
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 March 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.612826
This article is part of the Research TopicDeciphering the Microbiome-Immunity-Cancer AxisView all 9 articles
 Michelle Hanus1
Michelle Hanus1 Daniela Parada-Venegas1
Daniela Parada-Venegas1 Glauben Landskron1
Glauben Landskron1 Ana Maria Wielandt2
Ana Maria Wielandt2 Claudia Hurtado2
Claudia Hurtado2 Karin Alvarez3
Karin Alvarez3 Marcela A. Hermoso1
Marcela A. Hermoso1 Francisco López-Köstner3*
Francisco López-Köstner3* Marjorie De la Fuente2*
Marjorie De la Fuente2*Colorectal cancer (CRC) is one of the most common cancers worldwide. As with other cancers, CRC is a multifactorial disease due to the combined effect of genetic and environmental factors. Most cases are sporadic, but a small proportion is hereditary, estimated at around 5-10%. In both, the tumor interacts with heterogeneous cell populations, such as endothelial, stromal, and immune cells, secreting different signals (cytokines, chemokines or growth factors) to generate a favorable tumor microenvironment for cancer cell invasion and metastasis. There is ample evidence that inflammatory processes have a role in carcinogenesis and tumor progression in CCR. Different profiles of cell activation of the tumor microenvironment can promote pro or anti-tumor pathways; hence they are studied as a key target for the control of cancer progression. Additionally, the intestinal mucosa is in close contact with a microorganism community, including bacteria, bacteriophages, viruses, archaea, and fungi composing the gut microbiota. Aberrant composition of this microbiota, together with alteration in the diet‐derived microbial metabolites content (such as butyrate and polyamines) and environmental compounds has been related to CRC. Some bacteria, such as pks+ Escherichia coli or Fusobacterium nucleatum, are involved in colorectal carcinogenesis through different pathomechanisms including the induction of genetic mutations in epithelial cells and modulation of tumor microenvironment. Epithelial and immune cells from intestinal mucosa have Pattern-recognition receptors and G-protein coupled receptors (receptor of butyrate), suggesting that their activation can be regulated by intestinal microbiota and metabolites. In this review, we discuss how dynamics in the gut microbiota, their metabolites, and tumor microenvironment interplays in sporadic and hereditary CRC, modulating tumor progression.
Colorectal cancer (CRC) is the third most common cancer in both sex worldwide with an estimated 1.8 million new cases and mortality rates in 2018 over 800,000 (1). The vast majority of CRC cases are classified as sporadic and occur in average risk patients, with no family history or an apparent genetic predisposition demonstrated, mostly affecting people older than 50 (2). However, heredity represents a significant cause of CRC, in which 20-30% are familial cases, with 5-10% linked to inherited variants in cancer-predisposing genes (3). The most common hereditary CRC is Lynch syndrome, accounting for 2-3% of cases, followed by familial adenomatous polyposis (FAP) responsible for 0.5-1% of cases, and other hereditary variants such as MUTYH-associated polyposis (MAP), Peutz-Jeghers syndrome, among others, represent <1% of cases (3).
Diverse studies have highlighted the role of gut commensal microbiota in host physiology and diseases such as cancer by modulating the immune response, genetic damage, and apoptosis (4–6). The intestinal tract is an extensive surface in contact with lumen antigens, including more than 100 trillion microorganisms, predominating the phyla Firmicutes and Bacteroidetes (7). Accordingly, the mucosa has a complex immune system maintaining the homeostasis in steady conditions but swiftly responding to an insult (8). Additionally, alteration in the gut microbiota disrupts the epithelial barrier promoting inflammation and tumorigenesis-associated pathways (9).
Cancer, rather than being formed by homogeneous malignant cells, contains a heterogeneous cell population, such as endothelial, stromal, and immune cells, secreting soluble signals (cytokines, chemokines, or growth factors), interacting with tumor cells, generating a favorable microenvironment to support tumor growth and progression (10, 11). Moreover, tumor and microenvironment cells respond to signals from microbiota, regulating multiple host pathways related to carcinogenic processes, such as the induction of mutations in infected cells by a pathogenic E. coli strain or epithelial invasion by Fusobacterium nucleatum (12, 13). Additionally, microbiota, through the production of metabolites, such as short-chain fatty acids (SCFAs) or polyamines (4, 14), controls tumor cell function and microenvironment.
Here we aim to review the role of tumor microenvironment components in sporadic and hereditary CRC, with an emphasis in interactions between microbiota, immune system, and diet-derived metabolites.
The tumor microenvironment (TME) has a wide diversity of molecules and cell types, including immune cells, fibroblast, adipocytes, endothelial cell and microbiome (6, 15). The immune system is particularly composed of innate immune cells such as neutrophils, macrophages, dendritic cells (DCs), mast cells and natural killer cells (NK), and adaptive immune cells such as T and B lymphocytes (16), participating in prevention and promotion of tumor development, having pro and anti-tumor functions (17).
Immunosurveillance refers to the role of the immune system in recognizing antigens from transformed cells, thus generating memory and effector cells, which seek out and control generation of new tumor cells (18).
Tumor cells undergo a selection process called immunoediting consisting of 3 stages: elimination, equilibrium, and escape. In the first phase, the antitumor immune response eliminates initiating tumor cells; the tumor then evolves into a static phase (equilibrium) in which some malignant cells avoid the immune response, without tumor elimination. Finally, the resistant clones manage to evade the immune system acquiring pro-tumorigenic properties and reducing their immunogenicity, allowing tumor development and clinical manifestations (19).
Lymphocytes are key cells in the tumor microenvironment, and according to their profile have different functions in cancer progression: CD8+ T cells lyse tumor cells and release cytokines enhancing cytotoxic responses, such as IFNγ; CD4+ Th1 T cells release cytokines improving lymphocyte cytotoxic function; regulatory CD4+ T cells (Treg) are immunosuppressive cells, preventing chronic inflammation and maintaining immune tolerance, by suppressing effector T cell proliferation and activation (17, 20). B cells recognize tumor antigens and produce specific antibodies against the tumor with the cooperation of helper T cells, decreasing tumor progression (21).
Currently, the use of tumor infiltrated lymphocyte quantification to stratify patients and predict survival is rising, with a score system called Immunoscore™, based on the CD3+ CD8+ T cells and memory T (CD45RO+) density in CRC (22–24). Alternatively, high density of FOXP3+ cells has been described in CRC (25), even though the relation of FOXP3+ Treg with CRC prognosis is still controversial (favorable prognosis (25–28); poor prognosis (29, 30). These differences can account for a heterogeneous FOXP3+ population, and not necessarily with a suppressor T cell phenotype (31, 32) therefore deeper studies on the function of infiltrating FOXP3+ subpopulations and their interaction with other cells of the tumor microenvironment are required to understand their role in CRC.
NK cells eliminate tumor cells lacking major histocompatibility complex class I (MHC-I) expression, by Fas/FasL pathway, by releasing perforin and granzyme, or DCs and macrophages-activating cytokines (33). MHC-I loss of expression is a common alteration in CRC (34). However, CRC tissue shows low NK cell content compared to adjacent normal tissue, independent of MHC-I expression (35), proposing poor NK infiltration to the TME as a cancer evasion mechanism in immune surveillance. Non-classical MHC class I molecule expression avoid NK antitumor function, such as human leukocyte antigen HLA-E, binding to NK cell inhibitory receptors and suppressing cytotoxic activity in CRC (36). Furthermore, peripheral NK cells from CRC patients have a dysregulated phenotype with decreased cytotoxic function, thus allowing tumor cells dissemination (37).
Tumor-associated macrophages (TAMs) are usually associated with M2-profile macrophages, possess immunosuppressive properties, promote tumor progression, and, M2-TAMs marker expression is a poor prognostic factor in CRC (38).
Other essential cells in the tumor microenvironment are myeloid-derived suppressor cells (MDSCs), that probably through T reg cell induction, in addition to NK and T effector cell suppression, inhibit antitumor responses (39). In CRC their increase in both tumor and peripheral blood correlates with stage and metastasis (39, 40), revealing their immunosuppressive activity.
Additionally, tumor associated neutrophils (TAN) appear to be an important component of tumor-infiltrating cell populations in CRC. Indeed, high TAN content is associated with improved survival in CRC patients (41–43) possibly indicating a positive response to 5-FU-based chemotherapy (44). Neutrophils in the tumor microenvironment can also promote anti-tumor responses mediated by macrophages (45) or CD8+ T cells (42), although contradictory results have been reported in mouse models. In mice with inducible colon adenoma, T‐cell suppression is mediated by neutrophil‐secreted metalloproteinase activation, thus considered an immunosuppressive mechanism in CRC (46). Moreover, neutrophils promote hepatic metastasis growth and angiogenesis mediated by fibroblast growth factor 2 (FGF2) (47). Additionally, in a model of colitis-associated colorectal cancer (CAC), an anti-neutrophil antibody reduced the number of tumors and intracolonic neutrophils infiltration along with MMP-9 mRNA expression, suggesting TANs promote tumor development (48).
Contradictions between murine models and human sample data possibly associate with differing neutrophil functions and biology, along with tumor formation, as mice form faster and do not fully mirror human tumor development stages and cellular interactions (49). Consequently, this consideration should be taken with studies from murine-human models.
There exists vast information regarding immunity in CRC, revealing a complex interaction between components of the immune system and the tumor. The immune system plays a double role, at the beginning and in the development of CRC, where immune-tumor interactions present opportunities for creating treatments preventing tumor development or improving current treatments.
A healthy gut microbiome varies according to the high grade of interindividual differences, influenced by diet, lifestyle, age, gender, and geography. In 2018, the International Life Sciences Institute (ILSI) of North America workshop concluded that “mechanistic links of specific changes in gut microbiome structure with function or markers of human health are not yet established” (50). However, there is consensus that the healthy gut microbiome goes through a stable and resilient equilibrium state (51) with high species diversity (50–52) predominantly composed of Bacteroidetes, Firmicutes, and Actinobacteria, exhibiting differences in distribution between mucosal-to-luminal and proximal-to-distal (53–55). The gut microbiota is influenced by the birth mode, breastfeeding (56), gender (hormones) (57, 58), pregnancy (59, 60), lifestyle (e.g. sedentary or sports practice) (61), diet (52), age (62), among others. Furthermore, diet patterns enriched in animal protein/fat or carbohydrates are associated with Bacteroides and Prevotella enterotypes, respectively (63). The above was confirmed by the fecal microbiome characterization in healthy subjects (children and adults) from Amazonas of Venezuela, rural Malawi, and the US metropolitan area revealed prominent differences between age groups and between rural and urban cohorts (52).
The first reports revealing CRC-associated dysbiosis date from 2011, which is related to the expansion of next generation sequencing (NGS) techniques (64–66) and highlighting the overrepresentation of the anaerobic bacterium Fusobacterium nucleatum in CRC (65, 66).
CRC patient gut microbiota differs from healthy individuals, highlighting a reduced diversity (67) at fecal and tumor levels (68), with the phyla Proteobacteria, Fusobacteria, and Lentisphaerae enrichment in fecal samples, and Firmicutes and Actinobacteria reduced (67). The bacterial genera Fusobacterium, Peptostreptococcus, Porphyromonas, Prevotella, Parvimonas, Bacteroides, and Gemella, are prominently enriched in CRC (69) and in contrast, the genera Roseburia, Clostridium, Faecalibacterium and Bifidobacterium decreased (68).
A metagenomics study of fecal samples from patients with CRC showed a stage-dependent microbiota variation, with some species increasing in abundance through tumor development (Fusobacterium nucleatum, Solobacterium moorei, Peptostreptococcus stomatis, Peptostreptococcus anaerobius, Lactobacillus sanfranciscensis, Parvimonas micra and Gemella morbillorum), and some species stage-specific. The Atopobium parvulum, Actinomyces odontolyticus, Desulfovibrio longreachensis and Phascolarctobacterium succinatutens species increased only in early stage S0, in contrast, Colinsella aerofaciens, Porphyromonas uenonis and Dorea longicatena increased only in late stages III/IV (70). Similar studies with tissue samples demonstrated that early stages have an enrichment of Fusobacterium, Leptotrichia, Gemella and Parvimonas, and reduction of Blautia and Bacteroides. Furthermore, in CRC pre-tumor lesions an enrichment of Pseudomonas veronii and E. coli exists (71), suggesting these species have oncogenic potential.
A reduction of some bacterial species during gut tumorigenesis has also been reported. One example is Faecalibaculum rodentium and its human equivalent Holdemanella biformis belonging to the Erysipelotrichaceae family, showing a reduced fecal content in early phases of tumorigenesis from ApcMin/+ model and patients with large colorectal adenoma, respectively (72). These bacteria block tumor proliferation by mediating the production of SCFAs (see later in Short-Chain Fatty Acids (SCFAs) section), (especially butyrate) inhibiting HDAC in adenomas, by increasing H3 histone acetylation and downmodulating the calcineurin-NFATc3 pathway (72). Additionally, gut microbiota relates to tumor immune cell infiltration, where specific bacteria genera, such as Alloprevotella, Treponema, and Desulfovibrio, are enriched in tumors with high T cell marker (CD3) content and accompany prolonged CRC patient survival. Whereas, Prevotella, Bacteroides, and Fretibacterium, are overrepresented in CD3 low tumors by regulating chemokine expression from tumor cells (73).
