 Te-Sheng Lien1
Te-Sheng Lien1- 1Department of Molecular Biology and Human Genetics, Tzu-Chi University, Hualien, Taiwan
- 2Institute of Medical Sciences, Tzu-Chi University, Hualien, Taiwan
- 3Division of General Surgery, Department of Surgery, Hualien Tzu Chi Hospital, Buddhist Tzu Chi Medical Foundation, Hualien, Taiwan
- 4School of Medicine, Tzu Chi University, Hualien, Taiwan
Abnormal immune responses and cytokine storm are involved in the development of severe dengue, a life-threatening disease with high mortality. Dengue virus-induced neutrophil NETosis response is associated with cytokine storm; while the role of viral factors on the elicitation of excessive inflammation mains unclear. Here we found that treatments of dengue virus envelope protein domain III (EIII), cellular binding moiety of virion, is sufficient to induce neutrophil NETosis processes in vitro and in vivo. Challenges of EIII in inflammasome Nlrp3−/− and Casp1−/− mutant mice resulted in less inflammation and NETosis responses, as compared to the wild type controls. Blockages of EIII-neutrophil interaction using cell-binding competitive inhibitor or selective Nlrp3 inflammasome inhibitors OLT1177 and Z-WHED-FMK can suppress EIII-induced NETosis response. These results collectively suggest that Nlrp3 inflammsome is a molecular target for treating dengue-elicited inflammatory pathogenesis.
Introduction
Dengue is one the most important mosquito borne diseases in the tropical and subtropical areas of the world (1, 2), while specific treatments and effective vaccines are currently unavailable (3–8). Infections with dengue viruses (DENV) can lead to a wide range of clinical manifestations and disease severity. Severe dengue (also known as dengue hemorrhage fever, DHF) is characterized by plasma leakage and abnormal bleeding that can lead to shock and high mortality. Because DHF typically occurs during secondary infections with DENVs, abnormal adaptive immune responses are considered as part of the pathophysiology. For example, reports have suggested that antibody-dependent enhancement (9), original antigenic sin (10), autoantibody production (11) may be involved. However, detrimental innate immune responses such as excessive inflammation and cytokine storm are likely the critical pathological changes that lead to exacerbated disease, tissue injuries and ultimate death in DHF (9, 10, 12–15).
The mechanism underlying dengue-induced unregulated inflammation remains elusive (9, 10, 12, 13). In the innate immune system, neutrophils are first line of defense against infection through engulfment of microbes, secretion of anti-microbials and induction of neutrophil extracellular traps (NETs)-releasing cell death process termed NETosis (16, 17). NETs are extracellular DNA-protein complexed networks, which bind pathogens and modulate inflammation (16, 18). Pathogenic roles of NETosis have been found in non-infectious diseases, such as autoimmunity, coagulation, acute injuries and cancer (19). In addition, NETosis has been reported associating with cytokine storm in various infectious diseases, including dengue (14, 20, 21). Interleukin (IL)-1β, a potent proinflammatory cytokine released by DENV-infected leukocytes, has been considered as a critical component in cytokine storm (22–24). Inflammasomes, cellular sensors for pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs), are critical for IL-1 activation (14, 25), and the cell death process pyroptosis (26). Reports revealed that the elevated levels of circulating IL-1β and gene expression in DHF patients suggesting the involvement of IL-1β in the disease severity (27, 28). IL-1β enhances the vascular permeability, particularly in association with other proinflammatory cytokines such as tumor necrosis factor (TNF)-α and interferon in clinical profiles of DF and DHF (14, 29–31).
In our previous reports, in the two-hit model, we found that sequential injections of DENV (32) or DENV-envelope protein domain III (rEIII) (33) (1st hits) plus anti-DENV non-structural protein NS1 antibody (anti-NS1 Ig; 2nd hit) to simulate the disease progression of DHF, induced hemorrhage pathogenesis recapitulate certain disease-signatures of DHF, including thrombocytopenia, plasma leakage, vascular injury, hemorrhage, liver damage, and high mortality (32, 33). In addition to these manifestations, we also found that circulating proinflammatory cytokine levels such as TNF, IL-1, and IL-6, are greatly increased (32, 33). Intriguingly, IL-1 receptor antagonist (IL-1RA) treatments greatly ameliorated such 2-hit induced pathogenesis (32, 33). In addition, Nlrp3 deficiencies as observed in the Nlrp3−/− and Casp1−/− mutant mice, also greatly reduced 2-hit induced pathogenesis (32, 33). These results collectively suggested that Nlrp3 inflammasome-IL-1 axis is involved in dengue induced pathogenesis in this DENV-, and EIII-induced hemorrhage mouse models.
The DENV viral factor that contributes to NETosis remains unclear. Plasma EIII levels could be detected in acute DENV infection (34). Evidences have shown that EIII treatments induced inflammasome activation and inflammation of macrophages (35). Our previous study revealed that challenges with the DHF-viral-load-equivalent levels of EIII can suppress megakaryocyte, and endothelial cell function through initiating cell death (33, 36). Accordingly, EIII may be a cytotoxic virulence factor of DENV to cause NETosis and initiate downstream inflammation. As a result, in this present study, we would like to investigate whether DENV and EIII can directly initiate NETosis, and whether Nlrp3 inflammasome is involved. In addition, whether we can ameliorate EIII-mediated inflammation through suppression of Nlrp3 inflammasome and NETosis pathways is also addressed. Relevant implications and applications are discussed.
Materials and Methods
DENV, Recombinant Protein, and Antibodies
Mosquito C6/36 cell line (ATCC CRL-1660) and DENV-2 (PL046) were maintained and amplified using standard cell culture methods (36–38). Soluble recombinant proteins glutathione-S transferase (rGST), and EIII (rEIII) were obtained from cultured bacteria (Escherichia coli) (39), after isopropyl β-D-1-thiogalactopyranoside induction, and were purified as previously described (32, 33, 36). To reduce endotoxin (lipopolysaccharide; LPS) contamination to a desired level (<1 EU/mg protein), the lysate- and resin-packed column was washed with a buffer (8 M urea, 100 mM NaH2PO4, and 10 mM Tris-HCl; pH = 6.3) with the addition of 1% Triton X-114 (Sigma–Aldrich, St. Louis, MO, USA). The rEIII was eluted with a buffer (8 M urea, 100 mM NaH2PO4, and 10 mM Tris-HCl; pH = 4.5) and refolded using a linear 4–0 M urea gradient in a dialysis buffer (2 mM reduced glutathione, 0.2 mM oxidized glutathione, 80 mM glycine, 1 mM EDTA, 50 mM Tris-HCl, 50 mM NaCl, and 0.1 mM phenylmethylsulfonyl fluoride) at 4°C for 2–3 h, as previously described (36). The purity of the rEIII protein can reach ~90%. The LPS contamination was monitored with a Limulus Amoebocyte Lysate QCL-1000 kit (Lonza, Walkersville, MD, USA) (36, 37, 40). Batches of purified recombinant proteins with an LPS contamination level of <1 EU/mg of protein were used. The pre-immune control Ig, anti-NS1 Ig, and anti-EIII Ig from experimental rabbits (New Zealand White; Oryctolagus cuniculus) were obtained before and after rNS1- and rEIII-immunizations according to previously described methods (41). According to previously described methods (33), recombinant proteins (50 μg/mL) were used to block rEIII-cell (neutrophil) binding, including recombinant mouse dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN; CD209), DC-SIGNR, C-type lectin domain family 5 member A (CLEC5A) and CLEC2 (R&D Systems, Indianapolis, IN, USA). To analyze the binding properties of rEIII proteins on protein-coated beads (latex, 1.1 μm, Sigma-Aldrich) and mouse neutrophils, rEIII protein were conjugated with biotin by using an EZ-Link™ Sulfo-NHS-Biotinylation kit (Thermo Fisher Scientific). The rEIII-beads binding experiments were performed using biotin-labeled rEIII proteins (300 μg/mL, 20 μL) incubated with protein-precoated beads [2 μg protein coating with 1 mg/mL beads 1 h in total 50 μL phosphate buffered saline PBS, blocking with 1% bovine serum albumin (BSA, Sigma-Aldrich)/PBS, 30 min, resuspended in 20 μL PBS after wash]. The levels of biotin-labeled rEIII proteins bound to beads or neutrophils [50 μg/mL competing protein + (2 × 105) cells/mL in culture medium for 30 min] were determined through flow cytometry by PE/Cy5 avidin (Biolegend, San Diego, CA, USA) staining. DENV-2 envelope protein fragment (32 kDa, domain I + domain II; ProSpec-Tany TechnoGene, Ness-Ziona, Israel) was used as a control protein. The rEIII-competitive inhibitor chondroitin sulfate B (CSB, 10 μg/mL; Sigma-Aldrich) was used to suppress rEIII-induced neutrophil binding and cell death. Anti-citrullinated histone H3 (CitH3; citrulline R8), anti-histone H2A family member X (H2AX), and anti-gasdermin D (GSDMD) antibodies (Abcam, Cambridge, UK) were used for flow cytometry NETosis analysis.
Experimental Mice
Wild-type mice of ages 8–12 weeks in C57BL/6J background were purchased from the National Laboratory Animal Center (Taipei, Taiwan) (38, 42–46). Gene knockout mice with a C57BL/6J background, including Nlrp3−/− and Casp1−/− (32), were obtained from the Center National de Recherche Scientifique (Orléans, France) (32, 33, 47). All experimental animals were housed in the Animal Center of Tzu-Chi University in a specific-pathogen-free, temperature-, and lighting-controlled environment with free access to filtered water and food. All genetic knockout strains were backcrossed with the wild-type C57Bl/6J mice for at least 6 generations. After challenged with vehicle (saline), BSA (a control protein, 2 mg/kg), DENV (1.2 × 107 PFU /kg; DHF viral load) and rEIII (2 mg/kg; a dosage equivalent to 1.2 × 107 PFU/kg), experimental mice were immediately rescued with or without Nlrp3 inhibitor OLT1177 treatments (50 mg/kg). Plasma levels of IL-1β, TNF-α, and CitH3 of the experimental mice were determined through enzyme-linked immunosorbent assay (ELISA) (IL-1β, TNF-α, Biolegend; CitH3, Cayman Chemical, Ann Arbor, MI, USA) 1 d after rEIII treatments; neutrophils were isolated and analyzed (see following “Analyses of neutrophils”).
