 Sara R. Moore
Sara R. Moore Smrithi S. Menon
Smrithi S. Menon Claudio Cortes
Claudio Cortes Viviana P. Ferreira
Viviana P. Ferreira- 1Department of Medical Microbiology and Immunology, University of Toledo College of Medicine and Life Sciences, Toledo, OH, United States
- 2Department of Foundational Medical Sciences, Oakland University William Beaumont School of Medicine, Rochester, MI, United States
The complement system is an essential player in innate and adaptive immunity. It consists of three pathways (alternative, classical, and lectin) that initiate either spontaneously (alternative) or in response to danger (all pathways). Complement leads to numerous outcomes detrimental to invaders, including direct killing by formation of the pore-forming membrane attack complex, recruitment of immune cells to sites of invasion, facilitation of phagocytosis, and enhancement of cellular immune responses. Pathogens must overcome the complement system to survive in the host. A common strategy used by pathogens to evade complement is hijacking host complement regulators. Complement regulators prevent attack of host cells and include a collection of membrane-bound and fluid phase proteins. Factor H (FH), a fluid phase complement regulatory protein, controls the alternative pathway (AP) both in the fluid phase of the human body and on cell surfaces. In order to prevent complement activation and amplification on host cells and tissues, FH recognizes host cell-specific polyanionic markers in combination with complement C3 fragments. FH suppresses AP complement-mediated attack by accelerating decay of convertases and by helping to inactivate C3 fragments on host cells. Pathogens, most of which do not have polyanionic markers, are not recognized by FH. Numerous pathogens, including certain bacteria, viruses, protozoa, helminths, and fungi, can recruit FH to protect themselves against host-mediated complement attack, using either specific receptors and/or molecular mimicry to appear more like a host cell. This review will explore pathogen complement evasion mechanisms involving FH recruitment with an emphasis on: (a) characterizing the structural properties and expression patterns of pathogen FH binding proteins, as well as other strategies used by pathogens to capture FH; (b) classifying domains of FH important in pathogen interaction; and (c) discussing existing and potential treatment strategies that target FH interactions with pathogens. Overall, many pathogens use FH to avoid complement attack and appreciating the commonalities across these diverse microorganisms deepens the understanding of complement in microbiology.
The Complement System
Complement activates through a domino-like cascade comprising over 50 proteins, resulting in outcomes essential for innate and adaptive immunity. Activation of complement occurs through three pathways: classical, lectin, and alternative (Figure 1A), which converge on the cleavage of the central component, C3 [reviewed in (1)]. The classical pathway (CP) activates when C1q of the C1 complex (C1q, C1r, C1s) recognizes and binds pathogen- or cell-bound immunoglobulins, circulating immune complexes, or to pentraxins (e.g., C-reactive protein, pentraxin 3, serum amyloid P). When C1q binds a ligand, C1r is activated, which then activates C1s. C1s sequentially cleaves C4 and C2, resulting in the CP C3 convertase, C4bC2b [reviewed in (2)]. The lectin pathway (LP) activates when mannose-binding lectin (MBL), ficolins, or collectins recognize molecular patterns such as carbohydrates and other ligands on foreign surfaces. This leads to activation of MBL-associated serine proteases (MASPs), whereby MASP-2 cleaves C4 and C2 to form the LP C3 convertase, C4bC2b [reviewed in (3)].
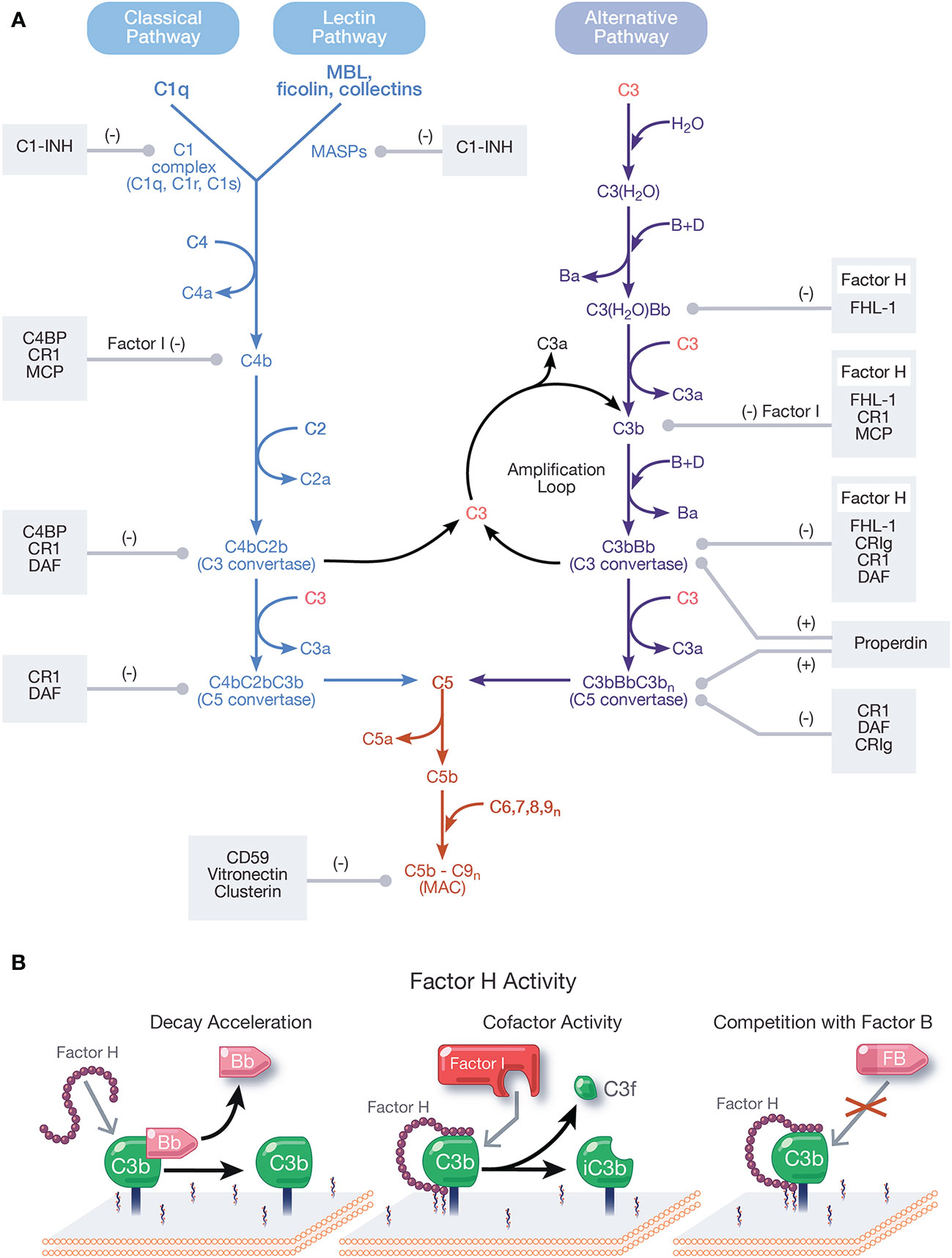
Figure 1 Overview and regulation of the complement system. (A) Complement is activated by three pathways: the classical, lectin, and alternative pathways. The classical pathway (CP) activates when the C1 complex (C1q, C1r, C1s) recognizes and binds pathogen- or cell-bound immunoglobulins, circulating immune complexes, or pentraxins. The lectin pathway (LP) activates when mannose-binding lectin (MBL) ficolins, or collectins recognize molecular patterns such as carbohydrates and other ligands on foreign surfaces. CP and LP activation results in cleavage of C4, followed by cleavage of C2, forming the CP/LP surface bound C3 convertase, C4bC2b. The alternative pathway (AP) is spontaneously activated when soluble C3 hydrolyzes to C3(H2O). C3(H2O) can bind FB (labeled B) and recruit Factor D (labeled D) which cleaves FB to Bb (and Ba), resulting in the fluid phase AP C3 convertase, C3(H2O)Bb. C3(H2O)Bb cleaves C3 to C3a and C3b. C3b then binds covalently to nearby surfaces to form membrane-bound C3 convertase, C3bBb. C3 convertases derived from all pathways cleave C3 to C3a and C3b. C3b combines with formed C3 convertases to form the CP/LP C5 convertase (C4bC2bC3b) and AP C5 convertase (C3bBbC3bn). C5 is cleaved by C5 convertases to initiate the common, terminal pathway, which culminates in the formation of the membrane attack complex (MAC). C3b produced from the cleavage of C3 by C3 convertases from all pathways forms an amplification loop that contributes to the generation of additional AP C3 convertases. Positive (labeled +) and negative (labeled −) regulators of all complement pathways are shown. Membrane-bound complement negative regulators include complement receptor 1 (CR1/CD35), decay accelerating factor (DAF/CD55), membrane cofactor protein (MCP/CD46), CD59, and complement receptor of the immunoglobulin family (CRIg). Soluble negative regulators include Factor H (FH), FI, C4 binding protein (C4BP), C1 inhibitor (C1-INH), clusterin, vitronectin, and Factor-H like protein 1 (FHL-1). Positive regulators of complement include FH-related proteins (FHRs) -1, -4, and -5, and properdin. (B) Function of FH. FH accelerates the decay of AP C3 convertase by dissociating Bb (decay acceleration function); acts as a cofactor for Factor I-mediated cleavage of C3b into iC3b, an inactivated form that does not allow complement activity to progress (cofactor activity function); and competes with FB for binding to C3b to form the AP C3 convertase.
Unlike the CP and LP, which are triggered upon recognition of distinct ligands, the alternative pathway (AP) is continuously active and initiates spontaneously on surfaces not protected by complement regulatory proteins. In blood, low levels of C3 undergo spontaneous hydrolysis (“tick-over”) to form C3(H2O). C3(H2O) binds Factor B (FB) and circulating Factor D cleaves FB to Bb and Ba, resulting in formation of the fluid phase AP C3 convertase, C3(H2O)Bb. C3(H2O)Bb cleaves C3 to C3b and C3b binds covalently to nearby surfaces to form membrane-bound C3 convertase, C3bBb. The AP also contributes to a powerful amplification loop through activation of C3b, which in some cases contributes up to 80% of the total complement response, even after initiation by the CP and LP (4, 5). In fact, it is argued the AP is mainly an amplification mechanism with minimal contributions from the tick-over of C3 [reviewed in (6)].
C3 convertases derived from each pathway converge to cleave C3, generating C3a and C3b, which complexes on or near C3 convertases to form C5 convertases. Cleavage of C5 by C5 convertases generates C5a and C5b to initiate the terminal pathway. Sequential binding to C5b by C6, C7, C8, and multiple copies of C9 form the membrane attack complex (MAC, C5b-9). Outcomes of complement are numerous and include generation of pro-inflammatory mediators C3a and C5a, C3 fragments involved in opsonization and immune modulation, and cell lysis by the pore-forming MAC [reviewed in (7, 8)].
Regulation of all complement pathways protects the host from unwarranted complement-mediated attack. Complement negative regulators circulate in blood and include FH, Factor I (FI), C4 binding protein (C4BP), C1 inhibitor (C1-INH), clusterin, vitronectin, and Factor-H like protein 1 (FHL-1). Membrane-bound complement negative regulators include complement receptor 1 (CR1/CD35), decay accelerating factor (DAF/CD55), membrane cofactor protein (MCP/CD46), CD59, and complement receptor of the immunoglobulin family (CRIg) [reviewed in (9)]. Importantly, FH is the primary regulator of the AP in the fluid phase and on cell surfaces and is essential for protecting the host from AP attack. Pathogens have developed survival strategies to evade the immune response including coopting FH from the host to avoid the AP. This phenomenon will be described in detail in this review.
Factor H in the Host
FH, formerly known as β1H (10, 11), is abundantly found in plasma with a wide concentration range between 116 and 810 μg/ml [reviewed in (12, 13)]. However, recent studies reveal an average FH concentration of ~230 μg/ml, when measurement of FH family proteins FHL-1 and Factor-H related proteins (FHRs) are excluded [reviewed in (14)]. FH is constitutively expressed by hepatocytes (15, 16) and also produced by monocytes, fibroblasts, endothelial cells, platelets, retinal pigment epithelial cells, peripheral blood lymphocytes, myoblasts, rhabdomyosarcoma cells, glomerular mesangial cells, neurons, and glial cells [reviewed in (12, 17)]. FH is a 155-kDa glycoprotein (18, 19) encoded from a single gene, HF1/CFH, found within the regulator of complement activation gene cluster on chromosome 1q32 [reviewed in (20)]. FH consists of 20 homologous complement control protein modules (CCP) (21, 22), with each module containing ~60 amino acid residues (22) connected by short spaces of three to eight amino acid residues [reviewed in (17)]. Structural studies indicate FH may adopt a flexible folded back conformation in solution (23–25).
FH is the primary negative regulator of the AP in the fluid phase and at the cell surface. The three functions of FH include: (a) competing with FB for C3b binding (26); (b) accelerating the decay of surface-bound C3 and C5 convertases (10, 27, 28) and to a lesser extent, fluid-phase C3 convertase (29), by disassociating Bb from convertases; and (c) acting as a cofactor for FI-mediated cleavage of C3b into the inactive form, iC3b (28, 30, 31) (Figure 1B). These regulatory functions are carried out through CCPs 1–4 (32–35).
Upon binding to a surface, FH protects against AP activity. FH simultaneously recognizes C3 fragments and host cell markers to discriminate self (host, non-activators) from non-self (activators), which lack or have very low levels of surface polyanions. Several FH domains participate in binding C3 fragments and/or polyanions (Figure 2, top panel). CCPs 1–6 bind C3 and C3b and a weak binding site for C3b is recognized at CCPs 13–15 (36). CCPs 19–20 recognize iC3b, C3b, and C3d (36, 37). Host cell polyanions, such as sialic acids (38, 39) and glycosaminoglycans (GAGs), which include heparins (40, 41) and dextran sulfate (41, 42), serve as recognition markers for FH regulation on host cells. CCP 20 is the only known site to recognize sialic acid (43). Heparin binding sites include CCPs 6–8 and 18–20, with a possible weak binding site on CCPs 11–13 (36). CCPs 19–20 are the most important region of FH for binding to cell surfaces by recognizing both C3b and polyanions [reviewed in (44)]. FH has a 10-fold increase in affinity towards C3b in the presence of host sialic acid (38, 39, 41). In addition to recognizing host cell markers, FH acts as a ligand for annexin II, DNA, C-reactive protein, and pentraxin-3 to limit excessive complement activation during apoptosis [reviewed in (44)].
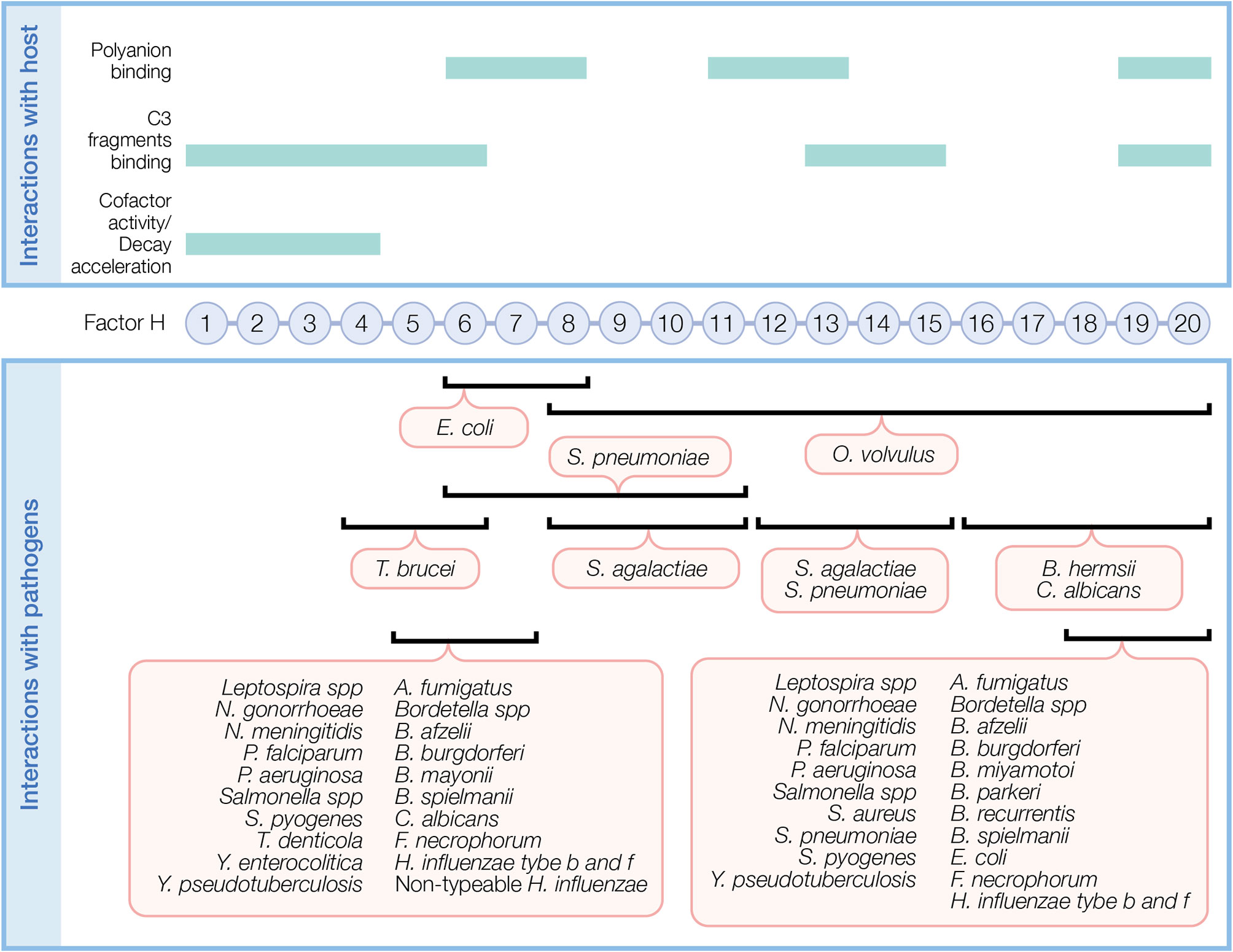
Figure 2 Factor H domains involved in binding to the host and pathogens. FH contains 20 complement control protein modules (CCP) each with about 60 amino acids linked by three to five amino acids. In the top panel, FH domains necessary for its regulatory function and domains interacting with C3 fragments and polyanions are indicated. In the bottom panel, overlapping FH domains involved in binding pathogens are indicated. Refer to Table 1 for specific FH domains involved in binding to individual pathogens and for pathogens binding FH through undefined domains.
