
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 25 January 2021
Sec. Autoimmune and Autoinflammatory Disorders
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.631743
This article is part of the Research TopicRecent Advances in Potential Biomarkers for Rheumatic Diseases and in Cell-based Therapies in the Management of Inflammatory Rheumatic DiseasesView all 28 articles
 Cristina Municio*
Cristina Municio* Gabriel Criado*
Gabriel Criado*The concept of trained immunity has recently emerged as a mechanism contributing to several immune mediated inflammatory conditions. Trained immunity is defined by the immunological memory developed in innate immune cells after a primary non-specific stimulus that, in turn, promotes a heightened inflammatory response upon a secondary challenge. The most characteristic changes associated to this process involve the rewiring of cell metabolism and epigenetic reprogramming. Under physiological conditions, the role of trained immune cells ensures a prompt response. This action is limited by effective resolution of inflammation and tissue repair in order to restore homeostasis. However, unrestrained activation of innate immune cells contributes to the development of chronic inflammation and tissue destruction through the secretion of inflammatory cytokines, proteases and growth factors. Therefore, interventions aimed at reversing the changes induced by trained immunity provide potential therapeutic approaches to treat inflammatory and autoimmune diseases like rheumatoid arthritis (RA). We review cellular approaches that target metabolism and the epigenetic reprogramming of dendritic cells, macrophages, natural killer cells, and other trained cells in the context of autoimmune inflammatory diseases.
Vertebrate immunity is coordinated by a complex interplay of physical and chemical barriers (epithelia and antimicrobial substances), humoral factors and different cell types that reacts to the continuous exposure to diverse internal and external stimuli. Traditionally, the responses to these challenges have been classified as two independent systems, innate and adaptive immunity.
Innate immunity has been classically defined as a rapid and nonspecific response that comprises biochemical and cellular mechanisms that are present before infection and are considered the first line of defense. It is triggered within minutes after infection but does not generate immune memory because their effectors are germline-encoded. Its main components are the complement system, myeloid cells (neutrophils, monocytes, dendritic cells, and macrophages), natural killer (NK) cells or innate lymphoid cells (ILCs), responsible for molecular recognition and antigen presentation, phagocytosis and elimination of pathogens. In contrast, adaptive immunity is carried out by B and T lymphocytes and has been described as slow and specific. It takes days or weeks to generate an adequate humoral and cellular response, mediated by genetic rearrangement and clonal selection. This results in antigen-specific responses that can induce lasting immune memory.
This dichotomy has changed in recent years and both immune arms are currently considered highly intertwined and collaborative. The dogma establishing the innate system as nonspecific and incapable of adaption or develop immunological memory has been replaced by a model where phagocytosis, microorganism removal or lysis of infected cells are considered as one more specific response. This has been due, at least in part, to the discovery of Pathogen Associated Molecular Patterns (PAMPs), essential microbial components, and the endogenous signs of damage (Damage-Associated Molecular Patterns, DAMPs), which are recognized through the constitutive expression of different families of Pattern-Recognition Receptors (PRRs) (e.g., Toll-like receptors, NOD-like receptors, C-type lectin receptors, RIG-I-like receptors) thus allowing the implementation of an adequate response depending on the type of molecules that are recognized (1–4).
Mackaness, G.B (5). described in mice that, in addition to generating specific B and T cell memory upon exposure to one pathogen, there is an increase in the innate response that can generate cross-protection against a second pathogen, regardless of its phylogenetic origin. This finding was then attributed solely to CD8 memory lymphocytes. Recently, the issue has sparked renewed interest and multiple works have focused on the activation state of innate immunity against a stimulus and the cross-protection that is generated against a second challenge. This type of “memory” against past inflammatory events is well established in plants and is known as Systemic Acquired Resistance (SAR) (6, 7). In that case, plants that are inoculated with attenuated microorganisms develop lasting protection against a wide spectrum of plant pathogens (8). Therefore, SAR is considered a form of innate memory in plants equivalent to immunization in vertebrates. It is also been described in a variety of invertebrates, ideal animal models to study innate immunity as they do not present adaptive immunity: insects such as Anopheles, Drosophila, and the mealworm beetle T. molitor (9, 10), nematodes such as C. elegans (11); or corals (12). This situation allows us to be prepared for future challenges and supposes an ancestral form of “immune memory”.
Similarly, “a heightened response to a secondary infection that can be exerted both toward the same microorganism and a different one (cross-protection)” has been termed “innate immune memory” or “trained innate immunity” in vertebrates (13).
Exposure of innate immune cells to a stimulus through PRRs, promotes a series of long-term modifications that involve rewiring of cell metabolism and epigenetic reprogramming. Since several metabolites function as signalling molecules or cofactors for the enzymes responsible of epigenetic changes, these two processes are closely related (14). Depending on the type and concentration of PAMPs, this immunological imprint can lead to two opposite outcomes: trained innate immunity or innate immune tolerance. In the case of trained immunity, the “training” generates a greater response to a second challenge, while innate immune tolerance is aimed at attenuating or reducing this response (14). Thus, strategies aimed at potentiating the latter can be very useful in regulating physiological processes to avoid harmful reactions to allergens, the microbiota or autoimmune inflammation. However, there must be a balance between the pro- and anti-inflammatory responses to avoid situations of chronic inflammation or immunoparalysis and increased sensitivity to secondary infections.
