 Bram Van Den Eeckhout
Bram Van Den Eeckhout Jan Tavernier
Jan Tavernier Sarah Gerlo
Sarah Gerlo
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 27 January 2021
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.621931
The three-signal paradigm tries to capture how the innate immune system instructs adaptive immune responses in three well-defined actions: (1) presentation of antigenic peptides in the context of MHC molecules, which allows for a specific T cell response; (2) T cell co-stimulation, which breaks T cell tolerance; and (3) secretion of polarizing cytokines in the priming environment, thereby specializing T cell immunity. The three-signal model provides an empirical framework for innate instruction of adaptive immunity, but mainly discusses STAT-dependent cytokines in T cell activation and differentiation, while the multi-faceted roles of type I IFNs and IL-1 cytokine superfamily members are often neglected. IL-1α and IL-1β are pro-inflammatory cytokines, produced following damage to the host (release of DAMPs) or upon innate recognition of PAMPs. IL-1 activity on both DCs and T cells can further shape the adaptive immune response with variable outcomes. IL-1 signaling in DCs promotes their ability to induce T cell activation, but also direct action of IL-1 on both CD4+ and CD8+ T cells, either alone or in synergy with prototypical polarizing cytokines, influences T cell differentiation under different conditions. The activities of IL-1 form a direct bridge between innate and adaptive immunity and could therefore be clinically translatable in the context of prophylactic and therapeutic strategies to empower the formation of T cell immunity. Understanding the modalities of IL-1 activity during T cell activation thus could hold major implications for rational development of the next generation of vaccine adjuvants.
Innate immune cells represent the first line of defense in response to an injury or a pathogen encounter. Next to this, specialized innate immune cells called antigen-presenting cells (APCs), including dendritic cells (DCs), also collect peripheral information on the source of the antigen and transfer this data to cells of the adaptive immune system. This information transfer happens over three major routes: (1) presentation of antigenic peptides to antigen-specific T cells, resulting in an antigen-specific response; (2) co-stimulation, which can be perceived as qualitative information on the context of the antigen and allows for breaking tolerance; and (3) release of priming cytokines that polarize the T cell response, as such raising a tailor-made reaction to the encountered threat. This so-called “three-signal paradigm” places activation of the adaptive immune system under innate control and is currently a well-established paradigm in immunobiology (1). In order to perform these essential functions, innate immune cells are equipped with germline-encoded extra- and intracellular pathogen-recognition receptors (PRRs). These allow the innate immune system to recognize evolutionary conserved “non-self” microbial components (pathogen-associated molecular patterns (PAMPs), e.g. bacterial cell wall components or viral nucleic acids) and host-derived damage-associated molecular patterns (DAMPs), e.g. extracellular ATP and monosodium urate crystals) (2, 3).
The T cell reaction that results from this three-leveled instruction can be regarded as “productive” adaptive immunity: a measurable and antigen-specific response that is characterized by activated T cells that undergo clonal expansion and produce effector cytokines, but which is not necessarily “protective” upon pathogen encounter (4). Vaccines aim to mimic natural infections, but progression from productive to actually protective T cell responses remains a major challenge for the vaccine field, where most strategies are still dependent on the generation of long-lasting neutralizing antibodies (4, 5). Nevertheless, there is a dire need for T cell-inducing vaccines, as both CD4+ and CD8+ T cell responses have been deemed crucial for protection against different pathogens, such as Plasmodium falciparum, HIV and Mycobacterium tuberculosis, and for successful treatment of cancer. Although a lot of research has already been dedicated to develop vaccines that provide protective T cell immunity, the majority of these efforts were found to only result in low-level and non-protective responses (5, 6).
A limitation of the three-signal model, as it stands today, is its oversimplification of how the innate immune system controls and sculpts T cell responses. Indeed, innate immune cells provide extra clues on the origin of the antigen outside of the three major information routes addressed above. In their 2017 opinion piece (4), Jain and Pasare discuss different limitations of the current three-signal model and argue that its revisitation is needed in order to successfully translate these principles for the generation of protective T cell responses in different contexts. For instance, the fact that pathogens carry ligands for multiple PRRs infers cross-talk between different PRRs in innate immune cells, which can alter the outcome of their activation. Eventual occurrence of co-infection should also be considered, as this was found to possibly influence the T cell reaction mounted in response to the initial infection. Recent work has also demonstrated that the host’s history of prior infection, which is often neglected in mouse models, and the composition of the commensal microbiota and the metabolites derived thereof can sculpt the behavior of innate and adaptive immune cells (4).
Another issue is that during T cell priming, the cytokine environments that are formed are probably more complex and include other players next to the prototypical priming cytokines. The innate cytokines produced during the initiation of T cell immunity can be roughly categorized based on their dependence on STATs or MyD88 for signaling. Cytokines that signal via STATs are well-studied as these induce the expression of lineage-specific transcription factors and as such drive differentiation of CD4+ T cells towards different phenotypes (Table 1). This plasticity is not restricted to CD4+ T cells as also naive CD8+ T cells can acquire different phenotypes during priming, not all of which with cytotoxic functionality and the capacity to produce IFN-γ. These so-called TC subsets mirror the different CD4+ T cell phenotypes and are also shaped by the same environmental STAT-signaling cytokines as for their CD4+ counterparts (30). The roles of MyD88-dependent cytokines in the priming environment, including interleukin-1 (IL-1) cytokine superfamily members, are rather neglected as the focus of the three-signal model primarily lies on STAT-signaling cytokines that polarize the T cell response (4). However, the pro-inflammatory activities of the IL-1 cytokines IL-1α and IL-1β take on a central role during activation of the innate immune system and innate instruction of adaptive immunity (1). IL-1α is released in the extracellular environment as a consequence of cellular damage and locally acts as an alarmin. On the other hand, innate immune cells integrate PAMP and DAMP signals and respond by controlled production of IL-1β (31).
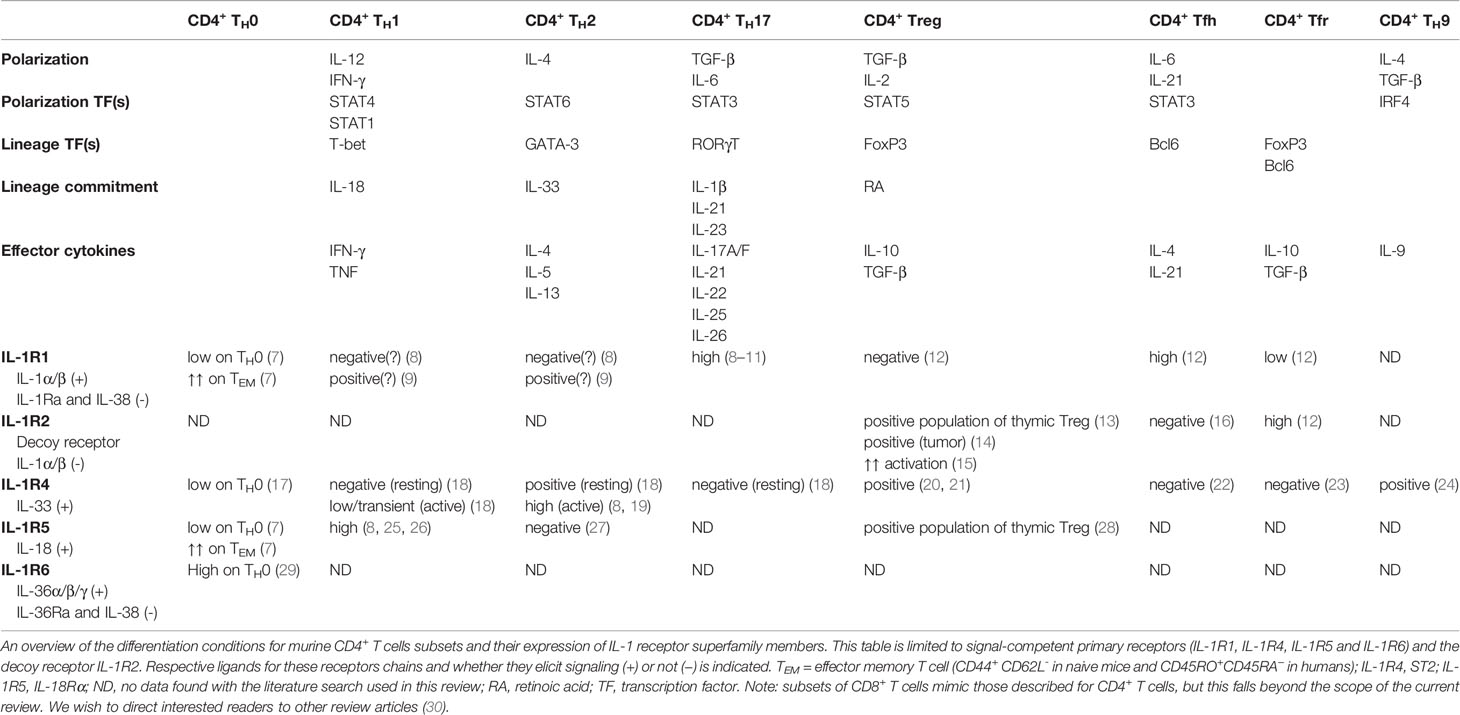
Table 1 Overview of the described subsets of CD4+ T cells discussed in this review.
Here, we will review established and new literature in support of a role for IL-1 as innate instructor of adaptive immune responses and elaborate on how IL-1 activity influences three protagonists of the three-signal model: DCs, CD4+ and CD8+ T cells. It has become clear that IL-1 activity on both the antigen-presenting DC and the T cell, either alone or in synergy with priming cytokines, can contribute to the formation of protective T cell immunity. As such, IL-1α and IL-1β might play a prominent role as innate instructors of adaptive immune responses. Gaining more insight in the environmental context wherein T cell priming takes place may help to advance prophylactic and therapeutic applications (4).
The IL-1 superfamily of ligands and receptors currently comprises 21 members: 11 soluble factors and 10 receptor molecules. Among these soluble factors, seven perform pro-inflammatory actions (IL-1α, IL-1β, IL-18, IL-33, IL-36α, IL-36β, and IL-36γ) and four display anti-inflammatory activities. Two of these anti-inflammatory mediators are receptor agonists (IL-37 and IL-38) and two are receptor antagonists (IL-1Ra and IL-36Ra). The IL-1 receptor superfamily is organized in several (overlapping) subgroups: ligand-binding receptor proteins (IL-1R1, IL-1R2, IL-1R4, IL-1R5, and IL-1R6), accessory chains (IL-1R3 and IL-1R7), molecules that inhibit signaling (IL-1R2, IL-1R8, and IL-18 binding protein) and orphan receptors (IL-1R9 and IL-1R10). In this review, we solely discuss the IL-1 cytokines IL-1α and IL-1β. These cytokines exert their biology via the same binary receptor complex that contains the primary receptor IL-1R1 and the accessory chain IL-1R3 (31–33). From here on, this is the IL-1 receptor (IL-1R) complex referred to in this review.
IL-1α is constitutively expressed as a 31 kDa precursor protein (pro-IL-1α) with a basal level of pro-inflammatory activity (31, 34). Expression of pro-IL-1α varies over different cell types, but cells lining body barriers, such as epithelial and endothelial cells, contain relatively high cytoplasmic levels of this protein under homeostatic conditions (35). Alternatively, expression of the IL-1α precursor protein is inducible and potential triggers include pro-inflammatory mediators (e.g. IL-1α itself and IL-1β) and growth or stress-associated factors (e.g. TLR agonists, such as LPS) (35, 36). Pro-IL-1α comprises an N-terminal precursor part, which is linked to the mature and fully biologically active IL-1α cytokine (31, 34). This N-terminus contains the LKKRRL nuclear localization sequence (NLS) that allows for translocation of pro-IL-1α to the nucleus (37). Nuclear translocation is regulated by interaction with HCLS1-associated protein X (HAX)-1 (38). In the nucleus, pro-IL-1α can directly regulate the expression of pro-inflammatory chemokines and interact with histone modifying enzymes, such as the acetyltransferases p300, p300/CBP-associated factor (PCAF) and GCN5 (39, 40). Pro-IL-1α continuously shuttles between the nucleus and the cytosol within nanoseconds and its commitment to a subcellular compartment is determined by environmental cues. When the cell receives a pro-apoptotic trigger, pro-IL-1α rapidly migrates into the nucleus and binds tightly to the chromatin, while upon necrotic cell death, the precursor is found exclusively in the cytosol (41). As such, accidental cell death results in far more pro-inflammatory lysates and under these conditions, pro-IL-1α performs the role of a classical DAMP, hence its “alarmin” alias (35, 41). Pro-IL-1α can be further processed intra- and extracellularly by different proteases, including calpain and neutrophil elastases. These cleave the N-terminal precursor from the mature IL-1α protein, which augments biological activity (34). Next to a nuclear and an extracellular role, pro-IL-1α can also appear bound on the cell surface following stimulation of hematopoietic and non-hematopoietic cells, including fibroblasts and endothelial cells, with pro-inflammatory mediators (35, 42). Pro-IL-1α probably becomes glycosylated intracellularly and subsequently anchors to the cell membrane via a lectin-like interaction. This membrane-associated pro-IL-1α has the ability to interact with IL-1R complexes on neighboring cells and as such mediate paracrine signaling (34, 35). Membrane-bound pro-IL-1α can be shaved from the cell surface by extracellular proteases, as such allowing for its release in the environment (35, 43). Figure 1 summarizes the expression, post-translational modification and subsequent processing of (pro-)IL-1α.
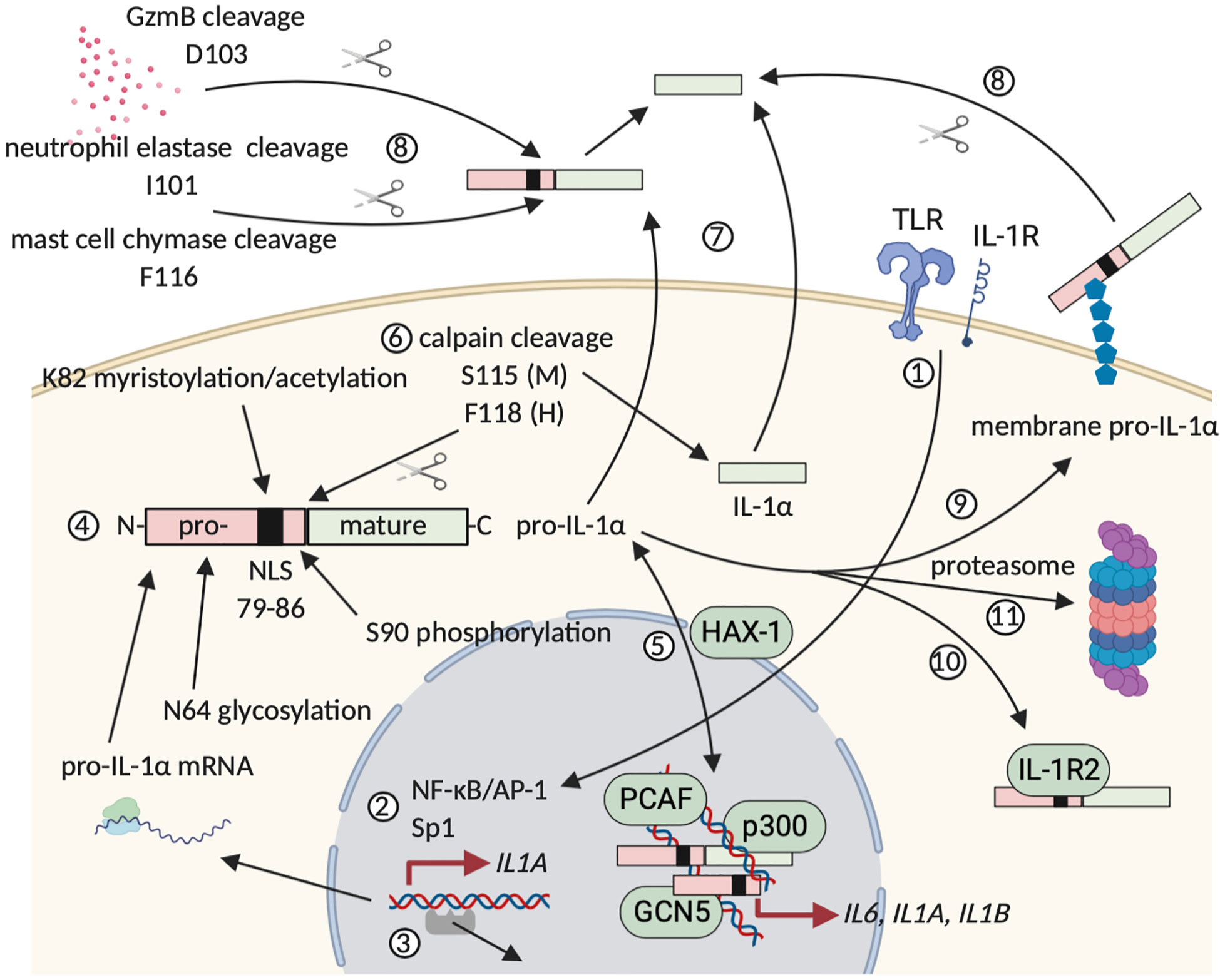
Figure 1 Synthesis, modification and extracellular release of human (pro-)IL-1α. (1) and (2) Inflammatory triggers can induce pro-IL-1α expression, whereas homeostatic expression is regulated by housekeeping transcription factors, such as Sp1. (3) Alternatively, expression can be initiated following DNA release of negative regulators. (4) Pro-IL-1α is modified post-translationally. (5) Pro-IL-1α continuously shuttles between cytosol and nucleus and nuclear translocation is mediated via HAX-1, which binds to the pro-IL-1α NLS. Binding of pro-IL-1α to chromatin sequesters the cytokine in the nucleus, whereas interaction with histone modifying enzymes mediates pro-inflammatory gene expression. (6) Calpain cleaves pro-IL-1α in the cytosol, leading to the formation of mature and fully biologically active IL-1α. (7) and (8) Following accidental cell death or damage, (pro-)IL-1α is released extracellularly, where maturation by cleavage is mediated by granzyme B, neutrophil elastases and mast cell chymases. (9) Post-translational modification can allow for extracellular membrane anchorage of pro-IL-1α. (10) A unique intracellularly occurring form of IL-1R2 can neutralize and sequester pro-IL-1α in the cytosol, possibly by masking its NLS. (11) Turnover of (pro-)IL-1α and the cleaved N-terminal pro-piece is mediated in the proteasome. Figure adapted from (35). Created with BioRender.com.
IL-1β is a circulating factor, for which the expression is not constitutive, but tightly regulated. A major source of IL-1β are cells of myeloid origin, which express the cytokine as a cytoplasmic 31 kDa precursor protein (pro-IL-1β) that needs to be converted into a mature form to allow for activity (31, 44, 45). The conventional mechanism leading to activation of pro-IL-1β is proteolytic cleavage in the N-terminal precursor region by caspase 1 (46, 47). Caspase 1 houses in the cytoplasm as an inactive protease that depends on further proteolytic processing to become activated, which takes place in multimolecular protein complexes called inflammasomes (34, 48, 49). Processing of inactive pro-caspase 1 into fully biologically active caspase 1 can be mediated by different canonical inflammasomes, the NLRP3 inflammasome unarguably being the most extensively studied (48–50). Assembly and activation of the NLRP3 inflammasome depends on two distinct signals: (1) an NF-κB-activating stimulus, such as a cytokine or TLR agonist (PAMP), which induces the expression of pro-IL-1β, NLRP3 and caspase 1 in the cytosol (“priming”); and (2) an NLRP3-activating agent that leads to the actual assembly of the functional inflammasome complex (“activation”) (50, 51). This signal two can be rendered by different bacterial, viral and fungal PAMPs (e.g. peptidoglycan, single and double stranded RNA and zymosan) and a multitude of DAMPs (e.g. extracellular ATP and monosodium urate crystals) (50, 52–56). Alternatively, cleavage of pro-IL-1β can be mediated by caspase 8, a caspase family protease that is commonly associated with pro-apoptotic signaling via TNF receptor family members, such as Fas and TRAIL (57). Although unprocessed IL-1β is usually not found in the extracellular space, it can be found outside of the cell as a result of uncontrolled cell death following a stressful event or a strong inflammatory trigger (34). Different cleavage sites for neutrophil proteases and mast cell chymases are present within the N-terminal region of pro-IL-1β. Despite the fact that these proteases can individually yield mature and biologically active IL-1β (58, 59), recent work found that protease mixtures from phorbol 12-myristate 13-acetate (PMA)-activated neutrophils can activate pro-IL-1α, but surprisingly fail to activate pro-IL-1β (60). As IL-1β contains no leader sequence, the matured cytokine is released in the environment via an unconventional secretion mechanism that is independent of the endoplasmic reticulum (ER) and Golgi network (61). Multiple theories on how such a “leaderless” protein is released in the environment have been proposed, one of the earlier working models suggesting that IL-1β could be secreted via vesicular mechanisms, such as secretory lysosomes, multivesicular bodies and exosomes, or via secretory autophagy (61–64). Recent research into the secretion mechanism of IL-1β focuses on pyroptosis, a rapid and inflammatory form of programmed cell death induced following NLRP3 inflammasome activation. The pore-forming protein gasdermin D (GSDMD) was recently identified as the primary mediator of pyroptosis and its activity was found to depend on maturation by caspase 1 cleavage. Pyroptosis ultimately leads to cell death, which allows for the release of mature IL-1β in the process (65–67). However, GSDMD pores can perform an additional non-pyroptotic role that allows for IL-1β release from viable cells with intact, but porous cell membranes (68–70). In Figure 2, the controlled expression of (pro-)IL-1β is illustrated graphically.
