 Natalia Zeber-Lubecka
Natalia Zeber-Lubecka Ewa E. Hennig
Ewa E. Hennig
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 February 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.606620
Polycystic ovary syndrome (PCOS) and Hashimoto’s thyroiditis (HT) are endocrine disorders that commonly occur among young women. A higher prevalence of HT in women with PCOS, relative to healthy individuals, is observed consistently. Combined occurrence of both diseases is associated with a higher risk of severe metabolic and reproductive complications. Genetic factors strongly impact the pathogenesis of both PCOS and HT and several susceptibility loci associated with a higher risk of both disorders have been identified. Furthermore, some candidate gene polymorphisms are thought to be functionally relevant; however, few genetic variants are proposed to be causally associated with the incidence of both disorders together.
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder among women of reproductive age (1). Similarly, thyroid dysfunction, including Hashimoto’s thyroiditis (HT) as the principal autoimmune thyroid disease (AITD), is frequently observed among young women (2, 3). Both PCOS and HT can result in a wide range of metabolic syndrome features, including weight gain, impaired glucose tolerance, insulin resistance (IR), and dyslipidemia, which can lead to obesity, diabetes, or cardiovascular disease over a lifetime (4–6). Moreover, both disorders are major causes of infertility, a medical problem of a growing prevalence that is associated with strong physical, emotional, and socioeconomic consequences.
There is growing evidence that there may be mutual interaction between PCOS and HT. More importantly, combined occurrence of both diseases is associated with a higher risk of severe metabolic and reproductive complications than either PCOS or HT alone, and the severity of disease symptoms depends on the duration of the thyroid dysfunction (7–10). In addition, co-occurrence of thyroid autoimmune disease is associated with poorer response to treatment in infertile women with PCOS (11). The strong genetic influence on the development of these two diseases is well documented. Many candidate gene variants and susceptibility loci have been identified that are associated with a higher risk of PCOS or HT; however, the mechanisms underlying the close relationship between these two diseases, and common components to their genetic backgrounds, remain uncertain. Here, we comprehensively review a wide range of reports on genetic discoveries related to PCOS, HT, and their combined occurrence.
PCOS is a heterogeneous endocrinopathy that affects 5–20% of women of reproductive age (12). It is characterized mainly by clinical and/or biochemical hyperandrogenism (HA), ovary dysfunction (OD), often reflected by chronic oligo- or anovulatory menstrual cycles, and polycystic ovarian morphology (PCOM) on pelvic ultrasound; for diagnosis, two of these three criteria must be present (13). Consequently, four main PCOS phenotypes have been proposed: phenotype A, with HA, OD, and PCOM; phenotype B, with HA and OD but without PCOM; phenotype C, with HA and PCOM but without OD; and phenotype D, with OD and PCOM but without HA; with phenotypes A and B comprising an estimated > 40% of cases (14, 15). PCOS is characterized by symptoms of hypothalamic-pituitary-ovarian axis dysfunction, including increased hypothalamic gonadotropin-releasing hormone (GnRH) pulse frequency and GnRH quantity, as well as an elevated ratio of luteinizing hormone (LH) to follicle-stimulating hormone (FSH), which contribute to excessive ovarian androgen production and ovulatory dysfunction (16, 17).
Depending on the phenotype, POCS is associated with variable degrees of reproductive and metabolic dysfunction. Dyslipidemia occurs with a particularly high prevalence of up to 70% (14, 18). Also, defects in insulin activity and secretion, which may lead to IR, hyperinsulinemia, and impaired glucose tolerance, are observed very frequently (19, 20). PCOS is a low-grade inflammatory state (21); women with PCOS have higher levels of inflammatory markers such as C reactive protein (CRP), tumor necrosis factor α (TNF-α), and interleukin 6 (IL6), independent of associated obesity (22). Over a lifetime, PCOS is associated with a higher risk of menstrual irregularities, hirsutism, anovulatory infertility, spontaneous abortion, obesity, and obesity-linked co-morbidities such as nonalcoholic fatty liver disease and metabolic syndrome, type 2 diabetes mellitus, and cardiovascular pathologies, in addition to endometrial adenocarcinoma (20, 23–27). PCOS reduces quality of life, resulting in more frequent depressive episodes and suicide in women affected by this disease (28).
The etiology of PCOS is not fully elucidated. It is a complex disorder that might be represented by a common clinical phenotype of different processes. An IR or intrinsic ovarian dysfunction, possibly stimulated by an imbalance of female sex hormones, are considered inciting factors, which primarily results in increased androgen biosynthesis and secretion by the ovary (29, 30). Resulted hyperandrogenism can be further modulated by impaired insulin action (14). Insulin can also directly stimulate androgen production by amplifying theca cell responses to LH (31).
PCOS is a complex, multifactorial disorder with a strong heritable component, as indicated by familial clustering and twin studies, which accounts for as much as 72% of the risk of syndrome development (32, 33). Until recently, the genomic approach to PCOS research was dominated by candidate gene association studies, including more than 100 genes whose alterations may be functionally involved in PCOS development. These genes are mainly related to the insulin signaling pathway and IR, chronic inflammation, reproductive hormone disorders, and biosynthesis of androgens; such as those encoding insulin (INS), INS receptor (INSR), INSR substrates (IRS), methylenetetrahydrofolate reductase (MTHFR), IL family members, TNF-α, toll-like receptor 2 (TLR2), fat mass and obesity-associated protein (FTO), paraoxonase 1 (PON1), calpain 10 (CAPN10), peroxisome proliferator-activated receptor γ (PPAR-γ), FSH receptor (FSHR), LH/choriogonadotropin receptor (LHCGR), LH-β, sex hormone-binding globulin (SHBG), androgen receptor, transforming growth factor β (TGF-β), fibrillin 3 (FBN3), and vitamin D receptor (VDR) (17, 33–37). Nevertheless, the vast majority of candidate gene variants associated with PCOS in previous studies have not been replicated, indicating the possibility of substantial genetic heterogeneity across various ethnic populations or different genetic architectures underlying specific PCOS phenotypes (38–49).
To more deeply investigate genetic predisposition to PCOS, several genome-wide association studies (GWAS) have been carried out (Table 1) (50–54, 59, 66). The first two studies were conducted in China, and together identified 11 susceptibility loci located within or near genes implicated in: 1) gonadotropin activity, ovulation, and erectile dysfunction [FSHR and LHCGR, both at 2p16.3, and the zinc aminopeptidase gene (C9ORF3) at 9q22.32]; 2) insulin signaling and diabetes mellitus [the thyroid adenoma-associated protein (THADA) at 2p21, high mobility group AT-hook protein (HMGA2) at 12q14.3, and INSR at 19p13.3]; 3) organ growth control, cell proliferation, and apoptosis [yes-associated protein (YAP1) at 11q22.1 and zinc finger protein 217 (ZNF217) at 20q13.2]; 4) endosomal membrane trafficking and exocytosis of gonadotropins [DENN domain-containing protein 1A (DENND1A) at 9q33.3 and RAS-related protein (RAB5B) at 12q13.2]; and 5) modification of chromatin structure [TOX high mobility group box family member 3 protein (TOX3) at 16q12.1] (51, 52). Subsequently, associations between the single nucleotide polymorphism (SNP) rs13425728 in LHCGR with higher levels of testosterone, triglycerides, and low density lipoprotein (LDL) were detected among Hui Chinese subjects (56), and the two SNPs rs10818854 and rs10986105 in DENND1A were associated with increased HOMA-IR and anti-Müllerian hormone (AMH) levels in Arab women with PCOS (68).
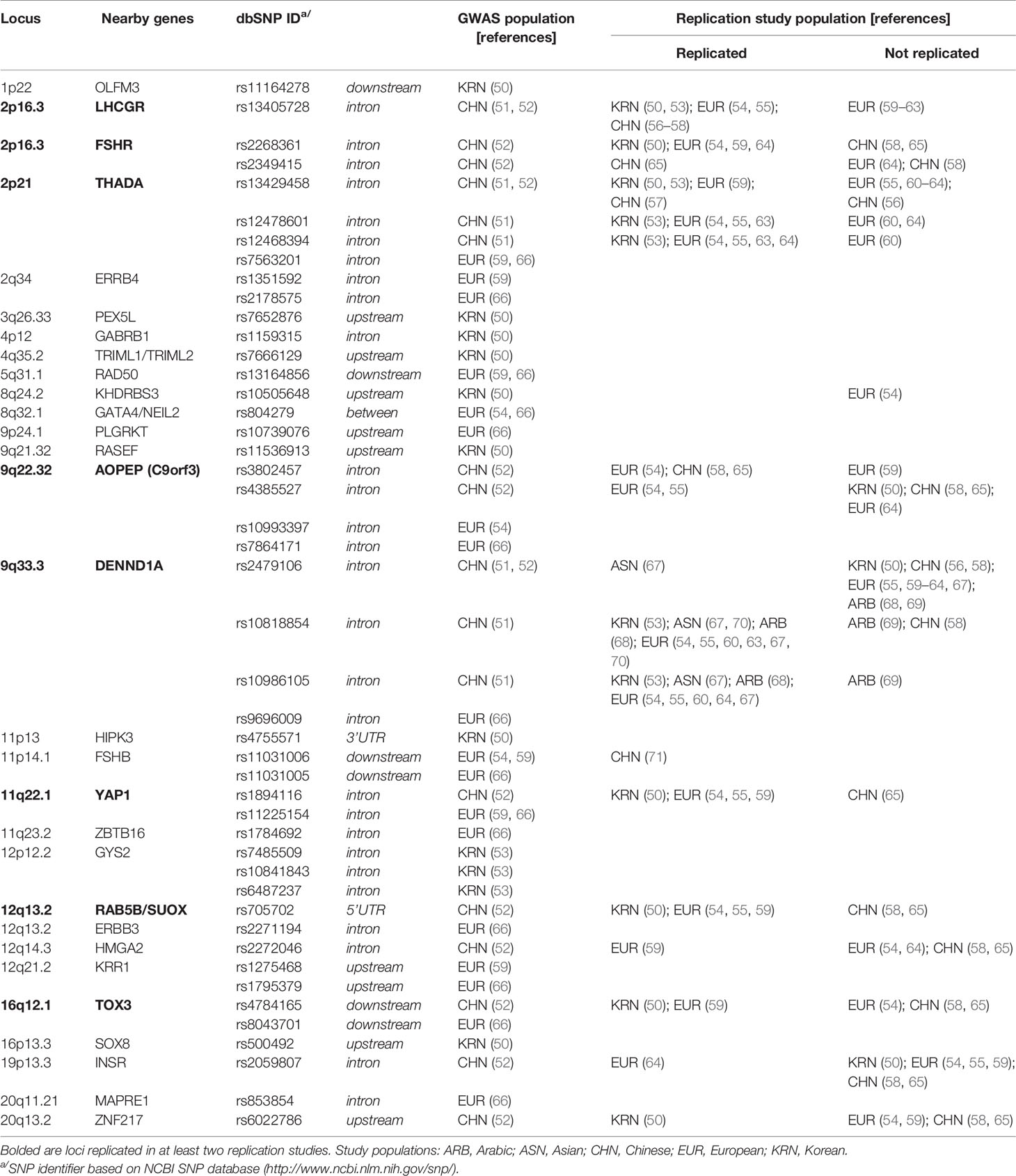
Table 1 GWAS and replication studies for PCOS susceptibility.
Later, two GWAS were conducted in Korean populations (50, 53). In the first, three novel SNPs in the glycogen synthase 2 (GYS2) gene on chromosome 12p12.2 were identified as being associated with PCOS through investigation of individuals with obesity-related condition (53); however, none of these variants reached the genome-wide significance threshold. In the second study, eight new susceptibility loci were suggested, including a main association signal at rs10505648 on chromosome 8q24.2, close to KH RNA-binding domain-containing, signal transduction associated 3 (KHDRBS3), which is associated with regulation of telomerase activity and variant splicing (72), and seven moderate signals at 1p22, 3q26.33, 4p12, 4q35.2, 9q21.32, 11p13, and 16p13.3 (50). In addition, eight of the Chinese susceptibility loci were replicated in Korean women (Table 1).
Two GWAS were recently conducted in populations of European ancestry, confirming the association of the three Asian susceptibility loci (YAP1, THADA, and C9ORF3), as well as identifying five novel loci corresponding to the epidermal growth factor receptor (ERBB4/HER4) gene at 2q34, double strand break repair protein (RAD50) at 5q31.1, the GATA4/NEIL2 region encoding zinc finger transcription factor and endonuclease VIII-like 2 at 8q23.1, FSH β polypeptide (FSHB) at 11p14.1, and KRR1 at 12q21.2, which encodes a ribosome assembly factor (Table 1) (54, 59). The same rs11031006 variant in the FSHB region, which was identified independently in both European GWAS, was associated with lower level of FSH and higher levels of LH and the LH/FSH ratio, suggesting that it may act by affecting gonadotropin secretion (54, 59). Together with the Chinese findings implicating FSHR variants, these results strongly suggest an etiologic role for gonadotropins in PCOS development. Additionally, the identified associations point to gonadal development, ovarian folliculogenesis, and follicular maturation, as well as repair of DNA damage, as processes involved in PCOS pathogenesis in European woman.
Recently, a large-scale genome-wide meta-analysis including 10,074 PCOS cases and 103,164 controls of European ancestry (66), confirmed 11 previously reported loci and identified three additional novel susceptibility loci: ZBTB16 (zinc finger and BTB domain-containing 16) at 11q23.2, which is involved in cell cycle progression, control of the early stages of spermatogenesis, and endometrial stromal cell decidualization (73, 74); MAPRE1 (microtubule associated protein RP/EB family member 1) at 20q11.20, which is involved in the regulation of microtubule structures and chromosome stability, and may participate in follicle development; and the plasminogen receptor (PLGRKT) gene at 9p24.1, which contributes to regulation of inflammatory responses and matrix metalloproteinase activation (Table 1). These newly identified loci provide additional evidence for the involvement of neuroendocrine, metabolic, and reproductive factors in PCOS. Additionally, the data suggest that the genetic architecture underlying the different PCOS phenotypes does not differ in terms of common susceptibility variants (66).
A number of gene polymorphisms discovered in Chinese populations, including LHCGR, FSHR, THADA, DENND1A, C9ORF3, YAP1, and RAB5B/SUOX, have been confirmed in at least two replication studies or meta-analyses for different Caucasian populations, and can be considered general PCOS susceptibility variants (Table 1) (55–58, 60–65, 67–71). However, GWAS have identified < 10% of the estimated heritability of PCOS (36), and there is relatively little overlap between the gene loci identified by candidate gene studies and those found by GWAS, likely because polymorphisms in gene coding regions are usually rare genetic variants that have a large impact on disease risk, whereas GWAS is designed to focus primarily on common variants with a low impact on genetic risk.
The greatest challenge for genomic studies is to identify causative mechanisms and determine the functional relevance of loci identified as risk factors by GWAS. A pathway-based approach has been proposed to increase the power to determine the biological meaning of GWAS results. Application of meta-analysis gene-set enrichment of variant associations (MAGENTA) to the PCOS GWAS dataset revealed oocyte meiosis and regulation of insulin secretion by acetylcholine and free fatty acids as biological pathways significantly associated with the syndrome (75). Among significantly associated genes, the INS gene was indicated in all three pathways. However, some caution should be taken with regard to these results as the analysis included variants of lower significance level than generally accepted for GWAS.
The genetic loci identified by GWAS are often named according to the nearest gene. While the nearby location of a gene does not necessarily mean that it is an effector gene at a given locus, several putative susceptibility genes located within GWAS-discovered risk loci for PCOS were found to be abnormally expressed in women with this condition. PCOS subtypes have been proposed based on phenotypes, expression quantitative trait loci, and biological pathway analyses, in which genes related to gonadotropin levels, metabolic mechanisms, or inflammation and its consequences, have independent impacts on the risk of syndrome development (76). Similarly, expression analysis of genes from 11 GWAS-identified PCOS risk loci revealed that different mechanisms may be involved in the pathogenesis of clinically diverse PCOS subtypes in relation to obesity, since LHCGR is over-expressed in non-obese women and INSR is under-expressed only in obese women with PCOS. A causal role was hypothesized for SNPs in the LHCGR, INSR, and RAB5B regions, suggesting that they may affect gene expression directly or indirectly through epigenetic mechanisms (77). Also, lower expression levels of the RAB5A gene in granulosa cells of obese patients with PCOS were observed relative to those in obese women without the syndrome, which may explain the high FSHR levels and FSH-FSHR signaling pathway disorders in PCOS (78). In turn, HMGA2, another gene identified by GWAS as a candidate, is expressed at high levels in granulosa cells of women with PCOS, where the HMGA2/IMP2 pathway is suggested to be responsible for granulosa cell proliferation (79).
One of the most extensively studied GWAS-identified PCOS candidate genes is DENND1A, which encodes a clathrin binding protein (80). The PCOS risk SNP, rs10986105, in DENND1A was also found to increase the risk of hyperandrogenism in women without PCOS (60). DENND1A variant 2 (DENND1A.V2), a truncated splice isoform of DENND1A with a unique C-terminal sequence, is up-regulated in PCOS theca cells, and compelled expression of V2 in normal theca cells results in increased expression of genes encoding cytochrome P450 isoforms (CYP17A1 and CYP11A1) and higher androgen production (81, 82). Recently, a functional network, including miR-130b-3p, DENND1A.V.2, LHCGR, RSB5B, and the signaling pathways they target, was proposed to potentially mediate PCOS-related hyperandrogenemia. The functional significance of this network is strongly supported by the discovery of the co-localization of LHCGR, DENND1A.V.2, and RAB5B proteins in theca cells (82), as well as the fact that decreased expression of miR-130b-3p, which is predicted to target DENND1A, correlates with increased V2 mRNA levels and androgen synthesis in PCOS theca cells (83).
Despite the clear and strong influence of genetic factors in the pathogenesis of PCOS, common genetic risk loci identified to date explain only a small percentage of estimated PCOS heritability. Therefore, next-generation sequencing studies to investigate entire genomic regions have been conducted to determine whether rare genetic variants with large effect size may contribute to PCOS pathogenesis. Potentially deleterious variants in AMH were identified using whole-genome and targeted sequencing of AMH in a case-control PCOS study (84). Seventeen PCOS-specific rare AMH coding variants resulted in significant reduction of AMH-mediated signaling in dual luciferase reporter tests. Therefore, AMH mutations in PCOS are suggested to result in decreased AMH-mediated inhibition of CYP17 expression and androgen biosynthesis, leading to syndrome-specific hyperandrogenism. Furthermore, the missense variant rs104893836 in the first exon of the GnRH receptor gene (GNRHR) was detected by the whole-exome sequencing (85). Functional analyses revealed significantly reduced GnRH binding by the resulting variant GnRHR containing a Gln106Arg substitution; however, since this is a fairly common variant affecting the first extracellular receptor loop, its effect in vivo is likely to be mild (86). It is worth noting that an increase rather than a decrease in GnRH signaling is a hallmark of PCOS. Recently, whole-genome sequencing revealed several rare non-coding variants in DENND1A associated with reproductive and metabolic traits in PCOS families, suggesting their contribution to disease pathogenesis and providing additional evidence for the central role of DENND1A in PCOS (87).
