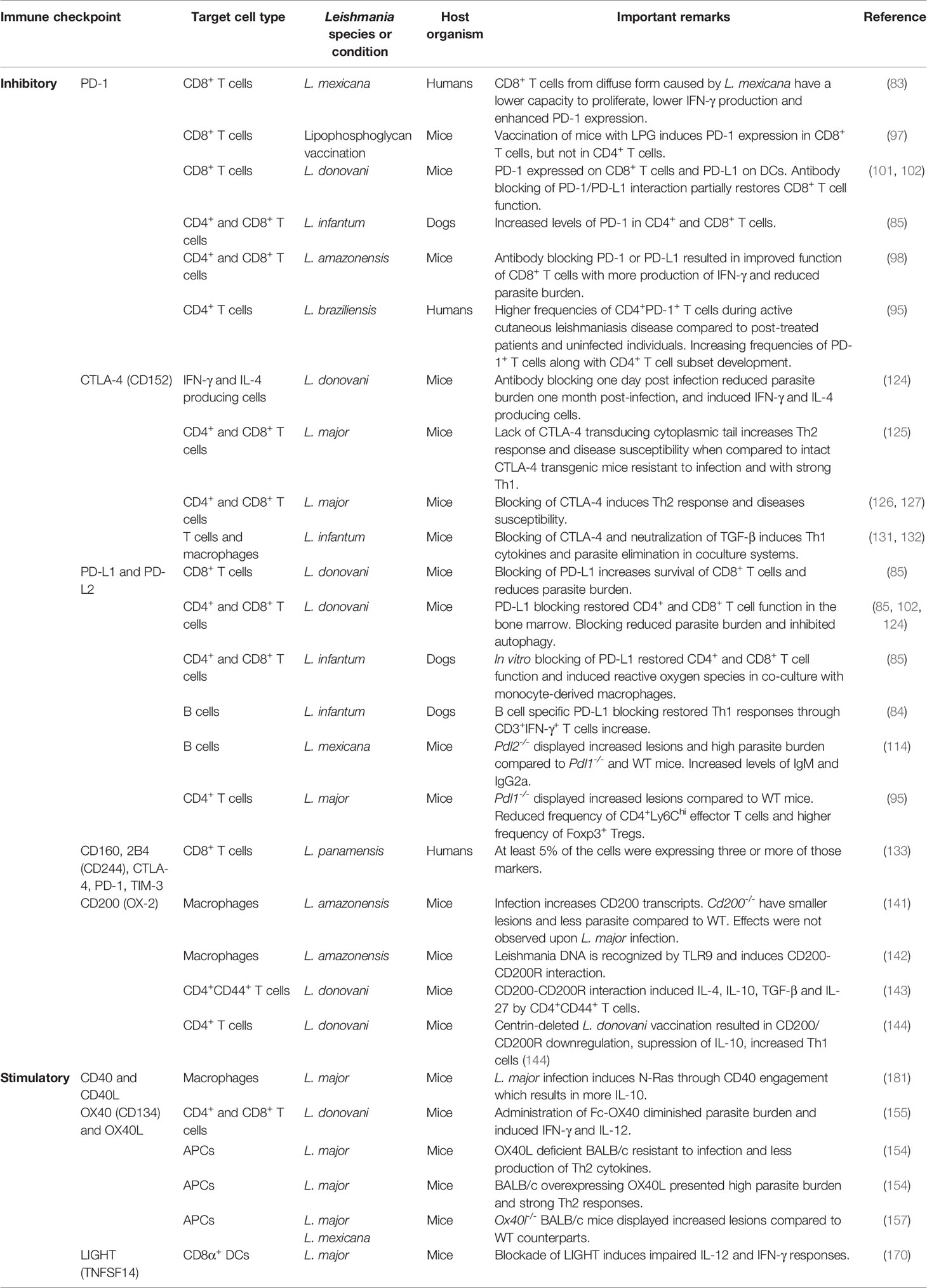
Table 1 Role of main immune checkpoints investigated during Leishmania infection.
Potential Checkpoints to be Investigated in Leishmaniasis
2B4 (CD244)
2B4 is a member of the signaling lymphocyte activation molecule (SLAM) family and is found on T cells, NK cells, γδ T cells, basophils, monocytes and a subset of memory-phenotype CD8+ αβ T cells (182, 183). 2B4 binds CD48 (also known as BCM-1, BLAST-1) in APCs resulting in T cell stimulus (184–187). Egui et al. observed a higher frequency of blood circulating CD8+ T cells expressing 2B4 in patients with active leishmaniasis caused by L. panamensis (132).
T cell Immunoglobulin and Mucin Domain-Containing Protein 3
TIM-3 is another co-inhibitory receptor that was initially identified in IFN-γ producing CD4+ and CD8+ T cells (188, 189). TIM-3 can also be found in other cell types namely Treg, myeloid cells, NK cells and mast cells (190–193). More recently, TIM-3 has been identified as an important part of a complex of co-inhibitory receptors present in dysfunctional or exhausted T cells (194). Briefly, models indicate that TIM-3 associates with HLA-B-associated transcript 3 (BAT3) to keep cells active through recruitment of tyrosine kinase LCK (195, 196). However, upon binding of either Galectin-9 or CEACAM1 on APCs releases BAT3 and there is recruitment of tyrosine kinase FYN and disruption of immune synapse, phosphatases recruitment and apoptosis (197–199). Therefore, TIM-3 together with other co-inhibitory receptors are potential blocking targets to restore T cell activity (200).
Limitations of the Available Data
One point to consider is that murine models are important to mechanistically understand the disease and its immunity, however, they cannot mimic all clinical outcomes of human leishmaniasis, such as DCL, MCL, and PKDL (Post-Kala Azar dermal leishmaniasis). Thus, for those clinical forms most of what we know relies on human data. So far, the information on the role of checkpoint molecules during CL is very scarce, and the majority of the work has been done studying human VL.
The role of checkpoint molecules has been extensively described in chronic viral infections in which these receptors are up regulated and, usually, suppress the activity of CD8+ T cells. Their capacity to suppress other cell subsets has been correlated with the co-expression of different checkpoint molecules and the production of key cytokines such as IL-10 (201). However, the role of CD8+ T cells during Leishmania infections is still not clear yet and depending on the level of its activity, CD8+ T cells might be involved with the production of local IFN-γ and activation of macrophages to kill parasites or might be involved with immunopathology and worse tissue outcome.
On the other hand, the role of CD4+ T helper 1 (Th1) cells has been better described due to its multifunctional production of cytokines, such as IFN-γ and TNF-α, and, consequently, activation of macrophages to kill parasites. However, the role of checkpoint molecules being expressed on CD4+ T cells is not clear yet, whether these molecules indicate an activated cell phenotype or depending on the degree of co-expression an exhausted cell (94, 202–206). So far, data suggests that during CL caused by distinct species of Leishmania there is an important upregulation of immune checkpoints in CD4+ and, especially, CD8+ T cells. However, the role those pathways play during infection is still inconclusive and the parasites might use some of them to bypass immunity.
Leishmaniasis imposes a complex scenario in which different species of one parasite united by the same genus can cause a broad spectrum of clinical manifestations and thus different immune responses. Checkpoint molecules appear to have a distinct role for immunity elicited by the different species of Leishmania or the different clinical outcomes.
Translational Implications
One central question to ask is how much of this knowledge can be translated to human leishmaniasis? Therapies using anti-PD-1, anti-PD-L1, and anti-CTLA-4 blocking antibodies were approved by the United States Food and Drug Administration (FDA) and became an important treatment for different types of cancer (207–212). Even though there is evidence that in VL the inhibition of some of those co-inhibitory receptors can restore T cell function and control parasite burden, no clinical studies have been performed.
Lack of translational implications brings two facts for reflection: recombinant antibody administrations are still expensive and therefore far from the governments and the main populations affected by leishmaniasis; and there are not enough scientists and funding to sponsor those clinical studies. Biological treatments are still costly and restricted to populations who can afford that (213, 214). It is still unclear whether these treatments are going to expand and consequently drop in price so that treatment of other diseases may benefit as well. Although Leishmania infections cause disease in one million individuals every year, the research in the field is still far beyond necessary for the magnitude and complexity of the problem. In this scenario, it is difficult to foresee translational studies in leishmaniasis patients needed in the upcoming future. As a result of global warming and population growth, substantial funding is need for research in leishmaniasis.
