 Camille Keenan
Camille Keenan Kim E. Nichols
Kim E. Nichols Sabrin Albeituni
Sabrin Albeituni
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 16 February 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.614704
This article is part of the Research TopicNovel Therapeutic Approaches to Primary ImmunodeficienciesView all 14 articles
Hemophagocytic lymphohistiocytosis (HLH) is a rare hyperinflammatory syndrome driven by overactive T cells and macrophages that abundantly secrete numerous pro-inflammatory cytokines, including interferon (IFN)-gamma, interleukin (IL)-1-beta, IL-2, IL-6, IL-10, IL-18, and tumor necrosis factor (TNF). The release of these and other cytokines underlies many of the clinical and pathologic manifestations of HLH, which if left untreated, can lead to multi-organ failure and death. The advent of etoposide-based regimens, such as the Histiocyte Society HLH-94 and HLH-2004 protocols, has substantially decreased the mortality associated with HLH. Nevertheless, the 5-year survival remains low at ~60%. To improve upon these results, studies have focused on the use of novel cytokine-directed therapies to dampen inflammation in HLH. Among the agents being tested is ruxolitinib, a potent inhibitor of the Janus Kinase (JAK) and Signal Transducer and Activation of Transcription (STAT) pathway, which functions downstream of many HLH-associated cytokines. Here, we review the basic biology of HLH, including the role of cytokines in disease pathogenesis, and discuss the use of ruxolitinib in the treatment of HLH.
Hemophagocytic lymphohistiocytosis (HLH) is a rare and life-threatening hyperinflammatory syndrome resulting from inherited (primary HLH) or acquired (secondary HLH) immune dysregulation. The excessive immune response is associated with T and myeloid cell infiltration of numerous organs, such as the liver, spleen, and central nervous system, where activated immune cells profusely secrete cytokines and mediate significant tissue damage. If not promptly treated, the exuberant immune response can lead to multiorgan failure and death (1). The diagnosis of HLH is based on criteria established by the Histiocyte Society in 1994 and revised in 2004, and should be strongly suspected in individuals exhibiting features such as fever, splenomegaly, hyperferritinemia, bi- or tri-lineage cytopenias, hypofibrinogenemia or hypertriglyceridemia, elevated soluble CD25, hemophagocytosis, and reduced or absent NK cell cytotoxicity (2). HLH thus constitutes a fascinating syndrome wherein familial or acquired defects in immunity result in hyperinflammation, as opposed to the phenotype of reduced immune function leading to chronic or rare infections as is seen with most other immunodeficiency syndromes (3).
Primary HLH is estimated to affect one in every 50,000–100,000 children (4, 5) and is caused by pathogenic germline variants that negatively impact CD8+ T and natural killer (NK) cell cytotoxicity. Primary HLH is autosomal recessive and caused by homozygous or compound heterozygous mutations in the genes PRF1, UNC13D, STX11, and STXBP2, which are required for the proper function and extrusion of perforin-containing cytolytic granules. Patients with the autosomal recessive syndromes of pigmentary dilution and impaired lysosomal formation or lytic granule secretion such as Chediak-Higashi syndrome (due to pathogenic variants in LYST) and Griscelli syndrome (RAB27A) are also at risk to develop HLH, as are patients with the X-linked disorders X-linked lymphoproliferative syndrome 1 (SH2D1A) and 2 (XIAP) (6).
Although its true incidence is not known, secondary HLH occurs in patients without bi-allelic HLH gene variants, a familial pattern of HLH, or evidence of prior immunodeficiency, who develop a hyperinflammatory phenotype following exposure to a strong immunologic trigger. Such triggers include a variety of infections (viruses being most common), malignancies (generally T or NK cell lymphomas), and autoimmune disorders [such as systemic juvenile idiopathic arthritis (SJIA), where the HLH-like manifestations are often referred to as macrophage activation syndrome (MAS)] (7). Rare patients with secondary HLH have been identified to harbor monoallelic pathogenic germline variants in the genes known to be associated with primary HLH; however, it remains questionable whether these variants play a role in the development of disease (8, 9).
Initial insights into the pathogenesis of primary HLH were provided in 1999 with the seminal discovery of the first primary HLH gene, PRF1, which encodes the pore forming protein perforin (10). Over the next decade, several additional primary HLH genes were identified, each of which plays an essential role in granule-mediated lymphocyte cytotoxicity. Studies using mouse genetic models have revealed that defects in these genes impair lymphocyte cytotoxicity leading to poor clearance of pathogens and activated antigen presenting cells and resulting in an ever-spiraling feed forward loop of immune cell activation without the ability to end this cycle.
The pathogenesis of secondary HLH is less well understood. One proposed mechanism is that aberrant stimulation of Toll like receptors (TLR) leads to excessive activation and cytokine production by cells of the innate immune system (11). This possibility is intriguing since Epstein Barr virus (EBV), one of the most common infectious triggers of secondary HLH, engages TLR9 to stimulate myeloid cells. Another possible mechanism involves the inflammasome-dependent overproduction of IL-18 by activated myeloid and epithelial cells. Excess free IL-18 amplifies lymphocyte production of IFN-gamma and is proposed to contribute to MAS (12).
Regardless of their underlying differences in pathogenesis, one of the features shared by primary and secondary HLH is the excessive production of pro-inflammatory cytokines with IFN-gamma, TNF, IL-1, IL-2, IL-6, IL-10, IL-12, IL-18 and granulocyte macrophage-colony stimulating factor (GM-CSF) representing some of the cytokines most notably elevated (13–16). Based on these findings, several studies have sought to determine which of these cytokines is most required for the development and/or propagation of HLH. Toward this end, a report by Binder et al. in 1998 (1 year before perforin-deficiency was identified as a cause for primary HLH) described the development of an HLH-like syndrome in perforin-deficient mice following infection with Lymphocytic Choriomeningitis Virus (LCMV) (17). Disease manifestations were similar to those observed in humans with HLH including expansion of activated CD8+ T cells, pancytopenia and excessive production of TNF and IFN-gamma (17). In this report, depletion of CD8+ T cells, genetic ablation of TNF, and/or antibody-mediated neutralization of IFN-gamma, each significantly improved bone marrow function and enhanced survival of LCMV-infected animals. Subsequent studies using perforin or RAB27A-deficient mice have confirmed these initial findings, with depletion of CD8+ T cells or blockade of IFN-gamma (but not TNF, IL-12, IL-18 or GM-CSF) improving survival and lessening the manifestations of disease (18, 19).