Ethics Statement
The animal experiments in this report were conducted in agreement with National (Taiwan Animal Protection Act, 2008) directive for protection of laboratory animals. All experimental protocols for examining the experimental animals were approved by the Animal Care and Use Committee of Tzu-Chi University, Hualien, Taiwan (approval ID: 101019).
Analyses of Neutrophils
Blood samples of mice were collected via the retro-orbital venous plexus using plain capillary tubes (Thermo Fisher Scientific, Waltham, MA, USA), and then transferred into polypropylene tubes (Eppendorf; Thermo Fisher Scientific) containing anticoagulant acid-citrate-dextrose solution (ACD; 38 mM citric acid, 75 mM sodium citrate, 100 mM dextrose) (48, 49). Following previously described methods (50), mouse neutrophils were purified from mouse blood samples using Ficoll-Paque (Ficoll-Paque Plus, 1.077 g/mL, GE Healthcare, Chicago, IL, USA) and dextran (Sigma-Aldrich) sedimentation (3% w/v) density gradient centrifugation and red blood cell lysis. To obtain fluorescent NET images, mouse neutrophils (1 × 105) were treated with vehicle (the diluent, normal saline, 0.9 % NaCl), rEIII (50 μg/mL or an equivalent dose 0.6 μM), DENV (1 × 105 PFU/mL, an equivalent dose of rEIII is 0.6 μM) or 12-O-tetradecanoylphorbol-13-acetate (TPA, 2 nM, Sigma-Aldrich) for 2 h at 37C. After fixation by 4% paraformaldehyde on coverslips, these neutrophils were then stained with rabbit anti-mouse citrulline Histone H3 antibody (1:1000) and 4',6-diamidino-2-phenylindole (DAPI, 5 μl/ml). A fluorescence microscope (Nikon Eclipse E800; Nikon Taiwan, Taipei, Taiwan) (51) was used for obtaining the NETosis images. To analyze mitochondria membrane potential, superoxide, and membrane potential levels, mitochondria labeling reagents MitoTracker™ Green FM (Thermo Fisher Scientific, Waltham, MA, USA), MitoSOX™ Red mitochondrial superoxide indicator (Thermo Fisher Scientific), MitoTracker™ Red CMXRos (Thermo Fisher Scientific) were used according to the manufacturer's instructions (33, 52). A flow cytometer (FACSCalibur; BD Biosciences, San Jose, CA, USA) (36, 38) was used in this study to analyze RCD, cell live/death and mitochondria metabolic activities with or without rEIII challenges and RCD or signaling pathway (e.g., PAD4, Nlrp3 inflammasome) inhibitor treatments (33). Levels of cellular reactive oxygen species (ROS) were analyzed using 2′,7′-dichlorofluorescin diacetate (Sigma-Aldrich) staining-flow cytometry analysis.
Analyses of Regulated Cell Death
To analyze DENV or rEIII induced neutrophil cell death, mouse neutrophils were incubated with DENV or rEIII for 1 h and then subjected to flow cytometry analyses after washed with PBS. Various regulated cell death (RCD) responses, including apoptosis (CaspGLOWTM Red Active Caspase-3 Staining Kit, #K193, BioVision, Milpitas, CA, USA), autophagy (Cyto-ID™ Autophagy Detection Kit, Enzo Life Sciences, #ENZ51031, Farmingdale, NY, USA), ferroptosis (C11 BODIPY 581/591, #27086, Cayman Chemical), necroptosis (RIP3/B-2 alexa Fluor 488, Santa Cruz Biotechnology, #sc-374639 AF488, Santa Cruz, CA, USA), pyroptosis (Caspase-1 Assay, Green, #9146, ImmunoChemistry Technologies, MI, USA), and live/dead cell labeling (Zombie NIR™ Fixable Viability Kit, #423105, Biolegend), were analyzed using respective cell labeling reagents (30 min in PBS). Treatments (1 h) of cell death inducers were used as positive controls for various type of regulated cell death (RCD; apoptosis: doxorubicin, 2.5 μg/mL, Nang Kuang Pharmaceutical, Taipei, Taiwan; autophagy: rapamycin, 250 nM, #R0395, Sigma-Aldrich; ferroptosis: erastin, 10 μM, #17754, Cayman Chemical; necroptosis, TNF-α, 2 ng/mL, #575202, Biolegend; pyroptosis: nigericin, 3.5 μM, #6698, ImmunoChemistry Technologies, Minnesota, USA; NETosis, TPA, #P8139, 2 nM Sigma-Aldrich) (30 min in PBS). Inhibitors were used to address the involvements of specific RCD pathways (apoptosis: Z-DEVD-FMK, 10 μM, #FMK004, R&D Systems, Indianapolis, IN, USA; autophagy: Chloroquine diphosphate, 60 μM, #C6628, Sigma-Aldrich; ferroptosis: Ferrostatin-1 2.5 μM, #17729, Cayman Chemical; necroptosis: Necrostatin-1, 50μM, #11658, Cayman Chemical; pyroptosis: Z-WHED-FMK 10 μM, #FMK002, R&D Systems; pyroptosis/GSDMD: dimethyl fumarate (DMF), 25–100 μM, #14714, Sigma–Aldrich; NETosis: peptidyl arginine deiminase 4 (PAD4) inhibitor GSK484, 10 μM, #17488, Sigma–Aldrich; Nlrp3: OLT1177, 10 μM, #24671, Cayman Chemical; EIII competitive blocker: Chondroitin sulfate B, CSB, 10 μg/mL, #C3788, Sigma–Aldrich; 30 min pretreatments before addition of DENV, rEIII and cell-death inducers according to the manufacturer's instructions).
Statistical Analyses
In this report, the means, standard deviations, and statistics of the quantifiable data were calculated using the SigmaPlot 10 and SPSS 17 software packages. Significance of the data was examined using one-way ANOVA, followed by the post hoc Bonferroni-corrected t-test. The probability of type-1 error α = 0.05 was recognized as the threshold of statistical significance.
Results
Nlrp3 Inflammasome Is Involved in rEIII Induced Neutrophil NETosis
Flow cytometry analysis of NETosis markers citrullinated histone H3 (CitH3) (53, 54) and histone H2A family member X (H2AX) (55, 56) revealed that rEIII is sufficient to initiate NETosis in neutrophils; the potency of rEIII is comparable to the classical NETosis inducer phorbol ester (18, 57) (Figure 1A, Supplementary Figure 1, flow cytometry gating; Figure 1B). In addition, such induction of NETosis can be suppressed by treatments of inflammasome/caspase 1 inhibitor Z-WHED-FMK (Figure 1). In agreement with this, we found that, when compared to the neutrophils from wild type mice, Nlrp3 inflammasome-deficient (Nlrp3−/− and Casp1−/−) neutrophils displayed relatively low NETosis levels after treated with rEIII and TPA (Figures 2A–C, cell images; green channels: CitH3, a NETosis marker; Figure 2D, quantified results). These data suggest that Nlrp3 inflammasome plays critical role in rEIII-induced NETosis.
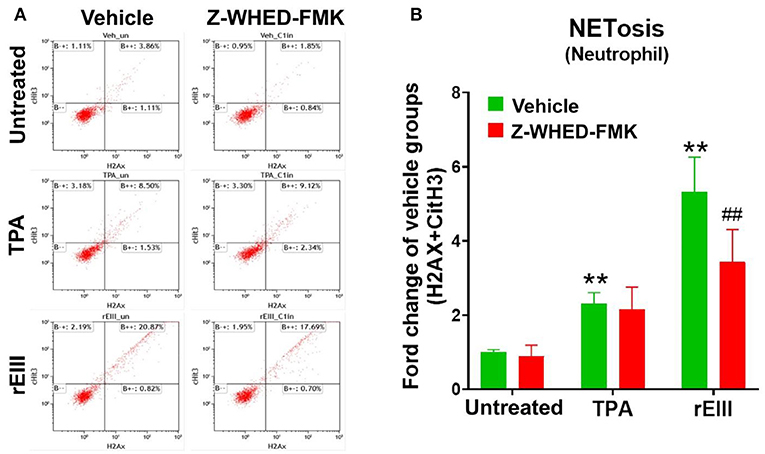
Figure 1. Essential role of caspase 1 in DENV rEIII-induced NETosis. The gating of flow cytometry analysis of vehicle, 12-O-tetradecanoylphorbol-13-acetate (TPA, a positive control NETosis inducer; 2 nM) and DENV rEIII (0.6 μM) challenged (1 h) wild mice neutrophils (1 × 105) with or without caspase 1 inhibitor Z-WHED-FMK pretreatments (30 min) (A). The quantified results of flow cytometry analysis, which reveal that TPA and rEIII treatments markedly induced NETosis formation; by contrast, caspase 1 inhibitor Z-WHED-FMK treatments can only considerably suppress rEIII-induced NETosis, but not TPA-induced NETosis (B). n = 6, **P < 0.01, vs. untreated controls; ##P < 0.01 vs. vehicle groups.
