 Wei Liang
Wei Liang Napoleone Ferrara
Napoleone Ferrara
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 12 February 2021
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.626812
This article is part of the Research Topic Iron Metabolism at the Crossroad of Innate Immune Response and Cancer Progression View all 10 articles
Cells of the innate immune system are a major component of the tumor microenvironment. They play complex and multifaceted roles in the regulation of cancer initiation, growth, metastasis and responses to therapeutics. Innate immune cells like neutrophils and macrophages are recruited to cancerous tissues by chemotactic molecules released by cancer cells and cancer-associated stromal cells. Once they reach the tumor, they can be instructed by a network of proteins, nucleic acids and metabolites to exert protumoral or antitumoral functions. Altered iron metabolism is a feature of cancer. Epidemiological studies suggest that increased presence of iron and/or iron binding proteins is associated with increased risks of cancer development. It has been shown that iron metabolism is involved in shaping the immune landscapes in inflammatory/infectious diseases and cancer-associated inflammation. In this article, we will dissect the contribution of macrophages and neutrophils to dysregulated iron metabolism in malignant cells and its impact on cancer growth and metastasis. The mechanisms involved in regulating the actions of macrophages and neutrophils will also be discussed. Moreover, we will examine the effects of iron metabolism on the phenotypes of innate immune cells. Both iron chelating and overloading agents are being explored in cancer treatment. This review highlights alternative strategies for management of iron content in cancer cells by targeting the iron donation and modulation properties of macrophages and neutrophils in the tumor microenvironment.
Innate immune cells such as neutrophils and macrophages are the host’s first line of defense against invading pathogens and are responsible for initiating inflammatory responses. Recruitment and activation of adaptive immune cells to/at the infection sites are crucial steps. Cancer-associated inflammation is correlated with poor patient survival and therapeutic outcomes and is listed among the hallmarks of cancer (1). Indeed, extensive efforts have been devoted to elucidating the contribution of the innate immune system in these events. Previous studies have shown that innate immune cells, such as tumor-associated macrophages (TAM) and neutrophils, can facilitate cancer cell growth and metastasis, induce tumor angiogenesis, suppress antitumor immune response and modulate response to anticancer therapies (2, 3). These complex effects are mediated by a network of cytokines, chemokines, growth factors and enzymes released by innate immune cells that act directly on cancer cells and components of the tumor microenvironment (TME) or through cell-cell contacts between innate immune cells and cancer cells or other stromal cells (2, 3).
Recent evidence has revealed a remarkable plasticity in the metabolism of innate immune cells recruited to the TME (4). These findings perhaps are not surprising given the unique features (e.g., low pH, limited supplies of nutrients and oxygen) concomitant with aberrant accumulation of metabolism-modulating molecules in the TME. Interestingly, cancer cells can utilize metabolic byproducts from innate immune cells and other stromal cells to support cancer growth and promote drug resistance (5–8). Further understanding of how dysregulated cell metabolism in innate immune cells affects cancer behaviors is of great interest and may uncover new therapeutic avenues.
Iron is an essential element for all organisms. Its ability to be oxidized and reduced makes it ideal for transporting electrons and functioning as a co-factor in a variety of biochemical reactions in DNA synthesis (9), mitochondria respiration (10), host defenses (11) and cell signaling (12). On the other hand, this unique property may result in formation of reactive oxygen species (ROS) that have detrimental effects on genomic stability and may induce malignant transformation (13). As a result, iron metabolism is one of the key factors deciding the fates of normal and malignant cells.
This review examines the crosstalk between cancer cells and innate immune cells from the perspective of iron metabolism. We will review the evidence on how innate immune cells contribute to the dysregulated iron metabolism in cancer cells and how this process is regulated by iron metabolism and signaling molecules in the TME. We will also analyze the potential clinical benefits for therapeutic targeting the iron donation and modulation properties of macrophages and neutrophils.
Iron metabolism and homeostasis under physiological conditions have been reviewed in details elsewhere (12, 14). Briefly, dietary iron enters the body through absorption by divalent metal transporter 1 (DMT1) expressed on enterocytes at the duodenum of the small intestine. It can then be released to circulation through the only known iron exporter ferroportin. In the systemic circulation, the majority of the iron is bound by an iron-transporting protein named transferrin that is mainly synthesized by hepatocytes. The iron-bound transferrin (holo-transferrin) recognizes the ubiquitously expressed transferrin receptor 1 (Tfr1) or tissue-specific transferrin receptor 2 (Tfr2) and enters cells through clathrin-mediated endocytosis. Non-transferrin bound iron also exists in extracellular spaces and can be taken up by cells through transferrin receptor-independent mechanisms (e.g., DMT1). In endosomes, iron is dissociated from transferrin, reduced by six-transmembrane epithelial antigen of the prostate (STEAP) proteins and released to cytoplasm by DMT1, while transferrin receptors are mostly recycled back to the plasma membrane. Once inside the cell, iron may enter mitochondria and nucleus to participate in a series of biochemical reactions. It may also be stored in ferritin and a labile iron pool (LIP) or exported by ferroportin. Hepcidin, an iron-regulatory peptide mainly synthesized by liver, directly binds to ferroportin, resulting in the internalization and degradation of ferroportin and reduced iron export. Cellular iron homeostasis is regulated through binding of the iron regulatory proteins (IRP1 and IRP2) to the iron-responsive element (IRE) located in the untranslated region (UTR) of target mRNAs involved in iron metabolism. When intracellular iron is low, IRPs binds to IRE of Tfr1, DMT1, ferritin and ferroportin mRNAs, resulting in increased expression of Tfr1 and DMT1 and decreased expression of ferritin and ferroportin. When intracellular iron is high, IRPs is dissociated from IRE, which suppresses expression of Tfr1 and DMT1 yet permits expression of ferritin and ferroportin.
Due to increased proliferation rate and synthetic/metabolic activities commonly associated with malignancy, iron demand in cancer cells is high. To ensure ample supply, the iron uptake machinery in cancer cells is usually enhanced, while export is repressed. Transferrin synthesis acts as an autocrine mechanism supporting iron supply and growth of cancer cells (15). Overexpression of Tfr1 is frequently found in malignant tissues and is associated with worse patient survival (16–18). Inhibition of Tfr1 expression significantly blocks tumor growth and metastasis (19, 20). Moreover, upregulation of other proteins involved in iron import, such as DMT1 and duodenal cytochrome b (DCYTB), has been reported in cancer cells (21, 22). Furthermore, STEAP family members, which are highly expressed in a variety of cancer cells, can facilitate iron uptake (23–26). Conversely, expression of ferroportin is reduced in cancer cells, relative to their normal counterparts and is associated with poor patient survival (27, 28). Overexpression of ferroportin results in suppressed tumor growth (27). Elevated levels of hepcidin are found in different cancer types, further restricting ferroportin-mediated iron export and favoring iron sequestering in cancer cells (28–31). In addition, high IRP2 levels contribute to cancer cell proliferation and survival and correlate with poor patient survival (32–35).
Emerging evidence suggests that innate immune cells such as macrophages and neutrophils contribute to dysregulated iron metabolism in cancer cells. Specifically, macrophages and neutrophils residing in the TME can either serve as sources of iron and iron-related proteins (Figure 1) or release factors that activate signaling pathways in control of iron metabolism in cancer cells (Figure 2).
