 Colleen S. Netherby-Winslow
Colleen S. Netherby-Winslow Katelyn N. Ayers
Katelyn N. Ayers Aron E. Lukacher
Aron E. Lukacher
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 27 January 2021
Sec. Immunological Memory
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.624144
This article is part of the Research TopicResident Memory T Cells: Guardians of the Balance of Local Immunity and PathologyView all 11 articles
Tissue-resident memory (TRM) CD8 T cells provide early frontline defense against regional pathogen reencounter. CD8 TRM are predominantly parked in nonlymphoid tissues and do not circulate. In addition to this anatomic difference, TRM are transcriptionally and phenotypically distinct from central-memory T cells (TCM) and effector-memory T cells (TEM). Moreover, TRM differ phenotypically, functionally, and transcriptionally across barrier tissues (e.g., gastrointestinal tract, respiratory tract, urogenital tract, and skin) and in non-barrier organs (e.g., brain, liver, kidney). In the brain, TRM are governed by a contextual milieu that balances TRM activation and preservation of essential post-mitotic neurons. Factors contributing to the development and maintenance of brain TRM, of which T cell receptor (TCR) signal strength and duration is a central determinant, vary depending on the infectious agent and modulation of TCR signaling by inhibitory markers that quell potentially pathogenic inflammation. This review will explore our current understanding of the context-dependent factors that drive the acquisition of brain (b)TRM phenotype and function, and discuss the contribution of TRM to promoting protective immune responses in situ while maintaining tissue homeostasis.
Development of long-lived T cell memory is vital to protection against microbial pathogens and cancer, and a goal of vaccination efforts. Initial work identified TCM which, like naive T cells, survey secondary lymphoid organs, and TEM, which circulate in the blood and non-lymphoid tissues. Because of their increased numbers over naïve T cell precursors to a particular antigen, and their lower threshold for activation and reduced dependence on costimulation, TCM and TEM respond rapidly to pathogen reencounter (1, 2). Nearly 20 years ago, evidence emerged supporting the idea that a population of memory T cells poised with an effector arsenal resided in non-lymphoid tissues (3). More recent evidence suggests that TRM, like TCM, are derived from a common naive T cell precursor after local antigen exposure (4). While sharing many effector capabilities with TEM, TRM differed from TEM in expression of trafficking molecules and having a distinct gene expression signature (5). The classification of TRM as a separate subset of CD8 T cell memory prompted new investigations to define the factors that contribute to TRM development and maintenance, how TRM-mediated immunity contributes to the dynamic immune response to microbial pathogens, and if TRM function can be harnessed for a multimodal therapeutic approach to treat or prevent infection and cancer.
An additional layer of complexity is that TRM are not a homogeneous subset, because tissue environments themselves impose tissue-specific heterogeneity to TRM. Most TRM characterization has been done in barrier tissues; far less is understood how TRM establish themselves in non-barrier sites. In particular, the brain and spinal cord are especially sensitive to tissue injury and loss from pro-inflammatory mediators. Mouse models of CNS infection, including by vesicular stomatitis virus (VSV), lymphocytic choriomeningitis virus (LCMV), Toxoplasma gondii, murine cytomegalovirus (MCMV), and mouse polyomavirus (MuPyV), have identified TRM in the brain that confer antigen-specific protection against reinfection (5–9). It is likely that brain-specific factors contribute to formation of TRM and their functional attributes due to the exquisite need to balance immune activation and tissue preservation in the CNS.
The trajectory of T cell differentiation is initiated by TCR engagement, then modified by costimulation and inflammation (10). The integration and duration of these signals directs a naïve T cell toward effector or memory fates, with peptide:MHC (pMHC) ligand-TCR interaction being the critical first step that guides the memory response. The strength of signal transduction events orchestrated after TCR binding with its cognate pMHC regulates induction of transcription factors, tissue-trafficking adhesion molecules, and cytokine receptors required for TRM generation. Thus, TCR signal strength per se dictates the quality and abundance of the resulting TRM population (11, 12). Additionally, regulating TCR signaling via inhibitory receptors, such as programmed cell death protein-1 [PD-1(CD279)], may be essential for TRM maintenance in particular tissues by operating as a rheostat to fine tune T cell activation and effector function. This review will focus on how TCR signaling shapes the TRM pool and how inhibitory receptor signaling drives the balance between effector function and long-term maintenance in tissues, an issue of especial importance in the CNS.
TRM are distinguished from circulating memory T cells by the expression of the integrins CD103 (αE subunit of the αEβ7 heterodimer) and CD49a (alpha subunit of the CD49a/CD29 heterodimer), as well as the C-type lectin CD69; these molecules act to direct and retain T cells in tissues (Figure 1). Additionally, TRM are phenotyped by the absence of cell surface sphingosine-1-phosphate receptor 1 (S1P1), the CCR7 chemokine receptor, and CD62L (L-selectin); these molecules contribute to T cell homing to (CCR7, CD62L) and egress from (S1P1) lymph nodes (13). The activating transcription factor Kruppel-like factor 2 (Klf2) targets the S1P1 gene and Klf2 downregulation is also used to define TRM (14). CD103 is a common marker for TRM due to its association with epithelial localization and tissue retention (15), but the requirements for CD103 expression for TRM development or maintenance is a topic of some debate (16).
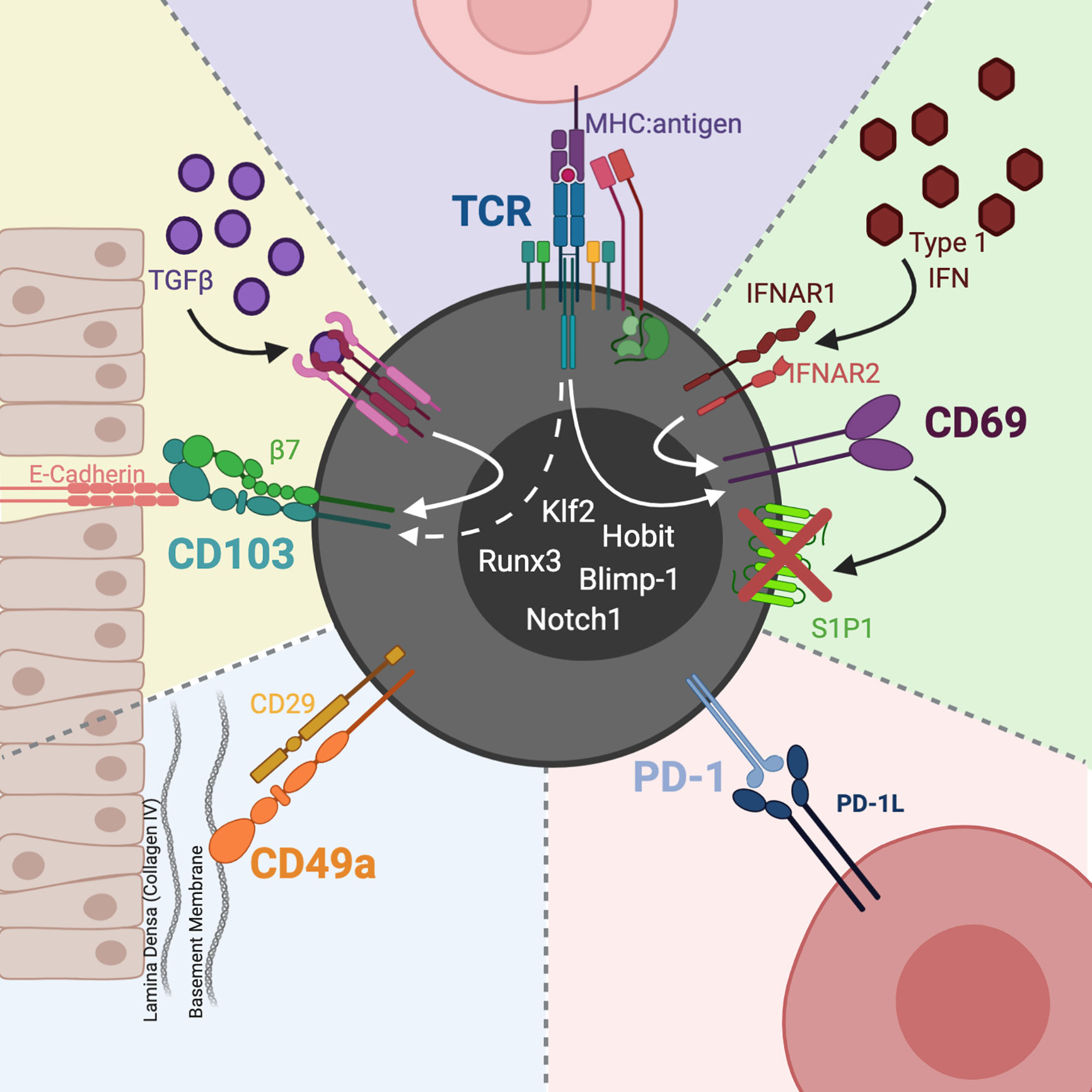
Figure 1 CD8 TRM phenotype and heterogeneity. CD103 is the receptor for the epithelial junction protein, E-cadherin. The CD103:E-cadherin interaction moors the T cell to the epithelial mucosa. TGFβ induces expression of CD103, whose levels may also be affected by TCR activation. CD49a partners with CD29 (integrin β1) to constitute the heterodimer VLA-1. VLA-1 binds collagen, with a predilection for Col IV in epithelial basement membranes. CD69 is a C-type lectin upregulated by type 1 IFNs as well as TCR activation. Once expressed, CD69 hinders TRM egress by complexing with S1P1, leading to S1P1s internalization and degradation. In particular sites, such as the CNS, TRM express PD-1 which acts to maintain functional TRM and preserve tissue homeostasis. Downregulation of Klf2 and upregulation of Blimp-1, Runx3, Notch1, and Hobit transcription factors have also been used to define TRM. Image created with BioRender.com.
