 Philip McMillan
Philip McMillan Thomas Dexhiemer2
Thomas Dexhiemer2 Bruce D. Uhal
Bruce D. Uhal- 1Doncaster and Bassetlaw National Health Service (NHS) Trust, Doncaster, United Kingdom
- 2Department of Pharmacology and Toxicology, Michigan State University, East Lansing, MI, United States
- 3Nicholas V. Perricone, M.D., Division of Dermatology, Department of Medicine, Michigan State University, East Lansing, MI, United States
- 4Department of Physiology, Michigan State University, East Lansing, MI, United States
The COVID-19 pandemic caused by the coronavirus SARS-COV-2 has cost many lives worldwide. In dealing with affected patients, the physician is faced with a very unusual pattern of organ damage that is not easily explained on the basis of prior knowledge of viral-induced pathogenesis. It is established that the main receptor for viral entry into tissues is the protein angiotensin-converting enzyme-2 [“ACE-2”, (1)]. In a recent publication (2), a theory of autoimmunity against ACE-2, and/or against the ACE-2/SARS-COV-2 spike protein complex or degradation products thereof, was proposed as a possible explanation for the unusual pattern of organ damage seen in COVID-19. In the light of more recent information, this manuscript expands on the earlier proposed theory and offers additional, testable hypotheses that could explain both the pattern and timeline of organ dysfunction most often observed in COVID-19.
Background of COVID-19
The first epidemic involving Severe Acute Respiratory Syndrome (“SARS”) was identified in 2003 as a novel clinical entity (3). This SARS-COV infection was primarily clustered in Asia (4) with some international spread, and ~770 deaths and over 8,000 people affected worldwide (5). SARS-COV belongs to the family Coronaviridae, which comprises enveloped RNA viruses in the order Nidovirales (6), and is the largest known non-segmented genome among RNA viruses (7) with ~15 spike proteins on the surface of each virion (8). These spike proteins represent the attachment point for entry of the virus into the cell. The reason for the disappearance of this dangerous infection is unclear, but effective preventative measures and an antigenic shift may be possible explanations (9).
The current COVID-19 pandemic was triggered by the spread of a novel coronavirus SARS-COV-2 (10), with the earliest reports coming out of Wuhan, China, in late 2019. Although it has an 80% similarity to SARS-COV, there are specific differences within the receptor-binding domain of the spike protein which impact on infectivity (1, 11). This variation has a combination of high infectivity and spread through undocumented infection, promoting rapid dissemination across the world (12). The severe organ damage, which occurs in a subset of people affected by the virus, is similar to that caused by SARS-COV, but occurs on a much larger scale because of the higher number of people infected. People at risk of severe disease represent ~2.3% of those who contract the virus, and are primarily the elderly and those with comorbidities (13).
Current Understanding of the Disease
ACE-2 is a Type 1 membrane-bound glycoprotein with 42% homology to angiotensin-converting enzyme (“ACE”) (14). The primary purpose of ACE-2 is to convert angiotensin II to angiotensin (1–7) which has an impact on arterial pressures and inflammation (15). This role is counter regulatory to ACE and serves to balance the renin-angiotensin system (“RAS”). It is relevant in COVID-19 because ACE-2 is also the entry receptor for SARS-COV-2.
The current thinking is that SARS-COV-2 targets the ACE-2 entry receptor (16), causing direct viral cellular damage, with the release of excessive immune mediators and coagulation abnormalities (17). Identification of viral particles in the lung tissue supports this mechanism for damage (18). Release of immune factors (19) appears to be part of the trigger for an associated cytokine storm. Elevated levels of interleukins, tumor necrosis factor and interferons have been observed (20). Immune-mediated disseminated intravascular coagulation with lung micro-thrombosis is an observed pattern across patients (21, 22). There is also the perspective that damage to the ACE-2 receptor could impact on the ability of RAS to function effectively (23). The presence of ACE-2 receptors in the kidney may offer an explanation of the extent of renal dysfunction in more severely affected patients (24).
These hypotheses are, however, unable to explain the severity of the disease and involvement of multiple organs with vasculitic-type responses in SARS-COV-2.
What Is Soluble ACE-2?
Soluble ACE-2, also called serum or plasma ACE-2, refers to the ACE-2 enzyme ectodomain that has been cleaved from the cell surface, a process called shedding (25). The purpose of shedding is unclear (26), but it seems to occur more frequently in hypertension and heart disease (27). Clinically relevant stimuli, such as supplemental oxygen at levels routinely used in neonatal medicine (FiO2 0.95), have also been shown to result in ACE-2 shedding from human lung cells in culture (28). This process reduces the amount of ACE-2 localized to the cells, but increases that free to enter other tissue compartments. To date, the functions and fate(s) of this soluble ACE-2 in different organs has not been rigorously studied.
Soluble ACE-2 has been shown to bind efficiently to the spike protein of SARS-COV (29) and, although not confirmed by direct measurements as of the time of this writing, such binding is assumed to occur with SARS-COV-2 as well. Indeed, recent modeling studies based on the sequence of soluble ACE-2 and the SARS-CoV2 spike protein (30) strongly predicted that SARS-CoV-2 can not only bind to soluble ACE-2, but also suggest that the strength of binding might be negatively impacted by nicotine, a finding which is being considered for potential therapeutic value.
