
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 05 March 2021
Sec. Primary Immunodeficiencies
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.612583
This article is part of the Research Topic Advances in Primary Immunodeficiencies in India View all 12 articles
 Snehal Shabrish1†
Snehal Shabrish1† Madhura Kelkar1†
Madhura Kelkar1† Reetika Malik Yadav1
Reetika Malik Yadav1 Umair Ahmed Bargir1
Umair Ahmed Bargir1 Maya Gupta1
Maya Gupta1 Aparna Dalvi1
Aparna Dalvi1 Jahnavi Aluri1
Jahnavi Aluri1 Manasi Kulkarni1Shweta Shinde1
Manasi Kulkarni1Shweta Shinde1 Sneha Sawant-Desai1
Sneha Sawant-Desai1 Priyanka Kambli1Gouri Hule1Priyanka Setia1
Priyanka Kambli1Gouri Hule1Priyanka Setia1 Neha Jodhawat1
Neha Jodhawat1 Pallavi Gaikwad1
Pallavi Gaikwad1 Amruta Dhawale1Nayana Nambiar1
Amruta Dhawale1Nayana Nambiar1 Vijaya Gowri2
Vijaya Gowri2 Ambreen Pandrowala3
Ambreen Pandrowala3 Prasad Taur2
Prasad Taur2 Revathi Raj4
Revathi Raj4 Ramya Uppuluri4Ratna Sharma5Pranoti Kini5
Ramya Uppuluri4Ratna Sharma5Pranoti Kini5 Meena Sivasankaran6
Meena Sivasankaran6 Deenadayalan Munirathnam6Ramprasad Vedam7
Deenadayalan Munirathnam6Ramprasad Vedam7 Pandiarajan Vignesh8
Pandiarajan Vignesh8 Aaqib Banday8
Aaqib Banday8 Amit Rawat8
Amit Rawat8 Amita Aggarwal9
Amita Aggarwal9 Ujjal Poddar9Meenakshi Girish10Abhijit Chaudhary10Abhilasha Sampagar11
Ujjal Poddar9Meenakshi Girish10Abhijit Chaudhary10Abhilasha Sampagar11 Dharani Jayaraman12
Dharani Jayaraman12 Narendra Chaudhary13Nitin Shah14Farah Jijina14S. Chandrakla15Swati Kanakia16Brijesh Arora17
Narendra Chaudhary13Nitin Shah14Farah Jijina14S. Chandrakla15Swati Kanakia16Brijesh Arora17 Santanu Sen18Madhukar Lokeshwar19
Santanu Sen18Madhukar Lokeshwar19 Mukesh Desai2
Mukesh Desai2 Manisha Madkaikar1*
Manisha Madkaikar1*Hemophagocytic lymphohistiocytosis (HLH) is a syndrome of immune dysregulation characterized by hyperactivation of the immune system, excessive cytokine secretion and severe systemic inflammation. HLH is classified as familial (FHL) when associated with mutations in PRF1, UNC13D, STX11, and STXBP2 genes. There is limited information available about the clinical and mutational spectrum of FHL patients in Indian population. This study is a retrospective analysis of 101 molecularly characterized FHL patients over the last 10 years from 20 different referral centers in India. FHL2 and FHL3 together accounted for 84% of cases of FHL in our cohort. Patients belonging to different FHL subtypes were indistinguishable based on clinical and biochemical parameters. However, flow cytometry-based assays viz. perforin expression and degranulation assay were found to be specific and sensitive in diagnosis and classification of FHL patients. Molecular characterization of respective genes revealed 76 different disease-causing mutations including 39 (51%) novel mutations in PRF1, UNC13D, STX11, and STXBP2 genes. Overall, survival was poor (28%) irrespective of the age of onset or the type of mutation in our cohort. Altogether, this article sheds light on the current scenario of FHL in India. Our data reveal a wide genetic heterogeneity of FHL in the Indian population and confirms the poor prognosis of FHL. This study also emphasizes that though mutational analysis is important for diagnostic confirmation of FHL, flow cytometry based assays help significantly in rapid diagnosis and functional validation of novel variants identified.
Familial hemophagocytic lymphohistiocytosis (FHL) is a disorder of immune dysregulation characterized by persistent high-grade fever, progressive cytopenias, hepatosplenomegaly and systemic inflammation. It is an autosomal recessive disorder and affects mostly infants and young children, but has also been reported in adolescents and adults (1, 2). So far, based on the gene mutations observed, FHL is categorized as FHL2 (PRF1), FHL3 (UNC13D), FHL4 (STX11), and FHL5 (STXBP2) encoding for Perforin, Munc13-4, Syntaxin11, and Syntaxin binding protein 2, respectively (3). These proteins play a fundamental role in lymphocyte cytotoxicity. FHL1 (9q21.3-22) was identified by homozygosity mapping of four inbred families of Pakistani origin (4); though, the disease-causing gene in this locus is yet been unknown.
As the clinical features are similar to those with various infections and inflammatory disorders, diagnosis can often be missed. Though, Histiocyte Society has established HLH diagnosis criteria, which includes clinical manifestations and laboratory findings; it does not help in classifying FHL patients and identifying the underline genetic defect.
Impaired function of NK cells and cytotoxic T lymphocytes (CTLs) due to inherited defect in the granule mediated cytotoxicity is the hallmark of FHL patients (5). Thus, for the diagnosis of these patients, evaluating the function of NK cell and CTLs is crucial. In recent years, assays based on flow cytometry have been developed for evaluating NK cell and CTL cell functions viz. measurement of intracellular perforin expression and degranulation assay determined by upregulation of CD107a expression (6, 7). Granule release assay (GRA) is a screening test for detection of FHL3, FHL4, and FHL5 patients. Measurement of intracellular perforin levels serves as a phenotypic assay in identifying FHL2 patients. These tests serve as rapid screening tests for identifying FHL patients. However, molecular characterization of the respective genes is essential for the final diagnostic confirmation. Identifying underlying genetic defect is also important for offering genetic counseling and prenatal diagnosis in affected families.
The incidence of the four FHL subtypes varies significantly in different ethnic groups; also certain mutations are unique or commonly seen in a particular population (1, 8–10). Understanding this pattern of mutation in a particular population not only helps in cost-effectively designing strategies for mutation screening but may also have epidemiological implications. However, very limited data is available on FHL from India. Thus, in this retrospective study, we report one of the largest series on general clinical features; immunological and molecular findings and outcome of FHL in 101 patients from 20 different referral centers of India over the last 10 years.
This study was approved by Institutional Ethics committee (IEC) for Human subjects of ICMR-National Institute of Immunohematology (NIIH). Ethical clearance was obtained at each center and patients received research information and provided written informed consent to participate in this study. Patients fitting into HLH criteria of the Histiocyte Society (5) referred to ICMR-NIIH or collaborating FPID centers or other tertiary care centers in India from 2010 to 2020 were included in this study. Detailed clinical and family history was recorded for these patients. All the procedures involving human subjects were performed in accordance with the international ethical standards.