A role for CD103 integrins in TRM retention in epithelial sites, like skin, lungs, salivary glands, and intestinal and female reproductive tract mucosa makes intuitive sense, due to its binding to the epithelial junction protein, E-cadherin. CD103 expressing T cells, however, can also be found in locations distant from epithelium, such as the brain and other non-barrier tissues; the function of CD103 expressed by TRM in these locations is unclear. Using peripheral infection and dendritic cell-mediated immunization, Urban et al. recently demonstrated that non-CNS infections generated CD8 bTRM. Notably, few of these CD8 bTRM expressed CD103 and donor CD103−/− CD8 T cells yielded CD8 bTRM at the same levels as donor WT cells (17). These data indicate that CD103 is dispensable for generating CD8 bTRM, which contrasts with the apparent requirement for CD103 for establishment of intestinal CD8 TRM (18).
To this point, CD103- TRM in the brain retain TRM migratory and phenotypic properties (e.g., being tissue-sessile, CD69+, and CD49a+) as well as TRM gene expression signatures (19). During persistent infection with MuPyV, a natural mouse pathogen, CD103+ bTRM are more efficient effectors (7), which is consistent with evidence of signaling from CD103:E-cadherin interactions enhancing CD8 TRM function, cytoskeleton reorganization, migration, cytokine release, and cytotoxicity (20–22). Although members of the cadherin family have been implicated in regulating neuron synaptic plasticity and flow cytometric analysis has shown E-cadherin expression on certain immune cells like dendritic cells and even some TRM (23–27), E-cadherin is predominantly expressed in epithelial tissues. With regard to CD103+ CD8 bTRM, however, there is little published data on E-cadherin expression in the brain, but it has been proposed that perhaps CD103+ brain CD8 T cells are interacting with E-cadherin-expressing immune cells rather than epithelial cells (16, 28). Aberrant expression of E-cadherin has also been associated with a more aggressive tumor subtype (28), but whether chronic inflammation or cancer alters E-cadherin expression in neural tissue is an open question. Alternatively, another ligand in the CNS may bind CD103 integrins expressed by CD8 bTRM. TGFβ is a well-documented inducer of CD103 on TRM (18). TGFβ receptor signaling acting concomitantly with TCR stimulation may modulate CD103 expression levels. This possibility raises the broader issue of whether TGFβ and pMHC availability act together or independently to affect TRM development, location, and function. Although CD103 expression seems to be specific to TRM, it is variably expressed by TRM in different tissues and is arguably dispensable for TRM functions. For example, CD103 blocking antibody does not negate the ability of lung CD8 TRM to protect mice from lethal influenza infection (29). Thus, the requirements for CD103 for CD8 bTRM maintenance, and the precise role TCR signaling plays in regulating CD103 expression warrants investigation. CD49a’s role in TRM development is less well defined than for CD103. CD49a does not directly attach to epithelia like CD103, but collagen IV (ColIV), its primary ligand, is positioned in the lamina densa layers of epithelial basement membranes (16, 30). The CD49a:ColIV interaction could then result in TRM localization to the epithelium and subsequent tethering to CD103:E-cadherin. Furthermore, in influenza infection CD49a protects lung CD8 TRM from apoptosis in part via interactions with collagen IV (31). A recent study shows that CD49a is required for TRM-mediated protection from lethal influenza pulmonary infection (29). In the skin, however, CD49a seems to influence the effector function of TRM, with CD49a+ CD8 TRM producing IFN-λ and CD8+ CD49a- TRM producing interleukin (IL)-17 (32). Although CD69 is often used as a marker of recent T cell activation, it is expressed by TRM in most tissues including those of the CNS (33). CD69 is also upregulated by type I interferons independent of TCR engagement (34). CD69 binds to and induces degradation of S1P1, which enables T cells to migrate along sphingosine-1-phosphate (S1P) gradients (SIP is higher in lymphatics than tissues). The expression profile for CD69, CD103, and CD49a, however, is not exclusive to nor is it uniform across TRM; disappointingly, there is no cleanly defined TRM phenotype (15).
Identifying TRM is made more challenging by evidence that TRM can be phenotypically heterogeneous even in the same organ (15). In mice intracranially (i.c.) inoculation with an attenuated LCMV variant, only ~50% of the bTRM are CD103+ (9). During persistent infection with MuPyV, the vast majority of virus-specific CD8 T cells in the brain are CD69+, but only ~40% expressed CD103 (19). In addition, the fraction of CD103- cells co-expressing CXCR5hi and TCF-1hi cells was higher than the CD103+ subpopulation. Elevation of both the transcription factor TCF-1 and the chemokine receptor CXCR5 on memory CD8 T cells has been linked to increased functional capability during chronic infection (35). This is noteworthy since in chronic viral infections TCF-1 and CXCR5 aid in establishing a population of proliferation-competent memory CD8 T cell precursors to maintain a pipeline leading to end-stage exhausted T cells (TEX) (36). The CD103+ and CD103- subsets, interestingly, expressed similar levels of Ki67 expression and antigen-stimulation IFN-γ production, indicating comparable proliferative and functional capabilities, respectively; however, the CD103+ subpopulation displayed higher effector activity (7, 19). A strategy to help reconcile these apparent discrepancies is to further stratify TRM by overlaying expression of additional transcriptome molecules and cytokine receptors linked to TRM differentiation, including Runx3, Notch, Hobit, and Blimp-1, as well as the receptors for IL-15, Type I IFN, TGF-β, and IL-12 (13, 37). Due to the phenotypic heterogeneity across TRM populations and shared markers with other CD8 T cell subsets, more in-depth “clustering” of these molecules may help not only to ensure that a T cell is a bona fide TRM but also to uncover additional breadth of TRM diversity between and within tissues.
