 Nina Pilat
Nina Pilat Jonathan Sprent
Jonathan Sprent
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
PERSPECTIVE article
Front. Immunol. , 28 January 2021
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.622810
This article is part of the Research Topic Emerging Therapeutics for Immune Tolerance View all 24 articles
Induction of immune tolerance is the Holy Grail in transplantation medicine and autoimmunity. Currently, patients are required to use immunosuppressive drugs for the rest of their lives, resulting in unwanted side effects and complication from global suppression of the immune response. It is well established that regulatory T cells (Tregs) are critical for the maintenance of immune tolerance towards self-antigens by several mechanisms of immune regulation, in parallel with intrathymic deletion of self-reactive T cells during ontogeny. Therefore, approaches for increasing Treg numbers or function in vivo could provide an all-purpose solution for tolerance induction. Currently, most state-of-the-art therapeutics for treating autoimmune diseases or preventing allograft rejection work either by general immunosuppression or blocking inflammatory reactions and are non-specific. Hence, these approaches cannot provide satisfactory long-term results, let alone a cure. However, in animal models the therapeutic potential of Treg expansion for inducing effective tolerance has now been demonstrated in various models of autoimmunity and allogeneic transplantation. Here, we focus on therapies for increasing the size of the Treg pool by expanding endogenous Treg numbers in vivo or by adoptive transfer of Tregs. In particular, we discuss IL-2 based approaches (low dose IL-2, IL-2 complexes) for inducing Treg expansion in vivo as well as cell-based approaches (polyclonal, antigen specific, or cell engineered) for adoptive Treg therapy. We also mention new questions arising from the first clinical studies on Treg therapy in the fields of transplantation and autoimmunity.
Foxp3+ CD4+ regulatory T cells (Tregs) are the key players in the maintenance of peripheral self-tolerance (1, 2). Here, the discovery of FoxP3 as a master regulator for Treg development and function was critical for characterization of these cells and in-depth analysis of Treg biology. A key finding was that mutations in the FoxP3 gene lead to the development of dysfunctional Treg cells, resulting in severe autoimmunity with early onset of uncontrolled lymphoproliferation in both mice (scurfy mutant) and man (IPEX syndrome). Furthermore, Treg cells have since been demonstrated to be impaired in a variety of autoimmune settings as manifested by a reduction in Treg cell numbers, function, or survival (3).
In healthy individuals, most immature self-reactive T cells are purged during their development in the thymus via negative selection (4). However, especially for tissue-specific antigens, not all self-antigens are displayed in the thymus, with the result that small numbers of self-reactive T cell clones escape into the periphery. Under normal conditions, autoreactivity of these tissue-specific T cells is suppressed by the activity of Tregs. In addition, by multiple mechanisms including synthesis of inhibitory cytokines and reducing the expression of costimulatory molecules on dendritic cells, Tregs play an important role in limiting the intensity of all immune responses, both to self and foreign antigens, thereby preventing immunopathology (5–8). For this reason, amplifying the suppressive function of Tregs is an attractive method for inducing transplantation tolerance. Here, both for autoimmune disease and organ transplantation, several approaches have been described for increasing the Treg : Teff cell ratio to favor tolerance, both in preclinical models and clinical trials (Figure 1).
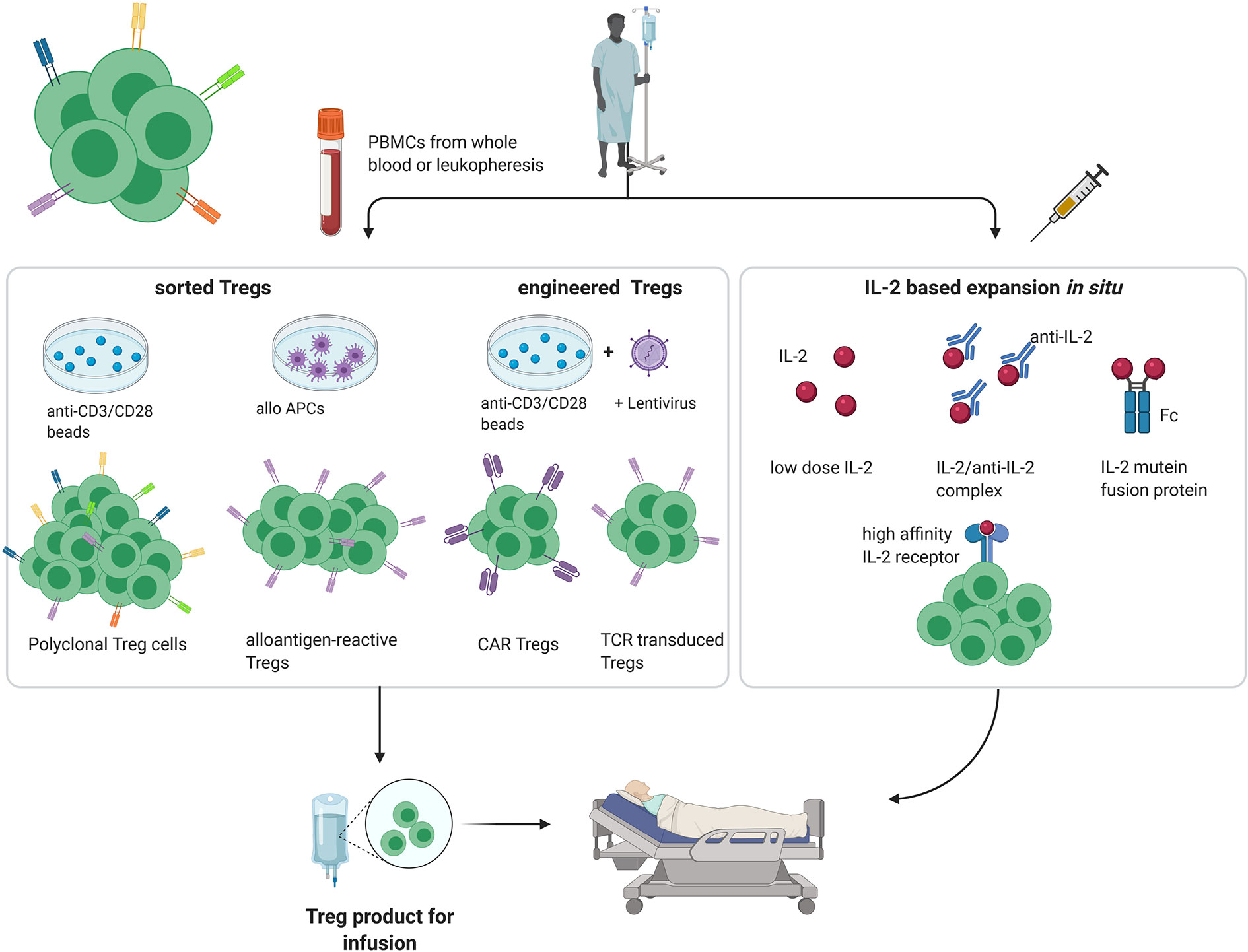
Figure 1 Different approaches for Treg therapy in transplantation and autoimmunity. (PBMC, peripheral blood mononuclear cell; APC, antigen-presenting cell; created with BioRender.com).
