 Haidy A. Saleh1,2
Haidy A. Saleh1,2 Mohamed H. Yousef
Mohamed H. Yousef Anwar Abdelnaser
Anwar Abdelnaser
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 26 March 2021
Sec. Nutritional Immunology
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.606069
This article is part of the Research TopicImmune Modulation by FlavonoidsView all 5 articles
Innate immune response induces positive inflammatory transducers and regulators in order to attack pathogens, while simultaneously negative signaling regulators are transcribed to maintain innate immune homeostasis and to avoid persistent inflammatory immune responses. The gene expression of many of these regulators is controlled by different epigenetic modifications. The remarkable impact of epigenetic changes in inducing or suppressing inflammatory signaling is being increasingly recognized. Several studies have highlighted the interplay of histone modification, DNA methylation, and post-transcriptional miRNA-mediated modifications in inflammatory diseases, and inflammation-mediated tumorigenesis. Targeting these epigenetic alterations affords the opportunity of attenuating different inflammatory dysregulations. In this regard, many studies have identified the significant anti-inflammatory properties of distinct naturally-derived phytochemicals, and revealed their regulatory capacity. In the current review, we demonstrate the signaling cascade during the immune response and the epigenetic modifications that take place during inflammation. Moreover, we also provide an updated overview of phytochemicals that target these mechanisms in macrophages and other experimental models, and go on to illustrate the effects of these phytochemicals in regulating epigenetic mechanisms and attenuating aberrant inflammation.
The innate immune system is the non-specific, inherited immune defense mechanism encoded in the germ-line genes of the host (1). It initiates a rapid response and recruits immune cells promptly to the site of infection or inflammation through cytokines and chemokines production (2, 3). The most important cellular components of the innate immune system are neutrophils and macrophages, which are effective phagocytes that act as the first line of defense against foreign bodies (3, 4). These cells are known as antigen-presenting cells (APCs) that usually recognize pathogens through their surface-expressed receptors, known as pattern recognition receptors (PRRs), which bind to molecular patterns expressed on the surfaces of invading microbes (5). Upon engulfment of a pathogen, antigen presenting macrophages express antigen peptides derived from the engulfed pathogen on the immune cell surface via the major histocompatibility complex (MHC) class II, in order to recruit CD4+ T cells, one type of adaptive immune cells (6, 7). This connection between phagocytic immune cells and T-lymphocytes, therefore, shapes the link between innate and adaptive immunity.
The innate immune response begins when pattern recognition receptors (PRRs) expressed on immune cells detect either pathogen-associated molecular patterns (PAMPs), such as microbial nucleic acids, lipoproteins, and carbohydrates, or damage-associated molecular patterns (DAMPs) released from damaged cells (5, 8). Then oligomerization of the receptor ensues, followed by the assembly of the activated PRRs subunits, which initiates signaling cascades leading to the activation of mediators that attract leukocytes to the site of infection or injury (5, 8). Subsequently, these leukocytes, including macrophages, neutrophils, and dendritic cells phagocytose microbial elements and release more proinflammatory cytokines, such as TNF-α, IL-6, IL-12, and type I and II interferons (IFNs), which collaboratively attempt to contain the pathogen until highly specific, activated cells of the adaptive immune response are recruited to completely eliminate the infection (6, 8).
A hallmark of the innate immune response is inflammation, which is a complex set of defense mechanisms acting in concert to restore homeostasis in body systems after injuries or infections (8, 9). Inflammatory reactions are mainly a result of the vasodilation due to the release of histamine, prostaglandins (PGs), and nitric oxide (NO) that leads to a noticeable increase in blood flow and accumulation of circulating leukocytes (8). Additionally, proinflammatory cytokines secreted from activated immune cells, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 (IL-1) and interleukin-6 (IL-6) enhance the vascular permeability of leukocytes through raising the levels of leukocyte adhesion molecules on endothelial cells (5, 10). Usually, inflammation is triggered to restore homeostasis and repair tissues; however, prolonged inflammation could lead to serious problems, including cellular dysregulation as observed with cell senescence, impaired proteolysis and apoptosis, and further tissue dysfunction (11). Recently, many studies correlated inflammatory disturbance with epigenetic modifications (11).
Epigenetic mechanisms modulate differential gene expression with no alteration to the DNA sequences (12). Epigenetic processes include modifications to DNA, such as DNA methylation or histone proteins, including histone methylation, acetylation and acylation, and they also involve microRNAs (13). Epigenetic modifications are potentially reversible, and, therefore, an in-depth understanding of these changes may help identifying new therapeutic targets (14). A plethora of reviews covered distinct pathways of innate immunity, inflammation and highlighted the relationship between TLR4 signaling and inflammatory diseases and cancer (15, 16). The current review focuses on the various regulatory epigenetic mechanisms involved in inflammation and summarizes the recent findings of targeted phytotherapy.
One of the essential and first-identified members of PRRs are TLRs (5, 17). They are expressed on various immune cells, including macrophages, dendritic cells (DCs), B cells, specific types of T cells, and even on non-immune cells such as cardiac cells, fibroblasts and epithelial cells (1). TLRs are type I transmembrane proteins characterized by the extracellular leucine-rich repeats (LRRs) that recognize different microbial epitopes and has a cytoplasmic signaling domain similar to that of the interleukin 1 receptor (IL-1R), called Toll/Interleukin 1 receptor (TIR) domain, which is responsible for signal transduction (1, 4, 5, 18). The TLR family was first identified in Drosophila flies, and now twelve members of the TLR family have been identified in mammals (5, 18). Grouped as subfamilies, TLR1, TLR2, and TLR6 recognize lipids, while TLR7, TLR8, and TLR9 recognize nucleic acids (6, 8). Some receptor/ligand pairs are commonly known, such as TLR4 and LPS, TLR5 and flagellin, TLR 1, 2, and 6 with lipoproteins (18). In addition, TLRs are either expressed on the cell-surface, e.g. TLRs 1, 2, 4, 5, and 6, or internalized to the endosome, e.g. TLRs 3, 7, 8, and 9 (5, 6, 18). Together with phagocytic-antigen presentation, the activation of TLRs leads to the expression of inflammatory cytokines, which further recruits antigen-specific cells (18).
When activated, TLRs, in turn, activate various genes that function to moderate host defense, including inflammatory cytokines, chemokines, MHC and co-stimulatory molecules (18). Mammalian TLRs also induce multiple effector molecules, such as iNOS and antimicrobial peptides that can directly destroy microbial pathogens (8). Depending on TIR domains, TLRs activate NF-κB and MAPK (mitogen activated protein kinases) and induce target genes (5, 18). Upon ligand binding (e.g. LPS), TLR4 dimerizes and induces the recruitment of intracellular adaptor proteins that trigger two standard models of signaling cascades: myeloid differentiation primary response gene 88 (MyD88)-dependent and Toll-interleukin-1 receptor domain-containing adaptor inducing interferon-beta (TRIF)-dependent pathways (8, 18).
The MyD88-dependent pathway originates from the cytoplasmic TIR domain (19). The activation of MyD88 causes the autophosphorylation of interleukin-1 receptor-associated kinases (IRAK), namely IRAK1, IRAK2, and IRAK4, which associate temporarily with TNF receptor-associated factor 6 (TRAF6) (8, 18). This autophosphorylation and oligomerization for IRAK and TRAF6, respectively, finally leads to the activation of IκB kinase (IKK) (in response to TAK1/TAB complex activation) and mitogen-activated protein kinase (MAPK), namely ERK, JNK, p38 (10, 18). Then, ensuing signal dissemination results in the activation and the translocation of nuclear factor kappa B (NF-κB) to the nucleus and the subsequent activation of the activator protein-1 (AP-1) transcriptional program (8). Both NF-κB and AP-1 control inflammatory responses through the induction of inflammatory cytokines, such as TNF-α, IL-12, and others (8, 18). The TRIF-dependent pathway, which mediates the late phase activation of NF-κB, primarily recruits TRIF and this results in the ubiquitination of TNF receptor-associated factor 3 (TRAF3) which induces TANK-binding kinase 1 (TBK1) binding to IκB (inhibitor of NF-κB) kinase epsilon (IKKϵ)) (8, 20–24). Thenceforth, the TBK1-IKKϵ complex phosphorylates the transcription factor interferon regulatory factor 3 (IRF3), ultimately driving the expression of interferon-beta (IFN-β), which induces STAT1-dependent genes encoding monocyte chemoattractant protein 5 (MCP-5), IFN-inducible protein 10 (IP-10) and iNOS (25, 26) (Figures 1 and 2).
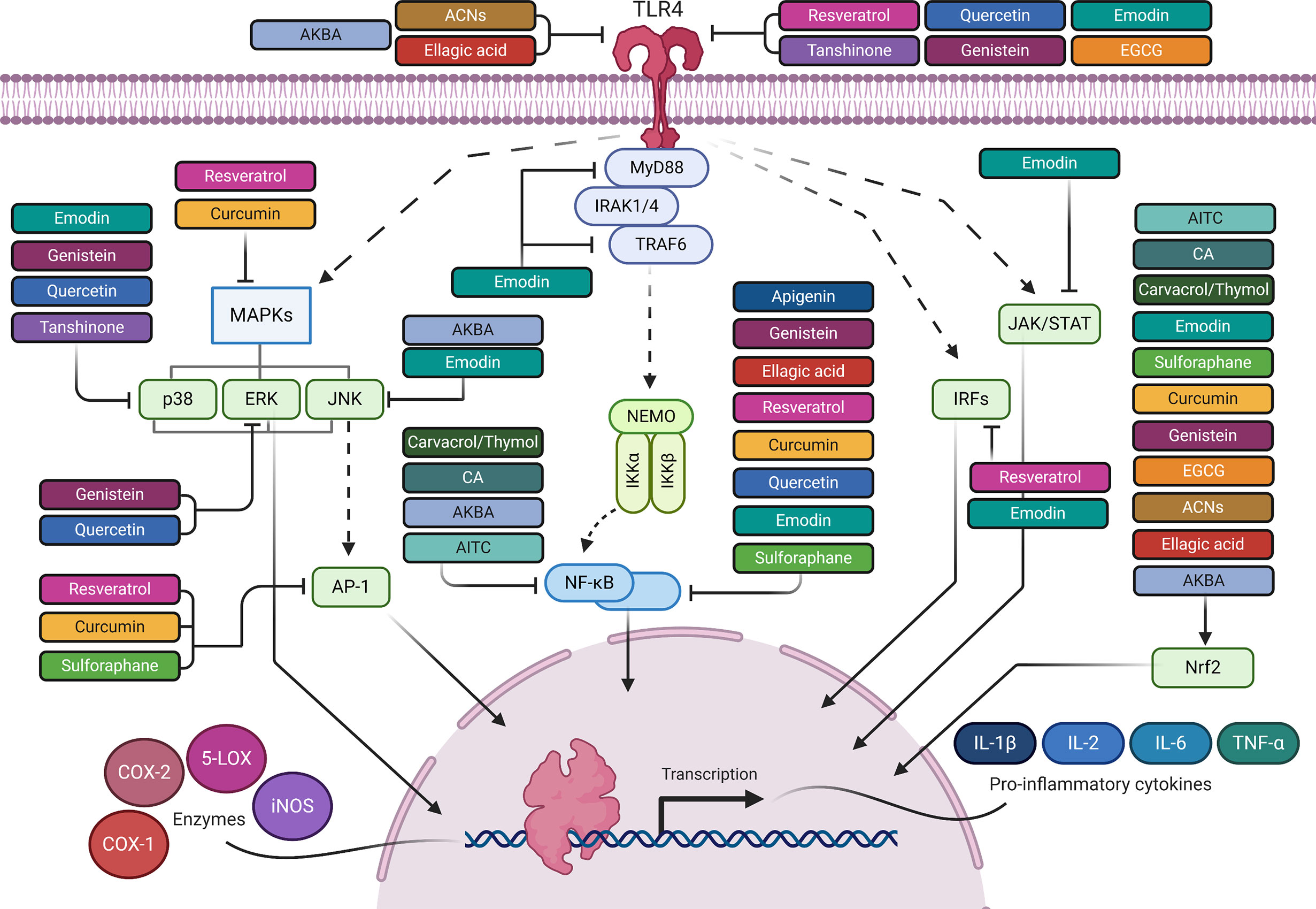
Figure 1 The anti-inflammatory mediated effects of phytochemicals along the TLR4 signaling pathway. Created with Biorender.
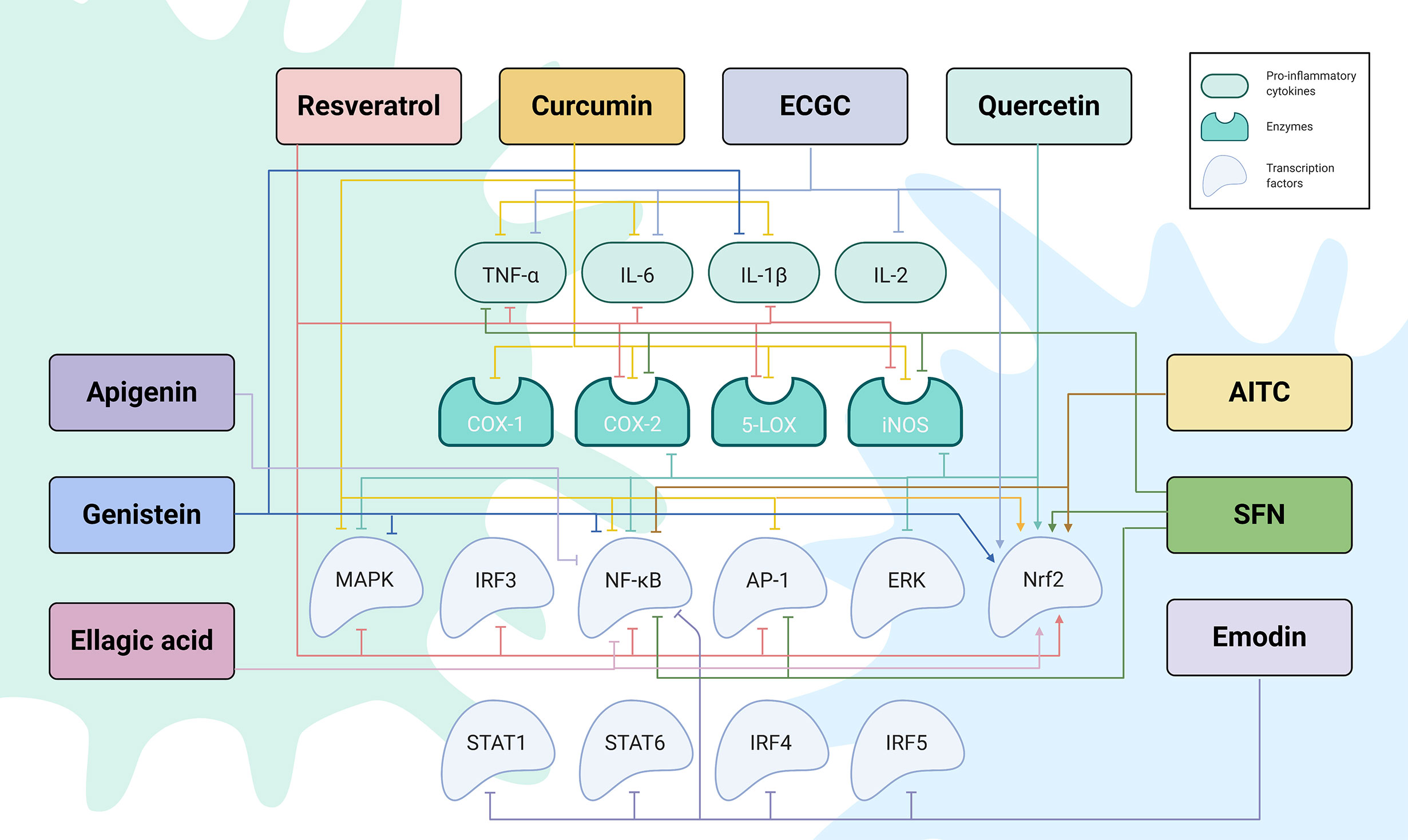
Figure 2 The regulatory network by which phytochemicals affect TLR4-mediated inflammatory responses. Created with Biorender.
Despite the great progress in understanding the molecular mechanisms underlying inflammation-related signaling events of TLRs, particularly TLR4, less data is available about the role of epigenetics in regulating the transcriptional responses downstream of the TLR system (27). Recent studies have demonstrated that TLR-induced epigenetic alterations forms a solid platform in both positive and negative regulation of activated inflammatory genes (27).
A balanced interplay of the chromatin machinery is pivotal in regulating the transient induction of TLR-related genes (28). Changes in chromatin play an essential role in both physiological and pathological processes through responding to environmental signals and regulating gene transcription (29). Epigenetic regulations mainly include DNA modifications, histone post-translational modifications (PTMs), chromatin remodeling and microRNAs (miRNAs), which together function in a coordinated manner (30). These epigenetic modifications occur with the aid of several catalytic enzymes that add or remove chemical moieties, including DNA methyltransferases (DNMTs), histone methyltransferases (HMTs), histone demethylases (HDMs), histone acetyltransferases (HATs), and histone deacetylases (HDACs) (13).
Histone modifications, such as acetylation and methylation, are critical regulators of gene transcription (31). These modifications can regulate the binding of effector molecules to DNA and, therefore, control transcription, repair, and replication processes (31). Acetylation of histones is associated with an “open” chromatin conformation that activates transcription (14). Histone acetylation of the lysine residues is regulated by histone acetyltransferases (HATs), which add an acetyl group and mediate histone hyperacetylation towards euchromatin and gene upregulation, whereas histone deacetyl transferases (HDATs), which remove the acetyl group, induce histone hypoacetylation towards heterochromatin and silencing of inflammatory genes (14, 32). Villagra et al. showed that promotors of several proinflammatory cytokines are acetylated and they, usually, display reduced HDAC activity, leading to transcriptional activation (33). Not only this, but also histone H3 acetylation (H3Ac) at the promoters of many cytokines and chemokines increases NF-κB recruitment to these regions, following inflammation (34). In this context, Hargreaves and colleagues stated that H3Ac modification primarily occurs at promotors of primary response genes (PRG), which are rapidly induced by TLR4 following macrophage activation (35). Furthermore, Wu and Chiang showed that upon TLR ligation, an inducible addition of histone H4Ac at lysine 5, 8, and 12 (H4K5, H4K8, and H4K12 Ac) at PRG promoters takes place, facilitating the binding of an epigenetic reader, called BRD4 that recognizes inducible acetylation marks on histones and recruits positive transcription factors for active transcription (36, 37). These chromatin modifiers are employed in regulating innate signaling pathways induced by infection, and regulate the expression of PRR signal transducers and inflammatory genes through initiating various histone modifications (29). TLR4 is one of the most important PRR signal transducers. It has been implicated in the epigenetic regulation of the TNF-α promoter, where LPS stimulation increased the H3 and H4 acetylation of the TNF-α locus, and thus induced gene transcription (38).
On the other side, Histone deacetylases (HDACs) reverse the activity of HAT by removing acetyl groups from histones, making chromatin more condensed and thus, promoting gene inactivation (39). It has been reported that HDAC recruitment is associated with the pathogenesis of diseases, including inflammation and cancer, and is targeted by therapeutic agents in patients (40). Inhibitors of HDACs have pleiotropic effects on the immune response, especially on TLR signaling; they are known to suppress different cytokines, such as IL-6, IL-10, TNF-α, IL-12, and IL-23 (41). Moreover, inhibitors of class I, II, and IV HDACs are found to inhibit LPS-induced upregulation of TLRs, CD14, and MD-2 genes (40). In this respect, reduced expression and activity of HDAC2, in particular, was noticed with macrophage tolerance, which was reported in several inflammatory models, such as lung macrophages, biopsies, and blood cells from patients with COPD, severe asthma, and smoking-induced asthma (42).
In contrast to histone acetylation that occurs primarily on lysine residues, histone methylation takes place on lysine, arginine and other amino acid residues in histone protein tails, and eventually modifies chromatin in either its activated or repressed states (43). For example, tri-methylation of histone H3 on lysine 4 and 36 (H3K4me3 and H3K36me3) are commonly referred to as activators of transcription (44). Nonetheless, histone methylation on lysines 9 and 27 (H3K9me3 and H3K27me3) facilitates gene silencing (45). Histone methyltransferases (HMT) are histone-modifying enzymes that regulate gene expression by catalyzing the transfer of methyl groups to lysine and arginine residues of histones (H3 and H4) at specific sites (46). The number of methyl groups transferred and the histone residues involved determine whether gene transcription is suppressed or activated (47). Multiple reports demonstrated the key role of H3K4me3 in positively regulating the TLR4 signaling pathway in macrophages during the LPS response. For example, increased levels of H3K4me3 at the gene promotor induced the transcription of PIGP, a product required for proper membrane anchoring of CD14 in primary macrophages for TLR4-mediated signaling (48). Another report showed that elevated levels of H3K4me3 at Socs1 promoter resulted in increased expression of suppressor of cytokine signaling 1 (SOCS1), which acts as a negative regulator of TLR4-induced inflammation (49). Similarly, increased H3K4me3 levels on Tnfaip3 promotor upon LPS stimulation in macrophages results in transcribing the Tnfaip3-encoded ubiquitin-editing enzyme A20, which in turn mediates K63 deubiquitylation of TRAF6 and NF-κB essential modulator (NEMO), therefore suppressing TLR4-mediated signal transduction (50). Hargreaves and colleagues conducted a ChIP analysis of RNA pol II-associated genes that shows the elevated basal levels of H3K4me3 modification in the promotors of many primary response genes (PRGs) to be usually characteristic of active transcription (35). On the contrary, the secondary response genes (SRGs), which have delayed induction, showed remarkable H3K4me3 modification only after TLR ligation, and not at the basal level like PRGs (35).
On the contrary, other histone methylations, such as H3K27me3 possess a clear suppressive effect on gene transcription. For instance, induced H3K27me3 levels at the promoter region of Tollip gene (a negative regulator of TLR pathway) inhibited Tollip transcription and, therefore, activation of the TLR signaling cascade (51). Moreover, the relationship between epigenome reprogramming and inflammation might also be expressed in macrophage polarization. Following NF-κB activation, macrophages induce a protein called Jmjd3 that binds to PcG target genes (a Polycomb Group proteins, which are involved in development and differentiation of macrophages) and regulates their H3K27me3 levels and, thus their transcriptional activity (52). Taken together, these interesting differences show the significant influence of chromatin epigenetic modifiers on different immune responses and time-dependent production of TLR-responsive genes.