FH belongs to a family of proteins including FHL-1 and five FHRs, reviewed elsewhere (14, 45), that are present at significantly lower concentrations than FH in blood [reviewed in (14)]. Briefly, FHL-1 is a result of alternative splicing of CFH and contains seven domains homologous to FH CCPs 1–7 plus four C-terminal amino acids. FHL-1 is a negative regulator of complement in the fluid phase [reviewed in (14, 45, 46)]. In contrast, FHRs do not share a gene with FH or FHL-1, but instead, have domains homologous to the center and C-terminal FH domains [reviewed in (14)]. Thus, due to a non-functional N-terminus, FHRs compete with FH, thereby acting as potential positive regulators of the AP, yet the roles of FHRs are not well defined [reviewed in (14, 45)].
In addition to the prototypical role of FH as a negative regulator of the AP, non-canonical roles of FH have emerged [reviewed in (45)]. FH is a ligand for complement receptor 3 (CR3; CD11b/CD18) on neutrophils and upon binding, results in release of hydrogen peroxide and lactoferrin (47). FH interaction with neutrophil CR3 also results in release of IL-8, prevention of neutrophil extracellular traps (NETs) formation, and production of reactive oxygen species (ROS) (48). Thus, FH may reduce host damage by inhibition of NETs and ROS (48). In particular, FH bound to Candida albicans modulates neutrophil function by interacting primarily with CR3, and to a lesser extent to complement receptor 4 (CR4; CD11c/CD18), leading to more effective killing of the pathogen (49). FH is also known as adrenomedullin-binding protein-1 and binds adrenomedullin (50), a vasodilator peptide hormone widely expressed in many human tissues [reviewed in (51)]. FH may protect adrenomedullin from proteolytic degradation and thus has therapeutic value in disease models of sepsis, wound healing, and hemorrhage [reviewed in (52)]. In addition to acting as a ligand, FH is internalized by early apoptotic cells, resulting in enhancement of intracellular C3 cleavage and increased iC3b surface opsonization to promote uptake by monocytes [reviewed in (45)].
Factor H Interactions With Pathogens
Evasion of complement attack is key to pathogen survival in the host. In general, pathogens evade complement through numerous strategies, which include: (a) expression of proteins that mimic host surface-bound complement regulators; (b) secretion of proteases to digest complement fragments; (c) exploitation of complement opsonization to promote intracellular invasion; (d) secretion of complement inhibitory proteins; and (e) recruitment of fluid phase complement regulators, which includes FH family proteins [reviewed in (53–55)].
Pathogen evasion of the complement system has been described [reviewed in (54–58)], including strategies particular to bacteria [reviewed in (53, 59)]; fungi [reviewed in (60)]; parasites [reviewed in (61)]; viruses [reviewed in (62)]; and evasion mechanisms involving FH family proteins [reviewed in (17, 19, 63)]. Herein, we review how pathogens steal FH to outsmart the immune system. Specifically, we describe the implications of FH binding in pathogen evasion, mechanisms of binding, and therapeutic targeting of the FH-pathogen interface.
Binding of Factor H to Pathogens to Control Alternative Pathway Activity
FH is the primary target of pathogens for AP evasion. When sequestered from blood to the pathogen surface, FH retains its function as a negative regulator of complement, thus circumventing lysis by MAC, opsonization by C3 fragments, and pro-inflammatory consequences of complement cleavage products such as C3a and C5a [reviewed in (7, 8)].
Binding of Factor H to Pathogens for Purposes Other Than Alternative Pathway Evasion
Though FH is primarily a negative regulator of the AP, it has also been shown to regulate the CP [reviewed in (64)]. Moreover, FH binding to certain pathogens facilitates host cellular adherence and invasion. Binding of FH to pneumococcal surface protein C (PspC) on Streptococcus pneumoniae increases attachment to, and invasion of, host cells (65). Likewise, in Mycoplasma hyopneumoniae, FH binding increases adherence to epithelial cells (66) while FH interaction with influenza A virus promotes viral cellular invasion. As opposed to binding FH to evade complement attack, pathogens also cleave FH. Example of pathogens that cleave and inactivate FH include Salmonella enterica (67), Yersinia pestis (67), C. albicans (68), and Treponema denticola (68, 69). Inactivation of FH by pathogen proteases may result in unchecked AP activation, consequentially depleting complement proteins surrounding the pathogens, thus protecting them from attack. In addition, FH inactivation leads to complement dysregulation on host cells, which, as suggested by Riva et al. and Miler et al., may compromise tissue integrity to facilitate pathogen invasion (67, 70).
Binding of Factor H Family Members to Pathogens
FHL-1 and FHRs are also capable of binding pathogens (Table 1). Pathogen recruitment of FHL-1 serves the same purpose in complement evasion as binding to full-length FH. However, as suggested by Kunert et al., low serum FHL-1 titers, along with the increased binding affinity of full-length FH because of its additional C-terminal binding domains, may limit pathogen binding to FHL-1 (132). Interestingly, although most Borrelia species bind FH, B. burgdorferi Complement Regulator-Acquiring Surface Proteins (CRASP)-2 (Csp-Z) is shown to preferentially bind FHL-1 (183).

Table 1 Pathogen interactions with Factor H family proteins.
The benefits of pathogens binding FHRs are shown when Scl of S. pyogenes binds FHR-1 to inhibit the formation of the terminal complement pathway (157). However, to date, most studies suggest pathogen binding to FHRs are disadvantageous, because it outcompetes full-length FH for binding to pathogen surfaces and can enhance AP activity [reviewed in (45)]. In S. pyogenes (157) and P. falciparum (184), FHR-1 outcompetes FH for binding and impairs FH cofactor activity. Moreover, in the case of P. falciparum, FHR-1 binding also leads to decreased parasite viability (184). In N. meningitidis, FHR-3 outcompetes FH binding to result in complement-mediated bacteria lysis (185). Borrelia CRASP-3 (ErpP) and -5 (ErpA) bind FHR-1, -2, and -5 while only weakly binding FH (186). However, since FHRs binding does not provide any complement evasion benefits to Borrelia (186), the consequences of FHRs binding are unclear.
Mechanisms of How Pathogens Bind Factor H to Evade the Alternative Pathway
Pathogens capture FH by adapting their surfaces to mimic host cells (i.e., expressing host cell markers) and/or expressing specific FH binding receptors (FH binding proteins). Pathogens must bind FH using accessible domains that do not interfere with the N-terminal functions of FH. The sections below describe mechanisms used by pathogens to bind FH. Figure 2 (bottom panel) illustrates regions of FH recognized by pathogens. Moreover, Table 1 summarizes pathogens known to bind FH, FHL-1, and FHRs.
The Role of Sialylation in Factor H Recruitment
Pathogens evade complement by protecting their surfaces from complement attack using host cell markers. In the host, FH recognizes specific sialic acid capped glycans on host surfaces to distinguish self from non-self, which importantly restricts AP activation on self surfaces, while allowing AP activity to continue on non-self (pathogen) surfaces. However, pathogens bypass AP attack by expressing host glycans (187) which go on to capture FH.
α-N-acetylneuraminic acid (Neu5Ac) is a sialic acid species present on pathogens and humans. FH exclusively binds Neu5Ac species with α(2,3) linkages (43), discriminating against pathogens expressing other Neu5Ac linkages including α(2,6) and α(2,8) [reviewed in (188, 189)]. Neisseria meningitidis and Neisseria gonorrhoeae are Gram-negative bacteria restricted to humans that express α(2,3) linked Neu5Ac which participate in FH binding and other complement evasion tactics [reviewed in (188–190)].
On the activated host surface, FH domain CCP 20 interacts with GAGs and CCP 19 binds C3b (191). Sialylated N. meningitidis likely recreates this interaction on the pathogen surface in order to bind FH. To bind FH, N. meningitidis and N. gonorrhoeae sialylate lacto-N-neotetraose (LNnT) branches of lipooligosaccharides (LOS) with Neu5Ac [reviewed in (188)]. Neu5Ac sialylation of N. meningitidis LOS enhances FH binding to pathogen surfaces (127) (Figure 3A). This is postulated to occur when sialylation on the pathogen surface replaces host GAGs as the ligand for CCP 20, while maintaining the interaction between CCP 19 and deposited C3 fragments [reviewed in (188)]. Similarly, binding of bovine FH to Histophilus somni increases when bacteria are sialylated with Neu5Ac (104). While pathogen sialylation promotes FH binding to the cell surface, it still renders pathogens vulnerable to complement-mediated damage because a portion of the deposited C3 fragments involved in binding FH will likely form C3 convertases as opposed to binding FH [reviewed in (190)].
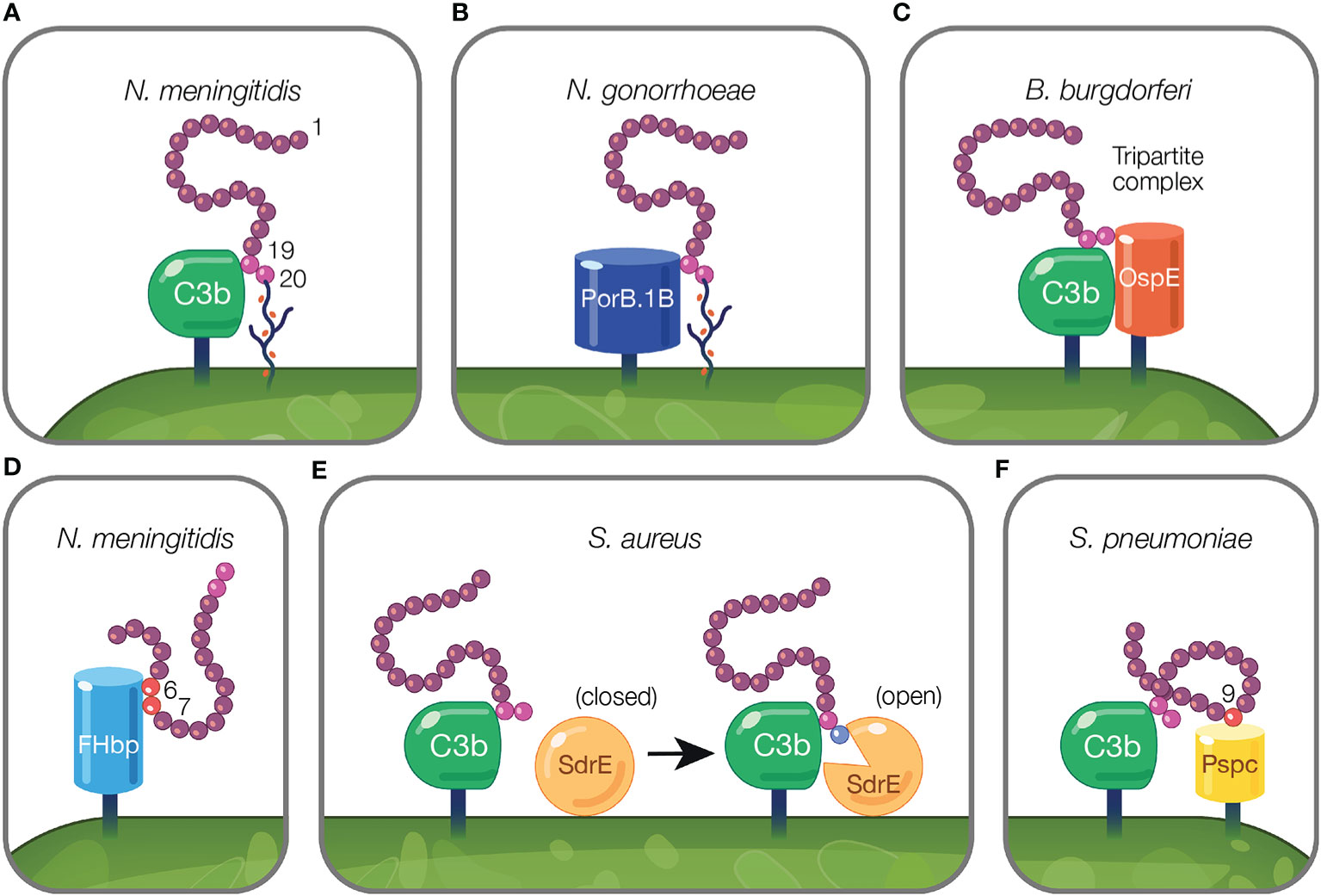
Figure 3 Mechanisms of pathogens binding Factor H to evade the alternative pathway. (A) Pathogens evade complement by protecting their surfaces from complement attack using host cell markers. Neu5Ac sialylation of N. meningitidis replaces host GAGs and binds FH CCP 20 domain while maintaining the interaction between CCP 19 and deposited C3 fragments (127). (B) Pathogen sialylation promotes FH binding in the absence of complement fragments. N. gonorrhoeae FH binding protein, PorB.1B, binds FH CCP 19 domain replacing the interaction of C3b with FH CCP 19 [reviewed in (189)]. (C, D) Pathogen FH binding proteins mimic interactions between host and FH. (C) B. burgdorferi FH binding protein, OspE, forms a tripartite complex with FH where CCP 19 binds to C3 fragments and CCP 20 interacts with OspE. (D) N. meningitidis binds FH through its FH binding protein, FHbp, via FH CCPs 6–7. (E, F) Pathogen FH interactions that do not mimic mechanisms utilized by the host. (E) FH binding to deposited C3b on the pathogen surface results in a transformational change in S. aureus FH binding protein, SdrE, from a closed to open state. In the open state, SdrE facilitates the docking of CCP 20 into a ligand binding groove (136). (F) S. pneumoniae FH binding protein, PspC, via its tyrosine 90 residue acts like a key inserting into a hydrophobic lock formed by four hydrophobic residues of FH CCP 9 (146).
Pathogen sialylation also promotes FH binding in the absence of complement fragments. Increased binding of FH to N. gonorrhoeae occurs with sialylation of LNnT LOS (120, 192) only with the concomitant presence of gonococcal FH binding protein, porin B.1B (PorB.1B) (192) (Figure 3B). This phenomenon is speculated to occur through an interaction similar to what is described for N. meningitidis, but with PorB.1B replacing the interaction of C3b with FH CCP 19 [reviewed in (189)], thus eliminating the need for deposited C3 fragments.
Sialylation also increases binding of FH CCPs 6-7 to Neisserial surface protein A (NspA) of N. meningitidis (124). Here, bacterial sialic acid potentially acts as a docking station for FH, perhaps through binding CCP 20, but this has yet to be shown experimentally and may be structurally impossible, as suggested by Lewis et al. (124). In contrast, in N. gonorrhoeae, LOS sialylation impedes binding of FH CCPs 6-7 to NspA (121).
α-N-glycolylneuraminic acid (Neu5Gc) is a sialic acid variant not expressed by humans, but present in mice (193) and on the surface of sheep erythrocytes (194). Human FH binds Neu5Gc in a similar manner as Neu5Ac (195) and N. gonorrhoeae LNnT-LOS incorporation of Neu5Gc results in FH binding and serum resistance (196). As noted by Schmidt et al., this phenomenon may skew conclusions from animal research, including infection models using humanized FH transgenic mice, as FH binding may be affected in a manner not possible in humans (195).
Pathogen Factor H Binding Proteins
In addition to decorating their surfaces with host markers, certain pathogens capture FH directly using binding proteins alone. While many FH binding proteins have been identified along with the regions to which they bind on FH, the mechanisms explaining many of these interactions remain unresolved. Below, we describe two strategies employed by pathogen FH binding proteins to capture FH. In the first mechanism, FH binding proteins mimic binding interactions present in the host, but using protein-protein interactions instead of binding to polyanions on host surfaces. In these instances, FH binding proteins bind to the same domains on FH (CCPs 6–7 and 19–20) that are used to bind to the host. In the second mechanism, FH binding proteins capture FH via domains (i.e., amino acid residues) on FH that do not bind to the host.
Pathogen Factor H Binding Proteins Mimic Interactions Between Host and Factor H
In this section, we describe how FH binding proteins capture FH by binding to the regions on FH that would normally bind to the host. However, unlike host surfaces, which bind FH through expression of host cell markers (i.e., polyanions) in combination with C3 fragment, the binding of FH to the FH binding protein on the pathogen, constitutes solely a protein-protein interaction instead.
An excellent example of FH interactions with a FH binding protein was demonstrated for the FH binding protein of N. meningitidis, FHbp (Figure 3D). Crystallography studies revealed FHbp binds FH through extensive interactions between β-barrels of FHbp and CCP 6, and through minor contacts with CCP 7 (122). The site within CCP 6 utilized by FHbp (122) overlaps with a previously described binding location for sucrose octasulphate (SOS), a highly sulphated analog of GAGs (197). Similarly, the binding domain for FhbB from T. denticola also overlaps with the SOS binding domain (70). Together, these examples demonstrate how FH binding proteins mimic interactions between FH and host charged sugars through amino acid side chains (122).
Protein motif mimicry also involve pathogen interactions with FH C-terminal domains. FH binds host surfaces through dual recognition when CCP 20 binds GAGs and CCP 19 binds the C3d part of C3b (191). Similarly, in the process of mimicking host surfaces, FH binding proteins form a tripartite complex when FH CCP 19 binds C3 fragments and CCP 20 interacts with a FH binding protein (Figure 3C). This phenomena has been described for FH binding proteins of Pseudomonas aeruginosa (Tuf) (74), Borrelia burgdorferi (OspE) (74, 85, 198), Borrelia hermsii (FhBA) (74), and Staphylococcus aureus (Sbi) (137).
In B. burgdorferi, crystallography studies indicate OspE (a paralog of CRASP-3) forms a tripartite complex between FH and C3 fragments. This occurs when loops β2–4 and the interface between loops β5–6 interact with CCP 20 which includes FH amino acids overlapping with those involved in heparin binding, while deposited C3dg interacts with CCP 19 (85, 198). The majority of amino acid residues utilized by OspE for binding FH are conserved across the OspE protein family (85). Interestingly, ectopic expression of B. burgdorferi CRASP-3 in a serum sensitive Borrelia strain bound minute amounts of FH and did not confer serum protection (186). As suggested by Kolodziejczyk et al., the FH tripartite complex may not form when OspE is ectopically expressed, leading to inadequate complement protection (198). Alternatively, according to Siegel et al., additional complement binding proteins may be required to unfold FH to a conformation sufficient for interacting with binding proteins, which does not occur when ectopically expressed (186). While this study examined tripartite complex formation under conditions in which the complex is not surface-bound, this formation is possible under physiological conditions because the domains responsible for tethering OspE to the surface do not interfere with the binding site for FH and bound FH is orientated toward the surface to which C3b is bound via the thioester bond (85).
