 Qi Ke
Qi Ke Charles J. Kroger
Charles J. Kroger Matthew Clark1
Matthew Clark1 Roland M. Tisch
Roland M. Tisch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 18 February 2021
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.624568
This article is part of the Research TopicEmerging Therapeutics for Immune ToleranceView all 24 articles
Type 1 diabetes (T1D) is widely considered to be a T cell driven autoimmune disease resulting in reduced insulin production due to dysfunction/destruction of pancreatic β cells. Currently, there continues to be a need for immunotherapies that selectively reestablish persistent β cell-specific self-tolerance for the prevention and remission of T1D in the clinic. The utilization of monoclonal antibodies (mAb) is one strategy to target specific immune cell populations inducing autoimmune-driven pathology. Several mAb have proven to be clinically safe and exhibit varying degrees of efficacy in modulating autoimmunity, including T1D. Traditionally, mAb therapies have been used to deplete a targeted cell population regardless of antigenic specificity. However, this treatment strategy can prove detrimental resulting in the loss of acquired protective immunity. Nondepleting mAb have also been applied to modulate the function of immune effector cells. Recent studies have begun to define novel mechanisms associated with mAb-based immunotherapy that alter the function of targeted effector cell pools. These results suggest short course mAb therapies may have persistent effects for regaining and maintaining self-tolerance. Furthermore, the flexibility to manipulate mAb properties permits the development of novel strategies to target multiple antigens and/or deliver therapeutic drugs by a single mAb molecule. Here, we discuss current and potential future therapeutic mAb treatment strategies for T1D, and T cell-mediated autoimmunity.
Type 1 diabetes (T1D) is an autoimmune disease defined by the immune-mediated destruction and/or dysfunction of the insulin producing β cells within the pancreatic islets of Langerhans (1–11). Both genetic and ill-defined environmental factors (e.g. viral infection, diet) influence T1D susceptibility (4–6, 12–16). Typically, it takes a number of years from the initiation of autoimmunity to diagnosis of clinical diabetes (5–9). When the functional β cell mass is reduced by ~80%, production of insulin becomes insufficient to regulate the body’s glucose levels. Currently there is no established curative treatment, and T1D is managed via daily exogenous insulin treatment and monitoring of blood glucose levels. Insufficient control of daily glucose levels can lead to severe complications including blindness, atherosclerosis, and neuropathy (6, 7).
T1D is a consequence of the breakdown of peripheral tolerance to β cell antigens, such as proinsulin, insulin, and glutamic acid decarboxylase (GAD65). The triggering event of T1D is poorly understood, and likely involves an environmental insult. CD4+ and CD8+ T cells are generally considered to be the primary drivers of β cell destruction in T1D patients. For instance, the strongest genetic risk factor for T1D is associated with specific alleles of HLA class II and class I molecules, and CD4+ and CD8+ T cells are found infiltrating the islets of T1D subjects (5, 6, 9, 13–33). Furthermore, the more aggressive childhood versus adult T1D onset is marked by an expanded effector T cell (Teff) response to proinsulin and insulin (20–22). However, examples of human islets lacking a T cell infiltrate have also been reported (24, 34, 35). Other adaptive immune cell populations such a B cells, and various innate effectors such as dendritic cells (DC), macrophages (MΦ), and natural killer (NK) cells reside in the islets of T1D subjects as well (24, 34, 35). Autoantibodies to islet proteins are also detected prior to clinical T1D diagnosis, and have been used to establish the risk of individuals progressing to overt diabetes (36–41).
Studies using the non-obese diabetic mouse (NOD), a model of spontaneous T1D have provided important information regarding disease progression and prevention (10, 11). Genetically manipulated NOD mice and adoptive transfer strategies have shown a direct role for CD4+ and CD8+ T cells as well as B cells in mediating β cell destruction. For example, in the absence of T or B cells, overt diabetes fails to develop (10, 11, 42–44). β cell-specific T cell reactivity is initiated by DC that ferry islet antigens from the pancreas into the draining pancreatic lymph node (PLN) (Figure 1) (45–49). In the PLN, naïve CD4+ and CD8+ T cells preferentially differentiate into proinflammatory Teff subsets, based on the cytokine milieu (Figure 1) (50–55). Release of IL-12 by DC induces the generation of type 1 CD4+ and CD8+ Teff, Th1 and Tc1, respectively, marked by expression of the transcription factor T-bet and the cytokine IFNγ (52, 56). Th1 and Tc1 cells have been closely linked to T1D development in both NOD mice and T1D patients (20, 52, 57, 58). However, IL-17A and IL-21-secreting Th17 cells, and IL-21-secreting T follicular helper (Tfh) cells also contribute to β cell destruction (50–52, 59–61). Th17 differentiation is driven by an IL-1β, IL-6, TGFβ, and IL-23 cytokine milieu (50, 52, 62), whereas IL-6 and IL-21 favor Tfh differentiation (51, 53–55). After APC-antigen encounter, self-reactive Teff migrate into the islets and promote β cell damage via direct cytolysis, and indirectly through production of proinflammatory cytokines, such as IFNγ, IL-1β and TNFα (Figure 1) (63–65). β cell damage and induced stress further exposes autoantigens, which leads to epitope spread and expansion of the pool of β cell-specific T cells (66, 67). Islet resident DC, MΦ and NK cells further promote β cell damage by maintaining the proinflammatory environment (5, 6, 9, 11, 24, 34, 45, 46, 57, 68–70). As islet inflammation or insulitis progresses, functional β cell mass declines until insulin production can no longer be sustained at sufficient levels to maintain appropriate blood glucose levels and overt diabetes is diagnosed (Figure 1) (5, 8, 9).
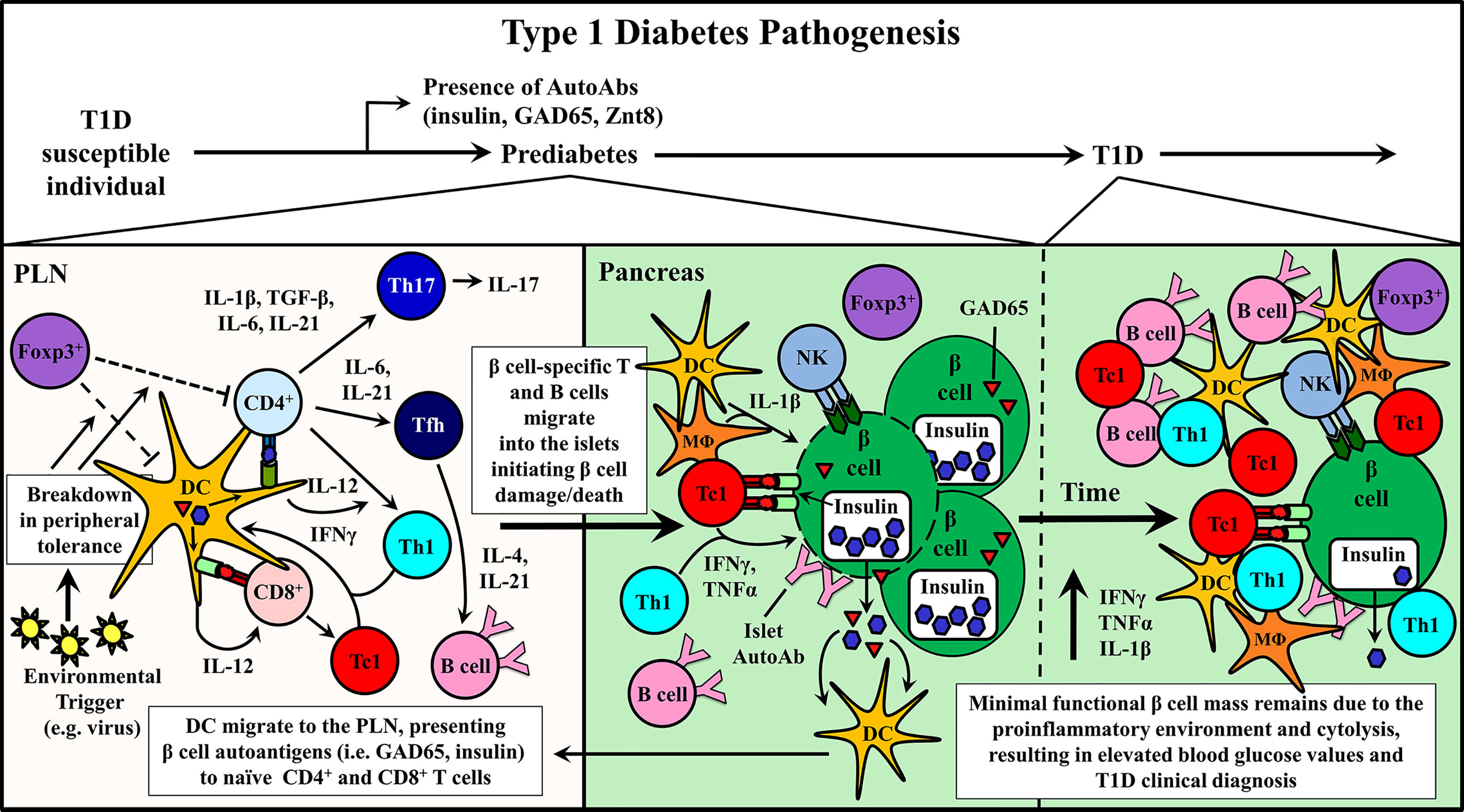
Figure 1 Type 1 diabetes (T1D) pathogenesis. Cellular events associated with driving T cell-mediated T1D are depicted within the pancreatic lymph node (PLN) and pancreas. Upon initiation of β cell autoimmunity via an ill-defined event, dendritic cells (DC) migrate from the pancreas ferrying islet autoantigens into the PLN. Here, naïve β cell-specific CD4+ and CD8+ T cells are activated and differentiate into distinct Teff subsets associated with T1D progression including CD8+ Tc1, CD4+ Th1, Th17, and Tfh. Early indication of ongoing autoimmunity is marked by the detection islet-specific autoantibodies (AutoAbs). Teff traffic into the pancreas and initiate β cell damage, which gradually increases over time prompting nominal insulin production.
Based on findings made in NOD mice and T1D patients, T cells, and to a lesser extent B cells, have been the focus of most immunotherapy strategies (10, 11, 42–44, 71). Nevertheless, due to the heterogeneity and complexity inherent with the diabetogenic response, designing effective immunotherapies to prevent and/or treat T1D has been challenging. Numerous therapeutic strategies to prevent and/or reverse T1D have been met with varying degrees of clinical success and disappointment (72).
The use of monoclonal antibodies (mAb) has been one approach clinically tested to prevent and/or treat T1D and other autoimmune diseases (73–75). The development of therapeutic mAb involves a number of key steps including: mAb generation, screening/selection, humanization, affinity maturation, molecule optimization, and engineering for commercial production (73, 74). Notably, advances in in vivo and in vitro generation of antigen-specific mAb, and engineering of immunoglobulin (Ig) molecules have greatly aided the production and application of mAb for therapeutic use. Clinically applied mAb and related molecules have provided safe and selective therapeutic targeting of biologically relevant proteins for the treatment of several diseases ranging from cancer to autoimmunity (73–75). For instance, mAb therapy targeting TNFα is being used in rheumatoid arthritis (RA) to mitigate disease severity (76, 77).
In T1D, mAb treatment must suppress ongoing β cell destruction while reestablishing long-term self-tolerance. Maintenance of long-lasting self-tolerance is largely mediated by various subsets of regulatory T cells (Treg). The timing of T1D immunotherapy is believed to be a critical factor impacting clinical efficacy. Intervention with mAb at early stages of β cell autoimmunity, when the frequency of pathogenic immune effectors infiltrating the islets is relatively low and the functional β cell mass high, is expected to be the most effective time to modulate the autoimmune response. Alternatively, if treatment is started later, it may be necessary to couple mAb therapy with strategies that enhance the expansion and function of the residual β cell mass in recent onset and long-standing diabetic individuals. Therapeutic mAb typically function via two general mechanisms: i) depletion of target cell populations, and ii) blockade of cell receptor function (Figure 2). However, advancements in mAb development have provided novel uses for therapeutic mAb such as inducing select receptor signaling and the delivery of therapeutic drugs to a target cell. This review will discuss strategies applied and advancements made in mAb therapies for T1D prevention and treatment.