DNA methylation involves a covalent attachment of a methyl group to the cytosine residue at cytosine-phosphate-guanine (CpG) site by DNA methyltransferase enzymes (DNMTs) (47). These CpG sites are present in about 70% of human gene promoters, and are essential modulators of gene transcription (53). Usually, DNA hyper-methylation induces chromatin condensation (heterochromatin) and enhance gene repression, inhibiting the binding of transcription factors at promoter sites on genomic DNA (54, 55). DNA methylation by DNMTs also induces coupling with other gene repressing proteins such as HDACs (56). Indeed, DNA methylation is highly imperative in regulating inflammatory genes. For example, epigenome-wide association studies (EWAS) linked DNA hypomethylation with increased inflammation (57, 58). In addition, DNA hypomethylation with aging has been suggested to be the cause of chronic inflammation and cancer (59). While, another study by Hahn et al. reported that atypical DNA methylation of some Polycomb group (PcG) protein targets in mammalian genome results in their efficient downregulation after chronic inflammation (60). Cooperatively, modifications in both DNA methylation and histone acetylation of genome status can regulate TLR4 signaling (61). Of interest, DNMT3A, the highly expressed DNA methyltransferase in macrophages, has been shown to indirectly induce the expression of type I IFNs during virus infection via maintaining increased HDAC9 expression, which, in turn, maintained the deacetylation status of the key TLR4 signaling molecule TBK1 and, thus, enhanced its kinase activity (29, 62).
As DNA methylation is a common epigenetic modification in inflammation-related diseases, it would be interesting to examine the effects of individual natural products on DNA methylation and inflammation-associated cascades in humans (27). Notably, the post-transcriptional modifications of inflammatory genes is suggested to be mediated by two distinct regulation levels of TLR4 signaling pathway (63). The first level is mediated by epigenetic alterations, and the second level is regulated by the differential expression of TLR4-responsive miRNAs and, specifically, miR-146a, miR-155, and miR-21 (14).
Recently, a plethora of research studies have focused on deciphering the role of miRNAs in the regulation of inflammatory gene expression (64). MiRNAs are short double-stranded, non-coding RNA molecules that are ~22 nucleotides in length (65). They bind to the 3` untranslated region (UTR) of a target mRNA sequence causing gene down-regulation at the post-transcriptional level, and inducing translational arrest (66). miRNAs are currently viewed as essential regulators in key immune responses, for example regulation of maturation, proliferation, differentiation and activation of both innate and adaptive immune cells (67). TLR-responsive miRNAs are either up-regulated or downregulated after LPS treatment (67). Although they are heritable, miRNAs are also inducible and reversible (68). This flexible nature of miRNA expression is obvious in inflammatory reactions, which are primarily dependent on the surrounding environment. For instance, the expression of most miRNAs is triggered in an NF-κB-dependent manner after TLR stimulus (65). The expression of miRNAs is dependent on TLR stimulation, highlighting the cause/effect relationship between LPS stimulation and the upregulation of miR-146a and miR-155 in human monocytes (69–73). In terms of response to TLR4 pathway activation, miRNAs are classified as either “early response miRNAs”, expressed rapidly after LPS stimulation such as miR-146 and miR-155 or “late response miRNAs”, expressed in macrophages at a later time after LPS treatment such as miR-21 (69–73). miR-146a-5p, miR-155-5p, and miR-21 are also involved in the regulation of TLR downstream signaling through TLR-induced transcriptional factors (74, 75). They target cytokines such as type I IFNs, TNF-α, IL-6, IL-12, and IL-10, and this has been indicated by the presence of binding sites for miRNAs on the mRNAs encoding these cytokines and chemokines (76–78). The miRNAs, miR-21, miR-146a, and miR-155 are particularly predominant in the majority of inflammation-related studies, because of their expression succeeding TLR stimulation, especially in macrophages (67) (Figure 3).
miR-146a is a pivotal repressor of NF-κB inflammatory signaling in several cell types. It is one of the miR-146 family that is present in chromosome 5 and 10 (67, 79–81). Upregulation of miR-146a has been reported in inflammatory diseases, such as osteoarthritis and rheumatoid arthritis (82). A recent study illustrated that miR-146a negatively regulated TLR4 signaling through blocking TRAF6 and IRAK1, which activates downstream transcription factors NF-κB and AP-1 (14, 71, 83–86). It has been reported that increased expression of TRAF6 was observed in 5q chromosomal deficient models, leading to impaired innate immune signaling and causing leukemia and bone marrow failure (87). In addition, IRAK2 and IRAK4 have been recognized as targets of miR-146a, which results in decreasing inflammatory cytokines (71, 83, 88). Another study illustrated the role of miR-146a as a negative regulator of type1 IFN response in human peripheral blood mononuclear cells (PBMCs) (89). Moreover, in LPS-treated human monocytes, miR-146a has been shown to degrade mRNA transcripts of IRF3, a transcriptional factor responsible for type1 IFN production (90). miR-146a has also been reported to be involved in regulating cytokines release and apoptosis in human dendritic cells (71, 91). From this angle, a recent study showed that by targeting IRAK1, IRAK2, and TRAF6 in LPS-stimulated macrophages, miR-146a sequentially suppresses the production of type I IFNs, and the cytokines TNF-α, IL-1β, and IL6 (83, 85)
Second, miR-21 is a cancer-associated miRNA that is induced by NF-κB activation, and acts as a negative regulator of TLR signaling inflammatory responses (67, 92). It has been revealed that miR-21 inhibits MyD88 and IRAK1 expression during hepatitis C viral infection in PBMCs (93). Furthermore, Sheedy et al. highlighted the role of miR-21 in downregulating the expression of programmed cell death protein 4 expression (PDCD4) in LPS-induced RAW 264.7 macrophages (94). PDCD4 acts as a tumor suppressor protein that activates the proinflammatory mediators NF-κB and IL-6, and suppresses the anti-inflammatory cytokine IL-10, which inhibits of the microRNA, miR-155 (94, 95). MiR-21, therefore, has anti-inflammatory effects by increasing the production of IL-10 and suppressing NF-κB activity (67, 94).
Another tumor-associated miRNA is miR-155. miR-155 has a significant role in the TLR-mediated immune response and can target related signaling proteins of the NF-κB pathway (65). A positive correlation exists between miR-155 overexpression and NF-κB activation as evidenced by Baltimore et al. in their study, which linked miR-155 upregulation with mammalian inflammatory reactions (71, 96). Another study by Tili et al. showed that miR-155 expression can have both positive and negative effects on NF-κB signaling proteins however, they supported a positive regulatory role of miR-155 in NF-κB pathway, which was evidenced by increased serum TNF-α in miR-155 transgenic mice (97).
As a positive regulator of TLR4 signaling pathway, miR-155 suppresses two negative regulators of TLR4-induced inflammation, SOCS1 and SH2 (Src homology 2)-containing inositol phosphatise-1 (SHIP-1) (98, 99). This suppression of these two important negative regulators of TLR4 signaling results in boosting MAPK activity and stimulating the expression of inflammatory cytokines in primary macrophages and dendritic cells isolated from mice (67, 100, 101). On the other hand, Ceppi et al. showed that, sometimes, miR-155 exerts an anti-inflammatory effect through targeting TAB2, thereby inhibiting TAK1-dependent stimulation, and further NF-κB and MAPK activation in human monocyte-derived DCs (102). On account of the above findings, miR-155 remains an interesting and significant player in downstream inflammatory pathways (67).
As demonstrated hereinabove, the significance of miRNAs in inflammatory processes has directed research towards a better understanding of their functions and interactions in TLR4 signaling. That being said, plant-derived bioactive compounds have ushered in a fresher phase of research in this area. These naturally-occurring agents have been proven to be a promising therapeutic recourse that could interfere, at the genomic level, with TLR4 signaling and modulating miRNA upregulation after NF-κB activation in multiple inflammatory reactions. In this regard, a rundown of bioactive phytochemicals and their proposed mechanism(s) of action within the TLR4 pathway are included in this review, highlighting their potential for post-transcriptional and epigenetic modulation. In brief, we considered three main epigenetic processes of gene repression: DNA methylation, histone modifications, and microRNAs targeting. However, alterations in histone modifications, DNA methylation, and miRNA regulation still need future investigations to provide a better understanding of the molecular basis for various chronic inflammatory diseases. It has to be highlighted that understanding the role of epigenetic modifications is becoming fundamental in human diseases, and since these epigenetic alterations can regulate several inflammatory signaling cascades, they indeed do play an integral part in inflammation (103–106). Therefore, knowing more about epigenetic events during inflammatory responses, and how epigenetic regulation is mediated by TLR signaling during inflammation is worth further exploration, since it opens up opportunities for developing therapeutic interventions. For example, histone deacetylase inhibitors and demethylating agents are currently being proposed for epigenetic therapy (14).
Originally known as secondary metabolites, phytochemicals are plant-synthesized compounds possessing health effects (107). Plant-derived bioactive compounds can be classified as phenolic compounds, including flavonoids and tannins, glucosinolates, alkaloids, and terpenoids (108). When consumed by humans, they get involved in different biological processes inside the body, such as redox processes, cell signaling and, inflammation (109–112). Nowadays, natural products are becoming a promising source for the treatment of several inflammatory conditions (113, 114). Interestingly, the most promising anti-inflammatory herbal extracts were identified to influence key TLR4 signaling pathways and macrophage repolarization (115). Evidence suggests that phytochemicals can attenuate the expression of proinflammatory genes, and promote anti-inflammatory genes; this differential gene expression is regulated by epigenetic modifications (116, 117). For instance, epigenome-wide association studies (EWAS) have demonstrated that differential methylation of inflammatory genes in peripheral white blood cells was associated with diets rich in phytochemicals (27, 118, 119).
In addition, many plant-derived compounds were recently suggested to exert their anti-inflammatory effect through regulating the expression of proinflammatory miRNAs, especially those upregulated after NF-κB activation (120). Although a few reports indicate the precise mechanisms that regulate or deregulate the expression of miRNAs, growing evidence suggests that phytochemicals regulate the expression of miRNAs by interfering with the processes associated with the miRNAs processing and maturation, and these phytochemical-mediated alterations in miRNAs biosynthesis machinery could in part contribute to the miRNAs dysregulation by either increasing or decreasing their levels (121, 122).
In this section, we will discuss, in detail, how some natural compounds can epigenetically alter inflammatory genes, recapitulating on current studies that link phytochemicals-mediated epigenetic modifications to inflammation-associated diseases.
Phenolic compounds are involved in numerous signaling pathways, and most importantly, in regulating the redox system and modulating the immune response through inhibiting inflammatory cytokines synthesis (123). Unfortunately, polyphenols, in general, are limited by their pharmacokinetics, such as their poor bioavailability and rapid metabolism (124–131).
A natural phenolic stilbene derivative, resveratrol (RES, 3, 4′,5-trihydroxystilbene) is a phytoalexin that acts as a plant defense mechanism against infection. It is found in grape skins, berries and peanuts (124, 132). This phenolic compound exists in both trans- and cis- isoforms, but the trans-isomer is more stable (132). It is usually taken at a daily dose of 50 to 500 mg (132). Fortunately, no significant adverse effects have been reported for RES, except for an antiplatelet activity that should be monitored, especially if taken with another prescribed antiplatelet or anticoagulants (132–139). Over 40 clinical trials were published in PubMed on the applications of RES in inflammatory disorders, including diabetes, obesity, and coronary artery disease (124). Pharmacologically, RES has been widely recognized for its remarkable anti-mutation, anti-inflammatory, and antioxidant activities (124, 132). By virtue of its anti-inflammatory effects, RES also has neuroprotective, cardioprotective and chemotherapeutic properties (131, 132, 140, 141).
Numerous studies have been conducted to provide in-depth insights into the powerful antioxidant and anti-inflammatory function of RES. In a dose-effect relationship, RES exerts its effects at multiple levels (Table 1, Figures 1 and 2). It inhibits TLR4 and MyD88 expression in activated RAW 264.7 macrophages (142, 143) (Table 1 and Figure 1). Going downstream, RES inhibits NF-κB, MAPK, IRF-3 and AP-1 transcription factors, as well as, iNOS, COX-2, and 5-LOX enzymes (124, 143–147) (Table 1 and Figures 1 and 2). This, in turn, reduces NF-κB-induced proinflammatory cytokines, including TNF-α, IL-6, and IL-1β, and the free radicals, NO and ROS, and LTs and PGs levels (124, 132, 140, 142, 147, 148, 301) (Table 1 and Figure 2). These results were confirmed in LPS-stimulated RAW 264.7 cells, and macrophages isolated from C57BL/6 and BALB/c mice (302). In addition to studies performed on macrophages, others were conducted on heart tissues of rats to investigate the anti-inflammatory effects of RES in response to TLR4/NF-κB-mediated cardiac inflammation (303). These studies showed the cardioprotective effect of RES, which was manifest in lowered left ventricular peroxidation and enhanced antioxidant production, such as GSH and SOD, as well as, reduced TNF-α levels (304) (Table 1 and Figure 2). Another study showed RES inhibition of TLR4/NF-κB signaling in an ischemic injured rat heart model, which is confirmed by TLR4 and NF-κB downregulation, and reduced myocardial TNF-α production (303) (Table 1 and Figures 1 and 2).
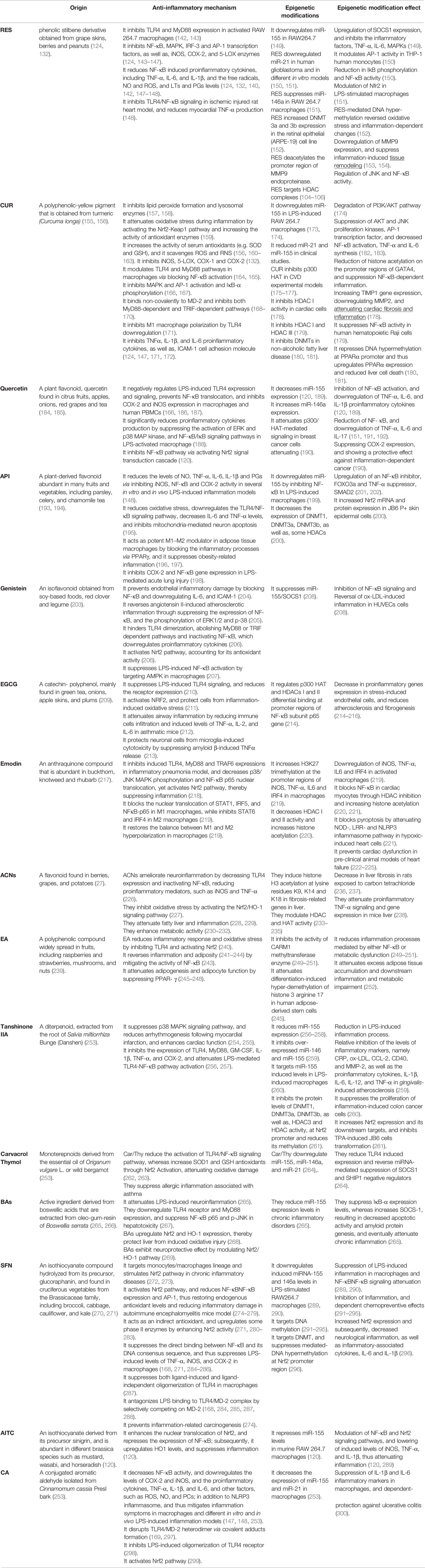
Table 1 The anti-inflammatory mechanism(s) of phytochemicals and their associated epigenetic modifications/effect.
Through epigenetic mechanisms (Table 1 and Figure 3), RES attenuated LPS-mediated inflammation in RAW264.7 macrophages through downregulating miR-155 and concurrently boosting, SOCS1 expression, leading to the inhibition of the inflammatory factors, TNF-α, IL-6, MAPKs (149). Additionally, Tili et al. showed another mechanism by which RES modulates AP-1 activity in THP-1 human monocytes through downregulating miR-155 (150). RES also downregulated miR-21 in different in vitro models (151). Li et al. introduced the inhibitory effect of RES on miR-21 expression in human glioblastoma (U251) cells, leading to a reduction in IκB phosphorylation and NF-κB activity (305). In RAW 264.7 murine macrophages, Bigagli et al. depicted the suppressive effect of RES on miR-146a, which targets the transcription factor nuclear factor (erythroid-derived 2)-like 2 (Nrf2) responsible for inhibiting proinflammatory mediators. Nfr2 was also positively modulated by the RES in LPS-stimulated macrophages (306). These findings indicate that RES modulatory effect on these miRNAs can be regarded as a buffering effect against physiological imbalance. Besides miRNAs, RES caused an increased DNMT activity, especially in DNMT3a and DNMT3b expression in the retinal epithelial cell line, ARPE-19 (152). This RES-mediated DNA hypermethylation results in a reversal of oxidative stress and inflammation-dependent changes (152). In addition to regulation of NF-κB and MAPKs signaling cascades, RES could inhibit inflammation through regulating histone deacetylation-dependent gene expression (153, 307). RES deacetylates the promoter region of matrix metalloproteinase 9 (MMP9), which in turn downregulates the expression of MMP9, an endoproteinase that is involved in inflammation-induced tissue remodeling and is activated by MAPK, c-Jun N-terminal kinases (JNK), and NF-κB binding (153, 154). Furthermore, independent of its histone deacetylase activity, RES could attenuate MMP9 activity, because this stilbene is known to inhibit JNK and NF-κB (308, 309). Moreover, JNK and NF-κB have been shown to be regulated by HDAC complexes, which suggests that RES can also regulate the activity of these molecules by acting on HDACs (104–106). Although RES showed positive outcomes in animal studies time and again, these results have not been well translated in clinical trials (27).
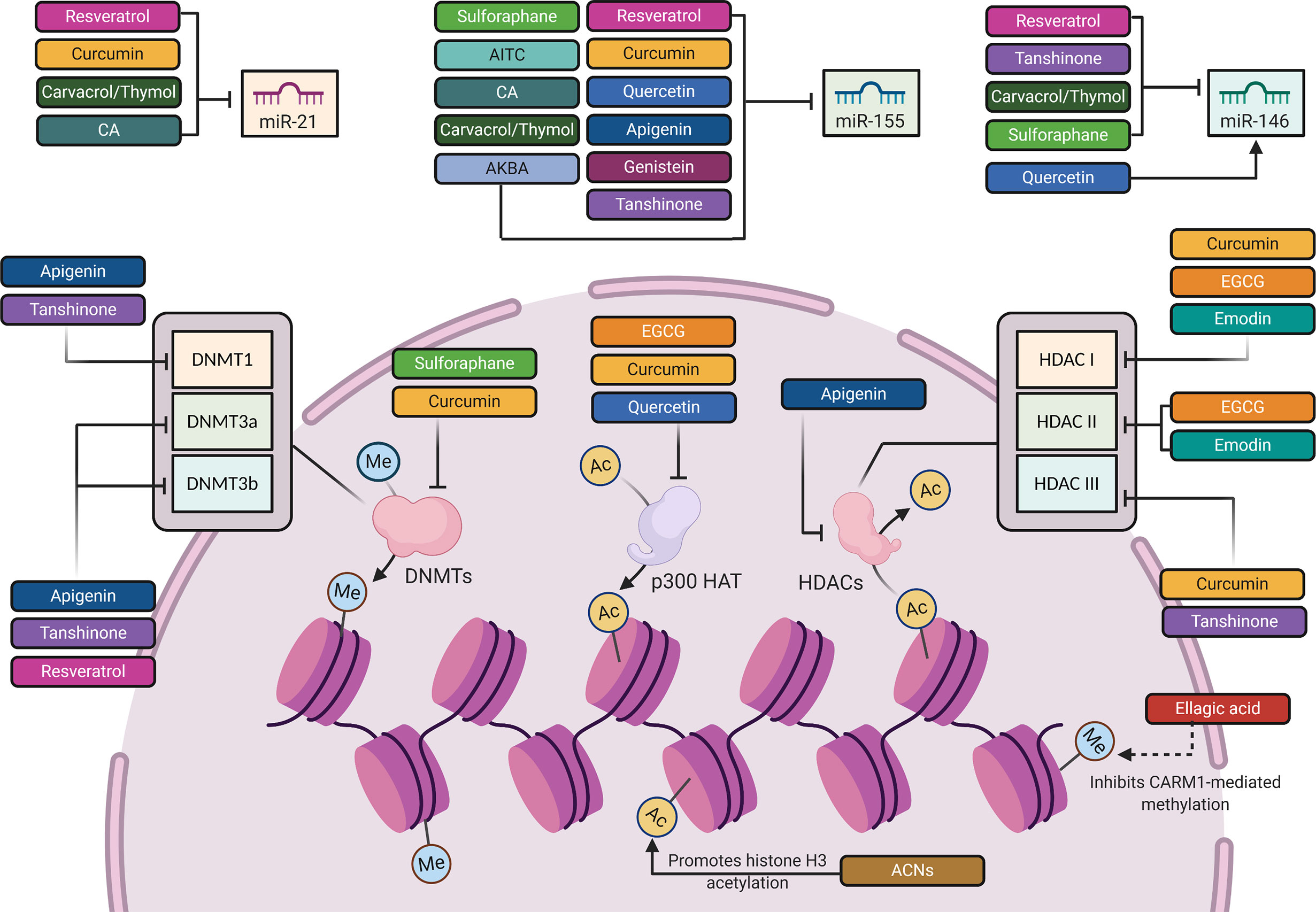
Figure 3 The epigenetic mechanisms involved in the regulation of TLR4 signaling pathway by phytochemicals. Created with Biorender.
Curcumin (CUR) is another naturally polyphenol. It is found as a yellow pigment that is obtained from turmeric (Curcuma longa), a flowering plant of the ginger family (155, 156). Traditionally, CUR is widely used in food as a curry coloring or flavoring spice (132). The phenolic compound exerts diverse bioactive effects; it has anti-bacterial, anti-inflammatory, antioxidant and anticancer functions (124, 310–313). Although CUR has a great many therapeutic benefits. Its main advantage lies in its antioxidant and anti-inflammatory effects (314, 315). CUR supplements are usually taken three times per day, with a dosage of 400–600 mg, and even with no side effects on kidney or liver when taken up to 12 g/day (316, 317). Only at very high doses, it may cause stomach upset with extended use, and gastric ulcers (132). CUR is usually combined with enhancing agents, such as piperine, to overcome its poor absorption and low bioavailability (318–320). Over 100 clinical trials on the anti-inflammatory activity of CUR have been published in PubMed, showing the significance of CUR in multiple inflammatory disorders, such as rheumatoid arthritis, inflammatory bowel diseases, nephropathies, and some cancers (124, 314).
