
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 26 February 2021
Sec. Immunological Tolerance and Regulation
Volume 12 - 2021 | https://doi.org/10.3389/fimmu.2021.597761
 Yun-Shiuan Olivia Hsu1,2
Yun-Shiuan Olivia Hsu1,2 Kun-Lin Lu2,3,4,5Yun Fu2,3,4,5Chuang-Wei Wang2,6
Kun-Lin Lu2,3,4,5Yun Fu2,3,4,5Chuang-Wei Wang2,6 Chun-Wei Lu2,3,4,7Yu-Fen Lin7,8Wen-Cheng Chang2,7,9Kun-Yun Yeh2,10Shuen-Iu Hung2,6
Chun-Wei Lu2,3,4,7Yu-Fen Lin7,8Wen-Cheng Chang2,7,9Kun-Yun Yeh2,10Shuen-Iu Hung2,6 Wen-Hung Chung2,3,4,5,6,7,11,12,13,14*
Wen-Hung Chung2,3,4,5,6,7,11,12,13,14* Chun-Bing Chen2,3,4,5,6,7,11,14,15*
Chun-Bing Chen2,3,4,5,6,7,11,14,15*The immunomodulatory effects of regulatory T cells (Tregs) and co-signaling receptors have gained much attention, as they help balance immunogenic and immunotolerant responses that may be disrupted in autoimmune and infectious diseases. Drug hypersensitivity has a myriad of manifestations, which ranges from the mild maculopapular exanthema to the severe Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms/drug-induced hypersensitivity syndrome (DRESS/DIHS). While studies have identified high-risk human leukocyte antigen (HLA) allotypes, the presence of the HLA allotype at risk is not sufficient to elicit drug hypersensitivity. Recent studies have suggested that insufficient regulation by Tregs may play a role in severe hypersensitivity reactions. Furthermore, immune checkpoint inhibitors, such as anti-CTLA-4 or anti-PD-1, in cancer treatment also induce hypersensitivity reactions including SJS/TEN and DRESS/DIHS. Taken together, mechanisms involving both Tregs as well as coinhibitory and costimulatory receptors may be crucial in the pathogenesis of drug hypersensitivity. In this review, we summarize the currently implicated roles of co-signaling receptors and Tregs in delayed-type drug hypersensitivity in the hope of identifying potential pharmacologic targets.
Hypersensitivity reactions refer to undesirable immune reactions that are exaggerated or inappropriate against an antigen or allergen. Generally, hypersensitivity reactions are classified into four types, which differ in pathogenesis, clinical manifestation, and prognosis (1). And when it comes to the treatment of these allergic reactions, type IV hypersensitivity remains the most challenging. Type IV hypersensitivity reaction is also called delayed-type hypersensitivity (DTH) because it reaches its peak 48 to 72 h after exposure to drugs, allergens, or toxins (2). DTH is mediated by direct cytotoxicity of CD8+ T cells or by the release of cytokines from CD4+ cells, which act through antigen-presenting cells (APC) such as macrophages and dendritic cells (DCs) to stimulate chronic inflammatory reactions. Therefore, unlike other types of hypersensitivity reactions that are antibody-dependent, DTH is majorly a T cell-mediated response, which accounts for a variety of drug eruptions including severe cutaneous adverse reactions (SCARs), and contact hypersensitivity (CHS) or allergic contact dermatitis (ACD) (3–5).
SCARs include syndromes such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms/drug-induced hypersensitivity syndrome (DRESS/DIHS) (5). SJS/TEN is a rare but life-threatening drug-induced cutaneous reaction characterized by extensive necrosis and detachment of epidermis and mucosal epithelium (6). SJS and TEN share the same clinical pattern, histopathologic features, and mechanisms; though SJS is defined by the involvement of less than 10% of total body surface area (TBSA) whereas TEN indicates detachment of more than 30% of TBSA (7, 8). On the other hand, DRESS/DIHS is also a potentially fatal drug-induced systemic hypersensitivity syndrome which mainly causes skin and internal organ damage (9–11). Patients with uncontrolled DRESS/DIHS or complications such as cytomegalovirus (CMV) reactivation are at high risk of death (12). Aside from drug eruptions, CHS or ACD is also an inflammatory disease of the skin that is primarily T cell-mediated (13). ACD results from exposure and sensitization of hosts to specific allergens, followed by subsequent exposure that initiates an inflammatory response, causing skin damage and inflammation (3).
Since the treatment of the DTH entity remains challenging, there is a need for physicians to look into the detailed interactions between immune cells and explore novel therapeutic targets (14–17). Fortunately, current evidence has demonstrated that both the dysregulations of regulatory T cells (Tregs) and the dysfunction of certain co-signaling axes are associated with the development of cutaneous inflammatory disease (18, 19). In this review, we summarize the established pathogenesis of clinically-relevant DTHs then focus on currently suggested concepts regarding how Tregs and various costimulatory and coinhibitory receptors regulate different components and cytokines in DTH in the hope of clarifying potential targets for future treatment.
Delayed-type hypersensitivities ranging from CHS to SCARs remain as major therapeutic challenges in clinical scenarios (20, 21). Upon exposure to a certain drug, not only does the drug itself but also its metabolites and drug-modified peptides are often present systemically (22, 23). Importantly, some of them could bind to human leukocyte antigen (HLA) and activate T cells, potentiating hypersensitivity (24, 25). Previous studies on drug hypersensitivity had identified several models explaining the pairwise associations between specific HLA class I alleles and the susceptibility to certain drug hypersensitivity, namely the hapten/pro-hapten model, the pharmacologic interaction model, and the altered peptide repertoire model, which have been thoroughly reviewed (26, 27).
After activating certain drug-specific T lymphocytes, cytotoxic molecules ranging from granulysin to granzyme B (GzmB) and a variety of proinflammatory cytokines including but not limited to tumor necrosis factor (TNF), interferon (IFN)-γ, and interleukin (IL)-2, are released, which collectively lead to unwanted inflammatory damages (28–31). In addition to cytotoxic T cells, activated natural killer (NK) cells, T helper 17 cells (Th17), and APCs also contribute to the release of IL-5, IL-6, IL-12, IL-15, IL-17, IL-18, further exacerbating the extensive collateral damages and result in SJS/TEN (27, 32). On the other hand, when the activation of certain drug-specific T helper 2 (Th2) lymphocytes occurs with the reactivation of human herpes virus (HHV)-6, HHV-7, Epstein–Barr virus (EBV), or CMV, these T cells are thought to further release IL-4, IL-5, and IL-13 which work together with eotaxin, thymus activation-regulated chemokine (TARC), and pro-inflammatory cytokines such as IFN-γ, TNF, IL-6, and IL-15, promoting systemic inflammation with eosinophilia and resulting in DRESS/DIHS (15, 33–36). Thanks to the high negative predictive values of genetic screenings for HLA, physicians could achieve early prevention of these fatal drug hypersensitivities prior to drug use (37–39). However, there are only a few HLA-typing (e.g., HLA-B*57:01 for abacavir, HLA-B*58:01 for allopurinol, and HLA-B*15:02 for carbamazepine) that have been recommended to prevent drug hypersensitivity in practice, and more attention is warranted to explore the applicability of this strategy in other dozens of drug-related DTH.
Besides the HLA alleles, the T cell receptor (TCR) repertoire also plays an important role in the pathogenesis of SCAR. Recently, we identified a public TCR from carbamazepine-induced SJS/TEN patients of Asians and Europeans. This observation may explain why patients of different ethnicities with different HLA alleles develop the same hypersensitivity. This public TCR shows drug-specificity and phenotype-specificity in an HLA allele-specific (HLA-B*15:02-favored) manner (40). Intriguingly, despite the presence of the culprit molecules systemically as well as the corresponding HLA type and TCR, many people remain unaffected while others develop severe drug hypersensitivity, implying that binding to drug-specific T cells per se is not sufficient to be pathogenic. The missing pieces to explain the unpredictability here could partially be attributed to both the activity of the Treg and the collateral co-signaling axes that largely determine the activation and effector functions of T cells, which had been extensively explored in fields targeting tumors as well as autoimmune diseases (41, 42).
Besides the pathophysiology of different drug hypersensitivity reactions identified previously, insufficient inhibitory mechanisms and/or enhanced costimulatory signals to T cells have also been gaining attention in the pathomechanism of SJS/TEN and DRESS/DIHS as outlined in Figures 1 and 2, respectively. Furthermore, since previous research on DTH has largely focused on CHS, reviewing costimulatory/coinhibitory signaling in CHS may help guide future drug hypersensitivity research; thus, the co-signaling pathways suggested in the pathogenesis of CHS are also depicted (Figure 3). Clarifying the immunological crosstalk may open the therapeutic window to further prevent and control potentially fatal drug hypersensitivity.
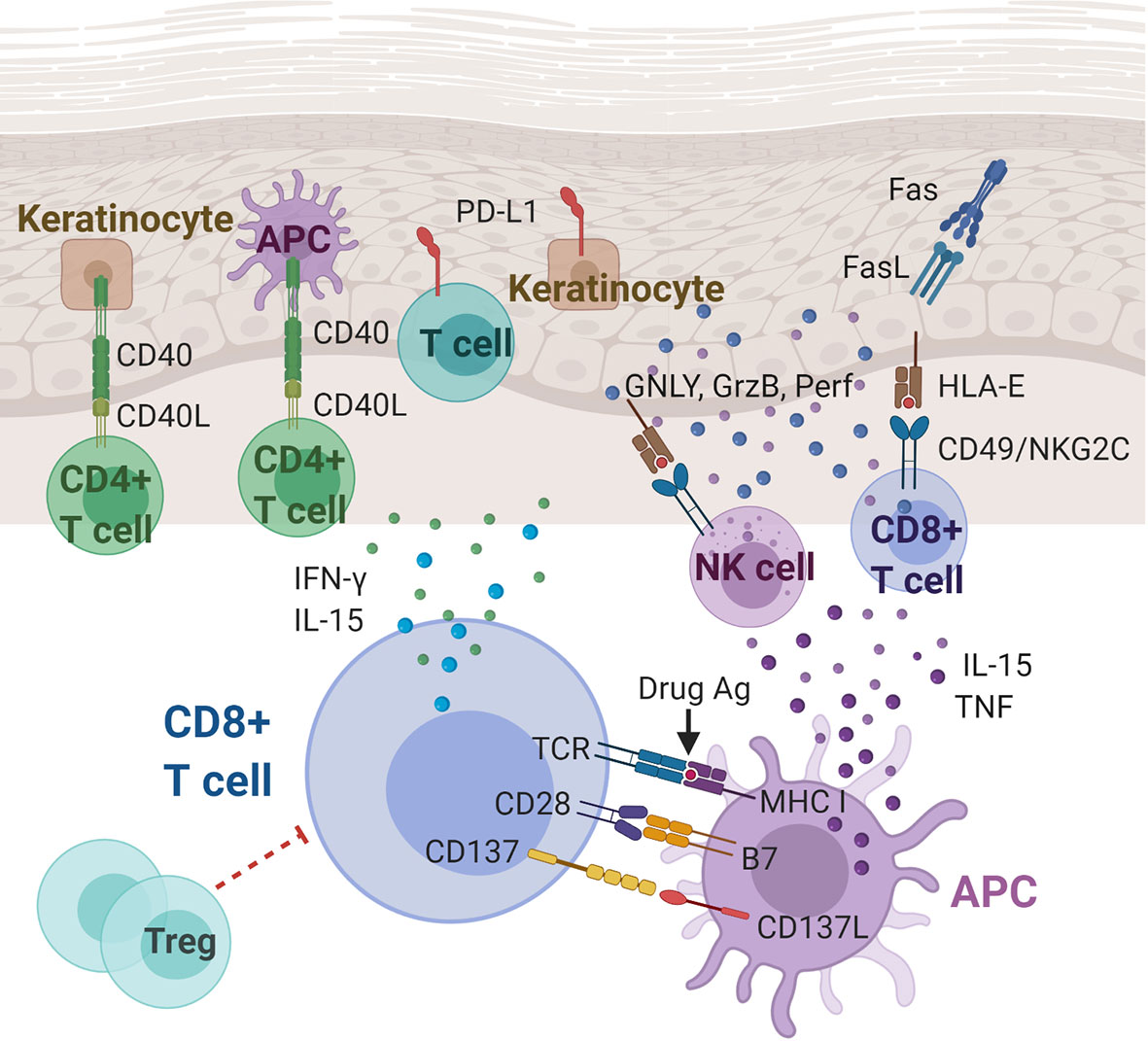
Figure 1 Immunoregulatory molecules and cytokines implicated in Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN). CD8+ cytotoxic T lymphocytes (CTLs) play a central role in the pathogenesis of SJS/TEN. Cytokines (including IL-15 and TNF) and costimulatory molecules of APCs can stimulate the CTLs, which in turn produce cytokines, including IFN-γ and IL-15. CTL and NK cell degranulation that induce keratinocyte apoptosis may be mediated, at least partially, by the interaction between CD49/NKG2C and HLA-E. Other players in SJS/TEN include Fas/FasL interactions, T cells and keratinocytes expressing PD-L1, and CD40/CD40L interactions at the dermal-epidermal junction. Ag, antigen; APC, antigen presenting cell; CD40(L), cluster of differentiation 40 (ligand); FasL, Fas ligand; GNLY, granulysin; GzmB, granzyme B; HLA-E, HLA class I histocompatibility antigen, alpha chain E; IFN-γ, interferon gamma; IL-15, Interleukin 15; MHC, major histocompatibility complex; NK cell, natural killer cell; Perf, perforin; PD-L1, programmed death ligand-1; TCR, T cell receptor; TNF, tumor necrosis factor; Treg: regulatory T cell.
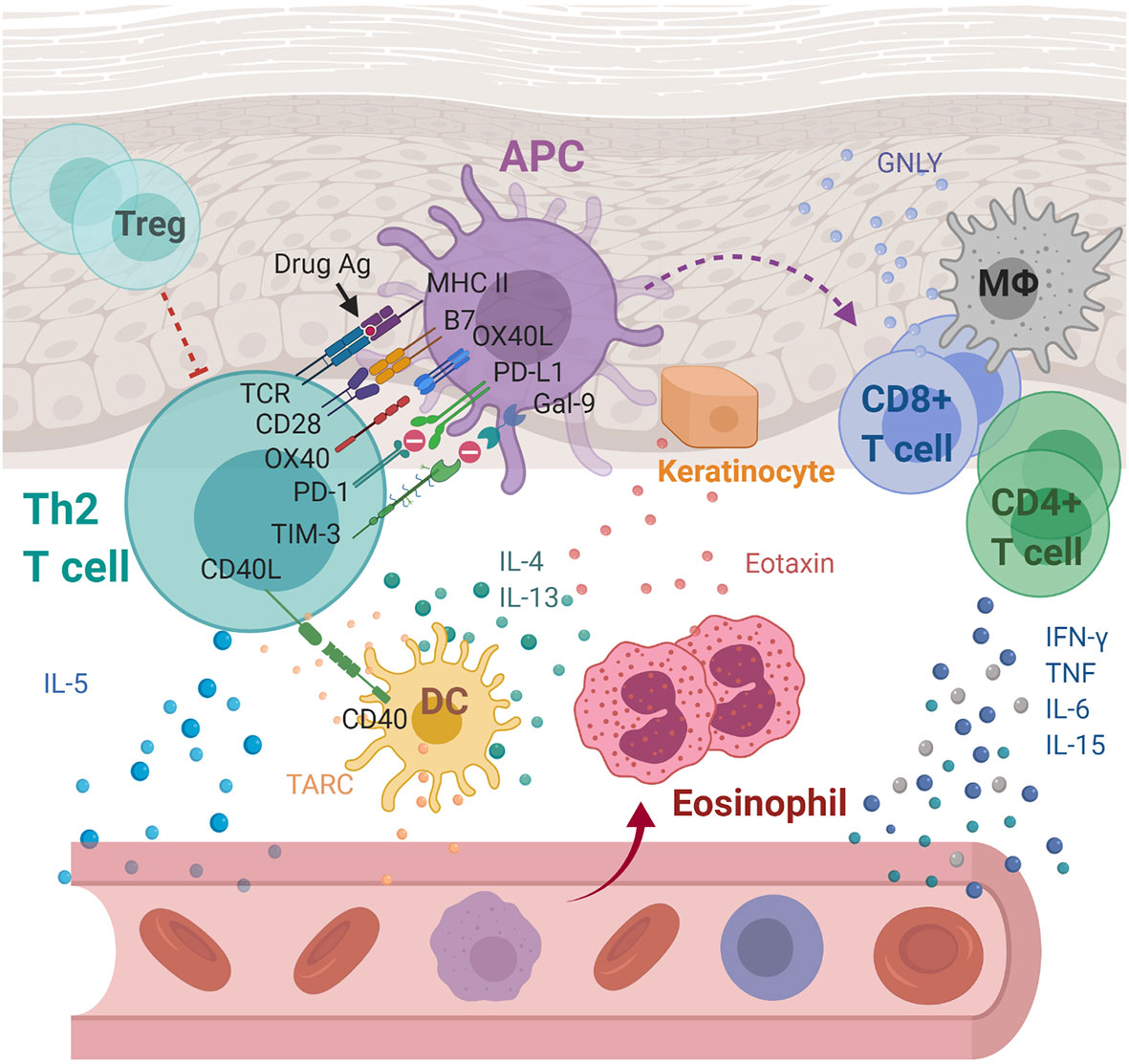
Figure 2 Immunoregulatory molecules and cytokines implicated in drug reaction with eosinophilia and systemic symptoms/drug-induced hypersensitivity syndrome (DRESS/DIHS) and maculopapular exanthema (MPE). Through cell-cell interaction, APCs are thought to activate drug-specific T cells with the aid of OX40 costimulation which prevent T cells from being inhibited by Tregs. Activated Th2 secretes cytokines including IL-4, IL-5, and IL-13, inducing eosinophilia. In addition, eotaxin and TARC produced by keratinocytes and DCs, respectively, promote the local accumulation of harmful eosinophils. Together with the elevated levels of pro-inflammatory cytokines, including IFN-γ, TNF, IL-6, and IL-15, they cause systemic inflammation characterized as DRESS/DIHS. In the case of MPE, the augmented immune responses through the CD40 axis along with the compromised inhibitory mechanisms of both PD-1 and TIM-3 axes were further identified. APC, antigen presenting cell; DC, dendritic cells; Gal-9, galectin-9; GNLY, granulysin; IFN-γ, interferon gamma; IL-4, interleukin 4; IL-5, interleukin 5; IL-6, interleukin 6; IL-13, interleukin 13; IL-15, interleukin 15; MΦ, macrophage; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand-1; TARC, thymus activation-regulated chemokine; TCR, T cell receptor; Th2, T helper 2 cell; TIM-3, T-cell immunoglobulin mucin-3; TNF, tumor necrosis factor; Treg, Regulatory T cell.