Concluding Remarks
Immune checkpoints either stimulatory or inhibitory are induced during the development of immunity against Leishmania (Figure 3). Stronger data for VL indicates that both PD-1 and PD-L1 are upregulated by CD4+ and CD8+ T cells. Moreover, in this setting the blocking strategy under the course of infection specially restored CD8+ T cell function and promoted a stronger Type I responses with IFN-γ activating macrophages and reducing parasite burden. The mechanisms behind the function of immune checkpoints during dermatotropic Leishmania infections is even more unclear, but data suggests that LIGHT and CD40 have important roles in maintaining immunity against Leishmania species inducing CL. The most promising data obtained with L. major suggested that LIGHT blockade impairs IL-12 secretion and Type I responses. Now it will be important to deeply characterize those pathways dissecting important questions: which are the cell types participating in this regulatory process and how those mechanisms act at the molecular, cellular and tissue levels. Translational application of those studies are still far from the field; however, they are shedding light on important mechanisms of the human system to control intracellular pathogens such as Leishmania.
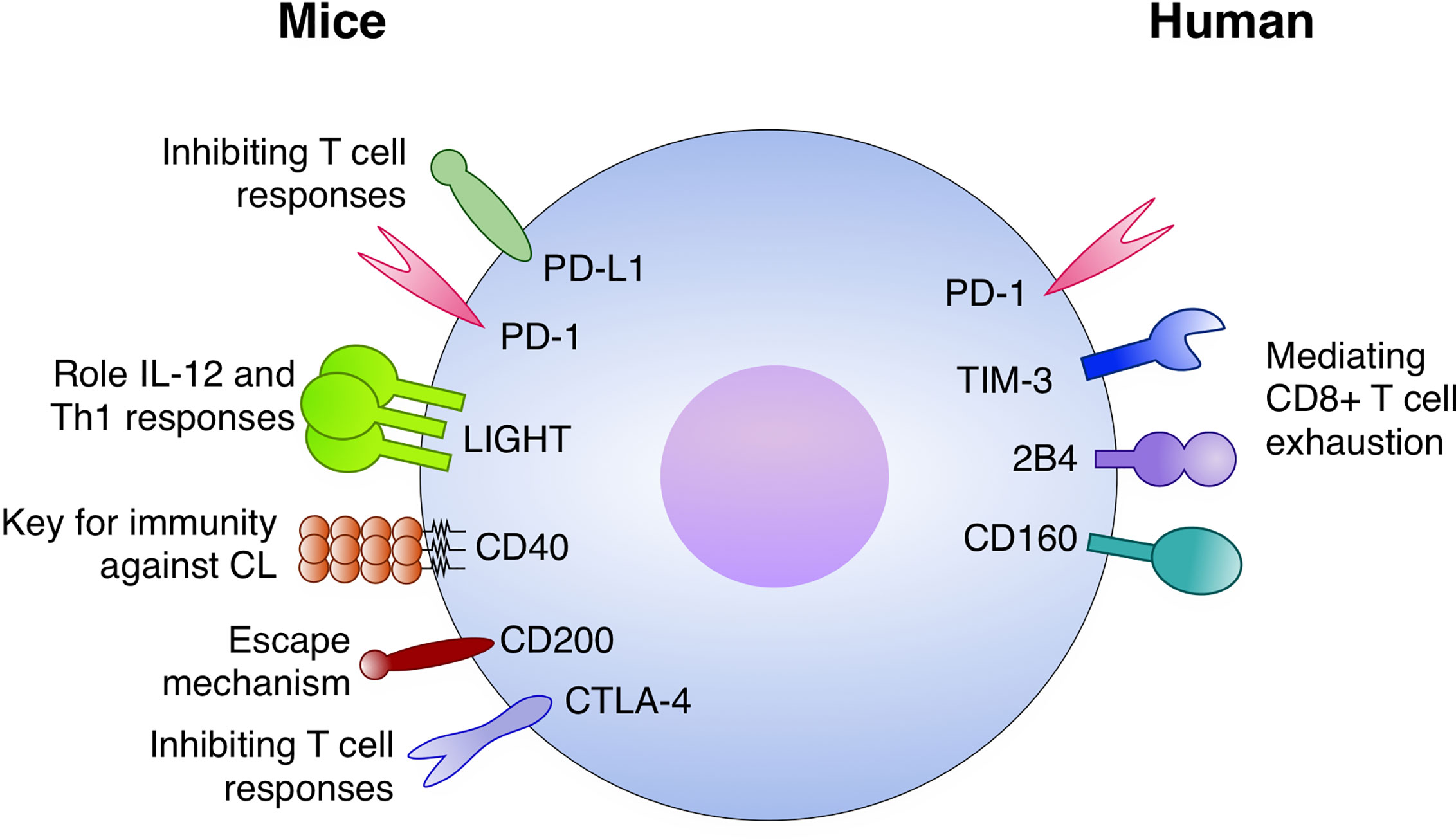
Figure 3 Immune checkpoints with a role in Leishmaniasis. In mice models: The PD-1/PD-L1 axis inhibits T cell responses, both CD4+ and CD8+, during infection. LIGHT has an important role for IL-12 and Th1 responses. CD40 is a key factor for immunity against cutaneous leishmaniasis (CL). CD200 expression is implicated with escape mechanisms by the parasite and CTLA-4 is an important inhibitor of T cell responses against Leismania. In humans, PD-1 TIM-3, 2B4, and CD160 have been potentially mediating CD8+ T cell exhaustion.
Author Contributions
RS wrote the paper with input from ES. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by the Universal-CNPq (Processo: 424889/2018-8) to RS and SFB 1292 (Deutsche Forschungsgemeinschaf, DFG) and the Center for Molecular Medicine Cologne (CMMC) to ES.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Dr. Roberto Lins from the Department of Virology at Aggeu Magalhães Institute (FIOCRUZ/Recife-Brazil) for kindly reviewing the first draft of the manuscript.
References
1. Casadevall A. Climate change brings the specter of new infectious diseases. J Clin Invest (2020) 130:553–5. doi: 10.1172/JCI135003
2. Filho WL, Scheday S, Boenecke J, Gogoi A, Maharaj A, Korovou S. Climate Change, Health and Mosquito-Borne Diseases: Trends and Implications to the Pacific Region. Int J Environ Res Public Health (2019) 16(24):1154. doi: 10.3390/ijerph16245114
3. Giesen C, Roche J, Redondo-Bravo L, Ruiz-Huerta C, Gomez-Barroso D, Benito A, et al. The impact of climate change on mosquito-borne diseases in Africa. Pathog Glob Health (2020) 114(6):1–15. doi: 10.1080/20477724.2020.1783865
4. Burza S, Croft SL, Boelaert M. Leishmaniasis. Lancet (London England) (2018) 392:951–70. doi: 10.1016/S0140-6736(18)31204-2
5. WHO. Leishmaniasis. WHO. Available at: https://www.who.int/gho/neglected_diseases/leishmaniasis/en/ (Accessed July 25, 2016).