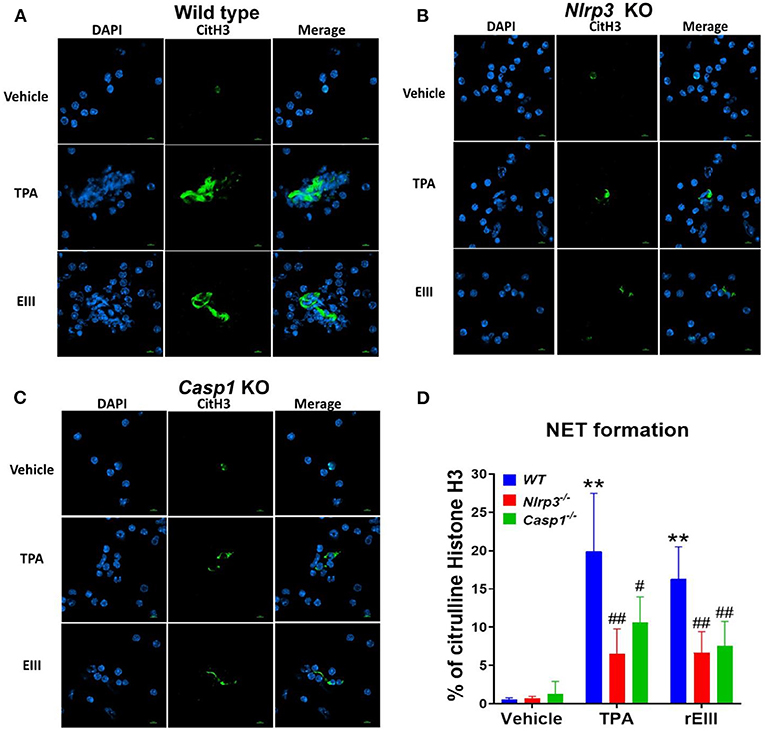
Figure 2. Essential role of Nlrp3 inflammasome in DENV rEIII-induced NETosis. After challenged (1 h) by TPA (2 nM) and DENV rEIII (0.6 μM), the images of DAPI (nucleus DNA staining) and citrullinated histone H3 (CitH3; NETosis marker) staining of NETosis formation of neutrophils from wild type (A), Nlrp3 null (Nlrp3−/−) (B), and caspase 1 null (Casp1−/−) (C) mouse were showed. The quantified results from flow cytometry are also indicted (D). n = 6, ** P < 0.01, vs. vehicle controls; #P < 0.05, ##P < 0.01 vs. wild type (WT) groups. Scale bars: 5 μm.
DENV and rEIII Induce Multiple Regulated Cell Death Pathways of Neutrophils
NETosis is a type of regulated cell death (RCD) (58). Previous reports suggested that neutrophil RCD exacerbate pathogenesis in infectious diseases (59, 60). As a result, we would like to investigate whether various RCD pathways are also involved in DENV- and rEIII-induced neutrophil cell death.
We first found that DENV and rEIII induced neutrophil cell death in a dose dependent manner (Figure 3A). Overall, various cell death inducers, including doxorubicin (apoptosis) (61, 62), rapamycin (autophagy) (63), erastin (ferroptosis) (64), TNF-α (necroptosis) (65, 66), nigericin (pyroptosis) (67), served as positive control agents to induce respective RCD pathways of the tested neutrophils (Figures 3B,C, dead cell population adjusted to 100%; Supplementary Figure 2, flow cytometry gating and calculation). Notably, when compared with cell death agonists, DENV and rEIII treatment induced considerable pyroptosis, necroptosis, autophagy and NETosis responses in the neutrophils, while only minor or no ferroptosis and apoptosis levels (Figure 3B, % of total cells; Figure 3C, % of total dead cell). In addition, the cell type specific RCD patterns/profiles (CTS-RCDPs) (33) of neutrophil in the DENV-, and rEIII-treated groups were somewhat similar, with pyroptosis exhibiting the highest levels in both groups among all tested RCD pathways (Figure 3B; ~40%), suggesting that DENV-induced CTS-RCDP in the neutrophils is likely mediated through EIII on the DENV virion. In case one dead cell may display multiple RCDs, here we defined CTS-RCD as a detection ratio of RCDs in 1 cell type at a specific condition.
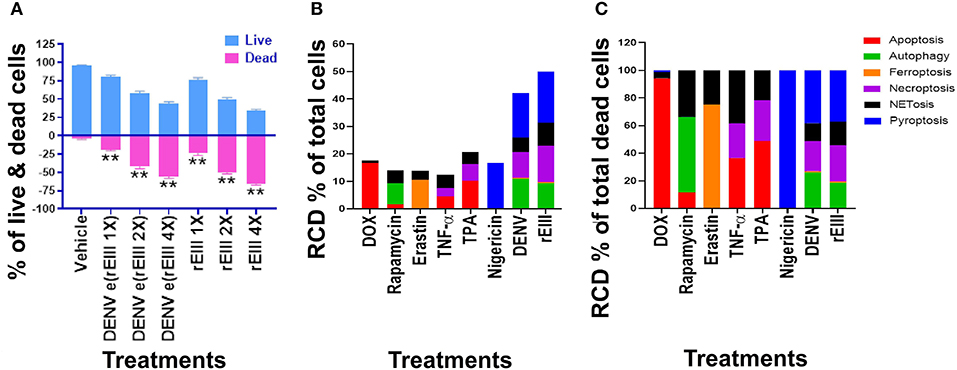
Figure 3. DENV- and rEIII-induced regulated cell death in neutrophils. (A) Wild type mouse neutrophils treated (1 h) with vehicle and various doses of DENV and rEIII; the live and death cell populations were revealed by Zombie-NIR Kit labeling and flow cytometry analysis. [rEIII 1× = 0.3 μM, 2× = 0.6 μM, 4× = 1.2 μM,; DENV e(rEIII 1×) is a DENV level equivalent to 0.3 μM rEIII, as indicted by the methods described elsewhere (33)]. (B) Treatments (1 h) of regulated cell death (RCD) inducers, doxorubicin (DOX; apoptosis) (2.5 μg/mL), rapamycin (autophagy) (0.5 μM), erastin (ferroptosis) (10 μM), TNF-α (necroptosis) (2.5 ng/mL), TPA (NETosis) (2 nM), and nigericin (pyroptosis) (3.5 μM) induced relatively simple RCD patterns. By contrast, DENV and rEIII induced multiple RCD pathways, in which pyroptosis is the major RCD response, counts ~40% of total RCD and the NETosis response displays only ~20% total RCD. (C) If the respective RCDs are normalized by the population of death cells (dead cell population normalized to 100%), we can obtain a more similar RCD pattern in DENV and rEIII groups (flow cytometry gating and calculation methods described in Supplementary Figure 2). DENV 1× = 4.2 × 104 PFU/mL, 2× = 8.4 × 104 PFU/mL, 4× = 1.7 × 105 PFU/mL; **P < 0.01 vs. vehicle groups.
An unexpected finding is that the DENV and rEIII induced NETosis only displayed approximately 20% of total RCDs (Figure 3B); and a classical NETosis inducer TPA (a phorbol ester) also induced NETosis about only 40% of total RCDs (Figure 3B, TPA groups). This let us wondered whether DENV and EIII-mediated induction of such a low percentage of NETosis in total RCD, could sufficiently lead to neutrophil dysfunction. In addition, we would like to investigate whether Nlrp3 inflammasome is involved in rEIII-induced neutrophil death. Accordingly, Nlrp3 inhibitor OLT1177 and inflammasome/caspase1 inhibitor Z-WHED-FMK were used to further characterizations of whether Nlrp3 inflammasome is involved in respective RCD responses.
We found that treatments with inflammasome inhibitors OLT1177 and Z-WHED-FMK both suppressed EIII-induced neutrophil cell death (Figures 4A,B; Supplementary Figure 3, percentage pie charts of OLT1177 and Z-WHED-FMK treatments; equivalent to some data in Figures 4A,B,I). In addition, OLT1177 and Z-WHED-FMK suppressed pyroptosis (Figures 4C,J), necroptosis (Figures 4D,K), autophagy (Figures 4G,N), and NETosis (Figures 4H,O), with (Figures 4I–O) or without (Figures 4B–H) normalization of dead cell population. These results suggested that pyroptosis is the major RCD of rEIII-induced neutrophil death, and which can be rescued by selective inhibitors against Nlrp3 inflammasome. In addition, Nlrp3 likely involved in neutrophil NETosis as treatments of selective inhibitor OLT1177 rescued total neutrophil death (Figure 4A) and NETosis (Figures 4H,O). Consistently, treatments of pyroptosis/GSDMD inhibitor DMF (68) suppressed EIII-induced neutrophil pyroptosis, NETosis, cellular ROS, surface GSDMD levels (Supplementary Figure 4, flow cytometry analyses), and caspase-1 activation (Supplementary Figure 5, colorimetric assay). Furthermore, despite NETosis displayed only ~20% cell death (Figures 4B,I), PAD4 inhibitor GSK484 suppressed considerable levels of total neutrophil death (Figure 4A) and RCDs including pyroptosis, necroptosis, autophagy, and NETosis (Figure 4). These evidences collectively suggesting that that RCDs pyroptosis, necroptosis, and autophagy are likely associated to NETosis, and NETosis is still a critical RCD pathway of neutrophil in response to rEIII exposure.