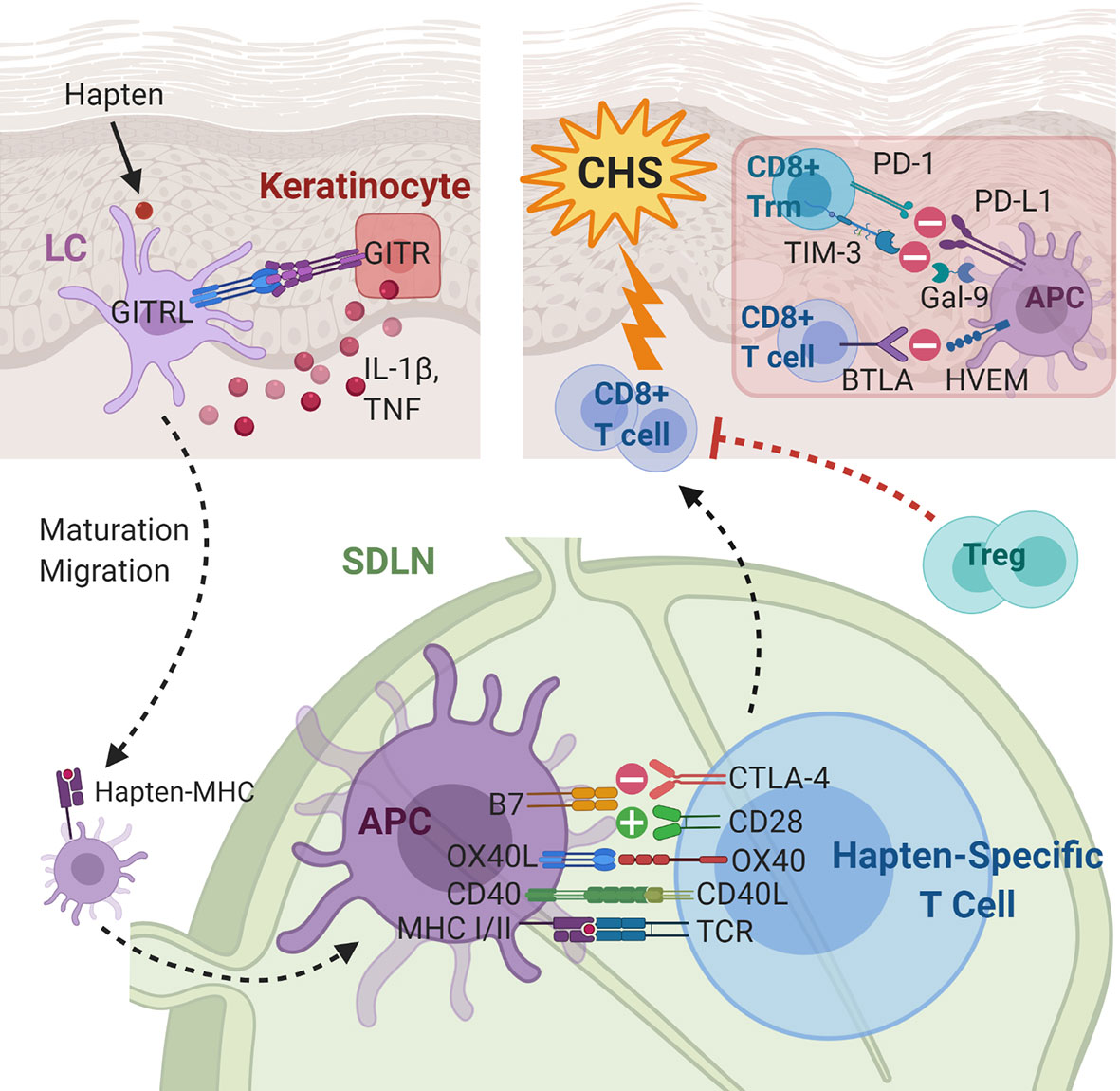
Figure 3 Immunoregulatory molecules and cytokines implicated in contact hypersensitivity (CHS). During sensitization, the skin-penetrating hapten is processed by Langerhans cells (LCs) or dermal dendritic cells. GITR-GTRL interactions between keratinocytes and LCs enhance keratinocyte secretion of cytokines, which aid maturation and migration of LCs to the skin-draining lymph node. In the SDLN, in addition to hapten presentation, several costimulatory and coinhibitory interactions, e.g., CD40-CD40L, OX40L-OX40, B7-CD28, and B7-CTLA-4, between the APC and the naïve T cell take place. The hapten-specific effector T cells, predominantly CD8+ cytotoxic T cells, infiltrate the skin and elicit the CHS response upon hapten re-exposure. The presence of Tregs and several coinhibitory receptors on CD8+ T cells and CD8+ Trm cells are associated with decreased CHS response. APC, antigen presenting cell; BTLA, B- and T-lymphocyte attenuator; CD40L, CD40 ligand; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; Gal-9, galectin-9; GITR(L), glucocorticoid induced TNF receptor (ligand); HEVM, herpes virus entry mediator; IL-1β, interleukin-1 beta; LC, Langerhans cell; MHC, major histocompatibility complex; OX40L, OX40 ligand; PD-1, programmed cell death protein 1; PD-L1, programmed death ligand-1; SDLN, skin-draining lymph nodes; TCR, T cell receptor; TIM-3, T-cell immunoglobulin mucin-3; TNF, tumor necrosis factor; Treg, regulatory T cell; Trm, tissue-resident memory T cell.
Different HLA alleles represent important risk factors for drug hypersensitivity by specific drugs in patients of specific genetic ancestry. However, evidence thus far seems to suggest that the commonly discussed variations of the HLA genotypes are not sufficient to fully predict susceptibility to DTH (43–47). It is known that alongside the TCR/peptide-MHC interaction, “signal 2” is crucial for proper activation of T cells by antigen-presenting cells. Aside from the prototypic costimulatory receptor, CD28, that is involved in this process (48), various costimulatory receptors have also been demonstrated to facilitate the immune responses in autoimmune diseases, cancer, and infectious diseases (49–51). Costimulatory receptors implicated in DTH are summarized in Table 1.
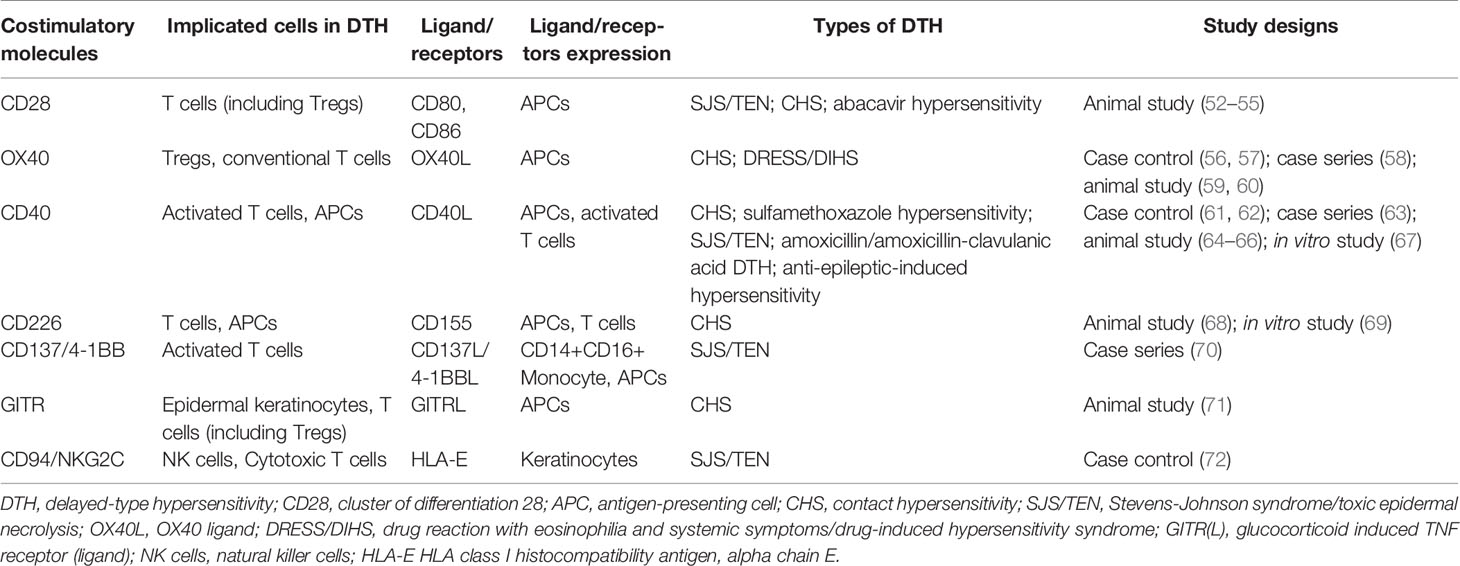
Table 1 Costimulatory molecules involved in delayed-type hypersensitivity.
The costimulatory molecules B7-1 (CD80) and B7-2 (CD86) on APCs interact with their cognate receptor CD28 on T cells to augment and sustain a T cell response (73). CD28 costimulation promotes T cell proliferation, cytokine production, and T cell survival (74). In a transgenic mouse model for HLA-B*57:01–linked abacavir hypersensitivity, CD80 on DCs are upregulated upon CD4+ T cell depletion (likely including Tregs), resulting in an adverse immune response (52, 53). To further assess the effect of blocking this pathway on DTH, mice with antibodies targeting CD28 or CD80 inhibited the accumulation of reactive CD8+PD-1+ T cells in the lymph nodes, dampening the immune response to abacavir. In addition, CHS to dinitrofluorobenzene (DNFB) and oxazolone is significantly decreased in CD28 −/− mutant mice (54). CD28 antagonist has also been suggested prevent Aldara-induced skin inflammation in non-human primates, as shown by the inhibition of T cell and macrophage infiltration at the epidermal interface (55). Since studies on the role of CD28 in DTH have largely been animal-based, future studies to investigate the role of CD28 in DTH in humans are warranted before assessing the applicability of CD28 manipulation for therapeutic purposes.
The costimulatory receptor OX40 (also known as CD134) is a member of the TNF receptor superfamily (TNFRSF) that is expressed on both activated CD4+ and CD8+ T cells, neutrophils, and NK cells (75). OX40 has important costimulatory functions in the activation, survival, and expansion of both CD4+ and CD8+ T cells as demonstrated in animal models of autoimmune disease, infectious disease, and cancer (76). Additionally, activation of T cells by the OX40 ligand (OX40L) makes them less responsive to the inhibitory signals from Tregs (77). One study demonstrated that mice lacking OX40L exhibited an impaired CHS response, due to defects in T cell priming and cytokine production (59). In another murine model of CHS, OX40L-deficient mice displayed a significant reduction in both hapten-induced ear swelling and hapten-specific T cell response. Conversely, these responses were markedly increased in mice with OX40L overexpression (60). In a comparison between eight patients of acute stage DRESS/DIHS and seven healthy controls, OX40 was found to be significantly upregulated on CD4+ T cells of DRESS/DIHS patients (56). In their recent follow-up study of 12 DRESS/DIHS patients, Miyagawa et al. have found that OX40L was also preferentially expressed on their peripheral blood mononuclear cells (PBMCs). Moreover, the percentage of OX40-expressing CD4+ T cells correlated with parameters observed in Th2-type immune responses (57). Additionally, during slow desensitization of five patients with previous imatinib-induced DTH, including maculopapular exanthema (MPE) and DRESS/DIHS, decreased imatinib-induced CD4+CD25+OX40+ T-cell percentages were observed (58), suggesting that decreased OX40+ drug-specific T cells may be associated with immune tolerance. Since co-expression of OX40 and CD25 after 48 h upon antigen stimulation has been described as a marker for antigen-specific CD4+ T cells (78), this result hints at the important role of OX40 in the T cell hypersensitivity response. Since studies on OX40 have largely focused on CHS in mice and DRESS/DIHS in humans, a comprehensive understanding of the role of OX40/OX40L in DTH may require future investigation of other types of DTH and the interactions between OX40 and other competing players in the immune response. On another note, though the potential therapeutic application of the OX40–OX40L interaction has not been explored in DTH, it has already been considered in autoimmune and cancer treatments (79).
The CD40/CD40 ligand (CD40L or CD154) costimulatory axis also belongs to the tumor necrosis factor receptor superfamily (TNFRSF) and amplifies the immune response and promotes inflammation. While both are expressed on the surface of a variety of cell types, CD40L expression mostly requires induction whereas CD40 expression is often constitutive. Cells expressing CD40L include, but are not limited to, activated T cells, DCs, NK cells, platelets, and non-immune cells. Similarly, CD40 is expressed on B cells, T cells, DCs, and many other cell types (80). An in vitro study of T-cell-mediated hypersensitivity to sulfamethoxazole (SMX) showed increased CD40 expression with DC surfaces exposed to SMX and its metabolite, nitroso SMX (67). CD40L blockade inhibited nitroso SMX-induced T cell activation in mice. Additionally, CD40 stimulation enhanced the drug-specific response. In mice, blockade of the CD40-CD40L pathway was associated with defective CHS response, possibly due to impaired migration of antigen-bearing DCs from the skin to draining lymph nodes, while injection of the agonist anti-CD40 monoclonal antibody “corrected” the CHS response (64, 65). However, CD40-CD40L interactions may not be mandatory for the development of the effector CD8+ or the regulatory CD4+ T cells during DNFB sensitization for CHS in mice (66). In order to study the role of CD40 in human diseases, the cellular infiltrates of the skin of eight patients with erythema multiforme and six with SJS/TEN had been analyzed. While CD40+ cells were similarly represented in erythema multiforme and SJS/TEN, CD40L+CD4+ T cells which may bind to CD40 on APCs were strongly represented in the perivascular and subjunctional dermis of SJS/TEN specimens (63). This study also revealed CD40, Fas, and Fas ligand expressions on keratinocytes in SJS/TEN patients. Recently, the potential of using CD40L to detect activated drug-specific T cells in DTH to antibiotics and anti-epileptics has been demonstrated in case control studies of small sample sizes, but its overall accuracy and application in SCARs remain to be further explored. For instance, among 14 patients clinically diagnosed with drug eruptions, only 8 of their causative drug-activated PBMCs were tested positive for CD40L (61, 62).
The costimulatory receptor CD226 (DNAM-1) is a glycoprotein expressed on the majority of NK cells, T cells, monocytes and platelets, and a subset of B lymphocytes. CD226 competes with the coinhibitory T cell immunoglobulin and ITIM domain (TIGIT) for the same ligands, mediating positive stimulatory signaling and inducing NK and T cell‐mediated cytotoxicity (81, 82). Ligands for CD226 include CD155 (poliovirus receptor, PVR) and CD112 (nectin-2 or poliovirus receptor-related 2, PVRL2), which are expressed on epithelial cells, endothelial cells, APCs, and tumor cells; thus, the interaction between CD226 and its ligands is more often discussed in the context of tumor immunity (83, 84). A previous in vitro study had suggested that CD226 on CD4+ naive T cells mediated an activating signal for T helper 1 cell (Th1)/Th17 differentiation as a result of ligation with CD155 (69). Somewhat surprisingly, one murine study has demonstrated that CD155-mediated signaling in CD4+ T cells triggered by the interaction with CD226 on APCs was involved in Th1 development and CHS (68). CD155, normally regarded as the “ligand” in this costimulatory couple, was suggested to serve as the “receptor” when present on T cells. In the same study, anti-CD155 monoclonal antibody administration was able to dampen the CHS response. Whether the same manner of CD226/CD155 interaction is involved in human CHS has yet to be demonstrated.
Another costimulatory receptor 4-1BB (CD137) is expressed on a vast array of cell types within the hematopoietic system, including both activated CD4+ and CD8+ T cells, DCs, B cells, monocytes, NK cells, neutrophils, and mast cells (85). The 4-1BB ligand (4-1BBL or CD137L) is expressed on most leukocytes, including APCs, and some non-immune cells. Interestingly, 4-1BBL can also transmit signals into the APC on which it is expressed (reverse signaling) and strengthen type 1, cell-mediated immune responses (86–88). The ability to potentiate effector responses by 4-1BB signaling in activated T cells makes 4-1BB an appealing target for cancer immunotherapy (89, 90). In one study, CD14+CD16+ cells of the monocyte lineage co-expressing CD80, CD86, and CD137L were found in the skin of all 11 SJS/TEN patients while interaction with CD8+CD137+ cells was also demonstrated on immunostaining (70). The interaction between cells bearing CD137 and cells bearing CD137L was suggested to contribute to epidermal damage most probably by enhancing the cytotoxicity of CD8+ T cell. Definitive conclusions cannot be drawn regarding CD137’s role in DTH due to limited evidence; further supporting studies on this costimulatory receptor are still needed.
The costimulatory receptor GITR (glucocorticoid-induced TNF receptor, CD357) is a member of the TNFRSF that is constitutively expressed at high levels on Tregs, at lower levels on conventional T cells and innate immune cells, and on non-immune cells, e.g., keratinocytes (91, 92). The GITR ligand (GITRL) is predominantly expressed on activated APCs and endothelial cells (91, 93). Some functions of GITR include lowering the threshold of CD8+ T cell activation by CD28 and abrogating Treg-mediated suppression of T cells (94, 95). Studies on agonistic GITR in the context of cancer immunotherapy similarly demonstrated its dual ability to enhance CD8+ T cell proliferation and cytokine production while dampening Treg suppressive function (96, 97). In the CHS mouse model, the anti-GITRL monoclonal antibody inhibited hapten-specific CD8+ T cell activation and CHS. However, Tregs and plasmacytoid DCs, known to express high levels of GITR and GITRL, respectively, were not the main players in GITRL-mediated CHS. Rather, their results suggested that the binding of GITRL on Langerhans cells to the GITR on epidermal keratinocytes induced the expression of proinflammatory cytokines by the keratinocytes and the migration of Langerhans cells to the draining lymph nodes to initiate the CHS response (71). Since similar results have not been reported by other studies, this mechanism in CHS mandates further investigation.