AITD is the most common autoimmune disorder, affecting 5%–20% of the female population of fertile age (88). Its prevalence varies depending on age, geographical origin, and iodine intake (2). AITD is classified as an organ-specific autoimmune disorder mediated by T cells (89). An autoimmune attack, directed against components of the thyroid gland, may lead to clinically heterogeneous conditions, manifested by either thyroid hormone excess (hyperthyroidism), as in the case of Graves’ disease (GD), or reduced hormone production (hypothyroidism), a typical feature of HT, on which this review will mainly focus.
HT is considered the principal autoimmune disease among young women and the most frequent form of AITD, affecting 4%–9.5% of the adult population (90); its occurrence is eight times more common in women than in men (88). As a result of autoimmunity to self-antigens, approximately 60%–80% of patients with HT have serum antibodies against thyroglobulin (Tg) and 90%–95% have antibodies against thyroid peroxidase (TPO) (91, 92). HT is characterized by infiltration of lymphocytes and chronic inflammation of the thyroid gland, which promotes T cell-induced apoptosis of thyroid follicular cells. The Fas/Fas ligand (FasL) cascade is the main signaling pathway leading to apoptosis of thyroid cells in response to infiltration by pro-inflammatory cytokines, such as interferon (IFN)-γ, TNF-α, and IL1β (93). It is proposed that the mechanism of apoptosis leading to the characteristic thyroid destruction in HT may be a consequence of abnormal Fas/FasL regulation and decreased expression of the apoptosis regulator Bcl-2 (94, 95).
The progressive destruction, and finally fibrosis, of the glandular parenchyma often leads to hypothyroidism. AITD is considered the most frequent cause of hypothyroidism, although it can go unnoticed for years, without overt thyroid dysfunction. Subclinical hypothyroidism (SCH), defined as a serum thyroid-stimulating hormone (TSH) above the defined upper limit of the reference range, in combination with a normal level of free thyroxine (fT4), is more common than overt hypothyroidism, in which fT4 levels are reduced below the normal lower limit and TSH levels are further increased (96). HT has clinically heterogeneous presentation, from the presence of thyroid antibodies but normal thyroid function, to SCH or overt hypothyroidism. The prevalence of hypothyroidism among women with HT increases with age, and in most of cases it eventually develops despite patients initially being euthyroid (97). The diagnostic criteria for HT are based on detection of elevated levels of circulating anti-thyroid antibodies (anti-TPO and/or anti-Tg) and a typical hypoechogenic pattern of the thyroid gland on ultrasound examination (3). Although the presence of thyroid antibodies is a marker for thyroid damage, they are unlikely to play a role in the pathogenesis of HT (98).
Hypothyroidism may be associated with serious pregnancy complications, such as spontaneous abortion, preterm delivery, and placental abruption, as well as reduced fertility, ovulation disorders, insufficient endometrial thickness, and excessive, irregular menstrual bleeding (99–101). A recently published meta-analysis showed that SCH significantly increases the risk of miscarriage before 20 weeks of pregnancy and that early treatment can reduce the miscarriage rate (102). In hypothyroid states, lower levels of several hormones related to the ovarian axis, including SHBG, 17-β-estradiol (E2), testosterone, and androstenedione, were observed (103). As a result of increased secretion of thyrotropin-releasing hormone (TRH), prolactin levels can increase, while LH and FSH concentrations remain normal (104). Underactive thyroid is also associated with the formation of ovarian cysts, which were normalized after T4 replacement (105, 106).
Metabolic changes are also common in hypothyroidism, particularly dyslipidemia and IR, and the severity of these disorders correlates with TSH concentration, even within its normal range (107–109). A link between autoimmunity and obesity has been noted, with leptin as a factor linking the two conditions (110). Being overweight during childhood is positively associated with anti-TPO antibody levels in women at 60–64 years-of-age (111). Since thyroid hormones have a wide impact, the symptoms of thyroid dysfunction can be systemic, affecting almost every physiological system, in addition to significant reproductive or metabolic defects (3). Nevertheless, the pathogenesis of HT remains largely unknown.
As in the case of PCOS, a strong genetic predisposition to AITD has been established, suggesting that over 70% of the susceptibility to the development of antibodies directed against thyroid antigens can be attributed to genetic factors (112). Among first-degree relatives of patients with HT, the risk of developing HT is increased by 20- to 30-fold; in 60% of first-degree relatives of patients with HT, anti-thyroid antibody positivity is observed (27). Also, the high concordance of HT in monozygotic twins (55%) strongly supports a genetic component in AITD predisposition (113).
Candidate gene case-control studies identified several putative susceptibility variants associated with AITD development or progression. These included variants in genes encoding proteins related to inflammation, modulation of immune responses, or specific for the thyroid, such as: human leukocyte antigen (HLA) class I and class II, forkhead box P3 (FOXP3), cytotoxic T-lymphocyte-associated protein 4 (CTLA4), cluster of differentiation 40 (CD40), protein tyrosine phosphatase, non-receptor type 22 (PTPN22), selenoprotein S (SEPS1), IL4, IL2 receptor α (IL2RA), VDR, Tg, TSH receptor (TSHR), signal transducer and activator of transcription 3 (STAT3), and STAT4, with HLA-DR3 carrying the highest risk (2, 114–120). Among non-HLA genes, CTLA4 and PTPN22 were most consistently identified as predisposing to both HT and GD (121–124), while the TSHR locus appears to be specific for GD, but not HT, suggesting some genetic differences between these two types of AITD (125, 126); however, similar to PCOS, candidate gene screening for HT susceptibility mainly generated controversial and non-replicable findings (127–132). Limited understanding of disease pathogenesis and the consequent lack of comprehensive datasets on candidate genes, small sample sizes, and low statistical power, were some of the main limitations of candidate gene studies for both disorders. The era of GWAS and subsequent replication studies have brought further data on genetic susceptibility to HT.
Several GWAS for AITD (HT or GD), hypothyroidism, positivity for anti-thyroid (anti-TPO or anti-Tg) antibodies, and thyroid function parameters (including TSH or fT4 levels), have been undertaken (Table 2) (133–135, 139–146, 148, 154, 155, 157–160). Some GWAS-identified AITD susceptibility loci have been replicated in different populations (Table 2) (136–138, 147, 149–153, 156); however, HT is rather poorly represented among GWAS. Numerous studies have been performed using small cohorts or patients with HT and GD grouped together (128, 161). In total, six GWAS included separate groups of patients with HT (134, 140, 146, 147, 154, 159), while three included patients with hypothyroidism, a typical feature of HT (133, 135, 158). Together, 16 putative HT susceptibility loci have been identified; however, the most convincing evidence for associations is limited to HLA (6p21), CTLA4 (2q33.2), PTPN22 (1q13.2), and FOXE1 (9q22.33) variants. Additionally, two loci, TPO (2p25.3) and ATXN2 (ataxin2; 12p24.12), are convincingly associated with the presence of anti-TPO antibodies, and three loci, CAPZB (capping actin protein of muscle Z-line subunit beta; 1p36.13), phosphodiesterase PDE8B (5q13.3), and PDE10A (6q27), are associated with TSH levels (Table 2).
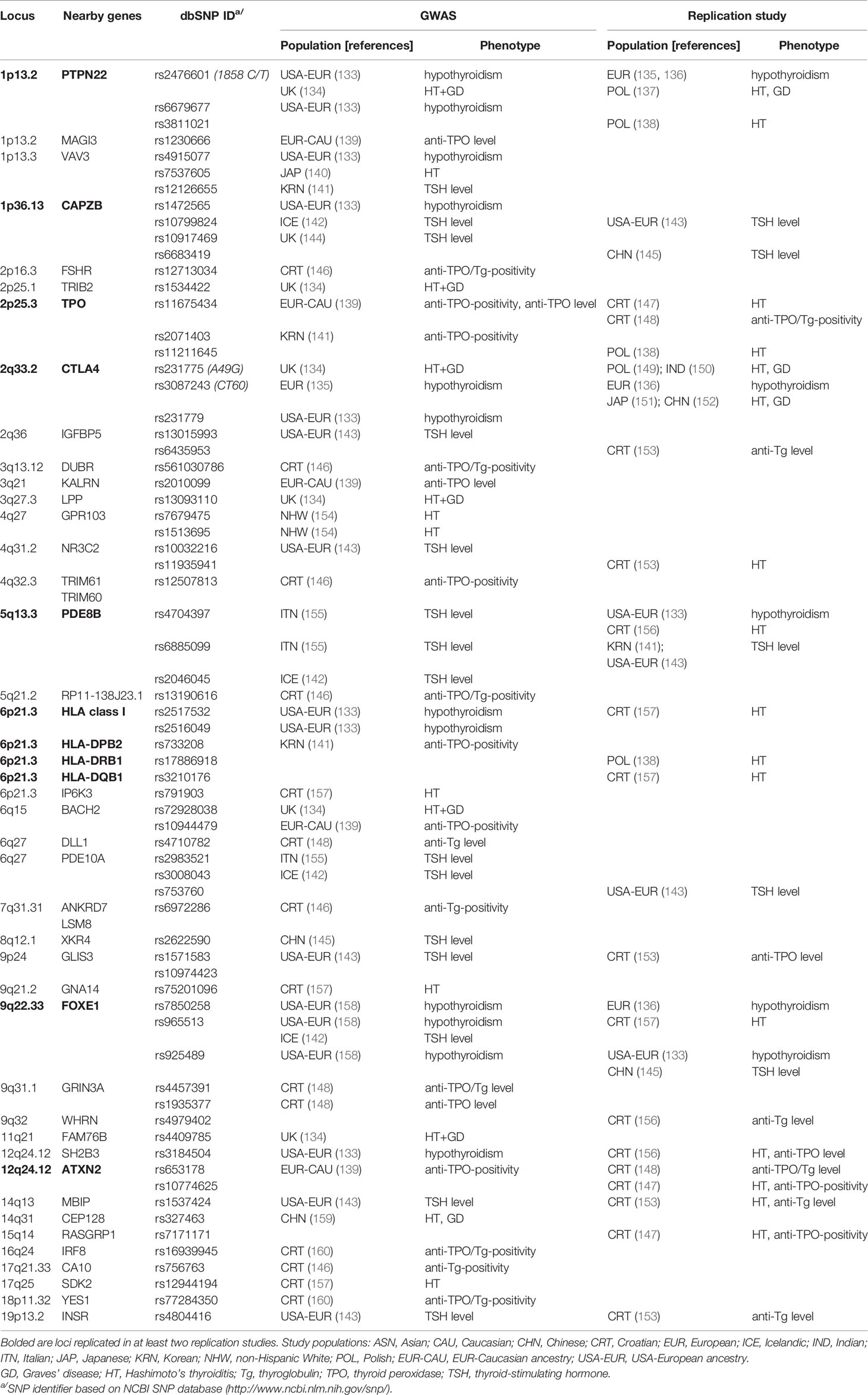
Table 2 GWAS and replication studies for AITD and thyroid dysfunction traits susceptibility.
A genetic overlap was observed between susceptibility loci identified in GWAS for AITD or hypothyroidism and GWAS for thyroid function, including FOXE1, CAPZB, and PDE8B. FOXE1, also known as TTF-2 (thyroid transcription factor 2), is associated with hypothyroidism, and with TSH and fT4 levels, as well as with thyroid cancer (133, 143, 158, 162). This factor is involved in thyroid gland development and differentiation (163, 164). FOXE1 regulates TG and TPO transcription by binding to response elements in their promoter regions (165), and is necessary for synthesis of thyroid hormones. Both PDE8B and CAPZB also have strong links to thyroid function. PDE8B encodes a phosphodiesterase that catalyzes the hydrolysis of cAMP and is primarily expressed in the thyroid gland (166). It is proposed that PDE8B may affect TSH release from the pituitary and mediate the effects of TSH in the thyroid by altering cAMP levels (155). A role for PDE8B in cAMP-dependent generation of triiodothyronine (T3) and T4 has also been proposed (143). Similarly, CAPZB, a member of the F-actin-capping protein family, is highly expressed in thyroid tissue. As a protein associated with cytoskeleton remodeling and assembly of cytoplasmic microtubules, it may contribute to the disturbance of thyroid follicular architecture commonly observed in AITD.
Interestingly, three independent GWAS connected VAV3 (vav guanine nucleotide exchange factor 3) variants with HT, hypothyroidism, and TSH levels (133, 140, 141). VAV3 activates Rho and Rac GTPases and is important for both thyroid and immune function (167). VAV3 is also necessary for B-cell receptor endocytosis and antigen presentation by major histocompatibility complex (MHC) class II molecules (168).
Notably, the MHC region, CTLA4, and PTPN22, which were identified by GWAS as most significantly associated with HT, were previously described as AITD susceptibility loci, based on a systematic review of candidate genes (128). All of these loci are related to autoimmune responses and involved in antigen presentation and T cell receptor signaling.
The highly polymorphic MHC region of chromosome 6p21, encoding the HLA glycoproteins, is the most intensively studied region of the genome in the search for associations with development of AITD. Numerous HLA alleles have been identified as genetic risk factors for AITD; however, the data on HLA haplotypes in HT are less definitive than that in GD. Although, some HLA alleles associated with HT are common to GD, others appear to be unique to each disease (119, 169, 170), suggesting that HLA genotypes may contribute, at least in part, to differences in HT and GD immunopathogenic mechanisms.
Among the HLA class I alleles, HLA-A*02:07 was associated with HT susceptibility in a Japanese population (169, 170). Of note, this allele was the strongest and most significantly associated susceptibility allele in HT, in contrast to GD, where alleles from the MHC class II region were more strongly associated (169). Furthermore, it was estimated that approximately 58% of patients with HT carried at least one of four HLA class I alleles (HLA-A*02:07, B*35:01, B*40:02, or B*40:06) (169). Conversely, in Caucasian populations, only a few studies have suggested associations between HLA class I alleles and HT risk (171, 172). Hence, it is hypothesized that ethnic differences may play a role in the immunological mechanisms involved in thyrocyte destruction (169).
Numerous associations between HLA class II alleles and HT have been demonstrated (particularly in Caucasian populations), including DQA1*03, DQB1*02, DQB1*03, DRB1*03, and DRB1*04 alleles, or DR3, DR4, and DR5 haplotypes (173–176). In Japanese population, DRB1*04 and DRB4*01 were suggested as susceptibility alleles, while DQA1*01 and DQB1*06 were protective (169). However, all of these studies included relatively small cohorts and generated inconsistent results. Some of these previous observations have been confirmed in a large group of UK Caucasian patients with HT; DRB1*04, DQB1*03:01, and DQA1*03:01 showed the most significant predisposing effect, while DQA1*01:02, DQA1*02:01, and DQB1*06 had the most significant protective effect. Overall, a strong predisposing association between DR4 or DR8 haplotypes and HT has been found, as well as a borderline association with DR3, whereas protective effects were detected for DR13, DR7, and DR15 (119). Similarly, DRB1*04:05, DQB1*02:01, DQB1*03:02, and DQA1*03:01 allele frequencies were higher in Greek patients with HT, while those of DRB1*07 were lower, than in the control group (177).
In recent studies including non-Caucasian populations, the HLA-DRB4*53:01 allele was identified as associated with HT susceptibility in Japanese patients (170) and the DR8 haplotype in Koreans (178). Additionally, HLA alleles in the HP-2 haplotype (HLA-A*33:03-C*14:03-B*44:03-DRB1*13:02-DQB1*06:04-DPB1*04:01) and the HP-2 haplotype itself were associated with protective effects against GD and HT in a Japanese population (169, 170), with OR = 0.36 for HT (169). Furthermore, the HLA-DRB1*03 allele was shown to be predisposing in Kayseri Turkish patients with HT, in contrast to DRB1*01, which was associated with a protective effect (179). Moreover, in an Indian population, DRB1*12 and DRB1*10 exhibited the strongest predisposing and protective effects, respectively, among different HLA-DRB1 alleles (150); however, in contrast to previous reports, a decreased frequency of the DRB1*03 allele was observed in HT patients in this study.
Due to its crucial role in the presentation of peptide antigens to T cells, particular attention has been paid to the molecular structure of the peptide binding pocket in HLA class II molecules. Arginine at position 74 of the HLA-DRβ1 chain (DRβ1-Arg74) was identified as a critical pocket amino acid signature that confers susceptibility to both GD and HT (180, 181). Conversely, the presence of glutamine at position 74 of the DRβ1 chain conferred a protective effect. It was suggested that a specific HLA-DR peptide binding pocket structure may predispose to AITD by enabling presentation of certain autoantigens, such as Tg, TPO, or TSHR pathogenic peptides. Indeed, the non-synonymous TG variant, W1999R, can interact with HLA-DRβ1-Arg74, and together these two variants confer a high risk for AITD (182). Recently, among Polish patients, HLA-DRB1 with phenylalanine at position 67 (encoded by the rs17886918T variant) was significantly associated with HT (138).
GWAS identified several CTLA4 genetic variants as being associated with AITD (both HT and GD) or hypothyroidism, and these associations were widely replicated in various ethnic groups (Table 2). The most consistent associations with HT were a SNP at position 49 (A49G, rs231775) in the CTLA4 leader peptide, resulting in an alanine to threonine substitution, and a SNP located near to the 3’ UTR (CT60, rs3087243) in both the Caucasian and Asian populations (Table 2). Several subsequent meta-analyses have confirmed these associations (183–187). CTLA4 is suggested to contribute to AITD susceptibility by interacting with other loci (188) and a strong synergy has been demonstrated for the CTLA4 and HLA genes in AITD (189). In an Indian population, associations with both susceptibility (for a combination of the G allele of CTLA4, A49G, and the HLA-DR5 allele) and protection (for a combination of the A allele of CTLA4, A49G, and the HLA-DR3, DR10, and DR15 alleles) have been established (150).
CTLA4 is a transmembrane protein expressed on activated T cells that functions as a negative regulator of their activation by competing with CD28 for binding to the ligand B7 on antigen-presenting cells (190). Therefore, it is likely that polymorphisms that reduce CTLA4 expression or activity may cause excessive activation and proliferation of T cells, predisposing to autoimmunity (114, 191). Consistent with this, SNPs in CTLA4 are risk variants for various autoimmune disorders (192); however, it is unclear which CTLA4 variants are causative and by what mechanism they confer susceptibility to autoimmunity.
Similar to CTLA4, PTPN22 is a powerful inhibitor of T cell activity; it belongs to a family of protein tyrosine phosphatases expressed in both mature and immature B and T cells. PTPN22 binds to the SH3 domain of the C-terminal Src kinase (Csk), thereby suppressing kinases that mediate T cell signaling (193). PTPN22 also functions as a negative regulator of T cell activation through its interaction with the Grb2 adaptor molecule (194).
