
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Immunol. , 29 March 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.605930
This article is part of the Research Topic Autoimmunity and Chronic Inflammation in Early Life View all 13 articles
 Mona Motwani1
Mona Motwani1 Jason McGowan1Jennifer Antonovitch1
Jason McGowan1Jennifer Antonovitch1 Kevin MingJie Gao1Zhaozhao Jiang1
Kevin MingJie Gao1Zhaozhao Jiang1 Shruti Sharma1Gretchen A. Baltus2
Shruti Sharma1Gretchen A. Baltus2 Kevin M. Nickerson3
Kevin M. Nickerson3 Ann Marshak-Rothstein4†
Ann Marshak-Rothstein4† Katherine A. Fitzgerald1*†
Katherine A. Fitzgerald1*†Detection of DNA is an important determinant of host-defense but also a driver of autoinflammatory and autoimmune diseases. Failure to degrade self-DNA in DNAseII or III(TREX1)-deficient mice results in activation of the cGAS-STING pathway. Deficiency of cGAS or STING in these models ameliorates disease manifestations. However, the contribution of the cGAS-STING pathway, relative to endosomal TLRs, in systemic lupus erythematosus (SLE) is controversial. In fact, STING deficiency failed to rescue, and actually exacerbated, disease manifestations in Fas-deficient SLE-prone mice. We have now extended these observations to a chronic model of SLE induced by the i.p. injection of TMPD (pristane). We found that both cGAS- and STING-deficiency not only failed to rescue mice from TMPD-induced SLE, but resulted in increased autoantibody production and higher proteinuria levels compared to cGAS STING sufficient mice. Further, we generated cGASKOFaslpr mice on a pure MRL/Faslpr background using Crispr/Cas9 and found slightly exacerbated, and not attenuated, disease. We hypothesized that the cGAS-STING pathway constrains TLR activation, and thereby limits autoimmune manifestations in these two models. Consistent with this premise, mice lacking cGAS and Unc93B1 or STING and Unc93B1 developed minimal systemic autoimmunity as compared to cGAS or STING single knock out animals. Nevertheless, TMPD-driven lupus in B6 mice was abrogated upon AAV-delivery of DNAse I, implicating a DNA trigger. Overall, this study demonstrated that the cGAS-STING pathway does not promote systemic autoimmunity in murine models of SLE. These data have important implications for cGAS-STING-directed therapies being developed for the treatment of systemic autoimmunity.
Systemic lupus erythematosus (SLE) is a heterogeneous autoimmune disease with a number of clinical manifestations including systemic inflammation, development of pathogenic antibodies, deposition of immune complexes and finally end organ damage (1). Genetic predisposition, environmental factors as well as both innate and adaptive arms of the immune system play an important role in initiation and amplification of this disease. Innate immune sensors present in endosomal compartments and in the cytosol, such as TLR9 and cGAS, respectively, can detect both microbial and host nucleic acids. It's been established in numerous murine models, that the endosomal TLRs, TLR7, and TLR9, play a critical role in SLE and related systemic autoimmune diseases (2, 3), while the role of STING in SLE is more controversial. Importantly, loss of function (LOF) mutations in the extracellular DNAse, DNAse1L3, originally identified in SLE patients (4–6), results in the accrual of DNA/RNA-associated microparticles in the blood (7–9). In mice, genetic deletion of DNAse1L3 promotes an SLE-like disease through a mechanism that is dependent on TLR7 and TLR9 and not STING (7, 9).
By contrast, LOF mutations of cytosolic DNAses such as DNAse II or DNAse III (Trex1) in patient populations are associated with systemic monogenic autoinflammatory diseases (10, 11). Genetic deletion of these cytosolic nucleases in mice, leads to embryonic lethality (DNAse II) (12) or myocarditis (TrexI) (13) through mechanisms dependent on the cGAS-STING pathway (14, 15). These data suggest that SLE is driven by extracellular DNA that is delivered to endosomal TLRs through receptors such as the BCR, LL37, or FcγRs while monogenic autoinflammatory diseases are driven by the aberrant accumulation of DNA in the cytosol that is detected by the cGAS/STING pathway. Autoantibody production has been detected in both Trex1 and DNAse II/IFNAR double deficient mice however, autoantibodies in DNAse II/IFNAR double defiicent mice have been shown to be Unc93B1 dependent (16). In addition, autoimmunity in DNAseIL3-deficient mice is dependent on endosomal MyD88 and endosomal TLRs (17).
Nevertheless, the cGAS/STING pathway has been postulated as a driver of lupus pathogenesis, although direct evidence for this is still limited. STING was recenlty implicated in the autoimmune phenotypes that develop in FcγR2-/- mice on a 129 background. However, since these mice were intercrossed with B6 STINGgt mice, the potential contribution of 129-associated risk alleles linked to the STING locus need to be considered (18). cGAMP, the cyclic dinucleotide generated by cGAS that activates STING, has been shown to be modestly elevated in the serum of a limited number of SLE patients (19). Most recently, UVB exposure which can drive lupus flares leads to an elevated type I IFN-I gene signature in the skin of mice which is dependent on the cGAS pathway and enhanced when cGAMP hydrolysis is blocked (20). Elevated STING activity has further been associated with increased expression of type I IFNs and ISGs (21, 22) and the degradation of damaged mitochondrial DNA by autophagy in patient populations (23). Moreover, while point mutations in human TREX1, that eliminate catalytic activity, cause the autoinflammatory disease Aicardi-Goutieres, point mutations in the N-terminus of TREX1, that retain DNAse activity, are associated with SLE and other unrelated conditions (24). The N-terminal mutations disrupt STING localization, and also disrupt the activity of the ER-resident enzyme oligosaccharyltransferase (OST) (25). In the absence of OST activity, immunogenic glycans accumulate in the ER and are the likely explanation for the association between the N-terminal mutations and SLE (24).
Murine models of C-terminal and N-terminal TREX1 mutations also develop distinct phenotypes. Patients heterozygous for a mutation that abrogates TREX1 catalytic activity, D18N, develop a skin disease that resembles familial chilblain lupus, while mice homozygous for this mutation develop myocarditis, multi-organ inflammation and autoantibody production, similar to TREX1−/− mice (26). By contrast, mice homozygous for C-terminal frameshift mutation, D272fs, previously associated with SLE (27), exhibit no clinical manifestations of disease but do develop high autoantibody titers primarily directed against non-nuclear antigens, and thus distinct from the autoantibody specificities associated with SLE (28). Therefore, the association between TREX1 mutations and murine lupus is weak.
We previously generated STING/Fas double deficient autoimmune prone mice and found that STING deficient mice developed more severe, not less severe, disease compared to Fas-deficient autoimmune prone controls (29). Mechanistically, we showed that STING-deficient macrophages expressed decreased levels of negative regulators of immune activation and therefore were hyper-responsive to TLR ligands (29). Together, our data indicated that the STING pathway does not promote, but rather constrains, TLR-driven lupus-like diseases. However, our prior work used STING-deficient autoimmune mice that were generated by intercrossing STING-deficient B6/129 and MRL/Faslpr mice; a limitation to these F2 studies was the potential contribution of risk alleles derived from the B6/129 background, that could be linked to the STING locus in the STING-deficient mice. Moreover, a critical question that remained unexplored was whether the regulatory effects of STING were dependent on the upstream DNA sensor cGAS. To address these issues, we have now further defined the role of STING and cGAS by directly targeting MRL/lpr mice using CRISPR/Cas9 genome editing. There has also been some concerns in the literature regarding the relevance of the MRL/lpr model to human disease. To address this concern, we have also expanded our analysis to the chronic model of TMPD-induced SLE in both cGAS- and STING-deficient B6 mice. We found that neither cGAS- nor STING- deficiency protected MRL/lpr mice. We also found that neither cGAS- nor STING-deficiency prevented TMPD-induced lupus. These studies further confirm our original observations and demonstrate that STING does not promote murine SLE in either genetically programmed or inducible models of SLE.