All these antecedents suggest that intestinal microbiota can give both beneficial and adverse effects on gut physiology contributing to health or disease susceptibility. This may therefore lead to novel strategies enriching specific kinds of bacteria or metabolite production favoring anti-tumor immune cell recruitment or tumor proliferation blocking.
The evidence of the microbiota role in cancer development of hereditary CRC patients is scarce.
In the inherited condition FAP (caused mainly by a germline mutation of the APC tumor suppressor gene) (3), intestinal mucosa with precursor lesions (polyps) presents bacterial biofilms composed predominantly by pks+ Escherichia coli and enterotoxigenic Bacteroides fragilis. Additionally, co-colonization of both bacteria in a CRC murine model accelerates tumor development and increased mortality (associated with high IL-17 levels and DNA damage), suggesting pks+ Escherichia coli and enterotoxigenic Bacteroides fragilis act as protumorigenic bacteria in early colonic tumor development in genetically susceptible patients (74).
Fecal microbial patterns demonstrate that FAP patients carrying pathogenic APC mutations showing increased abundance of Fusobacterium mortiferum and a decreased representation of Faecalibacterium prausnitzii and Bifidofacterium pseudocatenulatur, compared to patients without an identified mutation (75). Moreover, increased seric metabolites (R)-3-Hydroxybutryric acid and 2-Hydroxyphenethylamine exists in patients carrying APC mutation, together with lower levels of 7-Ketocholesterol, DL-lactate, L-Pyroglutamic acid (75). Among other relationships, a positive correlation between Faecalibacterium prausnitzii and cortisol exists; however, more evidence is needed to clarify the association between metabolites and gut microbiota (75).
In characterizing Lynch syndrome patients’ microbiome, an fecal over-representation of Faecalibacterium prausnitzii, Parabacteroides distasonis, Ruminococcus bromii, Bacteroides plebeius, Bacteroides fragilis and Bacteroides uniformis was identified in both postoperative LS female patients with colorectal syndrome or LS extracolonic cancer, however distinct to controls (76). Microbiota’s role in early stages of carcinogenesis, was prospectively evaluated in fecal and mucosa samples from LS patients (carriers of pathogenic germline MMR mutation) and demonstrated: 1) colectomy and CRC history has the largest effect on microbiome profiles; 2) microbial changes are similar in Lynch adenoma and CRC; 3) fecal microbial transcriptional activity is a weak predictor of adenomas development. Although microbiome monitoring does not appear to be effective in early prediction of adenomas, the possibility exists of early microbiota changes in LS neoplasia (77).
Bacteria are involved in colorectal carcinogenesis through pathomechanisms such as: tissue invasion, local immune response modulation and metabolites or toxins secretion (Table 1). The most relevant CRC-associated bacteria are Fusobacterium nucleatum, Peptostreptococcus ssp., Porphyromonas ssp., Prevotella intermedia, Parvimonas micra, Bacteroides fragilis and Gemella morbillorum (69).

Table 1 Bacteria involved in colorectal carcinogenesis.
Next, the role of pks+ Escherichia coli and Fusobacterium nucleatum is clarified in colorectal cancer, both being examples of bacteria thoroughly studied with mechanisms associated to CRC carcinogenesis.
E. coli is a commensal organism widely distributed along the gut, comprised by four phylogenetic groups (A, B1, B2 and D), with strains possibly linked to CRC as they produce inflammation and secrete toxins, such as cyclomodulins (toxins interfering with the eukaryotic cell cycle) (97). Among them, colibactin, is synthesized by 19 encoded genes within the 54-kilobase genomic island polyketide synthase (pks) (85), and relevantly pks+ E. coli are abundant in CRC and IBD patients (98).
Azoxymethane (AOM) administered to IL10-/- germ-free mice colonized with pks+ E. coli or E. faecalis induces aggressive inflammation; however, only pks+ E. coli developed invasive adenocarcinoma, demonstrating that E. coli-specific factors promote colitis-associated cancer (98), as colibactin induces DNA double-strand breaks and chromosomal instability in human cells (85, 86, 99). Additionally, prolonged exposure to pks+ E. coli in human intestinal organoids induces a DNA mutational signature characterized by random single-base substitutions, deletions and insertions (100) (Figure 1).
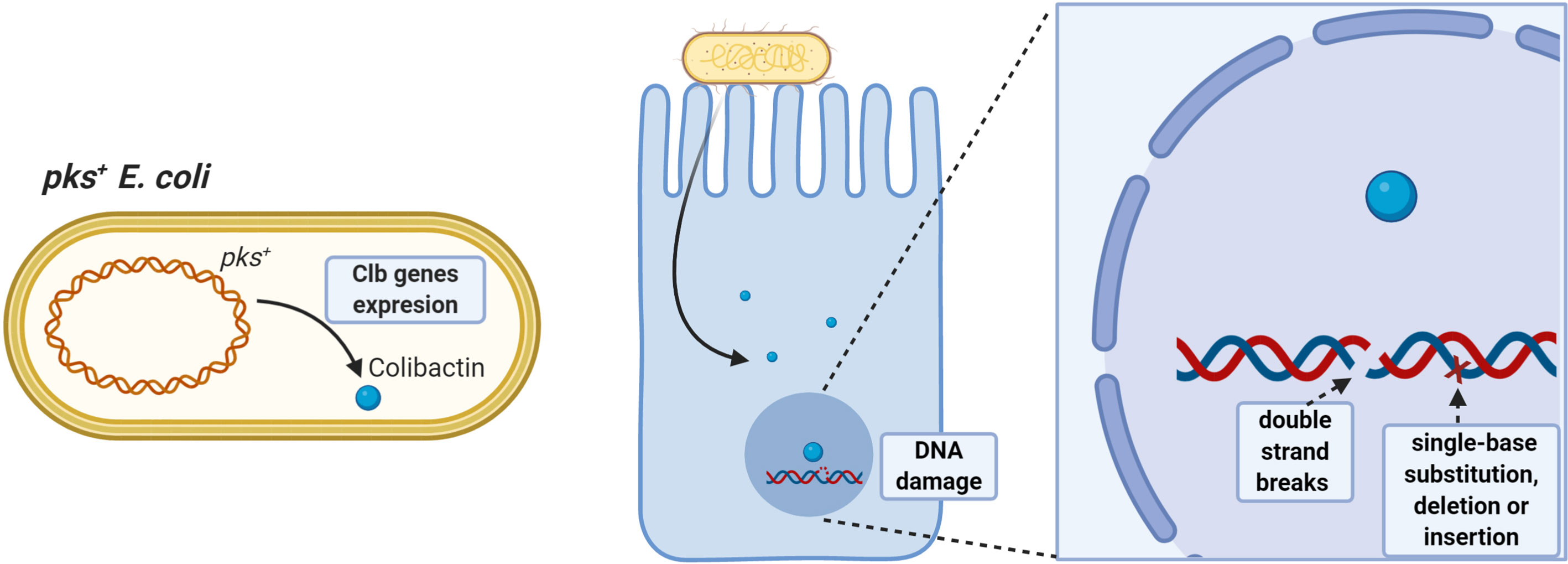
Figure 1 Pro-tumorigenic effects of pks+ E. coli. Strains having the pathogenicity island pks can synthesize colibactin toxin having oncogenic potential. Colibactin damages colonocyte DNA by inducing double-stranded DNA breaks and single-base substitution, deletion, and insertion mutations, favoring accumulation of damage and increasing the risk of malignant cell transformation.
Decreased tumor-infiltrating lymphocytes (TILs) in the invasive CRC margin is associated with pks+ E. coli (101). Moreover, APCMin/+ mice exposed to colibactin-producing E. coli exhibit more polyps and decreased CD3+ CD8+ T-cells than noninfected animals or infected with pks-lacking E. coli strains, suggesting colibactin induces a carcinogenic microenvironment (101).
Interestingly, microbial diversity is altered in mice receiving pks+ E. coli strain compared to non genotoxic-strain exposed mice, suggesting colibactin exert a direct effect on gut microbiota, additional to its other host effects (102).
Fusobacteria are a Gram-negative anaerobic bacilli, with Fusobacterium nucleatum a component of oral microbiota; although associated with oral, extraoral diseases (103), and colorectal adenomas and adenocarcinoma (65, 66, 104). Although detection rates of F. nucleatum in CRC patients differ widely due to methods used and samples analyzed (105) correlation between bacteria abundance and poor cancer-specific survival (106, 107) or lymph node metastasis (66) have been described, and is suggested as a prognostic marker.
F. nucleatum participates in carcinogenic processes through virulent factors (Figure 2), highlighting the adhesion protein FadA and the autotransporter protein Fap2. FadA allows bacteria attachment and invasion of E-cadherin-expressing cells (89) and induction of human CRC cell proliferation in a FadA-dependent ß-catenin signaling activation and proinflammatory response associated with NF-kB2 upregulation (89). Another FadA function is vascular endothelial VE-cadherin removal from cell-cell junctions, increasing endothelial cell permeability, thus allowing bacteria to cross junctions (108).
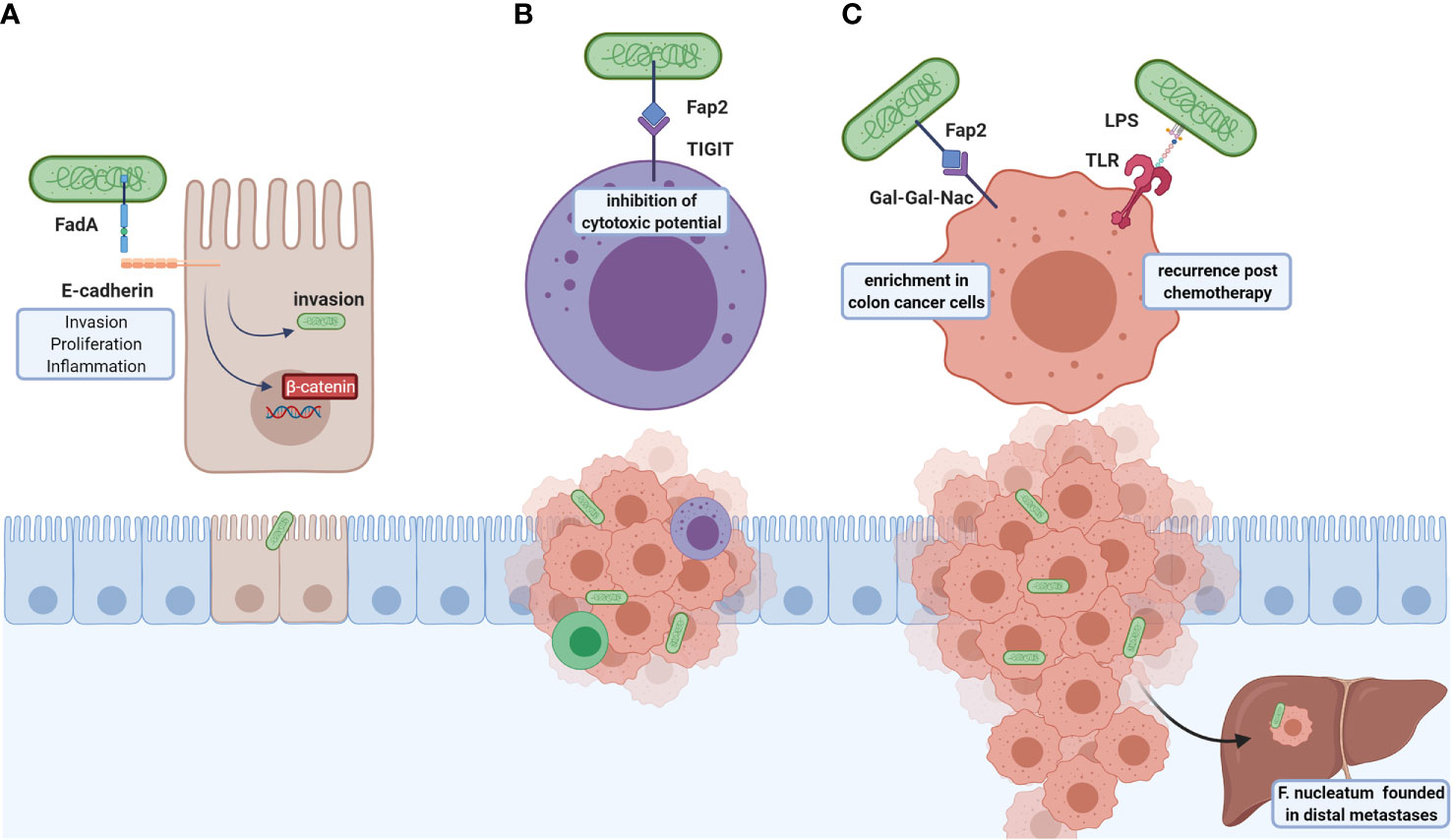
Figure 2 Roles of F. nucleatum in CRC tumoral development and metastasis. F. nucleatum virulence factors are FadA and Fap2: (A) FadA is an adhesin that binds to E-cadherin and allows bacterial invasion, which also induces the colonocyte proliferation through ß-catenin signaling and NFkB2-associated pro-inflammatory response. (B) Fap2 interacts with the TIGIT inhibitory receptor of NK cells resulting in cytotoxic inhibition, leading to immune evasion. (C) This bacterium associates with post-chemotherapy recurrence, suggested through LPS-TLR4 interaction activating autophagy, and altering chemotherapy response. Furthermore, Fap2 recognizes and binds to Gal-GalNac expressed in colorectal tumor cells; high F. nucleatum content found in distal metastases. The above-mentioned effects indicate that this bacterium participates in both carcinogenic and metastatic processes and may be a potential therapeutic target.