As a part of diagnostic workup of HLH, perforin expression and degranulation assay on NK cells were performed as previously described (6, 7, 11). Lymphocyte subset analysis of patient samples was performed using Multitest 6-color TBNK reagent (Becton Dickinson).
Further, confirmation of molecular diagnosis was done either by direct Sanger sequencing (in perforin deficient patients) or by targeted Next-Generation sequencing (NGS) or clinical whole-exome sequencing (WES) (in patients with abnormal degranulation).
The sequences obtained were compared with the reported gene structures (PRF1, UNC13D, STX11, STXBP2: NCBI) using the BLASTN program (http://www.ncbi.nlm.nih.gov/BLAST). Nature of any novel sequence variant was analyzed by using different prediction software i.e., PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), SIFT (http://www.sift.jcvi.org/). For splice region variants, Human Splicing Finder tool is used (https://hsf.genomnis.com/).
Data was presented in terms of median and percentages. For a comparison of >2 groups, One-way analysis of variance (ANOVA) test was used. Mann-Whitney U-test was used for comparing groups with non-parametric data. The p-values ≤0.05 were considered statistically significant. GraphPad Prism (Chicago, IL, USA) version 5 was used for statistical calculations.
In this study, we have included 101 molecularly characterized FHL patients from the year 2010–2020. Of these, 50 patients were identified as FHL2, 35 were FHL3, seven were FHL4 and nine were FHL5 based on the defective genes. Thus, in our Indian cohort, perforin deficiency was found to be the commonest (49%) followed by Munc13-4 (35%) deficiency.
The baseline characteristics of these patients are detailed in Table 1, Supplementary Table 1, which also defines the patients according to their genotype. Of these patients, 67% of patients were male, with a male: female ratio of 2:1. The median age of diagnosis for the entire cohort was 12 months (range: 8 days−33 years) with around 55% of the patients presenting within 1 year of age. Consanguinity was observed in 43% (37/86) patients and 28% (22/80) patients had a family history of an affected sibling. No significant difference in age of presentation was observed amongst the FHL groups.
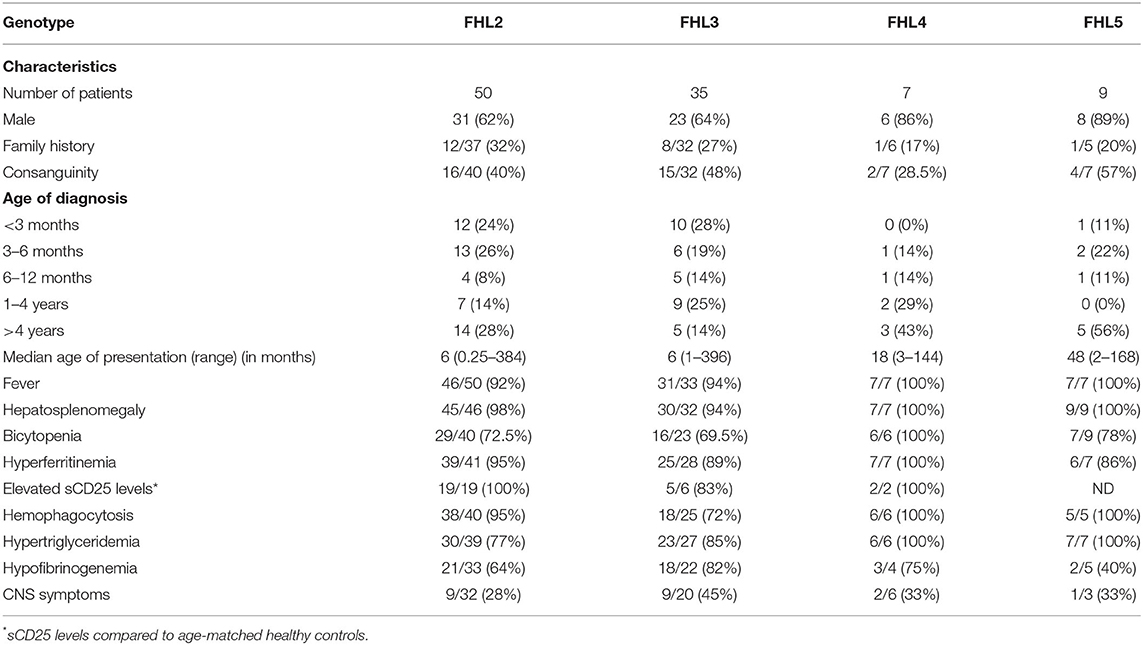
Table 1. Comparison of patient characteristics in different groups of FHL patients.
In our cohort, trigger for FHL development could be identified in 18 patients. Viral infection was found to be the most common trigger in nine patients while bacterial infections are associated with FHL in four patients. Parasitic and fungal infection was seen in one patient each. Three FHL patients had hematological malignancy. However, in rest of the patients, information of triggers for FHL development was not available. 74% of patients fulfilled at least 5/8 HLH diagnostic criteria; whereas 22% of patients fulfilled 4/8 criteria. A pre-symptomatic diagnosis in view of strong family history was achieved in 4% patients. The common clinical presentations seen in all groups of FHL were fever (94%) and hepatosplenomegaly (97%). Hyperferritinemia was observed in 93% cases while 88% patients presented with hemophagocytosis on bone marrow examination. 83.5 and 69% of the patients had hypertriglyceridemia and hypofibrinogenemia, respectively. Deranged liver enzymes were observed in 69% patients. CNS manifestations were seen in 32% patients. Seizure disorder was the commonest CNS manifestation while cerebral palsy and peripheral neuropathy was observed in one patient each. Erythematous skin rash was observed in 19% patients. No significant difference in clinical features was observed between the different subtypes of FHL patients (Table 1).
All FHL patients, except six patients (two FHL2, three FHL3, and one FHL5), had serum ferritin levels >500 ng/ml. Both the FHL2 patients had a family history and they were asymptomatic when referred to our laboratory. Three FHL3 patients (P58, P73, and P74) with <500 ng/ml ferritin had borderline ferritin levels (471, 474, and 488 ng/ml, respectively). Overall, 65% FHL patients had ferritin levels between 500 and 10,000 ng/ml while 17% of FHL2 and 11% of FHL3 patients had ferritin levels >20,000 ng/ml. Ferritin levels did not differ amongst the FHL subtypes. sCD25 levels were available in 27 patients and were elevated in 96% patients.
Lymphocyte subset analysis (enumerating T cells, Tc cells, Th cells, NK cells, and B cells) was performed in 37 patients (18 FHL2, 11 FHL3, 2 FHL4, and 6 FHL5). Though lymphopenia was observed in 49% of the patients, the frequency and absolute counts of lymphocyte subsets did not differ amongst the subsets of FHL (Supplementary Figure 1).