After the HLA system, PTPN22 polymorphisms may be the most important genetic risk factor for autoimmune diseases (195). Variations in PTPN22 are associated with various autoimmune disorders, including AITD (191). The most extensively studied and widely confirmed is the association of the C1858T missense mutation (rs2476601), which results in a substitution of arginine (R) to tryptophan (W) at position 620 (R620W) of the encoded protein. The PTPN22 C1858T variant is associated with an increased risk of both HT and GD (196, 197); however, significant differences in this association have been observed across various ethnic groups. A significant association between the PTPN22 C1858T polymorphism and susceptibility to AITD was demonstrated specifically in Caucasians, but it was hardly detected in non-Caucasian populations (198–201). It is suggested that this could be due to the ancestral effects and different prevalence rates of rare variants in the studied populations. Indeed, the susceptibility allele (T) of rs2476601 is generally extremely rare in Asian and African populations (198, 202). Also, a decreasing frequency of this allele was observed from the north to the south of the Europe (203). It is suggested that AITD susceptibility in different ethnic populations may be related to distinct risk loci. Notably, an extremely rare predisposing variant of PTPN22 (missense A77G mutation) has recently been identified in a Chinese HT pedigree using whole-exome sequencing (204).
Although the exact mechanism by which the R620W variant predisposes to autoimmunity remains largely unknown, PTPN22 with a tryptophan residue at position 620 (Trp620) binds Csk less efficiently than the variant with an arginine at this position (205). Consequently, the capacity of PTPN22 to downregulate T cell responses may be reduced, thereby increasing susceptibility to autoimmunity. By contrast, PTPN22 Trp620 is more active and a more potent negative regulator of T cell signaling, suggesting a role for R620W in positive selection of autoreactive T cells (206). Furthermore, PTPN22 Trp620 was associated with increased frequencies of thymically-derived T regulatory (Treg) cells and with increased expression of programmed cell death protein (PD-1) on both Treg and T effector cells (207). Currently, the role of the PTPN22 C1858T polymorphism in autoimmunity is suggested to be alteration of both the innate and adaptive immune responses (206, 208).
Overall, current knowledge of HT genetics is rather limited. The majority of identified associations relate to general immune-regulatory genes involved in the development of central and peripheral tolerance and antigen presentation, which are important for achieving the proper balance between an adequate immune response against foreign antigens and maintaining autoantigenic tolerance. Moreover, all susceptibility loci identified to date together account for only a small proportion of the heritability of HT (209). It is estimated that < 5.5% of total HT variance can be explained by common genetic variants (138, 157), indicating that a substantial number of HT predisposing factors remain to be discovered.
Considering the similar elements in the pathogenic mechanisms and high prevalence rates of both AITD and PCOS among women of reproductive age, the interesting question of whether thyroid dysfunction is significantly more frequent in women with PCOS arises. An increasing number of studies indicate a higher prevalence of AITD, and particularly hypothyroidism, in patients with PCOS (Table 3 and relevant references therein). In most of these studies, although not all, higher levels of anti-TPO and/or anti-Tg antibodies were observed, exceeding the upper limit of the normal range in an average of 22.3% of patients with PCOS (ranging from 4.8% to 37.9%, depending on the study), compared with an average of 8.5% in healthy women (range 3.3% to 15.3%). Also, decreased echogenicity of the thyroid gland, a characteristic ultrasound pattern typical of HT, was observed more frequently in women with PCOS than in those without the condition (mean, 29% vs. 9%, respectively). The prevalence of SCH at TSH levels between 4.2 and 10 mIU/L ranged from 11.3% to 30.3% (mean, 20.3%) among patients with PCOS, which was more than 3 times higher than that reported for women without this condition (mean, 6.2%; range, 1.9% to 8.75%). Altogether, the estimated prevalence of AITD among women with PCOS was nearly threefold higher than that in healthy women (mean, 28% vs. 10%, respectively), although it should be noted that the diagnostic criteria for AITD differed among studies (Table 3).
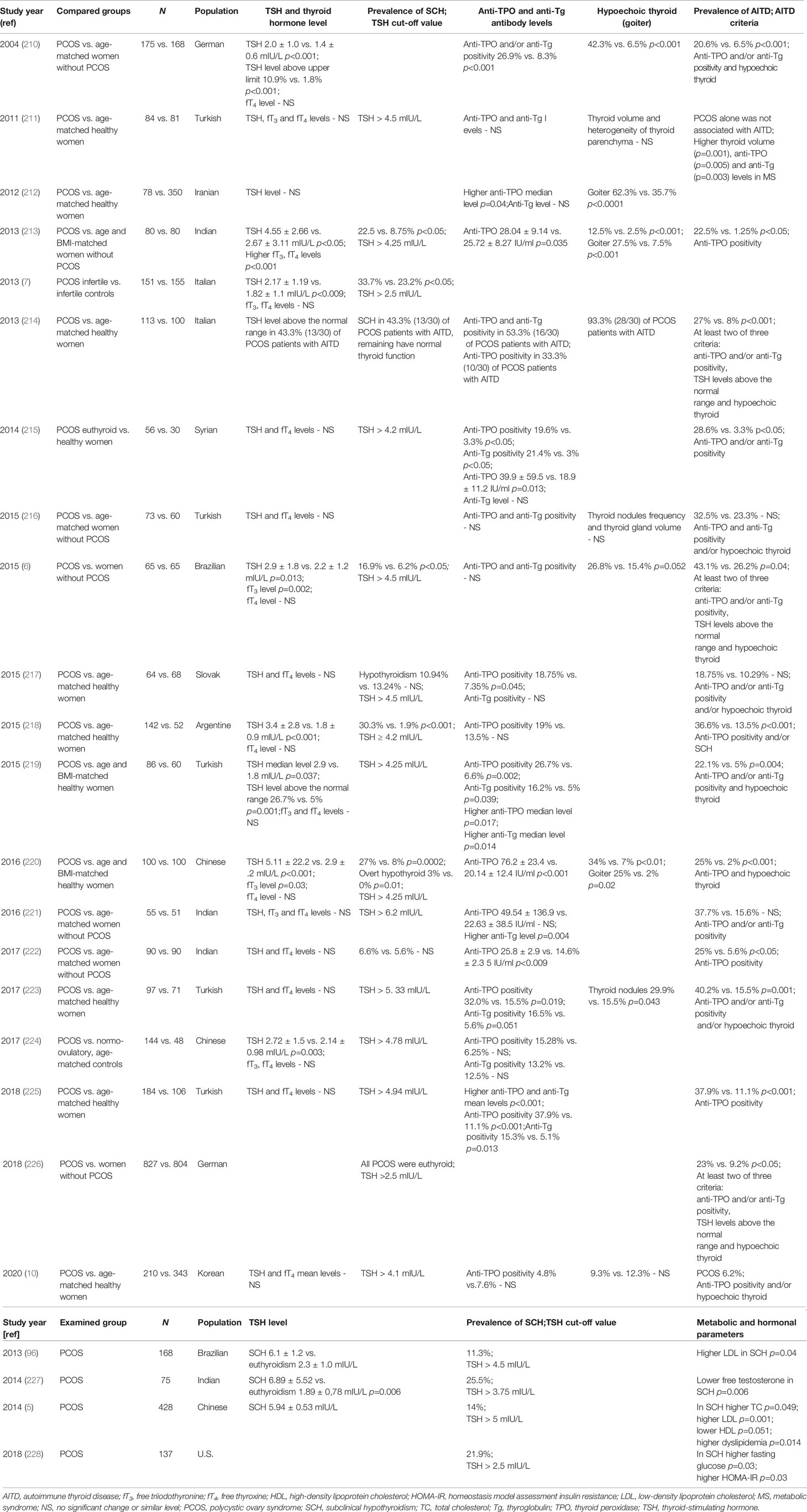
Table 3 Joint prevalence of SCH and AITD in women with PCOS.
In women with PCOS, compared with those without the condition, higher TSH levels, anti-thyroid antibody positivity rate, and prevalence of thyroid disorders, particularly HT, have been demonstrated in three independent meta-analyses conducted to date (9, 229, 230). Based on six studies, the combined OR of SCH risk for women with PCOS (compared with healthy women) was 2.87 (95% confidence interval (CI), 1.82–9.92; p < 10-6), assuming a TSH cut-off level of > 2.5 mIU/L, and 3.59 (95% CI, 2.25–5.73; p < 10-6) when limiting TSH to ≥ 4 mIU/L (229). Similarly, two meta-analyses indicated a significant association between PCOS and the presence of AITD: one included six studies (OR = 4.81; 95% CI, 2.88–8.04; p < 10-5) (9) and the other included 13 studies (OR = 3.27; 95% CI, 2.32–4.63; p < 10-4) (230). The higher risk of AITD among women with PCOS also persisted after geographical stratification of the study populations (230).
Although the association between PCOS and AITD is generally uncontested, its cause remains unclear. Both syndromes share a number of common clinical and pathological features; however, whether mutual interrelationships are present, or whether one condition predisposes an individual to another disorder, remains speculative (8). Some evidence suggests that PCOS may have an autoimmune background. Abnormally elevated levels of systemic autoimmune markers such as anti-histone, anti-double stranded DNA (anti-dsDNA), and anti-nuclear antibodies, which are considered classic features of autoimmune disease, have been observed in women with PCOS (231, 232); however, the presence of anti-ovarian antibodies in PCOS remains controversial (231).
The most obvious association between PCOS and HT is the increased metabolic risk of obesity, IR, and dyslipidemia (27, 233). Overweight and obese patients with PCOS showed a higher tendency toward thyroid dysfunction; TSH levels > 2.5 mIU/L were significantly more common in patients with BMI > 25 kg/m2 (56%) than in those with BMI ≤ 25 (25.8%, p < 0.005) (7). Patients with PCOS and AITD were more obese by an average of 2 kg/m² (226). Higher TSH levels (as well as a higher frequency of nodular goiter and thyroid volume) were observed in patients with PCOS, and these parameters correlated with IR (7, 223). Furthermore, fasting glucose and homeostasis model assessment (HOMA)-IR levels among patients with PCOS and SCH were higher than those in euthyroid PCOS patients, independent of BMI (10, 228, 234). Notably, ethnic diversity among euthyroid PCOS patients and patients with PCOS and SCH was suggested with respect to IR and lipid profiles (227). By contrast, SCH is not an independent risk factor for PCOS among obese women of reproductive age (235).
In particular, combined occurrence of PCOS and SCH increases the risk of impaired lipid profiles. Compared with euthyroid PCOS patients, PCOS patients with elevated TSH levels show a trend toward higher triglyceride and LDL cholesterol levels, as well as lower high-density lipoprotein (HDL) cholesterol levels (5, 96, 220). A positive correlation was found between TSH and LDL cholesterol levels, with the optimal TSH cut-off point for elevated LDL cholesterol risk defined as 4.07 mIU/L (5). Furthermore, two recent meta-analyses covering 12 and nine studies demonstrate that the presence of SCH in women with PCOS is associated with an increase in metabolic disorders, particularly dyslipidemia, which affect triglyceride, LDL, HDL, and total cholesterol levels (234). Taken together, current data suggest that the combined effect of PCOS and HT is associated with a higher risk of more pronounced metabolic disorders than either of these syndromes alone.
It has been hypothesized that increased IR in obesity and secretion of pro-inflammatory mediators can lead to elevated TSH levels through one of two pathways: decreased deiodinase-2 activity or increased levels of leptin hormone, which act directly to stimulate increased TRH secretion by the hypothalamus (110, 236). In addition, increased leptin, as a result of weight gain, can mediate autoimmunity by preferential up-regulation of autoreactive T cells and down-regulation of Treg cells, mediating a suppressive effect on the immune system (8). It is suggested that PCOS exacerbates development of SCH, likely via obesity and IR (237). Moreover, hypothyroidism may aggravate IR.
One possible explanation for the high prevalence of HT in PCOS assumes that changes in the fetal thymus, and resulting alterations in immune tolerance, may predispose to combined PCOS and HT in adulthood (27). Estrogen or adrenal steroids, such as corticosterone, injected into female mice early in life (before the final stage of thymus development) results in anovulation and follicular cyst formation (238). It is also suggested that estrogen can damage the thymus during its development, and that the resulting absence of Treg cells is a prerequisite for formation of ovarian cysts (238). Similar to PCOS, hypothyroidism can result in ovarian cyst formation (8) and, notably, they are normalized in response to T4 replacement (106). CD4+ CD25+ FOXP3+ Treg cells protected against autoimmunity in a murine model of HT (239). The transcription factor, FOXP3, which is involved in Treg cell formation and consequently FOXP3 expression, is dependent on production of the cytokine TGF-β (239). Since lower levels of TGF-β1 are reported in HT than in healthy women (240), lower levels of Treg cells are also expected. TGF-β1 inhibits proliferation, differentiation, and apoptosis of T cells, as well as increases the growth of naive T cells (241); hence, it is thought to be involved in development of autoimmunity. Furthermore, TGF-β signaling appears to be important for the fetal origin of PCOS and folliculogenesis (242).
The impact of sex hormones on development of autoimmunity appears obvious given the clear predominance of women among patients affected by autoimmune diseases; the ratio of women to men is 15:1 in the case of AITD (243). Estrogen can stimulate the immune system and increase Treg cell formation (244). In fertile, non-pregnant women, the number of CD4+ CD25+ FOXP3+ Treg cells increases in the late follicular phase of the menstrual cycle and decreases in the luteal phase, which correlates with E2 levels (245). Unlike estrogen, progesterone levels correlate inversely with dendritic cell secretion of pro-inflammatory cytokine IL6, which in turn inhibits FOXP3 expression and Treg cell generation (246, 247). Furthermore, progesterone may suppress CD4+ T cell proliferation and Th1 responses (248). In women with normal menstrual periods, the immunosuppressive action of progesterone and androgens counteracts the stimulating effect of estrogen (248, 249), while in women with PCOS, a decrease in progesterone levels is usually detected due to irregular menses and oligo- or anovulatory cycles (250). Also, a significantly higher E2 level and estrogen-to-progesterone ratio were observed in anti-TPO-positive compared with anti-TPO-negative women with PCOS (219). The resulting imbalance between estrogen and progesterone is thought to be associated with an excessive inflammatory response that promotes autoimmune disorders in PCOS (8). Consistent with this assumption, patients with PCOS exhibit low-grade inflammation, characterized by elevated levels of CRP and independent of obesity (251). PCOS is characterized by an excess of androgens; however, it is speculated that the suppressive effect of androgens on the immune system at the levels observed in PCOS is probably insufficient to prevent autoimmunity (27). In addition, lower testosterone levels, a lower free androgen index, and less severe hyperandrogenemia are observed in patients with PCOS and HT relative to those with PCOS alone (226), although, the results have not been confirmed in patients with PCOS and SCH (234, 252).
Low levels of vitamin D are associated with both PCOS and HT (253, 254) and provide another possible link between these syndromes and autoimmunity. Vitamin D is considered to be protective against autoimmune diseases (255). A strong association has been reported between the severity of vitamin D deficiency and HT, as well as anti-thyroid antibody and thyroid hormone levels (256). Nevertheless, concerns remain that the effects of vitamin D may not be direct, but rather secondary to estrogen dysregulation (257). Consistent with this hypothesis, low levels of 25 hydroxyvitamin D3 (25(OH)D3) are associated with AITD in pre-menopausal, but not post-menopausal, women (258). A genetic variant of CYP27B1 hydroxylase, which is responsible for production of an active form of vitamin D from 25(OH)D3, is associated with HT (259). Furthermore, polymorphisms in the VDR gene causing a decrease in vitamin D levels are associated with both HT and several metabolic syndrome features in women with PCOS (27). Moreover, levels of 25(OH)D3 are significantly lower in women with PCOS and AITD than those without AITD (253). Among overweight and obese persons, a significantly higher frequency of vitamin D deficiency was observed in patients with HT than without HT (69% vs. 52%, p = 0.042) (260), suggesting that low vitamin D level is not merely a marker of obesity.
As stated above, a strong genetic impact on inheritance and susceptibility to disease development is present in both PCOS and HT. Although a functional relevance of several candidate gene polymorphisms has been suggested, no common genetic background has been established. To date, only a few genetic variants have been proposed to be causally associated with the joint incidence of both disorders. The most convincingly described are three genetic polymorphisms that can contribute to both PCOS and HT. These are polymorphisms in FBN3, a gene related to TGF-β activity and Treg cell levels; CYP1B1, a gene involved in E2 metabolism; and GNRHR. In addition, there are two susceptibility loci identified in GWAS that are common for both diseases (Tables 1 and 2): FSHR (2p16.3) and INSR (19p13.3); however, no functional overlap between the two diseases has yet been confirmed.
The CYP1B1 gene encodes an enzyme belonging to the multi-gene-encoded cytochrome P450 enzyme family, which oxidizes E2 to 4-hydroxyestradiol (261). CYP1B1 is expressed at a level 3 times lower in PCOS ovaries than in control ovaries (262). The pathogenic polymorphism, L432V (rs1056836), in CYP1B1 is associated with serum T4, fT4, and fT3 concentrations among patients with PCOS (263). Although none of the studied polymorphisms in this gene are associated with disease risk, suggesting that CYP1B1 may not have a causative role in the etiology of PCOS, the CYP1B1 L432V polymorphism provides a potential link between PCOS and HT.
GnRH is a key hypothalamic peptide that, after binding to a specific receptor, GnRHR, stimulates a release of LH and FSH from pituitary gonadotropic cells. Increased levels of LH and a higher frequency of LH pulses (264), as well as a higher prevalence of increased TSH levels (8), were observed in women with PCOS. GnRH was shown to enhance the release of TSH (265). Furthermore, a 3’-UTR polymorphism in the GNRHR gene, rs1038426, was found to affect GNRHR expression, with a variant allele-dose effect, and was associated with the concentration of serum TSH as well as insulin secretion and insulin sensitivity in women with PCOS (266). An association between TSH and fasting insulin levels and insulin sensitivity was also reported (267). While GNRHR polymorphism did not contribute to the risk of PCOS (266), it links gonadotropin action-related dysfunction with IR and possibly also with thyroid function disorders in PCOS. Several PCOS risk variants were identified in the INSR gene, although replication studies produced mostly inconsistent results. The most convincing seems to be an association of rs2252673 in intron 11, identified by the entire gene examination with a tagging approach (268), and silent SNP rs1799817 at exon 17, encoding the tyrosine kinase domain of the INSR (80); especially among lean women with PCOS. The intronic INSR variants were also independently identified by the GWAS for PCOS and for the level of TSH (Tables 1 and 2); however, the causal variant of this loci and the impact on INSR expression remained unknown. Nevertheless, pronounced IR and defects in insulin secretion are commonly associated with PCOS. In women with PCOS, an increase in beta-cell function was observed when compared to the age and BMI matched controls, which was correlated with the intensity of IR (269).