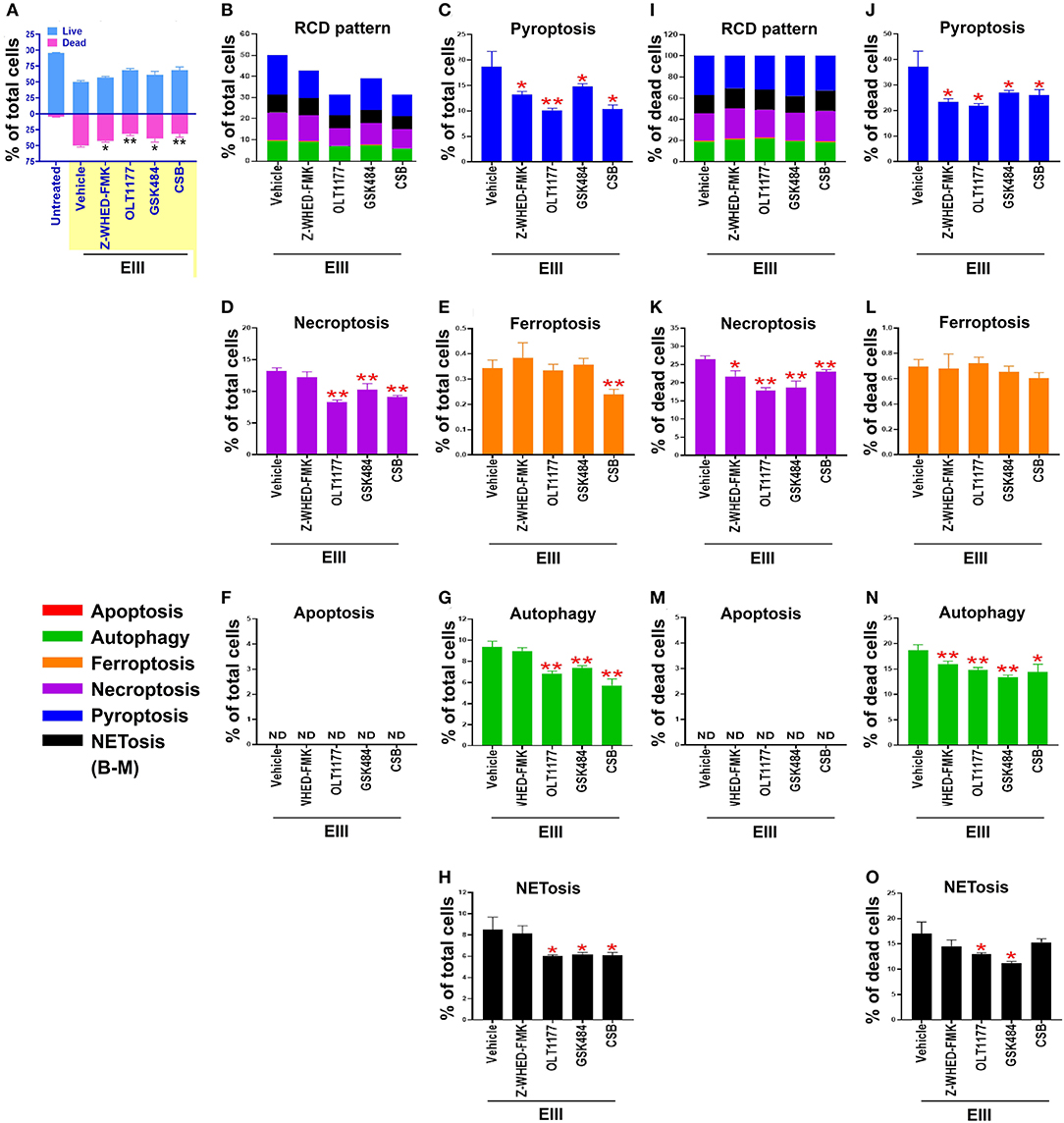
Figure 4. Protection of neutrophils from rEIII-induced pyroptosis under treatment with Nlrp3 inflammasome inhibitors. Pre-treatments (30 min) with NETosis inhibitor GSK484 (2 nM), Nlrp3 inhibitor OLT1177 (10 μM), and caspase 1 inhibitor Z-WHED-FMK (10 μM) on the rescue of rEIII-induced neutrophil total cell death (A). Pretreatments (30 min) with Nlrp3 inflammasome inhibitors OLT1177, Z-WHED-FMK, and NETosis inhibitor GSK484 rescued rEIII-induced (1 h) neutrophil pyroptosis (B,C), necroptosis (D), autophagy (G) and NETosis (H), but not ferroptosis and apoptosis (E,F). If the respective RCD% was normalized by the population of death cells [(I): dead cell population normalized to 100%], we found that CSB, OLT1177 and Z-WHED-FMK still display rescue effects on pyroptosis (J), necroptosis (K), autophagy (N), and NETosis (O), but not ferroptosis and apoptosis (L,M). Chondroitin sulfate B (CSB, 10 μg/mL), a competitive inhibitor against rEIII binding (33), serving as a positive inhibitor control to inhibit cell death. n = 6, *P < 0.05, **P < 0.01, vs. respective vehicle groups. ND, not detected.
Nlrp3 Inflammasome Deficiency and Inhibitor Treatments Rescue DENV- and rEIII- Exacerbated Mitochondria Metabolic Burden and Inflammation of Neutrophils
Because inflammasome-mediated pyroptosis is a major RCD involving in rEIII-induced neutrophil defect, here we would like to further investigate whether suppression of neutrophil Nlrp3 inflammasome through inhibitor treatments is sufficient to ameliorate DENV rEIII-induced neutrophil defects. Here we found that treatments of rEIII-increased mitochondria mass (Figure 5A), membrane potential (Figure 5C) and superoxide (Figure 5E) levels in a dose dependent manner, while treatments of Nlrp3 inflammasome inhibitors OLT1177 and Z-WHED-FMK ameliorated such metabolic burden of neutrophil mitochondria (Figures 5B,D,F). Levels of caspase-1 activation in the neutrophils were analyzed and confirmed in parallel using colorimetric assay (Supplementary Figure 5). In agreement with this, mouse experiments further revealed that, treatments of Nlrp3 inflammasome inhibitors OLT1177 markedly ameliorated rEIII- induced elevation of circulating soluble CitH3 (Figure 6A), IL-1β (Figure 6B), and TNF-α (Figure 6C) levels in mice. Consistently, compared to wild type mice, neutrophils from Nlrp3−/− and Casp1−/− mutant mice displayed markedly reduced levels of total mitochondria ROS (Figure 7A), hydrogen peroxide (Figure 7B), superoxide (Figure 7C) after in vitro treatments of rEIII. Similarly, rEIII treatments markedly induced circulating CitH3 in wild type mice, but not in Nlrp3−/− and Casp1−/− mutant mice (Figure 7D). These results collective suggest that EIII is a virulence factor to induce neutrophil defects, and Nlrp3 inflammasome is a critical target for DENV and EIII to induce neutrophil dysfunction, NETosis, and inflammation.
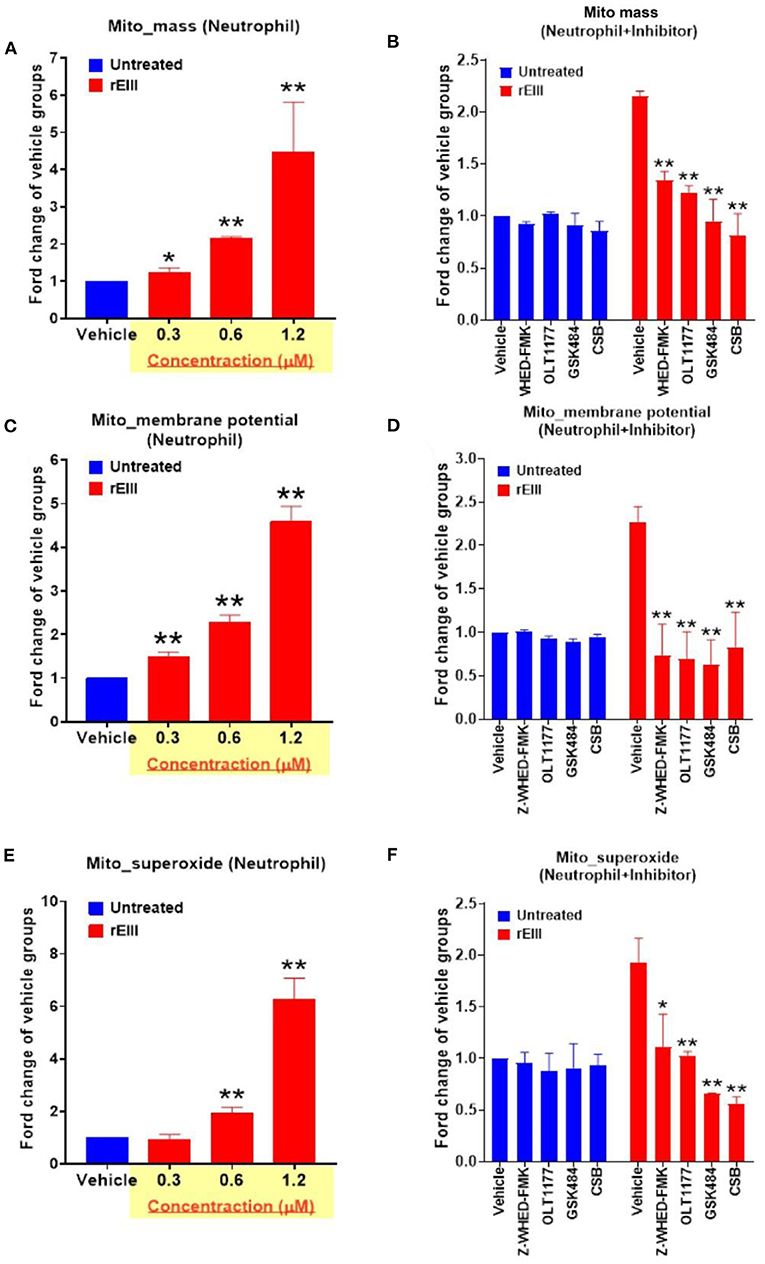
Figure 5. DENV rEIII-induced mitochondria ROS production is ameliorated by Nlrp3 inflammasome and NETosis inhibitor. The rEIII (0.6 μM)-induced (1 h) elevations of neutrophil mitochondria mass (A) membrane potential (C) and superoxide (E) levels in a dose dependent manner. The induction of these mitochondria metabolic burdens in the neutrophils could be suppressed by treatments of Nlrp3 and caspase 1 inhibitors Z-WHED-FMK (10 μM) and OLT1177 (10 μM), NETosis inhibitor GSK484 (10 μM), and the cell-binding competitive inhibitor chondroitin sulfate B (CSB, 10 μg/mL), respectively (B,D,F). n = 6, *P < 0.05, **P < 0.01, vs. respective untreated vehicle groups (A,C,E); *P < 0.05, **P < 0.01, vs. respective vehicle groups (B,D,F).