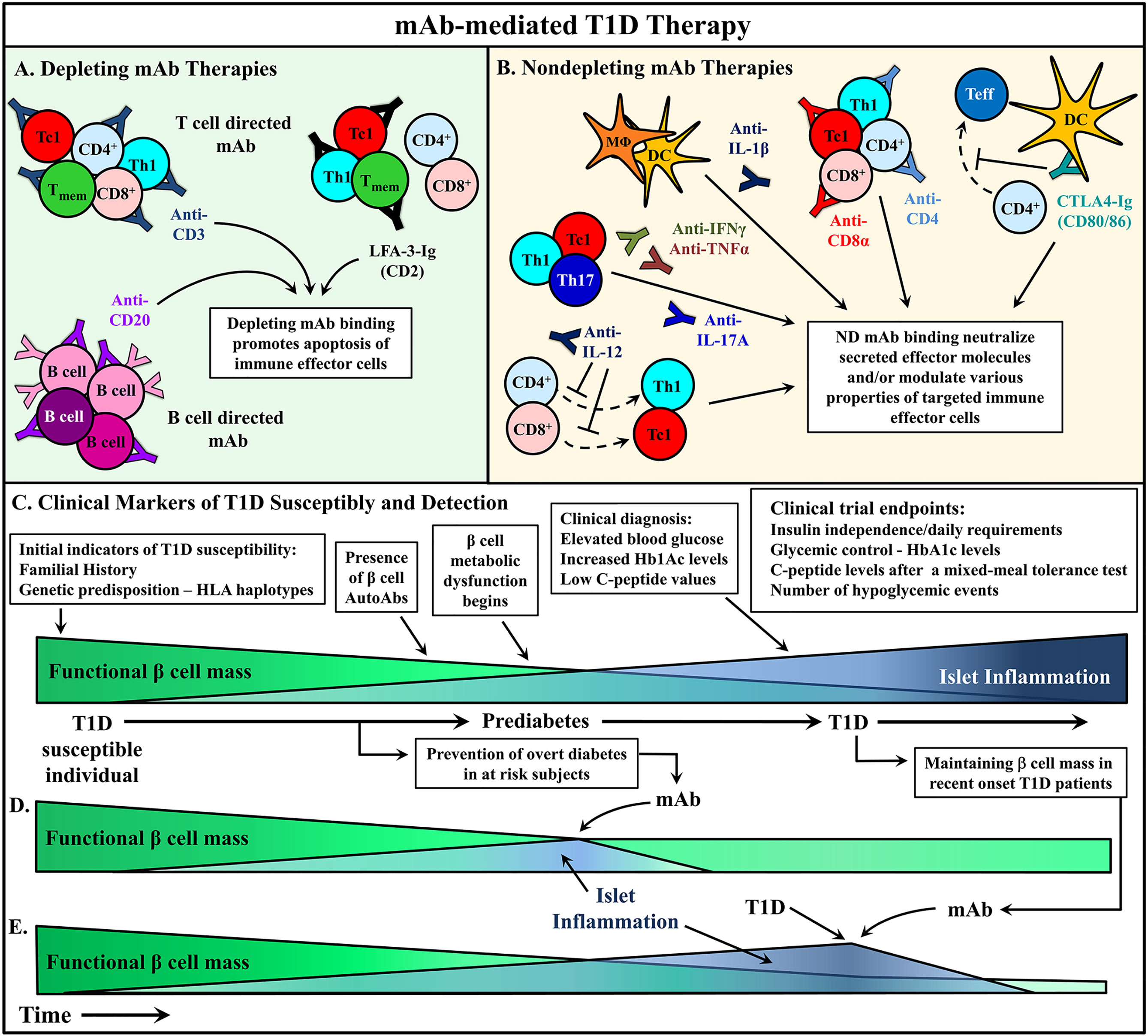
Figure 2 mAb therapies to ameliorate type 1 diabetes (T1D). (A, B) Monoclonal antibodies (mAb) treatments can be broadly divided into two categories: depleting mAb or nondepleting (ND)/neutralizing mAb. Some promising mAb treatments are depicted that have been used in either animal models or clinical trials to alter T1D progression. (A) Depleting mAb have been used to target T and B cells in the clinic. Transient depletion of T and B cells delays the progression of β cell autoimmunity. (B) ND mAb have been applied to neutralize cytokines to suppress the proinflammatory milieu of the pancreatic lymph node (PLN) and islets, as well as modulate the properties and activity of various immune effector cells. (C) The relationship between functional β cell mass versus islet inflammation is characterized. Over time, increased chronic islet inflammation results in decreased functional β cell mass, first detected via metabolic abnormalities, and ultimately leading to deficient insulin production, prompting clinical diagnosis of T1D. Individuals at different stages of T1D progression have treated with mAb therapies to alter T1D progression, and in turn (D) prevent diabetes onset, or (E) rescue residual β cell mass after clinical T1D diagnosis. Typically, these clinical trials have used metabolic readouts for β cell function as primary endpoints to determine therapeutic efficacy (C).
The advent of B cell hybridoma technology in the mid 1970’s provided the means to generate antigen-specific mAb, and in turn jump-started the field of mAb immunotherapy (78). The approach entails harvesting Ab-producing B cells from antigen immunized mice that are immortalized via fusion with myeloma cells to generate hybridoma cell lines (79). Although still a standard protocol for mAb production, the hybridoma method is generally time consuming and labor intensive (80). Accordingly, a variety of other approaches have been developed to provide more rapid production and expand the repertoire of antigen-specific mAb.
One such technique is phage display pioneered in the mid 1980’s (81). The general approach entails cloning a gene into gene III of filamentous phage, and having the encoded protein/peptide displayed on the surface of the phage (82). The engineered phages are then exposed to a protein that binds the ectopic protein/peptide, the protein-bound phages expanded in bacteria, and subjected to additional cycles of screening. This method has been adapted for Ab phage display (APD) to screen libraries of variable regions of antigen binding fragments (Fab) or recombinant single-chain variable fragments (scFv) expressed on the surface of phages (83). With large human Ig libraries readily available, the process of generating and screening sequences of complementary-determining regions (CDR) of human Ig is rapid (84). In addition, human Ig sequences negate the need for humanization (see below). Adalimumab (D2E7) was the first fully human anti-TNFα mAb developed using APD technology. Adalimumab exhibited comparable inhibitory efficiency to a murine anti-human TNFα mAb (MAK195), which was used as a template (85). APD can also be used for optimization of mAb generation and production (86, 87). This method allows immunization steps to be bypassed, which is a significant advantage for developing mAb against non-immunogenic, toxic, or self-antigens. Despite numerous benefits, a key drawback of APD is that the selection of heavy and light chains is based on random selection events that may not represent a functional Ig in vivo (88, 89). Nevertheless, APD provides an accelerated mAb discovery and screening method compared to the classic hybridoma mAb technique.
A murine mAb targeting the CD3 epsilon polypeptide of the human T cell receptor (TCR) complex was the first developed and approved for treatment in patients as an immunosuppressant drug to prevent acute allograft rejection after organ transplantation (90). Despite observed therapeutic benefits, severe side-effects emerged that limited clinical application (91). Patients treated with the murine mAb rapidly developed a human anti-mouse Ab (HAMA) response that ranged from the development of rashes to lethal kidney failure. In addition to significant safety issues, immune reactivity also reduces mAb efficacy and half-life. To overcome immune responses to mAb produced in non-human species, the Ig molecules undergo a “humanization” process. Here, non-human portions of the Ig molecule are reduced to minimize immunogenicity without compromising antigen binding. Initially, chimeric Ig molecules were generated consisting of a murine variable region coupled with a human constant region. However, the murine portion, making up ~30% of the Ig molecule, is still sufficient to elicit immune reactivity (92–94). Further mAb humanization, increasing the human content to ~85%, is accomplished by grafting the non-human CDR into similar human frameworks. However, this grafting can lead to the loss of antigen-binding affinity due to conformational alteration of the CDR loops (95, 96). The first FDA-approved humanized mAb daclizumab, an anti-CD25 mAb, was generated by grafting murine CDR, which resulted in markedly reduced binding affinity (97). Distinct residues in framework regions, known as vernier zone residues, are responsible for maintaining Ig binding affinity and need to be retained during humanization (98–100). However, murine residues found in vernier zones still can elicit HAMA responses (101). Therefore, efforts continue to preserve antigen binding while limiting HAMA responses for mAb engineered with a non-human binding domain on a human Ab backbone.
The use of transgenic rodents that express human (Hu)-Ig is one approach to generate bona fida human mAb following antigen immunization (102, 103). Transgenic mice, lacking endogenous Ig expression, have been established that express human light chain genes coupled with a germline human Vκ region. Human heavy chain genes encoding μ and γ1 are also expressed to allow class switching. Transgenic Hu-Ig mice have been used in combination with conventional hybridoma technology to produce several human mAb applied in the clinic including: zanolimumab (anti-CD4), canakinumab (anti-IL-1β), ustekinumab (anti-IL-12/23p40), and golimumab (anti-TNFα) (104–108).
In addition to epitope binding, mAb elicit a wide range of effector functions that are dependent on the Ig Fc region. Effector functions include Ab-dependent cell-mediated cytotoxicity (ADCC), Ab-dependent cell-mediated phagocytosis (ADCP), and complement-dependent cytotoxicity (CDC) (109). Glycosylation of the Fc region impacts binding to Fc receptors (FcR) on effector cells and subsequent ADCC and ADCP responses. Accordingly, mAb effector function can be manipulated via modification of glycosylation of the Fc region. For example, enhanced ADCC by tumor resident NK cells is seen with a human IgG1 lacking fucosylated glycan at Asn297 in the Fc region, leading to increased tumor rejection (110, 111). Modification of the Fc region has also been important in reducing unwanted adverse events associated with mAb effector function. Initial clinical trials using anti-human CD3 OKT3 IgG1 for the treatment of T1D resulted in cytokine release syndrome (CRS), driven by FcRγ binding (112, 113). Alanine substitutions at amino acid positions 234 and 235 were introduced into the CH2 Fc region of the γ1 backbone that reduce glycosylation and FcRγ binding. Consequently, the resulting mAb, teplizumab (huOKT3γ1(Ala-Ala)), exhibits only minimal CRS (114). A similar approach has been used with the anti-human CD3 otelixizumab, a humanized aglycosylated IgG1 tested in T1D clinical trials (115, 116). The Fc region, however, is important for structural stability and reduced Fc binding to the neonatal receptor, FcRn, leads to shortened Ab serum half-life (117, 118). Therefore, engineering of the Fc region is important for mAb development that needs to be optimized for both drug safety and pharmacokinetics.
mAb targeting cellular antigens are typically depleting due to ADCC, ADCP, and CDC responses. The goal of using a depleting mAb in the context of autoimmunity is to eliminate the pathogenic immune effectors preventing further tissue damage. In T1D, depleting mAb treatments in preclinical or clinical settings have targeted various cell populations. Transient depletion of T and B cells via mAb for example, have shown at least short-term benefits in recent onset T1D subjects (Figure 2) (119–123).
Arguably, the most successful clinical immunotherapy for T1D to date has been administration of anti-CD3 mAb. The first murine IgG2a specific for the human CD3 epsilon-subunit (OKT3) was developed in 1979, and approved by FDA as the first human mAb immunotherapy in 1986 (124). Treatment successfully prevented acute graft rejection and graft-versus-host-disease (GvHD) in organ transplant patients (124, 125).
In 1994, Chatenoud and Bach showed that anti-CD3 therapy reversed new onset diabetes in NOD mice, and established long-term remission and β-cell-specific tolerance (126). The mechanisms of protection induced by anti-CD3 mAb therapy have been extensively studied in mice (127–129). Following anti-CD3 mAb binding, increased TCR signaling promotes T cell activation-induced cell death (AICD) (130, 131). Interestingly, AICD by anti-CD3 mAb is selective for conventional T (Tconv) cells with limited effects on FoxP3-expressing CD4+ Treg (Foxp3+Treg) (132). In NOD mice, the depletion of islet infiltrating Teff by anti-CD3 suppresses ongoing β cell destruction, albeit at the expense of transient systemic depletion of Tconv (126, 133). Additionally, the ingestion of apoptotic T cells enhances TGFβ production by MΦ, which promotes Foxp3+Treg differentiation (128, 129, 134). This increased pool of Foxp3+Treg plays a critical role in maintenance of diabetes remission in NOD mice (128).
During a randomized, controlled, open-label phase I/II clinical trial, newly diagnosed T1D patients were given a 14-day course of treatment of teplizumab (120). Although diabetes reversal was not observed, the teplizumab-treated group had several promising metrics. Over a 2 year period C-peptide responses and insulin production were sustained, which correlated with decreased acetylated hemoglobin (HbA1c) and insulin dependency (119). Otelixizumab also was shown to preserve β cell function and reduce insulin use for 4 years in a phase II placebo-controlled trial (135). The beneficial effects of otelixizumab were most pronounced in patients with higher residual β cell function, but the therapeutic effects diminished by 24 months, suggesting overall efficacy was limited. In addition, otelixizumab treatment resulted reactivation of Epstein Barr virus in some subjects (121). Although the effects of otelixizumab were transient, this study indicated that intervening at an earlier time post-diagnosis enhanced efficacy.
Patients who received anti-CD3 mAb experienced significant reduction of peripheral T cells, which rebounded within a month after therapy (119, 120, 123, 136, 137). This reduction in numbers is in part believed to be due to T cell egress from the circulation (138). Evidence also indicated that anti-CD3 affected the T cell phenotype in treated T1D subjects. For example, the frequency of circulating central memory CD8+ T cells and exhausted islet-specific CD8+ T cells (TIGIT+KLRG1+PD-1+) were increased (136, 139, 140). Interestingly, recent studies show that aggressive T1D correlates with the presence of activated islet-specific HELIOS+ CD8+ memory T cells (Tmem) found in peripheral blood (141). On the other hand, slower progressing T1D is marked by peripheral blood islet-specific CD8+ Tmem exhibiting an exhausted phenotype characterized by upregulation of EOMES, 2B4, PD-1, TIGIT, and CD160 (141). These results suggest that anti-CD3 induced T cell exhaustion plays a role in the protective effect.
The phase III trials using either teplizumab or otelixizumab did not meet the primary endpoint goal (142–144). Nevertheless, post-hoc analyses showed a reduced loss of C-peptide in a subset of patients receiving teplizumab (142). On the other hand, the otelixizumab phase III trial was terminated after an untested and reduced dose of the mAb failed to significantly improve C-peptide levels compared to the control groups. A phase I/IIa repeat dose escalation study of otelixizumab has shown a dose-dependent relationship between anti-CD3-TCR engagement and TCR downregulation [NCT02000817 (145)]. The dosing of otelixizumab was also found to be well tolerated and preserved β cell function over 18 months (146). A new randomized, double-blind, placebo-controlled teplizumab phase III trial addresses the safety and tolerability of the treatment in recent-onset T1D young patients as well as the effect on β cell preservation using the most effective dosage and modified primary outcome based on earlier findings (NCT03875729).