Perforin expression could be checked by flow cytometry in 40 out of 50 FHL2 and 30 out of 51 FHL3/4/5 patients. 38/40 FHL2 (95%) had significantly lower expression of perforin on NK cells (median 2%; reference range 72 ± 2%) than the healthy controls (median 92%) and FHL patients in other groups (median 88%) (p < 0.001) (Figure 1A). 92.5% of the FHL2 patients had ≤10% perforin expression on NK cells with 62.5% having ≤2% expression on NK cells. The cut-off of <10% perforin expression on NK cells to identify patients with biallelic mutations compared to patients with normal sequencing results yielded a sensitivity of 92.5% the specificity of 100%, the positive predictive value of 100%, and negative predictive value of 93.75% with an accuracy of 97% (AUC of 0.9852) (Supplementary Figure 2A).
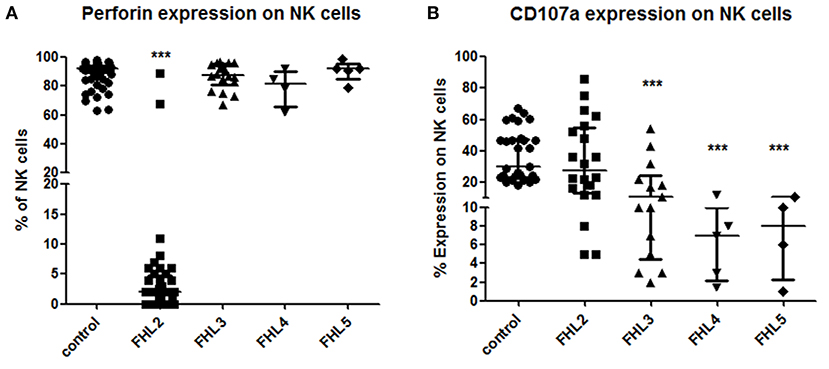
Figure 1. Comparison of Perforin expression and CD107a expression on stimulated NK cells in different groups of FHL patients (A) Perforin expression (B) CD107a expression. One way ANOVA test was used to evaluate differences between activation markers in different groups. ***P < 0.001.
CD107a degranulation assay could be performed in 20 out of 50 FHL2, 18 out of 35 FHL3, five out of seven FHL4 and four out of nine FHL5 patients. Defective degranulation was defined as ≤10% CD107a expression on stimulated NK cells. All FHL4 and FHL5 (100%) and 12/18 FHL3 patients (79%) patients had defective degranulation assay (Figure 1B). Three FHL3 patients (P56, P59, and P61) had borderline CD107a expression on NK cells (22, 18, and 17%, respectively), while three FHL3 patients (P62, P63, and P69) had degranulation within a normal range (Median 37.5%; 22–54%; reference range 28 ± 8%). Additionally, three FHL2 patients (P27, P36, and P39) had defective degranulation on NK cells (Figure 1B). The cut-off of <10% CD107a expression on NK cells for diagnosis of FHL patients with degranulation defect yielded a sensitivity of 73.91%, the specificity of 88.24%, the positive predictive value of 89%, and negative predictive value of 81% with an accuracy of 85% (AUC, 0.8939) (Supplementary Figure 2B).
Out of 102 molecularly characterized FHL patients, details of the mutation spectrum were available in 88 FHL patients (44 FHL2, 29 FHL3, six FHL4, and nine FHL5). This manuscript involves retrospective data from multiple centers all over India of last 10years. Thus, retrieving data of exact annotation of mutations of very old patients (six FHL2, seven FHL3, and one FHL4) was difficult. However, on records, these patients have proven mutations with details of underlying defective gene mentioned. Also for these patients' transplant details and outcome could be traced and hence are included in this study.
Total of 76 different disease-causing mutations were identified in four HLH-related genes (Table 2). Of these 76 mutations identified; 42 (55%) were missense mutations, 7 (9%) were nonsense mutations, 18 (24%) were frameshift mutations and 9 (12%) were intronic and splice site mutations (Table 2). 63 (72%) patients were detected to have homozygous mutations, 12 (14%) had compound heterozygous variants and 12 (14%) had a single heterozygous mutation (Figure 3A). In total, 39 (51%) novel mutations were identified that included 15 missense mutations, four nonsense mutations, 16 frameshift mutations and four splice site and intronic mutation.

Table 2. Different mutations identified in patients with FHL.
In the 44 FHL2 patients, we identified 33 different PRF1 mutations with a total of 17 novel variants (Table 2). Homozygous mutations were found in 62% of FHL2 patients. The most frequent mutations were c.386G>C (p.W129S) which was present in five patients; c.1349C>T (p.T450M) which was present in five patients; c.528_529delinsAA (p. C176X) which was present in three patients; and c.673C>T (p.R225W) which was also seen in three patients. Overall, missense mutations were commonly found in FHL2 patients (48%) in our cohort.
In the 28 patients with FHL3, 30 different mutations with 18 novel variants were identified (Table 2). Homozygous mutations were seen 62% of FHL3 patients. Frameshift, intronic and splice site mutations were more frequently observed in our FHL3 patients (59%). The most frequent mutations were c.762delC (p.C255AfsX73) (frameshift deletion); and c.1822del (V608CfsX16) (frameshift deletion); and c.858+1G>A (intronic splice site mutation) which were present in two patients, respectively.
Also, in the six FHL4 and nine FHL5 patients, we identified five and eight different mutations, respectively (Table 2). c.173T>C (L58P) was found in two FHL4 patients. Homozygous intronic splice site variant 1247_1G>C in STXBP2 gene was observed in three FHL5 patients.
Irrespective of FHL subtype, 53% patients harboring homozygous mutations presented below 1 year of age (median age of presentation was 10 months) while patients with compound heterozygous mutations had later onset of disease (median age of presentation was 3 years). Similarly, 12 patients with a monoallelic mutation in FHL genes had a median age of presentation of 10 months (Figures 2, 3A,B).
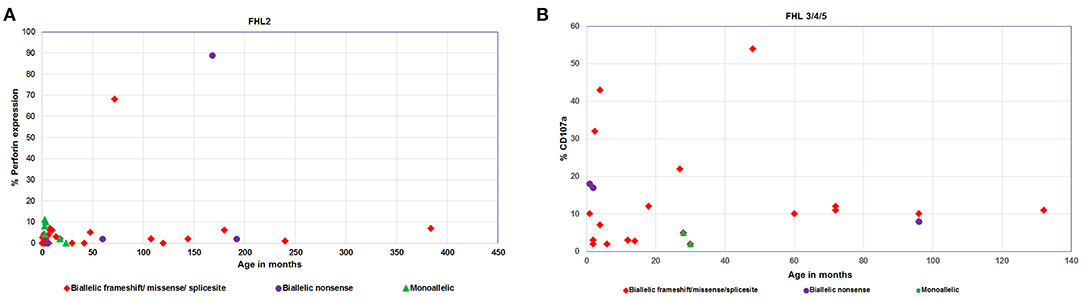
Figure 2. (A) Perforin expression and (B) CD107a degranulation in FHL patients with different age categories and genetic background.