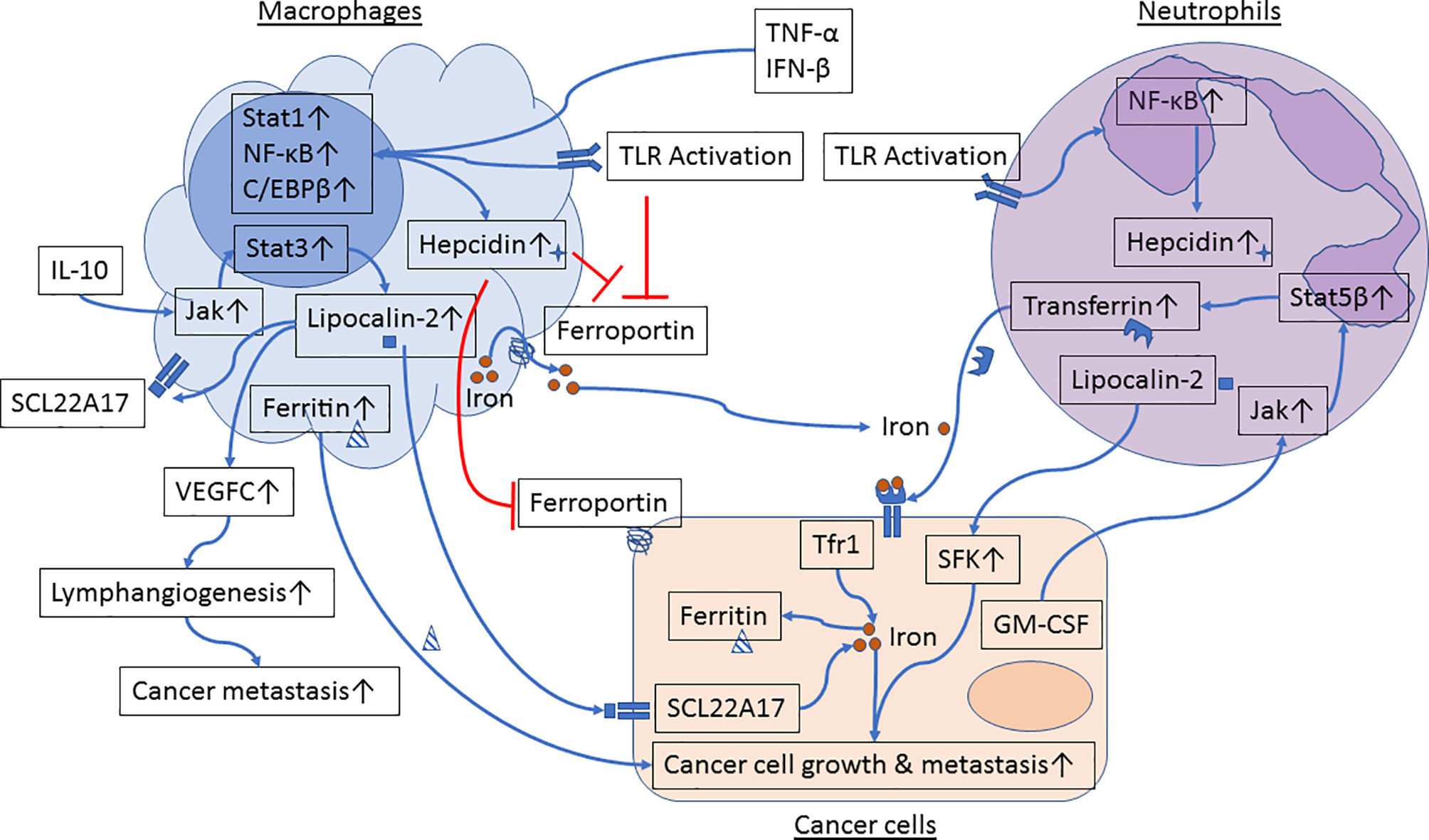
Figure 1 Iron metabolism in the crosstalk between cancer cells and macrophages or neutrophils. Proinflammatory cytokines and Toll-like receptor (TLR) agonists induce activation of Stat1, NF-κB and C/EBPβ in macrophages, resulting in upregulation of hepcidin and inhibition of ferroportin. In parallel, engagement of TLRs further inhibits ferroportin expression. Reduced ferroportin levels in macrophages limit iron transport from macrophages to cancer cells. IL-10 activates the Jak/Stat3 signaling pathway and upregulates expression of lipocalin-2 in macrophages. Lipocalin-2 released from macrophages can bind to its receptor in cancer cells and in macrophages to stimulate cancer cell growth and M2 polarization, respectively. Lipocalin-2 can also induce VEGFC expression, resulting in promotion of lymphangiogenesis and cancer metastasis. Moreover, macrophage-secreted ferritin directly stimulates cancer cell growth. TLR engagement induces activation of NF-κB and hepcidin expression in neutrophils. GM-CSF, produced by metastatic tumor cells, induces activation of the Jak/Stat5β signaling pathway and transferrin synthesis in neutrophils. Neutrophil-derived transferrin can promote growth of metastatic tumor cells. In addition, lipocalin-2 released by neutrophils induces activation of Src family kinases (SFK) in prostate cancer cells and enhances cancer cell migration and metastasis.
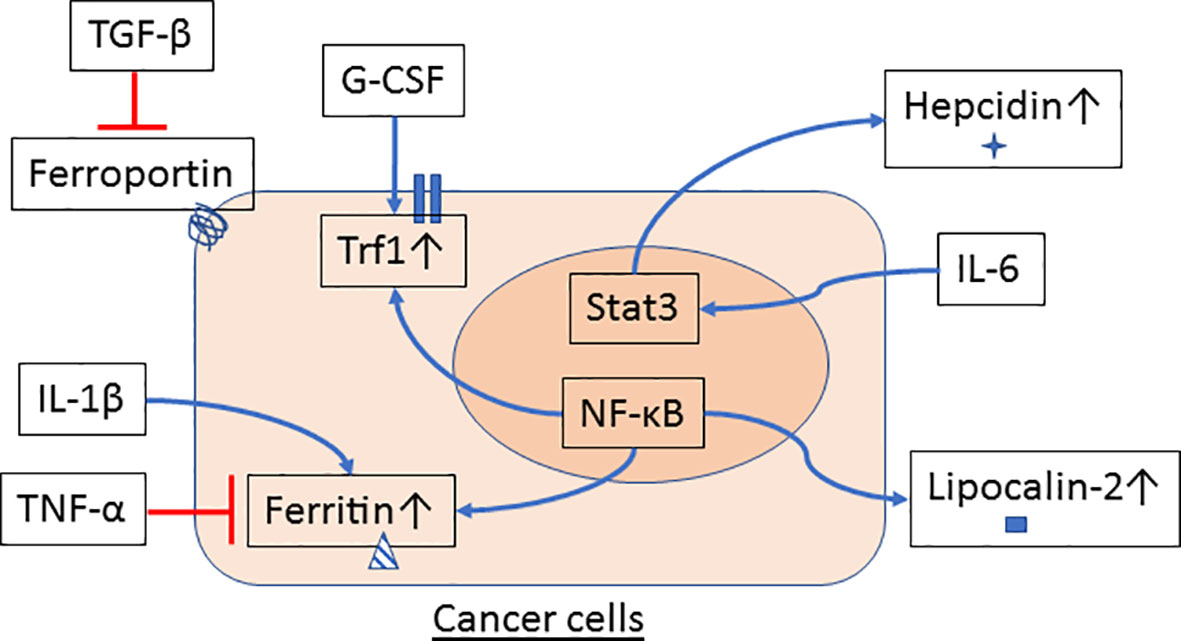
Figure 2 Innate immune cells signal iron metabolism in cancer cells. Cytokines and growth factors are released by macrophages and neutrophils in the tumor microenvironment (TME) and may affect the regulation of iron metabolism in cancer cells. TGF-β reduces ferroportin while G-CSF induces Tfr1 gene expression. Expression of ferritin is inhibited by TNF-α but increased by IL-1β. IL-6 stimulates Stat3 activation and hepcidin expression in hepatocytes and hepatoma cells. NF-κB, a transcription factor that can be activated by a variety of proinflammatory cytokines and factors, also participates in the regulation of gene expression of ferritin, Tfr1 and lipocalin-2 in cancer cells.
Macrophages are a group of innate immune system cells with high plasticity and are frequently found in the TME. TAM can stimulate cancer cell growth, angiogenesis and metastasis, suppress anticancer immunity and render cancer resistance to therapies (36, 37). In response to different stimuli, macrophages can be polarized to M1 (classically activated, proinflammatory) or M2 (alternatively activated, anti-inflammatory) subtypes (38). Interestingly, it has been reported that M1 macrophages display high expression of ferritin and low expression of ferroportin, favoring iron sequestration in macrophages (39, 40). M2 macrophages, on the other hand, show low expression of ferritin and high expression of ferroportin, representing an iron-releasing phenotype (39, 40). Since TAM shares many features with M2 macrophages (37), it is plausible that TAM acts as a source of iron in the TME and promotes cancer growth through an iron-dependent mechanism. In fact, analysis of primary tumors and axillary lymph nodes from breast cancer patients revealed a substantial iron reservoir in stromal inflammatory cells (41). Concomitantly, while breast cancer cells display an “iron-utilization” phenotype characterized by increased expression of hepcidin and Tfr1 and decreased expression of ferritin, macrophages in primary tumors and metastasized lymph nodes manifest an “iron-donor” phenotype characterized by increased expression of ferroportin and ferritin (41). Further supporting this notion, another group examined the mitogenic effects of M1 and M2 macrophages on cancer cells and found that compared to M1 macrophages, M2 macrophage-conditioned medium has significantly greater ability to stimulate cancer cell proliferation (40). Incubation of macrophages with an iron chelator inhibits M2 macrophage conditioned medium-induced cancer cell proliferation (40). Interestingly, comparison of the mitogenic response toward M1 and M2 macrophage-conditioned media from a patient with “loss of function” ferroportin mutation showed no differences in cancer cell proliferation (40). These results suggest that the greater mitogenic activity of M2 macrophages on cancer cells is at least in part mediated by ferroportin-controlled iron release from macrophages. Additionally, M2 macrophages stimulate MCF-7 breast cancer cell growth and migration by an iron-dependent mechanism (42). Coculture with iron-loaded monocytes recapitulates the effects of iron treatment on rendering myeloma cells resistance to bortezomib (43).