A recent phase II study in high-risk, nondiabetic relatives of T1D patients, investigated the efficacy of teplizumab to prevent diabetes onset (NCT01030861). Subjects receiving a single 14-day course of teplizumab exhibited an average delay of 24 months in the onset of diabetes (136). Notably, the largest response to teplizumab was seen in subjects with reduced median levels of C peptide at the time of intervention, indicative of a later stage of disease progression. In addition, HLA haplotype was found to influence the efficacy of teplizumab. This latter result suggests that the TCR repertoire, likely reflecting the size of the activated Teff pool, are parameters influencing the response to teplizumab. Importantly, this study provides the first evidence that anti-CD3 treatment can delay T1D onset in at-risk individuals, as well as further substantiate targeting the T cell compartment as a general means to modulate the human disease process. Consequently, teplizumab has been granted PRIority MEdicines (PRIME) designation by European Medicines Agency and Breakthrough therapy designation by the U.S. Food and Drug Administration (147, 148).
The route of administration of anti-CD3 is also being assessed to enhance efficacy and safety. Initial studies with oral anti-CD3 treatment in murine experimental autoimmune encephalitis have demonstrated decreased side effects and increased efficacy at lower dosages (149). Of note, oral administration of anti-CD3 failed to alter the CD3/TCR complex or induce pronounced downstream TCR related signaling events such as depletion or release of proinflammatory cytokines (150). Instead tolerance was achieved by induction of TGFβ1-expressing Th3 cells (149, 151). However, the therapeutic efficacy of oral anti-CD3 treatment in T1D has yet to be clinically tested.
B cells are critical to the pathogenesis of T1D, and are consistently detected within the pancreatic islet infiltrate (9, 24, 42–44, 70). Development of diabetes is prevented in NOD mice treated with a B cell depleting anti-CD20 at a preclinical stage of T1D (Figure 2) (152). Similarly, anti-CD22 mediated B cell depletion in NOD mice prevents diabetes, and has been reported to induce remission in new onset animals (153).
Studies suggest that B cells are also key drivers in the progression of human T1D. For example, the aggressive, early onset of diabetes correlates with high numbers of islet infiltrating CD20-expressing B cells (57, 154). Rituximab, a mouse-human chimeric IgG1 mAb specific for human CD20, has been studied in recent onset T1D patients (122). One year after treatment, rituximab versus control treated subjects exhibited an improvement in the levels of HbA1c and C-peptide, as well as the requirement for insulin indicating preserved β cell function. However, CD19+ B cells slowly repopulated the periphery and no long-term benefit was detected after two years (155). Previous studies have indicated that disease promoting autoreactive B cells may not be completely deleted after anti-CD20 treatment (156, 157). Further studies regarding the timing, potential as a preventative treatment, and dosage of rituximab are needed to optimize this therapeutic potential for T1D.
CD2 is a surface adhesion molecule expressed by a variety of cell populations including T cells, NK cells and DC (158–160). Notably, CD2 levels are increased on Tmem (158, 159). Alefacept, a fusion protein consisting of the CD2-binding domain of LFA3 fused to the Fc region of human IgG1 (IgG1Fc), has been utilized to treat psoriasis (88, 161). The fusion protein preferentially binds to Tmem, and induces apoptosis mediated by IgG1Fc binding to FcRγ expressed by NK cells (162). Accordingly, individuals treated with alefacept have reduced levels of Tmem. Regarding T1D, a 12 and 24 month clinical trial with recent onset patients demonstrated a reduced frequency of activated T cells and increased ratio of Treg to Tmem in blood. Additionally, C-peptide levels were improved after a mixed meal test (MMT) in the alefacept versus placebo group [NCT00965458 (163, 164)]. This correlated with reduced exogenous insulin requirements, and suggested that alefacept prolongs β cell function (163, 164). Interestingly, in psoriasis patients, alefacept reduced activated CD11c+/CD83+ DC subsets and inflammatory gene expression levels (165). Therefore, this strategy may restore peripheral tolerance via targeted deletion of activated self-reactive T cells and APC plus increasing Treg to dampen the ongoing autoimmune response.
Overall, depletion of immune populations via mAb has been effective in influencing β cell autoimmunity in both mice and humans. Nevertheless, there is the risk of limiting protective immunity following broad depletion of a given immune cell type, particularly if treatment requires continued mAb administration.
In addition to depletion, mAb have the capacity to block and/or modulate intercellular and effector molecule interactions. Naturally occurring or engineered nondepleting (ND) mAb have been clinically used to block cytokine/chemokine-receptor interactions and to inhibit cell surface receptor-ligand engagement to affect an immune response (Figure 2). Human IgG4 for instance, has a low affinity for most FcRγ, which limits ADCC, ADCP, and CDC (166, 167). As noted above, modifying glycosylation patterns can also be used to block the depleting function of mAb, as well as enhance therapeutic efficacy, and Ig half-life (168, 169). Overall, ND mAb have the therapeutic advantage of preserving the pool of targeted immune effector cells while disrupting an ongoing autoimmune response.
The combination of the proinflammatory cytokines IL-1β, IFNγ, and TNFα is cytotoxic to β cells (63, 64). IFNα also enhances CD8+ T cell-mediated destruction of β cells via upregulation of MHC class I by β cells (170–173). Therefore, mAb therapies have been used to neutralize the proinflammatory environment within the islets and preserve functional β cell mass (Figure 2). Two different therapies to inhibit IL-1β, an anti-IL-β mAb (canakinumab) and an antagonist of the IL-1R (anakinra), have been ineffective at maintaining β cell function in recent onset T1D patients [NCT00947427, NCT00711503 (174)]. In contrast, etanercept, an anti-TNFα fusion protein that binds to and removes TNFα from circulation, has demonstrated efficacy based on reduced Hb1Ac levels in recently diagnosed children [NCT00730392 (175)]. An ongoing clinical phase II trial is testing the tolerability and effects on β cell autoimmunity of etanercept in combination with vitamin D plus GAD65 prepared in Alum adjuvant (NCT02464033). The goal here is to suppress islet inflammation while inducing GAD65-specific Treg. Simponi, a neutralizing Ab that binds to both soluble and membrane bound TNFα, is also being investigated to maintain β cell mass (NCT02846545). Early evidence indicates that simponi may prolong insulin production, but long-term efficacy still needs to be determined (176).
Studies in NOD mice have shown that blocking IFNα or its receptor reduces T1D incidence (177, 178). Furthermore, mAb neutralization of IFNγ can prevent the progression of β cell autoimmunity in adoptive transfer models of T1D when administered at distinct treatment windows (179–181). In contrast, IFNγ-deficient NOD mice continue to develop diabetes, suggesting potential redundancy among proinflammatory cytokines and highlighting the importance how timing of mAb intervention can impact therapeutic efficacy (182–184). Overall, neutralizing a single proinflammatory cytokine has generally had limited success. This may in part reflect the relative role of a given cytokine in general and/or at a particular stage in the disease process. Targeting multiple cytokines that affect β cell viability and function may be needed to enhance the efficacy of the approach.
mAb targeting of cytokines to modulate T cell subset differentiation and effector function has also been a strategy to alter the progression of β cell autoimmunity. Neutralizing IL-12, which induces type 1 (e.g. Th1/Tc1) subset differentiation, limits insulitis and prevents the onset of diabetes in NOD mice (10). However, efficacy is dependent on continuous and frequent anti-IL-12 administration (185).
Cytokine-specific mAb have also been used to block the function and/or differentiation of other CD4+ T cell subsets involved in β cell autoimmunity, such as Th17 and Tfh cells (50, 52, 54, 55, 186). For instance, IL-21 secreted by Th17 and Tfh cells, has been targeted. A role for IL-21 in T1D was initially demonstrated in NOD mice lacking IL-21R, which exhibit minimal insulitis and reduced diabetes incidence (60). Ectopic expression of IL-21 by β cells also induces diabetes in non-autoimmune prone C57BL/6 mice (187). Furthermore, blockade of IL-21 reverses established T1D in NOD mice making IL-21 a promising therapeutic candidate in the treatment of T1D (188). Indeed, a phase II clinical trial of newly diagnosed T1D patients treated with anti-IL-21 and liraglutide (a glucagon-like-peptide-1 agonist), designed to curtail autoimmunity and boost insulin production, is underway (NCT02443155).
Diabetes incidence is reduced in NOD mice treated with anti-IL-17A starting at 10 weeks of age, a relatively late preclinical stage of T1D (59). Interestingly, protection correlates with an increased frequency of Foxp3+Treg in the islets and PLN, likely mediated in part by a dampened proinflammatory milieu. Both Foxp3+Treg and Th17 cells differentiate in the presence of TGFβ, however in the absence of additional proinflammatory cytokines such as IL-1β and IL-6, CD4+ T cell differentiation is skewed toward the Foxp3+Treg subset (189, 190). Notably, anti-IL-17A treatment initiated at an earlier stage of T1D progression is ineffective at preventing diabetes onset in NOD mice (59). These results suggest that interfering with Th17 differentiation and function is effective when insulitis is well established. Similarly, treatment of NOD mice at 10 weeks of age with recombinant IL-25, which antagonizes Th17 differentiation, also decreases T1D incidence (59). Thus, mAb therapies targeting Th17 and Tfh subsets have yielded promising results in murine T1D that may lead to beneficial clinical outcomes for human T1D treatment.
In addition to targeting soluble mediators regulating autoreactive Teff differentiation and function, ND mAb therapies have been applied to directly modulate Teff activity via binding to surface molecules. Naïve T cells have the plasticity to differentiate into various Teff subsets defined by unique transcription factor and cytokine profiles. One key factor driving Th1/Tc1 subset differentiation is a strong TCR signal, defined as the culmination of TCR (signal 1), co-stimulatory molecule (signal 2) and cytokine (signal 3) signaling pathways (191–193). Furthermore, continued TCR signaling is required to maintain Teff function. Therefore, strategies to dampen these signaling pathways are expected to prevent expansion and function of pathogenic Teff. Blocking the T cell co-stimulatory molecule pathway using abatacept, an anti-CTLA4-Ig fusion protein, slows β cell functional decline and improves Hb1Ac values in new-onset T1D patients, although insulin independence is not achieved [NCT00505375 (194, 195)]. Interestingly, recent studies have indicated clinical responsiveness to abatacept in recent-onset T1D subjects is dependent on suppression of Tfh cells, indicating that the therapy modulates the T cell pool (61).
Short-term ND mAb treatment targeting the T cell co-receptors CD4 and CD8α both prevents and reverses diabetes in NOD mice while establishing long-term β cell-specific tolerance (196–200). T cells examined shortly after mAb treatment exhibit reduced TCR signaling and suppressed production of proinflammatory cytokines (196, 197). Importantly, mAb-bound T cells rapidly egress from the islets and PLN (196–198). The latter is dependent on mAb-mediated co-receptor cross-linking, and is marked by distinct changes in T cell transcriptional activity, decreased sensitivity to local retention cues, and enhanced responsiveness to tissue egress-inducing chemokines (196, 197). Long-term maintenance of tolerance is tissue-specific, mediated by an induced β cell-specific Foxp3+Treg, while protective immunity is unperturbed (198). Interestingly, a ND humanized anti-human CD4 IgG1 mAb, tregalizumab, has been reported to preferentially activate and enhance the suppressor activity of FOXP3+Treg in vitro (201). Short-term ND mAb treatment strategies targeting T cell co-stimulatory molecules can inhibit β cell-specific T cell reactivity long-term via changes in T cell transcriptional profiles.
mAb that recognize β cell-peptide-MHC complexes may provide an additional strategy to alter TCR signaling, and enhance targeting of autoreactive Teff. For example, a mAb (mAb287) recognizing the peptide:MHC class II complex insulin B:9-23 peptide in the context of IAg7 blocks IL-2 cytokine secretion and tetramer binding by an insulin specific T cell hybridoma. Additionally, NOD mice treated weekly starting at 4 weeks of age with mAb287 significantly delays T1D onset (202).
mAb-mediated blockade of CD127, the IL-7Rα, both prevents and reverses diabetes in NOD mice (203, 204). CD127 is expressed by naïve T cells and Tmem, and is critical for maintaining T cell homeostasis. Short-term anti-CD127 treatment induces increased PD-1 expression and diminished proinflammatory cytokine production by Teff, consistent with an exhausted T cell phenotype (203, 204). Indeed, the protective effect induced by anti-CD127 mAb in NOD mice is reversed by treatment with a PD-1 blocking mAb, known to rescue exhausted T cells (203). T1D patients treated with the anti-IL-7Rα mAb RN168 show a reduction in Tmem and activated T cells while the FOXP3+Treg pool is maintained (205). Nevertheless, C-peptide levels are not markedly altered, which may reflect the dose and/or duration of RN168 administered.
ND mAb therapy has also been employed to alter NK cell activity. Specifically, studies have examined the role of NK cell activating receptor NKp46 or the mouse orthologue NCR1 in affecting the diabetogenic response (68, 206, 207). Anti-NCR1 mAb (NCR1.15) treatment initiated early in disease progression decreased diabetes incidence in NOD mice (206). This protective effect correlated with a pool of NK cells with reduced NCR1 surface expression, activation and degranulation (206). Although the ligand for NKp46 has yet to be identified, NCR1-Ig and NKp46-Ig fusion proteins bind to murine and human β cells respectively, indicating that β cells express a NK cell activating ligand (68, 208). Recently a humanized anti-NKp46 (hNKp46.02) has been shown to also reduce NK cell degranulation and internalization of the NKp46 activating ligand (207). Therefore, future studies are poised to investigate the efficacy of targeting NK cells for T1D treatment.
Central to the development of T1D is the ability of pathogenic autoreactive immune cells to traffic into the islets (209). T and B cells require various adhesion molecules to facilitate extravasation from circulation into sites of inflammation. Notably, NOD mice deficient in the T cell adhesion molecule ICAM-1 are protected from diabetes (209, 210). Accordingly, mAb-mediated blockade of adhesion molecules has been assessed in NOD mice as a means to prevent T and B cell trafficking into the islets (209). NOD mice treated with anti-ICAM-1 mAb exhibit reduced diabetes onset (209, 211). Similarly, blockade of MADCAM-1 in young NOD mice also reduces diabetes incidence. However, once islet infiltration has been established, MADCAM-1 blockade is ineffective (209, 212). These findings suggest that the timing of mAb blockade of adhesion molecules is critical, and that the approach is more effective at earlier stages of β cell autoimmunity when only a limited number if islets are infiltrated.