CUR has a pleiotropic mechanism of action against inflammation (157) (Table 1, Figures 1 and 2). Firstly, it has powerful antioxidant activity that inhibits lipid peroxide formation and lysosomal enzymes, such as acid phosphatase and cathepsin D (157, 158). CUR attenuates oxidative stress during inflammation by activating the Nrf2-Keap1 pathway and increasing the activity of antioxidant enzymes (159) (T1 & Figures 1 and 2). By increasing the activity of serum antioxidants such as superoxide dismutase (SOD) and glutathione peroxidase (GSH), CUR scavenges different free radicals, including reactive oxygen and nitrogen species (ROS and RNS), and peroxyl radicals (156, 160–163). CUR can also inhibit different ROS-generating enzymes, such as iNOS, COX system (COX-1 and COX-2), and 5-LOX, and suppresses the activity of several PGs (132) (Table 1 and Figure 2). Therefore, CUR can be considered a natural alternative to NSAIDs for inflammation (132, 140, 156). Secondly, CUR has a potent anti-inflammatory effect that derives from its modulation of TLR4 and MyD88 pathways in macrophages and evidenced by blocking NF-κB activation (164, 165) (Table 1 and Figures 1 and 2). CUR inhibits the activation of MAPK and AP-1 transcription factors, and also IκB-α phosphorylation and degradation (166, 167) (Table 1 and Figures 1 and 2). On top of that, CUR has been suggested to non-covalently bind MD-2 (a lymphocyte antigen responsible for LPS binding to TLR-4), which results in a competition with LPS for the TLR4/MD-2 complex that leads to the inhibition of both MyD88-dependent and TRIF-dependent pathways (168–170) (Table 1 and Figures 1 and 2). Interestingly, CUR inhibited M1 macrophage polarization in a dose-dependent manner through downregulating the expression of TLR4 (171) (Table 1 and Figure 1). These antagonistic effects to TLR4 signaling pathways and its downstream mediators are followed by an inhibition of proinflammatory cytokines, including TNFα, IL-1β, and IL-6 (124, 147, 171, 172) (Table 1 and Figure 2). Research studies suggest that CUR pretreatment protects against T cell-mediated hepatitis in mice and that the significant effect of CUR may be partly through inhibiting the expression levels of TLR2, TLR4 and TLR9 in the liver (321). Furthermore, CUR blocks the expression of cell adhesion molecules, such as ICAM-1, which are involved in the interaction between leukocytes and endothelial cells (124, 322) (Table 1).
Post-transcriptionally (Table 1 and Figure 3), on the miRNA level, several studies on RAW 264.7 macrophages revealed the significant inhibitory effect of CUR on miR-155, which is a key transcriptional regulator of TLR4-mediated inflammatory reactions (173, 174). In vivo and in vitro reports confirmed the downregulation of miR-155 expression by CUR through degrading phosphoinositide 3-kinase PI3K/AKT pathway after LPS stimulation (174). In clinical studies, daily intake of CUR decreased miR-21 and miR-155 expression. This decrease was followed by a suppression of AKT and JNK proliferation kinases, and the transcription factor AP-1, which attenuated inflammation via decreased NF-κB activation, TNF-α and IL-6 synthesis (182, 183).
At the epigenetic level, CUR exhibits pleiotropic mechanisms to regulate multiple molecular targets (27) (Table 1 and Figure 3). Knowing that NF-κB activity is regulated, in part, through p300 HAT-dependent actions (104). One target involves the regulation of histone acetylation by CUR-mediated p300 inhibition in CVD experimental models (175–177). This inhibition reduces histone acetylation on the promoter regions of the cardio pro-hypertrophic gene, GATA binding protein (GATA4), a protein which mediates inflammation through NF-κB -dependent mechanisms (177, 323, 324). Besides its HAT inhibitory actions, CUR has also been involved in the inhibition of inflammation-induced cardiac remodeling through inhibiting HDACs (178). Through inhibiting HDAC1 activity and thus, increasing histone acetylation at the promoter region of tissue inhibitor of metalloproteinase 1 (TIMP1), CUR increased TIMP1 gene expression and, therefore, attenuated cardiac fibrosis and inflammation (178). By increasing the expression of TIMP1, CUR reduced the expression of the TIMP1 inhibitory target, called metalloproteinase 2 (MMP2), which contributes to inflammatory signaling (178, 325). Another study by Marquardt and colleagues revealed that CUR inhibits the activity of NF-κB in human hematopoietic Raji cells via inhibition of histone deacetylase, HDAC1 and HDAC3 (179). In addition to histone acetylation, CUR was shown to repress DNA hypermethylation at CpG sites within the promoter region of peroxisome proliferator-activated receptor-alpha (PPARα) by inhibiting DNMTs in a non-alcoholic fatty liver disease, which results in upregulated PPARα expression and reduced liver cell death (180, 181)., PPARα, a transcription factor that predominates in the liver, regulates the expression of genes involved in inflammation and several metabolic processes (326–330).
A plant flavonoid, quercetin (3, 3′, 4′, 5, 7-pentahydroxyflvanone) is found in citrus fruits, apples, onions, red grapes and tea (184, 185). This phenolic compound exhibits an anti-inflammatory, antioxidant, chemopreventive and neuroprotective properties (331). The estimated dosage ranges from 50 to 800 mg/day, which is mainly dependent on dietary habits (332). The anti-inflammatory potential of quercetin can be discerned in different cell types of both animal and human models (333). Quercetin plays a modulatory, biphasic and regulatory action in inflammation and immunity, and possesses an immunosuppressive effect on dendritic cell function (334, 335).
Quercetin negatively regulates LPS-induced TLR4 signaling (Table 1, Figures 1 and 2). It reduces TLR4 expression and prevents NF-κB translocation to the nucleus in macrophages and human PBMCs, thereby ameliorating the inflammatory response (166, 186, 187, 336). Also, it inhibits COX-2 and iNOS gene expression in vitro, and significantly reduces the production of proinflammatory cytokines via MAP kinases and NF-κB pathway in LPS-activated macrophages (188). In this highlight, Cho et al. show that this flavonoid suppressed the activation of phosphorylated ERK kinase and p38 MAP kinase (not JNK MAP kinase), and inhibited NF-κB activation by stabilizing the NF-κB/IκB complex in LPS-treated RAW 264.7 macrophages (188).
Few reports have addressed the modulation of inflammatory-related miRNAs by quercetin (184) (Table 1 and Figure 3). The effect of quercetin and its main metabolites on miR-155 have been evaluated in LPS-stimulated macrophages. It has been reported that quercetin and its metabolite, known as isorhamnetin, downregulate the mRNA and protein levels of the proinflammatory mediators, including TNF-α, IL-6, and IL-1β by decreasing the expression of miR-155, which represents a mechanism by which this polyphenol may inhibit the activation of NF-κB, contributing to the containment of the inflammatory process (120, 189). These two compounds can also indirectly inhibit the NF-κB pathway through activating the Nrf2 signal transduction cascade (120) (Table 1 and Figure 2). Nonetheless, the direct activity of miR-155 on Nrf2 signaling remains a promising area for further investigations in order to determine whether Nrf2 signaling might be directly affected by miR-155. In this context, Saadatmandi et al. revealed that miR-155 targets Bach1, a Nrf2 signaling repressor (189). Furthermore, quercetin has been shown to upregulate miR-146a, which is followed by a reduction in the levels of NF-κB and the downregulation of TNF-α, IL-6 and IL-17 (151, 191, 192). Additionally, quercetin’s protective effect on inflammation-dependent cancer was evident in its suppression of COX-2 in breast cancer cells via its attenuation of p300/HAT-mediated signaling (190) (Table 1 and Figure 3).
Apigenin (API, 4’,5,7-dihydroxyflavone) is a plant-derived flavonoid abundant in many fruits and vegetables, including parsley, celery, and chamomile tea (193, 194). This compound is widely used as an anti-inflammatory agent, and it also has anticancer and cardioprotective properties (337, 338).
In different in vitro and in vivo inflammation models induced by LPS, API causes a reduction in the levels of NO, TNF-α, IL-6, IL-1β and PGs through the inhibition of iNOS, NF-κB and COX-2 activities (148) (Table 1 and Figures 1 and 2). A number of recent studies have highlighted the importance of API as a potent M1/M2 modulator, downregulating NO production and proinflammatory cytokines (197, 339). Favoring M2 polarization, API can also block the inflammatory processes in adipose tissue macrophages through PPARγ (196) (Table 1). In obese animal models, it plays a role in suppressing obesity-related inflammation (197). Balex et al. showed that API exerts its anti-inflammatory activity in LPS-mediated acute lung injury through inhibiting the gene expression of COX-2 and NF-κB (198) (Table 1 and Figures 1 and 2). Whereas, Zhao et al. revealed API effect in neuro-inflammation; it reduces oxidative stress, downregulates the TLR4/NF-κB signaling pathway, decreases the levels of IL-6 and TNF-α, and inhibits mitochondria-mediated neuron apoptosis (195) (Table 1 and Figures 1 and 2). In LPS-induced macrophages, miR-155 is downregulated by API treatment through inhibiting NF-κB (199) (Table 1 and Figure 3). Another study showed that succeeding miR-155 suppression by API, an observed upregulation of two miR-155 targets, namely FOXO3a (Forkhead Box O3a), an inhibitor of NF-κB, and SMAD2 (smooth-muscle-actin and MAD-related proteins 2), a suppressor of TNF-α and iNOS inflammatory molecules (199, 201, 202). In addition to miRNA-related epigenetic modifications, API successfully demethylated the promotor region of Nrf2, resulting in an increased Nrf2 mRNA and protein expression in JB6 P+ skin epidermal cells. This effect was mediated through a reduction in the expression of the epigenetic proteins, DNMT1, DNMT3a, DNMT3b, as well as some HDACs (200) (Table 1 and Figure 3).
An isoflavonoid, Genistein is primarily obtained from soy-based foods, red clover and legumes (203). Amongst many isoflavones, genistein is widely recognized for its antioxidant and anti-inflammatory functions, as well as, its anticancer and anti-proliferative activity (340). This compound has been successfully utilized as an immunosuppressive agent in vitro and in vivo (341). Unfortunately, the oral bioavailability of genistein and its plasma concentrations were very low in vivo, which might affect its efficacy, and interfere with the consistency of its pharmacological results in clinical trials (342–345).
Genistein prevented endothelial inflammatory damage by blocking NF-κB and the expression of the proinflammatory cytokine, IL-6, and adhesion molecule, ICAM-1 (204) (Table 1 and Figures 1 and 2). A recent study by Xu et al. highlighted the inhibitory effect of genistein on angiotensin II-induced vascular smooth muscle cell inflammation, in which angiotensin II induced the expression of NF-κB, C-reactive protein (CRP), and the phosphorylation of ERK1/2 and p-38, leading to atherosclerotic inflammation, which is reversed after genistein treatment (205) (Table 1 and Figures 1 and 2). Moreover, this isoflavonoid compound enhanced PPAR-γ expression, displaying a cardiovascular protective property via the regulatory crosstalk between p38/ERK1/2-PPARγ-NFκB signaling pathways (205). Another study linked the inhibitory effect of Genistein on LPS-mediated NF-κB activation in macrophages to the activation of adenosine monophosphate kinase (AMPK), which lead to repression of inflammation (207) (Table 1). Besides, Genistein is involved in hindering TLR4 dimerization, and thus abolishes MyD88 or TRIF dependent pathways, inactivating NF-κB and inhibiting its translocation into the nucleus, which, in turn, prevents proinflammatory cytokines transcription (206) (Table 1 and Figures 1 and 2). In line with its anti-inflammatory properties, Genistein possesses notable anti-oxidant activities via activating Nrf2/NQO1 pathway (206) (Table 1 and Figure 2).
Similar to the previously mentioned polyphenols, genistein plays a role in modulating TLR4-responsive miRNAs. Through miR-155/SOCS1-mediated suppression of the NF-κB signaling, genistein reversed ox-LDL-induced inflammation in human umbilical vein endothelial cells (HUVECs) (208) (Table 1 and Figure 3).
One of the most popular polyphenolic catechinsis EGCG, which is mainly found in green tea, onions, apple skin, and plums (209). There is an emerging group of evidence on its biological activity where it was found to exert remarkable anti-inflammatory, antioxidant, anticancer, and antiangiogenetic effects (346–350). A number of reports introduced the prominent anti-inflammatory effect of EGCG (351).
EGCG has been reported to suppress LPS-induced TLR4 signaling, and to reduce the receptor expression (210) (Table 1 and Figures 1 and 2). A potent activator of Nrf2, EGCG plays a critical role in inflammation-induced oxidative stress (211) (Table 1 and Figure 2). A recent study showed that EGCG attenuated airway inflammation in asthmatic mice by significantly reducing asthmatic symptoms, including inflammatory cell infiltration, and inflammatory induced levels of TNF-α, IL-2, and IL-6 (212). Another report highlighted the cytoprotective role of EGCG on neuronal cells against microglia-induced cytotoxicity and in suppressing amyloid β-induced TNF-α release (213).
Epigenetically, EGCG was also reported to mediate inflammation by regulating histone acetylation (214) (Table 1 and Figure 3). For example, a study by Liu and colleagues showed that EGCG regulated p300 and HDACs I and II differential binding at promoter regions of the NF-κB subunit p65 gene, and, consequently, decreased proinflammatory gene expression in stress-induced endothelial cells (214). It should be duly noted that endothelial cells may become impaired due to persistent inflammation, resulting in atherosclerosis and fibrogenesis (215, 216). Since NF-κB inflammatory action is dependent on p300-, HDAC1- and HDAC2-mediated actions (103, 104), EGCG, therefore, successfully prevents inflammation in endothelial cells via regulating histone acetylation at proinflammatory gene promoters (214). Collectively, this data outlines the anti-inflammatory activity of EGCG in regulating both inflammation homeostasis and cardiac function via histone acetylation-dependent mechanisms.
Emodin is an anthraquinone that is abundant in plants, such as buckthorn, knotweed and rhubarb (217). As a traditional Chinese medicine, emodin has been used for viral and bacterial infections, and for kidney and gastrointestinal disorders (27).
Emodin significantly inhibits induced TLR4, MyD88 and TRAF6 expressions in inflammatory pneumonia model, and decreases p38/JNK MAPK phosphorylation and NF-κB p65 nuclear translocation, whereas it activates Nrf2 pathway, thereby suppressing inflammation (218) (Table 1 and Figure 1). Emodin also blocks the nuclear translocation of signal transducer and activator of transcription 1 (STAT1), IRF5, and NF-κB-p65 in M1 macrophages, whereas it inhibits STAT6 and IRF4 in M2 macrophages (219). Macrophage hyperpolarization into either M1 or M2 phenotype is detrimental (352, 353). Most phytochemicals usually target M1 hyperpolarized macrophages. However, emodin is able to restore the balance between M1 and M2 hyperpolarization through epigenetic balancing of macrophage activation (219).
At the epigenetic level (Table 1 and Figure 3), recent reports showed that emodin increases H3K27 trimethylation in activated macrophages (219). This was evident at the promoter regions of the inflammatory signaling genes, iNOS, TNF-α, IL-6 and IRF4 in macrophages, to the end result of a reduction in their expression (219). Pertaining to its chelating properties, emodin is considered an HDAC inhibitor that chelates zinc ions within HDAC catalytic domains, thus regulating proinflammatory signaling cascades (354). It, therefore, increases histone acetylation by decreasing HDAC I and II activities (220). For instance, emodin was recently shown to block the proinflammatory signaling molecule, NF-κB, and pyroptosis in cardiac myocytes through HDAC inhibition, since NF-κB activity is regulated by HDACs (220, 221). In this regard, HDAC inhibition has been shown to prevent/reverse cardiac dysfunction in pre-clinical animal models of heart failure (222–225). Through this mechanism, emodin successfully attenuated NOD-, LRR- and pyrin-domain containing protein 3 (NLRP3) inflammasome pathway in hypoxic-induced heart cells (221), noting that NLRP3 inflammasome is involved in the synthesis of proinflammatory byproducts, and mediating inflammation-induced cell death or pyroptosis (355).
Flavonoids with three phenolic rings, anthocyanins(ACNs) are involved in food pigmentation, and are found in berries, grapes, and potatoes (27). Recently, anthocyanin-rich foods have proved their metabolic efficacy in humans (230–232), and have been effectively used to alleviate fatty liver and inflammation (228, 229).
Notably, ACNs ameliorate neuroinflammation by decreasing TLR4 expression and inactivating NF-κB, reducing proinflammatory mediators, such as iNOS and TNF-α (226). They also inhibit oxidative stress by activating the Nrf2/HO-1 signaling pathway (227) (Table 1 and Figure 1).
Not only this, but also ACNs exhibit epigenetic modulation capacity (Table 1 and Figure 3). Protecting the liver by mediating changes in histone acetylation, anthocyanin-rich extract induced histone H3 acetylation at lysine residues K9, K14 and K18 and decreased liver fibrosis in rats exposed to carbon tetrachloride (236, 237). Indeed, K9 and K14 acetylation is essential for proper liver function (356). Therefore, anthocyanins were able to decrease liver fibrosis by regulating gene expression through histone acetylation. Another study showed that ACNs modulate HDAC and HAT activities (233–235). However, the link between anthocyanin-mediated histone acetylation and HDAC or HAT activities remains unclear (236, 237). Notably, increased HAT activity and therefore, hyperacetylation of H3K9/14 at the promotor site of TNF-α, was shown to be associated with liver inflammation, leading to fibrosis in obesity-induced mice (357). That’s why most phytochemicals exhibiting hepatoprotective properties are dependent on HAT inhibition (358). In this context, ACNs have been reported to attenuate proinflammatory TNF-α signaling and gene expression in murine livers (238). Not only this, but also they decrease HAT activity and, hence, TNF-α signaling outside the liver (233). Nonetheless, future research should be directed towards ACNs blocking effects of inflammation-dependent liver fibrosis via HAT inhibition.
A polyphenolic hydroxybenzoic acid derivative, ellagic acid (EA) is widely present in fruits, like raspberries and strawberries, as well as in mushrooms, and nuts (239). Indeed, EA treatment has a potent anti-inflammatory activity; it has been shown to reverse inflammation and adiposity (241–244).
EA is known to reduce inflammatory response and oxidative stress by inhibiting TLR4 and activating Nrf2 (240) (Table 1 and Figure 1). Besides, EA has been previously reported to inhibit coactivator-associated arginine methyltransferase-1 (CARM1) activity, which is a methyltransferase enzyme involved in metabolic dysfunction and NF-κB-mediated inflammation (249–251) (Table 1 and Figure 3). Aside from CARM1 expression, EA remarkably attenuates differentiation-induced hyperdimethylation of histone 3 arginine 17 in human adipose-derived stem cells (245) (Table 1 and Figure 3). Obesity and excessive adipose tissue accumulation are common triggers of downstream inflammation and metabolic impairment (252). In differentiated adipocytes, Kang et al. showed that EA treatment suppressed PPAR-γ, a CARM1 target and an important regulator of adipogenesis and adipocyte function that is partially coactivated by CARM1-mediated histone methylation (245–248). Therefore, the role of EA treatment in repressing PPAR-γ may be beneficial in the long term. In this regard, some reports have shown that the anti-inflammatory activity of EA is, in part, PPAR-γ-dependent (359). From this perspective, further research is required to determine its molecular targets, and to assess the extent of EA effectiveness in chronic inflammation. It is also imperative if we are to better understand how this polyphenolic compound exerts its anti-inflammatory actions epigenetically.
A lipophilic diterpenoid, Tanshinone IIA (Tan IIA) is extracted from the root of Salvia miltiorrhiza Bunge (Danshen) (253). Traditionally, this herb was used to promote blood circulation, and a study by Shang et al. illustrated its cardioprotective actions (360, 361). Injections of sodium Tan IIA sulfonate was as an adjuvant in cardiovascular diseases in China (362).
Tan IIA treatment has been indicated to suppress the p38 MAPK signaling pathway, thus reducing arrhythmogenesis and mortality incidences following myocardial infarction, and enhancing cardiac function (254, 255) (Table 1 and Figure 1). Additionally, this diterpenoid significantly inhibits the expression of several inflammatory mediators, such as TLR4, MyD88, GM-CSF, and proinflammatory cytokines, including IL-1β, TNF-α, and COX-2, thereby attenuating LPS-mediated TLR4/NF-κB pathway activation (256, 257). One of the most important mechanisms for inhibiting inflammation by Tan IIA is through reducing miR-155 expression, which is an upstream regulator in the process of inflammation (256–258) (T1 & Figure 3). Tan IIA successfully downregulated the levels of inflammatory factors, namely, CRP, ox-LDL, C-C Motif Chemokine Ligand 2 (CCL-2), cluster of differentiation 40 (CD40), and matrix metalloproteinase-2 (MMP-2), as well as the proinflammatory cytokines, IL-1β, IL-6, IL-12, and TNF-α that were induced by Porphyromonas gingivalis infection (259). This inhibitory effect of Tan IIA has been associated with a relative inhibition of over-expressed miR-146 and miR-155, thereby exerting significant anti-inflammatory and antioxidant properties, and ameliorating P. gingivalis-induced atherosclerosis (259). Another study illustrated that treatment with Tan IIA suppressed the proliferation of inflammation-induced colon cancer cells through inhibiting miR-155 levels in macrophages (260). This confirms the anti-inflammatory activity of Tan IIA via miRNAs, especially miR-155, which is suggested by the aforementioned studies to be a target of Tan IIA in inflammation. Nonetheless, the pharmacological activity of Tan IIA is not limited to miRNAs (363). Wang and colleagues showed that Tan IIA successfully inhibits murine skin epidermal JB6 cells transformation induced by TPA (12-O-tetradecanoylphorbol-13-acetate); this inhibition has been made possible through epigenetic regulation of the Nrf2 signaling pathway (261). Tan IIA treatment decreased methylation at Nrf2 promoter by inhibiting the protein levels of DNMT1, DNMT3a, DNMT3b, as well as, HDAC3 and HDAC activities, thus increasing Nrf2 expression and its downstream targets (261) (Table 1 and Figure 3).
Monoterepnoids, carvacrol and thymol are isomers derived from the essential oil of Origanum vulgare L. or wild bergamot (253). The essential oil of Origanum vulgare L. was used, initially, for treating cold and heatstroke, and the bergamot was used as an anesthetic and antiemetic (253). Further research studied the anti-inflammatory activity of these two bioactive ingredients, carvacol and thymol (253).
Car and Thy reduce the activation of TLR4/NF-κB signaling pathway in vivo and in vitro in inflammatory reactions, whereas increase the expression of antioxidants, such as SOD1 and GSH through Nrf2 Activation and attenuates oxidative damage (262, 263) (Table 1 and Figure 1).
Furthermore, by regulating miRNAs and inflammatory factors, Car and Thy showed significant suppression of the allergic inflammation associated with asthma (Table 1 and Figure 3). As illustrated by Khosravi and colleagues study, the inflammation-associated miRNAs, including miR-155, miR- 146a and miR-21 were overexpressed in a chitin-induced model, whereas SOCS1 and SHIP1 (miR-155 targets and negative regulators of TLR-mediated inflammation) are inhibited by chitin (264). Car/Thy reversed the induced expression of TLR4, as well as, miR-155, miR-146a, and miR-21, and reversed their effects on the negative regulators (SOCS1 and SHIP1) (264) (Table 1 and Figures 1 and 3). According to these findings, the anti-inflammatory effects of Car/Thy have been linked to targeting TLR-responsive miRNAs. However, the direct targets of Car/Thy still need further investigations to be determined, since they are not reported clearly (253).
Extracted from the oleo-gum-resin of Boswellia serrata, boswellic acids (BAs) are traditionally known to promote blood circulation and relieve pain. Boswellic acids contain various ingredients, among which is 3-acetyl-11-keto-β-boswellic acid (AKBA), which possesses a potent anti-inflammatory activity (265, 266).