The assumption that soluble ACE-2 binds SARS-CoV-2 is central to the hypothesis of autoimmunity to ACE-2. To begin making direct measurements to test this assumption, a preparation of recombinant human ACE-2 (rhACE-2, Acrobiosystems, amino acids 18-740) that is of similar length to that of soluble ACE-2 [18-708, (30)] was analyzed by SPR (surface plasmon resonance) assay for binding to a recombinant SARS-CoV-2 spike protein receptor binding domain (RBD, amino acids 321-591, Acrobiosystems). As shown in Figure 1A, increasing concentrations of rhACE-2 showed proportional increases in binding to the immobilized SARS-2 protein spike RBD. This enabled estimation of a binding affinity (kD) of ~74 nM (Figure 1B), which is in the range predicted by modeling studies and is greater than the binding affinity of SARS-CoV spike protein to ACE-2 (31). Figure 2 shows concentration-dependent inhibition of immobilized rhACE-2 binding to immobilized recombinant spike RBD, in this case determined by a bead-based Alpha-Assay (32), by soluble rhACE-2 added to the bead-based assay system at the concentrations indicated. Thus, the data in Figures 1, 2 strongly support the theory that a complex of SARS-CoV-2 and soluble ACE-2 is formed and circulating in the blood of infected patients.
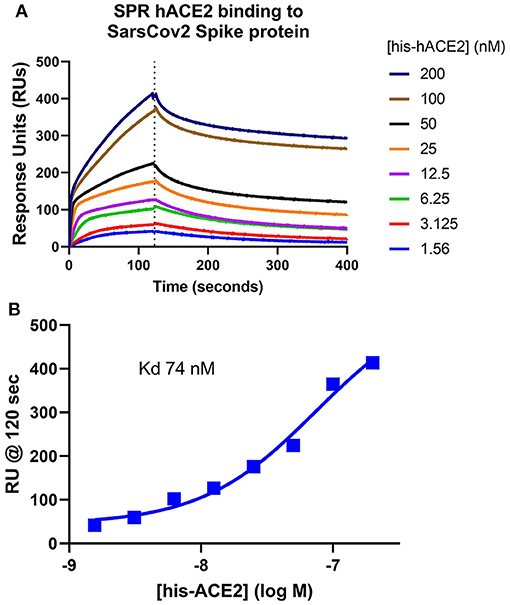
Figure 1. Surface Plasmon Resonance (SPR) determination of soluble ACE-2 binding affinity to SARS-CoV-2 spike protein. (A) Recombinant human ACE-2 (rhACE-2, Acrobiosystems), was analyzed by SPR assay for binding to a recombinant SARS-CoV-2 spike protein receptor binding domain (RBD, Acrobiosystems) immobilized in the flow chamber. As shown, increasing concentrations of the soluble rhACE-2 (in nM) showed proportional increases in binding to immobilized spike protein RBD. (B) These data allowed estimation of kD ~74 nM.
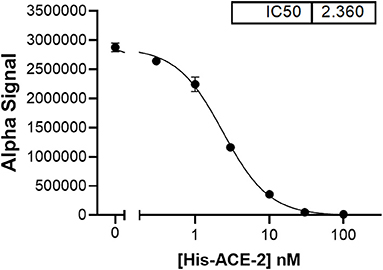
Figure 2. In a bead-based assay system [AlphaLISA proximity assay, (32)], which uses both rhACE-2 and spike RBD immobilized to beads, soluble rhACE-2 added to the assay buffer showed concentration-dependent inhibition of bead-bound spike RBD binding to bead-bound rhACE-2, with an IC50 ~2.4 nM.
Exploring differences in ACE-2 binding with the virus has not been straightforward, with some research suggesting similar binding affinities to SARS-COV (33) and other research inferring stronger complexes (34). The primary difference is the mutation in the spike protein which causes a 10-fold increase in the binding of SARS-COV-2 over that of SARS-CoV (31). Further, the same authors used cryo-EM studies to show that binding of ACE-2 to SARS-CoV-2 spike protein induces conformational changes in both proteins, which we postulate may form new epitopes that provide targets for autoantibody formation (35). This plays an important role in the hypothesis of autoimmunity.
There appears to be genetic polymorphism of the ACE-2 with increased risk of specific comorbidities—hypertension, cardiovascular disease, and diabetes (36, 37). The impact of allelic variants was reviewed in a computerized model and it was demonstrated that it is likely that some variations of ACE-2 will bind more tightly to the SARS-COV-2 spike protein (38). The current hypothesis of autoimmunity postulates that higher levels of soluble ACE-2, or augmented conformational binding to the spike protein, increases the probability that the combined entity will be processed by an antigen-presenting cell as part of the virus. This may lead to antibody production against ACE-2, which triggers Type 2 and 3 hypersensitivity responses, and Type 4 cellular immune targeting after the viral particles with attached soluble ACE-2 are processed by antigen-presenting cells.
Although most infectious diseases target both ends of the age spectrum because of either poorly-developed or impaired immune responses, COVID-19 disproportionately impacts the elderly. Soluble ACE-2 can explain the paradox of high mortality in the elderly without a similar raised infant mortality rate. Elevated levels of soluble ACE-2 have been noted in comorbidities associated with higher mortality in COVID-19 (39). There are undetectable levels in the serum of healthy individuals (40) and a correlation exists between the occurrence of soluble ACE-2 and an individual's age (41). Recent research has indicated that soluble ACE-2 is the most significant risk factor for cardiometabolic mortality and could be relevant in COVID-19 (42).