Taken together, ND mAb offer a promising therapeutic approach for prevention and treatment of T1D. Initial clinical and a substantial number of preclinical studies demonstrate that the function of effector molecules and properties of various immune cell types driving β cell autoimmunity can be modulated by ND mAb and fusion molecules. This is achieved without significant changes in systemic numbers of immune effectors or disruption of protective immunity.
Cytokine-based therapy has been used to modulate immune-mediated pathology, including autoimmunity and T1D. However, systemic delivery of cytokines is problematic due to pleiotropic effects, and non-specific cell signaling that leads to potentially severe adverse effects. To overcome these obstacles, mAb-cytokine complexes are being developed and applied to target specific cell populations, and in this way enhance efficacy and safety. An example is the use of IL-2-Ab complexes (213).
IL-2 is predominantly produced by activated T cells and promotes expansion and survival of Teff (214). Additionally, IL-2 is essential for Foxp3+Treg differentiation, fitness, and maintenance (214). Development of murine T1D has been linked to reduced IL-2 production by T cells leading to Foxp3+Treg dysfunction and heightened β-cell-specific Teff responses (215–221). Similarly, polymorphisms in the CD25 gene are associated with human T1D susceptibility, and in part, result in reduced sensitivity of FOXP3+Treg to IL-2 (215–222). Upon activation the IL-2 receptor complex, consisting of CD25, CD122, and CD132, is upregulated on Tconv. In contrast, Foxp3+Treg constitutively express elevated levels of the high affinity IL-2 receptor component CD25 (223). Increased CD25 expression by Foxp3+Treg provides a competitive advantage to acquire local IL-2 and therefore prevent Teff expansion and function (221, 223). Two murine anti-IL-2 mAb have been developed that exhibit distinct biological functions in vivo when bound to recombinant IL-2 (213, 224). The anti-IL-2 clone S4B6 establishes an IL-2 complex that preferentially binds to Teff. S4B6 binding to IL-2 prevents IL-2 interaction with CD25 causing selective binding of IL-2 to CD122 on Teff (213, 224). In contrast, the JES-61A2 anti-IL-2 clone establishes an IL-2 complex that is preferentially bound by Foxp3+Treg (213, 224). Here JES-61A2 binding to IL-2 prevents interaction with CD122 causing IL-2 to signal through CD25 (213, 224). Co-treatment of NOD mice with β cell peptide loaded tetramers and IL-2-JES-61A2 mAb complexes selectively expands β cell-specific Foxp3+Treg reducing diabetes incidence (225). A key benefit of mAb-IL-2 complexes is that the half-life of IL-2 is extended which aids pharmacokinetics (226). Low dosage IgG-IL-2 complexes significantly enhances FOXP3+Treg numbers and function in human peripheral blood as well as in cynomolgus monkeys for the treatment of GvHD (227).
Bispecific Ab (bsAb) contain two distinct antigen binding sites. Structurally bsAb typically consist of two different Fab arms, or two unique Ab linked by a common Fc region (228). bsAb can be used in autoimmunity to: i) neutralize multiple cytokines or receptors simultaneously, ii) force cell-cell interactions of different immune populations, and iii) initiate receptor co-localization on the cell surface (228, 229). The first clinically applied bsAb was blinatumomab, a CD19- and CD3-specific recombinant, for the treatment of non-Hodgkins B cell lymphoma. Blinatumomab forces an interaction between B cells and cytotoxic T cells. The result of this interaction is efficient elimination of the B lymphoma cells, expansion of protective T cells, and an increased life expectancy in the majority of patients (228, 230).
A bsAb specific for the β cell specific glucose transporter 2 molecule (Glut2) and the T cell inhibitory receptor CTLA-4 has been tested in NOD mice (231). This bsAb binds to β cells via Glut2 and engages CTLA-4 on Teff to suppress function. Glut2-CTLA-4-specific bsAb treatment of NOD mice results in reduced T cell proliferation and proinflammatory cytokine production, and decreased diabetes incidence (231). While the application of bsAb for the treatment of T1D has been limited to date, several enticing therapeutic strategies exist. bsAb that promote interactions between Foxp3+Treg and Teff, such as a CD25 and CD122, would be one approach. Another therapeutic option would be to use bsAb to establish “dual” anti-inflammatory cytokine complexes. An IL-12-IL-2 mAb fusion protein for example, has been used to simultaneously deliver both IL-2 and IL-12 to enhance Teff and NK function for cancer treatment (232). In the context of autoimmunity, dual cytokine fusion complexes of IL-2 and TGFβ may prove to be an effective strategy to induce and expand adaptive Foxp3+Treg. As autoimmune diseases are driven by several events, bsAb provide a novel therapeutic avenue to modulate multiple drivers of autoimmunity simultaneously.
The ultimate goal of an immunotherapy for T1D is to suppress ongoing β cell autoimmunity by restoring peripheral tolerance without affecting protective immunity, and preserve β cell function. The complexity of the disease process, marked by multiple immune effectors (Figure 1), and varying kinetics of disease progression among individuals, however, has made the development of effective immunotherapies highly challenging to date (5–8, 72).
In general, mAb therapies have been applied to alter disease progression by deleting immune effector cells, altering effector cell phenotype/function or blocking soluble/cell-surface protein interactions (Figure 2). Clinical therapies targeting T and B cells via anti-CD3 and anti-CD20, respectively, have demonstrated safety and efficacy in maintaining β cell mass in newly-diagnosed patients (Figure 2) (136, 233, 234). However, these therapies and others fail to reestablish self-tolerance long-term. This may in part be due to insufficient induction/expansion of FOXP3+Treg or adaptive Treg subsets, and/or failure to adequately tolerize relevant pools of pathogenic Teff and Tmem. In this regard, it is noteworthy that efficacy of anti-CD3 to delay diabetes onset in at risk subjects is in part dependent on HLA haplotype (136). This finding suggests that parameters such as TCR repertoire, the avidity/affinity of the Teff, and/or the size of the pathogenic Teff/Tmem pool contribute to therapeutic outcome. The results are intriguing and further underscore the complexity associated with effectively manipulating the complete autoimmune response.
One key variable that influences the efficacy of mAb in T1D is the timing of intervention in relationship to disease progression (7, 8, 72). The diabetogenic response in human T1D can be viewed as a succession of stages marked by: 1) the initiation of autoimmunity detected by presentation of multiple islet autoantibodies, 2) ongoing autoimmunity with the presentation of metabolic abnormalities that indicate aberrant stress on β cell mass, and 3) the onset of overt diabetes indicating loss of function of the majority of β cell mass (Figure 2). The majority of clinical trial interventions have been applied at the second and third stages of disease progression. It is well established by preclinical studies that a given mAb treatment may only be effective at a particular stage of T1D progression (72). Recent clinical results indicating that the efficacy of anti-CD3 therapy to delay diabetes onset is dependent on ongoing autoimmunity, further highlight this key aspect of T1D immunotherapy. Notably, a similar temporal effect is seen in NOD mice in which anti-CD3 therapy fails to prevent diabetes onset when given to NOD mice at an early stage in disease progression (133). The nature of the effector cells and molecules being targeted will ultimately determine clinical efficacy at a given stage of T1D progression. Here, it is critical that the mechanism by which a therapeutic mAb induces tolerance be fully understood to help better predict efficacy when administered at a given stage of disease progression, as well as the likelihood of induction of long-term tolerance without the need of persistent intervention. At earlier and less stringent stages of T1D, strategies that limit trafficking of effectors into the islets and/or activation and/or differentiation of pathogenic effectors are expected to be effective. In contrast, during late preclinical T1D stages or at the onset of diabetes the therapy must be sufficiently robust to rapidly tolerize an established and sizable pool of islet resident pathogenic effectors. In both settings induction and/or expansion of Treg subsets is needed to maintain tolerance by limiting subsequent differentiation, expansion and/or function of pathogenic Teff. In an attempt to minimize temporal effects of disease progression, mAb therapies that establish long-term, β cell-specific tolerance broadly over preclinical and clinical T1D stages in NOD mice and other rodent autoimmune models, need to be identified and prioritized. In this regard, the use of ND mAb specific for CD4 and CD8 is noteworthy. A short course of ND anti-CD4 and CD8α mAb prevents diabetes onset when administered to young NOD mice and results in rapid reversal of diabetes and tissue-specific long-term tolerance in NOD mice (196–199, 235).
In view of the complexities of the diabetogenic response in general, and the varied parameters linked to effectively tolerizing immune effectors, it is likely that multiple cell types and/or effector molecules will need to be targeted with combinations of mAb (Figure 1). For example, expansion of β cell-specific Foxp3+Treg may be enhanced by combining mAb-IL-2 complexes with mAb that quench the proinflammatory milieu of the PLN and islets and/or block Teff differentiation and function (225). Alternatively, β cell-specific Treg subsets can be induced and/or expanded via more traditional antigen-specific based strategies following “broad” tolerization of Teff via mAb therapy. The application of bsAb may be particularly advantageous for combinatorial strategies. The simultaneous targeting of multiple proteins (e.g. proinflammatory cytokines) by a single therapeutic agent simplifies treatment dosing and regimen, and limits potential drug-drug interactions (228, 229). Importantly, regardless of the mAb strategy, the ability to restore lasting peripheral tolerance to prevent further β cell destruction is necessary for clinical success.
An important consideration in selecting a given set of mAb strategies is whether β cell autoimmunity is driven by T cells versus innate cells, and/or β cell intrinsic defects. Currently, evidence indicates that the rapid, aggressive disease developing in children is largely T cell-mediated. However, adult onset T1D may be driven by T cells, and/or innate effectors and/or β cell intrinsic defects leading to dysregulation of insulin production (236). Needed are sensitive disease readouts and biomarkers that distinguish between the respective scenarios or endotypes, to ensure that the appropriate therapeutic strategy is being applied (237). Consideration is also needed in defining successful endpoints for testing a therapeutic approach in the clinic (237). The ideal scenario is that a given mAb immunotherapy protects or rescues β cell function measured in part by metabolic indicators (Figure 2). Nevertheless, in the absence of a successful metabolic outcome, there is much value in determining whether an immunotherapy has induced tolerance within the targeted effector cell pool. As alluded to above, establishing tolerance in one compartment of the disease process may be insufficient to achieve a therapeutic benefit. However, determining that tolerance is indeed established would provide justification to combine in a rational manner, appropriate complementary strategies to enhance therapeutic efficacy. In the case of T cells, single cell transcriptome analysis of T cells bound by β cell-specific multimers and sorted from the blood of test and control subjects would be one approach sufficiently sensitive to detect changes within the T cell compartment.
To date, mAb therapies have provided intriguing results in affecting the progression of T1D. Nevertheless, an effective strategy to reestablish self-tolerance long-term is still required. Ongoing T1D research continues to characterize novel genes and potential targets involved in T1D disease susceptibility and progression. The ability to customize the mAb target and respective effector function provides immense flexibility to discover and develop a successful mAb treatment for T1D.
QK, CK, MC, and RT contributed to the preparation of the review article. All authors approved the submitted version.
This work was supported by National Institutes of Health grants R01DK100256, R01AI139475, R01AI141631, R21AI115752 (RT), and T32AI007273 (MC), and the American Diabetes Association 1-18-PDF-108 (MC).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Clark M, Kroger CJ, Tisch RM. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front Immunol (2017) 8:1898. doi: 10.3389/fimmu.2017.01898
