
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 15 March 2021
Sec. Cancer Immunity and Immunotherapy
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.608625
This article is part of the Research Topic Immune Drivers of Malignancy View all 7 articles
 Lisa Rohrbacher1,2
Lisa Rohrbacher1,2 Bettina Brauchle1,2
Bettina Brauchle1,2 Ana Ogrinc Wagner1,2
Ana Ogrinc Wagner1,2 Michael von Bergwelt-Baildon1,2,3
Michael von Bergwelt-Baildon1,2,3 Veit L. Bücklein1,2
Veit L. Bücklein1,2 Marion Subklewe1,2,3*
Marion Subklewe1,2,3*B-cell receptors, multiple receptor tyrosine kinases, and downstream effectors are constitutively active in chronic lymphocytic leukemia (CLL) B cells. Activation of these pathways results in resistance to apoptosis and enhanced survival of the leukemic cells. Idelalisib is a highly selective inhibitor of the PI3K p110∂ isoform and is approved for the treatment of CLL in patients with relapsed/refractory disease or in those harboring 17p deletions or tp53 mutations. Despite the initial excitement centered around high response rates in clinical trials of idelalisib, its therapeutic success has been hindered by the incidence of severe opportunistic infections. To examine the potential contribution of idelalisib to the increased risk of infection, we investigated the effects of idelalisib on the immune cell compartments of healthy donors (HDs) and CLL patients. PI3K∂ blockade by idelalisib reduced the expression levels of inhibitory checkpoint molecules in T cells isolated from both HDs and CLL patients. In addition, the presence of idelalisib in cultures significantly decreased T-cell-mediated cytotoxicity and granzyme B secretion, as well as cytokine secretion levels in both cohorts. Furthermore, idelalisib reduced the proliferation and cytotoxicity of HD NK cells. Collectively, our data demonstrate that both human T and NK cells are highly sensitive to PI3K∂ inhibition. Idelalisib interfered with the functions of T and NK cell cells from both HDs and CLL patients. Therefore, idelalisib might contribute to an increased risk of infections regardless of the underlying B-cell malignancy.
Chronic lymphocytic leukemia (CLL) is characterized by impairment of the immune system and is therefore associated with an increased susceptibility to opportunistic infections (1–5). Several factors contribute to this increased risk profile: CLL cells compromise the development of healthy B cells, cause immunosuppression due to their close proximity to effector cells, modulate T-cell function, and cause immunoglobulin deficiency (6–9). Therefore, immunotherapeutic approaches are indicated and first-line treatments consist of rituximab or obinutuzumab as part of either chemoimmunotherapy or targeted therapy with ibrutinib and venetoclax (10). The latter target B-cell receptor (BCR) signaling and downstream receptor tyrosine kinases, which play a key role in the pathogenesis of CLL (11–17).
Idelalisib is a potent small-molecule inhibitor of phosphoinositide 3-kinases (18, 19). PI3K is one of the most commonly activated kinases in the BCR signaling cascade (20–23). Class I PI3Ks are comprised of a regulatory subunit and one of four catalytic subunits (p110 α, β, γ, and ∂)(24–27). These isoforms differ in tissue expression: PI3Kα and PI3Kβ are ubiquitously expressed, whereas PI3Kγ and PI3K∂ are highly enriched in the hematopoietic compartment (19, 21, 25, 28–30). Mechanistically, PI3K∂ activates the serine/threonine kinases AKT and mammalian target of rapamycin (mTOR), which leads to proliferation, differentiation, and enhanced survival of the cancer cells (19, 24, 25, 31–33). Idelalisib binds to the ATP-binding pocket of the catalytic subunit of PI3K, thereby specifically abrogating downstream PI3K∂/AKT signaling and inducing apoptosis of malignant cells (34, 35). Idelalisib, has been evaluated in conjunction with rituximab in a randomized, double-blind, phase III study in patients with CLL (ClinicalTrials.gov Identifier: NCT01539512). Due to high response rates of 81%, idelalisib was approved by the US Food and Drug Administration for the first-line treatment of CLL patients with the 17p deletion or TP53 mutation, and also in the relapsed or refractory (r/r) setting (35, 36).
However, after the initial excitement, three clinical trials involving idelalisib reported a high rate of adverse events, including severe diarrhea, liver toxicity, pneumonitis, severe colitis, and serious infections (37–42) (ClinicalTrials.gov Identifiers: NCT01539512, NCT01732913, NCT01569295). Although a high rate of opportunistic infections in CLL patients is well-documented (43–45), idelalisib treatment not only increased the incidence but also added other immune-related adverse events (46). In a randomized phase III trial in r/r CLL, patients were treated with Rituximab plus Idelalisib (IDELA/R-to-IDELA) vs. Rituximab monotherapy (placebo/R). The group with IDELA/R-to-IDELA had a higher incidence of infection or infestation: 53.6 vs. 23.1%, with lower respiratory tract infection in 23.6 vs. 11.1% (47). Opportunistic infections were a common cause with 5 vs. 1 patient presenting with pneumocystis jirovecii pneumonia, 2 vs. 0 patient presenting with cytomegaly virus infection and 22 patients with fungal infection in the IDELA/R-to-IDELA group. Confirmatory data were reported from another phase III trial comparing ofatumumab with and without Idelalisib in pretreated CLL patients with serious infections being more common in the ofatumumab plus idelalisib group: pneumonia was reported in 23 vs. 8 patients, sepsis in 11 vs. 1 patient, and pneumocystis jirovecii pneumonia in 8 vs. 1 patient (48). This resulted in 22-treatment-related deaths in the ofatumumab plus Idelalisib vs. only 6-treatment-related deaths in the ofatumumab group.
These observations suggest for an additive negative impact of idelalisib on immune effector cells. Although many studies have focused on the influence of idelalisib on CLL cells, less is known about the impact of PI3K∂ blockade on healthy immune cells.
In this study, we investigated the immunomodulatory influence of idelalisib on the adaptive cellular immune compartment. To start dissecting the effect of idelalisib on cellular immune responses independently of its impact on CLL cells, we isolated T and natural killer (NK) cells from healthy donor (HD) peripheral blood (PB). We analyzed changes in the proliferative behavior, cytokine secretion, and cytotoxic capacity of cocultures in the presence of idelalisib. In a second step, we sought to replicate these findings with samples from CLL patients. Our data suggest that idelalisib interferes with T- and NK-cell function, thereby adding to the already increased rate of opportunistic infections in CLL patients.
Idelalisib was provided by Gilead Sciences. A 10 mM stock solution of idelalisib was prepared in dimethyl sulfoxide (DMSO; Serva, 20385.01) and stored at −20°C. The used concentrations of 0.05, 0.5, and 1 μM were chosen to mimic the peak plasma concentrations observed in patients after 150 mg twice daily administration of idelalisib (49). Sources of all key reagents are listed in Supplementary Table 1.
PB samples from HDs and patients with CLL were collected with written consent in accordance with the Declaration of Helsinki and approval by the Institutional Review Board of the Ludwig-Maximilian University of Munich. Patient characteristics are summarized in Table 1. The patients had a median age of 64.5 years with a percentage of female patients of 46.7%.