Summary of Principle of Autoimmunity
It has been proposed that autoimmunity to ACE-2 (43) and ACE (2) is triggered by the viral infection in SARS-COV-2. It was postulated that in people with high levels of soluble ACE-2, the viral spike protein binding tightly together with ACE-2 could be endocytosed by macrophages which would function as antigen-presenting cells. The whole viral protein could be proteolytically cleaved, including ACE-2, tightly attached to the viral spike protein, possibly producing new epitopes for antibody generation. ACE-2 has a 42% homology with ACE, which could mean that autoantibodies that were formed against ACE-2 might cross- react with ACE as well. In this circumstance, the antibodies would target both ACE and ACE-2 cellular attached enzymes, leading to severe inflammation throughout the body, especially on lung endothelial cells. Consistent with this hypothesis, very recent data documented higher levels of soluble ACE-2 in the serum of critically ill CoVid-19 patients (44).
Types of Hypersensitivity Immune Responses
There are four types of immune responses in the body, with the first three being antibody dependent (45) and the fourth being cellular (46).
Type 1 Hypersensitivity
This is the typical anaphylaxis response to an external antigen after the body has been previously sensitized (47). This response is typically mediated by IgE after pre-sensitization to an antigen. The most important examples are that of pollen and nut allergies. In the context of autoimmunity to ACE-2, this is not considered to be particularly relevant as no significant difference with IgE was noted in COVID-19 (48).
Type 2 Hypersensitivity
This represents the production of IgM or IgG antibodies to cellular or extracellular matrix proteins. An example of the extracellular antibody response is seen in Goodpasture's syndrome where Type 4 collagen is the antigen (49) which subsequently activates complement and the phagocytic system with associated kidney damage. Another example is immune thrombocytopenia which demonstrates a cellular immune response to platelets (50) with subsequent platelet destruction by this antibody response. In this case, the antigen is on the surface of platelets and the antibody will target the platelets with complement activation and increased phagocytosis. This is likely to be an early type of immune response in SARS-COV-2, with IgM produced against ACE and ACE-2, primarily targeting the endothelial blood vessels in the lung and small intestines. This was also demonstrated with specific B cell activation in the serum of patients with COVID-19 along a similar pattern to Systemic Lupus Erythematosus (51).
Type 3 Hypersensitivity
The most common example is serum sickness where antigen and antibody complexes are formed in the blood and become deposited in tissues with associated inflammation (52). There is typically a systemic vasculitic response with endothelial swelling, inflammatory infiltrates, and fibrinoid necrosis of the arterial wall (53). This may be an early response in SARS-COV-2, with immune complexes deposited in the liver and lungs. In some of the limited autopsies in COVID-19, core lung biopsies revealed fibrinoid necrosis within pulmonary vessels (54), pathognomonic of Type III hypersensitivity.
Type 4 Hypersensitivity
In this form of immune response, the antigen triggers the activation of CD8 lymphocytes which target cells that are infected with virus. This is an essential aspect of immune regulation as it allows the immune system to target virally-infected cells selectively (46). An early immune response to the viral complex with ACE-2 would also stimulate CD8 lymphocytes to target ACE-2 and, potentially, ACE. This is also likely to create immune depletion of lymphocytes as the immune system becomes overwhelmed. To date, the characteristic Type 4 hypersensitivity responses observed in COVID-19 include perivascular T-cell infiltrates (55, 56) with evidence of distal organ involvement in the adrenals (57) and giant cell pathology (58, 59).
The principle of autoimmunity can therefore explain the cytokine storm. It is likely to be a combination of three types of immune hypersensitivity—Types 2, 3, and 4. This condition has not previously been described in any other disease.
Ferritin in COVID-19
Elevated levels of ferritin are a significant observation in COVID-19, with an increased risk of mortality (60). This hyperferritinemia is much worse than the typical liver function abnormalities that are also noted early in the course of the disease (61). Elevation of serum ferritin is a pattern in severe viral infections as well as septic shock (62). Whilst this association is observed, it is unclear if elevated ferritin is the cause of inflammation (63). Ferritin has been shown to have immune modulating functions (64, 65) and is also elevated in several autoimmune conditions (66). Significantly elevated serum ferritin levels also occur in hemochromatosis, a genetic disease, without a severe inflammatory response (67). This suggests that elevated ferritin, on its own, may not be a direct mediator of cellular damage. The presence of autoantibodies to hepatocytes and gastric chief cells, unique to serum from COVID-19 patients, is strongly suggestive of a Type II autoimmune response (68).
Proposed Timeline of Symptoms
The following timeline and associated symptoms are based on the hypothesis of autoimmunity as the primary cause of disease:
Approximately 70% of people with COVID-19 are asymptomatic (69). This raises the question about whether the symptoms are linked primarily to the viral infection or to an autoimmune response. Based on this autoimmune theory, all symptoms, from the mild to more severe, can be positioned along the autoimmune spectrum.
• Fever and mild coryzal symptoms occur in 43.8%/50% on admission (70) and represents the immune viremic phase which is typically of relatively short duration (20, 71). It could be assumed that only viremia would cause these symptoms as an upper airway infection tends to be asymptomatic or have mild coryzal symptoms. The viremic phase occurs when viral particles are in the bloodstream and represents the starting point of symptoms. Those people with elevated levels of soluble ACE-2 or with genetic variations in the binding of the spike protein could be most prone to form IgM antibodies to ACE-2.