Neutrophils are the most abundant immune cells in humans. Cytokines and growth factors secreted by tumor cells can stimulate neutrophil differentiation, proliferation, mobilization and release from the bone marrow (2, 44). Elevated neutrophil numbers in the systemic circulation and in tumor tissues are frequently reported in tumor patients and often correlate with poor survival (2). Like macrophages, neutrophils can be programed by tumor-released factors to exert a variety of protumoral functions by acting on cancer cells, endothelial cells, other immune cells and the extracellular matrix (2). Proteins involved in iron uptake, storage and export are expressed not only in macrophages but also in neutrophils (45). In fact, iron plays an important role in neutrophils’ defense against invading pathogens (45). Iron participates in generating the oxidative burst that is required to kill phagocytosed microbes in neutrophils (45). Moreover, neutrophils produce large amounts of lipocalin-2 and lactoferrin, both of which are iron-scavenging proteins and thus limit microbial growth (45).
Direct evidence that neutrophils are a source of iron in the TME is lacking. Yet, previous work showed that in a rat model mimicking human Alpha-1-antitrypsin (A1AT) deficiency, intratracheal administering of neutrophil elastase (NE) (a serine protease mainly synthesized by neutrophils), increases iron content in the bronchoalveolar lavage (46). NE degrades iron-containing proteins like ferritin in the extracellular space, increasing iron availability and uptake into human airway epithelial cells (46). Whether such mechanism exists in the TME and whether it contributes to increases iron uptake by tumor cells warrant further investigation.
Lipocalin-2 is an acute phase protein that can be synthesized by a variety of cell types including epithelial cells, macrophages and neutrophils (45). Lipocalin-2 binds to bacterial or mammalian siderophores loaded with iron (47), which has two potential consequences, possibly depending on the stages of inflammation. At early stages of inflammation, binding of lipocalin-2 to the iron-siderophore complex sequesters iron from uptake by bacteria, limiting bacterial growth and thus mediating the antimicrobial function of lipocalin-2. On the other hand, the iron-siderophore-lipocalin-2 complex can serve as an iron donor and stimulate epithelial cell proliferation, an event likely occurring during the resolution phase of inflammation when epithelial cell proliferation is needed to mediate tissue repair. Given the resemblance of wound healing to cancer development, it is tempting to speculate that lipocalin-2 derived from TAM or neutrophils stimulates cancer cell growth by an iron-dependent mechanism. In fact, enhanced expression of lipocalin-2 has been documented in multiple cancer types and was associated with poor patient survival (48–52). In vitro, iron-loaded lipocalin-2 promotes spheroid growth of cancer cells, whereas iron-free lipocalin-2 inhibits it (48).
Macrophage-derived lipocalin-2 induces proliferation, epithelial-mesenchymal transition and metastatic potential in MCF-7 human breast cancer cells (53, 54). Besides cancer cells, lipocalin-2 can enhance the protumoral functions of tumor-associated stromal cells. Lipocalin-2, released by macrophages, induces VEGFC production, lymphangiogenesis and metastasis (55). Moreover, apoptotic tumor cells stimulate expression of lipocalin-2 in macrophages and polarization of these macrophages to M2 phenotypes (56). It remains to be determined whether the aforementioned effects are dependent on the iron donation function of lipocalin-2. In another study, lipocalin-2 was found to be predominantly expressed in TAM and to act as a paracrine factor that supplies iron, thus stimulating proliferation of cancer cells (57). Lipocalin-2 deficiency in TAM inhibits tumor growth, which can be reversed by iron supplement (58). According to a published report, iron demand increased as tumors progressed to the metastatic stage, and this was met by TAM-derived lipocalin-2 and inhibited by lipocalin-2 antibody neutralization (57). Together, these results identify TAM-derived lipocalin-2 as a promising therapeutic target for inhibiting the tumor-supporting functions of TAM. Lipocalin-2 is also secreted by neutrophils as a component of secondary granules and deficiency of lipocalin-2 impairs the chemotaxis ability of neutrophils (59). A recent report indicates that CXCL1 secreted by myofibroblasts recruits neutrophils to the TME. Neutrophil-derived lipocalin-2 induces activation of Src family kinases in prostate cancer cells and promotes cancer cell migration and metastasis (60). Whether such action is dependent on the iron-binding function of lipocalin-2 remains to be determined. Lipocalin-2 can be expressed by other cell types including cancer cells. Interestingly, a recent study reports that in the metastatic microenvironment of cerebrospinal fluid, macrophages do not express lipocalin-2 but rather produce inflammatory cytokines that induce lipocalin-2 expression in cancer cells (61). Expression of lipocalin-2 and its receptor, SCL22A17, by cancer cells was essential for cancer cell growth at metastatic sites (61).
Though typically viewed as an intracellular iron storage protein, ferritin is present in the serum. Indeed, serum ferritin has been reported to be a diagnostic and prognostic marker for inflammatory diseases and cancer (62–66). Both hepatocytes and macrophages have been implicated as sources of serum ferritin (67, 68). The releasing mechanisms and the roles of serum ferritin in inflammatory diseases and cancer are largely uncharacterized. It was shown that ferritin released by erythrophagocytosing Kupffer cells is loaded with iron and can mediate iron transport from Kupffer cells to hepatocytes (69). Moreover, in the absence of transferrin, ferritin synthesized and secreted by macrophages may serve as a source of iron for co-cultured erythroid precursors (68). These results corroborate the potential of macrophage-secreted ferritin as an iron donor for other cell types. Nonetheless, whether such hypothesis can be extended to the TME where TAM and cancer cells co-exist remains to be determined. Interestingly, TAM has been shown to be a predominant source of extracellular ferritin and TAM-secreted ferritin can act as paracrine factor that promotes cancer cell proliferation, angiogenesis and immunosuppression (70). Still, it should be pointed out that TAM-secreted ferritin can stimulate cancer cell proliferation via iron-independent mechanisms (71). Further studies are warranted to dissect which functions mediated by TAM-secreted ferritin are iron-dependent or independent.
Hepcidin expression in macrophages and neutrophils can be further enhanced by proinflammatory cytokines like TNF-α and IFN-β, depending on the context; or by engagement of Toll-like receptors (TLR) (72–75). Inflammation-induced hepcidin expression in macrophages is mediated by activation of the Stat1 and NF-κB pathways and induction of C/EBPβ expression (73). Secreted hepcidin can then act as an autocrine factor that inhibits ferroportin function and iron release from these innate immune cells (76). Given the abundance of endogenous TLR agonists in the TME, it will be of great interest to determine whether hepcidin released by macrophages and neutrophils in the TME, in response to endogenous TLR agonists, inhibits ferroportin on cancer cells as a paracrine factor and increases intracellular iron content and thereby cancer cell proliferation.
Transferrin and its receptor Tfr1 are the major route for cellular iron uptake. We previously reported that transferrin is expressed by neutrophils, but not cancer cells, in the metastatic microenvironment and that it mediates neutrophil-dependent mitogenic effects on cancer cells (19). Depletion of neutrophils reduced transferrin levels in the metastatic microenvironment and inhibited metastasis in mouse models (19). GM-CSF, derived mainly from metastatic tumor cells, selectively induces transferrin gene expression in neutrophils through the Jak/Stat5β pathway (19). Blockade of GM-CSF or inhibition of Jak kinases inhibits neutrophil transferrin expression and disrupted the paracrine loop between metastatic cancer cells and neutrophils, resulting in reduced metastasis (19). This work highlighted the potential of neutrophils in modulating iron metabolism in cancer cells and validated the targeting strategies for blocking prometastatic functions of neutrophils.