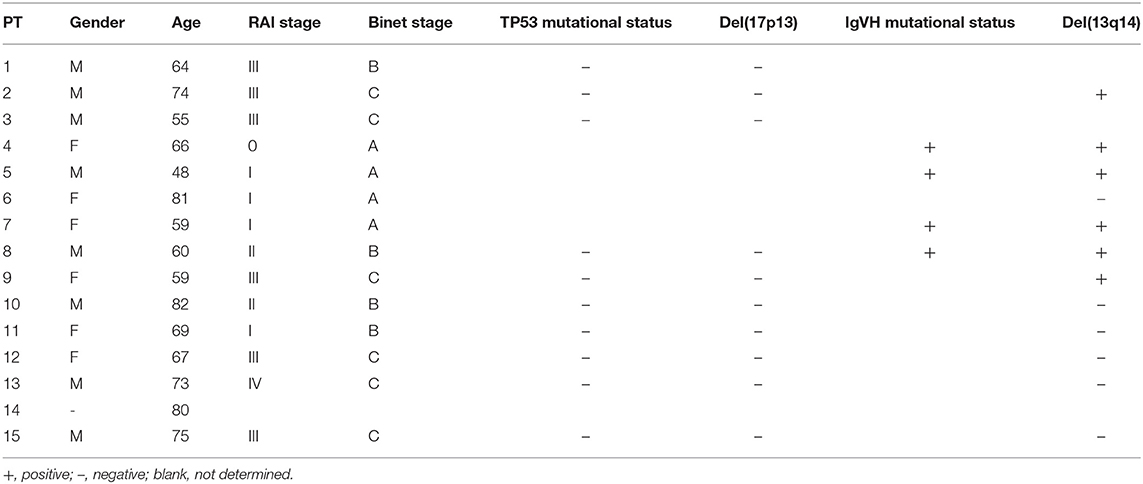
Table 1. Patient characteristics.
Cell lines were obtained from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) and were authenticated by their short tandem repeat profile. All cell lines were tested monthly for Mycoplasma contamination with the MycoAlert Mycoplasma Detection Kit (Lonza, LT07-705) according to the manufacturer's instructions. Cells were passaged twice a week and cultured in RPMI 1640 medium (PAN Biotech, P04-16500) media supplemented with 10% fetal calf serum (Thermo Fisher Scientific, 10270106), 1% HEPES (Carl Roth, HN78.1) and 1% penicillin-streptomycin-glutamine (PSG) (Thermo Fisher Scientific, 10378016) at 37°C in a 5% CO2 atmosphere. Cells were used within 2 months of thawing.
HDs had a median age of 28.4 years with a percentage of females of 48.3%. To overcome the age discrepancy, we extended the trial on an elderly HD cohort (n = 4, Supplementary Figure 2). The median age of this cohort was 62 years with a percentage of 50% females. PB mononuclear cells (PBMCs) from HD were isolated by density gradient centrifugation (using Biochrom separating solution, L6115) from PB and either cryopreserved at < -80°C in cell culture medium (described above) containing 10% DMSO, or directly used for experiments. T cells were negatively isolated from frozen HD PBMCs with the human Pan T Cell Isolation Kit (Miltenyi Biotec, 130-096-535) and cultured in cell culture medium. NK cells were either isolated negatively with the human NK Cell Isolation Kit (Miltenyi Biotec, 130-092-657) or with the EasySep Human CD56 Positive Selection Kit II (Stemcell Technologies, 17815) from fresh HD PBMCs and cultured in NK MACS Medium (Miltenyi Biotec, 130-114-429) supplemented with 5% human serum (Sigma–Aldrich, H6914). Monocytes were positively isolated from fresh HD PBMCs with human CD14 Microbeads (Miltenyi Biotec, 130-050-201). Neutrophils were negatively isolated from fresh HD PB with the EasySep Direct Human Neutrophil Isolation Kit (Stemcell Technologies, 19666).
HD-derived immune cells were cultured with 0.05, 0.5, or 1 μM idelalisib or with the DMSO concentration that matches the DMSO concentration of the drug-treated cultures as vehicle controls.
All measurements were conducted on a CytoFLEX flow cytometer (Beckman Coulter) and analyzed using FlowJo software (BD Biosciences, version 10; RRID: SCR_008520). All antibodies used in the following experiments are listed in Supplementary Table 1. Median fluorescence intensity (MFI) was determined, and the MFI ratio (MFI sample/MFI isotype control) was calculated (Supplementary Figure 1).
HD T cells were stained using the CellTrace CFSE Proliferation Kit (Thermo Fisher Scientific, C34554) according to the manufacturer's instructions. For stimulation, CD3/CD28 Dynabeads (Thermo Fisher Scientific, 11131D) at a bead-to-cell ratio of 1:2 and 30 U/mL interleukin-2 (IL-2; R&D Systems, 202-IL-010/CF) were added to the culture for 5 days. T cell subsets were discriminated by the expression of the chemokine receptor CCR7 in combination with the naïve cell marker CD45Ra (Supplementary Figure 2). NK cells were stimulated by addition of 500 U/mL IL-2 for 10 days. Fold change was calculated as: “Viable NK cell count day 10”/“Viable NK cell count day 0.”
HD T cells were stimulated as described above. After 3 days, the secretion of interferon-γ (IFN-γ), tumor necrosis factor (TNF), IL-2 and interleukin 10 (IL-10) was measured by analyzing the cell culture supernatant in a Th1/Th2 cytometric bead array (BD Biosciences, 551809) (50, 51). The assay was performed according to the manufacturer's instructions using flow cytometry.
The cytotoxic capacity of T cells was assessed in two different assays. In the first assay, HD T cells were activated as described above. After 3 days the secretion of the cytolytic molecules perforin and granzyme B was analyzed by multiparameter flow cytometry. In the second assay, the T-cell recruiting antibody-mediated lysis of the target cell line HL-60 at an effector– to–target ratio (E:T) of 1:3 was measured. Either an anti-CD33 bispecific T-cell-recruiting antibody (5 ng/mL) (52), recognizing CD3 on T cells and CD33 on target cells, or no antibody was added to the coculture. After 72 h, target and T cell counts were analyzed by flow cytometry. Lysis was calculated according to the formula %lysis = 100 – [#target cells (with antibody)]/[#target cells (without antibody)]*100.
The cytotoxic capacity of NK cells was assessed in two different assays. In the first assay, the cell line K562 was stained with calcein AM (Thermo Fisher Scientific, C3100MP) according to the manufacturer's instructions. Freshly isolated HD NK cells were cocultured with the stained K562 cells at an E:T ratio of 10:1 in presence of idelalisib or DMSO. After 4 h, the fluorescence intensity of the co-culture supernatant was measured on a microplate reader (excitation: 485 nm; emission: 535 nm). Lysis was calculated according to the formula [(Ftest) – (Fspontaneous)/(Fmaximum) – (Fspontaneous)] × 100, where Fspontaneous represents the fluorescence intensity of calcein released from target cells in medium alone, and Fmaximum is the fluorescence intensity of calcein released from target cells lysed in medium containing 2% Triton X-100 (Sigma–Aldrich, X100-5ML), each measured in at least three replicate wells. In the second assay, freshly isolated HD NK cells were cocultured with K562 or Jurkat cells at an E:T ratio of 5:1 for 20 h in presence of idelalisib or DMSO. After 20 h, specific lysis was analyzed by multiparameter flow cytometry (Beckman Coulter CytoFLEX S flow cytometer). The percentage of lysis was determined as the target cell count of idelalisib-treated relative to the control cultures.
Monocyte function was assessed by measuring phagocytosis of pHrodo Green E.coli BioParticles (Thermo Fisher Scientific, P35366). Freshly isolated monocytes were incubated with the pHrodo particles for 2 h, then phagocytosis was analyzed by flow cytometry.
Neutrophil function was analyzed over 4 h in a standard Seahorse XF neutrophil activation assay according to the manufacturer's instructions.
Statistical analyses were performed using GraphPad Prism Software Version 8.4.2. As statistical test to compare the two treatment groups Wilcoxon matched signed-rank test was used. P-values and the number of replicates performed to derive the data are indicated in the figure legends.
As PI3K∂ was previously shown to be important for T-cell signaling, we evaluated the effect of PI3K∂ blockade on T-cell proliferation. We found that proliferation of CD4+ and CD8+ T cells (Figure 1A) and the respective subsets (Figures 1B,C) was not affected by idelalisib. Next, we analyzed the expression levels of several inhibitory checkpoint molecules in T cells and Tregs after stimulation for 72 or 120 h in the presence of idelalisib. The expression of cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), lymphocyte activation gene 3 (LAG-3), and programmed cell death protein 1 (PD-1) was significantly downregulated in idelalisib-treated T cells and Tregs in comparison to the DMSO vehicle control cells (Figures 1D–G). Our results demonstrate that proliferation of T cells is not solely dependent on signaling through PI3K∂ and thus is not susceptible to inhibition of PI3K∂ by idelalisib. Furthermore, our data demonstrate that blockade of PI3K∂ signaling reduced expression of inhibitory checkpoint molecules in T cells and Tregs.