6. Pigott DM, Bhatt S, Golding N, Duda KA, Battle KE, Brady OJ, et al. Global distribution maps of the leishmaniases. Elife (2014) 3:e02851. doi: 10.7554/eLife.02851
7. Ghatee MA, Taylor WR, Karamian M. The Geographical Distribution of Cutaneous Leishmaniasis Causative Agents in Iran and Its Neighboring Countries, A Review. Front Public Heal (2020) 8:11. doi: 10.3389/fpubh.2020.00011
8. Hotez PJ, Fenwick A, Savioli L, Molyneux DH. Rescuing the bottom billion through control of neglected tropical diseases. Lancet (London England) (2009) 373:1570–5. doi: 10.1016/S0140-6736(09)60233-6
9. Heyde S, Philipsen L, Formaglio P, Fu Y, Baars I, Höbbel G, et al. CD11c-expressing Ly6C+CCR2+ monocytes constitute a reservoir for efficient Leishmania proliferation and cell-to-cell transmission. PloS Pathog (2018) 14:e1007374. doi: 10.1371/journal.ppat.1007374
10. Hotez PJ, Bottazzi ME, Strych U, Chang LY, Lim YAL, Goodenow MM, et al. Neglected Tropical Diseases among the Association of Southeast Asian Nations (ASEAN): Overview and Update. PloS Negl Trop Dis (2015) 9:1–15. doi: 10.1371/journal.pntd.0003575
12. Kobets T, Grekov I, Lipoldova M. Leishmaniasis: prevention, parasite detection and treatment. Curr Med Chem (2012) 19:1443–74. doi: 10.2174/092986712799828300
13. Selvapandiyan A, Croft SL, Rijal S, Nakhasi HL, Ganguly NK. Innovations for the elimination and control of visceral leishmaniasis. PloS Negl Trop Dis (2019) 13:e0007616. doi: 10.1371/journal.pntd.0007616
14. Alvar J, Alves F, Bucheton B, Burrows L, Büscher P, Carrillo E, et al. Implications of asymptomatic infection for the natural history of selected parasitic tropical diseases. Semin Immunopathol (2020) 42(3):321–46. doi: 10.1007/s00281-020-00796-y
15. Zabala-Peñafiel A, Todd D, Daneshvar H, Burchmore R. The potential of live attenuated vaccines against Cutaneous Leishmaniasis. Exp Parasitol (2020) 210:107849. doi: 10.1016/j.exppara.2020.107849
16. Zutshi S, Kumar S, Chauhan P, Bansode Y, Nair A, Roy S, et al. Anti-Leishmanial Vaccines: Assumptions, Approaches, and Annulments. Vaccines (2019) 7:156. doi: 10.3390/vaccines7040156
17. Sacks DL. C O M M E N TA RY Vaccines against tropical parasitic diseases : a persisting answer to a persisting problem. Nat Publ Gr (2014) 15:403–5. doi: 10.1038/ni.2853
18. Fenwick C, Joo V, Jacquier P, Noto A, Banga R, Perreau M, et al. T-cell exhaustion in HIV infection. Immunol Rev (2019) 292:149–63. doi: 10.1111/imr.12823
19. Ruffo E, Wu RC, Bruno TC, Workman CJ, Vignali DAA. Lymphocyte-activation gene 3 (LAG3): The next immune checkpoint receptor. Semin Immunol (2019) 42:101305. doi: 10.1016/j.smim.2019.101305
20. Winkler F, Bengsch B. Use of Mass Cytometry to Profile Human T Cell Exhaustion. Front Immunol (2020) 10:3039. doi: 10.3389/fimmu.2019.03039
21. Keir ME, Freeman GJ, Sharpe AH. PD-1 Regulates Self-Reactive CD8+ T Cell Responses to Antigen in Lymph Nodes and Tissues. J Immunol (2007) 179:5064–70. doi: 10.4049/JIMMUNOL.179.8.5064
22. Pauken KE, Juneja V, McGuire K, LaFleur M, Kuchroo J, Sage P, et al. CD8+ T cell-independent mechanisms of PD-1-mediated suppression of anti-tumor immunity in mice. J Immunol (2017) 198(1 Supplement):205.11.
23. Klechevsky E, Flamar A-L, Cao Y, Blanck J-P, Liu M, O’Bar A, et al. Cross-priming CD8+ T cells by targeting antigens to human dendritic cells through DCIR. Blood (2010) 116:1685–97. doi: 10.1182/blood-2010-01-264960
24. Murphy TL, Murphy KM. Slow Down and Survive: Enigmatic Immunoregulation by BTLA and HVEM. Annu Rev Immunol (2010) 28:389–411. doi: 10.1146/annurev-immunol-030409-101202
25. Hashimoto M, Kamphorst AO, Im SJ, Kissick HT, Pillai RN, Ramalingam SS, et al. CD8 T Cell Exhaustion in Chronic Infection and Cancer: Opportunities for Interventions. Annu Rev Med (2018) 69:301–18. doi: 10.1146/annurev-med-012017-043208
26. Salmaninejad A, Valilou SF, Shabgah AG, Aslani S, Alimardani M, Pasdar A, et al. PD-1/PD-L1 pathway: Basic biology and role in cancer immunotherapy. J Cell Physiol (2019) 234(10):16824–37. doi: 10.1002/jcp.28358
27. Afrin F, Khan I, Hemeg HA. Leishmania-Host Interactions—An Epigenetic Paradigm. Front Immunol (2019) 10:492. doi: 10.3389/fimmu.2019.00492
28. Gabriel Á, Valério-Bolas A, Palma-Marques J, Mourata-Gonçalves P, Ruas P, Dias-Guerreiro T, et al. Cutaneous Leishmaniasis: The Complexity of Host’s Effective Immune Response against a Polymorphic Parasitic Disease. J Immunol Res (2019) 2019:1–16. doi: 10.1155/2019/2603730
29. Misra P, Singh S. Site specific microbiome of Leishmania parasite and its cross-talk with immune milieu. Immunol Lett (2019) 216:79–88. doi: 10.1016/j.imlet.2019.10.004
30. León B, López-Bravo M, Ardavín C. Monocyte-derived dendritic cells formed at the infection site control the induction of protective T helper 1 responses against Leishmania. Immunity (2007) 26:519–31. doi: 10.1016/j.immuni.2007.01.017
31. Brittingham A, Morrison CJ, McMaster WR, McGwire BS, Chang KP, Mosser DM. Role of the Leishmania surface protease gp63 in complement fixation, cell adhesion, and resistance to complement-mediated lysis. J Immunol (1995) 155:3102–11. doi: 10.1016/0169-4758(95)80054-9
32. Esch KJ, Schaut RG, Lamb IM, Clay G, Morais Lima ÁL, do Nascimento PRP, et al. Activation of Autophagy and Nucleotide-Binding Domain Leucine-Rich Repeat–Containing-Like Receptor Family, Pyrin Domain–Containing 3 Inflammasome during Leishmania infantum–Associated Glomerulonephritis. Am J Pathol (2015) 185:2105–17. doi: 10.1016/j.ajpath.2015.04.017
33. Da Silva RP, Hall BF, Joiner KA, Sacks DL. CR1, the C3b receptor, mediates binding of infective Leishmania major metacyclic promastigotes to human macrophages. J Immunol (1989) 143:617–22.
34. Charmoy M, Brunner-Agten S, Aebischer D, Auderset F, Launois P, Milon G, et al. Neutrophil-Derived CCL3 Is Essential for the Rapid Recruitment of Dendritic Cells to the Site of Leishmania major Inoculation in Resistant Mice. PloS Pathog (2010) 6:e1000755. doi: 10.1371/journal.ppat.1000755
35. Ribeiro-Gomes FL, Sacks D. The influence of early neutrophil-Leishmania interactions on the host immune response to infection. Front Cell Infect Microbiol (2012) 2:59. doi: 10.3389/fcimb.2012.00059
36. Ritter U, Korner H. Divergent expression of inflammatory dermal chemokines in cutaneous leishmaniasis*. Parasite Immunol (2002) 24:295–301. doi: 10.1046/j.1365-3024.2002.00467.x
37. Conrad SM, Strauss-Ayali D, Field AE, Mack M, Mosser DM. Leishmania-derived murine monocyte chemoattractant protein 1 enhances the recruitment of a restrictive population of CC chemokine receptor 2-positive macrophages. Infect Immun (2007) 75:653–65. doi: 10.1128/IAI.01314-06
38. Costa DL, Lima-Júnior DS, Nascimento MS, Sacramento LA, Almeida RP, Carregaro V, et al. CCR2 signaling contributes to the differentiation of protective inflammatory dendritic cells in Leishmania braziliensis infection. J Leukoc Biol (2016) 100:423–32. doi: 10.1189/jlb.4A0715-288R
39. Romano A, Carneiro MBH, Doria NA, Roma EH, Ribeiro-Gomes FL, Inbar E, et al. Divergent roles for Ly6C+CCR2+CX3CR1+ inflammatory monocytes during primary or secondary infection of the skin with the intra-phagosomal pathogen Leishmania major. PloS Pathog (2017) 13:e1006479. doi: 10.1371/journal.ppat.1006479
40. von Stebut E, Belkaid Y, Jakob T, Sacks DL, Udey MC. Uptake of Leishmania major amastigotes results in activation and interleukin 12 release from murine skin-derived dendritic cells: implications for the initiation of anti-Leishmania immunity. J Exp Med (1998) 188:1547–52. doi: 10.1084/jem.188.8.1547
41. Nigg AP, Zahn S, Rückerl D, Hölscher C, Yoshimoto T, Ehrchen JM, et al. Stebut E von. Dendritic Cell-Derived IL-12p40 Homodimer Contributes to Susceptibility in Cutaneous Leishmaniasis in BALB/c Mice. J Immunol (2007) 178:7251–8. doi: 10.4049/JIMMUNOL.178.11.7251
42. von Stebut E, Belkaid Y, Nguyen BV, Cushing M, Sacks DL, Udey MC. Leishmania major-infected murine Langerhans cell-like dendritic cells from susceptible mice release IL-12 after infection and vaccinate against experimental cutaneous Leishmaniasis. Eur J Immunol (2000) 30:3498–506. doi: 10.1002/1521-4141(2000012)30:12<3498::AID-IMMU3498>3.0.CO;2-6
43. Reiner SL, Locksley RM. The regulation of immunity to Leishmania major. Annu Rev Immunol (1995) 13:151–77. doi: 10.1146/annurev.iy.13.040195.001055
44. Scott P, Pearce E, Cheever AW, Coffman RL, Sher A. Role of cytokines and CD4+ T-cell subsets in the regulation of parasite immunity and disease. Immunol Rev (1989) 112:161–82. doi: 10.1111/j.1600-065x.1989.tb00557.x
45. Boussoffara T, Chelif S, Ben Ahmed M, Mokni M, Ben Salah A, Dellagi K, et al. Immunity Against Leishmania major Infection: Parasite-Specific Granzyme B Induction as a Correlate of Protection. Front Cell Infect Microbiol (2018) 8:397. doi: 10.3389/fcimb.2018.00397
46. Olekhnovitch R, Bousso P. Induction, Propagation, and Activity of Host Nitric Oxide: Lessons from Leishmania Infection. Trends Parasitol (2015) 31:653–64. doi: 10.1016/j.pt.2015.08.001
47. Green SJ, Crawford RM, Hockmeyer JT, Meltzer MS, Nacy CA. Leishmania major amastigotes initiate the L-arginine-dependent killing mechanism in IFN-gamma-stimulated macrophages by induction of tumor necrosis factor-alpha. J Immunol (1990) 145:4290–7.