Additionally, F. nucleatum invasion promotes a proinflammatory response mediated by p38 or MAPK signaling in HEK293T cells (87), and mediated by ROS in Caco-2 cell line (88), both pathways involved in early tumorigenesis (109). Indeed, bacterium abundance is positively correlated to local cytokine gene expression, such as TNFα and IL10, in colorectal adenomas (110). In APCMin/+ mouse model, F. nucleatum exacerbates tumorigenesis and recruits tumor-infiltrating myeloid cells (granulocytes, macrophages, DCs, and MDSCs), and shares a proinflammatory signature with Fusobacterium-associated human colorectal cancer, suggesting these bacteria promotes a tumor microenvironment favoring neoplasia progression (111). F. nucleatum content inversely correlates with CD3+ T cells density in tumor, showing modulation of immune response is another disease mechanism (112). Additionally, F. nucleatum is associated to immune evasion in cancer, avoiding NK-mediated tumor cell lysis via FAP2 interaction with the inhibitory NK-receptor TIGIT (90, 91).
F. nucleatum is associated with a high degree of microsatellite instability (MSI-high) and CpG island methylator phenotype in colorectal carcinoma, suggesting that this bacteria is involved in a molecular specific CRC subtype (113, 114). MSI-high CRCs tumors with high F. nucleatum load are more invasive, show low FOXP3+ density and elevated CD163+ cells (M2 macrophages), supporting the bacterial pro-tumoral role (115).
F. nucleatum is more abundant in CRC tissues in patients with recurrence post-chemotherapy, associated with TLR4/MyD88 signaling blocking and microRNAs (miRNA-4802 and miRNA-18a*) activating the autophagy pathway in response to chemotherapy (116). Moreover, F. nucleatum appears in distal metastases, demonstrating microbiota stability between primary tumors and metastasis. Likewise, mouse xenograft models retain viable F. nucleatum through successive passages, while metronidazole treatment reduces bacterial load, cancer cell proliferation and tumor growth (117). In summary, F. nucleatum induces tumorigenesis through several mechanisms including epithelial cell adhesion and tumor microenvironment modulation, becoming an attractive CRC prognosis biomarker.
Gut bacteria modulates the host biology through direct cell interaction, as well as microbial-derived metabolites. Therefore, we focus on the role of SCFAs and Polyamines and their functional impact on colon carcinogenesis and TME, illustrating microbiota–metabolite–cell interactions.
Microbiota communicates with the host through the generation of metabolites, being the most well-studied the short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, corresponding to fermentation products of complex polysaccharides, such as starches and fiber (118). The Bacteroidetes phylum mainly produces acetate and propionate, whereas the Firmicutes phylum butyrate (119).
SCFAs are produced in the colon and absorbed into the bloodstream to be delivered to target tissues, with host cells responding to SCFAs through receptors (GPR41/FFAR3, GPR43/FFAR2, GPR109A/HCAR2) or transporters (MCT1/SLC16A1, SMCT1/SLC5A8) (4). Butyrate is relevant in intestinal function being the primary energy source for colonocytes, corresponding to about 70% of total energy consumption (120).
Administration of SCFAs mixture restores intestinal epithelial cell turnover in antibiotic-treated SPF mice, previously having reduced amounts of acetate, propionate, and butyrate (121), demonstrating that SCFAs are essential in maintaining intestinal homeostasis. Accordingly, butyrate enhances M2-macrophage polarization (122), inhibits lipopolysaccharide (LPS)-induced proinflammatory cytokine expression in dendritic cells (123) and lamina propria (LP) mouse macrophages (124), and increases Treg lymphocytes in a murine model (125, 126).
Intestinal microbiota imbalance possibly relates to changes in SCFAs levels and loss of gut homeostasis (5). Patients with CRC and adenoma have reduced SCFAs stool levels (127), possibly related to reduced butyrate-producing bacteria such as Bacteroides vulgaris, Bacteroides uniformis, Roseburia, and the Lachnospiracea family (128–130).
Additionally, decreased butyrate receptors GPR109A and GPR43 content in colon cancer samples (131, 132) suggests variations of microbiota or substrate impact on receptor expression on normal epithelial or transformed cells. Butyrate and propionate, but not acetate, induces crypt proliferation obtained from healthy cecal biopsies ex vivo (133), although, butyrate inhibits cell growth and induces colon adenoma and carcinoma cell apoptosis (134–137). These butyrate roles are associated with the Warburg effect (Figure 3), where butyrate generates energy by metabolizing to acetyl-CoA via the Krebs cycle in the normal colonocyte. However, anaerobic glycolysis is the primary energy source in cancerous colonocytes, and butyrate is available to modulate gene expression through Histone deacetylase (HDAC) inhibition and interrupting cell cycle (138) thus demonstrating how a metabolite differentially affects a cell according to its metabolic activity.
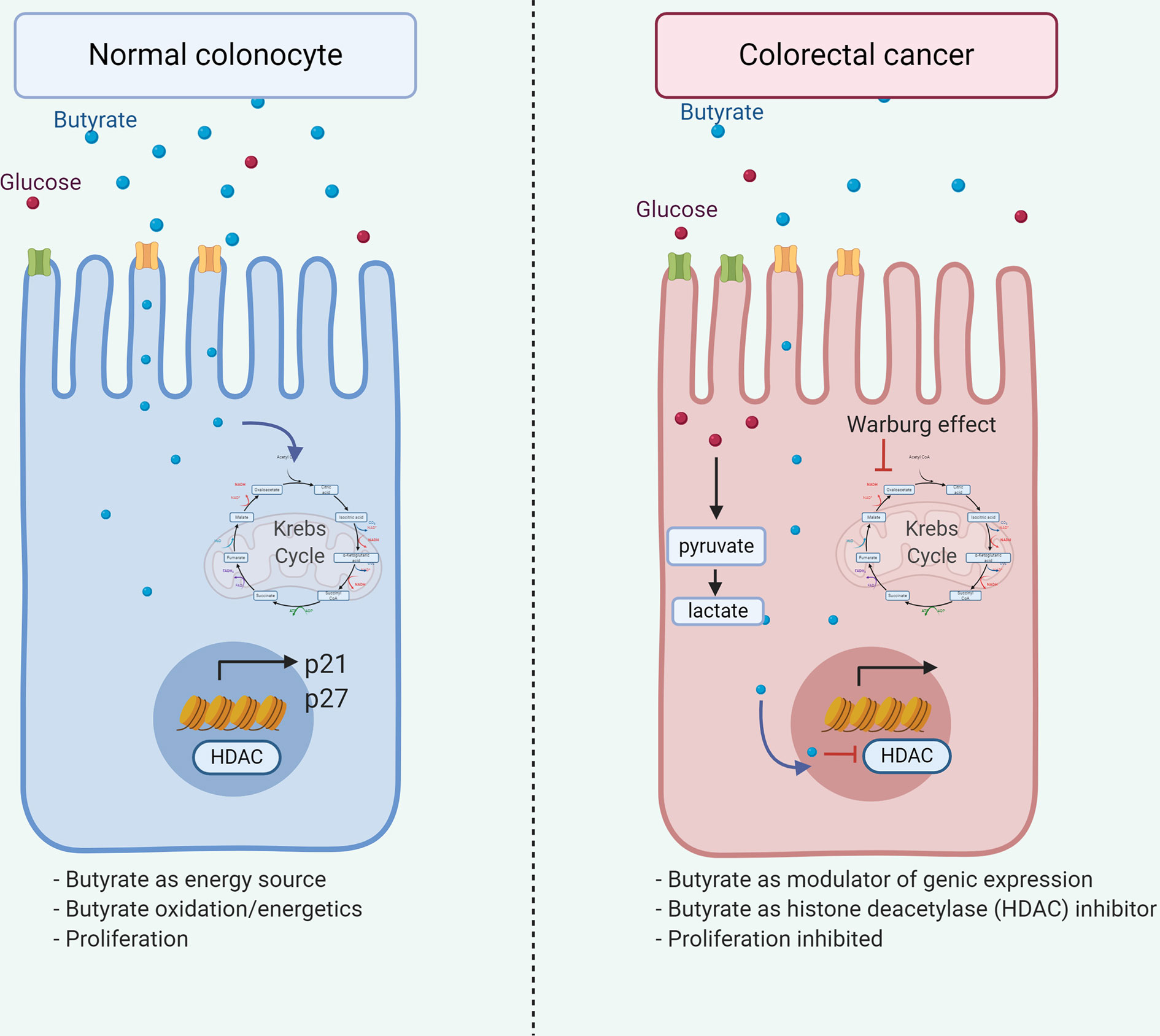
Figure 3 Dual role of butyrate in colorectal cancer. Butyrate exerts dual effects on normal and tumor colonocytes. In normal colonocytes, it functions as an energy source, being metabolized to acetyl-CoA in the Krebs cycle, allowing cell proliferation. Alternatively, in tumor colonocytes due to the Warburg effect, anaerobic glycolysis is the main energetic source, therefore, butyrate does not enter the Krebs cycle and available to the nucleus, modulating gene expression through the HDAC inhibition, leading to p21 and p27 downregulation and inhibiting cell proliferation, thus explaining the beneficial effect associated in cancer.
Other butyrate antitumoral effects are inhibition of epithelial-mesenchymal transition, cell proliferation, and migration in colon cancer cell lines, mediated by increased ROS levels associated with the small redox protein Thioredoxin-1 (Trx-1) downregulation (139), suggesting sodium butyrate exerts its role through Trx-1 downregulation, becoming a possible therapeutic target.
Generally, butyrate inhibits inflammation and carcinogenesis, reducing NF-kB and Wnt signaling, both active pathways in CRC (140, 141). Additionally, LPS-induced proinflammatory markers and chemokines, including CCL3 in dendritic cells is reduced after butyrate treatment (123), suggesting butyrate influences tumor microenvironment and tumor progression.
Polyamines are organic polycations involved in cell proliferation and differentiation, tissue repair, apoptosis, angiogenesis, immune response, signal transduction, and gene expression (14, 142–144). The principal polyamines are putrescine, spermidine, spermine, and cadaverine (145), participating in gut barrier function and epithelial turnover. Factors such as biosynthesis, catabolism, and transport finely control intracellular polyamines levels, maintaining a total concentration at the mM range, with free spermidine and spermine intracellular concentration corresponding respectively to 7-15% and 2-5% of the total (146).
Polyamines derive from endogenous synthesis, diet, and gut microbiota metabolism of amino-acids such as ornithine, methionine, and arginine (147), suggesting that diet influences its levels. The first step in polyamine synthesis is ornithine decarboxylation into putrescine, by Ornithine decarboxylase (ODC) (14), with oncogenes such as Myc (148) transcriptionally regulating ODC content and activity increased in cancer (147). Altered polyamine levels are related to cancer (146), with increased levels in CRC (149), possibly by uptake pathway and enzymatic synthesis induction (150).
Spermidine directly affects colibactin toxin synthesis by E. coli strains, suggesting a role of spermidine in bacterial pathogenicity and carcinogenesis (151). Polyamine levels increase throughout tumor development in the APCMin/+ murine CRC model, demonstrating dysregulation of its metabolic pathway could be involved in CRC development (152). Additionally, biofilms formation in colon cancer increases polyamine metabolites, suggesting polyamines produced by biofilm bacteria or the host, enhance tumor development (153). Moreover, fecal metabolomic analysis demonstrated increased amino-acids and polyamines, principally cadaverine and putrescine in CRC patients (67).
Regarding the immune response, polyamines are essential for B and T cells’ activation (154), synthesis being required to induce cytotoxic activity and T-cell proliferation in vitro and in vivo (155, 156), However, polyamines possibly have opposing roles depending on their concentrations, since increased levels in CRC interfere with anti-tumor immune function, associated to decreased adhesion molecule expression, such as CD44 and LFA-1 (157–159) and reduced cytokine production, such as IFN-γ and TNF (160–162), contributing to TME immunosuppression (163). Polyamine blocking therapy (PBT) targeting their synthesis and transport activates adaptive-dependent antitumor immune response in murine models, characterized by increased proinflammatory cytokine production, cytotoxic CD8+ T cell function, and decreased immunosuppressive cell levels. Consequently, increased tumor cell apoptosis leads to decreased tumor growth, indicating that blocking polyamines signaling could generate an anti-tumor immune memory and effector response, conferring protection against tumors (144, 148, 150). Moreover, polyamines inhibit macrophage polarization toward a proinflammatory M1 phenotype through ODC enzyme-dependent putrescine synthesis altering chromatin structure and preventing inflammatory gene transcription (164), thereby affecting anti-tumor responses (165).
Polyamines anti-inflammatory properties contribute to TME immunosuppression, (Figure 4), with PBT effective in inhibiting tumorigenesis, and dietary polyamine supplementation possibly benefiting aging-associated diseases (148). In TAMs, spermidine favors M1 polarization while spermine favors M2 (165), suggesting each polyamine having a unique role in normal and tumoral cells, although, exogenous spermidine treatment inhibits endogenous polyamines accumulation, tumor cell growth (166, 167) and promotes autophagy-mediated apoptosis (166). Furthermore, spermine-modified pullulan reduces immunosuppressive TME, contributing to inhibiting both tumor progression and metastasis (168). As mentioned above, endogenous polyamines could increase tumorigenesis, therefore endogenous synthesis inhibition, triggered by exogenous polyamines intake, could be a beneficial cancer treatment regulating polyamine metabolism with similar effects to PBT in the TME.