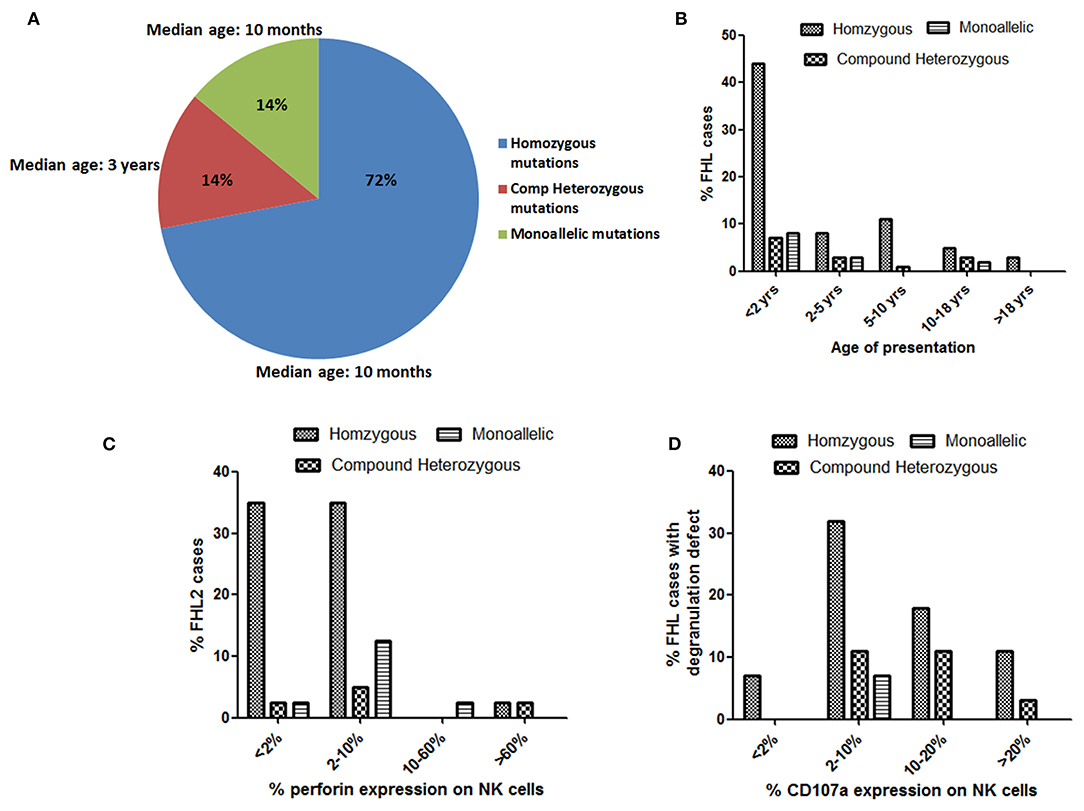
Figure 3. Evaluation of age of presentation of disease (A,B), Perforin expression (C) and CD107a expression (D) on stimulated NK cells in different groups of FHL patients with associated genetic profiles.
Irrespective of the genetic background i.e., presence of homozygous/compound heterozygous/monoallelic mutations, FHL2 patients had perforin expression ≤10% except for patient P34 (with homozygous mutation) and patient P41 (with compound heterozygous mutation). P34 and P41 had 68 and 89% perforin expression on NK cells, respectively (Figures 2A, 3C). However, in P41, mean fluorescence intensity (MFI) of perforin expression was very low (1.83) as compared to healthy control (19.83); indicating that not only expression but MFI of staining also has to be considered during interpretation of the results.
From 43 patients of FHL3/4/5 for whom mutation details were available; 32 patients had homozygous mutations in the corresponding genes (Figures 2B, 3D). Seven patients of FHL3/4/5 had compound heterozygous mutations in the respective genes while the remaining four patients had a monoallelic mutation in the respective genes. Five out of these seven patients with compound heterozygous variants had CD107a degranulation on NK cells ≤10%, one patient P61 had borderline degranulation and one patient P69 had degranulation in the normal range. Additionally, two FHL3 patients (P62 and P63) with homozygous frameshift mutations had normal degranulation assay. Amongst the four patients with monoallelic mutations, CD107a degranulation could be performed in two patients and it was found to be abnormal.
Although majority of the patients had typical HLH manifestations, four FHL patients (three FHL2 i.e., P6, P7, P14, and one FHL5 i.e., P93) are identified with atypical clinical presentations like lymphoma, leukemia, or autoimmune diseases. All of these patients received treatment for the respective pathological conditions and then were diagnosed with HLH. As there was a delay in diagnosis of FHL due to the unusual clinical presentations, all these patients succumbed to the disease.
Of the 101 cases of FHL, 13 patients were lost to follow up and the outcome and remission status could be reported in 88 patients. Seven patients expired before starting the HLH treatment, while 51 patients expired even after receiving HLH-2004/94 protocol. Of the latter, 43 patients did not achieve remission and expired; another eight showed a relapse of the disease post-remission and could not be salvaged. Hematopoietic cell transplantation (HCT) being the definitive treatment, 18 patients underwent BMT. Thirteen are currently doing well while five patients expired due to post-transplant complications. Twelve patients are currently in remission and awaiting transplantation. The treatment and outcome of HLH patients in our study are summarized in Table 3.
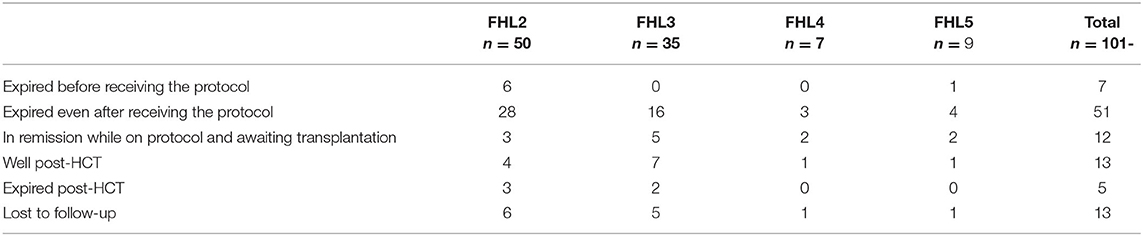
Table 3. Outcome of therapy seen in different FHL groups.