A strong association was observed in candidate gene studies between linked to each other missense FSHR gene variants: Thr307Ala (rs6165) and Asn680Ser (rs6166), and intensity of some PCOS clinical traits. The Ser680 allele was associated with higher serum gonadotrophic hormones concentrations and higher frequency of hyperandrogenism presence (44). Longer follicular phase and lower E2 levels after exogenous FSH stimulation were also observed, suggesting lower sensitivity of this FSHR variant (270). Several other FSHR polymorphisms have been shown to correlate with ovarian function, including SNPs identified by GWAS in Han Chinese population (Table 1) and by fine mapping of 2p16.3 region in a population of European ancestry (271); however, they seem to be associated with FSH levels and the PCOM phenotype rather than with disease risk (33, 47). Recent GWAS among patients with HT identified SNP rs12713034 in the FSHR gene that is associated with the presence of anti-thyroid antibodies (146). In addition, hypothyroidism has been found to decrease FSH and E2 levels and alter FSHR-mediated expression of CYP51, a key enzyme involved in sterols and steroids biosynthesis during folliculogenesis and oocyte maturation, which is regulated by FSH (272); thus providing a further link between the functions of the ovaries and thyroid gland.
FBN3 is a member of the fibrillin/LTBP (latent TGF-β binding protein) family of ubiquitously expressed extracellular matrix proteins that regulate the bioavailability and activity of TGF-β, providing binding sites for its sequestration (273). The FBN3 genetic variant, D19S884 allele 8 (A8), a dinucleotide repeat microsatellite marker in intron 55, is the variant most strongly associated with PCOS susceptibility at the 19p13.2 locus (274). A rare missense variant (Asp911Val) in FBN3 was also found by the whole-exome sequencing approach (275). Although the results were not consistent, further studies suggest that D19S884 is likely a causal variant for PCOS susceptibility (275, 276). It was suggested that the A8 allele may affect splicing of FBN3 transcript (277). Women with PCOS carrying the A8 allele had significantly lower circulating TGF-β1 levels, and higher inhibin B and aldosterone levels, as well as higher levels of fasting INS and HOMA-IR than women with PCOS without the A8 allele (277). It is hypothesized that women with PCOS and the A8 variant, and therefore lower TGF-β levels, are more susceptible to HT than those without this allele (27). Some findings suggest that expression of the FBN3 gene in fetal ovaries may predispose to PCOS development in later life, supporting the existing hypothesis of the fetal origin of PCOS (278).
As outlined in this review, our current knowledge of the genetic basis of the joint occurrence of PCOS and HT leads us to believe that there may be no strong shared genetic variants associated with the risk of both diseases. Rather, it is a specific combination of risk factors for individual diseases that predisposes them to occur together. Moreover, due to the complex background of both disorders, this combination may be specific not so much to the risk of the disease itself as to the expression of its individual phenotypes. In line with this assumption, it has been recently indicated that the use of combined polygenic and phenotypic risk prediction may improve the accuracy of PCOS diagnosis (279).
As shown in Figure 1, which simplifies the possible cross-linkages between PCOS and HT, the multi-directional link seems to be the best explanation for the predisposition to joint occurrence of both diseases. Treg cells dysregulation emerges as a critical point in the genetic and functional network connecting the two diseases. In PCOS, genetic variation emphasizes the contribution of both hormonal imbalance (gonadotropins, androgens, and female sex hormones) and metabolic factors (IR and INS secretion), often interacting through a feedback loop (230, 248). It can be assumed that the favorable hormonal and metabolic background in women with PCOS may predispose them to thyroid autoimmunity and aggravate the disease symptoms through the HT susceptibility factors (8, 219).
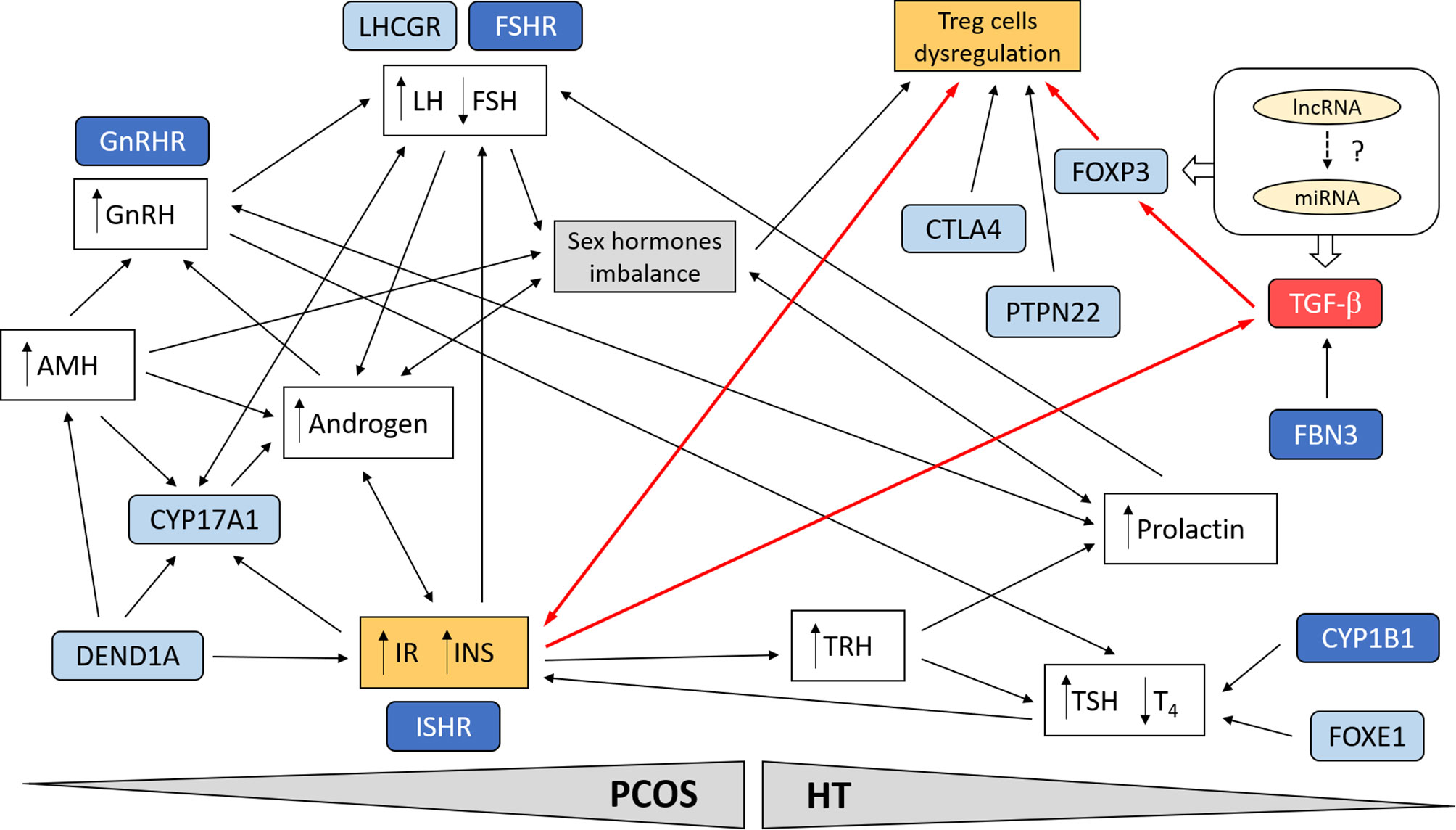
Figure 1 Schematic presentation of possible cross-linkages between PCOS and HT. Involvement of the most important genetic factors and molecular pathways. Treg cell dysregulation emerges as a critical point in the genetic and functional network linking the two diseases. In PCOS, genetic variation emphasizes the contribution of both hormonal imbalance (gonadotropins, androgens, and female sex hormones) and metabolic factors (IR and INS secretion), often interacting through a feedback loop. Suppression of TGF-β signaling pathway in combination with IR may lead to dysregulation of Treg cells and promotion of autoimmunity in women with PCOS (red line connections). Possible interaction of ncRNA (miRNA, lncRNA) with TGF-β signaling. The most important genes are indicated in blue; darker blue designates susceptibility genes for both diseases.
The best documented genetic association between PCOS and HT relates to the TGF-β signaling pathway. Factors involved in this pathway are clearly good candidate susceptibility genes for both syndromes, since they have key roles in the immune system, hormone regulation, inflammation, cell proliferation, tissue differentiation, apoptosis, and related metabolic consequences such as IR. In PCOS, the inflammation of visceral adipose tissue resulting in chronic release of pro-inflammatory cytokines is a major contributor to IR (280). Treg cells suppress pro-inflammatory effects of autoreactive T cells (281). Depletion of CD4+CD25+Foxp3+ Treg cells and increased inflammation in visceral adipose tissues was found to contribute to IR in HT (282). We are hypothesizing that the suppression of TGF-β signaling pathway in combination with IR may lead to Treg cells dysregulation and promotion of autoimmunity in women with PCOS (Figure 1). Beside FBN3, no members of the TGF-β signaling have been shown to be among the top GWAS associations for PCOS or HT. However, rs4803457 polymorphism in TGFB1 gene was associated with PCOS susceptibility in candidate gene studies in Chinese Han women (273). The genetic variation in transcription factor FOXP3 has also been reported in candidate gene studies for AITD (2). FOXP3 is a crucial regulator of Treg cells differentiation and function and its expression is induced by TGF-β, which activity, in turn, is regulated by FBNs (33).
In recent years, there has been an increase in evidence of the role of non-coding RNA (ncRNA) in the development of various diseases. Differentially expressed microRNA (miRNA) and long ncRNA (lncRNA) were identified both in PCOS and HT (283, 284). The ncRNAs were shown to be involved in regulating T cell production and differentiation (285). Furthermore, several ncRNAs were involved in regulation by the TGF-β signaling pathway. In HT, the expression of miR-141 was downregulated due to its involvement in TGF-β pathway and regulation through IL2 (286). In turn, lncRNA-IFNG-AS1 was upregulated in HT patients, and it was associated with IFN-γ expression in human CD4+ T cells and with the frequency of circulating Th1 cells (287). The lncRNA TGFB2-AS1 was shown to be transcriptionally regulated by TGF-β and to suppress TGF-β/BMP-mediated response of target genes (288). In granulosa cells from patients with PCOS, expression of miR-423 was downregulated, and both miR-33b and miR-142 were upregulated, compared to controls; miR-423 directly suppressed SMAD7, while miR-33b and miR-142 targeted TGF-β receptor 1 (TGFBR1) (289). Several lncRNA have been reported to positively regulate TGF-β/SMAD signaling (290). The lncRNAs can act directly or as miRNA sponges, thus reducing their regulatory effect on target mRNAs (291). In granulosa cells, lncRNA-NORFA directly interacts with miR-126 and prevents it from binding to TGFBR2 3’UTR (292). Another lncRNA, MALAT1, has been found to regulate TGF-β signaling through sponging miR-125b and miR-203a, TGF-β negative regulators by targeting TGFBR1 and TGFBR2 (293). Considering the above-mentioned examples of the role of ncRNA in modulating of TGF-β signaling, the involvement of ncRNA in the processes leading to the joint occurrence of PCOS and HT seems very likely; however, there are no strong data supporting this hypothesis and it requires confirmation in further studies.
PCOS and HT are two of the most common endocrine disorders among young women worldwide. Both syndromes are causally related to the risk of severe metabolic and reproductive disorders and are significant social problems. Thyroid dysfunction in PCOS appears to enhance the clinical symptoms of the disease and the severity of its consequences. The pathogenesis of the association of PCOS with HT is not clear, although the relationship between thyroid hormones and ovary function is indisputable. There is good evidence for a strong genetic impact on the development of both diseases; hence, a common genetic predisposition is possible. However, to date, it is not apparent. Furthermore, the question of whether PCOS predisposes to development HT, or whether HT is a forerunner of PCOS, remains open.
All authors contributed to the article and approved the submitted version. NZ-L wrote the original draft. EH wrote and critically revised the manuscript.
This work was supported by the Polish National Science Centre (grant number 2016/23/B/NZ2/00696) and the Centre of Postgraduate Medical Education (grant number 501-1009-12-20).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Ben-Shlomo I, Younis JS. Basic research in PCOS: are we reaching new frontiers? Reprod BioMed Online (2014) 28:669–83. doi: 10.1016/j.rbmo.2014.02.011
2. Lee HJ, Li CW, Hammerstad SS, Stefan M, Tomer Y. Immunogenetics of autoimmune thyroid diseases: A comprehensive review. J Autoimmun (2015) 64:82–90. doi: 10.1016/j.jaut.2015.07.009
3. Caturegli P, De Remigis A, Rose NR. Hashimoto thyroiditis: clinical and diagnostic criteria. Autoimmun Rev (2014) 13:391–7. doi: 10.1016/j.autrev.2014.01.007
4. Lim SS, Davies MJ, Norman RJ, Moran LJ. Overweight, obesity and central obesity in women with polycystic ovary syndrome: a systematic review and meta-analysis. Hum Reprod Update (2012) 18:618–37. doi: 10.1093/humupd/dms030
5. Huang R, Zheng J, Li S, Tao T, Liu W. Subclinical hypothyroidism in patients with polycystic ovary syndrome: distribution and its association with lipid profiles. Eur J Obstet Gynecol Reprod Biol (2014) 177:52–6. doi: 10.1016/j.ejogrb.2014.04.013
6. Novais J de SM, Benetti-Pinto CL, Garmes HM, Jales RM, Juliato CRT. Polycystic ovary syndrome and chronic autoimmune thyroiditis. Gynecol Endocrinol (2015) 31:48–51. doi: 10.3109/09513590.2014.958990
7. Morgante G, Musacchio MC, Orvieto R, Massaro MG, De Leo V. Alterations in thyroid function among the different polycystic ovary syndrome phenotypes. Gynecol Endocrinol (2013) 29:967–9. doi: 10.3109/09513590.2013.829445
8. Singla R, Gupta Y, Khemani M, Aggarwal S. Thyroid disorders and polycystic ovary syndrome: An emerging relationship. Indian J Endocrinol Metab (2015) 19:25–9. doi: 10.4103/2230-8210.146860
9. Du D, Li X. The relationship between thyroiditis and polycystic ovary syndrome: a meta-analysis. Int J Clin Exp Med (2013) 6:880–9.