While these data point to a central role for IFN-gamma in the pathogenesis of primary HLH, a recent report by Burn et al. reveals that mice lacking expression of both perforin and IFN-gamma develop a severe HLH-like illness following LCMV infection, although the characteristics of this illness differ from those observed in animals lacking expression only of perforin. Specifically, perforin- and IFN-gamma-double deficient mice develop a marked neutrophilia and a serum cytokine pattern notable for increased IL-6, IL-1-beta and GM-CSF but not increased IFN-gamma (20). These data strongly suggest that cytokines in addition to IFN-gamma likely also play important roles in the development and/or progression of HLH-associated hyperinflammation.
The mainstay of primary HLH treatment involves generalized suppression of the immune system, usually with steroids and chemotherapy. Without such treatment, primary HLH is almost always fatal. Notably, cure can only be obtained via allogeneic hematopoietic stem cell transplantation (HSCT) in which the defective immune system is replaced with a healthy one. Like hematologic malignancies, active HLH at the time of transplant may portend a poorer outcome (21, 22).
The advent of etoposide-based regimens, such as the HLH-94 and HLH-2004 protocols, has substantially increased the survival for patients with newly diagnosed primary HLH (1). Indeed, use of the HLH-94 protocol resulted in a 54% 5-year probability of survival following treatment with etoposide and dexamethasone, delayed cyclosporine, and intrathecal (IT) methotrexate for patients with central nervous system (CNS) involvement, followed by HSCT (21). Nevertheless, 29% of patients died before HSCT and 19% developed late neurologic sequelae. To reduce pre-transplant mortality and neurologic complications, the HLH-2004 protocol added up-front cyclosporine and included IT corticosteroids. Although HLH-2004 resulted in a 5-year estimated survival of 62%, neither treatment modification significantly improved overall outcome (23). Therefore, the HLH-94 regimen remains the current standard of care in most centers.
For patients with relapsed/refractory primary HLH, there are limited data on the utility of specific salvage therapies. Case reports and retrospective case series describe the use of the anti-CD52 antibody alemtuzumab and anti-thymocyte globulin (ATG); however, there are no published prospective trials using these agents. A retrospective study of alemtuzumab revealed that 64% of patients with relapsed/refractory HLH responded and 77% survived to HSCT (24). These less-than-perfect outcomes have prompted investigation of alternative therapeutic strategies. Toward this end, a recent report describes 27 patients with relapsed/refractory primary HLH treated with dexamethasone, the IFN-gamma neutralizing antibody emapalumab, and varying combinations of other agents (25). The overall response for these patients was 63% with 70.4% proceeding to HSCT. In this study, seven patients received emapalumab as first line therapy with none achieving a complete response (CR) despite IFN-gamma being a prominent cytokine in primary HLH.
When possible, the treatment of secondary HLH should always begin by targeting the underlying trigger using agents to combat infection, malignancy or auto-immune disease. However, in some cases it may also be necessary to treat the associated hyperinflammation. Various agents have been used, including dexamethasone and, in the most severe cases, etoposide. Agents that block the effects of individual cytokines have also been employed, with this approach particularly appealing for patients whose HLH is triggered by infection, where the administration of cytotoxic or other immunosuppressive drugs could compromise clearance of the inciting pathogen.
One such study retrospectively examined 44 patients with secondary HLH/MAS who received treatment with recombinant IL-1 receptor antagonist, anakinra. This study revealed a 73% survival with treatment most successful when given within the first 5 days of hospitalization and to those with underlying rheumatologic diseases such as SJIA (26). A current trial is ongoing to assess the efficacy of the recombinant IL-18 binding protein, Tadekinig alfa, in patients with MAS who harbor germline NLRC4 pathogenic variants or patients with XIAP deficiency (NCT03113760). The anti-IL-6R antibody, tocilizumab, has been reported to mitigate the signs and symptoms of cytokine overproduction [a condition known as cytokine release syndrome (CRS)] in cancer patients receiving treatment with chimeric antigen receptor transduced T (CAR-T) cells (27). Since the cytokine patterns in CAR-T-treated patients are similar to those observed in patients with HLH, IL-6 blockade might prove beneficial in the treatment of HLH. Supporting this notion, there is emerging evidence that tocilizumab can induce remission in adults with secondary HLH (28, 29).
Despite the use of currently available therapies, there are many patients with HLH who succumb to the disease. It is thus noteworthy that many of the cytokines elevated in HLH, such as IFN-gamma, IL-2, IL-6, IL-10, IL-12, and GM-CSF signal through a pathway involving the Janus kinases (JAKs) and Signal Transducers and Activators of Transcription (STATs) (Figure 1) (30). By dampening signaling downstream of numerous HLH-associated cytokines, the interruption of the JAK-STAT pathway holds promise to more effectively lessen HLH-associated immunopathology. Numerous JAK inhibitors, such as ruxolitinib, tofacitinib, baricitinib, and oclacitinib have been used for the treatment of inflammatory disorders (31). In this review, we will focus on ruxolitinib as it is the JAK inhibitor for which there are the most published data on its use in the treatment of patients with HLH.