CD94 expressed on NK cells can form heterodimers with NKG2A (CD94/NKG2A) or with NKG2C (CD94/NKG2C). Binding of the heterodimer CD94/NKG2C to its ligand, HLA‐E, delivers an activating signal in NK cells (98). Interestingly, CD94/NKG2C is also expressed by highly differentiated CD8+ effector T cells and co-stimulates TCR‐mediated cytotoxicity (99). Keratinocytes from affected skin in SJS/TEN express HLA-E, which sensitizes them to killing by CD94/NKG2C-expressing cytotoxic T lymphocytes (CTLs) and NK cells. Moreover, in a case control study of SJS/TEN patients, activated blister T and NK cells expressing CD94/NKG2C degranulate in response to HLA-E+ cells in an NKG2C-dependent manner (72). And the cytolytic granules secreted by CTLs and NK cells have been previously demonstrated to include perforin, GzmB, soluble Fas ligand, and granulysin, which are important for keratinocyte killing of patients with SJS/TEN (30). Thus, the interaction between CD94/NKG2C on CTLs and NK cells and HLA-E on keratinocytes seems to contribute to the pathogenesis of SJS/TEN.
Since costimulatory signals boost the activation of T cells and are suggested to play a role in drug hypersensitivity, developing targeting therapies against them may be a plausible strategy against SCARs. Contrariwise, when it comes to the immunological regulations that hold back unwanted immune responses which we would like to augment, it is indispensable to discuss Treg and its multifaceted immunosuppressive abilities that could largely change the landscape of drug hypersensitivity (100, 101).
Activation of the immune system is crucial for fending off foreign pathogens or annihilating endogenous malignancies. However, autoimmune diseases and hypersensitivity reactions are important illustrations of exaggerated immune responses to self and non-self antigens, respectively, that can be life-threatening. Maintaining immunologic self-tolerance and control requires a special subset of CD4+ T cells, the Tregs (102, 103). Tregs regulate immune system through multiple mechanisms, including the release of inhibitory cytokines, the initiation of cytolysis, and the modulation of DC function (104). Regulation through checkpoint receptors cytotoxic T lymphocyte-associated antigen-4 (CTLA-4) and programmed cell death 1 (PD-1) are also pivotal to the Treg function. By modulating CD4+ T cells, CD8+ T cells, B cells, and DCs, Tregs play a crucial role in the homeostasis of the immune response.
The constitutively expressed forkhead box protein 3 (FOXP3) is the lineage-defining transcription factor indispensable for Treg development and suppressive function (105, 106). Mutations in FOXP3 renders the scurfy transgenic mice and patients with IPEX (immunodysregulation polyendocrinopathy enteropathy X-linked) syndrome susceptible to spontaneous inflammation in different tissues, including the skin, and traits characteristic of connective tissue diseases (107–110). Recently, mapping regulation of gene expression in Tregs has revealed dysregulation of key Treg pathways in immune diseases, including many that display skin inflammation, providing hints to future drug targets for treatment (111).
More Tregs than non-Tregs accumulate in the skin during cutaneous immune response in mice. Furthermore, mRNA expression profiles of cytokines in Tregs contain significantly higher amounts of the inhibitory Il10, Tgfb1, and Ctla4 transcripts (112). Depletion of FOXP3+ Tregs in the PBMCs of healthy human donors allows the detection of CD4+ and CD8+ T cell responses to drugs and haptenic chemical, suggesting Treg’s canonical suppressive role (113). Not surprisingly, the reintroduction of Tregs blocks drug activation of naïve T cells in a cell concentration-dependent manner (114). In fact, the dysfunction of Tregs does contribute to the pathogenesis of drug hypersensitivity in clinical scenarios. For instance, the incidence and severity of drug hypersensitivity were increased in human immunodeficiency virus (115)-infected patients (116–118), whose CD4 counts and CD4/CD8 ratios decreased dramatically (118). Moreover, it was also observed that cancer patients receiving anti-CTLA4 and/or anti-PD-1 immunotherapy suffered from more drug hypersensitivity adverse events (119).
Treg dysfunction has been suggested in the pathogenesis of SJS/TEN. In mice, Tregs prevent experimentally induced epidermal injury mimicking TEN (120). In humans, non-responders to carbamazepine re-exposure have a statistically significant expansion of Tregs compared to responders (121). In skin lesions of HIV patients with TEN, the decrease in the number of skin-directed CD4+ cells and the increase in the CD8+/CD4+ cell ratio likely contribute to an increased risk of developing drug reactions because of the loss of skin-protective regulatory T cells (116, 122). In another patient using the PD-1 inhibitor nivolumab, sequential mRNA expression of FOXP3 and the CD4+/CD8+ ratio reached its nadir at the onset of TEN in peripheral blood cells (123). Additionally, the percentage of Tregs in peripheral blood continued to drop in the course of TEN (123). As expected, Treg-mediated suppression of drug-specific T-cells in humans is seen with the addition of Tregs in a concentration-dependent manner (124). A comparison of cell composition in the skin of patients with erythema multiforme and SJS/TEN shows that Tregs are less abundant in the latter (125). Qualitative deficits of Tregs also seem to contribute to the pathogenesis of SJS/TEN. Circulating Tregs of SJS/TEN patients display impaired suppressor function that can be restored after clinical resolution (126). Notably, our recent study has also found that the TNF-α antagonist, etanercept, successfully relieves SJS/TEN symptoms with an increase in frequencies of CD4+CD25+FOXP3+ Tregs after treatment (127).
Patients with DRESS/DIHS at the acute stage show significantly increased frequencies of Tregs in total CD4+ T cells compared with healthy controls in blood, which is not observed in TEN or MPE (126). A larger fraction of Tregs is likely to have a more potent ability to migrate into the skin (126). Indeed, increased FOXP3+ T cells in skin lesions of DRESS/DIHS were observed and appear significantly higher when compared to those in SJS/TEN (32, 128). Evidence suggests that functional Tregs expand at the acute stage of DRESS/DIHS and contract with the resolution of the disease while becoming functionally impaired (126). Intriguingly, a shift from the Treg response to the Th17 response due to CD16+ monocytes producing IL-6 can partly explain the observed decrease in Tregs upon resolution (129). Furthermore, Tregs seem important in ameliorating inflammatory responses while preventing the subsequent development of CMV infection or autoimmune diseases in DRESS/DIHS (11).
The modulatory role of Treg has also been demonstrated in CHS. In vivo Treg expansion in mice produces prolonged CHS suppression manifesting as a sustained reduction of hapten-specific CD8+ T cells and a decrease in effector cell influx in inflamed tissue (130). CD4+ T cells isolated from the peripheral blood of healthy nonallergic humans show a limited capacity to proliferate in response to nickel in vitro, but responsiveness is strongly augmented when cells are depleted of Tregs (131).
Although insufficient regulation by Tregs, whether quantitative or qualitative, appears relevant in DTH, the exact mechanisms leading to the pathogenesis of these diseases are still nascent. In the next sections, we attempt to highlight the currently proposed roles of Treg in DTH.
Tregs can regulate participants of the immune system to achieve tolerance through a variety of mechanisms. These mechanisms include suppression by inhibitory cytokines, suppression by metabolic interruption, suppression by checkpoint receptor modulation of DC maturation or function, and suppression by cytolysis (Figure 4) (132). Aside from these mechanisms, a novel mechanism involving enhanced binding of Tregs to DC to deplete peptide-MHC II from the surface has been proposed recently (133).
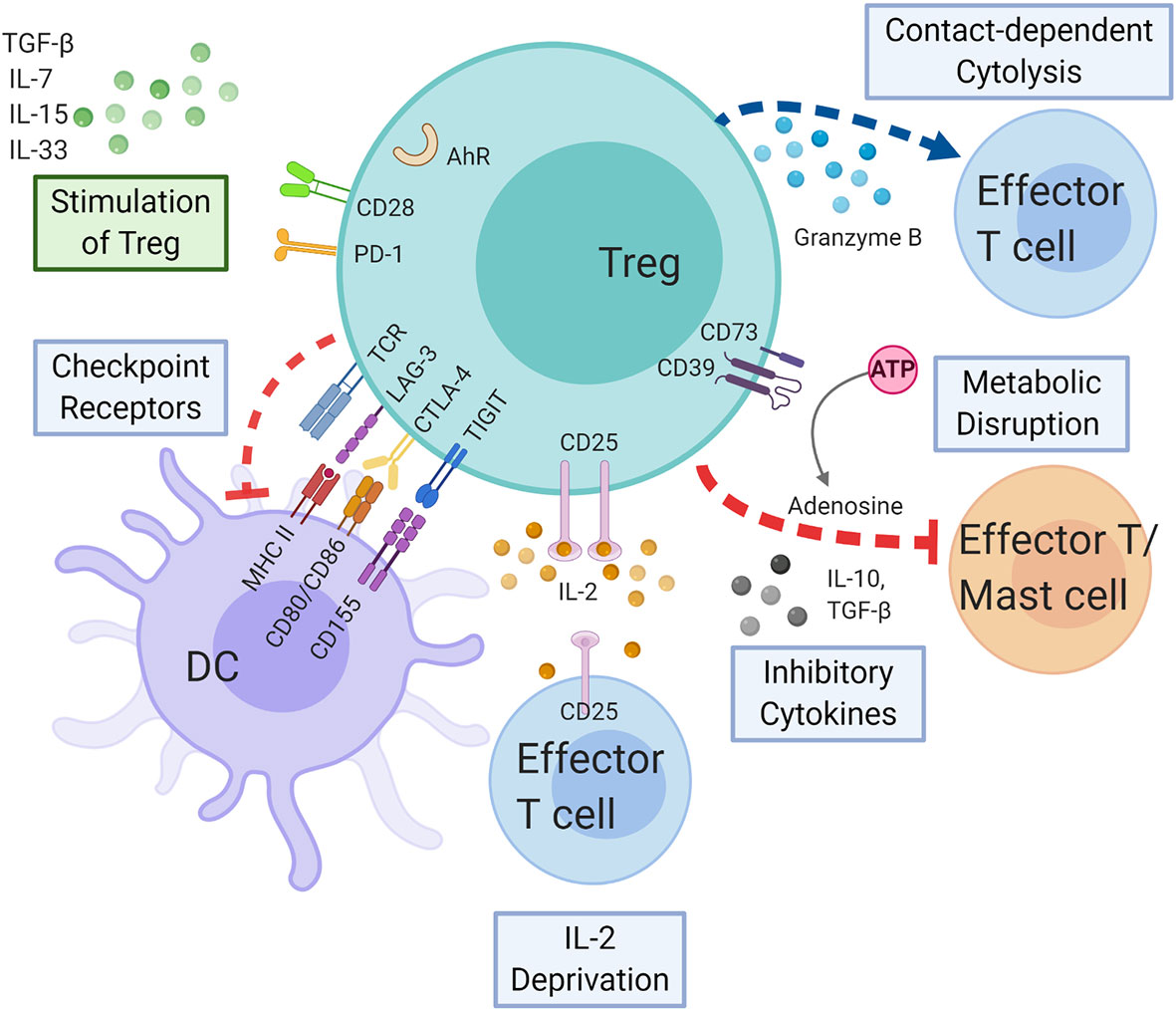
Figure 4 Regulatory T cells in delayed-type hypersensitivity. Tregs may exert immunosuppressive effect on APCs, effector T cells, and mast cells by the following mechanisms: coinhibitory receptors binding to cognate molecules on dendritic cells, secretion of inhibitory cytokines, e.g., IL-10 and TGF-β, metabolic disruption by depriving IL-2 binding and increasing adenosine binding to effector T cells, and contact-dependent cytolysis by granzyme B secretion. Likewise, cytokines and immunoregulatory molecules also modulate the Treg function. AHR, aryl hydrocarbon receptor; ATP, adenosine triphosphate; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; DC, dendritic cell; IL-2, interleukin 2; IL-7, interleukin 7; IL-10, interleukin 10; IL-15, interleukin 15; IL-33, interleukin 33; LAG-3, lymphocyte-activation gene 3; MHC, major histocompatibility complex; PD-1, programmed cell death protein 1; TCR, T cell receptor; TGF-β, transforming growth factor beta; TIGIT, T cell immunoreceptor with Ig and ITIM domains; Treg, regulatory T cell.
The suppression of various cells, including CD4+ T cells, by Tregs is mediated through inhibitory cytokines including TGF-β, IL-10, and IL-35 (134–137). An early study of mice with disrupted IL-10 gene mounted an exaggerated CHS response (138). Another murine model has demonstrated IL-10 production by Treg upon stimulation and that addition of anti-IL-10 antibodies abrogated the suppressive effects of Treg in CHS. Moreover, CD4+CD25+ T cells isolated from IL-10−/− mice were unable to suppress the immune response (139). In a study of humans with drug hypersensitivity, IL-10 was increased in successful drug desensitization regardless of the hypersensitivity reaction type (140). In patients with MPE, the frequency of IL‐10–producing Tregs was significantly lower compared to controls in both acute and resolution phases (141). Interestingly, the source of IL-10 appears to be important to have suppressive functions. In mice, CD8+ T cell‐derived IL‐10 does not contribute significantly to the resolution of CHS responses (142). One study treated CHS mice with induced Tregs (iTregs) that had been exposed to TGF-β1 small interfering RNA (siRNA) or control siRNA. Mast cell density and inflammatory cytokines messenger RNA expression suppression was compromised in the TGF-β1 siRNA-treated group, suggesting that TGF-β1 derived from iTreg cells might contribute to iTreg–mediated reversal of established CHS (143).
CD39 is essential for Treg immunosuppressive activity via the CD39/adenosinergic axis since adenosine generated by CD39 and CD73 on Tregs can bind the A2A adenosine receptors on effector T cells and enhance intracellular cAMP levels to suppress their function (144, 145). A murine CHS model has found that CD39 on Tregs depleted ATP in the extracellular environment, which downregulated ATP-induced shedding of CD62L on CD8+ T cells. This in effect interfered with the trafficking of CD8+ T cells in and out of skin-draining lymph nodes and dampened the immune response. Therefore, Tregs likely suppress CHS through a CD39, adenosine-dependent mechanism in vivo (146, 147). Higher frequencies of CD39+ Tregs have also been correlated with lower production of IFN-γ, an important cytokine detected in SJS/TEN blisters (121, 148). Furthermore, individuals originally unresponsive to carbamazepine re-exposure converted to an IFN-γ-producing state after treatment with the CD39 inhibitor, POM-1 (121). These data suggest that targeting the CD39/adenosinergic axis has therapeutic potential in SJS/TEN and CHS.
Interestingly, Tregs consistently express high levels of IL-2 receptor α chain, CD25, which implies a higher affinity to IL-2 than that of conventional T cells. By competing for IL-2, Tregs deplete IL-2 from other T cells, causing cytokine deprivation-induced apoptosis in these cells; however, this concept may require further validation (149, 150).
Downstream signaling of checkpoint receptors or coinhibitory receptors on Tregs has been shown to converge to the same pathway leading to Treg stability. Furthermore, the coinhibitory receptors may regulate immune and non-immune cells that express their corresponding ligands (151). CTLA-4 in conventional T cells rises only after activation but is constitutively expressed on Tregs (144). Treg-specific CTLA-4 deficiency shows impaired Treg suppressive function, especially in in vitro and in vivo downregulation of CD80 and CD86 expression on dendritic cells (152). Furthermore, CTLA-4 has been shown to bind to CD80/CD86 with higher affinity than its opponent CD28, thereby depriving conventional T cells of costimulatory signaling through CD28 (153). Tregs may also secrete soluble CTLA-4 that block CD80/CD86 or deplete CD80/CD86 from the APC surface by transendocytosis (154, 155). Additionally, Tregs use CTLA-4 to induce the co-inhibitory molecule PD-L2 (one of the ligand for PD-1) expression on dendritic cells (156).
PD-1 is mainly expressed on activated CD4+ T cells, activated CD8+ T cells, and B cells in the periphery. Just like CTLA-4, PD-1 delivers a negative signal when interacting with its ligands (144). Both PD-1 and PD-1 ligand 1 (PD-L1) are expressed on Tregs under resting and activated conditions (157). As for chronic viral infection, either PD-1 blockade on chronic Tregs or PD-L1 deficiency on CD8+ T cells dramatically diminishes the suppression of T cell immune response, demonstrating the importance of interaction between PD-1 on Tregs and PD-L1 on CD8+ T cells (158). Increasing cases of SJS/TEN secondary to immune checkpoint inhibitors of CTLA-4 and PD-1/PD-L1 suggests checkpoint receptors’ roles in suppressing DTH (159).
The coinhibitory receptor LAG-3 (lymphocyte activation gene-3) is a CD4-related protein that has been demonstrated to modulate both the in vitro and in vivo suppressive function of induced Tregs (160). MHC II cross-linking by LAG-3 on Tregs induces an inhibitory signaling pathway to suppress dendritic cell maturation and immunostimulatory capacity on conventional T cells (161). Of note, rather opposing functions of LAG-3 have been postulated in a study of DTH in primates: depleting LAG-3+ T cells may eliminate effector T cells to block inflammation but prevent Treg inhibition of immune responses in the draining lymph node (162). Whether LAG-3 plays a role in modulating Tregs’ suppressive function during DTH requires further elucidation.
TIGIT is another coinhibitory receptor that has been shown to contribute to Treg’s suppressive function (163). One of its corresponding ligands CD155 is expressed by dendritic cells and activated CD4+ effector T cells (68, 164). Though both TIGIT and its costimulatory counterpart CD226 bind to CD155, TIGIT binds its ligands with much higher affinity (164, 165). In a murine model, Treg has been suggested to inhibit keyhole limpet hemocyanin-induced delayed-type hypersensitivity reactions by inducing tolerogenic DC through the TIGIT-CD155 interaction (166), which we will discuss later in detail.
Tregs can also produce a serine protease called GzmB, which can induce apoptosis in effector T cells in a contact-dependent, perforin-independent manner (167). In a murine CHS model, single-gene profiling identified an IL-10/GzmB-expressing Treg population in the draining lymph node with skin-tropic chemokine receptors, which allowed retention of Tregs in inflamed skin and downregulation of the cutaneous immune response (168). However, GzmB is more widely known for its pathogenic role in DTH. The levels of GzmB, along with TNF-α, perforin, and Fas ligand, are markedly increased in PBMCs and blister fluids of SJS/TEN patients (28). GzmB is released to the cytosol of target cells upon cytotoxic T lymphocyte degranulation in SJS/TEN and promotes apoptosis either by direct cleavage of caspase-3 or by increasing the permeability of the mitochondrial outer membrane (intrinsic pathway) (15). Therefore, evidence suggests that GzmB molecules could either regulate or enhance DTH responses depending on their source, such that more caution should be paid when interpreting their role in DTH.