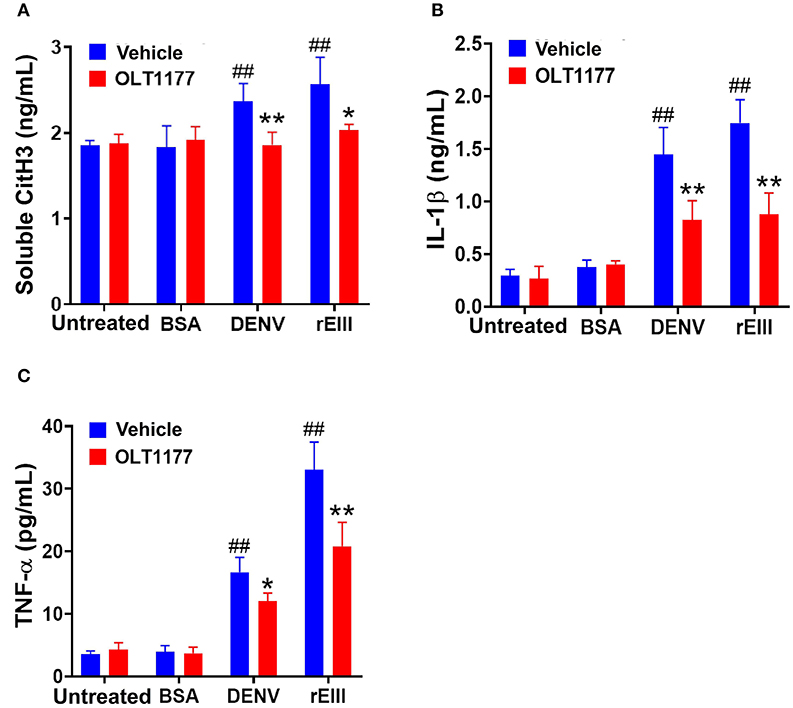
Figure 6. Nlrp3 inhibitor OLT1177 protects mice from DENV- and rEIII-induced NETosis and IL-1β, TNF-α production in mice. After challenged with vehicle (saline), BSA (a control protein) 2 mg/kg), DENV (1.2 × 107 PFU /kg; DHF viral load) and rEIII (2 mg/kg; a dosage equivalent to DENV 1.2 × 107 PFU/kg), experimental mice were immediately rescued with or without Nlrp3 inhibitor OLT1177 treatments (50 mg/kg). The ELISA analyzed plasma levels of circulating soluble citrullinated histone H3 (CitH3; a NETosis marker) (A), circulating IL-1β levels (B), and TNF-α levels (C) after 1 d treatments were showed. n = 6, ##P < 0.01, vs. respective untreated groups; *P < 0.05, **P < 0.01, vs. respective vehicle groups.
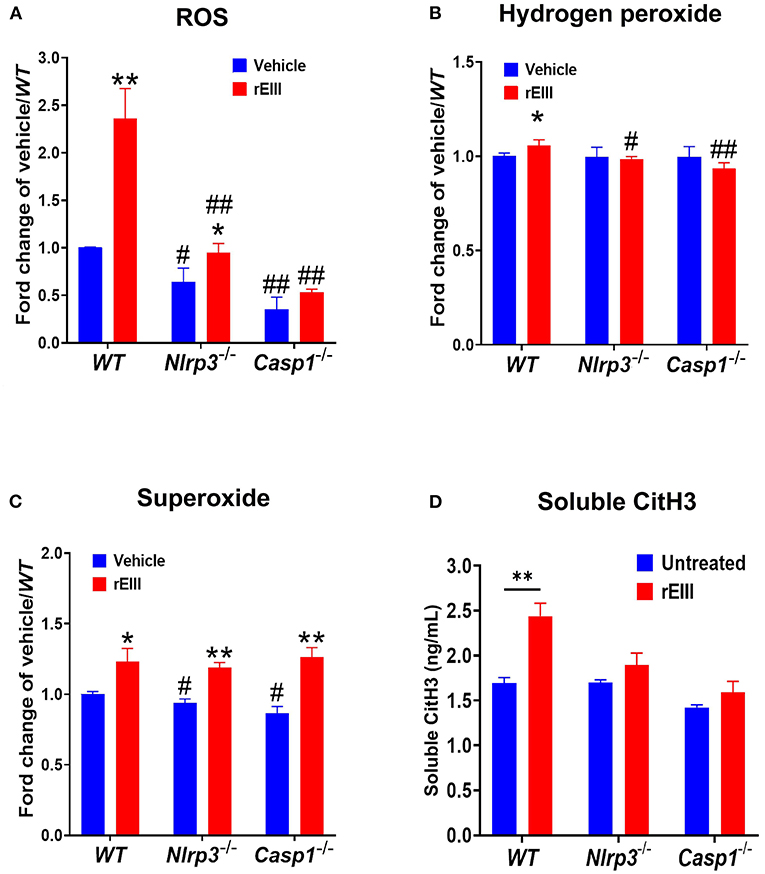
Figure 7. Nlrp3 and caspase 1 deficiencies protest rEIII-induced neutrophil ROS production and NETosis. Treatments (1 h) of rEIII (0.6 μM)-induced elevations of neutrophil cellular reactive oxygen species (ROS) (A) hydrogen peroxide (B), superoxide (C) in vitro. Treatments (1 d) of rEIII (2 mg/kg)-induced elevations of circulating CitH3 (D) levels in wild type mice. By contrast, such rEIII (0.6 μM in vitro, 2 mg/kg in mice)-induced stimulations are markedly reduced in Nlrp3−/− and Casp1−/− gene knockout mice in both in vitro and in vivo experiments (A–D). n = 6, #P < 0.05, ##P < 0.01, vs. respective wild type (WT) groups; *P < 0.05, **P < 0.01, vs. respective vehicle groups.
Discussion
Inflammasomes, cellular molecular sensors for PAMPs and DAMPs, are multimeric protein complexes comprising of NLRs [nucleotide-binding domain (NBD) and leucine-rich-repeat-(LRR)-containing], the absent in melanoma-2 (AIM2)-like receptors (ALRs), an adaptor molecule ASC (apoptosis-associated speck-like protein containing a CARD), and procaspase-1 (14, 25, 69, 70). Activation of inflammasomes leads to protein-cleavage processes, turning pro-caspase 1 into active form caspase 1, which converts pro-IL-1β and pro-IL-18 into respective active forms (IL-1β and IL-18) (69). Uncontrolled inflammasome activation exacerbates autoimmune and excessive inflammatory pathogenesis in infectious diseases, despite the adequate levels of inflammasome activation can help defending the pathogens (71). As a result, selective inhibitors against inflammasomes have been developed for treating various inflammatory and infectious diseases, and have got promising advancements (70, 72). High circulating levels of IL-1β and IL-18 have been associated DHF (27, 73), suggesting a critical role of inflammasomes in DENV-induced pathogenesis. Mouse experiments further revealed that DENV induced NET formation and inflammasome activation was impaired in neutrophils from CLEC5A−/− and TLR2−/− mutants (74). DENV-induced NET leads to endothelial cell damage and vascular leakage. Evidences have suggested that NET formation contributes significantly in disease pathogenesis, and NET components were more predominantly displayed in serum samples of DHF patients (75). Despite of these findings, the mechanism underlining DENV-induced NETosis formation is not fully understood; particularly, the viral factor leading to NETosis remains elusive. Here we found that, the DENV EIII could be a potential virulence fact that elicits abnormal neutrophil responses and NETosis. Because DENV and EIII induced similar RCD patterns of neutrophils (Figure 3B, DENV and rEIII groups), suggesting that, during the speak viremia stage, high levels of DENV virion conduct the cytotoxicity to enhance NETosis through EIII moiety. This is partly consistent with the findings that NET formation is markedly increased in neutrophils isolated from dengue patients during the acute phase of the infection (75). Additionally, our data suggests that such cell-type-specific RCD patterns may be a useful analysis method on the functional characterization of biologically drugs and hazardous materials at the cellular levels.
Inflammasomes regulate and interact with various RCDs (72, 76–78). The inhibitors of either Nlrp3 inflammasome/pyroptosis (inhibitor OLT1177) or NETosis (inhibitor GSK484) can suppress to each other (Figure 4), suggests there could exist a cross-talk between these 2 pathways. First, such cross-talk could be regulated at an intracellular signaling level; for example, GSDMD, an effector protein of pyroptosis, plays a critical role in the generation of NET (79). Consistently, here we found that treatments of GSDMD inhibitor DMF drastically reduced EIII-induced pyroptosis and NETosis in neutrophils (Supplementary Figure 4). Accordingly, if Nlrp3 inflammasome-activated GSDMD can further enhance NET formation, it is reasonable to observe an inhibition of NETosis using inhibitors from a pyroptosis pathway. Second, the cross-talk may also explain by a model of intercellular regulations between macrophage and neutrophil. For example, the NET is able to prime macrophages to produce IL-1 and IL-18 through the Nlrp3 inflammasome, thus amplifying the inflammatory response (80). At the same time, IL-18 effectively stimulated NET release and caspase-1 activation in primed macrophages compared to IL-18 alone (80). This suggests a feed-forward loop that NET increase the production of IL-1β and IL-18 in macrophages, which in turn can stimulate NET formation in neutrophils (80). These evidences may explain our observations that treatments of Nlrp3 inhibitor OLT1177 can markedly suppressed rEIII-induced neutrophil pyroptosis and NETosis in vitro (Figure 4), and markedly suppressed rEIII-induced NETosis and inflammation in mice (Figure 6). However, as these regulations still not been clearly demonstrated in dengue, more detailed mechanism is worth of further investigations.
As a glycosaminoglycan (GAG) binding lectin (81), EIII could have multiple neutrophil surface targets. Recent evidences have suggested that lectin DC-SIGN mediated DENV infection in dendritic cells (82); lectins CLEC2 and CLEC5A plays critical roles on DENV-induced inflammation (24, 74, 83, 84). CLEC5A and DC-SIGN are expressed by leukocyte subpopulations (82, 84); while their respective isoforms CLEC2 and DC-SIGNR preferentially expressed by the other cell types (84, 85). Here we used recombinant soluble CLEC2, CLEC5A, DC-SIGN, DC-SIGNR, to perform EIII competition experiments. Analysis results revealed that CLEC5A-, and DC-SIGN-, but not CLEC2-, and DC-SIGNR-coated beads can bind to rEIII (Supplementary Figure 6). Consistently, CLEC5A and DC-SIGN, but not CLEC2 and DC-SIGNR can block rEIII-neutrophil binding (Supplementary Figure 6). In addition, CLEC5A and DC-SIGN can reduce EIII-induced increased ROS and NETosis levels in neutrophils (Supplementary Figure 6). These evidences collectively suggested that CLEC5A and DC-SIGN are cellular targets of EIII on neutrophils. As a critical inflammatory regulator, the role of CLEC5A and DC-SIGN in EIII- mediated pathogenesis is worthy of further investigations.