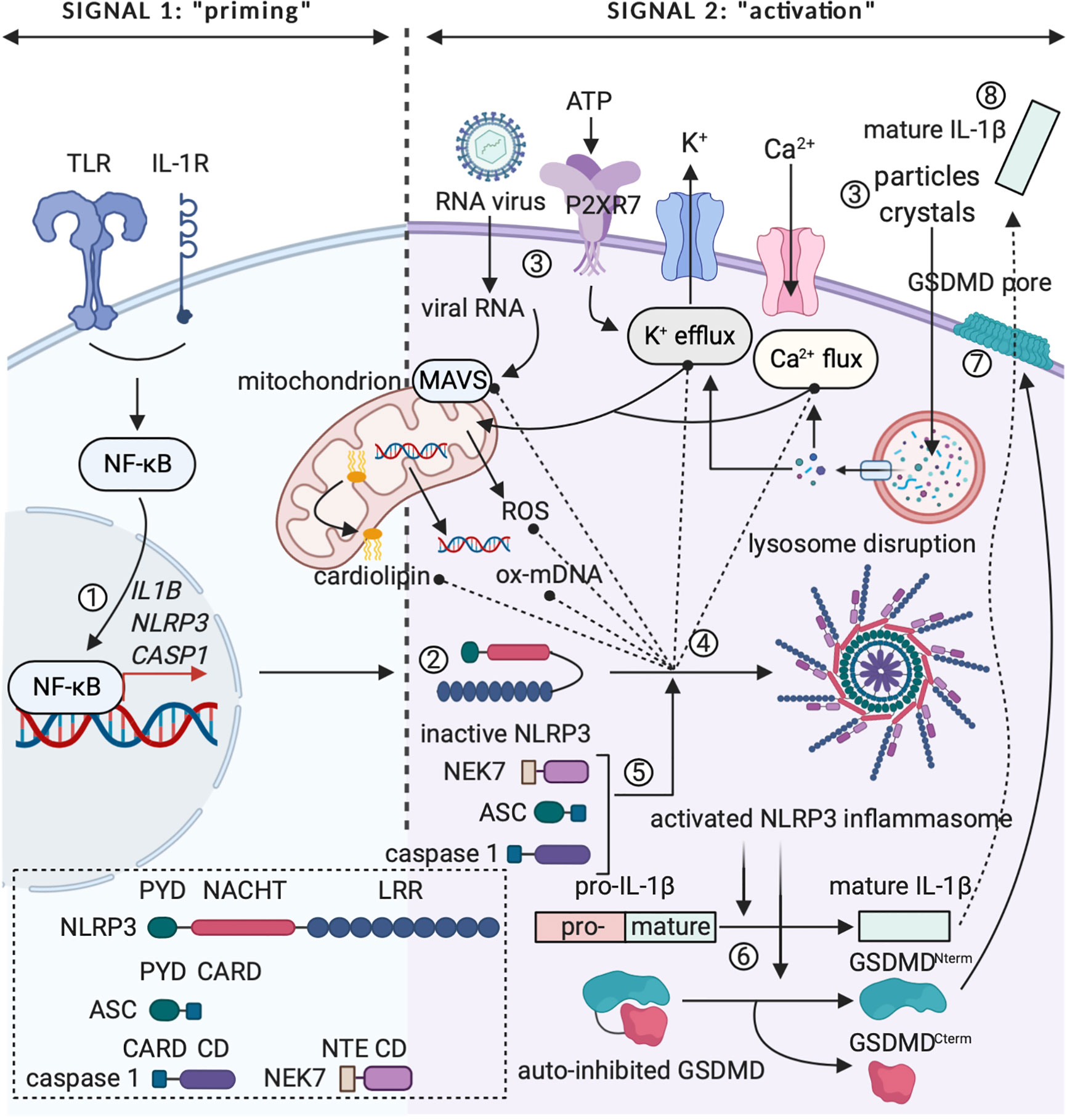
Figure 2 The two-step paradigm of controlled IL-1β release. (1) Signal 1 or “priming” involves expression of pro-IL-1β, NLRP3 and caspase 1 following triggering of pro-inflammatory receptor complexes (e.g. TLR4 or IL-1R) and activation of the transcription factor NF-κB. (2) NLRP3’s LRR falls back onto the NACHT domain, rendering the protein inactive in the cytoplasm. (3) Signal 2 or “activation” can be mediated by PAMPs (e.g. viral RNA) or DAMPs (e.g. extracellular ATP, extracellular particles and crystal complexes). Viral RNA can activate mitochondrial antiviral signaling (MAVS) protein. ATP is detected via the P2RX7 receptor and extracellular particles and crystal complexes can induce intracellular lysosome disruption, leading to changes in Ca2+ flux and K+ efflux. Intracellular changes in ion concentration can influence mitochondria, as such inducing release of ROS and oxidized mtDNA or relocalization of cardiolipin to the outer mitochondrial membrane. (4) and (5) Relocalized cardiolipin, oxidized mDNA, activated MAVS protein and changes in cytosolic ion concentrations can induce conformational changes in NLRP3 and lead to formation of the activated NLRP3 inflammasome after recruitment of NEK7, ASC and caspase 1. (6) Activated caspase 1 cleaves immature pro-IL-1β to mature and fully biologically active IL-1β. In addition, caspase 1 can cleave the GSDMD protein. (7) GSDMDNterm integrates in the membrane as a pore, which induces pyroptosis and represents a putative way for IL-1β release. CD, catalytic domain; NTE, N-terminal extension. Figure adapted from (50). Created with BioRender.com.
Receptors within the receptor superfamily generally have a strongly conserved, homologous molecular structure. The extracellular part of these molecules is “question mark”- or “grasping hand”-like shaped and comprises three immunoglobulin (Ig)-like domains (D1, D2 and D3). The transmembrane domain is a single helix that anchors the extracellular portion in the plasma membrane. The intracellular part contains a Toll-IL-1R (TIR) domain, which is responsible for signal transduction (32, 33). Agonists of the IL-1 cytokine superfamily mediate their signaling via a three-step mechanism: (1) formation of a binary complex upon binding to a ligand-binding receptor subunit; (2) formation of a ternary complex after inducing a conformational change in the ligand-binding receptor subunit, which allows for recruitment and binding to an accessory chain; (3) juxtaposing the TIR domains of both receptor subunits (if present), which allows for signal transduction (31–33). As mentioned above, IL-1α and IL-1β signal via a receptor complex composed of the primary ligand-binding subunit IL-1R1 and the accessory chain IL-1R3. The three extracellular Ig-like domains of IL-1R1 provide two comparably-sized cytokine interaction interfaces. D1 and D2, which are relatively rigid and positioned tightly next to each other, form a first binding interface, while a second contact area is provided by the more flexible D3 (71). The D1/D2 interface has been described to be sufficient for capturing the ligand (72), but the interaction provided by the D3 domain is responsible for the conformational changes in IL-1R1 that allow for recruitment and binding to IL-1R3 (73, 74). The accessory protein binds with its backside to ligand-bound IL-1R1/2, making contacts with D2 and D3 (73, 74). Upon formation of the ternary complex, the intracellular TIR domains of the ligand-binding subunit and the accessory chain juxtapose, as such forming a scaffold structure that allows for recruitment and binding of a sequence of adaptor molecules to the intracellular site of activated receptor complexes, which initiates downstream signal transduction and activation of different transcription factors (32, 33). Downstream IL-1R signaling events are extensively reviewed elsewhere (32, 36) and we summarized the main modalities in Figure 3.
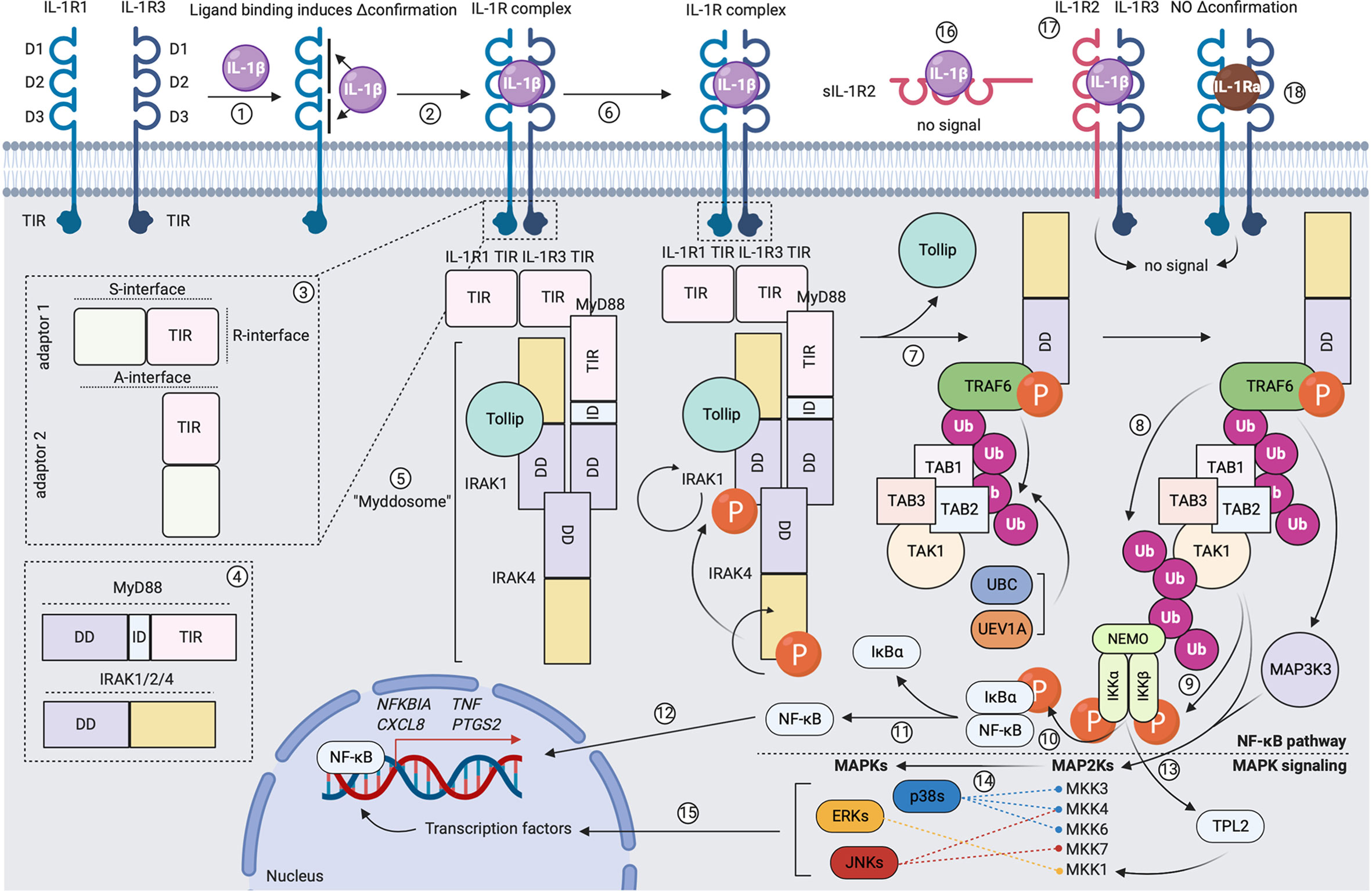
Figure 3 Formation of the signaling-competent IL-1R complex and downstream IL-1 signaling. (1) Extracellular IL-1β makes two contacts with IL-1R1 and induces a conformational change in the primary receptor. (2) This allows IL-1R1 to recruit and bind to co-receptor IL-1R3. (3) Receptor dimerization leads to scaffolding of intracellular receptor TIR domains via the R-interface. (4) and (5) Formation of an intracellular TIR scaffold allows for recruitment of the MyD88 adaptor molecule via S-interface interactions. MyD88 comprises an N-terminal death domain (DD), an intermediate domain (ID) and a C-terminal TIR domain. IRAK-1, IRAK-4 and/or IRAK-2 are recruited. These molecules comprise an N-terminal DD and a C-terminal catalytic domain. Under steady-state, IRAK-1 is bound to Tollip. As such, the Myddosome is formed. (6) IRAK-4 auto-phosphorylates and gains full activity, in turn activating IRAK-1 by phosphorylation. This induces rapid IRAK-1 auto-phosphorylation. (7) A conformational change in IRAK-1 allows its dissociation from Tollip. Different IRAK-1 oligomers form a scaffold for TRAF6, which becomes activated upon oligomerization. UBC13 and UEV1A facilitate TRAF6 self-poly-ubiquitination (K63-linked). TAB2 and TAB3 recruit TAB1 and the MAP3K TAK1 to the K63-linked poly-ubiquitin chain on TRAF6. (8) TRAF6 poly-ubiquitinates TAK1. (9) Initiation of the canonical NF-κB pathway: NEMO binds to the poly-ubiquitin chain on TAK1 and recruits IKKα and IKKβ. IKKα and IKKβ become activated following phosphorylation by TAK1. (10) and (11) IKKα and IKKβ phosphorylate IκBα, which allows for its dissociation from p50/p65 NF-κB. (12) Free p50/p65 NF-κB translocates to the nucleus and binds to specific κB sites on DNA, as such inducing gene expression. (13) Initiation of the MAPK pathway: the MAP3K MAP3K3 binds the poly-ubiquitin chain on TAK1. MAP3K3 becomes activated following phosphorylation by TRAF6. IKKα and IKKβ activate TPL2 by phosphorylation. MAP3K3, TAK1 and TPL2 activate different MAP2Ks by phosphorylation. (14) MAP2Ks activate the p38, JNK and ERK MAPKs by phosphorylation. (15) In turn, MAPKs activate different transcription factors by phosphorylation, as such inducing gene expression. (16) Binding of IL-1β to sIL-1R2 does not induce signal transduction. (17) Binding of IL-1β to the signaling-incompetent IL-1R complex does not induce signal transduction. (18) Binding of IL-1Ra to the signaling-competent IL-1R complex does not induce signal transduction. Created with BioRender.com.
A hallmark within the IL-1 superfamily is the tight control over cytokine activity, in particular for IL-1α and IL-1β (see also Figure 3). A first level of IL-1 activity regulation is the membrane-bound decoy receptor IL-1R2, which lacks an intracellular TIR domain and is therefore unable to transduce the signal (32, 33). However, cytokine-bound IL-1R2 is still able to recruit and interact with IL-1R3, as such decreasing the availability of this accessory chain on the cell membrane. IL-1R3 sequestering is an important regulatory mechanism, as the availability of surface-expressed IL-1R3 is the rate-limiting step in the formation of IL-1R complexes due to its relatively low abundance compared to the primary receptor chains (75). A second level of regulation is the availability of soluble decoy receptors with the capacity to bind and neutralize circulating cytokines, which can be generated by alternative splicing of the encoding mRNA or cleavage of the receptor protein from the membrane by metalloproteinases (32, 76). Receptor antagonists form a third level of IL-1 activity regulation. IL-1Ra shows a very homologous molecular configuration compared to IL-1α and IL-1β, but carries shorter peptide loops between β-strands 4/5 and 5/6. As such, IL-1Ra cannot bind to D3 of IL-1R1/2 and is therefore unable to induce the conformational change in the receptor subunit that is essential to recruit and interact with the accessory chain (33, 77, 78).
Functional maturation of DCs is required for successful priming of naive T cells and initiation of potent antigen-specific adaptive immune responses. Understanding the determinants of efficient DC maturation is therefore critical for the development of clinical applications, which becomes evident from advances made in the field of DC therapy for the treatment of different types of cancer (79). The first generation of such immunotherapies relied on the generation of monocyte-derived DCs (moDCs) from PBMCs after ex vivo culture in the presence of GM-CSF and IL-4 (80, 81). These moDCs were subsequently loaded with tumor lysates, tumor-associated antigens (TAAs) or synthetic peptides and administered to patients. However, DC therapy as such only showed a disappointing tumor-regression rate of around 3.3% (82–84). Second-generation therapies expanded on this concept by maturing the moDCs by treatment with LPS, CD40L or cocktails that included TNF, IL-6, IL-1β, PGE2 and polyinosinic:polycytidylic acid (poly(I:C)) or combinations thereof. The use of matured DCs further increased the clinical efficacy of these therapies up to 8-15%, depending on the tumor type (79, 85, 86). An alternative approach that bypasses the need for cytokine maturation cocktails involves ex vivo electroporation of moDCs with mRNA encoding CD40L, CD70 and constitutively active TLR4, which is referred to as the TriMix formula. Co-delivery of mRNA that encodes for tumor antigens was found to empower T cell responses and trials in melanoma patients revealed that this therapy is safe and establishes durable immunogenic clinical responses (87). Nowadays, the most recent efforts in the field focus on further combinations of these second-generation DC therapies with checkpoint inhibition molecules (e.g. anti-CTLA-4 and anti-PD-(L)1), which shows increased overall survival compared with monotherapies in stage III and IV melanoma patients, or chemotherapy (e.g. carboplatin and paclitaxel) (79, 88–90).
Maturation of DCs can thus be initiated by triggering PRRs, such as TLR4, or following activation of pro-inflammatory cytokine receptors, including the IL-1R complex. The overlapping effect of LPS and IL-1 on DC maturation results from the fact that both ligands signal via TIR domain-containing receptor complexes that share multiple downstream signaling mediators, many of these with a known involvement in the regulation of DC maturation and their capacity to present antigen (91). Signaling downstream of IL-1R activation is graphically represented in Figure 3. MyD88−/− mice, for example, fail to mount adaptive immune responses against Listeria monocytogenes and are therefore highly susceptible to infection (92, 93). However, restoration of MyD88 signaling in DCs only is sufficient to regain control over the infection (94). Conversely, upon hyperactivation of MyD88 in DCs, mice suffer from severe autoimmune effects, characterized by spontaneous activation of B and T cells, production of autoantibodies and systemic inflammation (95). Also, TRAF6-deficient DCs fail to upregulate CD86 and MHC-II, produce pro-inflammatory cytokines (e.g. IL-12 and IL-6) and prime naive T cells following stimulation with CD40L or LPS (96). Further downstream, canonical NF-κB signaling plays a prominent role during maturation of murine and human DCs as NF-κB drives the expression of co-stimulatory molecules and antigen presentation (97–100). Intriguingly, NF-κB signaling in DCs has been suggested to allow for self-tolerance, as steady-state migratory DCs display an enriched alternatively regulated network of NF-κB-instructed genes (101, 102). Among the MAPKs, p38 is described the most extensively in the context of DC activation and maturation. p38 inhibition impairs clustering of DCs with T cells and effector T cell activation (103). Phosphorylation of p38 in DCs correlates with improved uptake and presentation of antigens via MHC-I and MHC-II and the upregulation of co-stimulatory molecules (104–106). Studies on the role of JNK and ERK during DC maturation and antigen presentation are more limited. In human DCs, steady-state levels of JNK are notably lower than those of p38 (103). Although p38 and JNK are known to be redundant for many cellular functions, different reports propose that JNK signaling could be negatively regulating presentation of antigens via MHC-I and CD1d (107–109). ERK signaling, on the other hand, positively regulates CD1d-mediated presentation of lipid antigens and promotes the survival of matured DCs (91, 97).
The capacity of IL-1 to drive DC maturation (Figure 4A) was first suggested in 1987 by the group of Ralph Steinman, who demonstrated that DCs pre-treated with low doses of recombinant IL-1 were more potent in stimulating helper T cells compared to unconditioned DCs. In fact, IL-1 was the first cytokine reported to empower the activation of T cells by interfering at the level of the APC (110). Over the decades, different studies have shown that IL-1 signaling is indeed involved during the maturation of different DC subsets by driving the upregulation of several co-stimulatory molecules (e.g. CD80, CD86, CD40 and SLAM), release of pro-inflammatory cytokines (e.g. IL-12, IL-1α/β and IL-6) and expression of surface MHC (111–114). During steady state, lower numbers of peripheral CD103+ DCs are observed in IL-1R1−/− mice compared to wild-type (WT) animals and these cells additionally display a less mature phenotype, indicated by lower surface CD86 expression. Upon infection with influenza A virus (IAV), IL-1R1−/− CD103+ DCs in the lung fail to upregulate CCR7 and show an impaired capacity to migrate and accumulate in lung-draining lymph nodes (LNs) (115). Conversely, IL-1Ra−/− DCs are more matured under steady state conditions and release of IL-1Ra by epithelial cells suppresses the activation of moDCs (116, 117).
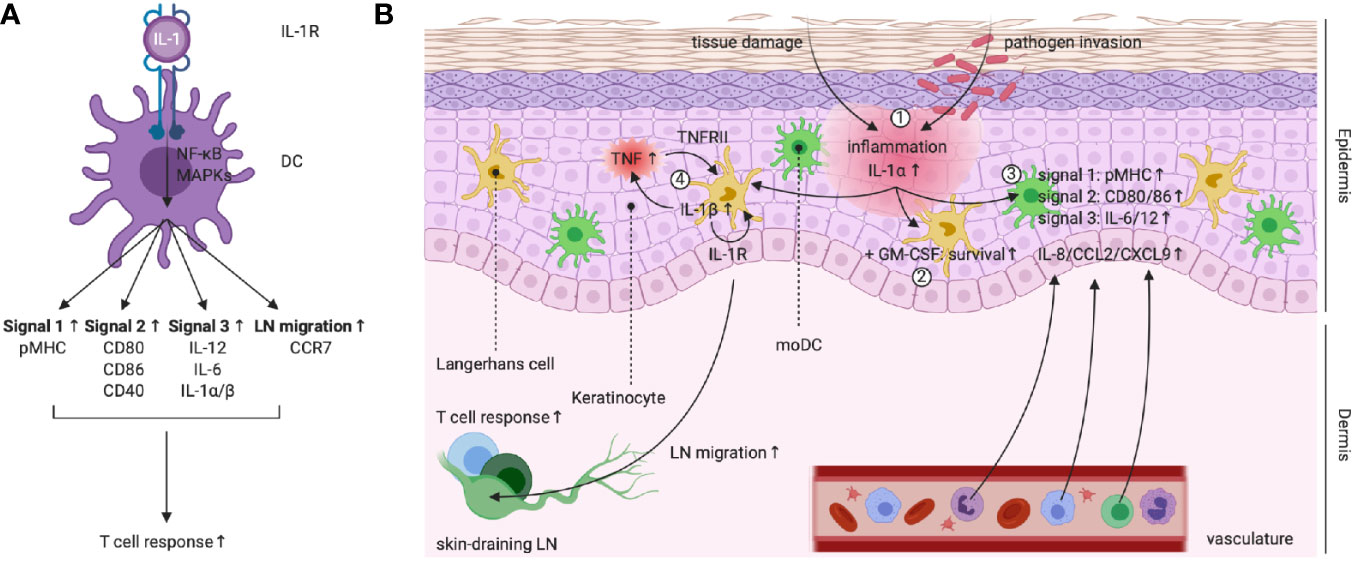
Figure 4 (A) Left scheme: IL-1R triggering in DCs empowers their capacity to promote T cell responses. (B) IL-1 activity plays a central role during the keratinocyte-LC cross-talk in the skin. (1) Damage to keratinocytes following tissue damage or pathogen invasion leads to the release of (pro-)IL-1α. (2) In synergy with tissue-derived GM-CSF, IL-1R signaling promotes LC survival. (3) IL-1R signaling in moDCs upregulates pMHC (signal 1), expression of co-stimulatory molecules (signal 2), production of priming cytokines (signal 3) and release of chemokines, leading to attraction of neutrophils, monocytes and lymphocytes. (4) PRR and IL-1R signaling promotes LC IL-1β production. IL-1β acts on keratinocytes and induces TNF release, which in turns signals via TNFRII on LCs. TNFRII and autocrine IL-1R signaling in LCs enable migration to the skin-draining LNs, where T cell responses can be initiated. Created with BioRender.com.
