
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Immunol., 17 February 2021
Sec. Molecular Innate Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.623071
This article is part of the Research TopicInnate Cells in the Pathogenesis of Food AllergyView all 17 articles
 Rosa Muñoz-Cano1,2,3,4*
Rosa Muñoz-Cano1,2,3,4* Clara San Bartolome2,3,5Rocío Casas-Saucedo1,2,3Giovanna Araujo1,3
Clara San Bartolome2,3,5Rocío Casas-Saucedo1,2,3Giovanna Araujo1,3 Sonia Gelis1,3
Sonia Gelis1,3 Maria Ruano-Zaragoza1,2,3Jordi Roca-Ferrer3,4,6Francis Palomares2,7
Maria Ruano-Zaragoza1,2,3Jordi Roca-Ferrer3,4,6Francis Palomares2,7 Margarita Martin2,3,8
Margarita Martin2,3,8 Joan Bartra1,2,3,4†
Joan Bartra1,2,3,4† Mariona Pascal2,3,5†
Mariona Pascal2,3,5†Cofactors may explain why in some cases food ingestion leads to anaphylaxis while in others elicits a milder reaction or tolerance. With cofactors, reactions become more severe and/or have a lower allergen threshold. Cofactors are present in up to 58% of food anaphylaxis (FAn). Exercise, NSAIDs, and alcohol are the most frequently described, although the underlying mechanisms are poorly known. Several hypotheses have suggested the influence of these cofactors on basophils and mast cells (MCs). Exercise has been suggested to enhance MC activation by increasing plasma osmolarity, redistributing blood flow, and activating adenosine and eicosanoid metabolism. NSAIDs’ cofactor effect has been related with cyclooxygenase inhibition and therefore, prostaglandin E2 (PGE2) production. Indeed, overexpression of adenosine receptor 3 (A3) gene has been described in NSAID-dependent FAn; A3 activation potentiates FcϵRI-induced MC degranulation. Finally, alcohol has been related with an increase of histamine levels by inhibition of diamino oxidase (DAO) and also with and increase of extracellular adenosine by inhibition of its uptake. However, most of these mechanisms have limited evidence, and further studies are urgently needed. In conclusion, the study of the immune-related mechanisms involved in food allergic reactions enhanced by cofactors is of the utmost interest. This knowledge will help to design both tailored treatments and prophylactic strategies that, nowadays, are non-existent.
Food allergy is the main cause of anaphylaxis in children and in some series, also in adults (1). However, only some food allergic reactions end up being an anaphylaxis, ranging from very low percentages (0.4%) up to 40% of the reactions in some series (2). This disparity may be related with differences in age, food trigger, and geographic area. Interestingly, food allergy in adults usually debuts in the second-third decade of life and, in up to 50% of the cases, as an anaphylaxis (1).
The severity of an allergic reaction is unpredictable. The same individual may present reactions with different severity, even with the same food. The presence of cofactors, such as NSAIDs, exercise or alcohol, may explain this heterogeneity. Patients with cofactor-dependent reactions may have no or mild symptoms with the food alone and a more severe reaction (generalized urticaria or anaphylaxis) when associating a cofactor. Indeed, the same patient may have reactions with different cofactors or even need more than one cofactor to develop the severe reaction. Cofactors may increase the severity of the reaction or decreasing the reactivity threshold, meaning that lower doses of allergen are able to induce a more severe reaction (from two to six times depending on the series) (2–6). Cofactors are identified in up to 58% of food anaphylaxis (FAn) in some series and related with greater severity (3, 4, 7, 8), but the underlying mechanisms are poorly understood.
Their role in anaphylaxis has been more widely studied in adult patients, being not yet optimally studied in children. The high frequency of cofactor-related reactions highlights the clinical impact of recognizing and including cofactors into the routine diagnostic workup. Indeed, the understanding of the underlying mechanisms will help in developing tailored prophylactic treatments and identifying risk biomarkers. Hereby we report the main evidence reported regarding the major cofactors involved in food allergy.
Exercise is described in up to 10% of anaphylactic reactions (3, 4). Exercise-induced anaphylaxis is a syndrome that may occur in food allergic patients but also independently of food ingestion (9). Considering the number of published studies, for the purpose of this review we will focus on food-dependent exercise-induced anaphylaxis (FDEIA). Several mechanisms have been postulated, although the evidence supporting each of them is scarce and limited by the low number of patients evaluated and the limited quality of the studies, as stated by a recent position paper from the European Academy of Allergy and Clinical Immunology (EAACI) (9).
The increase of both gastrointestinal permeability and tissue transglutaminase activity, two of the proposed mechanisms in the gut mucosa, is splendidly reviewed elsewhere (9). Other suggested mechanisms are related with the direct effect of exercise on basophils and mast cells (MCs) by modifying the cell count and histamine release (HR), redistributing the blood flow and increasing plasma osmolarity.
Acute exercise is related with the transient increase of blood circulating neutrophils, monocytes, dendritic and NK cells, although it remains unknown whether these changes may be also related with an altered immune function (10). Some in vivo studies have shown an increase of basophils count and HR after exercise, being more important in atopic individuals (11). In others, only an increase in HR has been demonstrated with no modification of basophil count (12). Interestingly, some authors have not found differences in HR when comparing allergic patients with controls, regardless of a significant basophil count increase in the atopic group (11). Indeed, increase in HR after in vitro IgE activation was only observed in highly trained athletes compared with non-trained ones, although both groups were non-atopic (13). Thus, these observations suggest that the atopic status together with the training level might be conditioning factors of HR and therefore, of exercise-induced basophil- and MC-activation. However, the interpretation and comparison of these findings are often complicated due to differences in experimental design of the studies (i.e., measurement times and methods, samples types and exercise intensity/duration).