The simplest method for increasing the Treg/Teff ratio in vivo is to infuse purified populations of Tregs (9). Indeed, pre-clinical studies as well as initial clinical trials have shown that adoptive Treg therapy is a promising therapeutic tool in the treatment of autoimmune diseases and in the induction of tolerance in the field of organ transplantation. However, for routine clinical application of adoptive Treg therapy, two key questions arise:
What is the best source of Tregs for infusion? In most clinical studies, Tregs prepared from peripheral blood mononuclear cells (PBMCs) are used as the starting population for ex vivo Treg cell expansion; for prevention of graft-versus-host disease (GvHD) after haematopoietic stem cell engraftment (HSCT), Tregs have also been prepared from umbilical cord blood (10). For HSCT, donor-derived Tregs are most effective in preventing GvHD, whereas for autoimmunity and organ transplantation, recipient-derived Tregs seem to be superior (11).
Do transferred Tregs need to be antigen specific? Although Tregs have to be activated to express their suppressor function, once activated their function is largely non-specific (12). For treatment of autoimmune disease, preclinical data suggest that antigen specific Tregs are superior to polyclonal Tregs in terms of their efficacy and lower risk of pan-immunosuppression (13, 14). However, in preclinical models of autoimmunity, the target antigens vary in their potency to prevent an unwanted immune response (15). Moreover, in human autoimmune diseases, Tregs seem to lack auto-antigen specificity and suppressive phenotype is limited (16). For transplantation tolerance, suppression by polyclonal Tregs can be effective, although Tregs with indirect specificity toward alloantigens have been shown to be preferable (17).
Several reports suggest, that the superiority of antigen specific Tregs is due to specific homing and activation in lymph nodes (15). However, polyclonal Tregs have been shown to prevent effector T cell homing to the graft by modulation of effector T cell trafficking (18). On this point, recent studies suggest that tailoring Treg homing efficiency might be the key to superior suppressor function (19).
The current methods for preparing polyclonal and antigen specific Tregs are considered below.
The potency of polyclonal Tregs for tolerance induction by adoptive cell therapy was shown many years ago by Sakaguchi and co-workers who showed that injections of sorted CD4+CD25+ T cells could rescue mice from organ-specific autoimmune diseases and GvHD-like wasting disease (20). These findings paved the way for comparable studies on Treg therapy for pre-clinical models of GvHD, autoimmune diseases and organ transplantation. For human studies, the first clinical trial of adoptive Treg therapy used purified fresh Tregs taken directly ex vivo for the treatment of GvHD after allogeneic HSCT (21). Thereafter, however, most clinical trials have used ex-vivo expanded Tregs; such expansion increases not only Treg cell numbers, but also their potency (22). In our hands, in vitro expanded polyclonal Tregs are superior to freshly isolated Tregs for induction of chimerism and tolerance in a murine mixed chimerism model (unpublished data of NP).
Polyclonal expansion of Tregs is generally induced by culturing purified CD4+ CD25+ cells with cross-linked anti-CD3/CD28 antibodies and IL-2 in vitro for 1–6 weeks, with most protocols using rapamycin to prevent Teff proliferation [The ONE study (23, 24)]. For efficient suppression of autoimmune disease and allograft survival, the expanded polyclonal Tregs must be injected in large numbers. Here, the main side effect is that increasing the total size of the Treg pool can lead to generalized immunosuppression (25). However, this effect is generally quite minor and substantial “off-target” immunosuppressive effects have not been reported so far.
Because of the risk of non-specific immunosuppression, there is increasing interest in the use of antigen-specific Tregs for tolerance induction, especially for solid organ transplantation. Hence, the transplant community is currently placing a lot of effort in the development of efficient approaches for expansion of alloantigen-reactive Tregs (26–29). Various cell culture protocols differing in the type and concentration of stimulator cells (donor derived PBMCs, CD40L activated donor B cells, B cell lines or DCs), the growth factors used and the duration of culture are under intensive investigation. Comparing these methods is difficult, however, because quantitating antigen-specific Tregs is still imprecise and to date has been shown only for freshly isolated Tregs (30).
Importantly, antigen-specific Tregs have been shown to play a major role in HLA-mediated susceptibility and protection of autoimmune diseases (31).
Despite the therapeutic potential of selectively expanding antigen-specific Tregs, cell culture protocols are complex, and the low frequency of the precursor cells tend to limit enthusiasm for this approach. Hence, there is parallel interest in developing methods for engineering Tregs to express antigen-specific TCRs or chimeric antigen receptors (CARs) (32).
TCR-transduced Tregs can be rapidly generated from polyclonal Tregs in large numbers. This is clearly an advantage over expanding Tregs from rare antigen-specific precursor cells, which can take up to 6 weeks of tissue culture. Moreover, TCR-transduced Tregs have been shown to migrate to the target tissue where they exert both antigen-specific and non-specific suppressive effects (33). As mentioned earlier, indirect bystander suppression by Tregs can be highly efficient at promoting allograft survival (17).