48. Liew FY, Li Y, Moss D, Parkinson C, Rogers MV, Moncada S. Resistance to Leishmania major infection correlates with the induction of nitric oxide synthase in murine macrophages. Eur J Immunol (1991) 21:3009–14. doi: 10.1002/eji.1830211216
49. Sacks D, Noben-Trauth N. The immunology of susceptibility and resistance to Leishmania major in mice. Nat Rev Immunol (2002) 2:845–58. doi: 10.1038/nri933
50. Himmelrich H, Parra-Lopez C, Tacchini-Cottier F, Louis JA, Launois P. The IL-4 rapidly produced in BALB/c mice after infection with Leishmania major down-regulates IL-12 receptor beta 2-chain expression on CD4+ T cells resulting in a state of unresponsiveness to IL-12. J Immunol (1998) 161:6156–63.
51. Jones D, Elloso MM, Showe L, Williams D, Trinchieri G, Scott P. Differential regulation of the interleukin-12 receptor during the innate immune response to Leishmania major. Infect Immun (1998) 66:3818–24. doi: 10.1128/IAI.66.8.3818-3824.1998
52. Louis JA, Conceiçao-Silva F, Himmelrich H, Tacchini-Cottier F, Launois P. Anti-Leishmania Effector Functions of CD4+ Th1 Cells and Early Events Instructing Th2 Cell Development and Susceptibility to Leishmania Major in BALB/c Mice. In: Gupta S., Sher A., Ahmed R. (eds) Mechanisms of Lymphocyte Activation and Immune Regulation VII. Advances in Experimental Medicine and Biology. Boston, MA: Springer (1998) vol 452. 53–60. doi: 10.1007/978-1-4615-5355-7_7
53. Kolls JK, Lindén A. Interleukin-17 family members and inflammation. Immunity (2004) 21:467–76. doi: 10.1016/j.immuni.2004.08.018
54. Gonzalez-Lombana C, Gimblet C, Bacellar O, Oliveira WW, Passos S, Carvalho LP, et al. IL-17 Mediates Immunopathology in the Absence of IL-10 Following Leishmania major Infection. PloS Pathog (2013) 9:e1003243. doi: 10.1371/journal.ppat.1003243
55. Lopez Kostka S, Dinges S, Griewank K, Iwakura Y, Udey MC, von Stebut E. IL-17 promotes progression of cutaneous leishmaniasis in susceptible mice. J Immunol (2009) 182:3039–46. doi: 10.4049/jimmunol.0713598
56. Dietze-Schwonberg K, Lopez Kostka S, Lorenz B, Regen T, Waisman A, von Stebut E. IL-17A/F in Leishmania major -resistant C57BL/6 mice. Exp Dermatol (2019) 28:321–3. doi: 10.1111/exd.13896
57. Campos TM, Novais FO, Saldanha M, Costa R, Lordelo M, Celestino D, et al. Granzyme B Produced by Natural Killer Cells Enhances Inflammatory Response and Contributes to the Immunopathology of Cutaneous Leishmaniasis. J Infect Dis (2020) 221:973–82. doi: 10.1093/infdis/jiz538
58. Novais FO, Nguyen BT, Scott P. Granzyme B inhibition by tofacitinib blocks pathology induced by CD8 T cells in cutaneous leishmaniasis. J Invest Dermatol (2020) S0022-202X(20)31942-4. doi: 10.1016/J.JID.2020.07.011
59. Novais FO, Carvalho AM, Clark ML, Carvalho LP, Beiting DP, Brodsky IE, et al. CD8+ T cell cytotoxicity mediates pathology in the skin by inflammasome activation and IL-1β production. PloS Pathog (2017) 13:e1006196. doi: 10.1371/journal.ppat.1006196
60. Belkaid Y, Von Stebut E, Mendez S, Lira R, Caler E, Bertholet S, et al. CD8+ T cells are required for primary immunity in C57BL/6 mice following low-dose, intradermal challenge with Leishmania major. J Immunol (2002) 168:3992–4000. doi: 10.4049/jimmunol.168.8.3992
61. Okwor IB, Jia P, Mou Z, Onyilagha C, Uzonna JE. CD8+ T cells Are Preferentially Activated during Primary Low Dose Leishmania major Infection but Are Completely Dispensable during Secondary Anti-Leishmania Immunity. PloS Negl Trop Dis (2014) 8:e3300. doi: 10.1371/journal.pntd.0003300
62. Savage PA, Klawon DEJ, Miller CH. Regulatory T Cell Development. Annu Rev Immunol (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
63. Belkaid Y, Hoffmann KF, Mendez S, Kamhawi S, Udey MC, Wynn TA, et al. The role of interleukin (IL)-10 in the persistence of Leishmania major in the skin after healing and the therapeutic potential of anti-IL-10 receptor antibody for sterile cure. J Exp Med (2001) 194:1497–506. doi: 10.1084/jem.194.10.1497
64. Belkaid Y, Piccirillo CA, Mendez S, Shevach EM, Sacks DL. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature (2002) 420:502–7. doi: 10.1038/nature01152
65. Ehrlich A, Castilho TM, Goldsmith-Pestana K, Chae W-J, Bothwell ALM, Sparwasser T, et al. The immunotherapeutic role of regulatory T cells in Leishmania (Viannia) panamensis infection. J Immunol (2014) 193:2961–70. doi: 10.4049/jimmunol.1400728
66. Dietze-Schwonberg K, Kautz-Neu K, Lorenz B, Clausen BE, von Stebut E. In cutaneous leishmaniasis, induction of retinoic acid in skin-derived Langerhans cells is not sufficient for induction of parasite persistence-mediating regulatory T cells. J Dermatol Sci (2017) 87:307–9. doi: 10.1016/j.jdermsci.2017.06.014
67. Kautz-Neu K, Noordegraaf M, Dinges S, Bennett CL, John D, Clausen BE, et al. Langerhans cells are negative regulators of the anti-Leishmania response. J Exp Med (2011) 208:885–91. doi: 10.1084/jem.20102318
68. Masopust D, Sivula CP, Jameson SC. Of Mice, Dirty Mice, and Men: Using Mice To Understand Human Immunology. J Immunol (2017) 199:383–8. doi: 10.4049/jimmunol.1700453
69. Nalleli L, José A-NEZF. REVIEW ANIMAL MODELS FOR THE STUDY OF LEISHMANIASIS IMMUNOLOGY. Rev Inst Med Trop Sao Paulo (2014) 56:1–11. doi: 10.1590/S0036-46652014000100001
70. Arango Duque G, Descoteaux A. Leishmania survival in the macrophage: where the ends justify the means. Curr Opin Microbiol (2015) 26:32–40. doi: 10.1016/j.mib.2015.04.007
71. Scott P, Novais FO. Cutaneous leishmaniasis: immune responses in protection and pathogenesis. Nat Publ Gr (2016) 16(9):581–92. doi: 10.1038/nri.2016.72
72. Kumar R, Engwerda C. Vaccines to prevent leishmaniasis. Clin Transl Immunol (2014) 3:e13. doi: 10.1038/cti.2014.4
74. Control of the leishmaniasis: report of a meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva. (2010). pp. 22–26.