It is well known that during exercise, blood flow is redistributed, being diverted from the viscera to the skeletal muscle, heart, and skin (14). Mounting evidence supports that HR during exercise is part of the physiological mechanism of recovery (15). Histamine acts as a vasodilator and is involved in post-exercise hypotension and hyperemia (15, 16). Therefore, one hypothesis that may need further exploration is whether this exercise recovery system in FDEIA patients is somehow damaged and, therefore, exercise is inducing “excessive” basophil/MC activation.
Some authors (17) have hypothesized that as a consequence of the blood flow redistribution, food allergens are transported away from the gut mucosa where resident MCs tolerate them to other tissues as skin or skeletal muscles, where MCs with a different phenotype react. For this reason, FDEIA patients tolerate the food at rest but have an allergic reaction when doing exercise. This is an interesting hypothesis and biologically plausible, although there is no experimental evidence yet.
Another effect of exercise is plasma osmolarity increase, which magnitude depends on exercise intensity and the resulting dehydration (9). Several in vitro studies have demonstrated that hyperosmolar environments induce MC and basophil activation. Torres-Atencio et al. (18) showed that mannitol, as a hyperosmolar stimulus, induced significant activation in MC from LAD2 cell line and healthy individuals (MC derived from CD34+ peripheral blood cells and primary lung MC). Other authors, in three patients (one FDEIA and two food allergic) and three healthy controls, showed that changes in osmolarity increase basophil activation only in FDEIA, but not in controls or food allergic patients (19). However, the in vitro osmolarity achieved in that study (340 and 450 mOsm) seems difficult to be reached during exercise or dehydration in physiological conditions (293–305 mOsm) (9, 20).
Adenosine is produced under conditions of increased energy consumption such as hypoxia or stress, rapidly increasing its circulating levels (21). Adenosine induces opposite effects in MC activation depending on the binding receptor (22). Whereas A2A ligation results in an increase of cAMP, and therefore, the inhibition of MC mediator release, A2B enhances MC activation through PLC. Finally, A3 seems to be involved in the potentiation of IgE-mediated MC activation in mouse and human models (22). Like the observations in PGE2–EP axis, the expression profile of adenosine receptors in the cell’s surface may condition the final effect of adenosine. Gomez et al. (23) demonstrated that adenosine enhanced IgE-mediated degranulation via A3 in human lung derived MC but not in skin MC. Interestingly, lung MCs were shown to express three-fold more A3 mRNA than the skin ones.
Adenosine is released into the venous efflux from skeletal muscle fibers in response to muscle contraction during exercise. Accumulated evidence shows that it is partially responsible for muscle hyperemia at submaximal and maximal workloads due to its effect on A2A that results in vasodilation (24). Indeed, one of the most important factors regulating exercise capacity is the vasodilatation of the exercising muscle (25). In vivo studies performed in chronic heart failure patients have shown that adenosine release is impaired, partially explaining the reduced exercise capacity observed in these patients (25). Interestingly, some studies have shown that trained athletes have higher adenosine baseline plasma levels when compared with recreational ones (26). A previous publication of Muñoz-Cano et al. (27) showed that cofactor-related FAn patients (NSAIDs and exercise) overexpressed A3 gene (ADORA3) and others related with adenosine metabolism. Interestingly, although A3 activation has been linked to anti-inflammatory effects in several models of inflammation (28), it has also been related with the enhancement of IgE-mediated degranulation in human MC and, thus contributing to allergic inflammation (29, 30). Therefore, we hypothesized that the adenosine released during exercise in FDEIA patients would preferably bind A3 with no deleterious effect in the absence of allergen. However, in the presence of allergen, adenosine would have a synergistic effect on MC activation, favoring the allergic reaction. However, further studies need to be conducted to confirm this theory.
Finally, another potential underlying mechanism in FDEIA may be related with the eicosanoid metabolism. Exercise is related with an increase in serum of products from the eicosanoid metabolism, as well as, cyclooxygenase (COX)-1 and 2-derived prostanoids (TXB2, PGE2, PGD2,…) and lipoxygenase (5-LOX, 12-LOX, 15-LOX) as a physiological response (31).
Different models have demonstrated that PGE2 abrogates IgE-mediated MC activation (32–34). Particularly misoprostol, a PGE1 analog, has shown to suppress symptoms in wheat-dependent exercise-induced anaphylaxis and IgE-mediated histamine release in both allergic rhinitis and healthy individuals (35–37). Rastogi et al. (38) have shown that patients with hymenoptera anaphylaxis had lower baseline PGE2 serum levels, suggesting that PGE2 may protect from anaphylaxis. Conversely, a very recent publication of Muñoz-Cano et al. (39) did not find any differences in plasma PGE2 at baseline in a series of FAn patients. Differences in the sample type and cause of anaphylaxis may account for this discrepancy.
Although there is no data regarding PGE2 levels at baseline or otherwise in FDEIA, we could suggest, as a hypothesis, that these patients may have a deficient production of PGE2 during exercise that would be predisposing to anaphylaxis in the presence of the allergen.
Non-steroidal anti-inflammatory drugs (NSAIDs) constitute a heterogenous group of widely used drugs with analgesic, anti-pyretic, and anti-inflammatory properties. Their main mechanism of action, despite the differences in their chemical structure, depends on prostanoid (prostaglandins and thromboxane) inhibition by blocking COX activity (40). However, some NSAIDs have COX-independent effects, such as the ability to modulate several transcription factors that control the expression of genes involved in inflammation (f.i. nuclear factor-kappa B) or signaling pathways (MAPK or PI3k/Akt) (41).