FHL is a genetically heterogeneous disorder of immune dysregulation affecting the cytotoxic function of lymphocytes (3, 5). The pattern of FHL has been reported to vary significantly in different ethnic groups (8–10, 12), however, its prevalence in Indian population is not known. In this study, we have comprehensively evaluated clinical, immunological and molecular findings in 101 molecularly characterized FHL patients from 20 different centers of India. To the best of our knowledge, this is one of the largest studies on the spectrum of FHL in Indian population. This study also evaluates the utility of flow cytometry based assays for efficient diagnosis of FHL patients.
In accordance with previous studies (1, 3, 9, 10, 13), 84% of our FHL patients were either FHL2 or FHL3 (50 and 35%, respectively); while the remaining 16% were FHL4 and FHL5 patients. Also as reported in various retrospective studies in other populations, in our cohort as well prolonged fever, hepatosplenomegaly, hemophagocytosis and hyperferritinemia were the most common clinical presentations (3, 9, 12, 14). CNS manifestations were more common in our FHL3 patients, which is consistent with that reported in literature (15, 16). Although FHL has widely been considered as a disease of infancy (3, 10, 14), 43% of our FHL patients had onset beyond 1year of age, with 24% of FHL2 patients presenting beyond 4 years of age and had severe disease. This suggests the importance of screening for underlying genetic defect in all HLH patients irrespective of their age.
As reported in previous literature (5, 12, 14), 22% of the patients in our cohort did not fulfill the Histiocyte society criteria (5) suggesting certain limitations of existing diagnostic criteria. Many patients may not meet these criteria early during the disease, and some patients may never meet criteria including those with atypical clinical presentations like isolated central nervous system disease (17–19). In some patients, information on levels of sCD25 or NK-cell cytotoxicity may not be available. So in such scenario, measurement of additional screening markers like flow cytometric detection of perforin, CD107a degranulation, T cell upregulation of HLA-DR, serum levels of CXCL9, IL18 are gaining attention for HLH diagnosis and differentiating HLH patients from patients with rheumatological diseases (20).
Nowadays, molecular analysis using targeted NGS or clinical WES is the preferred mode of FHL diagnosis (21). But such molecular diagnosis can be challenging in terms of cost, genetic heterogeneity, analytic difficulty, turnaround time and availability. Thus, in this scenario performing functional screening of cytotoxic cells by flow cytometry is complementary, as the results are available within 72 h. Currently, there are only two Centers of Excellence (COE) in India which provide Immunoassays like flow cytometry based detection of perforin expression and CD107a degranulation on NK cells. Immunoassays (at least one) could be performed in 70 patients in our cohort. In majority of the cases, results are available within 48 h except for the few traveled samples where additional time is required for transport. There are several peripheral centers which send the samples simultaneously for phenotypic and molecular analysis. In recent years, assays based on flow cytometry for evaluating NK cell and CTL cell functions are developed (6, 7, 22–24). In addition to cytotoxicity assays, quantification of perforin, SAP and XIAP proteins which are involved in granule-mediated exocytosis are useful in the diagnosis of primary HLH patients (22, 25–27). Along with NK cells, evaluation of CD107a degranulation on cytotoxic T lymphocytes (CTL) is recently reported in the diagnosis of FHL (28, 29). In our cohort, perforin expression and degranulation assay both had high specificity and sensitivity for distinguishing patients with HLH-associated mutations, as seen in previous studies (11, 23, 24).
Molecular characterization of patients in our cohort, revealed 39 novel variants (51%), highlighting the diverse molecular findings in Indian population compared to other populations reported in the literature (1, 12, 30, 31). The mutations identified in HLH-related genes viz. PRF1, UNC13D, STX11, and STXBP2 genes were widely spread across the coding regions of the respective genes and no founder mutation could be identified in our population. As consanguinity is mainly seen (43%) in our Indian cohort, homozygous mutations in FHL genes are majorly found in the study.
The minimal or complete absence of perforin protein is commonly associated with the most detrimental PRF1 gene mutations while, compound heterozygous PRF1 gene missense mutations may encode partially active perforin and are predominantly identified in older patients with milder clinical manifestations (1, 32, 33). In our cohort, most patients (82.5%) harboring homozygous mutation had perforin expression <10% (Figures 3B,C). In two patients viz. P34 and P41, perforin expression was found to be 68 and 89%, respectively, and had later onset of disease. Patient P34 had c.136G>A (p.E46K) which is a reported missense mutation. This mutation leads to a defect in the MACPF domain of perforin protein which consists of ~349 amino acids stretching in the middle of the protein and is thought to be involved in pore formation. In P41, though perforin expression was 89%, MFI of staining was low. This patient carried compound heterozygous mutation including missense mutation (c.1349 C>T; p.T450M) and nonsense mutation (c.1519G>T; p.E507X) affecting the C2 domain of perforin protein which is involved in Ca2+ and membrane binding. Both these patients had late onset of the disease (6 and 14 years, respectively) probably attributing to residual perforin expression. Despite the presence of perforin expression on NK cells and late-onset of disease, these patients presented with severe clinical manifestations. Mutations identified in these patients might be affecting the function or structure of perforin protein but not its expression, thus highlighting the fact that presence of a protein does not rule out its functional or structural defect. So, in case of high clinical suspicion, molecular analysis along with functional assays should be considered.
All FHL2 patients had normal degranulation pattern except patient P27, P36, and P39. P27 was only 45 days old and abnormal degranulation pattern could be attributed to the immature immune system at this early age (34). Unfortunately, degranulation assay could not be repeated in any of these patients. Majority of the patients with a homozygous mutation in either UNC13D or STX11 or STXBP2 had either defective (≤10%) degranulation on stimulated NK cells (Figure 3D). Borderline NK degranulation (22, 18, 10, and 17%) was observed in patients 56, 59, 60, and 61, respectively. Three of these patients (56, 59, and 61) harbored novel frame-shift mutation in homozygous state in UNC13D gene leading to premature stop codon causing truncated protein formation while patient 60 harbored compound heterozygous missense mutation (one novel and one reported mutation) in UNC13D gene. Although these patients had degranulation ≥10%, it was lower than the healthy controls (22–54%). Thus, indicating that these mutations definitely had effect on the NK cell degranulation pattern. Amongst the FHL3 patients, three patients (62, 63, and 69) harboring novel frameshift mutations in UNC13D gene had NK cell degranulation within normal range. NK cell degranulation assay though has high sensitivity and specificity, it is a screening test and hence can have false negative results. Similar findings were observed in study by Bryceson et al. (11) wherein degranulation assay was evaluated for diagnosis of FHL and was found that 4% of the patients with genetically determined degranulation defects had NK cell degranulation >10% while 7% had above 20% (11). Thus, indicating that, in case of high suspension or family history, one has to go ahead with molecular analysis irrespective of the outcome of functional assays. Evaluation of parents' status in these patients and in vitro functional assays in future may help in assigning pathogenicity in such novel variants.