Cell population is heterogeneous even in one cell line. This could be a reason that reports have revealed treatments of pathogens and cytotoxic agents leading to multiple types of RCDs simultaneously (86–92). For example, when cellular stress can activate both receptor-induced lysosomal-dependent, and mitochondrial-mediated cell death pathways, which will lead to both programmed necrosis and apoptosis (88). Similarly, as DENV and EIII been reported to have multiple cellular targets, it is reasonable to detect multiple RCDs after DENV and EIII challenges. Here, we found that DENV and EIII but not the other RCD inducers, induced a similar RCD pattern in neutrophils (Figures 3B,C). Previous reports have suggested that, upon stimulation by PAMP, neutrophil activation results in necrotic RCDs (93, 94); by contrast, neutrophil apoptosis contributes to the resolution of inflammation (93–96). Consistently, here we found that DENV and EIII (pathogen, PAMP) preferentially induced necrotic RCDs with almost no detectable apoptosis levels in neutrophils (Figure 4). Although further investigations are needed, our data suggested that the neutrophil CTS-RCDPs are useful on the characterization of cellular tendency on the induction of RCDs in particular conditions.
In addition to EIII, DENV membrane protein (M) and non-structural proteins NS2A, NS2B were also shown to induce inflammasome activation (14, 97, 98), and NS1 was demonstrated to enhance inflammation through a toll-like receptor 4 (99, 100). NS2A, NS2B have shown to serve as viroporins to increase cell-membrane permeability and activate inflammasome (14, 101, 102). Viroporins primarily affect virus-infected cells (103), and clinical course of DHF occurs specifically when the viremia markedly decreased (104, 105); these evidences suggest that NS2A, NS2B-mediated inflammasome activations may be not timely associated with the clinical course of DHF. DENV virus particle-expressed EIII and soluble NS1 are detected at high levels prior to the acute phase of DHF (104, 105), and may be considered as two virulence factors for severe dengue. Here we found that both EIII and NS1 treatments can induce increased NETosis levels in neutrophils, and EIII has a relative higher activity (Supplementary Figure 7; DENV envelop domain I, domain II protein fragment, served as a control protein). Because the induction of virion-expressed EIII and soluble NS1 are induced in a similar, but not a same time course (105), the respective pathogenic role of EIII and NS1 on the elicitation of DHF-related pathogenesis remains to be further studied. However, data obtained in the study suggested that virion-associated EIII is a candidate virulence factor that contributes to dengue-elicited NET formation.
In this present study, we found that treatments of both DENV and EIII, a cell-surface-binding and cytotoxic protein, can induce multiple cell death pathways in neutrophils with a similar cell-type-specific RCD pattern (Figure 3B). NETosis inhibitor GSK484 treatments sufficiently suppressed EIII-induced cell death (Figure 4A), NETosis (Figure 4G), and mitochondria metabolic burden (Figure 5) of neutrophils. This suggested that, despite the NETosis seems to be a relatively minor response that counts approximately 20% of total neutrophil RCDs, the EIII-stimulated NETosis is sufficiently leading to an abnormal neutrophil activation and cell death. In addition, our data revealed that blockage of Nlrp3 inflammasome, through treatments of inhibitor OLT1177 or gene deficiencies in Nlrp3 and caspase 1 expression, protects neutrophils from rEIII-induced NETosis (Figures 4, 7) and proinflammatory cytokines TNF and IL-1 secretion in mice (Figure 6). These results collectively suggest that Nlrp3 inflammasome is a promising target for treating DENV-induced inflammation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Ethics Statement
The animal study was reviewed and approved by Animal Care and Use Committee of Tzu-Chi University, Hualien, Taiwan (approval ID: 101019).
Author Contributions
H-HC conceptualized and supervised this project and wrote this manuscript. T-SL, D-SS, S-CH, and W-SW performed experiments and analyzed the data. All authors contributed to the article and approved the submitted version.
Funding
Ministry of Science and Technology, Taiwan (101-2320-B-320-004-MY3, 105-2923-B-320-001-MY3, and 107-2311-B-320-002-MY3), Tzu-Chi University (TCIRP95002; TCIRP98001; and TCIRP101001) and Tzu-Chi Medical Foundation (TC-NHRI105-02; TCMMP104-06; TCMMP108-04; and TCAS-108-01).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to thank Professor Yi-Ling Lin, academia sinica, for kindly provided DENV-EIII plasmid.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.618577/full#supplementary-material
References
1. Pang T, Mak TK, Gubler DJ. Prevention and control of dengue-the light at the end of the tunnel. Lancet Infect Dis. (2017) 17:e79–87. doi: 10.1016/S1473-3099(16)30471-6
2. Gubler DJ. Dengue and dengue hemorrhagic fever. Clin Microbiol Rev. (1998) 11:480–96. doi: 10.1128/CMR.11.3.480
3. Izmirly AM, Alturki SO, Alturki SO, Connors J, Haddad EK. Challenges in dengue vaccines development: pre-existing infections and cross-reactivity. Front Immunol. (2020) 11:1055. doi: 10.3389/fimmu.2020.01055
4. Roth C, Cantaert T, Colas C, Prot M, Casademont I, Levillayer L, et al. A modified mRNA vaccine targeting immunodominant NS epitopes protects against dengue virus infection in HLA class I transgenic mice. Front Immunol. (2019) 10:1424. doi: 10.3389/fimmu.2019.01424
5. Waickman AT, Friberg H, Gargulak M, Kong A, Polhemus M, Endy T, et al. Assessing the diversity and stability of cellular immunity generated in response to the candidate live-attenuated dengue virus vaccine TAK-003. Front Immunol. (2019) 10:1778. doi: 10.3389/fimmu.2019.01778
6. Pinto PBA, Assis ML, Vallochi AL, Pacheco AR, Lima LM, Quaresma KRL, et al. T cell responses Induced by DNA vaccines based on the DENV2 E and NS1 proteins in mice: importance in protection and immunodominant epitope identification. Front Immunol. (2019) 10:1522. doi: 10.3389/fimmu.2019.01522
7. Graham N, Eisenhauer P, Diehl SA, Pierce KK, Whitehead SS, Durbin AP, et al. Rapid induction and maintenance of virus-specific CD8(+) TEMRA and CD4(+) TEM cells following protective vaccination against dengue virus challenge in humans. Front Immunol. (2020) 11:479. doi: 10.3389/fimmu.2020.00479
8. Subramaniam KS, Lant S, Goodwin L, Grifoni A, Weiskopf D, Turtle L. Two is better than one: evidence for T-cell cross-protection between dengue and zika and implications on vaccine design. Front Immunol. (2020) 11:517. doi: 10.3389/fimmu.2020.00517
9. Kuczera D, Assolini JP, Tomiotto-Pellissier F, Pavanelli WR, Silveira GF. Highlights for dengue immunopathogenesis: antibody-dependent enhancement, cytokine storm, and beyond. J Interferon Cytokine Res. (2018) 38:69–80. doi: 10.1089/jir.2017.0037
10. Rothman AL. Immunity to dengue virus: a tale of original antigenic sin and tropical cytokine storms. Nat Rev Immunol. (2011) 11:532–43. doi: 10.1038/nri3014
11. Chuang YC, Wang SY, Lin YS, Chen HR, Yeh TM. Re-evaluation of the pathogenic roles of nonstructural protein 1 and its antibodies during dengue virus infection. J Biomed Sci. (2013) 20:42. doi: 10.1186/1423-0127-20-42
12. Srikiatkhachorn A, Mathew A, Rothman AL. Immune-mediated cytokine storm and its role in severe dengue. Semin Immunopathol. (2017) 39:563–74. doi: 10.1007/s00281-017-0625-1
13. Mahmud-Al-Rafat A, Majumder A, Taufiqur Rahman KM, Mahedi Hasan AM, Didarul Islam KM, Taylor-Robinson AW, et al. Decoding the enigma of antiviral crisis: Does one target molecule regulate all? Cytokine. (2019) 115:13–23. doi: 10.1016/j.cyto.2018.12.008
14. Shrivastava G, Valenzuela Leon PC, Calvo E. Inflammasome fuels dengue severity. Front Cell Infect Microbiol. (2020) 10:489. doi: 10.3389/fcimb.2020.00489
15. Cloherty APM, Olmstead AD, Ribeiro CMS, Jean F. Hijacking of lipid droplets by hepatitis C, dengue and zika viruses-from viral protein moonlighting to extracellular release. Int J Mol Sci. (2020) 21:7901. doi: 10.3390/ijms21217901
16. Papayannopoulos V. Neutrophil extracellular traps in immunity and disease. Nat Rev Immunol. (2018) 18:134–47. doi: 10.1038/nri.2017.105
17. Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. (2007) 176:231–41. doi: 10.1083/jcb.200606027
18. Yipp BG, Kubes P. NETosis: how vital is it? Blood. (2013) 122:2784–94. doi: 10.1182/blood-2013-04-457671
19. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