Besides directly supplying iron and iron-related proteins, macrophages and neutrophils may affect iron metabolism in cancer cells through release of cytokines, chemokines and growth factors that act on cancer cells and induce changes in signaling events that regulate iron metabolism. Treatment of breast cancer cells with TGF-β, a cytokine known to be produced by TAM, reduces ferroportin expression in cancer cells (77). G-CSF, a hematopoietic growth factor that can be expressed by macrophages, induces Tfr1 expression in human myeloid leukemia cell lines (78). TNF-α can be expressed by TAM and neutrophils and treatment with TNF-α results in reduced ferritin expression in prostate cancer cells (79). TAM and neutrophils are known to produce IL-1β. Treatment of hepatoma cells with IL-1β increases ferritin expression (80). IL-6 is expressed by TAM and neutrophils and stimulates hepcidin expression in hepatocytes (81, 82). Augmented levels of hepcidin inhibits ferroportin on cancer cells and increases iron content in cancer cells (31, 83).
Numerous reports have shown that NF-κB and Stat3, two essential transcription factors regulating the inflammatory responses and immune landscape in the TME, also play key roles in controlling gene expression of iron metabolism proteins (84–88). In fact, many of the products from TAM and neutrophils in the TME are known to upregulate or downregulate activation of NF-κB and Stat3 in cancer cells (89, 90), further highlighting the crosstalk between tumor-associated innate immunity and dysregulated iron metabolism in cancer cells.
Iron metabolism is thought to contribute to macrophage polarization, although there are conflicting reports on the precise effects. For example, Agoro et al (91) reported that iron-rich diet induces M2 polarization in liver and peritoneal macrophages and suppresses the proinflammatory M1 phenotypes in mice. Addition of iron to cultured macrophages inhibited expression of M1 costimulatory proteins and prevented LPS-induced NF-κB p65 nuclear translocation and expression of iNOS, IL-1β, IL-6, IL-12 and TNFα (91). The notion that iron overload favors M2 over M1 polarization was also supported by other studies (92, 93). Yet, opposite conclusions were reached by others as it was reported that treatment of bone marrow-derived macrophages with iron induces expression of M1 markers and reduces IL-4-induced M2 markers (94). Dietary iron overload in mice leads to M1 polarization of hepatic macrophages (94). Consistent with these findings, another group reported that iron supplement promotes M1 polarization in mechanisms depending on ROS production and p53 acetylation (95). Exposure of TAM to hemolytic red blood cells or iron nanoparticles increases iron content, M1 marker expression and antitumor activities of macrophages (96, 97). Moreover, lung cancer patients with positive iron staining in tumor tissues had better overall survival associated with higher expression of markers of M1 macrophages (98). These conflicting findings highlight the complex nature of macrophage polarization even in the context of iron metabolism. They also raise some concern for the use of iron overloading or chelating strategies to switch macrophage polarization for treatment of inflammatory diseases and cancer.
The markedly elevated demand of iron by cancer cells suggests that the use of iron chelators may be an effective anti-cancer strategy. Indeed, in preclinical models iron chelators inhibited activation of signaling pathways important for cell proliferation and survival and suppress tumor growth and metastasis (99). However, these agents demonstrated only modest therapeutic benefits when tested in cancer patients (99). While continuing efforts are ongoing to improve the bioavailability of and design combination therapies with the iron chelators, different iron-modulating strategies are warranted. The fact that innate immune cells can serve as a source of iron and iron-related proteins in the TME provided a clear rationale for target discovery and validation.
Despite the fact that iron is critically required by cancer cells at different stages of tumorigenesis, altering iron levels and cellular uptake/storage/utilization/export machineries at systemic levels is in principle detrimental, since maintaining iron homeostasis is essential for normal physiological processes. One alternative could be to identify local sources of iron and iron-related proteins in the TME that can be pharmacologically targeted.
Our laboratory has been investigating the role of neutrophils in tumor angiogenesis and in resistance to anti-angiogenic therapies with VEGF inhibitors and these efforts led to the identification of neutrophil-derived angiogenic factors such as Bv8/PROK2 (100–102). We next sought to elucidate the nature of neutrophil-derived factors that directly promote growth of metastatic tumor cells. By employing a proteomic/functional approach, we unexpectedly identified transferrin as the major mitogen for tumor cells secreted by neutrophils (19). Depletion of neutrophils inhibited lung metastasis and transferrin production in the metastatic microenvironment. Transferrin expression by neutrophils was induced by tumor derived GM-CSF (19). The mechanism (Jak/Stat5β) by which GM-CSF induces transferrin expression is unique to neutrophils and is not shared by other cell types (19). In this case, one can expect that therapeutic agents targeting the GM-CSF/Jak/Stat5β signaling pathway (e.g., GM-CSF neutralizing antibodies or Jak kinase inhibitors) lowers transferrin levels specifically in the neutrophil-dominant metastatic microenvironment and inhibits cancer metastasis. Such strategies should spare transferrin production by other cellular sources and leave transferrin-mediated physiological iron homeostasis untouched.
Evidence reviewed above indicates that expression of lipocalin-2 in macrophages or neutrophils can promote cancer cell growth, induce M2 polarization of macrophages and is required for neutrophil chemotaxis. Therefore, blocking lipocalin-2 production from macrophages and neutrophils might simultaneously target cancer cells and cancer-associated myeloid cells. Previous work found that IL-10, an immunosuppressive cytokine that enhances the tumor-supporting functions of macrophages, induces lipocalin-2 expression in macrophages via activation of the Jak/Stat3 signaling pathway (53). Pharmacological inhibition of Jak or Stat3 suppresses IL-10-induced lipocalin-2 mRNA and protein levels (53). A variety of inhibitors of Jak kinases or Stat3 have entered clinical trials for cancer treatment, owing primary to the multifaceted functions of the Jak/Stat3 pathway in facilitating cancer cell proliferation, survival and metastasis, tumor angiogenesis and immunosuppression (89). It will be of great interest to determine whether such inhibitors suppress lipocalin-2 production from patient tumor-associated macrophages and neutrophils and reduce iron content in patient tumor cells and whether these effects contribute to the anticancer efficacy.
Iron exiting macrophages through ferroportin can become available for uptake by cancer cells in the TME. Reducing ferroportin expression levels in TAM thus represents a promising strategy for cancer treatment. Numerous studies have shown that stimulation of various TLRs decreases ferroportin expression, associated with increased intracellular iron concentrations in macrophages (103–105). Currently, TLR agonists are being tested in clinical trials for their ability to orchestrate anticancer immunity (106). The anticancer activities are largely attributed to TLR-mediated maturation and activation of antigen-presenting cells and follow-up activation of adaptive immune responses against cancer cells (107). It remains to be determined, in both preclinical and clinical settings, whether TLR agonists modulate the iron-donation phenotypes of TAM and reduce iron contents in cancer cells and whether such effects contribute to their anticancer activities. Moreover, TLR activation induces hepcidin expression in macrophages and neutrophils (103, 104, 108). Therefore, treatment with TLR agonist might pack a “one-two punch” by decreasing ferroportin expression and inducing hepcidin release to act as an autocrine/paracrine factor to further inhibit ferroportin-mediated iron release from macrophages and neutrophils.
Ferroptosis is a newly-identified, nonapoptotic form of regulated cell death, the hallmark of which is the iron‐dependent accumulation of lipid hydroperoxides (109, 110). High iron levels, inhibition of glutathione peroxidase 4 (GPX4) and glutathione synthesis, starvation of cysteine, chemotherapy and targeted therapy are known to induce ferroptosis (109, 110). It has been hypothesized that cancer cells, compared to their normal counterparts, are more susceptible to ferroptosis for the following reasons: 1) Intracellular iron levels are higher in cancer cells due to upregulated iron uptake and downregulated export mechanisms. 2) Oxidative stress is more severe in cancer cells due to activation and/or mutation of oncogenic pathways, increased metabolic activities and hypoxia in the TME. As such, therapeutic approaches that induce ferroptosis are being actively investigated for cancer treatment. A recent study, using a hyperactivated transcriptional coactivator with PDZ-binding motif-driven mouse glioblastoma model, indicates that tumor-associated neutrophils induce ferroptosis in cocultured tumor cells and necrosis in tumor tissues, leading to the enhanced cancer aggressiveness and reduced mouse survival (111). Rescue of ferroptosis through GPX4 overexpression or ACSL4 silencing in tumor cells was reported to alleviate neutrophil-induced tumor cell killing and necrosis, inhibited tumor aggressiveness and improved mouse survival (111).