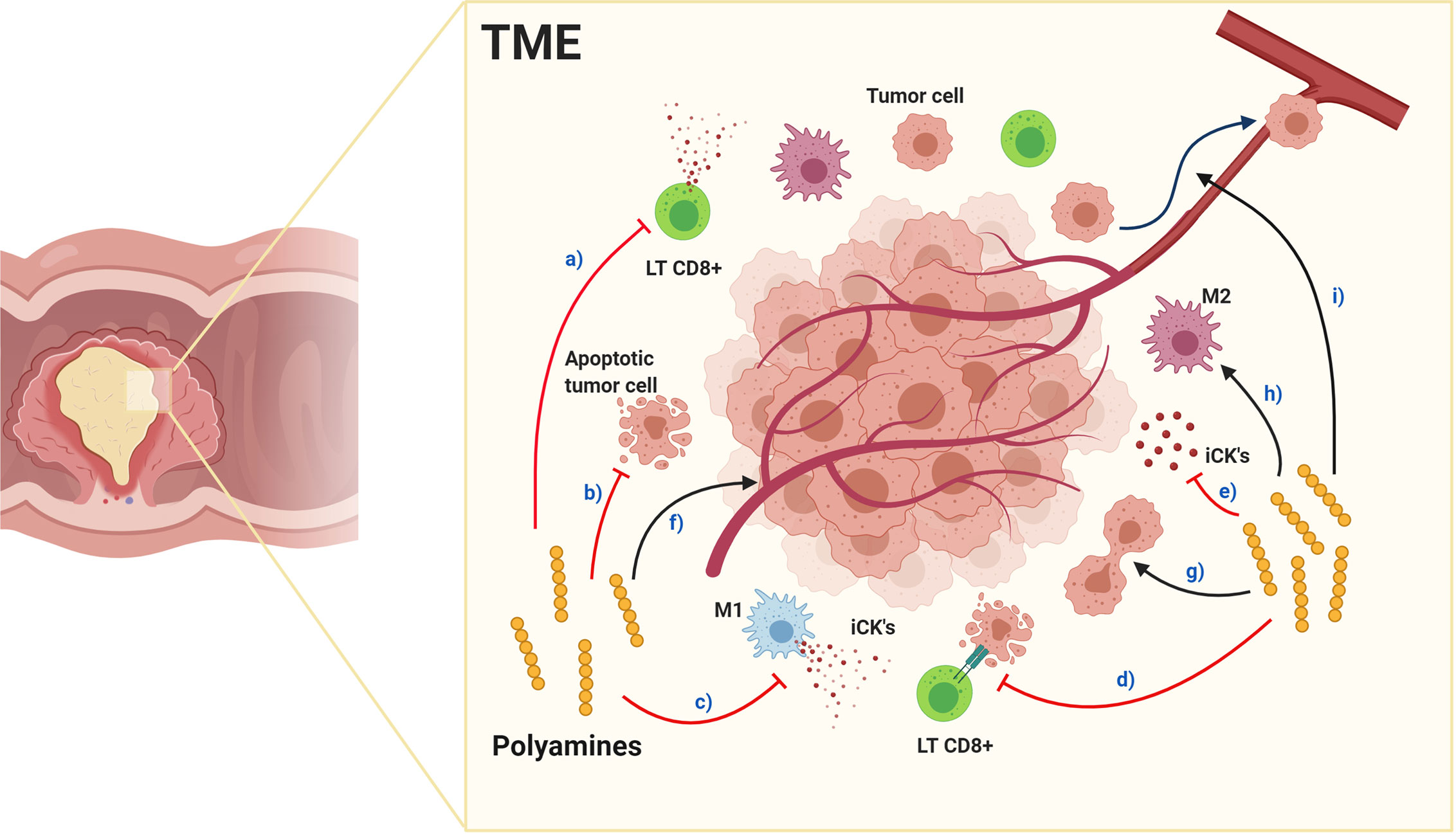
Figure 4 Effects of polyamines in the tumor microenvironment. Red arrows indicate inhibition and black arrows enhancement. Polyamines in TME: a) inhibit cytotoxic CD8+ LTs function and decrease quantity, inhibit: b) tumor cell apoptosis, c) macrophage polarization toward M1 pro-inflammatory phenotype, d) antitumor responses and e) pro-inflammatory cytokine production. Alternatively, polyamines enhance: f) tumor angiogenesis, g) tumor cell proliferation, h) macrophage polarization toward M2 immunosuppressive phenotype and i) tumor cell metastasis. Together, their effects produce an immunosuppressed environment facilitating tumor progression and metastasis.
Further investigation is merited on the complex interaction between polyamine metabolism and cancer, as their targeting undoubtedly offers promising results. In conclusion, polyamines are metabolites related to microbiota and carcinogenesis, substantially contributing to TME immunosuppression, and are becoming potential therapeutic targets in CRC. Additionally, exogenous polyamine supplements could be very beneficial in combination with conventional cancer treatments.
In 1971, Denis P. Burkitt analyzed the relation between bowel cancer frequency and fiber intake by different populations, highlighting notable differences between Western diet, where bowel cancer is more prevalent than less-developed communities. The proportion of ingested unabsorbable fiber and refined carbohydrate suggested that the diet in developed countries affected intestinal transit time, stool consistency, and bacterial microbiota-related to cancer incidence (169).
Furthermore, when comparing gut microbiota and metabolites in fecal samples from African Americans vs. native Africans, differences were found (170). Native Africans showed more abundant total bacteria, including starch fermenters and butyrate producers, along with higher SCFAs levels (171). In contrast, African Americans showed a higher abundance of microbial genes encoding for secondary bile acid production, generally associated with carcinogenic properties (172). African Americans diet is based on higher consumption of meat and fat, with lower consumption of fiber and less complex carbohydrate (170), thus suggesting the dietary influence in the production of beneficial or potentially carcinogenic metabolites.
Diet directly influences intestinal microbiota composition and metabolic activity, contributing to growing chronic diseases in the developed world, including obesity, cardiovascular disease, IBD, and cancer (173, 174). The high fat and high sugar (HF/HS) western diet have crucial implications on CRC; with a higher risk associated with red meat intake, opposed to high dietary fiber intake decreasing CRC risk (175, 176). Additionally, diet-induced intestinal inflammation (based on high plasma IL6, CRP, and TNFRSF1B levels) is associated with F. nucleatum-containing colorectal carcinoma in patients (177), indicating that diet alters microbiota balance.
The HF/HS diet in a mouse model induced intestinal inflammation and dysbiosis with an increase of Proteobacteria, such as E. coli, and decreased protective bacteria and SCFA levels (178). Furthermore, this diet gut abolishes SCFA effects on host chromatin states (histone acetylation and methylation) in colon, and extraintestinal tissues, such as liver and white adipose tissue (179). Interestingly, SCFA supplementation in germ-free mice recovers homeostatic epigenetic regulation associated with gut colonization (179).
Dietary interventions have potential effects in CRC prevention or treatment, as an adjuvant therapy. In healthy individuals, non-digestible carbohydrate (substrate for SCFA production)-enriched diets (whole grain rye flour bread/rye kernels bread + resistant starch) (180), or cooked barley kernels (181) showed beneficial effects as: a) decreased glucose and postprandial insulin levels (180, 181), b) reduce concentration of IL-6 and TNF-α (181), and c) increased satiety-inducing intestinal hormone PYY levels (180), evidencing a potential preventive effect of fiber supplementation in decreasing intestinal inflammation.
Studies of microbial interventions, using probiotics, were beneficial for CRC treatment (e.g. reducing F. nucleatum) (182), together with a combination of physical activity and improved dietary habits (e.g. reduction of red and processed meat or refined grain intake) in CRC survivors (183). Protocols defining the beneficial impacts of diet and physical activity in CRC patients during and after conventional therapies, and in the prevention of recurrence in CRC survivors (184) will confer benefits and should be further explored.
Considering the evidence of the influence of the diet in protecting and decreasing the risk of cancer, the 4th edition of the European Code against Cancer recommends that “people have a healthy diet to reduce their risk of cancer: eating plenty of whole grains, pulses, vegetables and fruits; limit high-calorie foods (high in sugar or fat); avoid sugary drinks and processed meat; and limit red meat and foods high in salt” (185).
In health, microbiota participates in functions such as immunity regulation, digestion, and nutrition. As dysbiosis has been associated with diverse pathologies including CRC, microbiota could play a role in carcinogenesis, as previously demonstrated.
The immune system, microbiota and their metabolites participate in the carcinogenic process, acting differently in each tumor development stage. In sporadic CRC, specific strains have been involved in its initiation and development. Specifically, carcinogenic effects of synthesized toxins and virulence factors from pks+ E. coli and F. nucleatum, cause DNA damage, increased epithelial cell proliferation, and immune system modulation, thus altering the microenvironment favoring tumorigenesis. Moreover, hereditary CRC reflects dysbiosis, suggesting microbiota changes during CRC progression occur in genetically susceptible subjects.
Some diet and microbiota metabolites associated with CRC, such as SCFAs and Polyamines, directly affect tumors and TME cells, thus becoming potential therapeutic targets.
Diet modifications, such as increasing dietary fiber intake help reduce the risk of developing CRC (186). In CRC animal models, prebiotics (oligofructose-maltrodextrin) in combination with probiotics (Lactobacillus acidophilus, Bifidobacteria bifidum) appears beneficial, reducing tumor growth and potentially carcinogenic bacteria, while increasing butyrate concentration and NK and NKT cell number (187–190).
Natural compounds, such as curcumin, genistein (an isoflavone found in soybeans) and apigenin (present in fruits and vegetables) possibly reduce aberrant crypt foci numbers (191), increasing intestinal T and B cells (192), and reducing ODC activity and polyamine levels in CRC cell lines (193, 194).
Due to polyamines and their metabolites participating in cell proliferation, they become interesting therapeutic targets for clinical intervention of CRC. As the use of nonsteroidal anti-inflammatory Sulindac plus DMFO (an inhibitor of polyamine biosynthesis) in patients reduces adenoma recurrence (195), PBT would be a promising antitumor response alternative (144). Alternatively, nanoparticles conjugated with a polyamine analog, which increase polyamine catabolism, are a possible innovative intervention, as they induce HCT116 cell apoptosis in vitro and inhibit xenograft tumor growth (196).
Another option in CRC carcinogenesis is blocking-cytokine antibodies, since IL-17 and IL-23 receptor blockade inhibits colorectal tumor formation (83).
Lastly, phages are novel interventions, selectively killing potentially carcinogenic bacteria, such as F. nucleatum in APCMin/+ mice, and revert chemotherapy resistance in CRC cell lines (197).
The above therapies demonstrate a vital relationship between the microbiota, metabolites, and the immune system, highlighting the relevance of studies evaluating their interaction, and offering great potential in CRC prevention and treatment. However, more clinical trials are needed to verify their efficacy and safety.
Additional to local properties in the intestinal mucosa, the gut microbiome systemically modulate other organs (198). Likewise, gut dysbiosis has been related to obesity (199), allergy (200, 201) and extra-intestinal cancer, such as lung and pancreatic adenocarcinoma (202, 203). Enrichment of phylum Proteobacteria is seen as a common hallmark in diverse cancer types (204, 205), as well as a potential diagnostic dysbiosis signature and chronic metabolic disease risk, such as Type 2 diabetes mellitus and cancer (206).
Another extra-intestinal effect of gut microbiota, is seen in the gut-lung axis, where SCFAs modulate the immune system (198) and are positively associated with immunotherapy response in lung cancer patients (207). Alternatively, the gut microbiota colonizing pancreatic tumors modulate tumor growth, immune responses and thus influence patient outcome (203, 204). Therefore, the modulation of the intestinal microbiota is an interesting target to control other pathologies, not only limited to the intestine.
As can be seen from the above, delving deeper into the interactions between metabolites, the immune system, and microbiota in CRC and other pathologies, will elucidate novel therapeutic targets. Accordingly, approaches such as diet modifications, supplementation with metabolites, specific antibiotics, immune system modulation for anti-tumor response are suitable and applicable resolutions.
MH and MF wrote most of the review. DP-V and GL contributed to writing and correcting the manuscript. AW, CH, KA, MAH, and FL-K participated reviewing and critically correcting the manuscript. FL-K and MF contributed to manuscript structure and supervised the work. All authors contributed to the article and approved the submitted version.