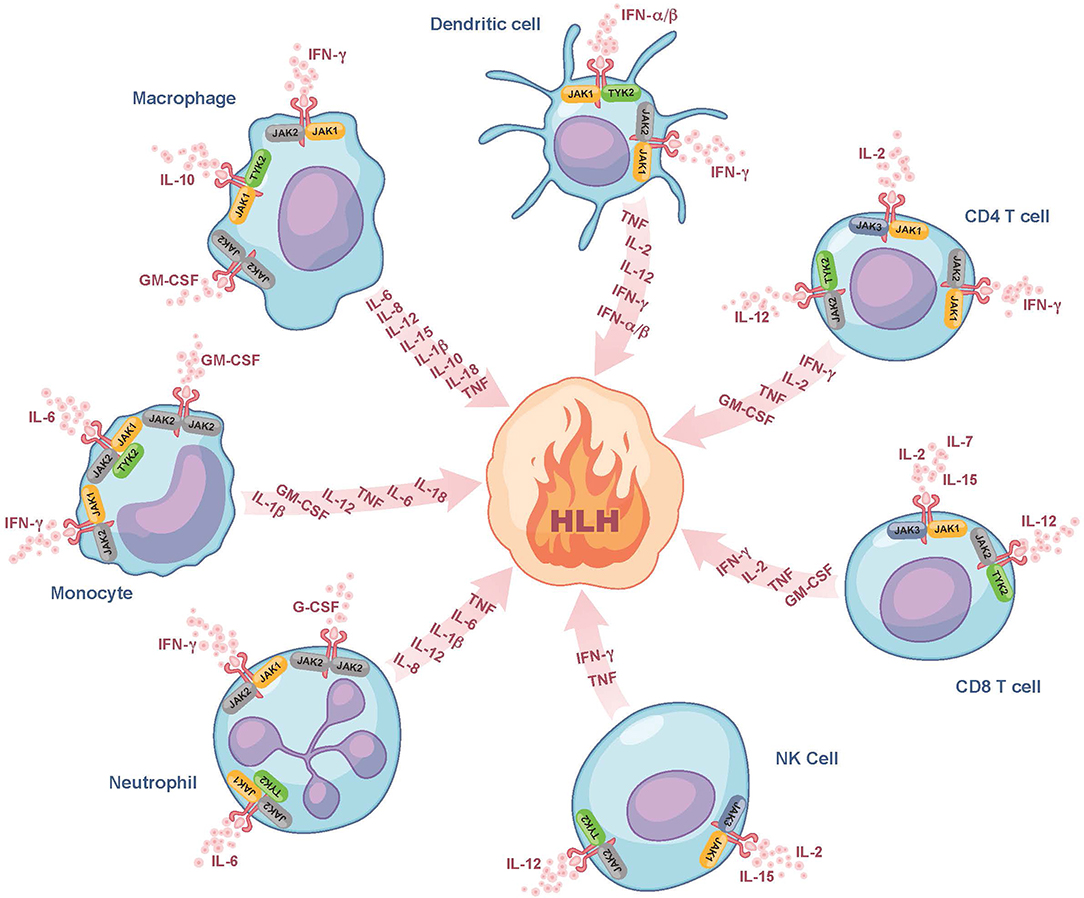
Figure 1. Role of JAK/STAT signaling in fueling the fire of HLH. Here we show several of the cytokines that fuel the fire of HLH and their associated cellular sources. We also illustrate which of these cytokines bind to receptors associated with JAK-family kinases (JAK1, JAK2, JAK3, or TYK2) and are expressed on the surface of dendritic cells, CD4 and CD8 T cells, NK cells, neutrophils, monocytes, and macrophages. These JAK-dependent receptor-mediated signaling pathways represent potential targets of ruxolitinib. Note that many of the cytokines produced are also those that trigger cell activation and hence part of the feedforward loop. For simplicity, all receptors are drawn similarly, but it is noted that there exist some receptors that typically form complexes (e.g., GM-CSF), which are not depicted in this Figure. G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte-macrophage colony-stimulating factor; HLH, hemophagocytic lymphohistiocytosis; IFN-α/β, interferon alpha and beta; IFN-γ, interferon gamma; IL, interleukin; JAK, Janus kinase; NK cell, natural killer cell; TNF, tumor necrosis factor; TYK2, tyrosine-protein kinase 2.
Ruxolitinib (Jakafi®) is an oral, potent, highly bioavailable JAK1/2 inhibitor that is approved by the Food and Drug Administration (FDA) for use in patients with myeloproliferative neoplasms and steroid refractory graft-vs.-host-disease (GVHD), where it has shown to induce responses, even in those with the most advanced disease (32–34). Currently, ruxolitinib is being trialed as GVHD prophylaxis for patients undergoing allogeneic HSCT (35, 36). In addition, ruxolitinib has been reported to lessen inflammation in patients with pathogenic germline STAT1 or STAT3 gain-of-function (GOF) variants. In one patient with a STAT3 GOF variant, combined treatment with ruxolitinib and tocilizumab led to HLH remission (37). Given the efficacy of ruxolitinib in these other disorders, we and Maschalidi et al. independently examined its activity in mouse models of HLH. Specifically, we used ruxolitinib to treat LCMV-infected perforin- or RAB27A-deficient mice (models of primary HLH), as well wild-type mice exposed to repeated injections of CpG DNA (model of secondary HLH) (11, 38). Through these approaches, we demonstrated that ruxolitinib reversed many HLH manifestations, including splenomegaly, cytopenias, hypercytokinemias, peripheral organ and CNS inflammation, and it significantly prolonged survival (39, 40).
Ruxolitinib inhibits both JAK1 and 2, which function downstream of IFN-gamma as well as several other HLH-associated cytokines, such as IL-2 IL-6, IL-10, IL-12, and GM-CSF. As a consequence, it remained unknown whether the beneficial effects of ruxolitinib resulted simply from its targeting of IFN-gamma, or instead from targeting of other cytokine signaling pathways. To gain further insights, we directly compared the effects resulting from ruxolitinib treatment vs. those obtained following administration of an equipotent dose of IFN-gamma-blocking antibody in the mouse models of HLH. Through this study, we observed that IFN-gamma was the main driver of anemia, but cytokines other than IFN-gamma promoted HLH-associated immunopathology. Supporting this observation, both ruxolitinib and the anti-IFN-gamma blocking antibody improved hemoglobin levels, but only ruxolitinib significantly reduced the number and activation status of immune cells, as well as their infiltration into various tissues (41).
Although single agent ruxolitinib greatly reduces inflammation in mouse pre-clinical models of HLH, disease manifestations are never completely eliminated. Therefore, we most recently examined whether treatment with dexamethasone and ruxolitinib might exert superior control of disease. In this study, we also show that elevated levels of STAT5-dependent cytokines such as IL-2, IL-7 and IL-15 induce resistance of activated CD8+ T cells to glucocorticoid-induced cell death. Notably, by blocking cytokine signaling with ruxolitinib, we could sensitize CD8+ T cells to dexamethasone-induced apoptosis in vitro and with combination therapy significantly reducing tissue immunopathology and HLH disease manifestations in vivo (42).