One interesting question is whether cancer cell death by ferroptosis is immunogenic and whether it can modulate the phenotypes of TAM and neutrophils in the TAM. Whereas direct evidence for such a link is lacking, it was recently shown that cancer cells undergoing ferroptosis release HMGB1 (112), an endogenous TLR agonist and a known activator of immunogenic cell death (113). In fact, ferroptotic cell death after heart transplantation stimulates neutrophil recruitment through a TLR4-dependent mechanism (114).
Ferroptosis also takes place in macrophages. Numerous reports confirmed that iron overload induces macrophage ferroptosis in vitro and in vivo (115–117). Interestingly, M1 macrophages, compared to M2 macrophages that resemble TAM, are more resistant to ferroptosis (117). Therefore, ferroptosis-inducing agents may selectively target the tumor-supporting TAM, while sparing the tumor-suppressive M1 macrophages. Inhibition of GPX4 induces ferroptosis in a variety of cell types including cancer cells (118). However, a recent study found that GPX4 deficiency does not affect survival of macrophages and neutrophils in mice (119). Instead, GPX4 deficiency induces oxidative stress and H2O2 release in myeloid cells, which promotes tumorigenesis by triggering genome-wide mutations in intestinal epithelial cells (119). This work argues against using GPX4 inhibitors, at least by itself, for inducing ferroptosis in macrophage and neutrophils. Future studies are warranted to determine the signaling pathways in regulation of ferroptosis in TAM and neutrophils in the context of cancer and whether other therapeutic approaches, alone or in combination, can eliminate TAM and neutrophils by a ferroptosis-dependent mechanism.
The evidence reviewed in this article supports the notion that innate immune cells like macrophages and neutrophils promote cancer development and progression through multiple iron-dependent mechanisms. The dysregulated iron metabolism in the TME may also affect the phenotypes of innate immune cells. Though somewhat speculative, it is possible that cancer cells, equipped with hyperfunctional iron uptake and downregulated export machineries, deprive the TME of iron and produce iron metabolism-related byproducts (e.g., ROS), to boost the protumoral functions or to suppress the anticancer activities of innate immune cells.
Questions to be addressed by future studies are: what is the cause of the inconsistent results on iron-induced macrophage polarization? Given largely shared expression of iron-related proteins with macrophages, can neutrophils contribute to the release of iron and iron-related proteins and aberrantly accumulated iron levels in cancer cells, to the same extent as macrophages? Does release of iron or iron-related proteins from innate immune cells act on other tumor-associated stromal cells like T cells, fibroblasts and endothelial cells and affect their functions? What are the resistance mechanisms of innate immune cells to GPX4 inhibitor-induced ferroptosis? Does iron metabolism in TAM and neutrophils mediate response or resistance of tumor cells to chemo-, targeted- and immunotherapies; are also highly warranted.
The gained knowledge will deepen our understanding of the role of iron metabolism in the crosstalk between macrophages/neutrophils and cancer cells or other stromal cells in the TME and should facilitate the identification of novel targets for disrupting such crosstalk.
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
WL was employed by the company BioDuro LLC.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
2. Liang W, Ferrara N. The Complex Role of Neutrophils in Tumor Angiogenesis and Metastasis. Cancer Immunol Res (2016) 4(2):83–91. doi: 10.1158/2326-6066.CIR-15-0313
3. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141(1):39–51. doi: 10.1016/j.cell.2010.03.014
4. Wu D. Innate and Adaptive Immune Cell Metabolism in Tumor Microenvironment. Adv Exp Med Biol (2017) 2017(1011):211–23. doi: 10.1007/978-94-024-1170-6_7
5. Halbrook CJ, Pontious C, Kovalenko I, Lapienyte L, Dreyer S, Lee HJ, et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab (2019) 29(6):1390–99.e6. doi: 10.1016/j.cmet.2019.02.001
6. Littlewood-Evans A, Sarret S, Apfel V, Loesle P, Dawson J, Zhang J, et al. GPR91 senses extracellular succinate released from inflammatory macrophages and exacerbates rheumatoid arthritis. J Exp Med (2016) 213(9):1655–62. doi: 10.1084/jem.20160061
7. Zhunussova A, Sen B, Friedman L, Tuleukhanov S, Brooks AD, Sensenig R, et al. Tumor microenvironment promotes dicarboxylic acid carrier-mediated transport of succinate to fuel prostate cancer mitochondria. Am J Cancer Res (2015) 5(5):1665–79.
8. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer (2019) 18(1):70. doi: 10.1186/s12943-019-0994-2
9. Puig S, Ramos-Alonso L, Romero AM, Martínez-Pastor MT. The elemental role of iron in DNA synthesis and repair. Metallomics (2017) 9(11):1483–500. doi: 10.1039/c7mt00116a
10. Paul BT, Manz DH, Torti FM, Torti SV. Mitochondria and Iron: current questions. Expert Rev Hematol (2017) 10(1):65–79. doi: 10.1080/17474086.2016.1268047
11. Nairz M, Weiss G. Iron in infection and immunity. Mol Aspects Med (2020) 75:100864. doi: 10.1016/j.mam.2020.100864
12. Muckenthaler MU, Rivella S, Hentze MW. Galy B. A Red Carpet for Iron Metabolism. Cell (2017) 168(3):344–61. doi: 10.1016/j.cell.2016.12.034
13. Bystrom LM, Guzman ML, Rivella S. Iron and reactive oxygen species: friends or foes of cancer cells? Antioxid Redox Signal (2014) 20(12):1917–24. doi: 10.1089/ars.2012.5014
14. Chifman J, Laubenbacher R, Torti SV. A systems biology approach to iron metabolism. Adv Exp Med Biol (2014) 844:201–25. doi: 10.1007/978-1-4939-2095-2_10
15. Vostrejs M, Moran PL, Seligman PA. Transferrin synthesis by small cell lung cancer cells acts as an autocrine regulator of cellular proliferation. J Clin Invest (1988) 82(1):331–9. doi: 10.1172/JCI113591
16. Adachi M, Kai K, Yamaji K, Ide T, Noshiro H, Kawaguchi A, et al. Transferrin receptor 1 overexpression is associated with tumour de-differentiation and acts as a potential prognostic indicator of hepatocellular carcinoma. Histopathology (2019) 75(1):63–73. doi: 10.1111/his.13847
17. Greene CJ, Attwood K, Sharma NJ, Gross KW, Smith GJ, Xu B, et al. Transferrin receptor 1 upregulation in primary tumor and downregulation in benign kidney is associated with progression and mortality in renal cell carcinoma patients. Oncotarget (2017) 8(63):107052–75. doi: 10.18632/oncotarget.22323
18. Wu H, Zhang J, Dai R, Xu J, Feng H. Transferrin receptor-1 and VEGF are prognostic factors for osteosarcoma. J Orthop Surg Res (2019) 14(1):296. doi: 10.1186/s13018-019-1301-z
19. Liang W, Li Q, Ferrara N. Metastatic growth instructed by neutrophil-derived transferrin. Proc Natl Acad Sci USA (2018) 115(43):11060–5. doi: 10.1073/pnas.1811717115
20. O’Donnell KA, Yu D, Zeller KI, Kim JW, Racke F, Thomas-Tikhonenko A, et al. Activation of transferrin receptor 1 by c-Myc enhances cellular proliferation and tumorigenesis. Mol Cell Biol (2006) 26(6):2373–86. doi: 10.1128/MCB.26.6.2373-2386.2006
21. Xue X, Ramakrishnan SK, Weisz K, Triner D, Xie L, Attili D, et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab (2016) 24(3):447–61. doi: 10.1016/j.cmet.2016.07.015
22. Brookes MJ, Hughes S, Turner FE, Reynolds G, Sharma N, Ismail T, et al. Modulation of iron transport proteins in human colorectal carcinogenesis. Gut (2006) 55(10):1449–60. doi: 10.1136/gut.2006.094060