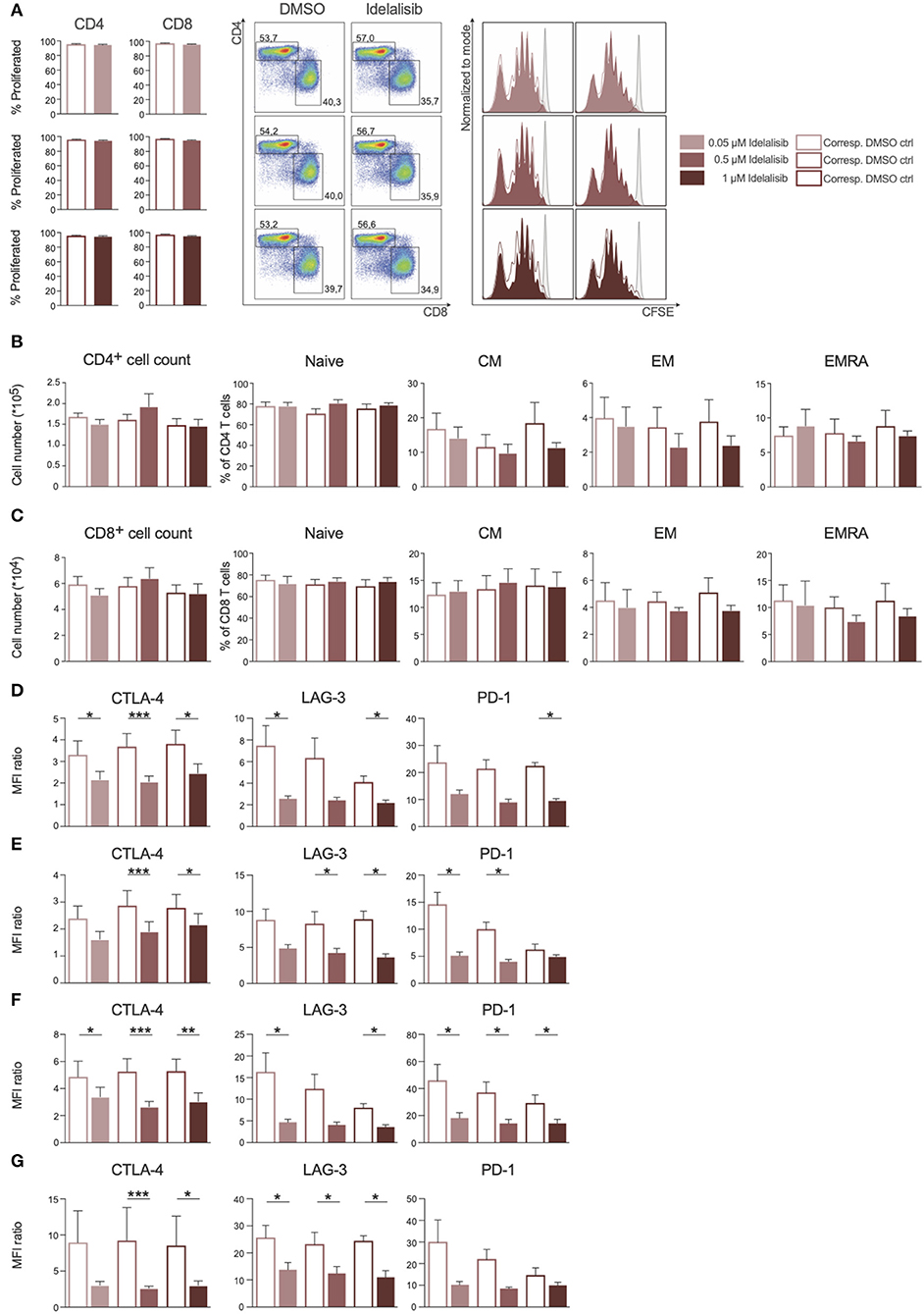
Figure 1. Inhibition of the PI3K∂ signaling pathway by Idelalisib reduces the expression levels of inhibitory checkpoint molecules in CD3+ T cells and Treg cells. (A) Compiled data showing the percentage of proliferated CD4+ and CD8+ T cells in the presence of various concentrations of idelalisib or DMSO, and representative flow cytometry dot plots and histograms. (B,C) Bar charts depicting the development of CD4+ and CD8+ T-cell subsets: naïve, central memory (CM), effector memory (EM), and terminally differentiated effector memory cells re-expressing CD45RA (ERMA) after 6 days of coculture, with different concentrations of idelalisib or DMSO as vehicle control. (D–G) Bar graphs of MFI ratios showing the expression levels of checkpoint molecules CTLA-4, LAG-3, and PD-1 in CD4+ T cells, CD8+ T cells, CD4+ Tregs and CD8+ Tregs after stimulation for 3 or 5 days with IL-2 and CD3/CD28 activation beads. Error bars represent mean ± SEM; *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005; n = 6–12 HDs.
To test whether the downregulation of inhibitory checkpoint molecules in T cells also mirrors reduced T-cell effector function, we analyzed the effect of PI3K∂ blockade on T-cell cytotoxicity and cytokine secretion. The cytotoxicity of T cells against target HL−60 cells was significantly reduced in a coculture in the presence of idelalisib (Figure 2A). To further investigate if this reduction in the cytolytic capacity was due to a decrease in secretion of cytolytic molecules, T cells were analyzed for perforin and granzyme B secretion after 72 h stimulation in the presence of idelalisib. There was a significantly lower degranulation of perforin and granzyme B in the cultures containing idelalisib (Figure 2B). Next, we evaluated the effect of PI3K∂ blockade on the secretion of different cytokines after stimulation for 72 h. Secretion levels of IL-10, TNF, and IFN-γ were significantly reduced in the presence of idelalisib. In contrast, secretion of IL-2 was slightly increased (Figure 2C). Taken together, these results demonstrate that blockade of PI3K∂ signaling with idelalisib has a negative impact on the effector functions of T cells, such as cytotoxicity and cytokine secretion.