With their capacity to recognize native antigen while retaining typical T cytolytic activity, CAR T cells are an invaluable tool for treating hematologic malignancies (34). With further engineering, CAR T cells have been modified to suppress rather than kill target tissues by generating (non-MHC restricted) antigen-specific CAR Tregs from sorted natural Tregs (32, 35). Such CAR Tregs have been shown to prevent xenogeneic GvHD in a humanized murine HSCT model (35). The efficacy and suppressive stability of CAR Tregs has been demonstrated in colitis and skin transplantation models (36); several mechanisms of CAR Treg-mediated suppression have been suggested.
It should be noted that, for both TCR-transduced and CAR Tregs, their production requires retroviral transduction techniques. Hence, their safety for therapy is receiving scrutiny.
As an alternative to adoptive Treg therapy, Tregs can be selectively expanded in vivo by various methods. This approach increases the Treg : Teff cell ratio and allows polyclonally-expanded Tregs to mediate non-specific immunosuppression. This method is simpler and less expensive than adoptive Treg therapy. For organ transplantation, both methods are generally limited to situations where the organ concerned is taken from a living donor.
Several strategies for the in vivo induction of Tregs have been described and date back to initial studies on the capacity of anti-CD3 antibody to prolong allograft survival. Thus, treatment with anti-CD3 antibody is now known to induce Treg cell expansion while selectively depleting T effector cells, both in pre-clinical murine (37) and clinical studies (38, 39).
Another approach for Treg induction is to inhibit mTOR function in vivo. Thus, it has been shown that injection of rapamycin, an mTOR antagonist, selectively increases endogenous Treg frequency, in parallel with promoting TCR-induced anergy of conventional T cells (40, 41). Moreover, rapamycin has been shown to stabilize the suppressor function and gene expression profile of Tregs, both for endogenous and adoptively-transferred Tregs (42). Although failing as monotherapy, rapamycin was shown to promote long-term persistence of adoptively-transferred Tregs in combination with therapeutic IL-2 in a non-human primate model (43).
As for conventional T cells, Treg expansion and survival depend critically on contact with IL-2 (44). Moreover, manipulating how IL-2 is presented under in vivo conditions can be used to either reduce or augment the Treg : Teff ratio, and thereby either augment immunity or induce tolerance. For the latter, below we discuss several approaches that utilize IL-2 for Treg induction in vivo.
IL-2 therapy was originally developed for cancer immunotherapy because of its potency to enhance the growth of CD8+ T cell and NK cells (45). Despite conspicuous success in some patients, however, therapy with unmodified IL-2 has fallen into disfavor because of severe problems with toxicity. When given in low doses, IL-2 is much less toxic but loses its capacity to stimulate typical cytotoxic cells. Nevertheless, low-dose IL-2 does retain the capacity to stimulate Tregs, reflecting the fact that these cells, unlike CD8 T cells and NK cells, express CD25, the α-chain of the IL-2R. Thus, constitutive expression of the high-affinity IL-2Rαβγ makes Tregs more sensitive to IL-2 than conventional T cells, most of which express low-affinity IL-2Rβγ. For this reason, low-dose IL-2 therapy has emerged as a convenient method for inducing selective expansion of Tregs in vivo (46). Tolerance induction by low-dose IL-2 therapy is used mainly for treatment of autoimmunity (47) but is also showing promise for organ transplantation (48) and treatment of GvHD (49).
The major limitation of low-dose IL-2 therapy is that being small, IL-2 is rapidly excreted in the urine and so has a relatively short half-life (<30 min). As discussed below, this problem can be avoided by using IL-2/antibody complexes or IL-2 fusion proteins.
Another problem with IL-2 therapy is that CD25 expression is not unique to Tregs and is also expressed at a lower level on activated T cells. Hence, even in low doses, IL-2 based therapies may cause some level of effector T cell stimulation in addition to Treg expansion.
Studies on the effects of anti-IL-2 monoclonal antibodies (mabs) in vivo showed that injecting mice with IL-2 bound to particular IL-2 mabs, notably JES6-1, led to selective expansion of Tregs and the onset of immunosuppression (50). The capacity of IL-2/mab complexes to enhance allograft survival was demonstrated in a murine model of islet transplantation (51). Thus, short-term IL-2/mab complex treatment led to indefinite survival of >80% of islet allografts without any immunosuppression. When combined with rapamycin, these complexes were also potent at preventing induction of experimental autoimmune encephalomyelitis (EAE) and other autoimmune diseases (51, 52).
Despite prolonging the survival of islet allografts, IL-2 mab complexes failed to augment the survival of skin allografts as monotherapy (53). However, later experiments showed that modification of the treatment protocol led to prolonged skin allograft survival. Thus, when IL-2/mab complexes were supplemented with rapamycin and injected for 30 days, accompanied initially with short-term anti-inflammatory treatment with anti-IL-6 mab, survival of fully-mismatched murine skin allografts was improved from 15 days to 85 days (53). Treg levels returned to near-normal levels in the lymphoid tissues soon after the injections were stopped; however, intragraft Treg levels remained elevated for several weeks (53). Notably, although the grafts were eventually rejected, donor antibody formation was minimal. This and other findings suggested that prolonged graft survival was largely a reflection of Treg-meditated immunological ignorance. With regard to clinical relevance, the efficacy of IL-2/mab therapy is currently being investigated in a murine model of cardiac transplantation; in preliminary experiments, the results have shown long-term allograft survival and prevention of chronic allograft vasculopathy (unpublished data of NP).
As with pegylation and binding to albumin, fusing IL-2 with the Fc portion of IgG (IL-2-Fc) retards excretion and thereby considerably increases the half-life of IL-2. For low-dose IL-2 therapy, IL-2 fused to non-FcRγ binding human IgG1 has been shown to be superior to unmodified IL-2 for the induction of Tregs in a non-human primate model (54).
Based on the binding interaction of IL-2 with IL-2 mabs, IL-2 can be mutated to selectively stimulate either CD8 T cells and NK cells or Tregs (45). For Treg stimulation, IL-2 is mutated to bind poorly to IL-2R γ or β chains with retention of normal or above-normal binding to the α chain (55). These IL-2 muteins thus resemble IL-2/JES6-1 mab complexes in preferentially stimulating Tregs. When prepared as IL-2-Fc fusion proteins to increase half-life, these IL-2 muteins were superior to wild-type IL-2-Fc in the treatment of type I diabetes (T1D) in a pre-clinical mouse model (56).