20. Wang J, Jiang M, Chen X, Montaner LJ. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J Leukoc Biol. (2020) 108:17–41. doi: 10.1002/JLB.3COVR0520-272R
21. Mozzini C, Girelli D. The role of neutrophil extracellular traps in covid-19: only an hypothesis or a potential new field of research? Thromb Res. (2020) 191:26–7. doi: 10.1016/j.thromres.2020.04.031
22. Chang DM, Shaio MF. Production of interleukin-1 (IL-1) and IL-1 inhibitor by human monocytes exposed to dengue virus. J Infect Dis. (1994) 170:811–7. doi: 10.1093/infdis/170.4.811
23. Dinarello CA. Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol. (2009) 27:519–50. doi: 10.1146/annurev.immunol.021908.132612
24. Wu MF, Chen ST, Yang AH, Lin WW, Lin YL, Chen NJ, et al. CLEC5A is critical for dengue virus-induced inflammasome activation in human macrophages. Blood. (2013) 121:95–106. doi: 10.1182/blood-2012-05-430090
25. Schroder K, Tschopp J. The inflammasomes. Cell. (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
26. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. (2009) 7:99–109. doi: 10.1038/nrmicro2070
27. Bozza FA, Cruz OG, Zagne SM, Azeredo EL, Nogueira RM, Assis EF, et al. Multiplex cytokine profile from dengue patients: MIP-1beta and IFN-gamma as predictive factors for severity. BMC Infect Dis. (2008) 8:86. doi: 10.1186/1471-2334-8-86
28. Jaiyen Y, Masrinoul P, Kalayanarooj S, Pulmanausahakul R, Ubol S. Characteristics of dengue virus-infected peripheral blood mononuclear cell death that correlates with the severity of illness. Microbiol Immunol. (2009) 53:442–50. doi: 10.1111/j.1348-0421.2009.00148.x
29. Netea MG, Kullberg BJ, Van der Meer JW. Circulating cytokines as mediators of fever. Clin Infect Dis. (2000) 31 (Suppl. 5):S178–84. doi: 10.1086/317513
30. Dinarello CA. Infection, fever, and exogenous and endogenous pyrogens: some concepts have changed. J Endotoxin Res. (2004) 10:201–22. doi: 10.1179/096805104225006129
31. Pan P, Zhang Q, Liu W, Wang W, Yu Z, Lao Z, et al. dengue virus infection activates interleukin-1beta to induce tissue injury and vascular leakage. Front Microbiol. (2019) 10:2637. doi: 10.3389/fmicb.2019.02637
32. Lien TS, Sun DS, Chang CM, Wu CY, Dai MS, Chan H, et al. Dengue virus and antiplatelet autoantibodies synergistically induce haemorrhage through Nlrp3-inflammasome and FcgammaRIII. Thromb Haemost. (2015) 113:1060–70. doi: 10.1160/TH14-07-0637
33. Lien TS, Sun DS, Wu CY, Chang HH. Exposure to dengue envelope protein domain III induces Nlrp3 inflammasome-dependent endothelial dysfunction and hemorrhage in mice. Front Immunol. (2021) 12:7251. doi: 10.3389/fimmu.2021.617251
34. Nguyen NM, Duong BT, Azam M, Phuong TT, Park H, Thuy PTB, et al. Diagnostic performance of dengue virus envelope domain III in acute dengue infection. Int J Mol Sci. (2019) 20:3464. doi: 10.3390/ijms20143464
35. Khan RA, Afroz S, Minhas G, Battu S, Khan N. Dengue virus envelope protein domain III induces pro-inflammatory signature and triggers activation of inflammasome. Cytokine. (2019) 123:154780. doi: 10.1016/j.cyto.2019.154780
36. Lin GL, Chang HH, Lien TS, Chen PK, Chan H, Su MT, et al. Suppressive effect of dengue virus envelope protein domain III on megakaryopoiesis. Virulence. (2017) 8:1719–31. doi: 10.1080/21505594.2017.1343769
37. Chang HH, Shyu HF, Wang YM, Sun DS, Shyu RH, Tang SS, et al. Facilitation of cell adhesion by immobilized dengue viral nonstructural protein 1 (NS1): arginine-glycine-aspartic acid structural mimicry within the dengue viral NS1 antigen. J Infect Dis. (2002) 186:743–51. doi: 10.1086/342600
38. Tsai CL, Sun DS, Su MT, Lien TS, Chen YH, Lin CY, et al. Suppressed humoral immunity is associated with dengue nonstructural protein NS1-elicited anti-death receptor antibody fractions in mice. Sci Rep. (2020) 10:6294. doi: 10.1038/s41598-020-62958-0
39. Wong MS, Sun MT, Sun DS, Chang HH. Visible-Light-Responsive antibacterial property of boron-doped titania films. Catalysts. (2020) 10:1349. doi: 10.3390/catal10111349
40. Sun DS, Kau JH, Huang HH, Tseng YH, Wu WS, Chang HH. Antibacterial properties of visible-light-responsive carbon-containing titanium dioxide photocatalytic nanoparticles against anthrax. Nanomaterials. (2016) 6:237. doi: 10.3390/nano6120237
41. Sun DS, King CC, Huang HS, Shih YL, Lee CC, Tsai WJ, et al. Antiplatelet autoantibodies elicited by dengue virus non-structural protein 1 cause thrombocytopenia and mortality in mice. J Thromb Haemost. (2007) 5:2291–9. doi: 10.1111/j.1538-7836.2007.02754.x
42. Ho YY, Sun DS, Chang HH. Silver nanoparticles protect skin from ultraviolet B-induced damage in mice. Int J Mol Sci. (2020) 21:7082. doi: 10.3390/ijms21197082
43. Lin YY, Hu CT, Sun DS, Lien TS, Chang HH. Thioacetamide-induced liver damage and thrombocytopenia is associated with induction of antiplatelet autoantibody in mice. Sci Rep. (2019) 9:17497. doi: 10.1038/s41598-019-53977-7
44. Perevedentseva E, Krivokharchenko A, Karmenyan AV, Chang HH, Cheng CL. Raman spectroscopy on live mouse early embryo while it continues to develop into blastocyst in vitro. Sci Rep. (2019) 9:6636. doi: 10.1038/s41598-019-42958-5
45. Chan H, Huang HS, Sun DS, Lee CJ, Lien TS, Chang HH. TRPM8 and RAAS-mediated hypertension is critical for cold-induced immunosuppression in mice. Oncotarget. (2018) 9:12781–95. doi: 10.18632/oncotarget.24356
46. Huang CY, Yu WS, Liu GC, Hung SC, Chang JH, Chang JC, et al. Opportunistic gill infection is associated with TiO2 nanoparticle-induced mortality in zebrafish. PLoS ONE. (2021) 16:e0247859. doi: 10.1371/journal.pone.0245356
47. Chang YS, Ko BH, Ju JC, Chang HH, Huang SH, Lin CW. SARS unique domain (SUD) of severe acute respiratory syndrome coronavirus induces NLRP3 inflammasome-dependent CXCL10-mediated pulmonary inflammation. Int J Mol Sci. (2020) 21:3179. doi: 10.3390/ijms21093179
48. Sun DS, Ho PH, Chang HH. Soluble P-selectin rescues viper venom-induced mortality through anti-inflammatory properties and PSGL-1 pathway-mediated correction of hemostasis. Sci Rep. (2016) 6:35868. doi: 10.1038/srep35868
49. Sun DS, Chang YW, Kau JH, Huang HH, Ho PH, Tzeng YJ, et al. Soluble P-selectin rescues mice from anthrax lethal toxin-induced mortality through PSGL-1 pathway-mediated correction of hemostasis. Virulence. (2017) 8:1216–28. doi: 10.1080/21505594.2017.1282027
50. Quach A, Ferrante A. The application of dextran sedimentation as an initial step in neutrophil purification promotes their stimulation, due to the presence of monocytes. J Immunol Res. (2017) 2017:1254792. doi: 10.1155/2017/1254792
51. Tseng PH, Sie ZL, Liu MC, Lin HS, Yang WY, Lin TY, et al. Identification of two novel small compounds that inhibit liver cancer formation in zebrafish and analysis of their conjugation to nanodiamonds to further reduce toxicity. Adv Ther. (2019) 2:1900105. doi: 10.1002/adtp.201900105
52. Mandal JP, Shiue CN, Chen YC, Lee MC, Yang HH, Chang HH, et al. PKCdelta mediates mitochondrial ROS generation and oxidation of HSP60 to relieve RKIP inhibition on MAPK pathway for HCC progression. Free Radic Biol Med. (2020) 163:69–87. doi: 10.1016/j.freeradbiomed.2020.12.003
53. Masuda S, Nakazawa D, Shida H, Miyoshi A, Kusunoki Y, Tomaru U, et al. NETosis markers: Quest for specific, objective, and quantitative markers. Clin Chim Acta. (2016) 459:89–93. doi: 10.1016/j.cca.2016.05.029
54. Kuczia P, Zuk J, Iwaniec T, Soja J, Dropinski J, Malesa-Wlodzik M, et al. Citrullinated histone H3, a marker of extracellular trap formation, is increased in blood of stable asthma patients. Clin Transl Allergy. (2020) 10:31. doi: 10.1186/s13601-020-00337-8
55. Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. (2013) 123:3446–58. doi: 10.1172/JCI67484
56. Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. (2016) 76:1367–80. doi: 10.1158/0008-5472.CAN-15-1591
57. Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. (1996) 59:229–40. doi: 10.1002/jlb.59.2.229
58. Tang D, Kang R, Berghe TV, Vandenabeele P, Kroemer G. The molecular machinery of regulated cell death. Cell Res. (2019) 29:347–64. doi: 10.1038/s41422-019-0164-5
59. Smith H, Rogers SL, Smith HV, Gillis D, Siskind V, Smith JA. Virus-associated apoptosis of blood neutrophils as a risk factor for invasive meningococcal disease. J Clin Pathol. (2013) 66:976–81. doi: 10.1136/jclinpath-2013-201579
60. Colamussi ML, White MR, Crouch E, Hartshorn KL. Influenza A virus accelerates neutrophil apoptosis and markedly potentiates apoptotic effects of bacteria. Blood. (1999) 93:2395–403. doi: 10.1182/blood.V93.7.2395
61. Chen YL, Tsai YT, Lee CY, Lee CH, Chen CY, Liu CM, et al. Urotensin II inhibits doxorubicin-induced human umbilical vein endothelial cell death by modulating ATF expression and via the ERK and Akt pathway. PLoS ONE. (2014) 9:e106812. doi: 10.1371/journal.pone.0106812