One caveat to the TMPD model is that it is thought to be driven predominantly by the RNA-sensing TLR, TLR7 (30, 31) bringing into question the actual role of DNA sensors in these mice. To evaluate the potential role of DNA sensors in the TMPD model, we overexpressed DNAseI using an AAV9 gene therapy approach by injecting an AAV9-DNAse I vector i.p., prior to the TMPD inoculation. The DNAseI expressing mice developed much less severe clinical manifestations than mice injected with the vector control. Thus, DNA contributes to systemic inflammation in TMPD injected mice, along with TLR7 ligands. Overall, our study shows that the cGAS-STING pathway is not a driver of disease in TLR-dependent models of SLE, but instead constrains disease activity, presumably by limiting TLR activation. Nonetheless, DNA sensing still plays an important role in mediating autoimmune pathologies and depleting extracellular DNA can rescue TMPD-injected mice from SLE.
Wild-type C57BL/6 mice were purchased from the Jackson Laboratory. STINGKO mice, fully backcrossed to the C57BL/6 background, were kindly provided by Dr. Daniel Stetson University of Washington, Seattle, WA (32). cGASKO mice on a B6 background were generated using cryopreserved embryos obtained from the European Conditional Mouse Mutagenesis Program (EUCOMM). cGASKO MRL/MpJ-Faslpr mice were generated via CRISPR/Cas9 genome editing at Merck & Co., Inc., Kenilworth, NJ using two gRNA sequences “AGGACCAGAACACCTTGTAG” and “TGACCGCACGACTTACCCTG” targeting exon 2. The deletion of exon 2, resulting in 581bp deletion, was confirmed using common forward primer 5′CCTAGCCTTGGCTATGTGGT3′ and reverse primer 5′AACAGTTCTAAATAACCGCTTTCG3′ for WT and 5′GAGCTGTAGATGCCCAAGTG3′ reverse primer for cGASKO. Unc93B1KO mice were provided by Eicke Latz, University of Massachusetts Medical School, Worcester, MA (33). All mice were bred and maintained at the Department of Animal Medicine of the University of Massachusetts Medical School in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care, and all protocols were approved by the Institutional Animal Care and Use Committee.
12–16-week-old mice received a single intraperitoneal injection of 500 ul of 2,6,10,14-Tetramethylpentadecane (TMPD, Sigma) and were analyzed 5–6 months after TMPD injection (34). In some experiments, C57BL/6 mice were injected with either AAV9-GFP or AAV9-DNAse1 10 weeks prior to TMPD injections (35). Each mouse received 1011 virus particles in 200 uL PBS delivered by intraperitoneal injection and AAV expression was measured by qPCR.
Peritoneal exudate was collected in Hanks' Balanced Salt Solution (HBSS) media. Single cell suspensions obtained from the peritoneal cavity were stained with the following antibodies: anti-CD11b PercpCy5.5, anti-F4/80 PECy7, anti-Ly-6C APC, anti-Tim4 PE and anti-Ly-6G FITC. Flow cytometric analysis was carried out using a BD LSRII with Diva software (BD) and FlowJo Software (Tree Star). Live and single cells were gated to identify CD11b+ cells which were then gated to identify Ly6Chi monocytes and Ly6Ghi granulocytes. Macrophages post TMPD injections were identified as live and single cells that were Ly6C− Ly6G− Tim4− CD11b+ and F4/80hi expressing cells. Splenocytes were stained with anti-CD11c pacific blue, SiglecH PE, anti-Bst2 APC, anti-CD3 PerCPCy5.5, anti-B220 Pacific blue, anti-CD4 PECy7 and anti-CD8 PE.
Bone marrow extracts were differentiated in vitro into bone marrow derived macrophages (BMDMs) in the presence of L929 supernatants for 7 days. The rested BMDMs were then transfected with ISD (interferon stimulatory DNA) (5 μM) overnight using Lipofectamine 2000 from Invitrogen.
HEp-2 human tissue culture substrate slides (MBL Code # ANK-120) were incubated with serum samples and bound antibodies were detected with DyLight 488-coupled detecting reagents as described in Christensen et al. (2). Anti–nucleosome concentrations were measured by ELISA with absorbance at 405/630 nm and compared with PL2-3 (in-house) as previously described (2). Autoantibodies reactive with dsDNA were measured by ELISA. Calf thymus DNA (Sigma) treated with S1 nuclease (ThermoFisher) was incubated for 1 h at room temperature on to poly-L-lysine treated ELISA plates. 1:50 diluted mouse sera was used and goat anti-mouse IgG H&L (HRP) (ab205719) was used as a secondary antibody. Anti-ds DNA antibody (ab27156) was used as standard.
An anti-mouse albumin ELISA kit was used to measure urine protein as per manufacturer's protocol (Bethyl Laboratories). Serum cytokine levels were determined using ELISA kits as per manufacturer's protocol: anti-mIFNγ and anti-mIL-17 (BD Biosciences), anti-mIL-10 (eBioscience) and anti-mTNFa (R&Dsystems). The IFN-β level in media supernatant was determined as previously described (36).
Total RNA was isolated (Qiazol; Qiagen) and quantitated via a Nanodrop ND-1000 spectrophotometer (Thermo Scientific). Next, 50 ng of RNA was hybridized and quantified with a custom probe set using the NanoString nCounter analysis system (NanoString Technologies). Gene-expression data were normalized to housekeeping genes. All values were scaled by a log2(x – min(x) + 1) function and a heatmap generated using R-based software.
Cells were lysed in 50 μL of ice-cold Pierce RIPA lysis buffer (ThermoFisher Scientific) supplemented with 1× complete protease and phosphatase mixture inhibitor (Sigma). Protein concentration was measured using a protein DC assay kit. Whole-cell lysates were denatured for 5 min at 85 °C in presence of 1× Sample Buffer and reducing agent (Invitrogen). Fifty micrograms of samples were separated by SDS/PAGE on 10% gels. Each gel was run initially for 15 min at 80 V and then at 120 V. Transfer onto nitrocellulose membranes (Bio-Rad) was done using a Trans-Blot Turbo Transfer system for 10 min. Membranes were blocked for 1 h with 5% skim-milk (Sigma Aldrich) at room temperature in PBS supplemented with 0.05% Tween-20 (PBST). Membranes were probed overnight at 4°C with the following primary antibodies: anti-cGAS (31659; CST) and anti–beta-actin Peroxidase (A3854; Sigma-Aldrich). All membranes were washed with PBST and exposed using the SuperSignal West Pico PLUS chemiluminescent substrate (ThermoScientific) on ImageQuant LAS4000 mini Imager (GE Healthcare).
All tissues were fixed in 10% neutral buffered formalin for 24–48 h before being processed and paraffin-embedded. Five-micrometer-thin sections were stained by H&E in an automated stainer (Leica Autostainer XL). Histomorphology of each H&E slides was evaluated by Applied Pathology Systems at low and high-power field on Olympus BX40 microscope, and the images were captured with Olympus cellSens Entry software at indicated magnifications.
All data were analyzed by non-parametric Mann-Whitney test using GraphPad Prism Software (GraphPad Software, San Diego, CA). Experiments are reported as mean +/– SEM. Differences are designated as one asterisk if p < 0.05, as two asterisks if p < 0.01 and three asterisks if p < 0.001.