Pathogen Factor H Binding Proteins Sequester Factor H Using Interactions Not Found in the Host
Pathogen interactions with FH have been described that do not reflect mechanisms utilized by the host. In the model proposed by Zhang and colleagues, S. aureus surface protein serine–aspartate repeat protein E (SdrE) tightly binds a 21 amino acid region of CCP 20 not involved in the binding of C3d or of host cell markers (i.e., heparin, GAGs, and sialic acid) (85, 136). Upon FH binding to deposited C3b on the pathogen surface, SdrE undergoes a transformational change from a close to open state, which facilitates the docking of CCP 20 into a ligand binding groove (136). SdrE functions as a “clamp” to stabilize the SdrE-FH complex by locking and latching the FH tail into its ligand binding groove (136). The “close, dock, lock, and latch” mechanism is similar to strategies utilized by members of the microbial surface components recognizing adhesive matrix molecules, to which SdrE belongs (136). The described mechanism of SdrE is unique in the field of complement immune evasion [reviewed in (199)] (Figure 3E).
S. pneumoniae FH binding protein, PspC, also binds FH through a domain less utilized by other pathogens. PspC binds CCP 9 when the tyrosine 90 residue of PspC inserts like a key into a hydrophobic lock formed by four hydrophobic residues of FH (146) (Figure 3F). Studies using PspC lacking the residue important for interacting with the hydrophobic lock, showed PspC forms stable complexes with FH CCPs 8–10 (145). Here, PspC induces a functionally enhanced FH conformation that accelerates C3bBb decay 5-fold compared to FH alone, and increases the ability of FH to bind C3b 2-fold (145). Herbert et al. suggest a similar process may occur in the host in which host cell markers on host surfaces bind FH, and result in configurations of FH with enhanced regulatory activity.
Role of Factor H Binding Proteins in Pathogen Virulence
The necessity of FH binding proteins in pathogen virulence is questionable. While most of the described interactions between pathogens and FH demonstrate the advantage of binding FH in AP evasion through in vitro assays, in vivo studies are essential for further characterizing the role of FH binding in pathogen virulence. The importance of FH in pathogen virulence varies according to pathogen. For example, inhibition of FH binding is protective in several pathogens as supported by studies in which pathogen FH binding is blocked with recombinant FH proteins, or through vaccination with FH binding proteins (discussed in Therapeutic and Preventive Strategies Targeting the Pathogen-Factor H Interface). In addition, in vivo studies involving the FH binding protein, plasmodial transmembrane protein gliding associated protein 50 (PfGAP50) of Plasmodium falciparum was shown to be important in pathogen virulence when antibody neutralization of PfGAP50 reduced parasite transmission to the mosquito vector (172).
In vitro serum sensitivity of Borrelia species is associated with FH binding potential, considering almost all serum-resistant species recruit FH [reviewed in (200)]. However, in vivo studies demonstrate FH binding to pathogens is not required for infection. Numerous studies in Borrelia indicate FH may be dispensable for infection. For instance, FH-deficient mice, and mice without FB or C3 (no AP activity), were infected with Borrelia at levels similar to wild type animals (201). This finding suggests additional complement-dependent and -independent immune evasion strategies of Borrelia replace the benefits of FH capture for survival in the host (201). B. garinii is responsible for causing Lyme disease, but does not bind FH, highlighting that FH may be dispensable for infection (202). The mechanisms by which B. garinii evades complement to establish infection in the host remain unknown [reviewed in (200)]. Similarly, CspA (CRASP-1) variants are not required for spirochete survival at the mouse tick bite site, though it is expressed at this location (203). Additionally, FH binding protein, FhbA of B. hermsii is not required for murine infection or human serum resistance (204). B. hermsii does not express additional FH binding proteins, suggesting the presence of additional complement evasion strategies (204). While in vitro studies demonstrate BclA, a FH binding protein of Bacillus anthracis, downregulates complement activation and protects against cell lysis, inoculation of mice with a lethal dose of B. anthracis spores lacking BclA, did not affect animal survival or bacterial burden compared to inoculation with an isogenic wild type strain (72).
While some studies convincingly demonstrate FH binding is dispensable for pathogen virulence, others have led to inconclusive findings. An example of disputable findings regarding the effect of FH binding in pathogen virulence involves studies of S. pyogenes. S. pyogenes is a human-specific pathogen that binds human FH through three proteins, including M protein (153). To study the effects of S. pyogenes complement evasion in vivo, transgenic mice expressing human FH and C4BP were generated. The human FH and C4BP in the serum from these animals, bound to S. pyogenes in vitro (205). Compared to wild type animals, transgenic mice expressing FH and C4BP were more susceptible to fatal infection by a S. pyogenes strain (AP1 strain) that binds human FH and C4BP through protein H (205), a member of the M protein family (206, 207). This demonstrates the additive virulence effect of binding more than one complement regulator (205). However, infection with a different S. pyogenes strain that does not bind FH or C4BP resulted in the same degree of mortality between transgenic and wildtype animals (205). Furthermore, pathogen inoculation with an isogenic S. pyogenes strain that does not express protein H, did not affect animal survival (205). Altogether, these results demonstrate recruitment of complement inhibitors by S. pyogenes exacerbates infection when FH and C4BP are present, but have no effect on disease if the strain cannot capture the regulatory proteins (205). However, in another study, FH binding to S. pyogenes did not modulate pathogen virulence. In this study, transgenic mice expressing chimeric FH (containing human FH CCPs 6–8) did not show an increased susceptibility to infection with a S. pyogenes strain capable of binding FH when compared to wild type animals (155). As suggested by Ermert et al., the discrepancy in findings may be due to differences in mouse strains, bacterial strains, and infection route (205).
Factors Affecting Pathogen Factor H Binding Protein Expression
FH binding proteins are not always constitutively expressed, and instead, expression is governed by numerous regulatory mechanisms described in the following section.
Environmental Stimuli Influence Factor H Binding Protein Expression
Pathogens are exposed to a wide variety of conditions within the host including transitions between hosts, nutrient supplies, and temperature, all of which have been shown to influence expression of FH binding protein.
FH binding protein expression is associated with nutrient availability within the pathogen environment. For example, expression of the FH binding protein, high-affinity glucose transporter 1 (Hgt1p) in C. albicans is highest at low, physiological, glucose concentrations compared to high glucose concentrations (168), which is likely due to the canonical role of Hgt1p in glucose metabolism (208). In Streptococcus agalactiae, zinc availability influences expression of FH binding proteins, Sht and ShtII (141). Purified Sht and ShtII proteins bind FH, suggesting a potential role in complement evasion. However, when bacteria survival was assessed in whole blood assays, no protective effect from Sht family proteins was observed. Though Sht and ShtII can bind FH, the primary role of these proteins is bacterial zinc acquisition. Thus, under conditions of low bioavailable zinc, Sht and ShtII expression is activated. In serum, adequate zinc levels repress Sht family gene expression, thus FH binding proteins may only participate in complement evasion under conditions of low zinc.
In some instances, FH binding protein expression is temperature dependent. In N. meningitidis, FHbp functions as a thermosensor when its expression increases with temperature (209, 210). N. meningitidis resides as harmless flora in the upper airway and enters the bloodstream during invasive disease where it encounters the complement system. Bloodstream temperature is warmer than that of the upper airway which, as suggested by Loh et al., may explain the upregulation of FHbp expression under conditions of increased temperature (210). Likewise, according to Kraiczy et al., temperature dependent upregulation of CRASPs occurs in cultured Borrelia (77), which may reflect differential regulation of CRASPs between mammals (higher temperatures) and ticks (lower temperatures) (77).
Similarly, binding outcomes of FH differ between N. meningitidis and N. gonorrhoeae to accommodate their respective environmental niches [reviewed in (211)]. N. meningitidis is an encapsulated organism, which enables high-level complement evasion (212). In contrast, N. gonorrhoeae, residing in the genitourinary tract, is not capsulated, and thus relies on host complement regulators for serum resistance. N. gonorrhoeae limits complement activity on its surface more effectively via FH binding through PorB.1B compared to FHbp of N. meningitidis (123). As noted by Shaughnessy et al., the functional differences in binding FH may, in part, be explained by the distinct niches in which these organisms inhabit. FH is very abundant in blood and therefore, likely easily recruited by N. meningitidis. In comparison, FH is present in low levels in the genitourinary tract, demanding efficient recruitment by N. gonorrhoeae (123).
Pathogen Life Cycle Affects Factor H Binding Protein Expression
Some pathogens reside in numerous hosts as part of a life cycle and must adapt accordingly to the changing host environment by modulating gene expression, including regulation of FH binding proteins. For example, Borrelia participate in an enzootic life cycle between Ixodes tick vectors, and mammal, bird, and reptile hosts [reviewed in (213)]. Interestingly, expression of CRASPs reflects the needs of the spirochete in response to its changing environment. CspA (CRASP-1) is produced by Borrelia during tick-to-mammal and mammal-to-tick transmission, but is not expressed during established infection (214) or when Borrelia is cultured in environments mimicking host conditions (215). In contrast, CspZ (CRASP-2) expression increases in Borrelia at the tick bite site in the mammal and is maintained throughout infection, but is undetectable in the tick (214). CspZ gene and protein expression is also induced when Borrelia are treated with human blood and its expression is required for Borrelia bacteremia and tissue colonization in mice (216). In this particular study, even though mice were infected using spirochetes previously subjected to human blood, which does not reflect the natural life cycle of Borrelia, it nevertheless exemplifies the influence of human blood on Borrelia gene expression (217).
In another example, Borrelia turicatae mRNA expression of the gene for the FH binding protein, FhbA, was lower in Borrelia from fed ticks compared to in vitro culture; however the functional role of FhbA in infection remains to be determined (218). Finally, OspE paralogs (CRASPs 3–5) are expressed during all stages of mammalian infection [reviewed in (83, 219)]. Concurrent expression of CRASPs during the spirochete infection cycle confounds interpretation of the role each protein plays in complement evasion [reviewed in (83)]. Instead of contributing separately to complement evasion, CRASPs may work synergistically to carry out their functions as suggested by Bykowski et al. (214).
Some pathogens including protozoans differentiate into unique morphological life stages and FH binding capabilities vary concordantly. P. falciparum, a protozoan parasite, recruits FH using unique FH binding proteins according to the parasite life stage (172, 173). The invasive blood-stage merozoite found in humans uses Pf92 to bind FH (173), while extracellular gametes residing in the mosquito midgut, recruit human FH from a blood meal using a different FH binding protein, PfGAP50 (172). In Trypanosoma brucei, the proliferating slender form expresses the lowest levels of FH binding protein, FH receptor (FHR), followed by the quiescent stumpy form, and finally, the procyclic form expresses the highest levels of FHR (169). Both the slender and stumpy forms are present in mammalian blood, whereas the procyclic form is present in the midgut of the tsetse fly vector [reviewed in (220)]. In a related example, the FH binding proteins of C. albicans, Pra1, is upregulated upon switching from yeast to hyphal growth, which may explain, in part, why the hyphal form is more invasive than the yeast (164).
Pathogen Strain and Passage Number Affects Factor H Binding Protein Expression
Expression of FH binding proteins is subject to variability amongst pathogen strains. For example, PspC expressed by S. pneumoniae undergoes sequence variation leading to changes in FH binding capacity (221). Antibodies raised against a particular PspC variant successfully prevented FH binding and in vivo opsonophagocytic bacterial killing, but did not cross-react with S. pneumoniae that contained different sequence variations of PspC (221). Similarly, in S. pneumoniae, serotype invasiveness correlates with FH binding (222). Additionally, S. pyogenes bind FH through expression of M proteins. M proteins exhibit sequence variation between strains, and hence, vary in the ability to bind FH (155). Furthermore, analysis of FHbp expression across a panel of serogroup B meningococcal strains revealed the level of FHbp expression varies by at least 15-fold, and that variant 1, which is a component of meningococcus vaccines (discussed later), expresses significantly more protein than variant 2 or 3 strains (223). Expression of FH binding proteins Pra1 and Gpm1 is conserved across clinical isolates of C. albicans; however, expression levels vary, and higher expression levels correlate with FH binding (224). Moreover, FH binding varies from 5-42% across patient strains of Fusobacterium necrophorum, and strains which bound FH strongly result in more severe infections (101).
Culture passage number also influences FH binding expression. Leptospira strains with few passages display higher FH binding expression compared to culture attenuated strains, suggesting pathogens lose the ability to bind FH in culture (225). Similarly, passage number also decreases expression of CRASP-1, -2, and -5 in Borrelia afzelii (77). Passage number is postulated to suppress CRASPs expression in B. garinii because isolates from patients with neuroborreliosis bind FH, whereas strains with prolonged growth in vitro, do not (226).
Localization of Factor H Binding Proteins
FH binding proteins assume strategic spatial positioning to promote efficient evasion of the AP. This is evidenced by PspC of S. pneumoniae. Pneumococci divisional septa are insufficiently protected by the bacterial capsule, especially at the site where cell separation is initiated, permitting entry of serum factors and allowing complement activity (227). PspC compensates for the breach of protection by localizing to the cell septa to control lateral complement amplification (227).
While most FH binding proteins are surface bound, some are secreted. For example, Pra1 of C. albicans is secreted and regulates FH in the fluid phase (164). Pra1 is also surface expressed and localizes primarily at the tip of cells, which suggests an important role of Pra1 upon contact with host tissues and surfaces during infection (164). Likewise, Hgt1p of C. albicans is present on the cell wall, cell membrane, and intracellular and extracellular vesicles (168).
Therapeutic and Preventive Strategies Targeting the Pathogen-Factor H Interface
Acquiring host complement negative regulators, such as FH, allow pathogens to bypass complement attack and persist in the host. Hence, strategies directed against the FH-pathogen interface hold therapeutic value. The following section describes existing and potential approaches to target pathogen binding of FH (Figure 4).
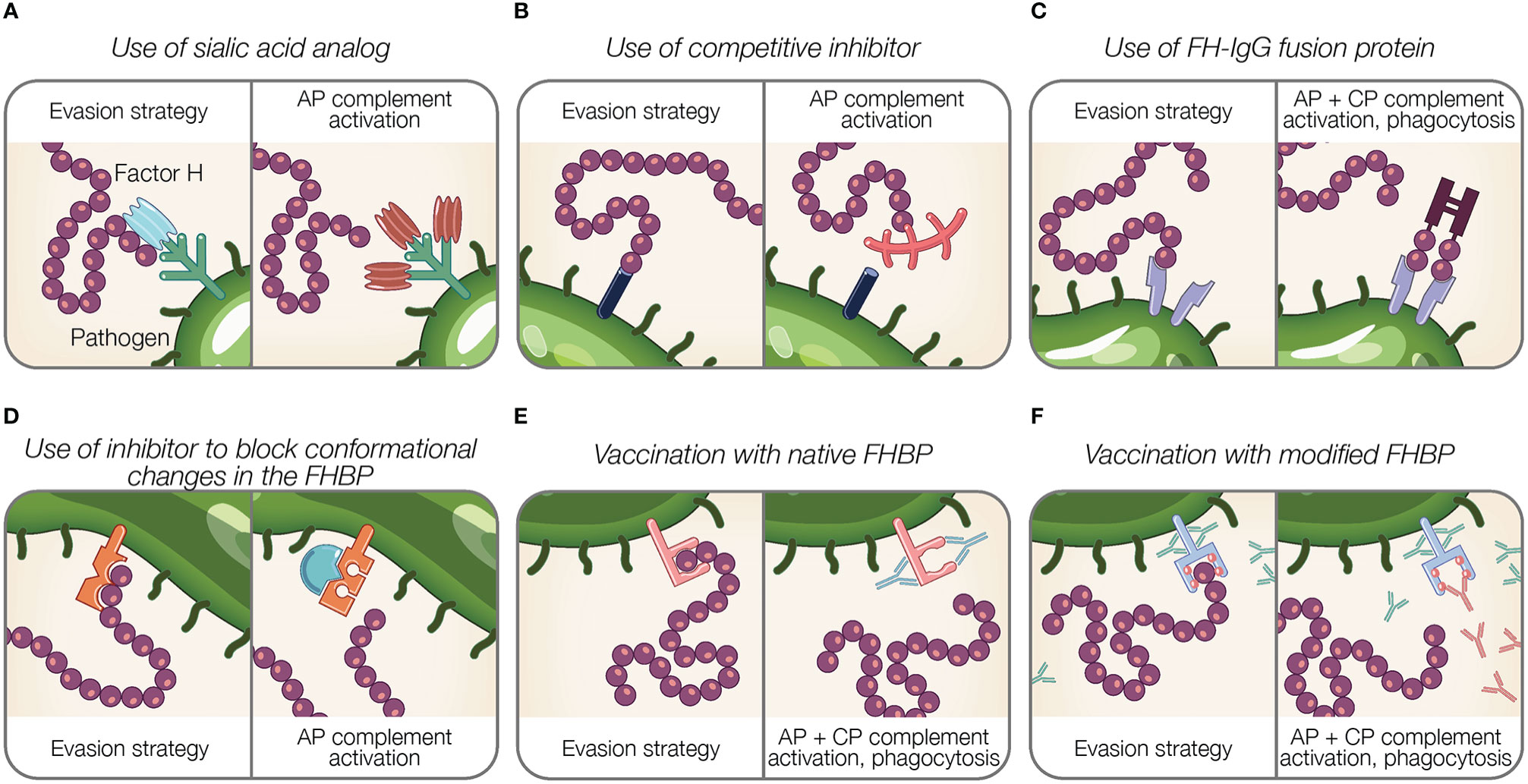
Figure 4 Potential therapeutic and preventive strategies targeting the interaction between Factor H and pathogens. (A–F) Theoretical representations of pathogen evasion strategies targeted for therapy and prevention are shown. For each box, the left panel illustrates a pathogen evasion strategy and the right panel represents the intended result of therapeutic intervention, which includes AP and CP activation, and phagocytosis. (A) Sialic acid species (shown in light blue) are replaced by analogs unable to recognize FH (shown in brown). (B) Competitive inhibitors for FH binding proteins (FHBP) prevent FH interaction with the pathogen surface. (C) FH fusion proteins containing the Fc portion of IgG fused to FH binding fragments (CCPs 6–7, 19–20) bind FHBP to activate the CP via antibody recognition, facilitate Fcγ receptor-mediated phagocytosis, and prevent FH binding to the pathogen surface. (D) Inhibitors prevent conformational change of FHBP required to enhance their binding to FH. (E) Vaccination with native FHBP. (F) Vaccination with modified FHBP unable to bind FH, reveals FHBP epitopes (red balls) to improve immunogenicity.
Interference of Pathogen Sialic Acid Expression
In addition to the role of sialylation in pathogen FH acquisition, it also modulates other pathways of complement, as well as phagocytosis and epithelial invasion [reviewed in (189)]. The importance of sialylation in pathogenesis renders it an enticing therapeutic target (Figure 4A). In the case of gonococci that are becoming increasingly antibiotic resistant, this approach is timely and reviewed elsewhere (188, 189, 228).