Since the initial reports by Das et al. and Maschalidi et al. describing use of ruxolitinib in mouse HLH models, there has been an increasing number of publications describing the use of this drug in patients with HLH. At the time of writing of this review, 18 independent studies describing 115 unique patients have been published, with most studies including adults with secondary HLH for whom ruxolitinib was incorporated as part of a salvage regimen (Table 1). In many cases, ruxolitinib improved clinical manifestations, such as fever, shock, renal failure, and respiratory depression within as little as 24–48 h following initiation of drug. In addition, serum levels of ferritin, soluble IL-2 receptor, and fibrinogen often normalized within seven to 30 days. Overall, ruxolitinib is described as being well-tolerated at doses ranging from 2.5 to 25 mg with the medication administered twice daily. Below, we describe several studies that used ruxolitinib for patients with primary or secondary HLH (focusing here on secondary HLH due to infections or rheumatologic diseases). Malignancy-associated HLH will not be discussed; however, the reader is referred to reports describing ruxolitinib use in this population, as outlined in Table 1.
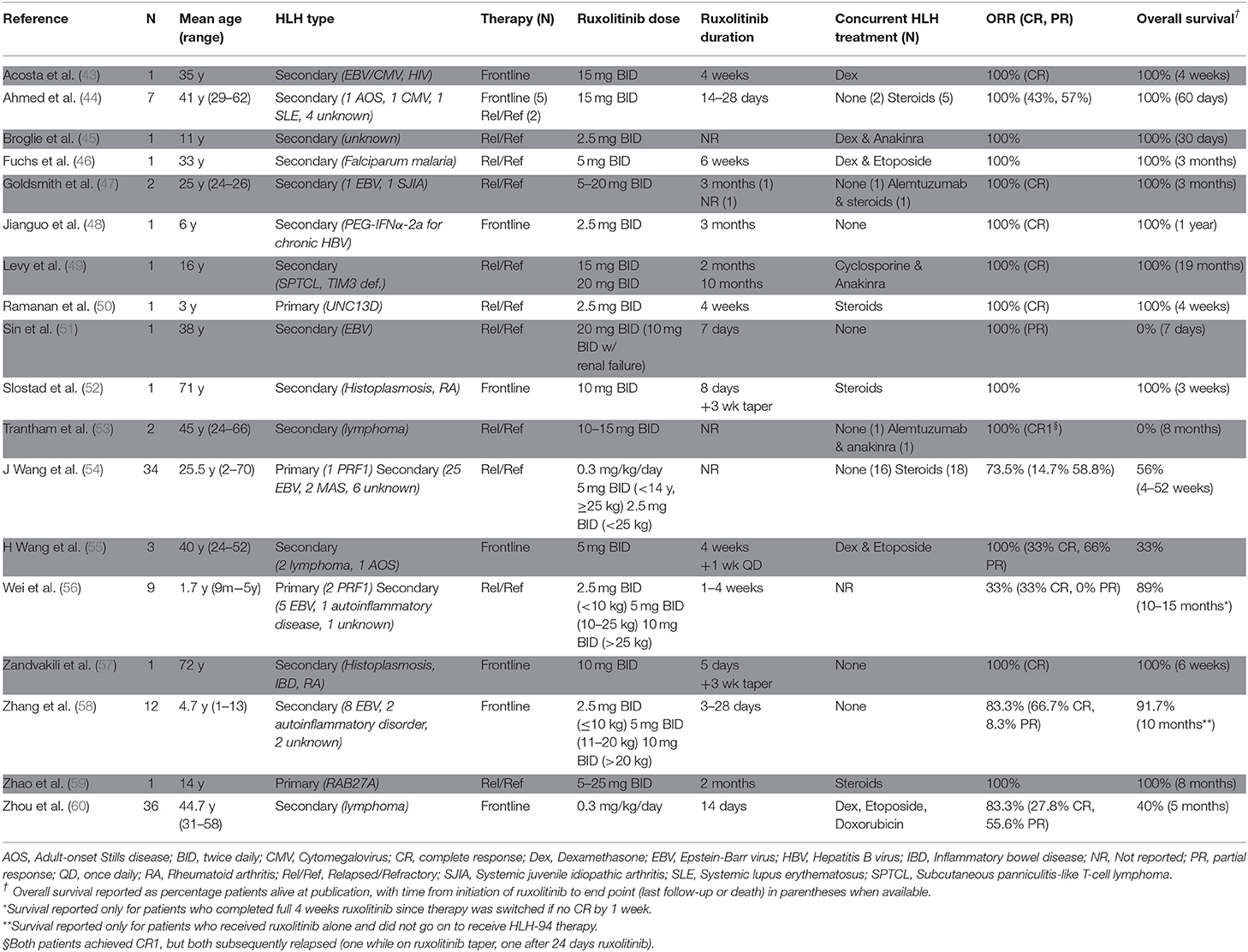
Table 1. Summary of publications describing use of ruxolitinib in patients with HLH.
The first report describing use of ruxolitinib as a treatment for HLH included an 11-year old boy with refractory disease and no identifiable trigger (45). Despite treatment with etoposide and dexamethasone, his HLH did not go into remission and his status deteriorated. The subsequent addition of ruxolitinib induced a rapid clinical response and within 24 h he had no fever and exhibited improved respiratory and liver function. The levels of ferritin and soluble IL-2 dropped within a month and the patient ultimately underwent a successful allogeneic HSCT.
Several additional studies have further described favorable responses to ruxolitinib when given as a component of salvage therapy. For example, Goldsmith et al. reported that treatment with ruxolitinib in conjunction with alemtuzumab induced remission in a patient with SJIA whose HLH/MAS failed to respond to treatment with HLH-94 therapy (47). One report from China describing use of ruxolitinib and corticosteroids to treat 34 children and adults with secondary HLH revealed a response rate of 74%, with 14.7% complete responses (CR) and 58.8% partial responses (PR), and 56% overall survival (54). Within 24 h of starting ruxolitinib, 30 patients (88%) became afebrile and ferritin and soluble CD25 levels were significantly lower after 2 weeks. In this study, complete or partial responses correlated with reduced levels of serum cytokines such as IFN-gamma, IL-18, Macrophage Inflammatory Protein (MIP)-1-alpha, and IFN-gamma-Inducible Protein (IP)-10. Several patients in this study relapsed, most notably those with EBV-related HLH, suggesting that early allogeneic HSCT is required for this population despite an initial positive clinical response to ruxolitinib (55). Ruxolitinib has been used in six pediatric patients with primary HLH whose disease relapsed before or after allogeneic HSCT, with all six patients exhibiting a complete or partial response (50, 56, 59). Overall, these and other studies suggest that ruxolitinib can effectively dampen inflammation in this challenging patient population. Nevertheless, as many patients ultimately relapse, allogeneic HSCT should still be considered the ultimate curative therapy for those who are able to tolerate the procedure. Given the profound immune suppression that accompanies repeated courses of therapy, patients require careful monitoring for and prophylaxis against infection.