2. Tisch R, McDevitt H. Insulin-dependent diabetes mellitus. Cell (1996) 85(3):291–7. doi: 10.1016/s0092-8674(00)81106-x
3. Eisenbarth GS. Type 1 diabetes: molecular, cellular and clinical immunology. Adv Exp Med Biol (2004) 552:306–10.
4. Bach JF. Insulin-dependent diabetes mellitus as an autoimmune disease. Endocr Rev (1994) 15(4):516–42. doi: 10.1210/edrv-15-4-516
5. van Belle TL, Coppieters KT, von Herrath MG. Type 1 diabetes: etiology, immunology, and therapeutic strategies. Physiol Rev (2011) 91(1):79–118. doi: 10.1152/physrev.00003.2010
6. Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, et al. Type 1 diabetes mellitus. Nat Rev Dis Primers (2017) 3:17016. doi: 10.1038/nrdp.2017.16
7. DiMeglio LA, Evans-Molina C, Oram RA. Type 1 diabetes. Lancet (2018) 391(10138):2449–62. doi: 10.1016/S0140-6736(18)31320-5
8. von Herrath M, Sanda S, Herold K. Type 1 diabetes as a relapsing-remitting disease? Nat Rev Immunol (2007) 7(12):988–94. doi: 10.1038/nri2192
9. Campbell-Thompson M, Fu A, Kaddis JS, Wasserfall C, Schatz DA, Pugliese A, et al. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes (2016) 65(3):719–31. doi: 10.2337/db15-0779
10. Anderson MS, Bluestone JA. The NOD mouse: a model of immune dysregulation. Annu Rev Immunol (2005) 23:447–85. doi: 10.1146/annurev.immunol.23.021704.115643
11. Driver JP, Serreze DV, Chen YG. Mouse models for the study of autoimmune type 1 diabetes: a NOD to similarities and differences to human disease. Semin Immunopathol (2011) 33(1):67–87. doi: 10.1007/s00281-010-0204-1
12. Barrett JC, Clayton DG, Concannon P, Akolkar B, Cooper JD, Erlich HA, et al. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet (2009) 41(6):703–7. doi: 10.1038/ng.381
13. Concannon P, Rich SS, Nepom GT. Genetics of type 1A diabetes. N Engl J Med (2009) 360(16):1646–54. doi: 10.1056/NEJMra0808284
14. Noble JA, Erlich HA. Genetics of type 1 diabetes. Cold Spring Harb Perspect Med (2012) 2(1):a007732. doi: 10.1101/cshperspect.a007732
15. Pociot F, Akolkar B, Concannon P, Erlich HA, Julier C, Morahan G, et al. Genetics of type 1 diabetes: what’s next? Diabetes (2010) 59(7):1561–71. doi: 10.2337/db10-0076
16. Pociot F, McDermott MF. Genetics of type 1 diabetes mellitus. Genes Immun (2002) 3(5):235–49. doi: 10.1038/sj.gene.6363875
17. Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, et al. Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J Exp Med (2012) 209(1):51–60. doi: 10.1084/jem.20111187
18. Coppieters KT, Roep BO, von Herrath MG. Beta cells under attack: toward a better understanding of type 1 diabetes immunopathology. Semin Immunopathol (2011) 33(1):1–7. doi: 10.1007/s00281-010-0236-6
19. Atkinson MA, von Herrath M, Powers AC, Clare-Salzler M. Current concepts on the pathogenesis of type 1 diabetes–considerations for attempts to prevent and reverse the disease. Diabetes Care (2015) 38(6):979–88. doi: 10.2337/dc15-0144
20. Arif S, Gibson VB, Nguyen V, Bingley PJ, Todd JA, Guy C, et al. beta-cell specific T-lymphocyte response has a distinct inflammatory phenotype in children with Type 1 diabetes compared with adults. Diabetes Med (2017) 34(3):419–25. doi: 10.1111/dme.13153
21. Michels AW, Landry LG, McDaniel KA, Yu L, Campbell-Thompson M, Kwok WW, et al. Islet-Derived CD4 T Cells Targeting Proinsulin in Human Autoimmune Diabetes. Diabetes (2017) 66(3):722–34. doi: 10.2337/db16-1025
22. Kent SC, Mannering SI, Michels AW, Babon JAB. Deciphering the Pathogenesis of Human Type 1 Diabetes (T1D) by Interrogating T Cells from the “Scene of the Crime”. Curr Diabetes Rep (2017) 17(10):95. doi: 10.1007/s11892-017-0915-y
23. Heninger AK, Eugster A, Kuehn D, Buettner F, Kuhn M, Lindner A, et al. A divergent population of autoantigen-responsive CD4+ T cells in infants prior to beta cell autoimmunity. Sci Transl Med (2017) 9(378). doi: 10.1126/scitranslmed.aaf8848
24. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Analysis of islet inflammation in human type 1 diabetes. Clin Exp Immunol (2009) 155(2):173–81. doi: 10.1111/j.1365-2249.2008.03860.x
25. Richardson SJ, Willcox A, Bone AJ, Morgan NG, Foulis AK. Immunopathology of the human pancreas in type-I diabetes. Semin Immunopathol (2011) 33(1):9–21. doi: 10.1007/s00281-010-0205-0
26. Babon JA, DeNicola ME, Blodgett DM, Crevecoeur I, Buttrick TS, Maehr R, et al. Analysis of self-antigen specificity of islet-infiltrating T cells from human donors with type 1 diabetes. Nat Med (2016) 22(12):1482–7. doi: 10.1038/nm.4203
27. Pugliese A. Autoreactive T cells in type 1 diabetes. J Clin Invest (2017) 127(8):2881–91. doi: 10.1172/JCI94549
28. Gomez-Tourino I, Arif S, Eichmann M, Peakman M. T cells in type 1 diabetes: Instructors, regulators and effectors: A comprehensive review. J Autoimmun (2016) 66:7–16. doi: 10.1016/j.jaut.2015.08.012
29. Mallone R, Brezar V, Boitard C. T cell recognition of autoantigens in human type 1 diabetes: clinical perspectives. Clin Dev Immunol (2011) 2011:513210. doi: 10.1155/2011/513210
30. Roep BO. The role of T-cells in the pathogenesis of Type 1 diabetes: from cause to cure. Diabetologia (2003) 46(3):305–21. doi: 10.1007/s00125-003-1089-5
31. Krishnamurthy B, Selck C, Chee J, Jhala G, Kay TW. Analysis of antigen specific T cells in diabetes - Lessons from pre-clinical studies and early clinical trials. J Autoimmun (2016) 71:35–43. doi: 10.1016/j.jaut.2016.03.018
32. Nerup J, Platz P, Andersen OO, Christy M, Lyngsoe J, Poulsen JE, et al. HL-A antigens and diabetes mellitus. Lancet (1974) 2(7885):864–6. doi: 10.1016/s0140-6736(74)90930-1
33. Singal DP, Blajchman MA. Histocompatibility (HL-A) antigens, lymphocytotoxic antibodies and tissue antibodies in patients with diabetes mellitus. Diabetes (1973) 22(6):429–32. doi: 10.2337/diab.22.6.429
34. In’t Veld P. Insulitis in human type 1 diabetes: The quest for an elusive lesion. Islets (2011) 3(4):131–8. doi: 10.4161/isl.3.4.15728
35. Skog O, Korsgren S, Melhus A, Korsgren O. Revisiting the notion of type 1 diabetes being a T-cell-mediated autoimmune disease. Curr Opin Endocrinol Diabetes Obes (2013) 20(2):118–23. doi: 10.1097/MED.0b013e32835edb89
36. Yu L, Rewers M, Gianani R, Kawasaki E, Zhang Y, Verge C, et al. Antiislet autoantibodies usually develop sequentially rather than simultaneously. J Clin Endocrinol Metab (1996) 81(12):4264–7. doi: 10.1210/jcem.81.12.8954025
37. Yu L, Robles DT, Abiru N, Kaur P, Rewers M, Kelemen K, et al. Early expression of antiinsulin autoantibodies of humans and the NOD mouse: evidence for early determination of subsequent diabetes. Proc Natl Acad Sci U S A (2000) 97(4):1701–6. doi: 10.1073/pnas.040556697
38. Simmons KM, Michels AW. Type 1 diabetes: A predictable disease. World J Diabetes (2015) 6(3):380–90. doi: 10.4239/wjd.v6.i3.380
39. Chmiel R, Giannopoulou EZ, Winkler C, Achenbach P, Ziegler AG, Bonifacio E. Progression from single to multiple islet autoantibodies often occurs soon after seroconversion: implications for early screening. Diabetologia (2015) 58(2):411–3. doi: 10.1007/s00125-014-3443-1
40. Ehlers MR. Strategies for clinical trials in type 1 diabetes. J Autoimmun (2016) 71:88–96. doi: 10.1016/j.jaut.2016.03.008
41. Ziegler AG, Rewers M, Simell O, Simell T, Lempainen J, Steck A, et al. Seroconversion to multiple islet autoantibodies and risk of progression to diabetes in children. JAMA (2013) 309(23):2473–9. doi: 10.1001/jama.2013.6285
42. Serreze DV, Chapman HD, Varnum DS, Hanson MS, Reifsnyder PC, Richard SD, et al. B lymphocytes are essential for the initiation of T cell-mediated autoimmune diabetes: analysis of a new “speed congenic” stock of NOD.Ig mu null mice. J Exp Med (1996) 184(5):2049–53. doi: 10.1084/jem.184.5.2049
43. Serreze DV, Fleming SA, Chapman HD, Richard SD, Leiter EH. Tisch RM. B lymphocytes are critical antigen-presenting cells for the initiation of T cell-mediated autoimmune diabetes in nonobese diabetic mice. J Immunol (1998) 161(8):3912–8.
44. Akashi T, Nagafuchi S, Anzai K, Kondo S, Kitamura D, Wakana S, et al. Direct evidence for the contribution of B cells to the progression of insulitis and the development of diabetes in non-obese diabetic mice. Int Immunol (1997) 9(8):1159–64. doi: 10.1093/intimm/9.8.1159
45. Ferris ST, Carrero JA, Unanue ER. Antigen presentation events during the initiation of autoimmune diabetes in the NOD mouse. J Autoimmun (2016) 71:19–25. doi: 10.1016/j.jaut.2016.03.007
46. Unanue ER, Ferris ST, Carrero JA. The role of islet antigen presenting cells and the presentation of insulin in the initiation of autoimmune diabetes in the NOD mouse. Immunol Rev (2016) 272(1):183–201. doi: 10.1111/imr.12430
47. Gagnerault MC, Luan JJ, Lotton C, Lepault F. Pancreatic lymph nodes are required for priming of beta cell reactive T cells in NOD mice. J Exp Med (2002) 196(3):369–77. doi: 10.1084/jem.20011353
48. Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med (2003) 198(10):1527–37. doi: 10.1084/jem.20030966
49. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol (2009) 5(4):219–26. doi: 10.1038/nrendo.2009.21
50. Shao S, He F, Yang Y, Yuan G, Zhang M, Yu X. Th17 cells in type 1 diabetes. Cell Immunol (2012) 280(1):16–21. doi: 10.1016/j.cellimm.2012.11.001
51. Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest (2015) 125(1):292–303. doi: 10.1172/JCI76238
52. Walker LS, von Herrath M. CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol (2016) 183(1):16–29. doi: 10.1111/cei.12672
53. Crotty S. Follicular helper CD4 T cells (TFH). Annu Rev Immunol (2011) 29:621–63. doi: 10.1146/annurev-immunol-031210-101400
54. Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity (2014) 41(4):529–42. doi: 10.1016/j.immuni.2014.10.004
55. Crotty S. T Follicular Helper Cell Biology: A Decade of Discovery and Diseases. Immunity (2019) 50(5):1132–48. doi: 10.1016/j.immuni.2019.04.011
56. Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol (2003) 3(2):133–46. doi: 10.1038/nri1001
57. Arif S, Leete P, Nguyen V, Marks K, Nor NM, Estorninho M, et al. Blood and islet phenotypes indicate immunological heterogeneity in type 1 diabetes. Diabetes (2014) 63(11):3835–45. doi: 10.2337/db14-0365
58. Arif S, Tree TI, Astill TP, Tremble JM, Bishop AJ, Dayan CM, et al. Autoreactive T cell responses show proinflammatory polarization in diabetes but a regulatory phenotype in health. J Clin Invest (2004) 113(3):451–63. doi: 10.1172/JCI19585
59. Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, et al. Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes (2009) 58(6):1302–11. doi: 10.2337/db08-1113
60. Spolski R, Kashyap M, Robinson C, Yu Z, Leonard WJ. IL-21 signaling is critical for the development of type I diabetes in the NOD mouse. Proc Natl Acad Sci U S A (2008) 105(37):14028–33. doi: 10.1073/pnas.0804358105
61. Edner NM, Heuts F, Thomas N, Wang CJ, Petersone L, Kenefeck R, et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat Immunol (2020) 21(10):1244–55. doi: 10.1038/s41590-020-0744-z
62. Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol (2009) 27:485–517. doi: 10.1146/annurev.immunol.021908.132710
63. Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes (2005) 54 Suppl 2:S97–107. doi: 10.2337/diabetes.54.suppl_2.s97
64. Wilcox NS, Rui J, Hebrok M, Herold KC. Life and death of beta cells in Type 1 diabetes: A comprehensive review. J Autoimmun (2016) 71:51–8. doi: 10.1016/j.jaut.2016.02.001
65. Rotondi M, Chiovato L, Romagnani S, Serio M, Romagnani P. Role of chemokines in endocrine autoimmune diseases. Endocr Rev (2007) 28(5):492–520. doi: 10.1210/er.2006-0044
66. Roep BO, Kracht MJ, van Lummel M, Zaldumbide A. A roadmap of the generation of neoantigens as targets of the immune system in type 1 diabetes. Curr Opin Immunol (2016) 43:67–73. doi: 10.1016/j.coi.2016.09.007
67. Delong T, Wiles TA, Baker RL, Bradley B, Barbour G, Reisdorph R, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science (2016) 351(6274):711–4. doi: 10.1126/science.aad2791
68. Gur C, Porgador A, Elboim M, Gazit R, Mizrahi S, Stern-Ginossar N, et al. The activating receptor NKp46 is essential for the development of type 1 diabetes. Nat Immunol (2010) 11(2):121–8. doi: 10.1038/ni.1834
69. Uno S, Imagawa A, Okita K, Sayama K, Moriwaki M, Iwahashi H, et al. Macrophages and dendritic cells infiltrating islets with or without beta cells produce tumour necrosis factor-alpha in patients with recent-onset type 1 diabetes. Diabetologia (2007) 50(3):596–601. doi: 10.1007/s00125-006-0569-9
70. Diana J, Simoni Y, Furio L, Beaudoin L, Agerberth B, Barrat F, et al. Crosstalk between neutrophils, B-1a cells and plasmacytoid dendritic cells initiates autoimmune diabetes. Nat Med (2013) 19(1):65–73. doi: 10.1038/nm.3042
71. Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes (1993) 42(1):44–55. doi: 10.2337/diab.42.1.44
72. Staeva TP, Chatenoud L, Insel R, Atkinson MA. Recent lessons learned from prevention and recent-onset type 1 diabetes immunotherapy trials. Diabetes (2013) 62(1):9–17. doi: 10.2337/db12-0562
73. Yasunaga M. Antibody therapeutics and immunoregulation in cancer and autoimmune disease. Semin Cancer Biol (2020) 64:1–12. doi: 10.1016/j.semcancer.2019.06.001
74. Weiner LM, Surana R, Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat Rev Immunol (2010) 10(5):317–27. doi: 10.1038/nri2744
75. Bruno V, Battaglia G, Nicoletti F. The advent of monoclonal antibodies in the treatment of chronic autoimmune diseases. Neurol Sci (2011) 31 Suppl 3:283–8. doi: 10.1007/s10072-010-0382-6
76. Tanaka T, Hishitani Y, Ogata A. Monoclonal antibodies in rheumatoid arthritis: comparative effectiveness of tocilizumab with tumor necrosis factor inhibitors. Biologics (2014) 8:141–53. doi: 10.2147/BTT.S37509
77. Croft M. The role of TNF superfamily members in T-cell function and diseases. Nat Rev Immunol (2009) 9(4):271–85. doi: 10.1038/nri2526
78. Kaplon H, Muralidharan M, Schneider Z, Reichert JM. Antibodies to watch in 2020. MAbs (2020) 12(1):1703531. doi: 10.1080/19420862.2019.1703531
79. Hnasko RM, Stanker LH. Hybridoma Technology. Methods Mol Biol (2015) 1318:15–28. doi: 10.1007/978-1-4939-2742-5_2
80. Hanack K, Messerschmidt K, Listek M. Antibodies and Selection of Monoclonal Antibodies. Adv Exp Med Biol (2016) 917:11–22. doi: 10.1007/978-3-319-32805-8_2
81. Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science (1985) 228(4705):1315–7. doi: 10.1126/science.4001944
82. Cho W, Fowler JD, Furst EM. Targeted binding of the M13 bacteriophage to thiamethoxam organic crystals. Langmuir (2012) 28(14):6013–20. doi: 10.1021/la300522g
83. Hoogenboom HR. Selecting and screening recombinant antibody libraries. Nat Biotechnol (2005) 23(9):1105–16. doi: 10.1038/nbt1126
84. Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol (1994) 12:433–55. doi: 10.1146/annurev.iy.12.040194.002245
85. Osbourn J, Groves M, Vaughan T. From rodent reagents to human therapeutics using antibody guided selection. Methods (2005) 36(1):61–8. doi: 10.1016/j.ymeth.2005.01.006
86. Prassler J, Thiel S, Pracht C, Polzer A, Peters S, Bauer M, et al. HuCAL PLATINUM, a synthetic Fab library optimized for sequence diversity and superior performance in mammalian expression systems. J Mol Biol (2011) 413(1):261–78. doi: 10.1016/j.jmb.2011.08.012
87. Hu D, Hu S, Wan W, Xu M, Du R, Zhao W, et al. Effective Optimization of Antibody Affinity by Phage Display Integrated with High-Throughput DNA Synthesis and Sequencing Technologies. PLoS One (2015) 10(6):e0129125. doi: 10.1371/journal.pone.0129125
88. Azzazy HM, Highsmith WE Jr. Phage display technology: clinical applications and recent innovations. Clin Biochem (2002) 35(6):425–45. doi: 10.1016/s0009-9120(02)00343-0
89. Imai S, Mukai Y, Takeda T, Abe Y, Nagano K, Kamada H, et al. Effect of protein properties on display efficiency using the M13 phage display system. Pharmazie (2008) 63(10):760–4.