Interestingly, in our cohort 12 patients (eight FHL2, four FHL3) though harbored monoallelic mutations, had severe clinical manifestations. Phenotypic assays could be performed in nine patients and were abnormal in eight of them. Clinical follow up was available in seven patients. Six of them expired while one required HCT because of relapse and currently well. Of these patients, mutations in three patients (P76–P78) were identified by Whole exome sequencing thus ruling out the possibility of compound heterozygous mutations in these patients. Presence of monoallelic mutations has also been observed in previous studies and there is a growing evidence that monoallelic variants can also contribute to FHL (16). Though involved in the pathogenesis of the disease, monoallelic variants may not be sufficient to initiate the disease phenotype alone. Additional unidentified genetic defects or possibly even environmental factors may contribute to the development of HLH (2, 35). The digenic mode of inheritance (35) and deep intronic variants [c.118-308C>T, c.118-307G>A and 253-kb inversion] in UNC13D gene (36, 37) are described in FHL patients carrying monoallelic variants support this inference. The simple possible explanation is that the variants like promoter mutation, deep intronic variants and big deletions can be missed with the commonly used molecular techniques. Another possibility is that some patients (especially with degranulation defects) might possess pathogenic variants in additional genes that contribute to the development of HLH (35, 38). On the other hand, few monoallelic mutations may confer dominant-negative function to the encoded protein interfering with the cytotoxic function of lymphocytes although the exact mechanism and clinical relevance of these monoallelic mutations need to be explored (39).
Since cellular cytotoxicity plays a crucial role in immune surveillance and tolerance, it gives rise to an association of FHL with cancer, autoimmune susceptibility and other manifestations (16). In our cohort, we identified four FHL patients with atypical initial presentations, namely, two FHL2 patients with malignancy (P6 and P7), one FHL5 patient with ALPS (P93), and one FHL2 patient with isolated neurological relapse (P14). Particularly, isolated CNS manifestations in FHL can be challenging to diagnose (17, 40, 41). These findings highlight some of the atypical manifestations in FHL which may delay diagnosis in selected cases.
Management of HLH is mainly focused on treating the infectious trigger, suppression of hyperactive immune system and correction of an underlying genetic defect. Prompt initiation of immunosuppressive chemotherapy is essential for improved outcome. This can be achieved by increasing the awareness about the availability of immunoassays for diagnosis of HLH helping in early initiation of therapy and preparation for HCT. The underlying triggering infections must be extensively evaluated as the disease activity can be improved by managing the etiology. Some biomarkers like sCD25 levels and sCD163 levels are available at only in limited centers. Awareness to incorporate these biomarkers along with the fall in ferritin levels must be used actively in the management of these patients. In cases of refractory HLH, other novel therapies like Emapalumab or Alemtuzumab are recommended but they are currently not easily available in India (20, 42). In our study cohort, 63 patients expired either before initiating therapy or when on therapy or post-HCT. Thirteen patients are well post-HCT while twelve patients are in remission on HLH protocol and awaiting bone marrow transplantation. Two FHL2 patients having late onset of disease, relapsed when treatment was tapered and hence, emphasizing on the fact that even patients with adult-onset FHL need HCT for better outcome (13). Thus, though early initiation of treatment is necessary for the survival of HLH patients; HCT is the only curative therapy for patients with a known genetic defect and also for patients with recurrence/relapse of the disease despite adequate therapy (though the genetic cause is not known). HLA-matched sibling donor is the preferred donor for FHL patients due to the minimized risk of GVHD. However, various reports on variation in the age at onset within each family, case studies reporting the adult FHL patients and also symptomatic patients harboring monoallelic mutations add to the dilemma of choosing a donor for HCT (43, 44). The studies from our lab have shown that parents and siblings of few FHL2 patients were asymptomatic carriers for the respective mutations in a heterozygous state and had partial (<50%) perforin expression (unpublished data) as has been reported previously (16). Thus, in such cases, the decision of whether to go ahead with HCT from these potential donors is a dilemma especially, in a country like India where transplantation procedures are often not feasible either due to unavailability of HLA-matched donor or the unaffordable cost of therapy.
Thus, to summarize, this is one of the largest studies of retrospective data on the clinical, immunological and molecular spectrum of FHL patients in Indian population. Availability of flow cytometry based assays and NGS have significantly improved the diagnosis and management of FHL patients. Both flow cytometry based assays and molecular studies are complementary to each other and aid in key management decisions. Forthcoming advances in FHL syndrome recognition to minimize delays in diagnosis and modified treatment and transplant approaches will continue to improve patient outcomes.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Institutional Ethics Committee (IEC) for human subjects of ICMR-National Institute of Immunohematology (NIIH). Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin.
SS and MKe analyzed the data and wrote the manuscript. RM and UB wrote the manuscript and obtained clinical details. MGu, ADa, JA, MKu, SSh, GH, PS, NJ, and NN performed the laboratory investigations. RV, SS-D, PKa, ADh, and PG involved the molecular investigations. PT involved in collection of samples and maintaining clinical details. VG, AP, RR, RU, RS, PKi, MS, DM, PV, AB, AR, AA, UP, MGi, AC, AS, DJ, NC, NS, FJ, SC, SK, BA, SSe, and ML supervised the clinical care and management of patients. MD and MM supervised the study and reviewed the manuscript. All authors contributed to the article and approved the submitted version.
Support from Indian Council of Medical Research (ICMR) to NIIH is gratefully acknowledged.
RV was employed by the company Medgenome, Pvt. Labs, Bangalore, India.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank the Foundation of Primary Immunodeficiency (FPID) for establishing FPID centers across India, which has significantly improved awareness, diagnostic facilities, and management of PIDs.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.612583/full#supplementary-material
1. Trizzino A, zur Stadt U, Ueda I, Risma K, Janka G, Ishii E, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis due to perforin mutations. J Med Genet. (2008) 45:15–21. doi: 10.1136/jmg.2007.052670
2. Clementi R, Emmi L, Maccario R, Liotta F, Moretta L, Danesino C, et al. Adult onset and atypical presentation of hemophagocytic lymphohistiocytosis in siblings carrying PRF1 mutations. Blood. (2002) 100:2266–7. doi: 10.1182/blood-2002-04-1030
3. Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Annu Rev Med. (2012) 63:233–46. doi: 10.1146/annurev-med-041610-134208
4. Ohadi M, Lalloz MR, Sham P, Zhao J, Dearlove AM, Shiach C, et al. Localization of a gene for familial hemophagocytic lymphohistiocytosis at chromosome 9q21.3-22 by homozygosity mapping. Am J Hum Genet. (1999) 64:165–71. doi: 10.1086/302187
5. Filipovich AH. Hemophagocytic lymphohistiocytosis (HLH) and related disorders. Hematology. (2009) 2009:127–31. doi: 10.1182/asheducation-2009.1.127
6. Shabrish S, Gupta M, Madkaikar M. A modified NK cell degranulation assay applicable for routine evaluation of NK cell function. J Immunol Res. (2016) 2016:3769590. doi: 10.1155/2016/3769590
7. Kogawa K, Lee SM, Villanueva J, Marmer D, Sumegi J, Filipovich AH. Perforin expression in cytotoxic lymphocytes from patients with hemophagocytic lymphohistiocytosis and their family members. Blood. (2002) 99:61–6. doi: 10.1182/blood.V99.1.61
8. Ishii E, Ohga S, Imashuku S, Kimura N, Ueda I, Morimoto A, et al. Review of hemophagocytic lymphohistiocytosis (HLH) in children with focus on Japanese experiences. Crit Rev Oncol Hematol. (2005) 53:209–23. doi: 10.1016/j.critrevonc.2004.11.002
9. Aricò M, Janka G, Fischer A, Henter JI, Blanche S, Elinder G, et al. Hemophagocytic lymphohistiocytosis. Report of 122 children from the International Registry. FHL Study Group of the Histiocyte Society. Leukemia. (1996) 10:197–203.