Gonococci incorporate sialic acid in surface glycans (LOS) resulting in FH binding [reviewed in (189)]. A strategy that manipulates pathogen sialylation to disable complement evasion, involves supplying gonococci with sialic acid analogs that do not bind FH. When such analogs are incorporated into pathogen glycans, it renders bacteria susceptible to complement and substantially reduces bacterial survival in normal human serum (196, 229). Intravaginal administration of a sialic acid analog (CMP-Leg5,7Ac2) to transgenic mice capable of expressing sialic acid compounds found in humans protects against multidrug-resistant gonococci (229). Moreover, this sialic acid analog was stable at vaginal pH and temperature and did not incorporate into glycans on host surfaces, which is noteworthy because incorporation into host glycans may cause an immunogenic response against host tissue (229). The stability and limited side effects of sialic acid analog indicate an effective treatment option (229).
Inhibitors to Functionally Disable Factor H Binding Proteins or Its Interaction With Factor H
Inhibitors that block the interaction between FH and FH binding proteins can be considered as potential therapeutics (Figures 4B, D). As described previously, FHbp of N. meningitidis binds FH using a site shared by SOS, a highly sulphated analog of GAGs (122). The addition of SOS inhibits FH interaction with FHbp in vitro, thus as suggested by Schneider et al., provides support for small molecule competitive inhibitors in disabling pathogen evasion (122). However, competitive inhibitors binding FH may also interfere with host regulation of complement activity.
The binding of FH to SdrE of S. aureus is enhanced after a conformational change in the binding protein from a resting, locked state, to an open state (136). Small molecules inhibitors may have a potential in preventing conformational changes of FH binding proteins required to bind FH, similar to what occurs for SdrE [reviewed in (199)].
Factor H-Fc Fusion Proteins
Targeting the pathogen-FH interface is accomplished using fusion proteins in which FH domains essential for surface recognition (CCPs 18–20 or 6–7) are fused to the Fc region of IgG. Fusion proteins are designated FHX-X/Fc, where X-X refers to the FH domains used in the fusion protein. The proposed mechanisms of fusion proteins are threefold: (a) prevent FH binding to the microbial surface; (b) activate the CP via antibody recognition; and (c) facilitate Fcγ receptor-mediated phagocytosis [reviewed in (189)] (Figure 4C). Fusion proteins are advantageous because identifying pathogen FH binding proteins or ligands is not required. However, determining FH domains involved in pathogen binding is necessary to design applicable fusion proteins.
Animal model studies demonstrate the efficacy of fusion proteins using FH CCP 6–7. In infant rats, administration of FH6-7/Fc prior to intraperitoneal inoculation of a serogroup C strain of N. meningitidis dose-dependently reduced bacterial blood burden (230). Similarly, concurrent intranasal administration of FH6-7/Fc with inoculation of non-typeable H. influenzae reduced mouse lung bacterial burden (129). In a model of S. pyogenes sepsis, intranasal FH6-7/Fc treatment of human FH transgenic mice resulted in decreased animal mortality (231). Additionally, fusion proteins with CCPs 18–20 bound to gonococci and resulted in complement-dependent bactericidal activity (119).
Given the importance of CCP 6–8 (232–236) and CCP 19–20 (237, 238) in discriminating self from non-self surfaces, fusion proteins containing these domains may outcompete full-length FH for binding host cells, resulting in unwanted complement-mediated attack. In order for fusion proteins utilizing CCP 19–20 to be effective therapeutic solutions, these domains must be modified to prevent host cell interaction. This was accomplished by the generation of a CCP 19–20 fusion protein containing a point mutation in CCP 19 (D1119G). This mutation results in lower affinity for C3b binding compared to wild type CCP 19–20 restricting its ability to bind human cells, even while maintaining normal binding affinity for polyanions (238). D1119G bound to highly sialylated gonococci in the presence of serum, resulting in a robust in vitro complement response (239). Moreover, therapeutic administration of D1119G reduced infection duration and burden in a mouse vaginal colonization model of N. gonorrhoeae without affecting lysis of human erythrocytes (239). Similarly, FH CCP 19–20 fragments containing the D1119G mutation also bound to whole P. aeruginosa, H. influenzae, Bordetella pertussis, S. pneumoniae, and C. albicans organisms (74), suggesting the therapeutic relevance of mutant FH fusion proteins against numerous pathogens.
Factor H Binding Proteins as Vaccines
A suitable vaccine candidate is (a) immunogenic; (b) conserved across different strains and genospecies of the respective pathogen; (c) expressed during human infection; (d) necessary for development of a clinical infection; (e) surface exposed; and (f) raises an immune response that neutralizes an important virulence determinant as proposed by Bhattacharjee et al. and Meri et al. (85, 240). FH binding proteins meet many of these requirements, and thus are viable targets for vaccine development. This section describes clinically approved (Figure 4E) and emerging vaccines (Figure 4F) utilizing FH binding proteins.
Clinically Approved Vaccines Utilizing Factor H Binding Proteins
FHbp is a successful vaccine target against N. meningitidis, the causative agent of invasive meningococcal disease. Five of the six serogroups of N. meningitidis express capsular polysaccharides that are effective vaccine targets. However, because the capsule of serogroup B is poorly immunogenic, alternative vaccine targets are necessary to protect against the high mortality and morbidity associated with meningococcus disease [reviewed in (241)]. FHbp is a viable therapeutic target because it is widely expressed across numerous meningococcus B isolates [reviewed in (241)]. Two licensed vaccines, 4CMenB and MenB-FHbp, both containing FHbp, are approved to protect 10–25 year old individuals against serogroup B meningococcus [reviewed in (241, 242)].
FHbp displays sequence variability with 3 main variants belonging to subfamilies A (variant 2 and variant 3) and B (variant 1) (243, 244). Consequentially, immunization with vaccines representing single variants do not offer protection against strains containing other variants (243–245). MenB-FHbp is a bivalent vaccine containing recombinant lipidated FHbp from subfamily A variant A05 and subfamily B variant B01 [reviewed in (242)]. Because FHbp from two variants are represented in this vaccine, MenB-FHbp offers broad coverage against multiple N. meningitidis FHbp variants (246). In contrast, 4CMenB is a multi-component vaccine where one of the components is a FHbp that represents only one variant (subfamily B) [reviewed in (247)].
Sequence variability limits the degree of protection against FHbp variants. For example, post immunization sera from mice injected with a single variant did not offer in vitro cross protection against other variants (245). Thus, sequence variability of FHbp poses difficulties in vaccine development. However, recent studies have introduced alternative vaccine candidates that elicit antibody responses against FHbp variants 1, 2, and 3 to offer broad protection against meningococcal disease. One such promising candidate is the Gonococcal homologue of meningococcal FHbp (Ghfp) that protects against N. meningitidis expressing any of the 3 FHbp variants (248). Vaccination with chimeric FHbp antigens is an additional approach for offering broad protection. Scarselli et al. developed a chimeric FHbp antigen in which the surface is modified to confer specificity against all three variants, resulting in cross protection in mice (249). Another example includes chimeric vaccines developed by fusing domains conserved across all FHbp variants and selective portions of domains from variant 1 FHbp and domains of variant 2 FHbp (250). These molecules were found to elicit bactericidal activity against strains expressing the different variants (250). Chimeric antigens can also be generated by fusing FHbp with other immunogenic antigens such as the VR2 epitope from the integral membrane protein PorA that induces an immunogenic response against N. meningitidis in mice (251).
Though approved for clinical use, understanding the scope of FHbp-based antibodies remains an area of active research. One such area involves long-term risks of vaccination, such as the development of anti-FH autoantibodies. Interaction of FHbp (delivered as a vaccine) with FH may result in the development of autoantibodies against FH. A few studies have investigated this potential outcome. Sharkey and colleagues noted that 2.5% of individuals vaccinated with 4CMenB show an increase in anti-FH autoantibodies (a low level of anti-FH autoantibodies are present in some individuals (252)). However, this increase was transient, and no adverse effects were reported in individuals with higher anti-FH autoantibodies (253). In another study, 4CMenB immunization resulted in the development of anti-FH autoantibodies in Rhesus macaques (254). Since the long-term effects of FH autoantibodies are still not clear, further investigation is required to assess the risk of autoimmune disease in response to meningococcal vaccines, as suggested by Sharkey et al. (253).
Other avenues of research into FHbp vaccines include expanding applications and improving efficacy. Recently, antibodies raised in response to MenB-FHbp were shown to be effective against non-serogroup B meningococci in serum bactericidal assays (255), suggesting this vaccine may provide broad protection against meningococcal disease. Additionally, vaccine delivery with polyhydroxybutyrate beads engineered to display FHbp antigens, show promise in preclinical studies as means to overcome limitations of recombinant protein vaccines such as poor immunogenicity and adjuvant requirements (256). Finally, recent studies using vaccines containing mutant FHbp with less ability to bind FH combined with native outer membrane vesicles showed higher serum bactericidal activity than 4CMenB vaccination (254). This vaccination approach also generated less FH autoantibodies than 4CMenB (254).
Emerging Vaccines Utilizing Factor H Binding Proteins
Vaccination With Native Factor H Binding Proteins
While vaccines against N. meningitidis are the only clinically approved therapies targeting the pathogen FH interface, other examples are emerging. The FH binding protein, PspC of S. pneumoniae, is a promising vaccine target. Mice immunized with a PspC fragment containing the FH binding domain conferred protection when challenged with the same strain used for immunization (257). Antibodies from immunized animals enhanced CP activity, but also competed for human FH binding, suggesting interference with the AP (257). However, antibodies generated against PspC from immunized human sera do not recognize the FH binding site, suggesting FH masks epitope recognition (258).
Vaccination with FH binding proteins is also effective against Leptospira interrogans. Immunization with a multi-subunit, adjuvant vaccine comprising multiple FH binding proteins from L. interrogans had similar protective efficacy and survival rate in hamsters challenged with L. interrogans compared to monovalent vaccine administration containing only LigAc (259). Though results indicate LigAc is likely a superior vaccine antigen in the multi-subunit vaccine, only the multi-subunit vaccine reduced leptospiral renal colonization in surviving animals (259). Further investigation is needed to identify the FH binding proteins in this observed effect from multi-subunit vaccination (259).
Vaccination With Modified Factor H Binding Proteins
An innovative approach towards applying FH binding proteins for vaccination involves manipulating FH binding proteins. Modification of FH binding proteins from several organisms has been shown to disable FH binding (Figure 4F).
Murine immunization with a non-binding FH mutant of N. meningitidis FHbp resulted in antibodies with a higher bactericidal activity than native FHbp vaccination (260). This finding suggests binding of FH to the native FHbp vaccine can decrease a protective antibody response and that mutant FH binding proteins may serve as a superior vaccine (260). Importantly, these studies utilized transgenic mice expressing human FH because FHbp only binds human FH (261).
As discussed above, PspC vaccination fails to generate antibodies against the FH binding site (258). Thus, as suggested by Glennie et al., generation of a modified PspC unable to bind FH presents a viable vaccine opportunity given the role of this protein in complement evasion and host cell invasion (258).
A recent study in Borrelia eloquently demonstrates the ability of mutated FH binding proteins to confer protective immunization. CspZ (CRASP-2) belongs to a family of lipoproteins utilized by B. burgdorferi, a causative agent of Lyme disease, to evade complement. CspZ binds FH (77) though the degree of binding varies across an extensive panel of human Lyme disease isolates (262). CspZ is an enticing therapeutic target because it is immunogenic in humans (though titers vary between individuals (262)) and its expression is highly conserved among species associated with Lyme disease [reviewed in (219)]. However, CspZ on its own is not protective against B. burgdorferi infection. Immunization with recombinant CspZ did not impact B. burgdorferi infection (262, 263) or prevent Lyme disease pathology in a mouse model (263).
Nonetheless, studies by Marcinkiewicz and colleagues revealed the potential of CspZ as a therapeutic target by using a nonbinding mutant of CspZ (CspZ-YA). Because CspZ-YA cannot bind FH, new epitopes previously cloaked by FH binding are revealed (216). As a result, the immunogenicity of CspZ increases and prevents Borrelia colonization (216). CspZ-YA conjugated to virus-like particles (VLP-CspZ-YA) protects passively immunized mice from Lyme infection (216). Additional work in mice infected by Ixodes scapularis nymphal ticks carrying Borrelia demonstrated VLP-CspZ-YA protects against spirochete tissue colonization and arthritis development (264). These authors suggest anti-CspZ-YA antibodies bind CspZ at epitopes within the FH binding site, thus competing with FH binding (264). Furthermore, CspZ-YA vaccination protected mice against a strain of B. burgdorferi that does not bind FH and immunized sera eliminated this strain in vitro, suggesting resulting antibodies may target CspZ through the CP (264).
Potential Targeting of Factor H in Other Infectious Diseases
While FH interactions among bacteria are well-described and applied in clinical therapy, further research in this field is warranted for other classes of pathogens including protozoa, helminths, viruses, and fungi. Closing this gap is relevant for developing treatments for diseases caused by pathogens known to evade complement. The following sections provides preliminary support for pathogens that interact with FH, but require further work to harness these findings for therapy.
Factor H Interactions With Protozoa
The protozoan, Trypanosoma cruzi is the causative agent of Chagas disease, which progresses from an acute, often undiagnosed, asymptomatic stage, to a deadly chronic stage of which 30–40% of chronically infected individuals develop cardiomyopathy, mega-syndromes, etc. [reviewed in (265)]. Currently, there are no vaccines for T. cruzi nor treatments available for the chronic, fatal stage of the disease. T. cruzi adopts various strategies to evade complement, including the AP [reviewed in (266)]. When the infective form of T. cruzi (trypomastigotes), which is normally resistant to complement-mediated killing, is treated with enzymes including sialidase and trypsin, the parasites become susceptible to complement-mediated lysis in human serum (267), suggesting parasites may be protected from the AP by FH that has been hijacked by surface-bound sialic acid. This notion is also supported by data showing trypomastigotes that are pre-opsonized with C3b bind FH (170). However, the molecular mechanisms involved in binding remain unknown. Likewise, as suggested by Sikorski et al., the mechanism by which Toxoplasma gondii captures FH may be through expression of sialic acid or heparan sulfated proteoglycans (174). In addition, P. falciparum, the causative agent of malaria, has developed resistance to all antimalarial drugs used against it [reviewed in (268)]. FH has been shown to bind P. falciparum, though not all receptors have been identified (171) and doing so holds promise for therapeutic avenues against drug resistance. Overall, interfering with the ability of infectious protozoa to bind FH represents a possible treatment strategy against diseases caused by these pathogens; however, more work is required to understand the molecular mechanisms involved in these interactions with FH.
Factor H Interactions With Helminths
Echinococcus granulosus, a zoonotic cestode, causes cystic echinococcosis in humans when larvae develop cysts in organs including liver, lungs, brain, and bones [reviewed in (269)]. FH binds E. granulosus through interactions involving the neutral, sugar-rich, laminated layer of the cyst wall (176). Myoinositol hexakisphosphate (InsP6) is a protein abundantly expressed in the laminated layer and is shown to bind FH through in vitro studies with purified protein (270). However, studies representing physiological conditions suggest InsP6 does not protect against AP-mediated attack and may instead, activate the CP through C1q recognition (270). Thus, the mechanism for FH interaction with E. granulosus remains largely unresolved and likely involves other components on the laminated layer other than InsP6 [reviewed in (271)].
Onchocerca volvulus is a parasitic nematode causing onchocerciasis, or river blindness. Novel treatment options are necessary for this disease given the threat of drug resistance to the standard therapy, ivermectin [reviewed in (272)]. The pathogenic stage of O. volvulus (microfilariae) bind FH using CCPs 8–20 with preservation of cofactor activity, but the binding ligand remains unknown (175). Loa Loa is another parasitic nematode and the causative agent of loiasis, characterized by subconjunctival eye passage of the adult worm, angioedema, and pathology associated with the lungs, brain, heart, and kidneys [reviewed in (273)]. Patients with a high infectious load are at risk for severe complications from ivermectin treatment (274), thus alternative treatment options are important. Larval L. Loa acquires FH on the outermost sheath layer and retains its cofactor activity (177). However, FH CCPs involved in this reaction are not yet described, but likely does not involve heparin binding domains (177).
Factor H Interactions With Viruses
There are many examples of viruses evading complement [reviewed in (62)], but few associate complement evasion to pathogen interactions with FH. West Nile virus binds FH using non-structural protein (NS1); however, the mechanisms of this interaction is unknown (178). Interestingly, serum levels of NS1 correlate with disease severity and viremia [reviewed in (275)], which may be due in part, to the ability of NS1 to capture FH.
Human immunodeficiency virus-1 (HIV-1) is resistant to complement lysis in circulation even though HIV-1 and anti-HIV-1 antibodies activate complement [reviewed in (276)]. FH binds the HIV-1 envelope proteins gp41 and gp120 (179–182). More robust studies are required to understand the role of FH binding in HIV-1 complement evasion.
Conclusion
Numerous pathogens exploit host FH, the primary negative regulator of the AP of complement, as part of a robust complement evasion strategy. Characterizing interactions between pathogens and FH provides insight into the failure of FH to discriminate self from non-self, and/or the impressive resourcefulness of pathogens to outsmart complement regulation. The biological relevance of binding FH is demonstrated by established and emerging vaccines targeting the pathogen-FH interface and evidenced by the number of pathogens utilizing FH and expressing multiple FH binding proteins. Additionally, studying pathogen interactions with FH may reveal more information about how FH operates in the host. The selective pressure of pathogens to maintain FH binding capabilities suggest undiscovered details about the role of complement in immune defense against pathogens.
Author Contributions
SRM, SSM, and VF conceived and wrote the paper. CC critically reviewed and edited the paper. SRM, SSM, and CC designed the figures. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by National Institute of Health R01HL112937 (VF), The University of Toledo Biomedical Research Innovation Program (VF), and The American Association of Immunologists Careers in Immunology Fellowship Program (VF and SSM).
Conflict of Interest
VF serves as a consultant for, and receives grant funding from, Apellis Pharmaceuticals.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Audrey Bell for figure illustrations.