90. Jaffers GJ, Fuller TC, Cosimi AB, Russell PS, Winn HJ, Colvin RB. Monoclonal antibody therapy. Anti-idiotypic and non-anti-idiotypic antibodies to OKT3 arising despite intense immunosuppression. Transplantation (1986) 41(5):572–8. doi: 10.1097/00007890-198605000-00004
91. Kimball JA, Norman DJ, Shield CF, Schroeder TJ, Lisi P, Garovoy M, et al. The OKT3 Antibody Response Study: a multicentre study of human anti-mouse antibody (HAMA) production following OKT3 use in solid organ transplantation. Transpl Immunol (1995) 3(3):212–21. doi: 10.1016/0966-3274(95)80027-1
92. LoBuglio AF, Wheeler RH, Trang J, Haynes A, Rogers K, Harvey EB, et al. Mouse/human chimeric monoclonal antibody in man: kinetics and immune response. Proc Natl Acad Sci U S A (1989) 86(11):4220–4. doi: 10.1073/pnas.86.11.4220
93. Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods (2005) 36(1):3–10. doi: 10.1016/j.ymeth.2005.01.001
94. Baert F, Noman M, Vermeire S, Van Assche G, DH G, Carbonez A, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn’s disease. N Engl J Med (2003) 348(7):601–8. doi: 10.1056/NEJMoa020888
95. Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature (1986) 321(6069):522–5. doi: 10.1038/321522a0
96. Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, et al. Humanization of an anti-p185HER2 antibody for human cancer therapy. Proc Natl Acad Sci U S A (1992) 89(10):4285–9. doi: 10.1073/pnas.89.10.4285
97. Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, et al. A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci U S A (1989) 86(24):10029–33. doi: 10.1073/pnas.86.24.10029
98. Studnicka GM, Soares S, Better M, Williams RE, Nadell R, Horwitz AH. Human-engineered monoclonal antibodies retain full specific binding activity by preserving non-CDR complementarity-modulating residues. Protein Eng (1994) 7(6):805–14. doi: 10.1093/protein/7.6.805
99. de Haard H, Kazemier B, van der Bent A, Oudshoorn P, Boender P, Arends JW, et al. Vernier zone residue 4 of mouse subgroup II kappa light chains is a critical determinant for antigen recognition. Immunotechnology (1999) 4(3-4):203–15. doi: 10.1016/s1380-2933(98)00021-9
100. Makabe K, Nakanishi T, Tsumoto K, Tanaka Y, Kondo H, Umetsu M, et al. Thermodynamic consequences of mutations in vernier zone residues of a humanized anti-human epidermal growth factor receptor murine antibody, 528. J Biol Chem (2008) 283(2):1156–66. doi: 10.1074/jbc.M706190200
101. Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: residual immunogenicity resides in the CDR regions. MAbs (2010) 2(3):256–65. doi: 10.4161/mabs.2.3.11641
102. Jakobovits A, Amado RG, Yang X, Roskos L, Schwab G. From XenoMouse technology to panitumumab, the first fully human antibody product from transgenic mice. Nat Biotechnol (2007) 25(10):1134–43. doi: 10.1038/nbt1337
103. Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol (2005) 23(9):1117–25. doi: 10.1038/nbt1135
104. Lonberg N, Taylor LD, Harding FA, Trounstine M, Higgins KM, Schramm SR, et al. Antigen-specific human antibodies from mice comprising four distinct genetic modifications. Nature (1994) 368(6474):856–9. doi: 10.1038/368856a0
105. Fishwild DM, O’Donnell SL, Bengoechea T, Hudson DV, Harding F, Bernhard SL, et al. High-avidity human IgG kappa monoclonal antibodies from a novel strain of minilocus transgenic mice. Nat Biotechnol (1996) 14(7):845–51. doi: 10.1038/nbt0796-845
107. Krueger GG, Langley RG, Leonardi C, Yeilding N, Guzzo C, Wang Y, et al. A human interleukin-12/23 monoclonal antibody for the treatment of psoriasis. N Engl J Med (2007) 356(6):580–92. doi: 10.1056/NEJMoa062382
108. Hirohata S. [Fully human anti TNF-alpha monoclonal antibodies (adalimumab, golimumab)]. Nihon Rinsho (2007) 65(7):1202–8.
109. Vidarsson G, Dekkers G, Rispens T. IgG subclasses and allotypes: from structure to effector functions. Front Immunol (2014) 5:520. doi: 10.3389/fimmu.2014.00520
110. Shields RL, Lai J, Keck R, O’Connell LY, Hong K, Meng YG, et al. Lack of fucose on human IgG1 N-linked oligosaccharide improves binding to human Fcgamma RIII and antibody-dependent cellular toxicity. J Biol Chem (2002) 277(30):26733–40. doi: 10.1074/jbc.M202069200
111. Liu SD, Chalouni C, Young JC, Junttila TT, Sliwkowski MX, Lowe JB. Afucosylated antibodies increase activation of FcgammaRIIIa-dependent signaling components to intensify processes promoting ADCC. Cancer Immunol Res (2015) 3(2):173–83. doi: 10.1158/2326-6066.CIR-14-0125
112. Chatenoud L, Ferran C, Legendre C, Thouard I, Merite S, Reuter A, et al. In vivo cell activation following OKT3 administration. Systemic cytokine release and modulation by corticosteroids. Transplantation (1990) 49(4):697–702. doi: 10.1097/00007890-199004000-00009
113. Ceuppens JL, Bloemmen FJ, Van Wauwe JP. T cell unresponsiveness to the mitogenic activity of OKT3 antibody results from a deficiency of monocyte Fc gamma receptors for murine IgG2a and inability to cross-link the T3-Ti complex. J Immunol (1985) 135(6):3882–6.
114. Xu D, Alegre ML, Varga SS, Rothermel AL, Collins AM, Pulito VL, et al. In vitro characterization of five humanized OKT3 effector function variant antibodies. Cell Immunol (2000) 200(1):16–26. doi: 10.1006/cimm.2000.1617
115. Friend PJ, Hale G, Chatenoud L, Rebello P, Bradley J, Thiru S, et al. Phase I study of an engineered aglycosylated humanized CD3 antibody in renal transplant rejection. Transplantation (1999) 68(11):1632–7. doi: 10.1097/00007890-199912150-00005
116. Hale G, Rebello P, Al Bakir I, Bolam E, Wiczling P, Jusko WJ, et al. Pharmacokinetics and antibody responses to the CD3 antibody otelixizumab used in the treatment of type 1 diabetes. J Clin Pharmacol (2010) 50(11):1238–48. doi: 10.1177/0091270009356299
117. Tao MH, Morrison SL. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J Immunol (1989) 143(8):2595–601.
118. Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol (2007) 7(9):715–25. doi: 10.1038/nri2155
119. Herold KC, Gitelman SE, Masharani U, Hagopian W, Bisikirska B, Donaldson D, et al. A single course of anti-CD3 monoclonal antibody hOKT3gamma1(Ala-Ala) results in improvement in C-peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes (2005) 54(6):1763–9. doi: 10.2337/diabetes.54.6.1763
120. Herold KC, Hagopian W, Auger JA, Poumian-Ruiz E, Taylor L, Donaldson D, et al. Anti-CD3 monoclonal antibody in new-onset type 1 diabetes mellitus. N Engl J Med (2002) 346(22):1692–8. doi: 10.1056/NEJMoa012864
121. Keymeulen B, Candon S, Fafi-Kremer S, Ziegler A, Leruez-Ville M, Mathieu C, et al. Transient Epstein-Barr virus reactivation in CD3 monoclonal antibody-treated patients. Blood (2010) 115(6):1145–55. doi: 10.1182/blood-2009-02-204875
122. Pescovitz MD, Greenbaum CJ, Krause-Steinrauf H, Becker DJ, Gitelman SE, Goland R, et al. Rituximab, B-lymphocyte depletion, and preservation of beta-cell function. N Engl J Med (2009) 361(22):2143–52. doi: 10.1056/NEJMoa0904452
123. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352(25):2598–608. doi: 10.1056/NEJMoa043980
124. Reinherz EL, Hussey RE, Schlossman SF. A monoclonal antibody blocking human T cell function. Eur J Immunol (1980) 10(10):758–62. doi: 10.1002/eji.1830101006
125. Herve P, Flesch M, Cahn JY, Racadot E, Plouvier E, Lamy B, et al. Removal of marrow T cells with OKT3-OKT11 monoclonal antibodies and complement to prevent acute graft-versus-host disease. A pilot study in ten patients. Transplantation (1985) 39(2):138–43. doi: 10.1097/00007890-198502000-00006
126. Chatenoud L, Thervet E, Primo J, Bach JF. Anti-CD3 antibody induces long-term remission of overt autoimmunity in nonobese diabetic mice. Proc Natl Acad Sci U S A (1994) 91(1):123–7. doi: 10.1073/pnas.91.1.123
127. Chatenoud L, Bluestone JA. CD3-specific antibodies: a portal to the treatment of autoimmunity. Nat Rev Immunol (2007) 7(8):622–32. doi: 10.1038/nri2134
128. Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med (2003) 9(9):1202–8. doi: 10.1038/nm924
129. Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med (2008) 14(5):528–35. doi: 10.1038/nm1749
130. Carpenter PA, Pavlovic S, Tso JY, Press OW, Gooley T, Yu XZ, et al. Non-Fc receptor-binding humanized anti-CD3 antibodies induce apoptosis of activated human T cells. J Immunol (2000) 165(11):6205–13. doi: 10.4049/jimmunol.165.11.6205
131. Yu XZ, Anasetti C. Enhancement of susceptibility to Fas-mediated apoptosis of TH1 cells by nonmitogenic anti-CD3epsilon F(ab’)2. Transplantation (2000) 69(1):104–12. doi: 10.1097/00007890-200001150-00019
132. Penaranda C, Tang Q, Bluestone JA. Anti-CD3 therapy promotes tolerance by selectively depleting pathogenic cells while preserving regulatory T cells. J Immunol (2011) 187(4):2015–22. doi: 10.4049/jimmunol.1100713
133. Chatenoud L, Primo J, Bach JF. CD3 antibody-induced dominant self tolerance in overtly diabetic NOD mice. J Immunol (1997) 158(6):2947–54.