TMPD (2,6,10,14-Tetramethylpentadecane) or pristane is a naturally occurring hydrocarbon oil, which when introduced into the peritoneal cavity induces features of SLE in non-autoimmune prone mice (34). These include systemic inflammation, autoantibody production and glomerulonephritis. TMPD-driven lupus in B6 mice is highly dependent on TLR7 (30, 31), while the role of TLR9 is less straightforward. In general, TLR9 is required for the production of anti-dsDNA autoantibodies (2), but TLR9−/− SLE-prone mice invariably develop more severe autoimmunity, and TLR9−/− TMPD-injected BALB/c mice develop much more severe renal disease than their TLR9-sufficient littermates (37). Since TMPD induces cell death, DNA- and RNA-associated-cellular debris is likely to accumulate in the peritoneal cavity and other tissues throughout the body (34, 38). In addition, dysregulation of extracellular enzymes like DNAseI and DNAse1L3 that degrade DNA and DNA-associated complexes (microparticles) released from dying cells have also been implicated in SLE (9). DNAse1 is the most abundant secreted endonuclease, that is primarily expressed in the salivary glands, kidneys and gut. To explore the role of extracellular DNA in TMPD-induced lupus, we overexpressed DNase I using AAV9 that leads to widespread expression in order to test whether it could limit DNA uptake and thereby prevent or enhance clinical manifestations of SLE.
B6 mice were first inoculated with AAV-9 expressing either GFP or DNaseI, and then injected with TMPD 10 weeks later. 6 months post-TMPD injection, these mice were evaluated for both DNAseI expression and features of systemic autoimmunity. The levels of DNAseI were examined in multiple tissues and found to be expressed at high levels in both the kidney and peritoneal lining in mice that were injected with AAV-9 expressing DNaseI as compared to mice that received AAV-GFP. Similarly, high level of GFP expression was detected only in mice that received AAV-GFP and not in AAV-DNAseI injected mice (Figure 1A). TMPD leads to an inflammatory infiltrate in the peritoneal cavity that includes both CD11b+ Ly6Chi monocytes and CD11b+ Ly6Ghi granulocytes. We found that the frequency and total number of both CD11b+ Ly6Chi inflammatory monocytes and CD11b+ Ly6Ghi granulocytes in the peritoneal exudate was dramatically decreased in the AAV/DNAseI+TMPD mice compared to the AAV/GFP+TMPD mice (Figure 1B). We also evaluated the spleen of these mice and observed reduced frequency and number of CD11b+ Ly6Chi inflammatory monocytes in AAVDNaseI treated mice that received TMPD (Figure 1C). We next evaluated the autoantibody profiles of TMPD treated mice and observed that the mice expressing AAV DNAseI showed very different ANA patterns when compared to the AAV GFP treated mice. Fifty percent of the AAV/GFP+TMPD sera gave a homogenous nuclear staining pattern, indicative of autoantibodies reactive with dsDNA, while the sera from the AAV/DNAseI+TMPD mice gave either a cytoplasmic and/or a speckled nuclear pattern (Figure 1D). Finally, the AAV/DNAseI+TMPD mice developed less severe renal disease as indicated by proteinuria levels (Figure 1E). These results highlight the importance of DNA sensing in TMPD mediated systemic autoimmunity.
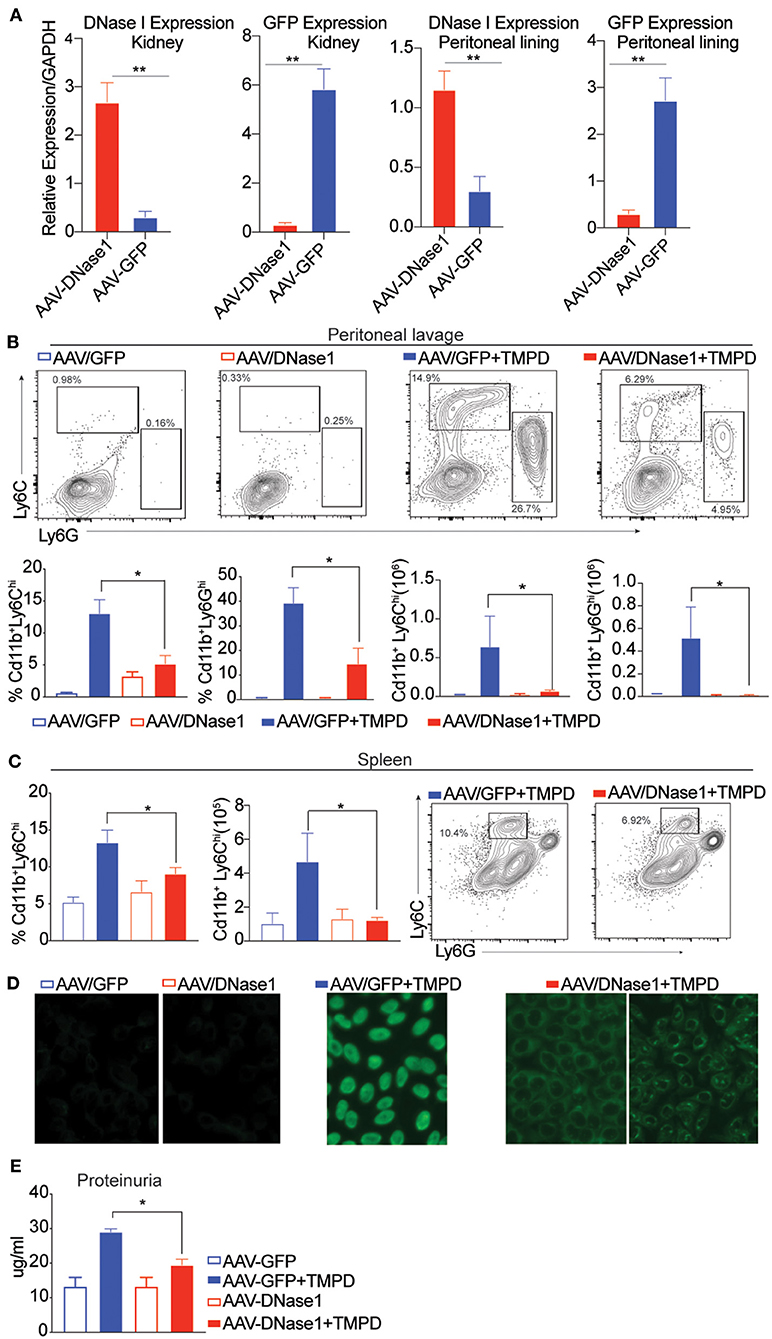
Figure 1. Overexpression of DNAse I ameliorates inflammation in chronic TMPD mediated peritonitis. 8–10 weeks old female C57BL6/J WT mice were either injected i.p. with 1011 AAV/GFP or AAV/DNAseI particles in 200 uL PBS. 10 weeks later mice were either injected with 500 ul TMPD or un-injected and analyzed from two independent experiments 6 months after TMPD injection. All the experiments were performed using the following N's of mice- AAV/GFP (blue open bars) AAV/DNAseI (red open bars) are uninjected mice n = 3. AAV/GFP+TMPD (blue closed bars) n = 4 and AAV/DNAseI +TMPD (red closed bars) are injected mice n = 8. (A) qPCR was performed to measure DNAse1 and GFP gene expression in total tissue, peritoneal lining and kidney. AAV/DNAseI injected mice are indicated in red bars and AAV/GFP mice are indicated in blue. (B) CD11b+ peritoneal exudate cells stained to identify Ly6Chi monocytes and Ly6Ghi granulocytes. AAV/GFP (blue) and AAV/DNAse I (red) uninjected mice are indicated by open bars and TMPD injected mice are indicated as closed bars. The top panel shows representative flow plots and frequency/ numbers are graphed in the bottom panel. (C) CD11b+ splenic cells stained to identify Ly6Chi monocytes. AAV/GFP (blue) and AAV/DNAseI (red) uninjected mice are indicated by open bars and TMPD injected mice are indicated as closed bars. The right panel shows representative flow plots and frequency/numbers are graphed in the left panel (D) Sera was collected 6 months after TMPD injection and ANA was measured using HEp2 substrate slides. Representative image from uninjected mice and AAV/GFP +TMPD injected mice is shown. Representative image is shown for two patterns observed in AAV DNAseI TMPD injected mice at 20X magnification. (E) Urine samples collected 6 months after TMPD injection were screened for proteinuria using an albumin ELISA assay. AAV/GFP (blue) and AAV/DNAseI (red) uninjected mice are indicated by open bars and TMPD injected mice are indicated as closed bars. Statistical significance is represented by *P < 0.05, **P < 0.01.