BAs downregulate the expression of hepatic TLR4 receptor and MyD88, and suppress that of NF-κB p65 and p-JNK in hepatotoxicity model (267) (Table 1 and Figure 1). Additionally, BAs upregulate Nrf2 and HO-1 expression, thereby protect liver from DOX-induced oxidative injury (268). Likewise, BAs also exhibit neuroprotective effect by modulating Nrf2/HO-1 pathway (269) (Table 1 and Figure 1). Moreover, BAs have been reported to attenuate LPS-induced neuroinflammation, with the same effect as that of dexamethasone (265). They reduce miR-155 and IκB-α expression levels, while increasing SOCS-1, resulting in decreased apoptotic activity and amyloid protein genesis, which is, when accumulated, responsible for chronic inflammation (265) (Table 1 and Figure 3). It’s worth mentioning that miR-155 regulation by BAs has been suggested to be a possible mechanism underlying the effective role of BAs in neuroinflammatory disorders. But, the exact targets of BAs remain an open question that requires further verification (253).
SFN is one of the highly studied plant-derived isothiocyanate organosulfur compounds (270). Characterized by the presence of a sulfocyanate group (N=C=S), SFN is found in cruciferous vegetables from the Brassicaceae family, including broccoli, cabbage, cauliflower, and kale (270, 271). SFN precursor, glucoraphanin is hydrolyzed to isothiocyanate by myrosinases enzymes (270, 364). Usually, SFN is used as a synthetic racemic mixture of D, L-SFN in research studies (271, 365, 366). In contrast to polyphenols, this isothiocyanate compound was reported to have relatively high bioavailability (around 80%), with oral dosage ranging from 20 to 40 mg in clinical trials (123, 274, 342, 367). Several reports have revealed its potential anti-inflammatory and antioxidant properties (274). In chronic inflammatory diseases, SFN exerts its potent immunomodulatory effect through targeting monocytes/macrophages and stimulating the nuclear factor erythroid-derived 2-like 2 (Nrf2) antioxidant defense pathway (272, 273). Clinically, over 1900 trials on SFN are published in PubMed (123).
SFN has a dual action in modulating the redox system and immune imbalance, through interacting with Nrf2 and NF-κB signaling pathways (123) (Table 1 and Figures 1 and 2). These two key transcription factors (Nrf2 and NF-κB) act both independently and dependently via their “cross talk”, which is not yet fully understood (123, 368). Several studies have pinpointed the crosstalk between Nrf2 and NF-κB pathways (271). For example, Li et al. showed that SFN activated the Nrf2 pathway through inhibiting Nrf2 ubiquitination, and concomitantly reduced NF-κB and AP-1 expression, thus restoring endogenous antioxidant levels and reducing inflammatory damage in an experimental autoimmune encephalomyelitis mice model (274–279). SFN is considered an indirect antioxidant, because it is not involved in quenching free radicals and ROS but, it upregulates some phase II enzymes by enhancing Nrf2 activity (271, 280–283). Moreover, the anti-inflammatory effects of SFN have been demonstrated in the form of reduced levels of LPS-induced proinflammatory mediators, such as TNF-α, iNOS, and COX-2 (168, 271, 284, 285). A recent study demonstrated, for the first time, the ability of SFN to suppress the direct binding between NF-κB and its consensus sequence in DNA via its thiol groups, therefore suppressing LPS-induced proinflammatory mediators in macrophages (286). Nonetheless, further mechanistic studies are recommended to investigate this cross-talk machinery in more details in different inflammatory models. In addition, one of the novel underlying anti-inflammatory mechanisms of SFN is its ability to suppress TLR4 oligomerization in a thiol-dependent manner in macrophages, where SFN formed adducts with cysteine residues in the extracellular domain of TLR4 (123, 168, 284, 285, 287). SFN suppressed both ligand-induced and ligand-independent oligomerization of TLR4 (287). Oligomerization is an important step for TLR4 activation and recruitment of adaptor molecules; therefore, the reactivity of SFN to the sulfhydryl moiety contributes to its inhibitory activities and subsequent downregulation of NF-κB activation (287). Similar to CUR, SFN antagonizes LPS binding to the TLR4/MD-2 complex by selectively competing on MD-2, a large hydrophobic pocket where LPS binds and mediates TLR4 dimerization (168, 284, 285, 287, 288). Furthermore, previous studies have shown that SFN successfully prevented carcinogenesis, which is partly attributed to its potent anti-inflammatory properties (274).
Importantly, studies on the miRNA level indicated that SFN significantly attenuated miRNA-155 and miRNA-146a levels in LPS-stimulated RAW264.7 macrophages in a dose-dependent manner (289, 290) (Table 1 and Figure 3). Additionally, SFN exhibited chemo preventive effects that have been regulated, in part, through inhibiting inflammation via changes in DNA methylation (291–295) (Table 1 and Figure 3). SFN attenuated DNMT-mediated DNA hypermethylation at the promoter region of Nrf2, thus increasing Nrf2 expression and subsequently, decreasing neurological inflammation, and inflammatory-associated cytokines, IL-6 and IL-1β (296) (Table 1 and Figure 3). Hence, SFN regulates Nrf2 activity through DNA hypomethylation, resulting in blocking proinflammatory signaling.
Another aliphatic isothiocyanate, is allyl-isothiocyanate (AITC), which is obtained from its precursor sinigrin, and is abundant in different brassica species such as mustard, wasabi, and horseradish (120).
Recent reports show that AITC enhances the nuclear translocation of Nrf2, which is known to repress the expression of NF-κB; subsequently, these actions upregulate HO1 (Nrf2 target gene) that further suppresses inflammation (120) (Table 1 and Figures 1 and 2).
Post-transcriptionally, AITC was reported to attenuate inflammation in murine RAW 264.7 macrophages by repressing miR-155 levels, and thus lowering target proinflammatory mediators, such as iNOS, TNF-α, and IL-1β (120) (Table 1 and Figure 3). Another study by Wagner et al. suggested that AITC exerted its anti-inflammatory actions via targeting miR-155, which acts on NF-κB and Nrf2 signaling pathways (289) (Table 1 and Figure 3). In spite of this, the link between the miR-155/NF-κB/Nrf2 signaling pathway and AITC treatment remains questionable, therefore requiring future verification (289). Although Nrf2 can also be regulated by other miRNAs, including miR-27a, miR-142-5p, miR-153, miR-144, miR-93, and miR-28, the effect of these miRNAs in AITC-mediated macrophage regulation is still not clear to date (369–371). Collectively, based on miRNA-regulation, the mechanism of naturally-derived isothiocyanates on stimulated macrophages can be a potential breakthrough in the inflammation research arena.
A conjugated aromatic aldehyde, cinnamaldehyde is isolated from the barks of Cinnamomum cassia Presl (253). Traditionally, this plant is often used to resolve symptoms of weakness, but recently, this bioactive compound has been used in cancer, cerebrovascular diseases, and ulcerative colitis (300, 372, 373). This action has been attributed to its anti-inflammatory properties, as well as, non-coding RNA regulatory functions.
CA mitigates inflammation symptoms by decreasing the levels of ROS, COX-2, and the proinflammatory cytokines, such as TNF-α, IL-1β, and IL-6, in addition to NLRP3 inflammasome, as well as miR-155 and miR-21 in macrophages (253) (Table 1 and Figures 1 and 3). Further studies linked the suppression of IL-1β and IL-6 to miR-21 or miR-155 inhibition, revealing that these inflammatory factors are positively regulated by miR-21 or miR-155 (300). Through this suppressive activity, CA performs its protective effect in ulcerative colitis (300). Similar to the aforementioned plant-derived polyphenols, CA downregulated several proinflammatory cytokines, including IL-1β, IL-6, TNF-α, and other inflammatory factors, such as NO and PGs, as well as COX-2, iNOS and NF-κB (148). Such effects of CA have been indicated in a plethora of studies using in vitro and in vivo models of LPS-induced inflammation (147). When CA was compared with the other aforementioned natural TLR4 modulators, some differences in the underlying molecular mechanisms could be highlighted. For example, CA contains α, β-unsaturated carbonyl moieties that act as electrophilic carbon and react with the nucleophilic thiol of a cysteine, subsequently forming Michael adducts with protein targets, thus disrupting the TLR4/MD-2 heterodimer. In contrast to curcumin, CA disrupts the TLR4/MD-2 heterodimer through the formation of covalent adducts (169, 297). In comparison to SFN, CA contains a different electrophilic group, an α,β-unsaturated carbonyl moiety, instead of the isothiocyanate moiety, which also forms covalent adducts with cysteine thiols of MD-2 (288). The pharmacological properties of the abovementioned phytochemicals on TLR4 signaling have been determined by mass analysis experiments on purified receptors (298). CA inhibition of LPS-induced NF-κB and IRF3 is the main molecular mechanism underlying its anti-inflammatory function (374). CA, however, does not show a significant action when NF-κB is activated by MyD88 and IKKβ downstream effectors, which confirms its upstream activity on the TLR4/MD-2 extracellular complex (374). Moreover, similar to SFN, CA inhibits LPS-induced TLR4 receptor oligomerization and activates Nrf2 pathway (298, 299) (Table 1 and Figure 1).
In a nutshell, this review focuses on the mechanisms of different plant-derived compounds involved in regulating inflammation within different cell types, such as macrophages, cardiac myocytes, adipose tissue and epithelial cells. While these mechanisms are pleiotropic and usually target multiple sites of action in the TLR4 pathway, some of them are common between different phytochemicals, and are significant to their established anti-inflammatory effects. In this context, the mentioned phytocompounds, collectively, inhibit the expression of TLR4 receptor, and block the activation of NF-κB transcription factor, thus inhibiting the generation of the downstream pro-inflammatory cytokines, such as TNF-α, IL-6 and IL-1β, and the free radicals, such as NO and ROS. Furthermore, unlike their inhibitory effect on TLR4/NF-κB cascade, these aforementioned phytochemicals activate Nrf-2 signaling pathway, which in turn, inhibits oxidative stress. On the other hand, there are also specialized specific mechanisms that are more relevant to some phytochemicals over others. For example, genisten and CA hinder the ligand-induced oligomerization of TLR4 receptor, while SFN suppresses both ligand-induced and ligand-independent oligomerization of TLR4. Moreover, CUR, SFN and CA competitively antagonize LPS binding to MD-2 binding site, and thus disrupt the TLR4/MD-2 heterodimer. This leads to the inhibition of both MyD88 and TRIF-dependent pathways, which are involved in TLR4 cascade activation. At the epigenetic level, plant-derived compounds undergo noteworthy epigenetic modifications that fine tune their anti-inflammatory functions. In this review, the majority of the mentioned phytochemicals significantly downregulated miR-155 and miR-21, both reduce NF-κB activity and suppress inflammatory factors, such as TNF-α, IL-6 and MAPKs, attenuating the induced-inflammation. Beside the regulation of miRNA, the mentioned phytochemicals play a critical role in decreasing the expression of pro-inflammatory genes and suppressing NF- κB-dependent inflammation in different cells through inhibiting HATs (e.g., p300 HAT) and HDACs (e.g., HDAC I, II and III). Furthermore, some phytochemicals, such as CUR, API, tanshinone IIA, and SFN downregulate DNMTs expression (e.g., DNMT 1, 3a, and 3b), and repress DNA hypermethylation at the promoter region of some genes, such as Nrf2, increasing their expression. To the contrary, RES, for instance, upregulates DNMT 3a and 3b expression, and thus mediates DNA hypermethylation at inflammatory genes, and therefore attenuates inflammation. Collectively, these stated similarities and differences between phytochemicals are what substantiate, in general, their remarkable anti-inflammatory effects, and in specific, their differential therapeutic potential and efficacy against certain inflammatory diseases in different cell types. Last but not least, as noticed, TLR4/NF-κB and Nrf2 pathways are crucial mechanisms that are targeted by almost all the mentioned phytochemicals. Taken together, future research should further investigate the crosstalk between the two pathways.
In conclusion, an evolving group of evidence has shown how plant-bioactive compounds significantly influence both health and disease via epigenetic modification. Although this report covered different phytochemicals used in distinct inflammatory experimental models and focused, in particular, on their epigenetic regulatory mechanisms, few studies have translated the epigenetic-mediated actions of these plant derivatives to human models and little is understood about gene regulation mediated by natural products in health and disease (27). Thus, it will be of great benefit if future research is directed to revealing the most effective phytochemicals in attenuating inflammatory-associated dysregulations, neurodegenerative and cardiovascular diseases. To our knowledge, how epigenetic regulation targets certain genes, in specific, is still elusive. For instance, the mechanism underlying histone modifications or DNA methylation in determining the patterns of transcription of PRRs and its downstream signaling mediators throughout infection and lineage differentiation remains unclear, and requires deep mechanistic investigations (29). As such, future research on innate immune cells should focus on identifying specific epitranscriptomes. Furthermore, post-transcriptional modifications in RNA have been known to modulate different signal transductions (29). In this regard, a mechanistic study on the effect(s) of the currently reported phytochemicals on an array of inflammatory-associated miRNAs would be of great benefit to understand how these natural compounds differentially modulate these miRNAs, and how they ultimately attenuate inflammatory processes. For instance, previous reports on the regulation of miR-155 by polyphenols give a clear insight into flavonoid mechanism in alleviating inflammation (120). There are still other miRNAs, which are involved in macrophage inflammatory responses, that have not been studied yet with phytochemicals, among which are miR-467b, miR-33s, and miR-125a (120). Also, studies have not revealed the effect of phytochemicals on histone acylation, in specific (27). This lack of information shed the light on the inevitability of investigating the mechanisms by which phytochemicals modulate histone acylation, and whether they could mitigate inflammation. In addition to epigenetic regulations, only few studies tackled the interactions taking place between phytochemicals (27); future work should therefore address phytochemical-phytochemical interactions, and elucidate the combined effect of these interactions during inflammation, particularly at the epigenetic level. Assessing phytochemical interactions sheds light on understanding the pharmacokinetics of these compounds, because these compounds usually vary in absorption, distribution, metabolism and excretion (ADME) inside the body (375, 376). Subsequently, studying pharmacokinetics variations between these phytochemicals is inevitable. This could unveil natural compounds with similar absorption rates and distribution sites to be further explored in combinations, and enable examining any potentiation of effect resulting from such combinations. Another consideration is the metabolic activity of these phytochemicals; whether the metabolites of parent phytochemicals possess a therapeutic activity or not. For example, in spite of emodin’s poor distribution to heart cells, its cardioprotective effect has been reported (221, 375). This observation has been explained in terms of metabolomics, in which blood-circulating metabolites of parent compounds exert, an efficacious activity that is observed in tissues having undetectable concentrations of the parent compounds (27). Nonetheless, most phytochemical metabolites are usually non-bioavailable (376, 377). A fact that raises the necessity to delve deeper into the area of phytochemical-related metabolomics. The poor bioavailability of several phytochemicals, especially polyphenols, is another issue to be tackled in future research. Increasing the dose of these phytochemicals is not always safe; it has to be mentioned that they, like other treatments, may exhibit toxic side effects at higher concentrations (378–380). This, therefore, brings up questions as to the phytochemical-associated toxidrome at high concentrations, and should engender explorative efforts to search for new technologies that enhance bioavailability at lower doses, such as solubilizers and targeted drug-delivery systems (381). Multiple reports suggest that co-administering plant-bioactive compounds with other pharmaceutical treatments resulted in differential gene expression, which might imply that their actions are epigenetically-related (27, 382). For example, when curcumin is co-administered with reinstate (SAHA; an FDA-approved HDAC inhibitor), an enhanced effect has been observed in ameliorating antibody-dependent neurodegeneration than that of single treatments (382). Another report showed that resveratrol co-treatment with metformin (anti-diabetic drug) successfully ameliorated inflammation, and other metabolic dysregulations in diabetic mice (383, 384). Therefore, there is a need to understand the exact epigenetic mechanism(s) that cause such synergism, and further explore approved, commercially available medications in conjunction with natural compounds.
HS and AA designed the outline of the review. MY and AA edited and proofread the review. HS, MY, and AA contributed to the writing, proof reading, and creating the table of this review. All authors contributed to the article and approved the submitted version.
This work is supported by AUC graduate research grant to HAS and MHY and AUC internal grant [FY19-RG (1-18)], Egyptian Academy of Scientific Research and Technology Grants (JESOR-2019-5305), and (ASRT-2019-4903), a Bartlett Fund for Critical Challenges Grant and an AUC COVID-19 Pandemic Research & Innovation Initiative Grant to AA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol (2010) 125(2):S3–S23. doi: 10.1016/j.jaci.2009.12.980
2. Marshall JS, Warrington R, Watson W, Kim HL. An introduction to immunology and immunopathology. Allergy Asthma Clin Immunol (2018) 14(2):49. doi: 10.1186/s13223-018-0278-1
4. Turvey SE, Broide DH. Innate immunity. J Allergy Clin Immunol (2010) 125(2):S24–32. doi: 10.1016/j.jaci.2009.07.016
5. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
6. Cronkite DA, Strutt TM. The regulation of inflammation by innate and adaptive lymphocytes. J Immunol Res (2018) 2018. doi: 10.1155/2018/1467538
7. Bonilla FA, Oettgen HC. Adaptive immunity. J Allergy Clin Immunol (2010) 125(2):S33–40. doi: 10.1016/j.jaci.2009.09.017
8. Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol (2012) 4(3):a006049. doi: 10.1101/cshperspect.a006049
9. Kizil C, Kyritsis N, Brand M. Effects of inflammation on stem cells: together they strive? EMBO Rep (2015) 16(4):416–26. doi: 10.15252/embr.201439702
10. Cignarella A, Bolego C. Innate Immunity in Inflammation. In: Riccardi C, Levi-Schaffer F, Tiligada E, editors. Immunopharmacology and Inflammation. Cham: Springer International Publishing (2018). p. 179–90.
11. Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, Epel ES, et al. Geroscience: linking aging to chronic disease. Cell (2014) 159(4):709–13. doi: 10.1016/j.cell.2014.10.039
12. Cole J, Morris P, Dickman MJ, Dockrell DH. The therapeutic potential of epigenetic manipulation during infectious diseases. Pharmacol Ther (2016) 167:85–99. doi: 10.1016/j.pharmthera.2016.07.013
13. Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation (2011) 123(19):2145–56. doi: 10.1161/CIRCULATIONAHA.110.956839
14. Bayarsaihan D. Epigenetic mechanisms in inflammation. J Dental Res (2011) 90(1):9–17. doi: 10.1177/0022034510378683
15. Chen C-Y, Kao C-L, Liu C-M. The cancer prevention, anti-inflammatory and anti-oxidation of bioactive phytochemicals targeting the TLR4 signaling pathway. Int J Mol Sci (2018) 19(9):2729. doi: 10.3390/ijms19092729
16. Cen X, Liu S, Cheng K. The role of toll-like receptor in inflammation and tumor immunity. Front Pharmacol (2018) 9:878. doi: 10.3389/fphar.2018.00878
17. Frantz S, Falcao-Pires I, Balligand JL, Bauersachs J, Brutsaert D, Ciccarelli M, et al. The innate immune system in chronic cardiomyopathy: a European Society of Cardiology (ESC) scientific statement from the Working Group on Myocardial Function of the ESC. Eur J Heart Fail (2018) 20(3):445–59. doi: 10.1002/ejhf.1138
18. Takeda K, Akira S. Toll-like receptors in innate immunity. Int Immunol (2005) 17(1):1–14. doi: 10.1093/intimm/dxh186
19. Guven-Maiorov E, Keskin O, Gursoy A, Nussinov R. A structural view of negative regulation of the toll-like receptor-mediated inflammatory pathway. Biophys J (2015) 109(6):1214–26. doi: 10.1016/j.bpj.2015.06.048
20. Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, et al. RIP1 is an essential mediator of Toll-like receptor 3–induced NF-κB activation. Nat Immunol (2004) 5(5):503. doi: 10.1038/ni1061
21. Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, et al. IKKϵ and TBK1 are essential components of the IRF3 signaling pathway. Nat Immunol (2003) 4(5):491. doi: 10.1038/ni921
22. Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, et al. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF-κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol (2003) 171(8):4304–10. doi: 10.4049/jimmunol.171.8.4304
23. Häcker H, Redecke V, Blagoev B, Kratchmarova I, Hsu L-C, Wang GG, et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature (2006) 439(7073):204. doi: 10.1038/nature04369
24. Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and-independent antiviral response. Nature (2006) 439(7073):208. doi: 10.1038/nature04374
25. Li H, Sun B. Toll-like receptor 4 in atherosclerosis. J Cell Mol Med (2007) 11(1):88–95. doi: 10.1111/j.1582-4934.2007.00011.x
26. Toshchakov V, Jones BW, Perera P-Y, Thomas K, Cody MJ, Zhang S, et al. TLR4, but not TLR2, mediates IFN-β–induced STAT1α/β-dependent gene expression in macrophages. Nat Immunol (2002) 3(4):392. doi: 10.1038/ni774
27. Evans LW, Stratton MS, Ferguson BS. Dietary natural products as epigenetic modifiers in aging-associated inflammation and disease. Natural Prod Rep (2020) 37(5):653–76. doi: 10.1039/C9NP00057G
28. Perkins DJ, Patel MC, Blanco JC, Vogel SN. Epigenetic mechanisms governing innate inflammatory responses. J Interferon Cytokine Res (2016) 36(7):454–61. doi: 10.1089/jir.2016.0003
29. Zhang Q, Cao X. Epigenetic regulation of the innate immune response to infection. Nat Rev Immunol (2019) 19(7):417–32. doi: 10.1038/s41577-019-0151-6
30. Allis CD, Jenuwein T. The molecular hallmarks of epigenetic control. Nat Rev Genet (2016) 17(8):487. doi: 10.1038/nrg.2016.59
31. Kouzarides T. Chromatin modifications and their function. Cell (2007) 128(4):693–705. doi: 10.1016/j.cell.2007.02.005
32. Eberharter A, Becker PB. Histone acetylation: a switch between repressive and permissive chromatin. EMBO Rep (2002) 3(3):224–9. doi: 10.1093/embo-reports/kvf053
33. Villagra A, Sotomayor E, Seto E. Histone deacetylases and the immunological network: implications in cancer and inflammation. Oncogene (2010) 29(2):157–73. doi: 10.1038/onc.2009.334
34. Barnes PJ. Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc (2009) 6(8):693–6. doi: 10.1513/pats.200907-071DP
35. Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell (2009) 138(1):129–45. doi: 10.1016/j.cell.2009.05.047
36. Wu S-Y, Chiang C-M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J Biol Chem (2007) 282(18):13141–5. doi: 10.1074/jbc.R700001200
37. Huang B, Yang X-D, Zhou M-M, Ozato K, Chen L-F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol Cell Biol (2009) 29(5):1375–87. doi: 10.1128/MCB.01365-08
38. Sullivan K, Reddy A, Dietzmann K, Suriano A, Kocieda V, Stewart M, et al. Epigenetic regulation of tumor necrosis factor alpha. Mol Cell Biol (2007) 27(14):5147–60. doi: 10.1128/MCB.02429-06
39. Yasmin R, Siraj S, Hassan A, Khan AR, Abbasi R, Ahmad N. Epigenetic regulation of inflammatory cytokines and associated genes in human malignancies. Mediators Inflamm (2015) 2015. doi: 10.1155/2015/201703
40. Roger T, Lugrin J, Le Roy D, Goy G, Mombelli M, Koessler T, et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood J Am Soc Hematol (2011) 117(4):1205–17. doi: 10.1182/blood-2010-05-284711
41. Song W, Tai Y, Tian Z, Hideshima T, Chauhan D, Nanjappa P, et al. HDAC inhibition by LBH589 affects the phenotype and function of human myeloid dendritic cells. Leukemia (2011) 25(1):161–8. doi: 10.1038/leu.2010.244
42. Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airway inflammation. Curr Opin Immunol (2007) 19(6):694–700. doi: 10.1016/j.coi.2007.07.016
43. Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet (2012) 13(5):343–57. doi: 10.1038/nrg3173
44. Barski A, Cuddapah S, Cui K, Roh T-Y, Schones DE, Wang Z, et al. High-resolution profiling of histone methylations in the human genome. Cell (2007) 129(4):823–37. doi: 10.1016/j.cell.2007.05.009
45. Lan F, Shi Y. Epigenetic regulation: methylation of histone and non-histone proteins. Sci China C Life Sci (2009) 52(4):311–22. doi: 10.1007/s11427-009-0054-z
46. Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun Z-W, Schmid M, et al. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature (2000) 406(6796):593–9. doi: 10.1038/35020506
47. Kouzarides T. Histone methylation in transcriptional control. Curr Opin Genet Dev (2002) 12(2):198–209. doi: 10.1016/S0959-437X(02)00287-3
48. Austenaa L, Barozzi I, Chronowska A, Termanini A, Ostuni R, Prosperini E, et al. The histone methyltransferase Wbp7 controls macrophage function through GPI glycolipid anchor synthesis. Immunity (2012) 36(4):572–85. doi: 10.1016/j.immuni.2012.02.016
49. Zhao D, Zhang Q, Liu Y, Li X, Zhao K, Ding Y, et al. H3K4me3 demethylase Kdm5a is required for NK cell activation by associating with p50 to suppress SOCS1. Cell Rep (2016) 15(2):288–99. doi: 10.1016/j.celrep.2016.03.035
50. Xia M, Liu J, Wu X, Liu S, Li G, Han C, et al. Histone methyltransferase Ash1l suppresses interleukin-6 production and inflammatory autoimmune diseases by inducing the ubiquitin-editing enzyme A20. Immunity (2013) 39(3):470–81. doi: 10.1016/j.immuni.2013.08.016
51. Liu Y, Zhang Q, Ding Y, Li X, Zhao D, Zhao K, et al. Histone lysine methyltransferase Ezh1 promotes TLR-triggered inflammatory cytokine production by suppressing Tollip. J Immunol (2015) 194(6):2838–46. doi: 10.4049/jimmunol.1402087
52. De Santa F, Totaro MG, Prosperini E, Notarbartolo S, Testa G, Natoli G. The histone H3 lysine-27 demethylase Jmjd3 links inflammation to inhibition of polycomb-mediated gene silencing. Cell (2007) 130(6):1083–94. doi: 10.1016/j.cell.2007.08.019
53. Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev (2011) 25(10):1010–22. doi: 10.1101/gad.2037511
54. Boyes J, Bird A. DNA methylation inhibits transcription indirectly via a methyl-CpG binding protein. Cell (1991) 64(6):1123–34. doi: 10.1016/0092-8674(91)90267-3
55. Serra RW, Fang M, Park SM, Hutchinson L, Green MR. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife (2014) 3:e02313. doi: 10.7554/eLife.02313
56. Rountree MR, Bachman KE, Baylin SB. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nat Genet (2000) 25(3):269–77. doi: 10.1038/77023
57. Jylhävä J, Nevalainen T, Marttila S, Jylhä M, Hervonen A, Hurme M. Characterization of the role of distinct plasma cell-free DNA species in age-associated inflammation and frailty. Aging Cell (2013) 12(3):388–97. doi: 10.1111/acel.12058
58. Bellizzi D, D’Aquila P, Montesanto A, Corsonello A, Mari V, Mazzei B, et al. Global DNA methylation in old subjects is correlated with frailty. Age (2012) 34(1):169–79. doi: 10.1007/s11357-011-9216-6
59. Agrawal A, Tay J, Yang G-E, Agrawal S, Gupta S. Age-associated epigenetic modifications in human DNA increase its immunogenicity. Aging (Albany NY) (2010) 2(2):93. doi: 10.18632/aging.100121
60. Hahn MA, Hahn T, Lee D-H, Esworthy RS, Kim B-w, Riggs AD, et al. Methylation of polycomb target genes in intestinal cancer is mediated by inflammation. Cancer Res (2008) 68(24):10280–9. doi: 10.1158/0008-5472.CAN-08-1957
61. Takahashi K, Sugi Y, Hosono A, Kaminogawa S. Epigenetic regulation of TLR4 gene expression in intestinal epithelial cells for the maintenance of intestinal homeostasis. J Immunol (2009) 183(10):6522–9. doi: 10.4049/jimmunol.0901271
62. Li X, Zhang Q, Ding Y, Liu Y, Zhao D, Zhao K, et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat Immunol (2016) 17(7):806–15. doi: 10.1038/ni.3464
63. El Gazzar M, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem (2010) 285(27):20940–51. doi: 10.1074/jbc.M110.115063
64. Guo Y, Su Z-Y, Kong A-NT. Current perspectives on epigenetic modifications by dietary chemopreventive and herbal phytochemicals. Curr Pharmacol Rep (2015) 1(4):245–57. doi: 10.1007/s40495-015-0023-0
65. He X, Jing Z, Cheng G. MicroRNAs: new regulators of Toll-like receptor signalling pathways. BioMed Res Int (2014) 2014. doi: 10.1155/2014/945169
66. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature (2010) 466(7308):835–40. doi: 10.1038/nature09267
67. Quinn SR, O’Neill LA. A trio of microRNAs that control Toll-like receptor signalling. Int Immunol (2011) 23(7):421–5. doi: 10.1093/intimm/dxr034
68. Epanchintsev A, Jung P, Menssen A, Hermeking H. Inducible microRNA expression by an all-in-one episomal vector system. Nucleic Acids Res (2006) 34(18):e119–e. doi: 10.1093/nar/gkl624
69. O’neill LA, Sheedy FJ, McCoy CE. MicroRNAs: the fine-tuners of Toll-like receptor signalling. Nat Rev Immunol (2011) 11(3):163. doi: 10.1038/nri2957
70. Li Y, Shi X. MicroRNAs in the regulation of TLR and RIG-I pathways. Cell Mol Immunol (2013) 10(1):65. doi: 10.1038/cmi.2012.55
71. Taganov KD, Boldin MP, Chang K-J, Baltimore D. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc Natl Acad Sci (2006) 103(33):12481–6. doi: 10.1073/pnas.0605298103
72. Jurkin J, Schichl YM, Koeffel R, Bauer T, Richter S, Konradi S, et al. miR-146a is differentially expressed by myeloid dendritic cell subsets and desensitizes cells to TLR2-dependent activation. J Immunol (2010) 184(9):4955–65. doi: 10.4049/jimmunol.0903021
73. Boldin MP, Taganov KD, Rao DS, Yang L, Zhao JL, Kalwani M, et al. miR-146a is a significant brake on autoimmunity, myeloproliferation, and cancer in mice. J Exp Med (2011) 208(6):1189–201. doi: 10.1084/jem.20101823
74. Doxaki C, Kampranis SC, Eliopoulos AG, Spilianakis C, Tsatsanis C. Coordinated regulation of miR-155 and miR-146a genes during induction of endotoxin tolerance in macrophages. J Immunol (2015) 195(12):5750–61. doi: 10.4049/jimmunol.1500615
75. Schulte LN, Westermann AJ, Vogel J. Differential activation and functional specialization of miR-146 and miR-155 in innate immune sensing. Nucleic Acids Res (2012) 41(1):542–53. doi: 10.1093/nar/gks1030
76. Asirvatham AJ, Gregorie CJ, Hu Z, Magner WJ, Tomasi TB. MicroRNA targets in immune genes and the Dicer/Argonaute and ARE machinery components. Mol Immunol (2008) 45(7):1995–2006. doi: 10.1016/j.molimm.2007.10.035
77. Asirvatham AJ, Magner WJ, Tomasi TB. miRNA regulation of cytokine genes. Cytokine (2009) 45(2):58–69. doi: 10.1016/j.cyto.2008.11.010
78. Liu X, Rennard SI. MicroRNA and cytokines. Mol Cell Pharmacol (2011) 3(3):143–51. doi: 10.1201/b11037
79. Bhaumik D, Scott G, Schokrpur S, Patil C, Campisi J, Benz C. Expression of microRNA-146 suppresses NF-κB activity with reduction of metastatic potential in breast cancer cells. Oncogene (2008) 27(42):5643. doi: 10.1038/onc.2008.171
80. Nakasa T, Miyaki S, Okubo A, Hashimoto M, Nishida K, Ochi M, et al. Expression of microRNA-146 in rheumatoid arthritis synovial tissue. Arthritis Rheum (2008) 58(5):1284–92. doi: 10.1002/art.23429
81. Sonkoly E, Ståhle M, Pivarcsi A. MicroRNAs and immunity: novel players in the regulation of normal immune function and inflammation. Semin Cancer Biol (2008) 18(2):131–40. doi: 10.1016/j.semcancer.2008.01.005
82. Chatzikyriakidou A, Voulgari PV, Georgiou I, Drosos AA. A polymorphism in the 3’-UTR of interleukin-1 receptor-associated kinase (IRAK1), a target gene of miR-146a, is associated with rheumatoid arthritis susceptibility. Joint Bone Spine (2010) 77(5):411–3. doi: 10.1016/j.jbspin.2010.05.013
83. Hou J, Wang P, Lin L, Liu X, Ma F, An H, et al. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J Immunol (2009) 183(3):2150–8. doi: 10.4049/jimmunol.0900707
84. Nahid MA, Satoh M, Chan EK. MicroRNA in TLR signaling and endotoxin tolerance. Cell Mol Immunol (2011) 8(5):388. doi: 10.1038/cmi.2011.26
85. Li S, Yue Y, Xu W, Xiong S. MicroRNA-146a represses mycobacteria-induced inflammatory response and facilitates bacterial replication via targeting IRAK-1 and TRAF-6. PloS One (2013) 8(12):e81438. doi: 10.1371/journal.pone.0081438
86. Lin L, Hou J, Ma F, Wang P, Liu X, Li N, et al. Type I IFN inhibits innate IL-10 production in macrophages through histone deacetylase 11 by downregulating microRNA-145. J Immunol (2013) 191(7):3896–904. doi: 10.4049/jimmunol.1203450
87. Starczynowski DT, Kuchenbauer F, Argiropoulos B, Sung S, Morin R, Muranyi A, et al. Identification of miR-145 and miR-146a as mediators of the 5q–syndrome phenotype. Nat Med (2010) 16(1):49. doi: 10.1038/nm.2054
88. Nahid MA, Yao B, Dominguez-Gutierrez PR, Kesavalu L, Satoh M, Chan EK. Regulation of TLR2-mediated tolerance and cross-tolerance through IRAK4 modulation by miR-132 and miR-212. J Immunol (2013) 190(3):1250–63. doi: 10.4049/jimmunol.1103060
89. Tang Y, Luo X, Cui H, Ni X, Yuan M, Guo Y, et al. MicroRNA-146a contributes to abnormal activation of the type I interferon pathway in human lupus by targeting the key signaling proteins. Arthritis Rheum (2009) 60(4):1065–75. doi: 10.1002/art.24436
90. Loubaki L, Chabot D, Paré I, Drouin M, Bazin R. MiR-146a potentially promotes IVIg-mediated inhibition of TLR4 signaling in LPS-activated human monocytes. Immunol Lett (2017) 185:64–73. doi: 10.1016/j.imlet.2017.02.015
91. Park H, Huang X, Lu C, Cairo MS, Zhou X. MicroRNA-146a and microRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J Biol Chem (2015) 290(5):2831–41. doi: 10.1074/jbc.M114.591420
92. Shin VY, Jin H, Ng EK, Cheng AS, Chong WW, Wong CY, et al. NF-κB targets miR-16 and miR-21 in gastric cancer: involvement of prostaglandin E receptors. Carcinogenesis (2010) 32(2):240–5. doi: 10.1093/carcin/bgq240
93. Chen Y, Chen J, Wang H, Shi J, Wu K, Liu S, et al. HCV-induced miR-21 contributes to evasion of host immune system by targeting MyD88 and IRAK1. PloS Pathog (2013) 9(4):e1003248. doi: 10.1371/journal.ppat.1003248
94. Sheedy FJ, Palsson-McDermott E, Hennessy EJ, Martin C, O’leary JJ, Ruan Q, et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat Immunol (2010) 11(2):141. doi: 10.1038/ni.1828
95. Cheung ST, So EY, Chang D, Ming-Lum A, Mui AL. Interleukin-10 inhibits lipopolysaccharide induced miR-155 precursor stability and maturation. PloS One (2013) 8(8):e71336. doi: 10.1371/journal.pone.0071336
96. O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci (2007) 104(5):1604–9. doi: 10.1073/pnas.0610731104
97. Tili E, Michaille J-J, Cimino A, Costinean S, Dumitru CD, Adair B, et al. Modulation of miR-155 and miR-125b levels following lipopolysaccharide/TNF-α stimulation and their possible roles in regulating the response to endotoxin shock. J Immunol (2007) 179(8):5082–9. doi: 10.4049/jimmunol.179.8.5082
98. Alivernini S, Gremese E, McSharry C, Tolusso B, Ferraccioli G, McInnes IB, et al. MicroRNA-155—at the critical interface of innate and adaptive immunity in arthritis. Front Immunol (2018) 8:1932. doi: 10.3389/fimmu.2017.01932
99. Strassheim D, Kim J-Y, Park J-S, Mitra S, Abraham E. Involvement of SHIP in TLR2-induced neutrophil activation and acute lung injury. J Immunol (2005) 174(12):8064–71. doi: 10.4049/jimmunol.174.12.8064
100. O’Connell RM, Kahn D, Gibson WS, Round JL, Scholz RL, Chaudhuri AA, et al. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity (2010) 33(4):607–19. doi: 10.1016/j.immuni.2010.09.009
101. Androulidaki A, Iliopoulos D, Arranz A, Doxaki C, Schworer S, Zacharioudaki V, et al. The kinase Akt1 controls macrophage response to lipopolysaccharide by regulating microRNAs. Immunity (2009) 31(2):220–31. doi: 10.1016/j.immuni.2009.06.024
102. Ceppi M, Pereira PM, Dunand-Sauthier I, Barras E, Reith W, Santos MA, et al. MicroRNA-155 modulates the interleukin-1 signaling pathway in activated human monocyte-derived dendritic cells. Proc Natl Acad Sci (2009) 106(8):2735–40. doi: 10.1073/pnas.0811073106
103. Ashburner BP, Westerheide SD, Baldwin AS. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol Cell Biol (2001) 21(20):7065–77. doi: 10.1128/MCB.21.20.7065-7077.2001
104. Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell (2002) 9(3):625–36. doi: 10.1016/S1097-2765(02)00477-X
105. Wei H, Wang B, Miyagi M, She Y, Gopalan B, Huang D-B, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc Natl Acad Sci (2013) 110(33):13516–21. doi: 10.1073/pnas.1311784110
106. Ferguson BS, Harrison BC, Jeong MY, Reid BG, Wempe MF, Wagner FF, et al. Signal-dependent repression of DUSP5 by class I HDACs controls nuclear ERK activity and cardiomyocyte hypertrophy. Proc Natl Acad Sci (2013) 110(24):9806–11. doi: 10.1073/pnas.1301509110
107. Yoo S, Kim K, Nam H, Lee D. Discovering health benefits of phytochemicals with integrated analysis of the molecular network, chemical properties and ethnopharmacological evidence. Nutrients (2018) 10(8):1042. doi: 10.3390/nu10081042
108. Mohammed MS, Osman WJ, Garelnabi EA, Osman Z, Osman B, Khalid HS, et al. Secondary metabolites as anti-inflammatory agents. J Phytopharmacol (2014) 3(4):275–85.
109. Egert S, Bosy-Westphal A, Seiberl J, Kürbitz C, Settler U, Plachta-Danielzik S, et al. Quercetin reduces systolic blood pressure and plasma oxidised low-density lipoprotein concentrations in overweight subjects with a high-cardiovascular disease risk phenotype: a double-blinded, placebo-controlled cross-over study. Br J Nutr (2009) 102:1065–74. doi: 10.1017/S0007114509359127
110. Seifried HE, Anderson DE, Fisher EI, Milner JA. A review of the interaction among dietary antioxidants and reactive oxygen species. J Nutr Biochem (2007) 18(9):567–79. doi: 10.1016/j.jnutbio.2006.10.007
111. Islam M, Alam F, Solayman M, Khalil M, Kamal MA, Gan SH. Dietary phytochemicals: natural swords combating inflammation and oxidation-mediated degenerative diseases. Oxid Med Cell Longevity (2016) 2016. doi: 10.1155/2016/5137431
112. Perez-Gregorio R, Simal-Gandara J. A critical review of bioactive food components, and of their functional mechanisms, biological effects and health outcomes. Curr Pharm Des (2017) 23(19):2731–41. doi: 10.2174/1381612823666170317122913
113. Bagul M, Srinivasa H, Kanaki N, Rajani M. Antiinflammatory activity of two Ayurvedic formulations containing guggul. Indian J Pharmacol (2005) 37(6):399. doi: 10.4103/0253-7613.19080
114. Gautam R, Jachak SM. Recent developments in anti-inflammatory natural products. Med Res Rev (2009) 29(5):767–820. doi: 10.1002/med.20156
115. Schink A, Neumann J, Leifke AL, Ziegler K, Fröhlich-Nowoisky J, Cremer C, et al. Screening of herbal extracts for TLR2-and TLR4-dependent anti-inflammatory effects. PloS One (2018) 13(10):e0203907. doi: 10.1371/journal.pone.0203907
116. Dong B, Liu C, Xue R, Wang Y, Sun Y, Liang Z, et al. Fisetin inhibits cardiac hypertrophy by suppressing oxidative stress. J Nutr Biochem (2018) 62:221–9. doi: 10.1016/j.jnutbio.2018.08.010
117. Strunz CMC, Roggerio A, Cruz PL, Pacanaro AP, Salemi VMC, Benvenuti LA, et al. Down-regulation of fibroblast growth factor 2 and its co-receptors heparan sulfate proteoglycans by resveratrol underlies the improvement of cardiac dysfunction in experimental diabetes. J Nutr Biochem (2017) 40:219–27. doi: 10.1016/j.jnutbio.2016.11.015
118. Arpón A, Milagro FI, Razquin C, Corella D, Estruch R, Fitó M, et al. Impact of consuming extra-virgin olive oil or nuts within a Mediterranean diet on DNA methylation in peripheral white blood cells within the PREDIMED-Navarra randomized controlled trial: a role for dietary lipids. Nutrients (2018) 10(1):15. doi: 10.3390/nu10010015
119. Arpon A, Riezu-Boj JI, Milagro F, Marti A, Razquin C, Martínez-González M, et al. Adherence to Mediterranean diet is associated with methylation changes in inflammation-related genes in peripheral blood cells. J Physiol Biochem (2016) 73(3):445–55. doi: 10.1007/s13105-017-0552-6
120. Shao D, Lian Z, Di Y, Zhang L, shahid riaz Rajoka M, Zhang Y, et al. Dietary compounds have potential in controlling atherosclerosis by modulating macrophage cholesterol metabolism and inflammation via miRNA. NPJ Sci Food (2018) 2(1):1–9. doi: 10.1038/s41538-018-0022-8
121. Slezak-Prochazka I, Durmus S, Kroesen B-J, van den Berg A. MicroRNAs, macrocontrol: regulation of miRNA processing. Rna (2010) 16(6):1087–95. doi: 10.1261/rna.1804410
122. Srivastava SK, Arora S, Averett C, Singh S, Singh AP. Modulation of microRNAs by phytochemicals in cancer: underlying mechanisms and translational significance. BioMed Res Int (2015) 2015. doi: 10.1155/2015/848710
123. Houghton CA. Sulforaphane: Its “Coming of Age” as a Clinically Relevant Nutraceutical in the Prevention and Treatment of Chronic Disease. Oxid Med Cell Longev (2019) 2019. doi: 10.1155/2019/2716870
124. Fürst R, Zündorf I. Plant-derived anti-inflammatory compounds: hopes and disappointments regarding the translation of preclinical knowledge into clinical progress. Mediators Inflamm (2014) 2014. doi: 10.1155/2014/146832
125. Amri A, Chaumeil J, Sfar S, Charrueau C. Administration of resveratrol: what formulation solutions to bioavailability limitations? J Control Release (2012) 158(2):182–93. doi: 10.1016/j.jconrel.2011.09.083
126. Augustin MA, Sanguansri L, Lockett T. Nano-and micro-encapsulated systems for enhancing the delivery of resveratrol. Ann N Y Acad Sci (2013) 1290(1):107–12. doi: 10.1111/nyas.12130
127. Neves A R, Lucio M LC, Lima J, Reis S. Resveratrol in medicinal chemistry: a critical review of its pharmacokinetics, drug-delivery, and membrane interactions. Curr Med Chem (2012) 19(11):1663–81. doi: 10.2174/092986712799945085
128. Santos AC, Veiga F, Ribeiro AJ. New delivery systems to improve the bioavailability of resveratrol. Expert Opin Drug Deliv (2011) 8(8):973–90. doi: 10.1517/17425247.2011.581655
129. Walle T. Bioavailability of resveratrol. Ann N Y Acad Sci (2011) 1215(1):9–15. doi: 10.1111/j.1749-6632.2010.05842.x
130. Wenzel E, Somoza V. Metabolism and bioavailability of trans-resveratrol. Mol Nutr Food Res (2005) 49(5):472–81. doi: 10.1002/mnfr.200500010
131. Singh AP, Singh R, Verma SS, Rai V, Kaschula CH, Maiti P, et al. Health benefits of resveratrol: Evidence from clinical studies. Med Res Rev (2019) 39(5):1851–91. doi: 10.1002/med.21565
132. Maroon JC, Bost JW, Maroon A. Natural anti-inflammatory agents for pain relief. Surg Neurol Int (2010) 1(80):80–9. doi: 10.4103/2152-7806.73804
133. Elmali N, Baysal O, Harma A, Esenkaya I, Mizrak B. Effects of resveratrol in inflammatory arthritis. Inflammation (2007) 30(1-2):1–6. doi: 10.1007/s10753-006-9012-0
134. Hollman PC, Feskens EJ, Katan MB. Tea flavonols in cardiovascular disease and cancer epidemiology. Proc Soc Exp Biol Med (1999) 220(4):198–202. doi: 10.3181/00379727-220-44365
135. Jancśo G, Király E, Jancsó-Gábor A. Direct evidence for an axonal site of action of capsaicin. Naunyn Schmiedebergs Arch Pharmacol (1980) 313(1):91–4. doi: 10.1007/BF00505809
136. Malmstrom K, Daniels S, Kotey P, Seidenberg BC, Desjardins PJ. Comparison of rofecoxib and celecoxib, two cyclooxygenase-2 inhibitors, in postoperative dental pain: a randomized, placebo-and active-comparator-controlled clinical trial. Clin Ther (1999) 21(10):1653–63. doi: 10.1016/S0149-2918(99)80045-9
137. Maroon JC, Bost JW. ω-3 Fatty acids (fish oil) as an anti-inflammatory: an alternative to nonsteroidal anti-inflammatory drugs for discogenic pain. Surg Neurol (2006) 65(4):326–31. doi: 10.1016/j.surneu.2005.10.023
138. Stanbury RM, Graham EM. Systemic corticosteroid therapy—side effects and their management. Br J Ophthalmol (1998) 82(6):704–8. doi: 10.1136/bjo.82.6.704
139. Sumpio BE, Cordova AC, Berke-Schlessel DW, Qin F, Chen QH. Green tea, the “Asian paradox,” and cardiovascular disease. J Am Coll Surg (2006) 202(5):813–25. doi: 10.1016/j.jamcollsurg.2006.01.018
140. Dutartre P. Inflammasomes and natural ingredients towards new anti-inflammatory agents. Molecules (2016) 21(11):1492. doi: 10.3390/molecules21111492
141. Tili E, Michaille J-J. Resveratrol, microRNAs, inflammation, and cancer. J Nucleic Acids (2011) 2011. doi: 10.4061/2011/102431
142. Yang Y, Li S, Yang Q, Shi Y, Zheng M, Liu Y, et al. Resveratrol reduces the proinflammatory effects and lipopolysaccharide-induced expression of HMGB1 and TLR4 in RAW264. 7 cells. Cell Physiol Biochem (2014) 33(5):1283–92. doi: 10.1159/000358696
143. Tong W, Chen X, Song X, Chen Y, Jia R, Zou Y, et al. Resveratrol inhibits LPS−induced inflammation through suppressing the signaling cascades of TLR4−NF−κB/MAPKs/IRF3. Exp Ther Med (2020) 19(3):1824–34. doi: 10.3892/etm.2019.8396
144. Alarcon De La Lastra C, Villegas I. Resveratrol as an anti-inflammatory and anti-aging agent: Mechanisms and clinical implications. Mol Nutr Food Res (2005) 49(5):405–30. doi: 10.1002/mnfr.200500022
145. Kundu JK, Shin YK, Surh Y-J. Resveratrol modulates phorbol ester-induced pro-inflammatory signal transduction pathways in mouse skin in vivo: NF-κB and AP-1 as prime targets. Biochem Pharmacol (2006) 72(11):1506–15. doi: 10.1016/j.bcp.2006.08.005
146. Surh Y-J, Chun K-S, Cha H-H, Han SS, Keum Y-S, Park K-K, et al. Molecular mechanisms underlying chemopreventive activities of anti-inflammatory phytochemicals: down-regulation of COX-2 and iNOS through suppression of NF-κB activation. Mutat Res Fundam Mol Mech Mutagen (2001) 480:243–68. doi: 10.1016/S0027-5107(01)00183-X
147. Azab A, Nassar A, Azab AN. Anti-inflammatory activity of natural products. Molecules (2016) 21(10):1321. doi: 10.3390/molecules21101321
148. Karunaweera N, Raju R, Gyengesi E, Münch G. Plant polyphenols as inhibitors of NF-κB induced cytokine production—a potential anti-inflammatory treatment for Alzheimer’s disease? Front Mol Neurosci (2015) 8:24. doi: 10.3389/fnmol.2015.00024
149. Ma C, Wang Y, Shen A, Cai W. Resveratrol upregulates SOCS1 production by lipopolysaccharide-stimulated RAW264. 7 macrophages by inhibiting miR-155. Int J Mol Med (2017) 39(1):231–7. doi: 10.3892/ijmm.2016.2802
150. Tili E, Michaille J-J, Adair B, Alder H, Limagne E, Taccioli C, et al. Resveratrol decreases the levels of miR-155 by upregulating miR-663, a microRNA targeting JunB and JunD. Carcinogenesis (2010) 31(9):1561–6. doi: 10.1093/carcin/bgq143
151. Kocic H, Damiani G, Stamenkovic B, Tirant M, Jovic A, Tiodorovic D, et al. Dietary compounds as potential modulators of microRNA expression in psoriasis. Ther Adv Chronic Dis (2019) 10:2040622319864805. doi: 10.1177/2040622319864805
152. Maugeri A, Barchitta M, Mazzone MG, Giuliano F, Basile G, Agodi A. Resveratrol modulates SIRT1 and DNMT functions and restores LINE-1 methylation levels in ARPE-19 cells under oxidative stress and inflammation. Int J Mol Sci (2018) 19(7):2118. doi: 10.3390/ijms19072118
153. Gao Z, Ye J. Inhibition of transcriptional activity of c-JUN by SIRT1. Biochem Biophys Res Commun (2008) 376(4):793–6. doi: 10.1016/j.bbrc.2008.09.079
154. Cheng C-Y, Hsieh H-L, Hsiao L-D, Yang C-M. PI3-K/Akt/JNK/NF-κB is essential for MMP-9 expression and outgrowth in human limbal epithelial cells on intact amniotic membrane. Stem Cell Res (2012) 9(1):9–23. doi: 10.1016/j.scr.2012.02.005
155. Aggarwal BB, Kumar A, Bharti AC. Anticancer potential of curcumin: preclinical and clinical studies. Anticancer Res (2003) 23(1/A):363–98.