134. You S, Leforban B, Garcia C, Bach JF, Bluestone JA, Chatenoud L. Adaptive TGF-beta-dependent regulatory T cells control autoimmune diabetes and are a privileged target of anti-CD3 antibody treatment. Proc Natl Acad Sci U S A (2007) 104(15):6335–40. doi: 10.1073/pnas.0701171104
135. Keymeulen B, Walter M, Mathieu C, Kaufman L, Gorus F, Hilbrands R, et al. Four-year metabolic outcome of a randomised controlled CD3-antibody trial in recent-onset type 1 diabetic patients depends on their age and baseline residual beta cell mass. Diabetologia (2010) 53(4):614–23. doi: 10.1007/s00125-009-1644-9
136. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
137. Hagopian W, Ferry RJ Jr., Sherry N, Carlin D, Bonvini E, Johnson S, et al. Teplizumab preserves C-peptide in recent-onset type 1 diabetes: two-year results from the randomized, placebo-controlled Protege trial. Diabetes (2013) 62(11):3901–8. doi: 10.2337/db13-0236
138. Waldron-Lynch F, Henegariu O, Deng S, Preston-Hurlburt P, Tooley J, Flavell R, et al. Teplizumab induces human gut-tropic regulatory cells in humanized mice and patients. Sci Transl Med (2012) 4(118):118ra12. doi: 10.1126/scitranslmed.3003401
139. Long SA, Thorpe J, Herold KC, Ehlers M, Sanda S, Lim N, et al. Remodeling T cell compartments during anti-CD3 immunotherapy of type 1 diabetes. Cell Immunol (2017) 319:3–9. doi: 10.1016/j.cellimm.2017.07.007
140. Long SA, Thorpe J, DeBerg HA, Gersuk V, Eddy J, Harris KM, et al. Partial exhaustion of CD8 T cells and clinical response to teplizumab in new-onset type 1 diabetes. Sci Immunol (2016) 1(5). doi: 10.1126/sciimmunol.aai7793
141. Wiedeman AE, Muir VS, Rosasco MG, DeBerg HA, Presnell S, Haas B, et al. Autoreactive CD8+ T cell exhaustion distinguishes subjects with slow type 1 diabetes progression. J Clin Invest (2020) 130(1):480–90. doi: 10.1172/JCI126595
142. Sherry N, Hagopian W, Ludvigsson J, Jain SM, Wahlen J, Ferry RJ Jr., et al. Teplizumab for treatment of type 1 diabetes (Protege study): 1-year results from a randomised, placebo-controlled trial. Lancet (2011) 378(9790):487–97. doi: 10.1016/S0140-6736(11)60931-8
143. Aronson R, Gottlieb PA, Christiansen JS, Donner TW, Bosi E, Bode BW, et al. Low-dose otelixizumab anti-CD3 monoclonal antibody DEFEND-1 study: results of the randomized phase III study in recent-onset human type 1 diabetes. Diabetes Care (2014) 37(10):2746–54. doi: 10.2337/dc13-0327
144. Ambery P, Donner TW, Biswas N, Donaldson J, Parkin J, Dayan CM. Efficacy and safety of low-dose otelixizumab anti-CD3 monoclonal antibody in preserving C-peptide secretion in adolescent type 1 diabetes: DEFEND-2, a randomized, placebo-controlled, double-blind, multi-centre study. Diabetes Med (2014) 31(4):399–402. doi: 10.1111/dme.12361
145. Vlasakakis G, Napolitano A, Barnard R, Brown K, Bullman J, Inman D, et al. Target engagement and cellular fate of otelixizumab: a repeat dose escalation study of an anti-CD3epsilon mAb in new-onset type 1 diabetes mellitus patients. Br J Clin Pharmacol (2019) 85(4):704–14. doi: 10.1111/bcp.13842
146. Keymeulen B, van Maurik A, Inman D, Oliveira J, McLaughlin R, Gittelman RM, et al. A randomised, single-blind, placebo-controlled, dose-finding safety and tolerability study of the anti-CD3 monoclonal antibody otelixizumab in new-onset type 1 diabetes. Diabetologia (2020) 64(2):313–24. doi: 10.1007/s00125-020-05317-y
147. PR Newswire. Provention Bio Announces PRV-031 (Teplizumab) Granted PRIME Designation by the European Medicines Agency (2019). Available at: https://www.prnewswire.com/news-releases/provention-bio-announces-prv-031-teplizumab-granted-prime-designation-by-the-european-medicines-agency-300944591.html (Accessed December 14, 2020).
148. PR Newswire. Provention Bio Announces Breakthrough Therapy Designation for Teplizumab (PRV-031) for the Prevention or Delay of Clinical Type 1 Diabetes in At-Risk Individuals (2019). Available at: https://www.prnewswire.com/news-releases/provention-bio-announces-breakthrough-therapy-designation-for-teplizumab-prv-031-for-the-prevention-or-delay-of-clinical-type-1-diabetes-in-at-risk-individuals-300896064.html (Accessed December 14, 2020).
149. Ochi H, Abraham M, Ishikawa H, Frenkel D, Yang K, Basso AS, et al. Oral CD3-specific antibody suppresses autoimmune encephalomyelitis by inducing CD4+ CD25- LAP+ T cells. Nat Med (2006) 12(6):627–35. doi: 10.1038/nm1408
150. Ilan Y, Zigmond E, Lalazar G, Dembinsky A, Ben Ya’acov A, Hemed N, et al. Oral administration of OKT3 monoclonal antibody to human subjects induces a dose-dependent immunologic effect in T cells and dendritic cells. J Clin Immunol (2010) 30(1):167–77. doi: 10.1007/s10875-009-9323-7
151. Ishikawa H, Ochi H, Chen ML, Frenkel D, Maron R, Weiner HL. Inhibition of autoimmune diabetes by oral administration of anti-CD3 monoclonal antibody. Diabetes (2007) 56(8):2103–9. doi: 10.2337/db06-1632
152. Xiu Y, Wong CP, Bouaziz JD, Hamaguchi Y, Wang Y, Pop SM, et al. B lymphocyte depletion by CD20 monoclonal antibody prevents diabetes in nonobese diabetic mice despite isotype-specific differences in Fc gamma R effector functions. J Immunol (2008) 180(5):2863–75. doi: 10.4049/jimmunol.180.5.2863
153. Fiorina P, Vergani A, Dada S, Jurewicz M, Wong M, Law K, et al. Targeting CD22 reprograms B-cells and reverses autoimmune diabetes. Diabetes (2008) 57(11):3013–24. doi: 10.2337/db08-0420
154. Leete P, Willcox A, Krogvold L, Dahl-Jorgensen K, Foulis AK, Richardson SJ, et al. Differential Insulitic Profiles Determine the Extent of beta-Cell Destruction and the Age at Onset of Type 1 Diabetes. Diabetes (2016) 65(5):1362–9. doi: 10.2337/db15-1615
155. Pescovitz MD, Greenbaum CJ, Bundy B, Becker DJ, Gitelman SE, Goland R, et al. B-lymphocyte depletion with rituximab and beta-cell function: two-year results. Diabetes Care (2014) 37(2):453–9. doi: 10.2337/dc13-0626
156. Chamberlain N, Massad C, Oe T, Cantaert T, Herold KC, Meffre E. Rituximab does not reset defective early B cell tolerance checkpoints. J Clin Invest (2016) 126(1):282–7. doi: 10.1172/JCI83840
157. Serreze DV, Chapman HD, Niens M, Dunn R, Kehry MR, Driver JP, et al. Loss of intra-islet CD20 expression may complicate efficacy of B-cell-directed type 1 diabetes therapies. Diabetes (2011) 60(11):2914–21. doi: 10.2337/db11-0705
158. Binder C, Cvetkovski F, Sellberg F, Berg S, Paternina Visbal H, Sachs DH, et al. CD2 Immunobiology. Front Immunol (2020) 11:1090. doi: 10.3389/fimmu.2020.01090
159. Sanders ME, Makgoba MW, Sharrow SO, Stephany D, Springer TA, Young HA, et al. Human memory T lymphocytes express increased levels of three cell adhesion molecules (LFA-3, CD2, and LFA-1) and three other molecules (UCHL1, CDw29, and Pgp-1) and have enhanced IFN-gamma production. J Immunol (1988) 140(5):1401–7.
160. Crawford K, Gabuzda D, Pantazopoulos V, Xu J, Clement C, Reinherz E, et al. Circulating CD2+ monocytes are dendritic cells. J Immunol (1999) 163(11):5920–8.
161. Jenneck C, Novak N. The safety and efficacy of alefacept in the treatment of chronic plaque psoriasis. Ther Clin Risk Manage (2007) 3(3):411–20.
162. Cooper JC, Morgan G, Harding S, Subramanyam M, Majeau GR, Moulder K, et al. Alefacept selectively promotes NK cell-mediated deletion of CD45R0+ human T cells. Eur J Immunol (2003) 33(3):666–75. doi: 10.1002/eji.200323586
163. Rigby MR, DiMeglio LA, Rendell MS, Felner EI, Dostou JM, Gitelman SE, et al. Targeting of memory T cells with alefacept in new-onset type 1 diabetes (T1DAL study): 12 month results of a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Diabetes Endocrinol (2013) 1(4):284–94. doi: 10.1016/S2213-8587(13)70111-6
164. Rigby MR, Harris KM, Pinckney A, DiMeglio LA, Rendell MS, Felner EI, et al. Alefacept provides sustained clinical and immunological effects in new-onset type 1 diabetes patients. J Clin Invest (2015) 125(8):3285–96. doi: 10.1172/JCI81722
165. Chamian F, Lowes MA, Lin SL, Lee E, Kikuchi T, Gilleaudeau P, et al. Alefacept reduces infiltrating T cells, activated dendritic cells, and inflammatory genes in psoriasis vulgaris. Proc Natl Acad Sci U S A (2005) 102(6):2075–80. doi: 10.1073/pnas.0409569102
166. Bruhns P, Iannascoli B, England P, Mancardi DA, Fernandez N, Jorieux S, et al. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood (2009) 113(16):3716–25. doi: 10.1182/blood-2008-09-179754
167. Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol (2016) 40:62–9. doi: 10.1016/j.coi.2016.03.002
168. Sha S, Agarabi C, Brorson K, Lee DY. Yoon S. N-Glycosylation Design and Control of Therapeutic Monoclonal Antibodies. Trends Biotechnol (2016) 34(10):835–46. doi: 10.1016/j.tibtech.2016.02.013
169. Brezski RJ, Kinder M, Grugan KD, Soring KL, Carton J, Greenplate AR, et al. A monoclonal antibody against hinge-cleaved IgG restores effector function to proteolytically-inactivated IgGs in vitro and in vivo. MAbs (2014) 6(5):1265–73. doi: 10.4161/mabs.29825
170. Coomans de Brachene A, Dos Santos RS, Marroqui L, Colli ML, Marselli L, Mirmira RG, et al. IFN-alpha induces a preferential long-lasting expression of MHC class I in human pancreatic beta cells. Diabetologia (2018) 61(3):636–40. doi: 10.1007/s00125-017-4536-4
171. Marroqui L, Dos Santos RS, Op de Beeck A, Coomans de Brachene A, Marselli L, Marchetti P, et al. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia (2017) 60(4):656–67. doi: 10.1007/s00125-016-4201-3
172. Newby BN, Brusko TM, Zou B, Atkinson MA, Clare-Salzler M, Mathews CE. Type 1 Interferons Potentiate Human CD8(+) T-Cell Cytotoxicity Through a STAT4- and Granzyme B-Dependent Pathway. Diabetes (2017) 66(12):3061–71. doi: 10.2337/db17-0106
173. Newby BN, Mathews CE. Type I Interferon Is a Catastrophic Feature of the Diabetic Islet Microenvironment. Front Endocrinol (Lausanne) (2017) 8:232. doi: 10.3389/fendo.2017.00232
174. Moran A, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Interleukin-1 antagonism in type 1 diabetes of recent onset: two multicentre, randomised, double-blind, placebo-controlled trials. Lancet (2013) 381(9881):1905–15. doi: 10.1016/S0140-6736(13)60023-9
175. Mastrandrea L, Yu J, Behrens T, Buchlis J, Albini C, Fourtner S, et al. Etanercept treatment in children with new-onset type 1 diabetes: pilot randomized, placebo-controlled, double-blind study. Diabetes Care (2009) 32(7):1244–9. doi: 10.2337/dc09-0054
176. University at Buffalo. Newly diagnosed children, young adults with Type 1 diabetes preserve endogenous insulin production with anti-TNF drug (2020). Available at: http://www.buffalo.edu/news/releases/2020/06/023.html (Accessed December 14, 2020).