Ruxolitinib has been trialed as a first-line therapy for HLH, albeit to a lesser extent than as a salvage therapy. This was first reported by Slostad et al. (52) and Zandvakili et al. (57) who each described a single adult patient with secondary HLH presenting in the context of histoplasma infection. Given their older age and critical illness, including worsening cytopenias, septic shock, renal, hepatic, and respiratory failure, ruxolitinib was given as a first-line therapy along with anti-fungal agents. For both patients, fever, multi-organ failure and cytopenias improved with full recovery and normalization of HLH-related laboratory parameters within 6 weeks.
More recently, Ahmed et al. reported the results of an open-label, prospective, single-center study enrolling five adults with newly diagnosed HLH (44). Two additional patients were included in the report despite being previously treated. Six of these patients had secondary HLH and one with systemic lupus erythematosus was later found to harbor a homozygous germline STXBP2 mutation. In all treated patients, cytopenias and serum ferritin levels improved within the first one to 2 weeks of treatment. Notably, the 2-month overall survival was 100% with no deaths reported after a median follow up of 490 days. Adverse events possibly related to ruxolitinib included one patient with Grade 4 neutropenia, and single patients each with Grade 1 or 2 skin findings (maculopapular rash, intermittent candidiasis), nausea or extremity pain.
Extending these studies to children, Zhang et al. used ruxolitinib to treat 12 pediatric patients with newly diagnosed secondary HLH, including eight with EBV-triggered disease (58). Ten patients (83%) experienced a favorable response at 28 days, with eight exhibiting a CR. Among these eight, seven (88%) maintained the CR for more than 6 months. The remaining four patients did not respond or deteriorated after ruxolitinib treatment, requiring subsequent therapy according to the HLH-94 protocol. Together, these studies suggest that ruxolitinib may prove useful for the initial treatment of secondary HLH. Nevertheless, the optimal dose and schedule of ruxolitinib administration remain to be determined. In addition, studies examining regimens including ruxolitinib and other HLH-directed agents for the treatment of infants and young children with primary HLH are needed to better understand the utility and safety of ruxolitinib in these settings.
The treatment of primary HLH has advanced tremendously over the last 30 years from a disease that was uniformly fatal to one where the majority of patients can now be cured. Nevertheless, there are still many patients with HLH who succumb to the disease or to the complications of its treatment. Therefore, novel approaches to therapy are desperately needed. Thanks to increased understanding of the roles played by cytokines in fueling the fire of HLH, studies of cytokine-targeting agents such as ruxolitinib have been initiated with emerging data suggesting that some of these agents, including ruxolitinib, represent potentially effective means to lessen inflammation in this disease. However, it is not yet possible to discern the true efficacy of ruxolitinib for children or adults with HLH without prospective clinical trials, preferably using standardized definitions of disease activity and response and comparing with current gold-standard therapies.
The cytokine patterns observed in HLH closely resemble those seen in other hyperinflammatory conditions, such as the CRS that commonly follows treatment with T cell-based cancer immunotherapies (61). Notably, treatment with ruxolitinib in a pre-clinical xenograft model prevented the onset of CRS after administration of CD123-directed CAR-T cells and it did so without affecting the anti-tumor activity of the CAR-T cells (62). Therefore, it is possible that ruxolitinib or other JAK inhibitors could prove beneficial in ameliorating the signs and symptoms of immunotherapy-associated CRS.
Similarly, a cytokine storm has been observed in patients infected by the novel coronavirus SARS-CoV-2, where there is upregulation of cytokines including IL-1 and IL-6 (63). Accordingly, anakinra, tocilizumab, and ruxolitinib are being trialed for this disease (64, 65). Of late, studies have reported that treatment of COVID-19 patients with baricitinib, a structural analog of ruxolitinib, reduces serum cytokine levels, restores lymphocyte counts, increases the levels of antibodies reactive against the SARS-CoV2 spike protein, and lessens viral burden (66). In line with these findings, patients with COVID-19 and moderate pneumonia who receive treatment with baricitinib exhibit improved clinical status, reduced need for supportive care and ICU admission, and decreased recovery time (66–68). Based on these data, baricitinib was granted emergency use authorization by the FDA to be given in combination with the anti-viral agent remdesivir for the treatment of suspected or laboratory confirmed COVID-19 in hospitalized adults and pediatric patients 2 years of age or older who require supplemental oxygen, invasive mechanical ventilation, or extracorporeal membrane oxygenation (69). All told, the future is looking brighter for patients with HLH and related disorders of the immune system thanks to the availability of ruxolitinib and other inhibitors of the JAK-STAT pathway.
CK and SA wrote the manuscript. KN: reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by grants from the American Lebanese Syrian Associated Charities.
KN receives research funding from Incyte Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Joshua Stokes, Manager of Graphic Arts at St. Jude Biomedical Communications, for drawing Figure 1.