Special attention should be dedicated to the delicate role that IL-1 signaling plays during the cross-talk between keratinocytes and Langerhans cells (LCs) in the skin (Figure 4B), which is of fundamental importance during the defense against skin pathogens (118). For example, during development of the neonatal skin, IL-1 signaling serves as a host safeguard mechanism that prevents tolerance to Staphylococcus aureus (119). LCs reside in the epidermis, the outmost layer of the skin, where they are closely associated with keratinocytes. LCs are ontogenically different from dermal DCs and probably arise from a Mafb-expressing macrophage progenitor, whereas dermal DCs are more related to the different cDC subsets found in lymphoid tissues. LCs later express the transcription factor Zbtb46, which induces a shift towards the cDC identity (118, 120). Keratinocytes are the epidermis’ major cell type and form an important reservoir for IL-1α, which is constitutively expressed in the cytoplasm of these cells (31, 118). IL-1α released from keratinocytes synergizes with GM-CSF to enhance LC survival and capacity to stimulate naive T cells (121–124). Moreover, release of IL-1α following chemically induced keratinocyte damage induces the expression of co-stimulatory molecules (e.g. CD80 and CD86), MHC-I and inhibitory receptors (e.g. PD-L1/2) on the surface of moDCs. Furthermore, these IL-1α-conditioned moDCs release more pro-inflammatory cytokines (e.g. IL-6 and IL-12) and chemokines (e.g. IL-8, CCL2 and CXCL9) and appear to be superior in driving CD4+ T cell proliferation compared to untreated DCs (114). Next to a clear and well-established role in LC maturation, IL-1R signaling has a known involvement in LC migration to skin-draining LNs, for which the synergy with TNF is essential (125, 126). Indeed, within 2 – 4h after injection of recombinant IL-1β in both murine and human skin, LCs migrate and accumulate in skin-draining LNs (127, 128). The synergy between TNF and IL-1β has been elucidated by experiments that show inhibition of IL-1β-induced LC migration following administration of an anti-TNF antibody and vice versa (129). The current working model suggests that upon receiving a sensitizing trigger, LCs produce IL-1β and as such stimulate keratinocytes to release TNF. In turn, TNF signaling via TNFRII provides a first license for LC migration, while a necessary secondary stimulus is delivered via autocrine IL-1β signaling (126, 130, 131). Moreover, treatment of mice with an anti-IL-1R1 neutralizing antibody completely abrogates LC migration to skin-draining LNs, as such indicating the importance of this trigger for the information transfer from the innate to the adaptive immune system (132).
Beyond a role in driving maturation of DCs prior to T cell priming, IL-1 signaling additionally mediates DC activation further downstream, for instance by facilitating the CD40/CD40L-interaction during DC/T cell cross-talk. CD4+ T cells upregulate CD40L during their activation which is used to interact with CD40 on the DC surface, as such equipping the APC for successful CD8+ T cell priming (133). Nakae et al. reported that DC-released IL-1β acts on T cells and upregulates their CD40L and OX40 surface expression (134). In turn, different reports demonstrated that IL-1β facilitates DC activation following the CD40/CD40L-interaction, ultimately leading to DCs that release more pro-inflammatory cytokines (e.g. IL-12, IL-1α/β and IL-6) and generate stronger TH1 CD4+ and CD8+ T cell responses (e.g. IFN-γ release) (111, 135, 136). However, the ability of T cells to induce release of IL-1β by DCs could be a double-edged sword for the host. For example, during autoimmune myocarditis, IL-1R signaling in DCs promotes the expansion of autoreactive CD4+ T cells (137). More recent work demonstrated that during self-reactivity, effector CD4+ T cells instruct DCs to release IL-1β via an inflammasome-independent mechanism potentiated by TNF and FasL signaling. This TNFR/Fas/caspase 8-dependent pathway is responsible for the systemic inflammation and pathology commonly associated with T cell-driven autoimmune diseases, such as experimental autoimmune encephalitis (EAE) (138). How IL-1 signaling acts during the interplay between DCs and T cells after their initial stimulation, as such further shaping adaptive immune responses, is intriguing and a recent study showed that CD8+ T cells also participate in this cross-talk. After antigen presentation, CD8+ T cells activate the NLRP3 inflammasome in DCs via a perforin-dependent mechanism, which subsequently led to release of IL-1β and further amplification of adaptive immune responses (139). Comparably, maturation and release of fully biologically active IL-1β from bystander cells can be induced by macrophage-derived granzyme A upon recognition of Pasteurella multocida toxin (140).
Finally, IL-1 signaling on bystander DC is proposed to potentiate T cell immunity and mediate protective responses during infection with pathogens. In case that pathogen-infected DCs are unable to mount effective adaptive immune responses, successful T cell priming during encounter of live pathogens depends on activation of bystander DCs (141). DCs infected with Legionella pneumophila were shown to release IL-1 and IL-1R signaling on bystander cells is essential to overcome infection (142, 143). Data from Akiko Iwasaki’s lab shows that a normal IAV-specific CD8+ T cell response can be formed in TLR7−/− and MAVS−/− mice, but fails to develop in IL-1R1−/− mice, which suggests that activation of CD8+ T cells upon IAV infection is dependent on the inflammasome-IL-1R axis rather than TLR7 and RIG-I. The authors propose a model where a first DC responds to virus-induced damage to the host by NLRP3 and caspase 1-mediated maturation of IL-1β, which acts on a second bystander DC and promotes its activation, ability to migrate to the draining LN and prime the CD8+ T cell response. In this context, DAMP detection via the inflammasome/IL-1R axis can thus be perceived as a surrogate for the direct recognition of PAMPs (115). Moreover, IL-1β activity on Leishmania amazonensis-infected DCs, which are maintained in a suppressed state, enhances their activation and maturation, which leads to augmented parasite-specific CD4+ T cell responses (113).
Together, these combined works provide evidence that IL-1 signaling in DC subsets contributes to their initial maturation, migration and accumulation in lymphoid organs, but also for secondary activation during DC/T cell-crosstalk. During pathogen infection, signaling via the IL-1R complex can serve as a surrogate for PAMP recognition, which enables effector T cell responses by activating bystander cells. Next, we will discuss how direct signaling of IL-1 on T cells flavors their differentiation and activation status.
The ability of IL-1 to drive CD4+ T cell responses has been reported for decades (111, 135, 144–146). Moreover, defective adaptive immune responses following IAV infection were observed in mice deficient for ASC, caspase 1 and IL-1R1, which all failed to raise effector IAV-specific CD4+ T cells and produce neutralizing antibodies (147–149). As addressed above, a first important mode of T cell activation by IL-1 is indirect and depends on the cytokine’s activity on DCs. However, more recent work suggests an important contribution of direct action of IL-1 on CD4+ T cells to advance cellular immune responses. Ben-Sasson et al. demonstrated that IL-1α and IL-1β show the same potency to stimulate CD4+ T cell responses, both in adoptive T cell transfer (ATCT) as well as in endogenous models. These responses included enhanced CD4+ T cell expansion through proliferation, peripheral survival, establishment of memory and memory recall. Moreover, different subsets of CD4+ T cells, including TH1, TH2 and TH17, were found to be sensitive for this direct IL-1 activity (150). The innate immune system locally releases inflammatory cytokines as an extra cue beyond TCR triggering and co-stimulation to fine-tune the recall of antigen-experienced effector and memory T cells. A recent study by Jain et al. looked further into how IL-1 drives the reactivation of these subsets and found that IL-1 signaling in memory CD4+ T cells of different lineages can license cytokine production. Intriguingly, IL-1R1−/− CD4+ T cells differentiated normally into TH1, TH2 and TH17 phenotypes, but failed to produce lineage-specific effector cytokines (i.e. IFN-γ, IL-4/5/13, and IL-17A/F/22, respectively) following reactivation. Moreover, this study demonstrated that DCs are a major source of IL-1β, which was found to act directly on CD4+ T cells to stabilize different cytokine-encoding transcripts, probably via a mechanism dependent on p38 MAPK signaling (7).
Altogether, this indicates that while IL-1 activity clearly facilitates differentiation of naive CD4+ T cells following the first antigen encounter, the absolute necessity of IL-1 signaling presumably lies in activation of antigen-experienced effector and memory helper T cells of different lineages. Both TCR triggering and the ligation of co-stimulatory molecules remain the two indispensable instructions that allow for reactivation of these CD4+ T cells, but IL-1 signaling can be perceived as an extra signal for further regulation and fine-tuning of these responses. We will summarize how IL-1 signaling modulates differentiation and further activation of the different CD4+ T cell subsets.
IL-12 is the main cytokine that drives the differentiation of naive CD4+ T cells towards an early TH1 phenotype after TCR triggering and co-stimulation by inducing the expression of the lineage transcription factor T-bet. While the IL-1 cytokine superfamily member IL-18 cannot induce TH1 development itself, its presence in the environment further drives commitment to the TH1 lineage, mainly by promoting peripheral TH1 proliferation and secretion of IFN-γ, TNF and IL-2. TH1 cells also show a remarkably high expression of the IL-18Rα (IL-1R5) (Table 1). Generally, TH1 cells are well-known for their critical role as initiators of cellular immune responses and as mediators of protection against different intracellular pathogens and generation of anticancer immunity (151). Naive T cells can acquire an early TH2 phenotype when the priming environment contains IL-4, which drives expression of the lineage factor GATA3. GATA3 upregulates the expression of the IL-33 receptor ST2 (IL-1R4) on the surface of TH2 cells. IL-33 is another IL-1 superfamily member that allows for TH2 lineage commitment, proliferation and release of IL-5 and IL-13 (Table 1) (18, 151). TH2 cells play pivotal roles during infections with helminths and facilitate humoral immune responses, yet, this is now believed to be largely a function of follicular helper T (Tfh) cells (151). Both the study by Ben-Sasson et al. as well as the report by Jain et al. have demonstrated that IL-1 can act directly on both TH1 and TH2 cells and boost their activation, expansion and effector cytokine production (7, 150). Interestingly, Jain et al. found elevated IL-1R1 expression on antigen-experienced CD4+ T cells of different lineages versus their naive counterparts, making the case for a role of IL-1 signaling as inducer of cellular immune responses beyond CD4+ T cell priming (7).
IL-1-mediated modulation of the TH1/TH2 balance has been widely studied in the murine model of cutaneous Leishmaniasis. It is widely accepted that during infection with intracellular protozoan parasites from the Leishmania genus, TH1 responses are associated with resolution of cutaneous lesions and clearance of the pathogen, while TH2 responses rather contribute to disease progression (152). Intracellular replication of L. amazonensis in macrophages triggers the assembly and activation of the NLRP3 inflammasome, which leads to release of IL-1β and subsequent induction of NO production, a critical mechanism of defense against Leishmania species (153). Moreover, mice with deficiencies for several inflammasome components (e.g. NLRP3, ASC and caspase 1) fail to control multiplication of L. amazonensis, whereas TH1 responses against L. major are reduced in IL-1R1−/− mice, next to enhanced TH2 immunity (153, 154). In accordance with these observations, treatment of BALB/c mice with IL-1α or IL-1β following L. major infection induces dramatic reductions in lesion sizes or parasite load due to augmented TH1 responses. This IL-1 effect depends on the presence of IL-12 and significantly reduces TH2 immunity (155, 156). In addition, infection of C57BL/6 mice with L. major leads to enhanced release of the TH1-inducing cytokines IL-12, IL-1β and TNF in the infected skin (157).
Different studies employing Leishmania infection models unambiguously prove that IL-1 activity promotes TH1 responses, while it impairs TH2 immunity. However, the exact contribution of IL-1 signaling to the regulation of TH2 responses clearly depends on the experimental model system. Infection with the intestinal helminth Heligmosomoides polygyrus bakeri promotes the local release of IL-1β, leading to diminished production of IL-25 and IL-33, suboptimal TH2 responses and chronic infection (158). Conversely, blocking IL-1 signaling by treatment with the recombinant IL-1Ra in a mouse model of systemic sclerosis enhances TH2-mediated inflammation and worsens pulmonary fibrosis (159). Following infection with Cryptococcus neoformans, IL-1R1−/− mice show impaired TH1 and TH17 responses, next to augmented TH2 immunity (160). However, mice with an IL-1R1 deficiency on radioresistant lung epithelial cells fail to raise a TH2 immune response and do not develop house dust mite-induced asthma, whereas caspase 8-mediated IL-1 activity promotes TH2 immunity and contributes to the development of asthma pathogenesis (161, 162). Ovalbumin (OVA)-induced airway hypersensitivity, TH2 cytokine production (i.e. IL-4 and IL-5) and IgE and IgG1 levels are all reduced in mice with deficiencies in both IL-1α and IL-1β and IL-1R1-deficient mice display diminished CD4+ T cell priming in bronchial LNs and lungs (163, 164). On the other hand, NLRP3 activation and IL-1β release do not appear to be required for the activation of TH2 immunity by uric acid during asthma development, but this pathway is suggested to be important for skin TH2 responses (165, 166). In conclusion, the ability of IL-1 to influence TH2 immune responses is clearly context-dependent, as evidenced from studies in different model systems.
Recent work by Kuhn et al. demonstrated that antitumor CD4+ T cell responses, including proliferation, tumor infiltration and intratumoral IFN-γ and TNF production, can be promoted by administration of monosodium urate acid crystals in combination with Mycobacterium smegmatis and this response completely depends on IL-1R signaling in the host (167, 168). Tumor-specific effector TH1 cells establish a pro-inflammatory tumor microenvironment after tumor infiltration and their ability to mediate anti-cancer immune responses depends on IL-1 signaling, which can be delivered by both IL-1α and IL-1β (169, 170).
IL-1 is generally perceived as a cytokine that favors the differentiation of naive CD4+ T cells during priming, with a notorious role as a driver of murine and human TH17 development. TH17 cells are a subset of CD4+ T cells that exert potent pro-inflammatory activities, which need to be kept in balance by the dampening anti-inflammatory roles of regulatory T cells (Treg cells). TH17 cells express CCR6 and CD103 and preferentially home to mucosal tissues, including the gut (171). TH17 cells are essential mediators of mucosal immunity, as they offer local protection against extracellular pathogens, such as opportunistic infections with fungi, and mediate barrier integrity via production of IL-17 cytokines. Next to this, TH17 cells are strongly implicated during autoimmune responses against self-antigens (172). Intriguingly, TH17 and inducible Treg cells are believed to arise from a common progenitor CD4+ T cell (172). Upon priming, expression of the transcription factor FoxP3 induces the differentiation of naive CD4+ T cells into Treg cells, whereas expression of the transcription factor RORγT enables the generation of TH17 cells (173–175). In mice, transforming growth factor (TGF)-β drives the differentiation of CD4+ T cells away from TH1 and TH2 phenotypes by inducing the expression of both RORγT and FoxP3, meaning that a second tier of regulation is necessary to further select between the TH17 or the Treg transcriptional programs. Key for this differentiation are cytokines that signal via the JAK-STAT pathway: STAT3-inducing cytokines (e.g. IL-6, IL-21 and IL-23) drive differentiation towards the TH17 phenotype and cytokines that utilize STAT5 (e.g. IL-2) promote development of Treg cells (Table 1 and Figure 5) (178).
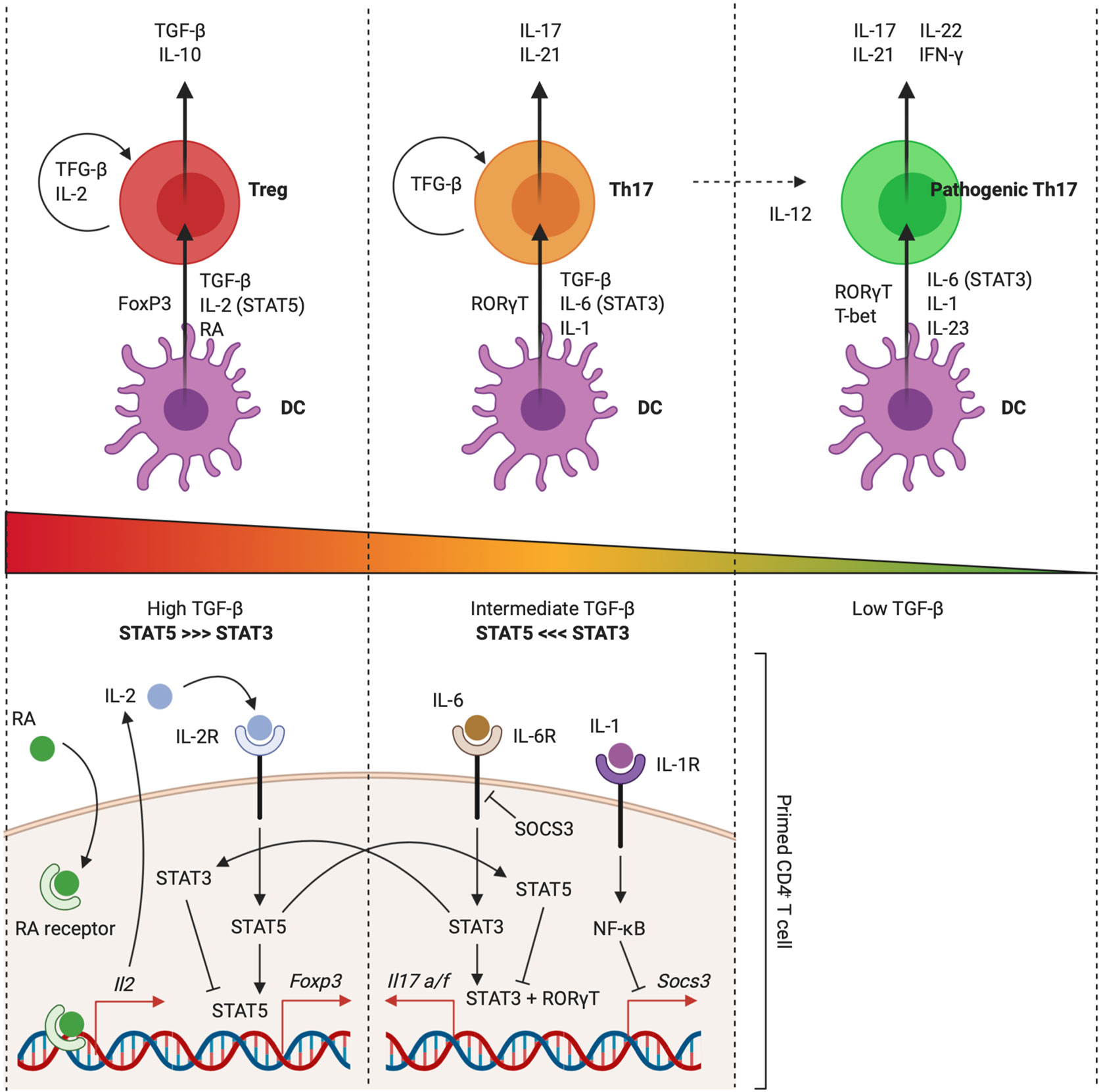
Figure 5 IL-1 signaling facilitates TH17 differentiation. TGF-β drives the expression of both FoxP3 and RORγT during CD4+ T cell priming. Commitment to a role as regulator or effector is further dependent on IL-2 (STAT5) and IL-6 (STAT3) activity, which antagonize each other. This process was found to be facilitated by other environmental factors, including the metabolite retinoic acid (RA) and the pro-inflammatory cytokine IL-1. Priming environments enriched in TGF-β concentrations and lacking STAT3-dependent cytokines drive Treg development. IL-2 activates STAT5, leading to sustained FoxP3 expression and inhibition of IL-17A and IL-17F production. Presence of RA empowers IL-2 expression and further pushes the balance towards the Treg phenotype. Priming environments that include IL-6 drive STAT3 activation, which mediates IL-17A and IL-17F expression and inhibits FoxP3 expression. IL-1 activates NF-κB, which inhibits expression of SOCS3. This further empowers STAT3 activity and pushes the balance towards the TH17 phenotype. In the absence of TGF-β, development of a TH1-like TH17 phenotype can be driven by IL-6, IL-23 (STAT3), and IL-1. This pathogenic phenotype is characterized by production of IL-17, IFN-γ, IL-21, and IL-22. Figure adapted from (176, 177). Created with BioRender.com.
The minimal requirement for naive murine CD4+ T cells to acquire the TH17 phenotype is the presence of IL-6 and TGF-β in the priming environment. Although it is generally not regarded as one of the typical polarizing cytokines for mouse TH17 differentiation, there is ample evidence in favor of a fundamental role for IL-1 as a facilitator of TH17 development in mice (179–181). Basu et al. demonstrated that IL-1 tips the balance between the TH17 and Treg transcriptional programs in favor of TH17 development by repressing the expression of SOCS3, which is an inhibitor of STAT3 phosphorylation, via an NF-κB-dependent mechanism. As such, IL-1 enhances the amplitude and duration of STAT3 phosphorylation downstream of TH17-polarizing cytokine signaling, without interfering with STAT5 phosphorylation (182). This leads to enrichment of STAT3 versus STAT5 at their shared consensus sequences, for example in the Foxp3 locus, which regulates FoxP3 expression, and the Il17a and Il17f loci, which regulate TH17 effector cytokine production (Figure 5) (178, 182, 183).