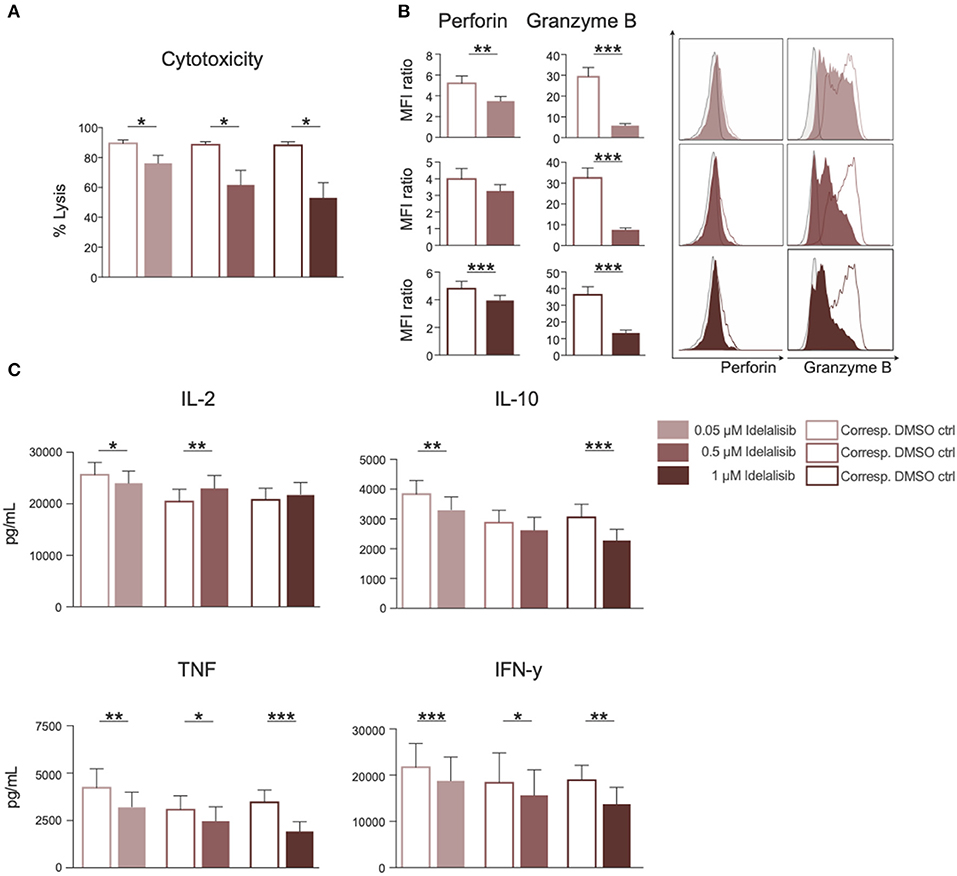
Figure 2. Inhibition of PI3K∂ by idelalisib leads to decreased T-cell-mediated cytotoxicity against target tumor cells and reduced secretion of IL-10, TNF, and IFNγ. (A) Cytolytic capacity of CD3+ T cells in coculture with target HL-60 cell line and various concentrations of idelalisib after 72 h. (B) Bar charts and representative overlaid flow cytometric histograms depicting expression levels of perforin and granzyme B in CD8 T cells after bead mediated activation for 72 h in the presence of different concentrations of idelalisib or DMSO. (C) Bar charts representing the levels of IL-2, IL-10, TNF, and IFNγ in the supernatant of the coculture after 72 h. Error bars represent mean ± SEM; *p ≤ 0.05, **p ≤ 0.005, ***p ≤ 0.0005, n = 12 HDs.
As we found that inhibition of PI3K∂ significantly impairs important functions of the T-cell compartment, we sought to evaluate if similar effects can be seen in NK cells, the cytotoxic lymphocytes of the innate immune system. The addition of idelalisib to IL-2 stimulated NK cells led to a decreased NK-cell expansion in comparison to the DMSO-treated control (Figure 3A). Furthermore, idelalisib affected the proliferation of the cytotoxic NK-cell population over the 10-day period. As the percentage of dead cells in both conditions is not significantly different, this observation is most likely due to a reduced proliferation instead of increased apoptosis in the idelalisib treated condition (media %dead cells 1 μM idelalisib vs. corresponding DMSO control: 24.3 vs. 25.5, n = 12, data not shown). The cytokine producing CD56brightCD16neg compartment showed no differences between cultures (Figure 3B). However, the proliferative capacity of the cytotoxic CD56dimCD16bright subset was reduced upon PI3K∂ blockade (Figure 3C).
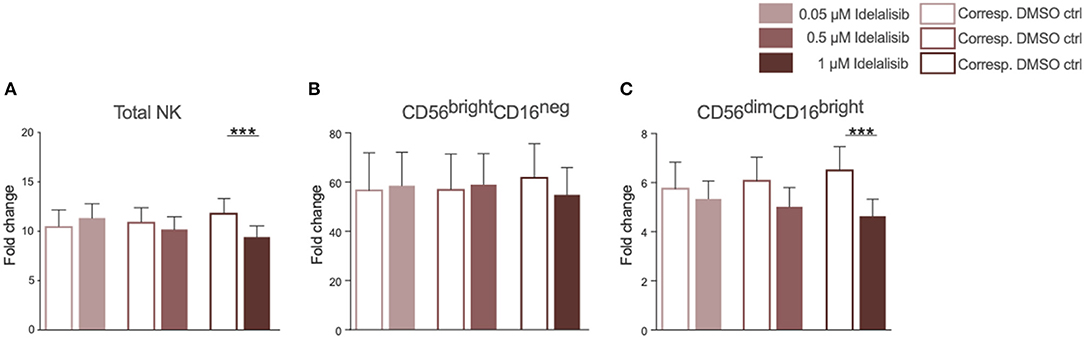
Figure 3. Inhibition of PI3K∂ by idelalisib reduces NK-cell proliferation and percentage of cytotoxic NK cells. (A) Compiled bar chart showing the proliferative capacity of total CD3−CD56+ NK cells after 10 days in the presence of IL-2 and different concentrations of Idelalisib or DMSO as vehicle control. (B,C) The shift in the NK-cell composition is depicted as the fold change between days 0 and day 10 of the subsets CD56brightCD16neg and CD56dimCD16bright. Error bars represent mean ± SEM; ***p ≤ 0.0005; n = 12 HDs.
To evaluate if this reduction translates into impaired cytotoxicity, we analyzed the cytolytic capacity of NK cells toward target cells in presence of idelalisib. NK cells are capable of killing target cells via two different apoptotic pathways, either through perforin- and granzyme- mediated lysis or through death receptor ligation with, for example, the Fas ligand (FasL). To investigate if idelalisib interferes with both apoptotic pathways, we separately cocultured two target cell lines with HD NK cells. The cytotoxicity of NK cells toward target K562 cells, which are lacking the MHC class I antigen and are thus killed via granzyme B-mediated lysis, was significantly reduced (Figure 4A). Lysis of target Jurkat cells, which are killed in a Fas–FasL-dependent manner, was also significantly decreased in the presence of idelalisib (Figure 4B). Together our data show that PI3K∂ signaling is important for NK cell-mediated lysis of cancer cells in vitro and that blockade by idelalisib significantly reduces the cytolytic capability of NK cells.
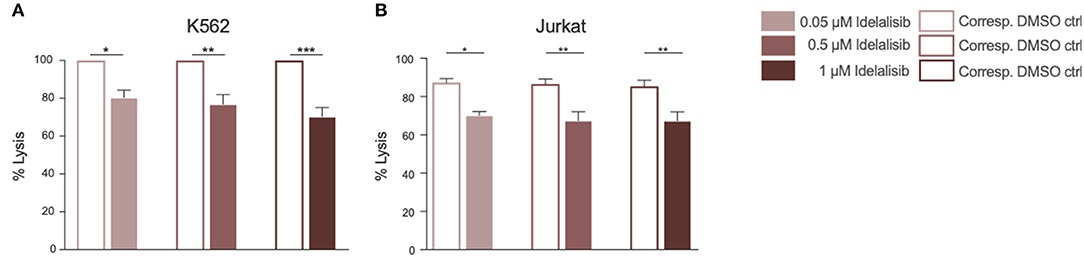
Figure 4. Inhibition of PI3K∂ by idelalisib decreases NK-cell cytotoxicity. (A,B) Cytotoxicity of CD3negCD56pos NK cells against the K562 cell line in a standard calcein-release assay and against the Jurkat cell line in a flow cytometry-based coculture assay. Error bars represent mean ± SEM; *p ≤ 0.05, **p ≤ 0.005; n = 12 HDs.
To further investigate the impact of idelalisib on other innate immune cells, we studied the effect of PI3K∂ blockade on the phagocytic capacity of monocytes and neutrophil activation in two independent short-term assays. No differences in the phagocytosis rates of monocytes were detected (Figure 5A). Neutrophil activation was measured via the generation of reactive oxygen species, termed an “oxidative burst,” in a Seahorse XF neutrophil activation assay. Neutrophils in the idelalisib-treated group did not show a significant difference in oxygen consumption rates in comparison to the vehicle-treated group (Figure 5B). These data indicate that PI3K∂ blockade does not have a direct impact on monocyte phagocytic capacity and neutrophil activation in these short-term assessments.