National Agency of Research and Development (ANID) (Fondecyt iniciación #11190990).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We want to thank the National Agency of Research and Development (ANID) (Fondecyt iniciación #11190990) and David Cox for his editing. Figures were created with BioRender.com.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Kuipers EJ, Grady WM, Lieberman D, Seufferlein T, Sung JJ, Boelens PG, et al. Colorectal cancer. Nat Rev Dis Prim (2015) 1:15065. doi: 10.1038/nrdp.2015.65
3. Wells K, Wise PE. Hereditary Colorectal Cancer Syndromes. Surg Clin North Am (2017) 97:605–25. doi: 10.1016/j.suc.2017.01.009
4. Natarajan N, Pluznick JL. From microbe to man: the role of microbial short chain fatty acid metabolites in host cell biology. Am J Physiol Physiol (2014) 307:C979–85. doi: 10.1152/ajpcell.00228.2014
5. Kim D, Yoo S-A, Kim W-U. Gut microbiota in autoimmunity: potential for clinical applications. Arch Pharm Res (2016) 39:1565–76. doi: 10.1007/s12272-016-0796-7
6. Kovács T, Mikó E, Ujlaki G, Sári Z, Bai P. The Microbiome as a Component of the Tumor Microenvironment. Adv Exp Med Biol (2020) 1225:137–53. doi: 10.1007/978-3-030-35727-6_10
7. Rinninella E, Raoul P, Cintoni M, Franceschi F, Miggiano G, Gasbarrini A, et al. What is the Healthy Gut Microbiota Composition? A Changing Ecosystem across Age, Environment, Diet, and Diseases. Microorganisms (2019) 7:14. doi: 10.3390/microorganisms7010014
8. Lazar V, Ditu L-M, Pircalabioru GG, Gheorghe I, Curutiu C, Holban AM, et al. Aspects of Gut Microbiota and Immune System Interactions in Infectious Diseases, Immunopathology, and Cancer. Front Immunol (2018) 9:1830. doi: 10.3389/fimmu.2018.01830
9. Levy M, Blacher E, Elinav E. Microbiome, metabolites and host immunity. Curr Opin Microbiol (2017) 35:8–15. doi: 10.1016/j.mib.2016.10.003
10. Burkholder B, Huang RY, Burgess R, Luo S, Jones VS, Zhang W, et al. Tumor-induced perturbations of cytokines and immune cell networks. Biochim Biophys Acta Rev Cancer (2014) 1845:182–201. doi: 10.1016/j.bbcan.2014.01.004
11. Aivaliotis IL, Pateras IS, Papaioannou M, Glytsou C, Kontzoglou K, Johnson EO, et al. How do cytokines trigger genomic instability? J BioMed Biotechnol (2012) 2012:1–12. doi: 10.1155/2012/536761
12. Dahmus JD, Kotler DL, Kastenberg DM, Kistler CA. The gut microbiome and colorectal cancer: A review of bacterial pathogenesis. J Gastrointest Oncol (2018) 9:769–77. doi: 10.21037/jgo.2018.04.07
13. Jahani-Sherafat S, Alebouyeh M, Moghim S, Amoli HA, Ghasemian-Safaei H. Role of gut microbiota in the pathogenesis of colorectal cancer; A review article. Gastroenterol Hepatol Bed Bench (2018) 11:101–9. doi: 10.22037/ghfbb.v0i0.1052
14. Gerner EW, Meyskens FL. Polyamines and cancer: old molecules, new understanding. Nat Rev Cancer (2004) 4:781–92. doi: 10.1038/nrc1454
16. Terzić J, Grivennikov S, Karin E, Karin M. Inflammation and Colon Cancer. Gastroenterology (2010) 138:2101–14.e5. doi: 10.1053/j.gastro.2010.01.058
17. Markman JL, Shiao SL. Impact of the immune system and immunotherapy in colorectal cancer. J Gastrointest Oncol (2015) 6:208–23. doi: 10.3978/j.issn.2078-6891.2014.077
18. Grivennikov SI, Greten FR, Karin M. Immunity, Inflammation, and Cancer. Cell (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
19. Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol (2004) 22:329–60. doi: 10.1146/annurev.immunol.22.012703.104803
20. de Vries N, Swets M, Vahrmeijer A, Hokland M, Kuppen P. The Immunogenicity of Colorectal Cancer in Relation to Tumor Development and Treatment. Int J Mol Sci (2016) 17:1030. doi: 10.3390/ijms17071030
21. Xiong Y, Wang Y, Tiruthani K. Tumor immune microenvironment and nano-immunotherapeutics in colorectal cancer. Nanomedicine (2019) 21:102034. doi: 10.1016/j.nano.2019.102034
22. Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (2006) 313:1960–4. doi: 10.1126/science.1129139
23. Angell HK, Bruni D, Carl Barrett J, Herbst R, Galon J. The immunoscore: Colon cancer and beyond a C. Clin Cancer Res (2020) 26:332–9. doi: 10.1158/1078-0432.CCR-18-1851
24. Anitei M-G, Zeitoun G, Mlecnik B, Marliot F, Haicheur N, Todosi A-M, et al. Prognostic and predictive values of the immunoscore in patients with rectal cancer. Clin Cancer Res (2014) 20:1891–9. doi: 10.1158/1078-0432.CCR-13-2830
25. Salama P, Phillips M, Grieu F, Morris M, Zeps N, Joseph D, et al. Tumor-Infiltrating FOXP3 + T Regulatory Cells Show Strong Prognostic Significance in Colorectal Cancer. J Clin Oncol (2009) 27:186–92. doi: 10.1200/JCO.2008.18.7229
26. Frey DM, Droeser RA, Viehl CT, Zlobec I, Lugli A, Zingg U, et al. High frequency of tumor-infiltrating FOXP3 + regulatory T cells predicts improved survival in mismatch repair-proficient colorectal cancer patients. Int J Cancer (2010) 126:NA–A. doi: 10.1002/ijc.24989
27. Kuwahara T, Hazama S, Suzuki N, Yoshida S, Tomochika S, Nakagami Y, et al. Intratumoural-infiltrating CD4 + and FOXP3 + T cells as strong positive predictive markers for the prognosis of resectable colorectal cancer. Br J Cancer (2019) 121:659–65. doi: 10.1038/s41416-019-0559-6
28. Cavalleri T, Bianchi P, Basso G, Celesti G, Grizzi F, Bossi P, et al. Combined Low Densities of FoxP3 + and CD3 + Tumor-Infiltrating Lymphocytes Identify Stage II Colorectal Cancer at High Risk of Progression. Cancer Immunol Res (2019) 7:751–8. doi: 10.1158/2326-6066.CIR-18-0661
29. Khoon LL, Pratap SE, Bates GJ, Singh B, Mortensen NJ, George BD, et al. Increased frequency of regulatory T cells in peripheral blood and tumour infiltrating lymphocytes in colorectal cancer patients. Cancer Immun (2007) 7:7.
30. Saito T, Nishikawa H, Wada H, Nagano Y, Sugiyama D, Atarashi K, et al. Two FOXP3+CD4+ T cell subpopulations distinctly control the prognosis of colorectal cancers. Nat Med (2016) 22:679–84. doi: 10.1038/nm.4086
31. Műzes G, Molnár B, Sipos F. Regulatory T cells in inflammatory bowel diseases and colorectal cancer. World J Gastroenterol (2012) 18:5688–94. doi: 10.3748/wjg.v18.i40.5688
32. Scurr M, Ladell K, Besneux M, Christian A, Hockey T, Smart K, et al. Highly prevalent colorectal cancer-infiltrating LAP+ Foxp3– T cells exhibit more potent immunosuppressive activity than Foxp3+ regulatory T cells. Mucosal Immunol (2014) 7:428–39. doi: 10.1038/mi.2013.62
33. Tougeron D, Fauquembergue É, Latouche JB. Réponse immunitaire et cancers colorectaux. Bull Cancer (2013) 100:283–94. doi: 10.1684/bdc.2013.1716
34. Browning M, Petronzelli F, Bicknell D, Krausa P, Rowan A, Tonks S, et al. Mechanisms of loss of HLA class I expression on colorectal tumor cells. Tissue Antigens (1996) 47:364–71. doi: 10.1111/j.1399-0039.1996.tb02571.x
35. Halama N, Braun M, Kahlert C, Spille A, Quack C, Rahbari N, et al. Natural killer cells are scarce in colorectal carcinoma tissue despite high levels of chemokines and cytokines. Clin Cancer Res (2011) 17:678–89. doi: 10.1158/1078-0432.CCR-10-2173
36. Levy EM, Bianchini M, Von Euw EM, Barrio MM, Bravo AI, Furman D, et al. Human leukocyte antigen-E protein is overexpressed in primary human colorectal cancer. Int J Oncol (2008) 32:633–41. doi: 10.3892/ijo.32.3.633
37. Rocca YS, Roberti MP, Arriaga JM, Amat M, Bruno L, Pampena MB, et al. Altered phenotype in peripheral blood and tumor-associated NK cells from colorectal cancer patients. Innate Immun (2013) 19:76–85. doi: 10.1177/1753425912453187
38. Herrera M, Herrera A, Domínguez G, Silva J, García V, García JM, et al. Cancer-associated fibroblast and M2 macrophage markers together predict outcome in colorectal cancer patients. Cancer Sci (2013) 104:437–44. doi: 10.1111/cas.12096
39. Zhang B, Wang Z, Wu L, Zhang M, Li W, Ding J, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PloS One (2013) 8:e57114. doi: 10.1371/journal.pone.0057114
40. OuYang L-Y, Wu X-J, Ye S-B, Zhang R, Li Z-L, Liao W, et al. Tumor-induced myeloid-derived suppressor cells promote tumor progression through oxidative metabolism in human colorectal cancer. J Transl Med (2015) 13:47. doi: 10.1186/s12967-015-0410-7
41. Berry RS, Xiong M-J, Greenbaum A, Mortaji P, Nofchissey RA, Schultz F, et al. High levels of tumor-associated neutrophils are associated with improved overall survival in patients with stage II colorectal cancer. PloS One (2017) 12:e0188799. doi: 10.1371/journal.pone.0188799
42. Governa V, Trella E, Mele V, Tornillo L, Amicarella F, Cremonesi E, et al. The Interplay Between Neutrophils and CD8 + T Cells Improves Survival in Human Colorectal Cancer. Clin Cancer Res (2017) 23:3847–58. doi: 10.1158/1078-0432.CCR-16-2047
43. Wikberg ML, Ling A, Li X, Öberg Å, Edin S, Palmqvist R. Neutrophil infiltration is a favorable prognostic factor in early stages of colon cancer. Hum Pathol (2017) 68:193–202. doi: 10.1016/j.humpath.2017.08.028
44. Galdiero MR, Bianchi P, Grizzi F, Di Caro G, Basso G, Ponzetta A, et al. Occurrence and significance of tumor-associated neutrophils in patients with colorectal cancer. Int J Cancer (2016) 139:446–56. doi: 10.1002/ijc.30076
45. Ponzetta A, Carriero R, Carnevale S, Barbagallo M, Molgora M, Perucchini C, et al. Neutrophils Driving Unconventional T Cells Mediate Resistance against Murine Sarcomas and Selected Human Tumors. Cell (2019) 178:346–360.e24. doi: 10.1016/j.cell.2019.05.047
46. Germann M, Zangger N, Sauvain M, Sempoux C, Bowler AD, Wirapati P, et al. Neutrophils suppress tumor-infiltrating T cells in colon cancer via matrix metalloproteinase-mediated activation of TGF β. EMBO Mol Med (2020) 12:e10681. doi: 10.15252/emmm.201910681
47. Gordon-Weeks AN, Lim SY, Yuzhalin AE, Jones K, Markelc B, Kim KJ, et al. Neutrophils promote hepatic metastasis growth through fibroblast growth factor 2-dependent angiogenesis in mice. Hepatology (2017) 65:1920–35. doi: 10.1002/hep.29088
48. Shang K, Bai Y-P, Wang C, Wang Z, Gu H-Y, Du X, et al. Crucial Involvement of Tumor-Associated Neutrophils in the Regulation of Chronic Colitis-Associated Carcinogenesis in Mice. PloS One (2012) 7:e51848. doi: 10.1371/journal.pone.0051848
49. Eruslanov EB, Singhal S, Albelda SM. Mouse versus Human Neutrophils in Cancer: A Major Knowledge Gap. Trends Cancer (2017) 3:149–60. doi: 10.1016/j.trecan.2016.12.006
50. McBurney MI, Davis C, Fraser CM, Schneeman BO, Huttenhower C, Verbeke K, et al. Establishing What Constitutes a Healthy Human Gut Microbiome: State of the Science, Regulatory Considerations, and Future Directions. J Nutr (2019) 149:1882–95. doi: 10.1093/jn/nxz154
51. Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature (2012) 489:220–30. doi: 10.1038/nature11550
52. Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, et al. Human gut microbiome viewed across age and geography. Nature (2012) 486:222–7. doi: 10.1038/nature11053
53. Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, Chinwalla AT, et al. Structure, function and diversity of the healthy human microbiome. Nature (2012) 486:207–14. doi: 10.1038/nature11234
54. Maukonen J, Saarela M. Human gut microbiota: does diet matter? Proc Nutr Soc (2015) 74:23–36. doi: 10.1017/S0029665114000688