75. Carrillo E, Moreno J. Editorial: Biomarkers in Leishmaniasis. Front Cell Infect Microbiol (2019) 9:388. doi: 10.3389/fcimb.2019.00388
76. dos Santos Meira C, Gedamu L. Protective or Detrimental? Understanding the Role of Host Immunity in Leishmaniasis. Microorganisms (2019) 7:695. doi: 10.3390/microorganisms7120695
77. Breloer M, Hartmann W, Blankenhaus B, Eschbach M-L, Pfeffer K, Jacobs T. Cutting Edge: The BTLA–HVEM Regulatory Pathway Interferes with Protective Immunity to Intestinal Helminth Infection. J Immunol (2015) 194:1413–6. doi: 10.4049/jimmunol.1402510
78. Gannavaram S, Bhattacharya P, Ismail N, Kaul A, Singh R, Nakhasi HL. Modulation of Innate Immune Mechanisms to Enhance Leishmania Vaccine-Induced Immunity: Role of Coinhibitory Molecules. Front Immunol (2016) 7:187. doi: 10.3389/fimmu.2016.00187
79. Silva LA, Romero HD, Nascentes GAN, Costa RT, Rodrigues V, Prata A. Antileishmania immunological tests for asymptomatic subjects living in a visceral leishmaniasis-endemic area in Brazil. Am J Trop Med Hyg (2011) 84:261–6. doi: 10.4269/ajtmh.2011.10-0092
80. Lepenies B, Pfeffer K, Hurchla MA, Murphy TL, Murphy KM, Oetzel J, et al. Ligation of B and T lymphocyte attenuator prevents the genesis of experimental cerebral malaria. J Immunol (2007) 179(6):4093–100. doi: 10.4049/jimmunol.179.6.4093
81. Mackroth MS, Abel A, Steeg C, Schulze zur Wiesch J, Jacobs T. Acute Malaria Induces PD1+CTLA4+ Effector T Cells with Cell-Extrinsic Suppressor Function. PloS Pathog (2016) 12:e1005909. doi: 10.1371/journal.ppat.1005909
82. Zhao Y, Lee CK, Lin CH, Gassen RB, Xu X, Huang Z, et al. PD-L1:CD80 Cis-Heterodimer Triggers the Co-stimulatory Receptor CD28 While Repressing the Inhibitory PD-1 and CTLA-4 Pathways. Immunity (2019) 51:1059–1073.e9. doi: 10.1016/j.immuni.2019.11.003
83. Hernández-Ruiz J, Salaiza-Suazo N, Carrada G, Escoto S, Ruiz-Remigio A, Rosenstein Y, et al. CD8 cells of patients with diffuse cutaneous leishmaniasis display functional exhaustion: The latter is reversed, in vitro, by TLR2 agonists. PloS Negl Trop Dis (2010) 4(11):e871. doi: 10.1371/journal.pntd.0000871
84. Schaut RG, Lamb IM, Toepp AJ, Scott B, Mendes-Aguiar CO, Coutinho JFV, et al. Regulatory IgD hi B Cells Suppress T Cell Function via IL-10 and PD-L1 during Progressive Visceral Leishmaniasis. J Immunol (2016) 196:4100–9. doi: 10.4049/jimmunol.1502678
85. Esch KJ, Juelsgaard R, Martinez PA, Jones DE, Petersen CA. Programmed Death 1-Mediated T Cell Exhaustion during Visceral Leishmaniasis Impairs Phagocyte Function. J Immunol (2013) 191:5542–50. doi: 10.4049/jimmunol.1301810
86. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc Natl Acad Sci USA (2001) 98:13866–71. doi: 10.1073/pnas.231486598
87. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol (2004) 173:945–54. doi: 10.4049/jimmunol.173.2.945
88. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med (2012) 209:1201–17. doi: 10.1084/jem.20112741
89. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science (2017) 355:1428–33. doi: 10.1126/science.aaf1292
90. Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, et al. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol (2009) 10:1185–92. doi: 10.1038/ni.1790
91. Dong Y, Sun Q, Zhang X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget (2017) 8:2171–86. doi: 10.18632/oncotarget.13895
92. DeLong JH, O’Hara Hall A, Rausch M, Moodley D, Perry J, Park J, et al. IL-27 and TCR Stimulation Promote T Cell Expression of Multiple Inhibitory Receptors. ImmunoHorizons (2019) 3:13–25. doi: 10.4049/immunohorizons.1800083
93. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med (2009) 206:3015–29. doi: 10.1084/jem.20090847
94. Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Lee CYQ, et al. Shankar EM. T-Cell Exhaustion in Chronic Infections: Reversing the State of Exhaustion and Reinvigorating Optimal Protective Immune Responses. Front Immunol (2018) 9:2569. doi: 10.3389/fimmu.2018.02569
95. de Freitas e Silva R, Gálvez RI, Pereira VRA, de Brito MEF, SL C, Lotter H, et al. Programmed Cell Death Ligand (PD-L)-1 Contributes to the Regulation of CD4+ T Effector and Regulatory T Cells in Cutaneous Leishmaniasis. Front Immunol (2020) 11:574491. doi: 10.3389/fimmu.2020.574491
96. Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, Sharpe AH. Dendritic Cell PD-L1 Limits Autoimmunity and Follicular T Cell Differentiation and Function. J Immunol (2018) 200:2592–602. doi: 10.4049/jimmunol.1701231
97. Martínez Salazar MB, Delgado Domínguez J, Silva Estrada J, González Bonilla C, Becker I. Vaccination with Leishmania mexicana LPG induces PD-1 in CD8+ and PD-L2 in macrophages thereby suppressing the immune response: A model to assess vaccine efficacy. Vaccine (2014) 32:1259–65. doi: 10.1016/j.vaccine.2014.01.016
98. da Fonseca-Martins AM, Ramos TD, Pratti JES, Firmino-Cruz L, Gomes DCO, Soong L, et al. Immunotherapy using anti-PD-1 and anti-PD-L1 in Leishmania amazonensis-infected BALB/c mice reduce parasite load. Sci Rep (2019) 9:20275. doi: 10.1038/s41598-019-56336-8
99. Habib S, El Andaloussi A, Elmasry K, Handoussa A, Azab M, Elsawey A, et al. PDL-1 Blockade Prevents T Cell Exhaustion, Inhibits Autophagy, and Promotes Clearance of Leishmania donovani. Infect Immun (2018) 86(6):e00019–8. doi: 10.1128/IAI.00019-18
100. Oliveira Silva KL, Marin Chiku V, Luvizotto Venturin G, Correa Leal AA, de Almeida BF, De Rezende Eugenio F, et al. PD-1 and PD-L1 regulate cellular immunity in canine visceral leishmaniasis. Comp Immunol Microbiol Infect Dis (2019) 62:76–87. doi: 10.1016/J.CIMID.2018.12.002
101. Bankoti R, Stäger S. Differential Regulation of the Immune Response in the Spleen and Liver of Mice Infected with Leishmania donovani. J Trop Med (2012) 2012:639304. doi: 10.1155/2012/639304
102. Joshi T, Rodriguez S, Perovic V, Cockburn IA, Stäger S. B7-H1 blockade increases survival of dysfunctional CD8+T cells and confers protection against Leishmania donovaniinfections. PloS Pathog (2009) 5:e1000431. doi: 10.1371/journal.ppat.1000431
103. Rodrigues V, Cordeiro-Da-Silva A, Laforge M, Silvestre R, Estaquier J. Regulation of immunity during visceral Leishmania infection. (2016) 9:118. doi: 10.1186/s13071-016-1412-x
104. Chiku VM, Silva KLO, de Almeida BFM, Venturin GL, Leal AAC, de Martini CC, et al. PD-1 function in apoptosis of T lymphocytes in canine visceral leishmaniasis. Immunobiology (2016) 221:879–88. doi: 10.1016/j.imbio.2016.03.007