It is now more than a decade since the first clinical trial using adoptive Treg therapy for the treatment of GvHD after allogeneic HSCT (21), which was followed by several early-phase trials focusing on safety, feasibility and tolerability of Treg infusions. Most of these phase I or phase I/II trials used polyclonally expanded Tregs from PBMCs and have been conducted in the setting of GvHD, new-onset T1D and solid organ transplantation (36).
Results of published and ongoing clinical trials in the setting of autoimmunity are summarized elsewhere (57). Notably, the first published trials using Treg therapy for T1D have reported cessation of disease progression or even remission of disease in some patients, promising not only safety and feasibility but also efficacy. Ongoing clinical trials suggest that effective tolerance protocols via Treg therapy will soon be available for several types of autoimmune disease. Data on long-term results are still very limited, however, and many questions remain open, including which cell product/expansion strategy is optimal in terms of cell yield/specificity/potency, and the number and frequency of Treg cell injections required for efficient tolerance induction. Hopefully, the ongoing clinical trials will resolve these questions.
For solid organ transplantation, a large number of clinical trials using Treg therapy for tolerance induction are ongoing. These trials are summarized in Table 1 and involve both polyclonal and antigen-specific Treg infusion.
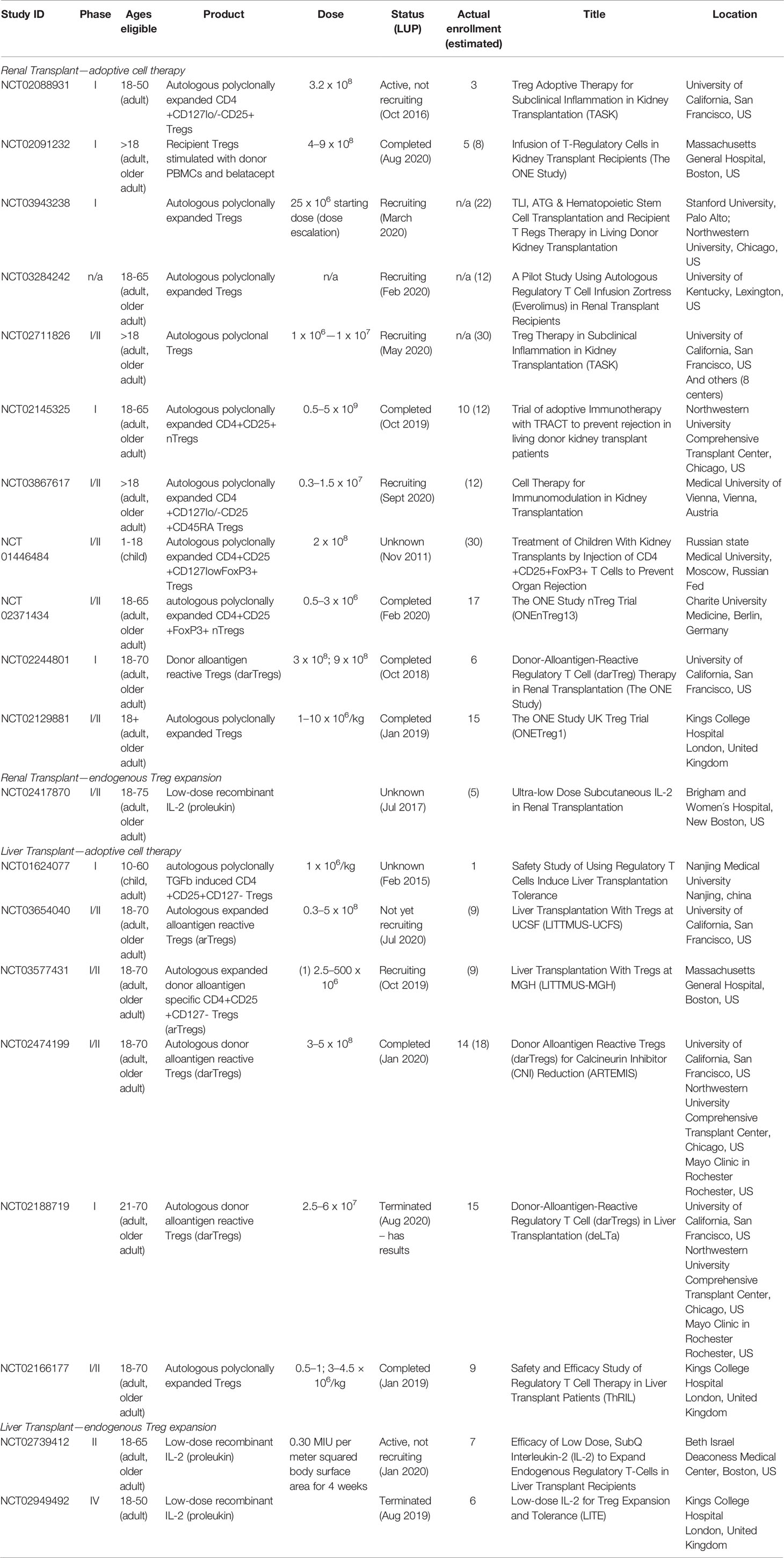
Table 1 Ongoing clinical trials adopting Tregs for tolerance induction in transplantation (search date Oct 1st 2020, listed in clinicaltrials.gov; LUP, last update posted).
In these studies, Treg isolation under GMP conditions was carried out with the “CliniMACS” system (CliniMACS TM Instruments, Miltenyi Biotec) which involves clinical-scale magnetic enrichment of cells (CD8 depletion followed by CD25 enrichment) in a closed and sterile system, or, outside European Union countries, by flow sorting of cells for expression of CD4, CD25, and CD127 (yielding populations of >99% purity). For preparation of antigen specific Tregs, magnetic bead isolation may be superior to flow-sorting of Tregs because contamination with residual antibodies impairs Treg expansion (26), perhaps by interfering with Treg : APC contact during culture (58).