An under-appreciated feature of TRM cells is the upregulation and maintenance of immune checkpoint molecules, particularly PD-1, in certain tissues and with particular viral infections (19, 38). TRM generated in the skin after HSV-1 infection or the brain following MuPyV infection have increased surface expression of multiple inhibitory receptors in addition to PD-1, but retain at least partial functionality (7, 39). PD-1 is transiently expressed by CD8 effector T cells after antigen receptor signaling, but even here PD-1 inhibits functionality (40). The appellation “persistent infection” as a catchall belies the complexity of lifecycles by viruses that co-reside long-term with their hosts, such as latency-reactivation by herpesviruses vs. smoldering infections by papillomavirus and polyomaviruses. Whether bona fide memory T cells develop in the setting of persistent infection is often debated. Often overlooked, however, is the nature of the persistent infection, which depending on level, location, and timing of epitope availability may allow co-habitation by both memory and effector T cells. Compounding this complexity is that some viruses previously thought to be completely cleared after acute infection (e.g., influenza, VSV) leave residual T cell epitope-bearing antigen-presenting cells (APCs) for several weeks (41–43). Unremitting strong TCR stimulation in neoplasia and chronic viremia arguably should be considered separately from transient/low-level persistent viral infections, as the former typically render CD8 T cells profoundly dysfunctional and direct them toward an adaptive state of differentiation termed TEX (44). Yet, even under these circumstances TEX exert antiviral activity as evidenced by the outgrowth of CD8 T cell epitope escape variants in HIV infection (45, 46). Although PD-1, as well as CTLA-4 and TIM-3, are upregulated and sustained on the surface of CD8 T cells infiltrating tumors and in chronically infected tissues, these T cells can express molecules and gene signatures shared with TRM (47, 48). Similar to its role in checking T cell-mediated autoimmunity, checkpoint inhibitors mitigate T cell-mediated immunopathology (19, 38, 49, 50). PD-1 expression as well as its role in the cell’s functional adaptivity may distinguish TRM from other memory CD8 T cell subsets that infiltrate the CNS (19, 38, 48).
TCR signaling has been implicated in the formation of a diverse memory pool. From its initial description in the early 1980s (51), extensive research has been conducted on how signals induced when the TCR engages the pMHC complex directs effector memory differentiation and function. The relative “strength” of the TCR signal is the composite of affinity of the pMHC ligand for its cognate TCR, the amount of antigen presented on the surface of the APC (i.e., pMHC epitope density), the number of cell surface TCRs, and the duration of the TCR:pMHC interactions (52–54). The prevailing model holds that activation through the TCR orchestrates an instructional program that directs CD8 T cell expansion, effector differentiation, contraction and memory formation (55). In addition, co-stimulation through CD28, CD27, CD40, 4-1BB, and/or ICOS during priming of naïve T cells further tailors T cell fate (56–60). Cytokine input complements TCR activation to select differentiation programs and T cell longevity. For example, IL-12 promotes effector function and survival (61, 62), and IL-15 supports homeostatic maintenance of memory T cells (63–65). Kaech and colleagues have shown that a critical determinant whether a naive T cell becomes a short-lived effector cell (SLEC) or a memory precursor effector cell (MPEC) is the amount of IL-12 present during naïve T cell priming (66). IL-12 was found to regulate the level of expression of the T-box transcription factor T-bet (Tbx21) in a dose-dependent manner; high levels of T-bet instructed cells to become SLECs, and low T-bet expression favored MPEC development. Together with strength of TCR signaling, a complex tapestry of inflammatory signals and co-stimulation coalesce to influence the size and durability of a T cell memory response.
TCR signal strength also quantitatively and qualitatively shapes memory T cell differentiation. Disruption in TCR proximal signaling in vivo by mutating SLP-76 caused impaired Ca2+ influx and dampened T cell activation, without disrupting the expansion of CD8 T cells in response to acute LCMV infection (67). Weaker TCR stimulation in SLP-76 mutant mice biased CD8 T cells toward memory differentiation, with weak TCR stimulation favoring the production of cells with a CD62Lhi TCM phenotype. Our group found that CD8 bTRM generated during persistent MuPyV infection possess high-affinity TCRs compared to counterparts in the spleen and kidney. Because virus-specific CD8 TEFF also express high-affinity TCRs, we suggested that these cells were the progeny of high-affinity effectors recruited to the brain during the acute stage of infection (68). Indeed, we observed that there is a window of opportunity for immune cells, and possibly virus, to breach a blood-brain barrier rendered permeable during acute MuPyV encephalitis (69). A plausible possibility is that high-affinity TCRs enable CD8 bTRM to detect low levels of viral antigen during persistent infection (68).
During MuPyV infection, our group reported that weaker TCR stimulation favored expansion of CD8 bTRM having superior ability to respond to homologous MuPyV i.c. re-infection (11). Using site-directed mutagenesis to alter a subdominant epitope in a nonstructural viral protein of MuPyV, Maru et al. generated a panel of viruses with non-synonymous mutations in a CD8 T cell epitope to assess in vivo the impact of TCR stimulation strength per se on bTRM differentiation. By using adoptively transferred CD8 T cells from a TCR transgenic mouse recognizing a subdominant epitope, these authors controlled the size, recruitment, and clonality of the naïve T cell response, and circumvented the confounding problems of changes in virus levels and inflammation over the course of infection. Although CD8 bTRM generated in a setting of suboptimal TCR stimulation enjoyed a more robust ability to expand upon pathogen reencounter, no impact on effector function was observed. Similarly, Langlois and colleagues reported an advantage in forming influenza-specific lung CD8 TRM after stimulation with low-affinity epitopes (12). Here, TCR transgenic OT-I CD8 T cells (specific for the H-2Kb-restricted SIINFEKL peptide from chicken ovalbumin residues 257–264) were adoptively transferred to mice infected with a recombinant influenza virus encoding native and altered OT-I epitopes. Although high- and low-affinity stimulated OT-I TRM had similar phenotype and function, transcriptional profiling revealed that TRM generated by low-affinity stimulation expressed increased pro-survival factors, which would favor long-term maintenance in tissues. CD8 bTRM having high-affinity TCRs would likely be selected by suboptimal TCR stimulation allowing them to engage low-density epitopes or epitopes modified to limit binding to TCRs (70). The level and duration of TCR stimulation, in concert with tissue-specific cytokines, may result in upregulation of inhibitory receptors on CD8 TRM to modulate their TCR signal strength, and thereby control their effector capabilities and survival (7, 71).