References
1. Ricklin D, Reis ES, Mastellos DC, Gros P, Lambris JD. Complement component C3 - The “Swiss Army Knife” of innate immunity and host defense. Immunol Rev (2016) 274:33–58. doi: 10.1111/imr.12500
2. Gaboriaud C, Ling WL, Thielens NM, Bally I, Rossi V. Deciphering the fine details of C1 assembly and activation mechanisms: “mission impossible”? Front Immunol (2014) 5:565. doi: 10.3389/fimmu.2014.00565
3. Garred P, Genster N, Pilely K, Bayarri-Olmos R, Rosbjerg A, Ma YJ, et al. A journey through the lectin pathway of complement—MBL and beyond. Immunol Rev (2016) 274:74–97. doi: 10.1111/imr.12468
4. Harboe M, Ulvund G, Vien L, Fung M, Mollnes TE. The quantitative role of alternative pathway amplification in classical pathway induced terminal complement activation. Clin Exp Immunol (2004) 138:439–46. doi: 10.1111/j.1365-2249.2004.02627.x
5. Harboe M, Garred P, Karlstrøm E, Lindstad JK, Stahl GL, Mollnes TE. The down-stream effects of mannan-induced lectin complement pathway activation depend quantitatively on alternative pathway amplification. Mol Immunol (2009) 47:373–80. doi: 10.1016/j.molimm.2009.09.005
6. Ekdahl KN, Mohlin C, Adler A, Åman A, Manivel VA, Sandholm K, et al. Is generation of C3(H2O) necessary for activation of the alternative pathway in real life? Mol Immunol (2019) 114:353–61. doi: 10.1016/j.molimm.2019.07.032
7. Merle NS, Church SE, Fremeaux-Bacchi V, Roumenina LT. Complement system part I - molecular mechanisms of activation and regulation. Front Immunol (2015) 6:262. doi: 10.3389/fimmu.2015.00262
8. Merle NS, Noe R, Halbwachs-Mecarelli L, Fremeaux-Bacchi V, Roumenina LT. Complement system part II: role in immunity. Front Immunol (2015) 6:257. doi: 10.3389/fimmu.2015.00257
9. Schmidt CQ, Lambris JD, Ricklin D. Protection of host cells by complement regulators. Immunol Rev (2016) 274:152–71. doi: 10.1111/imr.12475
10. Whaley K, Ruddy S. Modulation of C3b hemolytic activity by a plasma protein distinct from C3b inactivator. Science (1976) 193:1011–3. doi: 10.1126/science.948757
11. Nilsson UR, Mueller-Eberhard HJ. Isolation of beta 1f-globulin from human serum and its characterization as the fifth component of complement. J Exp Med (1965) 122:277–98. doi: 10.1084/jem.122.2.277
12. de Cordoba SR, de Jorge EG. Translational mini-review series on complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol (2008) 151:1–13. doi: 10.1111/j.1365-2249.2007.03552.x
13. Perkins SJ, Nan R, Li K, Khan S, Miller A. Complement factor H-ligand interactions: self-association, multivalency and dissociation constants. Immunobiology (2012) 217:281–97. doi: 10.1016/j.imbio.2011.10.003
14. Cserhalmi M, Papp A, Brandus B, Uzonyi B, Józsi M. Regulation of regulators: role of the complement factor H-related proteins. Semin Immunol (2019) 45:101341. doi: 10.1016/j.smim.2019.101341
15. Adinolfi M, Dobson NC, Bradwell AR. Synthesis of two components of human complement, beta 1H and C3bINA, during fetal life. Acta Paediatr Scand (1981) 70:705–10. doi: 10.1111/j.1651-2227.1981.tb05772.x
16. Schwaeble W, Zwirner J, Schulz TF, Linke RP, Dierich MP, Weiss EH. Human complement factor H: expression of an additional truncated gene product of 43 kDa in human liver. Eur J Immunol (1987) 17:1485–9. doi: 10.1002/eji.1830171015
17. Parente R, Clark SJ, Inforzato A, Day AJ. Complement factor H in host defense and immune evasion. Cell Mol Life Sci (2017) 74:1605–24. doi: 10.1007/s00018-016-2418-4
18. Sim RB, DiScipio RG. Purification and structural studies on the complement-system control protein beta 1H (Factor H). Biochem J (1982) 205:285–93. doi: 10.1042/bj2050285
19. Józsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol (2008) 29:380–7. doi: 10.1016/j.it.2008.04.008
20. Rodriguez de Cordoba S, Diaz-Guillen MA, Heine-Suner D. An integrated map of the human regulator of complement activation (RCA) gene cluster on 1q32. Mol Immunol (1999) 36:803–8. doi: 10.1016/s0161-5890(99)00100-5
21. Kristensen T, Tack BF. Murine protein H is comprised of 20 repeating units, 61 amino acids in length. Proc Natl Acad Sci USA (1986) 83:3963–7. doi: 10.1073/pnas.83.11.3963
22. Ripoche J, Day AJ, Harris TJ, Sim RB. The complete amino acid sequence of human complement factor H. Biochem J (1988) 249:593–602. doi: 10.1042/bj2490593
23. Aslam M, Perkins SJ. Folded-back solution structure of monomeric factor H of human complement by synchrotron X-ray and neutron scattering, analytical ultracentrifugation and constrained molecular modelling. J Mol Biol (2001) 309:1117–38. doi: 10.1006/jmbi.2001.4720
24. Osborne AJ, Nan R, Miller A, Bhatt JS, Gor J, Perkins SJ. Two distinct conformations of factor H regulate discrete complement-binding functions in the fluid phase and at cell surfaces. J Biol Chem (2018) 293:17166–87. doi: 10.1074/jbc.RA118.004767
25. Schmidt CQ, Herbert AP, Mertens HDT, Guariento M, Soares DC, Uhrin D, et al. The central portion of factor H (modules 10-15) is compact and contains a structurally deviant CCP module. J Mol Biol (2010) 395:105–22. doi: 10.1016/j.jmb.2009.10.010
26. Kazatchkine MD, Fearon DT, Austen KF. Human alternative complement pathway: membrane-associated sialic acid regulates the competition between B and beta1 H for cell-bound C3b. J Immunol (1979) 122:75–81.
27. Weiler JM, Daha MR, Austen KF, Fearon DT. Control of the amplification convertase of complement by the plasma protein beta1H. Proc Natl Acad Sci USA (1976) 73:3268–72. doi: 10.1073/pnas.73.9.3268
28. Whaley K, Ruddy S. Modulation of the alternative complement pathways by beta 1 H globulin. J Exp Med (1976) 144:1147–63. doi: 10.1084/jem.144.5.1147
29. Bexborn F, Andersson PO, Chen H, Nilsson B, Ekdahl KN. The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3(H2O)Bb). Mol Immunol (2008) 45:2370–9. doi: 10.1016/j.molimm.2007.11.003
30. Pangburn MK, Schreiber RD, Muller-Eberhard HJ. Human complement C3b inactivator: isolation, characterization, and demonstration of an absolute requirement for the serum protein beta1H for cleavage of C3b and C4b in solution. J Exp Med (1977) 146:257–70. doi: 10.1084/jem.146.1.257
31. Harrison RA, Lachmann PJ. The physiological breakdown of the third component of human complement. Mol Immunol (1980) 17:9–20. doi: 10.1016/0161-5890(80)90119-4
32. Alsenz J, Lambris JD, Schulz TF, Dierich MP. Localization of the complement-component-C3b-binding site and the cofactor activity for factor I in the 38kDa tryptic fragment of factor H. Biochem J (1984) 224:389–98. doi: 10.1042/bj2240389
33. Gordon DL, Kaufman RM, Blackmore TK, Kwong J, Lublin DM. Identification of complement regulatory domains in human factor H. J Immunol (1995) 155:348–56.
34. Kuhn S, Skerka C, Zipfel PF. Mapping of the complement regulatory domains in the human factor H-like protein 1 and in factor H1. J Immunol (1995) 155:5663–70.
35. Kuhn S, Zipfel PF. Mapping of the domains required for decay acceleration activity of the human factor H-like protein 1 and factor H. Eur J Immunol (1996) 26:2383–7. doi: 10.1002/eji.1830261017
36. Haque A, Cortes C, Alam MN, Sreedhar M, Ferreira VP, Pangburn MK. Characterization of binding properties of individual functional sites of human complement factor H. Front Immunol (2020) 11:1728. doi: 10.3389/fimmu.2020.01728
37. Jokiranta TS, Hellwage J, Koistinen V, Zipfel PF, Meri S. Each of the three binding sites on complement factor H interacts with a distinct site on C3b. J Biol Chem (2000) 275:27657–62. doi: 10.1074/jbc.M002903200
38. Fearon DT. Regulation by membrane sialic acid of beta1H-dependent decay-dissociation of amplification C3 convertase of the alternative complement pathway. Proc Natl Acad Sci USA (1978) 75:1971–5. doi: 10.1073/pnas.75.4.1971
39. Pangburn MK, Muller-Eberhard HJ. Complement C3 convertase: cell surface restriction of beta1H control and generation of restriction on neuraminidase-treated cells. Proc Natl Acad Sci USA (1978) 75:2416–20. doi: 10.1073/pnas.75.5.2416
40. Kazatchkine MD, Fearon DT, Silbert JE, Austen KF. Surface-associated heparin inhibits zymosan-induced activation of the human alternative complement pathway by augmenting the regulatory action of the control proteins on particle-bound C3b. J Exp Med (1979) 150:1202–15. doi: 10.1084/jem.150.5.1202
41. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA (1990) 87:3982–6. doi: 10.1073/pnas.87.10.3982
42. Carreno MP, Labarre D, Maillet F, Jozefowicz M, Kazatchkine MD. Regulation of the human alternative complement pathway: formation of a ternary complex between factor H, surface-bound C3b and chemical groups on nonactivating surfaces. Eur J Immunol (1989) 19:2145–50. doi: 10.1002/eji.1830191126
43. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrin D, Stehle T. Structural basis for sialic acid-mediated self-recognition by complement factor H. Nat Chem Biol (2015) 11:77–82. doi: 10.1038/nchembio.1696
44. Ferreira VP, Pangburn MK, Cortes C. Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol (2010) 47:2187–97. doi: 10.1016/j.molimm.2010.05.007
45. Józsi M, Schneider AE, Karpati E, Sandor N. Complement factor H family proteins in their non-canonical role as modulators of cellular functions. Semin Cell Dev Biol (2019) 85:122–31. doi: 10.1016/j.semcdb.2017.12.018
46. Zipfel PF, Skerka C. FHL-1/reconectin: a human complement and immune regulator with cell-adhesive function. Immunol Today (1999) 20:135–40. doi: 10.1016/s0167-5699(98)01432-7
47. DiScipio RG, Daffern PJ, Schraufstatter IU, Sriramarao P. Human polymorphonuclear leukocytes adhere to complement factor H through an interaction that involves alphaMbeta2 (CD11b/CD18). J Immunol (1998) 160:4057–66.
48. Schneider AE, Sándor N, Kárpáti É, Józsi M. Complement factor H modulates the activation of human neutrophil granulocytes and the generation of neutrophil extracellular traps. Mol Immunol (2016) 72:37–48. doi: 10.1016/j.molimm.2016.02.011
49. Losse J, Zipfel PF, Józsi M, Factor H. and factor H-related protein 1 bind to human neutrophils via complement receptor 3, mediate attachment to Candida albicans, and enhance neutrophil antimicrobial activity. J Immunol (2010) 184:912. doi: 10.4049/jimmunol.0901702
50. Pio R, Martinez A, Unsworth EJ, Kowalak JA, Bengoechea JA, Zipfel PF, et al. Complement factor H is a serum-binding protein for adrenomedullin, and the resulting complex modulates the bioactivities of both partners. J Biol Chem (2001) 276:12292–300. doi: 10.1074/jbc.M007822200
51. Geven C, Kox M, Pickkers P. Adrenomedullin and adrenomedullin-targeted therapy as treatment strategies relevant for sepsis. Front Immunol (2018) 9:292. doi: 10.3389/fimmu.2018.00292
52. Sim RB, Ferluga J, Al-Rashidi H, Abbow H, Schwaeble W, Kishore U. Complement factor H in its alternative identity as adrenomedullin-binding protein 1. Mol Immunol (2015) 68:45–8. doi: 10.1016/j.molimm.2015.06.006
53. Hovingh ES, van den Broek B, Jongerius I. Hijacking complement regulatory proteins for bacterial immune evasion. Front Microbiol (2006) 7:2004. doi: 10.3389/fmicb.2016.02004
54. Lambris JD, Ricklin D, Geisbrecht BV. Complement evasion by human pathogens. Nat Rev Microbiol (2008) 6:132–42. doi: 10.1038/nrmicro1824
55. Blom AM, Hallström T, Riesbeck K. Complement evasion strategies of pathogens-acquisition of inhibitors and beyond. Mol Immunol (2009) 46:2808–17. doi: 10.1016/j.molimm.2009.04.025
56. Ermert D, Ram S, Laabei M. The hijackers guide to escaping complement: Lessons learned from pathogens. Mol Immunol (2019) 114:49–61. doi: 10.1016/j.molimm.2019.07.018
57. Zipfel PF, Hallstrom T, Riesbeck K. Human complement control and complement evasion by pathogenic microbes–tipping the balance. Mol Immunol (2013) 56:152–60. doi: 10.1016/j.molimm.2013.05.222
58. Würzner R. Evasion of pathogens by avoiding recognition or eradication by complement, in part via molecular mimicry. Mol Immunol (1999) 36:249–60. doi: 10.1016/s0161-5890(99)00049-8
59. Sharma S, Bhatnagar R, Gaur D. Complement evasion strategies of human pathogenic bacteria. Indian J Microbiol (2020) 60:283–96. doi: 10.1007/s12088-020-00872-9
60. Harpf V, Rambach G, Würzner R, Lass-Flörl C, Speth C. Candida and complement: new aspects in an old battle. Front Immunol (2020) 11:1471. doi: 10.3389/fimmu.2020.01471
61. Shao S, Sun X, Chen Y, Zhan B, Zhu X. Complement evasion: an effective strategy that parasites utilize to survive in the host. Front Microbiol (2019) 10:532. doi: 10.3389/fmicb.2019.00532
62. Agrawal P, Nawadkar R, Ojha H, Kumar J, Sahu A. Complement evasion etrategies of viruses: an overview. Front Microbiol (2017) 8:1117. doi: 10.3389/fmicb.2017.01117
63. Józsi M. Factor H family proteins in complement evasion of microorganisms. Front Immunol (2017) 8:571. doi: 10.3389/fimmu.2017.00571
64. Kishore U, Sim RB. Factor H as a regulator of the classical pathway activation. Immunobiology (2012) 217:162–68. doi: 10.1016/j.imbio.2011.07.024
65. Hammerschmidt S, Agarwal V, Kunert A, Haelbich S, Skerka C, Zipfel PF. The host immune regulator factor H interacts via two contact sites with the PspC protein of Streptococcus pneumoniae and mediates adhesion to host epithelial cells. J Immunol (2007) 178:5848–58. doi: 10.4049/jimmunol.178.9.5848
66. Yu Y, Wang J, Han R, Wang L, Zhang L, Zhang AY, et al. Mycoplasma hyopneumoniae evades complement activation by binding to factor H via elongation factor thermo unstable (EF-Tu). Virulence (2020) 11:1059–74. doi: 10.1080/21505594.2020.1806664
67. Riva R, Korhonen TK, Meri S. The outer membrane protease PgtE of Salmonella enterica interferes with the alternative complement pathway by cleaving factors B and H. Front Microbiol (2015) 6:63. doi: 10.3389/fmicb.2015.00063
68. McDowell JV, Huang B, Fenno JC, Marconi RT. Analysis of a unique interaction between the complement regulatory protein factor H and the periodontal pathogen Treponema denticola. Infect Immun (2009) 77:1417–25. doi: 10.1128/iai.01544-08
69. Miller DP, Oliver LD Jr., Tegels BK, Reed LA, O’Bier NS, Kurniyati K, et al. The Treponema denticola FhbB protein is a dominant early antigen that elicits FhbB variant-specific antibodies that block factor H binding and cleavage by dentilisin. Infect Immun (2016) 84:2051–58. doi: 10.1128/iai.01542-15
70. Miller DP, Bell JK, McDowell JV, Conrad DH, Burgner JW, Héroux A, et al. Structure of factor H-binding protein B (FhbB) of the periopathogen, Treponema denticola: insights into progression of periodontal disease. J Biol Chem (2012) 287:12715–22. doi: 10.1074/jbc.M112.339721
71. Kim SW, Choi CH, Moon DC, Jin JS, Lee JH, Shin JH, et al. Serum resistance of Acinetobacter baumannii through the binding of factor H to outer membrane proteins. FEMS Microbiol Lett (2009) 301:224–31. doi: 10.1111/j.1574-6968.2009.01820.x
72. Wang Y, Jenkins SA, Gu C, Shree A, Martinez-Moczygemba M, Herold J, et al. Bacillus anthracis spore surface protein BclA mediates complement factor H binding to spores and promotes spore persistence. PloS Pathog (2016) 12:e1005678. doi: 10.1371/journal.ppat.1005678
73. Amdahl H, Jarva H, Haanperä M, Mertsola J, He Q, Jokiranta TS, et al. Interactions between Bordetella pertussis and the complement inhibitor factor H. Mol Immunol (2011) 48:697–705. doi: 10.1016/j.molimm.2010.11.015
74. Meri T, Amdahl H, Lehtinen MJ, Hyvarinen S, McDowell JV, Bhattacharjee A, et al. Microbes bind complement inhibitor factor H via a common site. PloS Pathog (2013) 9:e1003308. doi: 10.1371/journal.ppat.1003308
75. Kraiczy P, Skerka C, Kirschfink M, Brade V, Zipfel PF. Immune evasion of Borrelia burgdorferi by acquisition of human complement regulators FHL-1/reconectin and Factor H. Eur J Immunol (2001) 31:1674–84. doi: 10.1002/1521-4141(200106)31:6<1674::aid-immu1674>3.0.co;2-2
76. Wallich R, Pattathu J, Kitiratschky V, Brenner C, Zipfel PF, Brade V, et al. Identification and functional characterization of complement regulator-acquiring surface protein 1 of the Lyme disease spirochetes Borrelia afzelii and Borrelia garinii. Infect Immun (2005) 73:2351–9. doi: 10.1128/iai.73.4.2351-2359.2005
77. Kraiczy P, Skerka C, Brade V, Zipfel PF. Further characterization of complement regulator-acquiring surface proteins of Borrelia burgdorferi. Infect Immun (2001) 69:7800–9. doi: 10.1128/iai.69.12.7800-7809.2001
78. Rogers EA, Marconi RT. Delineation of species-specific binding properties of the CspZ protein (BBH06) of Lyme disease spirochetes: evidence for new contributions to the pathogenesis of Borrelia spp. Infect Immun (2007) 75:5272–81. doi: 10.1128/iai.00850-07
79. Dyer A, Brown G, Stejskal L, Laity PR, Bingham RJ. The Borrelia afzelii outer membrane protein BAPKO_0422 binds human factor-H and is predicted to form a membrane-spanning β-barrel. Biosci Rep (2015) 35:e00240. doi: 10.1042/BSR20150095