While FOXP3+ regulatory T cells have been described as the most physiologically relevant regulatory cell type (144), how other cells and molecules of the immune system regulate Treg development and function to maintain immune homeostasis may be equally important (Figure 4). Besides direct cell-cell contact through receptor-ligand interaction, as seen between dendritic cells and T cells, indirect interaction through cytokines also plays important roles in tolerogenic responses of Tregs (169, 170).
The CD28-CD80/CD86 interaction on T cells provides a second signal alongside the TCR ligation (signal 1) critical for regulatory T cell survival and the maintenance of immune homeostasis (48). In mice, Treg-specific deletion of CD28 demonstrates lower levels of CTLA-4, PD-1, and chemokine receptor 6 (CCR6) and systemic autoimmunity characterized by prominent skin inflammation (171). In a follow-up study, the upregulation of CCR6 upon CD28 costimulation suggests its role in Treg skin-homing in DTH (172). Interestingly, the percentage of CCR6+ cells among CD4+ T cells has been observed to be greater in SJS/TEN skin than in DRESS/DIHS skin (32).
IL-2 is required for sustaining the Treg population to maintain immune homeostasis and tolerance. Since Tregs scarcely produce IL-2, they are highly dependent on exogenous (paracrine) IL-2 for survival (173). As mentioned previously, Tregs express consistently high levels CD25 which serves as a “cytokine sink” to deplete IL-2 from other T cells (149, 150). In a murine model for CHS, IL-2 was required for CD4+CD25+ T cell restriction of the development of the hapten-reactive effector CD8+ T cells (174). Interestingly, by expanding allergen-specific Tregs and reducing pro-inflammatory effector T cells, microparticles (engineered to release TGF-β1, rapamycin, and IL-2) can inhibit hypersensitivity responses to subsequent allergen exposure in an allergen-specific manner, effectively preventing or reversing allergic contact dermatitis in previously sensitized mice (175). While low-dose IL-2 therapy has been recently suggested to have clinical efficacy in a phase I–IIa trial in patients with different autoimmune diseases (176, 177), its therapeutic potential in contact or drug hypersensitivity remains unexplored.
The transforming growth factor-β (TGF-β) family consists of pleiotropic cytokines with both proinflammatory and anti-inflammatory effects, contributing to immune system homeostasis (178). Regarding its connection to Tregs, different studies involving the Tgfb1−/− mice have disclosed a multiorgan autoimmunity phenotype and reduced the number of Tregs and expression of FOXP3 (179, 180). Conversion of CD4+ T cells into iTregs or peripheral Tregs (pTreg) is also dependent on TGF-β (181, 182). In mice, the lack of intact TGF-β signaling via Smad3 results in an increased proinflammatory, Th2, and Th17 type response in the skin and gastrointestinal tract (183, 184).
Th17s are a subtype of helper T cells known to cause autoimmunity and inflammation (185). Interestingly, Th17s and iTregs both depend on TGF-β for development. TGF-β can drive Th17 differentiation in the presence of other cytokines, such as IL-6 and IL-21 (186). Higher production of IL-17 in CD4+ T cells expanded from SJS/TEN lesions than those from DRESS/DIHS lesions suggests a role of the Th17/Treg axis in SJS/TEN (32). The percentages of Th17 also tend to be high in SJS/TEN (2–6 days after onset) as compared to normal subjects and MPE patients (187).
Accumulating evidence indicates that aryl hydrocarbon receptor (AhR) signaling has a specific impact on the generation of Treg. Naïve T cells isolated from AhR null mice inefficiently generate Tregs in vitro (188). The absence of AhR signaling leads to heightened inflammation and exacerbated skin pathology in a model of DTH skin reactions in AhR-deficient mice (189). It has been suggested that AhR ligands can regulate the differentiation of Tregs versus Th17 cells by expressing different microRNA signature profile in a model of DTH (190). In humans, the addition of an AhR ligand to naïve T cells differentiated in the presence of TGF-β induces suppressive FOXP3+ Tregs. Furthermore, AhR activation promotes the expression of CD39, an ectonucleotidase that hydrolyzes ATP and mediates the suppressive activity of Treg (191).
It appears that the regulatory effect of IL-7 is prominent on specialized subpopulations of Tregs rather than on the whole cell lineage. Therefore, contradictory results have been presented regarding the effects of IL-7 on Tregs in the periphery (170). Interestingly, in a murine skin inflammation model, high CD127 (IL-7 receptor α chain) expression correlated with Treg activation. Furthermore, two groups have demonstrated the critical role of IL-7 in the survival of CD127hi Tregs in the skin (192, 193). On the other hand, the percentage of CD4+CD25+CD127- cells, likely Treg cells, increased in T cells from DRESS/DIHS skin lesions compared with those from SJS/TEN (32). Thus, the role of IL-7 in Tregs may differ depending on the type of cutaneous adverse drug reaction and require further exploration.
Optimal Treg differentiation requires sensing of IL-2, with some compensatory contributions from IL-15 (194). A unique population of CD25-FOXP3+ T cells appears to depend on IL-15 for survival and maturation into Tregs in mice (195). Tregs in human skin proliferate when cultured in contact with dermal fibroblasts and IL-15 (196). However, our recent work on IL-15 has not only linked its elevation in serum to disease severity of SJS/TEN but also demonstrated its ability to induce the production of pathogenic TNF-α, granulysin and GzmB (197). Since IL-15 is vital for maintaining functions of APCs, memory T cells as well as NK cells (198–200), it may not be surprising that antagonizing against IL-15 has been found to inhibit DTH (201). Provided with the multifaceted influences of IL-15 on DTH, more research is warranted to approach the net effect of IL-15 under different circumstances.
IL-33 is an IL-1-like cytokine that is constitutively expressed by epithelial cells at barrier sites, which functions as an alarmin that is released in response to tissue damage. In a model of murine CHS, disruption of the epidermal barrier induced Tregs via IL-33 (202). Although its precise role is yet to be elucidated, serum IL-33 elevation has been observed in SJS/TEN (203).
The core of the CD28 gene family is composed of CD28, CTLA-4, and inducible T cell costimulator (ICOS) (204). ICOS is the most consistent marker for Treg activation which promotes the survival and improves the suppressive function of Tregs (193, 205). In a murine model of CHS, a population of CD4+CD25+FOXP3+ T cells upregulated ICOS on in vivo sensitization and specifically suppressed hapten-reactive CD8+ T cells both in vivo and in vitro (206). Langerhans cells also seem to prevent the development of CHS by activating ICOS+CD4+FOXP3+ Tregs (207). Intriguingly, two TNFRSF members, CD27 and OX40, are preferentially expressed by skin-resident Tregs. Both CD27 and OX40 signaling suppress the expression of Th17-associated genes from Tregs in vitro and in vivo. Tregs that lack both CD27 and OX40 are compromised in terms of controlling skin inflammation and expressed high levels of IL-17A (208). Therefore, the potential roles of CD27 and OX40 on Tregs and the Treg/Th17 axis in SCARs may deserve future investigation.
Besides the inhibitory Treg, various coinhibitory receptors also prevent T cells from being activated under different conditions. Thus, coinhibitory receptors involved in hypersensitivity immune responses also deserve detailed investigation to identify potential druggable targets (Table 2).
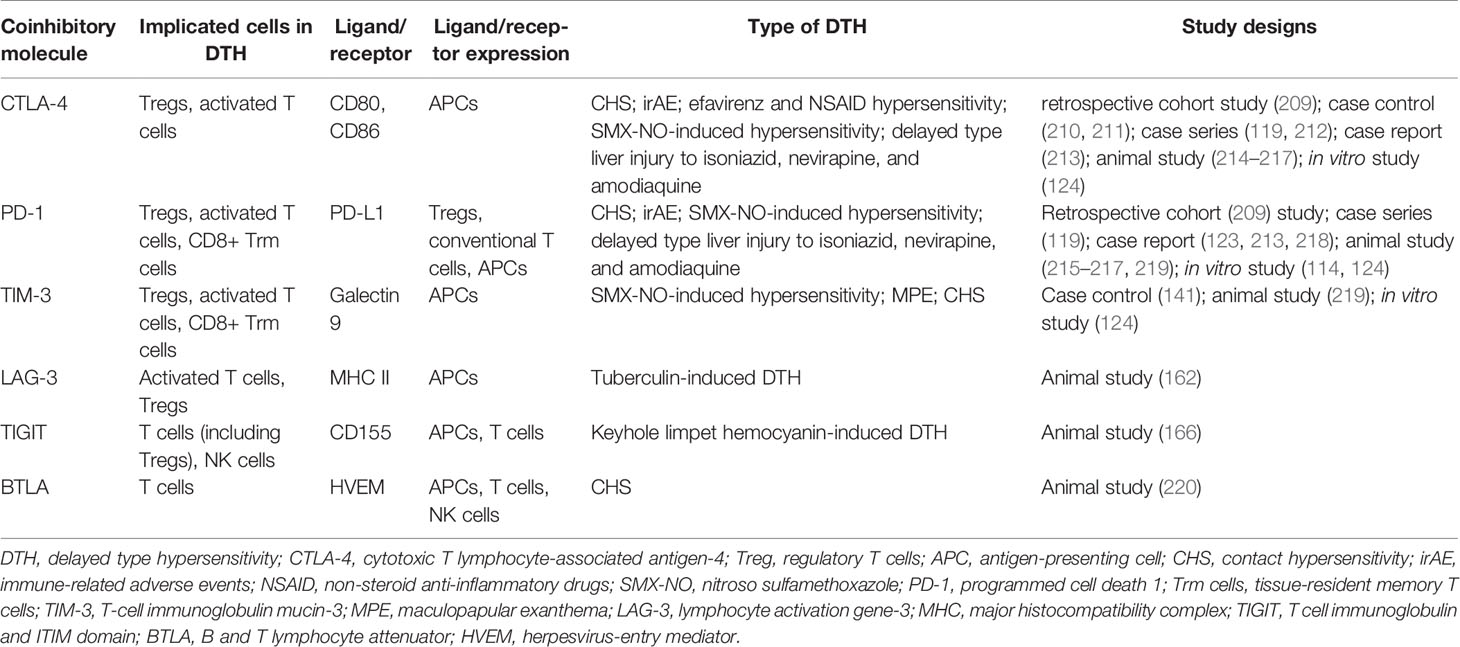
Table 2 Coinhibitory molecules involved in delayed-type hypersensitivity.
The most well-known coinhibitory receptors are perhaps CTLA-4 and PD-1 due to the application of checkpoint blockade in cancer immunotherapy (221, 222). CTLA-4 and CD28 are homologous receptors expressed on both CD4+ and CD8+ T cells that bind to CD80/CD86 on APCs; but unlike CD28, CTLA-4 serves to ultimately inhibit T-cell responses (223, 224). There have been several proposed cell-intrinsic and cell-extrinsic immunosuppressive mechanisms of CTLA-4, which suppress the cell carrying CTLA-4 or the cell carrying CD80/CD86, respectively (225). Thus, enhancing CTLA-4 activity may be beneficial in attenuating unwanted immune responses. Regarding the role of CTLA-4 in DTH, one murine study has found that CTLA-4-Immunoglobulin (Ig) provided long-term immunosuppression against both DNFB- and oxazolone-induced CHS in a dose-dependent manner. Inhibited activation of T cells in the draining lymph node and maturation of DCs and B cells, reduced infiltration of activated CD8+ T cells into inflamed tissue, and decreased cytokines and acute-phase proteins at the inflamed tissue and in circulation demonstrate the immunosuppressive function of CTLA-4-Ig both locally and systemically (214). The CTLA-4–Ig fusion protein, abatacept, has shown promise in treating rheumatoid arthritis in humans (226), while its use in various autoimmune diseases is being tested in clinical trials (227).
Not surprisingly, CTLA-4 mutations or polymorphisms have been observed in patients with autoimmune diseases and patients with Efavirenz and nonsteroidal anti-inflammatory drug hypersensitivity (210, 211, 228, 229), suggesting CTLA-4 alteration may disrupt homeostatic function, resulting in the observed disease phenotype. Indeed, deprivation of CTLA-4 leads to an enhanced immune response to drugs, as seen in the following studies. PD-1 knockout mice treated with amodiaquine and anti-CTLA-4 demonstrated liver injury similar to idiosyncratic drug-induced liver injury (IDILI) in humans with increased portal infiltration of lymphocytes and perforin and granzyme (215). Idiosyncratic drug reaction to isoniazid and nevirapine manifesting as delayed-type liver injury has been also been suggested by this model of PD-1 knockout mice treated with anti-CTLA-4 (216, 217). In the presence of CTLA-4 blocking antibodies, enhanced naïve T cell proliferative response and memory drug antigen-specific responses to SMX-NO are observed in an in vitro assay (124).
PD-1, as mentioned previously, is a checkpoint receptor mainly expressed on activated CD4+ T cells, activated CD8+ T cells, and B cells in the periphery, and it similarly transmits an immunosuppressive signal during ligation, inhibiting the proliferation, cytokine generation and release, and cytotoxicity of T cells (144, 230). Its ligand PD-L1 is expressed on T cells, B cells, DCs, macrophages, and non-hematopoietic cells, while another of its ligand, PD-L2, is expressed mainly on APCs (231). PD-1 is known to regulate T cell effector functions during various physiological responses, including acute and chronic infection, cancer, autoimmunity, and in immune homeostasis (232). In fact, agonist antibodies to PD-1 and other coinhibitory receptors have demonstrated promise in treating animal models of autoimmune diseases (233).
The blockade of the PD-1/PD-L1 axis has also been demonstrated to play a role in contact and drug hypersensitivity. In a murine study using the experimental model of CHS to DNFB, epidermal CD8+ tissue-resident memory T (Trm) cells expressing PD-1 seem to ameliorate CHS. Blocking PD-1 in vitro with anti–PD-1 monoclonal antibodies increased the reactivity of epidermal CD8+ Trm cells and their capacity to produce IFN-γ and GzmB on ex vivo reactivation. Furthermore, when animals with DNFB allergy were depleted of circulating CD8+ T cells, blockade of PD-1 triggered severe flare-up CHS reactions after low-dose DNFB re-exposure (219). Another study using PBMCs from human donors demonstrated that priming of drug-naive CD4+ and CD8+ T cells against drug antigens was more effective when PD-L1 signaling was blocked (114). However, memory T cell responses to drug antigens were not similarly enhanced by PD-L1 blockade (124).
We previously discussed that checkpoint receptor blockade may lead to disequilibrium of the immune system manifesting as boosted T cell response. In a case series of four melanoma patients, increased risk of sulfasalazine-induced cutaneous hypersensitivity was observed after treatment with the anti-PD-1 inhibitor pembrolizumab and/or the anti-CTLA-4 inhibitor ipilimumab (119). One patient developed fever with erythematous maculopapular rash and liver injury while another developed fever with unspecified rash and facial edema, though skin biopsy was not obtained for suspected SCAR. In another recent study, one patient developed TEN one month after discontinuing nivolumab. Her lymphocyte stimulation tests were simultaneously positive for several concomitant drugs while elevated levels of autoantibodies were noted (123). These observations suggest that checkpoint inhibitors may render patients more vulnerable to developing hypersensitivity to other drugs and autoimmunity. On the other hand, checkpoint inhibitors, though quite successful in treating various cancers, have autoinflammatory side effects termed immune-related adverse events (irAEs), including the commonly seen cutaneous toxicities (234). Although pruritus and MPE are more common, severe forms of cutaneous manifestations such as SJS/TEN, DRESS/DIHS, and Sweet’s Syndrome can occur on rare occasions (209, 218, 235, 236). In a melanoma patient treated with ipilimumab and nivolumab, morbilliform eruption progressed to TEN over 3 months. During the course of treatment, an increase of CD8+ T cells within the dermal-epidermal junction and an increase of PD-L1 expression in both T cells and keratinocytes were noted on skin biopsy (213). Similarly, one case series reported that 22% of the 68 patients on pembrolizumab or nivolumab developed inflammatory skin lesions ranging from mild maculopapular rash to SJS-like lesions with expression of PD-1 on skin-infiltrating T cells and keratinocytes by immunohistochemistry. Further gene expression analysis of lesional skin revealed a profile resembling SJS/TEN (212). Together, these data suggest that proper PD-1/PD-L1 functions are important in maintaining epidermal integrity while T-cell, antibody, and cytokine responses are likely involved in irAE pathogenesis (237). Although the complex mechanism of severe cutaneous reactions in patients using checkpoint inhibitors has yet to be revealed, the pivotal role of CTLA-4 and PD-1/PD-L1 in their pathogenesis has been sufficiently demonstrated.
In addition to CTLA-4 and PD-1, “second-generation” coinhibitory receptors and ligands that belong to the B7 family are emerging as potential new targets in cancer immunotherapy (238). One of these receptors is T cell immunoglobulin-3 (TIM-3). TIM-3 mediates the T cell function through binding to galectin-9 (Gal-9), leading to the death of predominantly Th1-specific T-cells, which has been shown to be defective in patients with drug‐induced MPE (124, 141). These patients exhibited reduced levels of TIM-3 in Th1 cells and impaired expression of Gal-9 in PBMCs and dendritic cells, while the addition of exogenous Gal-9 significantly reduced Tim3+ Th1 proliferation. However, another study has found that blocking TIM-3 did not enhance the proliferative response of SMX-NO-primed naïve or memory T-cells from any human donors (124). In a murine model of CHS, in vitro TIM-3 blockade, similar to PD-1 blockade, increased the activity of epidermal CD8+ Trm cell activity and cytokine production, though the effect was more modest compared to that of PD-1 (219). It appears that the effect of blocking TIM-3 variably dampened the immune response depending on the type of DTH and the study species. Therefore, the net effect of TIM-3 on DTH in the interplay of different co-signaling receptors is still warranted, especially in the insufficiently explored area of SCARs.