Unchecked T cell activation can cause autoimmunity and immunopathology. To prevent this, inhibitory receptors constrain T cell effector functions and proliferation following TCR engagement and are upregulated in chronic infection and cancer, with the level of expression and number of inhibitory receptors dictated by the density and duration of cognate epitope (72). The importance of PD-1 and other inhibitory receptors in mitigating T cell function and prolonging longevity are well-established in animal models and humans, where blockade of PD-1 or PD-L1 reinvigorates T cell responses, reduces viral load, and/or boosts tumor control. PD-1 primarily regulates T cell activity by dampening intracellular stimulatory signals from the TCR/CD3 complex. When the PD-1 monomeric receptor engages its ligands PD-L1 (CD274)/PD-L2 (CD273), its cytoplasmic immunoreceptor tyrosine-based inhibition motif (ITIM) and immunoreceptor tyrosine-based switch motif (ITSM) domains are phosphorylated, resulting in binding by the Src homology 2 domain-containing phosphatase 2 (SHP2) (73). Subsequent SHP2 activation leads to tyrosine dephosphorylation of signaling molecules downstream of TCR and costimulatory receptors (74). PD-1 signaling can also result in metabolic reprograming; e.g., PD-1 signaling reduces Akt activity, suppressing mTOR (75). This effectively switches T cell metabolism from glycolysis to fatty acid oxidation (FAO). TRM have a dynamic metabolic profile, but predominantly utilize oxidative phosphorylation (76). Skin CD8 TRM make use of exogenous fatty acids for FAO (77). Whether CD8 bTRM share this metabolic pathway remains to be determined.
PD-1 expression by CD8 TRM appears to be dependent on the tissue environment and the nature of the viral infection. What governs the stability of PD-1 expression and its role in TRM function and maintenance is an area of active interest. In VSV infection, CD8 bTRM express low levels of PD-1 transcripts but no detectable PD-1 protein, whereas bTRM from mice infected with mouse cytomegalovirus (MCMV) or MuPyV are PD-1hi (5, 6, 19, 78). Youngblood et al. established that the PD-1 promoter is dynamically epigenetically regulated, with the extent of demethylation of the PD-1 promoter correlating with the strength and duration of TCR stimulation. During acute LCMV infection, the PD-1 promoter is extensively demethylated and then remethylated upon viral clearance. During chronic LCMV infection, the PD-1 promoter remains demethylated in viral antigen-specific CD8 T cells (79). In MuPyV encephalitis, the PD-1 promoter is likewise heavily demethylated in bTRM, and undergoes only a partial remethylation in virus-specific T cells in the spleen (19). Interestingly, maintenance of PD-1 expression on MuPyV-specific CD8 bTRM was found to be independent of cognate antigen or inflammation (19). In contrast, PD-1hi CD8 TRM in the lungs of influenza-infected mice are maintained by MHC class I signaling and CD80 and CD86 costimulation (80). PD-1 may serve to dampen the level of TCR signaling in CD8 bTRM, allowing them to exert some antiviral activity and avoid apoptosis.
Because antigen is required for CD8 bTRM formation but not PD-1 maintenance, it is possible that PD-1 is an important regulator of TRM function specifically in the brain microenvironment. Memory CD8 T cells in the eye, an immune privileged organ, also express PD-1 (81). In a mouse model of coronavirus CNS infection, PD-1 expression on CD8 T cells limits immune pathology and axonal damage (82, 83). The concept that PD-1 expression plays an important regulatory role in the brain is strengthened by evidence that splenic CD8 TRM lack PD-1 expression during persistent MuPyV infection and that PD-L1 blockade limits CD8 bTRM effector function. bTRM produce IFN-γ, which regulates microglial function (84). It is also possible that microglia in turn regulate TRM homeostasis through PD-1:PD-L1 interaction. A complete understanding how PD-1 regulates deleterious CD8 bTRM activation in the setting of persistent viral encephalitides or whether PD-1 may selectively inhibit neuropathological effector activities remains unclear.
CD8 T cells expressing a TRM phenotype (CD69, CD103) and PD-1 progressively accumulate in the brain parenchyma with aging. Cerebral ischemia promotes production of inflammatory mediators by these CD8 bTRM (85). Clonally expanded CD8 T cells with gene signatures for cytokine-producing effector memory cells expressing CD69 and VLA-1/-4 transcripts accumulate in the subventricular zone (SVZ) of aged brains, a neurogenic niche containing neural stem cells (NSC), neural progenitor cells (NPC) and microglia; notably, IFN-γ secreted by CD8 T cells inhibits proliferation of NSCs and NPCs (86). In MuPyV encephalitis, virus-specific CD8 T cells aggregate in the SVZ subjacent to infected ependyma and produce IFN-γ in situ (69, 87). It is tempting to speculate that SVZ-localized antiviral CD8 bTRM produce IFN-γ, which is deleterious to neurogenic niches and contributes to cognitive decline in survivors of the life-threatening brain demyelinating disease progressive multifocal leukoencephalopathy (PML) caused by the JC polyomavirus (JCPyV). Following recovery from neuropathic flavivirus infection, IFN-γ from CD8 bTRM has also been show to drive microglia to eliminate synapses in the hippocampus and cause spatial-learning defects (84). These findings raise the ominous spectre that activation of JCPyV-specific CD8 bTRM after PD-1 blockade may compromise learning and memory in PML survivors.
Although PD-1 is highly expressed by CD8 bTRM during encephalitis by MuPyV and MCMV (7, 19, 88, 89), these bTRM do not display a clear exhaustion profile (19, 90, 91). Rather, PD-1 appears to operate in the brain primarily to balance bystander- and virus-induced inflammation and tissue damage against virus control by antiviral bTRM cells (90, 91). In the pancreas, PD-1 ligand-expressing macrophages control the function of the PD-1+ CD8 TRM cells. PD-1 blockade of pancreatic CD8 TRM cells significantly augmented their ability to produce IFN-α, TNF-α, and IL-2 upon TCR stimulation (90). In the lung, PD-L1 blockade promoted the expansion of TRM and enhanced secondary protection to influenza infection, but also resulted in the development of inflammation-induced fibrotic injury (80). These results are mirrored in the brain. bTRM in MuPyV-infected PD-L1−/− mice had a higher frequency of IFN-γ-producing cells than bTRM from MuPyV-infected wild type (WT) mice (91). Furthermore, PD-1:PD-L1 interactions were found to quell inflammation in the pancreas and brain (90, 91). CD8 TRM are detected in brains of patients dying of non-neurological causes. Interestingly, these TRM are CD103+ CD69+ and highly express PD-1 and CTLA-4 (92). bTRM in healthy human brains may be telltale signs of long-resolved infections. These bTRM may also provide the “fertile field” for CNS autoimmune diseases, such as multiple sclerosis by secreting chemokines that attract circulating self-reactive T cells (93). Thus, expression of checkpoint inhibitory receptors, such as PD-1, may act to halt production of such chemokines and the potential for CNS autoimmune diseases. PD-L1 expression by MHC-I/II-expressing CNS-resident cells (e.g., microglia) may, in turn, be critical determinants of susceptibility to CNS autoimmunity. Collectively, these data support the likelihood that CD8 TRM in the brain retain expression of checkpoint inhibitory molecules to limit tissue-injurious inflammation and preserve CNS integrity.