The development of autoimmunity progresses over a 6-month time course following administration of TMPD. To evaluate the role of STING and/or cGAS in disease progression, we injected WT, STINGKO and cGASKO with TMPD and assessed them 6 months later for various features of autoimmune disease. While B6 mice do not develop kidney pathology, they do develop immune cell abnormalities and modest proteinurea. We evaluated several parameters of inflammation, focusing first on the inflammatory infiltrate in the peritoneal cavity. We found a comparable frequency and number of CD11b+ Ly6Chi monocytes and CD11b+ Ly6Ghi granulocytes in the TMPD-injected STING and cGAS deficient animals as in the TMPD-injected WT mice (Supplementary Figure S1A). However, within the cells that were CD11b+ Ly6C− Ly6G− we found a myeloid subset that was CD11b+ and F4/80hi (Figure 2A). This subset of cells was Tim4+ in uninjected mice but Tim4− in all TMPD injected mice indicating that these are not resident macrophages (Supplementary Figure S1B). This CD11b+F4/80hi macrophage subset was increased in STING and cGAS deficient mice as compared to the WT mice both in frequency and number (Figure 2A). Several studies have reported distinct macrophage subsets to be pathological and major contributors of renal disease in both SLE prone patients and mice (39, 40). We also analyzed different immune cell subsets in the spleen and found that the numbers of Cd11b+ Ly6Chi monocytes was increased in the spleen of STING and cGAS deficient pristane injected mice as compared to the WT. Moreover, we observed an increased percentage of CD3+ B220+ (CD8 CD4 double negative) T cells in the STING and cGAS deficient pristane injected mice as compared to the WT pristane injected mice (Figure 2B and Supplementary Figure S1C). These double negative cells are expanded in SLE patient populations and contribute to kidney disease (41, 42). Overall, we found increased frequency and number of cellular subsets that contribute to disease pathology both in the peritoneal lavage and in the spleen of the STING and cGAS deficient mice.
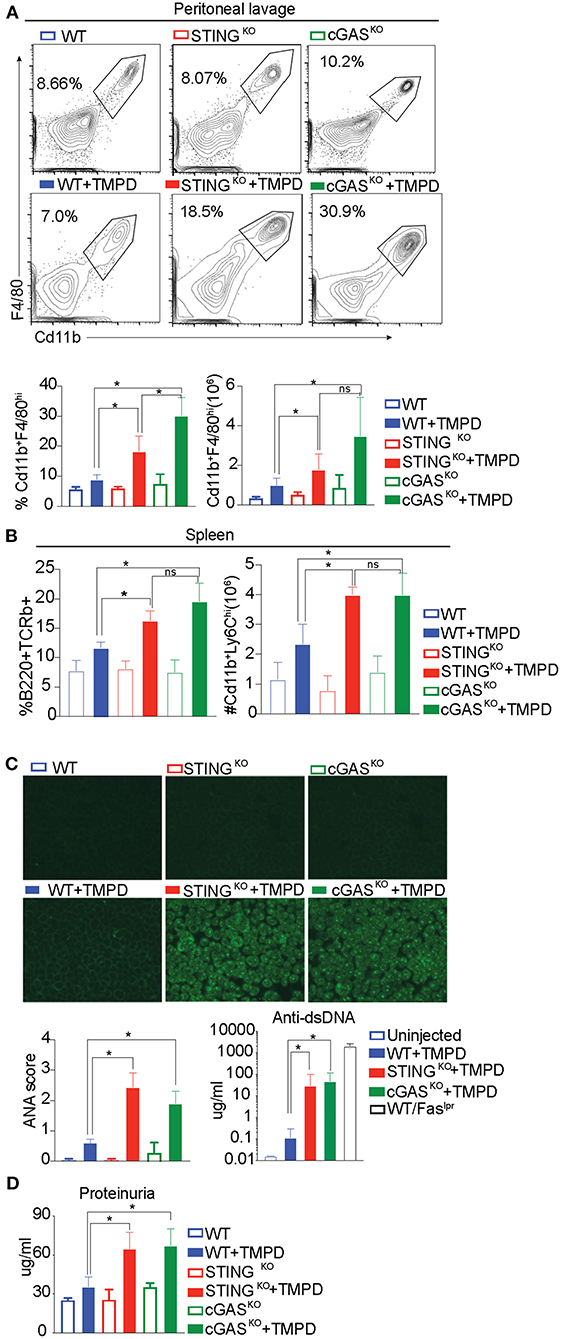
Figure 2. STING and cGAS deficiency exacerbate disease in chronic model of TMPD induced autoimmunity. 12–16 week-old female WT, STINGKO or cGASKO mice were either uninjected (open bars, n = 5) or injected with 500 ul of TMPD (closed bars, n = 15) were analyzed from two independent experiments. (A) 6 months later, peritoneal exudate cells were evaluated by flow cytometry. Live and single Ly6C−Ly6G− cells were gated for CD11b+ and F4/80hi. The top panel indicates representative flow plots from uninjected mice (WT = blue open bar, STINGKO = green open bar and cGASKO = red open bar). The middle panel shows representative flow plots from injected animals (closed bars) and the graphs for both frequency and numbers of CD11b+ and F4/80hi cells are shown in the bottom panel. (B) Flow cytometry was performed on splenocytes and double negative T cells (CD4− CD8−) were identified as CD3+ B220+ and CD11b+ and Ly6Chi cells were identified as inflammatory monocytes. The frequency of double negatives and cell numbers for inflammatory monocytes are graphed. (C) Sera was collected 3 months after TMPD injection and ANAs were detected using HEp2 substrate slides at 1:50 serum dilution. The representative images from each genotype and condition are shown at 20X magnification in the top panel. The slides were scored for fluorescence intensity and the scores are graphed at the bottom. Anti-ds DNA antibodies were measured in the serum at 3 months after TMPD injection (closed bars, n = 6). Sera from a WT/Faslpr strain was used as a positive control. (D) Proteinuria was assessed in urine collected 6 months after TMPD injection by using an albumin ELISA assay. Statistical significance is represented by *P < 0.05.
To further evaluate TMPD-injected WT, STINGKO and cGAS KO mice we examined serum for autoantibody production by immunofluorescent staining of HEp2 cells. Both STING- and cGAS-deficient mice developed detectable autoantibody levels by 12 weeks post TMPD injection, while the WT mice did not, as quantified in the bottom panel (Figure 2C left). We also quantified the production of anti-ds DNA antibodies in the serum and observed that the STINGKO and cGASKO TMPD injected animals had increased anti-dsDNA antibody titers as compared to WT injected mice (Figure 2C right). We also measured the status of serum cytokines in these animals and found increased levels of IL10, IL17, IFNγ and TNFα in STINGKO and cGASKO TMPD injected animals compared to WT mice (Supplementary Figure S1D).
Renal function was evaluated by assaying albumin titers in urine samples and both STINGKO and cGASKO injected mice showed evidence of increased proteinuria as compared to the WT injected mice (Figure 2D). Taken together, our data suggests that STING- or cGAS-deficient mice were not protected from TMPD-induced SLE, and even exhibited features of autoimmunity that were severe than their wild type counterparts.