Lymphocyte activation gene-3 (LAG-3) is expressed on activated CD4+ and CD8+ T cells, natural killer cells, and myeloid cells (239). A recent study has suggested that LAG-3 preferentially suppresses T cells responsive to stable complexes of peptide and MHC class II by transducing inhibitory signals via its intracellular region (240). LAG-3 suppresses T cell activation and cytokine secretion and seems to work synergistically with PD-1 to inhibit immune responses, making anti-LAG-3 an important player in cancer immunotherapy (241). A study has demonstrated that depleting activated T cells with a LAG-3 cytotoxic antibody prevented T cell-driven skin inflammation in a preclinical DTH model in non-human primates (162). The result from this study highlights that antibody-mediated depletion of LAG-3-activated T cells might have therapeutic potential in DTH, which deserves future validation in humans.
TIGIT a coinhibitory receptor highly expressed on Tregs, as mentioned previously in section 4.2.3. However, it is also expressed on other T cells and even NK cells (166). TIGIT expression varies with different diseases, and its expression has been shown to be upregulated in T cells and causes dysfunction independent of PD-1 and TIM-3 in leukemic patients (242, 243). While its potential as a therapeutic target in cancer is being explored, its role in DTH remains elusive. One murine study previously demonstrated that less ear swelling was seen in keyhole limpit hemocyanin-(KLH-) treated mice immunized with the TIGIT-Fc or CTLA-4–Fc fusion protein. However, TIGIT-Fc’s inhibition of DTH was not observed in IL-10 knockout mice, suggesting that IL-10 is involved in TIGIT function (166). Results from this study suggested the differential regulatory mechanisms of each coinhibitory receptor.
B and T lymphocyte attenuator (BTLA) is a coinhibitory receptor which belongs to the Ig superfamily and resembles PD-1 and CTLA-4 (244). BTLA is expressed on CD4+ and CD8+ T cells, B cells, and other APCs (245). The ligation of BTLA by the herpesvirus-entry mediator (HVEM) on APCs attenuates T cell proliferation (246). BTLA signaling pathway is also defective in T cells in autoimmune diseases, such as systemic lupus erythematosus (247). Regarding its potential role in CHS, BTLA−/− mice display enhanced DNFB-induced CHS and CD8+ T cell proliferation and IFN-γ production compared with the wild-type mice. Consequently, in vivo injection of agonist anti-BTLA antibody is able to suppress DNFB-induced CHS and IFN-γ production, suggesting that stimulation of BTLA has therapeutic potential in CHS (220). However, in the previously mentioned study by Gamradt et al. unlike PD-1 and TIM-3, BTLA expression was not detected in epidermal CD8+ Trm cells in mice with CHS (219). Therefore, the expression profile of BTLA deserves further investigation to verify its potential role in regulating the immune response to drug and contact antigen.
DTH manifesting as SJS/TEN and DRESS/DIHS are potentially lethal due to the involvement of internal organs, generating an overwhelming systemic inflammatory storm in addition to affecting the skin. It is therefore imperative to preceisly understand the complex pathogenesis of SCARs for appropriate treatment. As we have outlined in this review, a variety of costimulatory and coinhibitory receptors have been suggested by different studies to play a role in DTH. Unfortunately, the caveat is that research in this area is still in its early stage, such that much of what is currently known about co-signaling receptors in DTH is largely based on murine models, in vitro studies, or small retrospective studies. Additionally, while individual contributions of each co-signaling receptor or Treg has been suggested, the interplay between them in the formation of the disease phenotype is largely unexplored. All in all, future research should embrace a multi-faceted approach in order to explore the interplay between a variety of co-signaling pathways while also aim at clinical-based studies since animal models may not fully represent the pathogenesis in human.
Y-SH, K-LL, YF, W-HC, and C-BC conceptualized the review. W-HC and C-BC provided the resources. Y-SH, K-LL, YF, C-WW, C-WL, Y-FL, W-HC, and C-BC wrote the original draft. K-YY, W-CC, S-IH, W-HC, and C-BC reviewed and edited the draft. Y-SH, K-LL, and C-BC created the visualizations. W-HC and C-BC supervised. C-BC coordinated the project administration. W-HC and C-BC acquired funding. All authors contributed to the article and approved the submitted version.
This study was supported by research grants from the Ministry of Science and Technology, Taiwan (MOST 108-2314-B-182A-006-MY3 to CB Chen; MOST 103-2321-B-182-001, MOST 104-2314-B-182A-148-MY3, MOST 104-2325-B-182A-006, MOST 105-2325-B-182A-007, MOST 106-2314-B-182A-037-MY3, MOST 106-2622-B-182A-003-CC2, MOST 107-2622-B-182A-001-CC2, MOST 108-23 14-B-182A-104-MY3, MOST 108-2320-B-182A-023-MY3, MOST 108-2320-B-182A-024-MY2, MOST 109-2320-B-182A-008 -MY3, MOST-108-2314-B-182A-104 -MY3, MOST-109-2326-B-182A-001 to WH Chung), and Chang Gung Memorial Hospital, Taiwan (CMRPG2H0081, CMRPG2J0221, CMRPG2J0222, NMRPG2J6012, NMRPG2J6013, CIRPG2I0011, CIRPG2I0012, CIRPG2I0013 to CB Chen; CIRPG3I0022, CIRPG3I0023, CIRPG3I0042, CIRPG3I0043, CLRPG3E0036, CLRPG3J0012, CMRPG3I0382, CORPG3J0322, CLRPG2E0053, CMRPG3D0363, CORPG3F0042~3, NCRPG3G0023, NCRPG3GS023, NMRPG3G6293, NMRPG3J6062, NMRPG3J6063, NMRPG3K0521, OMRPG3E0041 to WH Chung).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
ACD, allergic contact dermatitis; APC, antigen-presenting cell; BTLA, B and T lymphocyte attenuator; CCR6, chemokine receptor 6; CHS, contact hypersensitivity; CMV, cytomegalovirus; CTL, cytotoxic T lymphocyte; CTLA-4, cytotoxic T lymphocyte-associated antigen-4; DC, dendritic cell; DIHS, drug-induced hypersensitivity syndrome; DNFB, dinitrofluorobenzene; DRESS, drug reaction with eosinophilia and systemic symptoms; DTH, delayed-type hypersensitivity; EBV, Epstein-Barr virus; FOXP3, Forkhead protein 3; Gal-9, galectin-9; GITR, glucocorticoid induced TNF receptor; GzmB, granzyme B; HHV, human herpes virus; HIV, human immunodeficiency virus; HLA, human leukocyte antigen; HVEM, herpesvirus-entry mediator; ICOS, inducible T cell costimulator; IFN, interferon; IL, interleukin; IPEX, immunodysregulation, polyendocrinopathy, enteropathy, X-linked; IrAE, immune-related adverse event; LAG-3, lymphocyte activation gene-3; MHC, Major histocompatibility complex; MPE, maculopapular exanthema; NK, natural killer; PBMC, peripheral blood mononuclear cell; PD-1, programmed cell death 1; SCAR, severe cutaneous adverse reaction; SJS, Stevens-Johnson syndrome; SMX, sulfamethoxazole; TARC, thymus activation-regulated chemokine; TBSA, total body surface area; TCR, T cell receptor; TEN, toxic epidermal necrolysis; TGF-β, transforming growth factor-beta; Th1, T helper 1 cell; Th17, T helper 17 cell; TIGIT, T cell immunoglobulin and ITIM domain; TIM-3, T-cell immunoglobulin mucin-3; TNF, tumor necrosis factor; TNFRSF, tumor necrosis factor receptor superfamily; Treg, regulatory T cell; Trm, tissue-resident memory T cell.
1. Uzzaman A, Cho SH. Chapter 28: Classification of hypersensitivity reactions. Allergy Asthma Proc (2012) 33 Suppl 1:96–9. doi: 10.2500/aap.2012.33.3561
2. Justiz Vaillant AA, Zulfiqar H, Ramphul K. Delayed Hypersensitivity Reactions. Treasure Island (FL: StatPearls (2020).
3. Nosbaum A, Vocanson M, Rozieres A, Hennino A, Nicolas JF. Allergic and irritant contact dermatitis. Eur J Dermatol (2009) 19(4):325–32. doi: 10.1684/ejd.2009.0686
4. Posadas SJ, Pichler WJ. Delayed drug hypersensitivity reactions - new concepts. Clin Exp Allergy (2007) 37(7):989–99. doi: 10.1111/j.1365-2222.2007.02742.x
5. Phillips EJ, Chung WH, Mockenhaupt M, Roujeau JC, Mallal SA. Drug hypersensitivity: pharmacogenetics and clinical syndromes. J Allergy Clin Immunol (2011) 127(3 Suppl):S60–6. doi: 10.1016/j.jaci.2010.11.046
6. Fritsch PO, Sidoroff A. Drug-induced Stevens-Johnson syndrome/toxic epidermal necrolysis. Am J Clin Dermatol (2000) 1(6):349–60. doi: 10.2165/00128071-200001060-00003
7. Roujeau JC, Stern RS. Severe adverse cutaneous reactions to drugs. N Engl J Med (1994) 331(19):1272–85. doi: 10.1056/NEJM199411103311906
8. Bastuji-Garin S, Rzany B, Stern RS, Shear NH, Naldi L, Roujeau JC. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol (1993) 129(1):92–6. doi: 10.1001/archderm.129.1.92
9. Cacoub P, Musette P, Descamps V, Meyer O, Speirs C, Finzi L, et al. The DRESS syndrome: a literature review. Am J Med (2011) 124(7):588–97. doi: 10.1016/j.amjmed.2011.01.017
10. Kardaun SH, Sekula P, Valeyrie-Allanore L, Liss Y, Chu CY, Creamer D, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS): an original multisystem adverse drug reaction. Results from the prospective RegiSCAR study. Br J Dermatol (2013) 169(5):1071–80. doi: 10.1111/bjd.12501
11. Shiohara T, Mizukawa Y. Drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS): An update in 2019. Allergol Int (2019) 68(3):301–8. doi: 10.1016/j.alit.2019.03.006
12. Mizukawa Y, Hirahara K, Kano Y, Shiohara T. Drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms severity score: A useful tool for assessing disease severity and predicting fatal cytomegalovirus disease. J Am Acad Dermatol (2019) 80(3):670–8 e2. doi: 10.1016/j.jaad.2018.08.052
13. Usatine RP, Riojas M. Diagnosis and management of contact dermatitis. Am Fam Physician (2010) 82(3):249–55.
14. Zimmermann S, Sekula P, Venhoff M, Motschall E, Knaus J, Schumacher M, et al. Systemic Immunomodulating Therapies for Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: A Systematic Review and Meta-analysis. JAMA Dermatol (2017) 153(6):514–22. doi: 10.1001/jamadermatol.2016.5668
15. Bellon T. Mechanisms of Severe Cutaneous Adverse Reactions: Recent Advances. Drug Saf (2019) 42(8):973–92. doi: 10.1007/s40264-019-00825-2
16. Chang WC, Abe R, Anderson P, Anderson W, Ardern-Jones MR, Beachkofsky TM, et al. SJS/TEN 2019: From science to translation. J Dermatol Sci (2020) 98(1):2–12. doi: 10.1016/j.jdermsci.2020.02.003
17. Dodiuk-Gad RP, Chung WH, Valeyrie-Allanore L, Shear NH. Stevens-Johnson Syndrome and Toxic Epidermal Necrolysis: An Update. Am J Clin Dermatol (2015) 16(6):475–93. doi: 10.1007/s40257-015-0158-0
18. Naisbitt DJ, Olsson-Brown A, Gibson A, Meng X, Ogese MO, Tailor A, et al. Immune dysregulation increases the incidence of delayed-type drug hypersensitivity reactions. Allergy (2020) 75(4):781–97. doi: 10.1111/all.14127
19. Ali N, Rosenblum MD. Regulatory T cells in skin. Immunology (2017) 152(3):372–81. doi: 10.1111/imm.12791
20. Park HJ, Yun J, Kang DY, Park J-W, Koh Y-I, Kim S, et al. Unique Clinical Characteristics and Prognosis of Allopurinol-Induced Severe Cutaneous Adverse Reactions. J Allergy Clin Immunol: In Pract (2019) 7(8):2739–49. doi: 10.1016/j.jaip.2019.05.047
21. Shiohara T, Kano Y, Hirahara K, Aoyama Y. Prediction and management of drug reaction with eosinophilia and systemic symptoms (DRESS). Taylor & Francis; (2017) 13(7):701–4. doi: 10.1080/17425255.2017.1297422
22. Lavergne SN, Park BK, Naisbitt DJ. The roles of drug metabolism in the pathogenesis of T-cell-mediated drug hypersensitivity. Curr Opin Allergy Clin Immunol (2008) 8(4):299–307. doi: 10.1097/ACI.0b013e3283079c64
23. Bohan KH, Mansuri TF, Wilson NM. Anticonvulsant hypersensitivity syndrome: implications for pharmaceutical care. Pharmacother: J Hum Pharmacol Drug Ther (2007) 27(10):1425–39. doi: 10.1592/phco.27.10.1425
24. Wei C-Y, Chung W-H, Huang H-W, Chen Y-T, Hung S-I. Direct interaction between HLA-B and carbamazepine activates T cells in patients with Stevens-Johnson syndrome. J Allergy Clin Immunol (2012) 129(6):1562–9. doi: 10.1016/j.jaci.2011.12.990
25. Yun J, Marcaida MJ, Eriksson KK, Jamin H, Fontana S, Pichler WJ, et al. Oxypurinol directly and immediately activates the drug-specific T cells via the preferential use of HLA-B* 58: 01. J Immunol (2014) 192(7):2984–93. doi: 10.4049/jimmunol.1302306
26. White KD, Chung W-H, Hung S-I, Mallal S, Phillips EJ. Evolving models of the immunopathogenesis of T cell–mediated drug allergy: The role of host, pathogens, and drug response. J Allergy Clin Immunol (2015) 136(2):219–34. doi: 10.1016/j.jaci.2015.05.050
27. Chen C-B, Abe R, Pan R-Y, Wang C-W, Hung S-I, Tsai Y-G, et al. An updated review of the molecular mechanisms in drug hypersensitivity. J Immunol Res (2018) 2018:6131694. doi: 10.1155/2018/6431694
28. Posadas SJ, Padial A, Torres MJ, Mayorga C, Leyva L, Sanchez E, et al. Delayed reactions to drugs show levels of perforin, granzyme B, and Fas-L to be related to disease severity. J Allergy Clin Immunol (2002) 109(1):155–61. doi: 10.1067/mai.2002.120563
29. Caproni M, Torchia D, Schincaglia E, Volpi W, Frezzolini A, Schena D, et al. Expression of cytokines and chemokine receptors in the cutaneous lesions of erythema multiforme and Stevens–Johnson syndrome/toxic epidermal necrolysis. Br J Dermatol (2006) 155(4):722–8. doi: 10.1111/j.1365-2133.2006.07398.x
30. Chung WH, Hung SI, Yang JY, Su SC, Huang SP, Wei CY, et al. Granulysin is a key mediator for disseminated keratinocyte death in Stevens-Johnson syndrome and toxic epidermal necrolysis. Nat Med (2008) 14(12):1343–50. doi: 10.1038/nm.1884
31. Abe R, Yoshioka N, Murata J, Fujita Y, Shimizu H. Granulysin as a marker for early diagnosis of the Stevens-Johnson syndrome. Ann Intern Med (2009) 151(7):514–5. doi: 10.7326/0003-4819-151-7-200910060-00016
32. Hashizume H, Fujiyama T, Tokura Y. Reciprocal contribution of Th17 and regulatory T cells in severe drug allergy. J Dermatol Sci (2016) 81(2):131. doi: 10.1016/j.jdermsci.2015.11.002
33. Yoshikawa T, Fujita A, Yagami A, Suzuki K, Matsunaga K, Ihira M, et al. Human herpesvirus 6 reactivation and inflammatory cytokine production in patients with drug-induced hypersensitivity syndrome. J Clin Virol (2006) 37:S92–S6. doi: 10.1016/S1386-6532(06)70019-1
34. Shiohara T, Kano Y. A complex interaction between drug allergy and viral infection. Clin Rev Allergy Immunol (2007) 33(1-2):124–33. doi: 10.1007/s12016-007-8010-9
35. Beeler A, Engler O, Gerber BO, Pichler WJ. Long-lasting reactivity and high frequency of drug-specific T cells after severe systemic drug hypersensitivity reactions. J Allergy Clin Immunol (2006) 117(2):455–62. doi: 10.1016/j.jaci.2005.10.030
36. Su S-C, Mockenhaupt M, Wolkenstein P, Dunant A, Le Gouvello S, Chen C-B, et al. Interleukin-15 is associated with severity and mortality in Stevens-Johnson syndrome/toxic epidermal necrolysis. J Invest Dermatol (2017) 137(5):1065–73. doi: 10.1016/j.jid.2016.11.034
37. Chen P, Lin J-J, Lu C-S, Ong C-T, Hsieh PF, Yang C-C, et al. Carbamazepine-induced toxic effects and HLA-B* 1502 screening in Taiwan. New Engl J Med (2011) 364(12):1126–33. doi: 10.1056/NEJMoa1009717
38. Mallal S, Phillips E, Carosi G, Molina J-M, Workman C, Tomažič J, et al. HLA-B* 5701 screening for hypersensitivity to abacavir. New Engl J Med (2008) 358(6):568–79. doi: 10.1056/NEJMoa0706135
39. Chung WH, Hung SI, Hong HS, Hsih MS, Yang LC, Ho HC, et al. Medical genetics: a marker for Stevens-Johnson syndrome. Nature (2004) 428(6982):486. doi: 10.1038/428486a
40. Pan RY, Chu MT, Wang CW, Lee YS, Lemonnier F, Michels AW, et al. Identification of drug-specific public TCR driving severe cutaneous adverse reactions. Nat Commun (2019) 10(1):3569. doi: 10.1038/s41467-019-11396-2
41. Shin DS, Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr Opin Immunol (2015) 33:23–35. doi: 10.1016/j.coi.2015.01.006
42. Lu K-L, Wu M-Y, Wang C-H, Wang C-W, Hung S-I, Chung W-H, et al. The Role of Immune Checkpoint Receptors in Regulating Immune Reactivity in Lupus. Cells (2019) 8(10):1213. doi: 10.3390/cells8101213
43. Mallal S, Phillips E, Carosi G, Molina J-M, Workman C, Tomazic J, et al. HLA-B*5701 Screening for Hypersensitivity to Abacavir. New Engl J Med (2008) 358(6):568–79. doi: 10.1056/NEJMoa0706135
44. Chung W-H, Hung S-I, Hong H-S, Hsih M-S, Yang L-C, Ho H-C, et al. A marker for Stevens–Johnson syndrome. Nature (2004) 428(6982):486–. doi: 10.1038/428486a
45. Kim S-H, Lee KW, Song W-J, Kim S-H, Jee Y-K, Lee S-M, et al. Carbamazepine-induced severe cutaneous adverse reactions and HLA genotypes in Koreans. Epilepsy Res (2011) 97(1):190–7. doi: 10.1016/j.eplepsyres.2011.08.010
46. Zhang FR, Liu H, Irwanto A, Fu XA, Li Y, Yu GQ, et al. HLA-B*13:01 and the Dapsone Hypersensitivity Syndrome. New Engl J Med (2013) 369(17):1620–8. doi: 10.1056/NEJMoa1213096
47. Fan W-L, Shiao M-S, Hui RC-Y, Su S-C, Wang C-W, Chang Y-C, et al. HLA Association with Drug-Induced Adverse Reactions. J Immunol Res (2017) 2017:3186328–. doi: 10.1155/2017/3186328
48. Esensten Jonathan H, Helou Ynes A, Chopra G, Weiss A. Bluestone Jeffrey A. CD28 Costimulation: From Mechanism to Therapy. Immunity (2016) 44(5):973–88. doi: 10.1016/j.immuni.2016.04.020
49. Huang C, Zhu H-X, Yao Y, Bian Z-H, Zheng Y-J, Li L, et al. Immune checkpoint molecules. Possible future therapeutic implications in autoimmune diseases. J Autoimmun (2019) 104:102333. doi: 10.1016/j.jaut.2019.102333
50. Davis RJ, Ferris RL, Schmitt NC. Costimulatory and coinhibitory immune checkpoint receptors in head and neck cancer: unleashing immune responses through therapeutic combinations. Cancers Head Neck (2016) 1(1):12. doi: 10.1186/s41199-016-0013-x
51. Attanasio J, Wherry EJ. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity (2016) 44(5):1052–68. doi: 10.1016/j.immuni.2016.04.022
52. Phillips EJ, Mallal SA. Active suppression rather than ignorance: tolerance to abacavir-induced HLA-B*57:01 peptide repertoire alteration. J Clin Invest (2018) 128(7):2746–9. doi: 10.1172/JCI121525
53. Cardone M, Garcia K, Tilahun ME, Boyd LF, Gebreyohannes S, Yano M, et al. A transgenic mouse model for HLA-B*57:01-linked abacavir drug tolerance and reactivity. J Clin Invest (2018) 128(7):2819–32. doi: 10.1172/JCI99321
54. Kondo S, Kooshesh F, Wang B, Fujisawa H, Sauder DN. Contribution of the CD28 molecule to allergic and irritant-induced skin reactions in CD28 -/- mice. J Immunol (1996) 157(11):4822.