With the heightened effector functionality of TRM consequent to interrupting PD-1 signaling, PD-1 or PD-L1 blockade could be anticipated to enhance TRM response against persistently infecting viral pathogens. In a small randomized and placebo-controlled study, 3 out of 6 patients with hepatitis C virus given a new humanized ligand-blocking PD-1 antibody exhibited 4-log reductions in viral load, but this was associated with immunologic adverse events, including autoimmune thyroiditis (94). In a phase Ib study of patients with chronic hepatitis B virus (HBV) infection, nearly all of the patients given a single infusion of the PD-1 blocking antibody nivolumab experienced a decrease in HBV surface antigen (HBsAg) titers (95). Finally, in individuals with PML, a significant number of patients receiving anti-PD-1 had fewer cerebrospinal fluid JCPyV genome copies, elevated JCPyV-specific CD4 and CD8 T cell responses, and importantly, clinical improvement or disease stabilization (96, 97). A likely critical variable in the success of PD-1 blockade therapy is the severity of infection at the time of therapy initiation, with higher viral burden being associated with greater risk of immune-mediated complications. Although these studies do not directly assign effects of the PD-1:PD-1L blockade to bTRM, they demonstrate the importance of checkpoint inhibitor blockade as an anti-viral therapy in humans. Knowing that bTRM have increased effectivity in mouse models lacking either PD-1 or PD-L1, a plausible hypothesis is that the antiviral effects of the PD-1:PD-1L blockade in humans could be due to resurrected effector activity by bTRM.
Beyond affecting the functional capabilities of TRM cells, recent reports suggest that PD-1 is involved in the development of TRM in different tissues, including those in the CNS. During MCMV infection, CD103+ CD69+ bTRM populations were sparse in PD-L1−/− and PD-1−/− mice compared to WT mice, implicating PD-1 signaling as a positive factor in development of bTRM (89). PD-1 is involved in governing T cell activation, fate, function, and tolerance as well as immune homeostasis (98). Therefore, using a global PD-1 knock-out system could have altered the fate of all T cell subsets and not just that of the bTRM. Conversely, in response to MuPyV, a higher frequency of CD103+ CD8 T cells populations were observed in brains of PD-L1−/− mice as well as in mice treated with PD-1 blocking antibodies compared to the WT mice (91). These conflicting findings raise the caveat that PD-1’s role in the CNS can differ between viral infections and highlight the need for caution in extrapolating conclusions of immune responses across infection models. By extension, understanding how PD-1 controls TRM development in different CNS viral infections should uncover novel insights in mechanisms of détente between viral control and collateral tissue injury by CD8 bTRM.
Accumulating evidence supports the concept that TRM progenitors are generated early in the course of effector differentiation. An intriguing possibility is that factors such as TCR signal strength or differential expression of inhibitory receptors contributes to a nuanced differentiation spectrum that guides development of TRM. Similar ideas hold true for TEX. Recent work reveals that TEX exist as a continuum from self-renewing “stem-like” progenitors that progress to a nonproliferative terminal state which is vulnerable to death. TEX at different stages vary in their ability to respond to immune checkpoint blockade therapy (36). MuPyV-specific CD8 bTRM heterogeneously express many molecules associated with TEX subsets (36, 87). Single-cell analysis of adaptive immune cells in ulcerative colitis patients suggests that transcriptional heterogeneity also exists in the TRM compartment and its demarcation into distinct differentiation stages (99). Similarly, lung CD8 TRM generated to influenza infection exhibit both exhausted and memory characteristics by phenotype, transcriptome, and function (80). The proportion of TRM in each stage of differentiation, however, will certainly be altered by disease processes and possibly by immunomodulatory regimens as well. Recent work also demonstrates that the quality of functional CD8 TRM responses in the influenza-infected lung is dependent on the type of cell presenting viral antigens (100). Furthermore, TRM can also egress from tissues, convert into other memory subsets, and change their migratory behavior depending on the inflammatory context (101, 102). Together these findings contribute to an increasingly multidimensional view of the factors that drive TRM formation, what constitutes tissue residence, and the role TRM play in antiviral defense. Particularly important for persistent neurotropic viruses is to develop a comprehensive understanding how bTRM balance virus control against neuropathology and to learn how this equilibrium is established for different viral infections.
CN-W wrote the original draft and revised the manuscript. KA wrote the original draft, revised the manuscript, and prepared the figure. AL revised this manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the National Institute of Neurological Disorders and Stroke and the National Cancer Institute grants R01NS088367 and R01NS092662 to AL, F32NS106730 to CN-W, and T32CA060395 to KA.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors thank Matthew Lauver and Sarah Carey for thoughtful comments and review of the manuscript.
APC, antigen-presenting cell; bTRM, brain tissue-resident memory CD8 T cell; CNS, central nervous system; ColIV, collagen IV; FAO, fatty acid oxidation; i.c., intracranial; ICOS, inducible T-cell costimulator; IL, interleukin; ITIM, immunoreceptor tyrosine-based inhibition motif; ITSM, immunoreceptor tyrosine-based switch motif; Klf2, Kruppel-like factor 2; LCMV, lymphocytic choriomeningitis virus; MCMV, mouse cytomegalovirus; MHC, major histocompatibility complex; MPEC, memory precursor effector cell; MuPyV, mouse polyomavirus; PD-1, programmed cell death protein-1; PD-L1, programmed death-ligand 1; pMHC, peptide:MHC complex; PML, progressive multifocal leukoencephalopathy; S1P, sphingosine-1-phosphate; S1P1, sphingosine-1-phosphate receptor 1; SHP2, Src homology 2 domain-containing phosphatase 2; SLEC, short lived effector cell; TCM, central memory T cell; TEM, effector memory T cell; TEX, exhausted T cell; TRM, tissue-resident memory T cell; TCR, T cell receptor; VSV, vesicular stomatitis virus; WT, wild type.
1. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature (1999) 401:708–12. doi: 10.1038/44385
2. von Andrian UH, Mackay CR. T-cell function and migration. Two sides of the same coin. N Engl J Med (2000) 343:1020–34. doi: 10.1056/NEJM200010053431407
3. Masopust D, Vezys V, Marzo AL, Lefrancois L. Preferential localization of effector memory cells in nonlymphoid tissue. Science (2001) 291:2413–7. doi: 10.1126/science.1058867
4. Gaide O, Emerson RO, Jiang X, Gulati N, Nizza S, Desmarais C, et al. Common clonal origin of central and resident memory T cells following skin immunization. Nat Med (2015) 21:647–53. doi: 10.1038/nm.3860
5. Wakim LM, Woodward-Davis A, Liu R, Hu Y, Villandangos J, Smyth G, et al. The molecular signature of tissue resident memory CD8 T cells isolated from the brain. J Immunol (2012) 189:3462–71. doi: 10.4049/jimmunol.1201305
6. Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A (2010) 107:17872–9. doi: 10.1073/pnas.1010201107
7. Mockus TE, Shwetank, Lauver MD, Ren HM, Netherby CS, Salameh T, et al. CD4 T cells control development and maintenance of brain-resident CD8 T cells during polyomavirus infection. PloS Pathog (2018) 14:e1007365. doi: 10.1371/journal.ppat.1007365
8. Landrith TA, Sureshchandra S, Rivera A, Jang JC, Rais M, Nair MG, et al. CD103+ CD8 T cells in the Toxoplasma-infected brain exhibit a tissue-resident memory transcriptional profile. Front Immunol (2017) 8:335. doi: 10.3389/fimmu.2017.00335
9. Steinbach K, Vincenti I, Kreutzfeldt M, Page N, Muschaweckh A, Wagner I, et al. Brain-resident memory T cells represent an autonomous cytotoxic barrier to viral infection. J Exp Med (2016) 213:1571–87. doi: 10.1084/jem.20151916
10. Cui W, Kaech SM. Generation of effector CD8+ T cells and their conversion to memory T cells. Immunol Rev (2010) 236:151–66. doi: 10.1111/j.1600-065X.2010.00926.x
11. Maru S, Jin G, Schell TD, Lukacher AE. TCR stimulation strength is inversely associated with establishment of functional brain-resident memory CD8 T cells during persistent viral infection. PloS Pathog (2017) 13:e1006318. doi: 10.1371/journal.ppat.1006318
12. Fiege JK, Stone IA, Fay EJ, Markman MW, Wijeyesinghe S, Macchietto MG, et al. The impact of TCR signal strength on resident memory T cell formation during influenza virus infection. J Immunol (2019) 203:936–45. doi: 10.4049/jimmunol.1900093
13. Mami-Chouaib F, Tartour E. Editorial: Tissue resident memory T cells. Front Immunol (2019) 10:1018. doi: 10.3389/fimmu.2019.01018
14. Skon CN, Lee JY, Anderson KG, Masopust D, Hogquist KA, Jameson SC. Transcriptional downregulation of S1pr1 is required for the establishment of resident memory CD8+ T cells. Nat Immunol (2013) 14:1285–93. doi: 10.1038/ni.2745
15. Steinbach K, Vincenti I, Merkler D. Resident-memory T cells in tissue-restricted immune Rresponses: for better or worse? Front Immunol (2018) 9:2827. doi: 10.3389/fimmu.2018.02827
16. Topham DJ, Reilly EC. Tissue-resident memory CD8+ T cells: From phenotype to function. Front Immunol (2018) 9:515. doi: 10.3389/fimmu.2018.00515
17. Urban SL, Jensen IJ, Shan Q, Pewe LL, X HH, Badovinac VP, et al. Peripherally induced brain tissue-resident memory CD8+ T cells mediate protection against CNS infection. Nat Immunol (2020) 21:938–49. doi: 10.1038/s41590-020-0711-8
18. Casey KA, Fraser KA, Schenkel JM, Moran A, Abt MC, Beura LK, et al. Antigen-independent differentiation and maintenance of effector-like resident memory T cells in tissues. J Immunol (2012) 188:4866–75. doi: 10.4049/jimmunol.1200402
19. Shwetank, Abdelsamed HA, Frost EL, Schmitz HM, Mockus TE, Youngblood BA, et al. Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol Cell Biol (2017) 95:953–9. doi: 10.1038/icb.2017.62
20. Corgnac S, Boutet M, Kfoury M, Naltet C, Mami-Chouaib F. The emerging role of CD8+ tissue resident memory T (TRM) cells in antitumor immunity: a unique functional contribution of the CD103 integrin. Front Immunol (2018) 9:1904. doi: 10.3389/fimmu.2018.01904
21. Le Floc’h A, Jalil A, Franciszkiewicz K, Validire P, Vergnon I, Mami-Chouaib F. Minimal engagement of CD103 on cytotoxic T lymphocytes with an E-cadherin-Fc molecule triggers lytic granule polarization via a phospholipase Cg-dependent pathway. Cancer Res (2011) 71:328–38. doi: 10.1158/0008-5472.CAN-10-2457
22. Le Floc’h A, Jalil A, Vergnon I, Le Maux Chansac B, Lazar V, Bismuth G, et al. aEb7 integrin interaction with E-cadherin promotes antitumor CTL activity by triggering lytic granule polarization and exocytosis. J Exp Med (2007) 204:559–70. doi: 10.1084/jem.20061524
23. Borkowski TA, Van Dyke BJ, Schwarzenberger K, McFarland VW, Farr AG, Udey MC. Expression of E-cadherin by murine dendritic cells: E-cadherin as a dendritic cell differentiation antigen characteristic of epidermal Langerhans cells and related cells. Eur J Immunol (1994) 24:2767–74. doi: 10.1002/eji.1830241129
24. Siddiqui KR, Laffont S, Powrie F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity (2010) 32:557–67. doi: 10.1016/j.immuni.2010.03.017
25. Hofmann M, Pircher H. E-cadherin promotes accumulation of a unique memory CD8 T-cell population in murine salivary glands. Proc Natl Acad Sci U S A (2011) 108:16741–6. doi: 10.1073/pnas.1107200108
26. Tang L, Hung CP, Schuman EM. A role for the cadherin family of cell adhesion molecules in hippocampal long-term potentiation. Neuron (1998) 20:1165–75. doi: 10.1016/S0896-6273(00)80497-3
27. Hirano S, Takeichi M. Cadherins in brain morphogenesis and wiring. Physiol Rev (2012) 92:597–634. doi: 10.1152/physrev.00014.2011
28. Lewis-Tuffin LJ, Rodriguez F, Giannini C, Scheithauer B, Necela BM, Sarkaria JN, et al. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PloS One (2010) 5:e13665. doi: 10.1371/journal.pone.0013665
29. Reilly EC, Emo KL, Buckley PM, Reilly NS, Smith I, Chaves FA, et al. TRM integrins CD103 and CD49a differentially support adherence and motility after resolution of influenza virus infection. Proc Natl Acad Sci U S A (2020) 117:12306–14. doi: 10.1073/pnas.1915681117
30. Kuhn K. Basement membrane (type IV) collagen. Matrix Biol (1995) 14:439–45. doi: 10.1016/0945-053X(95)90001-2
31. Ray SJ, Franki SN, Pierce RH, Dimitrova S, Koteliansky V, Sprague AG, et al. The collagen binding a1b1 integrin VLA-1 regulates CD8 T cell-mediated immune protection against heterologous influenza infection. Immunity (2004) 20:167–79. doi: 10.1016/S1074-7613(04)00021-4
32. Cheuk S, Schums H, Sérézal IG, Martini E, Chiang SC, Marquardt N, et al. CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity (2017) 46:287–300. doi: 10.1016/j.immuni.2017.01.009
33. Cibrian D, Sanchez-Madrid F. CD69: from activation marker to metabolic gatekeeper. Eur J Immunol (2017) 47:946–53. doi: 10.1002/eji.201646837
34. Shiow LR, Rosen H, Brdičková N, Xu Y, An J, Lanier LL, et al. CD69 acts downstream of interferon-a/b to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature (2006) 440:540–4. doi: 10.1038/nature04606
35. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol (2016) 1:eaai8593. doi: 10.1126/sciimmunol.aai8593
36. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental relationships of four exhausted CD8+ T cell subsets reveals underlying transcriptional and epigenetic landscape control mechanisms. Immunity (2020) 52:825–841 e828. doi: 10.1016/j.immuni.2020.04.014
37. Behr FM, Chuwonpad A, Stark R, van Gisbergen K. Armed and Ready: Transcriptional regulation of tissue-resident memory CD8 T cells. Front Immunol (2018) 9:1770. doi: 10.3389/fimmu.2018.01770
38. Clarke J, Panwar B, Madrigal A, Singh D, Gujar R, Wood O, et al. Single-cell transcriptomic analysis of tissue-resident memory T cells in human lung cancer. J Exp Med (2019) 216:2128–49. doi: 10.1084/jem.2019249
39. Park SL, Zaid A, Liang Hor J, Christo SN, Prier JE, Davies B, et al. Local proliferation maintains a stable pool of tissue-resident memory T cells after antiviral recall responses. Nat Immunol (2018) 19:183–91. doi: 10.1038/s41590-017-0027-5
40. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD-1 during effector CD8 T cell differentiation. Proc Natl Acad Sci U S A (2018) 115:4749–54. doi: 10.1073/pnas.1718217115
41. Jelley-Gibbs DM, Brown DM, Dibble JP, Haynes L, Eaton SM, Swain SL. Unexpected prolonged presentation of influenza antigens promotes CD4 T cell memory generation. J Exp Med (2005) 202:697–706. doi: 10.1084/jem.20050227
42. Kim TS, Hufford MM, Sun J, Fu YX, Braciale TJ. Antigen persistence and the control of local T cell memory by migrant respiratory dendritic cells after acute virus infection. J Exp Med (2010) 207:1161–72. doi: 10.1084/jem.20092017
43. Turner DL, Cauley LS, Khanna KM, Lefrancois L. Persistent antigen presentation after acute vesicular stomatitis virus infection. J Virol (2007) 81:2039–46. doi: 10.1128/JVI.02167-06
45. Allen TM, Altfeld M, Geer SC, Kalife ET, Moore C, O’Sullivan KM, et al. Selective escape from CD8+ T-cell responses represents a major driving force of human immunodeficiency virus type 1 (HIV-1) sequence diversity and reveals constraints on HIV-1 evolution. J Virol (2005) 79:13239–49. doi: 10.1128/JVI.79.21.13239-13249.2005
46. Bronke C, Almeida CAM, McKinnon E, Roberts SG, Keane NM, Chopra A, et al. HIV escape mutations occur preferentially at HLA-binding sites of CD8 T-cell epitopes. AIDS (2013) 27:899–905. doi: 10.1097/QAD.0b013e32835e1616
47. Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, et al. Human tissue-resident memory T cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep (2017) 20:2921–34. doi: 10.1016/j.celrep.2017.08.078
48. Petrelli A, Mijnheer G, Hoytema van Konijnenburg DP, van der Wal MM, Giovannone B, Mocholi E, et al. PD-1+ CD8+ T cells are clonally expanding effectors in human chronic inflammation. J Clin Invest (2018) 128:4669–81. doi: 10.1172/JCI96107
49. Francisco LM, Sage PT, Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev (2010) 236:219–42. doi: 10.1111/j.1600-065X.2010.00923.x
50. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol (2008) 26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331
51. Allison JP, McIntyre BW, Bloch D. Tumor-specific antigen of murine T-lymphoma defined with monoclonal antibody. J Immunol (1982) 129:2293–300.