80. Kraiczy P, Hellwage J, Skerka C, Becker H, Kirschfink M, Simon MM, et al. Complement resistance of Borrelia burgdorferi correlates with the expression of BbCRASP-1, a novel linear plasmid-encoded surface protein that interacts with human factor H and FHL-1 and is unrelated to Erp proteins. J Biol Chem (2004) 279:2421–9. doi: 10.1074/jbc.M308343200
81. Hartmann K, Corvey C, Skerka C, Kirschfink M, Karas M, Brade V, et al. Functional characterization of BbCRASP-2, a distinct outer membrane protein of Borrelia burgdorferi that binds host complement regulators factor H and FHL-1. Mol Microbiol (2006) 61:1220–36. doi: 10.1111/j.1365-2958.2006.05318.x
82. Kraiczy P, Hellwage J, Skerka C, Kirschfink M, Brade V, Zipfel PF, et al. Immune evasion of Borrelia burgdorferi: mapping of a complement-inhibitor factor H-binding site of BbCRASP-3, a novel member of the Erp protein family. Eur J Immunol (2003) 33:697–707. doi: 10.1002/eji.200323571
83. Lin Y-P, Frye AM, Nowak TA, Kraiczy P. New insights into CRASP-mediated complement evasion in the Lyme disease enzootic cycle. Front Cell Infect Microbiol (2020) 10:1. doi: 10.3389/fcimb.2020.00001
84. Hellwage J, Meri T, Heikkila T, Alitalo A, Panelius J, Lahdenne P, et al. The complement regulator factor H binds to the surface protein OspE of Borrelia burgdorferi. J Biol Chem (2001) 276:8427–35. doi: 10.1074/jbc.M007994200
85. Bhattacharjee A, Oeemig JS, Kolodziejczyk R, Meri T, Kajander T, Lehtinen MJ, et al. Structural basis for complement evasion by Lyme disease pathogen Borrelia burgdorferi. J Biol Chem (2013) 288:18685–95. doi: 10.1074/jbc.M113.459040
86. Hovis KM, McDowell JV, Griffin L, Marconi RT. Identification and characterization of a linear-plasmid-encoded factor H-binding protein (FhbA) of the relapsing fever spirochete Borrelia hermsii. J Bacteriol (2004) 186:2612–8. doi: 10.1128/jb.186.9.2612-2618.2004
87. Hovis KM, Jones JP, Sadlon T, Raval G, Gordon DL, Marconi RT. Molecular analyses of the interaction of Borrelia hermsii FhbA with the complement regulatory proteins factor H and factor H-like protein 1. Infect Immun (2006) 74:2007–14. doi: 10.1128/iai.74.4.2007-2014.2006
88. Rossmann E, Kraiczy P, Herzberger P, Skerka C, Kirschfink M, Simon MM, et al. Dual binding specificity of a Borrelia hermsii-associated complement regulator-acquiring surface protein for factor H and plasminogen discloses a putative virulence factor of relapsing fever spirochetes. J Immunol (2007) 178:7292–301. doi: 10.4049/jimmunol.178.11.7292
89. Walter L, Sürth V, Röttgerding F, Zipfel PF, Fritz-Wolf K, Kraiczy P. Elucidating the immune evasion mechanisms of Borrelia mayonii, the causative agent of Lyme disease. Front Immunol (2019) 10:2722. doi: 10.3389/fimmu.2019.02722
90. Röttgerding F, Wagemakers A, Koetsveld J, Fingerle V, Kirschfink M, Hovius JW, et al. Immune evasion of Borrelia miyamotoi: CbiA, a novel outer surface protein exhibiting complement binding and inactivating properties. Sci Rep (2017) 7:303. doi: 10.1038/s41598-017-00412-4
91. Schott M, Grosskinsky S, Brenner C, Kraiczy P, Wallich R. Molecular characterization of the interaction of Borrelia parkeri and Borrelia turicatae with human complement regulators. Infect Immun (2010) 78:2199–208. doi: 10.1128/iai.00089-10
92. Grosskinsky S, Schott M, Brenner C, Cutler SJ, Kraiczy P, Zipfel PF, et al. Borrelia recurrentis employs a novel multifunctional surface protein with anti-complement, anti-opsonic and invasive potential to escape innate immunity. PloS One (2009) 4:e4858. doi: 10.1371/journal.pone.0004858
93. Meri T, Cutler SJ, Blom AM, Meri S, Jokiranta TS. Relapsing fever spirochetes Borrelia recurrentis and B. duttonii acquire complement regulators C4b-binding protein and factor H. Infect Immun (2006) 74:4157–63. doi: 10.1128/iai.00007-06
94. Herzberger P, Siegel C, Skerka C, Fingerle V, Schulte-Spechtel U, van Dam A, et al. Human pathogenic Borrelia spielmanii sp. nov. resists complement-mediated killing by direct binding of immune regulators factor H and factor H-like protein 1. Infect Immun (2007) 75:4817–25. doi: 10.1128/iai.00532-07
95. Herzberger P, Siegel C, Skerka C, Fingerle V, Schulte-Spechtel U, Wilske B, et al. Identification and characterization of the factor H and FHL-1 binding complement regulator-acquiring surface protein 1 of the Lyme disease spirochete Borrelia spielmanii sp. nov. Int J Med Microbiol (2009) 299:141–54. doi: 10.1016/j.ijmm.2008.06.005
96. Seling A, Siegel C, Fingerle V, Jutras BL, Brissette CA, Skerka C, et al. Functional characterization of Borrelia spielmanii outer surface proteins that interact with distinct members of the human factor H protein family and with plasminogen. Infect Immun (2010) 78:39–48. doi: 10.1128/iai.00691-09
97. Orth D, Khan AB, Naim A, Grif K, Brockmeyer J, Karch H, et al. Shiga toxin activates complement and binds factor H: evidence for an active role of complement in hemolytic uremic syndrome. J Immunol (2009) 182:6394–400. doi: 10.4049/jimmunol.0900151
98. Poolpol K, Orth-Höller D, Speth C, Zipfel PF, Skerka C, de Córdoba SR, et al. Interaction of Shiga toxin 2 with complement regulators of the factor H protein family. Mol Immunol (2014) 58:77–84. doi: 10.1016/j.molimm.2013.11.009
99. Li W, Wen L, Li C, Chen R, Ye Z, Zhao J, et al. Contribution of the outer membrane protein OmpW in Escherichia coli to complement resistance from binding to factor H. Microb Pathog (2016) 98:57–62. doi: 10.1016/j.micpath.2016.06.024
100. Ben Nasr A, Klimpel GR. Subversion of complement activation at the bacterial surface promotes serum resistance and opsonophagocytosis of Francisella tularensis. J Leukoc Biol (2008) 84:77–85. doi: 10.1189/jlb.0807526
101. Friberg N, Carlson P, Kentala E, Mattila PS, Kuusela P, Meri S, et al. Factor H binding as a complement evasion mechanism for an anaerobic pathogen, Fusobacterium necrophorum. J Immunol (2008) 181:8624–32. doi: 10.4049/jimmunol.181.12.8624
102. Hallström T, Zipfel PF, Blom AM, Lauer N, Forsgren A, Riesbeck K. Haemophilus influenzae interacts with the human complement inhibitor factor H. J Immunol (2008) 181:537–45. doi: 10.4049/jimmunol.181.1.537
103. Fleury C, Su YC, Hallström T, Sandblad L, Zipfel PF, Riesbeck K. Identification of a Haemophilus influenzae factor H-Binding lipoprotein involved in serum resistance. J Immunol (2014) 192:5913–23. doi: 10.4049/jimmunol.1303449
104. Inzana TJ, Balyan R, Howard MD. Decoration of Histophilus somni lipooligosaccharide with N-acetyl-5-neuraminic acid enhances bacterial binding of complement factor H and resistance to killing by serum and polymorphonuclear leukocytes. Vet Microbiol (2012) 161:113–21. doi: 10.1016/j.vetmic.2012.07.008
105. Verma A, Hellwage J, Artiushin S, Zipfel PF, Kraiczy P, Timoney JF, et al. LfhA, a novel factor H-binding protein of Leptospira interrogans. Infect Immun (2006) 74:2659–66. doi: 10.1128/IAI.74.5.2659-2666.2006
106. Stevenson B, Choy HA, Pinne M, Rotondi ML, Miller MC, Demoll E, et al. Leptospira interrogans endostatin-like outer membrane proteins bind host fibronectin, laminin and regulators of complement. PloS One (2007) 2:e1188. doi: 10.1371/journal.pone.0001188
107. da Silva LB, Miragaia Ldos S, Breda LC, Abe CM, Schmidt MC, Moro AM, et al. Pathogenic Leptospira species acquire factor H and vitronectin via the surface protein LcpA. Infect Immun (2015) 83:888–97. doi: 10.1128/iai.02844-14
108. Castiblanco-Valencia MM, Fraga TR, Silva L, Monaris D, Abreu PAE, Strobel S, et al. Leptospiral immunoglobulin-like proteins interact with human complement regulators factor H, FHL-1, FHR-1, and C4BP. J Infect Dis (2012) 205:995–1004. doi: 10.1093/infdis/jir875
109. Castiblanco-Valencia MM, Fraga TR, Breda LC, Vasconcellos SA, Figueira CP, Picardeau M, et al. Acquisition of negative complement regulators by the saprophyte Leptospira biflexa expressing LigA or LigB confers enhanced survival in human serum. Immunol Lett (2016) 173:61–8. doi: 10.1016/j.imlet.2016.03.005
110. Siqueira GH, Atzingen MV, Alves IJ, de Morais ZM, Vasconcellos SA, Nascimento AL. Characterization of three novel adhesins of Leptospira interrogans. Am J Trop Med Hyg (2013) 89:1103–16. doi: 10.4269/ajtmh.13-0205
111. Siqueira GH, Atzingen MV, de Souza GO, Vasconcellos SA, Nascimento A. Leptospira interrogans Lsa23 protein recruits plasminogen, factor H and C4BP from normal human serum and mediates C3b and C4b degradation. Microbiology (2016) 162:295–308. doi: 10.1099/mic.0.000217
112. Ghosh KK, Prakash A, Dhara A, Hussain MS, Shrivastav P, Kumar P, et al. Role of supramolecule ErpY-like lpoprotein of Leptospira in thrombin-catalyzed fibrin clot inhibition and binding to complement factors H and I, and its diagnostic potential. Infect Immun (2019) 87:e00536–19. doi: 10.1128/iai.00536-19
113. Wolff DG, Castiblanco-Valencia MM, Abe CM, Monaris D, Morais ZM, Souza GO, et al. Interaction of Leptospira elongation factor Tu with plasminogen and complement factor H: a metabolic leptospiral protein with moonlighting activities. PloS One (2013) 8:e81818. doi: 10.1371/journal.pone.0081818
114. Meri T, Murgia R, Stefanel P, Meri S, Cinco M. Regulation of complement activation at the C3-level by serum resistant leptospires. Microb Pathog (2005) 39:139–47. doi: 10.1016/j.micpath.2005.07.003
115. Salazar N, Souza MC, Biasioli AG, Silva LB, Barbosa AS. The multifaceted roles of Leptospira enolase. Res Microbiol (2017) 168:157–64. doi: 10.1016/j.resmic.2016.10.005
116. Bernhard S, Fleury C, Su YC, Zipfel PF, Koske I, Nordstrom T, et al. Outer membrane protein OlpA contributes to Moraxella catarrhalis serum resistance via interaction with factor H and the alternative pathway. J Infect Dis (2014) 210:1306–10. doi: 10.1093/infdis/jiu241
117. Lavender H, Poncin K, Tang CM. Neisseria cinerea expresses a functional factor H binding protein which Is recognized by immune responses elicited by meningococcal vaccines. Infect Immun (2017) 85:e00305–17. doi: 10.1128/IAI.00305-17
118. Ngampasutadol J, Ram S, Gulati S, Agarwal S, Li C, Visintin A, et al. Human factor H interacts selectively with Neisseria gonorrhoeae and results in species-specific complement evasion. J Immunol (2008) 180:3426–35. doi: 10.4049/jimmunol.180.5.3426
119. Shaughnessy J, Ram S, Bhattacharjee A, Pedrosa J, Tran C, Horvath G, et al. Molecular characterization of the interaction between sialylated Neisseria gonorrhoeae and factor H. J Biol Chem (2011) 286:22235–42. doi: 10.1074/jbc.M111.225516
120. Ram S, Sharma AK, Simpson SD, Gulati S, McQuillen DP, Pangburn MK, et al. A novel sialic acid binding site on factor H mediates serum resistance of sialylated Neisseria gonorrhoeae. J Exp Med (1998) 187:743–52. doi: 10.1084/jem.187.5.743
121. Lewis LA, Rice PA, Ram S. Role of gonococcal neisserial surface protein A (NspA) in serum resistance and comparison of its factor H binding properties with those of its meningococcal counterpart. Infect Immunity (2019) 87:e00658–18. doi: 10.1128/IAI.00658-18
122. Schneider MC, Prosser BE, Caesar JJ, Kugelberg E, Li S, Zhang Q, et al. Neisseria meningitidis recruits factor H using protein mimicry of host carbohydrates. Nature (2009) 458:890–3. doi: 10.1038/nature07769
123. Shaughnessy J, Lewis LA, Jarva H, Ram S. Functional comparison of the binding of factor H short consensus repeat 6 (SCR 6) to factor H binding protein from Neisseria meningitidis and the binding of factor H SCR 18 to 20 to Neisseria gonorrhoeae porin. Infect Immun (2009) 77:2094–103. doi: 10.1128/IAI.01561-08
124. Lewis LA, Ngampasutadol J, Wallace R, Reid JEA, Vogel U, Ram S. The meningococcal vaccine candidate neisserial surface protein A (NspA) binds to factor H and enhances meningococcal resistance to complement. PloS Pathog (2010) 6:e1001027. doi: 10.1371/journal.ppat.1001027
125. Lewis LA, Vu DM, Vasudhev S, Shaughnessy J, Granoff DM, Ram S. Factor H-dependent alternative pathway inhibition mediated by porin B contributes to virulence of Neisseria meningitidis. mBio (2013) 4:e00339–13. doi: 10.1128/mBio.00339-13
126. Giuntini S, Pajon R, Ram S, Granoff DM. Binding of complement factor H to PorB3 and NspA enhances resistance of Neisseria meningitidis to anti-factor H binding protein bactericidal activity. Infect Immun (2015) 83:1536–45. doi: 10.1128/iai.02984-14
127. Lewis LA, Carter M, Ram S. The relative roles of factor H binding protein, neisserial surface protein A, and lipooligosaccharide sialylation in regulation of the alternative pathway of complement on meningococci. J Immunol (2012) 188:5063. doi: 10.4049/jimmunol.1103748
128. Langereis JD, de Jonge MI, Weiser JN. Binding of human factor H to outer membrane protein P5 of non-typeable Haemophilus influenzae contributes to complement resistance. Mol Microbiol (2014) 94:89–106. doi: 10.1111/mmi.12741
129. Wong SM, Shaughnessy J, Ram S, Akerley BJ. Defining the binding region in Factor H to develop a therapeutic factor H-Fc fusion protein against non-typeable Haemophilus influenzae. Front Cell Infect Microbiol (2016) 6:40. doi: 10.3389/fcimb.2016.00040
130. Sahagún-Ruiz A, Granados Martinez AP, Breda LCD, Fraga TR, Castiblanco Valencia MM, Barbosa AS, et al. Pasteurella pneumotropica evades the human complement system by acquisition of the complement regulators factor H and C4BP. PloS One (2014) 9:e111194–e94. doi: 10.1371/journal.pone.0111194
131. Hallström T, Mörgelin M, Barthel D, Raguse M, Kunert A, Hoffmann R, et al. Dihydrolipoamide dehydrogenase of Pseudomonas aeruginosa is a surface-exposed immune evasion protein that binds three members of the factor H family and plasminogen. J Immunol (2012) 189:4939. doi: 10.4049/jimmunol.1200386
132. Kunert A, Losse J, Gruszin C, Hühn M, Kaendler K, Mikkat S, et al. Immune evasion of the human pathogen Pseudomonas aeruginosa: elongation factor Tuf is a factor H and plasminogen binding protein. J Immunol (2007) 179:2979–88. doi: 10.4049/jimmunol.179.5.2979
133. Riley SP, Patterson JL, Martinez JJ. The rickettsial OmpB beta-peptide of Rickettsia conorii is sufficient to facilitate factor H-mediated serum resistance. Infect Immun (2012) 80:2735–43. doi: 10.1128/IAI.00349-12
134. Ho DK, Jarva H, Meri S. Human complement factor H binds to outer membrane protein Rck of Salmonella. J Immunol (2010) 185:1763–9. doi: 10.4049/jimmunol.1001244
135. Sharp JA, Echague CG, Hair PS, Ward MD, Nyalwidhe JO, Geoghegan JA, et al. Staphylococcus aureus surface protein SdrE binds complement regulator factor H as an immune evasion tactic. PloS One (2012) 7:e38407. doi: 10.1371/journal.pone.0038407
136. Zhang Y, Wu M, Hang T, Wang C, Yang Y, Pan W, et al. Staphylococcus aureus SdrE captures complement factor H’s C-terminus via a novel ‘close, dock, lock and latch’ mechanism for complement evasion. Biochem J (2017) 474:1619–31. doi: 10.1042/BCJ20170085
137. Haupt K, Reuter M, van den Elsen J, Burman J, Hälbich S, Richter J, et al. The Staphylococcus aureus protein Sbi acts as a complement inhibitor and forms a tripartite complex with host complement Factor H and C3b. PloS Pathog (2008) 4:e1000250. doi: 10.1371/journal.ppat.1000250
138. Areschoug T, Linse S, Stålhammar-Carlemalm M, Hedén L-O, Lindahl G. A proline-rich region with a highly periodic sequence in Streptococcal beta protein adopts the polyproline II structure and is exposed on the bacterial surface. J Bacteriol (2002) 184:6376–83. doi: 10.1128/jb.184.22.6376-6393.2002
139. Jarva H, Hellwage J, Jokiranta TS, Lehtinen MJ, Zipfel PF, Meri S. The group B streptococcal beta and pneumococcal Hic proteins are structurally related immune evasion molecules that bind the complement inhibitor factor H in an analogous fashion. J Immunol (2004) 172:3111–8. doi: 10.4049/jimmunol.172.5.3111
140. Maruvada R, Prasadarao NV, Rubens CE. Acquisition of factor H by a novel surface protein on group B Streptococcus promotes complement degradation. FASEB J (2009) 23:3967–77. doi: 10.1096/fj.09-138149
141. Moulin P, Rong V, Ribeiro ESA, Pederick VG, Camiade E, Mereghetti L, et al. Defining the role of the Streptococcus agalactiae Sht-family proteins in zinc acquisition and complement evasion. J Bacteriol (2019) 201:e00757–18. doi: 10.1128/jb.00757-18
142. Janulczyk R, Iannelli F, Sjoholm AG, Pozzi G, Bjorck L. Hic, a novel surface protein of Streptococcus pneumoniae that interferes with complement function. J Biol Chem (2000) 275:37257–63. doi: 10.1074/jbc.M004572200
143. Dave S, Brooks-Walter A, Pangburn MK, McDaniel LS. PspC, a pneumococcal surface protein, binds human factor H. Infect Immun (2001) 69:3435–7. doi: 10.1128/iai.69.5.3435-3437.2001
144. Dave S, Pangburn MK, Pruitt C, McDaniel LS. Interaction of human factor H with PspC of Streptococcus pneumoniae. Indian J Med Res (2004) 119(Suppl):66–73.