55. Poirier N, Chevalier M, Mary C, Hervouet J, Minault D, Le Bas-Bernardet S, et al. Selective CD28 antagonist prevents Aldara-induced skin inflammation in non-human primates. Exp Dermatol (2016) 25(3):233–4. doi: 10.1111/exd.12891
56. Miyagawa F, Nakamura Y, Miyashita K, Iioka H, Himuro Y, Ogawa K, et al. Preferential expression of CD134, an HHV-6 cellular receptor, on CD4T cells in drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic symptoms (DRESS). J Dermatol Sci (2016) 83(2):151–4. doi: 10.1016/j.jdermsci.2016.05.001
57. Miyagawa F, Nakamura-Nishimura Y, Kanatani Y, Asada H. Correlation Between Expression of CD134, a Human Herpesvirus 6 Cellular Receptor, on CD4+ T cells and Th2-type Immune Responses in Drug-induced Hypersensitivity Syndrome/Drug Reaction with Eosinophilia and Systemic Symptoms. Acta Derm Venereol (2020) 100(6):adv00102. doi: 10.2340/00015555-3465
58. Klaewsongkram J, Thantiworasit P, Sodsai P, Buranapraditkun S, Mongkolpathumrat P. Slow desensitization of imatinib-induced nonimmediate reactions and dynamic changes of drug-specific CD4+CD25+CD134+ lymphocytes. Ann Allergy Asthma Immunol (2016) 117(5):514–9. doi: 10.1016/j.anai.2016.08.035
59. Chen AI, McAdam AJ, Buhlmann JE, Scott S, Lupher ML, Greenfield EA, et al. Ox40-Ligand Has a Critical Costimulatory Role in Dendritic Cell:T Cell Interactions. Immunity (1999) 11(6):689–98. doi: 10.1016/S1074-7613(00)80143-0
60. Sato T, Ishii N, Murata K, Kikuchi K, Nakagawa S, Ndhlovu LC, et al. Consequences of OX40-OX40 ligand interactions in Langerhans cell function: enhanced contact hypersensitivity responses in OX40L-transgenic mice. Eur J Immunol (2002) 32(11):3326–35. doi: 10.1002/1521-4141(200211)32:11<3326::AID-IMMU3326>3.0.CO;2-9
61. Higuchi T, Satoh T, Yokozeki H. Using CD40 ligand expression to detect antigen-specific T cells in patients with drug eruptions. Acta Dermato Venereologica (2014) 94(1):86–7. doi: 10.2340/00015555-1617
62. Van Gasse AL, Ebo DG, Mertens CM, Bridts CH, Elst J, De Puysseleyr L, et al. CD154 (CD40L): A novel aid to document nonimmediate hypersensitivity to amoxicillin or amoxicillin clavulanic acid. Clin Exp Allergy (2020) 50(5):640–2. doi: 10.1111/cea.13581
63. Caproni M, Torchia D, Schincaglia E, Volpi W, Frezzolini A, Schena D, et al. The CD40/CD40 ligand system is expressed in the cutaneous lesions of erythema multiforme and Stevens–Johnson syndrome/toxic epidermal necrolysis spectrum. Br J Dermatol (2006) 154(2):319–24. doi: 10.1111/j.1365-2133.2005.07023.x
64. Tang A, Judge TA, Turka LA. Blockade of CD40-CD40 ligand pathway induces tolerance in murine contact hypersensitivity. Eur J Immunol (1997) 27(12):3143–50. doi: 10.1002/eji.1830271210
65. Moodycliffe AM, Shreedhar V, Ullrich SE, Walterscheid J, Bucana C, Kripke ML, et al. Cd40–Cd40 Ligand Interactions in Vivo Regulate Migration of Antigen-Bearing Dendritic Cells from the Skin to Draining Lymph Nodes. J Exp Med (1999) 191(11):2011–20. doi: 10.1084/jem.191.11.2011
66. Gorbachev AV, Heeger PS, Fairchild RL. CD4+ and CD8+ T cell priming for contact hypersensitivity occurs independently of CD40-CD154 interactions. J Immunol (2001) 166(4):2323. doi: 10.4049/jimmunol.166.4.2323
67. Sanderson JP, Naisbitt DJ, Farrell J, Ashby CA, Tucker MJ, Rieder MJ, et al. Sulfamethoxazole and Its Metabolite Nitroso Sulfamethoxazole Stimulate Dendritic Cell Costimulatory Signaling. J Immunol (2007) 178(9):5533. doi: 10.4049/jimmunol.178.9.5533
68. Yamashita-Kanemaru Y, Takahashi Y, Wang Y, Tahara-Hanaoka S, S-i H, Bernhardt G, et al. CD155 (PVR/Necl5) Mediates a Costimulatory Signal in CD4<sup<+</sup< T Cells and Regulates Allergic Inflammation. J Immunol (2015) 194(12):5644. doi: 10.4049/jimmunol.1401942
69. Lozano E, Joller N, Cao Y, Kuchroo VK, Hafler DA. The CD226/CD155 interaction regulates the proinflammatory (Th1/Th17)/anti-inflammatory (Th2) balance in humans. J Immunol (Baltimore Md 1950) (2013) 191(7):3673–80. doi: 10.4049/jimmunol.1300945
70. Tohyama M, Watanabe H, Murakami S, Shirakata Y, Sayama K, Iijima M, et al. Possible involvement of CD14+ CD16+ monocyte lineage cells in the epidermal damage of Stevens–Johnson syndrome and toxic epidermal necrolysis. Br J Dermatol (2012) 166(2):322–30. doi: 10.1111/j.1365-2133.2011.10649.x
71. Kamimura Y, Iwai H, Piao J, Hashiguchi M, Azuma M. The Glucocorticoid-Induced TNF Receptor-Related Protein (GITR)-GITR Ligand Pathway Acts As a Mediator of Cutaneous Dendritic Cell Migration and Promotes T Cell-Mediated Acquired Immunity. J Immunol (2009) 182(5):2708. doi: 10.4049/jimmunol.0803704
72. Morel E, Escamochero S, Cabañas R, Díaz R, Fiandor A, Bellón T. CD94/NKG2C is a killer effector molecule in patients with Stevens-Johnson syndrome and toxic epidermal necrolysis. J Allergy Clin Immunol (2010) 125(3):703–10.e8. doi: 10.1016/j.jaci.2009.10.030
73. Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol (1996) 14(1):233–58. doi: 10.1146/annurev.immunol.14.1.233
74. Beyersdorf N, Kerkau T, Hünig T. CD28 co-stimulation in T-cell homeostasis: a recent perspective. Immunotargets Ther (2015) 4:111–22. doi: 10.2147/ITT.S61647
75. Ishii N, Takahashi T, Soroosh P, Sugamura K. Chapter 3 - OX40–OX40 Ligand Interaction in T-Cell-Mediated Immunity and Immunopathology. In: Alt FW, editor. Adv Immunol (2010) 105:63–98. doi: 10.1016/S0065-2776(10)05003-0
76. Willoughby J, Griffiths J, Tews I, Cragg MS. OX40: Structure and function – What questions remain? Mol Immunol (2017) 83:13–22. doi: 10.1016/j.molimm.2017.01.006
77. Greisen S, Kunder R, Deleuran B. T cell co-stimulatory factors. Rheumatology (2016) 56(6):861–2. doi: 10.1093/rheumatology/kew276
78. Zaunders JJ, Munier ML, Seddiki N, Pett S, Ip S, Bailey M, et al. High levels of human antigen-specific CD4+ T cells in peripheral blood revealed by stimulated coexpression of CD25 and CD134 (OX40). J Immunol (2009) 183(4):2827–36. doi: 10.4049/jimmunol.0803548
79. Fu Y, Lin Q, Zhang Z, Zhang L. Therapeutic strategies for the costimulatory molecule OX40 in T-cell-mediated immunity. Acta Pharm Sin B (2020) 10(3):414–33. doi: 10.1016/j.apsb.2019.08.010
80. Schonbeck U, Libby P. The CD40/CD154 receptor/ligand dyad. Cell Mol Life Sci (2001) 58(1):4–43. doi: 10.1007/PL00000776
81. Tahara-Hanaoka S, Shibuya K, Onoda Y, Zhang H, Yamazaki S, Miyamoto A, et al. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112). Int Immunol (2004) 16(4):533–8. doi: 10.1093/intimm/dxh059
82. Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, et al. Identification of PVR (CD155) and Nectin-2 (CD112) as Cell Surface Ligands for the Human DNAM-1 (CD226) Activating Molecule. J Exp Med (2003) 198(4):557–67. doi: 10.1084/jem.20030788
83. Jeong S, Park S-H. Co-Stimulatory Receptors in Cancers and Their Implications for Cancer Immunotherapy. Immune Netw (2020) 20(1):e3–e. doi: 10.4110/in.2020.20.e3
84. Fourcade J, Sun Z, Chauvin J-M, Ka M, Davar D, Pagliano O, et al. CD226 opposes TIGIT to disrupt Tregs in melanoma. JCI Insight (2018) 3(14):e121157. doi: 10.1172/jci.insight.121157
85. Vinay DS, Kwon BS. 4-1BB signaling beyond T cells. Cell Mol Immunol (2011) 8(4):281–4. doi: 10.1038/cmi.2010.82
86. Shao Z, Schwarz H. CD137 ligand, a member of the tumor necrosis factor family, regulates immune responses via reverse signal transduction. J Leukocyte Biol (2011) 89(1):21–9. doi: 10.1189/jlb.0510315
87. Zeng Q, Zhou Y, Schwarz H. CD137L-DCs, Potent Immune-Stimulators—History, Characteristics, and Perspectives. Front Immunol (2019) 10:2216. doi: 10.3389/fimmu.2019.02216
88. Dharmadhikari B, Wu M, Abdullah NS, Rajendran S, Ishak ND, Nickles E, et al. CD137 and CD137L signals are main drivers of type 1, cell-mediated immune responses. Oncoimmunology (2015) 5(4):e1113367–e. doi: 10.1080/2162402X.2015.1113367
89. Bartkowiak T, Curran MA. 4-1BB Agonists: Multi-Potent Potentiators of Tumor Immunity. Front Oncol (2015) 5:117–. doi: 10.3389/fonc.2015.00117
90. Claus C, Ferrara C, Xu W, Sam J, Lang S, Uhlenbrock F, et al. Tumor-targeted 4-1BB agonists for combination with T cell bispecific antibodies as off-the-shelf therapy. Sci Trans Med (2019) 11(496):eaav5989. doi: 10.1126/scitranslmed.aav5989
91. Knee DA, Hewes B, Brogdon JL. Rationale for anti-GITR cancer immunotherapy. Eur J Cancer (2016) 67:1–10. doi: 10.1016/j.ejca.2016.06.028
92. Wang J, Devgan V, Corrado M, Prabhu NS, El-Deiry WS, Riccardi C, et al. Glucocorticoid-induced Tumor Necrosis Factor Receptor Is a p21Cip1/WAF1 Transcriptional Target Conferring Resistance of Keratinocytes to UV Light-induced Apoptosis. J Biol Chem (2005) 280(45):37725–31. doi: 10.1074/jbc.M507976200
93. Clouthier DL, Watts TH. Cell-specific and context-dependent effects of GITR in cancer, autoimmunity, and infection. Cytokine Growth Factor Rev (2014) 25(2):91–106. doi: 10.1016/j.cytogfr.2013.12.003
94. Ronchetti S, Nocentini G, Bianchini R, Krausz LT, Migliorati G, Riccardi C. Glucocorticoid-induced TNFR-related protein lowers the threshold of CD28 costimulation in CD8+ T cells. J Immunol (2007) 179(9):5916. doi: 10.4049/jimmunol.179.9.5916
95. Shimizu J, Yamazaki S, Takahashi T, Ishida Y, Sakaguchi S. Stimulation of CD25+CD4+ regulatory T cells through GITR breaks immunological self-tolerance. Nat Immunol (2002) 3(2):135–42. doi: 10.1038/ni759
96. Sabharwal SS, Rosen DB, Grein J, Tedesco D, Joyce-Shaikh B, Ueda R, et al. GITR agonism enhances cellular metabolism to support CD8+ T-cell proliferation and effector cytokine production in a mouse tumor model. Cancer Immunol Res (2018) 6(10):1199–211. doi: 10.1158/2326-6066.CIR-17-0632
97. Mahne AE, Mauze S, Joyce-Shaikh B, Xia J, Bowman EP, Beebe AM, et al. Dual Roles for Regulatory T-cell Depletion and Costimulatory Signaling in Agonistic GITR Targeting for Tumor Immunotherapy. Cancer Res (2017) 77(5):1108. doi: 10.1158/0008-5472.CAN-16-0797
98. Houchins JP, Lanier LL, Niemi EC, Phillips JH, Ryan JC. Natural killer cell cytolytic activity is inhibited by NKG2-A and activated by NKG2-C. J Immunol (1997) 158(8):3603.