177. Li Q, Xu B, Michie SA, Rubins KH, Schreriber RD, McDevitt HO. Interferon-alpha initiates type 1 diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A (2008) 105(34):12439–44. doi: 10.1073/pnas.0806439105
178. Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H, Oldstone MB. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-alpha signaling. Proc Natl Acad Sci U S A (2017) 114(14):3708–13. doi: 10.1073/pnas.1700878114
179. Debray-Sachs M, Carnaud C, Boitard C, Cohen H, Gresser I, Bedossa P, et al. Prevention of diabetes in NOD mice treated with antibody to murine IFN gamma. J Autoimmun (1991) 4(2):237–48. doi: 10.1016/0896-8411(91)90021-4
180. Prud’homme GJ, Chang Y. Prevention of autoimmune diabetes by intramuscular gene therapy with a nonviral vector encoding an interferon-gamma receptor/IgG1 fusion protein. Gene Ther (1999) 6(5):771–7. doi: 10.1038/sj.gt.3300879
181. Calderon B, Suri A, Pan XO, Mills JC, Unanue ER. IFN-gamma-dependent regulatory circuits in immune inflammation highlighted in diabetes. J Immunol (2008) 181(10):6964–74. doi: 10.4049/jimmunol.181.10.6964
182. Serreze DV, Chapman HD, Post CM, Johnson EA, Suarez-Pinzon WL, Rabinovitch A. Th1 to Th2 cytokine shifts in nonobese diabetic mice: sometimes an outcome, rather than the cause, of diabetes resistance elicited by immunostimulation. J Immunol (2001) 166(2):1352–9. doi: 10.4049/jimmunol.166.2.1352
183. Serreze DV, Post CM, Chapman HD, Johnson EA, Lu B, Rothman PB. Interferon-gamma receptor signaling is dispensable in the development of autoimmune type 1 diabetes in NOD mice. Diabetes (2000) 49(12):2007–11. doi: 10.2337/diabetes.49.12.2007
184. Hultgren B, Huang X, Dybdal N, Stewart TA. Genetic absence of gamma-interferon delays but does not prevent diabetes in NOD mice. Diabetes (1996) 45(6):812–7. doi: 10.2337/diab.45.6.812
185. Fujihira K, Nagata M, Moriyama H, Yasuda H, Arisawa K, Nakayama M, et al. Suppression and acceleration of autoimmune diabetes by neutralization of endogenous interleukin-12 in NOD mice. Diabetes (2000) 49(12):1998–2006. doi: 10.2337/diabetes.49.12.1998
186. Wu X, Tian J, Wang S. Insight Into Non-Pathogenic Th17 Cells in Autoimmune Diseases. Front Immunol (2018) 9:1112. doi: 10.3389/fimmu.2018.01112
187. Sutherland AP, Van Belle T, Wurster AL, Suto A, Michaud M, Zhang D, et al. Interleukin-21 is required for the development of type 1 diabetes in NOD mice. Diabetes (2009) 58(5):1144–55. doi: 10.2337/db08-0882
188. McGuire HM, Walters S, Vogelzang A, Lee CM, Webster KE, Sprent J, et al. Interleukin-21 is critically required in autoimmune and allogeneic responses to islet tissue in murine models. Diabetes (2011) 60(3):867–75. doi: 10.2337/db10-1157
189. Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, et al. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity (2008) 29(1):44–56. doi: 10.1016/j.immuni.2008.05.007
190. Astry B, Venkatesha SH, Moudgil KD. Involvement of the IL-23/IL-17 axis and the Th17/Treg balance in the pathogenesis and control of autoimmune arthritis. Cytokine (2015) 74(1):54–61. doi: 10.1016/j.cyto.2014.11.020
191. Tubo NJ, Jenkins MK. TCR signal quantity and quality in CD4(+) T cell differentiation. Trends Immunol (2014) 35(12):591–6. doi: 10.1016/j.it.2014.09.008
192. Bhattacharyya ND, Feng CG. Regulation of T Helper Cell Fate by TCR Signal Strength. Front Immunol (2020) 11:624:624. doi: 10.3389/fimmu.2020.00624
193. van Panhuys N. TCR Signal Strength Alters T-DC Activation and Interaction Times and Directs the Outcome of Differentiation. Front Immunol (2016) 7:6:6. doi: 10.3389/fimmu.2016.00006
194. Orban T, Bundy B, Becker DJ, DiMeglio LA, Gitelman SE, Goland R, et al. Co-stimulation modulation with abatacept in patients with recent-onset type 1 diabetes: a randomised, double-blind, placebo-controlled trial. Lancet (2011) 378(9789):412–9. doi: 10.1016/S0140-6736(11)60886-6
195. Orban T, Bundy B, Becker DJ, Dimeglio LA, Gitelman SE, Goland R, et al. Costimulation modulation with abatacept in patients with recent-onset type 1 diabetes: follow-up 1 year after cessation of treatment. Diabetes Care (2014) 37(4):1069–75. doi: 10.2337/dc13-0604
196. Martin AJ, Clark M, Gojanovich G, Manzoor F, Miller K, Kline DE, et al. Anti-coreceptor therapy drives selective T cell egress by suppressing inflammation-dependent chemotactic cues. JCI Insight (2016) 1(17):e87636. doi: 10.1172/jci.insight.87636
197. Morillon YM,2, Lessey-Morillon EC, Clark M, Zhang R, Wang B, Burridge K, et al. Antibody Binding to CD4 Induces Rac GTPase Activation and Alters T Cell Migration. J Immunol (2016) 197(9):3504–11. doi: 10.4049/jimmunol.1501600
198. Yi Z, Diz R, Martin AJ, Morillon YM, Kline DE, Li L, et al. Long-term remission of diabetes in NOD mice is induced by nondepleting anti-CD4 and anti-CD8 antibodies. Diabetes (2012) 61(11):2871–80. doi: 10.2337/db12-0098
199. Phillips JM, Parish NM, Raine T, Bland C, Sawyer Y, De La Pena H, et al. Type 1 diabetes development requires both CD4+ and CD8+ T cells and can be reversed by non-depleting antibodies targeting both T cell populations. Rev Diabetes Stud (2009) 6(2):97–103. doi: 10.1900/RDS.2009.6.97
200. Phillips JM, Harach SZ, Parish NM, Fehervari Z, Haskins K, Cooke A. Nondepleting anti-CD4 has an immediate action on diabetogenic effector cells, halting their destruction of pancreatic beta cells. J Immunol (2000) 165(4):1949–55. doi: 10.4049/jimmunol.165.4.1949
201. Konig M, Rharbaoui F, Aigner S, Dalken B, Schuttrumpf J. Tregalizumab - A Monoclonal Antibody to Target Regulatory T Cells. Front Immunol (2016) 7:11. doi: 10.3389/fimmu.2016.00011
202. Zhang L, Crawford F, Yu L, Michels A, Nakayama M, Davidson HW, et al. Monoclonal antibody blocking the recognition of an insulin peptide-MHC complex modulates type 1 diabetes. Proc Natl Acad Sci U S A (2014) 111(7):2656–61. doi: 10.1073/pnas.1323436111
203. Lee LF, Logronio K, Tu GH, Zhai W, Ni I, Mei L, et al. Anti-IL-7 receptor-alpha reverses established type 1 diabetes in nonobese diabetic mice by modulating effector T-cell function. Proc Natl Acad Sci U S A (2012) 109(31):12674–9. doi: 10.1073/pnas.1203795109
204. Penaranda C, Kuswanto W, Hofmann J, Kenefeck R, Narendran P, Walker LS, et al. IL-7 receptor blockade reverses autoimmune diabetes by promoting inhibition of effector/memory T cells. Proc Natl Acad Sci U S A (2012) 109(31):12668–73. doi: 10.1073/pnas.1203692109
205. Herold KC, Bucktrout SL, Wang X, Bode BW, Gitelman SE, Gottlieb PA, et al. Immunomodulatory activity of humanized anti-IL-7R monoclonal antibody RN168 in subjects with type 1 diabetes. JCI Insight (2019) 4(24). doi: 10.1172/jci.insight.126054
206. Yossef R, Gur C, Shemesh A, Guttman O, Hadad U, Nedvetzki S, et al. Targeting natural killer cell reactivity by employing antibody to NKp46: implications for type 1 diabetes. PloS One (2015) 10(2):e0118936. doi: 10.1371/journal.pone.0118936
207. Berhani O, Glasner A, Kahlon S, Duev-Cohen A, Yamin R, Horwitz E, et al. Human anti-NKp46 antibody for studies of NKp46-dependent NK cell function and its applications for type 1 diabetes and cancer research. Eur J Immunol (2019) 49(2):228–41. doi: 10.1002/eji.201847611
208. Gur C, Enk J, Kassem SA, Suissa Y, Magenheim J, Stolovich-Rain M, et al. Recognition and killing of human and murine pancreatic beta cells by the NK receptor NKp46. J Immunol (2011) 187(6):3096–103. doi: 10.4049/jimmunol.1101269
209. Sandor AM, Jacobelli J, Friedman RS. Immune cell trafficking to the islets during type 1 diabetes. Clin Exp Immunol (2019) 198(3):314–25. doi: 10.1111/cei.13353
210. Martin S, van den Engel NK, Vinke A, Heidenthal E, Schulte B, Kolb H. Dominant role of intercellular adhesion molecule-1 in the pathogenesis of autoimmune diabetes in non-obese diabetic mice. J Autoimmun (2001) 17(2):109–17. doi: 10.1006/jaut.2001.0526
211. Yang XD, Michie SA, Tisch R, Karin N, Steinman L, McDevitt HO. A predominant role of integrin alpha 4 in the spontaneous development of autoimmune diabetes in nonobese diabetic mice. Proc Natl Acad Sci U S A (1994) 91(26):12604–8. doi: 10.1073/pnas.91.26.12604
212. Hanninen A, Jaakkola I, Jalkanen S. Mucosal addressin is required for the development of diabetes in nonobese diabetic mice. J Immunol (1998) 160(12):6018–25.
213. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (2006) 311(5769):1924–7. doi: 10.1126/science.1122927
214. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi: 10.1038/nri3156
215. Brusko TM, Wasserfall CH, Clare-Salzler MJ, Schatz DA, Atkinson MA. Functional defects and the influence of age on the frequency of CD4+ CD25+ T-cells in type 1 diabetes. Diabetes (2005) 54(5):1407–14. doi: 10.2337/diabetes.54.5.1407
216. Lowe CE, Cooper JD, Brusko T, Walker NM, Smyth DJ, Bailey R, et al. Large-scale genetic fine mapping and genotype-phenotype associations implicate polymorphism in the IL2RA region in type 1 diabetes. Nat Genet (2007) 39(9):1074–82. doi: 10.1038/ng2102
217. Maier LM, Anderson DE, Severson CA, Baecher-Allan C, Healy B, Liu DV, et al. Soluble IL-2RA levels in multiple sclerosis subjects and the effect of soluble IL-2RA on immune responses. J Immunol (2009) 182(3):1541–7. doi: 10.4049/jimmunol.182.3.1541
218. Giordano C, Panto F, Caruso C, Modica MA, Zambito AM, Sapienza N, et al. Interleukin 2 and soluble interleukin 2-receptor secretion defect in vitro in newly diagnosed type I diabetic patients. Diabetes (1989) 38(3):310–5. doi: 10.2337/diab.38.3.310
219. Brusko T, Wasserfall C, McGrail K, Schatz R, Viener HL, Schatz D, et al. No alterations in the frequency of FOXP3+ regulatory T-cells in type 1 diabetes. Diabetes (2007) 56(3):604–12. doi: 10.2337/db06-1248
220. Qu HQ, Verlaan DJ, Ge B, Lu Y, Lam KC, Grabs R, et al. A cis-acting regulatory variant in the IL2RA locus. J Immunol (2009) 183(8):5158–62. doi: 10.4049/jimmunol.0901337
221. Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol (2008) 8(7):523–32. doi: 10.1038/nri2343
222. Hulme MA, Wasserfall CH, Atkinson MA, Brusko TM. Central role for interleukin-2 in type 1 diabetes. Diabetes (2012) 61(1):14–22. doi: 10.2337/db11-1213
223. Spolski R, Li P, Leonard WJ. Biology and regulation of IL-2: from molecular mechanisms to human therapy. Nat Rev Immunol (2018) 18(10):648–59. doi: 10.1038/s41577-018-0046-y
224. Spangler JB, Tomala J, Luca VC, Jude KM, Dong S, Ring AM, et al. Antibodies to Interleukin-2 Elicit Selective T Cell Subset Potentiation through Distinct Conformational Mechanisms. Immunity (2015) 42(5):815–25. doi: 10.1016/j.immuni.2015.04.015
225. Izquierdo C, Ortiz AZ, Presa M, Malo S, Montoya A, Garabatos N, et al. Treatment of T1D via optimized expansion of antigen-specific Tregs induced by IL-2/anti-IL-2 monoclonal antibody complexes and peptide/MHC tetramers. Sci Rep (2018) 8(1):8106. doi: 10.1038/s41598-018-26161-6
226. Tomala J, Chmelova H, Mrkvan T, Rihova B, Kovar M. In vivo expansion of activated naive CD8+ T cells and NK cells driven by complexes of IL-2 and anti-IL-2 monoclonal antibody as novel approach of cancer immunotherapy. J Immunol (2009) 183(8):4904–12. doi: 10.4049/jimmunol.0900284
227. Bell CJ, Sun Y, Nowak UM, Clark J, Howlett S, Pekalski ML, et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun (2015) 56:66–80. doi: 10.1016/j.jaut.2014.10.002
228. Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discovery (2019) 18(8):585–608. doi: 10.1038/s41573-019-0028-1
229. Kufer P, Lutterbuse R, Baeuerle PA. A revival of bispecific antibodies. Trends Biotechnol (2004) 22(5):238–44. doi: 10.1016/j.tibtech.2004.03.006
230. Topp MS, Stelljes M, Zugmaier G, Barnette P, Heffner LT Jr., Trippett T, et al. Blinatumomab retreatment after relapse in patients with relapsed/refractory B-precursor acute lymphoblastic leukemia. Leukemia (2018) 32(2):562–5. doi: 10.1038/leu.2017.306
231. Bhattacharya P, Fan J, Haddad C, Essani A, Gopisetty A, Elshabrawy HA, et al. A novel pancreatic beta-cell targeting bispecific-antibody (BsAb) can prevent the development of type 1 diabetes in NOD mice. Clin Immunol (2014) 153(1):187–98. doi: 10.1016/j.clim.2014.04.014
232. Jahn T, Zuther M, Friedrichs B, Heuser C, Guhlke S, Abken H, et al. An IL12-IL2-antibody fusion protein targeting Hodgkin’s lymphoma cells potentiates activation of NK and T cells for an anti-tumor attack. PLoS One (2012) 7(9):e44482. doi: 10.1371/journal.pone.0044482
233. Chatenoud L. A future for CD3 antibodies in immunotherapy of type 1 diabetes. Diabetologia (2019) 62(4):578–81. doi: 10.1007/s00125-018-4808-7
234. Hamad AR, Ahmed R, Donner T, Fousteri G. B cell-targeted immunotherapy for type 1 diabetes: What can make it work? Discovery Med (2016) 21(115):213–9.
235. Wang B, Gonzalez A, Benoist C, Mathis D. The role of CD8+ T cells in the initiation of insulin-dependent diabetes mellitus. Eur J Immunol (1996) 26(8):1762–9. doi: 10.1002/eji.1830260815
236. Carre A, Richardson SJ, Larger E, Mallone R. Presumption of guilt for T cells in type 1 diabetes: lead culprits or partners in crime depending on age of onset? Diabetologia (2021) 64(1):15–25. doi: 10.1007/s00125-020-05298-y
Keywords: diabetes, immunotherapy, monoclonal antibodies, immunoregulation, self-tolerance
Citation: Ke Q, Kroger CJ, Clark M and Tisch RM (2021) Evolving Antibody Therapies for the Treatment of Type 1 Diabetes. Front. Immunol. 11:624568. doi: 10.3389/fimmu.2020.624568
Received: 31 October 2020; Accepted: 31 December 2020;
Published: 18 February 2021.
Edited by:
Thomas Malek, University of Miami, United StatesReviewed by:
Sylvaine You, Institut National de la Santé et de la Recherche Médicale (INSERM), FranceCopyright © 2021 Ke, Kroger, Clark and Tisch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roland M. Tisch, cm10aXNjaEBtZWQudW5jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.