NSAIDs, as a cofactor, are involved in up to 25% of food-induced anaphylaxis and are considered a risk factor with an odds ratio >11 (42). Several studies have shown that NSAIDs can also induce anaphylaxis in FDEIA patients despite that NSAIDs were not originally involved in previous reactions (43, 44). The underlying mechanisms of this synergistic effect are not completely understood, and two main theories have been suggested. One is related with the alteration of intestinal permeability by NSAIDs leading to an increase of allergen absorption (45) and the other suggesting a direct effect of NSAIDs on basophils and MC.
NSAIDs have shown to induce MC activation in certain human and animal models. In NSAID exacerbated respiratory disease (N-ERD) patients, Steinke et al. showed that aspirin induced MC activation by measuring calcium influx and PGD2 release (46). Interestingly, it has also been demonstrated that N-ERD patients have a decreased expression of PGE2 receptor 2 (EP2) that may contribute to reducing PGE2 capacity to mediate anti-proliferative and anti-inflammatory effects (47). Indeed, N-ERD has also decreased production of PGE2 (48).
Matsuo et al. (49) showed that aspirin did not induce HR by itself but enhanced IgE-mediated basophil activation. Interestingly, the authors suggested that this effect was not related with a COX-dependent mechanism but with Syk phosphorylation. On the contrary, Pascal et al. (50) showed that the ability of NSAID to enhance the IgE-mediated reactions in FAn patients may be COX1-dependent. Using a model of basophil activation test, these authors demonstrated that the activation with the allergen (peach lipid transfer protein) was enhanced by aspirin. However, this effect was not observed when co-stimulating with valdecoxib (selective COX-2 inhibitor). In the same line, Wojnar et al. showed that several chemically unrelated NSAIDs (non-selective COX inhibitors) enhanced HR induced by ragweed (51). Indeed, Matsukura (43) and Aihara (52) demonstrated a potentiation of the allergic reaction with aspirin but not with nimesulide and etodolac, both preferential COX-2 inhibitors, in FDEIA patients.
Finally, several authors have demonstrated that PGE2 prevents MC degranulation when acting through EP2 and induces a pro-inflammatory response when signaling through EP3 (18, 32, 53). Very recently, Rastogi et al. (38) have shown that anaphylaxis in mice can be prevented by blocking PGE2 degradation. They also showed that MC IgE-mediated degranulation is suppressed by PGE2 through EP4 in mouse MC and through both EP2 and EP4 ligation in human skin MC. It has been suggested that the ratio of EP receptors expressed on cell’s surface may be determinant in the final effect of PGE2. EP3 is considered to mediate pro-inflammatory effects, and EP2 and EP4 have anti-inflammatory activity (32). Also very recently, Muñoz-Cano et al. (39) showed that PGE2 reduced IgE-mediated basophil activation in patients with FAn. Furthermore, these authors showed a decreased expression of EP4 (anti-inflammatory) and increased expression of EP3 (pro-inflammatory) receptors in basophils. However, they did not find differences among EP pattern expression when comparing FDNIA and FAn, and all patients had a ratio EP3/EP4+EP2 favoring a pro-inflammatory activation upon PGE2 ligation.
All this suggests that eicosanoid metabolism may be involved in the development of anaphylaxis in general, and therefore, anything blocking PGE2 production, such as NSAIDs, may facilitate the development of a severe reaction. However, if that seems to be a universal mechanism in anaphylaxis, we must wonder why not all food allergic patients require a cofactor in order to have an anaphylaxis. In this line, Pascal et al. (50) showed that the synergistic effect of NSAID was present in both NSAID-dependent (FDNIA) and -independent FAn patients, and the main difference between them was the basophil sensitivity. Thus, FAn (NSAID-independent) patients had higher (about 148-fold) basophil sensitivity, requiring further less allergen concentration to elicit 50% of basophil maximal response compared to FDNIA patients. This suggests that enough allergen concentration could elicit an anaphylaxis in the absence of a cofactor in FDNIA. A similar observation was made in FDEIA patients in vivo, where the increase of the amount of allergen was enough to reproduce the anaphylaxis in the absence of exercise (43, 52, 54). Some other evidence of the differences in the pathogenic mechanism in cofactor-dependent and -independent anaphylaxis was provided by Muñoz-Cano et al. (27), who reported differences at transcriptome level. Thus, altered B-cell pathways, increased markers of neutrophil activation and reactive oxygen species levels were exclusively observed in FAn patients. However, adenosine metabolism related genes were differentially expressed only in FDNIA.
Altogether, these findings suggest that (1) eicosanoid metabolism may play a role in the development of any anaphylaxis; (2) NSAID may have a universal synergistic effect in any food allergic patient; (3) the right amount of allergen may induce an anaphylaxis in FDNIA even in the absence of a cofactor; and (4) there are other yet to be confirmed mechanisms that explain the differences between NSAID-dependent and -independent FAn.
Adenosine metabolism has been linked to some NSAID-exacerbated cutaneous and respiratory diseases. ADORA3 polymorphism has been identified in NSAID-exacerbated urticaria patients (55) and ADORA1 and ADORA2A in N-ERD patients (56). Cronstein et al. (57–59) showed in a series of studies with animal and human models that NSAIDs at pharmacologic concentrations increase the release of adenosine into the extracellular milieu by uncoupling oxidative phosphorylation and, therefore, increasing ATP catabolism. These authors have suggested that the anti-inflammatory effects of NSAIDs are partly COX-independent and mediated by adenosine. However, considering that the receptor expression profile in the cell’s surface may condition the final effect of adenosine, as in the PGE2–EP axis, the anti-inflammatory effect of adenosine in these models may be related with a particular expression pattern of the cells/mouse strains studied.