55. Bäckhed F, Fraser CM, Ringel Y, Sanders ME, Sartor RB, Sherman PM, et al. Defining a healthy human gut microbiome: current concepts, future directions, and clinical applications. Cell Host Microbe (2012) 12:611–22. doi: 10.1016/j.chom.2012.10.012
56. Stewart CJ, Ajami NJ, O’Brien JL, Hutchinson DS, Smith DP, Wong MC, et al. Temporal development of the gut microbiome in early childhood from the TEDDY study. Nature (2018) 562:583–8. doi: 10.1038/s41586-018-0617-x
57. Haro C, Rangel-Zúñiga OA, Alcalá-Díaz JF, Gómez-Delgado F, Pérez-Martínez P, Delgado-Lista J, et al. Intestinal microbiota is influenced by gender and body mass index. PloS One (2016) 11:1–16. doi: 10.1371/journal.pone.0154090
58. Santos-Marcos JA, Haro C, Vega-Rojas A, Alcala-Diaz JF, Molina-Abril H, Leon-Acuña A, et al. Sex Differences in the Gut Microbiota as Potential Determinants of Gender Predisposition to Disease. Mol Nutr Food Res (2019) 63:1–11. doi: 10.1002/mnfr.201800870
59. Mesa MD, Loureiro B, Iglesia I, Gonzalez SF, Olivé EL, Algar OG, et al. The evolving microbiome from pregnancy to early infancy: A comprehensive review. Nutrients (2020) 12:1–21. doi: 10.3390/nu12010133
60. Nuriel-Ohayon M, Neuman H, Koren O. Microbial changes during pregnancy, birth, and infancy. Front Microbiol (2016) 7:1031. doi: 10.3389/fmicb.2016.01031
61. Hughes RL. A Review of the Role of the Gut Microbiome in Personalized Sports Nutrition. Front Nutr (2020) 6:191. doi: 10.3389/fnut.2019.00191
62. Xu C, Zhu H, Qiu P. Aging progression of human gut microbiota. BMC Microbiol (2019) 19:1–10. doi: 10.1186/s12866-019-1616-2
63. Wu GD, Chen J, Hoffmann C, Bittinger K, Chen Y-Y, Keilbaugh SA, et al. Linking Long-Term Dietary Patterns with. Sci (80-) (2011) 334:105–9. doi: 10.1126/science.1208344
64. Sobhani I, Tap J, Roudot-Thoraval F, Roperch JP, Letulle S, Langella P, et al. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PloS One (2011) 6:e16393. doi: 10.1371/journal.pone.0016393
65. Kostic AD, Gevers D, Pedamallu CS, Michaud M, Duke F, Earl AM, et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res (2012) 22:292–8. doi: 10.1101/gr.126573.111
66. Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res (2012) 22:299–306. doi: 10.1101/gr.126516.111
67. Yang Y, Misra BB, Liang L, Bi D, Weng W, Wu W, et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics (2019) 9:4101–14. doi: 10.7150/thno.35186
68. Saus E, Iraola-Guzmán S, Willis JR, Brunet-Vega A, Gabaldón T. Microbiome and colorectal cancer: Roles in carcinogenesis and clinical potential. Mol Aspects Med (2019) 69:93–106. doi: 10.1016/j.mam.2019.05.001
69. Ternes D, Karta J, Tsenkova M, Wilmes P, Haan S, Letellier E. Microbiome in Colorectal Cancer: How to Get from Meta-omics to Mechanism? Trends Microbiol (2020) 28:401–23. doi: 10.1016/j.tim.2020.01.001
70. Yachida S, Mizutani S, Shiroma H, Shiba S, Nakajima T, Sakamoto T, et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat Med (2019) 25:968–76. doi: 10.1038/s41591-019-0458-7
71. Nakatsu G, Li X, Zhou H, Sheng J, Wong SH, Wu WKK, et al. Gut mucosal microbiome across stages of colorectal carcinogenesis. Nat Commun (2015) 6:8727. doi: 10.1038/ncomms9727
72. Zagato E, Pozzi C, Bertocchi A, Schioppa T, Saccheri F, Guglietta S, et al. Endogenous murine microbiota member Faecalibaculum rodentium and its human homologue protect from intestinal tumour growth. Nat Microbiol (2020) 5:511–24. doi: 10.1038/s41564-019-0649-5
73. Cremonesi E, Governa V, Garzon JFG, Mele V, Amicarella F, Muraro MG, et al. Gut microbiota modulate T cell trafficking into human colorectal cancer. Gut (2018) 67:1984–94. doi: 10.1136/gutjnl-2016-313498
74. Dejea CM, Fathi P, Craig JM, Boleij A, Taddese R, Geis AL, et al. Patients with familial adenomatous polyposis harbor colonic biofilms containing tumorigenic bacteria. Science (2018) 359:592–7. doi: 10.1126/science.aah3648
75. Liang S, Mao Y, Liao M, Xu Y, Chen Y, Huang X, et al. Gut microbiome associated with APC gene mutation in patients with intestinal adenomatous polyps. Int J Biol Sci (2020) 16:135–46. doi: 10.7150/ijbs.37399
76. Mori G, Orena BS, Cultrera I, Barbieri G, Albertini AM, Ranzani GN, et al. Gut Microbiota Analysis in Postoperative Lynch Syndrome Patients. Front Microbiol (2019) 10:1746:1746. doi: 10.3389/fmicb.2019.01746
77. Yan Y, Drew DA, Markowitz A, Lloyd-Price J, Abu-Ali G, Nguyen LH, et al. Structure of the Mucosal and Stool Microbiome in Lynch Syndrome. Cell Host Microbe (2020) 27:585–600.e4. doi: 10.1016/j.chom.2020.03.005
78. Pasquereau-Kotula E, Martins M, Aymeric L, Dramsi S. Significance of Streptococcus gallolyticus subsp. gallolyticus Association With Colorectal Cancer. Front Microbiol (2018) 9:614. doi: 10.3389/fmicb.2018.00614
79. Abdulamir AS, Hafidh RR, Bakar F. Molecular detection, quantification, and isolation of Streptococcus gallolyticus bacteria colonizing colorectal tumors: inflammation-driven potential of carcinogenesis via IL-1, COX-2, and IL-8. Mol Cancer (2010) 9:249. doi: 10.1186/1476-4598-9-249
80. Kumar R, Herold JL, Taylor J, Xu J, Xu Y. Variations among Streptococcus gallolyticus subsp. gallolyticus strains in connection with colorectal cancer. Sci Rep (2018) 8:1514. doi: 10.1038/s41598-018-19941-7
81. Rhee K-J, Wu S, Wu X, Huso DL, Karim B, Franco AA, et al. Induction of Persistent Colitis by a Human Commensal, Enterotoxigenic Bacteroides fragilis, in Wild-Type C57BL/6 Mice. Infect Immun (2009) 77:1708–18. doi: 10.1128/IAI.00814-08
82. Goodwin AC, Shields CED, Wu S, Huso DL, Wu X, Murray-Stewart TR, et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc Natl Acad Sci (2011) 108:15354–9. doi: 10.1073/pnas.1010203108
83. Wu S, Rhee K-J, Albesiano E, Rabizadeh S, Wu X, Yen H-R, et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med (2009) 15:1016–22. doi: 10.1038/nm.2015
84. Chung L, Orberg ET, Geis AL, Chan JL, Fu K, DeStefano Shields CE, et al. Bacteroides fragilis Toxin Coordinates a Pro-carcinogenic Inflammatory Cascade via Targeting of Colonic Epithelial Cells. Cell Host Microbe (2018) 23:421. doi: 10.1016/j.chom.2018.02.004
85. Cuevas-Ramos G, Petit CR, Marcq I, Boury M, Oswald E, Nougayrède J-P. Escherichia coli induces DNA damage in vivo and triggers genomic instability in mammalian cells. Proc Natl Acad Sci USA (2010) 107:11537–42. doi: 10.1073/pnas.1001261107
86. Nougayrède J-P, Homburg S, Taieb F, Boury M, Brzuszkiewicz E, Gottschalk G, et al. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science (2006) 313:848–51. doi: 10.1126/science.1127059
87. Quah SY, Bergenholtz G, Tan KS. Fusobacterium nucleatum induces cytokine production through Toll-like-receptor-independent mechanism. Int Endod J (2014) 47:550–9. doi: 10.1111/iej.12185
88. Tang B, Wang K, Jia Y-P, Zhu P, Fang Y, Zhang Z-J, et al. Fusobacterium nucleatum-Induced Impairment of Autophagic Flux Enhances the Expression of Proinflammatory Cytokines via ROS in Caco-2 Cells. PloS One (2016) 11:e0165701. doi: 10.1371/journal.pone.0165701
89. Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe (2013) 14:195–206. doi: 10.1016/j.chom.2013.07.012
90. Gur C, Ibrahim Y, Isaacson B, Yamin R, Abed J, Gamliel M, et al. Binding of the Fap2 protein of Fusobacterium nucleatum to human inhibitory receptor TIGIT protects tumors from immune cell attack. Immunity (2015) 42:344–55. doi: 10.1016/j.immuni.2015.01.010
91. Bashir A, Miskeen AY, Hazari YM, Asrafuzzaman S, Fazili KM. Fusobacterium nucleatum, inflammation, and immunity: the fire within human gut. Tumour Biol (2016) 37:2805–10. doi: 10.1007/s13277-015-4724-0
92. Tsoi H, Chu ESH, Zhang X, Sheng J, Nakatsu G, Ng SC, et al. Peptostreptococcus anaerobius Induces Intracellular Cholesterol Biosynthesis in Colon Cells to Induce Proliferation and Causes Dysplasia in Mice. Gastroenterology (2017) 152:1419–33.e5. doi: 10.1053/j.gastro.2017.01.009
93. Long X, Wong CC, Tong L, Chu ESH, Ho Szeto C, Go MYY, et al. Peptostreptococcus anaerobius promotes colorectal carcinogenesis and modulates tumour immunity. Nat Microbiol (2019) 4:2319–30. doi: 10.1038/s41564-019-0541-3
94. Boonanantanasarn K, Gill AL, Yap Y, Jayaprakash V, Sullivan MA, Gill SR. Enterococcus faecalis Enhances Cell Proliferation through Hydrogen Peroxide-Mediated Epidermal Growth Factor Receptor Activation. Infect Immun (2012) 80:3545–58. doi: 10.1128/IAI.00479-12
95. Huycke MM, Abrams V, Moore DR. Enterococcus faecalis produces extracellular superoxide and hydrogen peroxide that damages colonic epithelial cell DNA. Carcinogenesis (2002) 23:529–36. doi: 10.1093/carcin/23.3.529
96. Wang X, Allen TD, May RJ, Lightfoot S, Houchen CW, Huycke MM. Enterococcus faecalis Induces Aneuploidy and Tetraploidy in Colonic Epithelial Cells through a Bystander Effect. Cancer Res (2008) 68:9909–17. doi: 10.1158/0008-5472.CAN-08-1551
97. Buc E, Dubois D, Sauvanet P, Raisch J, Delmas J, Darfeuille-Michaud A, et al. High prevalence of mucosa-associated E. coli producing cyclomodulin and genotoxin in colon cancer. PloS One (2013) 8:e56964. doi: 10.1371/journal.pone.0056964
98. Arthur JC, Perez-Chanona E, Mühlbauer M, Tomkovich S, Uronis JM, Fan T, et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Sci (80-) (2012) 338:120–3. doi: 10.1126/science.1224820
99. Bonnet M, Buc E, Sauvanet P, Darcha C, Dubois D, Pereira B, et al. Colonization of the human gut by E. coli and colorectal cancer risk. Clin Cancer Res (2014) 20:859–67. doi: 10.1158/1078-0432.CCR-13-1343
100. Pleguezuelos-Manzano C, Puschhof J, Rosendahl Huber A, van Hoeck A, Wood HM, Nomburg J, et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature (2020) 580:269–73. doi: 10.1038/s41586-020-2080-8
101. Lopès A, Billard E, Casse AH, Villéger R, Veziant J, Roche G, et al. Colibactin-positive Escherichia coli induce a procarcinogenic immune environment leading to immunotherapy resistance in colorectal cancer. Int J Cancer (2020) 146:3147–59. doi: 10.1002/ijc.32920
102. Tronnet S, Floch P, Lucarelli L, Gaillard D, Martin P, Serino M, et al. The Genotoxin Colibactin Shapes Gut Microbiota in Mice. mSphere (2020) 5:1–11. doi: 10.1128/mSphere.00589-20
103. Brennan CA, Garrett WS. Fusobacterium nucleatum — symbiont, opportunist and oncobacterium. Nat Rev Microbiol (2019) 17:156–66. doi: 10.1038/s41579-018-0129-6
104. Ito M, Kanno S, Nosho K, Sukawa Y, Mitsuhashi K, Kurihara H, et al. Association of Fusobacterium nucleatum with clinical and molecular features in colorectal serrated pathway. Int J Cancer (2015) 137:1258–68. doi: 10.1002/ijc.29488
105. Shang F-M, Liu H-L. Fusobacterium nucleatum and colorectal cancer: A review. World J Gastrointest Oncol (2018) 10:71–81. doi: 10.4251/wjgo.v10.i3.71
106. Flanagan L, Schmid J, Ebert M, Soucek P, Kunicka T, Liska V, et al. Fusobacterium nucleatum associates with stages of colorectal neoplasia development, colorectal cancer and disease outcome. Eur J Clin Microbiol Infect Dis (2014) 33:1381–90. doi: 10.1007/s10096-014-2081-3
107. Mima K, Nishihara R, Rong Qian Z, Cao Y, Sukawa Y, Nowak JA, et al. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis were responsible for collection of tumour tissue, and acquisition of epidemiologic, clinical and tumour tissue data, including histopathological and immunohistochemical character. Gut (2016) 65:1973–80. doi: 10.1136/gutjnl-2015-310101