Our previous study showed that STING deficient macrophages when stimulated with TLR9 and TLR7 ligands CpGB and CL097, respectively, produced increased levels of the proinflammatory cytokines IL6 and TNFα as compared to STING sufficient cells (29). This hyper-responsiveness correlated with reduced expression of genes involved in negative regulation of TLR signaling such as A20, suppressor of cytokine signaling 1 (SOCS1) and 3 (SOCS3) (29). To determine whether the STING-exacerbated features of SLE were dependent on endosomal TLRs in vivo, we generated STINGKO Unc93B1KO and cGASKO Unc93B1KO double deficient lines. We then injected single Unc93B1KO, the double deficient mice, and their Unc93B1-sufficient counterparts with TMPD, and evaluated them for features of systemic autoimmunity 5 months later. As above, the number of CD11b+ F/480hi macrophages in the peritoneal cavity was increased in STINGKO and cGASKO mice as compared to WT mice and significantly reduced in cGASKO UNC93B1KO and STINGKO Unc93B1KO double-deficient mice (Figure 3A). We also found that again both STINGKO and cGASKO mice developed higher levels of autoantibody than the WT mice, but failed to produce autoantibodies if they were Unc93B1-deficient (Figure 3B). Moreover, the proteinuria levels of STING and cGAS deficient mice were significantly higher than the WT mice, but STING/Unc93B1 and cGAS/Unc93B1double deficient mice failed to develop detectable proteinuria (Figure 3C). Collectively, these findings indicate that the elevated features of autoimmunity observed in cGAS or STING-deficient mice was abrogated when endosomal TLR signaling was abolished by deletion of Unc93B1 in vivo.
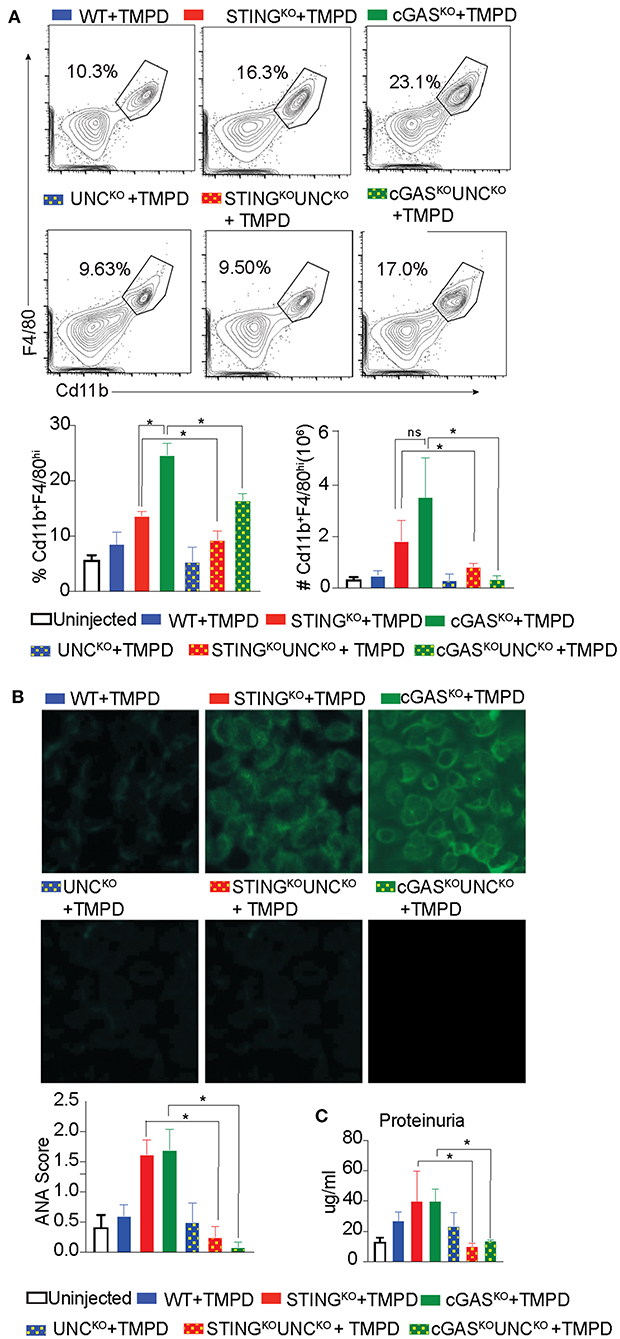
Figure 3. Exacerbation of TMPD-induced SLE in cGAS or STING deficient mice is dependent on endosomal TLRs. 12–16 week old female WT (blue), STINGKO (red), cGASKO (green), UNCKO (blue with dots), UNCKO STINGKO (red with dots), and UNCKOcGASKO (green with dots) mice were injected with 500 ul of TMPD (n = 10). Uninjected controls are indicated as open bars (n = 4 per genotype). The mice were analyzed 5 months post injection from two independent experiments. (A) Peritoneal exudate cells were evaluated by flow cytometry. Live and single Ly6C−Ly6G− cells were gated for CD11b+ and F4/80hi. Representative flow plots for WT STINGKO, cGASKO injected mice are shown in the top panel, the double deficient injected mice in the middle panel and the graphs for cell frequency and cell numbers are shown in bottom panel. (B) Sera was collected 3 months after TMPD injection and ANA were detected using HEp2 substrate slides at 1:50 serum dilution. The representative images from each genotype and condition are shown at 20X magnification in the top and middle panels and the scores for fluorescence intensity are graphed in the bottom. (C) Proteinuria was assessed in urine collected 5 months after TMPD injection by using an albumin ELISA assay. Statistical significance is represented by *P < 0.05.
We had previously reported exacerbation of autoimmune features in STING-deficient Faslpr mice generated by intercrossing B6/129 STING-deficient mice with MRL/Faslpr mice (29). To avoid any risk alleles that could be linked to the cGAS locus and modify disease outcome, we directly generated cGAS deficient MRL/Faslpr mice using a CRISPR/Cas9 genome editing strategy in MRL/lpr mice. We confirmed cGAS deficiency in the gene-edited strain by stimulating cGAS-deficient and control bone marrow derived macrophages (BMDMs) with ISD and measuring interferon production. cGASKO/ Fas lpr cells failed to produce Interferonβ (IFNβ) compared to WT/Faslpr cells in response to ISD, confirming that the CRISPR generated strain was functionally cGAS deficient (Figure 4A left). We also immunoblotted lysates obtained from WT/Faslpr and cGASKO/Fas lpr BMDMs. As expected, we found no detectable levels of cGAS protein in macrophages from cGASKO/Fas lpr mice whereas WT/Faslpr macrophages expressed cGAS protein (Figure 4A right). cGASKO/Faslpr and WT/Faslpr littermate controls were then evaluated for various SLE features and WT and cGAS deficient mice on a C57BL/6 background were used as negative controls since they do not develop autoimmunity spontaneously. Survival of the cGASKO/Faslpr mice was slightly compromised as compared to their WT/Faslpr littermates (Figure 4B). The cGASKO/Faslpr mice developed significantly higher levels anti-nucleosome antibodies in the serum as compared to WT/Faslpr littermate controls (Figure 4C). The cGASKO/Faslpr mice show increased proteinuria in the urine and increased cellular infiltrate in the kidneys as compared to the WT/Faslpr littermate controls (Figure 4D). However, splenomegaly or lymphadenopathy showed only trending increases in cGASKO/Faslpr as compared to littermate controls (Figure 4E). Similarly, the percentage of CD3+ B220+ (CD8 CD4 double negative) T cells or pDC were only modestly increased in the cGASKO/Faslpr mice compared to WT/Faslpr (Figure 4F). In addition, we also performed gene expression analysis in the total splenocytes obtained from these mice and we did not see any significant diffrences between the cGASKO/Faslpr mice compared to WT/Faslpr (Supplementary Figure S2A). Importantly, our data indicates that neither STING nor cGAS deficiency rescue MRL/lpr mice from SLE and are therefore unlikely to sense the extracellular DNA that accumulates in this model. Thus, the importance of the cGAS/STING pathway in the promotion of autoinflammation does not necessarily translate to the promotion of SLE.