Most cells use aerobic respiration as their main source of ATP under homeostatic conditions. In the case of the cells of the immune system, there are important metabolic differences depending on the cell type or its activation state (14). Whereas neutrophils have a high basal glycolytic metabolism, other cell types, such as pro-inflammatory macrophages or T cells, need a rapid increase in their glucose consumption and ATP generation when stimulated. This demands cause a metabolic shift from oxidative phosphorylation to aerobic glycolysis, allowing cells to quickly obtain energy and metabolites. Among the processes necessary for the induction of trained immunity, the following can be considered: the increase of the metabolic capacity of the cells, through the Akt/mTOR/HIF1α/pathway; the accumulation of certain metabolic intermediates of the tricarboxylic acid cycle (TCA) with immunomodulatory functions such as fumarate or succinate (15). Some of these metabolites control histone methylation and acetylation, and others are cofactors for histone and DNA methyltransferases and demethylases, as well as histone acetyltransferases and deacetylases (16).
Recent studies suggest that other metabolic pathways also play an important role in cell reprogramming, such as the fatty acid synthesis pathway, which produces cellular stress and activates innate immunity responses. For example, cellular accumulation of unsaturated fatty acids (oleic acid, linoleic acid) induces a pro-inflammatory phenotype in macrophages due to uncoupling of mitochondrial respiration and production of inflammasome components such as IL1-α (17). Likewise, accumulation of mevalonate derived from the pathway of cholesterol synthesis is related to epigenetic changes that promote trained immunity (18). A role for oxLDL in the induction of trained immunity through the activation of the NLPR3 in monocytes has also been shown by studies analysing the effect of western diet in systemic inflammatory diseases (19). This triggers an inflammatory response and the reprogramming of granulocyte monocyte precursor cells (GMPs) (18–20).
MiRNAs provide an additional layer of regulation in the maintenance of innate immune memory. Due to their stability and long half-life, once induced by a stimulus they are capable of maintaining gene expression programs that enhance the resistance of cells to subsequent insults (21). Some miRNAs, such as miR-146, decrease the activation of NF-κB by blocking TRAF6 and IRAK1 thus limiting the immune response (22). In contrast, miR-155, when activated via inflammatory cytokines or TLR ligands, rapidly increases its expression and acts as an activator of inflammation through the down regulation of phosphatases of various signalling pathways (23).
One of the main objections to substantiate the existence of innate immune memory has come from the short half-live of myeloid cells, between 5–7 days, that make difficult to explain how trained immunity is maintained for months or years. However, it has been recently documented that metabolic changes and epigenetic modifications also induce long-term phenotypic and functional reprogramming in the hematopoietic precursors of myeloid cells (HSPC) (24–26). This allows epigenetic reprogramming carried out in innate memory to be transferred through the hematopoietic pathway and its cellular progenitors. Likewise, acquisition of immune functions by fibroblasts and other resident cells can play a role in sustaining organ-specific trained immunity (27, 28).
The origin and development of autoimmune diseases is mainly attributed to an excessive and sustained response to autoantigens mediated by B and T cells. On the other hand, they are also characterized by excessive inflammation and there is strong evidence that innate immunity reprogramming is one contributing factor.
Under physiological conditions, the action of the innate immune response is curtailed by an effective resolution of inflammation and the induction of tissue repair but when the system is dysregulated and the cellular response gets uncontrolled chronic inflammation and tissue destruction, mediated by inflammatory mediators (cytokines, proteases, growth factors), ensue (29). In this context, it is relevant that, in addition to microbial products, trained immunity can be induced by endogenous stimuli and environmental agents, like tobacco smoke, microbiota, and diet, that contribute to the development of inflammatory and autoimmune diseases (19, 30, 31). Consequently, it has been suggested that strategies aimed at controlling the hallmarks of trained immunity, i.e., altered metabolism and epigenetic reprogramming, can provide potential treatments for the chronic inflammation associated to autoimmunity (Figure 1) (29).