108. Fardini Y, Wang X, Témoin S, Nithianantham S, Lee D, Shoham M, et al. Fusobacterium nucleatum adhesin FadA binds vascular endothelial cadherin and alters endothelial integrity. Mol Microbiol (2011) 82:1468–80. doi: 10.1111/j.1365-2958.2011.07905.x
109. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic Inflammation and Cytokines in the Tumor Microenvironment. J Immunol Res (2014) 2014:1–19. doi: 10.1155/2014/149185
110. McCoy AN, Araújo-Pérez F, Azcárate-Peril A, Yeh JJ, Sandler RS, Keku TO. Fusobacterium Is Associated with Colorectal Adenomas. PloS One (2013) 8:e53653. doi: 10.1371/journal.pone.0053653
111. Kostic AD, Chun E, Robertson L, Glickman JN, Gallini CA, Michaud M, et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe (2013) 14:207–15. doi: 10.1016/j.chom.2013.07.007
112. Mima K, Sukawa Y, Nishihara R, Qian ZR, Yamauchi M, Inamura K, et al. Fusobacterium nucleatum and T cells in colorectal carcinoma. JAMA Oncol (2015) 1:653–61. doi: 10.1001/jamaoncol.2015.1377
113. Tahara T, Yamamoto E, Suzuki H, Maruyama R, Chung W, Garriga J, et al. Fusobacterium in Colonic Flora and Molecular Features of Colorectal Carcinoma. Cancer Res (2014) 74:1311–8. doi: 10.1158/0008-5472.CAN-13-1865
114. Nosho K, Sukawa Y, Adachi Y, Ito M, Mitsuhashi K, Kurihara H, et al. Association of Fusobacterium nucleatum with immunity and molecular alterations in colorectal cancer. World J Gastroenterol (2016) 22:557–66. doi: 10.3748/wjg.v22.i2.557
115. Lee JA, Yoo S-Y, Oh HJ, Jeong S, Cho N-Y, Kang GH, et al. Differential immune microenvironmental features of microsatellite-unstable colorectal cancers according to Fusobacterium nucleatum status. Cancer Immunol Immunother (2021) 70:47–59. doi: 10.1007/s00262-020-02657-x
116. Yu T, Guo F, Yu Y, Sun T, Ma D, Han J, et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell (2017) 170:548–63.e16. doi: 10.1016/j.cell.2017.07.008
117. Bullman S, Pedamallu CS, Sicinska E, Clancy TE, Zhang X, Cai D, et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science (2017) 358:1443–8. doi: 10.1126/science.aal5240
118. Ríos-Covián D, Ruas-Madiedo P, Margolles A, Gueimonde M, de los Reyes-Gavilán CG, Salazar N. Intestinal Short Chain Fatty Acids and their Link with Diet and Human Health. Front Microbiol (2016) 7:185. doi: 10.3389/fmicb.2016.00185
119. den Besten G, van Eunen K, Groen AK, Venema K, Reijngoud D-J, Bakker BM. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J Lipid Res (2013) 54:2325–40. doi: 10.1194/jlr.R036012
120. Scheppach W. Effects of short chain fatty acids on gut morphology and function. Gut (1994) 35:S35–8. doi: 10.1136/gut.35.1_Suppl.S35
121. Park J, Kotani T, Konno T, Setiawan J, Kitamura Y, Imada S, et al. Promotion of Intestinal Epithelial Cell Turnover by Commensal Bacteria: Role of Short-Chain Fatty Acids. PloS One (2016) 11:e0156334. doi: 10.1371/journal.pone.0156334
122. Ji J, Shu D, Zheng M, Wang J, Luo C, Wang Y, et al. Microbial metabolite butyrate facilitates M2 macrophage polarization and function. Sci Rep (2016) 6:24838. doi: 10.1038/srep24838
123. Nastasi C, Candela M, Bonefeld CM, Geisler C, Hansen M, Krejsgaard T, et al. The effect of short-chain fatty acids on human monocyte-derived dendritic cells. Sci Rep (2015) 5:16148. doi: 10.1038/srep16148
124. Chang PV, Hao L, Offermanns S, Medzhitov R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci (2014) 111:2247–52. doi: 10.1073/pnas.1322269111
125. Arpaia N, Campbell C, Fan X, Dikiy S, Van Der Veeken J, Deroos P, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature (2013) 504:451–5. doi: 10.1038/nature12726
126. Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly --M, et al. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science (2013) 341:569–73. doi: 10.1126/science.1241165
127. Ohigashi S, Sudo K, Kobayashi D, Takahashi O, Takahashi T, Asahara T, et al. Changes of the Intestinal Microbiota, Short Chain Fatty Acids, and Fecal pH in Patients with Colorectal Cancer. Dig Dis Sci (2013) 58:1717–26. doi: 10.1007/s10620-012-2526-4
128. Wang T, Cai G, Qiu Y, Fei N, Zhang M, Pang X, et al. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J (2012) 6:320–9. doi: 10.1038/ismej.2011.109
129. Wu N, Yang X, Zhang R, Li J, Xiao X, Hu Y, et al. Dysbiosis Signature of Fecal Microbiota in Colorectal Cancer Patients. Microb Ecol (2013) 66:462–70. doi: 10.1007/s00248-013-0245-9
130. Weir TL, Manter DK, Sheflin AM, Barnett BA, Heuberger AL, Ryan EP. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PloS One (2013) 8:e70803. doi: 10.1371/journal.pone.0070803
131. Thangaraju M, Cresci GA, Liu K, Ananth S, Gnanaprakasam JP, Browning DD, et al. GPR109A Is a G-protein-Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res (2009) 69:2826–32. doi: 10.1158/0008-5472.CAN-08-4466
132. Tang Y, Chen Y, Jiang H, Robbins GT, Nie D. G-protein-coupled receptor for short-chain fatty acids suppresses colon cancer. Int J Cancer (2011) 128:847–56. doi: 10.1002/ijc.25638
133. Scheppach W, Bartram P, Richter A, Richter F, Liepold H, Dusel G, et al. Effect of Short-Chain Fatty Acids on the Human Colonic Mucosa in Vitro. J Parenter Enter Nutr (1992) 16:43–8. doi: 10.1177/014860719201600143
134. Arun KB, Madhavan A, Reshmitha TR, Thomas S, Nisha P. Short chain fatty acids enriched fermentation metabolites of soluble dietary fibre from Musa paradisiaca drives HT29 colon cancer cells to apoptosis. PloS One (2019) 14:1–20. doi: 10.1371/journal.pone.0216604
135. Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr (2002) 132:1012–7. doi: 10.1093/jn/132.5.1012
136. Hague A, Manning AM, Hanlon KA, Hart D, Paraskeva C, Huschtscha LI. Sodium butyrate induces apoptosis in human colonic tumour cell lines in a p53-independent pathway: Implications for the possible role of dietary fibre in the prevention of large-bowel cancer. Int J Cancer (1993) 55:498–505. doi: 10.1002/ijc.2910550329
137. Fu H, Shi YQ, Mo SJ. Effect of short-chain fatty acids on the proliferation and differentiation of the human colonic adenocarcinoma cell line Caco-2. Chin J Dig Dis (2004) 5:115–7. doi: 10.1111/j.1443-9573.2004.00167.x
138. Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg Effect Dictates the Mechanism of Butyrate-Mediated Histone Acetylation and Cell Proliferation. Mol Cell (2012) 48:612–26. doi: 10.1016/j.molcel.2012.08.033
139. Wang W, Fang D, Zhang H, Xue J, Wangchuk D, Du J, et al. Sodium Butyrate Selectively Kills Cancer Cells and Inhibits Migration in Colorectal Cancer by Targeting Thioredoxin-1. Onco Targets Ther (2020) 13:4691–704. doi: 10.2147/OTT.S235575
140. Inan MS, Rasoulpour RJ, Yin L, Hubbard AK, Rosenberg DW, Giardina C. The luminal short-chain fatty acid butyrate modulates NF-κB activity in a human colonic epithelial cell line. Gastroenterology (2000) 118:724–34. doi: 10.1016/S0016-5085(00)70142-9
141. Uchiyama K, Sakiyama T, Hasebe T, Musch MW, Miyoshi H, Nakagawa Y, et al. Butyrate and bioactive proteolytic form of Wnt-5a regulate colonic epithelial proliferation and spatial development. Sci Rep (2016) 6:32094. doi: 10.1038/srep32094
142. Murray Stewart T, Dunston TT, Woster PM, Casero RA. Polyamine catabolism and oxidative damage. J Biol Chem (2018) 293:18736–45. doi: 10.1074/jbc.TM118.003337
143. Cervelli M, Pietropaoli S, Signore F, Amendola R, Mariottini P. Polyamines metabolism and breast cancer: state of the art and perspectives. Breast Cancer Res Treat (2014) 148:233–48. doi: 10.1007/s10549-014-3156-7
144. Hayes CS, Shicora AC, Keough MP, Snook AE, Burns MR, Gilmour SK. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res (2014) 2:274–85. doi: 10.1158/2326-6066.CIR-13-0120-T
145. Coni S, Di Magno L, Serrao SM, Kanamori Y, Agostinelli E, Canettieri G. Polyamine Metabolism as a Therapeutic Target inHedgehog-Driven Basal Cell Carcinomaand Medulloblastoma. Cells (2019) 8:150. doi: 10.3390/cells8020150
146. Ramos-Molina B, Queipo-Ortuño MI, Lambertos A, Tinahones FJ, Peñafiel R. Dietary and Gut Microbiota Polyamines in Obesity- and Age-Related Diseases. Front Nutr (2019) 6:24. doi: 10.3389/fnut.2019.00024
147. Miller-Fleming L, Olin-Sandoval V, Campbell K, Ralser M. Remaining Mysteries of Molecular Biology: The Role of Polyamines in the Cell. J Mol Biol (2015) 427:3389–406. doi: 10.1016/j.jmb.2015.06.020
148. Fan J, Feng Z, Chen N. Spermidine as a target for cancer therapy. Pharmacol Res (2020) 159:104943. doi: 10.1016/j.phrs.2020.104943
149. Wallace HM, Caslake R. Polyamines and colon cancer. Eur J Gastroenterol Hepatol (2001) 13:1033–9. doi: 10.1097/00042737-200109000-00006
150. Alexander ET, Minton A, Peters MC, Phanstiel O, Gilmour SK. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget (2017) 8:84140–52. doi: 10.18632/oncotarget.20493
151. Chagneau CV, Garcie C, Bossuet-Greif N, Tronnet S, Brachmann AO, Piel J, et al. The Polyamine Spermidine Modulates the Production of the Bacterial Genotoxin Colibactin. mSphere (2019) 4:e00414–19. doi: 10.1128/mSphere.00414-19
152. Manna SK, Tanaka N, Krausz KW, Haznadar M, Xue X, Matsubara T, et al. Biomarkers of Coordinate Metabolic Reprogramming in Colorectal Tumors in Mice and Humans. Gastroenterology (2014) 146:1313–24. doi: 10.1053/j.gastro.2014.01.017
153. Johnson CH, Dejea CM, Edler D, Hoang LT, Santidrian AF, Felding BH, et al. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab (2015) 21:891–7. doi: 10.1016/j.cmet.2015.04.011
154. Hesterberg R, Cleveland J, Epling-Burnette P. Role of Polyamines in Immune Cell Functions. Med Sci (2018) 6:22. doi: 10.3390/medsci6010022
155. Bowlin TL, McKown BJ, Schroeder KK. Methyl-acetylenicputrescine (MAP), an inhibitor of polyamine biosynthesis, reduces the frequency and cytolytic activity of alloantigen-induced lyt 2.2 positive lymphocytes in vivo. Int J Immunopharmacol (1989) 11:259–65. doi: 10.1016/0192-0561(89)90163-X
156. Bowlin TL, McKown BJ, Sunkara PS. Increased ornithine decarboxylase activity and polyamine biosynthesis are required for optimal cytolytic T lymphocyte induction. Cell Immunol (1987) 105:110–7. doi: 10.1016/0008-8749(87)90060-8
157. Soda. Spermine accelerates hypoxia-initiated cancer cell migration. Int J Oncol (2011) 38:305–12. doi: 10.3892/ijo.2010.849
158. Soda K, Kano Y, Nakamura T, Kasono K, Kawakami M, Konishi F. Spermine, a Natural Polyamine, Suppresses LFA-1 Expression on Human Lymphocyte. J Immunol (2005) 175:237–45. doi: 10.4049/jimmunol.175.1.237
159. Kano Y, Soda K, Nakamura T, Saitoh M, Kawakami M, Konishi F. Increased blood spermine levels decrease the cytotoxic activity of lymphokine-activated killer cells: A novel mechanism of cancer evasion. Cancer Immunol Immunother (2007) 56:771–81. doi: 10.1007/s00262-006-0229-4
160. Soda K, Kano Y, Nakamura T, Kawakami M, Konishi F. Spermine And Spermidine Induce Some Of The Immune Suppression Observed In Cancer Patients. Ann Cancer Res Ther (2003) 11:243–53. doi: 10.4993/acrt1992.11.243