Muñoz-Cano et al. (27) observed that FDNIA patients had a unique transcriptome signature related with adenosine metabolism genes, particularly an overexpression of ADORA3 that may be having a dual effect in these patients. A3 agonists have also shown anti-inflammatory effects in several mouse models due to inhibition of IFN-γ (60, 61). Interestingly, these authors also showed that FDNIA patients had a repressed IFN-γ production and IFN-γ-regulated genes. Considering that FDNIA patients usually have no or mild reaction when exposed to the allergen alone, A3R may be exerting its protective (anti-inflammatory) effect in this scenario through IFN-γ repression. However, when the patient is exposed to the food allergen plus NSAID, the adenosine released by NSAIDs, via A3, would enhance the IgE-mediated reaction, resulting in a systemic reaction. Nevertheless, further studies are still needed to completely understand the specific role of adenosine metabolism in FAn.
Finally, Pouliot et al. (62) showed that adenosine up-regulates COX-2 expression, with a consequent increase of PGE2 production through A2A. These findings suggest that the inhibitory effect of A2A receptor depends on COX2-PGE2–EP axis. The potential connection between adenosine and PGE2 metabolism, both apparently involved in the development of FDNIA, opens an exciting research field that must still be developed.
Alcohol is one of the classic cofactors in FAn, present in up to 15% of cases in some series (2), although the evidence supporting the underlying mechanism of its effects is scarce. We propose some hypothesis based on evidence of alcohol effect on immune cells that we briefly review hereby.
Some authors have shown that alcohol modifies intestinal permeability due to local activation of MC and modification of the expression of tight junction-associated proteins by acetaldehyde, one of its metabolites (63, 64). Actually, acetaldehyde-induced MC activation is one of the suggested mechanisms involved in alcohol-induced asthma in Japanese patients. It is well known that this population has a defective alcohol catabolism (aldehyde dehydrogenase 2 decreased activity) that facilitates acetaldehyde accumulation (64). Alcohol has also been shown to increase histamine levels by inhibiting diamino oxidase (DAO), an enzyme that catabolizes histamine (65). It has also been described that alcohol induces pro-inflammatory mediator (such IL-6, IL-10, and IFN-γ) release and eicosanoid metabolite production, such as PGE2 (66). Similar to the hypothesis in FDEIA, food-dependent alcohol-induced anaphylaxis (FDAIA) patients could have a deficient production of PGE2. Further studies evaluating the productions of eicosanoid metabolites in these patients would shed light upon the underlying mechanism.
Finally, adenosine metabolism again, may be involved in FDAIA. Alcohol inhibits adenosine uptake, increasing its extracellular levels (67). However, this effect is only observed in acute consumption, and chronic intake does not modify adenosine transport (67). So, as suggested in FDNIA, adenosine released upon alcohol consumption may enhance the IgE-mediated reaction induced by food allergen. Conversely to the observation in FDNIA patients, no data regarding expression profile of adenosine receptors in FDAIA exists.
The limited knowledge about the mechanisms involved in cofactor-enhanced FAn (CEFA) makes exceedingly difficult the development of prophylactic strategies. Apparently, avoiding strategies in CEFA patients may seem straightforward. However, in those allergic to ubiquitous allergens (f.i. nuts) or panallergens such as lipid transfer proteins, avoiding strategies are quite complicated considering that cofactors are everyday common situations (f.i. physical activity).
Although the evidence in CEFA is limited, and we are currently working mostly based on hypothesis, the high complexity of the underlying mechanism seems evident. In the light of this data and other existent evidence not reviewed in this manuscript, several pathogenic mechanisms may be intertwined. The “cofactor effect” seems to be a universal phenomenon as demonstrated in in vivo and in vitro experiments not only in FAn patients but also in healthy individuals (43, 49, 50, 54). That means that (1) a personal predisposition may be required to develop a CEFA (or any anaphylaxis), and (2) most cofactors are interchangeable and capable of reproduce an anaphylaxis. Considering the unrelated nature of the cofactors, this observation may suggest that all of them may share, somehow, some common pathogenic mechanisms. One may suspect that all these cofactors are interfering with a compensatory system that is blocking (totally or partially) the allergic reaction induced by the food alone. The adenosine and eicosanoid metabolisms (Figure 1) and/or the disruption of intestinal permeability (not reviewed here) may be some of these mechanisms. In conclusion, and at risk of sounding cliché, further studies are needed to understand this “cofactor effect” and to identify risk biomarkers and prophylactic treatments.