• Mild shortness of breath with cough could be triggered by IgM at day 3–4, targeting the endothelial-attached ACE and ACE-2 leading to Type 2 and 3 hypersensitivity. IgM could then cause endothelial damage in the lungs with associated shortness of breath secondary to pulmonary vascular leakage as evidenced by bibasal consolidation on the chest x-ray. There could also be microthrombi in the lung vessels.
• Abnormal liver function tests (“LFTs”) on admission to hospital occur in over half of patients (72) and may relate to Type 3 hypersensitivity, with immune complexes being deposited in the liver and subsequent hepatic inflammation. The IgM antibodies may bind to ACE-2 surface receptors on enterocytes in the small intestine and a proportion of these complexes are shed into the portal circulation. These immune complexes may then become lodged in the liver if the hepatic reticuloendothelial system becomes overwhelmed, and cause a mild hepatitis as evidenced by the abnormal LFTs early in the disease. There is no specific immune targeting in this case; the ACE-2 receptors are located in the small intestine and the complexes of ACE-2 plus IgM have to traverse the portal system. The observation of elevated serum ferritin is likely to be related to increased release from hepatocytes as part of the inflammatory response (66, 73).
• Lymphopenia could result from immune exhaustion in Type 4 hypersensitivity at day 7–14 (74). Once the viremia has occurred and soluble ACE-2 has become part of the immune response, CD8 lymphocytes are sensitized to target the lung, heart, and kidney due to overlap between ACE and ACE-2. The large number of antigens being targeted causes rapid lymphocyte exhaustion and lymphopenia (75).
• CXR infiltrates occur in 41% of positive COVID-19 admissions (76) and could indicate early lung damage at day 7–10. It involves Type 2, 3, and 4 hypersensitivity responses and may explain why this organ is primarily affected in mortality. Initially this is an IgM antibody response, but the pentameric structure of IgM may limit the number of ACE and ACE-2 receptors that can be targeted. Worsening of lung inflammation at day 11 could be linked to formation of IgG antibodies against ACE and ACE-2. Lung endothelial damage increases pulmonary vascular leakage with loss of albumin in the tissues and associated hypoalbuminemia (77).
• Renal failure occurs on admission in 14.4% of patients (78) as IgM could initially target ACE and ACE-2 receptors in the kidney leading to Type 2 hypersensitivity with involvement of renal tubular cells (79). There is no ACE in the glomerulus, but ACE-2 is located on the podocytes of the basement membrane in the glomerulus. Nephritis is not commonly a part of SARS-COV-2 (24).
• Acute Respiratory Distress Syndrome (“ARDS”) represents a severe deterioration in lung function possibly caused by formation of IgG antibodies at day 11 (80), worsening the Type 2 and 3 hypersensitivity response and leading to the cytokine storm. Clonal expansion of plasma cells with high levels of IgG to ACE-2 could increase lung and kidney inflammation. Persistent severe lung inflammation initiates fibroblast expansion, and hyaluronic acid is part of the immune response (81) which may contribute to lung fibrosis.
• Kawasaki-like disease in children (82) is likely to be Type 3 serum sickness-related vasculitis with fibrinoid necrosis in the arterial vessel walls.
• Taste abnormalities are possibly related to the targeting of ACE-2 receptors on taste buds (83) by IgA antibodies to ACE-2.
• Myocarditis is known to occur as an autoimmune response to viral infection (84). In COVID-19, myocarditis (85) may be triggered by Type 2 hypersensitivity to ACE and ACE-2 receptors on the myocardium.
• Stroke could be secondary to myocarditis combined with increased levels of clotting factors released from the liver secondary to immune-mediated hepatitis. Although a number of patients may have had a stroke during the COVID-19 infection, it is not clear if this represents a higher risk (86).
Conclusion
The autoimmune hypothesis appears to explain a significant proportion of the symptoms in SARS-COV-2-induced organ damage, along the lines of similar autoimmune diseases. In the vast majority of people affected, the disease is mild or asymptomatic. Identification of significant risk factors including elevated serum ACE-2 is critical as it would allow vaccination to target the most vulnerable population. This theory of autoimmunity in SARS-COV-2 has been reinforced by the recent RECOVERY Trial (87) showing the benefit of the steroid dexamethasone for immunosuppression. Additionally, high-dose methylprednisolone has also been proposed as a rescue, second-line treatment for patients who did not respond well to tocilizumab (88). In both treatment methods, there appears to be a benefit which would support the hypothesis of immune dysregulation due to autoimmunity to ACE and ACE-2.
In the time since the original submission of this manuscript, autoimmunity to COVID-19 has now been confirmed, as autoantibodies to ACE-2 have been demonstrated in the serum of individuals with severe disease (89). Autoantibodies to Type I interferons have also been characterized in severe COVID-19 (90) and may represent a bystander effect, in keeping with our primary hypothesis of autoimmunity to ACE-2. Whilst these studies are supportive of the theory of autoimmunity, further research will have to be conducted to demonstrate conclusively that autoantibodies are the cause of the organ damage observed in COVID-19. We invite the research community to help explore these hypotheses for the betterment of clinical strategies for treating individuals suffering from this disease.
Author Contributions
PM conceived and drafted the original manuscript. TD and RN conceived and performed protein-protein interaction experiments to obtain the original data. All authors edited, revised and approved the final manuscript.
Funding
This study was supported by Department of Physiology, Michigan State University and Doncaster & Bassetlaw NHS Trust, and by the Department of Pharmacology and Toxicology, MSU.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the expert editorial and preparatory work of Jacqueline Elaine Allison, Ph.D. and Simran Atul Patel, B.S.