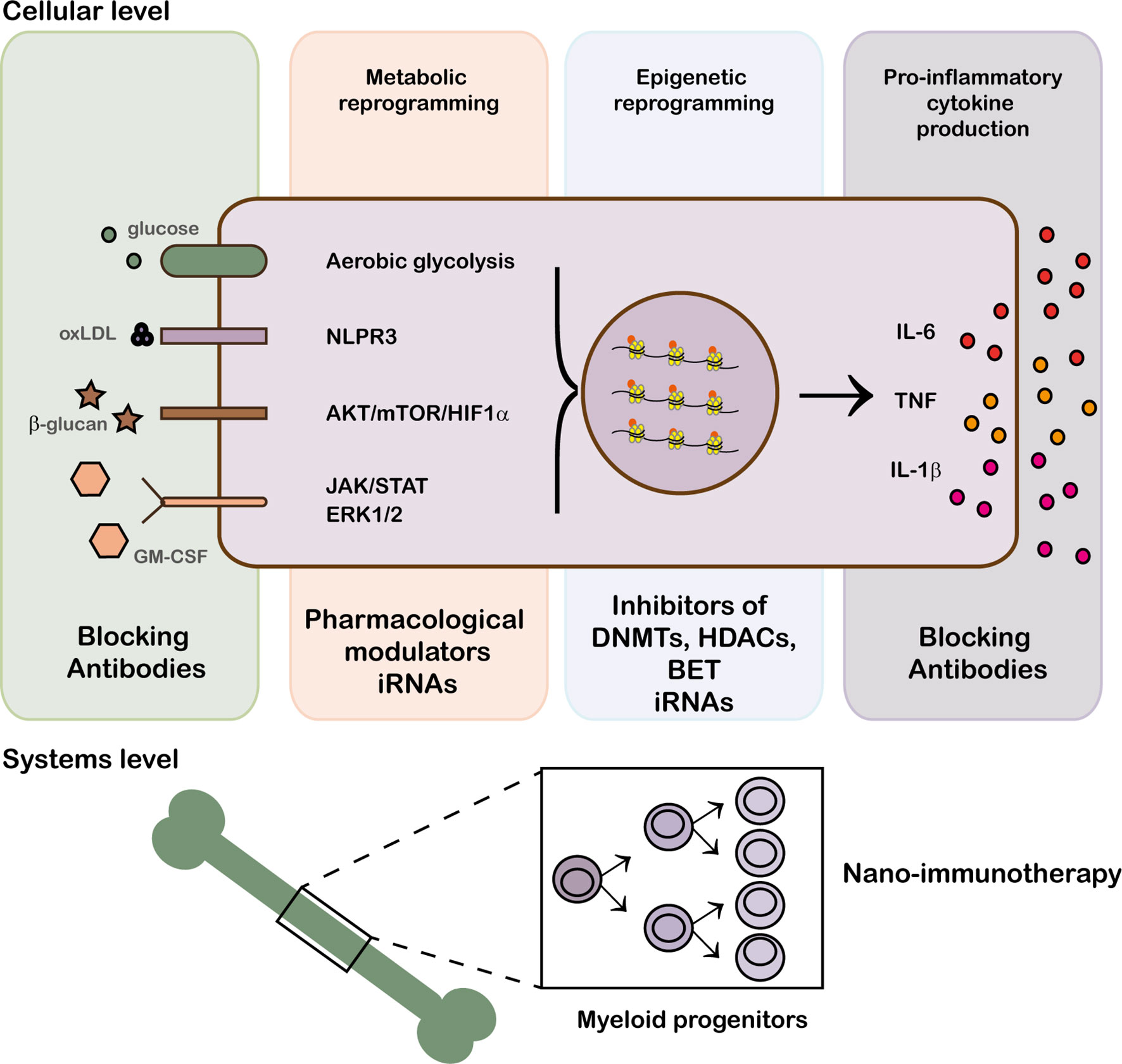
Figure 1 Modulation of trained immunity at epigenetic, cellular and system levels. Trained immunity can be modified at different levels: blockade of receptor recognition with biological therapies, drug modulation of metabolic pathways and epigenetic remodeling.
The shift from oxidative phosphorylation to aerobic glycolysis is a critical component of reprogramming trained immunity and there are several pharmacological modulators that target glucose metabolism. 2-DG (2-deoxy-D-glucose) and 3-BP (3-bromopyruvate), that block glycolysis by inhibiting pathway-limiting enzymes, have shown protection in animal models of arthritis and Systemic lupus erythematosus (SLE) (32, 33). Their effects are mainly attributed to the action on T cells and stromal cells, but they can also regulate the numbers and activity of myeloid cells in model of inflammatory arthritis (34, 35). In line with this observation, oxamate, an alternative inhibitor of glycolysis, reduced the proinflammatory polarization of human macrophages in vitro (36). Lack of tissue specificity and concerns about their toxicity limit the application of these glycolytic inhibitors.
Conversely, the fact that mTOR inhibitors, such as metformin and rapamycin, are already used in the clinic for the treatment of transplant rejection and glycemic control make them more attractive candidates to target trained immunity. Metformin synergizes with 2-DG in the treatment of lupus mice (33) and has shown protective effects in some models of arthritis and Sjöegren syndrome (37–39), but these studies are focussed on acquired immunity. In humans, clinical trials have shown that metformin ameliorates SLE activity, at least in part by reducing neutrophil activation and plasmacytoid dendritic cell function (40, 41).
As in the case of glycolytic inhibitors, the lack of cell specificity makes difficult to evaluate an intrinsic effect on trained immunity. To circumvent this limitation, the application of HDL-based nanobiologics has been used successfully to target mTOR inhibitors to macrophages in the context of transplantation (42).