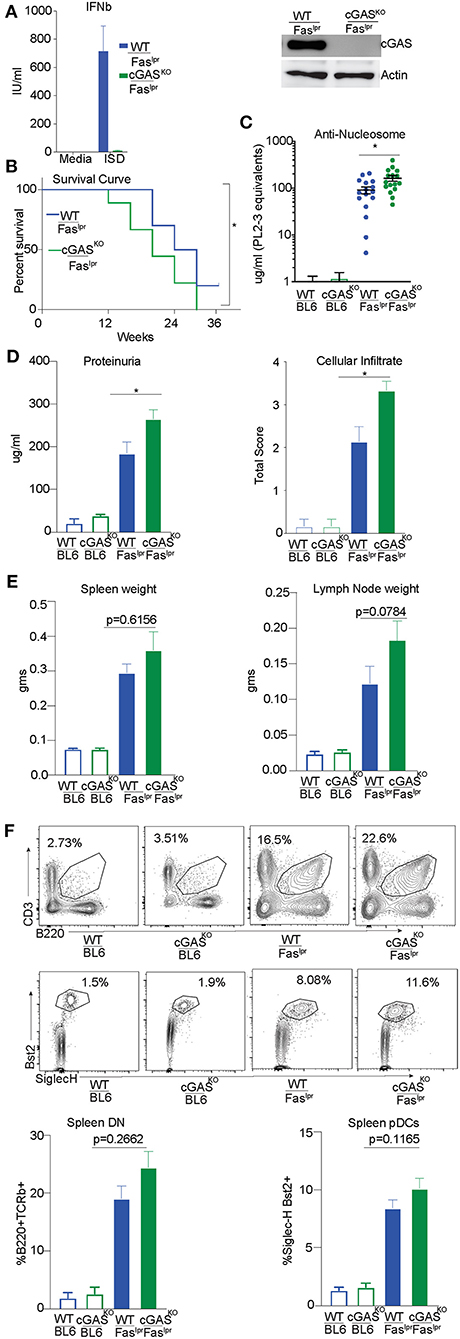
Figure 4. cGAS deficiency does not rescue SLE-prone MRL/Faslpr mice. (A) BMDMs were isolated from WT/ Faslpr and cGAS KO/ Faslpr mice. These cells were either transfected or un-transfected with ISD 5 uM and interferon β ELISA was performed after overnight treatments. (B) BMDM cell lysates were prepared and western blot was performed to detect cGAS levels. Actin was blotted as loading control. (B) WT/Faslpr (n = 10) and cGASKO/Faslpr (n = 18) mice were observed until the time of death and Kaplan Meier survival analysis was performed. (C) Anti-nucleosome ELISA was performed for 12–16 week old mice B6 WT and B6 cGASKO mice (open bars), WT/Faslpr littermates (blue bar, n = 16), cGASKO/Faslpr mice (green bar, n = 16). (D) Urine samples from 16 week old B6 WT and B6 cGASKO mice (open bars), WT/Faslpr littermates (blue bar, n = 16), cGASKO/Faslpr mice (green bar, n = 18) were screened for proteinuria using an albumin ELISA assay. H&E staining was performed to analyze cellular infiltrate WT/Faslpr littermates (blue bar, n = 7), cGASKO/Faslpr mice (green bar, n = 7). (E) Spleen and lymph nodes weights were determined at 12 weeks of age (n = 16 mice each for WT/Faslpr and cGASKO/Faslpr). (F) Flow cytometry was performed on splenocytes and double negative T cells (CD4− CD8−) were identified as CD3+ B220+. To enumerate pDCs, B220− CD3− CD11c+ splenocytes were gated for SiglecH+ and Bst2+ cells. Representative flow plot is shown for each cell type and corresponding genotype in the top/middle panels and the frequency of cells is graphed in the bottom. All the flow analysis was performed on 12 week old female mice, (n = 16 mice each for WT/Faslpr and cGASKO/Faslpr). Statistical significance is represented by *P < 0.05.
Engagement of cGAS and the endosomal DNA sensor TLR9, can lead to the production of type I IFNs and proinflammatory cytokines. Stressed, damaged, or dying cells can provide sources of endogenous ligands that can activate both pathways. The importance of TLRs in SLE disease pathogenesis has been demonstrated by both loss-of-function and gain-of-function mutations in multiple animal models of SLE (3, 43–45), whereas DNA sensing mediated by the cGAS-STING pathway has been implicated in several human monogenic autoinflammatory diseases (46–48), as well as in mouse models of type I interferonopathies (49–51). In contrast to autoinflammatory diseases, where STING activation amplifies inflammation, our initial study showed that the STING pathway can attenuate clinical manifestations of SLE (29). We have now extended our initial observation to a chronic inducible model of SLE (TMPD induced SLE) as well as gene-targeted MRL/lpr SLE-prone mice, where we can monitor the progression of the disease without the caveat of additional risk alleles due to mixed genetic backgrounds. Our findings show that STING or cGAS deficiency does not cure lupus in either case. Rather, consistent with our earlier work (29), STING or cGAS deficiency leads to an earlier onset of SLE as measured by autoantibody levels in the serum and proteinuria in the urine. These data provide a cautionary note for the use of cGAS-STING targeted therapies for the treatment of SLE.
We have now also shown that neither Unc93B1/STING nor Unc93B1/cGAS mice develop TMPD induced SLE. These data further demonstrate that even in mice that fail to express STING, SLE is still dependent on endosomal TLRs. To determine whether DNA plays a role as an instigator of TMPD-induced lupus, we generated mice with increased levels of extracellular DNAse I using a one-time i.p. injection of AAV encoding DNAse I. We initially set out to test the premise that the protective role of STING (and TLR9) was DNA-dependent, and if this were the case, depletion of DNA should have resulted in an amplified TLR7 signal and led to a more severe clinical outcome. Instead, DNAseI-AAV-treated mice did not make autoantibodies against dsDNA, as indicated by the ANA staining pattern, and did not even develop other clinical manifestations of SLE. These data point to a critical role for a DNA sensor, possibly TLR9, in promoting the development of TMPD-induced SLE, and suggest that there may be a bimodal interplay between TLR7 and TLR9 in disease initiation and then disease progression. Alternatively, a DNA sensor other than cGAS or TLR9 may be involved. DNAseI-treated mice injected with TMPD show an overall reduction in inflammation and proteinuria but a very different autoantibody profile which could be more RNA driven, a phenomenon similar to the impact of TLR9-deficiency. TLR9- and TLR7-deficient SLE prone mice fail to make autoantibodies reactive with DNA and RNA, respectively, while TLR7-deficient and TLR7/TLR9 double deficient mice develop much less severe clinical phenotypes and exhibit markedly prolonged survival (2). It is also important to mention, that although DNAse1 is the most abundant secreted endonuclease expressed in some tissues including the kidneys, the AAV gene delivery approach here could have led to a more widespread expression of DNAseI than that seen under physiological conditions.
Although the mechanisms remain unresolved, this study reinforces the importance of investigating both proinflammatory and anti-inflammatory activities of nucleic acid-sensors. Both the TLR9 and STING pathways promote the IFN- and NFκB-driven expression of proinflammatory cytokines and chemokines, but at the same time induce negative regulators of these pathways. Thus, the outcome of nucleic acid sensor engagement will depend on the balance between positive and negative regulators of inflammation and transcriptional regulation of the relevant genes which is likely to reflect activation status of the receptor expressing cells. The dual functionality for cytosolic pathways is not surprising since a published study showed that the cytosolic RNA sensor, RIG-I also dampens TLR responses in myeloid cells by suppressing TLR driven interleukin 12 (IL-12b) gene transcription (52). The evidence of increased autoantibody production due to STING deficiency is also seen in a DNAse1L3-deficient mouse model used to evaluate SLE, where DNAse1L3 another extracellular DNAse clears microvesicle-associated DNA. The DNAse1L3 knockout mice were crossed with STING-deficient mice and serum titers of anti-dsDNA IgG as determined by ELISA, were higher in DNAse1L3/ STING double knockout mice than in single DNAse1L3 knockout mice (7).