References
1. Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. (2020) 581:215–20. doi: 10.1038/s41586-020-2180-5
2. McMillan P, Uhal BD. COVID-19 a theory of autoimmunity to ACE-2. MOJ Immunol. (2020) 7:17–9. doi: 10.15406/moji.2020.07.002510.15406
3. Drosten C, Günther S, Preiser W, van der Werf S, Brodt HR, Becker S, et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. New Engl J Med. (2003) 348:1967–76. doi: 10.1056/NEJMoa030747
4. Tsang KW, Ho PL, Ooi GC, Yee WK, Wang T, Chan-Yeung M, et al. A cluster of cases of severe acute respiratory syndrome in Hong Kong. N Engl J Med. (2003) 348:1977–85. doi: 10.1056/NEJMoa030666
5. Wang B, Potter SJ, Lin Y, Cunningham AL, Dwyer DE, Su Y, et al. Rapid and sensitive detection of severe acute respiratory syndrome coronavirus by rolling circle amplification. J Clin Microbiol. (2005) 43:2339–44. doi: 10.1128/JCM.43.5.2339–2344.2005
6. Cavanagh D. Nidovirales: a new order comprising Coronaviridae and Arteriviridae. Arch Virol. (1997) 142:629–33.
7. Tong TR. Airborne severe acute respiratory syndrome coronavirus and its implications. J Infect Dis. (2005) 191:1401–2. doi: 10.1086/429637
8. Lin S, Lee CK, Lee SY, Kao CL, Lin CW, Wang AB, et al. Surface ultrastructure of SARS coronavirus revealed by atomic force microscopy. Cell Microbiol. (2005) 7:1763–70. doi: 10.1111/j.1462-5822.2005.00593.x
9. Guan WJ, Zheng XY, Zeng GQ, Zhong NS. Severe acute respiratory syndrome: a vanished evil? J Thorac Dis. (2013) 5(Suppl. 2):S87–9. doi: 10.3978/j.issn.2072-1439.2013.02.08
10. Huang Y, Chen S, Yang Z, Guan W, Liu D, Lin Z, et al. SARS-CoV-2 viral load in clinical samples of critically ill patients. Am J Respir Crit Care Med. (2020) 201:1435–8. doi: 10.1164/rccm.202003-0572LE
11. Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med. (2020) 26:450–2. doi: 10.1038/s41591-020-0820-9
12. Li H, Liu L, Zhang D, Xu J, Dai H, Tang N, et al. SARS-CoV-2 and viral sepsis: observations and hypotheses. Lancet. (2020) 395:1517–20. doi: 10.1016/S0140-6736(20)30920-X
13. Porcheddu R, Serra C, Kelvin N, Rubino S. Similarity in case fatality rates (CFR) of COVID-19/SARS-COV-2 in Italy and China. J Infect Dev Countr. (2020) 14:125–8. doi: 10.3855/jidc.12600
14. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, et al. A novel angiotensin-converting enzyme–related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. (2000) 87:E1–9. doi: 10.1161/01.res.87.5.e1
15. Hayashi N, Yamamoto K, Ohishi M, Tatara Y, Takeya Y, Shiota A, et al. The counterregulating role of ACE2 and ACE2-mediated angiotensin 1–7 signaling against angiotensin II stimulation in vascular cells. Hypertens Res. (2010) 33:1182–5. doi: 10.1038/hr.2010.147
16. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. (2004) 203:631–7. doi: 10.1002/path.1570
17. Li R, Pei S, Chen B, Song Y, Zhang T, Yang W, et al. Substantial undocumented infection facilitates the rapid dissemination of novel coronavirus (SARS-CoV-2). Science. (2020) 368:489–93. doi: 10.1126/science.abb3221
18. Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, et al. A pathological report of three COVID-19 cases by minimally invasive autopsies. Chinese J Pathol. (2020) 49:411–7. doi: 10.3760/cma.j.cn112151-20200312-00193
19. Liu J, Li S, Liu J, Liang B, Wang X, Wang H, et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine. (2020) 55:102763. doi: 10.1016/j.ebiom.2020.102763
20. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y, et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. (2020) 395:497–506. doi: 10.1016/S0140-6736(20)30183-5
21. McGonagle D, O'Donnell J, Sharif K, Emery P, Bridgewood C. Immune mechanisms of pulmonary intravascular coagulopathy in COVID-19 pneumonia. Lancet Rheumatol. (2020) 2:e437–45. doi: 10.1016/S2665-991330121-1
22. Tang N, Li D, Wang X, Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. (2020) 18:844–7. doi: 10.1111/jth.14768
23. South AM, Tomlinson L, Edmonston D, Hiremath S, Sparks M. Controversies of renin–angiotensin system inhibition during the COVID-19 pandemic. Nat Rev Nephrol. (2020) 16:305–7. doi: 10.1038/s41581-020-0279-4
24. Su H, Yang M, Wan C, Yi LX, Tang F, Zhu HY, et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. (2020) 98:219–27. doi: 10.1016/j.kint.2020.04.003
25. Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus ADME, et al. The emerging role of ACE2 in physiology and disease. J Pathol. (2007) 212:1–11. doi: 10.1002/path.2162