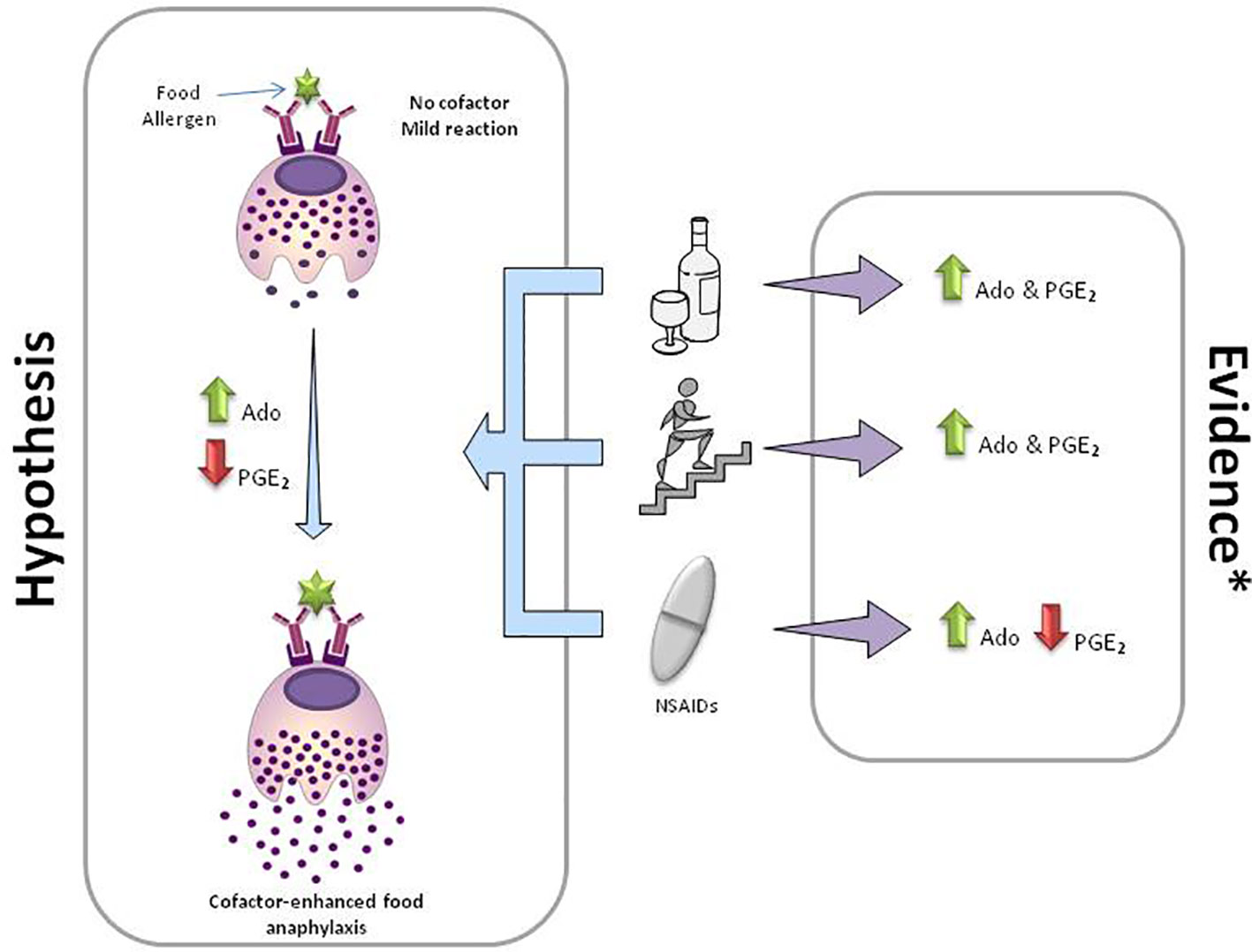
Figure 1 Mechanisms in cofactor-enhanced food anaphylaxis. Adenosine and eicosanoid metabolism hypothesis. The panel depicts the most frequent cofactors involved in food anaphylaxis (from top to bottom): alcohol, exercise, and non-steroidal anti-inflammatory drugs (NSAIDs). Mild reactions in the absence of a cofactor may end up in an anaphylaxis with the same amount of allergen together with the presence of a cofactor. Ado, adenosine; green arrows, increase; red arrows, decrease. *Evidence based on references (23, 26, 28, 29, 38, 39, 41, 50, 57–59, 66, 67).
RM-C has coordinated the different authors and written part of the manuscript. CS, RC-S, GA, MR-Z, JR-F, FP, and MM have written part of the manuscript. JB and MP have supervised the review and written part of the manuscript. All authors contributed to the article and approved the submitted version.
RC-S is a recipient of a Rio Hortega fellowship (Carlos III Health Institute CM19/00046).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Kamdar TA, Peterson S, Lau CH, Saltoun CA, Gupta RS, Bryce PJ. Prevalence and characteristics of adult-onset food allergy. J Allergy Clin Immunol Pract (2015) 3:114–15.e1. doi: 10.1016/j.jaip.2014.07.007
2. Muraro A, Roberts G, Worm M, Bilò MB, Brockow K, Fernández Rivas M, et al. Anaphylaxis: guidelines from the European Academy of Allergy and Clinical Immunology. Allergy (2014) 69:1026–45. doi: 10.1111/all.12437
3. Wölbing F, Fischer J, Köberle M, Kaesler S, Biedermann T. About the role and underlying mechanisms of cofactors in anaphylaxis. Allergy Eur J Allergy Clin Immunol (2013) 68:1085–92. doi: 10.1111/all.12193
4. Worm M, Francuzik W, Renaudin JM, Bilo MB, Cardona V, Scherer Hofmeier K, et al. Factors increasing the risk for a severe reaction in anaphylaxis: An analysis of data from The European Anaphylaxis Registry. Allergy (2018) 73:1322–30. doi: 10.1111/all.13380
5. Worm M, Moneret-Vautrin A, Scherer K, Lang R, Fernandez-Rivas M, Cardona V, et al. First European data from the network of severe allergic reactions (NORA). Allergy (2014) 69:1397–404. doi: 10.1111/all.12475
6. Panesar SS, Javad S, de Silva D, Nwaru BI, Hickstein L, Muraro A, et al. The epidemiology of anaphylaxis in Europe: A systematic review. Allergy (2013) 68(11):1353–61. doi: 10.1111/all.12272
7. Cardona V, Luengo O, Garriga T, Labrador-Horrillo M, Sala-Cunill A, Izquierdo A, et al. Co-factor-enhanced food allergy. Allergy (2012) 67:1316–8. doi: 10.1111/j.1398-9995.2012.02877.x.