10. Kim JJ, Yoon JW, Kim MJ, Kim SM, Hwang KR, Choi YM. Thyroid autoimmunity markers in women with polycystic ovary syndrome and controls. Hum Fertil Camb Engl (2020) 7:1–7. doi: 10.1080/14647273.2019.1709668
11. Ott J, Aust S, Kurz C, Nouri K, Wirth S, Huber JC, et al. Elevated antithyroid peroxidase antibodies indicating Hashimoto’s thyroiditis are associated with the treatment response in infertile women with polycystic ovary syndrome. Fertil Steril (2010) 94:2895–7. doi: 10.1016/j.fertnstert.2010.05.063
12. Azziz R, Carmina E, Chen Z, Dunaif A, Laven JSE, Legro RS, et al. Polycystic ovary syndrome. Nat Rev Dis Primers (2016) 2:16057. doi: 10.1038/nrdp.2016.57
13. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril (2004) 81:19–25. doi: 10.1016/j.fertnstert.2003.10.004
14. Crespo RP, Bachega TASS, Mendonça BB, Gomes LG. An update of genetic basis of PCOS pathogenesis. Arch Endocrinol Metab (2018) 62:352–61. doi: 10.20945/2359-3997000000049
15. Azziz R. Introduction: Determinants of polycystic ovary syndrome. Fertil Steril (2016) 106:4–5. doi: 10.1016/j.fertnstert.2016.05.009
16. McCartney CR, Marshall JC. CLINICAL PRACTICE. Polycystic Ovary Syndrome. N Engl J Med (2016) 375:54–64. doi: 10.1056/NEJMcp1514916
17. Li Y, Chen C, Ma Y, Xiao J, Luo G, Li Y, et al. Multi-system reproductive metabolic disorder: significance for the pathogenesis and therapy of polycystic ovary syndrome (PCOS). Life Sci (2019) 228:167–75. doi: 10.1016/j.lfs.2019.04.046
18. Wild RA, Rizzo M, Clifton S, Carmina E. Lipid levels in polycystic ovary syndrome: systematic review and meta-analysis. Fertil Steril (2011) 95:1073–1079.e11. doi: 10.1016/j.fertnstert.2010.12.027
19. Song DK, Lee H, Hong YS, Sung Y-A. Insulin resistance is associated with hirsutism in unselected reproductive-aged women. Clin Endocrinol (Oxf) (2019) 90:586–91. doi: 10.1111/cen.13936
20. Lim SS, Kakoly NS, Tan JWJ, Fitzgerald G, Bahri Khomami M, Joham AE, et al. Metabolic syndrome in polycystic ovary syndrome: a systematic review, meta-analysis and meta-regression. Obes Rev (2019) 20:339–52. doi: 10.1111/obr.12762
21. Spritzer PM, Lecke SB, Satler F, Morsch DM. Adipose tissue dysfunction, adipokines, and low-grade chronic inflammation in polycystic ovary syndrome. Reproduction (2015) 149:R219–27. doi: 10.1530/REP-14-0435
22. Xiong Y, Liang X, Yang X, Li Y, Wei L. Low-grade chronic inflammation in the peripheral blood and ovaries of women with polycystic ovarian syndrome. Eur J Obstet Gynecol Reprod Biol (2011) 159:148–50. doi: 10.1016/j.ejogrb.2011.07.012
23. Spritzer PM, Barone CR, Oliveira FB. Hirsutism in Polycystic Ovary Syndrome: Pathophysiology and Management. Curr Pharm Des (2016) 22:5603–13. doi: 10.2174/1381612822666160720151243
24. Romanowski MD, Parolin MB, Freitas ACT, Piazza MJ, Basso J, Urbanetz AA. Prevalence of non-alcoholic fatty liver disease in women with polycystic ovary syndrome and its correlation with metabolic syndrome. Arq Gastroenterol (2015) 52:117–23. doi: 10.1590/S0004-28032015000200008
25. Condorelli RA, Calogero AE, Mauro MD, Vignera SL. PCOS and diabetes mellitus: from insulin resistance to altered beta pancreatic function, a link in evolution. Gynecol Endocrinol (2017) 33:665–7. doi: 10.1080/09513590.2017.1342240
26. Gunning MN, Fauser BCJM. Are women with polycystic ovary syndrome at increased cardiovascular disease risk later in life? Climacteric (2017) 20:222–7. doi: 10.1080/13697137.2017.1316256
27. Gaberšček S, Zaletel K, Schwetz V, Pieber T, Obermayer-Pietsch B, Lerchbaum E. Mechanisms in endocrinology: thyroid and polycystic ovary syndrome. Eur J Endocrinol (2015) 172:R9–21. doi: 10.1530/EJE-14-0295
28. Kerchner A, Lester W, Stuart SP, Dokras A. Risk of depression and other mental health disorders in women with polycystic ovary syndrome: a longitudinal study. Fertil Steril (2009) 91:207–12. doi: 10.1016/j.fertnstert.2007.11.022
29. Abbott DH, Dumesic DA, Levine JE. Hyperandrogenic origins of polycystic ovary syndrome - implications for pathophysiology and therapy. Expert Rev Endocrinol Metab (2019) 14:131–43. doi: 10.1080/17446651.2019.1576522
30. Reid SP, Kao C-N, Pasch L, Shinkai K, Cedars MI, Huddleston HG. Ovarian morphology is associated with insulin resistance in women with polycystic ovary syndrome: a cross sectional study. Fertil Res Pract (2017) 3:8. doi: 10.1186/s40738-017-0035-z
31. Cadagan D, Khan R, Amer S. Thecal cell sensitivity to luteinizing hormone and insulin in polycystic ovarian syndrome. Reprod Biol (2016) 16:53–60. doi: 10.1016/j.repbio.2015.12.006
32. Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab (2006) 91:2100–4. doi: 10.1210/jc.2005-1494
33. Jones MR, Goodarzi MO. Genetic determinants of polycystic ovary syndrome: progress and future directions. Fertil Steril (2016) 106:25–32. doi: 10.1016/j.fertnstert.2016.04.040
34. Deswal R, Yadav A, Dang AS. Sex hormone binding globulin - an important biomarker for predicting PCOS risk: A systematic review and meta-analysis. Syst Biol Reprod Med (2018) 64:12–24. doi: 10.1080/19396368.2017.1410591
35. Patel S. Polycystic ovary syndrome (PCOS), an inflammatory, systemic, lifestyle endocrinopathy. J Steroid Biochem Mol Biol (2018) 182:27–36. doi: 10.1016/j.jsbmb.2018.04.008
36. Hiam D, Moreno-Asso A, Teede HJ, Laven JSE, Stepto NK, Moran LJ, et al. The Genetics of Polycystic Ovary Syndrome: An Overview of Candidate Gene Systematic Reviews and Genome-Wide Association Studies. J Clin Med (2019) 8:1606. doi: 10.3390/jcm8101606
37. Zhao H, Lv Y, Li L, Chen Z-J. Genetic Studies on Polycystic Ovary Syndrome. Best Pract Res Clin Obstet Gynaecol (2016) 37:56–65. doi: 10.1016/j.bpobgyn.2016.04.002
38. Chen D-J, Ding R, Cao J-Y, Zhai J-X, Zhang J-X, Ye D-Q. Two follicle-stimulating hormone receptor polymorphisms and polycystic ovary syndrome risk: a meta-analysis. Eur J Obstet Gynecol Reprod Biol (2014) 182:27–32. doi: 10.1016/j.ejogrb.2014.08.014
39. Liu XB, Deng XH, Zhou B, Zhang L, Niu XM. Meta-analysis of the correlation between the TNF-α308G/A polymorphism and polycystic ovary syndrome. Genet Mol Res (2016) 15:1–9. doi: 10.4238/gmr.15027923
40. Shi X, Xie X, Jia Y, Li S. Associations of insulin receptor and insulin receptor substrates genetic polymorphisms with polycystic ovary syndrome: A systematic review and meta-analysis. J Obstet Gynaecol Res (2016) 42:844–54. doi: 10.1111/jog.13002
41. Liu AL, Xie HJ, Xie HY, Liu J, Yin J, Hu JS, et al. Association between fat mass and obesity associated (FTO) gene rs9939609 A/T polymorphism and polycystic ovary syndrome: a systematic review and meta-analysis. BMC Med Genet (2017) 18:89. doi: 10.1186/s12881-017-0452-1
42. Liao D, Yu H, Han L, Zhong C, Ran X, Wang D, et al. Association of PON1 gene polymorphisms with polycystic ovarian syndrome risk: a meta-analysis of case–control studies. J Endocrinol Invest (2018) 41(11):1289–300. doi: 10.1007/s40618-018-0866-4
43. Li Y, Zhu H, Liu M, Zeng Z, Zeng Y, Xu X, et al. Significant association between methylenetetrahydrofolate reductase gene C677T polymorphism with polycystic ovary syndrome risk: A meta-analysis update. Medicine (Baltimore) (2020) 99(4):e18720. doi: 10.1097/MD.0000000000018720
44. Valkenburg O, Uitterlinden AG, Piersma D, Hofman A, Themmen APN, de Jong FH, et al. Genetic polymorphisms of GnRH and gonadotrophic hormone receptors affect the phenotype of polycystic ovary syndrome. Hum Reprod Oxf Engl (2009) 24:2014–22. doi: 10.1093/humrep/dep113
45. Wang F, Niu W-B, Kong H-J, Guo Y-H, Sun Y-P. The role of AMH and its receptor SNP in the pathogenesis of PCOS. Mol Cell Endocrinol (2017) 439:363–8. doi: 10.1016/j.mce.2016.09.023
46. Chen L, Zhang Z, Huang J, Jin M. Association between rs1800795 polymorphism in the interleukin-6 gene and the risk of polycystic ovary syndrome. Medicine (Baltimore) (2018) 97:e11558. doi: 10.1097/MD.0000000000011558
47. Laven JSE. Follicle Stimulating Hormone Receptor (FSHR) Polymorphisms and Polycystic Ovary Syndrome (PCOS). Front Endocrinol (2019) 10:23. doi: 10.3389/fendo.2019.00023
48. Shi X-Y, Huang A-P, Xie D-W, Yu X-L. Association of vitamin D receptor gene variants with polycystic ovary syndrome: a meta-analysis. BMC Med Genet (2019) 20:32. doi: 10.1186/s12881-019-0763-5
49. Wu H, Yu K, Yang Z. Associations between TNF-α and interleukin gene polymorphisms with polycystic ovary syndrome risk: a systematic review and meta-analysis. J Assist Reprod Genet (2015) 32:625–34. doi: 10.1007/s10815-015-0449-7
50. Lee H, Oh J-Y, Sung Y-A, Chung H, Kim H-L, Kim GS, et al. Genome-wide association study identified new susceptibility loci for polycystic ovary syndrome. Hum Reprod (2015) 30:723–31. doi: 10.1093/humrep/deu352
51. Chen Z-J, Zhao H, He L, Shi Y, Qin Y, Shi Y, et al. Genome-wide association study identifies susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21 and 9q33.3. Nat Genet (2011) 43:55–9. doi: 10.1038/ng.732
52. Shi Y, Zhao H, Shi Y, Cao Y, Yang D, Li Z, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet (2012) 44:1020–5. doi: 10.1038/ng.2384
53. Hwang J-Y, Lee E-J, Jin Go M, Sung Y-A, Lee HJ, Heon Kwak S, et al. Genome-wide association study identifies GYS2 as a novel genetic factor for polycystic ovary syndrome through obesity-related condition. J Hum Genet (2012) 57:660–4. doi: 10.1038/jhg.2012.92
54. Hayes MG, Urbanek M, Ehrmann DA, Armstrong LL, Lee JY, Sisk R, et al. Genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun (2015) 6:7502. doi: 10.1038/ncomms8502
55. Louwers YV, Stolk L, Uitterlinden AG, Laven JSE. Cross-ethnic meta-analysis of genetic variants for polycystic ovary syndrome. J Clin Endocrinol Metab (2013) 98:E2006–2012. doi: 10.1210/jc.2013-2495
56. Ha L, Shi Y, Zhao J, Li T, Chen Z-J. Association Study between Polycystic Ovarian Syndrome and the Susceptibility Genes Polymorphisms in Hui Chinese Women. PLoS One (2015) 10:e0126505. doi: 10.1371/journal.pone.0126505
57. Xia J-Y, Tian W, Yin G-H, Yan H. Association of Rs13405728, Rs12478601, and Rs2479106 single nucleotide polymorphisms and in vitro fertilization and embryo transfer efficacy in patients with polycystic ovarian syndrome: A case control genome-wide association study. Kaohsiung J Med Sci (2019) 35:49–55. doi: 10.1002/kjm2.12008
58. Xu Y, Li Z, Ai F, Chen J, Xing Q, Zhou P, et al. Systematic Evaluation of Genetic Variants for Polycystic Ovary Syndrome in a Chinese Population. PLoS One (2015) 10:e0140695. doi: 10.1371/journal.pone.0140695
59. Day FR, Hinds DA, Tung JY, Stolk L, Styrkarsdottir U, Saxena R, et al. Causal mechanisms and balancing selection inferred from genetic associations with polycystic ovary syndrome. Nat Commun (2015) 6:8464. doi: 10.1038/ncomms9464
60. Welt CK, Styrkarsdottir U, Ehrmann DA, Thorleifsson G, Arason G, Gudmundsson JA, et al. Variants in DENND1A are associated with polycystic ovary syndrome in women of European ancestry. J Clin Endocrinol Metab (2012) 97:E1342–1347. doi: 10.1210/jc.2011-3478
61. Lerchbaum E, Trummer O, Giuliani A, Gruber H-J, Pieber TR, Obermayer-Pietsch B. Susceptibility loci for polycystic ovary syndrome on chromosome 2p16.3, 2p21, and 9q33.3 in a cohort of Caucasian women. Horm Metab Res (2011) 43:743–7. doi: 10.1055/s-0031-1286279
62. Eriksen MB, Brusgaard K, Andersen M, Tan Q, Altinok ML, Gaster M, et al. Association of polycystic ovary syndrome susceptibility single nucleotide polymorphism rs2479106 and PCOS in Caucasian patients with PCOS or hirsutism as referral diagnosis. Eur J Obstet Gynecol Reprod Biol (2012) 163:39–42. doi: 10.1016/j.ejogrb.2012.03.020
63. Goodarzi MO, Jones MR, Li X, Chua AK, Garcia OA, Chen Y-DI, et al. Replication of association of DENND1A and THADA variants with polycystic ovary syndrome in European cohorts. J Med Genet (2012) 49:90–5. doi: 10.1136/jmedgenet-2011-100427
64. Brower MA, Jones MR, Rotter JI, Krauss RM, Legro RS, Azziz R, et al. Further investigation in europeans of susceptibility variants for polycystic ovary syndrome discovered in genome-wide association studies of Chinese individuals. J Clin Endocrinol Metab (2015) 100:E182–186. doi: 10.1210/jc.2014-2689
65. Zhao S, Tian Y, Gao X, Zhang X, Liu H, You L, et al. Family-based analysis of eight susceptibility loci in polycystic ovary syndrome. Sci Rep (2015) 5:12619. doi: 10.1038/srep12619
66. Day F, Karaderi T, Jones MR, Meun C, He C, Drong A, et al. Large-scale genome-wide meta-analysis of polycystic ovary syndrome suggests shared genetic architecture for different diagnosis criteria. PLoS Genet (2018) 14:e1007813. doi: 10.1371/journal.pgen.1007813
67. Gao J, Xue J-D, Li Z-C, Zhou L, Chen C. The association of DENND1A gene polymorphisms and polycystic ovary syndrome risk: a systematic review and meta-analysis. Arch Gynecol Obstet (2016) 294:1073–80. doi: 10.1007/s00404-016-4159-x
68. Dallel M, Sarray S, Douma Z, Hachani F, Al-Ansari AK, Letaifa DB, et al. Differential association of DENND1A genetic variants with polycystic ovary syndrome in Tunisian but not Bahraini Arab women. Gene (2018) 647:79–84. doi: 10.1016/j.gene.2018.01.028
69. Gammoh E, Arekat MR, Saldhana FL, Madan S, Ebrahim BH, Almawi WY. DENND1A gene variants in Bahraini Arab women with polycystic ovary syndrome. Gene (2015) 560:30–3. doi: 10.1016/j.gene.2015.01.034
70. Bao S, Cai JH, Yang SY, Ren Y, Feng T, Jin T, et al. Association of DENND1A Gene Polymorphisms with Polycystic Ovary Syndrome: A Meta-Analysis. J Clin Res Pediatr Endocrinol (2016) 8:135–43. doi: 10.4274/jcrpe.2259
71. Tian Y, Zhao H, Chen H, Peng Y, Cui L, Du Y, et al. Variants in FSHB Are Associated With Polycystic Ovary Syndrome and Luteinizing Hormone Level in Han Chinese Women. J Clin Endocrinol Metab (2016) 101:2178–84. doi: 10.1210/jc.2015-3776
72. Venables JP, Dalgliesh C, Paronetto MP, Skitt L, Thornton JK, Saunders PT, et al. SIAH1 targets the alternative splicing factor T-STAR for degradation by the proteasome. Hum Mol Genet (2004) 13:1525–34. doi: 10.1093/hmg/ddh165
73. Lovelace DL, Gao Z, Mutoji K, Song YC, Ruan J, Hermann BP. The regulatory repertoire of PLZF and SALL4 in undifferentiated spermatogonia. Development Camb Engl (2016) 143:1893–906. doi: 10.1242/dev.132761
74. Kommagani R, Szwarc MM, Vasquez YM, Peavey MC, Mazur EC, Gibbons WE, et al. The Promyelocytic Leukemia Zinc Finger Transcription Factor Is Critical for Human Endometrial Stromal Cell Decidualization. PLoS Genet (2016) 12:e1005937. doi: 10.1371/journal.pgen.1005937
75. Shim U, Kim H-N, Lee H, Oh J-Y, Sung Y-A, Kim H-L. Pathway Analysis Based on a Genome-Wide Association Study of Polycystic Ovary Syndrome. PLoS One (2015) 10:e0136609. doi: 10.1371/journal.pone.0136609
76. Pau CT, Mosbruger T, Saxena R, Welt CK. Phenotype and Tissue Expression as a Function of Genetic Risk in Polycystic Ovary Syndrome. PLoS One (2017) 12:e0168870. doi: 10.1371/journal.pone.0168870
77. Jones MR, Brower MA, Xu N, Cui J, Mengesha E, Chen Y-DI, et al. Systems Genetics Reveals the Functional Context of PCOS Loci and Identifies Genetic and Molecular Mechanisms of Disease Heterogeneity. PLoS Genet (2015) 11:e1005455. doi: 10.1371/journal.pgen.1005455
78. Zhu K, Li S, Liu J, Hong Y, Chen Z-J, Du Y. Role of RAB5A in FSHR-mediated signal transduction in human granulosa cells. Reproduction Camb Engl (2018) 155:505–14. doi: 10.1530/REP-18-0015
79. Li M, Zhao H, Zhao S-G, Wei D-M, Zhao Y-R, Huang T, et al. The HMGA2-IMP2 Pathway Promotes Granulosa Cell Proliferation in Polycystic Ovary Syndrome. J Clin Endocrinol Metab (2019) 104:1049–59. doi: 10.1210/jc.2018-00544
80. McAllister JM, Legro RS, Modi BP, Strauss JF. Functional genomics of PCOS: from GWAS to molecular mechanisms. Trends Endocrinol Metab (2015) 26:118–24. doi: 10.1016/j.tem.2014.12.004
81. McAllister JM, Modi B, Miller BA, Biegler J, Bruggeman R, Legro RS, et al. Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc Natl Acad Sci U S A (2014) 111:E1519–1527. doi: 10.1073/pnas.1400574111
82. Kulkarni R, Teves ME, Han AX, McAllister JM, Strauss JF. Colocalization of Polycystic Ovary Syndrome Candidate Gene Products in Theca Cells Suggests Novel Signaling Pathways. J Endocr Soc (2019) 3:2204–23. doi: 10.1210/js.2019-00169
83. McAllister JM, Han AX, Modi BP, Teves ME, Mavodza GR, Anderson ZL, et al. miRNA Profiling Reveals miRNA-130b-3p Mediates DENND1A Variant 2 Expression and Androgen Biosynthesis. Endocrinology (2019) 160:1964–81. doi: 10.1210/en.2019-00013