The lipid metabolism that is involved in trained immunity can be regulated at different levels: treatment with cytochalasin D to block the internalization of CD36, receptor for oxLDL (which induces trained immunity) (20); treatment with methyl-β-cyclodextrin to prevent the formation of cholesterol crystals; inhibition of the enzyme HMG-CoA reductase with fluvastatin to block cholesterol synthesis (18). The inflammasome pathway and the production of IL-1β can be targeted by suppressing NLRP3 activation with Z-VAD-FMK (19), the inhibitor MCC950o, and the ketone metabolite β-hydroxybutyrate (43). To prevent epigenetic changes orchestrated by mevalonate, the enzyme 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase can be inhibited with statins. Several studies have proposed the use of eicosanoid precursors such as omega-3 polyunsaturated fatty acids (PUFAs) as candidates for the treatment of type 1 diabetes, SLE or RA. These molecules have anti-inflammatory properties (44, 45). Also modulation of lipoxins, resolvins and protectins with aspirin is used in the treatment of SLE (46). In vitro and in vivo studies show how the administration of products of arachidonic acid metabolism (EET, epoxyeicosatrienoic acids) can serve as a therapeutic strategy in those diseases where osteoclastogenesis is deregulated, such as rheumatoid arthritis (RA) (47).
Reverting the epigenetic modifications that occurred during the immunity training to normal values using different inhibitors can potentially be used for the treatment of inflammatory and autoimmune diseases (48). Inhibitors of DNMTs (DNA methyltransferases) such as azacytidine and decitabine have been used in the field of oncology for 50 years as cytostatics, and recently, as inhibitors of DNMTs, but little is known about their effect outside this field. A wide variety of proteins capable of lysine methylation, a mark associated with transcriptionally silenced chromatin, are potential pharmacological targets of small inhibitory molecules (49). There is also a large number of compounds used as HDACs (histone deacetylase) inhibitors: from molecules pan-HDAC inhibitors, such as trichostatin A (TSA) or vorinostat (SAHA), to other family-specific, such as valproic acid (VPA), givinostat (ITF2357), or etinostat. Regardless of the mechanism, HDAC inhibitors modify the immune response by increasing and decreasing gene expression (48). The effect of multiple HDAC inhibitors in reducing systemic inflammation and pro-inflammatory cytokines has been investigated in various animal models such as arthritis, diabetes, sepsis, asthma. Most of the studies in humans have focused on the context of the RA. The production of proinflammatory cytokines derived from the macrophages of the inflamed synovium can be inhibited by TSA, vorinostat and “sodium phenylbutyrate” (50). Both TSA and MI192 inhibit IL-6 production in LPS-stimulated PBMCs (51). Furthermore, TSA and givinostat (ITF2357) interfere with the stability of IL-6 mRNA, reducing its production in synovial fibroblasts and macrophages (52) and inducing RA synovial fibroblasts to a TRAIL-induced apoptosis (53). Other molecules such as romidepsin (FK228) or MPT0G009 inhibit the proliferation of synovial fibroblasts (54, 55) and FK228 inhibits angiogenesis in synovial tissue (56). Preliminary studies in human monocytes and macrophages show that the small inhibitory molecules of BET bormodomain-containig proteins have great therapeutic potential in the treatment of immune-mediated diseases (48).
Biologics currently used in the clinic for the treatment of autoimmune diseases can also have an impact on trained immunity. For example, Lin and colleagues have described that TNF inhibitors etanercept and adalimumab suppress the expression of CC-chemokine ligand 2 (CCL2) in monocytes by regulating histone acetylation and trimethylation, changes that correlate with RA activity (57).
IL-1β associated with increased inflammasome activity may also serve as a target to actively suppress trained immunity. The observation that monocytes from patients with autoimmune and autoinflammatory diseases showed increased released of IL-1β than healthy individuals (58, 59) promoted IL-1β neutralization as a potential therapy for some chronic diseases. Although the IL-1Ra anakinra is a marginal treatment in RA (60), both anakinra and the IL-1 blocking antibody canakinumab are effective in suppressing symptoms and keeping the disease under control in systemic autoinflammatory syndromes (61, 62). In the case of the “cryopyrin-associated periodic syndrome” (CASP) where there is a mutation in an amino acid that codes for the cryopyrin protein (currently known as NLRP3), early diagnosis along with treatment with an IL-1 blocker is essential to prevent future disabilities or complications (59, 63). It is also approved for use in the treatment of Hyper-IgD syndrome (HIDS). This disease presents a defect in the enzyme mevalonate kinase that favors the AKT/mTOR pathway and the consequent change to glycolytic metabolism. This, together with attacks of sterile inflammation, is a clear example of uncontrolled trained immunity (18, 64, 65) and IL-1 blockers can reduce the frequency and severity of flares (66, 67).