23. Hubert RS, Vivanco I, Chen E, Rastegar S, Leong K, Mitchell SC, et al. STEAP: a prostate-specific cell-surface antigen highly expressed in human prostate tumors. Proc Natl Acad Sci USA (1999) 96(25):14523–8. doi: 10.1073/pnas.96.25.14523
24. Yamamoto T, Tamura Y, Kobayashi J, Kamiguchi K, Hirohashi Y, Miyazaki A, et al. Six-transmembrane epithelial antigen of the prostate-1 plays a role for in vivo tumor growth via intercellular communication. Exp Cell Res (2013) 319(17):2617–26. doi: 10.1016/j.yexcr.2013.07.025
25. Whiteland H, Spencer-Harty S, Morgan C, Kynaston H, Thomas DH, Bose P, et al. A role for STEAP2 in prostate cancer progression. Clin Exp Metastasis (2014) 31(8):909–20. doi: 10.1007/s10585-014-9679-9
26. Challita-Eid PM, Morrison K, Etessami S, An Z, Morrison KJ, Perez-Villar JJ, et al. Monoclonal antibodies to six-transmembrane epithelial antigen of the prostate-1 inhibit intercellular communication in vitro and growth of human tumor xenografts in vivo. Cancer Res (2007) 67(12):5798–805. doi: 10.1158/0008-5472.CAN-06-3849
27. Pinnix ZK, Miller LD, Wang W, D'Agostino R, Kute T, Willingham MC, et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci Transl Med (2010) 2(43):43ra56. doi: 10.1126/scisignal.3001127
28. Toshiyama R, Konno M, Eguchi H, Asai A, Noda T, Koseki J, et al. Association of iron metabolic enzyme hepcidin expression levels with the prognosis of patients with pancreatic cancer. Oncol Lett (2018) 15(5):8125–33. doi: 10.3892/ol.2018.8357
29. Zhou Q, Chen J, Feng J, Wang J. E4BP4 promotes thyroid cancer proliferation by modulating iron homeostasis through repression of hepcidin. Cell Death Dis (09) 20189(10):987. doi: 10.1038/s41419-018-1001-3
30. Tesfay L, Clausen KA, Kim JW, Hegde P, Wang X, Miller LD, et al. Hepcidin regulation in prostate and its disruption in prostate cancer. Cancer Res (2015) 75(11):2254–63. doi: 10.1158/0008-5472.CAN-14-2465
31. Zhang S, Chen Y, Guo W, Yuan L, Zhang D, Xu Y, et al. Disordered hepcidin-ferroportin signaling promotes breast cancer growth. Cell Signal (2014) 26(11):2539–50. doi: 10.1016/j.cellsig.2014.07.029
32. Khiroya H, Moore JS, Ahmad N, Kay J, Woolnough K, Langman G, et al. IRP2 as a potential modulator of cell proliferation, apoptosis and prognosis in nonsmall cell lung cancer. Eur Respir J (2017) 49(4). doi: 10.1183/13993003.00711-2016
33. Wang W, Deng Z, Hatcher H, Miller LD, Di X, Tesfay L, et al. IRP2 regulates breast tumor growth. Cancer Res (2014) 74(2):497–507. doi: 10.1158/0008-5472.CAN-13-1224
34. Deng Z, Manz DH, Torti SV, Torti FM. Iron-responsive element-binding protein 2 plays an essential role in regulating prostate cancer cell growth. Oncotarget (2017) 8(47):82231–43. doi: 10.18632/oncotarget.19288
35. Miyazawa M, Bogdan AR, Tsuji Y. Perturbation of Iron Metabolism by Cisplatin through Inhibition of Iron Regulatory Protein 2. Cell Chem Biol (2019) 26(1):85–97.e4. doi: 10.1016/j.chembiol.2018.10.009
36. Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov (2018) 17(12):887–904. doi: 10.1038/nrd.2018.169
37. Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev (2006) 25(3):315–22. doi: 10.1007/s10555-006-9001-7
38. Murray PJ. Macrophage Polarization. Annu Rev Physiol (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
39. Corna G, Campana L, Pignatti E, Castiglioni A, Tagliafico E, Bosurgi L, et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica (2010) 95(11):1814–22. doi: 10.3324/haematol.2010.023879
40. Recalcati S, Locati M, Marini A, Santambrogio P, Zaninotto F, De Pizzol M, et al. Differential regulation of iron homeostasis during human macrophage polarized activation. Eur J Immunol (2010) 40(3):824–35. doi: 10.1002/eji.200939889
41. Marques O, Porto G, Rêma A, Faria F, Cruz Paula A, Gomez-Lazaro M, et al. Local iron homeostasis in the breast ductal carcinoma microenvironment. BMC Cancer (2016) 16:187. doi: 10.1186/s12885-016-2228-y
42. Mertens C, Akam EA, Rehwald C, Brüne B, Tomat E, Jung M. Intracellular Iron Chelation Modulates the Macrophage Iron Phenotype with Consequences on Tumor Progression. PLoS One (2016) 11(11):e0166164. doi: 10.1371/journal.pone.0166164
43. Camiolo G, Barbato A, Giallongo C, Vicario N, Romano A, Parrinello NL, et al. Iron regulates myeloma cell/macrophage interaction and drives resistance to bortezomib. Redox Biol (2020) 36:101611. doi: 10.1016/j.redox.2020.101611
44. Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA (2010) 107(50):21248–55. doi: 10.1073/pnas.1015855107
45. Cronin SJF, Woolf CJ, Weiss G, Penninger JM. The Role of Iron Regulation in Immunometabolism and Immune-Related Disease. Front Mol Biosci (2019) 6:116. doi: 10.3389/fmolb.2019.00116
46. Fischer BM, Domowicz DA, Zheng S, Carter JL, McElvaney NG, Taggart C, et al. Neutrophil elastase increases airway epithelial nonheme iron levels. Clin Transl Sci (2009) 2(5):333–9. doi: 10.1111/j.1752-8062.2009.00151.x
47. Wilson BR, Bogdan AR, Miyazawa M, Hashimoto K, Tsuji Y. Siderophores in Iron Metabolism: From Mechanism to Therapy Potential. Trends Mol Med (2016) 22(12):1077–90. doi: 10.1016/j.molmed.2016.10.005
48. Rehwald C, Schnetz M, Urbschat A, Mertens C, Meier JK, Bauer R, et al. The iron load of lipocalin-2 (LCN-2) defines its pro-tumour function in clear-cell renal cell carcinoma. Br J Cancer (02) 2020122(3):421–33. doi: 10.1038/s41416-019-0655-7
49. Tai J, Wang S, Zhang J, Ge W, Liu Y, Li X, et al. Up-regulated lipocalin-2 in pediatric thyroid cancer correlated with poor clinical characteristics. Eur Arch Otorhinolaryngol (2018) 275(11):2823–8. doi: 10.1007/s00405-018-5118-x
50. Srdelić Mihalj S, Kuzmić-Prusac I, Zekić-Tomaš S, Šamija-Projić I, Čapkun V. Lipocalin-2 and matrix metalloproteinase-9 expression in high-grade endometrial cancer and their prognostic value. Histopathology (2015) 67(2):206–15. doi: 10.1111/his.12633
51. Maier HT, Aigner F, Trenkwalder B, Zitt M, Vallant N, Perathoner A, et al. Up-regulation of neutrophil gelatinase-associated lipocalin in colorectal cancer predicts poor patient survival. World J Surg (2014) 38(8):2160–7. doi: 10.1007/s00268-014-2499-x
52. Mannelqvist M, Stefansson IM, Wik E, Kusonmano K, Raeder MB, Øyan E, et al. Lipocalin 2 expression is associated with aggressive features of endometrial cancer. BMC Cancer (2012) 12:169. doi: 10.1186/1471-2407-12-169
53. Jung M, Weigert A, Tausendschön M, Mora J, Ören B, Sola A, et al. Interleukin-10-induced neutrophil gelatinase-associated lipocalin production in macrophages with consequences for tumor growth. Mol Cell Biol (2012) 32(19):3938–48. doi: 10.1128/MCB.00413-12
54. Ören B, Urosevic J, Mertens C, Mora J, Guiu M, Gomis RR, et al. Tumour stroma-derived lipocalin-2 promotes breast cancer metastasis. J Pathol (2016) 239(3):274–85. doi: 10.1002/path.4724
55. Jung M, Ören B, Mora J, Merten C, Dziumbla S, Popp R, et al. Lipocalin 2 from macrophages stimulated by tumor cell-derived sphingosine 1-phosphate promotes lymphangiogenesis and tumor metastasis. Sci Signal (2016) 9(434):ra64. doi: 10.1126/scisignal.aaf3241
56. Sola A, Weigert A, Jung M, Vinuesa E, Brecht K, Weis N, et al. Sphingosine-1-phosphate signalling induces the production of Lcn-2 by macrophages to promote kidney regeneration. J Pathol (2011) 225(4):597–608. doi: 10.1002/path.2982
57. Duan X, He K, Li J, Cheng M, Song H, Liu J, et al. Tumor associated macrophages deliver iron to tumor cells via Lcn2. Int J Physiol Pathophysiol Pharmacol (2018) 10(2):105–14.