52. Corse E, Gottschalk RA, Allison JP. Strength of TCR-peptide/MHC interactions and in vivo T cell responses. J Immunol (2011) 186:5039–45. doi: 10.4049/jimmunol.1003650
53. Kuhns MS, Davis MM. TCR signaling emerges from the sum of many parts. Front Immunol (2012) 3:159. doi: 10.3389/fimmu.2012.00159
54. Daniels MA, Teixeiro E. TCR signaling in T cell memory. Front Immunol (2015) 6:617. doi: 10.3389/fimmu.2015.00617
55. Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr Opin Immunol (2004) 16:217–25. doi: 10.1016/j.coi.2004.02.005
56. Hendriks J, Gravestein LA, Tesselaar K, van Lier RAW, Schumacher TNM, Borst J. CD27 is required for generation and long-term maintenance of T cell immunity. Nat Immunol (2000) 1:433–40. doi: 10.1038/80877
57. Hendriks J, Xiao Y, Borst J. CD27 promotes survival of activated T cells and complements CD28 in generation and establishment of the effector T cell pool. J Exp Med (2003) 198:1369–80. doi: 10.1084/jem.20030916
58. Takahashi C, Mittler RS, Vella AT. Cutting edge: 4-1BB is a bona fide CD8 T cell survival signal. J Immunol (1999) 162:5037–40.
59. Wallin JJ, Liang L, Bakardjiev A, Sha WC. Enhancement of CD8+ T cell responses by ICOS/B7h costimulation. J Immunol (2001) 167:132–9. doi: 10.4049/jimmunol.167.1.132
60. Liu X, Bai XF, Wen J, Gao JX, Liu J, Lu P, et al. B7H costimulates clonal expansion of, and cognate destruction of tumor cells by, CD8+ T lymphocytes in vivo. J Exp Med (2001) 194:1339–48. doi: 10.1084/jem.194.9.1339
61. Curtsinger JM, Lins DC, Mescher MF. Signal 3 determines tolerance versus full activation of naive CD8 T cells: dissociating proliferation and development of effector function. J Exp Med (2003) 197:1141–51. doi: 10.1084/jem.20021910
62. Curtsinger JM, Johnson CM, Mescher MF. CD8 T cell clonal expansion and development of effector function require prolonged exposure to antigen, costimulation, and signal 3 cytokine. J Immunol (2003) 171:5165–71. doi: 10.4049/jimmunol.171.10.5165
63. Goldrath AW, Sivakumar PV, Glaccum M, Kennedy MK, Bevan MJ, Benoist C, et al. Cytokine requirements for acute and basal homeostatic proliferation of naive and memory CD8+ T cells. J Exp Med (2002) 195:1515–22. doi: 10.1084/jem.20020033
64. Becker TC, Wherry EJ, Boone D, Murali-Krishna K, Antia R, Ma A, et al. Interleukin 15 is required for proliferative renewal of virus-specific memory CD8 T cells. J Exp Med (2002) 195:1541–8. doi: 10.1084/jem.20020369
65. Judge AD, Zhang X, Fujii H, Surh CD, Sprent J. Interleukin 15 controls both proliferation and survival of a subset of memory-phenotype CD8+ T cells. J Exp Med (2002) 196:935–46. doi: 10.1084/jem.20020772
66. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation directs memory precursor and short-lived effector CD8+ T cell fates via the graded expression of T-bet transcription factor. Immunity (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
67. Smith-Garvin JE, Burns JC, Gohil M, Zou T, Kim JS, Maltxman JS, et al. T-cell receptor signals direct the composition and function of the memory CD8+ T-cell pool. Blood (2010) 116:5548–59. doi: 10.1182/blood-2010-06-292748
68. Frost EL, Kersh AE, Evavold BD, Lukacher AE. Cutting Edge: Resident memory CD8 T cells express high-affinity TCRs. J Immunol (2015) 195:3520–4. doi: 10.4049/jimmunol.1501521
69. Mockus TE, Netherby-Winslow CS, Atkins HM, Lauver MD, Jin G, Ren HM, et al. CD8 T cells and STAT1 signaling are essential codeterminants in protection from polyomavirus encephalopathy. J Virol (2020) 94. doi: 10.1128/JVI.02038-19
70. Martinez RJ, Evavold BD. Lower affinity T cells are critical components and active participants of the immune response. Front Immunol (2015) 6:468. doi: 10.3389/fimmu.2015.00468
71. Bally AP, Austin JW, Boss JM. Genetic and epigenetic regulation of PD-1 expression. J Immunol (2016) 196:2431–7. doi: 10.4049/jimmunol.1502643
72. Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol (2015) 15:486–99. doi: 10.1038/nri3862
73. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular mechanism of SHP2 activation by PD-1 stimulation. Sci Adv (2020) 6:eaay4458. doi: 10.1126/sciadv.aay4458
74. Fernandes RA, Su L, Nishiga Y, Ren J, Bhuiyan AM, Cheng N, et al. Immune receptor inhibition through enforced phosphatase recruitment. Nature (2020) 586:779–84. doi: 10.1038/s41586-020-2851-2
75. Saeidi A, Zandi K, Cheok YY, Saeidi H, Wong WF, Lee CYQ, et al. T-cell exhaustion in chronic infections: reversing the state of exhaustion and reinvigorating optimal protective immune responses. Front Immunol (2018) 9:2569. doi: 10.3389/fimmu.2018.02569
76. Konjar S, Veldhoen M. Dynamic metabolic state of tissue resident CD8 T cells. Front Immunol (2019) 10:1683. doi: 10.3389/fimmu.2019.01683
77. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu S, et al. Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature (2017) 543:252–6. doi: 10.1038/nature21379
78. Schachtele SJ, Hu S, Sheng WS, Mutnal MB, Lokensgard JR. Glial cells suppress postencephalitic CD8+ T lymphocytes through PD-L1. Glia (2014) 62:1582–94. doi: 10.1002/glia.22701
79. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, et al. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity (2011) 35:400–12. doi: 10.1016/j.immuni.2011.06.015
80. Wang Z, Wang S, Goplen NP, Li C, Cheon IS, Dai Q, et al. PD-1hi CD8+ resident memory T cells balance immunity and fibrotic sequelae. Sci Immunol (2019) 4. doi: 10.1126/sciimmunol.aaw1217