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a major cytokine in the development of trained immunity (26). Several studies in patients with inflammatory diseases show elevated levels of GM-CSF in blood and synovial fluid, as well as expression of GM-CSFR in inflamed synovial tissues (68, 69). GM-CSF has a main effect in promoting inflammation and therapies aimed at inhibiting its activity are expected to impact trained immunity. Currently, clinical trials addressing the effect of monoclonal antibodies against GM-CSF (namilumab, MOR10) and against GM-CSFR (Mavrilimumab) are underway in patients with RA (NCT02393378; NCT01023256) or psoriatic arthritis (NCT02129777).
It has been proposed that trained immunity can be beneficial against SARS-CoV2 infection (70). In this context, trained immunity induced by BCG vaccination protects from several viral infections (71, 72). However, attempts to find an association between BCG vaccination status and COVID-19 severity have yielded inconclusive results (73, 74).
Conversely, the presence of previous inflammatory diseases and the excessive inflammatory response triggered by SARS-CoV2 infection are poor prognostic factors in COVID-19 progression (75) and, given the role of trained immunity in the development of both processes, its responses can contribute to greater COVID-19 severity (70).
The reprogramming of the innate immune memory can provide an excellent therapeutic target in autoimmune inflammatory diseases, allowing to restore the balance between hyperinflammation and immunodepression and to achieve therapeutic benefits. However, there are limitations to the in vivo use of molecules that target myeloid cells and their progenitors. The compounds used to regulate trained immunity show toxicity, low bioavailability and some adverse effects related to immunity. More suitable approaches are needed to reduce side effects and increase specific targeting.
GC and CM conceived the review, searched databases, and drafted the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Ministerio de Ciencia e Innovación to CM (Programa Juan de la Cierva IJCI-2016-27666).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Medzhitov R, Janeway CA. Innate immunity: impact on the adaptive immune response. Curr Opin Immunol (1997) 9(1):4–9. doi: 10.1016/S0952-7915(97)80152-5
2. Janeway CA, Medzhitov R. Innate immune recognition. Annu Rev Immunol (2002) 20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359
3. Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol (2004) 4(7):499–511. doi: 10.1038/nri1391
4. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell (2006) 124(4):783–801. doi: 10.1016/j.cell.2006.02.015
5. Mackaness GB. The influence of immunologically committed lymphoid cells on macrophage activity in vivo. J Exp Med (1969) 129(5):973–92. doi: 10.1084/jem.129.5.973
6. Chester KS. The Problem of Acquired Physiological Immunity in Plants. Q Rev Biol (1933) 8(3):275–324. doi: 10.1086/394440
7. Durrant WE, Dong X. Systemic acquired resistance. Annu Rev Phytopathol (2004) 42:185–209. doi: 10.1146/annurev.phyto.42.040803.140421
8. Penkov S, Mitroulis I, Hajishengallis G, Chavakis T. Immunometabolic Crosstalk: An Ancestral Principle of Trained Immunity? Trends Immunol (2019) 40(1):1–11. doi: 10.1016/j.it.2018.11.002
9. Rodrigues J, Brayner FA, Alves LC, Dixit R, Barillas-Mury C. Hemocyte differentiation mediates innate immune memory in Anopheles gambiae mosquitoes. Science (2010) 329(5997):1353–5. doi: 10.1126/science.1190689
10. Moret Y, Siva-Jothy MT. Adaptive innate immunity? Responsive-mode prophylaxis in the mealworm beetle, Tenebrio molitor. Proc Biol Sci (2003) 270(1532):2475–80. doi: 10.1098/rspb.2003.2511
11. Anyanful A, Easley KA, Benian GM, Kalman D. Conditioning protects C. elegans from lethal effects of enteropathogenic E. coli by activating genes that regulate lifespan and innate immunity. Cell Host Microbe (2009) 5(5):450–62. doi: 10.1016/j.chom.2009.04.012
12. Hildemann WH, Raison RL, Cheung G, Hull CJ, Akaka L, Okamoto J. Immunological specificity and memory in a scleractinian coral. Nature (1977) 270(5634):219–23. doi: 10.1038/270219a0
13. Netea MG, Quintin J, van der Meer JWM. Trained immunity: a memory for innate host defense. Cell Host Microbe (2011) 9(5):355–61. doi: 10.1016/j.chom.2011.04.006