99. Arlettaz L, Villard J, de Rham C, Degermann S, Chapuis B, Huard B, et al. Activating CD94:NKG2C and inhibitory CD94:NKG2A receptors are expressed by distinct subsets of committed CD8+ TCR αβ lymphocytes. Eur J Immunol (2004) 34(12):3456–64. doi: 10.1002/eji.200425210
100. Wing K, Sakaguchi S. Regulatory T cells exert checks and balances on self tolerance and autoimmunity. Nat Immunol (2010) 11(1):7. doi: 10.1038/ni.1818
101. Brunet J-F, Denizot F, Luciani M-F, Roux-Dosseto M, Suzan M, Mattei M-G, et al. A new member of the immunoglobulin superfamily—CTLA-4. Nature (1987) 328(6127):267–70. doi: 10.1038/328267a0
102. Sakaguchi S. Naturally Arising CD4+ Regulatory T Cells for Immunologic Self-Tolerance and Negative Control of Immune Responses. Annu Rev Immunol (2004) 22(1):531–62. doi: 10.1146/annurev.immunol.21.120601.141122
103. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133(5):775–87. doi: 10.1016/j.cell.2008.05.009
104. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
105. Lu L, Barbi J, Pan F. The regulation of immune tolerance by FOXP3. Nat Rev Immunol (2017) 17(11):703–17. doi: 10.1038/nri.2017.75
106. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
107. Godfrey VL, Wilkinson JE, Rinchik EM, Russell LB. Fatal lymphoreticular disease in the scurfy (sf) mouse requires T cells that mature in a sf thymic environment: potential model for thymic education. Proc Natl Acad Sci (1991) 88(13):5528. doi: 10.1073/pnas.88.13.5528
108. Hadaschik EN, Wei X, Leiss H, Heckmann B, Niederreiter B, Steiner G, et al. Regulatory T cell-deficient scurfy mice develop systemic autoimmune features resembling lupus-like disease. Arthritis Res Ther (2015) 17(1):35–. doi: 10.1186/s13075-015-0538-0
109. Yilmaz OK, Haeberle S, Zhang M, Fritzler MJ, Enk AH, Hadaschik EN. Scurfy Mice Develop Features of Connective Tissue Disease Overlap Syndrome and Mixed Connective Tissue Disease in the Absence of Regulatory T Cells. Front Immunol (2019) 10:881. doi: 10.3389/fimmu.2019.00881
110. Wildin RS, Smyk-Pearson S, Filipovich AH. Clinical and molecular features of the immunodysregulation, polyendocrinopathy, enteropathy, X linked (IPEX) syndrome. J Med Genet (2002) 39(8):537–45. doi: 10.1136/jmg.39.8.537
111. Bossini-Castillo L, Glinos DA, Kunowska N, Golda G, Lamikanra A, Spitzer M, et al. Immune disease variants modulate gene expression in regulatory CD4+ T cells and inform drug targets. bioRxiv (2019> ) 2019:654632. doi: 10.1101/654632
112. Tomura M, Honda T, Tanizaki H, Otsuka A, Egawa G, Tokura Y, et al. Activated regulatory T cells are the major T cell type emigrating from the skin during a cutaneous immune response in mice. J Clin Invest (2010) 120(3):883–93. doi: 10.1172/JCI40926
113. Vocanson M, Cluzel-Tailhardat M, Poyet G, Valeyrie M, Chavagnac C, Levarlet B, et al. Depletion of Human Peripheral Blood Lymphocytes in CD25+ Cells Allows for the Sensitive “In Vitro” Screening of Contact Allergens. J Invest Dermatol (2008) 128(8):2119–22. doi: 10.1038/jid.2008.15
114. Gibson A, Ogese M, Sullivan A, Wang E, Saide K, Whitaker P, et al. Negative Regulation by PD-L1 during Drug-Specific Priming of IL-22–Secreting T Cells and the Influence of PD-1 on Effector T Cell Function. J Immunol (2014) 192(6):2611. doi: 10.4049/jimmunol.1302720
115. Abdeladhim M, Zhang A-H, Kropp LE, Lindrose AR, Venkatesha SH, Mitre E, et al. Engineered ovalbumin-expressing regulatory T cells protect against anaphylaxis in ovalbumin-sensitized mice. Clin Immunol (2019) 207:49–54. doi: 10.1016/j.clim.2019.07.009
116. Yang C, Mosam A, Mankahla A, Dlova N, Saavedra A. HIV infection predisposes skin to toxic epidermal necrolysis via depletion of skin-directed CD4 (+) T cells. J Am Acad Dermatol (2014) 70(6):1096–102. doi: 10.1016/j.jaad.2013.12.025
117. Rzany B, Mockenhaupt M, Stocker U, Hamouda O, Schopf E. Incidence of Stevens-Johnson syndrome and toxic epidermal necrolysis in patients with the acquired immunodeficiency syndrome in Germany. Arch Dermatol (1993) 129(8):1059. doi: 10.1001/archderm.1993.01680290135026
118. Smith KJ, Skelton HG, Yeager J, Ledsky R, Ng TH, Wagner KF. Increased drug reactions in HIV-1-positive patients: a possible explanation based on patterns of immune dysregulation seen in HIV-1 disease. The Military Medical Consortium for the Advancement of Retroviral Research (MMCARR). Clin Exp Dermatol (1997) 22(3):118–23. doi: 10.1046/j.1365-2230.1997.2130642.x
119. Ford M, Sahbudin I, Filer A, Steven N, Fisher BA. High proportion of drug hypersensitivity reactions to sulfasalazine following its use in anti-PD-1-associated inflammatory arthritis. Rheumatology (2018) 57(12):2244–6. doi: 10.1093/rheumatology/key234
120. Azukizawa H, Sano S, Kosaka H, Sumikawa Y, Itami S. Prevention of toxic epidermal necrolysis by regulatory T cells. Eur J Immunol (2005) 35(6):1722–30. doi: 10.1002/eji.200425773
121. Shen M, Lim JME, Chia C, Ren EC. CD39+ regulatory T cells modulate the immune response to carbamazepine in HLA-B*15:02 carriers. Immunobiology (2019) 225(1):151686. doi: 10.1016/j.imbio.2019.11.003
122. Sharma P, Sachdeva S, Dorjay K, Choudhry D. Fluconazole induced Stevens-Johnson Syndrome Leading to the Diagnosis of HIV Infection. J Clin Diagn Res (2019) 13(12):WD01-3. doi: 10.7860/JCDR/2019/42126.13338
123. Watanabe Y, Yamaguchi Y, Takamura N, Takahashi Y, Aihara M. Toxic epidermal necrolysis accompanied by several immune-related adverse events developed after discontinuation of nivolumab. Eur J Cancer (2020) 131:1–4. doi: 10.1016/j.ejca.2020.02.044
124. Gibson A, Faulkner L, Lichtenfels M, Ogese M, Al-Attar Z, Alfirevic A, et al. The Effect of Inhibitory Signals on the Priming of Drug Hapten-Specific T Cells That Express Distinct Vβ Receptors. J Immunol (Baltimore Md 1950) (2017) 199(4):1223–37. doi: 10.4049/jimmunol.1602029
125. Iwai S, Sueki H, Watanabe H, Sasaki Y, Suzuki T, Iijima M. Distinguishing between erythema multiforme major and Stevens–Johnson syndrome/toxic epidermal necrolysis immunopathologically. J Dermatol (2012) 39(9):781–6. doi: 10.1111/j.1346-8138.2012.01532.x
126. Takahashi R, Kano Y, Yamazaki Y, Kimishima M, Mizukawa Y, Shiohara T. Defective Regulatory T Cells In Patients with Severe Drug Eruptions: Timing of the Dysfunction Is Associated with the Pathological Phenotype and Outcome. J Immunol (2009) 182(12):8071. doi: 10.4049/jimmunol.0804002
127. Wang C-W, Yang L-Y, Chen C-B, Ho H-C, Hung S-I, Yang C-H, et al. Randomized, controlled trial of TNF-α antagonist in CTL-mediated severe cutaneous adverse reactions. J Clin Invest (2018) 128(3):985–96. doi: 10.1172/JCI93349
128. Morito H, Ogawa K, Fukumoto T, Kobayashi N, Morii T, Kasai T, et al. Increased ratio of FoxP3+ regulatory T cells/CD3+ T cells in skin lesions in drug-induced hypersensitivity syndrome/drug rash with eosinophilia and systemic symptoms. Clin Exp Dermatol (2014) 39(3):284–91. doi: 10.1111/ced.12246
129. Ushigome Y, Mizukawa Y, Kimishima M, Yamazaki Y, Takahashi R, Kano Y, et al. Monocytes are involved in the balance between regulatory T cells and Th17 cells in severe drug eruptions. Clin Exp Allergy (2018) 48(11):1453–63. doi: 10.1111/cea.13252
130. El Beidaq A, Link CWM, Hofmann K, Frehse B, Hartmann K, Bieber K, et al. In Vivo Expansion of Endogenous Regulatory T Cell Populations Induces Long-Term Suppression of Contact Hypersensitivity. J Immunol (2016) 197(5):1567. doi: 10.4049/jimmunol.1600508
131. Cavani A, Nasorri F, Ottaviani C, Sebastiani S, De Pità O, Girolomoni G. Human CD25+ Regulatory T Cells Maintain Immune Tolerance to Nickel in Healthy, Nonallergic Individuals. J Immunol (2003) 171(11):5760. doi: 10.4049/jimmunol.171.11.5760
132. Arce-Sillas A, Álvarez-Luquín DD, Tamaya-Domínguez B, Gomez-Fuentes S, Trejo-García A, Melo-Salas M, et al. Regulatory T Cells: Molecular Actions on Effector Cells in Immune Regulation. J Immunol Res (2016) 2016:1720827–. doi: 10.1155/2016/1720827
133. Akkaya B, Oya Y, Akkaya M, Al Souz J, Holstein AH, Kamenyeva O, et al. Regulatory T cells mediate specific suppression by depleting peptide–MHC class II from dendritic cells. Nat Immunol (2019) 20(2):218–31. doi: 10.1038/s41590-018-0280-2
134. Ito T, Hanabuchi S, Wang Y-H, Park WR, Arima K, Bover L, et al. Two Functional Subsets of FOXP3+ Regulatory T Cells in Human Thymus and Periphery. Immunity (2008) 28(6):870–80. doi: 10.1016/j.immuni.2008.03.018
135. Collison LW, Delgoffe GM, Guy CS, Vignali KM, Chaturvedi V, Fairweather D, et al. The composition and signaling of the IL-35 receptor are unconventional. Nat Immunol (2012) 13(3):290–9. doi: 10.1038/ni.2227
136. Palomares O, Martín-Fontecha M, Lauener R, Traidl-Hoffmann C, Cavkaytar O, Akdis M, et al. Regulatory T cells and immune regulation of allergic diseases: roles of IL-10 and TGF-β. Genes Immunity (2014) 15(8):511–20. doi: 10.1038/gene.2014.45
137. Gianchecchi E, Fierabracci A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front Immunol (2018) 9:2374–. doi: 10.3389/fimmu.2018.02374
138. Berg DJ, Leach MW, Kühn R, Rajewsky K, Müller W, Davidson NJ, et al. Interleukin 10 but not interleukin 4 is a natural suppressant of cutaneous inflammatory responses. J Exp Med (1995) 182(1):99–108. doi: 10.1084/jem.182.1.99
139. Ring S, Schäfer SC, Mahnke K, Lehr H-A, Enk AH. CD4+CD25+ regulatory T cells suppress contact hypersensitivity reactions by blocking influx of effector T cells into inflamed tissue. Eur J Immunol (2006) 36(11):2981–92. doi: 10.1002/eji.200636207
140. Gelincik A, Demir S, Şen F, Bozbey UH, Olgaç M, Ünal D, et al. Interleukin-10 is increased in successful drug desensitization regardless of the hypersensitivity reaction type. Asia Pacific Allergy (2019) 9(1):e9. doi: 10.5415/apallergy.2019.9.e9
141. Fernandez-Santamaría R, Palomares F, Salas M, Doña I, Bogas G, Ariza A, et al. Expression of the Tim3-galectin-9 axis is altered in drug-induced maculopapular exanthema. Allergy (2019) 74(9):1769–79. doi: 10.1111/all.13847
142. Dolch A, Kunz S, Dorn B, Roers A, Martin SF, Jakob T. Contact allergens induce CD8+ T cell-derived interleukin 10 that appears dispensable for regulation of contact hypersensitivity. Exp Dermatol (2017) 26(5):449–51. doi: 10.1111/exd.13237
143. Su W, Fan H, Chen M, Wang J, Brand D, He X, et al. Induced CD4+ forkhead box protein–positive T cells inhibit mast cell function and established contact hypersensitivity through TGF-β1. J Allergy Clin Immunol (2012) 130(2):444–52.e7. doi: 10.1016/j.jaci.2012.05.011
144. Zhao H, Liao X, Kang Y. Tregs: Where We Are and What Comes Next? Front Immunol (2017) 8:1578. doi: 10.3389/fimmu.2017.01578
145. Takenaka MC, Robson S, Quintana FJ. Regulation of the T Cell Response by CD39. Trends Immunol (2016) 37(7):427–39. doi: 10.1016/j.it.2016.04.009
146. Ring S, Oliver SJ, Cronstein BN, Enk AH, Mahnke K. CD4+CD25+ regulatory T cells suppress contact hypersensitivity reactions through a CD39, adenosine-dependent mechanism. J Allergy Clin Immunol (2009) 123(6):1287–96.e2. doi: 10.1016/j.jaci.2009.03.022
147. Mahnke K, Useliene J, Ring S, Kage P, Jendrossek V, Robson SC, et al. Down-Regulation of CD62L Shedding in T Cells by CD39+ Regulatory T Cells Leads to Defective Sensitization in Contact Hypersensitivity Reactions. J Invest Dermatol (2017) 137(1):106–14. doi: 10.1016/j.jid.2016.08.023
148. Shen M, Lim JME, Chia C, Ren EC. CD39+ regulatory T cells modulate the immune response to carbamazepine in HLA-B* 15: 02 carriers. Immunobiology (2020) 225(1):151868. doi: 10.1016/j.imbio.2019.11.003
149. Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation–mediated apoptosis of effector CD4+ T cells. Nat Immunol (2007) 8(12):1353–62. doi: 10.1038/ni1536
150. Chinen T, Kannan AK, Levine AG, Fan X, Klein U, Zheng Y, et al. An essential role for the IL-2 receptor in T(reg) cell function. Nat Immunol (2016) 17(11):1322–33. doi: 10.1038/ni.3540
151. Lucca LE, Dominguez-Villar M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat Rev Immunol (2020) 20(11):680–93. doi: 10.1038/s41577-020-0296-3
152. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 Control over Foxp3+ Regulatory T Cell Function. Science (2008) 322(5899):271. doi: 10.1126/science.1160062
153. Collins AV, Brodie DW, Gilbert RJC, Iaboni A, Manso-Sancho R, Walse B, et al. The Interaction Properties of Costimulatory Molecules Revisited. Immunity (2002) 17(2):201–10. doi: 10.1016/S1074-7613(02)00362-X
154. Sakaguchi S, Mikami N, Wing JB, Tanaka A, Ichiyama K, Ohkura N. Regulatory T Cells and Human Disease. Annu Rev Immunol (2020) 38:541–66. doi: 10.1146/annurev-immunol-042718-041717
155. Qureshi OS, Zheng Y, Nakamura K, Attridge K, Manzotti C, Schmidt EM, et al. Trans-endocytosis of CD80 and CD86: a molecular basis for the cell-extrinsic function of CTLA-4. Sci (New York NY) (2011) 332(6029):600–3. doi: 10.1126/science.1202947
156. Seitz C, Liu S, Klocke K, Joly A-L, Czarnewski PV, Tibbitt CA, et al. Multi-faceted inhibition of dendritic cell function by CD4+Foxp3+ regulatory T cells. J Autoimmun (2019) 98:86–94. doi: 10.1016/j.jaut.2018.12.002
157. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236(1):219–42. doi: 10.1111/j.1600-065X.2010.00923.x
158. Park HJ, Park JS, Jeong YH, Son J, Ban YH, Lee B-H, et al. PD-1 Upregulated on Regulatory T Cells during Chronic Virus Infection Enhances the Suppression of CD8<sup<+</sup< T Cell Immune Response via the Interaction with PD-L1 Expressed on CD8<sup<+</sup< T Cells. J Immunol (2015) 194(12):5801. doi: 10.4049/jimmunol.1401936
159. White KD, Abe R, Ardern-Jones M, Beachkofsky T, Bouchard C, Carleton B, et al. SJS/TEN 2017: Building Multidisciplinary Networks to Drive Science and Translation. J Allergy Clin Immunol In Pract (2018) 6(1):38–69. doi: 10.1016/j.jaip.2017.11.028
160. Huang C-T, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, et al. Role of LAG-3 in Regulatory T Cells. Immunity (2004) 21(4):503–13. doi: 10.1016/j.immuni.2004.08.010
161. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T Cells Inhibit Dendritic Cells by Lymphocyte Activation Gene-3 Engagement of MHC Class II. J Immunol (2008) 180(9):5916. doi: 10.4049/jimmunol.180.9.5916
162. Poirier N, Haudebourg T, Brignone C, Dilek N, Hervouet J, Minault D, et al. Antibody-mediated depletion of lymphocyte-activation gene-3 (LAG-3(+) )-activated T lymphocytes prevents delayed-type hypersensitivity in non-human primates. Clin Exp Immunol (2011) 164(2):265–74. doi: 10.1111/j.1365-2249.2011.04329.x
163. Fuhrman CA, Yeh W-I, Seay HR, Saikumar Lakshmi P, Chopra G, Zhang L, et al. Divergent Phenotypes of Human Regulatory T Cells Expressing the Receptors TIGIT and CD226. J Immunol (2015) 195(1):145. doi: 10.4049/jimmunol.1402381
164. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48. doi: 10.1038/ni.1674
165. Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, et al. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc Natl Acad Sci (2009) 106(42):17858–63. doi: 10.1073/pnas.0903474106
166. Yu X, Harden K C, Gonzalez L, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol (2009) 10(1):48–57. doi: 10.1038/ni.1674
167. Gondek DC, Lu L-F, Quezada SA, Sakaguchi S, Noelle RJ. Cutting edge: contact-mediated suppression by CD4+ CD25+ regulatory cells involves a granzyme B-dependent, perforin-independent mechanism. J Immunol (2005) 174(4):1783–6. doi: 10.4049/jimmunol.174.4.1783
168. Ikebuchi R, Teraguchi S, Vandenbon A, Honda T, Shand FHW, Nakanishi Y, et al. A rare subset of skin-tropic regulatory T cells expressing Il10/Gzmb inhibits the cutaneous immune response. Sci Rep (2016) 6(1):35002. doi: 10.1038/srep35002
169. Kornete M, Piccirillo CA. Functional crosstalk between dendritic cells and Foxp3(+) regulatory T cells in the maintenance of immune tolerance. Front Immunol (2012) 3:165–. doi: 10.3389/fimmu.2012.00165
170. Toomer KH, Malek TR. Cytokine Signaling in the Development and Homeostasis of Regulatory T cells. Cold Spring Harbor Perspect Biol (2018) 10(3):a028597. doi: 10.1101/cshperspect.a028597
171. Zhang R, Huynh A, Whitcher G, Chang J, Maltzman JS, Turka LA. An obligate cell-intrinsic function for CD28 in Tregs. J Clin Invest (2013) 123(2):580–93. doi: 10.1172/JCI65013
172. Zhang R, Borges CM, Fan MY, Harris JE, Turka LA. Requirement for CD28 in Effector Regulatory T Cell Differentiation, CCR6 Induction, and Skin Homing. J Immunol (2015) 195(9):4154. doi: 10.4049/jimmunol.1500945
173. Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol (2005) 6(11):1142–51. doi: 10.1038/ni1263
174. Kish DD, Gorbachev AV, Fairchild RL. CD8+ T cells produce IL-2, which is required for CD4+CD25+ T cell regulation of effector CD8+ T cell development for contact hypersensitivity responses. J Leukocyte Biol (2005) 78(3):725–35. doi: 10.1189/jlb.0205069
175. Balmert SC, Donahue C, Vu JR, Erdos G, Falo LD, Little SR. In vivo induction of regulatory T cells promotes allergen tolerance and suppresses allergic contact dermatitis. J Controlled Release (2017) 261:223–33. doi: 10.1016/j.jconrel.2017.07.006
176. Collison J. Low-dose IL-2 therapy for autoimmune diseases. Nat Rev Rheumatol (2019) 15(1):2–. doi: 10.1038/s41584-018-0142-1
177. Rosenzwajg M, Lorenzon R, Cacoub P, Pham HP, Pitoiset F, El Soufi K, et al. Immunological and clinical effects of low-dose interleukin-2 across 11 autoimmune diseases in a single, open clinical trial. Ann Rheum Diseases (2019) 78(2):209. doi: 10.1136/annrheumdis-2018-214229
178. Weissler KA, Frischmeyer-Guerrerio PA. Genetic evidence for the role of transforming growth factor-β in atopic phenotypes. Curr Opin Immunol (2019) 60:54–62. doi: 10.1016/j.coi.2019.05.002
179. Kulkarni AB, Karlsson S. Transforming growth factor-beta 1 knockout mice. A mutation in one cytokine gene causes a dramatic inflammatory disease. Am J Pathol (1993) 143(1):3–9.