156. Menon VP, Sudheer AR. Antioxidant and anti-inflammatory properties of curcumin. In: . The molecular targets and therapeutic uses of curcumin in health and disease. Springer (2007). p. 105–25.
157. Perez G. Anti-inflammatory activity of compounds isolated from plants. Sci World J (2001) 1:713–84. doi: 10.1100/tsw.2001.77
158. IBRAHIM R, BARRON D. Phenylpropanoids. Methods in plant biochemistry. 1. Elsevier (1989) p. 75–111.
159. Lin X, Bai D, Wei Z, Zhang Y, Huang Y, Deng H, et al. Curcumin attenuates oxidative stress in RAW264. 7 cells by increasing the activity of antioxidant enzymes and activating the Nrf2-Keap1 pathway. PloS One (2019) 14(5):e0216711. doi: 10.1371/journal.pone.0216711
160. Sahebkar A, Serban M-C, Ursoniu S, Banach M. Effect of curcuminoids on oxidative stress: A systematic review and meta-analysis of randomized controlled trials. J Funct Foods (2015) 18:898–909. doi: 10.1016/j.jff.2015.01.005
161. Banach M, Serban C, Aronow WS, Rysz J, Dragan S, Lerma EV, et al. Lipid, blood pressure and kidney update 2013. Int Urol Nephrol (2014) 46(5):947–61. doi: 10.1007/s11255-014-0657-6
162. Panahi Y, Alishiri GH, Parvin S, Sahebkar A. Mitigation of systemic oxidative stress by curcuminoids in osteoarthritis: results of a randomized controlled trial. J Diet Suppl (2016) 13(2):209–20. doi: 10.3109/19390211.2015.1008611
163. Priyadarsini KI, Maity DK, Naik G, Kumar MS, Unnikrishnan M, Satav J, et al. Role of phenolic OH and methylene hydrogen on the free radical reactions and antioxidant activity of curcumin. Free Radic Biol Med (2003) 35(5):475–84. doi: 10.1016/S0891-5849(03)00325-3
164. Lubbad A, Oriowo M, Khan I. Curcumin attenuates inflammation through inhibition of TLR-4 receptor in experimental colitis. Mol Cell Biochem (2009) 322(1-2):127–35. doi: 10.1007/s11010-008-9949-4
165. Guimarães MR, Leite FRM, Spolidorio LC, Kirkwood KL, Rossa J. Curcumin abrogates LPS-induced pro-inflammatory cytokines in RAW 264.7 macrophages. Evidence for novel mechanisms involving SOCS-1,-3 and p38 MAPK. Arch Oral Biol (2013) 58(10):1309–17. doi: 10.1016/j.archoralbio.2013.07.005
166. Bastos DHM, Rogero MM, Arêas JAG. Mecanismos de ação de compostos bioativos dos alimentos no contexto de processos inflamatórios relacionados à obesidade. Arq Bras Endocrinol Metabol (2009) 53:646–56. doi: 10.1590/S0004-27302009000500017
167. Hatcher H, Planalp R, Cho J, Torti F, Torti S. Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci (2008) 65(11):1631–52. doi: 10.1007/s00018-008-7452-4
168. Molteni M, Bosi A, Rossetti C. Natural products with toll-like receptor 4 antagonist activity. Int J Inflam (2018) 2018. doi: 10.1155/2018/2859135
169. Gradišar H, Keber MM, Pristovšek P, Jerala R. MD-2 as the target of curcumin in the inhibition of response to LPS. J Leukoc Biol (2007) 82(4):968–74. doi: 10.1189/jlb.1206727
170. Youn HS, Saitoh SI, Miyake K, Hwang DH. Inhibition of homodimerization of Toll-like receptor 4 by curcumin. Biochem Pharmacol (2006) 72(1):62–9. doi: 10.1016/j.bcp.2006.03.022
171. Zhou Y, Zhang T, Wang X, Wei X, Chen Y, Guo L, et al. Curcumin modulates macrophage polarization through the inhibition of the toll-like receptor 4 expression and its signaling pathways. Cell Physiol Biochem (2015) 36(2):631–41. doi: 10.1159/000430126
172. Shah VO, Ferguson JE, Hunsaker LA, Deck LM, Vander Jagt DL. Natural products inhibit LPS-induced activation of pro-inflammatory cytokines in peripheral blood mononuclear cells. Natural Prod Res (2010) 24(12):1177–88. doi: 10.1080/14786410903112680
173. Tili E, Chiabai M, Palmieri D, Brown M, Cui R, Fernandes C, et al. Quaking and miR-155 interactions in inflammation and leukemogenesis. Oncotarget (2015) 6(28):24599. doi: 10.18632/oncotarget.5248
174. Ma F, Liu F, Ding L, You M, Yue H, Zhou Y, et al. Anti-inflammatory effects of curcumin are associated with down regulating microRNA-155 in LPS-treated macrophages and mice. Pharm Biol (2017) 55(1):1263–73. doi: 10.1080/13880209.2017.1297838
175. Morimoto T, Sunagawa Y, Kawamura T, Takaya T, Wada H, Nagasawa A, et al. The dietary compound curcumin inhibits p300 histone acetyltransferase activity and prevents heart failure in rats. J Clin Invest (2008) 118(3):868–78. doi: 10.1172/JCI33160
176. Feng B, Chen S, Chiu J, George B, Chakrabarti S. Regulation of cardiomyocyte hypertrophy in diabetes at the transcriptional level. Am J Physiol Endocrinol Metab (2008) 294(6):E1119–E26. doi: 10.1152/ajpendo.00029.2008
177. Sun H, Yang X, Zhu J, Lv T, Chen Y, Chen G, et al. Inhibition of p300-HAT results in a reduced histone acetylation and down-regulation of gene expression in cardiac myocytes. Life Sci (2010) 87(23-26):707–14. doi: 10.1016/j.lfs.2010.10.009
178. Hu J, Shen T, Xie J, Wang S, He Y, Zhu F. Curcumin modulates covalent histone modification and TIMP1 gene activation to protect against vascular injury in a hypertension rat model. Exp Ther Med (2017) 14(6):5896–902. doi: 10.3892/etm.2017.5318
179. Marquardt JU, Gomez-Quiroz L, Camacho LOA, Pinna F, Lee Y-H, Kitade M, et al. Curcumin effectively inhibits oncogenic NF-κB signaling and restrains stemness features in liver cancer. J Hepatol (2015) 63(3):661–9. doi: 10.1016/j.jhep.2015.04.018
180. Li Y, Tang D, Du Y, Cao C, Nie Y, Cao J, et al. Fatty liver mediated by PPAR-α DNA methylation can be reversed by a methylation inhibitor and curcumin. J Dig Dis (2018) 19:421–30. doi: 10.1111/1751-2980.12610
181. Maugeri A, Mazzone MG, Giuliano F, Vinciguerra M, Basile G, Barchitta M, et al. Curcumin modulates DNA methyltransferase functions in a cellular model of diabetic retinopathy. Oxid Med Cell Longev (2018) 2018. doi: 10.1155/2018/5407482
182. Reuter S, Gupta SC, Park B, Goel A, Aggarwal BB. Epigenetic changes induced by curcumin and other natural compounds. Genes Nutr (2011) 6(2):93–108. doi: 10.1007/s12263-011-0222-1
183. Esatbeyoglu T, Huebbe P, Ernst IM, Chin D, Wagner AE, Rimbach G. Curcumin—from molecule to biological function. Angew Chem Int Ed (2012) 51(22):5308–32. doi: 10.1002/anie.201107724
184. Quintanilha BJ, Reis BZ, Duarte GBS, Cozzolino SM, Rogero MM. Nutrimiromics: role of microRNAs and nutrition in modulating inflammation and chronic diseases. Nutrients (2017) 9(11):1168. doi: 10.3390/nu9111168
185. Oliveira TT, Campos KM, Cerqueira-Lima AT, Carneiro TCB, da Silva Velozo E, Melo ICAR, et al. Potential therapeutic effect of Allium cepa L. and quercetin in a murine model of Blomia tropicalis induced asthma. DARU J Pharm Sci (2015) 23(1):18. doi: 10.1186/s40199-015-0098-5
186. Marunaka Y, Marunaka R, Sun H, Yamamoto T, Kanamura N, Inui T, et al. Actions of quercetin, a polyphenol, on blood pressure. Molecules (2017) 22(2):209. doi: 10.3390/molecules22020209
187. Byun E-B, Yang M-S, Choi H-G, Sung N-Y, Song D-S, Sin S-J, et al. Quercetin negatively regulates TLR4 signaling induced by lipopolysaccharide through Tollip expression. Biochem Biophys Res Commun (2013) 431(4):698–705. doi: 10.1016/j.bbrc.2013.01.056
188. Cho S-Y, Park S-J, Kwon M-J, Jeong T-S, Bok S-H, Choi W-Y, et al. Quercetin suppresses proinflammatory cytokines production through MAP kinases and NF-κB pathway in lipopolysaccharide-stimulated macrophage. Mol Cell Biochem (2003) 243(1-2):153–60. doi: 10.1023/A:1021624520740
189. Boesch-Saadatmandi C, Loboda A, Wagner AE, Stachurska A, Jozkowicz A, Dulak J, et al. Effect of quercetin and its metabolites isorhamnetin and quercetin-3-glucuronide on inflammatory gene expression: role of miR-155. J Nutr Biochem (2011) 22(3):293–9. doi: 10.1016/j.jnutbio.2010.02.008
190. Xiao X, Shi D, Liu L, Wang J, Xie X, Kang T, et al. Quercetin suppresses cyclooxygenase-2 expression and angiogenesis through inactivation of P300 signaling. PloS One (2011) 6(8):e22934. doi: 10.1371/journal.pone.0022934
191. Lovendorf MB, Zibert JR, Hagedorn PH, Glue C, Odum N, Ropke MA, et al eds. Comparison of microRNA expression using different preservation methods of matched psoriatic skin samples. In: . JOURNAL OF INVESTIGATIVE DERMATOLOGY. 75 VARICK ST, 9TH FLR, NEW YORK, NY 10013-1917 USA: NATURE PUBLISHING GROUP.
192. Løvendorf MB, Zibert JR, Hagedorn PH, Glue C, Ødum N, Røpke MA, et al. Influence of quercetin-rich food intake on microRNA expression in lung cancer tissues. Cancer Epidemiol Prev Biomarkers (2012) 21(12):2176–84. doi: 10.1158/1055-9965.EPI-12-0745
193. Manach C, Scalbert A, Morand C, Rémésy C, Jiménez L. Polyphenols: food sources and bioavailability. Am J Clin Nutr (2004) 79(5):727–47. doi: 10.1093/ajcn/79.5.727
194. McKay DL, Blumberg JB. A review of the bioactivity and potential health benefits of chamomile tea (Matricaria recutita L.). Phytother Res: Int J Devoted to Pharmacol Toxicological Eval Natural Prod Derivatives. (2006) 20(7):519–30. doi: 10.1002/ptr.1900
195. Zhao F, Dang Y, Zhang R, Jing G, Liang W, Li Z. Apigenin attenuates acrylonitrile-induced neuro-inflammation in rats: Involved of inactivation of the TLR4/NF-κB signaling pathway. Int Immunopharmacol (2019) 75:105697. doi: 10.1016/j.intimp.2019.105697
196. Wang J, Liu Y-T, Xiao L, Zhu L, Wang Q, Yan T. Anti-inflammatory effects of apigenin in lipopolysaccharide-induced inflammatory in acute lung injury by suppressing COX-2 and NF-kB pathway. Inflammation (2014) 37(6):2085–90. doi: 10.1007/s10753-014-9942-x
197. Saqib U, Sarkar S, Suk K, Mohammad O, Baig MS, Savai R. Phytochemicals as modulators of M1-M2 macrophages in inflammation. Oncotarget (2018) 9(25):17937. doi: 10.18632/oncotarget.24788
198. Balez R, Steiner N, Engel M, Muñoz SS, Lum JS, Wu Y, et al. Neuroprotective effects of apigenin against inflammation, neuronal excitability and apoptosis in an induced pluripotent stem cell model of Alzheimer’s disease. Sci Rep (2016) 6:31450. doi: 10.1038/srep31450
199. Arango D, Diosa-Toro M, Rojas-Hernandez LS, Cooperstone JL, Schwartz SJ, Mo X, et al. Dietary apigenin reduces LPS-induced expression of miR-155 restoring immune balance during inflammation. Mol Nutr Food Res (2015) 59(4):763–72. doi: 10.1002/mnfr.201400705
200. Paredes-Gonzalez X, Fuentes F, Su Z-Y, Kong A-NT. Apigenin reactivates Nrf2 anti-oxidative stress signaling in mouse skin epidermal JB6 P+ cells through epigenetics modifications. AAPS J (2014) 16(4):727–35. doi: 10.1208/s12248-014-9613-8
201. Lin L, Hron JD, Peng SL. Regulation of NF-κB, Th activation, and autoinflammation by the forkhead transcription factor Foxo3a. Immunity (2004) 21(2):203–13. doi: 10.1016/j.immuni.2004.06.016
202. Sugiyama Y, Kakoi K, Kimura A, Takada I, Kashiwagi I, Wakabayashi Y, et al. Smad2 and Smad3 are redundantly essential for the suppression of iNOS synthesis in macrophages by regulating IRF3 and STAT1 pathways. Int Immunol (2012) 24(4):253–65. doi: 10.1093/intimm/dxr126
203. Desmawati D, Sulastri D. Phytoestrogens and their health effect. Open Access Maced J Med Sci (2019) 7(3):495. doi: 10.3889/oamjms.2019.086
204. Han S, Wu H, Li W, Gao P. Protective effects of genistein in homocysteine-induced endothelial cell inflammatory injury. Mol Cell Biochem (2015) 403(1-2):43–9. doi: 10.1007/s11010-015-2335-0
205. Xu L, Liu J-t, Li K, Wang S-y, Xu S. Genistein inhibits Ang II-induced CRP and MMP-9 generations via the ER-p38/ERK1/2-PPARγ-NF-κB signaling pathway in rat vascular smooth muscle cells. Life Sci (2019) 216:140–6. doi: 10.1016/j.lfs.2018.11.036
206. Tuli HS, Tuorkey MJ, Thakral F, Sak K, Kumar M, Sharma AK, et al. Molecular mechanisms of action of genistein in cancer: Recent advances. Front Pharmacol (2019) 10:1336–51. doi: 10.3389/fphar.2019.01336
207. Ji G, Zhang Y, Yang Q, Cheng S, Hao J, Zhao X, et al. Genistein suppresses LPS-induced inflammatory response through inhibiting NF-κB following AMP kinase activation in RAW 264.7 macrophages. PloS One (2012) 7(12):e53101. doi: 10.1371/journal.pone.0053101
208. Zhang H, Zhao Z, Pang X, Yang J, Yu H, Zhang Y, et al. Genistein protects against Ox-LDL-induced inflammation through microRNA-155/SOCS1-mediated repression of NF-ĸB signaling pathway in HUVECs. Inflammation (2017) 40(4):1450–9. doi: 10.1007/s10753-017-0588-3
209. Wang H, Bian S, Yang CS. Green tea polyphenol EGCG suppresses lung cancer cell growth through upregulating miR-210 expression caused by stabilizing HIF-1α. Carcinogenesis (2011) 32(12):1881–9. doi: 10.1093/carcin/bgr218
210. Byun EH, Fujimura Y, Yamada K, Tachibana H. TLR4 signaling inhibitory pathway induced by green tea polyphenol epigallocatechin-3-gallate through 67-kDa laminin receptor. J Immunol (2010) 185(1):33–45. doi: 10.4049/jimmunol.0903742
211. Sun W, Liu X, Zhang H, Song Y, Li T, Liu X, et al. Epigallocatechin gallate upregulates NRF2 to prevent diabetic nephropathy via disabling KEAP1. Free Radical Biol Med (2017) 108:840–57. doi: 10.1016/j.freeradbiomed.2017.04.365
212. Shan L, Kang X, Liu F, Cai X, Han X, Shang Y. Epigallocatechin gallate improves airway inflammation through TGF−β1 signaling pathway in asthmatic mice. Mol Med Rep (2018) 18(2):2088–96. doi: 10.3892/mmr.2018.9183
213. Wei JC-C, Huang H-C, Chen W-J, Huang C-N, Peng C-H, Lin C-L. Epigallocatechin gallate attenuates amyloid β-induced inflammation and neurotoxicity in EOC 13.31 microglia. Eur J Pharmacol (2016) 770:16–24. doi: 10.1016/j.ejphar.2015.11.048
214. Liu D, Perkins JT, Hennig B. EGCG prevents PCB-126-induced endothelial cell inflammation via epigenetic modifications of NF-κB target genes in human endothelial cells. J Nutr Biochem (2016) 28:164–70. doi: 10.1016/j.jnutbio.2015.10.003
215. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol (2007) 7(10):803–15. doi: 10.1038/nri2171
216. Ross R. Atheroslcerosis-an inflammatory disease. N Engl J Med (1999) 340:115–26. doi: 10.1056/NEJM199901143400207
217. Tu Y, Sun W, Wan Y-G, Gao K, Liu H, Yu B-Y, et al. Dahuang Fuzi Decoction ameliorates tubular epithelial apoptosis and renal damage via inhibiting TGF-β1-JNK signaling pathway activation in vivo. J Ethnopharmacol (2014) 156:115–24. doi: 10.1016/j.jep.2014.08.035
218. Dai J-P, Wang Q-W, Su Y, Gu L-M, Zhao Y, Chen X-X, et al. Emodin inhibition of influenza A virus replication and influenza viral pneumonia via the Nrf2, TLR4, p38/JNK and NF-kappaB pathways. Molecules (2017) 22(10):1754. doi: 10.3390/molecules22101754
219. Iwanowycz S, Wang J, Altomare D, Hui Y, Fan D. Emodin bidirectionally modulates macrophage polarization and epigenetically regulates macrophage memory. J Biol Chem (2016) 291(22):11491–503. doi: 10.1074/jbc.M115.702092
220. Godoy LD, Lucas JE, Bender AJ, Romanick SS, Ferguson BS. Targeting the epigenome: Screening bioactive compounds that regulate histone deacetylase activity. Mol Nutr Food Res (2017) 61(4):1600744. doi: 10.1002/mnfr.201600744
221. Ye B, Chen X, Dai S, Han J, Liang X, Lin S, et al. Emodin alleviates myocardial ischemia/reperfusion injury by inhibiting gasdermin D-mediated pyroptosis in cardiomyocytes. Drug Des Dev Ther (2019) 13:975. doi: 10.2147/DDDT.S195412
222. Antos CL, McKinsey TA, Dreitz M, Hollingsworth LM, Zhang C-L, Schreiber K, et al. Dose-dependent blockade to cardiomyocyte hypertrophy by histone deacetylase inhibitors. J Biol Chem (2003) 278(31):28930–7. doi: 10.1074/jbc.M303113200
223. Kong Y, Tannous P, Lu G, Berenji K, Rothermel BA, Olson EN, et al. Suppression of class I and II histone deacetylases blunts pressure-overload cardiac hypertrophy. Circulation (2006) 113(22):2579. doi: 10.1161/CIRCULATIONAHA.106.625467
224. Williams SM, Golden-Mason L, Ferguson BS, Schuetze KB, Cavasin MA, Demos-Davies K, et al. Class I HDACs regulate angiotensin II-dependent cardiac fibrosis via fibroblasts and circulating fibrocytes. J Mol Cell Cardiol (2014) 67:112–25. doi: 10.1016/j.yjmcc.2013.12.013
225. Demos-Davies KM, Ferguson BS, Cavasin MA, Mahaffey JH, Williams SM, Spiltoir JI, et al. HDAC6 contributes to pathological responses of heart and skeletal muscle to chronic angiotensin-II signaling. Am J Physiol Heart Circ Physiol (2014) 307(2):H252–H8. doi: 10.1152/ajpheart.00149.2014
226. Cui H-X, Chen J-H, Li J-W, Cheng F-R, Yuan K. Protection of anthocyanin from Myrica rubra against cerebral ischemia-reperfusion injury via modulation of the TLR4/NF-κB and NLRP3 pathways. Molecules (2018) 23(7):1788. doi: 10.3390/molecules23071788
227. Aboonabi A, Singh I. Chemopreventive role of anthocyanins in atherosclerosis via activation of Nrf2–ARE as an indicator and modulator of redox. Biomed Pharmacother (2015) 72:30–6. doi: 10.1016/j.biopha.2015.03.008
228. Zhang P-W, Chen F-X, Li D, Ling W-H, Guo H-H. A CONSORT-compliant, randomized, double-blind, placebo-controlled pilot trial of purified anthocyanin in patients with nonalcoholic fatty liver disease. Medicine (2015) 94(20):e758. doi: 10.1097/MD.0000000000000758
229. Habanova M, Saraiva JA, Haban M, Schwarzova M, Chlebo P, Predna L, et al. Intake of bilberries (Vaccinium myrtillus L.) reduced risk factors for cardiovascular disease by inducing favorable changes in lipoprotein profiles. Nutr Res (2016) 36(12):1415–22. doi: 10.1016/j.nutres.2016.11.010
230. Stote K, Corkum A, Sweeney M, Shakerley N, Kean T, Gottschall-Pass K. Postprandial effects of blueberry (Vaccinium angustifolium) consumption on glucose metabolism, gastrointestinal hormone response, and perceived appetite in healthy adults: a randomized, placebo-controlled crossover trial. Nutrients (2019) 11(1):202. doi: 10.3390/nu11010202
231. Yang L, Ling W, Yang Y, Chen Y, Tian Z, Du Z, et al. Role of purified anthocyanins in improving cardiometabolic risk factors in chinese men and women with prediabetes or early untreated diabetes—A randomized controlled trial. Nutrients (2017) 9(10):1104. doi: 10.3390/nu9101104
232. Castro-Acosta ML, Stone SG, Mok JE, Mhajan RK, Fu C-I, Lenihan-Geels GN, et al. Apple and blackcurrant polyphenol-rich drinks decrease postprandial glucose, insulin and incretin response to a high-carbohydrate meal in healthy men and women. J Nutr Biochem (2017) 49:53–62. doi: 10.1016/j.jnutbio.2017.07.013