26. Peron JPS, Helder N. Susceptibility of the elderly to SARS-CoV-2 infection: ACE-2 overexpression, shedding, and antibody-dependent enhancement (ADE). Clinics. (2020) 75:e1912. doi: 10.6061/clinics/2020/e1912
27. Epelman S, Tang WHW, Chen SY, Van Lente F, Francis GS, Sen S. Detection of soluble angiotensin-converting enzyme 2 in heart failure: insights into the endogenous counter-regulatory pathway of the renin-angiotensin-aldosterone system. J Am Coll Cardiol. (2008) 52:750–4. doi: 10.1016/j.jacc.2008.02.088
28. Oarhe C, Dang V, Dang M, Hguyen H, Gopallawa I, Gewolb I, et al. Hyperoxia downregulates angiotensin converting enzyme 2 (ACE-2) in human fetal lung fibroblasts. Pediatr Res. (2015) 77:656–62. doi: 10.1038/pr.2015.27
29. Wong SK, Li W, Moore MJ, Choe H, Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem. (2004) 279:3197–201. doi: 10.1074/jbc.C300520200
30. Kumar CS, Kumar SA, Wei H. Comparative docking studies to understand the binding affinity of nicotine with soluble ACE2 (sACE2)-SARS-CoV-2 complex over sACE2. Toxicol Rep. (2020) 7:1366–72. doi: 10.1016/j.toxrep.2020.10.002
31. Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL, Olubukola A, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. (2020) 367:1260–3. doi: 10.1126/science.abb2507
32. Hanson QM, Wilson KM, Shen M, Itkin Z, Eastman RT, Shinn P, et al. Targeting ACE2-RBD interaction as a platform for COVID19 therapeutics: development and drug repurposing screen of an AlphaLISA proximity assay. BioRxiv. (2020). doi: 10.1101/2020.06.16.154708
33. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. (2020) 181:281–92.e.6. doi: 10.1016/j.cell.2020.02.058
34. Chowdhury R, Maranas CD. Biophysical characterization of the SARS-CoV2 spike protein binding with the ACE2 receptor explains increased COVID-19 pathogenesis. BioRxiv. (2020). doi: 10.1101/2020.03.30.015891
35. Zhang Y, Zheng N, Hao P, Cao Y, Zhong Y. A molecular docking model of SARS-CoV S1 protein in complex with its receptor, human ACE2. Comput Biol Chem. (2005) 29:254–7. doi: 10.1016/j.compbiolchem.2005.04.008
36. Luo Y, Liu C, Guan T, Li Y, Lai Y, Li F, et al. Association of ACE2 genetic polymorphisms with hypertension-related target organ damages in south Xinjiang. Hypertens. Res. (2019) 42:681–9. doi: 10.1038/s41440-018-0166-6
37. Meng N, Zhang Y, Ma J, Li H, Zhou F, Qu Y. Association of polymorphisms of angiotensin I converting enzyme 2 with retinopathy in type 2 diabetes mellitus among Chinese individuals. Eye. (2015) 29:266–71. doi: 10.1038/eye.2014.254
38. Hussain M, Jabeen N, Raza F, Shabbir S, Baig AA, Amanullah A, et al. Structural variations in human ACE2 may influence its binding with SARS-CoV-2 spike protein. J Med Virol. (2020) 92:1580–6. doi: 10.1002/jmv.25832
39. Sama IE, Ravera A, Santema BT, van Goor H, Maaten JMT, Cleland JGF, et al. Circulating plasma concentrations of angiotensin-converting enzyme 2 in men and women with heart failure and effects of renin–angiotensin–aldosterone inhibitors. Eur Heart J. (2020) 41:1810–7. doi: 10.1093/eurheartj/ehaa373
40. Lew RA, Warner FJ, Hanchapola I, Yarski MA, Ramchand J, Burrell LM, et al. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp Physiol. (2008) 93:685–93. doi: 10.1113/expphysiol.2007.040352
41. Rice GI, Jones AL, Grant PJ, Carter AM, Turner AJ, Hooper NM. Circulating activities of angiotensin-converting enzyme, its homolog, angiotensin-converting enzyme 2, and neprilysin in a family study. Hypertension. (2006) 48:914–20. doi: 10.1161/01.HYP.0000244543.91937.79
42. Narula S, Yusuf S, Chong M, Ramasundarahettige C, Rangarajan S, Bangdiwala SI, et al. Plasma ACE2 and risk of death or cardiometabolic diseases: a case-cohort analysis. Lancet. (2020) 396:968–76. doi: 10.1016/S0140-673631964-4
43. Townsend A. Autoimmunity to ACE2 as a possible cause of tissue inflammation in Covid-19. Med Hypotheses. (2020) 144:110043. doi: 10.1016/j.mehy.2020.110043
44. van Lier D, Kox M, Santos K, van der Hoeven H, Pillay J, Pickkers P. Increased blood Angiotensin Converting Enzyme 2 activity in critically ill COVID-19 patients. ERJ Open Res. (2021) 7. doi: 10.1183/23120541.00848-2020
45. Vaillant AA, Vaillant J, Vashisht R, Zito PM. Immediate Hypersensitivity Reactions. Treasure Island, FL: StatPearls Publishing (2020).