14. Dominguez-Andres J, Netea MG. Long-term reprogramming of the innate immune system. J Leukoc Biol (2019) 105(2):329–38. doi: 10.1002/JLB.MR0318-104R
15. Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science (2014) 345(6204):1250684. doi: 10.1126/science.1250684
16. Etchegaray J-P, Mostoslavsky R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol Cell (2016) 62(5):695–711. doi: 10.1016/j.molcel.2016.05.029
17. Freigang S, Ampenberger F, Weiss A, Kanneganti T-D, Iwakura Y, Hersberger M, et al. Fatty acid-induced mitochondrial uncoupling elicits inflammasome-independent IL-1α and sterile vascular inflammation in atherosclerosis. Nat Immunol (2013) 14(10):1045–53. doi: 10.1038/ni.2704
18. Bekkering S, Arts RJW, Novakovic B, Kourtzelis I, van der Heijden CDCC, Li Y, et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell (2018) 172(1–2):135–46.e9. doi: 10.1016/j.cell.2017.11.025
19. Christ A, Günther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming. Cell (2018) 172(1–2):162–75.e14. doi: 10.1016/j.cell.2017.12.013
20. Bekkering S, Quintin J, Joosten LAB, van der Meer JWM, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol (2014) 34(8):1731–8. doi: 10.1161/ATVBAHA.114.303887
21. Monticelli S, Natoli G. Short-term memory of danger signals and environmental stimuli in immune cells. Nat Immunol (2013) 14(8):777–84. doi: 10.1038/ni.2636
22. Ma X, Becker Buscaglia LE, Barker JR, Li Y. MicroRNAs in NF- B signaling. J Mol Cell Biol (2011) 3(3):159–66. doi: 10.1093/jmcb/mjr007
23. O’Connell RM, Chaudhuri AA, Rao DS, Baltimore D. Inositol phosphatase SHIP1 is a primary target of miR-155. Proc Natl Acad Sci U S A (2009) 106(17):7113–8. doi: 10.1073/pnas.0902636106
24. Saeed S, Quintin J, Kerstens HHD, Rao NA, Aghajanirefah A, Matarese F, et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science (2014) 345(6204):1251086. doi: 10.1126/science.1251086
25. Kaufmann E, Sanz J, Dunn JL, Khan N, Mendonça LE, Pacis A, et al. BCG Educates Hematopoietic Stem Cells to Generate Protective Innate Immunity against Tuberculosis. Cell (2018) 172(1–2):176–90.e19. doi: 10.1016/j.cell.2017.12.031
26. Mitroulis I, Ruppova K, Wang B, Chen L-S, Grzybek M, Grinenko T, et al. Modulation of Myelopoiesis Progenitors Is an Integral Component of Trained Immunity. Cell (2018) 172(1–2):147–61.e12. doi: 10.1016/j.cell.2017.11.034
27. Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, et al. Defining trained immunity and its role in health and disease. Nat Rev Immunol (2020), 20(6):375–88. doi: 10.1038/s41577-020-0285-6
28. Krausgruber T, Fortelny N, Fife-Gernedl V, Senekowitsch M, Schuster LC, Lercher A, et al. Structural cells are key regulators of organ-specific immune responses. Nature (2020) 583(7815):296–302. doi: 10.1038/s41586-020-2424-4
29. Arts RJW, Joosten LAB, Netea MG. The Potential Role of Trained Immunity in Autoimmune and Autoinflammatory Disorders. Front Immunol (2018) 9:298. doi: 10.3389/fimmu.2018.00298
30. Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol (2010) 10(12):826–37. doi: 10.1038/nri2873
31. Brasacchio D, Okabe J, Tikellis C, Balcerczyk A, George P, Baker EK, et al. Hyperglycemia induces a dynamic cooperativity of histone methylase and demethylase enzymes associated with gene-activating epigenetic marks that coexist on the lysine tail. Diabetes (2009) 58(5):1229–36. doi: 10.2337/db08-1666
32. Garcia-Carbonell R, Divakaruni AS, Lodi A, Vicente-Suarez I, Saha A, Cheroutre H, et al. Critical Role of Glucose Metabolism in Rheumatoid Arthritis Fibroblast-like Synoviocytes. Arthritis Rheumatol (2016) 68(7):1614–26. doi: 10.1002/art.39608
33. Yin Y, Choi S-C, Xu Z, Perry DJ, Seay H, Croker BP, et al. Normalization of CD4+ T Cell Metabolism Reverses Lupus. Sci Transl Med (2015) 7(274):274ra18. doi: 10.1126/scitranslmed.aaa0835
34. Okano T, Saegusa J, Nishimura K, Takahashi S, Sendo S, Ueda Y, et al. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci Rep (2017) 7:42412. doi: 10.1038/srep42412
35. Abboud G, Choi S-C, Kanda N, Zeumer-Spataro L, Roopenian DC, Morel L. Inhibition of Glycolysis Reduces Disease Severity in an Autoimmune Model of Rheumatoid Arthritis. Front Immunol (2018) 9:1973 doi: 10.3389/fimmu.2018.01973
36. Izquierdo E, Cuevas VD, Fernandez-Arroyo S, Riera-Borrull M, Orta-Zavalza E, Joven J, et al. Reshaping of Human Macrophage Polarization through Modulation of Glucose Catabolic Pathways. J Immunol (2015) 195(5):2442–51. doi: 4049/jimmunol.1403045