8. Oropeza AR, Bindslev-Jensen C, Broesby-Olsen S, Kristensen T, Møller MB, Vestergaard H, et al. Patterns of anaphylaxis after diagnostic workup: A follow-up study of 226 patients with suspected anaphylaxis. Allergy (2017) 72:1944–52. doi: 10.1111/all.13207
9. Ansley L, Bonini M, Delgado L, Del Giacco S, Du Toit G, Khaitov M, et al. Pathophysiological mechanisms of exercise-induced anaphylaxis: an EAACI position statement. Allergy (2015) 70:1212–21. doi: 10.1111/all.12677
10. Nieman DC. Exercise immunology: practical applications. Int J Sports Med (1997) 18(Suppl 1):S91–100. doi: 10.1055/s-2007-972705
11. Morgan D, Moodley I, Phillips M, Thorax R--. 1983 U. Plasma histamine in asthmatic and control subjects following exercise: influence of circulating basophils and different assay techniques. Thorax (1983) 38:771–7. doi: 10.1136/thx.38.10.771
12. Harries MG, Burgue PS, O’Brien I, Cromwell O, Pepys J. Blood histamine levels after exercise testing. Clin Exp Allergy (1979) 9:437–41. doi: 10.1111/j.1365-2222.1979.tb02506.x
13. Mucci P, Anselme-Poujol F, Caillaud C, Couret I, Rossi M, Préfaut C. Basophil releasability in young highly trained and older athletes. Med Sci Sports Exerc (1999) 31:507–13. doi: 10.1097/00005768-199904000-00003
14. Sheel AW, Boushel R, Dempsey JA. Competition for blood flow distribution between respiratory and locomotor muscles: implications for muscle fatigue. J Appl Physiol (2018) 125:820–31. doi: 10.1152/japplphysiol.00189.2018
15. Luttrell MJ, Halliwill JR. The Intriguing Role of Histamine in Exercise Responses. Exerc Sport Sci Rev (2017) 45:16–23. doi: 10.1249/JES.0000000000000093
16. Halliwill JR, Buck TM, Lacewell AN, Romero SA. Postexercise hypotension and sustained postexercise vasodilatation: what happens after we exercise? Exp Physiol (2013) 98:7–18. doi: 10.1113/expphysiol.2011.058065
17. Robson-Ansley P, Du TG. Pathophysiology, diagnosis and management of exercise-induced anaphylaxis. Curr Opin Allergy Clin Immunol (2010) 10:312–7. doi: 10.1097/ACI.0b013e32833b9bb0
18. Torres-Atencio I, Ainsua-Enrich E, de Mora F, Picado C, Martín M. Prostaglandin E2 prevents hyperosmolar-induced human mast cell activation through prostanoid receptors EP2 and EP4. PloS One (2014) 9:e110870. doi: 10.1371/journal.pone.0110870
19. Barg W, Wolanczyk-Medrala A Obojski A, Wytrychowski K, B Panaszek WM. Food-dependent exercise-induced anaphylaxis: possible impact of increased basophil histamine releasability in hyperosmolar conditions. J Investig Allergol Clin Immunol (2008) 18:312–5.
20. Popowski LA, Oppliger RA, Patrick Lambert G, Johnson RF, Kim Johnson A, Gisolf CV. Blood and urinary measures of hydration status during progressive acute dehydration. Med Sci Sports Exerc (2001) 33:747–53. doi: 10.1097/00005768-200105000-00011
21. Bours MJL, Swennen ELR, Di Virgilio F, Cronstein BN, Dagnelie PC. Adenosine 5’-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther (2006) 112:358–404. doi: 10.1016/j.pharmthera.2005.04.013
22. Rudich N, Ravid K, Sagi-Eisenberg R. Mast cell adenosine receptors function: a focus on the a3 adenosine receptor and inflammation. Front Immunol (2012) 3:134. doi: 10.3389/fimmu.2012.00134
23. Gomez G, Zhao W, Schwartz LB. Disparity in FcϵRI-induced degranulation of primary human lung and skin mast cells exposed to adenosine. J Clin Immunol (2011) 31:479–87. doi: 10.1007/s10875-011-9517-7
24. Marshall JM. The roles of adenosine and related substances in exercise hyperaemia. In: Journal of Physiology. J Physiol (2007) 583(Pt 3):835–45. doi: 10.1113/jphysiol.2007.136416
25. Matsui S, Tamura N, Takekoshi N, Murakami E. Plasma adenosine concentration during and after dynamic exercise in patients with chronic heart failure. Int J Angiol (1995) 4:188–92. doi: 10.1007/BF02042918
26. Pospieszna B, Kusy K, Słomińska EM, Dudzinska W, Ciekot-Sołtysiak M, Zieliński J. The effect of training on erythrocyte energy status and plasma purine metabolites in athletes. Metabolites (2020) 10(1):5. doi: 10.3390/metabo10010005
27. Muñoz-Cano R, Pascal M, Bartra J, Picado C, Valero A, Kim D-K, et al. Distinct transcriptome profiles differentiate nonsteroidal anti-inflammatory drug-dependent from nonsteroidal anti-inflammatory drug-independent food-induced anaphylaxis. J Allergy Clin Immunol (2016) 137(1):137–46. doi: 10.1016/j.jaci.2015.05.042
28. Fishman P, Cohen S, Itzhak I, Amer J, Salhab A, Barer F, et al. The A3 adenosine receptor agonist, namodenoson, ameliorates non-alcoholic steatohepatitis in mice. Int J Mol Med (2019) 44:2256–64. doi: 10.3892/ijmm.2019.4364
29. Leung CT, Li A, Banerjee J, Gao ZG, Kambayashi T, Jacobson KA, et al. The role of activated adenosine receptors in degranulation of human LAD2 mast cells. Purinergic Signal (2014) 10:465–75. doi: 10.1007/s11302-014-9409-4
30. Jin X, Shepherd RK, Duling BR, Linden J. Inosine binds to A3 adenosine receptors and stimulates mast cell degranulation. J Clin Invest (1997) 100:2849–57. doi: 10.1172/JCI119833
31. Markworth JF, Vella L, Lingard BS, Tull DL, Rupasinghe TW, Sinclair AJ, et al. Human inflammatory and resolving lipid mediator responses to resistance exercise and ibuprofen treatment. Am J Physiol Regul Integr Comp Physiol (2013) 305(11):R1281–96. doi: 10.1152/ajpregu.00128.2013
32. Serra-Pages M, Olivera A, Torres R, Picado C, de Mora F, Rivera J. E-prostanoid 2 receptors dampen mast cell degranulation via cAMP/PKA-mediated suppression of IgE-dependent signaling. J Leukoc Biol (2012) 92:1155–65. doi: 10.1189/jlb.0212109
33. Gauvreau GM, Watson RM, O’Byrne PM. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med (1999) 159:31–6. doi: 10.1164/ajrccm.159.1.9804030
34. Torres R, Picado C, de Mora F. The PGE2-EP2-mast cell axis: An antiasthma mechanism. Mol Immunol (2015) 1:61–8. doi: 10.1016/j.molimm.2014.03.007
35. Inoue Y, Adachi A, Ueno M, Fukumoto T, Nishitani N, Fujiwara N, et al. The inhibition effect of a synthetic analogue of prostaglandin E1 to the provocation by aspirin in the patients of WDEIA. Arerugi (2009) 58:1418–25.