More recently, several groups have described a role for the STING pathway in the clearance of DNA by autophagy (53, 54), suggesting that STING deficiency could lead to reduced clearance of DNA. The ensuing accumulation of DNA in endosomal compartments of these cells could then enhance TLR dependent cytokine production through a cell intrinsic mechanism. DNAse II-deficient mice provide another example of excessive DNA accrual leading to the activation of multiple nucleic acid sensing pathways since the cGAS-STING, AIM2, and endosomal TLRs all have been shown to contribute to the development of autoimmunity or autoinflammation in this model (16, 55, 56). It is also possible that different DNA sensors may function in tissue specific manner for instance cGAS STING pathway can still contribute to certain aspects of disease such as the cutaneous lupus features as suggested by Skopelja-Gardner et al. (20). A better understanding of the interplay between cells responding to endogenous nucleic acid ligands will provide important insights for preventing the onset and progression of both autoinflammatory and autoimmune diseases in patients suffering from these conditions.
The original contributions generated for this study are included in the article/Supplementary Material and submitted to NCBI GEO (Gene Expression Omnibus) with accession number GSE169655 and linked to https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE169655. Further inquiries can be directed to the corresponding author/s.
All mice were bred and maintained at the Department of Animal Medicine of the University of Massachusetts Medical School in accordance with the regulations of the American Association for the Accreditation of Laboratory Animal Care and all protocols were approved by the Institutional Animal Care and Use Committee.
SS, AM-R, and KAF designed research. SS, MM, JM, JA, KMG, and ZJ performed research. GAB and KMN contributed new reagents and analytic tools. MM, AM-R, and KAF wrote the paper. All authors contributed to the article and approved the submitted version.
This work was supported by NIH grant AI128358 and AI132152. The funder was not involved in the study design, collection, analysis, interpretation of data, the writing of this article or the decision to submit it for publication.
GAB is employed by the company Merck and Co., Inc., Kenilworth, NJ, United States. The authors declare that this study received funding from sponsored research agreement from GSK (100251747). The funder had the following involvement with the study: decision to submit it for publication.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We would like to thank Sreya Ghosh for technical help and Dr. Mark Shlomchik, MD, PhD for his feedback on this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fimmu.2021.605930/full#supplementary-material
1. Pascual V, Farkas L, Banchereau J. Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol. (2006) 18:676–82. doi: 10.1016/j.coi.2006.09.014
2. Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. (2006) 25:417–28. doi: 10.1016/j.immuni.2006.07.013
3. Santiago-Raber ML, Baudino L, Izui S. Emerging roles of TLR7 and TLR9 in murine SLE. J Autoimmun. (2009) 33:231–8. doi: 10.1016/j.jaut.2009.10.001
4. Ueki M, Takeshita H, Fujihara J, Iida R, Yuasa I, Kato H, et al. Caucasian-specific allele in non-synonymous single nucleotide polymorphisms of the gene encoding deoxyribonuclease I-like 3, potentially relevant to autoimmunity, produces an inactive enzyme. Clin Chim Acta. (2009) 407:20–4. doi: 10.1016/j.cca.2009.06.022
5. Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet. (2011) 43:1186–8. doi: 10.1038/ng.975
6. Carbonella A, Mancano G, Gremese E, Alkuraya FS, Patel N, Gurrieri F, et al. An autosomal recessive DNASE1L3-related autoimmune disease with unusual clinical presentation mimicking systemic lupus erythematosus. Lupus. (2017) 26:768–72. doi: 10.1177/0961203316676382
7. Sisirak V, Sally B, D'Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, et al. Digestion of chromatin in apoptotic cell microparticles prevents autoimmunity. Cell. (2016) 166:88–101. doi: 10.1016/j.cell.2016.05.034
8. Mobarrez F, Fuzzi E, Gunnarsson I, Larsson A, Eketjall S, Pisetsky DS, et al. Microparticles in the blood of patients with SLE: size, content of mitochondria and role in circulating immune complexes. J Autoimmun. (2019) 102:142–9. doi: 10.1016/j.jaut.2019.05.003
9. Soni C, Reizis B. Self-DNA at the epicenter of SLE: immunogenic forms, regulation, and effects. Front Immunol. (2019) 10:1601. doi: 10.3389/fimmu.2019.01601
10. Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet. (2006) 38:917–20. doi: 10.1038/ng1845
11. Rodero MP, Tesser A, Bartok E, Rice GI, Della Mina E, Depp M, et al. Type I interferon-mediated autoinflammation due to DNase II deficiency. Nat Commun. (2017) 8:2176. doi: 10.1038/s41467-017-01932-3
12. Yoshida H, Okabe Y, Kawane K, Fukuyama H, Nagata S. Lethal anemia caused by interferon-beta produced in mouse embryos carrying undigested DNA. Nat Immunol. (2005) 6:49–56. doi: 10.1038/ni1146
13. Stetson DB, Ko JS, Heidmann T, Medzhitov R. Trex1 prevents cell-intrinsic initiation of autoimmunity. Cell. (2008) 134:587–98. doi: 10.1016/j.cell.2008.06.032
14. Ahn J, Gutman D, Saijo S, Barber GN. STING manifests self DNA-dependent inflammatory disease. Proc Natl Acad Sci U S A. (2012) 109:19386–91. doi: 10.1073/pnas.1215006109
15. Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr, Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity. (2012) 36:120–31. doi: 10.1016/j.immuni.2011.11.018
16. Baum R, Sharma S, Carpenter S, Li QZ, Busto P, Fitzgerald KA, et al. Cutting edge: AIM2 and endosomal TLRs differentially regulate arthritis and autoantibody production in DNase II-deficient mice. J Immunol. (2015) 194:873–7. doi: 10.4049/jimmunol.1402573
17. Serpas L, Chan RWY, Jiang P, Ni M, Sun K, Rashidfarrokhi A, et al. Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc Natl Acad Sci U S A. (2019) 116:641–9. doi: 10.1073/pnas.1815031116
18. Thim-Uam A, Prabakaran T, Tansakul M, Makjaroen J, Wongkongkathep P, Chantaravisoot N, et al. STING mediates lupus via the activation of conventional dendritic cell maturation and plasmacytoid dendritic cell differentiation. iScience. (2020) 23:101530. doi: 10.1016/j.isci.2020.101530
19. An J, Durcan L, Karr RM, Briggs TA, Rice GI, Teal TH, et al. Expression of cyclic GMP-AMP synthase in patients with systemic lupus erythematosus. Arthritis Rheumatol. (2017) 69:800–7. doi: 10.1002/art.40002
20. Skopelja-Gardner S, An J, Tai J, Tanaka L, Sun X, Hermanson P, et al. The early local and systemic Type I interferon responses to ultraviolet B light exposure are cGAS dependent. Sci Rep. (2020) 10:7908. doi: 10.1038/s41598-020-64865-w
21. Wang J, Dai M, Cui Y, Hou G, Deng J, Gao X, et al. Association of abnormal elevations in IFIT3 with overactive cyclic GMP-AMP synthase/stimulator of interferon genes signaling in human systemic lupus erythematosus monocytes. Arthritis Rheumatol. (2018) 70:2036–45. doi: 10.1002/art.40576
22. Murayama G, Chiba A, Kuga T, Makiyama A, Yamaji K, Tamura N, et al. Inhibition of mTOR suppresses IFNalpha production and the STING pathway in monocytes from systemic lupus erythematosus patients. Rheumatology (Oxford). (2020) 59:2992–3002. doi: 10.1093/rheumatology/keaa060
23. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNalpha impairs autophagic degradation of mtDNA promoting autoreactivity of SLE monocytes in a STING-dependent fashion. Cell Rep. (2018) 25:921–33 e925. doi: 10.1016/j.celrep.2018.09.001
24. Hasan M, Fermaintt CS, Gao N, Sakai T, Miyazaki T, Jiang S, et al. Cytosolic nuclease TREX1 regulates oligosaccharyltransferase activity independent of nuclease activity to suppress immune activation. Immunity. (2015) 43:463–74. doi: 10.1016/j.immuni.2015.07.022
25. Yan N. Immune diseases associated with TREX1 and STING dysfunction. J Interferon Cytokine Res. (2017) 37:198–206. doi: 10.1089/jir.2016.0086
26. Grieves JL, Fye JM, Harvey S, Grayson JM, Hollis T, Perrino FW. Exonuclease TREX1 degrades double-stranded DNA to prevent spontaneous lupus-like inflammatory disease. Proc Natl Acad Sci U S A. (2015) 112:5117–22. doi: 10.1073/pnas.1423804112
27. Lee-Kirsch MA, Gong M, Chowdhury D, Senenko L, Engel K, Lee YA, et al. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 are associated with systemic lupus erythematosus. Nat Genet. (2007) 39:1065–7. doi: 10.1038/ng2091
28. Sakai T, Miyazaki T, Shin DM, Kim YS, Qi CF, Fariss R, et al. DNase-active TREX1 frame-shift mutants induce serologic autoimmunity in mice. J Autoimmun. (2017) 81:13–23. doi: 10.1016/j.jaut.2017.03.001
29. Sharma S, Campbell AM, Chan J, Schattgen SA, Orlowski GM, Nayar R, et al. Suppression of systemic autoimmunity by the innate immune adaptor STING. Proc Natl Acad Sci U S A. (2015) 112:E710–7. doi: 10.1073/pnas.1420217112
30. Lee PY, Kumagai Y, Li Y, Takeuchi O, Yoshida H, Weinstein J, et al. TLR7-dependent and FcgammaR-independent production of type I interferon in experimental mouse lupus. J Exp Med. (2008) 205:2995–3006. doi: 10.1084/jem.20080462
31. Savarese E, Steinberg C, Pawar RD, Reindl W, Akira S, Anders HJ, et al. Requirement of Toll-like receptor 7 for pristane-induced production of autoantibodies and development of murine lupus nephritis. Arthritis Rheum. (2008) 58:1107–15. doi: 10.1002/art.23407
32. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
33. Pelka K, Bertheloot D, Reimer E, Phulphagar K, Schmidt SV, Christ A, et al. The Chaperone UNC93B1 regulates toll-like receptor stability independently of endosomal TLR transport. Immunity. (2018) 48:911–22.e917. doi: 10.1016/j.immuni.2018.04.011
34. Reeves WH, Lee PY, Weinstein JS, Satoh M, Lu L. Induction of autoimmunity by pristane and other naturally occurring hydrocarbons. Trends Immunol. (2009) 30:455–64. doi: 10.1016/j.it.2009.06.003
35. Schattgen SA, Gao G, Kurt-Jones EA, Fitzgerald KA. Cutting edge: DNA in the lung microenvironment during influenza virus infection tempers inflammation by engaging the DNA sensor AIM2. J Immunol. (2016) 196:29–33. doi: 10.4049/jimmunol.1501048
36. Roberts ZJ, Goutagny N, Perera PY, Kato H, Kumar H, Kawai T, et al. The chemotherapeutic agent DMXAA potently and specifically activates the TBK1-IRF-3 signaling axis. J Exp Med. (2007) 204:1559–69. doi: 10.1084/jem.20061845
37. Bossaller L, Christ A, Pelka K, Nundel K, Chiang PI, Pang C, et al. TLR9 deficiency leads to accelerated renal disease and myeloid lineage abnormalities in pristane-induced murine lupus. J Immunol. (2016) 197:1044–53. doi: 10.4049/jimmunol.1501943
38. Calvani N, Caricchio R, Tucci M, Sobel ES, Silvestris F, Tartaglia P, et al. Induction of apoptosis by the hydrocarbon oil pristane: implications for pristane-induced lupus. J Immunol. (2005) 175:4777–82. doi: 10.4049/jimmunol.175.7.4777
39. Chalmers SA, Chitu V, Herlitz LC, Sahu R, Stanley ER, Putterman C. Macrophage depletion ameliorates nephritis induced by pathogenic antibodies. J Autoimmun. (2015) 57:42–52. doi: 10.1016/j.jaut.2014.11.007
40. Ma WT, Gao F, Gu K, Chen DK. The role of monocytes and macrophages in autoimmune diseases: a comprehensive review. Front Immunol. (2019) 10:1140. doi: 10.3389/fimmu.2019.01140
41. Sieling PA, Porcelli SA, Duong BT, Spada F, Bloom BR, Diamond B, et al. Human double-negative T cells in systemic lupus erythematosus provide help for IgG and are restricted by CD1c. J Immunol. (2000) 165:5338–44. doi: 10.4049/jimmunol.165.9.5338
42. Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, et al. Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol. (2008) 181:8761–6. doi: 10.4049/jimmunol.181.12.8761
43. Marshak-Rothstein A. Toll-like receptors in systemic autoimmune disease. Nat Rev Immunol. (2006) 6:823–35. doi: 10.1038/nri1957
44. Fairhurst AM, Hwang SH, Wang A, Tian XH, Boudreaux C, Zhou XJ, et al. Yaa autoimmune phenotypes are conferred by overexpression of TLR7. Eur J Immunol. (2008) 38:1971–8. doi: 10.1002/eji.200838138
45. Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, et al. Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol. (2014) 192:4525–32. doi: 10.4049/jimmunol.1400098
46. Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med. (2014) 371:507–18. doi: 10.1056/NEJMoa1312625
47. Rodero MP, Crow YJ. Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J Exp Med. (2016) 213:2527–38. doi: 10.1084/jem.20161596
48. Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet. (2019) 20:657–74. doi: 10.1038/s41576-019-0151-1
49. Gao D, Li T, Li XD, Chen X, Li QZ, Wight-Carter M, et al. Activation of cyclic GMP-AMP synthase by self-DNA causes autoimmune diseases. Proc Natl Acad Sci U S A. (2015) 112:E5699–705. doi: 10.1073/pnas.1516465112
50. Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A. (2019) 116:7941–50. doi: 10.1073/pnas.1818281116
51. Xiao N, Wei J, Xu S, Du H, Huang M, Zhang S, et al. cGAS activation causes lupus-like autoimmune disorders in a TREX1 mutant mouse model. J Autoimmun. (2019) 100:84–94. doi: 10.1016/j.jaut.2019.03.001
52. Negishi H, Yanai H, Nakajima A, Koshiba R, Atarashi K, Matsuda A, et al. Cross-interference of RLR and TLR signaling pathways modulates antibacterial T cell responses. Nat Immunol. (2012) 13:659–66. doi: 10.1038/ni.2307
53. Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature. (2019) 567:262–6. doi: 10.1038/s41586-019-1006-9
54. Yang J, Tang X, Nandakumar KS, Cheng K. Autophagy induced by STING, an unnoticed and primordial function of cGAS. Cell Mol Immunol. (2019) 16:683–4. doi: 10.1038/s41423-019-0240-2
55. Pawaria S, Sharma S, Baum R, Nundel K, Busto P, Gravallese EM, et al. Taking the STING out of TLR-driven autoimmune diseases: good, bad, or indifferent? J Leukoc Biol. (2017) 101:121–6. doi: 10.1189/jlb.3MR0316-115R
Keywords: cGAS/STING, SLE, pristane, DNaseI, TLRs
Citation: Motwani M, McGowan J, Antonovitch J, Gao KM, Jiang Z, Sharma S, Baltus GA, Nickerson KM, Marshak-Rothstein A and Fitzgerald KA (2021) cGAS-STING Pathway Does Not Promote Autoimmunity in Murine Models of SLE. Front. Immunol. 12:605930. doi: 10.3389/fimmu.2021.605930
Received: 13 September 2020; Accepted: 22 February 2021;
Published: 29 March 2021.
Edited by:
Jennifer Konopa Mulligan, Medical University of South Carolina, United StatesReviewed by:
Onkar Prakash Kulkarni, Birla Institute of Technology and Science, IndiaCopyright © 2021 Motwani, McGowan, Antonovitch, Gao, Jiang, Sharma, Baltus, Nickerson, Marshak-Rothstein and Fitzgerald. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katherine A. Fitzgerald, a2F0ZS5maXR6Z2VyYWxkQHVtYXNzbWVkLmVkdQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.