46. Chaplin DD. Overview of the immune response. J Allergy Clin Immunol. (2010) 125:S3–23. doi: 10.1016/j.jaci.2009.12.980
47. Koike Y, Sato S, Yanagida N, Asaumi T, Ogura K, Ohtani K, et al. Predictors of persistent milk allergy in children: a retrospective cohort study. Int Arch Allergy Immunol. (2018) 175:177–80. doi: 10.1159/000486311
48. Dong X, Cao Y-Y, Lu X, Zhang J, Du H, Yan Y, et al. Eleven faces of coronavirus disease 2019. Allergy. (2020) 75:1699–709. doi: 10.1111/all.14289
49. Berends-De Vries T, Boerma S, Doornebal J, Dikkeschei B, Stegeman C, Veneman TF. Goodpasture's syndrome with negative anti-glomerular basement membrane antibodies. Eur J Case Rep Intern Med. (2017) 4:000687. doi: 10.12890/2017_000687
50. Lee E, Kim M, Jeon K, Lee J, Lee JS, Kim HS, et al. Mean platelet volume, platelet distribution width, and platelet count, in connection with immune thrombocytopenic purpura and essential thrombocytopenia. Lab Med. (2019) 50:279–85. doi: 10.1093/labmed/lmy082
51. Woodruff MC, Ramonell RP, Ngyuen DC, Cashman KS, Saini AS, Haddad NS, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. (2020) 21:1506–16. doi: 10.1038/s41590-020-00814-z
53. Bajema IM, Bruijn JA. What stuff is this! A historical perspective on fibrinoid necrosis. J Pathol. (2000) 191:235–8. doi: 10.1002/(SICI)1096-9896(0000)9999:9999<N/A::AID-PATH610>3.0.CO;2-I
54. Tian S, Xiong Y, Liu H, Niu L, Guo J, Liao M, et al. Pathological study of the 2019 novel coronavirus disease (COVID-19) through postmortem core biopsies. Mod Pathol. (2020) 33:1007–14. doi: 10.1038/s41379-020-0536-x
55. Konopka KE, Nguyen T, Jentzen JM, Rayes O, Schmidt CJ, Wilson AM, et al. Diffuse alveolar damage (DAD) resulting from coronavirus disease 2019 Infection is Morphologically Indistinguishable from Other Causes of DAD. Histopathology. (2020) 77:570–8. doi: 10.1111/his.14180
56. Ackermann M, Verleden SE, Kuehnel M, Haverich A, Welte T, Laenger F, et al. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. (2020) 383:120–8. doi: 10.1056/NEJMoa2015432
57. Zinserling VA, Semenova NY, Markov AG, Rybalchenko OV, Wang J, Rodionov RN, et al. Inflammatory cell infiltration of adrenals in COVID-19. Horm Metab Res. (2020) 52:639–41. doi: 10.1055/a-1191-8094
58. Zeng Z, Xu L, Xie XY, Yan HL, Xie BJ, Xu WZ, et al. Pulmonary pathology of early-phase COVID-19 pneumonia in a patient with a benign lung lesion. Histopathology. (2020) 77:823–31. doi: 10.1111/his.14138
59. Stadlmann S, Hein-Kuhnt R, Singer G. Viropathic multinuclear syncytial giant cells in bronchial fluid from a patient with COVID-19. J Clin Pathol. (2020) 73:607–8. doi: 10.1136/jclinpath-2020-206657
60. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. (2020) 395:1033–4. doi: 10.1016/S0140-673630628-0
61. Richardson S, Hirsch JS, Narasimhan M, Crawford J, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA. (2020) 323:2052–9. doi: 10.1001/jama.2020.6775
62. Lan P, Pan KH, Wang SJ, Shi QC, Yu YX, Fu Y, et al. High serum iron level is associated with increased mortality in patients with sepsis. Sci Rep. (2018) 8:11072. doi: 10.1038/s41598-018-29353-2
63. Rosário C, Zandman-Goddard G, Meyron-Holtz EG, D'Cruz DP, Shoenfeld Y. The Hyperferritinemic Syndrome: macrophage activation syndrome, Still's disease, septic shock and catastrophic antiphospholipid syndrome. BMC Med. (2013) 11:185. doi: 10.1186/1741-7015-11-185
64. Wigginton JM. Reversal of ferritin-mediated immunosuppression by levamisole: a rationale for its application to management of the acquired immune deficiency syndrome (AIDS). Med Hypotheses. (1995) 44:85–8. doi: 10.1016/0306-9877(95)90075-6
65. Gray CP, Franco AV, Arosio P, Hersey P. Immunosuppressive effects of melanoma-derived heavy-chain ferritin are dependent on stimulation of IL-10 production. Int J Cancer. (2001) 92:843–50. doi: 10.1002/ijc.1269