10. Zur Stadt U, Beutel K, Kolberg S, Schneppenheim R, Kabisch H, Janka G, et al. Mutation. Spectrum in children with primary hemophagocytic lymphohistiocytosis: molecular functional analyses of PRF1, UNC13D, STX11, RAB27A. Hum Mutat. (2006) 27:62–8. doi: 10.1002/humu.20274
11. Bryceson YT, Pende D, Maul-Pavicic A, Gilmour KC, Ufheil H, Vraetz T, et al. A prospective evaluation of degranulation assays in the rapid diagnosis of familial hemophagocytic syndromes. Blood. (2012) 119:2754–63. doi: 10.1182/blood-2011-08-374199
12. Ariffin H, Lum SH, Cheok SA, Shekhar K, Ariffin WA, Chan LL, et al. Haemophagocytic lymphohistiocytosis in Malaysian children. J Paediatr Child Health. (2005) 41:136–9. doi: 10.1111/j.1440-1754.2005.00564.x
13. Sieni E, Cetica V, Piccin A, Gherlinzoni F, Sasso FC, Rabusin M, et al. Familial hemophagocytic lymphohistiocytosis may present during adulthood: clinical and genetic features of a small series. PLoS ONE. (2012) 7:e44649. doi: 10.1371/journal.pone.0044649
14. Cetica V, Sieni E, Pende D, Danesino C, De Fusco C, Locatelli F, et al. Genetic predisposition to hemophagocytic lymphohistiocytosis: report on 500 patients from the Italian registry. J Allergy Clin Immunol. (2016) 137:188–96.e4. doi: 10.1016/j.jaci.2015.06.048
15. Sieni E, Cetica V, Santoro A, Beutel K, Mastrodicasa E, Meeths M, et al. Genotype-phenotype study of familial haemophagocytic lymphohistiocytosis type 3. J Med Genet. (2011) 48:343–52. doi: 10.1136/jmg.2010.085456
16. Sieni E, Cetica V, Hackmann Y, Coniglio ML, Da Ros M, Ciambotti B, et al. Familial hemophagocytic lymphohistiocytosis: when rare diseases shed light on immune system functioning. Front Immunol. (2014) 5:167. doi: 10.3389/fimmu.2014.00167
17. Madkaikar M, Gupta M, Dixit A, Patil V. Predominant neurologic manifestations seen in a patient with a biallelic perforin1 mutation (PRF1; p.R225W). J Pediatr Hematol Oncol. (2017) 39:143–6. doi: 10.1097/MPH.0000000000000597
18. Rohr J, Beutel K, Maul-Pavicic A, Vraetz T, Thiel J, Warnatz K, et al. Atypical familial hemophagocytic lymphohistiocytosis due to mutations in UNC13D and STXBP2 overlaps with primary immunodeficiency diseases. Haematologica. (2010) 95:2080–7. doi: 10.3324/haematol.2010.029389
19. Mhatre S, Madkaikar M, Jijina F, Ghosh K. Unusual clinical presentations of familial hemophagocytic lymphohistiocytosis type-2. J Pediatr Hematol Oncol. (2014) 36:e524–7. doi: 10.1097/MPH.0000000000000102
20. Marsh RA, Haddad E. How i treat primary haemophagocytic lymphohistiocytosis. Br J Haematol. (2018) 182:185–99. doi: 10.1111/bjh.15274
21. Ammann S, Lehmberg K, zur Stadt U, Klemann C, Bode SFN, Speckmann C, et al. Effective immunological guidance of genetic analyses including exome sequencing in patients evaluated for hemophagocytic lymphohistiocytosis. J Clin Immunol. (2017) 37:770–80. doi: 10.1007/s10875-017-0443-1
22. Madkaikar MR, Shabrish S, Kulkarni M, Aluri J, Dalvi A, Kelkar M, et al. Application of flow cytometry in primary immunodeficiencies: experience from India. Front Immunol. (2019) 10:1248. doi: 10.3389/fimmu.2019.01248
23. Rubin TS, Zhang K, Gifford C, Lane A, Choo S, Bleesing JJ, et al. Perforin and CD107a testing is superior to NK cell function testing for screening patients for genetic HLH. Blood. (2017) 129:2993–9. doi: 10.1182/blood-2016-12-753830
24. Abdalgani M, Filipovich AH, Choo S, Zhang K, Gifford C, Villanueva J, et al. Accuracy of flow cytometric perforin screening for detecting patients with FHL due to PRF1 mutations. Blood. (2015) 126:1858–9. doi: 10.1182/blood-2015-06-648659
25. Gifford CE, Weingartner E, Villanueva J, Johnson J, Zhang K, Filipovich AH, et al. Clinical flow cytometric screening of SAP and XIAP expression accurately identifies patients with SH2D1A and XIAP/BIRC4 mutations. Cytometry B Clin Cytom. (2014) 86:263–71. doi: 10.1002/cytob.21166
26. Chiang SCC, Bleesing JJ, Marsh RA. Current flow cytometric assays for the screening and diagnosis of primary HLH. Front Immunol. (2019) 10:1740. doi: 10.3389/fimmu.2019.01740
27. Ammann S, Elling R, Gyrd-Hansen M, Dückers G, Bredius R, Burns SO, et al. A new functional assay for the diagnosis of X-linked inhibitor of apoptosis (XIAP) deficiency. Clin Exp Immunol. (2014) 176:394–400. doi: 10.1111/cei.12306
28. Hori M, Yasumi T, Shimodera S, Shibata H, Hiejima E, Oda H, et al. A CD57+ CTL degranulation assay effectively identifies familial hemophagocytic lymphohistiocytosis type 3 patients. J Clin Immunol. (2017) 37:92–9. doi: 10.1007/s10875-016-0357-3
29. Chiang SCC, Theorell J, Entesarian M, Meeths M, Mastafa M, Al-Herz W, et al. Comparison of primary human cytotoxic T-cell and natural killer cell responses reveal similar molecular requirements for lytic granule exocytosis but differences in cytokine production. Blood. (2013) 121:1345–66. doi: 10.1182/blood-2012-07-442558
30. Molleran Lee S, Villanueva J, Sumegi J, Zhang K, Kogawa K, Davis J, et al. Characterisation of diverse PRF1 mutations leading to decreased natural killer cell activity in North American families with haemophagocytic lymphohistiocytosis. J Med Genet. (2004) 41:137–44. doi: 10.1136/jmg.2003.011528