161. Zhang M, Caragine T, Wang H, Cohen PS, Botchkina G, Soda K, et al. Spermine Inhibits Proinflammatory Cytokine Synthesis in Human Mononuclear Cells: A Counterregulatory Mechanism that Restrains the Immune Response. J Exp Med (1997) 185:1759–68. doi: 10.1084/jem.185.10.1759
162. Haskó G, Kuhel DG, Marton A, Nemeth ZH, Deitch EA, Szabó C. SPERMINE DIFFERENTIALLY REGULATES THE PRODUCTION OF INTERLEUKIN-12 P40 AND INTERLEUKIN-10 AND SUPPRESSES THE RELEASE OF THE T HELPER 1 CYTOKINE INTERFERON-γ. Shock (2000) 14:144–9. doi: 10.1097/00024382-200014020-00012
163. Soda K. The mechanisms by which polyamines accelerate tumor spread. J Exp Clin Cancer Res (2011) 30:95. doi: 10.1186/1756-9966-30-95
164. Hardbower DM, Asim M, Luis PB, Singh K, Barry DP, Yang C, et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc Natl Acad Sci (2017) 114:E751–60. doi: 10.1073/pnas.1614958114
165. Latour YL, Gobert AP, Wilson KT. The role of polyamines in the regulation of macrophage polarization and function. Amino Acids (2020) 52:151–60. doi: 10.1007/s00726-019-02719-0
166. Chen Y, Zhuang H, Chen X, Shi Z, Wang X. Spermidine−induced growth inhibition and apoptosis via autophagic activation in cervical cancer. Oncol Rep (2018) 39:2845–54. doi: 10.3892/or.2018.6377
167. Miao H, Ou J, Peng Y, Zhang X, Chen Y, Hao L, et al. Macrophage ABHD5 promotes colorectal cancer growth by suppressing spermidine production by SRM. Nat Commun (2016) 7:11716. doi: 10.1038/ncomms11716
168. Xie L, Yang Y, Meng J, Wen T, Liu J, Xu H. Cationic polysaccharide spermine-pullulan drives tumor associated macrophage towards M1 phenotype to inhibit tumor progression. Int J Biol Macromol (2019) 123:1012–9. doi: 10.1016/j.ijbiomac.2018.11.089
169. Burkitt DP. Epidemiology of cancer of the colon and rectum. Cancer (1971) 28:3–13. doi: 10.1002/1097-0142(197107)28:1<3::AID-CNCR2820280104>3.0.CO;2-N
170. Ou J, Carbonero F, Zoetendal EG, DeLany JP, Wang M, Newton K, et al. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am J Clin Nutr (2013) 98:111–20. doi: 10.3945/ajcn.112.056689
171. Waldecker M, Kautenburger T, Daumann H, Busch C, Schrenk D. Inhibition of histone-deacetylase activity by short-chain fatty acids and some polyphenol metabolites formed in the colon. J Nutr Biochem (2008) 19:587–93. doi: 10.1016/j.jnutbio.2007.08.002
172. Bernstein C, Holubec H, Bhattacharyya AK, Nguyen H, Payne CM, Zaitlin B, et al. Carcinogenicity of deoxycholate, a secondary bile acid. Arch Toxicol (2011) 85:863–71. doi: 10.1007/s00204-011-0648-7
173. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature (2006) 444:1027–31. doi: 10.1038/nature05414
174. Clemente JC, Ursell LK, Parfrey LW, Knight R. The impact of the gut microbiota on human health: An integrative view. Cell (2012) 148:1258–70. doi: 10.1016/j.cell.2012.01.035
175. Bradbury KE, Murphy N, Key TJ. Diet and colorectal cancer in UK Biobank: A prospective study. Int J Epidemiol (2020) 49:246–58. doi: 10.1093/ije/dyz064
176. Larsson SC, Bergkvist L, Wolk A. Processed meat consumption, dietary nitrosamines and stomach cancer risk in a cohort of Swedish women. Int J Cancer (2006) 119:915–9. doi: 10.1002/ijc.21925
177. Liu L, Tabung FK, Zhang X, Nowak JA, Qian ZR, Hamada T, et al. Diets That Promote Colon Inflammation Associate With Risk of Colorectal Carcinomas That Contain Fusobacterium nucleatum. Clin Gastroenterol Hepatol (2018) 16:1622–31.e3. doi: 10.1016/j.cgh.2018.04.030
178. Agus A, Denizot J, Thévenot J, Martinez-Medina M, Massier S, Sauvanet P, et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci Rep (2016) 6:19032. doi: 10.1038/srep19032
179. Krautkramer KA, Kreznar JH, Romano KA, Vivas EI, Barrett-Wilt GA, Rabaglia ME, et al. Diet-Microbiota Interactions Mediate Global Epigenetic Programming in Multiple Host Tissues. Mol Cell (2016) 64:982–92. doi: 10.1016/j.molcel.2016.10.025
180. Sandberg JC, Björck IME, Nilsson AC. Effects of whole grain rye, with and without resistant starch type 2 supplementation, on glucose tolerance, gut hormones, inflammation and appetite regulation in an 11-14.5 hour perspective; a randomized controlled study in healthy subjects. Nutr J (2017) 16:25. doi: 10.1186/s12937-017-0246-5
181. Priebe MG, Wang H, Weening D, Schepers M, Preston T, Vonk RJ. Factors related to colonic fermentation of nondigestible carbohydrates of a previous evening meal increase tissue glucose uptake and moderate glucose-associated inflammation. Am J Clin Nutr (2010) 91:90–7. doi: 10.3945/ajcn.2009.28521
182. Si H, Yang Q, Hu H, Ding C, Wang H, Lin X. Colorectal cancer occurrence and treatment based on changes in intestinal flora. Semin Cancer Biol (2020) 70:3–10. doi: 10.1016/j.semcancer.2020.05.004
183. Lee CF, Ho JWC, Fong DYT, MacFarlane DJ, Cerin E, Lee AM, et al. Dietary and Physical Activity Interventions for Colorectal Cancer Survivors: A Randomized Controlled Trial. Sci Rep (2018) 8:5731. doi: 10.1038/s41598-018-24042-6
184. Ho JWC, Lee AM, Macfarlane DJ, Fong DYT, Leung S, Cerin E, et al. Study protocol for “Moving Bright, Eating Smart”- A phase 2 clinical trial on the acceptability and feasibility of a diet and physical activity intervention to prevent recurrence in colorectal cancer survivors. BMC Public Health (2013) 13:487. doi: 10.1186/1471-2458-13-487
185. Norat T, Scoccianti C, Boutron-Ruault MC, Anderson A, Berrino F, Cecchini M, et al. European Code against Cancer 4th Edition: Diet and cancer. Cancer Epidemiol (2015) 39:S56–66. doi: 10.1016/j.canep.2014.12.016
186. Terry P, Giovannucci E, Michels KB, Bergkvist L, Hansen H, Holmberg L, et al. Fruit, Vegetables, Dietary Fiber, and Risk of Colorectal Cancer. JNCI J Natl Cancer Inst (2001) 93:525–33. doi: 10.1093/jnci/93.7.525
187. Kuugbee ED, Shang X, Gamallat Y, Bamba D, Awadasseid A, Suliman MA, et al. Structural Change in Microbiota by a Probiotic Cocktail Enhances the Gut Barrier and Reduces Cancer via TLR2 Signaling in a Rat Model of Colon Cancer. Dig Dis Sci (2016) 61:2908–20. doi: 10.1007/s10620-016-4238-7
188. Foo N-P, Ou Yang H, Chiu H-H, Chan H-Y, Liao C-C, Yu C-K, et al. Probiotics Prevent the Development of 1,2-Dimethylhydrazine (DMH)-Induced Colonic Tumorigenesis through Suppressed Colonic Mucosa Cellular Proliferation and Increased Stimulation of Macrophages. J Agric Food Chem (2011) 59:13337–45. doi: 10.1021/jf203444d
189. Bauer-Marinovic M. Dietary resistant starch type 3 prevents tumor induction by 1,2-dimethylhydrazine and alters proliferation, apoptosis and dedifferentiation in rat colon. Carcinogenesis (2006) 27:1849–59. doi: 10.1093/carcin/bgl025
190. Ohkawara S, Furuya H, Nagashima K, Asanuma N, Hino T. Oral Administration of Butyrivibrio fibrisolvens, a Butyrate-Producing Bacterium, Decreases the Formation of Aberrant Crypt Foci in the Colon and Rectum of Mice. J Nutr (2005) 135:2878–83. doi: 10.1093/jn/135.12.2878
191. Carroll RE, Benya RV, Turgeon DK, Vareed S, Neuman M, Rodriguez L, et al. Phase IIa Clinical Trial of Curcumin for the Prevention of Colorectal Neoplasia. Cancer Prev Res (2011) 4:354–64. doi: 10.1158/1940-6207.CAPR-10-0098
192. Churchill M, Chadburn A, Bilinski RT, Bertagnolli MM. Inhibition of Intestinal Tumors by Curcumin Is Associated with Changes in the Intestinal Immune Cell Profile. J Surg Res (2000) 89:169–75. doi: 10.1006/jsre.2000.5826
193. Linsalata M, Russo F, Notarnicola M, Guerra V, Cavallini A, Clemente C, et al. Effects of Genistein on the Polyamine Metabolism and Cell Growth in DLD-1 Human Colon Cancer Cells. Nutr Cancer (2005) 52:84–93. doi: 10.1207/s15327914nc5201_11
194. Au A, Li B, Wang W, Roy H, Koehler K, Birt D. Effect of Dietary Apigenin on Colonic Ornithine Decarboxylase Activity, Aberrant Crypt Foci Formation, and Tumorigenesis in Different Experimental Models. Nutr Cancer (2006) 54:243–51. doi: 10.1207/s15327914nc5402_11
195. Meyskens FL, McLaren CE, Pelot D, Fujikawa-Brooks S, Carpenter PM, Hawk E, et al. Difluoromethylornithine Plus Sulindac for the Prevention of Sporadic Colorectal Adenomas: A Randomized Placebo-Controlled, Double-Blind Trial. Cancer Prev Res (2008) 1:32–8. doi: 10.1158/1940-6207.CAPR-08-0042
196. Xie Y, Murray-Stewart T, Wang Y, Yu F, Li J, Marton LJ, et al. Self-immolative nanoparticles for simultaneous delivery of microRNA and targeting of polyamine metabolism in combination cancer therapy. J Control Release (2017) 246:110–9. doi: 10.1016/j.jconrel.2016.12.017
197. Zheng DW, Dong X, Pan P, Chen KW, Fan JX, Cheng SX, et al. Phage-guided modulation of the gut microbiota of mouse models of colorectal cancer augments their responses to chemotherapy. Nat BioMed Eng (2019) 3:717–28. doi: 10.1038/s41551-019-0423-2
198. Dang AT, Marsland BJ. Microbes, metabolites, and the gut–lung axis. Mucosal Immunol (2019) 12:843–50. doi: 10.1038/s41385-019-0160-6
199. Aoun A, Darwish F, Hamod N. The Influence of the Gut Microbiome on Obesity in Adults and the Role of Probiotics, Prebiotics, and Synbiotics for Weight Loss. Prev Nutr Food Sci (2020) 25:113–23. doi: 10.3746/pnf.2020.25.2.113
200. Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. Low gut microbiota diversity in early infancy precedes asthma at school age. Clin Exp Allergy (2014) 44:842–50. doi: 10.1111/cea.12253
201. Abrahamsson TR, Jakobsson HE, Andersson AF, Björkstén B, Engstrand L, Jenmalm MC. Low diversity of the gut microbiota in infants with atopic eczema. J Allergy Clin Immunol (2012) 129:434–440.e2. doi: 10.1016/j.jaci.2011.10.025
202. Zhuang H, Cheng L, Wang Y, Zhang Y-K, Zhao M-F, Liang G-D, et al. Dysbiosis of the Gut Microbiome in Lung Cancer. Front Cell Infect Microbiol (2019) 9:112. doi: 10.3389/fcimb.2019.00112
203. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell (2019) 178:795–806.e12. doi: 10.1016/j.cell.2019.07.008
204. Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discovery (2018) 8:403–16. doi: 10.1158/2159-8290.CD-17-1134
205. Ma J, Sun L, Liu Y, Ren H, Shen Y, Bi F, et al. Alter between gut bacteria and blood metabolites and the anti-tumor effects of Faecalibacterium prausnitzii in breast cancer. BMC Microbiol (2020) 20:82. doi: 10.1186/s12866-020-01739-1
206. Shin N-R, Whon TW, Bae J-W. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol (2015) 33:496–503. doi: 10.1016/j.tibtech.2015.06.011
Keywords: colorectal cancer, tumor micronvironment (TME), intestinal microbiota, diet-derived metabolites, immune system
Citation: Hanus M, Parada-Venegas D, Landskron G, Wielandt AM, Hurtado C, Alvarez K, Hermoso MA, López-Köstner F and De la Fuente M (2021) Immune System, Microbiota, and Microbial Metabolites: The Unresolved Triad in Colorectal Cancer Microenvironment. Front. Immunol. 12:612826. doi: 10.3389/fimmu.2021.612826
Received: 30 September 2020; Accepted: 02 March 2021;
Published: 26 March 2021.
Edited by:
Chengcheng Jin, University of Pennsylvania, United StatesReviewed by:
Luis De La Cruz-Merino, Virgen Macarena University Hospital, SpainCopyright © 2021 Hanus, Parada-Venegas, Landskron, Wielandt, Hurtado, Alvarez, Hermoso, López-Köstner and De la Fuente. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marjorie De la Fuente, bWRlbGFmdWVudGVsQGNsaW5pY2FsYXNjb25kZXMuY2w=; Francisco López-Köstner, ZmxvcGV6a0BjbGluaWNhdWFuZGVzLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.