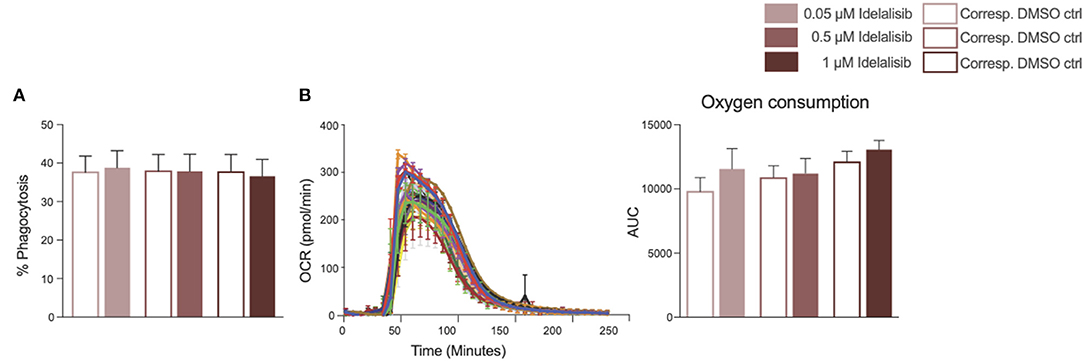
Figure 5. Inhibition of PI3K∂ by idelalisib alters neither monocyte nor neutrophil function. (A) Compiled data showing the phagocytic capacity of monocytes after 4 h in the presence of different concentrations of idelalisib or DMSO as vehicle control; n = 12 HDs. (B) Representative kinetic trace of the oxygen consumption rate (OCR) and compiled bar chart depicting the oxidative burst of neutrophils as areas under the curve (AUC) in a Seahorse XF neutrophil activation assay; n = 8 HDs.
Next, we analyzed if the previously described effects of PI3K∂ inhibition could also be seen if CLL patient cells were treated in culture with idelalisib. To this end, we isolated T cells from cryopreserved PBMCs of CLL patients at time of initial diagnosis. Again, patient samples treated with the vehicle DMSO served as controls. PI3K∂ blockade by idelalisib did not alter the proliferative capacity of T cells isolated from CLL PBMCs (Figure 6A). The cytotoxicity of CLL T cells against target HL-60 cells was significantly reduced in a coculture treated with idelalisib (Figure 6B). This could be further verified by a significant decrease in granzyme B secretion in a culture containing the inhibitor (Figure 6C). Furthermore, PD-1 expression was significantly decreased in the idelalisib-treated T-cell group (Figure 6D). Next, we evaluated the effect of PI3K∂ blockade on the secretion of different cytokines after coculture for 72 h with the target HL-60 cell line. Cytometric bead array analysis of CLL T cells revealed significantly reduced secretion levels of IL-10, IL-4, IL-6, and IFN-γ (Figure 6E). Taken together, these results demonstrate that blockade of PI3K∂ signaling with idelalisib has a negative impact on the effector functions of CLL T cells, such as cytotoxicity and cytokine secretion. However, differences in age are possible cofounding variables.
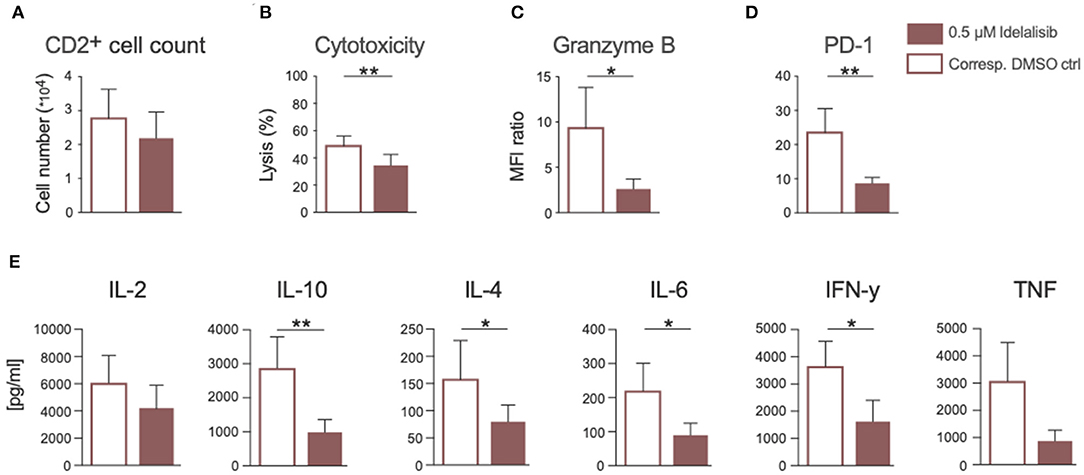
Figure 6. Inhibition of PI3K∂ by idelalisib impairs cytotoxicity and cytokine secretion of T cells isolated from CLL patient samples. (A) Compiled data showing the percentage of proliferated CD2+ T cells in the presence or absence of idelalisib (n = 15). (B) Cytolytic capacity of CD3+ T cells in coculture with target HL−60 cell line and idelalisib after 72 h. (C,D) Bar charts depicting expression levels of granzyme B and PD-1 in T cells after 72 h coculture with HL-60 cells in the presence of idelalisib or DMSO (n = 6). (E) Bar charts representing the levels of IL-2, IL-10, IL-4, IL-6, TNF, and IFNγ in the supernatant of the coculture after 72 h in the presence or absence of idelalisib (n = 15). Error bars represent mean ± SEM; *p ≤ 0.05, **p ≤ 0.005.
PI3K∂ inhibition by idelalisib has proven to be highly effective in the treatment of r/r CLL patients. However, this clinical success is somewhat diminished by the increased rate of opportunistic infections in these patients (37–42). The factors contributing to this observation are incompletely understood.
In the present study, we evaluated the effect of PI3K∂ blockade by idelalisib on the non-malignant human immune cell compartment of healthy individuals. First, we analyzed the effects of idelalisib on T cells and Tregs. PI3K∂ blockade by idelalisib did not have an impact on the proliferative capacity of HD T cells. However, we observed a significant decrease in the expression levels of the inhibitory checkpoint molecules PD-1, CTLA-4, and LAG-3 in both T cells and Tregs in cultures containing idelalisib. Our findings are supported by previous studies, which described PI3K∂ as the main transducer of PI3K signaling in human T cells (25, 53) and Tregs (54). Next, we evaluated if these findings correlated to impaired T-cell function. We demonstrated that secretion of IL-10, TNF, and IFNγ by idelalisib-treated HD T cells was significantly reduced. This is consistent with previous studies that looked at the impact of other PI3K∂-blocking agents on cytokine secretion of T cells in mice (25, 53, 55). Furthermore, we observed that PI3K∂ blockade significantly decreased the cytolytic capacity of HD T cells. This manifested through a significantly reduced secretion of the cytolytic molecules perforin and granzyme B, as well as a significant decrease in antibody-mediated target cell killing. Together, our data show that idelalisib severely impairs the functions of T cells and Tregs isolated from HDs. Combined with the findings of a previous study by Chellappa and colleagues (56), our data support the hypothesis that idelalisib leads to T and NK cell dysfunction. Therefore, our in vitro observations might reflect the increased rate of opportunistic infections in treated CLL patients. Idelalisib exposure led to a significant decrease in the expression of PD-1, which is often described as a marker for T-cell activation. This might be an indicator for reduced T-cell activity and in turn a dampened immune response, which might be a factor behind the increased rates of infections measured in idelalisib-treated CLL patients.
Emerging data from clinical trials suggests that the improved T-cell-mediated antitumor response and the impressive clinical outcome in CLL might be due to reduced Treg numbers and their reduced suppressive function in idelalisib-treated patients (57, 58). Conversely, inhibiting the suppressive activity of Tregs can also lead to severe adverse autoimmune effects (56, 59).