The results of these trials will clearly be awaited with great interest. The results are not easy to predict because, in contrast to mice, inducing tolerance in humans is proving to be especially difficult. This issue is discussed below.
For autoimmune diseases, Treg based therapies are promising and have shown initial success for treatment of T1D, SLE and inflammatory bowel disease [(59, 60) and reviewed in (61)]. For solid organ transplantation, however, the results are less clear, perhaps reflecting that the precursor frequency of T cells recognizing allo-MHC is very high (up to 10%) (62, 63). For this reason, employing Treg therapy alone to induce allograft tolerance may be a challenging task. Moreover, as discussed below, there is the additional problem that effective models for tolerance inductions in mice are not necessarily applicable to clinical transplantation. Thus, it has to be borne in mind that typical mouse studies are generally based entirely on a single inbred strain housed in a “clean” environment; also, memory T cells, which are more difficult to regulate than naïve cells, are less common in clean mice than humans (64).
Despite the importance of Tregs, long-term tolerance to all antigens, including alloantigens, requires efficient elimination of reactive T cells in the thymus during ontogeny (65). In pre-clinical models, efficient tolerance to organ allografts generally requires hematopoietic chimerism, which is induced by transfer of donor stem cells after myelosuppressive treatment of the host to remove donor-reactive mature T cells (66). Although the mixed chimerism approach is also successful in a clinical setting (67), the side effects of this procedure are considerable (68). For this reason, there is much interest in developing mixed-chimerism approaches that avoid heavy immunosuppression of the host (69). For the protocol for HLA-disparate renal allografts developed by investigators at Massachusetts General Hospital (MGH), the transplant hosts are conditioned with non-myeloablative immunosuppression followed by combined kidney and HSCT. The results of this approach are promising and have shown long-term graft acceptance in a proportion of patients, with no evidence of GvHD. These findings are surprising because donor cell chimerism is only transient, implying that long-term graft survival involves some form of immunoregulation. Indeed, ongoing studies have shown that persistence of tolerance when chimerism wanes depends crucially on continuous contact with the donor kidney (70), such contact causing progressive elimination of graft-reactive effector T cells in parallel with expansion of graft-specific Tregs (71, 72).
These clinical data are intriguing and are in line with comparable studies in various preclinical models (73, 74). Overall, the results highlight the view that induction and maintenance of tolerance is remarkably complex and involves the combined effects of clonal deletion (affecting both immature and mature T cells) and specific suppression as well as bystander immunoregulation by Tregs. In addition, it is important to bear in mind that, for organ transplantation, tolerance protocols designed for particular organs may not be successful for other transplants. Thus, it was mentioned earlier that polyclonal Treg expansion in mice is much more tolerogenic for islet than skin allografts. Similarly, non-human primate studies have shown that the MGH mixed-chimerism protocol for renal allografts is much less successful for transplantation of other organs (70). There is also the enigma that tolerance induction to allografts is intrinsically difficult for some organs such as skin or intestines but relatively easy for certain other organs, notably the liver (75, 76). Whether this difference is simply related to organ size or has other explanations is still unclear (77).
Despite several decades of research in tolerance induction and Treg therapy in animal models, clinical trials with these models are still uncommon. For autoimmune disease, there is reason for optimism because early trials with Treg therapy are encouraging in terms of both safety and efficacy; moreover, new methods of genetic engineering to prepare antigen-specific Tregs and modify IL-2 to promote their survival in vivo show considerable promise (61, 78). However, many questions remain, including the range of autoimmune diseases suitable for Treg therapy. Thus, therapies that work well for one disease, e.g. multiple sclerosis, might work poorly for other diseases, e.g. systemic lupus erythematodes (SLE). In addition, there is the problem that many patients with autoimmune disease are routinely treated with immunosuppressive drugs, making it challenging to evaluate the effect of Treg cell therapy in the presence of these drugs. For these reasons, progress in employing tolerance protocols to treat autoimmune disease is likely to be slow and involve studies on multiple types of disease, both in animal models and clinical trials.
In the field of organ transplantation, using tolerance protocols to prevent rejection is still largely experimental because the current approach of continuous maintenance on calcineurin inhibitors and/or other immunosuppressants is remarkably successful. Nevertheless, the increased incidence of malignancy with this treatment as well as uncontrollable chronic rejection continues to elicit interest in devising methods for long-term tolerance induction without immunosuppression, especially in younger patients. Considering the success of permanent tolerance induction via mixed chimerism in animal models, it is clearly disappointing that achieving permanent chimerism without the risk of GvHD in a clinical setting is still not possible. However, the finding that even transient chimerism can be followed by prolonged graft survival and operational tolerance with renal allografts is clearly encouraging. Here, we envisage that, although Treg therapy alone might be insufficient to allow long-term allograft survival, combining Treg therapy with other forms of alloimmune response suppression will significantly boost tolerance induction, especially with antigen-specific Tregs and the use of IL-2 muteins to maintain their survival. Clearly much more research is needed in this area.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
NP and JS designed the concept and wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Austrian Science Fund (P31186-B28 to NP) and grants from the National Health and Medical Research Council of Australia to JS.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (New York NY) (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
2. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4(4):330–6. doi: 10.1038/ni904
3. Miyara M, Gorochov G, Ehrenstein M, Musset L, Sakaguchi S, Amoura Z. Human FoxP3+ regulatory T cells in systemic autoimmune diseases. Autoimmun Rev (2011) 10(12):744–55. doi: 10.1016/j.autrev.2011.05.004
4. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat Rev Immunol (2014) 14(6):377–91. doi: 10.1038/nri3667