81. Boldison J, Chu CJ, Copland DA, Lait PJP, Khera TK, Dick AD, et al. Tissue-resident exhausted effector memory CD8+ T cells accumulate in the retina during chronic experimental autoimmune uveoretinitis. J Immunol (2014) 192:4541–50. doi: 10.4049/jimmunol.1301390
82. Phares TW, Stohlman SA, Hinton DR, Atkinson R, Bergmann CC. Enhanced antiviral T cell function in the absence of B7-H1 is insufficient to prevent persistence but exacerbates axonal bystander damage during viral encephalomyelitis. J Immunol (2010) 185:5607–18. doi: 10.4049/jimmunol.1001984
83. Phares TW, Ramakrishna C, Parra GI, Espstein A, Chen L, Atkinson R, et al. Target-dependent B7-H1 regulation contributes to clearance of central nervous system infection and dampens morbidity. J Immunol (2009) 182:5430–8. doi: 10.4049/jimmunol.0803557
84. Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, et al. T cells promote microglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. Nat Neurosci (2019) 22:1276–88. doi: 10.1038/s41593-019-0427-y
85. Ritzel RM, Crapser J, Patel AR, Verma R, Grenier JM, Chauhan A, et al. Age-associated resident memory CD8 T cells in the central nervous system are primed to potentiate inflammation after ischemic brain injury. J Immunol (2016) 196:3318–30. doi: 10.4049/jimmunol.1502021
86. Dulken BW, Buckley MT, Navarro Negredo P, Saligrama N, Cayrol R, Leeman DS, et al. Single-cell analysis reveals T cell infiltration in old neurogenic niches. Nature (2019) 571:205–10. doi: 10.1038/s41586-019-1362-5
87. Ren HM, Kolawole EM, Ren M, Jin G, Netherby-Winslow CS, Wade Quinn, et al. IL-21 from high-affinity CD4 T cells drives differentiation of brain-resident CD8 T cells during persistent viral infection. Sci Immunol (2020) 5:eabb5590. doi: 10.1126/sciimmunol.abb5590
88. Park SL, Mackay LK. PD-1: always on my mind. Immunol Cell Biol (2017) 95:857–8. doi: 10.1038/icb.2017.69
89. Prasad S, Hu S, Sheng WS, Chauhan P, Singh A, Lokensgard JR. The PD-1: PD-L1 pathway promotes development of brain-resident memory T cells following acute viral encephalitis. J Neuroinflammation (2017) 14:82. doi: 10.1186/s12974-017-0860-3
90. Weisberg SP, Carpenter DJ, Cait M, Dogra P, Gartrell-Corrado RD, Chen AX, et al. Tissue-resident memory T cells mediate immune homeostasis in the human pancreas through the PD-1/PD-L1 pathway. Cell Rep (2019) 29:3916–3932 e3915. doi: 10.1016/j.celrep.2019.11.056
91. Shwetank, Frost EL, Mockus TE, Ren HM, Toprak M, Lauver MD, et al. PD-1 dynamically regulates inflammation and development of brain-resident memory CD8 T cells during persistent viral encephalitis. Front Immunol (2019) 10:783. doi: 10.3389/fimmu.2019.00783
92. Smolders J, Heutinck KM, Fransen NL, Remmerswaal EBM, Hombrink P, ten Berge IJM, et al. Tissue-resident memory T cells populate the human brain. Nat Commun (2018) 9:4593. doi: 10.1038/s41467-018-07053-9
93. Steinbach K, Vincenti I, Egervari K, Kreutzfeldt M, van der Meer F, Page N, et al. Brain-resident memory T cells generated early in life predispose to autoimmune disease in mice. Sci Transl Med (2019) 11:eaav5519. doi: 10.1126/scitranslmed.aav5519
94. Gardiner D, Lalezari J, Lawitz E, DiMico M, Ghalib R, Reddy KR, et al. A randomized, double-blind, placebo-controlled assessment of BMS-936558, a fully human monoclonal antibody to programmed death-1 (PD-1), in patients with chronic hepatitis C virus infection. PloS One (2013) 8:e63818. doi: 10.1371/journal.pone.0063818
95. Gane E, Verdon DJ, Brooks AE, Gagger A, Hguyen AH, Subramanian GM, et al. Anti-PD-1 blockade with nivolumab with and without therapeutic vaccination for virally suppressed chronic hepatitis B: A pilot study. J Hepatol (2019) 71:900–7. doi: 10.1016/j.jhep.2019.06.028
96. Beck ES, Cortese I. Checkpoint inhibitors for the treatment of JC virus-related progressive multifocal leukoencephalopathy. Curr Opin Virol (2020) 40:19–27. doi: 10.1016/j.coviro.2020.02.005
97. Cortese I, Muranski P, Enose-Akahata Y, Ha SK, Smith B, Monaco MC, et al. Pembrolizumab treatment for progressive multifocal leukoencephalopathy. N Engl J Med (2019) 380:1597–605. doi: 10.1056/NEJMoa1815039
98. Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway. Nat Rev Immunol (2018) 18:153–67. doi: 10.1038/nri.2017.108
99. Boland BS, He Z, Tsai MS, Olvera JG, Omilusik KD, Duong HG, et al. Heterogeneity and clonal relationships of adaptive immune cells in ulcerative colitis revealed by single-cell analyses. Sci Immunol (2020) 5:eabb4432. doi: 10.1126/sciimmunol.abb4432
100. Low JS, Farsakoglu Y, Vesely MCA, Sefik E, Kelly JB, Harman CCD, et al. Tissue-resident memory T cell reactivation by diverse antigen-presenting cells imparts distinct functional responses. J Exp Med (2020) 217:e20192291. doi: 10.1084/jem.20192291
101. Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, et al. T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity (2018) 48:327–338 e325. doi: 10.1016/j.immuni.2018.01.015
Keywords: T cell receptor, PD-1, brain-resident memory CD8 T cells, virus, neuroinflammation
Citation: Netherby-Winslow CS, Ayers KN and Lukacher AE (2021) Balancing Inflammation and Central Nervous System Homeostasis: T Cell Receptor Signaling in Antiviral Brain TRM Formation and Function. Front. Immunol. 11:624144. doi: 10.3389/fimmu.2020.624144
Received: 30 October 2020; Accepted: 08 December 2020;
Published: 27 January 2021.
Edited by:
Jie Sun, Mayo Clinic, United StatesReviewed by:
Aaron Jon Johnson, Mayo Clinic, United StatesCopyright © 2021 Netherby-Winslow, Ayers and Lukacher. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aron E. Lukacher, YWx1a2FjaGVyQHBlbm5zdGF0ZWhlYWx0aC5wc3UuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.