62. Kotamraju S, Konorev EA, Joseph J, Kalyanaraman B. Doxorubicin-induced apoptosis in endothelial cells and cardiomyocytes is ameliorated by nitrone spin traps and ebselen. Role of reactive oxygen and nitrogen species. J Biol Chem. (2000) 275:33585–92. doi: 10.1074/jbc.M003890200
63. Chen HR, Chuang YC, Chao CH, Yeh TM. Macrophage migration inhibitory factor induces vascular leakage via autophagy. Biol Open. (2015) 4:244–52. doi: 10.1242/bio.201410322
64. Xiao FJ, Zhang D, Wu Y, Jia QH, Zhang L, Li YX, et al. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem Biophys Res Commun. (2019) 515:448–54. doi: 10.1016/j.bbrc.2019.05.147
65. Sawai H. Characterization of TNF-induced caspase-independent necroptosis. Leuk Res. (2014) 38:706–13. doi: 10.1016/j.leukres.2014.02.002
66. Choi ME, Price DR, Ryter SW, Choi AMK. Necroptosis: a crucial pathogenic mediator of human disease. JCI Insight. (2019) 4:e128834. doi: 10.1172/jci.insight.128834
67. Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X, et al. Caspase-1 inflammasome activation mediates homocysteine-induced pyrop-apoptosis in endothelial cells. Circ Res. (2016) 118:1525–39. doi: 10.1161/CIRCRESAHA.116.308501
68. Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. (2020) 369:1633–7. doi: 10.1126/science.abb9818
69. Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. (2002) 10:417–26. doi: 10.1016/S1097-2765(02)00599-3
70. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
71. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
72. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. (2019) 10:128. doi: 10.1038/s41419-019-1413-8
73. Mustafa AS, Elbishbishi EA, Agarwal R, Chaturvedi UC. Elevated levels of interleukin-13 and IL-18 in patients with dengue hemorrhagic fever. FEMS Immunol Med Microbiol. (2001) 30:229–33. doi: 10.1111/j.1574-695X.2001.tb01575.x
74. Sung PS, Huang TF, Hsieh SL. Extracellular vesicles from CLEC2-activated platelets enhance dengue virus-induced lethality via CLEC5A/TLR2. Nat Commun. (2019) 10:2402. doi: 10.1038/s41467-019-10360-4
75. Opasawatchai A, Amornsupawat P, Jiravejchakul N, Chan-In W, Spoerk NJ, Manopwisedjaroen K, et al. Neutrophil activation and early features of NET formation are associated with dengue virus infection in human. Front Immunol. (2018) 9:3007. doi: 10.3389/fimmu.2018.03007
76. Biasizzo M, Kopitar-Jerala N. Interplay Between NLRP3 Inflammasome and Autophagy. Front Immunol. (2020) 11:591803. doi: 10.3389/fimmu.2020.591803
77. Zhen Y, Zhang H. NLRP3 inflammasome and inflammatory bowel disease. Front Immunol. (2019) 10:276. doi: 10.3389/fimmu.2019.00276
78. Yuk JM, Silwal P, Jo EK. Inflammasome and mitophagy connection in health and disease. Int J Mol Sci. (2020) 21:4714. doi: 10.3390/ijms21134714
79. Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. (2018) 3:eaar6689. doi: 10.1126/sciimmunol.aar6689
80. Bonaventura A, Vecchie A, Abbate A, Montecucco F. Neutrophil extracellular traps and cardiovascular diseases: an update. Cells. (2020) 9:231. doi: 10.3390/cells9010231
81. Watterson D, Kobe B, Young PR. Residues in domain III of the dengue virus envelope glycoprotein involved in cell-surface glycosaminoglycan binding. J Gen Virol. (2012) 93:72–82. doi: 10.1099/vir.0.037317-0
82. Tassaneetrithep B, Burgess TH, Granelli-Piperno A, Trumpfheller C, Finke J, Sun W, et al. DC-SIGN (CD209) mediates dengue virus infection of human dendritic cells. J Exp Med. (2003) 197:823–9. doi: 10.1084/jem.20021840
83. Chen ST, Lin YL, Huang MT, Wu MF, Cheng SC, Lei HY, et al. CLEC5A is critical for dengue-virus-induced lethal disease. Nature. (2008) 453:672–6. doi: 10.1038/nature07013
84. Sung PS, Hsieh SL. CLEC2 and CLEC5A: pathogenic host factors in acute viral infections. Front Immunol. (2019) 10:2867. doi: 10.3389/fimmu.2019.02867
85. Soilleux EJ. DC-SIGN (dendritic cell-specific ICAM-grabbing non-integrin) and DC-SIGN-related (DC-SIGNR): friend or foe? Clin Sci. (2003) 104:437–46. doi: 10.1042/cs1040437
86. Chien H, Dix RD. Evidence for multiple cell death pathways during development of experimental cytomegalovirus retinitis in mice with retrovirus-induced immunosuppression: apoptosis, necroptosis, and pyroptosis. J Virol. (2012) 86:10961–78. doi: 10.1128/JVI.01275-12
87. Panzarini E, Inguscio V, Dini L. Timing the multiple cell death pathways initiated by rose Bengal acetate photodynamic therapy. Cell Death Dis. (2011) 2:e169. doi: 10.1038/cddis.2011.51
88. Moeckel GW. Hypertonic stress and cell death. Focus on “Multiple cell death pathways are independently activated by lethal hypertonicity in renal epithelial cells”. Am J Physiol Cell Physiol. (2013) 305:C1009–10. doi: 10.1152/ajpcell.00263.2013
89. Choi SY, Lee-Kwon W, Lee HH, Lee JH, Sanada S, Kwon HM. Multiple cell death pathways are independently activated by lethal hypertonicity in renal epithelial cells. Am J Physiol Cell Physiol. (2013) 305:C1011–20. doi: 10.1152/ajpcell.00384.2012
90. Chen Y, Hua Y, Li X, Arslan IM, Zhang W, Meng G. Distinct types of cell death and the implication in diabetic cardiomyopathy. Front Pharmacol. (2020) 11:42. doi: 10.3389/fphar.2020.00042
91. Korsnes MS. Yessotoxin as a tool to study induction of multiple cell death pathways. Toxins. (2012) 4:568–79. doi: 10.3390/toxins4070568
92. Miller C, Kennington L, Cooney R, Kohjimoto Y, Cao LC, Honeyman T, et al. Oxalate toxicity in renal epithelial cells: characteristics of apoptosis and necrosis. Toxicol Appl Pharmacol. (2000) 162:132–41. doi: 10.1006/taap.1999.8835
93. Lawrence SM, Corriden R, Nizet V. How neutrophils meet their end. Trends Immunol. (2020) 41:531–44. doi: 10.1016/j.it.2020.03.008
94. Brostjan C, Oehler R. The role of neutrophil death in chronic inflammation and cancer. Cell Death Discov. (2020) 6:26. doi: 10.1038/s41420-020-0255-6
95. Serhan CN, Savill J. Resolution of inflammation: the beginning programs the end. Nat Immunol. (2005) 6:1191–7. doi: 10.1038/ni1276
96. Iba T, Hashiguchi N, Nagaoka I, Tabe Y, Murai M. Neutrophil cell death in response to infection and its relation to coagulation. J Intensive Care. (2013) 1:13. doi: 10.1186/2052-0492-1-13
97. Pan P, Zhang Q, Liu W, Wang W, Lao Z, Zhang W, et al. Dengue virus m protein promotes NLRP3 inflammasome activation to induce vascular leakage in mice. J Virol. (2019) 93:e00996–19. doi: 10.1128/JVI.00996-19
98. Shrivastava G, Visoso-Carvajal G, Garcia-Cordero J, Leon-Juarez M, Chavez-Munguia B, Lopez T, et al. Dengue virus serotype 2 and its non-structural proteins 2A and 2B activate NLRP3 inflammasome. Front Immunol. (2020) 11:352. doi: 10.3389/fimmu.2020.00352
99. Beatty PR, Puerta-Guardo H, Killingbeck SS, Glasner DR, Hopkins K, Harris E. Dengue virus NS1 triggers endothelial permeability and vascular leak that is prevented by NS1 vaccination. Sci Transl Med. (2015) 7:304ra141. doi: 10.1126/scitranslmed.aaa3787
100. Modhiran N, Watterson D, Muller DA, Panetta AK, Sester DP, Liu L, et al. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci Transl Med. (2015) 7:304ra142. doi: 10.1126/scitranslmed.aaa3863
101. Shrivastava G, Garcia-Cordero J, Leon-Juarez M, Oza G, Tapia-Ramirez J, Villegas-Sepulveda N, et al. NS2A comprises a putative viroporin of dengue virus 2. Virulence. (2017) 8:1450–6. doi: 10.1080/21505594.2017.1356540
102. Leon-Juarez M, Martinez-Castillo M, Shrivastava G, Garcia-Cordero J, Villegas-Sepulveda N, Mondragon-Castelan M, et al. Recombinant Dengue virus protein NS2B alters membrane permeability in different membrane models. Virol J. (2016) 13:1. doi: 10.1186/s12985-015-0456-4
103. Nieva JL, Madan V, Carrasco L. Viroporins: structure and biological functions. Nat Rev Microbiol. (2012) 10:563–74. doi: 10.1038/nrmicro2820
Keywords: dengue envelope protein domain III, dengue hemorrhage fever, neutrophil, neutrophil extracellular traps, NEtosis, Nlrp3 inflammasome, pyroptosis
Citation: Lien T-S, Sun D-S, Hung S-C, Wu W-S and Chang H-H (2021) Dengue Virus Envelope Protein Domain III Induces Nlrp3 Inflammasome-Dependent NETosis-Mediated Inflammation in Mice. Front. Immunol. 12:618577. doi: 10.3389/fimmu.2021.618577
Received: 17 October 2020; Accepted: 22 February 2021;
Published: 17 March 2021.
Edited by:
Kuo-Feng Hua, National Ilan University, TaiwanReviewed by:
Leticia Cedillo-Barron, Instituto Politécnico Nacional de México (CINVESTAV), MexicoJianguo Wu, Wuhan University, China
Tadayoshi Karasawa, Jichi Medical University, Japan
Copyright © 2021 Lien, Sun, Hung, Wu and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hsin-Hou Chang, hhchang@mail.tcu.edu.tw