5. Yi J, Kawabe T, Sprent J. New insights on T-cell self-tolerance. Curr Opin Immunol (2020) 63:14–20. doi: 10.1016/j.coi.2019.10.002
6. Sakaguchi S, Ono M, Setoguchi R, Yagi H, Hori S, Fehervari Z, et al. Foxp3+ CD25+ CD4+ natural regulatory T cells in dominant self-tolerance and autoimmune disease. Immunol Rev (2006) 212:8–27. doi: 10.1111/j.0105-2896.2006.00427.x
7. Sakaguchi S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu Rev Immunol (2004) 22:531–62. doi: 10.1146/annurev.immunol.21.120601.141122
8. Ramsdell F, Rudensky AY. Foxp3: a genetic foundation for regulatory T cell differentiation and function. Nat Immunol (2020) 21(7):708–9. doi: 10.1038/s41590-020-0694-5
9. Riley JL, June CH, Blazar BR. Human T regulatory cell therapy: Take a billion or so and call me in the morning. Immunity (2009) 30(5):656–65. doi: 10.1016/j.immuni.2009.04.006
10. Brunstein CG, Miller JS, Cao Q, McKenna DH, Hippen KL, Curtsinger J, et al. Infusion of ex vivo expanded T regulatory cells in adults transplanted with umbilical cord blood: safety profile and detection kinetics. Blood (2011) 117(3):1061–70. doi: 10.1182/blood-2010-07-293795
11. Pilat N, Klaus C, Hock K, Baranyi U, Unger L, Mahr B, et al. Polyclonal Recipient nTregs Are Superior to Donor or Third-Party Tregs in the Induction of Transplantation Tolerance. J Immunol Res (2015) 2015:562935. doi: 10.1155/2015/562935
12. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
13. Daly A, McAfee S, Dey B, Colby C, Schulte L, Yeap B, et al. Nonmyeloablative bone marrow transplantation: Infectious complications in 65 recipients of HLA-identical and mismatched transplants. Biol Blood Marrow Transplant (2003) 9(6):373–82. doi: 10.1016/S1083-8791(03)00100-9
14. Masteller EL, Warner MR, Tang Q, Tarbell KV, McDevitt H, Bluestone JA. Expansion of functional endogenous antigen-specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol (2005) 175(5):3053–9. doi: 10.4049/jimmunol.175.5.3053
15. Jaeckel E, von Boehmer H, Manns MP. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes (2005) 54(2):306–10. doi: 10.2337/diabetes.54.2.306
16. Long SA, Buckner JH. CD4+FOXP3+ T regulatory cells in human autoimmunity: more than a numbers game. J Immunol (2011) 187(5):2061–6. doi: 10.4049/jimmunol.1003224
17. Tsang JY, Tanriver Y, Jiang S, Xue SA, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest (2008) 118(11):3619–28. doi: 10.1172/JCI33185
18. Davidson TS, Shevach EM. Polyclonal Treg cells modulate T effector cell trafficking. Eur J Immunol (2011) 41(10):2862–70. doi: 10.1002/eji.201141503
19. Hoeppli RE, MacDonald KN, Leclair P, Fung VCW, Mojibian M, Gillies J, et al. Tailoring the homing capacity of human Tregs for directed migration to sites of Th1-inflammation or intestinal regions. Am J Transplant (2019) 19(1):62–76. doi: 10.1111/ajt.14936
20. Sakaguchi S, Sakaguchi N, Asano M, Itoh M, Toda M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J Immunol (1995) 155(3):1151–64.
21. Trzonkowski P, Bieniaszewska M, Juscinska J, Dobyszuk A, Krzystyniak A, Marek N, et al. First-in-man clinical results of the treatment of patients with graft versus host disease with human ex vivo expanded CD4+CD25+CD127- T regulatory cells. Clin Immunol (2009) 133(1):22–6. doi: 10.1016/j.clim.2009.06.001
22. Chai JG, Coe D, Chen D, Simpson E, Dyson J, Scott D. In vitro expansion improves in vivo regulation by CD4+CD25+ regulatory T cells. J Immunol (2008) 180(2):858–69. doi: 10.4049/jimmunol.180.2.858
23. Fraser H, Safinia N, Grageda N, Thirkell S, Lowe K, Fry LJ, et al. A Rapamycin-Based GMP-Compatible Process for the Isolation and Expansion of Regulatory T Cells for Clinical Trials. Mol Ther Methods Clin Dev (2018) 8:198–209. doi: 10.1016/j.omtm.2018.01.006
24. Sawitzki B, Harden PN, Reinke P, Moreau A, Hutchinson JA, Game DS, et al. Regulatory cell therapy in kidney transplantation (The ONE Study): a harmonised design and analysis of seven non-randomised, single-arm, phase 1/2A trials. Lancet (2020) 395(10237):1627–39. doi: 10.1016/S0140-6736(20)30167-7
25. Alzhrani A, Bottomley M, Wood K, Hester J, Issa F. Identification, selection, and expansion of non-gene modified alloantigen-reactive Tregs for clinical therapeutic use. Cell Immunol (2020) 357:104214. doi: 10.1016/j.cellimm.2020.104214
26. Peters JH, Hilbrands LB, Koenen HJ, Joosten I. Ex vivo generation of human alloantigen-specific regulatory T cells from CD4(pos)CD25(high) T cells for immunotherapy. PLoS One (2008) 3(5):e2233. doi: 10.1371/journal.pone.0002233
27. Putnam AL, Safinia N, Medvec A, Laszkowska M, Wray M, Mintz MA, et al. Clinical grade manufacturing of human alloantigen-reactive regulatory T cells for use in transplantation. Am J Transplant (2013) 13(11):3010–20. doi: 10.1111/ajt.12433
28. Sagoo P, Ali N, Garg G, Nestle FO, Lechler RI, Lombardi G. Human regulatory T cells with alloantigen specificity are more potent inhibitors of alloimmune skin graft damage than polyclonal regulatory T cells. Sci Trans Med (2011) 3(83):3002076. doi: 10.1126/scitranslmed.3002076
29. Landwehr-Kenzel S, Issa F, Luu SH, Schmuck M, Lei H, Zobel A, et al. Novel GMP-compatible protocol employing an allogeneic B cell bank for clonal expansion of allospecific natural regulatory T cells. Am J Transplant (2014) 14(3):594–606. doi: 10.1111/ajt.12629
30. Lalfer M, Chappert P, Carpentier M, Urbain D, Davoust JM, Gross D-A. Foxp3(+) Regulatory and Conventional CD4(+) T Cells Display Similarly High Frequencies of Alloantigen-Reactive Cells. Front Immunol (2019) 10:521–. doi: 10.3389/fimmu.2019.00521
31. Ooi JD, Petersen J, Tan YH, Huynh M, Willett ZJ, Ramarathinam SH, et al. Dominant protection from HLA-linked autoimmunity by antigen-specific regulatory T cells. Nature (2017) 545(7653):243–7. doi: 10.1038/nature22329
32. Zhang Q, Lu W, Liang CL, Chen Y, Liu H, Qiu F, et al. Chimeric Antigen Receptor (CAR) Treg: A Promising Approach to Inducing Immunological Tolerance. Front Immunol (2018) 9:2359. doi: 10.3389/fimmu.2018.02359
33. Stephens LA, Malpass KH, Anderton SM. Curing CNS autoimmune disease with myelin-reactive Foxp3+ Treg. Eur J Immunol (2009) 39(4):1108–17. doi: 10.1002/eji.200839073
34. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi: 10.1038/leu.2015.247
35. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
36. Rana J, Biswas M. Regulatory T cell therapy: Current and future design perspectives. Cell Immunol (2020) 356:104193. doi: 10.1016/j.cellimm.2020.104193
37. Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med (2003) 9(9):1202–8. doi: 10.1038/nm924
38. Keymeulen B, Vandemeulebroucke E, Ziegler AG, Mathieu C, Kaufman L, Hale G, et al. Insulin needs after CD3-antibody therapy in new-onset type 1 diabetes. N Engl J Med (2005) 352(25):2598–608. doi: 10.1056/NEJMoa043980
39. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
40. Powell JD, Lerner CG, Schwartz RH. Inhibition of cell cycle progression by rapamycin induces T cell clonal anergy even in the presence of costimulation. J Immunol (1999) 162(5):2775–84.
41. Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood (2005) 105(12):4743–8. doi: 10.1182/blood-2004-10-3932
42. Singh K, Stempora L, Harvey RD, Kirk AD, Larsen CP, Blazar BR, et al. Superiority of rapamycin over tacrolimus in preserving nonhuman primate Treg half-life and phenotype after adoptive transfer. Am J Transplant Off J Am Soc Transplant Am Soc Transplant Surgeons (2014) 14(12):2691–703. doi: 10.1111/ajt.12934
43. Furlan SN, Singh K, Lopez C, Tkachev V, Hunt DJ, Hibbard J, et al. IL-2 enhances ex vivo-expanded regulatory T-cell persistence after adoptive transfer. Blood Adv (2020) 4(8):1594–605. doi: 10.1182/bloodadvances.2019001248
44. Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol (2012) 12(3):180–90. doi: 10.1038/nri3156
45. Liao W, Lin JX, Leonard WJ. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity (2013) 38(1):13–25. doi: 10.1016/j.immuni.2013.01.004
46. Klatzmann D, Abbas AK. The promise of low-dose interleukin-2 therapy for autoimmune and inflammatory diseases. Nat Rev Immunol (2015) 15(5):283–94. doi: 10.1038/nri3823
47. Rosenzwajg M, Churlaud G, Mallone R, Six A, Derian N, Chaara W, et al. Low-dose interleukin-2 fosters a dose-dependent regulatory T cell tuned milieu in T1D patients. J Autoimmun (2015) 58:48–58. doi: 10.1016/j.jaut.2015.01.001
48. Pilon CB, Petillon S, Naserian S, Martin GH, Badoual C, Lang P, et al. Administration of low doses of IL-2 combined to rapamycin promotes allogeneic skin graft survival in mice. Am J Transplant (2014) 14(12):2874–82. doi: 10.1111/ajt.12944
49. Matsuoka K, Koreth J, Kim HT, Bascug G, McDonough S, Kawano Y, et al. Low-dose interleukin-2 therapy restores regulatory T cell homeostasis in patients with chronic graft-versus-host disease. Sci Trans Med (2013) 5(179):179ra43. doi: 10.1126/scitranslmed.3005265
50. Boyman O, Kovar M, Rubinstein MP, Surh CD, Sprent J. Selective stimulation of T cell subsets with antibody-cytokine immune complexes. Science (New York NY (2006) 311(5769):1924–7. doi: 10.1126/science.1122927
51. Webster KE, Walters S, Kohler RE, Mrkvan T, Boyman O, Surh CD, et al. In vivo expansion of T reg cells with IL-2-mAb complexes: induction of resistance to EAE and long-term acceptance of islet allografts without immunosuppression. J Exp Med (2009) 206(4):751–60. doi: 10.1084/jem.20082824
52. Yokoyama Y, Iwasaki T, Kitano S, Satake A, Nomura S, Furukawa T, et al. IL-2-Anti-IL-2 Monoclonal Antibody Immune Complexes Inhibit Collagen-Induced Arthritis by Augmenting Regulatory T Cell Functions. J Immunol (2018) 201(7):1899–906. doi: 10.4049/jimmunol.1701502
53. Pilat N, Wiletel M, Weijler AM, Steiner R, Mahr B, Warren J, et al. Treg-mediated prolonged survival of skin allografts without immunosuppression. Proc Natl Acad Sci U S A (2019) 116(27):13508–16. doi: 10.1073/pnas.1903165116
54. Bell CJ, Sun Y, Nowak UM, Clark J, Howlett S, Pekalski ML, et al. Sustained in vivo signaling by long-lived IL-2 induces prolonged increases of regulatory T cells. J Autoimmun (2015) 56:66–80. doi: 10.1016/j.jaut.2014.10.002
55. Peterson LB, Bell CJM, Howlett SK, Pekalski ML, Brady K, Hinton H, et al. A long-lived IL-2 mutein that selectively activates and expands regulatory T cells as a therapy for autoimmune disease. J Autoimmun (2018) 95:1–14. doi: 10.1016/j.jaut.2018.10.017
56. Khoryati L, Pham MN, Sherve M, Kumari S, Cook K, Pearson J, et al. An IL-2 mutein engineered to promote expansion of regulatory T cells arrests ongoing autoimmunity in mice. Sci Immunol (2020) 5(50):eaba5264. doi: 10.1126/sciimmunol.aba5264
57. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
58. Cherai M, Hamel Y, Baillou C, Touil S, Guillot-Delost M, Charlotte F, et al. Generation of Human Alloantigen-Specific Regulatory T Cells Under Good Manufacturing Practice-Compliant Conditions for Cell Therapy. Cell Transplant (2015) 24(12):2527–40. doi: 10.3727/096368914X683566
59. Marek-Trzonkowska N, Myśliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juścińska J, et al. Therapy of type 1 diabetes with CD4+CD25highCD127-regulatory T cells prolongs survival of pancreatic islets — Results of one year follow-up. Clin Immunol (2014) 153(1):23–30. doi: 10.1016/j.clim.2014.03.016
60. Dall’Era M, Pauli ML, Remedios K, Taravati K, Sandova PM, Putnam AL, et al. Adoptive Treg Cell Therapy in a Patient With Systemic Lupus Erythematosus. Arthritis Rheumatol (2019) 71(3):431–40. doi: 10.1002/art.40737