2. Janka GE, Lehmberg K. Hemophagocytic syndromes–an update. Blood Rev. (2014) 28:135–42. doi: 10.1016/j.blre.2014.03.002
3. Sammer CE, Lykens K, Singh KP, Mains DA, Lackan NA. What is patient safety culture? A review of the literature. J Nurs Scholarsh. (2010) 42:156–65. doi: 10.1111/j.1547-5069.2009.01330.x
4. Niece JA, Rogers ZR, Ahmad N, Langevin AM, McClain KL. Hemophagocytic lymphohistiocytosis in Texas, observations on ethnicity and race. Pediatr Blood Cancer. (2010) 54:424–8. doi: 10.1002/pbc.22359
5. Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. (1991) 80:428–35. doi: 10.1111/j.1651-2227.1991.tb11878.x
6. Usmani GN, Woda BA, Newburger PE. Advances in understanding the pathogenesis of HLH. Br J Haematol. (2013) 161:609–22. doi: 10.1111/bjh.12293
7. Ramos-Casals M, Brito-Zeron P, Lopez-Guillermo A, Khamashta MA, Bosch X. Adult haemophagocytic syndrome. Lancet. (2014) 383:1503–16. doi: 10.1016/S0140-6736(13)61048-X
8. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–96 e4. doi: 10.1016/j.jaci.2015.06.048
9. Chen X, Wang F, Zhang Y, Teng W, Wang M, Nie D, et al. Genetic variant spectrum in 265 Chinese patients with hemophagocytic lymphohistiocytosis: molecular analyses of PRF1, UNC13D, STX11, STXBP2, SH2D1A, and XIAP. Clin Genet. (2018) 94:200–12. doi: 10.1111/cge.13363
10. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
11. Behrens EM, Canna SW, Slade K, Rao S, Kreiger PA, Paessler M, et al. Repeated TLR9 stimulation results in macrophage activation syndrome-like disease in mice. J Clin Invest. (2011) 121:2264–77. doi: 10.1172/JCI43157
12. Weiss ES, Girard-Guyonvarc'h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood. (2018) 131:1442–55. doi: 10.1182/blood-2017-12-820852
13. Morimoto A, Nakazawa Y, Ishii E. Hemophagocytic lymphohistiocytosis: pathogenesis, diagnosis, and management. Pediatr Int. (2016) 58:817–25. doi: 10.1111/ped.13064
14. Akashi K, Hayashi S, Gondo H, Mizuno S, Harada M, Tamura K, et al. Involvement of interferon-gamma and macrophage colony-stimulating factor in pathogenesis of haemophagocytic lymphohistiocytosis in adults. Br J Haematol. (1994) 87:243–50. doi: 10.1111/j.1365-2141.1994.tb04905.x
15. Osugi Y, Hara J, Tagawa S, Takai K, Hosoi G, Matsuda Y, et al. Cytokine production regulating Th1 and Th2 cytokines in hemophagocytic lymphohistiocytosis. Blood. (1997) 89:4100–3. doi: 10.1182/blood.V89.11.4100
16. Yang SL, Xu XJ, Tang YM, Song H, Xu WQ, Zhao FY, et al. Associations between inflammatory cytokines and organ damage in pediatric patients with hemophagocytic lymphohistiocytosis. Cytokine. (2016) 85:14–7. doi: 10.1016/j.cyto.2016.05.022
17. Binder D, van den Broek MF, Kagi D, Bluethmann H, Fehr J, Hengartner H, et al. Aplastic anemia rescued by exhaustion of cytokine-secreting CD8+ T cells in persistent infection with lymphocytic choriomeningitis virus. J Exp Med. (1998) 187:1903–20. doi: 10.1084/jem.187.11.1903
18. Jordan MB, Hildeman D, Kappler J, Marrack P. An animal model of hemophagocytic lymphohistiocytosis (HLH): CD8+ T cells and interferon gamma are essential for the disorder. Blood. (2004) 104:735–43. doi: 10.1182/blood-2003-10-3413
19. Pachlopnik Schmid J, Ho CH, Chretien F, Lefebvre JM, Pivert G, Kosco-Vilbois M, et al. Neutralization of IFNgamma defeats haemophagocytosis in LCMV-infected perforin- and Rab27a-deficient mice. EMBO Mol Med. (2009) 1:112–24. doi: 10.1002/emmm.200900009
20. Burn TN, Weaver L, Rood JE, Chu N, Bodansky A, Kreiger PA, et al. Genetic Deficiency of interferon-gamma reveals interferon-gamma-independent manifestations of murine hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. (2020) 72:335–47. doi: 10.1002/art.41076
21. Trottestam H, Horne A, Arico M, Egeler RM, Filipovich AH, Gadner H, et al. Chemoimmunotherapy for hemophagocytic lymphohistiocytosis: long-term results of the HLH-94 treatment protocol. Blood. (2011) 118:4577–84. doi: 10.1182/blood-2011-06-356261
22. Yanagisawa R, Nakazawa Y, Matsuda K, Yasumi T, Kanegane H, Ohga S, et al. Outcomes in children with hemophagocytic lymphohistiocytosis treated using HLH-2004 protocol in Japan. Int J Hematol. (2019) 109:206–13. doi: 10.1007/s12185-018-02572-z
23. Bergsten E, Horne A, Arico M, Astigarraga I, Egeler RM, Filipovich AH, et al. Confirmed efficacy of etoposide and dexamethasone in HLH treatment: long-term results of the cooperative HLH-2004 study. Blood. (2017) 130:2728–38. doi: 10.1182/blood-2017-06-788349
24. Marsh RA, Allen CE, McClain KL, Weinstein JL, Kanter J, Skiles J, et al. Salvage therapy of refractory hemophagocytic lymphohistiocytosis with alemtuzumab. Pediatr Blood Cancer. (2013) 60:101–9. doi: 10.1002/pbc.24188
25. Locatelli F, Jordan MB, Allen C, Cesaro S, Rizzari C, Rao A, et al. Emapalumab in children with primary hemophagocytic lymphohistiocytosis. N Engl J Med. (2020) 382:1811–22. doi: 10.1056/NEJMoa1911326
26. Eloseily EM, Weiser P, Crayne CB, Haines H, Mannion ML, Stoll ML, et al. Benefit of anakinra in treating pediatric secondary hemophagocytic lymphohistiocytosis. Arthritis Rheumatol. (2020) 72:326–34. doi: 10.1002/art.41103
27. Le RQ, Li L, Yuan W, Shord SS, Nie L, Habtemariam BA, et al. FDA approval summary: tocilizumab for treatment of chimeric antigen receptor T cell-induced severe or life-threatening cytokine release syndrome. Oncologist. (2018) 23:943–7. doi: 10.1634/theoncologist.2018-0028
28. Dufranc E, Del Bello A, Belliere J, Kamar N, Faguer S, Ts group. IL6-R blocking with tocilizumab in critically ill patients with hemophagocytic syndrome. Crit Care. (2020) 24:166. doi: 10.1186/s13054-020-02878-7
29. Ohmura S-I, Uehara K, Yamabe T, Tamechika S, Maeda S, Naniwa T. Successful use of short-term add-on tocilizumab for refractory adult-onset still's disease with macrophage activation syndrome despite treatment with high-dose glucocorticoids, cyclosporine, and etoposide. Mod Rheumatol Case Rep. (2020) 4:202–7. doi: 10.1080/24725625.2020.1741073
30. O'Shea JJ, Schwartz DM, Villarino AV, Gadina M, McInnes IB, Laurence A. The JAK-STAT pathway: impact on human disease and therapeutic intervention. Annu Rev Med. (2015) 66:311–28. doi: 10.1146/annurev-med-051113-024537
31. Gadina M, Le MT, Schwartz DM, Silvennoinen O, Nakayamada S, Yamaoka K, et al. Janus kinases to jakinibs: from basic insights to clinical practice. Rheumatology (Oxford). (2019) 58(Suppl. 1):i4–16. doi: 10.1093/rheumatology/key432
32. Mascarenhas J, Hoffman R. Ruxolitinib, the first FDA approved therapy for the treatment of myelofibrosis. Clin Cancer Res. (2012) 18:3008–14. doi: 10.1158/1078-0432.CCR-11-3145
33. Verstovsek S, Mesa RA, Gotlib J, Gupta V, DiPersio JF, Catalano JV, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol. (2017) 10:55. doi: 10.1186/s13045-017-0417-z
34. Risitano AM, Peffault de Latour R. Ruxolitinib for steroid-resistant acute GVH D. Blood. (2020) 135:1721–2. doi: 10.1182/blood.2020005364
35. Morozova EV, Barabanshikova MV, Moiseev IS, Shakirova AI, Barhatov IM, Ushal IE, et al. A prospective pilot study of graft-vs.-host disease prophylaxis with post-transplantation cyclophosphamide and ruxolitinib in patients with myelofibrosis. Acta Haematol. (2020). doi: 10.1159/000506758. [Epub ahead of print].