36. Babakhin AA, Nolte H, Dubuske LM. Effect of misoprostol on the secretion of histamine from basophils of whole blood. Ann Allergy Asthma Immunol (2000) 84:361–5. doi: 10.1016/S1081-1206(10)62787-1
37. Takahashi A, Nakajima K, Ikeda M SS. Pre-treatment with misoprostol prevents food-dependent exercise-induced anaphylaxis (FDEIA). Int J Dermatol (2011) 50:233–40. doi: 10.1111/j.1365-4632.2010.04314.x
38. Rastogi S, Willmes DM, Nassiri M, Babina M, Worm M. PGE2 deficiency predisposes to anaphylaxis by causing mast cell hyper-responsiveness. J Allergy Clin Immunol (2020) 11:S0091–6. doi: 10.1016/j.jaci.2020.03.046
39. Muñoz-Cano R, Casas-Saucedo R, Araujo G, Roca-Ferrer J, de la Cruz C, Martin M, et al. Prostaglandin E2 Decreases Basophil Activation In Patients With Food-Induced Anaphylaxis. Allergy (2020). doi: 10.1111/all.14615
40. Bacchi S, Palumbo P, Sponta A, Coppolino MF. Clinical Pharmacology of Non-Steroidal Anti-Inflammatory Drugs: A Review. Antiinflamm Antiallergy Agents Med Chem (2012) 11:52–64. doi: 10.2174/187152312803476255
41. Tegeder I, Pfeilschifter J, Geisslinger G. Cyclooxygenase-independent actions of cyclooxygenase inhibitors. FASEB J (2001) 15:2057–72. doi: 10.1096/fj.01-0390rev
42. Moneret-Vautrin DA. Drugs as risk factors of food anaphylaxis in adults. Medecine/Sciences (2010) 26:719–23. doi: 10.1051/medsci/2010268-9719
43. Matsukura S, Aihara M, Sugawara M, Kunimi Y, Matsuki M, Inoue Y, et al. Two cases of wheat-dependent anaphylaxis induced by aspirin administration but not by exercise. Clin Exp Dermatol (2010) 35:233–7. doi: 10.1111/j.1365-2230.2009.03709.x
44. Harada S, Horikawa T, Ashida M, Kamo T, Nishioka E, Ichihashi M. Aspirin enhances the induction of type I allergic symptoms when combined with food and exercise in patients with food-dependent exercise-induced anaphylaxis. Br J Dermatol (2001) 145:336–9. doi: 10.1046/j.1365-2133.2001.04329.x
45. Matsuo H, Kaneko S, Tsujino Y, Honda S, Kohno K, Takahashi H, et al. Effects of non-steroidal anti-inflammatory drugs (NSAIDs) on serum allergen levels after wheat ingestion. J Dermatol Sci (2009) 53:241–3. doi: 10.1016/j.jdermsci.2008.09.004
46. Steinke JW, Negri J, Liu L, Payne SC, Borish L. Aspirin activation of eosinophils and mast cells: implications in the pathogenesis of aspirin-exacerbated respiratory disease. J Immunol (2014) 193:41–7. doi: 10.4049/jimmunol.1301753
47. Machado-Carvalho L, Torres R, Perez-Gonzalez M, Alobid I, Mullol J, Pujols L, et al. Altered expression and signalling of EP2 receptor in nasal polyps of AERD patients: role in inflammation and remodelling. Rhinology (2016) 54:254–65. doi: 10.4193/Rhin15.207
48. Roca-Ferrer J, Pérez-Gonzalez M, Garcia-Garcia FJ, Pereda J, Pujols L, Alobid I, et al. Low prostaglandin E2 and cyclooxygenase expression in nasal mucosa fibroblasts of aspirin-intolerant asthmatics. Respirology (2013) 18:711–7. doi: 10.1111/resp.12076
49. Matsuo H, Yokooji T, Morita H, Ooi M, Urata K, Ishii K, et al. Aspirin Augments IgE-Mediated Histamine Release from Human Peripheral Basophils via Syk Kinase Activation. Allergol Int (2013) 62:503–11. doi: 10.2332/allergolint.13-OA-0536
50. Pascal M, Muñoz-Cano R, Milà J, Sanz ML, Diaz-Perales A, Sánchez-López J, et al. Nonsteroidal anti-inflammatory drugs enhance IgE-mediated activation of human basophils in patients with food anaphylaxis dependent on and independent of nonsteroidal anti-inflammatory drugs. Clin Exp Allergy (2016) 46:1111–9. doi: 10.1111/cea.12735
51. Wojnar RJ, Hearn T, Starkweather S. Augmentation of allergic histamine release from human leukocytes by nonsteroidal anti-inflammatory-analgesic agents. J Allergy Clin Immunol (1980) 66:37–45. doi: 10.1016/0091-6749(80)90136-0
52. Aihara M, Miyazawa M, Osuna H, Tsubaki K, Ikebe T, Aihara Y, et al. Food-dependent exercise-induced anaphylaxis: influence of concurrent aspirin administration on skin testing and provocation. Br J Dermatol (2002) 146:466–72. doi: 10.1046/j.1365-2133.2002.04601.x
53. Feng C, Beller EM, Bagga S, Boyce JA. Human mast cells express multiple EP receptors for prostaglandin E 2 that differentially modulate activation responses. Blood (2006) 107:3243–50. doi: 10.1182/blood-2005-07-2772