In both mice and human, TH17 cells are among the most sensitive immune cells for IL-1 activity due to their relatively high expression of IL-1R1 (10, 11). Abrogated TH17 responses and lower incidences of EAE are reported in mice deficient for different players involved in the biology of IL-1, including IL-1R1, IRAK-4, ASC and caspase 1 (184–190). Conversely, mice deficient for IL-1R8 (or SIGIRR), a negative regulator of signaling via the IL-1R complex, are more susceptible to EAE due to hyperactivation of TH17 cells following immunization with myelin oligodendrocyte glycoprotein peptide (191). Also in experimental arthritis models, IL-1 signaling drives the development of pathogenic TH17 cells (192, 193). Uncontrolled and excessive activity of IL-1 in IL-1Ra−/−IL-6−/− mice inhibits TGF-β-mediated expression of FoxP3 and induces a TH17 transcriptional program in CD4+ T cells, which even bypasses the need for IL-6 signaling (194). Interestingly, T cell-specific deletion of IL-1R1 in mouse does not impair TH17 development under steady state conditions, but strongly abrogates the potential of TH17 cells to migrate and proliferate in the anti-CD3 treatment model (195). Different studies have reported on the capacity of IL-1 to promote differentiation of TH17 cells and enhance their expansion, IL-17 production, peripheral survival and capacity to mediate vaccine-induced protection against pathogens, such as Coccidioides species and Blastomyces dermatitidis (196–200). Proliferation and persistence of TH17 cells following IL-1 signaling is suggested to be a consequence of Akt-mediated activation of the mTOR pathway, which has been linked with cell cycle progression. As a negative feedback switch, TH17 cells upregulate IL-1R8 after differentiation, which directly inhibits different signaling pathways induced by IL-1, including phosphorylation of mTOR and JNK (191, 196, 198, 201).
Human TH17 differentiation is substantially differently regulated compared to the murine situation, as IL-1 has been demonstrated to be a main polarizing cytokine for TH17 development by directly inducing the expression of the lineage transcription factor RORγT in human CD4+ T cells. In these studies, differentiation of naive human CD4+ T cells towards the TH17 phenotype is additionally facilitated by IL-23 or IL-6, but surprisingly antagonized by TGF-β (202, 203). The truth possibly lies somewhere in between and the local concentrations of both TGF-β as well as inflammatory mediators are proposed to be critical determinants of the outcome of TH17 development. High local levels of TGF-β can drive the differentiation of naive CD4+ T cells towards Treg phenotypes. TH17 development is favored in environments that contain pro-inflammatory cytokines, such as IL-6 and IL-1, and are less enriched in TGF-β. Absence of environmental TGF-β or local presence of IL-12 allows the formation of TH17 cells with pathogenic phenotypes that typically produce IFN-γ and express T-bet and RORγT (176, 204, 205). A recent study by Grandclaudon et al. demonstrated that IL-1β can synergize with the activity of IL-12 to drive differentiation of naive human CD4+ T cells following polyclonal stimulation with CD3/CD28 towards a mixed TH1/TH17 phenotype, which corresponded with enhanced IL-17F production and expression of RORc, T-bet and IL-23R (206). Both self- and commensal antigens are known as weak triggers of T cell and costimulatory receptors and low-strength TCR and CD28 signals have been reported to favor the differentiation of TH17 cells in the presence of cytokines. Revu et al. found that IL-1β synergizes with IL-23 to drive the development of human TH17 cells in the absence of CD28 by empowering glucose uptake and glycolytic activity, as such supporting TH17 expansion and avoiding anergy (207). Signaling of IL-1β on human Treg cells interferes with alternative splicing of the Foxp3 transcript by promoting excision of exon 7, which facilitates TH17 differentiation in vitro (208). In mice, IL-1 activity was found to attenuate the function of CD4+CD25+FoxP3+ Treg cells, allowing for autoreactive T cells to break tolerance and cause autoimmune disease (209). Administration of increasing amounts of exogenous IL-1β in mice blocks thymic development of Treg cells, which results in decreased amounts of CD4+CD25+FoxP3+ cells and enhanced levels of Treg precursor cells (13). Moreover, T cell-specific ablation of MyD88 abrogated TH1 and TH17 responses and memory CD4+ T cell formation, while no effect could be observed in the Treg subset (210). Besides acting directly on T cells, IL-1β also indirectly drives a TH17 instructional program in human immature DCs that leads to upregulation of CD14 and enhancement of a TH17-like phenotype in human CD4+ memory T cells (211). Human DCs are a major source of IL-1β and addition of NO to LPS-matured human DCs limits their IL-12 production, while enhancing the release of IL-1β, IL-6 and IL-23, thus favoring TH17 development (212).
Tfh cells aid during the differentiation of B lymphocytes into antibody-secreting plasma cells, a process that is negatively regulated by regulatory Tfh (Tfr) cells, which were demonstrated to descend from a Treg lineage (16, 213–216). This cellular interplay is organized in the germinal centers (GCs) of LNs, where Tfh and B cells are located in the middle and Tfr cells build up the surroundings (217). It has been demonstrated that IL-1β acts directly on Tfh cells to enhance their proliferation and induces the production of IL-4 and IL-21, as such initiating a B cell response. Recent work showed that this pathway is strongly maintained by Tfr cells, which express the IL-1 decoy receptor subunit IL-1R2 and actively release IL-1Ra (12). As such, Tfr cells employ a dual homeostatic regulation mechanism to keep IL-1β-mediated activation of Tfh cells in check (12, 16). Barbet et al. identified MHC-II+CD11b+CD11c+CX3CR1+ monocytes as the main source of IL-1β upon live vaccine administration. These cells express CCR7, localize to T cell zones in LNs and depend on IFN-β signaling to release IL-1β, which acts directly on Tfh cells and drives their differentiation in response to live bacteria (218). Reduced Tfh responses were also found after sensitization of IL-1R1−/− mice with peanut allergen (219). Also in human systems, the presence of IL-1β in the priming environment enhances Tfh cell development and these responses are diminished upon neutralization of IL-1 activity (220, 221).
Corroborating these recent findings, reduced antibody production following prime/boost immunization has been reported in BALB/c mice with combined deficiencies in IL-1α and IL-1β, while humoral immune responses were enhanced in IL-1Ra−/− counterparts. This work suggests that IL-1 produced by APCs upregulates OX40 and CD40L expression on T cells during priming, as such enhancing their capacity to activate B cells. Despite this, IL-1 appears not be involved in the humoral immune response to T cell-independent antigens (134, 222). In other studies, intact humoral immune responses were observed in C57BL/6 IL-1R1−/− mice following immunization with both T cell-dependent and -independent antigens (154, 223). The contribution of IL-1 activity in humoral immunity is thus controversial. Besides this, the influence of the mouse background, type of antigen used and its delivery method should not be underestimated.
The amount of tumor-infiltrating Tfh cells and B lymphocytes inversely correlates with the progression and recurrence of human colon carcinoma (224). In a recent paper, Roberti et al. demonstrated that in colon carcinoma models, the effect of chemotherapy with oxaliplatin (OXA) on intestinal epithelial cell (IEC) death is dictated by the gut microbiota, which determines the clinical response to anti-PD-1 immune checkpoint blockade. OXA-induced immunogenic IEC death is favored in environments dominated by Erysipelotrichaceae family members or Bacillus fragilis, which allow for superior responses to anti-PD-1 treatment. This effect relies on migration of CD103+CD11b-Batf3+ cross-presenting DCs to mesenteric LNs, where a Tfh response is primed, which depends on T cell-intrinsic IL-1 and IL-12 signaling. IL-1β-induced Tfh cells promote B cell maturation and class switch and stimulate effector CD8+ T cell responses against neo-antigens in the tumor microenvironment (225).
Approximately a decade ago, IL-9-producing CD4+ T cells were first identified and termed TH9 cells, which are formed following priming of naive CD4+ T cells in the presence of IL-4 and TGF-β (Table 1) (226, 227). Over the years, TH9 cells have been associated with protection against helminth parasites and were demonstrated to exert very potent antitumor immunity (228–230). In this latter context, a recent report has demonstrated that IL-4 and IL-1β act synergistically in the absence of TGF-β to empower a tumor-specific TH9 response, which mediates robust antitumor activity. Of note, the authors showed that direct IL-1 signaling on the CD4+ T cell is required for TH9 development and they identified the NF-κB pathway as a major signaling cascade that allows for pro-inflammatory IL-9 production (231). Furthermore, ATCT of TH9 cells in advanced B16-OVA melanoma tumor-bearing mice revealed that these cells exert potent effector functions, which are distinct from TH1 and TH17 CD4+ T cell subsets, and express less exhaustion markers (i.e. PD-1 and LAG3). The proliferative potential of TH9 cells appears to be NF-κB-driven, while TRAF6 and Eomes were identified as the main instructors of TH9 antitumor effector functions (230).
The importance of IL-1 signaling for CD8+ T cell activation becomes evident from the evolutionary pressure imposed on this pathway by viruses including different vaccinia strains, which encode a viral soluble protein that binds and incapacitates IL-1β. Remarkably, this binding appears to be specific for IL-1β, as IL-1α was found not to be subjected to this neutralization (232–234). Using a modified vaccinia virus Ankara (MVA) that lacks the gene encoding for the anti-IL-1β soluble protein, the lab of Gerd Sutter found enhanced virus-specific memory CD8+ T cell responses following prime/boost immunization, which correlated with improved long-lasting protection upon respiratory challenge infection (235, 236). Moreover, MVA-induced murine DCs were identified as the main source for IL-1β production and cytokine secretion following MVA infection is abrogated in DCs with a caspase 1 deficiency, indicating the importance of the inflammasome-IL-1R axis for potent induction of protective virus-specific CD8+ T cell immunity (235). In addition, the inflammasome-IL-1R axis appears to be important for successful priming of IAV-specific CD8+ T cells, as different reports have demonstrated defective CD8+ T cell responses in IL-1R1−/− mice following IAV infection (115, 147, 148). Activation of the NLRP3 inflammasome and subsequent release of IL-1β is also critical during control of infection with West Nile Virus (WNV) (237–239). CD11b+CD45high macrophages were identified as the predominant producers of IL-1β in the WNV-infected central nervous system (CNS). While Ramos et al. found less WNV-specific effector CD8+ T cells (i.e. producing TNF, IFN-γ, perforin and granzyme) in infected IL-1R1−/− mice, Durrant et al. could only link impaired effector CD4+ T cells responses to IL-1R1 deficiency during infection (237–239). A follow-up paper by Durrant et al. identified IL-1β as an essential regulator of CXCL12 expression and CXCR4-mediated retention of WNV-specific effector CD4+ and CD8+ T cells (i.e. expressing CD69, IFN-γ and granzyme B) to the blood-brain barrier, as such facilitating their migration into the parenchyma of the CNS (238).
Besides their pivotal work on the role of IL-1α and IL-1β signaling during the development, maintenance and recall of CD4+ T cell responses, Ben-Sasson et al. looked into the effects of IL-1 activity on the CD8+ T cell compartment (150, 240). Treatment of mice with IL-1β massively enhances the expansion of adoptively transferred OT-I CD8+ T cells in both lymphoid (i.e. spleen and LNs) and peripheral (i.e. lung and liver) organs in response to OVA antigen administration. Next to improved expansion and tissue localization, Ben-Sasson et al. showed that IL-1β empowers CD8+ T cell effector functions (i.e. production of IFN-γ/granzyme B and cytolytic activity) and memory recall. In addition, the protective capacity of inefficient experimental vaccines against L. monocytogenes, vaccinia virus and the lung epithelial TC1 carcinoma is enhanced upon inclusion of IL-1β in the immunization formula (240). Lee et al. combined adoptive transfer of tumor antigen-specific CD8+ T cells with repeated IL-1β administration in a murine melanoma model. IL-1β improves the antitumor effect of ATCT, which correlates with stronger infiltration of CD8+ T cells in the tumor, a more pronounced effector phenotype and enhanced peripheral survival (241). IL-1R1−/− mice fail to clear infection with lymphocytic choriomeningitis virus (LCMV) and LCMV-specific CD8+ T cells from IL-1R1-deficient mice do not synthesize granzymes and lack potent cytolytic activities (242). Despite the fact that CD8+ T cell-intrinsic MyD88 expression is essential for adequate expansion and accumulation of antiviral CD8+ T cells that control LCMV infection, two independent studies showed that this effect does not depend on signaling via IL-1R1 (243, 244). At the peak of the antiviral effector CD8+ T cell response to LCMV, impaired expansion, cytokine production (i.e. IFN-y, TNF and IL-2) and formation of a CD127+ memory precursor pool was observed in IL-1R1−/− CD8+ T cells with specificity towards different viral epitopes. Next to the importance of IL-1 signaling in programming potent effector CD8+ T cell polyfunctionality, defective formation of a functional LCMV-specific memory CD8+ T repertoire was found in IL-1R1−/− mice at 8 weeks after pathogen encounter (245).
The roles of the inflammasome-IL-1R axis and the process of pyroptosis, the form of inflammatory cell death initiated following the formation of membrane GSDM pores after inflammasome activation, have also been studied in the context of mounting anticancer CD8+ T cell immunity. A first report by Ghiringhelli et al. showed that the capacity of chemotherapy to induce CD8+ T cell responses against developing tumors depends on their ability to trigger the NLRP3 inflammasome in DCs and DC-mediated release of IL-1β. Moreover, the therapeutic response of two independent tumor models to treatment with doxorubicin diminishes upon treatment with an anti-IL-1β, but not an anti-IL-1α neutralizing antibody (246). Delivery of an anti-IL-1β neutralizing antibody also reduces the antitumor properties of other anthracycline chemotherapy in established tumor models (247). On the same note, a recent study by Wang et al. demonstrated a novel method to selectively deliver active GSDMA3 protein to murine tumor cells in vivo, as such inducing pro-inflammatory pyroptosis. Intriguingly, pyroptosis levels of less than 15% were found to be sufficient to clear a complete 4T1 mammary carcinoma graft and this effect depends on circulating IL-1β, as inhibition of IL-1β activity using a specific neutralizing antibody completely nullifies antitumor immunity (248). However, no contribution of IL-1β activity in GSDME-mediated immune responses against tumors was observed by Zhang et al. (249).
While literature provides ample evidence in favor of a stimulatory role of IL-1 for different aspects of CD8+ T cell biology, there is ongoing debate on whether IL-1 exerts these actions directly on CD8+ T cells or indirectly via an intermediate cell type (Figure 6). Sarkar et al. reported that intrinsic IL-1 signaling is necessary for the development of effector and memory CD8+ T cells responses against LCMV, as both numbers as well as cytokine synthesis were decimated in IL-1R1−/− LCMV-specific CD8+ T cells. This indicates that direct IL-1 activity regulates the size and functionality of effector and memory CD8+ T cell pools (245). The study by Ben-Sasson et al. shows that IL-1β-mediated expansion of CD8+ T cells and their migration into lymphoid organs only requires IL-1R1 expression on OT-I cells, advocating a role for direct IL-1β activity, whereas peripheral localization and cytokine production demands IL-1R1 expression in the host (240). Lee et al. demonstrated that IL-1R1 expression on both the transferred CD8+ T cells as well as the host environment is required for the enhancing effect of IL-1β on peripheral accumulation and survival of CD8+ T cells. However, IL-1β‘s ability to enhance granzyme B expression in CD8+ T cells surprisingly depends on IL-1R1 expression on vascular endothelial cells and host release of IL-2 and IL-15, but not IL-6 and IL-12, nor TCR stimulation. CD11b+Ly6G+Ly6Cint neutrophils in the spleen were additionally suggested as the predominant source of IL-15 (241). While Pang et al. showed that IL-1R1 expression in the hematopoietic cell lineage is an absolute requirement to mount potent anti-IAV CD8+ T cell responses, expression of the receptor complex on CD8+ T cells is not needed for their activation following infection. In fact, DC-intrinsic IL-1R signaling was found to mediate their upregulation of CCR7 upon IAV infection and their subsequent migration towards draining LNs, where they were shown to prime potent antiviral CD8+ T cell immunity (115). Also, in the vaccinia model, reduced CD8+ T cell responses were demonstrated in MyD88−/− mice following viral infection, yet, IL-1R1 expression on CD8+ T cells does not appear to play a role, according to a study by Zhao et al. (250). From these combined reports, we can at least conclude three important findings considering IL-1R signaling in the regulation of CD8+ T cell immunity: (1) both IL-1R1 expression on CD8+ T cells as well as peripheral expression of the receptor complex contribute to IL-1-mediated CD8+ T cell activation; (2) different bodies of evidence indicate a segregation in this contribution, meaning that the different cellular targets of IL-1 possibly regulate different aspects of the CD8+ T cell response; (3) the relative importance of IL-1R signaling in different cell populations could be context-dependent and thus variable in naive mice versus virus-infected or tumor-bearing backgrounds. The recent generation of Il1r1-floxed mice is therefore very exciting, as this will allow for detailed dissection of the importance of IL-1R signaling in different cell populations under specific conditions (251).
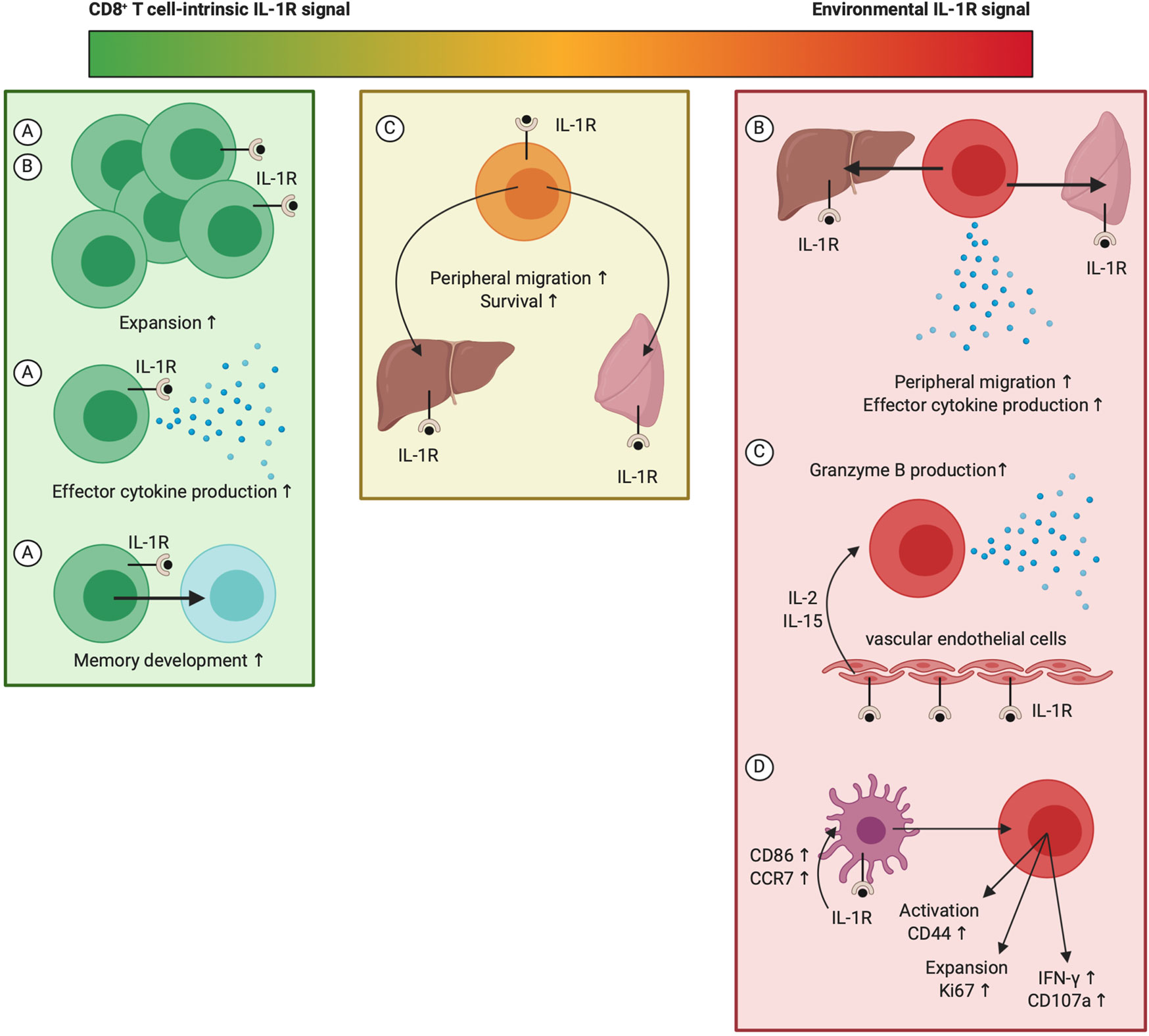
Figure 6 The whereabouts of IL-1R signaling during stimulation of CD8+ T cell responses. While IL-1 activity can potently promote CD8+ T cell responses at multiple levels, the exact whereabouts of IL-1R triggering for mediation of these effects remain under debate. This figure indicates the context-dependency of several key findings: (A) CD8+ T cell responses during LCMV-Armstrong infection; (B) Treatment of naive mice with antigen and IL-1β following ATCT of CD8+ T cells (240); (C) Treatment of B16 melanoma tumor-bearing mice that received ATCT of CD8+ T cells with IL-1β (241); (D) CD8+ T cell responses during IAV infection (115). Created with BioRender.com.
Prophylactic and therapeutic vaccination strategies are, considering the evidence above, the most obvious clinical applications where the activities of IL-1 could play meaningful roles. Prophylactic vaccination has been used with great success to (nearly) eradicate numerous infectious diseases across the globe, including smallpox, rubella and polio. These strategies depend on evoking potent immunological memory against pathogenic antigens, in such a way that invading pathogens are swiftly eliminated upon encounter (252, 253). The far more experimental therapeutic vaccines have been used primarily in the context of cancer and aim to stimulate T cells that recognize TAAs. TAAs can appear in different forms, which can be highly tumor-specific (e.g. neo-epitopes or aberrantly expressed germline antigen genes) or demonstrate rather low tumor specificity (e.g. tissue-specific antigens, such as melanocyte proteins, or overexpressed antigens) (254). This can impose an immunological challenge, as certain classes of TAAs are self-antigens for which high-affinity T cells have been eliminated during the immune system’s development, only leaving T cells carrying low-affinity TCRs. Next to this, established tumor masses and immunosuppressive environments are other major hurdles that have been limiting the success of therapeutic cancer vaccination (252).