84. Gorsic LK, Kosova G, Werstein B, Sisk R, Legro RS, Hayes MG, et al. Pathogenic Anti-Müllerian Hormone Variants in Polycystic Ovary Syndrome. J Clin Endocrinol Metab (2017) 102:2862–72. doi: 10.1210/jc.2017-00612
85. Caburet S, Fruchter RB, Legois B, Fellous M, Shalev S, Veitia RA. A homozygous mutation of GNRHR in a familial case diagnosed with polycystic ovary syndrome. Eur J Endocrinol (2017) 176:K9–K14. doi: 10.1530/EJE-16-0968
86. Bedecarrats GY, Linher KD, Kaiser UB. Two Common Naturally Occurring Mutations in the Human Gonadotropin-Releasing Hormone (GnRH) Receptor Have Differential Effects on Gonadotropin Gene Expression and on GnRH-Mediated Signal Transduction. J Clin Endocrinol Metab (2003) 88:834–43. doi: 10.1210/jc.2002-020806
87. Dapas M, Sisk R, Legro RS, Urbanek M, Dunaif A, Hayes MG. Family-Based Quantitative Trait Meta-Analysis Implicates Rare Noncoding Variants in DENND1A in Polycystic Ovary Syndrome. J Clin Endocrinol Metab (2019) 104:3835–50. doi: 10.1210/jc.2018-02496
88. McLeod DSA, Cooper DS. The incidence and prevalence of thyroid autoimmunity. Endocrine (2012) 42:252–65. doi: 10.1007/s12020-012-9703-2
89. Antonelli A, Ferrari SM, Corrado A, Di Domenicantonio A, Fallahi P. Autoimmune thyroid disorders. Autoimmun Rev (2015) 14:174–80. doi: 10.1016/j.autrev.2014.10.016
90. Hollowell JG, Staehling NW, Flanders WD, Hannon WH, Gunter EW, Spencer CA, et al. Serum TSH, T(4), and thyroid antibodies in the United States population (1988 to 1994): National Health and Nutrition Examination Survey (NHANES III). J Clin Endocrinol Metab (2002) 87:489–99. doi: 10.1210/jcem.87.2.8182
91. Chardès T, Chapal N, Bresson D, Bès C, Giudicelli V, Lefranc M-P, et al. The human anti-thyroid peroxidase autoantibody repertoire in Graves’ and Hashimoto’s autoimmune thyroid diseases. Immunogenetics (2002) 54:141–57. doi: 10.1007/s00251-002-0453-9
92. Fröhlich E, Wahl R. Thyroid Autoimmunity: Role of Anti-thyroid Antibodies in Thyroid and Extra-Thyroidal Diseases. Front Immunol (2017) 8:521. doi: 10.3389/fimmu.2017.00521
93. Eschler DC, Hasham A, Tomer Y. Cutting edge: the etiology of autoimmune thyroid diseases. Clin Rev Allergy Immunol (2011) 41:190–7. doi: 10.1007/s12016-010-8245-8
94. Ajjan RA, Weetman AP. The Pathogenesis of Hashimoto’s Thyroiditis: Further Developments in our Understanding. Horm Metab Res (2015) 47:702–10. doi: 10.1055/s-0035-1548832
95. Erdogan M, Kulaksizoglu M, Ganidagli S, Berdeli A. Fas/FasL gene polymorphism in patients with Hashimoto’s thyroiditis in Turkish population. J Endocrinol Invest (2017) 40:77–82. doi: 10.1007/s40618-016-0534-5
96. Benetti-Pinto CL, Piccolo VRSB, Garmes HM, Juliato CRT. Subclinical hypothyroidism in young women with polycystic ovary syndrome: an analysis of clinical, hormonal, and metabolic parameters. Fertil Steril (2013) 99:588–92. doi: 10.1016/j.fertnstert.2012.10.006
97. Canaris GJ, Manowitz NR, Mayor G, Ridgway EC. The Colorado thyroid disease prevalence study. Arch Intern Med (2000) 160:526–34. doi: 10.1001/archinte.160.4.526
98. Radetti G. Clinical aspects of Hashimoto’s thyroiditis. Endocr Dev (2014) 26:158–70. doi: 10.1159/000363162
99. Krassas GE, Poppe K, Glinoer D. Thyroid function and human reproductive health. Endocr Rev (2010) 31:702–55. doi: 10.1210/er.2009-0041
100. Verma I, Sood R, Juneja S, Kaur S. Prevalence of hypothyroidism in infertile women and evaluation of response of treatment for hypothyroidism on infertility. Int J Appl Basic Med Res (2012) 2:17–9. doi: 10.4103/2229-516X.96795
101. Moleti M, Sturniolo G, Mauro MD, Russo M, Vermiglio F. Autoimmune thyroid diseases and pregnancy. Ann Thyroid (2018) 3:1–12. doi: 10.21037/aot.2018.07.03
102. Zhang Y, Wang H, Pan X, Teng W, Shan Z. Patients with subclinical hypothyroidism before 20 weeks of pregnancy have a higher risk of miscarriage: A systematic review and meta-analysis. PLoS One (2017) 12:e0175708. doi: 10.1371/journal.pone.0175708
103. Saran S, Gupta BS, Philip R, Singh KS, Bende SA, Agroiya P, et al. Effect of hypothyroidism on female reproductive hormones. Indian J Endocrinol Metab (2016) 20:108–13. doi: 10.4103/2230-8210.172245
104. Goel P, Kahkasha, Narang S, Gupta BK, Goel K. Evaluation of Serum Prolactin Level in Patients of Subclinical and Overt Hypothyroidism. J Clin Diagn Res (2015) 9:BC15–7. doi: 10.7860/JCDR/2015/9982.5443
105. Dharmshaktu P, Kutiyal A, Dhanwal D. Vanishing large ovarian cyst with thyroxine therapy. Endocrinol Diabetes Metab Case Rep (2013) 2013:130050. doi: 10.1530/EDM-13-0050
106. Muderris II, Boztosun A, Oner G, Bayram F. Effect of thyroid hormone replacement therapy on ovarian volume and androgen hormones in patients with untreated primary hypothyroidism. Ann Saudi Med (2011) 31:145–51. doi: 10.4103/0256-4947.77500
107. Maratou E, Hadjidakis DJ, Kollias A, Tsegka K, Peppa M, Alevizaki M, et al. Studies of insulin resistance in patients with clinical and subclinical hypothyroidism. Eur J Endocrinol (2009) 160:785–90. doi: 10.1530/EJE-08-0797
108. Asvold BO, Bjøro T, Nilsen TIL, Vatten LJ. Association between blood pressure and serum thyroid-stimulating hormone concentration within the reference range: a population-based study. J Clin Endocrinol Metab (2007) 92:841–5. doi: 10.1210/jc.2006-2208
109. Pearce EN. Update in lipid alterations in subclinical hypothyroidism. J Clin Endocrinol Metab (2012) 97:326–33. doi: 10.1210/jc.2011-2532
110. Duntas LH, Biondi B. The interconnections between obesity, thyroid function, and autoimmunity: the multifold role of leptin. Thyroid (2013) 23:646–53. doi: 10.1089/thy.2011.0499
111. Ong KK, Kuh D, Pierce M, Franklyn JA. Childhood Weight Gain and Thyroid Autoimmunity at Age 60–64 Years: The 1946 British Birth Cohort Study. J Clin Endocrinol Metab (2013) 98:1435–42. doi: 10.1210/jc.2012-3761
112. Hansen PS, Brix TH, Bennedbæk FN, Bonnema SJ, Iachine I, Kyvik KO, et al. The relative importance of genetic and environmental factors in the aetiology of thyroid nodularity: a study of healthy Danish twins. Clin Endocrinol (Oxf) (2005) 62:380–6. doi: 10.1111/j.1365-2265.2005.02230.x
113. Brix TH, Kyvik KO, Hegedüs L. A population-based study of chronic autoimmune hypothyroidism in Danish twins. J Clin Endocrinol Metab (2000) 85:536–9. doi: 10.1210/jcem.85.2.6385
114. Zaletel K, Gaberšček S. Hashimoto’s Thyroiditis: From Genes to the Disease. Curr Genomics (2011) 12:576–88. doi: 10.2174/138920211798120763
115. Xiao L, Muhali F-S, Cai T, Song R, Hu R, Shi X, et al. Association of single-nucleotide polymorphisms in the STAT3 gene with autoimmune thyroid disease in Chinese individuals. Funct Integr Genomics (2013) 13:455–61. doi: 10.1007/s10142-013-0337-0
116. Ban Y, Taniyama M, Ban Y. Vitamin D receptor gene polymorphisms in Hashimoto’s thyroiditis. Thyroid (2001) 11:607–8. doi: 10.1089/105072501750302967
117. Santos LR, Durães C, Mendes A, Prazeres H, Alvelos MI, Moreira CS, et al. A polymorphism in the promoter region of the selenoprotein S gene (SEPS1) contributes to Hashimoto’s thyroiditis susceptibility. J Clin Endocrinol Metab (2014) 99:E719–723. doi: 10.1210/jc.2013-3539
118. Li M, Sun H, Liu S, Yu J, Li Q, Liu P, et al. CD40 C/T-1 polymorphism plays different roles in Graves’ disease and Hashimoto’s thyroiditis: a meta-analysis. Endocr J (2012) 59:1041–50. doi: 10.1507/endocrj.ej12-0126
119. Zeitlin AA, Heward JM, Newby PR, Carr-Smith JD, Franklyn JA, Gough SCL, et al. Analysis of HLA class II genes in Hashimoto’s thyroiditis reveals differences compared to Graves’ disease. Genes Immun (2008) 9:358–63. doi: 10.1038/gene.2008.26
120. Fujii A, Inoue N, Watanabe M, Kawakami C, Hidaka Y, Hayashizaki Y, et al. TSHR Gene Polymorphisms in the Enhancer Regions Are Most Strongly Associated with the Development of Graves’ Disease, Especially Intractable Disease, and of Hashimoto’s Disease. Thyroid (2017) 27:111–9. doi: 10.1089/thy.2016.0345
121. Ueda H, Howson JMM, Esposito L, Heward J, Snook H, Chamberlain G, et al. Association of the T-cell regulatory gene CTLA4 with susceptibility to autoimmune disease. Nature (2003) 423:506–11. doi: 10.1038/nature01621
122. Furugaki K, Shirasawa S, Ishikawa N, Ito K, Ito K, Kubota S, et al. Association of the T-cell regulatory gene CTLA4 with Graves’ disease and autoimmune thyroid disease in the Japanese. J Hum Genet (2004) 49:166–8. doi: 10.1007/s10038-003-0120-5
123. Smyth D, Cooper JD, Collins JE, Heward JM, Franklyn JA, Howson JMM, et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes (2004) 53:3020–3. doi: 10.2337/diabetes.53.11.3020
124. Heward JM, Brand OJ, Barrett JC, Carr-Smith JD, Franklyn JA, Gough SC. Association of PTPN22 haplotypes with Graves’ disease. J Clin Endocrinol Metab (2007) 92:685–90. doi: 10.1210/jc.2006-2064
125. Brand OJ, Barrett JC, Simmonds MJ, Newby PR, McCabe CJ, Bruce CK, et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves’ disease. Hum Mol Genet (2009) 18:1704–13. doi: 10.1093/hmg/ddp087
126. Dechairo BM, Zabaneh D, Collins J, Brand O, Dawson GJ, Green AP, et al. Association of the TSHR gene with Graves’ disease: the first disease specific locus. Eur J Hum Genet (2005) 13:1223–30. doi: 10.1038/sj.ejhg.5201485
127. Gao X, Wang J, Yu Y. The Association Between STAT4 rs7574865 Polymorphism and the Susceptibility of Autoimmune Thyroid Disease: A Meta-Analysis. Front Genet (2018) 9:708. doi: 10.3389/fgene.2018.00708
128. Simmonds MJ. GWAS in autoimmune thyroid disease: redefining our understanding of pathogenesis. Nat Rev Endocrinol (2013) 9:277–87. doi: 10.1038/nrendo.2013.56
129. Wang X, Cheng W, Ma Y, Zhu J. Vitamin D receptor gene FokI but not TaqI, ApaI, BsmI polymorphism is associated with Hashimoto’s thyroiditis: a meta-analysis. Sci Rep (2017) 7:1–11. doi: 10.1038/srep41540
130. Wang D, Chen J, Zhang H, Zhang F, Yang L, Mou Y. Role of Different CD40 Polymorphisms in Graves’ Disease and Hashimoto’s Thyroiditis. Immunol Invest (2017) 46:544–51. doi: 10.1080/08820139.2017.1319382
131. Ban Y, Tozaki T, Taniyama M, Nakano Y, Ban Y, Ban Y, et al. Association of the protein tyrosine phosphatase nonreceptor 22 haplotypes with autoimmune thyroid disease in the Japanese population. Thyroid (2010) 20:893–9. doi: 10.1089/thy.2010.0104
132. Shen X, Yan X, Xie B, Xu D, Wang K, Zhu J, et al. Genetic variants of interleukin-4 gene in autoimmune thyroid diseases: an updated meta-analysis. Autoimmunity (2015) 48:129–35. doi: 10.3109/08916934.2014.962025
133. Eriksson N, Tung JY, Kiefer AK, Hinds DA, Francke U, Mountain JL, et al. Novel Associations for Hypothyroidism Include Known Autoimmune Risk Loci. PLoS One (2012) 7:e34442. doi: 10.1371/journal.pone.0034442
134. Cooper JD, Simmonds MJ, Walker NM, Burren O, Brand OJ, Guo H, et al. Seven newly identified loci for autoimmune thyroid disease. Hum Mol Genet (2012) 21:5202–8. doi: 10.1093/hmg/dds357
135. Pickrell JK, Berisa T, Liu JZ, Ségurel L, Tung JY, Hinds DA. Detection and interpretation of shared genetic influences on 42 human traits. Nat Genet (2016) 48:709–17. doi: 10.1038/ng.3570
136. Kichaev G, Bhatia G, Loh P-R, Gazal S, Burch K, Freund MK, et al. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am J Hum Genet (2019) 104:65–75. doi: 10.1016/j.ajhg.2018.11.008
137. Rydzewska M, Góralczyk A, Gościk J, Wawrusiewicz-Kurylonek N, Bossowska A, Krętowski A, et al. Analysis of chosen polymorphisms rs2476601 a/G - PTPN22, rs1990760 C/T - IFIH1, rs179247 a/G - TSHR in pathogenesis of autoimmune thyroid diseases in children. Autoimmunity (2018) 51:183–90. doi: 10.1080/08916934.2018.1486824
138. Jabrocka-Hybel A, Skalniak A, Piątkowski J, Turek-Jabrocka R, Vyhouskaya P, Ludwig-Słomczyńska A, et al. How much of the predisposition to Hashimoto’s thyroiditis can be explained based on previously reported associations? J Endocrinol Invest (2018) 41:1409–16. doi: 10.1007/s40618-018-0910-4
139. Medici M, Porcu E, Pistis G, Teumer A, Brown SJ, Jensen RA, et al. Identification of novel genetic Loci associated with thyroid peroxidase antibodies and clinical thyroid disease. PLoS Genet (2014) 10:e1004123. doi: 10.1371/journal.pgen.1004123
140. Oryoji D, Ueda S, Yamamoto K, Yoshimura Noh J, Okamura K, Noda M, et al. Identification of a Hashimoto thyroiditis susceptibility locus via a genome-wide comparison with Graves’ disease. J Clin Endocrinol Metab (2015) 100:E319–324. doi: 10.1210/jc.2014-3431
141. Kwak SH, Park YJ, Go MJ, Lee KE, Kim S, Choi HS, et al. A genome-wide association study on thyroid function and anti-thyroid peroxidase antibodies in Koreans. Hum Mol Genet (2014) 23:4433–42. doi: 10.1093/hmg/ddu145
142. Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Masson G, He H, et al. Discovery of common variants associated with low TSH levels and thyroid cancer risk. Nat Genet (2012) 44:319–22. doi: 10.1038/ng.1046
143. Porcu E, Medici M, Pistis G, Volpato CB, Wilson SG, Cappola AR, et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. PLoS Genet (2013) 9:e1003266. doi: 10.1371/journal.pgen.1003266
144. Panicker V, Wilson SG, Walsh JP, Richards JB, Brown SJ, Beilby JP, et al. A locus on chromosome 1p36 is associated with thyrotropin and thyroid function as identified by genome-wide association study. Am J Hum Genet (2010) 87:430–5. doi: 10.1016/j.ajhg.2010.08.005
145. Zhan M, Chen G, Pan C-M, Gu Z-H, Zhao S-X, Liu W, et al. Genome-wide association study identifies a novel susceptibility gene for serum TSH levels in Chinese populations. Hum Mol Genet (2014) 23:5505–17. doi: 10.1093/hmg/ddu250
146. Brčić L, Barić A, Gračan S, Torlak V, Brekalo M, Škrabić V, et al. Genome-wide association analysis suggests novel loci underlying thyroid antibodies in Hashimoto’s thyroiditis. Sci Rep (2019) 9:1–10. doi: 10.1038/s41598-019-41850-6
147. Brčić L, Barić A, Gračan S, Brdar D, Torlak Lovrić V, Vidan N, et al. Association of established thyroid peroxidase autoantibody (TPOAb) genetic variants with Hashimoto’s thyroiditis. Autoimmunity (2016) 49:480–5. doi: 10.1080/08916934.2016.1191475
148. Matana A, Popović M, Boutin T, Torlak V, Brdar D, Gunjača I, et al. Genome-wide meta-analysis identifies novel gender specific loci associated with thyroid antibodies level in Croatians. Genomics (2019) 111:737–43. doi: 10.1016/j.ygeno.2018.04.012
149. Pastuszak-Lewandoska D, Sewerynek E, Domańska D, Gładyś A, Skrzypczak R, Brzeziańska E. CTLA-4 gene polymorphisms and their influence on predisposition to autoimmune thyroid diseases (Graves’ disease and Hashimoto’s thyroiditis). Arch Med Sci (2012) 8:415–21. doi: 10.5114/aoms.2012.28593
150. Ramgopal S, Rathika C, Padma MR, Murali V, Arun K, Kamaludeen MN, et al. Interaction of HLA-DRB1* alleles and CTLA4 (+49 AG) gene polymorphism in Autoimmune Thyroid Disease. Gene (2018) 642:430–8. doi: 10.1016/j.gene.2017.11.057
151. Ban Y, Tozaki T, Taniyama M, Tomita M, Ban Y. Association of a CTLA-4 3’ untranslated region (CT60) single nucleotide polymorphism with autoimmune thyroid disease in the Japanese population. Autoimmunity (2005) 38:151–3. doi: 10.1080/08916930500050319
152. Ting W-H, Chien M-N, Lo F-S, Wang C-H, Huang C-Y, Lin C-L, et al. Association of Cytotoxic T-Lymphocyte-Associated Protein 4 (CTLA4) Gene Polymorphisms with Autoimmune Thyroid Disease in Children and Adults: Case-Control Study. PLoS One (2016) 11:e0154394. doi: 10.1371/journal.pone.0154394
153. Brčić L, Gračan S, Barić A, Gunjača I, Torlak Lovrić V, Kolčić I, et al. Association of Established Thyroid-stimulating Hormone and Free Thyroxine Genetic Variants with Hashimoto’s Thyroiditis. Immunol Invest (2017) 46:625–38. doi: 10.1080/08820139.2017.1337785
154. Tomer Y, Dolan LM, Kahaly G, Divers J, D’Agostino RB, Imperatore G, et al. Genome wide identification of new genes and pathways in patients with both autoimmune thyroiditis and type 1 diabetes. J Autoimmun (2015) 60:32–9. doi: 10.1016/j.jaut.2015.03.006
155. Arnaud-Lopez L, Usala G, Ceresini G, Mitchell BD, Pilia MG, Piras MG, et al. Phosphodiesterase 8B Gene Variants Are Associated with Serum TSH Levels and Thyroid Function. Am J Hum Genet (2008) 82:1270–80. doi: 10.1016/j.ajhg.2008.04.019
156. Barić A, Brčić L, Gračan S, Torlak Lovrić V, Gunjača I, Šimunac M, et al. Association of established hypothyroidism-associated genetic variants with Hashimoto’s thyroiditis. J Endocrinol Invest (2017) 40:1061–7. doi: 10.1007/s40618-017-0660-8
157. Brčić L, Barić A, Gračan S, Brekalo M, Kaličanin D, Gunjača I, et al. Genome-wide association analysis suggests novel loci for Hashimoto’s thyroiditis. J Endocrinol Invest (2019) 42:567–76. doi: 10.1007/s40618-018-0955-4