233. Seong A-R, Yoo J-Y, Choi K, Lee M-H, Lee Y-H, Lee J, et al. Delphinidin, a specific inhibitor of histone acetyltransferase, suppresses inflammatory signaling via prevention of NF-κB acetylation in fibroblast-like synoviocyte MH7A cells. Biochem Biophys Res Commun (2011) 410(3):581–6. doi: 10.1016/j.bbrc.2011.06.029
234. Rahnasto-Rilla M, Tyni J, Huovinen M, Jarho E, Kulikowicz T, Ravichandran S, et al. Natural polyphenols as sirtuin 6 modulators. Sci Rep (2018) 8(1):1–11. doi: 10.1038/s41598-018-22388-5
235. Downing LE, Heidker RM, Caiozzi GC, Wong BS, Rodriguez K, Del Rey F, et al. A grape seed procyanidin extract ameliorates fructose-induced hypertriglyceridemia in rats via enhanced fecal bile acid and cholesterol excretion and inhibition of hepatic lipogenesis. PloS One (2015) 10(10):e0140267. doi: 10.1371/journal.pone.0140267
236. Zhan W, Liao X, Xie R-J, Tian T, Yu L, Liu X, et al. The effects of blueberry anthocyanins on histone acetylation in rat liver fibrosis. Oncotarget (2017) 8(57):96761. doi: 10.18632/oncotarget.17842
237. Zhan W, Liao X, Tian T, Yu L, Liu X, Li B, et al. Study on the effects of blueberry treatment on histone acetylation modification of CCl4-induced liver disease in rats. Genet Mol Res (2017) 16(1). doi: 10.4238/gmr16019188
238. Wei J, Zhang G, Zhang X, Xu D, Gao J, Fan J. Anthocyanins delay ageing-related degenerative changes in the liver. Plant Foods Hum Nutr (2017) 72(4):425–31. doi: 10.1007/s11130-017-0644-z
239. Ceci C, Lacal PM, Tentori L, De Martino MG, Miano R, Graziani G. Experimental evidence of the antitumor, antimetastatic and antiangiogenic activity of ellagic acid. Nutrients (2018) 10(11):1756. doi: 10.3390/nu10111756
240. Wang W, Yang L, Liu T, Wang J, Wen A, Ding Y. Ellagic acid protects mice against sleep deprivation-induced memory impairment and anxiety by inhibiting TLR4 and activating Nrf2. Aging (Albany NY) (2020) 12(11):10457. doi: 10.18632/aging.103270
241. Panchal SK, Ward L, Brown L. Ellagic acid attenuates high-carbohydrate, high-fat diet-induced metabolic syndrome in rats. Eur J Nutr (2013) 52(2):559–68. doi: 10.1007/s00394-012-0358-9
242. Marín M, Giner RM, Ríos J-L, Recio MC. Intestinal anti-inflammatory activity of ellagic acid in the acute and chronic dextrane sulfate sodium models of mice colitis. J Ethnopharmacol (2013) 150(3):925–34. doi: 10.1016/j.jep.2013.09.030
243. Rosillo MA, Sánchez-Hidalgo M, Cárdeno A, Aparicio-Soto M, Sánchez-Fidalgo S, Villegas I, et al. Dietary supplementation of an ellagic acid-enriched pomegranate extract attenuates chronic colonic inflammation in rats. Pharmacol Res (2012) 66(3):235–42. doi: 10.1016/j.phrs.2012.05.006
244. Gourineni V, Shay NF, Chung S, Sandhu AK, Gu L. Muscadine grape (Vitis rotundifolia) and wine phytochemicals prevented obesity-associated metabolic complications in C57BL/6J mice. J Agric Food Chem (2012) 60(31):7674–81. doi: 10.1021/jf3013663
245. Kang I, Okla M, Chung S. Ellagic acid inhibits adipocyte differentiation through coactivator-associated arginine methyltransferase 1-mediated chromatin modification. J Nutr Biochem (2014) 25(9):946–53. doi: 10.1016/j.jnutbio.2014.04.008
246. Yadav N, Cheng D, Richard S, Morel M, Iyer VR, Aldaz CM, et al. CARM1 promotes adipocyte differentiation by coactivating PPARγ. EMBO Rep (2008) 9(2):193–8. doi: 10.1038/sj.embor.7401151
247. Larsen T, Toubro S, Astrup A. PPARgamma agonists in the treatment of type II diabetes: is increased fatness commensurate with long-term efficacy? Int J Obes (2003) 27(2):147–61. doi: 10.1038/sj.ijo.802223
248. Wang M, Tafuri S. Modulation of PPARγ activity with pharmaceutical agents: treatment of insulin resistance and atherosclerosis. J Cell Biochem (2003) 89(1):38–47. doi: 10.1002/jcb.10492
249. Selvi BR, Batta K, Kishore AH, Mantelingu K, Varier RA, Balasubramanyam K, et al. Identification of a novel inhibitor of coactivator-associated arginine methyltransferase 1 (CARM1)-mediated methylation of histone H3 Arg-17. J Biol Chem (2010) 285(10):7143–52. doi: 10.1074/jbc.M109.063933
250. Porta M, Amione C, Barutta F, Fornengo P, Merlo S, Gruden G, et al. The co-activator-associated arginine methyltransferase 1 (CARM1) gene is overexpressed in type 2 diabetes. Endocrine (2019) 63(2):284–92. doi: 10.1007/s12020-018-1740-z
251. Miao F, Li S, Chavez V, Lanting L, Natarajan R. CARM1 enhances NF-{kappa} B mediated gene transcription through methylation of histone H3 at arginine 17. Mol Endocrinol (2006) 20:1562–73. doi: 10.1210/me.2005-0365
252. Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest (2000) 106(2):171–6. doi: 10.1172/JCI10583
253. Dong Y, Chen H, Gao J, Liu Y, Li J, Wang J. Bioactive ingredients in Chinese herbal medicines that target non-coding RNAs: promising new choices for disease treatment. Front Pharmacol (2019) 10:515. doi: 10.3389/fphar.2019.00515
254. Shan H, Li X, Pan Z, Zhang L, Cai B, Zhang Y, et al. Tanshinone IIA protects against sudden cardiac death induced by lethal arrhythmias via repression of microRNA-1. Br J Pharmacol (2009) 158(5):1227–35. doi: 10.1111/j.1476-5381.2009.00377.x
255. Zhang Y, Zhang L, Chu W, Wang B, Zhang J, Zhao M, et al. Tanshinone IIA inhibits miR-1 expression through p38 MAPK signal pathway in post-infarction rat cardiomyocytes. Cell Physiol Biochem (2010) 26(6):991–8. doi: 10.1159/000324012
256. Pan Y, Qian J-X, Lu S-Q, Chen J-W, Zhao X-D, Jiang Y, et al. Protective effects of tanshinone IIA sodium sulfonate on ischemia-reperfusion-induced myocardial injury in rats. Iran J Basic Med Sci (2017) 20(3):308.
257. Cheng J, Chen T, Li P, Wen J, Pang N, Zhang L, et al. Sodium tanshinone IIA sulfonate prevents lipopolysaccharide-induced inflammation via suppressing nuclear factor-κB signaling pathway in human umbilical vein endothelial cells. Can J Physiol Pharmacol (2018) 96(1):26–31. doi: 10.1139/cjpp-2017-0023
258. Fan G, Jiang X, Wu X, Fordjour PA, Miao L, Zhang H, et al. Anti-inflammatory activity of tanshinone IIA in LPS-stimulated RAW264. 7 macrophages via miRNAs and TLR4–NF-κB pathway. Inflammation (2016) 39(1):375–84. doi: 10.1007/s10753-015-0259-1
259. Xuan Y, Gao Y, Huang H, Wang X, Cai Y, Luan QX. Tanshinone IIA attenuates atherosclerosis in apolipoprotein E knockout mice infected with Porphyromonas gingivalis. Inflammation (2017) 40(5):1631–42. doi: 10.1007/s10753-017-0603-8
260. Tu J, Xing Y, Guo Y, Tang F, Guo L, Xi T. TanshinoneIIA ameliorates inflammatory microenvironment of colon cancer cells via repression of microRNA-155. Int Immunopharmacol (2012) 14(4):353–61. doi: 10.1016/j.intimp.2012.08.015
261. Wang L, Zhang C, Guo Y, Su Z-Y, Yang Y, Shu L, et al. Blocking of JB6 cell transformation by tanshinone IIA: epigenetic reactivation of Nrf2 antioxidative stress pathway. AAPS J (2014) 16(6):1214–25. doi: 10.1208/s12248-014-9666-8
262. Zhao W, Deng C, Han Q, Xu H, Chen Y. Carvacrol may alleviate vascular inflammation in diabetic db/db mice. Int J Mol Med (2020) 46(3):977–88. doi: 10.3892/ijmm.2020.4654
263. Zou Y, Wang J, Peng J, Wei H. Oregano essential oil induces SOD1 and GSH expression through Nrf2 activation and alleviates hydrogen peroxide-induced oxidative damage in IPEC-J2 cells. Oxid Med Cell Longev (2016) 2016. doi: 10.1155/2016/5987183
264. Khosravi AR, Erle DJ. Chitin-induced airway epithelial cell innate immune responses are inhibited by carvacrol/thymol. PloS One (2016) 11(7):e0159459. doi: 10.1371/journal.pone.0159459
265. Sayed AS, Gomaa IEO, Bader M, El Sayed NSED. Role of 3-acetyl-11-keto-beta-boswellic acid in counteracting LPS-induced neuroinflammation via modulation of miRNA-155. Mol Neurobiol (2018) 55(7):5798–808. doi: 10.1007/s12035-017-0801-2
266. Siddiqui M. Boswellia serrata, a potential antiinflammatory agent: an overview. Indian J Pharm Sci (2011) 73(3):255. doi: 10.4103/0250-474X.93507
267. Chen L-C, Hu L-H, Yin M-C. Alleviative effects from boswellic acid on acetaminophen-induced hepatic injury. Biomedicine (2016) 6(2):12–9. doi: 10.7603/s40681-016-0009-1
268. Barakat BM, Ahmed HI, Bahr HI, Elbahaie AM. Protective effect of boswellic acids against doxorubicin-induced hepatotoxicity: impact on Nrf2/HO-1 defense pathway. Oxid Med Cell Longev (2018) 2018. doi: 10.1155/2018/8296451
269. Ding Y, Chen M, Wang M, Wang M, Zhang T, Park J, et al. Neuroprotection by acetyl-11-keto-β-boswellic acid, in ischemic brain injury involves the Nrf2/HO-1 defense pathway. Sci Rep (2014) 4:7002. doi: 10.1038/srep07002
270. Kim JK, Park SU. Current potential health benefits of sulforaphane. Exp Clin Sci (2016) 15:571–7. doi: 10.15299/jk.2016.2.58.571
271. Briones-Herrera A, Eugenio-Pérez D, Reyes-Ocampo JG, Rivera-Mancía S, Pedraza-Chaverri J. New highlights on the health-improving effects of sulforaphane. Food Funct (2018) 9(5):2589–606. doi: 10.1039/C8FO00018B
272. Pal S, Konkimalla VB. Sulforaphane regulates phenotypic and functional switching of both induced and spontaneously differentiating human monocytes. Int Immunopharmacol (2016) 35:85–98. doi: 10.1016/j.intimp.2016.03.008
273. Zhao Z, Liao G, Zhou Q, Lv D, Holthfer H, Zou H. Sulforaphane attenuates contrast-induced nephropathy in rats via Nrf2/HO-1 pathway. Oxid Med Cell Longev (2016) 2016. doi: 10.1155/2016/9825623
274. Mazarakis N, Snibson K, Licciardi PV, Karagiannis TC. The potential use of l-sulforaphane for the treatment of chronic inflammatory diseases: a review of the clinical evidence. Clin Nutr (2020) 39(3):664–75. doi: 10.1016/j.clnu.2019.03.022
275. Li B, Cui W, Liu J, Li R, Liu Q, Xie X-H, et al. Sulforaphane ameliorates the development of experimental autoimmune encephalomyelitis by antagonizing oxidative stress and Th17-related inflammation in mice. Exp Neurol (2013) 250:239–49. doi: 10.1016/j.expneurol.2013.10.002
276. Li Y, Paonessa JD, Zhang Y. Mechanism of chemical activation of Nrf2. PloS One (2012) 7(4):e35122. doi: 10.1371/journal.pone.0035122
277. Brandenburg L-O, Kipp M, Lucius R, Pufe T, Wruck CJ. Sulforaphane suppresses LPS-induced inflammation in primary rat microglia. Inflammation Res (2010) 59(6):443–50. doi: 10.1007/s00011-009-0116-5
278. Eren E, Tufekci KU, Isci KB, Tastan B, Genc K, Genc S. Sulforaphane inhibits lipopolysaccharide-induced inflammation, cytotoxicity, oxidative stress, and miR-155 expression and switches to Mox phenotype through activating extracellular signal-regulated kinase 1/2–nuclear factor erythroid 2-related factor 2/antioxidant response element pathway in murine microglial cells. Front Immunol (2018) 9:s. doi: 10.3389/fimmu.2018.00036
279. Hung C-N, Huang H-P, Wang C-J, Liu K-L, Lii C-K. Sulforaphane inhibits TNF-α-induced adhesion molecule expression through the Rho A/ROCK/NF-κB signaling pathway. J Med Food (2014) 17(10):1095–102. doi: 10.1089/jmf.2013.2901
280. Gaona-Gaona L, Molina-Jijón E, Tapia E, Zazueta C, Hernández-Pando R, Calderón-Oliver M, et al. Protective effect of sulforaphane pretreatment against cisplatin-induced liver and mitochondrial oxidant damage in rats. Toxicology (2011) 286(1-3):20–7. doi: 10.1016/j.tox.2011.04.014
281. Mein JR, James DR, Lakkanna S. Induction of phase 2 antioxidant enzymes by broccoli sulforaphane: perspectives in maintaining the antioxidant activity of vitamins A, C, and E. Front Genet (2012) 3:7. doi: 10.3389/fgene.2012.00007
282. Riedl MA, Saxon A, Diaz-Sanchez D. Oral sulforaphane increases Phase II antioxidant enzymes in the human upper airway. Clin Immunol (2009) 130(3):244–51. doi: 10.1016/j.clim.2008.10.007
283. Thimmulappa RK, Mai KH, Srisuma S, Kensler TW, Yamamoto M, Biswal S. Identification of Nrf2-regulated genes induced by the chemopreventive agent sulforaphane by oligonucleotide microarray. Cancer Res (2002) 62:5196–203.
284. Heiss E, Herhaus C, Klimo K, Bartsch H, Gerhäuser C. Nuclear factor κB is a molecular target for sulforaphane-mediated anti-inflammatory mechanisms. J Biol Chem (2001) 276(34):32008–15. doi: 10.1074/jbc.M104794200
285. Killeen ME, Englert JA, Stolz DB, Song M, Han Y, Delude RL, et al. The phase 2 enzyme inducers ethacrynic acid, DL-sulforaphane, and oltipraz inhibit lipopolysaccharide-induced high-mobility group box 1 secretion by RAW 264.7 cells. J Pharmacol Exp Ther (2006) 316(3):1070–9. doi: 10.1124/jpet.105.092841
286. Checker R, Gambhir L, Thoh M, Sharma D, Sandur SK. Sulforaphane, a naturally occurring isothiocyanate, exhibits anti-inflammatory effects by targeting GSK3β/Nrf-2 and NF-κB pathways in T cells. J Funct Foods (2015) 19:426–38. doi: 10.1016/j.jff.2015.08.030
287. Youn HS, Kim YS, Park ZY, Kim SY, Choi NY, Joung SM, et al. Sulforaphane suppresses oligomerization of TLR4 in a thiol-dependent manner. J Immunol (2010) 184(1):411–9. doi: 10.4049/jimmunol.0803988
288. Koo JE, Park Z-Y, Kim ND, Lee JY. Sulforaphane inhibits the engagement of LPS with TLR4/MD2 complex by preferential binding to Cys133 in MD2. Biochem Biophys Res Commun (2013) 434(3):600–5. doi: 10.1016/j.bbrc.2013.03.123
289. Wagner AE, Boesch-Saadatmandi C, Dose J, Schultheiss G, Rimbach G. Anti-inflammatory potential of allyl-isothiocyanate–role of Nrf2, NF-κB and microRNA-155. J Cell Mol Med (2012) 16(4):836–43. doi: 10.1111/j.1582-4934.2011.01367.x
290. Deramaudt TB, Ali M, Vinit S, Bonay M. Sulforaphane reduces intracellular survival of Staphylococcus aureus in macrophages through inhibition of JNK and p38 MAPK−induced inflammation. Int J Mol Med (2020) 45(6):1927–41. doi: 10.3892/ijmm.2020.4563
291. Kaufman-Szymczyk A, Majewski G, Lubecka-Pietruszewska K, Fabianowska-Majewska K. The role of sulforaphane in epigenetic mechanisms, including interdependence between histone modification and DNA methylation. Int J Mol Sci (2015) 16(12):29732–43. doi: 10.3390/ijms161226195
292. Lubecka-Pietruszewska K, Kaufman-Szymczyk A, Stefanska B, Cebula-Obrzut B, Smolewski P, Fabianowska-Majewska K. Sulforaphane alone and in combination with clofarabine epigenetically regulates the expression of DNA methylation-silenced tumour suppressor genes in human breast cancer cells. Lifestyle Genomics (2015) 8(2):91–101. doi: 10.1159/000439111
293. Myzak MC, Hardin K, Wang R, Dashwood RH, Ho E. Sulforaphane inhibits histone deacetylase activity in BPH-1, LnCaP and PC-3 prostate epithelial cells. Carcinogenesis (2006) 27(4):811–9. doi: 10.1093/carcin/bgi265
294. Hsu A, Wong CP, Yu Z, Williams DE, Dashwood RH, Ho E. Promoter de-methylation of cyclin D2 by sulforaphane in prostate cancer cells. Clin Epigenet (2011) 3(1):1–9. doi: 10.1186/1868-7083-3-3
295. Senanayake GV, Banigesh A, Wu L, Lee P, Juurlink BH. The dietary phase 2 protein inducer sulforaphane can normalize the kidney epigenome and improve blood pressure in hypertensive rats. Am J Hypertens (2012) 25(2):229–35. doi: 10.1038/ajh.2011.200
296. Zhao F, Zhang J, Chang N. Epigenetic modification of Nrf2 by sulforaphane increases the antioxidative and anti-inflammatory capacity in a cellular model of Alzheimer’s disease. Eur J Pharmacol (2018) 824:1–10. doi: 10.1016/j.ejphar.2018.01.046
297. Park S-J, Youn H-S. Suppression of homodimerization of toll-like receptor 4 by isoliquiritigenin. Phytochemistry (2010) 71(14-15):1736–40. doi: 10.1016/j.phytochem.2010.07.008
298. Peri F, Calabrese V. Toll-like receptor 4 (TLR4) modulation by synthetic and natural compounds: an update: miniperspective. J Med Chem (2014) 57(9):3612–22. doi: 10.1021/jm401006s
299. Wang P, Yang Y, Wang D, Yang Q, Wan J, Liu S, et al. Cinnamaldehyde ameliorates vascular dysfunction in diabetic mice by activating Nrf2. Am J Hypertens (2020) 33:610–9. doi: 10.1093/ajh/hpaa024
300. Qu S, Shen Y, Wang M, Wang X, Yang Y. Suppression of miR-21 and miR-155 of macrophage by cinnamaldehyde ameliorates ulcerative colitis. Int Immunopharmacol (2019) 67:22–34. doi: 10.1016/j.intimp.2018.11.045
301. Huang T-T, Lai H-C, Chen Y-B, Chen L-G, Wu Y-H, Ko Y-F, et al. cis-Resveratrol produces anti-inflammatory effects by inhibiting canonical and non-canonical inflammasomes in macrophages. Innate Immun (2014) 20(7):735–50. doi: 10.1177/1753425913507096
302. Qureshi AA, Guan XQ, Reis JC, Papasian CJ, Jabre S, Morrison DC, et al. Inhibition of nitric oxide and inflammatory cytokines in LPS-stimulated murine macrophages by resveratrol, a potent proteasome inhibitor. Lipids Health Dis (2012) 11(1):76. doi: 10.1186/1476-511X-11-76
303. Li J, Xie C, Zhuang J, Li H, Yao Y, Shao C, et al. Resveratrol attenuates inflammation in the rat heart subjected to ischemia-reperfusion: Role of the TLR4/NF-κB signaling pathway. Mol Med Rep (2015) 11(2):1120–6. doi: 10.3892/mmr.2014.2955
304. Arafa MH, Mohammad NS, Atteia HH, Abd-Elaziz HR. Protective effect of resveratrol against doxorubicin-induced cardiac toxicity and fibrosis in male experimental rats. J Physiol Biochem (2014) 70(3):701–11. doi: 10.1007/s13105-014-0339-y
305. Li H, Jia Z, Li A, Jenkins G, Yang X, Hu J, et al. Resveratrol repressed viability of U251 cells by miR-21 inhibiting of NF-κB pathway. Mol Cell Biochem (2013) 382(1-2):137–43. doi: 10.1007/s11010-013-1728-1
306. Bigagli E, Cinci L, Paccosi S, Parenti A, D’Ambrosio M, Luceri C. Nutritionally relevant concentrations of resveratrol and hydroxytyrosol mitigate oxidative burst of human granulocytes and monocytes and the production of pro-inflammatory mediators in LPS-stimulated RAW 264.7 macrophages. Int Immunopharmacol (2017) 43:147–55. doi: 10.1016/j.intimp.2016.12.012
307. de Sá Coutinho D, Pacheco MT, Frozza RL, Bernardi A. Anti-inflammatory effects of resveratrol: Mechanistic insights. Int J Mol Sci (2018) 19(6):1812. doi: 10.3390/ijms19061812
308. Ren Z, Wang L, Cui J, Huoc Z, Xue J, Cui H, et al. Resveratrol inhibits NF-κB signaling through suppression of p65 and IB kinase activities. Pharmazie (2013) 68(8):689–94.