145. Herbert AP, Makou E, Chen ZA, Kerr H, Richards A, Rappsilber J, et al. Complement evasion mediated by enhancement of captured factor H: implications for protection of self-surfaces from complement. J Immunol (2015) 195:4986–98. doi: 10.4049/jimmunol.1501388
146. Achila D, Liu A, Banerjee R, Li Y, Martinez-Hackert E, Zhang JR, et al. Structural determinants of host specificity of complement Factor H recruitment by Streptococcus pneumoniae. Biochem J (2015) 465:325–35. doi: 10.1042/bj20141069
147. Jarva H, Janulczyk R, Hellwage J, Zipfel PF, Björck L, Meri S. Streptococcus pneumoniae evades complement attack and opsonophagocytosis by expressing the pspC locus-encoded Hic protein that binds to short consensus repeats 8-11 of factor H. J Immunol (2002) 168:1886. doi: 10.4049/jimmunol.168.4.1886
148. Duthy TG, Ormsby RJ, Giannakis E, Ogunniyi AD, Stroeher UH, Paton JC, et al. The human complement regulator factor H binds pneumococcal surface protein PspC via short consensus repeats 13 to 15. Infect Immun (2002) 70:5604–11. doi: 10.1128/iai.70.10.5604-5611.2002
149. Mohan S, Hertweck C, Dudda A, Hammerschmidt S, Skerka C, Hallström T, et al. Tuf of Streptococcus pneumoniae is a surface displayed human complement regulator binding protein. Mol Immunol (2014) 62:249–64. doi: 10.1016/j.molimm.2014.06.029
150. Ramos-Sevillano E, Urzainqui A, Campuzano S, Moscoso M, González-Camacho F, Domenech M, et al. Pleiotropic effects of cell wall amidase LytA on Streptococcus pneumoniae sensitivity to the host immune response. Infect Immun (2015) 83:591–603. doi: 10.1128/iai.02811-14
151. Pandiripally V, Gregory E, Cue D. Acquisition of regulators of complement activation by Streptococcus pyogenes serotype M1. Infect Immun (2002) 70:6206–14. doi: 10.1128/iai.70.11.6206-6214.2002
152. Pandiripally V, Wei L, Skerka C, Zipfel PF, Cue D. Recruitment of complement factor H-like protein 1 promotes intracellular invasion by group A streptococci. Infect Immun (2003) 71:7119. doi: 10.1128/IAI.71.12.7119-7128.2003
153. Horstmann RD, Sievertsen HJ, Knobloch J, Fischetti VA. Antiphagocytic activity of streptococcal M protein: selective binding of complement control protein factor H. Proc Natl Acad Sci USA (1988) 85:1657–61. doi: 10.1073/pnas.85.5.1657
154. Yu J, Wiita P, Kawaguchi R, Honda J, Jorgensen A, Zhang K, et al. Biochemical analysis of a common human polymorphism associated with age-related macular degeneration. Biochemistry (2007) 46:8451–61. doi: 10.1021/bi700459a
155. Gustafsson MC, Lannergård J, Nilsson OR, Kristensen BM, Olsen JE, Harris CL, et al. Factor H binds to the hypervariable region of many Streptococcus pyogenes M proteins but does not promote phagocytosis resistance or acute virulence. PloS Pathog (2013) 9:e1003323. doi: 10.1371/journal.ppat.1003323
156. Caswell CC, Han R, Hovis KM, Ciborowski P, Keene DR, Marconi RT, et al. The Scl1 protein of M6-type group A streptococcus binds the human complement regulatory protein, factor H, and inhibits the alternative pathway of complement. Mol Microbiol (2008) 67:584–96. doi: 10.1111/j.1365-2958.2007.06067.x
157. Reuter M, Caswell CC, Lukomski S, Zipfel PF. Binding of the human complement regulators CFHR1 and factor H by streptococcal collagen-like protein 1 (Scl1) via their conserved C termini allows control of the complement cascade at multiple levels. J Biol Chem (2010) 285:38473–85. doi: 10.1074/jbc.M110.143727
158. Pian Y, Gan S, Wang S, Guo J, Wang P, Zheng Y, et al. Fhb, a novel factor H-binding surface protein, contributes to the antiphagocytic ability and virulence of Streptococcus suis. Infect Immun (2012) 80:2402–13. doi: 10.1128/iai.06294-11
159. McDowell JV, Lankford J, Stamm L, Sadlon T, Gordon DL, Marconi RT. Demonstration of factor H-like Protein 1 binding to Treponema denticola, a pathogen associated with periodontal disease in humans. Infect Immun (2005) 73:7126–32. doi: 10.1128/iai.73.11.7126-7132.2005
160. Biedzka-Sarek M, Jarva H, Hyytiainen H, Meri S, Skurnik M. Characterization of complement factor H binding to Yersinia enterocolitica serotype O:3. Infect Immun (2008) 76:4100–9. doi: 10.1128/IAI.00313-08
161. Ho DK, Riva R, Skurnik M, Meri S. The Yersinia pseudotuberculosis outer membrane protein Ail recruits the human complement regulatory protein factor H. J Immunol (2012) 189:3593–9. doi: 10.4049/jimmunol.1201145
162. Dasari P, Koleci N, Shopova IA, Wartenberg D, Beyersdorf N, Dietrich S, et al. Enolase from Aspergillus fumigatus is a moonlighting protein that binds the human plasma complement proteins factor H, FHL-1, C4BP, and plasminogen. Front Immunol (2019) 10:2573. doi: 10.3389/fimmu.2019.02573
163. Dasari P, Shopova IA, Stroe M, Wartenberg D, Martin-Dahse H, Beyersdorf N, et al. Aspf2 From Aspergillus fumigatus recruits human immune regulators for immune evasion and cell damage. Front Immunol (2018) 9:1635. doi: 10.3389/fimmu.2018.01635
164. Luo S, Poltermann S, Kunert A, Rupp S, Zipfel PF. Immune evasion of the human pathogenic yeast Candida albicans: Pra1 is a Factor H, FHL-1 and plasminogen binding surface protein. Mol Immunol (2009) 47:541–50. doi: 10.1016/j.molimm.2009.07.017
165. Poltermann S, Kunert A, von der Heide M, Eck R, Hartmann A, Zipfel PF. Gpm1p is a factor H-, FHL-1-, and plasminogen-binding surface protein of Candida albicans. J Biol Chem (2007) 282:37537–44. doi: 10.1074/jbc.M707280200
166. Luo S, Hoffmann R, Skerka C, Zipfel PF. Glycerol-3-phosphate dehydrogenase 2 is a novel factor H–, factor H–like protein 1–, and plasminogen-binding surface protein of Candida albicans. J Infect Dis (2012) 207:594–603. doi: 10.1093/infdis/jis718
167. Lesiak-Markowicz I, Vogl G, Schwarzmüller T, Speth C, Lass-Flörl C, Dierich MP, et al. Candida albicans Hgt1p, a multifunctional evasion molecule: complement inhibitor, CR3 analogue, and human immunodeficiency virus–binding molecule. J Infect Dis (2011) 204:802–09. doi: 10.1093/infdis/jir455
168. Kenno S, Speth C, Rambach G, Binder U, Chatterjee S, Caramalho R, et al. Candida albicans factor H binding molecule Hgt1p – a low glucose-induced transmembrane protein is trafficked to the cell wall and impairs phagocytosis and killing by human neutrophils. Front Microbiol (2019) 9:3319. doi: 10.3389/fmicb.2018.03319
169. Macleod OJS, Bart JM, MacGregor P, Peacock L, Savill NJ, Hester S, et al. A receptor for the complement regulator factor H increases transmission of trypanosomes to tsetse flies. Nat Commun (2020) 11:1326. doi: 10.1038/s41467-020-15125-y
170. Joiner K, Sher A, Gaither T, Hammer C. Evasion of alternative complement pathway by Trypanosoma cruzi results from inefficient binding of factor B. Proc Natl Acad Sci USA (1986) 83:6593–7. doi: 10.1073/pnas.83.17.6593
171. Rosa TF, Flammersfeld A, Ngwa CJ, Kiesow M, Fischer R, Zipfel PF, et al. The Plasmodium falciparum blood stages acquire factor H family proteins to evade destruction by human complement. Cell Microbiol (2016) 18:573–90. doi: 10.1111/cmi.12535
172. Simon N, Lasonder E, Scheuermayer M, Kuehn A, Tews S, Fischer R, et al. Malaria parasites co-opt human factor H to prevent complement-mediated lysis in the mosquito midgut. Cell Host Microbe (2013) 13:29–41. doi: 10.1016/j.chom.2012.11.013
173. Kennedy AT, Schmidt CQ, Thompson JK, Weiss GE, Taechalertpaisarn T, Gilson PR, et al. Recruitment of factor H as a novel complement evasion strategy for blood-stage Plasmodium falciparum infection. J Immunol (2016) 196:1239–48. doi: 10.4049/jimmunol.1501581
174. Sikorski PM, Commodaro AG, Grigg ME. Toxoplasma gondii recruits factor H and C4b-binding protein to mediate resistance to serum killing and promote parasite persistence in vivo. Front Immunol (2020) 10:3105. doi: 10.3389/fimmu.2019.03105
175. Meri T, Jokiranta TS, Hellwage J, Bialonski A, Zipfel PF, Meri S. Onchocerca volvulus microfilariae avoid complement attack by direct binding of factor H. J Infect Dis (2002) 185:1786–93. doi: 10.1086/340649
176. Diaz A, Ferreira A, Sim RB. Complement evasion by Echinococcus granulosus: sequestration of host factor H in the hydatid cyst wall. J Immunol (1997) 158:3779–86.
177. Haapasalo K, Meri T, Jokiranta TS. Loa loa microfilariae evade complement attack in vivo by acquiring regulatory proteins from host plasma. Infect Immun (2009) 77:3886–93. doi: 10.1128/IAI.01583-08
178. Chung KM, Liszewski MK, Nybakken G, Davis AE, Townsend RR, Fremont DH, et al. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc Natl Acad Sci USA (2006) 103:19111–6. doi: 10.1073/pnas.0605668103
179. Pinter C, Siccardi AG, Longhi R, Clivio A. Direct interaction of complement factor H with the C1 domain of HIV type 1 glycoprotein 120. AIDS Res Hum Retroviruses (1995) 11:577–88. doi: 10.1089/aid.1995.11.577
180. Pinter C, Siccardi AG, Lopalco L, Longhi R, Clivio A. HIV glycoprotein 41 and complement factor H interact with each other and share functional as well as antigenic homology. AIDS Res Hum Retroviruses (1995) 11:971–80. doi: 10.1089/aid.1995.11.971
181. Stoiber H, Ebenbichler C, Schneider R, Janatova J, Dierich MP. Interaction of several complement proteins with gp120 and gp41, the two envelope glycoproteins of HIV-1. AIDS (1995) 9:19–26. doi: 10.1097/00002030-199501000-00003
182. Stoiber H, Schneider R, Janatova J, Dierich MP. Human complement proteins C3b, C4b, factor H and properdin react with specific sites in gp120 and gp41, the envelope proteins of HIV-1. Immunobiology (1995) 193:98–113. doi: 10.1016/s0171-2985(11)80158-0
183. Haupt K, Kraiczy P, Wallich R, Brade V, Skerka C, Zipfel PF. Binding of human factor H-related protein 1 to serum-resistant Borrelia burgdorferi is mediated by borrelial complement regulator-acquiring surface proteins. J Infect Dis (2007) 196:124–33. doi: 10.1086/518509
184. Reiss T, Rosa T, Blaesius K, Bobbert RP, Zipfel PF, Skerka C, et al. Cutting edge: FHR-1 binding impairs factor H-mediated complement evasion by the malaria parasite. Plasmodium Falciparum J Immunol (2018) 201:3497–502. doi: 10.4049/jimmunol.1800662
185. Caesar JJ, Lavender H, Ward PN, Exley RM, Eaton J, Chittock E, et al. Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. Elife (2014) 3:e04008. doi: 10.7554/eLife.04008
186. Siegel C, Hallström T, Skerka C, Eberhardt H, Uzonyi B, Beckhaus T, et al. Complement factor H-related proteins CFHR2 and CFHR5 represent novel ligands for the infection-associated CRASP proteins of Borrelia burgdorferi. PloS One (2010) 5:e13519. doi: 10.1371/journal.pone.0013519
187. Cress BF, Englaender JA, He W, Kasper D, Linhardt RJ, Koffas MAG. Masquerading microbial pathogens: capsular polysaccharides mimic host-tissue molecules. FEMS Microbiol Rev (2014) 38:660–97. doi: 10.1111/1574-6976.12056
188. Ram S, Shaughnessy J, DeOliveira RB, Lewis LA, Gulati S, Rice PA. Utilizing complement evasion strategies to design complement-based antibacterial immunotherapeutics: Lessons from the pathogenic Neisseriae. Immunobiology (2016) 221:1110–23. doi: 10.1016/j.imbio.2016.05.016
189. Ram S, Shaughnessy J, de Oliveira RB, Lewis LA, Gulati S, Rice PA. Gonococcal lipooligosaccharide sialylation: virulence factor and target for novel immunotherapeutics. Pathog Dis (2017) 75:ftx049. doi: 10.1093/femspd/ftx049
190. Lewis LA, Ram S. Complement interactions with the pathogenic Neisseriae: clinical features, deficiency states, and evasion mechanisms. FEBS Lett (2020) 594:2670–94. doi: 10.1002/1873-3468.13760
191. Kajander T, Lehtinen MJ, Hyvärinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci USA (2011) 108:2897–902. doi: 10.1073/pnas.1017087108
192. Madico G, Ngampasutadol J, Gulati S, Vogel U, Rice PA, Ram S. Factor H binding and function in sialylated pathogenic neisseriae is influenced by gonococcal, but not meningococcal, porin. J Immunol (2007) 178:4489. doi: 10.4049/jimmunol.178.7.4489
193. Hedlund M, Tangvoranuntakul P, Takematsu H, Long JM, Housley GD, Kozutsumi Y, et al. N-glycolylneuraminic acid deficiency in mice: implications for human biology and evolution. Mol Cell Biol (2007) 27:4340–6. doi: 10.1128/mcb.00379-07
194. Krotkiewski H. The structure of glycophorins of animal erythrocytes. Glycoconjugate J (1988) 5:35–48. doi: 10.1007/BF01048330
195. Schmidt CQ, Hipgrave Ederveen AL, Harder MJ, Wuhrer M, Stehle T, Blaum BS. Biophysical analysis of sialic acid recognition by the complement regulator Factor H. Glycobiology (2018) 28:765–73. doi: 10.1093/glycob/cwy061
196. Gulati S, Schoenhofen IC, Whitfield DM, Cox AD, Li J, St Michael F, et al. Utilizing CMP-sialic scid analogs to unravel Neisseria gonorrhoeae lipooligosaccharide-mediated complement resistance and design novel therapeutics. PloS Pathog (2015) 11:e1005290. doi: 10.1371/journal.ppat.1005290
197. Prosser BE, Johnson S, Roversi P, Herbert AP, Blaum BS, Tyrrell J, et al. Structural basis for complement factor H linked age-related macular degeneration. J Exp Med (2007) 204:2277–83. doi: 10.1084/jem.20071069
198. Kolodziejczyk R, Mikula KM, Kotila T, Postis VLG, Jokiranta TS, Goldman A, et al. Crystal structure of a tripartite complex between C3dg, C-terminal domains of factor H and OspE of Borrelia burgdorferi. PloS One (2017) 12:e0188127. doi: 10.1371/journal.pone.0188127
199. Herr AB, Thorman AW. Hiding in plain sight: immune evasion by the staphylococcal protein SdrE. Biochem J (2017) 474:1803–06. doi: 10.1042/bcj20170132
200. Kraiczy P. Hide and seek: how Lyme disease spirochetes overcome complement attack. Front Immunol (2016) 7:385. doi: 10.3389/fimmu.2016.00385
201. Woodman ME, Cooley AE, Miller JC, Lazarus JJ, Tucker K, Bykowski T, et al. Borrelia burgdorferi binding of host complement regulator factor H Is not required for efficient mammalian infection. Infect Immun (2007) 75:3131–9. doi: 10.1128/IAI.01923-06
202. Alitalo A, Meri T, Ramo L, Jokiranta TS, Heikkila T, Seppala IJ, et al. Complement evasion by Borrelia burgdorferi: serum-resistant strains promote C3b inactivation. Infect Immun (2001) 69:3685–91. doi: 10.1128/iai.69.6.3685-3691.2001
203. Hart T, Nguyen NTT, Nowak NA, Zhang F, Linhardt RJ, Diuk-Wasser M, et al. Polymorphic factor H-binding activity of CspA protects Lyme borreliae from the host complement in feeding ticks to facilitate tick-to-host transmission. PloS Pathog (2018) 14:e1007106. doi: 10.1371/journal.ppat.1007106
204. Fine LM, Miller DP, Mallory KL, Tegels BK, Earnhart CG, Marconi RT. The Borrelia hermsii factor H binding protein FhbA is not required for infectivity in mice or for resistance to human complement in vitro. Infect Immun (2014) 82:3324–32. doi: 10.1128/iai.01892-14
205. Ermert D, Shaughnessy J, Joeris T, Kaplan J, Pang CJ, Kurt-Jones EA, et al. Virulence of group A streptococci is enhanced by human complement inhibitors. PloS Pathog (2015) 11:e1005043. doi: 10.1371/journal.ppat.1005043
206. Johnsson E, Thern A, Dahlbäck B, Hedén LO, Wikström M, Lindahl G. A highly variable region in members of the streptococcal M protein family binds the human complement regulator C4BP. J Immunol (1996) 157:3021–9.