31. Yoon HS, Kim HJ, Yoo KH, Sung KW, Koo HH, Kang HJ, et al. UNC13D is the predominant causative gene with recurrent splicing mutations in Korean patients with familial hemophagocytic lymphohistiocytosis. Haematologica. (2010) 95:622–6. doi: 10.3324/haematol.2009.016949
32. Chia J, Yeo KP, Whisstock JC, Dunstone MA, Trapani JA, Voskoboinik I. Temperature sensitivity of human perforin mutants unmasks subtotal loss of cytotoxicity, delayed FHL, and a predisposition to cancer. Proc Natl Acad Sci USA. (2009) 106:9809–14. doi: 10.1073/pnas.0903815106
33. Stepp SE, Dufourcq-Lagelouse R, Le Deist F, Bhawan S, Certain S, Mathew PA, et al. Perforin gene defects in familial hemophagocytic lymphohistiocytosis. Science. (1999) 286:1957–9. doi: 10.1126/science.286.5446.1957
34. Strauss-Albee DM, Liang EC, Ranganath T, Aziz N, Blish CA. The newborn human NK cell repertoire is phenotypically formed but functionally reduced. Cytom B Clin Cytom. (2017) 92:33–41. doi: 10.1002/cyto.b.21485
35. Zhang K, Jordan MB, Marsh RA, Johnson JA, Kissell D, Meller J, et al. Hypomorphic mutations in PRF1, MUNC13-4, and STXBP2 are associated with adult-onset familial HLH. Blood. (2011) 118:5794–8. doi: 10.1182/blood-2011-07-370148
36. Entesarian M, Chiang SCC, Schlums H, Meeths M, Chan M-Y, Mya S-N, et al. Novel deep intronic and missense UNC13D mutations in familial haemophagocytic lymphohistiocytosis type 3. Br J Haematol. (2013) 162:415–8. doi: 10.1111/bjh.12371
37. Qian Y, Johnson JA, Connor JA, Valencia CA, Barasa N, Schubert J, et al. The 253-kb inversion and deep intronic mutations in UNC13D are present in North American patients with familial hemophagocytic lymphohistiocytosis 3. Pediatr Blood Cancer. (2014) 61:1034–40. doi: 10.1002/pbc.24955
38. Zhang K, Chandrakasan S, Chapman H, Valencia CA, Husami A, Kissell D, et al. Synergistic defects of different molecules in the cytotoxic pathway lead to clinical familial hemophagocytic lymphohistiocytosis. Blood. (2014) 124:1331–4. doi: 10.1182/blood-2014-05-573105
39. Spessott WA, Sanmillan ML, McCormick ME, Patel N, Villanueva J, Zhang K, et al. Hemophagocytic lymphohistiocytosis caused by dominant-negative mutations in STXBP2 that inhibit SNARE-mediated membrane fusion. Blood. (2015) 125:1566. doi: 10.1182/blood-2014-11-610816
40. Chiapparini L, Uziel G, Vallinoto C, Bruzzone MG, Rovelli A, Tricomi G, et al. Hemophagocytic lymphohistiocytosis with neurological presentation: MRI findings and a nearly miss diagnosis. Neurol Sci. (2011) 32:473–7. doi: 10.1007/s10072-010-0467-2
41. Khazal S, Polishchuk V, Soffer G, Prinzing S, Gill J, Mahadeo KM. Allogeneic hematopoietic stem cell transplantation is associated with cure and durable remission of late-onset primary isolated central nervous system hemophagocytic lymphohistiocytosis. Pediatr Transplant. (2018) 22:473–7. doi: 10.1111/petr.13101
42. Lounder DT, Bin Q, de Min C, Jordan MB. Treatment of refractory hemophagocytic lymphohistiocytosis with emapalumab despite severe concurrent infections. Blood Adv. (2019) 3:47–50. doi: 10.1182/bloodadvances.2018025858
43. Tesi B, Lagerstedt-Robinson K, Chiang SCC, Bdira E Ben, Abboud M, Belen B, et al. Targeted high-throughput sequencing for genetic diagnostics of hemophagocytic lymphohistiocytosis. Genome Med. (2015) 7:130. doi: 10.1186/s13073-015-0244-1
Keywords: familial hemophagocytic lymphohistocytosis, perforin, degranulation, HLH-targeted therapy, flow cytomertry, NGS
Citation: Shabrish S, Kelkar M, Yadav RM, Bargir UA, Gupta M, Dalvi A, Aluri J, Kulkarni M, Shinde S, Sawant-Desai S, Kambli P, Hule G, Setia P, Jodhawat N, Gaikwad P, Dhawale A, Nambiar N, Gowri V, Pandrowala A, Taur P, Raj R, Uppuluri R, Sharma R, Kini P, Sivasankaran M, Munirathnam D, Vedam R, Vignesh P, Banday A, Rawat A, Aggarwal A, Poddar U, Girish M, Chaudhary A, Sampagar A, Jayaraman D, Chaudhary N, Shah N, Jijina F, Chandrakla S, Kanakia S, Arora B, Sen S, Lokeshwar M, Desai M and Madkaikar M (2021) The Spectrum of Clinical, Immunological, and Molecular Findings in Familial Hemophagocytic Lymphohistiocytosis: Experience From India. Front. Immunol. 12:612583. doi: 10.3389/fimmu.2021.612583
Received: 30 September 2020; Accepted: 04 February 2021;
Published: 05 March 2021.
Edited by:
Sudhir Gupta, University of California, Irvine, United StatesReviewed by:
Kimberly Risma, Cincinnati Children's Hospital Medical Center, United StatesCopyright © 2021 Shabrish, Kelkar, Yadav, Bargir, Gupta, Dalvi, Aluri, Kulkarni, Shinde, Sawant-Desai, Kambli, Hule, Setia, Jodhawat, Gaikwad, Dhawale, Nambiar, Gowri, Pandrowala, Taur, Raj, Uppuluri, Sharma, Kini, Sivasankaran, Munirathnam, Vedam, Vignesh, Banday, Rawat, Aggarwal, Poddar, Girish, Chaudhary, Sampagar, Jayaraman, Chaudhary, Shah, Jijina, Chandrakla, Kanakia, Arora, Sen, Lokeshwar, Desai and Madkaikar. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manisha Madkaikar, bWFka2Fpa2FybWFuaXNoYUBnbWFpbC5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.