309. Kawabata T, Tokuda H, Fujita K, Kainuma S, Sakai G, Matsushima-Nishiwaki R, et al. Resveratrol Inhibits the Epidermal Growth Factor-Induced Migration of Osteoblasts: the Suppression of SAPK/JNK and Akt. Cell Physiol Biochem (2017) 43(3):1025–36. doi: 10.1159/000481700
310. Shao-Ling W, Ying L, Ying W, Yan-Feng C, Li-Xin N, Song-Tao L, et al. Curcumin, a potential inhibitor of up-regulation of TNF-alpha and IL-6 induced by palmitate in 3T3-L1 adipocytes through NF-kappaB and JNK pathway. Biomed Environ Sci (2009) 22(1):32–9. doi: 10.1016/S0895-3988(09)60019-2
311. Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol (2003) 171(7):3863–71. doi: 10.4049/jimmunol.171.7.3863
312. Hussain AR, Ahmed M, Al-Jomah NA, Khan AS, Manogaran P, Sultana M, et al. Curcumin suppresses constitutive activation of nuclear factor-κB and requires functional Bax to induce apoptosis in Burkitt’s lymphoma cell lines. Mol Cancer Ther (2008) 7(10):3318–29. doi: 10.1158/1535-7163.MCT-08-0541
313. Jurenka JS. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: a review of preclinical and clinical research. Altern Med Rev (2009) 14(2).:141–53
314. Gupta SC, Patchva S, Aggarwal BB. Therapeutic roles of curcumin: lessons learned from clinical trials. AAPS J (2013) 15(1):195–218. doi: 10.1208/s12248-012-9432-8
315. Aggarwal BB, Harikumar KB. Potential therapeutic effects of curcumin, the anti-inflammatory agent, against neurodegenerative, cardiovascular, pulmonary, metabolic, autoimmune and neoplastic diseases. Int J Biochem Cell Biol (2009) 41(1):40–59. doi: 10.1016/j.biocel.2008.06.010
316. Lichtenstein GR. Medical therapy of ulcerative colitis. Springer (2014). Available at: https://link.springer.com/book/10.1007%2F978-1-4939-1677-1
317. Rahmani AH, Alsahli MA, Aly SM, Khan MA, Aldebasi YH. Role of curcumin in disease prevention and treatment. Adv Biomed Res (2018) 7(38):38–46. doi: 10.4103/abr.abr_147_16
318. Anand P, Kunnumakkara AB, Newman RA, Aggarwal BB. Bioavailability of curcumin: problems and promises. Mol Pharm (2007) 4(6):807–18. doi: 10.1021/mp700113r
319. Hewlings SJ, Kalman DS. Curcumin: a review of its’ effects on human health. Foods (2017) 6(10):92. doi: 10.3390/foods6100092
320. Shoba G, Joy D, Joseph T, Majeed M, Rajendran R, Srinivas P. Influence of piperine on the pharmacokinetics of curcumin in animals and human volunteers. Planta Med (1998) 64(04):353–6. doi: 10.1055/s-2006-957450
321. Tu C-t, Han B, Yao Q-y, Zhang Y-a, Liu H-c, Zhang S-c. Curcumin attenuates Concanavalin A-induced liver injury in mice by inhibition of Toll-like receptor (TLR) 2, TLR4 and TLR9 expression. Int Immunopharmacol (2012) 12(1):151–7. doi: 10.1016/j.intimp.2011.11.005
322. Kumar A, Dhawan S, Hardegen NJ, Aggarwal BB. Curcumin (diferuloylmethane) inhibition of tumor necrosis factor (TNF)-mediated adhesion of monocytes to endothelial cells by suppression of cell surface expression of adhesion molecules and of nuclear factor-κB activation. Biochem Pharmacol (1998) 55(6):775–83. doi: 10.1016/S0006-2952(97)00557-1
323. Kim HN, Chang J, Shao L, Han L, Iyer S, Manolagas SC, et al. DNA damage and senescence in osteoprogenitors expressing Osx1 may cause their decrease with age. Aging Cell (2017) 16(4):693–703. doi: 10.1111/acel.12597
324. Kang C, Xu Q, Martin TD, Li MZ, Demaria M, Aron L, et al. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science (2015) 349(6255):aaa5612(1-13). doi: 10.1126/science.aaa5612
325. Manicone AM, McGuire JK eds. Matrix metalloproteinases as modulators of inflammation. Semin Cell Dev Biol (2007) 17(5):347–53.
326. Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator–activated receptor α mediates the adaptive response to fasting. J Clin Invest (1999) 103(11):1489–98. doi: 10.1172/JCI6223
327. Wang W, Lin Q, Lin R, Zhang J, Ren F, Zhang J, et al. PPARα agonist fenofibrate attenuates TNF-α-induced CD40 expression in 3T3-L1 adipocytes via the SIRT1-dependent signaling pathway. Exp Cell Res (2013) 319(10):1523–33. doi: 10.1016/j.yexcr.2013.04.007
328. Stienstra R, Duval C, Müller M, Kersten S. PPARs, obesity, and inflammation. PPAR Res (2007) 2007:1–7. doi: 10.1155/2007/95974
329. Mandard S, Muller M, Kersten S. Peroxisome proliferator-activated receptor alpha target genes. Cell Mol Life Sci (2004) 61:393–416. doi: 10.1007/s00018-003-3216-3
330. Patsouris D, Reddy JK, Müller M, Kersten S. Peroxisome proliferator-activated receptor α mediates the effects of high-fat diet on hepatic gene expression. Endocrinology (2006) 147(3):1508–16. doi: 10.1210/en.2005-1132
331. Fürst R, Zündorf I. Plant-derived anti-inflammatory compounds: hopes and disappointments regarding the translation of preclinical knowledge into clinical progress. Mediators Inflamm (2015) 2014. doi: 10.1155/2014/146832
332. Chun OK, Chung SJ, Song WO. Estimated dietary flavonoid intake and major food sources of US adults. J Nutr (2007) 137(5):1244–52. doi: 10.1093/jn/137.5.1244
333. Li Y, Yao J, Han C, Yang J, Chaudhry MT, Wang S, et al. Quercetin, inflammation and immunity. Nutrients (2016) 8(3):167. doi: 10.3390/nu8030167
334. Chirumbolo S. The role of quercetin, flavonols and flavones in modulating inflammatory cell function. Inflamm Allergy Drug Targets (Formerly Curr Drug Targets-Inflammation Allergy) (2010) 9(4):263–85. doi: 10.2174/187152810793358741
335. Huang R-Y, Yu Y-L, Cheng W-C, OuYang C-N, Fu E, Chu C-L. Immunosuppressive effect of quercetin on dendritic cell activation and function. J Immunol (2010) 184(12):6815–21. doi: 10.4049/jimmunol.0903991
336. Bhaskar S, Shalini V, Helen A. Quercetin regulates oxidized LDL induced inflammatory changes in human PBMCs by modulating the TLR-NF-κB signaling pathway. Immunobiology (2011) 216(3):367–73. doi: 10.1016/j.imbio.2010.07.011
337. Gates MA, Vitonis AF, Tworoger SS, Rosner B, Titus-Ernstoff L, Hankinson SE, et al. Flavonoid intake and ovarian cancer risk in a population-based case-control study. Int J Cancer (2009) 124(8):1918–25. doi: 10.1002/ijc.24151
338. Estruch R, Ros E, Salas-Salvadó J, Covas M-I, Corella D, Arós F, et al. Primary prevention of cardiovascular disease with a Mediterranean diet. N Engl J Med (2013) 368(14):1279–90. doi: 10.1056/NEJMoa1200303
339. Feng X, Weng D, Zhou F, Owen YD, Qin H, Zhao J, et al. Activation of PPARγ by a natural flavonoid modulator, apigenin ameliorates obesity-related inflammation via regulation of macrophage polarization. EBioMedicine (2016) 9:61–76. doi: 10.1016/j.ebiom.2016.06.017
340. Jefferson WN, Williams CJ. Circulating levels of genistein in the neonate, apart from dose and route, predict future adverse female reproductive outcomes. Reprod Toxicol (2011) 31(3):272–9. doi: 10.1016/j.reprotox.2010.10.001
341. Polkowski K, Mazurek AP. Biological properties of genistein. A review of in vitro and in vivo data. Acta Pol Pharm Drug Res (2000) 57(2):l35–55.
342. Manach C, Williamson G, Morand C, Scalbert A, Rémésy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr (2005) 81(1):230S–42S. doi: 10.1093/ajcn/81.1.230S
343. Kano M, Takayanagi T, Harada K, Sawada S, Ishikawa F. Bioavailability of isoflavones after ingestion of soy beverages in healthy adults. J Nutr (2006) 136(9):2291–6. doi: 10.1093/jn/136.9.2291
344. Setchell KD, Brown NM, Desai P, Zimmer-Nechemias L, Wolfe BE, Brashear WT, et al. Bioavailability of pure isoflavones in healthy humans and analysis of commercial soy isoflavone supplements. J Nutr (2001) 131(4):1362S–75S. doi: 10.1093/jn/131.4.1362S
345. Larkin T, Price WE, Astheimer L. The key importance of soy isoflavone bioavailability to understanding health benefits. Crit Rev Food Sci Nutr (2008) 48(6):538–52. doi: 10.1080/10408390701542716
346. Singh BN, Shankar S, Srivastava RK. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem Pharmacol (2011) 82(12):1807–21. doi: 10.1016/j.bcp.2011.07.093
347. Santo Domingo D, Camouse MM, Hsia AH, Matsui M, Maes D, Ward NL, et al. Anti-angiogenic effects of epigallocatechin-3-gallate in human skin. Int J Clin Exp Pathol (2010) 3(7):705.
348. Riegsecker S, Wiczynski D, Kaplan MJ, Ahmed S. Potential benefits of green tea polyphenol EGCG in the prevention and treatment of vascular inflammation in rheumatoid arthritis. Life Sci (2013) 93(8):307–12. doi: 10.1016/j.lfs.2013.07.006
349. Steinmann J, Buer J, Pietschmann T, Steinmann E. Anti-infective properties of epigallocatechin-3-gallate (EGCG), a component of green tea. Br J Pharmacol (2013) 168(5):1059–73. doi: 10.1111/bph.12009
350. Yang CS, Wang H, Li GX, Yang Z, Guan F, Jin H. Cancer prevention by tea: evidence from laboratory studies. Pharmacol Res (2011) 64(2):113–22. doi: 10.1016/j.phrs.2011.03.001
351. Oz HS. Chronic inflammatory diseases and green tea polyphenols. Nutrients (2017) 9(6):561. doi: 10.3390/nu9060561
352. Zhou D, Huang C, Lin Z, Zhan S, Kong L, Fang C, et al. Macrophage polarization and function with emphasis on the evolving roles of coordinated regulation of cellular signaling pathways. Cell Signal (2014) 26(2):192–7. doi: 10.1016/j.cellsig.2013.11.004
353. Wynn TA, Chawla A, Pollard JW. Macrophage biology in development, homeostasis and disease. Nature (2013) 496(7446):445–55. doi: 10.1038/nature12034
354. Pal T, Jana NR, Das PK. Spectrophotometric determination of magnesium (II) with emodin (1, 3, 8-trihydroxy-6-methylanthraquinone). Analyst (1992) 117(4):791–3. doi: 10.1039/an9921700791
355. Deng M, Guo H, Tam JW, Johnson BM, Brickey WJ, New JS, et al. Platelet-activating factor (PAF) mediates NLRP3-NEK7 inflammasome induction independently of PAFR. J Exp Med (2019) 216(12):2838–53. doi: 10.1084/jem.20190111
356. Farrall AJ, Wardlaw JM. Blood–brain barrier: ageing and microvascular disease–systematic review and meta-analysis. Neurobiol Aging (2009) 30(3):337–52. doi: 10.1016/j.neurobiolaging.2007.07.015
357. Mikula M, Majewska A, Ledwon JK, Dzwonek A, Ostrowski J. Obesity increases histone H3 lysine 9 and 18 acetylation at Tnfa and Ccl2 genes in mouse liver. Int J Mol Med (2014) 34(6):1647–54. doi: 10.3892/ijmm.2014.1958
358. Chung M-Y, Song J-H, Lee J, Shin EJ, Park JH, Lee S-H, et al. Tannic acid, a novel histone acetyltransferase inhibitor, prevents non-alcoholic fatty liver disease both in vivo and in vitro model. Mol Metab (2019) 19:34–48. doi: 10.1016/j.molmet.2018.11.001
359. Kuo M-Y, Ou H-C, Lee W-J, Kuo W-W, Hwang L-L, Song T-Y, et al. Ellagic acid inhibits oxidized low-density lipoprotein (OxLDL)-induced metalloproteinase (MMP) expression by modulating the protein kinase C-α/extracellular signal-regulated kinase/peroxisome proliferator-activated receptor γ/nuclear factor-κB (PKC-α/ERK/PPAR-γ/NF-κB) signaling pathway in endothelial cells. J Agric Food Chem (2011) 59(9):5100–8. doi: 10.1021/jf1041867
360. Shang Q, Xu H, Huang L. Tanshinone IIA: a promising natural cardioprotective agent. Evid Based Complement Altern Med (2012) 2012. doi: 10.1155/2012/716459
361. Feng J, Li S, Chen H. Tanshinone IIA inhibits myocardial remodeling induced by pressure overload via suppressing oxidative stress and inflammation: Possible role of silent information regulator 1. Eur J Pharmacol (2016) 791:632–9. doi: 10.1016/j.ejphar.2016.09.041
362. Yu M-l, Li S-m, Gao X, Li J-g, Xu H, Chen K-j. Sodium tanshinone II a sulfonate for coronary heart disease: a systematic review of randomized controlled trials. Chin J Integr Med (2018) 26:1–8. doi: 10.1007/s11655-018-2556-7
363. Zhu J, Xu Y, Ren G, Hu X, Wang C, Yang Z, et al. Tanshinone IIA Sodium sulfonate regulates antioxidant system, inflammation, and endothelial dysfunction in atherosclerosis by downregulation of CLIC1. Eur J Pharmacol (2017) 815:427–36. doi: 10.1016/j.ejphar.2017.09.047
364. Fahey JW, Holtzclaw WD, Wehage SL, Wade KL, Stephenson KK, Talalay P. Sulforaphane bioavailability from glucoraphanin-rich broccoli: Control by active endogenous myrosinase. PloS One (2015) 10(11):e0140963. doi: 10.1371/journal.pone.0140963
365. Fahey JW, Zalcmann AT, Talalay P. The chemical diversity and distribution of glucosinolates and isothiocyanates among plants. Phytochemistry (2001) 56(1):5–51. doi: 10.1016/S0031-9422(00)00316-2
366. Sehrawat A, Roy R, Pore SK, Hahm E-R, Samanta SK, Singh KB, et al. Mitochondrial dysfunction in cancer chemoprevention by phytochemicals from dietary and medicinal plants. Semin Cancer Biol (2017) 47:147–53. doi: 10.1016/j.semcancer.2016.11.009
367. Hanlon N, Coldham N, Gielbert A, Kuhnert N, Sauer MJ, King LJ, et al. Absolute bioavailability and dose-dependent pharmacokinetic behaviour of dietary doses of the chemopreventive isothiocyanate sulforaphane in rat. Br J Nutr (2008) 99(3):559–64. doi: 10.1017/S0007114507824093
368. Wardyn JD, Ponsford AH, Sanderson CM. Dissecting molecular cross-talk between Nrf2 and NF-κB response pathways. Biochem Soc Trans (2015) 43(4):621–6. doi: 10.1042/BST20150014
369. Nguyen T, Nioi P, Pickett CB. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J Biol Chem (2009) 284(20):13291–5. doi: 10.1074/jbc.R900010200
370. Singh B, Ronghe AM, Chatterjee A, Bhat NK, Bhat HK. MicroRNA-93 regulates NRF2 expression and is associated with breast carcinogenesis. Carcinogenesis (2013) 34(5):1165–72. doi: 10.1093/carcin/bgt026
371. Yang M, Yao Y, Eades G, Zhang Y, Zhou Q. MiR-28 regulates Nrf2 expression through a Keap1-independent mechanism. Breast Cancer Res Treat (2011) 129(3):983–91. doi: 10.1007/s10549-011-1604-1
372. Zhao J, Zhang X, Dong L, Wen Y, Zheng X, Zhang C, et al. Cinnamaldehyde inhibits inflammation and brain damage in a mouse model of permanent cerebral ischaemia. Br J Pharmacol (2015) 172(20):5009–23. doi: 10.1111/bph.13270
373. Tian F, Yu C, Ye W, Wang Q. Cinnamaldehyde induces cell apoptosis mediated by a novel circular RNA hsa_circ_0043256 in non-small cell lung cancer. Biochem Biophys Res Commun (2017) 493(3):1260–6. doi: 10.1016/j.bbrc.2017.09.136
374. Youn HS, Lee JK, Choi YJ, Saitoh SI, Miyake K, Hwang DH, et al. Cinnamaldehyde suppresses toll-like receptor 4 activation mediated through the inhibition of receptor oligomerization. Biochem Pharmacol (2008) 75(2):494–502. doi: 10.1016/j.bcp.2007.08.033
375. Lin S-P, Chu P-M, Tsai S-Y, Wu M-H, Hou Y-C. Pharmacokinetics and tissue distribution of resveratrol, emodin and their metabolites after intake of Polygonum cuspidatum in rats. J Ethnopharmacol (2012) 144(3):671–6. doi: 10.1016/j.jep.2012.10.009
376. Lampe JW, Chang J-L eds. Interindividual differences in phytochemical metabolism and disposition. In: . Seminars in cancer biology. Elsevier.
377. Rein MJ, Renouf M, Cruz-Hernandez C, Actis-Goretta L, Thakkar SK, da Silva Pinto M. Bioavailability of bioactive food compounds: a challenging journey to bioefficacy. Br J Clin Pharmacol (2013) 75(3):588–602. doi: 10.1111/j.1365-2125.2012.04425.x
378. Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res (2016) 30(8):1207–18. doi: 10.1002/ptr.5631
379. Wilk-Zasadna I, Bernasconi C, Pelkonen O, Coecke S. Biotransformation in vitro: An essential consideration in the quantitative in vitro-to-in vivo extrapolation (QIVIVE) of toxicity data. Toxicology (2015) 332:8–19. doi: 10.1016/j.tox.2014.10.006
380. Morgan S, Stair E, Martin T, Edwards W, Morgan G. Clinical, clinicopathologic, pathologic, and toxicologic alterations associated with gossypol toxicosis in feeder lambs. Am J Vet Res (1988) 49(4):493–9.
381. Allijn IE, Vaessen SF, Quarles van Ufford LC, Beukelman KJ, De Winther MP, Storm G, et al. Head-to-head comparison of anti-inflammatory performance of known natural products in vitro. PloS One (2016) 11(5):e0155325. doi: 10.1371/journal.pone.0155325
382. Meng J, Li Y, Camarillo C, Yao Y, Zhang Y, Xu C, et al. The anti-tumor histone deacetylase inhibitor SAHA and the natural flavonoid curcumin exhibit synergistic neuroprotection against amyloid-beta toxicity. PloS One (2014) 9(1):e85570. doi: 10.1371/journal.pone.0085570
383. Frendo-Cumbo S, MacPherson RE, Wright DC. Beneficial effects of combined resveratrol and metformin therapy in treating diet-induced insulin resistance. Physiol Rep (2016) 4(15):e12877. doi: 10.14814/phy2.12877
Keywords: inflammation, flavonoids, miRNAs, TLR4 signaling pathway, innate immunity
Citation: Saleh HA, Yousef MH and Abdelnaser A (2021) The Anti-Inflammatory Properties of Phytochemicals and Their Effects on Epigenetic Mechanisms Involved in TLR4/NF-κB-Mediated Inflammation. Front. Immunol. 12:606069. doi: 10.3389/fimmu.2021.606069
Received: 14 September 2020; Accepted: 08 March 2021;
Published: 26 March 2021.
Edited by:
Melanie Coombs, Acadia University, CanadaReviewed by:
Yaping Yang, Huazhong Agricultural University, ChinaCopyright © 2021 Saleh, Yousef and Abdelnaser. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anwar Abdelnaser, YW53YXIuYWJkZWxuYXNlckBhdWNlZ3lwdC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.