105. Butler NS, Moebius J, Pewe LL, Traore B, Doumbo OK, Tygrett LT, et al. Therapeutic blockade of PD-L1 and LAG-3 rapidly clears established blood-stage Plasmodium infection. Nat Immunol (2012) 13:188–95. doi: 10.1038/ni.2180
106. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
107. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol (2001) 2:261–8. doi: 10.1038/85330
108. Peng C, Hu Q, Yang F, Zhang H, Li F, Huang C. BCL6-Mediated Silencing of PD-1 Ligands in Germinal Center B Cells Maintains Follicular T Cell Population. J Immunol (2019) 202:704–13. doi: 10.4049/jimmunol.1800876
109. Pinchuk IV, Powell DW. Immunosuppression by Intestinal Stromal Cells. Adv Exp Med Biol (2018) 1060:115–29. doi: 10.1007/978-3-319-78127-3_7
110. Solinas C, Garaud S, De Silva P, Boisson A, Van den Eynden G, de Wind A, et al. Immune Checkpoint Molecules on Tumor-Infiltrating Lymphocytes and Their Association with Tertiary Lymphoid Structures in Human Breast Cancer. Front Immunol (2017) 8:1412. doi: 10.3389/fimmu.2017.01412
111. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-DC, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med (2001) 193:839–46. doi: 10.1084/jem.193.7.839
112. Loke P, Allison JP. PD-L1 and PD-L2 are differentially regulated by Th1 and Th2 cells. Proc Natl Acad Sci USA (2003) 100:5336–41. doi: 10.1073/pnas.0931259100
113. Selenko-Gebauer N, Majdic O, Szekeres A, Höfler G, Guthann E, Korthäuer U, et al. B7-H1 (programmed death-1 ligand) on dendritic cells is involved in the induction and maintenance of T cell anergy. J Immunol (2003) 170:3637–44. doi: 10.4049/jimmunol.170.7.3637
114. Liang SC, Greenwald RJ, Latchman YE, Rosas L, Satoskar A, Freeman GJ, et al. PD-L1 and PD-L2 have distinct roles in regulating host immunity to cutaneous leishmaniasis. Eur J Immunol (2006) 36:58–64. doi: 10.1002/eji.200535458
115. Brunet J-F, Denizot F, Luciani M-F, Roux-Dosseto M, Suzan M, Mattei M-G, et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature (1987) 328:267–70. doi: 10.1038/328267a0
116. Tivol EA, Borriello F, Nicola Schweitzer A, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 Leads to Massive Lymphoproliferation and Fatal Multiorgan Tissue Destruction, Revealing a Critical Negative Regulatory Role of CTLA-4. (1995) 3:541–7. doi: 10.1016/1074-7613(95)90125-6
117. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative Disorders with Early Lethality in Mice Deficient in Ctla-4. Sci (80 ) (1995) 270:985–8. doi: 10.1126/science.270.5238.985
118. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Science (2011) 332:600–3. doi: 10.1126/science.1202947
119. Paterson AM, Lovitch SB, Sage PT, Juneja VR, Lee Y, Trombley JD, et al. Deletion of CTLA-4 on regulatory T cells during adulthood leads to resistance to autoimmunity. J Exp Med (2015) 212:1603–21. doi: 10.1084/jem.20141030
120. Fallarino F, Grohmann U, Hwang KW, Orabona C, Vacca C, Bianchi R, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol (2003) 4:1206–12. doi: 10.1038/ni1003
121. Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med (1999) 189:1363–72. doi: 10.1084/jem.189.9.1363
122. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86. doi: 10.3389/fonc.2018.00086
123. Wei SC, Anang N-AAS, Sharma R, Andrews MC, Reuben A, Levine JH, et al. Combination anti-CTLA-4 plus anti-PD-1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci USA (2019) 116:22699–709. doi: 10.1073/pnas.1821218116
124. Murphy ML, Cotterell SE, Gorak PM, Engwerda CR, Kaye PM. Blockade of CTLA-4 enhances host resistance to the intracellular pathogen, Leishmania donovani. J Immunol (1998) 161:4153–60.
125. Masteller EL, Chuang E, Mullen AC, Reiner SL, Thompson CB, Abbas AK, et al. Structural analysis of CTLA-4 function in vivo. J Immunol (2000) 164:5319–27. doi: 10.4049/jimmunol.164.10.5319
126. Heinzel FP, Maier RA Jr. Interleukin-4-independent acceleration of cutaneous leishmaniasis in susceptible BALB/c mice following treatment with anti-CTLA4 antibody. Infect Immun (1999) 67:6454–60.
127. Saha B, Chattopadhyay S, Germond R, Harlan DM, Perrin PJ. CTLA4 (CD152) modulates the Th subset response and alters the course of experimentalLeishmania major infection. Eur J Immunol (1998) 28:4213–20. doi: 10.1002/(SICI)1521-4141(199812)28:12<4213::AID-IMMU4213>3.0.CO;2-C
128. Bluestone Roli Khattri JA, Auger JA, Griffin MD. CD28-Regulated Activation of Th2 Responses Knockout Mice Is Characterized by Lymphoproliferative Disorder in CTLA-4. (1999) 162(10):5784–91.
129. Chen W, Jin W, Wahl SM. Engagement of cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) induces transforming growth factor beta (TGF-beta) production by murine CD4(+) T cells. J Exp Med (1998) 188:1849–57. doi: 10.1084/jem.188.10.1849
130. Wilson ME, Recker TJ, Rodriguez NE, Young BM, Burnell KK, Streit JA, et al. The TGF-beta response to Leishmania chagasi in the absence of IL-12. Eur J Immunol (2002) 32:3556–65. doi: 10.1002/1521-4141(200212)32:12<3556::AID-IMMU3556>3.0.CO;2-Q
131. Gomes NA, Gattass CR, Barreto-De-Souza V, Wilson ME, DosReis GA, Roberts AB, et al. TGF-beta mediates CTLA-4 suppression of cellular immunity in murine kalaazar. J Immunol (2000) 164:2001–8. doi: 10.4049/jimmunol.164.4.2001
132. Gomes NA, Barreto-de-Souza V, Wilson ME, DosReis GA. Unresponsive CD4+ T lymphocytes from Leishmania chagasi-infected mice increase cytokine production and mediate parasite killing after blockade of B7-1/CTLA-4 molecular pathway. J Infect Dis (1998) 178:1847–51. doi: 10.1086/314520
133. Egui A, Ledesma D, Pérez-Antón E, Montoya A, Gómez I, Robledo SM, et al. Phenotypic and Functional Profiles of Antigen-Specific CD4+ and CD8+ T Cells Associated With Infection Control in Patients With Cutaneous Leishmaniasis. Front Cell Infect Microbiol (2018) 8:393. doi: 10.3389/fcimb.2018.00393
134. Chen Z, Kapus A, Khatri I, Kos O, Zhu F, Gorczynski RM. Cell membrane-bound CD200 signals both via an extracellular domain and following nuclear translocation of a cytoplasmic fragment. Leuk Res (2018) 69:72–80. doi: 10.1016/j.leukres.2018.04.007
135. Gorczynski RM, Zhu F. Checkpoint blockade in solid tumors and B-cell malignancies, with special consideration of the role of CD200. Cancer Manag Res (2017) 9:601–9. doi: 10.2147/CMAR.S147326
136. Ngwa C, Liu F. CD200-CD200R signaling and diseases: a potential therapeutic target? Int J Physiol Pathophysiol Pharmacol (2019) 11:297–309.