158. Denny JC, Crawford DC, Ritchie MD, Bielinski SJ, Basford MA, Bradford Y, et al. Variants near FOXE1 are associated with hypothyroidism and other thyroid conditions: using electronic medical records for genome- and phenome-wide studies. Am J Hum Genet (2011) 89:529–42. doi: 10.1016/j.ajhg.2011.09.008
159. Wang B, Jia X, Yao Q, Li Q, He W, Li L, et al. CEP128 is a crucial risk locus for autoimmune thyroid diseases. Mol Cell Endocrinol (2019) 480:97–106. doi: 10.1016/j.mce.2018.10.017
160. Matana A, Boutin T, Torlak V, Brdar D, Gunjača I, Kolčić I, et al. Genome-Wide Analysis Identifies Two Susceptibility Loci for Positive Thyroid Peroxidase and Thyroglobulin Antibodies. J Clin Endocrinol Metab (2020) 105:dgz239. doi: 10.1210/clinem/dgz239
161. Hwangbo Y, Park YJ. Genome-Wide Association Studies of Autoimmune Thyroid Diseases, Thyroid Function, and Thyroid Cancer. Endocrinol Metab (2018) 33:175–84. doi: 10.3803/EnM.2018.33.2.175
162. Gudmundsson J, Sulem P, Gudbjartsson DF, Jonasson JG, Sigurdsson A, Bergthorsson JT, et al. Common variants on 9q22.33 and 14q13.3 predispose to thyroid cancer in European populations. Nat Genet (2009) 41:460–4. doi: 10.1038/ng.339
163. Castanet M, Polak M. Spectrum of Human Foxe1/TTF2 Mutations. Horm Res Paediatr (2010) 73:423–9. doi: 10.1159/000281438
164. Fagman H, Nilsson M. Morphogenetics of early thyroid development. J Mol Endocrinol (2011) 46:R33–42. doi: 10.1677/jme-10-0084
165. Ortiz L, Aza-Blanc P, Zannini M, Cato AC, Santisteban P. The interaction between the forkhead thyroid transcription factor TTF-2 and the constitutive factor CTF/NF-1 is required for efficient hormonal regulation of the thyroperoxidase gene transcription. J Biol Chem (1999) 274:15213–21. doi: 10.1074/jbc.274.21.15213
166. Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev (2006) 58:488–520. doi: 10.1124/pr.58.3.5
167. Dumont C, Corsoni-Tadrzak A, Ruf S, de Boer J, Williams A, Turner M, et al. Rac GTPases play critical roles in early T-cell development. Blood (2009) 113:3990–8. doi: 10.1182/blood-2008-09-181180
168. Malhotra S, Kovats S, Zhang W, Coggeshall KM. B Cell Antigen Receptor Endocytosis and Antigen Presentation to T Cells Require Vav and Dynamin. J Biol Chem (2009) 284:24088–97. doi: 10.1074/jbc.M109.014209
169. Sasazuki T, Inoko H, Morishima S, Morishima Y. Gene Map of the HLA Region, Graves’ Disease and Hashimoto Thyroiditis, and Hematopoietic Stem Cell Transplantation. Adv Immunol (2016) 129:175–249. doi: 10.1016/bs.ai.2015.08.003
170. Ueda S, Oryoji D, Yamamoto K, Noh JY, Okamura K, Noda M, et al. Identification of independent susceptible and protective HLA alleles in Japanese autoimmune thyroid disease and their epistasis. J Clin Endocrinol Metab (2014) 99:E379–383. doi: 10.1210/jc.2013-2841
171. Bernecker C, Ostapczuk M, Vordenbäumen S, Ehlers M, Thiel A, Schinner S, et al. HLA-A2 phenotype may be protective against Graves’ disease but not against Hashimoto’s thyroiditis in Caucasians. Horm Metab Res (2013) 45:74–7. doi: 10.1055/s-0032-1323704
172. Tomer Y, Davies TF. Searching for the autoimmune thyroid disease susceptibility genes: from gene mapping to gene function. Endocr Rev (2003) 24:694–717. doi: 10.1210/er.2002-0030
173. Tendon N, Zhang L, Weetman AP. HLA Associations with Hashimoto’s thyroiditis. Clin Endocrinol (Oxf) (1991) 34:383–6. doi: 10.1111/j.1365-2265.1991.tb00309.x
174. Shi Y, Zou M, Robb D, Farid NR. Typing for major histocompatibility complex class II antigens in thyroid tissue blocks: association of Hashimoto’s thyroiditis with HLA-DQA0301 and DQB0201 alleles. J Clin Endocrinol Metab (1992) 75:943–6. doi: 10.1210/jcem.75.3.1517390
175. Hunt PJ, Marshall SE, Weetman AP, Bunce M, Bell JI, Wass JA, et al. Histocompatibility leucocyte antigens and closely linked immunomodulatory genes in autoimmune thyroid disease. Clin Endocrinol (Oxf) (2001) 55:491–9. doi: 10.1046/j.1365-2265.2001.01356.x
176. Petrone A, Giorgi G, Mesturino CA, Capizzi M, Cascino I, Nistico L, et al. Association of DRB1*04-DQB1*0301 haplotype and lack of association of two polymorphic sites at CTLA-4 gene with Hashimoto’s thyroiditis in an Italian population. Thyroid (2001) 11:171–5. doi: 10.1089/105072501300042901
177. Kokaraki G, Daniilidis M, Yiangou M, Arsenakis M, Karyotis N, Tsilipakou M, et al. Major histocompatibility complex class II (DRB1*, DQA1*, and DQB1*) and DRB1*04 subtypes’ associations of Hashimoto’s thyroiditis in a Greek population. Tissue Antigens (2009) 73:199–205. doi: 10.1111/j.1399-0039.2008.01182.x
178. Cho WK, Jung MH, Choi E-J, Choi H-B, Kim T-G, Suh B-K. Association of HLA Alleles with Autoimmune Thyroid Disease in Korean Children. Horm Res Paediatr (2011) 76:328–34. doi: 10.1159/000331134
179. Patiroğlu T, Akar HH, Akay A, Ok E. The distribution of HLA-DRB1 alleles in patients with hashimoto’s thyroiditis -. Şişli Etfal Tıp Bül (2015) 49:255–9. doi: 10.5350/SEMB.20151005010345
180. Ban Y, Davies TF, Greenberg DA, Concepcion ES, Osman R, Oashi T, et al. Arginine at position 74 of the HLA-DR beta1 chain is associated with Graves’ disease. Genes Immun (2004) 5:203–8. doi: 10.1038/sj.gene.6364059
181. Menconi F, Monti MC, Greenberg DA, Oashi T, Osman R, Davies TF, et al. Molecular amino acid signatures in the MHC class II peptide-binding pocket predispose to autoimmune thyroiditis in humans and in mice. Proc Natl Acad Sci U S A (2008) 105:14034–9. doi: 10.1073/pnas.0806584105
182. Hodge SE, Ban Y, Strug LJ, Greenberg DA, Davies TF, Concepcion ES, et al. Possible Interaction Between HLA-DRβ1 and Thyroglobulin Variants in Graves’ Disease. Thyroid (2006) 16:351–5. doi: 10.1089/thy.2006.16.351
183. Ni J, Qiu L-J, Zhang M, Wen P-F, Ye X-R, Liang Y, et al. CTLA-4 CT60 (rs3087243) polymorphism and autoimmune thyroid diseases susceptibility: a comprehensive meta-analysis. Endocr Res (2014) 39:180–8. doi: 10.3109/07435800.2013.879167
184. Hou H-F, Jin X, Sun T, Li C, Jiang B-F, Li Q-W. Cytotoxic T Lymphocyte-Associated Antigen 4 Gene Polymorphisms and Autoimmune Thyroid Diseases: An Updated Systematic Review and Cumulative Meta-Analysis. Int J Endocrinol (2015) 2015:747816. doi: 10.1155/2015/747816
185. Ji R, Feng Y, Zhan WW. Updated analysis of studies on the cytotoxic T-lymphocyte-associated antigen-4 gene A49G polymorphism and Hashimoto’s thyroiditis risk. Genet Mol Res (2013) 12:1421–30. doi: 10.4238/2013.April.26.4
186. Hu Y, Xu K, Jiang L, Zhang L, Shi H, Cui D. Associations Between Three CTLA-4 Polymorphisms and Hashimoto’s Thyroiditis Risk: An Updated Meta-Analysis with Trial Sequential Analysis. Genet Test Mol Biomarkers (2018) 22:224–36. doi: 10.1089/gtmb.2017.0243
187. Qiu H, Tang W, Yin P, Cheng F, Wang L. Cytotoxic T-lymphocyte associated antigen 4 polymorphism and Hashimoto’s thyroiditis susceptibility: a meta-analysis. Endocrine (2014) 45:198–205. doi: 10.1007/s12020-013-9985-z
188. Vieland VJ, Huang Y, Bartlett C, Davies TF, Tomer Y. A multilocus model of the genetic architecture of autoimmune thyroid disorder, with clinical implications. Am J Hum Genet (2008) 82:1349–56. doi: 10.1016/j.ajhg.2008.04.017
189. Einarsdottir E, Söderström I, Löfgren-Burström A, Haraldsson S, Nilsson-Ardnor S, Penha-Goncalves C, et al. The CTLA4 region as a general autoimmunity factor: an extended pedigree provides evidence for synergy with the HLA locus in the etiology of type 1 diabetes mellitus, Hashimoto’s thyroiditis and Graves’ disease. Eur J Hum Genet EJHG (2003) 11:81–4. doi: 10.1038/sj.ejhg.5200903
190. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol (2006) 24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535
191. Xiaoheng C, Yizhou M, Bei H, Huilong L, Xin W, Rui H, et al. General and Specific Genetic Polymorphism of Cytokines-Related Gene in AITD. Mediators Inflamm (2017) 2017:e3916395. doi: 10.1155/2017/3916395
192. Ban Y, Greenberg DA, Davies TF, Jacobson E, Concepcion E, Tomer Y. ‘Linkage Analysis of Thyroid Antibody Production: Evidence for Shared Susceptibility to Clinical Autoimmune Thyroid Disease. J Clin Endocrinol Metab (2008) 93:3589–96. doi: 10.1210/jc.2008-0364
193. Cloutier JF, Veillette A. Cooperative inhibition of T-cell antigen receptor signaling by a complex between a kinase and a phosphatase. J Exp Med (1999) 189:111–21. doi: 10.1084/jem.189.1.111
194. Hill RJ, Zozulya S, Lu Y-L, Ward K, Gishizky M, Jallal B. The lymphoid protein tyrosine phosphatase Lyp interacts with the adaptor molecule Grb2 and functions as a negative regulator of T-cell activation. Exp Hematol (2002) 30:237–44. doi: 10.1016/S0301-472X(01)00794-9
195. Burn GL, Svensson L, Sanchez-Blanco C, Saini M, Cope AP. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett (2011) 585:3689–98. doi: 10.1016/j.febslet.2011.04.032
196. Velaga MR, Wilson V, Jennings CE, Owen CJ, Herington S, Donaldson PT, et al. The codon 620 tryptophan allele of the lymphoid tyrosine phosphatase (LYP) gene is a major determinant of Graves’ disease. J Clin Endocrinol Metab (2004) 89:5862–5. doi: 10.1210/jc.2004-1108
197. Criswell LA, Pfeiffer KA, Lum RF, Gonzales B, Novitzke J, Kern M, et al. Analysis of families in the multiple autoimmune disease genetics consortium (MADGC) collection: the PTPN22 620W allele associates with multiple autoimmune phenotypes. Am J Hum Genet (2005) 76:561–71. doi: 10.1086/429096
198. Ban Y, Tozaki T, Taniyama M, Tomita M, Ban Y. The codon 620 single nucleotide polymorphism of the protein tyrosine phosphatase-22 gene does not contribute to autoimmune thyroid disease susceptibility in the Japanese. Thyroid (2005) 15:1115–8. doi: 10.1089/thy.2005.15.1115
199. Chabchoub G, Teixiera EP, Maalej A, Ben Hamad M, Bahloul Z, Cornelis F, et al. The R620W polymorphism of the protein tyrosine phosphatase 22 gene in autoimmune thyroid diseases and rheumatoid arthritis in the Tunisian population. Ann Hum Biol (2009) 36:342–9. doi: 10.1080/03014460902817968
200. Luo L, Cai B, Liu F, Hu X, Wang L. Association of Protein Tyrosine Phosphatase Nonreceptor 22 (PTPN22) C1858T gene polymorphism with susceptibility to autoimmune thyroid diseases: a meta-analysis. Endocr J (2012) 59:439–45. doi: 10.1507/endocrj.ej11-0381
201. Alkhateeb A, Marzouka NA-D, Tashtoush R. Variants in PTPN22 and SMOC2 genes and the risk of thyroid disease in the Jordanian Arab population. Endocrine (2013) 44:702–9. doi: 10.1007/s12020-013-9908-z
202. Mori M, Yamada R, Kobayashi K, Kawaida R, Yamamoto K. Ethnic differences in allele frequency of autoimmune-disease-associated SNPs. J Hum Genet (2005) 50:264–6. doi: 10.1007/s10038-005-0246-8
203. Reddy MVPL, Johansson M, Sturfelt G, Jönsen A, Gunnarsson I, Svenungsson E, et al. The R620W C/T polymorphism of the gene PTPN22 is associated with SLE independently of the association of PDCD1. Genes Immun (2005) 6:658–62. doi: 10.1038/sj.gene.6364252
204. Gong L, Liu B, Wang J, Pan H, Qi A, Zhang S, et al. Novel missense mutation in PTPN22 in a Chinese pedigree with Hashimoto’s thyroiditis. BMC Endocr Disord (2018) 18:1–6. doi: 10.1186/s12902-018-0305-8
205. Bottini N, Musumeci L, Alonso A, Rahmouni S, Nika K, Rostamkhani M, et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet (2004) 36:337–8. doi: 10.1038/ng1323
206. Gianchecchi E, Palombi M, Fierabracci A. The putative role of the C1858T polymorphism of protein tyrosine phosphatase PTPN22 gene in autoimmunity. Autoimmun Rev (2013) 12:717–25. doi: 10.1016/j.autrev.2012.12.003
207. Ferreira RC, Castro Dopico X, Oliveira JJ, Rainbow DB, Yang JH, Trzupek D, et al. Chronic Immune Activation in Systemic Lupus Erythematosus and the Autoimmune PTPN22 Trp620 Risk Allele Drive the Expansion of FOXP3+ Regulatory T Cells and PD-1 Expression. Front Immunol (2019) 10:2606. doi: 10.3389/fimmu.2019.02606
208. Rawlings DJ, Dai X, Buckner JH. The role of PTPN22 risk variant in the development of autoimmunity: Finding common ground between mouse and man. J Immunol Baltim Md 1950 (2015) 194:2977–84. doi: 10.4049/jimmunol.1403034
209. Weetman AP. The Immunopathogenesis of Chronic Autoimmune Thyroiditis One Century after Hashimoto. Eur Thyroid J (2013) 1:243–50. doi: 10.1159/000343834
210. Janssen OE, Mehlmauer N, Hahn S, Offner AH, Gartner R. High prevalence of autoimmune thyroiditis in patients with polycystic ovary syndrome. Eur J Endocrinol (2004) 150:363–9. doi: 10.1530/eje.0.1500363
211. Anaforoglu I, Topbas M, Algun E. Relative associations of polycystic ovarian syndrome vs metabolic syndrome with thyroid function, volume, nodularity and autoimmunity. J Endocrinol Invest (2011) 34:e259–264. doi: 10.3275/7681
212. Kachuei M, Jafari F, Kachuei A, Keshteli AH. Prevalence of autoimmune thyroiditis in patients with polycystic ovary syndrome. Arch Gynecol Obstet (2012) 285:853–6. doi: 10.1007/s00404-011-2040-5
213. Sinha U, Sinharay K, Saha S, Longkumer TA, Baul SN, Pal SK. Thyroid disorders in polycystic ovarian syndrome subjects: A tertiary hospital based cross-sectional study from Eastern India. Indian J Endocrinol Metab (2013) 17:304. doi: 10.4103/2230-8210.109714
214. Garelli S, Masiero S, Plebani M, Chen S, Furmaniak J, Armanini D, et al. High prevalence of chronic thyroiditis in patients with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol (2013) 169:248–51. doi: 10.1016/j.ejogrb.2013.03.003
215. Al-Saab R, Haddad S. Detection of Thyroid Autoimmunity Markers in Euthyroid Women With Polycystic Ovary Syndrome: A Case-Control Study From Syria. Int J Endocrinol Metab (2014) 12:e17954. doi: 10.5812/ijem.17954
216. Duran C, Basaran M, Kutlu O, Kucukaydin Z, Bakdik S, Burnik FS, et al. Frequency of nodular goiter and autoimmune thyroid disease in patients with polycystic ovary syndrome. Endocrine (2015) 49:464–9. doi: 10.1007/s12020-014-0504-7
217. Petrikova J, Lazurova I, Dravecka I, Vrbikova J, Kozakova D, Figurova J, et al. The prevalence of non organ specific and thyroid autoimmunity in patients with polycystic ovary syndrome. BioMed Pap Med Fac Univ Palacky Olomouc Czech Repub (2015) 159:302–6. doi: 10.5507/bp.2014.062
218. Calvar CE, Bengolea SV, Deutsch SI, Hermes R, Ramos G, Loyato M. [High frequency of thyroid abnormalities in polycystic ovary syndrome]. Medicina (Mex) (2015) 75:213–7.
219. Arduc A, Aycicek Dogan B, Bilmez S, Imga Nasiroglu N, Tuna MM, Isik S, et al. High prevalence of Hashimoto’s thyroiditis in patients with polycystic ovary syndrome: does the imbalance between estradiol and progesterone play a role? Endocr Res (2015) 40:204–10. doi: 10.3109/07435800.2015.1015730
220. Yu Q, Wang J-B. Subclinical Hypothyroidism in PCOS: Impact on Presentation, Insulin Resistance, and Cardiovascular Risk. BioMed Res Int (2016) 2016:2067087. doi: 10.1155/2016/2067087
221. Arora S, Sinha K, Kolte S, Mandal A. Endocrinal and autoimmune linkage: Evidences from a controlled study of subjects with polycystic ovarian syndrome. J Hum Reprod Sci (2016) 9:18–22. doi: 10.4103/0974-1208.178636
222. Menon M, Ramachandran V. Antithyroid Peroxidase Antibodies in Women with Polycystic Ovary Syndrome. J Obstet Gynaecol India (2017) 67:61–5. doi: 10.1007/s13224-016-0914-y
223. Karaköse M, Hepsen S, Çakal E, Saykı Arslan M, Tutal E, Akın Ş, et al. Frequency of nodular goiter and autoimmune thyroid disease and association of these disorders with insulin resistance in polycystic ovary syndrome. J Turk Ger Gynecol Assoc (2017) 18:85–9. doi: 10.4274/jtgga.2016.0217
224. Yin D, Ruan X, Tian X, Du J, Zhao Y, Cui Y, et al. The relationship between thyroid function and metabolic changes in Chinese women with polycystic ovary syndrome. Gynecol Endocrinol (2017) 33:332–5. doi: 10.1080/09513590.2016.1273895
225. Hepşen S, Karaköse M, Çakal E, Öztekin S, Ünsal İ, Akhanlı P, et al. The assessment of thyroid autoantibody levels in euthyroid patients with polycystic ovary syndrome. J Turk Ger Gynecol Assoc (2018) 19:215–9. doi: 10.4274/jtgga.2018.0001
226. Ulrich J, Goerges J, Keck C, Müller-Wieland D, Diederich S, Janssen OE. Impact of Autoimmune Thyroiditis on Reproductive and Metabolic Parameters in Patients with Polycystic Ovary Syndrome. Exp Clin Endocrinol Diabetes (2018) 126:198–204. doi: 10.1055/s-0043-110480
227. Enzevaei A, Salehpour S, Tohidi M, Saharkhiz N. Subclinical hypothyroidism and insulin resistance in polycystic ovary syndrome: is there a relationship? Iran J Reprod Med (2014) 12:481–6.