Vaccines need to display an exquisite safety profile, especially when used prophylactically in healthy subjects. For this reason, traditional vaccines that generally comprise live attenuated or inactivated pathogens have been gradually replaced with recombinant protein subunit formulations, which show an improved safety due to a reduction in their reactogenicity. In order to enhance the efficacy of subunit vaccines, these formulations have been supplemented with adjuvants (Lat. “adiuvare” or “to help”). Not only could IL-1 be an interesting candidate for a new vaccine adjuvant as such, but the activities of IL-1α, IL-1β, and other members of the IL-1 cytokine superfamily might be involved in the mode of action (MOA) of several vaccine adjuvants, as extensively summarized in a recent review by Muñoz-Wolf and Lavelle (253). Alum, for instance, which is an overarching denominator for different aluminum-based adjuvants, is known as a potent driver of TH2-biased immune responses and antibody production. However, despite being used for nearly a century, alum’s MOA is not completely resolved. Both in vitro and in vivo, alum has been found to activate the NLRP3 inflammasome and promote the release of IL-1β, but how this exactly influences alum’s adjuvanticity has not been clearly unraveled yet. The relative contribution of IL-1 superfamily cytokines in the alum-mediated immunity could be tissue-dependent and subjected to the susceptibility of resident cells to alum-induced necrosis (253, 255–258). Indeed, Kuroda et al. demonstrated that alum inhalation induces necrosis of CD11c+SiglecF+ alveolar macrophages and release of IL-1α, which evokes TH2-driven airway hypersensitivity. Upon subsequent OVA administration, host IL-1R1 expression was found to be essential in the formation of inducible bronchus-associated lymphoid tissue and antibody production (258). Others have shown that IL-1 superfamily cytokines are unlikely to be prominent regulators of the antibody response induced following alum administration (259–261). Muñoz-Wolf and Lavelle also mention that while different other vaccine adjuvants, including MF59, GLA-SE, AS03 and CAF01, trigger the inflammasome-IL-1 axis, the precise importance of this pathway in the MOA of these adjuvants is incompletely understood (253).
While most of the licensed adjuvant systems mentioned above undeniably raise potent TH2 and humoral immune responses, adequate protection against different infectious pathogens (e.g. M. tuberculosis, dengue virus or Plasmodium falciparum) and cancer requires potent TH1, TH17 and CD8+ T cell immunity (253). In the specific case of IAV, the moderate heterosubtypic immunity that occurs naturally following infection is most likely mediated by cross-reactive T cell responses aimed against highly conserved internal viral epitopes, which makes a case for vaccine adjuvants that promote cellular immunity in the search for a universal IAV vaccine (262, 263). In this context, Lapuente et al. designed an adenoviral vector-encoded IL-1β and co-delivered this intranasally as an adjuvant with vector-encoded IAV nucleoprotein (NP) and hemagglutinin (HA). Next to enhancing HA-specific antibody responses, IL-1β strongly increases both mucosal and systemic NP-specific CD4+ and CD8+ T cell immunity. Moreover, the CD8+ tissue-resident T (TRM) cell population induced in the lung was shown to be sufficient for heterosubtypic protection against other IAV strains (264). In a follow-up study, these authors evaluated whether DNA plasmid-encoded IL-1β could act as a cellular adjuvant to improve the efficiency of a DNA vaccine with plasmids encoding the IAV antigens HA and NP. However, while intramuscular co-delivery of IL-1β slightly improved the formation of cross-reactive anti-HA antibodies, no stimulatory effect on NP- and HA-specific CD8+ T cells could be observed, suggesting that IL-1β might act as a strictly mucosal adjuvant in IAV vaccination strategies (265).
TH17 cells do not only play detrimental roles during autoimmune disease, but also mediate mucosal protection against different pathogens, including extracellular bacteria, Toxoplasma gondii and fungi (266–268). Considering the global threat imposed by pathogens such as M. tuberculosis, vaccine adjuvants that drive protective mucosal immune responses thus could be of great value (269). The above mentioned CAF01 cationic liposome adjuvant, which is currently under clinical investigation, was found to induce long-lived TH17 responses that can be recalled to the lung parenchyma upon challenge infection with M. tuberculosis two years after their initial induction (270). Intriguingly, peripheral CAF01-induced TH17 cells were found to promote the induction of IgA-producing B lymphocytes in the lung parenchyma and enhanced the formation of GCs and Tfh cells in lung-draining mediastinal LNs (271). Moreover, IL-1R1 signaling appears to be essential for the induction of IFN-γ and IL-17-producing cells in mice following vaccination with CAF01, but not for the generation of an antibody response (272). In an effort to evaluate IL-1β as a possible TH17-driving adjuvant, Wüthrich et al. demonstrated that an experimental live attenuated B. dermatiditis vaccine supplemented with IL-1β improves TH17-mediated protection against subsequent infection (200).
In the context of cancer, the inflammasome-IL-1 axis can be perceived as a double-edged sword with both pro- and antitumorigenic properties, which is extensively summarized in a recent paper by Rébé and Ghiringhelli (273). There is ample evidence demonstrating that in many different types of cancer, including malignancies in skin, colon and lung, upregulated expression of IL-1β correlates with progression of disease. In addition, several common cancer-associated mutations, such as genetic alterations in KRAS and BRACA1, promote IL-1β expression. Cancer cells can also drive the production of IL-1β in tumor-associated inflammatory macrophages, as such installing an environment where immune responses are dampened, for instance via IL-1β-mediated accumulation of myeloid-derived suppressor cells (MDSCs) that produce IL-10 and recruit pro-tumorigenic neutrophils. Besides this, IL-1β has been described a promotor of angiogenesis and cancer cell metastasis (273). Consequentially, clinical cancer research strongly focuses on antagonizing IL-1 activity in patients, which can be achieved by treatment with NLRP3 inflammasome inhibitors, recombinant IL-1Ra or monoclonal antibodies that neutralize IL-1β (273–275). The CANTOS (for “canakinumab anti-inflammatory thrombosis outcome study”) trial evaluated the effect of treatment with canakinumab, an anti-IL-1β neutralizing monoclonal IgG1 antibody, in patients with a history of myocardial infarction and high circulating levels of C-reactive protein (CRP). The study found that treatment with canakinumab could dose-dependently reduce levels of CRP and the risk of recurrence of a cardiovascular event (276). Moreover, treatment with canakinumab significantly lowered the incidence and mortality of lung cancer, making a case for strategic inhibition of IL-1 activity in patients at risk (277).
In contrast to this, IL-1 activity can promote anti-tumor immune responses by influencing both DCs and T cells in different models and experimental settings. Therapeutic vaccination of mice with irradiated melanoma cells that secrete IL-1β impairs tumor growth, which correlates with enhanced T cell activity (i.e. more release of IL-2/IFN-γ and augmented cytolytic activity upon ex vivo restimulation) (278). IL-1R triggering activates bystander DCs, which could be of importance for the improvement of DC vaccination strategies (143). Indeed, different studies have shown that successful priming of CD8+ T cell responses during vaccination with ex vivo matured and antigen-loaded DCs is dependent on help from bystander DCs (279, 280). Allogeneic DCs that are matured ex vivo with a cocktail containing poly(I:C), R848 and IFN-γ in combination with an infection-enhanced adenoviral vector produce high amounts of IL-1β. This correlates with an enhanced potential to activate bystander DCs, which in turn cross-present antigen to CD8+ T cells and induce an antigen-specific adaptive immune response (281). In the CT26 colon carcinoma model, systemic administration of Salmonella typhimurium in mice enhances the release of IL-1β by DCs and inhibits tumor growth, which can be reverted upon treatment with an IL-1β-neutralizing antibody (282, 283). Work by Segovia et al. demonstrated that mice with a deficiency for the transmembrane protein 176B (TMEM176B), which inhibits activation of the NLRP3 inflammasome by controlling the cellular Ca2+ flux, efficiently control tumor growth in a caspase 1 and IL-1β-dependent manner. Moreover, pharmacological targeting of TMEM176B mediates inhibition of tumor growth and increases susceptibility to immune checkpoint inhibition (284). Recent work by Zhivaki et al. shows that hyperactive cDC1s produce mature IL-1β in a NLRP3-dependent, but pyroptosis-independent manner and display an enhanced ability to take up and present TAAs, migrate to tumor-draining LNs and promote antitumor effector CD8+ T cells, altogether establishing a durable antitumor response. Interestingly, neutralization of IL-1β activity with an antibody completely abrogates the antitumor response mediated by these hyperactive cDC1s (70).
IL-1 could be a strong candidate to improve the therapeutic efficacy of adoptive T cell transfer in cancer, which remains largely ineffective for the majority of patients carrying solid tumors. The general idea is that cancer patients receive a transfer of T cells with reactivity against different tumor antigens, which can be either inherent or acquired following genetic insertion of transgenic TCRs or chimeric antigen receptors (CARs) (285). In this context, Lee et al. showed that repeated administration of IL-1β improves the antitumor activity of ATCT in a mouse model of melanoma, while Sarkar et al. found that IL-1 supplementation enhances priming and expansion of anti-CD19 human CAR-T cells (241, 245).
In conclusion, while IL-1 activity primarily shows pro-tumorigenic properties, different bodies of evidence demonstrated that antitumor immune responses can be facilitated by IL-1. Therapeutic application of IL-1’s activity in the context of cancer will completely depend on the ability to clearly segregate its pro-tumorigenic from its antitumorigenic effects.
Efficient clinical translation of cytokine activity is hampered by a variety of problems. First, long-term and high-dose cytokine administration might raise detrimental side effects. In the case of IL-1α and IL-1β, these toxicities include fevers, rigor and headaches. Hypotension is the dose-limiting factor and the maximum tolerated dose (MTD) of intravenously administered IL-1β was found to range from 0.07 – 0.3 μg/kg body weight (286). A second problem that some cytokines face is the necessity of dose escalation to reach sufficient therapeutic efficacy, which is due their limited serum half-life (287). Third, cytokines are highly pleiotropic molecules and not all of their functionalities are desirable. The abovementioned tumor-promoting functions of IL-1 could therefore nullify the favorable promotion of an antitumor CD8+ T cell response (273). Isolation of the favorable immunostimulatory properties of IL-1 from its undesired side effects might pave the way for its safe and efficient clinical application. In this regard, structure/function analysis has revealed a region within IL-1β (i.e. the primary sequence VQGEESNDK located between β-strand four and five) that displays comparable adjuvanticity to complete IL-1β in the absence of inflammation and toxicity (288). An alternative solution for the abovementioned problems are so-called immunocytokines, which represent fusion proteins comprised of a WT cytokine and a targeting moiety directed against a cell type-specific surface molecule. Immunocytokines aim to widen the therapeutic window of cytokines by increasing their local concentration at carefully selected target sites. This empowers the local biological activity of the cytokine moiety and allows for the use of lower amounts of immunocytokine to achieve the same therapeutic effect compared with the WT cytokine only, as such reducing cytokine-related toxicities (289). As of today, one IL-1β-based immunocytokine has been preclinically evaluated. This immunocytokine delivers IL-1β activity to the tumor microenvironment and modestly slows down the subcutaneous growth of a murine B16 melanoma tumor (290). A limitation of immunocytokines is the fact that these still comprise WT cytokines that maintain the capacity to exert undesired cytokine activities on non-targeted cells (291, 292). A modification of the immunocytokine concept are AcTakines (for “Activity-on-Target cytokines”), which represent fusion molecules that carry a mutation in the cytokine moiety, as such rendering them essentially inactive. Upon delivery of the mutated cytokine to a cell type-specific surface molecule, cytokine activity can be restored up to WT level. As such, AcTakines are devised to remain inactive en route through the body and reveal their full agonist activity following binding to selected cell types only (293). AcTakines based on type I IFN (294, 295) and TNF (296) activity were previously demonstrated to mediate potent antitumor activity without raising the toxicities observed with WT cytokine administration. Recently, we developed an AcTakine molecule that delivers IL-1β activity to CD8+ T cells and safely promotes T cell activation, proliferation and memory differentiation. Inclusion of this AcTakine in an experimental prime/boost IAV vaccine established protection against a high influenza challenge dose, which correlated with the formation of a potent and durable IAV-specific cellular immune response (297). The use of this strategy could be expanded to other models, for instance to evaluate antitumor CD8+ T cell activation by IL-1β.
The pro-inflammatory cytokines IL-1α and IL-1β can strongly impact the outcome of adaptive immune responses. However, their pleiotropic activities are usually not included in the three-signal model, an empirical framework that places adaptive immunity under innate control. Successful progression from productive to actual protective T cell immunity requires a more holistic view, with additional attention for the biological effects of these pro-inflammatory IL-1 cytokine superfamily members. A revised model could be of major importance for more rational development of adjuvants that promote cellular immune responses, which are highly requested in vaccination against intracellular pathogens and the new generation of recombinant personalized tumor vaccines.
As IL-1α and IL-1β utilize the exact same receptor complex, their biological activities can be regarded as redundant. However, clear temporal and spatial factors distinguish these two pro-inflammatory mediators: whereas IL-1α performs a local role as an alarmin that is released upon cellular damage, the release of IL-1β, which can also appear in circulation, is strictly controlled. From this biological perspective, the activities of cytokine alarmins, including IL-1α and the IL-1 cytokine superfamily member IL-33, could be important to induce in mucosal vaccination strategies. As many pathogens enter the host via mucosal membranes of the respiratory, genital and gastro-intestinal tract, raising local immune responses at the sites where infection and transmission take place is highly requested and the success of mucosal vaccination to locally induce functional T cell responses will in part depend on the availability of powerful adjuvants that yield such responses.
The IL-1 cytokine superfamily contains other members besides IL-1α and IL-1β with functionalities that could be used to fine-tune adaptive immune responses. IL-33 is an established inducer of TH2 immunity and promotes the release of TH2-polarizing cytokines in vivo (298). IL-18 was originally described as “IFN-γ-releasing factor” due to its ability to promote TH1 responses and NK cell activation (299). Further research is requested on how these cytokine properties can be exploited to advance immunization strategies.
BVDE wrote the manuscript and assembled the figures. JT and SG revised the manuscript. All authors contributed to the article and approved the submitted version.
BVDE is a doctoral fellow receiving FWO-SB funding. This work was supported by an FWO grant (project G045318) awarded to JT and SG and a UGent Methusalem and an Advanced ERC grant (CYRE 340941) awarded to JT. The Receptor Research Laboratories headed by JT. received financial research support from Orionis Biosciences BV.
JT is affiliated with Orionis Biosciences BV as scientific advisor and holds equity interests in Orionis Biosciences BV.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The authors wish to thank the reviewers for their careful and thorough evaluation of our manuscript. We also would like to thank Ed Lavelle, Kenny Roose, Jo Van Ginderachter, Karim Vermaelen and Andy Wullaert for their valuable input that vastly improved the quality of the work presented here.
1. Iwasaki A, Medzhitov R. Control of adaptive immunity by the innate immune system. Nat Immunol (2015) 16:343–53. doi: 10.1038/ni.3123
2. Janeway CA. a. Pillars article: approaching the asymptote? Evolution and revolution in immunology. Cold spring harb symp quant biol. 1989. 54: 1-13. J Immunol (2013) 191:4475–87.
3. Matzinger P. Tolerance, danger and the extended family. Annu Rev Immunol (1994) 12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015
4. Jain A, Pasare C. Innate control of adaptive immunity: Beyond the three-signal paradigm. J Immunol (2017) 198:3791–800. doi: 10.4049/jimmunol.1602000
5. Gilbert SC. T-cell-inducing vaccines - what’s the future. Immunology (2012) 135:19–26. doi: 10.1111/j.1365-2567.2011.03517.x
6. Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity (2013) 39:1–10. doi: 10.1016/j.immuni.2013.07.012
7. Jain A, Song R, Wakeland EK, Pasare C. T cell-intrinsic IL-1R signaling licenses effector cytokine production by memory CD4 T cells. Nat Comm (2018) 9:3185. doi: 10.1038/s41467-018-05489-7
8. Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci (2009) 106:13463–8. doi: 10.1073/pnas.0906988106
9. Caucheteux SM, Hu-Li J, Guo L, Bhattacharyya N, Crank M, Collins MT, et al. IL-1β enhances inflammatory TH2 differentiation. J Allergy Clin Immunol (2016) 138:898–901.e4. doi: 10.1016/j.jaci.2016.02.033
10. Hirota K, Yoshitomi H, Hashimoto M, Maeda S, Teradaira S, Sugimoto N, et al. Preferential recruitment of CCR6-expressing Th17 cells to inflamed joints via CCL20 in rheumatoid arthritis and its animal model. J Exp Med (2007) 204:2803–12. doi: 10.1084/jem.20071397
11. Sha Y, Markovic-Plese S. Activated IL-1RI Signaling Pathway Induces Th17 Cell Differentiation via Interferon Regulatory Factor 4 Signaling in Patients with Relapsing-Remitting Multiple Sclerosis. Front Immunol (2016) 7:543. doi: 10.3389/fimmu.2016.00543
12. Ritvo P-GG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, et al. T(fr) cells lack IL-2Rα but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of T(fh) cells. Sci Immunol (2017) 2:eaan0368. doi: 10.1126/sciimmunol.aan0368
13. Nikolouli E, Elfaki Y, Herppich S, Schelmbauer C, Delacher M, Falk C, et al. Recirculating IL-1R2+ Tregs fine-tune intrathymic Treg development under inflammatory conditions. Cell Mol Immunol (2020) 18(1):182–93. doi: 10.1038/s41423-019-0352-8
14. De Simone M, Arrigoni A, Rossetti G, Gruarin P, Ranzani V, Politano C, et al. Transcriptional Landscape of Human Tissue Lymphocytes Unveils Uniqueness of Tumor-Infiltrating T Regulatory Cells. Immunity (2016) 45:1135–47. doi: 10.1016/j.immuni.2016.10.021
15. Mercer F, Kozhaya L, Unutmaz D. Expression and function of TNF and IL-1 receptors on human regulatory T cells. PLoS One (2010) 5:e8639. doi: 10.1371/journal.pone.0008639
16. Ritvo P-G, Klatzmann D. Interleukin-1 in the Response of Follicular Helper and Follicular Regulatory T Cells. Front Immunol (2019) 10:250. doi: 10.3389/fimmu.2019.00250
17. Alvarez F, Fritz JH, Piccirillo CA. Pleiotropic Effects of IL-33 on CD4+ T Cell Differentiation and Effector Functions. Front Immunol (2019) 10(522):1–14. doi: 10.3389/fimmu.2019.00522
18. Peine M, Marek RM, Löhning M. IL-33 in T Cell Differentiation, Function, and Immune Homeostasis. Trends Immunol (2016) 37:321–33. doi: 10.1016/j.it.2016.03.007
19. Bulek K, Swaidani S, Qin J, Lu Y, Gulen MF, Herjan T, et al. The essential role of single Ig IL-1 receptor-related molecule/Toll IL-1R8 in regulation of Th2 immune response. J Immunol (2009) 182:2601–9. doi: 10.4049/jimmunol.0802729
20. Schiering C, Krausgruber T, Chomka A, Fröhlich A, Adelmann K, Wohlfert EA, et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature (2014) 513:564–8. doi: 10.1038/nature13577
21. Siede J, Fröhlich A, Datsi A, Hegazy AN, Varga DV, Holecska V, et al. IL-33 Receptor-Expressing Regulatory T Cells Are Highly Activated, Th2 Biased and Suppress CD4 T Cell Proliferation through IL-10 and TGFβ Release. PLoS One (2016) 11:e0161507. doi: 10.1371/journal.pone.0161507
22. Kobayashi T, Iijima K, Dent AL, Kita H. Follicular helper T cells mediate IgE antibody response to airborne allergens. J Allergy Clin Immunol (2017) 139:300–13.e7. doi: 10.1016/j.jaci.2016.04.021
23. Koh B, Ulrich BJ, Nelson AS, Panangipalli G, Kharwadkar R, Wu W, et al. Bcl6 and Blimp1 reciprocally regulate ST2+ Treg–cell development in the context of allergic airway inflammation. J Allergy Clin Immunol (2020) 146:1121–36.e9. doi: 10.1016/j.jaci.2020.03.002
24. Ramadan A, Zhang J, Griesenauer B, Kapur R, Hanenberg H, Sun J, et al. IL-33/ST2 activation of IL-9-secreting T cells alters the balance of fatal immunity and tumor immunity (TRAN1P.926). J Immunol (2015) 194: 140.8 LP–140.8. doi: 10.1182/blood.V126.23.231.231
25. Yoshimoto T, Takeda K, Tanaka T, Ohkusu K, Kashiwamura S, Okamura H, et al. IL-12 up-regulates IL-18 receptor expression on T cells, Th1 cells, and B cells: synergism with IL-18 for IFN-γ production. J Immunol (1998) 161:3400–7.
26. Novick D, Kim SH, Fantuzzi G, Reznikov LL, Dinarello CA, Rubinstein M. Interleukin-18 binding protein: a novel modulator of the Th1 cytokine response. Immunity (1999) 10:127–36. doi: 10.1016/S1074-7613(00)80013-8
27. Xu D, Chan WL, Leung BP, Hunter D, Schulz K, Carter RW, et al. Selective expression and functions of interleukin 18 receptor on T helper (Th) type 1 but not Th2 cells. J Exp Med (1998) 188:1485–92. doi: 10.1084/jem.188.8.1485
28. Peligero-Cruz C, Givony T, Sebé-Pedrós A, Dobeš J, Kadouri N, Nevo S, et al. IL18 signaling promotes homing of mature Tregs into the thymus. Elife (2020) 9:e58213. doi: 10.7554/eLife.58213
29. Vigne S, Palmer G, Martin P, Lamacchia C, Strebel D, Rodriguez E. IL-36 signaling amplifies Th1 responses by enhancing proliferation and Th1 polarization of naive CD4+ T cells. Blood (2012) 120:3478–87. doi: 10.1182/blood-2012-06-439026
30. St. Paul M, Ohashi PS. The Roles of CD8+ T Cell Subsets in Antitumor Immunity. Trends Cell Biol (2020) 30:695–704. doi: 10.1016/j.tcb.2020.06.003
31. Mantovani A, Dinarello CA, Molgora M, Garlanda C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity (2019) 50:778–95. doi: 10.1016/j.immuni.2019.03.012
32. Boraschi D, Italiani P, Weil S, Martin MU. The family of the interleukin-1 receptors. Immunol Rev (2018) 281:197–232. doi: 10.1111/imr.12606
33. Fields JK, Günther S, Sundberg EJ. Structural Basis of IL-1 Family Cytokine Signaling. Front Immunol (2019) 10(1412):1–20. doi: 10.3389/fimmu.2019.01412
34. Afonina IS, uller CM, Martin SJ, Beyaert R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity (2015) 42:991–1004. doi: 10.1016/j.immuni.2015.06.003
35. Di Paolo NC, Shayakhmetov DM. Interleukin 1α and the inflammatory process. Nat Immunol (2016) 17:906–13. doi: 10.1038/ni.3503
36. Weber A, Wasiliew P, Kracht M. Interleukin-1 (IL-1) Pathway. Sci Signal (2010) 3:1–6. doi: 10.1126/scisignal.3105cm1
37. Wessendorf JH, Garfinkel S, Zhan X, Brown S, Maciag T. Identification of a nuclear localization sequence within the structure of the human interleukin-1 alpha precursor. J Biol Chem (1993) 268:22100–4.