36. Zhao Y, Shi J, Luo Y, Gao F, Tan Y, Lai X, et al. Calcineurin inhibitors replacement by ruxolitinib as graft-vs.-host disease prophylaxis for patients after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. (2020) 26:e128–33. doi: 10.1016/j.bbmt.2020.01.012
37. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Schussler E, Weinacht KG, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. (2018) 142:1665–9. doi: 10.1016/j.jaci.2018.07.020
38. Canna SW, Wrobel J, Chu N, Kreiger PA, Paessler M, Behrens EM. Interferon-gamma mediates anemia but is dispensable for fulminant toll-like receptor 9-induced macrophage activation syndrome and hemophagocytosis in mice. Arthritis Rheum. (2013) 65:1764–75. doi: 10.1002/art.37958
39. Das R, Guan P, Sprague L, Verbist K, Tedrick P, An QA, et al. Janus kinase inhibition lessens inflammation and ameliorates disease in murine models of hemophagocytic lymphohistiocytosis. Blood. (2016) 127:1666–75. doi: 10.1182/blood-2015-12-684399
40. Maschalidi S, Sepulveda FE, Garrigue A, Fischer A, de Saint Basile G. Therapeutic effect of JAK1/2 blockade on the manifestations of hemophagocytic lymphohistiocytosis in mice. Blood. (2016) 128:60–71. doi: 10.1182/blood-2016-02-700013
41. Albeituni S, Verbist KC, Tedrick PE, Tillman H, Picarsic J, Bassett R, et al. Mechanisms of action of ruxolitinib in murine models of hemophagocytic lymphohistiocytosis. Blood. (2019) 134:147–59. doi: 10.1182/blood.2019000761
42. Meyer LK, Verbist KC, Albeituni S, Scull BP, Bassett RC, Stroh AN, et al. JAK/STAT pathway inhibition sensitizes CD8 T cells to dexamethasone-induced apoptosis in hyperinflammation. Blood. (2020) 136:657–68. doi: 10.1182/blood.2020006075
43. Acosta SG, Rincon MJ. Ruxolitinib as first-line therapy in secondary hemophagocytic lymphohistiocytosis and HIV infection. Int J Hematol. (2020) 112:418–21. doi: 10.1007/s12185-020-02882-1
44. Ahmed A, Merrill SA, Alsawah F, Bockenstedt P, Campagnaro E, Devata S, et al. Ruxolitinib in adult patients with secondary haemophagocytic lymphohistiocytosis: an open-label, single-centre, pilot trial. Lancet Haematol. (2019) 6:e630–7. doi: 10.1016/S2352-3026(19)30156-5
45. Broglie L, Pommert L, Rao S, Thakar M, Phelan R, Margolis D, et al. Ruxolitinib for treatment of refractory hemophagocytic lymphohistiocytosis. Blood Adv. (2017) 1:1533–6. doi: 10.1182/bloodadvances.2017007526
46. Fuchs A, Orth HM, Germing U, Kondakci M, Holtfreter M, Feldt T, et al. Falciparum malaria-induced secondary hemophagocytic lymphohistiocytosis successfully treated with ruxolitinib. Int J Infect Dis. (2020) 100:382–5. doi: 10.1016/j.ijid.2020.07.062
47. Goldsmith SR, Saif Ur Rehman S, Shirai CL, Vij K, DiPersio JF. Resolution of secondary hemophagocytic lymphohistiocytosis after treatment with the JAK1/2 inhibitor ruxolitinib. Blood Adv. (2019) 3:4131–5. doi: 10.1182/bloodadvances.2019000898
48. Jianguo L, Zhixuan Z, Rong L, Xiaodong S. Ruxolitinib in alleviating the cytokine storm of hemophagocytic lymphohistiocytosis. Pediatrics. (2020) 146:e20191301. doi: 10.1542/peds.2019-1301
49. Levy R, Fusaro M, Guerin F, Chetouani A, Moshous D, Fischer A, et al. Efficacy of ruxolitinib in subcutaneous panniculitis-like T-cell lymphoma and hemophagocytic lymphohistiocytosis. Blood Adv. (2020) 4:1383–7. doi: 10.1182/bloodadvances.2020001497
50. Ramanan KM, Uppuluri R, Ravichandran N, Patel S, Swaminathan VV, Jayakumar I, et al. Successful remission induction in refractory familial hemophagocytic lymphohistiocytosis with ruxolitinib as a bridge to hematopoietic stem cell transplantation. Pediatr Blood Cancer. (2020) 67:e28071. doi: 10.1002/pbc.28071
51. Sin JH, Zangardi ML. Ruxolitinib for secondary hemophagocytic lymphohistiocytosis: first case report. Hematol Oncol Stem Cell Ther. (2019) 12:166–70. doi: 10.1016/j.hemonc.2017.07.002
52. Slostad J, Hoversten P, Haddox CL, Cisak K, Paludo J, Tefferi A. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a single patient experience. Am J Hematol. (2018) 93:E47–9. doi: 10.1002/ajh.24971
53. Trantham T, Auten J, Muluneh B, Van Deventer H. Ruxolitinib for the treatment of lymphoma-associated hemophagocytic lymphohistiocytosis: a cautionary tale. J Oncol Pharm Pract. (2020) 26:1005–8. doi: 10.1177/1078155219878774
54. Wang J, Wang Y, Wu L, Wang X, Jin Z, Gao Z, et al. Ruxolitinib for refractory/relapsed hemophagocytic lymphohistiocytosis. Haematologica. (2020) 105:e210–2. doi: 10.3324/haematol.2019.222471