54. Christensen MJ, Eller E, Mortz CG, Brockow K, Bindslev-Jensen C. Exercise Lowers Threshold and Increases Severity, but Wheat-Dependent, Exercise-Induced Anaphylaxis Can Be Elicited at Rest. J Allergy Clin Immunol Pract (2018) 6:514–20. doi: 10.1016/j.jaip.2017.12.023
55. Kim SH, Nam EJ, Kim YK, Ye YM, Park HS. Functional variability of the adenosine A3 receptor (ADORA3) gene polymorphism in aspirin-induced urticaria. Br J Dermatol (2010) 163:977–85. doi: 10.1111/j.1365-2133.2010.09983.x
56. Kim S-H, Kim Y-K, Park H-W, Kim S-H, Kim S-H, Ye Y-M, et al. Adenosine deaminase and adenosine receptor polymorphisms in aspirin-intolerant asthma. Respir Med (2009) 103:356–63. doi: 10.1016/j.rmed.2008.10.008
57. Cronstein BN, Montesinos MC, Weissmann G. Salicylates and sulfasalazine, but not glucocorticoids, inhibit leukocyte accumulation by an adenosine-dependent mechanism that is independent of inhibition of prostaglandin synthesis and p105 of NFkappaB. Proc Natl Acad Sci USA (1999) 96:6377–81. doi: 10.1073/pnas.96.11.6377
58. Cronstein BN, Vandestouwe M, Druska L, Levin RI, Weissmann G. Nonsteroidal Antiinflammatory Agents Inhibit Stimulated Neutrophil Adhesion to Endothelium - Adenosine-Dependent and Independent Mechanisms. Inflammation (1994) 18:323–35. doi: 10.1007/BF01534273
59. Cronstein BN, Montesinos MC, Weissmann G. Sites of action for future therapy: an adenosine-dependent mechanism by which aspirin retains its antiinflammatory activity in cyclooxygenase-2 and NFkappaB knockout mice. Osteoarthr Cartil (1999) 7:361–3. doi: 10.1053/joca.1998.0236
60. Fishman P, Bar-Yehuda S, Liang BT, Jacobson KA. Pharmacological and therapeutic effects of A3 adenosine receptor agonists. Drug Discovery Today (2012) 17:359–66. doi: 10.1016/j.drudis.2011.10.007
61. Cohen S, Barer F, Bar-Yehuda S, AP IJ, Jacobson KA, Fishman P. A(3) adenosine receptor allosteric modulator induces an anti-inflammatory effect: in vivo studies and molecular mechanism of action. Mediat Inflammation (2014) 2014:708746. doi: 10.1155/2014/708746
62. Pouliot M, Fiset M-E, Masse M, Naccache PH, Borgeat P. Adenosine Up-Regulates Cyclooxygenase-2 in Human Granulocytes: Impact on the Balance of Eicosanoid Generation. J Immunol (2002) 169:5279–86. doi: 10.4049/jimmunol.169.9.5279
63. Ferrier L, Bérard F, Debrauwer L, Chabo C, Langella P, Buéno L, et al. Impairment of the intestinal barrier by ethanol involves enteric microflora and mast cell activation in rodents. Am J Pathol (2006) 168:1148–54. doi: 10.2353/ajpath.2006.050617
64. Kawano T, Matsuse H, Kondo Y, Machida I, Saeki S, Tomari S, et al. Acetaldehyde induces histamine release from human airway mast cells to cause bronchoconstriction. Int Arch Allergy Immunol (2004) 134:233–9. doi: 10.1159/000078771
65. Zimatkin SM, Anichtchik OV. Alcohol-histamine interactions. Alcohol Alcohol (1999) 34:141–7. doi: 10.1093/alcalc/34.2.141
66. van de Loo AJAE, Mackus M, Kwon O, Krishnakumar IM, Garssen J, Kraneveld AD, et al. The Inflammatory Response to Alcohol Consumption and Its Role in the Pathology of Alcohol Hangover. J Clin Med (2020) 9:2081. doi: 10.3390/jcm9072081
Keywords: mast cell, basophil, adenosin, NSAID, cofactor, exercise, prostaglandin, food allergy
Citation: Muñoz-Cano R, San Bartolome C, Casas-Saucedo R, Araujo G, Gelis S, Ruano-Zaragoza M, Roca-Ferrer J, Palomares F, Martin M, Bartra J and Pascal M (2021) Immune-Mediated Mechanisms in Cofactor-Dependent Food Allergy and Anaphylaxis: Effect of Cofactors in Basophils and Mast Cells. Front. Immunol. 11:623071. doi: 10.3389/fimmu.2020.623071
Received: 29 October 2020; Accepted: 31 December 2020;
Published: 17 February 2021.
Edited by:
Simon Patrick Hogan, University of Michigan, United StatesReviewed by:
Glenn Cruse, North Carolina State University, United StatesCopyright © 2021 Muñoz-Cano, San Bartolome, Casas-Saucedo, Araujo, Gelis, Ruano-Zaragoza, Roca-Ferrer, Palomares, Martin, Bartra and Pascal. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rosa Muñoz-Cano, cm11bm96QGNsaW5pYy5jYXQ=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.