37. Son H-J, Lee J, Lee S-Y, Kim E-K, Park M-J, Kim K-W, et al. Metformin Attenuates Experimental Autoimmune Arthritis through Reciprocal Regulation of Th17/Treg Balance and Osteoclastogenesis. Mediators Inflammation (2014) 2014:973986. doi: 10.1155/2014/973986
38. Kang KY, Kim Y-K, Yi H, Kim J, Jung H-R, Kim IJ, et al. Metformin downregulates Th17 cells differentiation and attenuates murine autoimmune arthritis. Int Immunopharmacol (2013) 16(1):85–92. doi: 10.1016/j.intimp.2013.03.020
39. Kim J-W, Kim S-M, Park J-S, Hwang S-H, Choi J, Jung K-A, et al. Metformin improves salivary gland inflammation and hypofunction in murine Sjögren’s syndrome. Arthritis Res Ther (2019) 21(1):136. doi: 10.1186/s13075-019-1904-0
40. Sun F, Geng S, Wang H, Wang H, Liu Z, Wang X, et al. Effects of metformin on disease flares in patients with systemic lupus erythematosus: post hoc analyses from two randomised trials. Lupus Sci Med (2020) 7(1):e000429. doi: 10.1136/lupus-2020-000429
41. Wang H, Li T, Chen S, Gu Y, Ye S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol (2015) 67(12):3190–200. doi: 10.1002/art.39296
42. Braza MS, van Leent MMT, Lameijer M, Sanchez-Gaytan BL, Arts RJW, Pérez-Medina C, et al. Inhibiting Inflammation with Myeloid Cell-Specific Nanobiologics Promotes Organ Transplant Acceptance. Immunity (2018) 49(5):819–828.e6. doi: 10.1016/j.immuni.2018.09.008
43. Shao B-Z, Xu Z-Q, Han B-Z, Su D-F, Liu C. NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol (2015) 6:262. doi: 10.3389/fphar.2015.00262
44. Li X, Bi X, Wang S, Zhang Z, Li F, Zhao AZ. Therapeutic Potential of ω-3 Polyunsaturated Fatty Acids in Human Autoimmune Diseases. Front Immunol (2019) 10:2241 doi: 10.3389/fimmu.2019.02241
45. Cas MD, Roda G, Li F, Secundo F. Functional Lipids in Autoimmune Inflammatory Diseases. Int J Mol Sci (2020) 21(9):3074. doi: 10.3390/ijms21093074
46. Das UN. Current and emerging strategies for the treatment and management of systemic lupus erythematosus based on molecular signatures of acute and chronic inflammation. J Inflammation Res (2010) 3:143–70. doi: 10.2147/JIR.S9425
47. Guan H, Zhao L, Cao H, Chen A, Xiao J. Epoxyeicosanoids suppress osteoclastogenesis and prevent ovariectomy-induced bone loss. FASEB J (2015) 29(3):1092–101. doi: 10.1096/fj.14-262055
48. Tough DF, Tak PP, Tarakhovsky A, Prinjha RK. Epigenetic drug discovery: breaking through the immune barrier. Nat Rev Drug Discovery (2016) 15(12):835–53. doi: 10.1038/nrd.2016.185
49. Tough DF, Lewis HD, Rioja I, Lindon MJ, Prinjha RK. Epigenetic pathway targets for the treatment of disease: accelerating progress in the development of pharmacological tools: IUPHAR Review 11. Br J Pharmacol (2014) 171(22):4981–5010. doi: 10.1111/bph.12848
50. Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME, et al. Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J Immunol (2010) 184(5):2718–28. doi: 10.4049/jimmunol.0901467
51. Gillespie J, Savic S, Wong C, Hempshall A, Inman M, Emery P, et al. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheumatol (2012) 64(2):418–22. doi: 10.1002/art.33382
52. Grabiec AM, Korchynskyi O, Tak PP, Reedquist KA. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis (2012) 71(3):424–31. doi: 10.1136/ard.2011.154211
53. Jüngel A, Baresova V, Ospelt C, Simmen BR, Michel BA, Gay RE, et al. Trichostatin A sensitises rheumatoid arthritis synovial fibroblasts for TRAIL-induced apoptosis. Ann Rheum Dis (2006) 65(7):910–2. doi: 10.1136/ard.2005.044065
54. Nishida K, Komiyama T, Miyazawa S-I, Shen Z-N, Furumatsu T, Doi H, et al. Histone deacetylase inhibitor suppression of autoantibody-mediated arthritis in mice via regulation of p16INK4a and p21(WAF1/Cip1) expression. Arthritis Rheumatol (2004) 50(10):3365–76. doi: 10.1002/art.20709
55. Hsieh I-N, Liou J-P, Lee H-Y, Lai M-J, Li Y-H, Yang C-R. Preclinical anti-arthritic study and pharmacokinetic properties of a potent histone deacetylase inhibitor MPT0G009. Cell Death Dis (2014) 5:e1166. doi: 10.1038/cddis.2014.133
56. Grabiec AM, Tak PP, Reedquist KA. Targeting histone deacetylase activity in rheumatoid arthritis and asthma as prototypes of inflammatory disease: should we keep our HATs on? Arthritis Res Ther (2008) 10(5):226. doi: 10.1186/ar2489