228. Bedaiwy MA, Abdel-Rahman MY, Tan J, AbdelHafez FF, Abdelkareem AO, Henry D, et al. Clinical, Hormonal, and Metabolic Parameters in Women with Subclinical Hypothyroidism and Polycystic Ovary Syndrome: A Cross-Sectional Study. J Womens Health (2018) 27:659–64. doi: 10.1089/jwh.2017.6584
229. Ding X, Yang L, Wang J, Tang R, Chen Q, Pan J, et al. Subclinical Hypothyroidism in Polycystic Ovary Syndrome: A Systematic Review and Meta-Analysis. Front Endocrinol (2018) 9:700. doi: 10.3389/fendo.2018.00700
230. Romitti M, Fabris VC, Ziegelmann PK, Maia AL, Spritzer PM. Association between PCOS and autoimmune thyroid disease: a systematic review and meta-analysis. Endocr Connect (2018) 7:1158–67. doi: 10.1530/EC-18-0309
231. Hefler-Frischmuth K, Walch K, Huebl W, Baumuehlner K, Tempfer C, Hefler L. Serologic markers of autoimmunity in women with polycystic ovary syndrome. Fertil Steril (2010) 93:2291–4. doi: 10.1016/j.fertnstert.2009.01.056
232. Makled AK, Fathi HM, Gomaa MF, Bakr RM. Serologic markers of autoimmunity in women with polycystic ovary syndrome. Middle East Fertil Soc J (2015) 20:86–90. doi: 10.1016/j.mefs.2014.05.006
233. Kowalczyk K, Franik G, Kowalczyk D, Pluta D, Blukacz Ł, Madej P. Thyroid disorders in polycystic ovary syndrome. Eur Rev Med Pharmacol Sci (2017) 21:346–60.
234. de Medeiros SF, de Medeiros MAS, Ormond CM, Barbosa JS, Yamamoto MMW. Subclinical Hypothyroidism Impact on the Characteristics of Patients with Polycystic Ovary Syndrome. A Meta-Analysis of Observational Studies. Gynecol Obstet Invest (2018) 83:105–15. doi: 10.1159/000485619
235. Zhang B, Wang J, Shen S, Liu J, Sun J, Gu T, et al. Subclinical hypothyroidism is not a risk factor for polycystic ovary syndrome in obese women of reproductive age. Gynecol Endocrinol (2018) 34:875–9. doi: 10.1080/09513590.2018.1462319
236. Muscogiuri G, Sorice GP, Mezza T, Prioletta A, Lassandro AP, Pirronti T, et al. High-normal TSH values in obesity: is it insulin resistance or adipose tissue’s guilt? Obesity Silver Spring Md (2013) 21:101–6. doi: 10.1002/oby.20240
237. Tagliaferri V, Romualdi D, Guido M, Mancini A, De Cicco S, Di Florio C, et al. The link between metabolic features and TSH levels in polycystic ovary syndrome is modulated by the body weight: an euglycaemic-hyperinsulinaemic clamp study. Eur J Endocrinol (2016) 175:433–41. doi: 10.1530/EJE-16-0358
238. Chapman JC, Min SH, Freeh SM, Michael SD. The estrogen-injected female mouse: new insight into the etiology of PCOS. Reprod Biol Endocrinol (2009) 7:47. doi: 10.1186/1477-7827-7-47
239. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198:1875–86. doi: 10.1084/jem.20030152
240. Akinci B, Comlekci A, Yener S, Bayraktar F, Demir T, Ozcan MA, et al. Hashimoto’s thyroiditis, but not treatment of hypothyroidism, is associated with altered TGF-beta1 levels. Arch Med Res (2008) 39:397–401. doi: 10.1016/j.arcmed.2007.12.001
241. Govinden R, Bhoola KD. Genealogy, expression, and cellular function of transforming growth factor-β. Pharmacol Ther (2003) 98:257–65. doi: 10.1016/S0163-7258(03)00035-4
242. Raja-Khan N, Urbanek M, Rodgers RJ, Legro RS. The role of TGF-β in polycystic ovary syndrome. Reprod Sci Thousand Oaks Calif (2014) 21:20–31. doi: 10.1177/1933719113485294
243. Quintero OL, Amador-Patarroyo MJ, Montoya-Ortiz G, Rojas-Villarraga A, Anaya J-M. Autoimmune disease and gender: plausible mechanisms for the female predominance of autoimmunity. J Autoimmun (2012) 38:J109–119. doi: 10.1016/j.jaut.2011.10.003
244. Pennell LM, Galligan CL, Fish EN. Sex affects immunity. J Autoimmun (2012) 38:J282–291. doi: 10.1016/j.jaut.2011.11.013
245. Moulton VR. Sex Hormones in Acquired Immunity and Autoimmune Disease. Front Immunol (2018) 9:1–21. doi: 10.3389/fimmu.2018.02279
246. Angstwurm MW, Gärtner R, Ziegler-Heitbrock HW. Cyclic plasma IL-6 levels during normal menstrual cycle. Cytokine (1997) 9:370–4. doi: 10.1006/cyto.1996.0178
247. Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Plasticity of CD4(+) FoxP3(+) T cells. Curr Opin Immunol (2009) 21:281–5. doi: 10.1016/j.coi.2009.05.007
248. Hughes GC. Progesterone and autoimmune disease. Autoimmun Rev (2012) 11:A502–514. doi: 10.1016/j.autrev.2011.12.003
249. Gubbels Bupp MR, Jorgensen TN. Androgen-Induced Immunosuppression. Front Immunol (2018) 9:794. doi: 10.3389/fimmu.2018.00794
250. Petríková J, Lazúrová I, Yehuda S. Polycystic ovary syndrome and autoimmunity. Eur J Intern Med (2010) 21:369–71. doi: 10.1016/j.ejim.2010.06.008
251. Escobar-Morreale HF, Luque-Ramírez M, González F. Circulating Inflammatory Markers in Polycystic Ovary Syndrome: A Systematic Review and Meta-Analysis. Fertil Steril (2011) 95:1048–58.e1-2. doi: 10.1016/j.fertnstert.2010.11.036
252. Cai J, Zhang Y, Wang Y, Li S, Wang L, Zheng J, et al. High Thyroid Stimulating Hormone Level Is Associated With Hyperandrogenism in Euthyroid Polycystic Ovary Syndrome (PCOS) Women, Independent of Age, BMI, and Thyroid Autoimmunity: A Cross-Sectional Analysis. Front Endocrinol (2019) 10:222. doi: 10.3389/fendo.2019.00222
253. Muscogiuri G, Palomba S, Caggiano M, Tafuri D, Colao A, Orio F. Low 25 (OH) vitamin D levels are associated with autoimmune thyroid disease in polycystic ovary syndrome. Endocrine (2016) 53:538–42. doi: 10.1007/s12020-015-0745-0
254. Tamer G, Arik S, Tamer I, Coksert D. Relative vitamin D insufficiency in Hashimoto’s thyroiditis. Thyroid (2011) 21:891–6. doi: 10.1089/thy.2009.0200
255. Skaaby T, Husemoen LLN, Thuesen BH, Linneberg A. Prospective population-based study of the association between vitamin D status and incidence of autoimmune disease. Endocrine (2015) 50:231–8. doi: 10.1007/s12020-015-0547-4
256. Kim D. Low vitamin D status is associated with hypothyroid Hashimoto’s thyroiditis. Hormomes Athens Greece (2016) 15:385–93. doi: 10.14310/horm.2002.1681
257. Nandi A, Sinha N, Ong E, Sonmez H, Poretsky L. Is there a role for vitamin D in human reproduction? Horm Mol Biol Clin Investig (2016) 25:15–28. doi: 10.1515/hmbci-2015-0051
258. Kim C-Y, Lee YJ, Choi J-H, Lee SY, Lee HY, Jeong DH, et al. The Association between Low Vitamin D Status and Autoimmune Thyroid Disease in Korean Premenopausal Women: The 6th Korea National Health and Nutrition Examination Survey, 2013-2014. Korean J Fam Med (2019) 40:323–8. doi: 10.4082/kjfm.18.0075
259. Lopez ER, Zwermann O, Segni M, Meyer G, Reincke M, Seissler J, et al. A promoter polymorphism of the CYP27B1 gene is associated with Addison’s disease, Hashimoto’s thyroiditis, Graves’ disease and type 1 diabetes mellitus in Germans. Eur J Endocrinol (2004) 151:193–7. doi: 10.1530/eje.0.1510193
260. De Pergola G, Triggiani V, Bartolomeo N, Giagulli VA, Anelli M, Masiello M, et al. Low 25 Hydroxyvitamin D Levels are Independently Associated with Autoimmune Thyroiditis in a Cohort of Apparently Healthy Overweight and Obese Subjects. Endocr Metab Immune Disord Drug Targets (2018) 18:646–52. doi: 10.2174/1871530318666180406163426
261. Tsuchiya Y, Nakajima M, Kyo S, Kanaya T, Inoue M, Yokoi T. Human CYP1B1 is regulated by estradiol via estrogen receptor. Cancer Res (2004) 64:3119–25. doi: 10.1158/0008-5472.can-04-0166
262. Jansen E, Laven JSE, Dommerholt HBR, Polman J, van Rijt C, van den Hurk C, et al. Abnormal gene expression profiles in human ovaries from polycystic ovary syndrome patients. Mol Endocrinol Baltim Md (2004) 18:3050–63. doi: 10.1210/me.2004-0074
263. Zou S, Sang Q, Wang H, Feng R, Li Q, Zhao X, et al. Common genetic variation in CYP1B1 is associated with concentrations of T4, FT4 and FT4 in the sera of polycystic ovary syndrome patients. Mol Biol Rep (2013) 40:3315–20. doi: 10.1007/s11033-012-2406-1
264. Marshall JC, Eagleson CA. Neuroendocrine aspects of polycystic ovary syndrome. Endocrinol Metab Clin North Am (1999) 28:295–324. doi: 10.1016/s0889-8529(05)70071-2
265. Okada R, Kobayashi T, Yamamoto K, Nakakura T, Tanaka S, Vaudry H, et al. Neuroendocrine regulation of thyroid-stimulating hormone secretion in amphibians. Ann N Y Acad Sci (2009) 1163:262–70. doi: 10.1111/j.1749-6632.2008.03662.x
266. Li Q, Yang G, Wang Y, Zhang X, Sang Q, Wang H, et al. Common genetic variation in the 3’-untranslated region of gonadotropin-releasing hormone receptor regulates gene expression in cella and is associated with thyroid function, insulin secretion as well as insulin sensitivity in polycystic ovary syndrome patients. Hum Genet (2011) 129:553–61. doi: 10.1007/s00439-011-0954-4
267. Michalaki MA, Vagenakis AG, Leonardou AS, Argentou MN, Habeos IG, Makri MG, et al. Thyroid function in humans with morbid obesity. Thyroid (2006) 16:73–8. doi: 10.1089/thy.2006.16.73
268. Goodarzi MO, Louwers YV, Taylor KD, Jones MR, Cui J, Kwon S, et al. Replication of association of a novel insulin receptor gene polymorphism with polycystic ovary syndrome. Fertil Steril (2011) 95:1736–41.e1-11. doi: 10.1016/j.fertnstert.2011.01.015
269. Song DK, Hong YS, Sung Y-A, Lee H. Insulin resistance according to β-cell function in women with polycystic ovary syndrome and normal glucose tolerance. PLoS One (2017) 12:e0178120. doi: 10.1371/journal.pone.0178120
270. Behre HM, Greb RR, Mempel A, Sonntag B, Kiesel L, Kaltwasser P, et al. Significance of a common single nucleotide polymorphism in exon 10 of the follicle-stimulating hormone (FSH) receptor gene for the ovarian response to FSH: a pharmacogenetic approach to controlled ovarian hyperstimulation. Pharmacogenet Genomics (2005) 15:451–6. doi: 10.1097/01.fpc.0000167330.92786.5e
271. Mutharasan P, Galdones E, Peñalver Bernabé B, Garcia OA, Jafari N, Shea LD, et al. Evidence for chromosome 2p16.3 polycystic ovary syndrome susceptibility locus in affected women of European ancestry. J Clin Endocrinol Metab (2013) 98:E185–190. doi: 10.1210/jc.2012-2471
272. Weng X, Ma X, Wang Q, Xu K, Hu X, Liu W, et al. Effect of hypothyroidism on CYP51 and FSHR expression in rat ovary. Theriogenology (2019) 138:145–51. doi: 10.1016/j.theriogenology.2019.07.012
273. Yang J, Zhong T, Xiao G, Chen Y, Liu J, Xia C, et al. Polymorphisms and haplotypes of the TGF-β1 gene are associated with risk of polycystic ovary syndrome in Chinese Han women. Eur J Obstet Gynecol Reprod Biol (2015) 186:1–7. doi: 10.1016/j.ejogrb.2014.11.004
274. Urbanek M, Sam S, Legro RS, Dunaif A. Identification of a polycystic ovary syndrome susceptibility variant in fibrillin-3 and association with a metabolic phenotype. J Clin Endocrinol Metab (2007) 92:4191–8. doi: 10.1210/jc.2007-0761
275. Prodoehl MJ, Hatzirodos N, Irving-Rodgers HF, Zhao ZZ, Painter JN, Hickey TE, et al. Genetic and gene expression analyses of the polycystic ovary syndrome candidate gene fibrillin-3 and other fibrillin family members in human ovaries. Mol Hum Reprod (2009) 15:829–41. doi: 10.1093/molehr/gap072
276. Xie G, Xu P, Che Y, Xia Y, Cao Y, Wang W, et al. Microsatellite polymorphism in the fibrillin 3 gene and susceptibility to PCOS: a case-control study and meta-analysis. Reprod BioMed Online (2013) 26:168–74. doi: 10.1016/j.rbmo.2012.10.014
277. Raja-Khan N, Kunselman AR, Demers LM, Ewens KG, Spielman RS, Legro RS. A variant in the fibrillin-3 gene is associated with TGF-β and inhibin B levels in women with polycystic ovary syndrome. Fertil Steril (2010) 94:2916–9. doi: 10.1016/j.fertnstert.2010.05.047
278. Hatzirodos N, Bayne RA, Irving-Rodgers HF, Hummitzsch K, Sabatier L, Lee S, et al. Linkage of regulators of TGF-β activity in the fetal ovary to polycystic ovary syndrome. FASEB J (2011) 25:2256–65. doi: 10.1096/fj.11-181099
279. Joo YY, Actkins K, Pacheco JA, Basile AO, Carroll R, Crosslin DR, et al. A Polygenic and Phenotypic Risk Prediction for Polycystic Ovary Syndrome Evaluated by Phenome-Wide Association Studies. J Clin Endocrinol Metab (2020) 105:1918–36. doi: 10.1210/clinem/dgz326
280. Winer DA, Winer S, Shen L, Chng MHY, Engleman EG. B lymphocytes as emerging mediators of insulin resistance. Int J Obes Suppl (2012) 2:S4–7. doi: 10.1038/ijosup.2012.2
281. Shevach EM. Regulatory T cells in autoimmmunity*. Annu Rev Immunol (2000) 18:423–49. doi: 10.1146/annurev.immunol.18.1.423
282. Yang M, Su L, Tao Q, Zhang C, Wu Y, Liu J. Depletion of Regulatory T Cells in Visceral Adipose Tissues Contributes to Insulin Resistance in Hashimoto’s Thyroiditis. Front Physiol (2018) 9:136. doi: 10.3389/fphys.2018.00136
283. Yin L, Zeng C, Yao J, Shen J. Emerging Roles for Noncoding RNAs in Autoimmune Thyroid Disease. Endocrinology (2020) 161:bqaa053. doi: 10.1210/endocr/bqaa053
284. Lin L, Du T, Huang J, Huang L-L, Yang D-Z. Identification of differentially expressed microRNAs in the ovary of polycystic ovary syndrome with hyperandrogenism and insulin resistance. Chin Med J (Engl) (2015) 128:169–74. doi: 10.4103/0366-6999.149189
285. Wang B, Shao X, Song R, Xu D, Zhang J-A. The Emerging Role of Epigenetics in Autoimmune Thyroid Diseases. Front Immunol (2017) 8:396. doi: 10.3389/fimmu.2017.00396
286. Dorris ER, Smyth P, O’Leary JJ, Sheils O. MIR141 Expression Differentiates Hashimoto Thyroiditis from PTC and Benign Thyrocytes in Irish Archival Thyroid Tissues. Front Endocrinol (2012) 3:102. doi: 10.3389/fendo.2012.00102
287. Peng H, Liu Y, Tian J, Ma J, Tang X, Rui K, et al. The Long Noncoding RNA IFNG-AS1 Promotes T Helper Type 1 Cells Response in Patients with Hashimoto’s Thyroiditis. Sci Rep (2015) 5:17702. doi: 10.1038/srep17702
288. Papoutsoglou P, Tsubakihara Y, Caja L, Morén A, Pallis P, Ameur A, et al. The TGFB2-AS1 lncRNA Regulates TGF-β Signaling by Modulating Corepressor Activity. Cell Rep (2019) 28:3182–3198.e11. doi: 10.1016/j.celrep.2019.08.028
289. Li Y, Xiang Y, Song Y, Wan L, Yu G, Tan L. Dysregulated miR-142, -33b and -423 in granulosa cells target TGFBR1 and SMAD7: a possible role in polycystic ovary syndrome. Mol Hum Reprod (2019) 25:638–46. doi: 10.1093/molehr/gaz014
290. Farooqi AA, Attar R, Qureshi MZ, Fayyaz S, Sohail MI, Sabitaliyevich UY, et al. Interplay of long non-coding RNAs and TGF/SMAD signaling in different cancers. Cell Mol Biol Noisy–Gd Fr (2018) 64:1–6. doi: 10.14715/cmb/2017.64.15.1
291. Sun B, Liu C, Li H, Zhang L, Luo G, Liang S, et al. Research progress on the interactions between long non-coding RNAs and microRNAs in human cancer. Oncol Lett (2020) 19:595–605. doi: 10.3892/ol.2019.11182
292. Du X, Liu L, Li Q, Zhang L, Pan Z, Li Q. NORFA, long intergenic noncoding RNA, maintains sow fertility by inhibiting granulosa cell death. Commun Biol (2020) 3:131. doi: 10.1038/s42003-020-0864-x
Keywords: autoimmune thyroid disease, Hashimoto’s thyroiditis, polycystic ovary syndrome, genetic variants, association studies, susceptibility loci
Citation: Zeber-Lubecka N and Hennig EE (2021) Genetic Susceptibility to Joint Occurrence of Polycystic Ovary Syndrome and Hashimoto’s Thyroiditis: How Far Is Our Understanding? Front. Immunol. 12:606620. doi: 10.3389/fimmu.2021.606620
Received: 15 September 2020; Accepted: 07 January 2021;
Published: 26 February 2021.
Edited by:
José Carlos Crispín, Instituto Nacional de Ciencias Médicas y Nutrición Salvador Zubirán (INCMNSZ), MexicoReviewed by:
Trine N. Jorgensen, Case Western Reserve University, United StatesCopyright © 2021 Zeber-Lubecka and Hennig. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ewa E. Hennig, ZXdhLmhlbm5pZ0BwaWItbmlvLnBs
†ORCID: Natalia Zeber-Lubecka orcid.org/0000-0003-4036-3191
Ewa E. Hennig orcid.org/0000-0002-6388-2237
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.