137. Manich G, Recasens M, Valente T, Almolda B, González B, Castellano B. Role of the CD200-CD200R Axis During Homeostasis and Neuroinflammation. Neuroscience (2019) 405:118–36. doi: 10.1016/j.neuroscience.2018.10.030
138. Songyang Z, Yamanashi Y, Liu D, Baltimore D. Domain-dependent function of the rasGAP-binding protein p62Dok in cell signaling. J Biol Chem (2001) 276:2459–65. doi: 10.1074/jbc.M005504200
139. Lock P, Casagranda F, Dunn AR. Independent SH2-binding sites mediate interaction of Dok-related protein with RasGTPase-activating protein and Nck. J Biol Chem (1999) 274:22775–84. doi: 10.1074/jbc.274.32.22775
140. Chakrabarty K, Heumann R. Prospective of Ras signaling in stem cells. Biol Chem (2008) 389:791–8. doi: 10.1515/BC.2008.104
141. Cortez M, Huynh C, Fernandes MC, Kennedy KA, Aderem A, Andrews NW. Leishmania promotes its own virulence by inducing expression of the host immune inhibitory ligand CD200. Cell Host Microbe (2011) 9:463–71. doi: 10.1016/j.chom.2011.04.014
142. Sauter IP, Madrid KG, de Assis JB, Sá-Nunes A, Torrecilhas AC, Staquicini DI, et al. TLR9/MyD88/TRIF signaling activates host immune inhibitory CD200 in Leishmania infection. JCI Insight (2019) 4. doi: 10.1172/jci.insight.126207
143. Rawat AK, Pal K, Singh R, Anand A, Gupta S, Kishore D, et al. The CD200-CD200R cross-talk helps Leishmania donovani to down regulate macrophage and CD4+CD44+ T cells effector functions in an NFκB independent manner. Int J Biol Macromol (2020) 151:394–401. doi: 10.1016/j.ijbiomac.2020.02.189
144. Singh RK, Gannavaram S, Ismail N, Kaul A, Gedda MR, Nakhasi HL. Centrin-Deleted Leishmania donovani Parasites Help CD4+ T Cells to Acquire Th1 Phenotype and Multi-Functionality Through Downregulation of CD200–CD200R Immune Inhibitory Axis. Front Immunol (2018) 9:1176. doi: 10.3389/fimmu.2018.01176
145. Ma DY, Clark EA. The role of CD40 and CD154/CD40L in dendritic cells. Semin Immunol (2009) 21:265–72. doi: 10.1016/j.smim.2009.05.010
146. García-González P, Morales R, Hoyos L, Maggi J, Campos J, Pesce B, et al. A short protocol using dexamethasone and monophosphoryl lipid A generates tolerogenic dendritic cells that display a potent migratory capacity to lymphoid chemokines. J Transl Med (2013) 11:128. doi: 10.1186/1479-5876-11-128
147. Okwor I, Uzonna JE. Pathways leading to interleukin-12 production and protective immunity in cutaneous leishmaniasis. Cell Immunol (2016) 309:32–6. doi: 10.1016/j.cellimm.2016.06.004
148. Chakraborty D, Banerjee S, Sen A, Banerjee KK, Das P, Roy S. Leishmania donovani affects antigen presentation of macrophage by disrupting lipid rafts. J Immunol (2005) 175:3214–24. doi: 10.4049/jimmunol.175.5.3214
149. Alves Costa Silva C, Facchinetti F, Routy B, Derosa L. New pathways in immune stimulation: targeting OX40. ESMO Open (2020) 5(1):e000573. doi: 10.1136/esmoopen-2019-000573
150. Rogers PR, Song J, Gramaglia I, Killeen N, Croft M. OX40 Promotes Bcl-xL and Bcl-2 Expression and Is Essential for Long-Term Survival of CD4 T Cells. Immunity (2001) 15:445–55. doi: 10.1016/S1074-7613(01)00191-1
151. Song J, So T, Cheng M, Tang X, Croft M. Sustained Survivin Expression from OX40 Costimulatory Signals Drives T Cell Clonal Expansion. Immunity (2005) 22:621–31. doi: 10.1016/J.IMMUNI.2005.03.012
152. Jember AG, Zuberi R, Liu FT, Croft M. Development of allergic inflammation in a murine model of asthma is dependent on the costimulatory receptor OX40. J Exp Med (2001) 193:387–92. doi: 10.1084/jem.193.3.387
153. Tsukada N, Akiba H, Kobata T, Aizawa Y, Yagita H, Okumura K. Blockade of CD134 (OX40)-CD134L interaction ameliorates lethal acute graft-versus-host disease in a murine model of allogeneic bone marrow transplantation. Blood (2000) 95:2434–9. doi: 10.1182/blood.V95.7.2434
154. Ishii N, Ndhlovu LC, Murata K, Sato T, Kamanaka M, Sugamura K. OX40 (CD134) and OX40 ligand interaction plays an adjuvant role during in vivo Th2 responses. Eur J Immunol (2003) 33:2372–81. doi: 10.1002/eji.200324031
155. Zubairi S, Sanos SL, Hill S, Kaye PM. Immunotherapy with OX40L-Fc or anti-CTLA-4 enhances local tissue responses and killing of Leishmania donovani. Eur J Immunol (2004) 34:1433–40. doi: 10.1002/eji.200324021
156. Snider H, Reville P, Sharpe A, Satoskar A. OX40L mediates susceptibility to chronic Leishmania donovani infection (40.26). J Immunol (2010) 184(1 Supplement):40.26.
157. Tuladhar R, Oghumu S, Dong R, Peterson A, Sharpe AH, Satoskar AR. Ox40L–Ox40 pathway plays distinct roles in regulating Th2 responses but does not determine outcome of cutaneous leishmaniasis caused by Leishmania mexicana and Leishmania major. Exp Parasitol (2015) 148:49–55. doi: 10.1016/j.exppara.2014.11.002
158. Ward-Kavanagh LK, Lin WW, Šedý JR, Ware CF. The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity (2016) 44:1005–19. doi: 10.1016/j.immuni.2016.04.019
159. So T, Ishii N. The TNF–TNFR Family of Co-signal Molecules. Adv Exp Med Biol (2019) 1189:53–84. doi: 10.1007/978-981-32-9717-3_3
160. Wakeley ME, Shubin NJ, Monaghan SF, Gray CC, Ayala A, Heffernan DS. Herpes Virus Entry Mediator (HVEM): A Novel Potential Mediator of Trauma-Induced Immunosuppression. J Surg Res (2020) 245:610–8. doi: 10.1016/j.jss.2019.07.009
161. Kennedy R, Klein U. A T Cell-B Cell Tumor-Suppressive Axis in the Germinal Center. Immunity (2019) 51:204–6. doi: 10.1016/j.immuni.2019.07.006
162. Herrero-Cervera A, Vinué Á, Burks DJ, González-Navarro H. Genetic inactivation of the LIGHT (TNFSF14) cytokine in mice restores glucose homeostasis and diminishes hepatic steatosis. Diabetologia (2019) 62:2143–57. doi: 10.1007/s00125-019-4962-6
163. Liu W, Garrett SC, Fedorov EV, Ramagopal UA, Garforth SJ, Bonanno JB, et al. Structural Basis of CD160:HVEM Recognition. Structure (2019) 27:1286–1295.e4. doi: 10.1016/j.str.2019.05.010
164. Rodriguez-Barbosa JI, Schneider P, Weigert A, Lee K-M, Kim T-J, Perez-Simon J-A, et al. HVEM, a cosignaling molecular switch, and its interactions with BTLA, CD160 and LIGHT. Cell Mol Immunol (2019) 16:679–82. doi: 10.1038/s41423-019-0241-1
165. El-Far M, Pellerin C, Pilote L, Fortin J-F, Lessard IAD, Peretz Y, et al. CD160 isoforms and regulation of CD4 and CD8 T-cell responses. J Transl Med (2014) 12:217. doi: 10.1186/s12967-014-0217-y
166. Cheung TC, Steinberg MW, Oborne LM, Macauley MG, Fukuyama S, Sanjo H, et al. Unconventional ligand activation of herpesvirus entry mediator signals cell survival. Proc Natl Acad Sci (2009) 106:6244–9. doi: 10.1073/pnas.0902115106
167. Hsu H, Solovyev I, Colombero A, Elliott R, Kelley M, Boyle WJ. ATAR, a novel tumor necrosis factor receptor family member, signals through TRAF2 and TRAF5. J Biol Chem (1997) 272:13471–4. doi: 10.1074/jbc.272.21.13471
168. Shui J-W, Larange A, Kim G, Vela JL, Zahner S, Cheroutre H, et al. HVEM signalling at mucosal barriers provides host defence against pathogenic bacteria. Nature (2012) 488:222–5. doi: 10.1038/nature11242
169. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol (2005) 6:90–8. doi: 10.1038/ni1144
170. Okwor I, Xu G, Tang H, Liang Y, Fu Y-X, Uzonna JE. Deficiency of CD40 Reveals an Important Role for LIGHT in Anti- Leishmania Immunity. J Immunol (2015) 195:194–202. doi: 10.4049/jimmunol.1401892
171. Stanley AC, de Labastida Rivera F, Haque A, Sheel M, Zhou Y, Amante FH, et al. Critical Roles for LIGHT and Its Receptors in Generating T Cell-Mediated Immunity during Leishmania donovani Infection. PloS Pathog (2011) 7:e1002279. doi: 10.1371/journal.ppat.1002279
172. Steinberg MW, Cheung TC, Ware CF. The signaling networks of the herpesvirus entry mediator (TNFRSF14) in immune regulation. Immunol Rev (2011) 244:169–87. doi: 10.1111/j.1600-065X.2011.01064.x
173. Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, et al. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity (1998) 8:21–30. doi: 10.1016/s1074-7613(00)80455-0
174. Wang J, Lo JC, Foster A, Yu P, Chen HM, Wang Y, et al. The regulation of T cell homeostasis and autoimmunity by T cell-derived LIGHT. J Clin Invest (2001) 108:1771–80. doi: 10.1172/JCI13827
175. Rooney IA, Butrovich KD, Glass AA, Borboroglu S, Benedict CA, Whitbeck JC, et al. The lymphotoxin-beta receptor is necessary and sufficient for LIGHT-mediated apoptosis of tumor cells. J Biol Chem (2000) 275:14307–15. doi: 10.1074/jbc.275.19.14307