66. Zandman-Goddard G, Shoenfeld Y. Hyperferritinemia in autoimmunity. Israel Med Assoc J. (2008) 10:83–4.
67. Piperno A, Pelucchi S, Mariani R. Inherited iron overload disorders. Transl Gastroenterol Hepatol. (2020) 5:25. doi: 10.21037/tgh.2019.11.15
68. Schiaffino MT, Natale MD, García-Martínez E, Navarro J, Muñoz-Blanco JL, Demelo-Rodríguez P, et al. Immunoserologic detection and diagnostic relevance of cross-reactive autoantibodies in coronavirus disease 2019 patients. J Infect Dis. (2020) 222:1439–43. doi: 10.1093/infdis/jiaa485
69. Petersen I, Phillips A. Three quarters of people with SARS-CoV-2 infection are asymptomatic: analysis of english household survey data. Clin Epidemiol. (2020) 12:1039–43. doi: 10.2147/CLEP.S276825
70. Guan WJ, Ni ZY, Hu Y, Liang WH, Ou CQ, He JX, et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. (2020) 382:1708–20. doi: 10.1056/NEJMoa2002032
71. Wölfel R, Corman VM, Guggemos W, Seilmaier M, Zange S, Müller MA, et al. Virological assessment of hospitalized patients with COVID-2019. Nature. (2020) 581:465–9. doi: 10.1038/s41586-020-2196-x
72. Cai Q, Huang D, Yu H, Zhu Z, Xia Z, Su Y, et al. Characteristics of liver tests in COVID-19 patients. J Hepatol. (2020) 73:566–74. doi: 10.1016/j.jhep.2020.04.006
73. Han Y, Zhang H, Mu S, Wei W, Chaoyuan J, Chaoyang T, et al. Lactate dehydrogenase, a risk factor of severe COVID-19 patients: a retrospective and observational study. Aging. (2020) 12:11245–58. doi: 10.18632/aging.103372
74. Terpos E, Ntanasis-Stathopoulos I, Elalamy I, Kastritis E, Sergentanis TN, Politou M, et al. Hematological findings and complications of COVID-19. Am J Hematol. (2020) 95:834–47. doi: 10.1002/ajh.25829
75. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D, et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. (2020) 17:533–5. doi: 10.1038/s41423-020-0402-2
76. Wong HYF, Lam HYS, Fong AHT, Leung ST, Chin TWY, Lo CSY, et al. Frequency and distribution of chest radiographic findings in COVID-19 positive patients. Radiology. (2020) 296:E72–E78. doi: 10.1148/radiol.2020201160
77. Hennig B, Honchel R, Goldblum SE, McClain CJ. Tumor necrosis factor-mediated hypoalbuminemia in rabbits. J Nutr. (1988) 118:1586–90. doi: 10.1093/jn/118.12.1586
78. Cheng Y, Luo R, Wang K, Zhang M, Wang Z, Dong L, et al. Kidney disease is associated with in-hospital death of patients with COVID-19. Kidney Int. (2020) 97:829–38. doi: 10.1016/j.kint.2020.03.005
79. Batlle D, Soler MJ, Sparks MA, Hiremath S, South AM, Welling PA, et al. Acute kidney injury in COVID-19: emerging evidence of a distinct pathophysiology. J Am Soc Nephrol. (2020) 31:1380–3. doi: 10.1681/ASN.2020040419
80. Hsueh PR, Huang LM, Chen PJ, Kao CL, Yang PC. Chronological evolution of IgM, IgA, IgG and neutralisation antibodies after infection with SARS-associated coronavirus. Clin Microbiol Infect. (2004) 10:1062–6. doi: 10.1111/j.1469-0691.2004.01009.x
81. Shi Y, Wang Y, Shao C, Huang J, Gan J, Huang X, et al. COVID-19 infection: the perspectives on immune responses. Cell Death Differ. (2020) 27:1451–4. doi: 10.1038/s41418-020-0530-3
82. Viner RM, Whittaker E. Kawasaki-like disease: emerging complication during the COVID-19 pandemic. Lancet. (2020) 395:1741–3. doi: 10.1016/S0140-673631129-6
83. Fontenele M, Pedrosa M. Xerostomia and taste alterations in COVID-19. Ear Nose Throat. (2020) 3:145561320982686. doi: 10.1177/0145561320982686
84. Caforio AL, Marcolongo R, Jahns R, Fu M, Felix SB, Iliceto S. Immune-mediated and autoimmune myocarditis: clinical presentation, diagnosis and management. Heart Fail Rev. (2013) 18:715–32. doi: 10.1007/s10741-012-9364-5
85. Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac involvement in a patient with coronavirus disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:819–24. doi: 10.1001/jamacardio.2020.1096
86. Markus HS, Brainin M. COVID-19 and stroke—A global World Stroke Organization perspective. Int J Stroke. (2020) 15:361–4. doi: 10.1177/1747493020923472
87. Wilkinson E. RECOVERY trial: the UK covid-19 study resetting expectations for clinical trials. BMJ. (2020) 369:m1626. doi: 10.1136/bmj.m1626
88. Conticini E, Franchi F, Bennett D, Valente, Mazzei M, Bargagli E, et al. High dosage of methylprednisolone as a rescue, second-line treatment in COVID-19 patients who failed to respond to tocilizumab. Ann Rheum Dis. (2020). doi: 10.1136/annrheumdis-2020-218761
89. Casciola-Rosen L, Thiemann DR, Andrade F, Zambrano MIT, Hooper JE, Leonard EK, et al. IgM autoantibodies recognizing ACE2 are associated with severe COVID-19. medRxiv. (2020). doi: 10.1101/2020.10.13.20211664
Keywords: COVID-19, autoimmunity, lung, solubleACE-2, macrophage
Citation: McMillan P, Dexhiemer T, Neubig RR and Uhal BD (2021) COVID-19—A Theory of Autoimmunity Against ACE-2 Explained. Front. Immunol. 12:582166. doi: 10.3389/fimmu.2021.582166
Received: 14 July 2020; Accepted: 08 February 2021;
Published: 23 March 2021.
Edited by:
Xulin Chen, Jinan University, ChinaReviewed by:
Maria George Ioannou, University Hospital of Larissa, GreeceRuchi Tiwari, U.P. Pandit Deen Dayal Upadhyaya Veterinary University, India
Copyright © 2021 McMillan, Dexhiemer, Neubig and Uhal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bruce D. Uhal, YmR1aGFsQGdtYWlsLmNvbQ==