As a next step, we wanted to evaluate the effect of idelalisib on NK cells, the cytotoxic lymphocytes of the innate immune system. Previous studies in mice with defective PI3K∂ have suggested that PI3K plays a critical role in NK-cell effector function (60, 61). Furthermore, Zebedin and colleagues demonstrated that selective inhibition of PI3K∂ in mice leads to impaired degranulation and target cell killing by NK cells (62). Therefore, we wanted to evaluate whether PI3K∂ blockade also has a negative impact on the human NK-cell compartment. In our study, we observed that idelalisib reduced NK-cell proliferation. Moreover, PI3K∂ inhibition led to a decrease in the percentage of cytotoxic NK cells, which also translated into reduced target cell killing by NK cells. We show that two different apoptosis pathways are affected. Idelalisib impaired cell death through secretion of cytolytic molecules, as well as cell death via the Fas–FasL pathway. NK cells are an important part of the innate immune system and play a key role in the defense against infections. Taken together, our data demonstrate a decrease NK-cell proliferation and cytolytic activity by idelalisib and that this might contribute to the increased frequency of infectious events observed in clinical trials.
CLL has been associated with profound defects in T-cell function and synapse formation (63–66). These T-cell defects are thought to result in failure of antitumor immunity and increased susceptibility to infections (67, 68). In view of our findings on healthy T cells, PI3K∂ inhibition might have an even more pronounced effect on the immune response of CLL patients, contributing to an elevated risk of severe side effects such as opportunistic infections. To confirm the clinical relevance of our findings and that these effects might indeed contribute to the increased rate of infections during idelalisib therapy, we isolated T cells from cryopreserved PBMCs collected from CLL patients at the time of initial diagnosis. As expected, the proliferative capacity of these T cells was lower compared to healthy T cells. Idelalisib had only a minor impact on the proliferation on T cells from CLL patient. However, we observed a significant decrease in the cytolytic capacity of CLL T cells treated with idelalisib. This was supported by our finding of a significantly reduced level of secreted granzyme B. Consistent with our data from HDs, PD-1 expression in T cells was significantly decreased in the presence of idelalisib, most likely as a result of reduced T-cell activation. In line with these findings, we also observed a significantly reduced secretion of IL-10, IL-4, IL-6, and IFN-γ; secretion of IL-2 and TNF also appeared to be affected, albeit to a lesser extent. As proinflammatory cytokines are mainly secreted by Tregs, the decrease in IL-10 secretion might indicate impaired Treg suppressive function, which could also contribute to a higher risk of autoimmune diseases. Furthermore, reduced TNF and IFNγ secretion serves as an indicator of less activated, less functional, or even exhausted T cells.
Our data demonstrates that both human T and NK cells are highly sensitive to PI3K∂ inhibition. Idelalisib interfered with the functions of T and NK cells from both HDs and CLL patients. In summary our in vitro data suggest that idelalisib-induced impairment of T and NK-cell function contributes to an increased rate of infections regardless of the underlying B-cell malignancy.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by PB samples from HDs and patients with CLL were collected with written consent in accordance with the Declaration of Helsinki and approval by the Institutional Review Board of the Ludwig-Maximilian University of Munich. The patients/participants provided their written informed consent to participate in this study.
MS, VB, and MB-B: conceptualization and funding acquisition. LR, BB, and AO: methodology. LR, BB, and AO: investigation. LR: writing—original draft. AO, BB, VB, and MS: writing, review, and editing. LR, BB, and MS: review. MS: supervision. All authors read and approved the final manuscript.
This study was supported by funds from the international doctoral program iTarget: Immunotargeting of Cancer funded by the Elite Network of Bavaria (to LR), the German Research Council (DFG) within the Collaborative Research Center (SFB) 1243 Cancer Evolution (project A10: to BB), the Else Kröner-Fresenius-Stiftung (Cancer Immunotherapy, Clinician Scientist Program Munich, to VB), and Gilead Sciences Inc.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge the iFlow Core Facility of the university hospital Munich (INST 409/225-1 FUGG) for assistance with the generation of flow cytometry data. We also thank Prof. Dr. Sebastian Theurich for his support with the Seahorse Analyzer.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.608625/full#supplementary-material
Supplementary Figure 1. Descriptive Plots. (A) Flow cytometry histogram: calculation of the MFI ratio (Median fluorescence intensity ratio). (B) Gating strategy T-cell subset discrimination. The four subsets were discriminated as follows: naïve T cells: CD45Ra+/CCR7+, memory T cells: CD45Ra−/CCR7+, effector T cells: CD45Ra−/CCR7−, and terminally differentiated effector memory T cells: CD45Ra+/CCR7−. (C) Representative flow cytometry histograms, MFI ratio: CTLA-4 (idelalisib = 1.9; DMSO = 4.7), LAG-3 (MFI ratios: idelalisib =2.3; DMSO = 19.5), and PD-1 (MFI-ratios: idelalisib = 13.7; DMSO = 39.3).
Supplementary Figure 2. Inhibition of PI3K∂ by idelalisib in an older reference cohort. (A) Compiled data showing the percentage of proliferated CD4+ and CD8+ T cells in the presence of various concentrations of idelalisib or DMSO. (B–E) Bar graphs of median fluorescence intensity (MFI) ratios showing the expression levels of checkpoint molecules CTLA-4, LAG-3, and PD-1 in CD4+ T cells, CD8+ T cells, CD4+ Tregs, and CD8+ Tregs after stimulation for 3 days with IL-2 and CD3/CD28 activation beads. (F) Cytolytic capacity of CD3+ T cells in coculture with HL-60 cell line and various concentrations of idelalisib after 72 h. (G) Bar charts depicting expression levels of perforin and granzyme B in CD8+ T cells after stimulation with CD3/CD28 beads for 72 h in the presence of various concentrations of idelalisib or DMSO. (H) Cytotoxicity of CD3−CD56+ NK cells against the K562 cell line in a flow cytometry-based coculture assay. Error bars represent ± SEM, n = 4 HDs in all depicted assays.
Supplementary Table 1. Key Resources.
PI3K∂, Phosphoinositide 3-kinase delta; PB, Peripheral blood; PBMC, Peripheral blood mononuclear cell; CLL, Chronic lymphocytic leukemia; HD, Healthy donor; NK cell, Natural killer cells; AKT, Protein kinase B, or PKB; mTOR, mammalian target of rapamycin; CTLA-4, Cytotoxic T-lymphocyte-associated protein 4; PD-1, Programmed cell death protein 1; LAG-3, Lymphocyte activation gene 3; IL-2, Interleukin 2; IL-10, Interleukin 10; TNF, Tumor necrosis factor; IFN-γ, Interferon-γ; FasL, Fas ligand; MFI, Median fluorescence intensity; SEM, Standard error of mean.
1. Johnston PB, Kay NE. Pathogenesis of Impaired Cellular Immune Function in CLL. In: Faguet GB, editor. Chronic Lymphocytic Leukemia: Molecular Genetics, Biology, Diagnosis, and Management. Totowa, NJ: Humana Press (2004) p. 109–21. doi: 10.1007/978-1-59259-412-2_5