176. Giles DA, Zahner S, Krause P, Van Der Gracht E, Riffelmacher T, Morris V, et al. The Tumor Necrosis Factor Superfamily Members TNFSF14 (LIGHT), Lymphotoxin β and Lymphotoxin β Receptor Interact to Regulate Intestinal Inflammation. Front Immunol (2018) 9:2585. doi: 10.3389/fimmu.2018.02585
177. Tumanov AV, Grivennikov SI, Shakhov AN, Rybtsov SA, Koroleva EP, Takeda J, et al. Dissecting the role of lymphotoxin in lymphoid organs by conditional targeting. Immunol Rev (2003) 195:106–16. doi: 10.1034/j.1600-065x.2003.00071.x
178. Gommerman JL, Browning JL. Lymphotoxin/light, lymphoid microenvironments and autoimmune disease. Nat Rev Immunol (2003) 3:642–55. doi: 10.1038/nri1151
179. Scheu S, Alferink J, Pötzel T, Barchet W, Kalinke U, Pfeffer K. Targeted disruption of LIGHT causes defects in costimulatory T cell activation and reveals cooperation with lymphotoxin beta in mesenteric lymph node genesis. J Exp Med (2002) 195:1613–24. doi: 10.1084/jem.20020215
180. Xu G, Liu D, Okwor I, Wang Y, Korner H, Kung SKP, et al. LIGHT Is critical for IL-12 production by dendritic cells, optimal CD4+ Th1 cell response, and resistance to Leishmania major. J Immunol (2007) 179:6901–9. doi: 10.4049/jimmunol.179.10.6901
181. Chakraborty S, Srivastava A, Jha MK, Nair A, Pandey SP, Srivastava N, et al. Inhibition of CD40-induced N-Ras activation reduces leishmania major infection. J Immunol (2015) 194:3852–60. doi: 10.4049/jimmunol.1401996
182. Cannons JL, Tangye SG, Schwartzberg PL. SLAM Family Receptors and SAP Adaptors in Immunity. Annu Rev Immunol (2011) 29:665–705. doi: 10.1146/annurev-immunol-030409-101302
183. McNerney ME, Lee K-M, Kumar V. 2B4 (CD244) is a non-MHC binding receptor with multiple functions on natural killer cells and CD8+ T cells. Mol Immunol (2005) 42:489–94. doi: 10.1016/J.MOLIMM.2004.07.032
184. Chijioke O, Marcenaro E, Moretta A, Capaul R, Münz C. Role of the 2B4 Receptor in CD8 + T-Cell-Dependent Immune Control of Epstein-Barr Virus Infection in Mice With Reconstituted Human Immune System Components. J Infect Dis (2015) 212:803–7. doi: 10.1093/infdis/jiv114
185. Georgoudaki A-M, Khodabandeh S, Puiac S, Persson CM, Larsson MK, Lind M, et al. CD244 is expressed on dendritic cells and regulates their functions. Immunol Cell Biol (2015) 93:581–90. doi: 10.1038/icb.2014.124
186. Pahima H, Puzzovio PG, Levi-Schaffer F. 2B4 and CD48: A powerful couple of the immune system. Clin Immunol (2019) 204:64–8. doi: 10.1016/j.clim.2018.10.014
187. Pombo C, Wherry EJ, Gostick E, Price DA, Betts MR. Elevated Expression of CD160 and 2B4 Defines a Cytolytic HIV-Specific CD8 + T-Cell Population in Elite Controllers. J Infect Dis (2015) 212:1376–86. doi: 10.1093/infdis/jiv226
188. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1-specific cell surface protein Tim-3 regulates macrophage activation and severity of an autoimmune disease. Nature (2002) 415:536–41. doi: 10.1038/415536a
189. Wolf Y, Anderson AC, Kuchroo VK. TIM3 comes of age as an inhibitory receptor. Nat Rev Immunol (2020) 20:173–85. doi: 10.1038/s41577-019-0224-6
190. Phong BL, Avery L, Sumpter TL, Gorman JV, Watkins SC, Colgan JD, et al. Tim-3 enhances FcϵRI-proximal signaling to modulate mast cell activation. J Exp Med (2015) 212:2289–304. doi: 10.1084/jem.20150388
191. Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, et al. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood (2012) 119:3734–43. doi: 10.1182/blood-2011-11-392951
192. Anderson AC, Anderson DE, Bregoli L, Hastings WD, Kassam N, Lei C, et al. Promotion of tissue inflammation by the immune receptor Tim-3 expressed on innate immune cells. Science (2007) 318:1141–3. doi: 10.1126/science.1148536
193. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM-3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PloS One (2012) 7:e30676. doi: 10.1371/journal.pone.0030676
194. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and transcriptional regulation of the co-inhibitory gene module in T cells. Nature (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
195. Rangachari M, Zhu C, Sakuishi K, Xiao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim-3–mediated cell death and exhaustion. Nat Med (2012) 18:1394–400. doi: 10.1038/nm.2871
196. Lee J, Su EW, Zhu C, Hainline S, Phuah J, Moroco JA, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol (2011) 31:3963–74. doi: 10.1128/MCB.05297-11
197. Clayton KL, Haaland MS, Douglas-Vail MB, Mujib S, Chew GM, Ndhlovu LC, et al. T cell Ig and mucin domain-containing protein 3 is recruited to the immune synapse, disrupts stable synapse formation, and associates with receptor phosphatases. J Immunol (2014) 192:782–91. doi: 10.4049/jimmunol.1302663
198. van de Weyer PS, Muehlfeit M, Klose C, Bonventre JV, Walz G, Kuehn EW. A highly conserved tyrosine of Tim-3 is phosphorylated upon stimulation by its ligand galectin-9. Biochem Biophys Res Commun (2006) 351:571–6. doi: 10.1016/J.BBRC.2006.10.079
199. Huang Y-H, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature (2015) 517:386–90. doi: 10.1038/nature13848
200. Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity (2016) 44:989–1004. doi: 10.1016/j.immuni.2016.05.001
201. Brockmann L, Soukou S, Steglich B, Czarnewski P, Zhao L, Wende S, et al. Molecular and functional heterogeneity of IL-10-producing CD4+ T cells. Nat Commun (2018) 9:5457. doi: 10.1038/s41467-018-07581-4
202. De Sousa Linhares A, Leitner J, Grabmeier-Pfistershammer K, Steinberger P. Not All Immune Checkpoints Are Created Equal. Front Immunol (2018) 9:1909. doi: 10.3389/fimmu.2018.01909
203. Dong Y, Li X, Zhang L, Zhu Q, Chen C, Bao J, et al. CD4+ T cell exhaustion revealed by high PD-1 and LAG-3 expression and the loss of helper T cell function in chronic hepatitis B. BMC Immunol (2019) 20:27. doi: 10.1186/s12865-019-0309-9
204. Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and Transcriptional Basis of CD4+ T Cell Dysfunction during Chronic Infection. Immunity (2014) 40:289–302. doi: 10.1016/j.immuni.2014.01.005
205. Rad Pour S, Morikawa H, Kiani NA, Yang M, Azimi A, Shafi G, et al. Exhaustion of CD4+ T-cells mediated by the Kynurenine Pathway in Melanoma. Sci Rep (2019) 9:12150. doi: 10.1038/s41598-019-48635-x
206. Flemming A. What’s driving T cell dysfunction? Nat Rev Immunol (2019) 19:199–9. doi: 10.1038/s41577-019-0152-5
207. Akinleye A, Rasool Z. Immune checkpoint inhibitors of PD-L1 as cancer therapeutics. J Hematol Oncol (2019) 12:92. doi: 10.1186/s13045-019-0779-5
208. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
209. Powles T, O’Donnell PH, Massard C, Arkenau H-T, Friedlander TW, Hoimes CJ, et al. Efficacy and Safety of Durvalumab in Locally Advanced or Metastatic Urothelial Carcinoma. JAMA Oncol (2017) 3:e172411. doi: 10.1001/jamaoncol.2017.2411
210. Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature (2014) 515:558–62. doi: 10.1038/nature13904
211. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N Engl J Med (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
212. Syn NL, Teng MWL, Mok TSK, Soo RA. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol (2017) 18:e731–41. doi: 10.1016/S1470-2045(17)30607-1
213. Lu R-M, Hwang Y-C, Liu I-J, Lee C-C, Tsai H-Z, Li H-J, et al. Development of therapeutic antibodies for the treatment of diseases. J BioMed Sci (2020) 27:1. doi: 10.1186/s12929-019-0592-z
Keywords: Leishmania, Leishmaniasis, Immune Checkpoints, Co-inhibitory Receptors, Immunotherapeutic
Citation: de Freitas e Silva R and von Stebut E (2021) Unraveling the Role of Immune Checkpoints in Leishmaniasis. Front. Immunol. 12:620144. doi: 10.3389/fimmu.2021.620144
Received: 22 October 2020; Accepted: 13 January 2021;
Published: 11 March 2021.
Edited by:
Heinrich Korner, University of Tasmania, AustraliaReviewed by:
Hira Nakhasi, Center for Biologics Evaluation and Research (FDA), United StatesDidier Soulat, University Hospital Erlangen, Germany
Copyright © 2021 de Freitas e Silva and von Stebut. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael de Freitas e Silva, cmFmYWVsLnNpbHZhQHVwZS5icg==