58. Mertens C, Mora J, Ören B, Grein S, Winslow S, Scholich K, et al. Macrophage-derived lipocalin-2 transports iron in the tumor microenvironment. Oncoimmunology (2018) 7(3):e1408751. doi: 10.1080/2162402X.2017.1408751
59. Ye D, Yang K, Zang S, Lin Z, Chau HT, Wang Y, et al. Lipocalin-2 mediates non-alcoholic steatohepatitis by promoting neutrophil-macrophage crosstalk via the induction of CXCR2. J Hepatol (2016) 65(5):988–97. doi: 10.1016/j.jhep.2016.05.041
60. Lu Y, Dong B, Xu F, Xu Y, Pan J, Song J, et al. CXCL1-LCN2 paracrine axis promotes progression of prostate cancer via the Src activation and epithelial-mesenchymal transition. Cell Commun Signal (2019) 17(1):118. doi: 10.1186/s12964-019-0434-3
61. Chi Y, Remsik J, Kiseliovas V, Derderian C, Sener U, Alghader M, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Science (2020) 369(6501):276–82. doi: 10.1126/science.aaz2193
62. Lee S, Song A, Eo W. Serum Ferritin as a Prognostic Biomarker for Survival in Relapsed or Refractory Metastatic Colorectal Cancer. J Cancer (2016) 7(8):957–64. doi: 10.7150/jca.14797
63. Milman N, Pedersen LM. The serum ferritin concentration is a significant prognostic indicator of survival in primary lung cancer. Oncol Rep (2002) 9(1):193–8.
64. Lorenzi M, Lorenzi B, Vernillo R. Serum ferritin in colorectal cancer patients and its prognostic evaluation. Int J Biol Markers (2006) 21(4):235–41. doi: 10.5301/jbm.2008.2954
65. Bertoli S, Paubelle E, Bérard E, Saland E, Thomas X, Tavitian S, et al. Ferritin heavy/light chain (FTH1/FTL) expression, serum ferritin levels, and their functional as well as prognostic roles in acute myeloid leukemia. Eur J Haematol (2019) 102(2):131–42. doi: 10.1111/ejh.13183
66. Kalousová M, Krechler T, Jáchymová M, Kuběna AA, Zák A, Zima T. Ferritin as an independent mortality predictor in patients with pancreas cancer. Results of a pilot study. Tumour Biol (2012) 33(5):1695–700. doi: 10.1007/s13277-012-0426-z
67. Cohen LA, Gutierrez L, Weiss A, Leichtmann-Bardoogo Y, Zhang DL, Crooks DR, et al. Serum ferritin is derived primarily from macrophages through a nonclassical secretory pathway. Blood (2010) 116(9):1574–84. doi: 10.1182/blood-2009-11-253815
68. Leimberg MJ, Prus E, Konijn AM, Fibach E. Macrophages function as a ferritin iron source for cultured human erythroid precursors. J Cell Biochem (2008) 103(4):1211–8. doi: 10.1002/jcb.21499
69. Sibille JC, Kondo H, Aisen P. Interactions between isolated hepatocytes and Kupffer cells in iron metabolism: a possible role for ferritin as an iron carrier protein. Hepatology (1988) 8(2):296–301. doi: 10.1002/hep.1840080218
70. Alkhateeb AA, Connor JR. The significance of ferritin in cancer: anti-oxidation, inflammation and tumorigenesis. Biochim Biophys Acta (2013) 1836(2):245–54. doi: 10.1016/j.bbcan.2013.07.002
71. Alkhateeb AA, Han B, Connor JR. Ferritin stimulates breast cancer cells through an iron-independent mechanism and is localized within tumor-associated macrophages. Breast Cancer Res Treat (2013) 137(3):733–44. doi: 10.1007/s10549-012-2405-x
72. Wu S, Zhang K, Lv C, Wang H, Cheng B, Jin Y, et al. Nuclear factor-κB mediated lipopolysaccharide-induced mRNA expression of hepcidin in human peripheral blood leukocytes. Innate Immun (2012) 18(2):318–24. doi: 10.1177/1753425911405087
73. Sow FB, Alvarez GR, Gross RP, Satoskar AR, Schlesinger LS, Zwilling BS, et al. Role of STAT1, NF-kappaB, and C/EBPbeta in the macrophage transcriptional regulation of hepcidin by mycobacterial infection and IFN-gamma. J Leukoc Biol (2009) 86(5):1247–58. doi: 10.1189/jlb.1208719
74. Sow FB, Florence WC, Satoskar AR, Schlesinger LS, Zwilling BS, Lafuse WP. Expression and localization of hepcidin in macrophages: a role in host defense against tuberculosis. J Leukoc Biol (2007) 82(4):934–45. doi: 10.1189/jlb.0407216
75. Nguyen NB, Callaghan KD, Ghio AJ, Haile DJ, Yang F. Hepcidin expression and iron transport in alveolar macrophages. Am J Physiol Lung Cell Mol Physiol (2006) 291(3):L417–25. doi: 10.1152/ajplung.00484.2005
76. Theurl I, Theurl M, Seifert M, Mair S, Nairz M, Rumpold H, et al. Autocrine formation of hepcidin induces iron retention in human monocytes. Blood (2008) 111(4):2392–9. doi: 10.1182/blood-2007-05-090019
77. Shan Z, Wei Z, Shaikh ZA. Suppression of ferroportin expression by cadmium stimulates proliferation, EMT, and migration in triple-negative breast cancer cells. Toxicol Appl Pharmacol (2018) 356:36–43. doi: 10.1016/j.taap.2018.07.017
78. Morishita Y, Kataoka T, Towatari M, Ito T, Inoue H, Ogura M, et al. Up-regulation of transferrin receptor gene expression by granulocyte colony-stimulating factor in human myeloid leukemia cells. Cancer Res (1990) 50(24):7955–61.
79. Antosiewicz J, Ziolkowski W, Kaczor JJ, Herman-Antosiewicz A. Tumor necrosis factor-alpha-induced reactive oxygen species formation is mediated by JNK1-dependent ferritin degradation and elevation of labile iron pool. Free Radic Biol Med (2007) 43(2):265–70. doi: 10.1016/j.freeradbiomed.2007.04.023
80. Rogers JT, Bridges KR, Durmowicz GP, Glass J, Auron PE, Munro HN. Translational control during the acute phase response. Ferritin synthesis in response to interleukin-1. J Biol Chem (1990) 265(24):14572–8.