2. Goldman D. Chronic lymphocytic leukemia and its impact on the immune system. Clin J Oncol Nurs. (2000) 4:233–4.
3. Morrison VA. Infectious complications in patients with chronic lymphocytic leukemia: pathogenesis, spectrum of infection, and approaches to prophylaxis. Clin Lymphoma Myeloma. (2009) 9:365–70. doi: 10.3816/CLM.2009.n.071
4. Molica S. Infections in chronic lymphocytic leukemia: risk factors, and impact on survival, and treatment. Leukemia Lymphoma. (1994) 13:203–14. doi: 10.3109/10428199409056283
5. Itala M, Helenius H, Nikoskelainen J, Remes K. Infections and serum IgG levels in patients with chronic lymphocytic leukemia. Eur J Haematol. (1992) 48:266–70. doi: 10.1111/j.1600-0609.1992.tb01805.x
6. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
7. Dhalla F, Lucas M, Schuh A, Bhole M, Jain R, Patel SY, et al. Antibody deficiency secondary to chronic lymphocytic leukemia: should patients be treated with prophylactic replacement immunoglobulin? J Clin Immunol. (2014) 34:277–82. doi: 10.1007/s10875-014-9995-5
8. Freeman CL, Gribben JG. Immunotherapy in chronic lymphocytic leukaemia (CLL). Curr Hematol Malig Rep. (2016) 11:29–36. doi: 10.1007/s11899-015-0295-9
9. Wadhwa PD, Morrison VA. Infectious complications of chronic lymphocytic leukemia. Sem Oncol. (2006) 33:240–9. doi: 10.1053/j.seminoncol.2005.12.013
10. Hallek M. Chronic lymphocytic leukemia: 2020 update on diagnosis, risk stratification and treatment. Am J Hematol. (2019) 94:1266–87. doi: 10.1002/ajh.25595
11. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. (2005) 352:804–15. doi: 10.1056/NEJMra041720
12. Bernal A, Pastore RD, Asgary Z, Keller SA, Cesarman E, Liou HC, et al. Survival of leukemic B cells promoted by engagement of the antigen receptor. Blood. (2001) 98:3050–7. doi: 10.1182/blood.V98.10.3050
13. Chen L, Widhopf G, Huynh L, Rassenti L, Rai KR, Weiss A, et al. Expression of ZAP-70 is associated with increased B-cell receptor signaling in chronic lymphocytic leukemia. Blood. (2002) 100:4609–14. doi: 10.1182/blood-2002-06-1683
14. Herishanu Y, Perez-Galan P, Liu D, Biancotto A, Pittaluga S, Vire B, et al. The lymph node microenvironment promotes B-cell receptor signaling, NF-kappaB activation, and tumor proliferation in chronic lymphocytic leukemia. Blood. (2011) 117:563–74. doi: 10.1182/blood-2010-05-284984
15. Ferrer G, Montserrat E. Critical molecular pathways in CLL therapy. Mol Med. (2018) 24:9. doi: 10.1186/s10020-018-0001-1
16. Janku F, Yap TA, Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway? Nat Rev Clin Oncol. (2018) 15:273–91. doi: 10.1038/nrclinonc.2018.28
17. Randis TM, Puri KD, Zhou H, Diacovo TG. Role of PI3Kdelta and PI3Kgamma in inflammatory arthritis and tissue localization of neutrophils. Eur J Immunol. (2008) 38:1215–24. doi: 10.1002/eji.200838266
18. Zirlik K, Veelken H. Idelalisib. Recent results. Cancer Res. (2018) 212:243–64. doi: 10.1007/978-3-319-91439-8_12
19. Lannutti BJ, Meadows SA, Herman SE, Kashishian A, Steiner B, Johnson AJ, et al. CAL-101, a p110delta selective phosphatidylinositol-3-kinase inhibitor for the treatment of B-cell malignancies, inhibits PI3K signaling and cellular viability. Blood. (2011) 117:591–4. doi: 10.1182/blood-2010-03-275305
20. Fruman DA. Phosphoinositide 3-kinase and its targets in B-cell and T-cell signaling. Curr Opin Immunol. (2004) 16:314–20. doi: 10.1016/j.coi.2004.03.014
21. Herman SE, Gordon AL, Wagner AJ, Heerema NA, Zhao W, Flynn JM, et al. Phosphatidylinositol 3-kinase-delta inhibitor CAL-101 shows promising preclinical activity in chronic lymphocytic leukemia by antagonizing intrinsic and extrinsic cellular survival signals. Blood. (2010) 116:2078–88. doi: 10.1182/blood-2010-02-271171
22. Ringshausen I, Schneller F, Bogner C, Hipp S, Duyster J, Peschel C, et al. Constitutively activated phosphatidylinositol-3 kinase (PI-3K) is involved in the defect of apoptosis in B-CLL: association with protein kinase Cδ. Blood. (2002) 100:3741–8. doi: 10.1182/blood-2002-02-0539
23. Pauls S, Lafarge S, Landego I, Zhang T-t, Marshall A. The phosphoinositide 3-kinase signaling pathway in normal and malignant B cells: activation mechanisms, regulation and impact on cellular functions. Front Immunol. (2012) 3:224. doi: 10.3389/fimmu.2012.00224
24. Hawkins PT, Stephens LR. PI3K signalling in inflammation. Biochim Biophys Acta. (2015) 1851:882–97. doi: 10.1016/j.bbalip.2014.12.006
25. Koyasu S. The role of PI3K in immune cells. Nat Immunol. (2003) 4:313–9. doi: 10.1038/ni0403-313
26. Okkenhaug K. Signaling by the phosphoinositide 3-kinase family in immune cells. Annu Rev Immunol. (2013) 31:675–704. doi: 10.1146/annurev-immunol-032712-095946
27. Fruman DA, Cantley LC. Phosphoinositide 3-kinase in immunological systems. Semin Immunol. (2002) 14:7–18. doi: 10.1006/smim.2001.0337
28. Beer-Hammer S, Zebedin E, von Holleben M, Alferink J, Reis B, Dresing P, et al. The catalytic PI3K isoforms p110gamma and p110delta contribute to B cell development and maintenance, transformation, and proliferation. J Leukoc Biol. (2010) 87:1083–95. doi: 10.1189/jlb.0809585
29. Okkenhaug K, Vanhaesebroeck B. PI3K in lymphocyte development, differentiation and activation. Nat Rev Immunol. (2003) 3:317–30. doi: 10.1038/nri1056
30. Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci USA. (1997) 94:4330–5. doi: 10.1073/pnas.94.9.4330
31. Hoellenriegel J, Meadows SA, Sivina M, Wierda WG, Kantarjian H, Keating MJ, et al. The phosphoinositide 3'-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood. (2011) 118:3603–12. doi: 10.1182/blood-2011-05-352492
32. Berning P, Lenz G. The role of PI3K inhibitors in the treatment of malignant lymphomas. Leukemia Lymphoma. (2020) 2:1–11. doi: 10.1080/10428194.2020.1839654
33. Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. (2012) 4:a011189. doi: 10.1101/cshperspect.a011189
34. Somoza JR, Koditek D, Villaseñor AG, Novikov N, Wong MH, Liclican A, et al. Structural, biochemical, and biophysical characterization of idelalisib binding to phosphoinositide 3-kinase δ. J Biol Chem. (2015) 290:8439–46. doi: 10.1074/jbc.M114.634683
35. Yang Q, Modi P, Newcomb T, Quéva C, Gandhi V. Idelalisib: first-in-class PI3K delta inhibitor for the treatment of chronic lymphocytic leukemia, small lymphocytic leukemia, and follicular lymphoma. Clin Cancer Res. (2015) 21:1537–42. doi: 10.1158/1078-0432.CCR-14-2034
36. Markham A. Idelalisib: first global approval. Drugs. (2014) 74:1701–7. doi: 10.1007/s40265-014-0285-6
37. Okkenhaug K, Graupera M, Vanhaesebroeck B. Targeting PI3K in cancer: impact on tumor cells, their protective stroma, angiogenesis, and immunotherapy. Cancer Discov. (2016) 6:1090–105. doi: 10.1158/2159-8290.CD-16-0716
38. Louie CY, DiMaio MA, Matsukuma KE, Coutre SE, Berry GJ, Longacre TA. Idelalisib-associated enterocolitis: clinicopathologic features and distinction from other enterocolitides. Am J Surg Pathol. (2015) 39:1653–60. doi: 10.1097/PAS.0000000000000525