61. Esensten JH, Muller YD, Bluestone JA, Tang Q. Regulatory T-cell therapy for autoimmune and autoinflammatory diseases: The next frontier. J Allergy Clin Immunol (2018) 142(6):1710–8. doi: 10.1016/j.jaci.2018.10.015
62. Ford WL, Atkins RC. The proportion of lymphocytes capable of recognizing strong transplantation antigens in vivo. Adv Exp Med Biol (1973) 29(0):255–62. doi: 10.1007/978-1-4615-9017-0_37
63. Matzinger P, Bevan MJ. Hypothesis: why do so many lymphocytes respond to major histocompatibility antigens? Cell Immunol (1977) 29(1):1–5. doi: 10.1016/0008-8749(77)90269-6
64. Beura LK, Hamilton SE, Bi K, Schenkel JM, Odumade OA, Casey KA, et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature (2016) 532(7600):512–6. doi: 10.1038/nature17655
65. Griesemer AD, Sorenson EC, Hardy MA. The role of the thymus in tolerance. Transplantation (2010) 90(5):465–74. doi: 10.1097/TP.0b013e3181e7e54f
66. Pilat N, Wekerle T. Transplantation tolerance through mixed chimerism. Nat Rev (2010) 6(10):594–605. doi: 10.1038/nrneph.2010.110
67. Mahr B, Granofszky N, Muckenhuber M, Wekerle T. Transplantation Tolerance through Hematopoietic Chimerism: Progress and Challenges for Clinical Translation. Front Immunol (2017) 8:1762. doi: 10.3389/fimmu.2017.01762
68. Leventhal J, Abecassis M, Miller J, Gallon L, Tollerud D, Elliott MJ, et al. Tolerance Induction in HLA Disparate Living Donor Kidney Transplantation by Donor Stem Cell Infusion: Durable Chimerism Predicts Outcome. Transplantation (2013) 95(1):169–76. doi: 10.1097/TP.0b013e3182782fc1
69. Kawai T, Cosimi AB, Spitzer TR, Tolkoff-Rubin N, Suthanthiran M, Saidman SL, et al. HLA-mismatched renal transplantation without maintenance immunosuppression. N Engl J Med (2008) 358(4):353–61. doi: 10.1056/NEJMoa071074
70. Sasaki H, Oura T, Spitzer TR, Chen YB, Madsen JC, Allan J, et al. Preclinical and clinical studies for transplant tolerance via the mixed chimerism approach. Hum Immunol (2018) 79(5):258–65. doi: 10.1016/j.humimm.2017.11.008
71. Hotta K, Aoyama A, Oura T, Yamada Y, Tonsho M, Huh KH, et al. Induced regulatory T cells in allograft tolerance via transient mixed chimerism. JCI Insight (2016) 1(10):e86419. doi: 10.1172/jci.insight.86419
72. Pilat N, Mahr B, Unger L, Hock K, Schwarz C, Farkas A, et al. Incomplete clonal deletion as prerequisite for tissue-specific minor antigen tolerization. JCI Insight (2016) 1(7):e85911. doi: 10.1172/jci.insight.85911
73. Yamada Y, Nadazdin O, Boskovic S, Lee S, Zorn E, Smith RN, et al. Repeated Injections of IL-2 Break Renal Allograft Tolerance Induced via Mixed Hematopoietic Chimerism in Monkeys. Am J Transplant (2015) 15(12):3055–66. doi: 10.1111/ajt.13382
74. Pilat N, Farkas AM, Mahr B, Schwarz C, Unger L, Hock K, et al. T-regulatory cell treatment prevents chronic rejection of heart allografts in a murine mixed chimerism model. J Heart Lung Transplant (2014) 33(4):429–37. doi: 10.1016/j.healun.2013.11.004
75. Todo S, Yamashita K, Goto R, Zaitsu M, Nagatsu A, Oura T, et al. A pilot study of operational tolerance with a regulatory T-cell-based cell therapy in living donor liver transplantation. Hepatology (2016) 64(2):632–43. doi: 10.1002/hep.28459
76. Starzl TE, Zinkernagel RM. Transplantation tolerance from a historical perspective. Nat Rev Immunol (2001) 1(3):233–9. doi: 10.1038/35105088
77. Steger U, Denecke C, Sawitzki B, Karim M, Jones ND, Wood KJ. Exhaustive differentiation of alloreactive CD8+ T cells: critical for determination of graft acceptance or rejection. Transplantation (2008) 85(9):1339–47. doi: 10.1097/TP.0b013e31816dd64a
Keywords: regulatory T cells, tolerance, IL-2 complexes, cell therapy, immunotherapy, autoimmunity, transplantation
Citation: Pilat N and Sprent J (2021) Treg Therapies Revisited: Tolerance Beyond Deletion. Front. Immunol. 11:622810. doi: 10.3389/fimmu.2020.622810
Received: 29 October 2020; Accepted: 14 December 2020;
Published: 28 January 2021.
Edited by:
Byron Au-Yeung, Emory University, United StatesReviewed by:
Robert Ian Lechler, King’s College London, United KingdomCopyright © 2021 Pilat and Sprent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonathan Sprent, ai5zcHJlbnRAZ2FydmFuLm9yZy5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.