38. Kawaguchi Y, Nishimagi E, Tochimoto A, Kawamoto M, Katsumata Y, Soejima M, et al. Intracellular IL-1α-binding proteins contribute to biological functions of endogenous IL-1α in systemic sclerosis fibroblasts. Proc Natl Acad Sci (2006) 103:14501–6. doi: 10.1073/pnas.0603545103
39. Buryskova M, Pospisek M, Grothey A, Simmet T, Burysek L. Intracellular interleukin-1alpha functionally interacts with histone acetyltransferase complexes. J Biol Chem (2004) 279:4017–26. doi: 10.1074/jbc.M306342200
40. Zamostna B, Novak J, Vopalensky V, Masek T, Burysek L, Pospisek M. N-terminal domain of nuclear IL-1α shows structural similarity to the C-terminal domain of Snf1 and binds to the HAT/core module of the SAGA complex. PLoS One (2012) 7:e41801–1. doi: 10.1371/journal.pone.0041801
41. Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, et al. Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proc Natl Acad Sci USA (2010) 107:2574–9. doi: 10.1073/pnas.0915018107
42. Kurt-Jones EA, Virgin HW 4th, Unanue ER. In vivo and in vitro expression of macrophage membrane interleukin 1 in response to soluble and particulate stimuli. J Immunol (1986) 137:10–4.
43. Afonina IS, Tynan GA, Logue SE, Cullen SP, Bots M, Lüthi AU, et al. Granzyme B-dependent proteolysis acts as a switch to enhance the proinflammatory activity of IL-1α. Mol Cell (2011) 44:265–78. doi: 10.1016/j.molcel.2011.07.037
44. Hiscott J, Marois J, Garoufalis J, D’Addario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol (1993) 13:6231–40. doi: 10.1128/MCB.13.10.6231
45. Mosley B, Urdal DL, Prickett KS, Larsen A, Cosman D, Conlon PJ, et al. The interleukin-1 receptor binds the human interleukin-1 alpha precursor but not the interleukin-1 beta precursor. J Biol Chem (1987) 262:2941–4.
46. Kostura MJ, Tocci MJ, Limjuco G, Chin J, Cameron P, Hillman AG, et al. Identification of a monocyte specific pre-interleukin 1 beta convertase activity. Proc Natl Acad Sci USA (1989) 86:5227–31. doi: 10.1073/pnas.86.14.5227
47. Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature (1992) 356:768–74. doi: 10.1038/356768a0
48. Lamkanfi M, Dixit VM. Mechanisms and Functions of Inflammasomes. Cell (2014) 157:1013–22. doi: 10.1016/j.cell.2014.04.007
49. Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int J Mol Sci (2019) 20:3328. doi: 10.3390/ijms20133328
50. Swanson KV, Deng M. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 19:477–89. doi: 10.1038/s41577-019-0165-0
51. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol (2009) 183:787–91. doi: 10.4049/jimmunol.0901363
52. Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature (2006) 440:237–41. doi: 10.1038/nature04516
53. Perregaux D, Gabel CA. Interleukin-1 beta maturation and release in response to ATP and nigericin. Evidence that potassium depletion mediated by these agents is a necessary and common feature of their activity. J Biol Chem (1994) 269:15195–203.
54. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
55. Franchi L, Eigenbrod T, Muñoz-Planillo R, Ozkurede U, Kim Y-G, Arindam C, et al. Cytosolic double-stranded RNA activates the NLRP3 inflammasome via MAVS-induced membrane permeabilization and K+ efflux. J Immunol (2014) 193:4214–22. doi: 10.4049/jimmunol.1400582
56. Lamkanfi M, Malireddi RKS, Kanneganti T-D. Fungal zymosan and mannan activate the cryopyrin inflammasome. J Biol Chem (2009) 284:20574–81. doi: 10.1074/jbc.M109.023689
57. van Raam BJ, Salvesen GS. Proliferative versus apoptotic functions of caspase-8 Hetero or homo: the caspase-8 dimer controls cell fate. Biochim Biophys Acta (2012) 1824:113–22. doi: 10.1016/j.bbapap.2011.06.005
58. van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LAB. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol (2011) 32:110–6. doi: 10.1016/j.it.2011.01.003
59. Coeshott C, Ohnemus C, Pilyavskaya A, Ross S, Wieczorek M, Kroona H, et al. Converting enzyme-independent release of tumor necrosis factor alpha and IL-1beta from a stimulated human monocytic cell line in the presence of activated neutrophils or purified proteinase 3. Proc Natl Acad Sci USA (1999) 96:6261–6. doi: 10.1073/pnas.96.11.6261
60. Clancy DM, Sullivan GP, Moran HBT, Henry CM, Reeves EP, McElvaney NG, et al. Extracellular Neutrophil Proteases Are Efficient Regulators of IL-1, IL-33, and IL-36 Cytokine Activity but Poor Effectors of Microbial Killing. Cell Rep (2018) 22:2937–50. doi: 10.1016/j.celrep.2018.02.062
61. Monteleone M, Stow JL, Schroder K. Mechanisms of unconventional secretion of IL-1 family cytokines. Cytokine (2015) 74:213–8. doi: 10.1016/j.cyto.2015.03.022
62. Brough D, Rothwell NJ. Caspase-1-dependent processing of pro-interleukin-1β is cytosolic and precedes cell death. J Cell Sci (2007) 120:772– 81. doi: 10.1242/jcs.03377
63. Dupont N, Jiang S, Pilli M, Ornatowski W, Bhattacharya D, Deretic V. Autophagy-based unconventional secretory pathway for extracellular delivery of IL-1β. EMBO J (2011) 30:4701–11. doi: 10.1038/emboj.2011.398
64. Harris J, Hartman M, Roche C, Zeng SG, O’Shea A, Sharp FA, et al. Autophagy controls IL-1beta secretion by targeting pro-IL-1beta for degradation. J Biol Chem (2011) 286:9587–97. doi: 10.1074/jbc.M110.202911
65. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
66. He W, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1β secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
67. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
68. Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity (2018) 48:35–44.e6. doi: 10.1016/j.immuni.2017.11.013
69. Zanoni I, Tan Y, Di Gioia M, Broggi A, Ruan J, Shi J, et al. An endogenous caspase-11 ligand elicits interleukin-1 release from living dendritic cells. Science (80- ) (2016) 352:1232–6. doi: 10.1126/science.aaf3036
70. Zhivaki D, Borriello F, Chow OA, Doran B, Fleming I, Theisen DJ, et al. Inflammasomes within Hyperactive Murine Dendritic Cells Stimulate Long-Lived T Cell-Mediated Anti-tumor Immunity. Cell Rep (2020) 33:108381. doi: 10.1016/j.celrep.2020.108381
71. Vigers GP, Anderson LJ, Caffes P, Brandhuber BJ. Crystal structure of the type-I interleukin-1 receptor complexed with interleukin-1beta. Nature (1997) 386:190–4. doi: 10.1038/386190a0
72. Kollewe C, Neumann D, Martin MU. The first two N-terminal immunoglobulin-like domains of soluble human IL-1 receptor type II are sufficient to bind and neutralize IL-1β. FEBS Lett (2000) 29:189–93. doi: 10.1016/S0014-5793(00)02345-0
73. Thomas C, Bazan JF. Structure of the activating IL-1 receptor signaling complex. Nat Struct Mol Biol (2012) 19:455–7. doi: 10.1038/nsmb.2260
74. Wang D, Zhang S, Li L, Liu X, Mei K, Wang X. Structural insights into the assembly and activation of IL-1β with its receptors. Nat Immunol (2011) 11:905–11. doi: 10.1038/ni.1925
75. Malinowsky D, Lundkvist J, Laye S, Bartfai T. Interleukin-1 receptor accessory protein interacts with the type II interleukin-1 receptor. FEBS Lett (1998) 429:299–302. doi: 10.1016/S0014-5793(98)00467-0
76. Yamada M, Nishizawa M, Nakatake R, Habara K, Yoshida H, Ozaki T, et al. Characterization of alternatively spliced isoforms of the type I interleukin-1 receptor on iNOS induction in rat hepatocytes. Nitric Oxide (2007) 17:98–105. doi: 10.1016/j.niox.2007.06.003
77. Schreuder H, Tardif C, Trump-Kallmeyer S, Soffientini A, Sarubbi E, Akeson A, et al. A New Cytokine-Receptor Binding Mode Revealed by the Crystal Structure of the IL-1 Receptor With an Antagonist. Nature (1997) 386:194–200. doi: 10.1038/386194a0
78. Vigers GP, Caffes P, Evans RJ, Thompson RC, Eisenberg SP, Brandhuber BJ. X-ray structure of interleukin-1 receptor antagonist at 2.0-A resolution. J Biol Chem (1994) 269:12874–9.
79. Belderbos RA, Aerts JGJV, Vroman H. Enhancing Dendritic Cell Therapy in Solid Tumors with Immunomodulating Conventional Treatment. Mol Ther Oncolytics (2019) 13:67–81. doi: 10.1016/j.omto.2019.03.007
80. Mayordomo J I, Zorina T, Storkus WJ, Zitvogel L, Celluzzi C, Falo LD, et al. Bone marrow-derived dendritic cells pulsed with synthetic tumour peptides elicit protective and therapeutic antitumour immunity. Nat Med (1995) 1:1297–302. doi: 10.1038/nm1295-1297
81. Heystek HC, Mudde GC, Ohler R, Kalthoff FS. Granulocyte-macrophage colony-stimulating factor (GM-CSF) has opposing effects on the capacity of monocytes versus monocyte-derived dendritic cells to stimulate the antigen-specific proliferation of a human T cell clone. Clin Exp Immunol (2000) 120:440–7. doi: 10.1046/j.1365-2249.2000.01225.x
82. Hsu FJ, Benike C, Fagnoni F, Liles TM, Czerwinski D, Taidi B, et al. Vaccination of patients with B-cell lymphoma using autologous antigen-pulsed dendritic cells. Nat Med (1996) 2:52–8. doi: 10.1038/nm0196-52
83. Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med (1998) 4:328–32. doi: 10.1038/nm0398-328
84. Ahmed MS, Bae Y-S. Dendritic cell-based therapeutic cancer vaccines: past, present and future. Clin Exp Vaccine Res (2014) 3:113–6. doi: 10.7774/cevr.2014.3.2.113
85. Castiello L, Sabatino M, Jin P, Clayberger C, Marincola FM, Krensky AM, et al. Monocyte-derived DC maturation strategies and related pathways: a transcriptional view. Cancer Immunol Immunother (2011) 60:457–66. doi: 10.1007/s00262-010-0954-6
86. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol (2014) 15:e257–67. doi: 10.1016/S1470-2045(13)70585-0
87. Van Lint S, Wilgenhof S, Heirman C, Corthals J, Breckpot K, Bonehill A, et al. Optimized dendritic cell-based immunotherapy for melanoma: the TriMix-formula. Cancer Immunol Immunother (2014) 63:959–67. doi: 10.1007/s00262-014-1558-3
88. Boudewijns S, Westdorp H, Koornstra RHT, Aarntzen EHJG, Schreibelt G, Creemers JHA, et al. Immune-related Adverse Events of Dendritic Cell Vaccination Correlate With Immunologic and Clinical Outcome in Stage III and IV Melanoma Patients. J Immunother (2016) 39:241–8. doi: 10.1097/CJI.0000000000000127
89. Fukuda K, Funakoshi T, Sakurai T, Nakamura Y, Mori M, Tanese K, et al. Peptide-pulsed dendritic cell vaccine in combination with carboplatin and paclitaxel chemotherapy for stage IV melanoma. Melanoma Res (2017) 27:326–34. doi: 10.1097/CMR.0000000000000342
90. Koido S, Homma S, Okamoto M, Takakura K, Mori M, Yoshizaki S, et al. Treatment with chemotherapy and dendritic cells pulsed with multiple Wilms’ tumor 1 (WT1)-specific MHC class I/II-restricted epitopes for pancreatic cancer. Clin Cancer Res (2014) 20:4228–39. doi: 10.1158/1078-0432.CCR-14-0314
91. Brutkiewicz RR. Cell Signaling Pathways That Regulate Antigen Presentation. J Immunol (2015) 197:2971–9. doi: 10.4049/jimmunol.1600460
92. Edelson BT, Unanue ER. MyD88-Dependent but Toll-Like Receptor 2-Independent Innate Immunity to <em<Listeria</em<: No Role for Either in Macrophage Listericidal Activity. J Immunol (2002) 169:3869–75. doi: 10.4049/jimmunol.169.7.3869
93. Seki E, Tsutsui H, Tsuji NM, Hayashi N, Adachi K, Nakano H, et al. Critical Roles of Myeloid Differentiation Factor 88-Dependent Proinflammatory Cytokine Release in Early Phase Clearance of Listeria monocytogenes in Mice. J Immunol (2002) 169:3863–8. doi: 10.4049/jimmunol.169.7.3863
94. Arnold-Schrauf C, Dudek M, Dielmann A, Pace L, Swallow M, Kruse F, et al. Dendritic Cells Coordinate Innate Immunity via MyD88 Signaling to Control Listeria monocytogenes Infection. Cell Rep (2014) 6:698–708. doi: 10.1016/j.celrep.2014.01.023
95. Lamagna C, Scapini P, van Ziffle JA, DeFranco AL, Lowell CA. Hyperactivated MyD88 signaling in dendritic cells, through specific deletion of Lyn kinase, causes severe autoimmunity and inflammation. Proc Natl Acad Sci (2013) 110:E3311–20. doi: 10.1073/pnas.1300617110
96. Kobayashi T, Walsh PT, Walsh MC, Speirs KM, Chiffoleau E, King CG, et al. TRAF6 is a critical factor for dendritic cell maturation and development. Immunity (2003) 19:353–63. doi: 10.1016/S1074-7613(03)00230-9
97. Rescigno M, Martino M, Sutherland CL, Gold MR, Ricciardi-Castagnoli P. Dendritic cell survival and maturation are regulated by different signaling pathways. J Exp Med (1998) 188:2175–80. doi: 10.1084/jem.188.11.2175
98. Hernandez A, Burger M, Blomberg BB, Ross WA, Gaynor JJ, Lindner I, et al. Inhibition of NF-kappa B during human dendritic cell differentiation generates anergy and regulatory T-cell activity for one but not two human leukocyte antigen DR mismatches. Hum Immunol (2007) 68:715–29. doi: 10.1016/j.humimm.2007.05.010
99. Ade N, Antonios D, Kerdine-Romer S, Boisleve F, Rousset F, Pallardy M. NF-kappaB plays a major role in the maturation of human dendritic cells induced by NiSO(4) but not by DNCB. Toxicol Sci (2007) 99:488–501. doi: 10.1093/toxsci/kfm178
100. Ouaaz F, Arron J, Zheng Y, Choi Y, Beg AA. Dendritic Cell Development and Survival Require Distinct NF-κB Subunits. Immunity (2002) 16:257–70. doi: 10.1016/S1074-7613(02)00272-8
101. Baratin M, Foray C, Demaria O, Habbeddine M, Pollet E, Maurizio J, et al. Homeostatic NF-κB Signaling in Steady-State Migratory Dendritic Cells Regulates Immune Homeostasis and Tolerance. Immunity (2015) 42:627–39. doi: 10.1016/j.immuni.2015.03.003
102. Hägerbrand K, Westlund J, Yrlid U, Agace W, Johansson-Lindbom B. MyD88 Signaling Regulates Steady-State Migration of Intestinal CD103<sup<+</sup< Dendritic Cells Independently of TNF-α and the Gut Microbiota. J Immunol (2015) 195:2888–99. doi: 10.4049/jimmunol.1500210
103. Bunyard P, Handley M, Pollara G, Rutault K, Wood I, Chaudry M, et al. Ribotoxic stress activates p38 and JNK kinases and modulates the antigen-presenting activity of dendritic cells. Mol Immunol (2003) 39:815–27. doi: 10.1016/S0161-5890(02)00262-6
104. Bharadwaj U, Zhang R, Yang H, Li M, Doan LX, Chen C, et al. Effects of cyclophilin A on myeloblastic cell line KG-1 derived dendritic like cells (DLC) through p38 MAP kinase activation. J Surg Res (2005) 127:29–38. doi: 10.1016/j.jss.2005.02.020
105. Seshasayee D, Wang H, Lee WP, Gribling P, Ross J, Van Bruggen N, et al. A novel in vivo role for osteoprotegerin ligand in activation of monocyte effector function and inflammatory response. J Biol Chem (2004) 279:30202–9. doi: 10.1074/jbc.M403968200
106. Yu Q, Kovacs C, Yue FY, Ostrowski MA. The role of the p38 mitogen-activated protein kinase, extracellular signal-regulated kinase, and phosphoinositide-3-OH kinase signal transduction pathways in CD40 ligand-induced dendritic cell activation and expansion of virus-specific CD8+ T cell memory. J Immunol (2004) 172:6047–56. doi: 10.4049/jimmunol.172.10.6047
107. Arbour N, Naniche D, Homann D, Davis RJ, Flavell RA, Oldstone MBA, et al. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 signaling pathways have divergent roles in CD8(+) T cell-mediated antiviral immunity. J Exp Med (2002) 195:801–10. doi: 10.1084/jem.20011481
108. Conze D, Krahl T, Kennedy N, Weiss L, Lumsden J, Hess P, et al. c-Jun NH(2)-terminal kinase (JNK)1 and JNK2 have distinct roles in CD8(+) T cell activation. J Exp Med (2002) 195:811–23. doi: 10.1084/jem.20011508
109. Liu J, Gallo RM, Khan MA, Iyer AK, Kratzke IM, Brutkiewicz RR. JNK2 modulates the CD1d-dependent and -independent activation of iNKT cells. Eur J Immunol (2019) 49:255–65. doi: 10.1002/eji.201847755
110. Koide SL, Inaba K, Steinman RM. Interleukin 1 Enhances T-dependent Immune Responses by Amplifying the Function of Dendritic Cells. J Exp Med (1987) 165:515–30. doi: 10.1084/jem.165.2.515
111. Wesa A, Galy A. Increased production of pro-inflammatory cytokines and enhanced T cell responses after activation of human dendritic cells with IL-1 and CD40 ligand. BMC Immunol (2002) 3:14. doi: 10.1186/1471-2172-3-14
112. Kruse M, Meinl E, Henning G, Kuhnt C, Berchtold S, Berger T, et al. Signaling Lymphocytic Activation Molecule Is Expressed on Mature CD83<sup<+</sup< Dendritic Cells and Is Up-Regulated by IL-1β. J Immunol (2001) 167:1989–95. doi: 10.4049/jimmunol.167.4.1989
113. Xin L, Li Y, Soong L. Role of Interleukin-1β in Activating the CD11chigh CD45RB− Dendritic Cell Subset and Priming Leishmania amazonensis Specific CD4+ T Cells In Vitro and In Vivo. Infect Immun (2007) 75:5018–26. doi: 10.1128/IAI.00499-07
114. Matjeka T, Summerfield V, Noursadeghi M, Chain BM. Chemical toxicity to keratinocytes triggers dendritic cell activation via an IL-1α path. J Allergy Clin Immunol (2012) 129:247–50. doi: 10.1016/j.jaci.2011.08.018
115. Pang IK, Ichinohe T, Iwasaki A. IL-1R signaling in dendritic cells replaces pattern-recognition receptors in promoting CD8+ T cell responses to influenza A virus. Nat Immunol (2013) 14:246–53. doi: 10.1038/ni.2514
116. Kautz-Neu K, Kostka SL, Dinges S, Iwakura Y, Udey MC, von Stebut E. A Role for Leukocyte-Derived IL-1RA in DC Homeostasis Revealed by Increased Susceptibility of IL-1RA-Deficient Mice to Cutaneous Leishmaniasis. J Invest Dermatol (2011) 131:1650–9. doi: 10.1038/jid.2011.99
117. Sugita S, Kawazoe Y, Imai A, Usui Y, Iwakura Y, Isoda K, et al. Mature Dendritic Cell Suppression by IL-1 Receptor Antagonist on Retinal Pigment Epithelium Cells. Invest Ophthalmol Vis Sci (2013) 54:3240–9. doi: 10.1167/iovs.12-11483
118. Deckers J, Hammad H, Hoste E. Langerhans Cells: Sensing the Environment in Health and Disease. Front Immunol (2018) 9(93):1–14. doi: 10.3389/fimmu.2018.00093
119. Leech JM, Dhariwala MO, Lowe MM, Chu K, Merana GR, Cornuot C, et al. Toxin-Triggered Interleukin-1 Receptor Signaling Enables Early-Life Discrimination of Pathogenic versus Commensal Skin Bacteria. Cell Host Microbe (2019) 26:795–809.e5. doi: 10.1016/j.chom.2019.10.007
120. Wu X, Briseño CG, Durai V, Albring JC, Haldar M, Bagadia P, et al. Mafb lineage tracing to distinguish macrophages from other immune lineages reveals dual identity of Langerhans cells. J Exp Med (2016) 213:2553–65. doi: 10.1084/jem.20160600
121. Heufler C, Koch F, Schuler G. Granulocyte/macrophage colony-stimulating factor and interleukin 1 mediate the maturation of murine epidermal Langerhans cells into potent immunostimulatory dendritic cells. J Exp Med (1988) 167:700–5. doi: 10.1084/jem.167.2.700
122. Koch F, Heufler C, Kämpgen E, Schneeweiss D, Böck G, Schuler G. Tumor necrosis factor alpha maintains the viability of murine epidermal Langerhans cells in culture, but in contrast to granulocyte/macrophage colony-stimulating factor, without inducing their functional maturation. J Exp Med (1990) 171:159–71. doi: 10.1084/jem.171.1.159
123. Furue M, Chang CH, Tamaki K. Interleukin-1 but not tumour necrosis factor alpha synergistically upregulates the granulocyte-macrophage colony-stimulating factor-induced B7-1 expression of murine Langerhans cells. Br J Dermatol (1996) 135:194–8. doi: 10.1046/j.1365-2133.1996.d01-974.x
124. Ozawa H, Nakagawa S, Tagami H, Aiba S. Interleukin-1 beta and granulocyte-macrophage colony-stimulating factor mediate Langerhans cell maturation differently. J Invest Dermatol (1996) 106:441–5. doi: 10.1111/1523-1747.ep12343589
125. Enk AH, Angeloni VL, Udey MC, Katz S II. An essential role for Langerhans cell-derived IL-1 beta in the initiation of primary immune responses in skin. J Immunol (1993) 150:3698–704.