39. Weidner AS, Panarelli NC, Geyer JT, Bhavsar EB, Furman RR, Leonard JP, et al. Idelalisib-associated colitis: histologic findings in 14 patients. Am J Surg Pathol. (2015) 39:1661–7. doi: 10.1097/PAS.0000000000000522
40. Coutre SE, Barrientos JC, Brown JR, de Vos S, Furman RR, Keating MJ, et al. Management of adverse events associated with idelalisib treatment: expert panel opinion. Leuk Lymphoma. (2015) 56:2779–86. doi: 10.3109/10428194.2015.1022770
41. Cheah CY, Fowler NH. Idelalisib in the management of lymphoma. Blood. (2016) 128:331–6. doi: 10.1182/blood-2016-02-702761
42. Barr PM, Saylors GB, Spurgeon SE, Cheson BD, Greenwald DR, O'Brien SM, et al. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. (2016) 127:2411–5. doi: 10.1182/blood-2015-12-683516
43. Teh BW, Tam CS, Handunnetti S, Worth LJ, Slavin MA. Infections in patients with chronic lymphocytic leukaemia: mitigating risk in the era of targeted therapies. Blood Rev. (2018) 32:499–507. doi: 10.1016/j.blre.2018.04.007
44. Nosari A. Infectious complications in chronic lymphocytic leukemia. Mediterr J Hematol Infect Dis. (2012) 4:e2012070. doi: 10.4084/mjhid.2012.070
45. Williams AM, Baran AM, Meacham PJ, Herr M, Valencia HE, Newsom C, et al. High risk of infections in chronic lymphocytic leukemia patients treated with B-cell receptor inhibitors. Blood. (2016) 128:3203. doi: 10.1182/blood.V128.22.3203.3203
46. Hanlon A, Brander DM. Managing toxicities of phosphatidylinositol-3-kinase (PI3K) inhibitors. Hematology. (2020) 2020:346–56. doi: 10.1182/hematology.2020000119
47. Sharman JP, Coutre SE, Furman RR, Cheson BD, Pagel JM, Hillmen P, et al. Final results of a randomized, phase iii study of rituximab with or without idelalisib followed by open-label idelalisib in patients with relapsed chronic lymphocytic leukemia. J Clin Oncol. (2019) 37:1391–402. doi: 10.1200/JCO.18.01460
48. Jones JA, Robak T, Brown JR, Awan FT, Badoux X, Coutre S, et al. Efficacy and safety of idelalisib in combination with ofatumumab for previously treated chronic lymphocytic leukaemia: an open-label, randomised phase 3 trial. Lancet Haematol. (2017) 4:e114–e26. doi: 10.1016/S2352-3026(17)30019-4
50. Schmidt RLJ, Jutz S, Goldhahn K, Witzeneder N, Gerner MC, Trapin D, et al. Chloroquine inhibits human CD4+ T-cell activation by AP-1 signaling modulation. Sci Rep. (2017) 7:42191. doi: 10.1038/srep42191
51. Arnold KB, Szeto GL, Alter G, Irvine DJ, Lauffenburger DA. CD4+ T cell-dependent and CD4+ T cell-independent cytokine-chemokine network changes in the immune responses of HIV-infected individuals. Sci Signal. (2015) 8:ra104. doi: 10.1126/scisignal.aab0808
52. Herrmann M, Krupka C, Deiser K, Brauchle B, Marcinek A, Ogrinc Wagner A, et al. Bifunctional PD-1 × αCD3 × αCD33 fusion protein reverses adaptive immune escape in acute myeloid leukemia. Blood. (2018) 132:2484–94. doi: 10.1182/blood-2018-05-849802
53. Soond DR, Bjorgo E, Moltu K, Dale VQ, Patton DT, Torgersen KM, et al. PI3K p110delta regulates T-cell cytokine production during primary and secondary immune responses in mice and humans. Blood. (2010) 115:2203–13. doi: 10.1182/blood-2009-07-232330
54. Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. (2006) 177:6598–602. doi: 10.4049/jimmunol.177.10.6598
55. Okkenhaug K, Patton DT, Bilancio A, Garcon F, Rowan WC, Vanhaesebroeck B. The p110delta isoform of phosphoinositide 3-kinase controls clonal expansion and differentiation of Th cells. J Immunol. (2006) 177:5122–8. doi: 10.4049/jimmunol.177.8.5122
56. Chellappa S, Kushekhar K, Munthe LA, Tjonnfjord GE, Aandahl EM, Okkenhaug K, et al. The PI3K p110delta Isoform Inhibitor Idelalisib Preferentially Inhibits Human Regulatory T Cell Function. J Immunol. (2019) 202:1397–405. doi: 10.4049/jimmunol.1701703
57. D'Arena G, Laurenti L, Minervini MM, Deaglio S, Bonello L, De Martino L, et al. Regulatory T-cell number is increased in chronic lymphocytic leukemia patients and correlates with progressive disease. Leuk Res. (2011) 35:363–8. doi: 10.1016/j.leukres.2010.08.010
58. Hanna BS, Roessner PM, Scheffold A, Jebaraj BMC, Demerdash Y, Ozturk S, et al. PI3Kdelta inhibition modulates regulatory and effector T-cell differentiation and function in chronic lymphocytic leukemia. Leukemia. (2019) 33:1427–38. doi: 10.1038/s41375-018-0318-3
59. Lampson BL, Kasar SN, Matos TR, Morgan EA, Rassenti L, Davids MS, et al. Idelalisib given front-line for treatment of chronic lymphocytic leukemia causes frequent immune-mediated hepatotoxicity. Blood. (2016) 128:195–203. doi: 10.1182/blood-2016-03-707133
60. Guo H, Samarakoon A, Vanhaesebroeck B, Malarkannan S. The p110 delta of PI3K plays a critical role in NK cell terminal maturation and cytokine/chemokine generation. J Exp Med. (2008) 205:2419–35. doi: 10.1084/jem.20072327
61. Mace EM. Phosphoinositide-3-kinase signaling in human natural killer cells: new insights from primary immunodeficiency. Front Immunol. (2018) 9:445. doi: 10.3389/fimmu.2018.00445
62. Zebedin E, Simma O, Schuster C, Putz EM, Fajmann S, Warsch W, et al. Leukemic challenge unmasks a requirement for PI3Kdelta in NK cell-mediated tumor surveillance. Blood. (2008) 112:4655–64. doi: 10.1182/blood-2008-02-139105
63. Palma M, Gentilcore G, Heimersson K, Mozaffari F, Näsman-Glaser B, Young E, et al. T cells in chronic lymphocytic leukemia display dysregulated expression of immune checkpoints and activation markers. Haematologica. (2017) 102:562–72. doi: 10.3324/haematol.2016.151100
64. Görgün G, Holderried TAW, Zahrieh D, Neuberg D, Gribben JG. Chronic lymphocytic leukemia cells induce changes in gene expression of CD4 and CD8 T cells. J Clin Invest. (2005) 115:1797–805. doi: 10.1172/JCI24176
65. Vardi A, Stamatopoulos K, Hadzidimitriou A. T cells in chronic lymphocytic leukemia: can they fight? Oncotarget. (2017) 8:99209–10. doi: 10.18632/oncotarget.22277
66. Roessner PM, Seiffert M. T-cells in chronic lymphocytic leukemia: guardians or drivers of disease? Leukemia. (2020) 34:2012–24. doi: 10.1038/s41375-020-0873-2
67. Riches JC, Davies JK, McClanahan F, Fatah R, Iqbal S, Agrawal S, et al. T cells from CLL patients exhibit features of T-cell exhaustion but retain capacity for cytokine production. Blood. (2013) 121:1612–21. doi: 10.1182/blood-2012-09-457531
Keywords: cancer immunotherapy, chronic lymphocytic leukemia, idelalisib, immune effector cells, PI3K inhibition
Citation: Rohrbacher L, Brauchle B, Ogrinc Wagner A, von Bergwelt-Baildon M, Bücklein VL and Subklewe M (2021) The PI3K∂-Selective Inhibitor Idelalisib Induces T- and NK-Cell Dysfunction Independently of B-Cell Malignancy-Associated Immunosuppression. Front. Immunol. 12:608625. doi: 10.3389/fimmu.2021.608625
Received: 21 September 2020; Accepted: 11 February 2021;
Published: 15 March 2021.
Edited by:
Roberto Benelli, San Martino Hospital (IRCCS), ItalyReviewed by:
Aaron James Marshall, University of Manitoba, CanadaCopyright © 2021 Rohrbacher, Brauchle, Ogrinc Wagner, von Bergwelt-Baildon, Bücklein and Subklewe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marion Subklewe, bWFyaW9uLnN1YmtsZXdlQG1lZC51bmktbXVlbmNoZW4uZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.