57. Lin Y-C, Lin Y-C, Huang M-Y, Kuo P-L, Wu C-C, Lee M-S, et al. Tumor necrosis factor-alpha inhibitors suppress CCL2 chemokine in monocytes via epigenetic modification. Mol Immunol (2017) 83:82–91. doi: 10.1016/j.molimm.2017.01.009
58. Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med (2005) 201(9):1479–86. doi: 10.1084/jem.20050473
59. Goldbach-Mansky R. Current status of understanding the pathogenesis and management of patients with NOMID/CINCA. Curr Rheumatol Rep (2011) 13(2):123–31. doi: 10.1007/s11926-011-0165-y
60. Ramírez J, Cañete JD. Anakinra for the treatment of rheumatoid arthritis: a safety evaluation. Expert Opin Drug Saf (2018) 17(7):727–32. doi: 10.1080/14740338.2018.1486819
61. Gentileschi S, Rigante D, Vitale A, Sota J, Frediani B, Galeazzi M, et al. Efficacy and safety of anakinra in tumor necrosis factor receptor-associated periodic syndrome (TRAPS) complicated by severe renal failure: a report after long-term follow-up and review of the literature. Clin Rheumatol (2017) 36(7):1687–90. doi: 10.1007/s10067-017-3688-4
62. De Benedetti F, Gattorno M, Anton J, Ben-Chetrit E, Frenkel J, Hoffman HM, et al. Canakinumab for the Treatment of Autoinflammatory Recurrent Fever Syndromes. N Engl J Med (2018) 378(20):1908–19. doi: 10.1056/NEJMoa1706314
63. Hui A, Johnson LB, Greemberg R, Penney L, Ramsey SE. Severe cryopyrin-associated periodic syndrome first characterized by early childhood-onset sensorineural hearing loss - Case report and literature review. Int J Pediatr Otorhinolaryngol (2019) 120:68–72. doi: 10.1016/j.ijporl.2019.01.037
64. Stoffels M, Simon A. Hyper-IgD syndrome or mevalonate kinase deficiency. Curr Opin Rheumatol (2011) 23(5):419–23. doi: 10.1097/BOR.0b013e328349c3b1
65. Gosavi S, Chavez LL, Jennings PA, Onuchic JN. Topological frustration and the folding of interleukin-1 beta. J Mol Biol (2006) 357(3):986–96. doi: 10.1016/j.jmb.2005.11.074
66. Korppi M, Van Gijn ME, Antila K, Hyperimmunoglobulinemia D. and periodic fever syndrome in children. Rev Ther Biol Drugs Case Rep Acta Paediatr (2011) 100(1):21–5. doi: 10.1111/j.1651-2227.2010.01974.x
67. Bodar EJ, Kuijk LM, Drenth JPH, van der Meer JWM, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis (2011) 70(12):2155–8. doi: 10.1136/ard.2011.149922
68. Bell AL, Magill MK, McKane WR, Kirk F, Irvine AE. Measurement of colony-stimulating factors in synovial fluid: potential clinical value. Rheumatol Int (1995) 14(5):177–82. doi: 10.1007/BF00262295
69. Berenbaum F, Rajzbaum G, Amor B, Toubert A. Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur Cytokine Netw (1994) 5(1):43–6.
70. O’Neill LAJ, Netea MG. BCG-induced trained immunity: can it offer protection against COVID-19? Nat Rev Immunol (2020) 20(6):335–7. doi: 10.1038/s41577-020-0337-y
71. Arts RJW, Moorlag SJCFM, Novakovic B, Li Y, Wang S-Y, Oosting M, et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe (2018) 23(1):89–100.e5. doi: 10.1016/j.chom.2017.12.010
72. Giamarellos-Bourboulis EJ, Netea MG, Rovina N, Akinosoglou K, Antoniadou A, Antonakos N, et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe (2020) 27(6):992–1000.e3. doi: 10.1016/j.chom.2020.04.009
73. Noval Rivas M, Ebinger JE, Wu M, Sun N, Braun J, Sobhani K, et al. BCG vaccination history associates with decreased SARS-CoV-2 seroprevalence across a diverse cohort of healthcare workers. J Clin Invest (2020) 145157. doi: 10.1172/JCI145157
74. Hensel J, McAndrews KM, McGrail DJ, Dowlatshahi DP, LeBleu VS, Kalluri R. Protection against SARS-CoV-2 by BCG vaccination is not supported by epidemiological analyses. Sci Rep (2020) 10(1):18377. doi: 10.1038/s41598-020-75491-x
Keywords: trained immunity, autoimmune disease, inflammation, therapy, metabolism, epigenetics, COVID-19
Citation: Municio C and Criado G (2021) Therapies Targeting Trained Immune Cells in Inflammatory and Autoimmune Diseases. Front. Immunol. 11:631743. doi: 10.3389/fimmu.2020.631743
Received: 20 November 2020; Accepted: 21 December 2020;
Published: 25 January 2021.
Edited by:
Eric Toussirot, INSERM CIC1431 Centre d’Investigation Clinique Besançon, FranceReviewed by:
Richard Williams, University of Oxford, United KingdomCopyright © 2021 Municio and Criado. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gabriel Criado, Z2NyaWFkb0BoMTJvLmVz; Cristina Municio, Y211bmljaW8uaW1hczEyQGgxMm8uZXM=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.