126. Kimber I, Cumberbatch M, Dearman RJ, Bhushan M, Griffiths CEM. REVIEWCytokines and chemokines in the initiation and regulation of epidermal Langerhans cell mobilization. Br J Dermatol (2000) 142:401–12. doi: 10.1046/j.1365-2133.2000.03349.x
127. Cumberbatch M, Dearman RJ, Kimber I. Interleukin 1 beta and the stimulation of Langerhans cell migration: comparisons with tumour necrosis factor alpha. Arch Dermatol Res (1997) 289:277–84. doi: 10.1007/s004030050193
128. Cumberbatch M, Bhushan M, Dearman RJ, Kimber I, Griffiths CEM. IL-1beta-induced Langerhans’ cell migration and TNF-alpha production in human skin: regulation by lactoferrin. Clin Exp Immunol (2003) 132:352–9. doi: 10.1046/j.1365-2249.2003.02146.x
129. Cumberbatch M, Dearman RJ, Kimber I. Langerhans cells require signals from both tumour necrosis factor-alpha and interleukin-1 beta for migration. Immunology (1997) 92:388–95. doi: 10.1046/j.1365-2567.1997.00360.x
130. Cumberbatch M, Dearman RJ, Kimber I. Characteristics and regulation of the expression on interleukin 1 receptors by murine Langerhans cells and keratinocytes. Arch Dermatol Res (1998) 290:688–95. doi: 10.1007/s004030050374
131. Nishibu A, Ward BR, Boes M, Takashima A. Roles for IL-1 and TNFalpha in dynamic behavioral responses of Langerhans cells to topical hapten application. J Dermatol Sci (2007) 45:23–30. doi: 10.1016/j.jdermsci.2006.10.003
132. Cumberbatch M, Dearman RJ, Kimber I. Langerhans cell migration in mice requires intact type I interleukin 1 receptor (IL-1RI) function. Arch Dermatol Res (1999) 291:357–61. doi: 10.1007/s004030050422
133. Kawabe T, Matsushima M, Hashimoto N, Imaizumi K, Hasegawa Y. CD40/CD40 ligand interactions in immune responses and pulmonary immunity. Nagoya J Med Sci (2011) 73:69–78.
134. Nakae S, Asano M, Horai R, Sakaguchi N, Iwakura Y. IL-1 Enhances T Cell-Dependent Antibody Production Through Induction of CD40 Ligand and OX40 on T Cells. J Immunol (2001) 167:90–7. doi: 10.4049/jimmunol.167.1.90
135. Luft T, Jefford M, Luetjens P, Hochrein H, Masterman K-A, Maliszewski C, et al. IL-1 Beta Enhances CD40 Ligand-Mediated Cytokine Secretion by Human Dendritic Cells (DC): A Mechanism for T Cell-Independent DC Activation. J Immunol (2002) 168:713–22. doi: 10.4049/jimmunol.168.2.713
136. Wesa AK, Galy A. IL-1β induces dendritic cells to produce IL-12. Int Immunol (2001) 13:1053–61. doi: 10.1093/intimm/13.8.1053
137. Eriksson U, Kurrer MO, Sonderegger I, Iezzi G, Tafuri A, Hunziker L, et al. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med (2003) 197:323–31. doi: 10.1084/jem.20021788
138. Jain A, Irizarry-Caro RA, McDaniel MM, Chawla AS, Carroll KR, Overcast GR, et al. T cells instruct myeloid cells to produce inflammasome-independent IL-1β and cause autoimmunity. Nat Immunol (2020) 21:65–74. doi: 10.1038/s41590-019-0559-y
139. Yao Y, Chen S, Cao M, Fan X, Yang T, Huang Y, et al. Antigen-specific CD8+ T cell feedback activates NLRP3 inflammasome in antigen-presenting cells through perforin. Nat Commun (2017) 8:15402. doi: 10.1038/ncomms15402
140. Hildebrand D, Bode KA, Rieß D, Cerny D, Waldhuber A, Römmler F, et al. Granzyme A Produces Bioactive IL-1β through a Nonapoptotic Inflammasome-Independent Pathway. Cell Rep (2014) 9:910–7. doi: 10.1016/j.celrep.2014.10.003
141. Iwasaki A, Medzhitov R. Regulation of adaptive immunity by the innate immune system. Science (2010) 327:291–5. doi: 10.1126/science.1183021
142. Asrat S, Dugan AS, Isberg RR. The frustrated host response to Legionella pneumophila is bypassed by MyD88-dependent translation of pro-inflammatory cytokines. PLoS Pathog (2014) 10:e1004229. doi: 10.1371/journal.ppat.1004229
143. Copenhaver AM, Casson CN, Nguyen HT, Duda MM, Shin S. IL-1R signaling enables bystander cells to overcome bacterial blockade of host protein synthesis. Proc Natl Acad Sci (2015) 112:7557–62. doi: 10.1073/pnas.1501289112
144. Lichtman AH, Chin J, Schmidt JA, Abbas AK. Role of interleukin 1 in the activation of T lymphocytes. Proc Natl Acad Sci (1988) 85:9699–703. doi: 10.1073/pnas.85.24.9699
145. Khoruts A, Osness RE, Jenkins MK. IL-1 acts on antigen-presenting cells to enhance the in vivo proliferation of antigen-stimulated naive CD4 T cells via a CD28-dependent mechanism that does not involve increased expression of CD28 ligands. Eur J Immunol (2004) 34:1085–90. doi: 10.1002/eji.200324170
146. Pape KA, Khoruts A, Mondino A, Jenkins MK. Inflammatory cytokines enhance the in vivo clonal expansion and differentiation of antigen-activated CD4+ T cells. J Immunol (1997) 159:591–8.
147. Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J Exp Med (2009) 206:79–87. doi: 10.1084/jem.20081667
148. Schmitz N, Kurrer M, Bachmann MF, Kopf M. Interleukin-1 is responsible for acute lung immunopathology but increases survival of respiratory influenza virus infection. J Virol (2005) 79:6441–8. doi: 10.1128/JVI.79.10.6441-6448.2005
149. Szretter KJ, Gangappa S, Lu X, Smith C, Shieh W-J, Zaki SR, et al. Role of Host Cytokine Responses in the Pathogenesis of Avian H5N1 Influenza Viruses in Mice. J Virol (2007) 81:2736–44. doi: 10.1128/JVI.02336-06
150. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, et al. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci USA (2009) 106:7119–24. doi: 10.1073/pnas.0902745106
151. Zhu J, Yamane H, Paul WE. Differentiation of Effector CD4 T Cell Populations. Annu Rev Immunol (2010) 28:445–89. doi: 10.1146/annurev-immunol-030409-101212
152. Iwasaki A. The Importance of CD11b+ Dendritic Cells in CD4+ T Cell Activation In Vivo : With Help from Interleukin 1. J Exp Med (2003) 198:185–90. doi: 10.1084/jem.20030737
153. Lima-Junior DS, Costa DL, Carregaro V, Cunha LD, Silva ALN, Mineo TWP, et al. Inflammasome-derived IL-1β production induces nitric oxide–mediated resistance to Leishmania. Nat Med (2013) 19:909–15. doi: 10.1038/nm.3221
154. Satoskar AR, Okano M, Connaughton S, Raisanen-Sokolwski A, David JR, Labow M. Enhanced Th2-like responses in IL-1 type 1 receptor-deficient mice. Eur J Immunol (1998) 28:2066–74. doi: 10.1002/(SICI)1521-4141(199807)28:07<2066::AID-IMMU2066>3.0.CO;2-X
155. Von Stebut E, Ehrchen JM, Belkaid Y, Kostka SL, Molle K, Knop J. Interleukin 1alpha promotes Th1 differentiation and inhibits disease progression in Leishmania major-susceptible BALB/c mice. J Exp Med (2003) 198:191–9. doi: 10.1084/jem.20030159
156. Filippi C, Hugues S, Cazareth J, Julia V, Glaichenhaus N, Ugolini S. CD4+ T cell polarization in mice is modulated by strain-specific major histocompatibility complex-independent differences within dendritic cells. J Exp Med (2003) 198:201–9. doi: 10.1084/jem.20021893
157. Ehrchen JM, Roebrock K, Foell D, Nippe N, von Stebut E, Weiss JM, et al. Keratinocytes Determine Th1 Immunity during Early Experimental Leishmaniasis. PLoS Pathog (2010) 6:e1000871. doi: 10.1371/journal.ppat.1000871
158. Zaiss MM, Maslowski KM, Mosconi I, Guenat N, Marsland BJ, Harris NL. IL-1β Suppresses Innate IL-25 and IL-33 Production and Maintains Helminth Chronicity. PLoS Pathog (2013) 9:e1003531. doi: 10.1371/journal.ppat.1003531
159. Birnhuber A, Crnkovic S, Biasin V, Marsh LM, Odler B, Sahu-Osen A, et al. IL-1 receptor blockade skews inflammation towards Th2 in a mouse model of systemic sclerosis. Eur Respir J (2019) 54:1900154. doi: 10.1183/13993003.00154-2019
160. Shourian M, Ralph B, Angers I, Sheppard DC, Qureshi ST. Contribution of IL-1RI Signaling to Protection against Cryptococcus neoformans 52D in a Mouse Model of Infection. Front Immunol (2018) 8(1987):1–18. doi: 10.3389/fimmu.2017.01987
161. Willart MAM, Deswarte K, Pouliot P, Braun H, Beyaert R, Lambrecht BN, et al. Interleukin-1α controls allergic sensitization to inhaled house dust mite via the epithelial release of GM-CSF and IL-33. J Exp Med (2012) 209:1505–17. doi: 10.1084/jem.20112691
162. Qi X, Gurung P, Malireddi RKS, Karmaus PWF, Sharma D, Vogel P, et al. Critical role of caspase-8-mediated IL-1 signaling in promoting Th2 responses during asthma pathogenesis. Mucosal Immunol (2017) 10:128–38. doi: 10.1038/mi.2016.25
163. Nakae S, Komiyama Y, Yokoyama H, Nambu A, Umeda M, Iwase M, et al. IL-1 is required for allergen-specific Th2 cell activation and the development of airway hypersensitivity response. Int Immunol (2003) 15:483–90. doi: 10.1093/intimm/dxg054
164. Schmitz N, Kurrer M, Kopf M. The IL-1 receptor 1 is critical for Th2 cell type airway immune responses in a mild but not in a more severe asthma model. Eur J Immunol (2003) 33:991–1000. doi: 10.1002/eji.200323801
165. Kool M, Willart MAM, van Nimwegen M, Bergen I, Pouliot P, Virchow JC, et al. An Unexpected Role for Uric Acid as an Inducer of T Helper 2 Cell Immunity to Inhaled Antigens and Inflammatory Mediator of Allergic Asthma. Immunity (2011) 34:527–40. doi: 10.1016/j.immuni.2011.03.015
166. Dai X, Sayama K, Tohyama M, Shirakata Y, Hanakawa Y, Tokumaru S, et al. Mite allergen is a danger signal for the skin via activation of inflammasome in keratinocytes. J Allergy Clin Immunol (2011) 127:806–814.e4. doi: 10.1016/j.jaci.2010.12.006
167. Kuhn S, Hyde EJ, Yang J, Rich FJ, Harper JL, Kirman JR, et al. Increased numbers of monocyte-derived dendritic cells during successful tumor immunotherapy with immune-activating agents. J Immunol (2013) 191:1984–92. doi: 10.4049/jimmunol.1301135
168. Kuhn S, Yang J, Hyde EJ, Harper JL, Kirman JR, Ronchese F. IL-1βR-dependent priming of antitumor CD4+ T cells and sustained antitumor immunity after peri-tumoral treatment with MSU and mycobacteria. Oncoimmunology (2015) 4:e1042199. doi: 10.1080/2162402X.2015.1042199
169. Haabeth OAW, Lorvik KB, Hammarström C, Donaldson IM, Haraldsen G, Bogen B, et al. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun (2011) 2:240. doi: 10.1038/ncomms1239
170. Haabeth OAW, Lorvik KB, Yagita H, Bogen B, Corthay A. Interleukin-1 is required for cancer eradication mediated by tumor-specific Th1 cells. Oncoimmunology (2016) 5:e1039763. doi: 10.1080/2162402X.2015.1039763
171. Omenetti S, Bussi C, Metidji A, Iseppon A, Lee S, Tolaini M, et al. The Intestine Harbors Functionally Distinct Homeostatic Tissue-Resident and Inflammatory Th17 Cells. Immunity (2019) 51:77–89.e6. doi: 10.1016/j.immuni.2019.05.004
172. Basu R, Hatton RD, Weaver CT. The Th17 family: flexibility follows function. Immunol Rev (2013) 252:89–103. doi: 10.1111/imr.12035
173. Hori S, Nomura T, Sakaguchi S. Control of Regulatory T Cell Development by the Transcription Factor. Science (80- ) (2003) 299:1057–61. doi: 10.1126/science.1079490
174. Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol (2003) 4:330–6. doi: 10.1038/ni904
175. Ivanov I I, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, et al. The Orphan Nuclear Receptor RORγt Directs the Differentiation Program of Proinflammatory IL-17+ T Helper Cells. Cell (2006) 126:1121–33. doi: 10.1016/j.cell.2006.07.035
176. Hatton RD. TGF-β in Th17 cell development: the truth is out there. Immunity (2011) 34:288–90. doi: 10.1016/j.immuni.2011.03.009
177. Villarino AV, Laurence A. IL-1 watches the watchmen. Nat Immunol (2015) 16:226–7. doi: 10.1038/ni.3105
178. Yang X-P, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, et al. Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol (2011) 12:247–54. doi: 10.1038/ni.1995
179. Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature (2006) 441:235–8. doi: 10.1038/nature04753
180. Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity (2006) 24:179–89. doi: 10.1016/j.immuni.2006.01.001
181. Kryczek I, Wei S, Vatan L, Escara-Wilke J, Szeliga W, Keller ET, et al. Cutting edge: opposite effects of IL-1 and IL-2 on the regulation of IL-17+ T cell pool IL-1 subverts IL-2-mediated suppression. J Immunol (2007) 179:1423–6. doi: 10.4049/jimmunol.179.3.1423
182. Basu R, Whitley SK, Bhaumik S, Zindl CL, Schoeb TR, Benveniste EN, et al. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell–iTreg cell balance. Nat Immunol (2015) 16:286–95. doi: 10.1038/ni.3099
183. Whitley SK, Balasubramani A, Zindl CL, Sen R, Shibata Y, Crawford GE, et al. IL-1R signaling promotes STAT3 and NF-κB factor recruitment to distal cis-regulatory elements that regulate Il17a/f transcription. J Biol Chem (2018) 293:15790–800. doi: 10.1074/jbc.RA118.002721
184. Schiffenbauer J, Streit WJ, Butfiloski E, LaBow M, Edwards C 3rd, Moldawer LL. The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clin Immunol (2000) 95:117–23. doi: 10.1006/clim.2000.4851
185. Sutton C, Brereton C, Keogh B, Mills KHG, Lavelle EC. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med (2006) 203:1685–91. doi: 10.1084/jem.20060285
186. Ronchi F, Basso C, Preite S, Reboldi A, Baumjohann D, Perlini L, et al. Experimental priming of encephalitogenic Th1/Th17 cells requires pertussis toxin-driven IL-1β production by myeloid cells. Nat Commun (2016) 7:11541. doi: 10.1038/ncomms11541
187. Inoue M, Williams KL, Gunn MD, Shinohara ML. NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA (2012) 109:10480–5. doi: 10.1073/pnas.1201836109
188. Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti T-D. Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol (2010) 184:4610–4. doi: 10.4049/jimmunol.1000217
189. Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, et al. Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol (1999) 163:2403–9.
190. Staschke KA, Dong S, Saha J, Zhao J, Brooks NA, Hepburn DL, et al. IRAK4 kinase activity is required for Th17 differentiation and Th17-mediated disease. J Immunol (2009) 183:568–77. doi: 10.4049/jimmunol.0802361
191. Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity (2010) 32:54–66. doi: 10.1016/j.immuni.2009.12.003
192. Nakae S, Saijo S, Horai R, Sudo K, Mori S, Iwakura Y, et al. IL-17 production from activated T cells is required for the spontaneous development of destructive arthritis in mice deficient in IL-1 receptor antagonist. Proc Natl Acad Sci (2003) 100:5986–90. doi: 10.1073/pnas.1035999100
193. Koenders M I, Devesa I, Marijnissen RJ, Abdollahi-Roodsaz S, Boots AMH, Walgreen B, et al. Interleukin-1 drives pathogenic Th17 cells during spontaneous arthritis in interleukin-1 receptor antagonist-deficient mice. Arthritis Rheumatol (2008) 58:3461–70. doi: 10.1002/art.23957
194. Ikeda S, Saijo S, Murayama MA, Shimizu K, Akitsu A, Iwakura Y, et al. Excess IL-1 signaling enhances the development of Th17 cells by downregulating TGF-β-induced Foxp3 expression. J Immunol (2014) 192:1449–58. doi: 10.4049/jimmunol.1300387
195. Mufazalov IA, Regen T, Schelmbauer C, Kuschmann J, Muratova AM, Nikolaev A, et al. Generation of a Novel T Cell Specific Interleukin-1 Receptor Type 1 Conditional Knock Out Mouse Reveals Intrinsic Defects in Survival, Expansion and Cytokine Production of CD4 T Cells. PLoS One (2016) 11:e0161505–e0161505. doi: 10.1371/journal.pone.0161505
196. Chang J, Burkett PR, Borges CM, Kuchroo VK, Turka LA, Chang C-H. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc Natl Acad Sci USA (2013) 110:2270–5. doi: 10.1073/pnas.1206048110
197. Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity (2009) 30:576–87. doi: 10.1016/j.immuni.2009.02.007
198. Gulen MF, Bulek K, Xiao H, Yu M, Gao J, Sun L, et al. Inactivation of the enzyme GSK3α by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity (2012) 37:800–12. doi: 10.1016/j.immuni.2012.08.019
199. Hung C-Y, Jiménez-Alzate M del P, Gonzalez A, Wüthrich M, Klein BS, Cole GT. Interleukin-1 receptor but not Toll-like receptor 2 is essential for MyD88-dependent Th17 immunity to Coccidioides infection. Infect Immun (2014) 82:2106–14. doi: 10.1128/IAI.01579-13
200. Wüthrich M, LeBert V, Galles K, Hu-Li J, Ben-Sasson SZ, Paul WE, et al. Interleukin 1 enhances vaccine-induced antifungal T-helper 17 cells and resistance against Blastomyces dermatitidis infection. J Infect Dis (2013) 208:8713–9. doi: 10.1093/infdis/jit283
201. Deason K, Troutman TD, Jain A, Challa DK, Mandraju R, Brewer T, et al. BCAP links IL-1R to the PI3K-mTOR pathway and regulates pathogenic Th17 cell differentiation. J Exp Med (2018) 215:2413–28. doi: 10.1084/jem.20171810
202. Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F. Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol (2007) 8:942–9. doi: 10.1038/ni1496
203. Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol (2007) 8:950–7. doi: 10.1038/ni1497
204. Ghoreschi K, Laurence A, Yang X-P, Tato CM, McGeachy MJ, Konkel JE, et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature (2010) 467:967–71. doi: 10.1038/nature09447
205. Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO. Autocrine transforming growth factor-β1 promotes in vivo Th17 cell differentiation. Immunity (2011) 34:396–408. doi: 10.1016/j.immuni.2011.03.005
206. Grandclaudon M, Perrot-Dockès M, Trichot C, Karpf L, Abouzid O, Chauvin C, et al. A Quantitative Multivariate Model of Human Dendritic Cell-T Helper Cell Communication. Cell (2019) 179:432–47.e21. doi: 10.1016/j.cell.2019.09.012
207. Revu S, Wu J, Henkel M, Rittenhouse N, Menk A, Delgoffe GM, et al. IL-23 and IL-1β Drive Human Th17 Cell Differentiation and Metabolic Reprogramming in Absence of CD28 Costimulation. Cell Rep (2018) 22:2642–53. doi: 10.1016/j.celrep.2018.02.044
208. Mailer RKW, Joly A-L, Liu S, Elias S, Tegner J, Andersson J. IL-1β promotes Th17 differentiation by inducing alternative splicing of FOXP3. Sci Rep (2015) 5:14674. doi: 10.1038/srep14674
209. O’Sullivan BJ, Thomas HE, Pai S, Santamaria P, Iwakura Y, Steptoe RJ, et al. IL-1 beta breaks tolerance through expansion of CD25+ effector T cells. J Immunol (2006) 176:7278–87. doi: 10.4049/jimmunol.176.12.7278
210. Schenten D, Nish S a, Yu S, Yan X, Lee H, Brodsky I, et al. Signaling through the adaptor molecule MyD88 in CD4+ T cells is required to overcome suppression by regulatory T cells. Immunity (2014) 40:78–90. doi: 10.1016/j.immuni.2013.10.023
211. Ilarregui JM, Van Beelen AJ, Fehres CM, Bruijns SCM, García-vallejo JJ, Van Kooyk Y. New roles for CD14 and IL-[beta] linking inflammatory dendritic cells to IL-17 production in memory CD4+ T cells. Immunol Cell Biol (2016) 94:907–16. doi: 10.1038/icb.2016.66
212. Obregon C, Graf L, Chung KF, Cesson V, Nicod LP. Nitric Oxide Sustains IL-1β Expression in Human Dendritic Cells Enhancing Their Capacity to Induce IL-17–Producing T-Cells. PLoS One (2015) 10:e0120134. doi: 10.1371/journal.pone.0120134
213. Chung Y, Tanaka S, Chu F, Nurieva RI, Martinez GJ, Rawal S, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med (2011) 17:983–8. doi: 10.1038/nm.2426
214. Linterman MA, Pierson W, Lee SK, Kallies A, Kawamoto S, Rayner TF, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med (2011) 17:975–82. doi: 10.1038/nm.2425
215. Wollenberg I, Agua-Doce A, Hernández A, Almeida C, Oliveira VG, Faro J, et al. Regulation of the germinal center reaction by Foxp3+ follicular regulatory T cells. J Immunol (2011) 187:4553–60. doi: 10.4049/jimmunol.1101328
216. Aloulou M, Carr EJ, Gador M, Bignon A, Liblau RS, Fazilleau N, et al. Follicular regulatory T cells can be specific for the immunizing antigen and derive from naive T cells. Nat Commun (2016) 7:10579. doi: 10.1038/ncomms10579
217. Sayin I, Radtke AJ, Vella LA, Jin W, Wherry EJ, Buggert M, et al. Spatial distribution and function of T follicular regulatory cells in human lymph nodes. J Exp Med (2018) 215:1531–42. doi: 10.1084/jem.20171940
218. Barbet G, Sander LE, Geswell M, Leonardi I, Cerutti A, Iliev I, et al. Sensing Microbial Viability through Bacterial RNA Augments T Follicular Helper Cell and Antibody Responses. Immunity (2018) 48:584–98.e5. doi: 10.1016/j.immuni.2018.02.015
219. Dolence JJ, Kobayashi T, Iijima K, Krempski J, Drake LY, Dent AL, et al. Airway exposure initiates peanut allergy by involving the IL-1 pathway and T follicular helper cells in mice. J Allergy Clin Immunol (2018) 142:1144–58.e8. doi: 10.1016/j.jaci.2017.11.020
220. Schmitt N, Liu Y, Bentebibel S-E, Munagala I, Bourdery L, Venuprasad K, et al. The cytokine TGF-β co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol (2014) 15:856–65. doi: 10.1038/ni.2947
221. Ugolini M, Gerhard J, Burkert S, Jensen KJ, Georg P, Ebner F, et al. Recognition of microbial viability via TLR8 drives TFH cell differentiation and vaccine responses. Nat Immunol (2018) 19:386–96. doi: 10.1038/s41590-018-0068-4
222. Nakae S, Asano M, Horai R, Iwakura Y. Interleukin-1 beta, but not interleukin-1 alpha, is required for T-cell-dependent antibody production. Immunology (2001) 104:402–9. doi: 10.1046/j.1365-2567.2001.01337.x
223. Glaccum MB, Stocking KL, Charrier K, Smith JL, Willis CR, Maliszewski C, et al. Phenotypic and functional characterization of mice that lack the type I receptor for IL-1. J Immunol (1997) 159:3364–71.
224. Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, et al. Spatiotemporal Dynamics of Intratumoral Immune Cells Reveal the Immune Landscape in Human Cancer. Immunity (2013) 39:782–95. doi: 10.1016/j.immuni.2013.10.003
225. Roberti MP, Yonekura S, Duong CPM, Picard M, Ferrere G, Tidjani Alou M, et al. Chemotherapy-induced ileal crypt apoptosis and the ileal microbiome shape immunosurveillance and prognosis of proximal colon cancer. Nat Med (2020) 26:919–31. doi: 10.1038/s41591-020-0882-8
226. Dardalhon V, Awasthi A, Kwon H, Galileos G, Gao W, Sobel RA, et al. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3– effector T cells. Nat Immunol (2008) 9:1347–55. doi: 10.1038/ni.1677
227. Veldhoen M, Uyttenhove C, van Snick J, Helmby H, Westendorf A, Buer J, et al. Transforming growth factor-β ‘reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9–producing subset. Nat Immunol (2008) 9:1341–6. doi: 10.1038/ni.1659
228. Licona-Limón P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limón I, Ishigame H, et al. Th9 Cells Drive Host Immunity against Gastrointestinal Worm Infection. Immunity (2013) 39:744–57. doi: 10.1016/j.immuni.2013.07.020
229. Lu Y, Hong S, Li H, Park J, Hong B, Wang L, et al. Th9 cells promote antitumor immune responses in vivo. J Clin Invest (2012) 122:4160–71. doi: 10.1172/JCI65459
230. Lu Y, Wang Q, Xue G, Bi E, Ma X, Wang A, et al. Th9 Cells Represent a Unique Subset of CD4+ T Cells Endowed with the Ability to Eradicate Advanced Tumors. Cancer Cell (2018) 33:1048–60.e7. doi: 10.1016/j.ccell.2018.05.004
231. Xue G, Jin G, Fang J, Lu Y. IL-4 together with IL-1β induces antitumor Th9 cell differentiation in the absence of TGF-β signaling. Nat Commun (2019) 10:1376. doi: 10.1038/s41467-019-09401-9
232. Alcamí A, Smith GL. A soluble receptor for interleukin-1 beta encoded by vaccinia virus: a novel mechanism of virus modulation of the host response to infection. Cell (1992) 71:153–67. doi: 10.1016/0092-8674(92)90274-G
233. Alcamí A, Smith GL. A mechanism for the inhibition of fever by a virus. Proc Natl Acad Sci USA (1996) 93:11029–34. doi: 10.1073/pnas.93.20.11029
234. Blanchard TJ, Alcami A, Andrea P, Smith GL. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J Gen Virol (1998) 79:1159–67. doi: 10.1099/0022-1317-79-5-1159
235. Zimmerling S, Waibler Z, Resch T, Sutter G, Schwantes A. Interleukin-1β receptor expressed by modified vaccinia virus Ankara interferes with interleukin-1β activity produced in various virus-infected antigen-presenting cells. Virol J (2013) 10:34. doi: 10.1186/1743-422X-10-34
236. Staib C, Kisling S, Erfle V, Sutter G. Inactivation of the viral interleukin 1beta receptor improves CD8+ T-cell memory responses elicited upon immunization with modified vaccinia virus Ankara. J Gen Virol (2005) 86:1997–2006. doi: 10.1099/vir.0.80646-0
237. Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, et al. IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog (2012) 8:e1003039–e1003039. doi: 10.1371/journal.ppat.1003039
238. Durrant DM, Daniels BP, Klein RS. IL-1R1 Signaling Regulates CXCL12-Mediated T Cell Localization and Fate within the Central Nervous System during West Nile Virus Encephalitis. J Immunol (2014) 1401192. doi: 10.4049/jimmunol.1401192
239. Durrant DM, Robinette ML, Klein RS. IL-1R1 is required for dendritic cell–mediated T cell reactivation within the CNS during West Nile virus encephalitis. J Exp Med (2013) 210:503–16. doi: 10.1084/jem.20121897
240. Ben-Sasson SZ, Hogg A, Hu-Li J, Wingfield P, Chen X, Crank M, et al. IL-1 enhances expansion, effector function, tissue localization, and memory response of antigen-specific CD8 T cells. J Exp Med (2013) 210:491–502. doi: 10.1084/jem.20122006
241. Lee P-H, Yamamoto TN, Gurusamy D, Sukumar M, Yu Z, Hu-Li J, et al. Host conditioning with IL-1β improves the antitumor function of adoptively transferred T cells. J Exp Med (2019) 216:2619–34. doi: 10.1084/jem.20181218
242. Joeckel LT, Wallich R, Metkar SS, Froelich CJ, Simon MM, Borner C. Interleukin-1R Signaling Is Essential for Induction of Proapoptotic CD8 T Cells, Viral Clearance, and Pathology during Lymphocytic Choriomeningitis Virus Infection in Mice. J Virol (2012) 86:8713–9. doi: 10.1128/JVI.00682-12
243. Bartholdy C, Christensen JE, Grujic M, Christensen JP, Thomsen AR. T-cell intrinsic expression of MyD88 is required for sustained expansion of the virus-specific CD8+ T-cell population in LCMV-infected mice. J Gen Virol (2009) 90:423–31. doi: 10.1099/vir.0.004960-0
244. Rahman AH, Cui W, Larosa DF, Taylor DK, Zhang J, Goldstein DR, et al. MyD88 plays a critical T cell-intrinsic role in supporting CD8 T cell expansion during acute lymphocytic choriomeningitis virus infection. J Immunol (2008) 181:3804–10. doi: 10.4049/jimmunol.181.6.3804
245. Sarkar S, Yuzefpolskiy Y, Xiao H, Baumann FM, Yim S, Lee DJ, et al. Programming of CD8 T Cell Quantity and Polyfunctionality by Direct IL-1 Signals. J Immunol (2018) 201(12):3641–50:ji1800906. doi: 10.4049/jimmunol.1800906
246. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi: 10.1038/nm.2028
247. Mattarollo SR, Loi S, Duret H, Ma Y, Zitvogel L, Smyth MJ. Pivotal Role of Innate and Adaptive Immunity in Anthracycline Chemotherapy of Established Tumors. Cancer Res (2011) 71:4809–20. doi: 10.1158/0008-5472.CAN-11-0753
248. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature (2020) 579:421–6. doi: 10.1038/s41586-020-2079-1
249. Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature (2020) 579:415–20. doi: 10.1038/s41586-020-2071-9
250. Zhao Y, De Trez C, Flynn R, Ware CF, Croft M, Salek-Ardakani S. The adaptor molecule MyD88 directly promotes CD8 T cell responses to vaccinia virus. J Immunol (2009) 182:6278–86. doi: 10.4049/jimmunol.0803682
251. Pinteaux E, Abdulaal WH, Mufazalov IA, Humphreys NE, Simonsen-Jackson M, Francis S, et al. Cell-specific conditional deletion of interleukin-1 (IL-1) ligands and its receptors: a new toolbox to study the role of IL-1 in health and disease. J Mol Med (Berl) (2020) 98:923—930. doi: 10.1007/s00109-020-01928-5
252. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines (2019) 4:7. doi: 10.1038/s41541-019-0103-y
253. Muñoz-Wolf N. & Lavelle, E. C. A Guide to IL-1 family cytokines in adjuvanticity. FEBS J (2018) 285:2377–401. doi: 10.1111/febs.14467
254. Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T. Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nat Rev Cancer (2014) 14:135–46. doi: 10.1038/nrc3670
255. Li H, Nookala S, Re F. Aluminum hydroxide adjuvants activate caspase-1 and induce IL-1beta and IL-18 release. J Immunol (2007) 178:5271–6. doi: 10.4049/jimmunol.178.8.5271
256. Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature (2008) 453:1122–6. doi: 10.1038/nature06939
257. Li H, Willingham SB, Ting JP-Y, Re F. Cutting edge: inflammasome activation by alum and alum’s adjuvant effect are mediated by NLRP3. J Immunol (2008) 181:17–21. doi: 10.4049/jimmunol.181.1.17
258. Kuroda E, Ozasa K, Temizoz B, Ohata K, Koo CX, Kanuma T, et al. Inhaled Fine Particles Induce Alveolar Macrophage Death and Interleukin-1α Release to Promote Inducible Bronchus-Associated Lymphoid Tissue Formation. Immunity (2016) 45:1299–310. doi: 10.1016/j.immuni.2016.11.010
259. Gavin AL, Hoebe K, Duong B, Ota T, Martin C, Beutler B, et al. Adjuvant-enhanced antibody responses in the absence of toll-like receptor signaling. Science (2006) 314:1936–8. doi: 10.1126/science.1135299
260. Oleszycka E, Moran HBT, Tynan GA, Hearnden CH, Coutts G, Campbell M, et al. IL-1α and inflammasome-independent IL-1β promote neutrophil infiltration following alum vaccination. FEBS J (2016) 283:9–24. doi: 10.1111/febs.13546
261. Franchi L, Núñez G. The Nlrp3 inflammasome is critical for aluminium hydroxide-mediated IL-1beta secretion but dispensable for adjuvant activity. Eur J Immunol (2008) 38:2085–9. doi: 10.1002/eji.200838549
262. Gianfrani C, Oseroff C, Sidney J, Chesnut RW, Sette A. Human memory CTL response specific for influenza A virus is broad and multispecific. Hum Immunol (2000) 61:438–52. doi: 10.1016/S0198-8859(00)00105-1
263. Bui H-H, Peters B, Assarsson E, Mbawuike I, Sette A. Ab and T cell epitopes of influenza A virus, knowledge and opportunities. Proc Natl Acad Sci (2007) 104:246–51. doi: 10.1073/pnas.0609330104
264. Lapuente D, Storcksdieck Genannt Bonsmann M, Maaske A, Stab V, Heinecke V, Watzstedt K, et al. IL-1β as mucosal vaccine adjuvant: the specific induction of tissue-resident memory T cells improves the heterosubtypic immunity against influenza A viruses. Mucosal Immunol (2018) 11:1265–78. doi: 10.1038/s41385-018-0017-4
265. Lapuente D, Stab V, Storcksdieck Genannt Bonsmann M, Maaske A, Köster M, Xiao H, et al. Innate signalling molecules as genetic adjuvants do not alter the efficacy of a DNA-based influenza A vaccine. PLoS One (2020) 15:e0231138. doi: 10.1371/journal.pone.0231138
266. Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, et al. IL-22 mediates mucosal host defense against Gram-negative bacterial pneumonia. Nat Med (2008) 14:275–81. doi: 10.1038/nm1710
267. Kelly MN, Kolls JK, Happel K, Schwartzman JD, Schwarzenberger P, Combe C, et al. Interleukin-17/Interleukin-17 Receptor-Mediated Signaling Is Important for Generation of an Optimal Polymorphonuclear Response against <em<Toxoplasma gondii</em< Infection. Infect Immun (2005) 73:617–21. doi: 10.1128/IAI.73.1.617-621.2005
268. Huang W, Na L, Fidel PL, Schwarzenberger P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J Infect Dis (2004) 190:624–31. doi: 10.1086/422329
269. Hatherill M, White RG, Hawn TR. Clinical Development of New TB Vaccines: Recent Advances and Next Steps. Front Microbiol (2020) 10(3154):1–12. doi: 10.3389/fmicb.2019.03154
270. Lindenstrøm T, Woodworth J, Dietrich J, Aagaard C, Andersen P, Agger EM. Vaccine-induced th17 cells are maintained long-term postvaccination as a distinct and phenotypically stable memory subset. Infect Immun (2012) 80:3533–44. doi: 10.1128/IAI.00550-12
271. Christensen D, Mortensen R, Rosenkrands I, Dietrich J, Andersen P. Vaccine-induced Th17 cells are established as resident memory cells in the lung and promote local IgA responses. Mucosal Immunol (2017) 10:260–70. doi: 10.1038/mi.2016.28
272. Desel C, Werninghaus K, Ritter M, Jozefowski K, Wenzel J, Russkamp N, et al. The Mincle-Activating Adjuvant TDB Induces MyD88-Dependent Th1 and Th17 Responses through IL-1R Signaling. PLoS One (2013) 8:e53531. doi: 10.1371/journal.pone.0053531
273. Rébé C, Ghiringhelli F. Interleukin-1β and Cancer. Cancers (Basel) (2020) 12(7):1791. doi: 10.3390/cancers12071791
274. Isambert N, Hervieu A, Rébé C, Hennequin A, Borg C, Zanetta S, et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology (2018) 7:e1474319. doi: 10.1080/2162402X.2018.1474319
275. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
276. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
277. Ridker PM, MacFadyen JG, Thuren T, Everett BM, Libby P, Glynn RJ, et al. Effect of interleukin-1β inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet (2017) 390:1833–42. doi: 10.1016/S0140-6736(17)32247-X
278. Björkdahl O, Dohlsten M, Sjögren H-O. Vaccination with B16 melanoma cells expressing a secreted form of interleukin-1β induces tumor growth inhibition and an enhanced immunity against the wild-type B16 tumor. Cancer Gene Ther (2000) 7:1365–74. doi: 10.1038/sj.cgt.7700248
279. Yewdall AW, Drutman SB, Jinwala F, Bahjat KS, Bhardwaj N. CD8+ T cell priming by dendritic cell vaccines requires antigen transfer to endogenous antigen presenting cells. PLoS One (2010) 5:e11144. doi: 10.1371/journal.pone.0011144
280. Liu C, Lou Y, Lizée G, Qin H, Liu S, Rabinovich B, et al. Plasmacytoid dendritic cells induce NK cell-dependent, tumor antigen-specific T cell cross-priming and tumor regression in mice. J Clin Invest (2008) 118:1165–75. doi: 10.1172/JCI33583
281. Fotaki G, Jin C, Ramachandran M, Kerzeli IK, Karlsson-Parra A, Yu D, et al. Pro-inflammatory allogeneic DCs promote activation of bystander immune cells and thereby license antigen-specific T-cell responses. Oncoimmunology (2018) 7:e1395126. doi: 10.1080/2162402X.2017.1395126
282. Kim J-E, Phan TX, Nguyen VH, Dinh-Vu H-V, Zheng JH, Yun M, et al. Salmonella typhimurium Suppresses Tumor Growth via the Pro-Inflammatory Cytokine Interleukin-1β. Theranostics (2015) 5:1328–42. doi: 10.7150/thno.11432
283. Wu Y, Feng Z, Jiang S, Chen J, Zhan Y, Chen J. Secreting-lux/pT-ClyA engineered bacteria suppresses tumor growth via interleukin-1β in two pathways. AMB Express (2019) 9:189. doi: 10.1186/s13568-019-0910-6
284. Segovia M, Russo S, Jeldres M, Mahmoud YD, Perez V, Duhalde M, et al. Targeting TMEM176B Enhances Antitumor Immunity and Augments the Efficacy of Immune Checkpoint Blockers by Unleashing Inflammasome Activation. Cancer Cell (2019) 35:767–81.e6. doi: 10.1016/j.ccell.2019.04.003
285. Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol (2017) 18:255–62. doi: 10.1038/ni.3682
286. Veltri S, Smith II JW. Interleukin 1 Trials in Cancer Patients: A Review of the Toxicity, Antitumor and Hematopoietic Effects. Stem Cells (1996) 14:164–76. doi: 10.1002/stem.140164
287. Vazquez-Lombardi R, Roome B, Christ D. Molecular engineering of therapeutic cytokines. Antibodies (2013) 2:426–51. doi: 10.3390/antib2030426
288. Boraschi D, Tagliabue A. Interleukin-1 and interleukin-1 fragments as vaccine adjuvants. Methods (1999) 19:108–13. doi: 10.1006/meth.1999.0835
289. Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol (2016) 40:96–102. doi: 10.1016/j.coi.2016.03.006
290. Hess C, Neri D. Tumor-targeting properties of novel immunocytokines based on murine IL1β and IL6. Protein Eng Des Sel (2014) 27:207–13. doi: 10.1093/protein/gzu013
291. Uzé G, Tavernier J. High efficiency targeting of IFN-α activity: Possible applications in fighting tumours and infections. Cytokine Growth Factor Rev (2014) 26:179–82. doi: 10.1016/j.cytogfr.2014.10.006
292. Tzeng A, Kwan BH, Opel CF, Navaratna T, Wittrup KD. Antigen specificity can be irrelevant to immunocytokine efficacy and biodistribution. Proc Natl Acad Sci (2015) 112:3320–5. doi: 10.1073/pnas.1416159112
293. Garcin G, Paul F, Staufenbiel M, Bordat Y, Van der Heyden J, Wilmes S, et al. High efficiency cell-specific targeting of cytokine activity. Nat Commun (2014) 5:3016. doi: 10.1038/ncomms4016
294. Cauwels A, Van Lint S, Paul F, Garcin G, De Koker S, Van Parys A, et al. Delivering Type I Interferon to Dendritic Cells Empowers Tumor Eradication and Immune Combination Treatments. Cancer Res (2018) 78:463–74. doi: 10.1158/0008-5472.CAN-17-1980
295. Cauwels A, Van Lint S, Garcin G, Bultinck J, Paul F, Gerlo S, et al. A safe and highly efficient tumor-targeted type I interferon immunotherapy depends on the tumor microenvironment. Oncoimmunology (2018) 7:e1398876. doi: 10.1080/2162402X.2017.1398876
296. Huyghe L, Van Parys A, Cauwels A, Van Lint S, De Munter S, Bultinck J, et al. Safe eradication of large established tumors using neovasculature-targeted tumor necrosis factor-based therapies. EMBO Mol Med (2020) 12:e11223. doi: 10.15252/emmm.201911223
297. Van Den Eeckhout B, Van Hoecke L, Burg E, Van Lint S, Peelman F, Kley N, et al. Specific targeting of IL-1β activity to CD8+ T cells allows for safe use as a vaccine adjuvant. NPJ Vaccines (2020) 5:64. doi: 10.1038/s41541-020-00211-5
298. Schmitz J, Owyang A, Oldham E, Song Y, Murphy E, McClanahan TK, et al. IL-33, an Interleukin-1-like Cytokine that Signals via the IL-1 Receptor-Related Protein ST2 and Induces T Helper Type 2-Associated Cytokines. Immunity (2005) 23:479–90. doi: 10.1016/j.immuni.2005.09.015
Keywords: interleukin-1, dendritic cells, CD4+ T cells, CD8+ T cells, cellular adjuvant, vaccination, cancer immunotherapy
Citation: Van Den Eeckhout B, Tavernier J and Gerlo S (2021) Interleukin-1 as Innate Mediator of T Cell Immunity. Front. Immunol. 11:621931. doi: 10.3389/fimmu.2020.621931
Received: 27 October 2020; Accepted: 08 December 2020;
Published: 27 January 2021.
Edited by:
Diana Boraschi, National Research Council (CNR), ItalyReviewed by:
Klaus Ley, La Jolla Institute for Immunology (LJI), United StatesCopyright © 2021 Van Den Eeckhout, Tavernier and Gerlo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jan Tavernier, amFuLnRhdmVybmllckB2aWItdWdlbnQuYmU=; Sarah Gerlo, c2FyYWguZ2VybG9AdWdlbnQuYmU=
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.