55. Wang H, Gu J, Liang X, Mao X, Wang Z, Huang W. Low dose ruxolitinib plus HLH-94 protocol: A potential choice for secondary HLH. Semin Hematol. (2020) 57:26–30. doi: 10.1053/j.seminhematol.2018.07.006
56. Wei A, Ma H, Li Z, Zhang L, Zhang Q, Wang D, et al. Short-term effectiveness of ruxolitinib in the treatment of recurrent or refractory hemophagocytic lymphohistiocytosis in children. Int J Hematol. (2020) 112:568–76. doi: 10.1007/s12185-020-02936-4
57. Zandvakili I, Conboy CB, Ayed AO, Cathcart-Rake EJ, Tefferi A. Ruxolitinib as first-line treatment in secondary hemophagocytic lymphohistiocytosis: a second experience. Am J Hematol. (2018) 93:E123–5. doi: 10.1002/ajh.25063
58. Zhang Q, Wei A, Ma HH, Zhang L, Lian HY, Wang D, et al. A pilot study of ruxolitinib as a front-line therapy for 12 children with secondary hemophagocytic lymphohistiocytosis. Haematologica. (2020). doi: 10.3324/haematol.2020.253781. [Epub ahead of print].
59. Zhao Y, Shi J, Li X, Wang J, Sun J, Zhou J, et al. Salvage therapy with dose-escalating ruxolitinib as a bridge to allogeneic stem cell transplantation for refractory hemophagocytic lymphohistiocytosis. Bone Marrow Transplant. (2020) 55:824–6. doi: 10.1038/s41409-019-0577-2
60. Zhou L, Liu Y, Wen Z, Yang S, Li M, Zhu Q, et al. Ruxolitinib combined with doxorubicin, etoposide, and dexamethasone for the treatment of the lymphoma-associated hemophagocytic syndrome. J Cancer Res Clin Oncol. (2020) 146:3063–74. doi: 10.1007/s00432-020-03301-y
61. Hashmi H, Bachmeier C, Chavez JC, Song J, Hussaini M, Krivenko G, et al. Haemophagocytic lymphohistiocytosis has variable time to onset following CD19 chimeric antigen receptor T cell therapy. Br J Haematol. (2019) 187:e35–8. doi: 10.1111/bjh.16155
62. Saad Kenderian S, Shestova O, Kim M, Klichinsky M, Chen F, Kengle N, et al. 2 - ruxolitinib prevents cytokine release syndrome after car T-cell therapy without impairing the anti-tumor effect in a xenograft model. Biol Blood Marrow Transplant. (2017) 23:S19–20. doi: 10.1016/j.bbmt.2016.12.003
63. Buckley LF, Wohlford GF, Ting C, Alahmed A, Van Tassell BW, Abbate A, et al. Role for anti-cytokine therapies in severe coronavirus disease 2019. Crit Care Explor. (2020) 2:e0178. doi: 10.1097/CCE.0000000000000178
64. Capochiani E, Frediani B, Iervasi G, Paolicchi A, Sani S, Roncucci P, et al. Ruxolitinib rapidly reduces acute respiratory distress syndrome in COVID-19 disease. analysis of data collection from RESPIRE protocol. Front Med (Lausanne). (2020) 7:466. doi: 10.3389/fmed.2020.00466
65. Cao Y, Wei J, Zou L, Jiang T, Wang G, Chen L, et al. Ruxolitinib in treatment of severe coronavirus disease 2019 (COVID-19): a multicenter, single-blind, randomized controlled trial. J Allergy Clin Immunol. (2020) 146:137–46 e3. doi: 10.1016/j.jaci.2020.05.019
66. Bronte V, Ugel S, Tinazzi E, Vella A, De Sanctis F, Cane S, et al. Baricitinib restrains the immune dysregulation in patients with severe COVID-19. J Clin Invest. (2020) 130:6409–16. doi: 10.1172/JCI141772
67. Cantini F, Niccoli L, Nannini C, Matarrese D, Natale MED, Lotti P, et al. Beneficial impact of Baricitinib in COVID-19 moderate pneumonia; multicentre study. J Infect. (2020) 81:647–79. doi: 10.1016/j.jinf.2020.06.052
68. Kalil AC, Patterson TF, Mehta AK, Tomashek KM, Wolfe CR, Ghazaryan V, et al. Baricitinib plus remdesivir for hospitalized adults with Covid-19. N Engl J Med. (2020). doi: 10.1056/NEJMoa2031994. [Epub ahead of print].
69. US Food and Drug Administration. (2020). Available online at: https://www.fda.gov/media/143822/download (accessed December 15, 2020).
Keywords: Hemophagocytic lymphohistiocytosis, ruxolitinib, jak-stat, inflammation, cytokine storm
Citation: Keenan C, Nichols KE and Albeituni S (2021) Use of the JAK Inhibitor Ruxolitinib in the Treatment of Hemophagocytic Lymphohistiocytosis. Front. Immunol. 12:614704. doi: 10.3389/fimmu.2021.614704
Received: 06 October 2020; Accepted: 22 January 2021;
Published: 16 February 2021.
Edited by:
Siobhan Oisin Burns, University College London, United KingdomReviewed by:
Lisa Renee Forbes, Baylor College of Medicine, United StatesCopyright © 2021 Keenan, Nichols and Albeituni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sabrin Albeituni, c2FicmluLmFsYmVpdHVuaUBzdGp1ZGUub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.