207. Kihlberg BM, Collin M, Olsén A, Björck L. Protein H, an antiphagocytic surface protein in Streptococcus pyogenes. Infect Immun (1999) 67:1708–14. doi: 10.1128/IAI.67.4.1708-1714.1999
208. Fan J, Chaturvedi V, Shen SH. Identification and phylogenetic analysis of a glucose transporter gene family from the human pathogenic yeast Candida albicans. J Mol Evol (2002) 55:336–46. doi: 10.1007/s00239-002-2330-4
209. Loh E, Kugelberg E, Tracy A, Zhang Q, Gollan B, Ewles H, et al. Temperature triggers immune evasion by Neisseria meningitidis. Nature (2013) 502:237–40. doi: 10.1038/nature12616
210. Loh E, Lavender H, Tan F, Tracy A, Tang CM. Thermoregulation of meningococcal fHbp, an important virulence factor and vaccine antigen, is mediated by anti-ribosomal binding site sequences in the open reading frame. PloS Pathog (2016) 12:e1005794. doi: 10.1371/journal.ppat.1005794
211. Welsch JA, Ram S. Factor H and neisserial pathogenesis. Vaccine (2008) 26(Suppl 8):I40–5. doi: 10.1016/j.vaccine.2008.11.060
212. Kahler CM, Martin LE, Shih GC, Rahman MM, Carlson RW, Stephens DS. The (alpha2–>8)-linked polysialic acid capsule and lipooligosaccharide structure both contribute to the ability of serogroup B Neisseria meningitidis to resist the bactericidal activity of normal human serum. Infect Immun (1998) 66:5939–47. doi: 10.1128/iai.66.12.5939-5947.1998
213. Steere AC, Strle F, Wormser GP, Hu LT, Branda JA, Hovius JWR, et al. Lyme borreliosis. Nat Rev Dis Primers (2016) 2:16090. doi: 10.1038/nrdp.2016.90
214. Bykowski T, Woodman ME, Cooley AE, Brissette CA, Brade V, Wallich R, et al. Coordinated expression of Borrelia burgdorferi complement regulator-acquiring surface proteins during the Lyme disease spirochete’s mammal-tick infection cycle. Infect Immun (2007) 75:4227–36. doi: 10.1128/IAI.00604-07
215. Brooks CS, Hefty PS, Jolliff SE, Akins DR. Global analysis of Borrelia burgdorferi genes regulated by mammalian host-specific signals. Infect Immun (2003) 71:3371–83. doi: 10.1128/iai.71.6.3371-3383.2003
216. Marcinkiewicz AL, Dupuis AP,2, Zamba-Campero M, Nowak N, Kraiczy P, Ram S, et al. Blood treatment of Lyme borreliae demonstrates the mechanism of CspZ-mediated complement evasion to promote systemic infection in vertebrate hosts. Cell Microbiol (2019) 21:e12998. doi: 10.1111/cmi.12998
217. Tokarz R, Anderton JM, Katona LI, Benach JL. Combined effects of blood and temperature shift on Borrelia burgdorferi gene expression as determined by whole genome DNA array. Infect Immun (2004) 72:5419–32. doi: 10.1128/iai.72.9.5419-5432.2004
218. Neelakanta G, Sultana H, Sonenshine DE, Marconi RT. An in vitro blood-feeding method revealed differential Borrelia turicatae (Spirochaetales: Spirochaetaceae) gene expression after spirochete acquisition and colonization in the soft tick Ornithodoros turicata (Acari: Argasidae). J Med Entomol (2017) 54:441–49. doi: 10.1093/jme/tjw171
219. Kraiczy P, Stevenson B. Complement regulator-acquiring surface proteins of Borrelia burgdorferi: structure, function and regulation of gene expression. Ticks Tick Borne Dis (2013) 4:26–34. doi: 10.1016/j.ttbdis.2012.10.039
220. Silvester E, McWilliam KR, Matthews KR. The cytological events and molecular control of life cycle development of Trypanosoma brucei in the mammalian bloodstream. Pathogens (2017) 6:29. doi: 10.3390/pathogens6030029
221. Georgieva M, Kagedan L, Lu Y-J, Thompson CM, Lipsitch M. Antigenic variation in Streptococcus pneumoniae PspC promotes immune escape in the presence of variant-specific immunity. mBio (2018) 9:e00264–18. doi: 10.1128/mBio.00264-18
222. Hyams C, Trzcinski K, Camberlein E, Weinberger DM, Chimalapati S, Noursadeghi M, et al. Streptococcus pneumoniae capsular serotype invasiveness correlates with the degree of factor H binding and opsonization with C3b/iC3b. Infect Immun (2013) 81:354–63. doi: 10.1128/iai.00862-12
223. Biagini M, Spinsanti M, De Angelis G, Tomei S, Ferlenghi I, Scarselli M, et al. Expression of factor H binding protein in meningococcal strains can vary at least 15-fold and is genetically determined. Proc Natl Acad Sci USA (2016) 113:2714–9. doi: 10.1073/pnas.1521142113
224. Luo S, Hipler UC, Munzberg C, Skerka C, Zipfel PF. Sequence variations and protein expression levels of the two immune evasion proteins Gpm1 and Pra1 influence virulence of clinical Candida albicans isolates. PloS One (2015) 10:e0113192. doi: 10.1371/journal.pone.0113192
225. Moreno-Torres A, Malvido-Jimenez IR, de la Pena-Moctezuma A, Castillo Sanchez LO, Fraga TR, Barbosa AS, et al. Culture-attenuated pathogenic Leptospira lose the ability to survive to complement-mediated-killing due to lower expression of factor H binding proteins. Microbes Infect (2019) 21:377–85. doi: 10.1016/j.micinf.2019.03.001
226. Alitalo A, Meri T, Comstedt P, Jeffery L, Tornberg J, Strandin T, et al. Expression of complement factor H binding immunoevasion proteins in Borrelia garinii isolated from patients with neuroborreliosis. Eur J Immunol (2005) 35:3043–53. doi: 10.1002/eji.200526354
227. Pathak A, Bergstrand J, Sender V, Spelmink L, Aschtgen M-S, Muschiol S, et al. Factor H binding proteins protect division septa on encapsulated Streptococcus pneumoniae against complement C3b deposition and amplification. Nat Commun (2018) 9:3398. doi: 10.1038/s41467-018-05494-w
228. Gulati S, Shaughnessy J, Ram S, Rice PA. Targeting lipooligosaccharide (LOS) for a gonococcal vaccine. Front Immunol (2019) 10:321. doi: 10.3389/fimmu.2019.00321
229. Gulati S, Schoenhofen IC, Lindhout-Djukic T, Schur MJ, Landig CS, Saha S, et al. Therapeutic CMP-nonulosonates against multidrug-resistant Neisseria gonorrhoeae. J Immunol (2020), 204:3283–95. doi: 10.4049/jimmunol.1901398
230. Shaughnessy J, Vu DM, Punjabi R, Serra-Pladevall J, DeOliveira RB, Granoff DM, et al. Fusion protein comprising factor H domains 6 and 7 and human IgG1 Fc as an antibacterial immunotherapeutic. Clin Vaccine Immunol (2014) 21:1452–9. doi: 10.1128/CVI.00444-14
231. Blom AM, Magda M, Kohl L, Shaughnessy J, Lambris JD, Ram S, et al. Factor H-IgG chimeric proteins as a therapeutic approach against the gram-positive bacterial pathogen Streptococcus pyogenes. J Immunol (2017) 199:3828–39. doi: 10.4049/jimmunol.1700426
232. Edwards AO, Ritter R, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Science (2005) 308:421–4. doi: 10.1126/science.1110189
233. Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science (2005) 308:419–21. doi: 10.1126/science.1110359
234. Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science (2005) 308:385–9. doi: 10.1126/science.1109557
235. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA (2005) 102:7227–32. doi: 10.1073/pnas.0501536102
236. Clark SJ, Ridge LA, Herbert AP, Hakobyan S, Mulloy B, Lennon R, et al. Tissue-specific host recognition by complement factor H is mediated by differential activities of its glycosaminoglycan-binding regions. J Immunol (2013) 190:2049–57. doi: 10.4049/jimmunol.1201751
237. Ferreira VP, Herbert AP, Hocking HG, Barlow PN, Pangburn MK. Critical role of the C-terminal domains of factor H in regulating complement activation at cell surfaces. J Immunol (2006) 177:6308–16. doi: 10.4049/jimmunol.177.9.6308
238. Ferreira VP, Herbert AP, Cortes C, McKee KA, Blaum BS, Esswein ST, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol (2009) 182:7009–18. doi: 10.4049/jimmunol.0804031
239. Shaughnessy J, Gulati S, Agarwal S, Unemo M, Ohnishi M, Su X-H, et al. A novel factor H–Fc chimeric immunotherapeutic molecule against Neisseria gonorrhoeae. J Immunol (2016) 196:1732. doi: 10.4049/jimmunol.1500292
240. Meri S, Jördens M, Jarva H. Microbial complement inhibitors as vaccines. Vaccine (2008) 26(Suppl 8):I113–7. doi: 10.1016/j.vaccine.2008.11.058
241. Donald RG, Hawkins JC, Hao L, Liberator P, Jones TR, Harris SL, et al. Meningococcal serogroup B vaccines: estimating breadth of coverage. Hum Vaccin Immunother (2017) 13:255–65. doi: 10.1080/21645515.2017.1264750
242. Christodoulides M, Heckels J. Novel approaches to Neisseria meningitidis vaccine design. Pathog Dis (2017) 75:ftx033. doi: 10.1093/femspd/ftx033
243. Fletcher LD, Bernfield L, Barniak V, Farley JE, Howell A, Knauf M, et al. Vaccine potential of the Neisseria meningitidis 2086 lipoprotein. Infect Immun (2004) 72:2088–100. doi: 10.1128/iai.72.4.2088-2100.2004
244. Masignani V, Comanducci M, Giuliani MM, Bambini S, Adu-Bobie J, Arico B, et al. Vaccination against Neisseria meningitidis using three variants of the lipoprotein GNA1870. J Exp Med (2003) 197:789–99. doi: 10.1084/jem.20021911
245. Seib KL, Brunelli B, Brogioni B, Palumbo E, Bambini S, Muzzi A, et al. Characterization of diverse subvariants of the meningococcal factor H (fH) binding protein for their ability to bind fH, to mediate serum resistance, and to induce bactericidal antibodies. Infect Immun (2011) 79:970. doi: 10.1128/IAI.00891-10
246. Harris SL, Donald RG, Hawkins JC, Tan C, O’Neill R, McNeil LK, et al. Neisseria meningitidis serogroup B vaccine, bivalent rLP2086, induces broad serum bactericidal activity against diverse invasive disease strains including outbreak strains. Pediatr Infect Dis J (2017) 36:216–23. doi: 10.1097/inf.0000000000001399
247. Principato S, Pizza M, Rappuoli R. Meningococcal factor H binding protein as immune evasion factor and vaccine antigen. FEBS Lett (2020) 594:2657–69. doi: 10.1002/1873-3468.13793
248. Jongerius I, Lavender H, Tan L, Ruivo N, Exley RM, Caesar JJE, et al. Distinct binding and immunogenic properties of the gonococcal homologue of meningococcal factor h binding protein. PloS Pathog (2013) 9:e1003528–e28. doi: 10.1371/journal.ppat.1003528
249. Scarselli M, Arico B, Brunelli B, Savino S, Di Marcello F, Palumbo E, et al. Rational design of a meningococcal antigen inducing broad protective immunity. Sci Transl Med (2011) 3:91ra62. doi: 10.1126/scitranslmed.3002234
250. Beernink PT, Granoff DM. Bactericidal antibody responses induced by meningococcal recombinant chimeric factor H-binding protein vaccines. Infect Immun (2008) 76:2568–75. doi: 10.1128/iai.00033-08
251. Hollingshead S, Jongerius I, Exley RM, Johnson S, Lea SM, Tang CM. Structure-based design of chimeric antigens for multivalent protein vaccines. Nat Commun (2018) 9:1051. doi: 10.1038/s41467-018-03146-7
252. Dhillon B, Wright AF, Tufail A, Pappworth I, Hayward C, Moore I, et al. Complement factor H autoantibodies and age-related macular degeneration. Invest Ophthalmol Vis Sci (2010) 51:5858–63. doi: 10.1167/iovs.09-5124
253. Sharkey K, Beernink PT, Langley JM, Gantt S, Quach C, Dold C, et al. Anti-factor H antibody reactivity in young adults vaccinated with a meningococcal serogroup B vaccine containing factor H binding protein. mSphere (2019) 4:e00393–19. doi: 10.1128/mSphere.00393-19
254. Beernink PT, Vianzon V, Lewis LA, Moe GR, Granoff DM. A meningococcal outer membrane vesicle vaccine with overexpressed mutant FHbp elicits higher protective antibody responses in infant rhesus macaques than a licensed serogroup B vaccine. mBio (2019) 10:e01231–19. doi: 10.1128/mBio.01231-19
255. Harris SL, Tan C, Andrew L, Hao L, Liberator PA, Absalon J, et al. The bivalent factor H binding protein meningococcal serogroup B vaccine elicits bactericidal antibodies against representative non-serogroup B meningococci. Vaccine (2018) 36:6867–74. doi: 10.1016/j.vaccine.2018.05.081
256. González-Miró M, Rodríguez-Noda LM, Fariñas-Medina M, Cedré-Marrero B, Madariaga-Zarza S, Zayas-Vignier C, et al. Bioengineered polyester beads co-displaying protein and carbohydrate-based antigens induce protective immunity against bacterial infection. Sci Rep (2018) 8:1888. doi: 10.1038/s41598-018-20205-7
257. Ricci S, Janulczyk R, Gerlini A, Braione V, Colomba L, Iannelli F, et al. The factor H-binding fragment of PspC as a vaccine antigen for the induction of protective humoral immunity against experimental pneumococcal sepsis. Vaccine (2011) 29:8241–9. doi: 10.1016/j.vaccine.2011.08.119
258. Glennie S, Gritzfeld JF, Pennington SH, Garner-Jones M, Coombes N, Hopkins MJ, et al. Modulation of nasopharyngeal innate defenses by viral coinfection predisposes individuals to experimental pneumococcal carriage. Mucosal Immunol (2016) 9:56–67. doi: 10.1038/mi.2015.35
259. Techawiwattanaboon T, Barnier-Quer C, Palaga T, Jacquet A, Collin N, Sangjun N, et al. Reduced renal colonization and enhanced protection by Leptospiral factor H binding proteins as a multisubunit vaccine against Leptospirosis in hamsters. Vaccines (Basel) (2019) 7:95. doi: 10.3390/vaccines7030095
260. Beernink PT, Shaughnessy J, Braga EM, Liu Q, Rice PA, Ram S, et al. A meningococcal factor H binding protein mutant that eliminates factor H binding enhances protective antibody responses to vaccination. J Immunol (2011) 186:3606–14. doi: 10.4049/jimmunol.1003470
261. Granoff DM, Welsch JA, Ram S. Binding of complement factor H (fH) to Neisseria meningitidis is specific for human fH and inhibits complement activation by rat and rabbit sera. Infect Immun (2009) 77:764–9. doi: 10.1128/iai.01191-08
262. Rogers EA, Abdunnur SV, McDowell JV, Marconi RT. Comparative analysis of the properties and ligand binding characteristics of CspZ, a factor H binding protein, derived from Borrelia burgdorferi isolates of human origin. Infect Immun (2009) 77:4396–405. doi: 10.1128/IAI.00393-09
263. Coleman AS, Yang X, Kumar M, Zhang X, Promnares K, Shroder D, et al. Borrelia burgdorferi complement regulator-acquiring surface protein 2 does not contribute to complement resistance or host infectivity. PloS One (2008) 3:3010e. doi: 10.1371/journal.pone.0003010
264. Marcinkiewicz AL, Lieknina I, Yang X, Lederman PL, Hart TM, Yates J, et al. The factor H-binding site of CspZ as a protective target against multistrain, tick-transmitted Lyme disease. Infect Immun (2020) 88:e00956–19. doi: 10.1128/iai.00956-19
265. Perez-Molina JA, Molina I. Chagas disease. Lancet (2018) 391:82–94. doi: 10.1016/s0140-6736(17)31612-4
266. Lidani KCF, Bavia L, Ambrosio AR, de Messias-Reason IJ. The complement system: a prey of Trypanosoma cruzi. Front Microbiol (2017) 8:607. doi: 10.3389/fmicb.2017.00607
267. Kipnis TL, David JR, Alper CA, Sher A, da Silva WD. Enzymatic treatment transforms trypomastigotes of Trypanosoma cruzi into activators of alternative complement pathway and potentiates their uptake by macrophages. Proc Natl Acad Sci USA (1981) 78:602–05. doi: 10.1073/pnas.78.1.602
268. Thu AM, Phyo AP, Landier J, Parker DM, Nosten FH. Combating multidrug-resistant Plasmodium falciparum malaria. FEBS J (2017) 284:2569–78. doi: 10.1111/febs.14127
269. Wen H, Vuitton L, Tuxun T, Li J, Vuitton DA, Zhang W, et al. Echinococcosis: advances in the 21st Century. Clin Microbiol Rev (2019) 32:e00075–18. doi: 10.1128/CMR.00075-18
270. Irigoin F, Laich A, Ferreira AM, Fernandez C, Sim RB, Diaz A. Resistance of the Echinococcus granulosus cyst wall to complement activation: analysis of the role of InsP6 deposits. Parasite Immunol (2008) 30:354–64. doi: 10.1111/j.1365-3024.2008.01034.x
271. Díaz A, Casaravilla C, Allen JE, Sim RB, Ferreira AM. Understanding the laminated layer of larval Echinococcus II: immunology. Trends Parasitol (2011) 27:264–73. doi: 10.1016/j.pt.2011.01.008
272. Lustigman S, Makepeace BL, Klei TR, Babayan SA, Hotez P, Abraham D, et al. Onchocerca volvulus: the road from basic biology to a vaccine. Trends Parasitol (2018) 34:64–79. doi: 10.1016/j.pt.2017.08.011
273. Akue JP, Eyang-Assengone ER, Dieki R. Loa loa infection detection using biomarkers: current perspectives. Res Rep Trop Med (2018) 9:43–8. doi: 10.2147/RRTM.S132380
274. Chippaux JP, Boussinesq M, Gardon J, Gardon-Wendel N, Ernould JC. Severe adverse reaction risks during mass treatment with ivermectin in loiasis-endemic areas. Parasitol Today (1996) 12:448–50. doi: 10.1016/0169-4758(96)40006-0
275. Rastogi M, Sharma N, Singh SK. Flavivirus NS1: a multifaceted enigmatic viral protein. Virol J (2016) 13:131. doi: 10.1186/s12985-016-0590-7
Keywords: complement system, alternative pathway, Factor H, Factor H binding proteins, complement evasion, pathogen
Citation: Moore SR, Menon SS, Cortes C and Ferreira VP (2021) Hijacking Factor H for Complement Immune Evasion. Front. Immunol. 12:602277. doi: 10.3389/fimmu.2021.602277
Received: 02 September 2020; Accepted: 15 January 2021;
Published: 25 February 2021.
Edited by:
Seppo Meri, University of Helsinki, FinlandReviewed by:
Peter Kraiczy, Goethe University Frankfurt, GermanyIlse Jongerius, Sanquin Research, Netherlands
Copyright © 2021 Moore, Menon, Cortes and Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Viviana P. Ferreira, Vml2aWFuYS5GZXJyZWlyYUB1dG9sZWRvLmVkdQ==
†These authors have contributed equally to this work