81. Nemeth E, Rivera S, Gabayan V, Keller C, Taudorf S, Pedersen BK, et al. IL-6 mediates hypoferremia of inflammation by inducing the synthesis of the iron regulatory hormone hepcidin. J Clin Invest (2004) 113(9):1271–6. doi: 10.1172/JCI20945
82. Pietrangelo A, Dierssen U, Valli L, Garuti C, Rump A, Corradini E, et al. STAT3 is required for IL-6-gp130-dependent activation of hepcidin in vivo. Gastroenterology (2007) 132(1):294–300. doi: 10.1053/j.gastro.2006.10.018
83. Zhao B, Li R, Cheng G, Li Z, Zhang Z, Li J, et al. Role of hepcidin and iron metabolism in the onset of prostate cancer. Oncol Lett (2018) 15(6):9953–8. doi: 10.3892/ol.2018.8544
84. Chen SJ, Kuo CC, Pan HY, Tsou TC, Yeh SC, Chang JY. Desferal regulates hCtr1 and transferrin receptor expression through Sp1 and exhibits synergistic cytotoxicity with platinum drugs in oxaliplatin-resistant human cervical cancer cells in vitro and in vivo. Oncotarget (2016) 7(31):49310–21. doi: 10.18632/oncotarget.10336
85. Iannetti A, Pacifico F, Acquaviva R, Lavorgna A, Crescenzi E, Vascotto C, et al. The neutrophil gelatinase-associated lipocalin (NGAL), a NF-kappaB-regulated gene, is a survival factor for thyroid neoplastic cells. Proc Natl Acad Sci USA (2008) 105(37):14058–63. doi: 10.1073/pnas.0710846105
86. Kiessling MK, Klemke CD, Kaminski MM, Galani IE, Krammer PH, Gülow K. Inhibition of constitutively activated nuclear factor-kappaB induces reactive oxygen species- and iron-dependent cell death in cutaneous T-cell lymphoma. Cancer Res (2009) 69(6):2365–74. doi: 10.1158/0008-5472.CAN-08-3221
87. Verga Falzacappa MV, Vujic Spasic M, Kessler R, Stolte J, Hentze MW, Muckenthaler MU. STAT3 mediates hepatic hepcidin expression and its inflammatory stimulation. Blood (2007) 109(1):353–8. doi: 10.1182/blood-2006-07-033969
88. Pham CG, Bubici C, Zazzeroni F, Papa S, Jones J, Alvarez K, et al. Ferritin heavy chain upregulation by NF-kappaB inhibits TNFalpha-induced apoptosis by suppressing reactive oxygen species. Cell (2004) 119(4):529–42. doi: 10.1016/j.cell.2004.10.017
89. Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer (2009) 9(11):798–809. doi: 10.1038/nrc2734
90. Hoesel B, Schmid JA. The complexity of NF-κB signaling in inflammation and cancer. Mol Cancer (2013) 12:86. doi: 10.1186/1476-4598-12-86
91. Agoro R, Taleb M, Quesniaux VFJ, Mura C. Cell iron status influences macrophage polarization. PLoS One (2018) 13(5):e0196921. doi: 10.1371/journal.pone.0196921
92. Wilkinson HN, Roberts ER, Stafford AR, Banyard KL, Matteucci P, Mace KA, et al. Tissue Iron Promotes Wound Repair via M2 Macrophage Polarization and the Chemokine (C-C Motif) Ligands 17 and 22. Am J Pathol (2019) 189(11):2196–208. doi: 10.1016/j.ajpath.2019.07.015
93. Gan ZS, Wang QQ, Li JH, Wang XL, Wang YZ, Du HH. Iron Reduces M1 Macrophage Polarization in RAW264.7 Macrophages Associated with Inhibition of STAT1. Mediators Inflamm (2017) 2017:8570818. doi: 10.1155/2017/8570818
94. Handa P, Thomas S, Morgan-Stevenson V, Maliken BD, Gochanour E, Boukhar S, et al. Iron alters macrophage polarization status and leads to steatohepatitis and fibrogenesis. J Leukoc Biol (2019) 105(5):1015–26. doi: 10.1002/JLB.3A0318-108R
95. Zhou Y, Que KT, Zhang Z, Yi ZJ, Zhao PX, You Y, et al. Iron overloaded polarizes macrophage to proinflammation phenotype through ROS/acetyl-p53 pathway. Cancer Med (2018) 7(8):4012–22. doi: 10.1002/cam4.1670
96. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol (2016) 11(11):986–94. doi: 10.1038/nnano.2016.168
97. Costa da Silva M, Breckwoldt MO, Vinchi F, Correia MP, Stojanovic A, Thielmann CM, et al. Iron Induces Anti-tumor Activity in Tumor-Associated Macrophages. Front Immunol (2017) 8:1479. doi: 10.3389/fimmu.2017.01479
98. Thielmann CM, Costa da Silva M, Muley T, Meister M, Herpel E, Muckenthaler MU. Iron accumulation in tumor-associated macrophages marks an improved overall survival in patients with lung adenocarcinoma. Sci Rep (2019) 9(1):11326. doi: 10.1038/s41598-019-47833-x
99. Yu Y, Gutierrez E, Kovacevic Z, Saletta F, Obeidy P, Suryo Rahmanto Y, et al. Iron chelators for the treatment of cancer. Curr Med Chem (2012) 19(17):2689–702. doi: 10.2174/092986712800609706
100. Shojaei F, Wu X, Malik AK, Zhong C, Baldwin ME, Schanz S, et al. Tumor refractoriness to anti-VEGF treatment is mediated by CD11b+Gr1+ myeloid cells. Nat Biotechnol (2007) 25(8):911–20. doi: 10.1038/nbt1323
101. Shojaei F, Wu X, Zhong C, Yu L, Liang XH, Yao J, et al. Bv8 regulates myeloid-cell-dependent tumour angiogenesis. Nature (2007) 450(7171):825–31. doi: 10.1038/nature06348
102. Itatani Y, Yamamoto T, Zhong C, Molinolo AA, Ruppel J, Hegde P, et al. Suppressing neutrophil-dependent angiogenesis abrogates resistance to anti-VEGF antibody in a genetic model of colorectal cancer. Proc Natl Acad Sci USA (2020) 117(35):21598–608. doi: 10.1073/pnas.2008112117
103. Agoro R, Mura C. Inflammation-induced up-regulation of hepcidin and down-regulation of ferroportin transcription are dependent on macrophage polarization. Blood Cells Mol Dis (2016) 61:16–25. doi: 10.1016/j.bcmd.2016.07.006
104. Layoun A, Huang H, Calvé A, Santos MM. Toll-like receptor signal adaptor protein MyD88 is required for sustained endotoxin-induced acute hypoferremic response in mice. Am J Pathol (2012) 180(6):2340–50. doi: 10.1016/j.ajpath.2012.01.046
105. Verma S, Prescott R, Cherayil BJ. The commensal bacterium Bacteroides fragilis down-regulates ferroportin expression and alters iron homeostasis in macrophages. J Leukoc Biol (2019) 106(5):1079–88. doi: 10.1002/JLB.2A1018-408RR
106. Mikulandra M, Pavelic J, Glavan TM. Recent Findings on the Application of Toll-like Receptors Agonists in Cancer Therapy. Curr Med Chem (2017) 24(19):2011–32. doi: 10.2174/0929867324666170320114359
107. Dunne A, Marshall NA, Mills KH. TLR based therapeutics. Curr Opin Pharmacol (2011) 11(4):404–11. doi: 10.1016/j.coph.2011.03.004
108. Peyssonnaux C, Zinkernagel AS, Datta V, Lauth X, Johnson RS, Nizet V. TLR4-dependent hepcidin expression by myeloid cells in response to bacterial pathogens. Blood (2006) 107(9):3727–32. doi: 10.1182/blood-2005-06-2259
109. Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer (2019) 19(7):405–14. doi: 10.1038/s41568-019-0149-1
110. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell (2012) 149(5):1060–72. doi: 10.1016/j.cell.2012.03.042
111. Yee PP, Wei Y, Kim SY, Lu T, Chih SY, Lawson C, et al. Neutrophil-induced ferroptosis promotes tumor necrosis in glioblastoma progression. Nat Commun (2020) 11(1):5424. doi: 10.1038/s41467-020-19193-y
112. Wen Q, Liu J, Kang R, Zhou B, Tang D. The release and activity of HMGB1 in ferroptosis. Biochem Biophys Res Commun (2019) 510(2):278–83. doi: 10.1016/j.bbrc.2019.01.090
113. Adkins I, Fucikova J, Garg AD, Agostinis P, Špíšek R. Physical modalities inducing immunogenic tumor cell death for cancer immunotherapy. Oncoimmunology (2014) 3(12):e968434. doi: 10.4161/21624011.2014.968434
114. Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest (2019) 129(6):2293–304. doi: 10.1172/JCI126428
115. Youssef LA, Rebbaa A, Pampou S, Weisberg SP, Stockwell BR, Hod EA, et al. Increased erythrophagocytosis induces ferroptosis in red pulp macrophages in a mouse model of transfusion. Blood (2018) 131(23):2581–93. doi: 10.1182/blood-2017-12-822619
116. Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology (2017) 66(2):449–65. doi: 10.1002/hep.29117
117. Kapralov AA, Yang Q, Dar HH, Tyurina YY, Anthonymuthu TS, Kim R, et al. Redox lipid reprogramming commands susceptibility of macrophages and microglia to ferroptotic death. Nat Chem Biol (2020) 16(3):278–90. doi: 10.1038/s41589-019-0462-8
118. Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell (2014) 156(1-2):317–31. doi: 10.1016/j.cell.2013.12.010
Keywords: iron, neutrophils, macrophage, cancer, metastasis
Citation: Liang W and Ferrara N (2021) Iron Metabolism in the Tumor Microenvironment: Contributions of Innate Immune Cells. Front. Immunol. 11:626812. doi: 10.3389/fimmu.2020.626812
Received: 06 November 2020; Accepted: 30 December 2020;
Published: 12 February 2021.
Edited by:
Stefania Recalcati, University of Milan, ItalyReviewed by:
Keehoon Jung, Seoul National University, South KoreaCopyright © 2021 Liang and Ferrara. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Napoleone Ferrara, bmZlcnJhcmFAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.