180. Marie JC, Letterio JJ, Gavin M, Rudensky AY. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J Exp Med (2005) 201(7):1061–7. doi: 10.1084/jem.20042276
181. Chen W, Jin W, Hardegen N, Lei K-J, Li L, Marinos N, et al. Conversion of peripheral CD4+CD25- naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med (2003) 198(12):1875–86. doi: 10.1084/jem.20030152
182. Kanamori M, Nakatsukasa H, Okada M, Lu Q, Yoshimura A. Induced Regulatory T Cells: Their Development, Stability, and Applications. Trends Immunol (2016) 37(11):803–11. doi: 10.1016/j.it.2016.08.012
183. Anthoni M, Fyhrquist-Vanni N, Wolff H, Alenius H, Lauerma A. Transforming growth factor-beta/Smad3 signalling regulates inflammatory responses in a murine model of contact hypersensitivity. Br J Dermatol (2008) 159(3):546–54. doi: 10.1111/j.1365-2133.2008.08696.x
184. Konkel JE, Zhang D, Zanvit P, Chia C, Zangarle-Murray T, Jin W, et al. Transforming growth factor-β signaling in regulatory T cells controls T helper-17 cells and tissue-specific immune responses. Immunity (2017) 46(4):660–74. doi: 10.1016/j.immuni.2017.03.015
185. Lee GR. The Balance of Th17 versus Treg Cells in Autoimmunity. Int J Mol Sci (2018) 19(3):730. doi: 10.3390/ijms19030730
186. Yamazaki T, Yang XO, Chung Y, Fukunaga A, Nurieva R, Pappu B, et al. CCR6 regulates the migration of inflammatory and regulatory T cells. J Immunol (Baltimore Md 1950) (2008) 181(12):8391–401. doi: 10.4049/jimmunol.181.12.8391
187. Fujiyama T, Kawakami C, Sugita K, Kubo-Kabashima R, Sawada Y, Hino R, et al. Increased frequencies of Th17 cells in drug eruptions. J Dermatol Sci (2014) 73(1):85–8. doi: 10.1016/j.jdermsci.2013.08.008
188. Kimura A, Naka T, Nohara K, Fujii-Kuriyama Y, Kishimoto T. Aryl hydrocarbon receptor regulates Stat1 activation and participates in the development of Th17 cells. Proc Natl Acad Sci (2008) 105(28):9721. doi: 10.1073/pnas.0804231105
189. Di Meglio P, Duarte JH, Ahlfors H, Owens ND, Li Y, Villanova F, et al. Activation of the aryl hydrocarbon receptor dampens the severity of inflammatory skin conditions. Immunity (2014) 40(6):989–1001. doi: 10.1016/j.immuni.2014.04.019
190. Singh NP, Singh UP, Rouse M, Zhang J, Chatterjee S, Nagarkatti PS, et al. Dietary Indoles Suppress Delayed-Type Hypersensitivity by Inducing a Switch from Proinflammatory Th17 Cells to Anti-Inflammatory Regulatory T Cells through Regulation of MicroRNA. J Immunol (2016) 196(3):1108. doi: 10.4049/jimmunol.1501727
191. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol (2010) 11(9):846–53. doi: 10.1038/ni.1915
192. Gratz IK, Truong H-A, Yang SH-Y, Maurano MM, Lee K, Abbas AK, et al. Cutting Edge: memory regulatory t cells require IL-7 and not IL-2 for their maintenance in peripheral tissues. J Immunol (Baltimore Md 1950) (2013) 190(9):4483–7. doi: 10.4049/jimmunol.1300212
193. Simonetta F, Chiali A, Cordier C, Urrutia A, Girault I, Bloquet S, et al. Increased CD127 expression on activated FOXP3+CD4+ regulatory T cells. Eur J Immunol (2010) 40(9):2528–38. doi: 10.1002/eji.201040531
194. Savage PA, Klawon DEJ, Miller CH. Regulatory T Cell Development. Annu Rev Immunol (2020) 38:421–53. doi: 10.1146/annurev-immunol-100219-020937
195. Marshall D, Sinclair C, Tung S, Seddon B. Differential requirement for IL-2 and IL-15 during bifurcated development of thymic regulatory T cells. J Immunol (Baltimore Md 1950) (2014) 193(11):5525–33. doi: 10.4049/jimmunol.1402144
196. Clark RA, Kupper TS. IL-15 and dermal fibroblasts induce proliferation of natural regulatory T cells isolated from human skin. Blood (2007) 109(1):194–202. doi: 10.1182/blood-2006-02-002873
197. Su S-C, Mockenhaupt M, Wolkenstein P, Dunant A, Le Gouvello S, Chen C-B, et al. Interleukin-15 Is Associated with Severity and Mortality in Stevens-Johnson Syndrome/Toxic Epidermal Necrolysis. J Invest Dermatol (2017) 137(5):1065–73. doi: 10.1016/j.jid.2016.11.034
198. Ku CC, Murakami M, Sakamoto A, Kappler J, Marrack P. Control of homeostasis of CD8+ memory T cells by opposing cytokines. Science (2000) 288(5466):675–8. doi: 10.1126/science.288.5466.675
199. Carson WE, Fehniger TA, Haldar S, Eckhert K, Lindemann MJ, Lai C-F, et al. A potential role for interleukin-15 in the regulation of human natural killer cell survival. J Clin Invest (1997) 99(5):937–43. doi: 10.1172/JCI119258
200. Abdel-Salam B, Ebaid H. Upregulation of major histocompatibility complex class II, CD83, CD64, and CD14 on polymorphonuclear neutrophils stimulated with interleukin-15. J Microbiol Immunol Infect (2008) 41(6):462–8.
201. Kim YS, Maslinski W, Zheng XX, Stevens AC, Li XC, Tesch GH, et al. Targeting the IL-15 receptor with an antagonist IL-15 mutant/Fcγ2a protein blocks delayed-type hypersensitivity. J Immunol (1998) 160(12):5742–8. doi: 10.1097/00007890-199805131-00709
202. Bruhs A, Proksch E, Schwarz T, Schwarz A. Disruption of the Epidermal Barrier Induces Regulatory T Cells via IL-33 in Mice. J Invest Dermatol (2018) 138(3):570–9. doi: 10.1016/j.jid.2017.09.032
203. Adachi A, Komine M, Tsuda H, Nakajima S, Kabashima K, Ohtsuki M. Differential expression of alarmins: IL-33 as a candidate marker for early diagnosis of toxic epidermal necrolysis. J Allergy Clin Immunol: In Pract (2019) 7(1):325–7. doi: 10.1016/j.jaip.2018.05.037
204. Schildberg FA, Klein SR, Freeman GJ, Sharpe AH. Coinhibitory Pathways in the B7-CD28 Ligand-Receptor Family. Immunity (2016) 44(5):955–72. doi: 10.1016/j.immuni.2016.05.002
205. Chen Q, Mo L, Cai X, Wei L, Xie Z, Li H, et al. ICOS signal facilitates Foxp3 transcription to favor suppressive function of regulatory T cells. Int J Med Sci (2018) 15(7):666–73. doi: 10.7150/ijms.23940
206. Vocanson M, Rozieres A, Hennino A, Poyet G, Gaillard V, Renaudineau S, et al. Inducible costimulator (ICOS) is a marker for highly suppressive antigen-specific T cells sharing features of TH17/TH1 and regulatory T cells. J Allergy Clin Immunol (2010) 126(2):280–9.e7. doi: 10.1016/j.jaci.2010.05.022
207. Gomez de Agüero M, Vocanson M, Hacini-Rachinel F, Taillardet M, Sparwasser T, Kissenpfennig A, et al. Langerhans cells protect from allergic contact dermatitis in mice by tolerizing CD8(+) T cells and activating Foxp3(+) regulatory T cells. J Clin Invest (2012) 122(5):1700–11. doi: 10.1172/JCI59725
208. Remedios KA, Zirak B, Sandoval PM, Lowe MM, Boda D, Henley E, et al. The TNFRSF members CD27 and OX40 coordinately limit TH17 differentiation in regulatory T cells. Sci Immunol (2018) 3(30):eaau2042. doi: 10.1126/sciimmunol.aau2042
209. Phillips GS, Wu J, Hellmann MD, Postow MA, Rizvi NA, Freites-Martinez A, et al. Treatment Outcomes of Immune-Related Cutaneous Adverse Events. J Clin Oncol (2019) 37(30):2746–58. doi: 10.1200/JCO.18.02141
210. de Oliveira Rodrigues R, Helena Barem Rabenhorst S, Germano de Carvalho P, Luri Sasahara G, Mabel Ferreira Vasconcelos L, Antônio Gomes de Arruda É, et al. Association of IL10, IL4, IFNG, and CTLA4 gene polymorphisms with efavirenz hypersensitivity reaction in patients infected with human immunodeficiency virus. Japanese J Infect Diseases (2017) 70(4):430–6. doi: 10.7883/yoken.JJID.2016.075
211. Ferreira Vasconcelos LM, Rodrigues RDO, Albuquerque AA, Barroso GD, Sasahara GL, Severo Ferreira JF, et al. Polymorphism of IL10, IL4, CTLA4, and DAO Genes in Cross-Reactive Nonsteroidal Anti-inflammatory Drug Hypersensitivity. J Clin Pharmacol (2018) 58(1):107–13. doi: 10.1002/jcph.986
212. Goldinger SM, Stieger P, Meier B, Micaletto S, Contassot E, French LE, et al. Cytotoxic Cutaneous Adverse Drug Reactions during Anti-PD-1 Therapy. Clin Cancer Res (2016) 22(16):4023. doi: 10.1158/1078-0432.CCR-15-2872
213. Vivar KL, Deschaine M, Messina J, Divine JM, Rabionet A, Patel N, et al. Epidermal programmed cell death-ligand 1 expression in TEN associated with nivolumab therapy. J Cutaneous Pathol (2017) 44(4):381–4. doi: 10.1111/cup.12876
214. Christensen AD, Skov S, Haase C. Local and systemic effects of co-stimulatory blockade using cytotoxic T lymphocyte antigen-4-immunoglobulin in dinitrofluorobenzene- and oxazolone-induced contact hypersensitivity in mice. Clin Exp Immunol (2013) 171(2):220–30. doi: 10.1111/cei.12005
215. Metushi IG, Hayes MA, Uetrecht J. Treatment of PD-1(-/-) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology (2015) 61(4):1332–42. doi: 10.1002/hep.27549
216. Cho T, Uetrecht J. How Reactive Metabolites Induce an Immune Response That Sometimes Leads to an Idiosyncratic Drug Reaction. Chem Res Toxicol (2017) 30(1):295–314. doi: 10.1021/acs.chemrestox.6b00357
217. Mak A, Uetrecht J. The Combination of Anti-CTLA-4 and PD1-/- Mice Unmasks the Potential of Isoniazid and Nevirapine To Cause Liver Injury. Chem Res Toxicol (2015) 28(12):2287–91. doi: 10.1021/acs.chemrestox.5b00305
218. Lu J, Thuraisingam T, Chergui M, Nguyen K. Nivolumab-associated DRESS syndrome: A case report. JAAD Case Rep (2019) 5(3):216–8. doi: 10.1016/j.jdcr.2018.11.017
219. Gamradt P, Laoubi L, Nosbaum A, Mutez V, Lenief V, Grande S, et al. Inhibitory checkpoint receptors control CD8+ resident memory T cells to prevent skin allergy. J Allergy Clin Immunol (2019) 143(6):2147–57.e9. doi: 10.1016/j.jaci.2018.11.048
220. Nakagomi D, Suzuki K, Hosokawa J, Kobayashi Y, Suto A, Takatori H, et al. Therapeutic Potential of B and T Lymphocyte Attenuator Expressed on CD8+ T Cells for Contact Hypersensitivity. J Invest Dermatol (2013) 133(3):702–11. doi: 10.1038/jid.2012.396
221. Seidel JA, Otsuka A, Kabashima K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front Oncol (2018) 8:86–. doi: 10.3389/fonc.2018.00086
222. Wei SC, Duffy CR, Allison JP. Fundamental Mechanisms of Immune Checkpoint Blockade Therapy. Cancer Discov (2018) 8(9):1069. doi: 10.1158/2159-8290.CD-18-0367
223. Rowshanravan B, Halliday N, Sansom DM. CTLA-4: a moving target in immunotherapy. Blood (2018) 131(1):58–67. doi: 10.1182/blood-2017-06-741033
224. Stamper CC, Zhang Y, Tobin JF, Erbe DV, Ikemizu S, Davis SJ, et al. Crystal structure of the B7-1/CTLA-4 complex that inhibits human immune responses. Nature (2001) 410(6828):608–11. doi: 10.1038/35069118
225. Walker LSK, Sansom DM. The emerging role of CTLA4 as a cell-extrinsic regulator of T cell responses. Nat Rev Immunol (2011) 11(12):852–63. doi: 10.1038/nri3108
226. Blair HA, Deeks ED. Abatacept: A Review in Rheumatoid Arthritis. Drugs (2017) 77(11):1221–33. doi: 10.1007/s40265-017-0775-4
227. Hosseini A, Gharibi T, Marofi F, Babaloo Z, Baradaran B. CTLA-4: From mechanism to autoimmune therapy. Int Immunopharmacol (2020) 80:106221. doi: 10.1016/j.intimp.2020.106221
228. Chang M-C, Chang Y-T, Tien Y-W, Liang P-C, Jan IS, Wei S-C, et al. T-Cell Regulatory Gene CTLA-4 Polymorphism/Haplotype Association with Autoimmune Pancreatitis. Clin Chem (2007) 53(9):1700–5. doi: 10.1373/clinchem.2007.085951
229. Verma N, Burns SO, Walker LSK, Sansom DM. Immune deficiency and autoimmunity in patients with CTLA-4 (CD152) mutations. Clin Exp Immunol (2017) 190(1):1–7. doi: 10.1111/cei.12997
230. Wu Y, Chen W, Xu ZP, Gu W. PD-L1 Distribution and Perspective for Cancer Immunotherapy—Blockade, Knockdown, or Inhibition. Front Immunol (2019) 10:2022. doi: 10.3389/fimmu.2019.02022
231. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annu Rev Immunol (2008) 26(1):677–704. doi: 10.1146/annurev.immunol.26.021607.090331
232. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18(3):153–67. doi: 10.1038/nri.2017.108
233. Paluch C, Santos AM, Anzilotti C, Cornall RJ, Davis SJ. Immune Checkpoints as Therapeutic Targets in Autoimmunity. Front Immunol (2018) 9:2306–. doi: 10.3389/fimmu.2018.02306
234. Sibaud V. Dermatologic Reactions to Immune Checkpoint Inhibitors. Am J Clin Dermatol (2018) 19(3):345–61. doi: 10.1007/s40257-017-0336-3
235. Pintova S, Sidhu H, Friedlander PA, Holcombe RF. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res (2013) 23(6):498–501. doi: 10.1097/CMR.0000000000000017
236. Maloney NJ, Ravi V, Cheng K, Bach DQ, Worswick S. Stevens-Johnson syndrome and toxic epidermal necrolysis-like reactions to checkpoint inhibitors: a systematic review. Int J Dermatol (2020) 59(6):e183–8. doi: 10.1111/ijd.14811
237. Postow MA, Sidlow R, Hellmann MD. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. New Engl J Med (2018) 378(2):158–68. doi: 10.1056/NEJMra1703481
238. Andrews LP, Yano H, Vignali DAA. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol (2019) 20(11):1425–34. doi: 10.1038/s41590-019-0512-0
239. Le Mercier I, Lines JL, Noelle RJ. Beyond CTLA-4 and PD-1, the Generation Z of Negative Checkpoint Regulators. Front Immunol (2015) 6:418). doi: 10.3389/fimmu.2015.00418
240. Maruhashi T, Okazaki I-M, Sugiura D, Takahashi S, Maeda TK, Shimizu K, et al. LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat Immunol (2018) 19(12):1415–26. doi: 10.1038/s41590-018-0217-9
241. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer (2018) 9(5-6):176–89. doi: 10.18632/genesandcancer.180
242. Blessin NC, Simon R, Kluth M, Fischer K, Hube-Magg C, Li W, et al. Patterns of TIGIT Expression in Lymphatic Tissue, Inflammation, and Cancer. Dis Markers (2019) 2019:5160565. doi: 10.1155/2019/5160565
243. Meng F, Li L, Lu F, Yue J, Liu Z, Zhang W, et al. Overexpression of TIGIT in NK and T Cells Contributes to Tumor Immune Escape in Myelodysplastic Syndromes. Front Oncol (2020) 10:1595. doi: 10.3389/fonc.2020.01595
244. Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, et al. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol (2003) 4(7):670–9. doi: 10.1038/ni944
245. Han P, Goularte OD, Rufner K, Wilkinson B, Kaye J. An Inhibitory Ig Superfamily Protein Expressed by Lymphocytes and APCs Is Also an Early Marker of Thymocyte Positive Selection. J Immunol (2004) 172(10):5931. doi: 10.4049/jimmunol.172.10.5931
246. Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, et al. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol (2005) 6(1):90–8. doi: 10.1038/ni1144
Keywords: contact dermatitis, delayed type hypersensitivity, drug reaction with eosinophilia and systemic symptoms, cosignaling pathways, immune checkpoints, regulatory T cells, Stevens-Johnson Syndrome, toxic epidermal necrolysis
Citation: Hsu Y-SO, Lu K-L, Fu Y, Wang C-W, Lu C-W, Lin Y-F, Chang W-C, Yeh K-Y, Hung S-I, Chung W-H and Chen C-B (2021) The Roles of Immunoregulatory Networks in Severe Drug Hypersensitivity. Front. Immunol. 12:597761. doi: 10.3389/fimmu.2021.597761
Received: 21 August 2020; Accepted: 18 January 2021;
Published: 26 February 2021.
Edited by:
Stephen Robert Daley, Queensland University of Technology, AustraliaReviewed by:
Nicole Andrea Mifsud, Monash University, AustraliaCopyright © 2021 Hsu, Lu, Fu, Wang, Lu, Lin, Chang, Yeh, Hung, Chung and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Hung Chung, d2VuaHVuZ2NodW5nQHlhaG9vLmNvbQ==; Chun-Bing Chen, Y2h1bmJpbmcuY2hlbkBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.