 Dieter Stevens
Dieter Stevens Joline Ingels3
Joline Ingels3 Bart Vandekerckhove
Bart Vandekerckhove Karim Vermaelen
Karim Vermaelen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 12 February 2021
Sec. Vaccines and Molecular Therapeutics
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.620374
This article is part of the Research TopicCancer Vaccines: Time to Think Differently!View all 17 articles
Lung cancer remains the leading cause of cancer-related death worldwide. The advent of immune checkpoint inhibitors has led to a paradigm shift in the treatment of metastatic non-small cell and small cell lung cancer. However, despite prolonged overall survival, only a minority of the patients derive clinical benefit from these treatments suggesting that the full anti-tumoral potential of the immune system is not being harnessed yet. One way to overcome this problem is to combine immune checkpoint blockade with different strategies aimed at inducing or restoring cellular immunity in a tumor-specific, robust, and durable way. Owing to their unique capacity to initiate and regulate T cell responses, dendritic cells have been extensively explored as tools for immunotherapy in many tumors, including lung cancer. In this review, we provide an update on the nearly twenty years of experience with dendritic cell-based immunotherapy in lung cancer. We summarize the main results from the early phase trials and give an overview of the future perspectives within this field.
Lung cancer is the leading cause of cancer-related mortality worldwide, with 1.8 million deaths estimated in 2018 (1). Non-small cell lung cancer (NSCLC) represents 85% of all cases, while small cell lung cancer (SCLC) accounts for approximately 15% of all lung cancers. Treatment depends on tumor stage at diagnosis and comprises surgery, radiotherapy and chemotherapy in early stages, and palliative treatments in metastatic disease. Since almost three quarter of the patients are diagnosed with stage III or IV disease and a significant number of patients relapse systemically after a curative treatment, prognosis remains poor with an estimated 5-year overall survival (OS) of only 18% (2).
Immune checkpoint blockade with drugs that target the programmed cell death protein pathway (PD-1/PD-L1) has changed the therapeutic landscape of locally advanced and metastatic lung cancer. Several randomized controlled trials have shown promising results with checkpoint inhibitors alone (3), or in combination with chemotherapy (4–7). PD-1/PD-L1 inhibitors such as nivolumab, pembrolizumab and atezolizumab are now approved for the first and second line treatment of metastatic NSCLC (without actionable driver mutations) and SCLC, and as a maintenance treatment after chemoradiotherapy in inoperable stage III NSCLC (durvalumab). In addition, trials with checkpoint inhibitors as adjuvant or neo-adjuvant therapy in resectable lung cancer are now underway and the results are eagerly awaited.
Despite better outcomes in terms of OS, only a minority of the patients derive clinical benefit from these treatments. In metastatic NSCLC, more than 40% to 50% of the subjects do not respond to immune checkpoint blockade when given in the first line (3, 4, 6). In the second or higher line setting, the objective response rate (ORR) is even lower (< 20%) (8–11). These results suggest that the full anti-tumoral potential of the immune system is not being harnessed yet, possibly explained by immune evasion mechanisms developed by the tumor to escape from immune destruction (12, 13). One way to overcome this problem is to combine immune checkpoint inhibition with other strategies aimed at inducing or restoring cellular immunity such as cancer vaccination (14, 15).
The goal of therapeutic cancer vaccines is to instruct the patient’s own immune system to kill cancer cells and to induce immunological memory against later disease relapse (16–18). In contrast to immune checkpoint blockade, which impacts the full T cell repertoire including self-reactive lymphocytes which translates into substantial toxicity, cancer vaccines expose the patient’s immune system to a unique selection of relevant antigenic targets resulting in a highly tumor-focused immune response (17, 18). A limitation of this strategy is that the ability of such vaccines to activate patient’s T cells depends on the characteristics and level of activation of local dendritic cells (DCs), which are frequently dysfunctional in patients with advanced cancer (17). Hence, vaccines by themselves have failed to show any clinical benefit in NSCLC so far (19–23).
Cell-based approaches that involve patient’s ex vivo-generated antigen presenting cells (APCs) such as DC-based vaccines avoid the reliance on endogenous APCs and are nowadays one of the most advanced forms of cancer immunotherapy (17). DCs, first identified by Ralph Steinman in 1973 (24), are recognized as the most potent APCs and play a pivotal role in the initiation, programming, and regulation of tumor-specific immune responses (25, 26). They are seeded in all tissues and continuously sample their environment for danger signals and antigens such as those derived from evolving cancer cells. DCs are unique in initiating de novo immune responses by processing the captured antigen to peptides and presenting these peptides to naive T cells in lymphoid tissues on major histocompatibility complex (MHC) molecules (26, 27).
Classical DC-based “vaccines” consist of DCs derived in vitro from autologous peripheral blood monocytes (PBMCs), exposed to activating factors for maturation and subsequently loaded with tumor-associated antigens (TAAs) (Figure 1) (28). These cells are then injected into the patient, a process that has been repeatedly shown to be safe and feasible (27, 29). Alternatively, naturally circulating DCs can be isolated and activated thereby avoiding lengthy ex vivo culture periods (30). The selection of tumor antigens for loading onto DCs is crucial to maximize the likelihood of eliciting a strong and tumor-directed immune response. Different sources of TAAs can be used and include cancer cell line lysate, whole tumor lysate, tumor-derived peptides, (synthetic) protein antigen(s), mRNA(s) encoding selected tumor antigen(s), autologous whole-tumor-derived mRNA or antigens packaged within viral vectors (18, 29, 31).
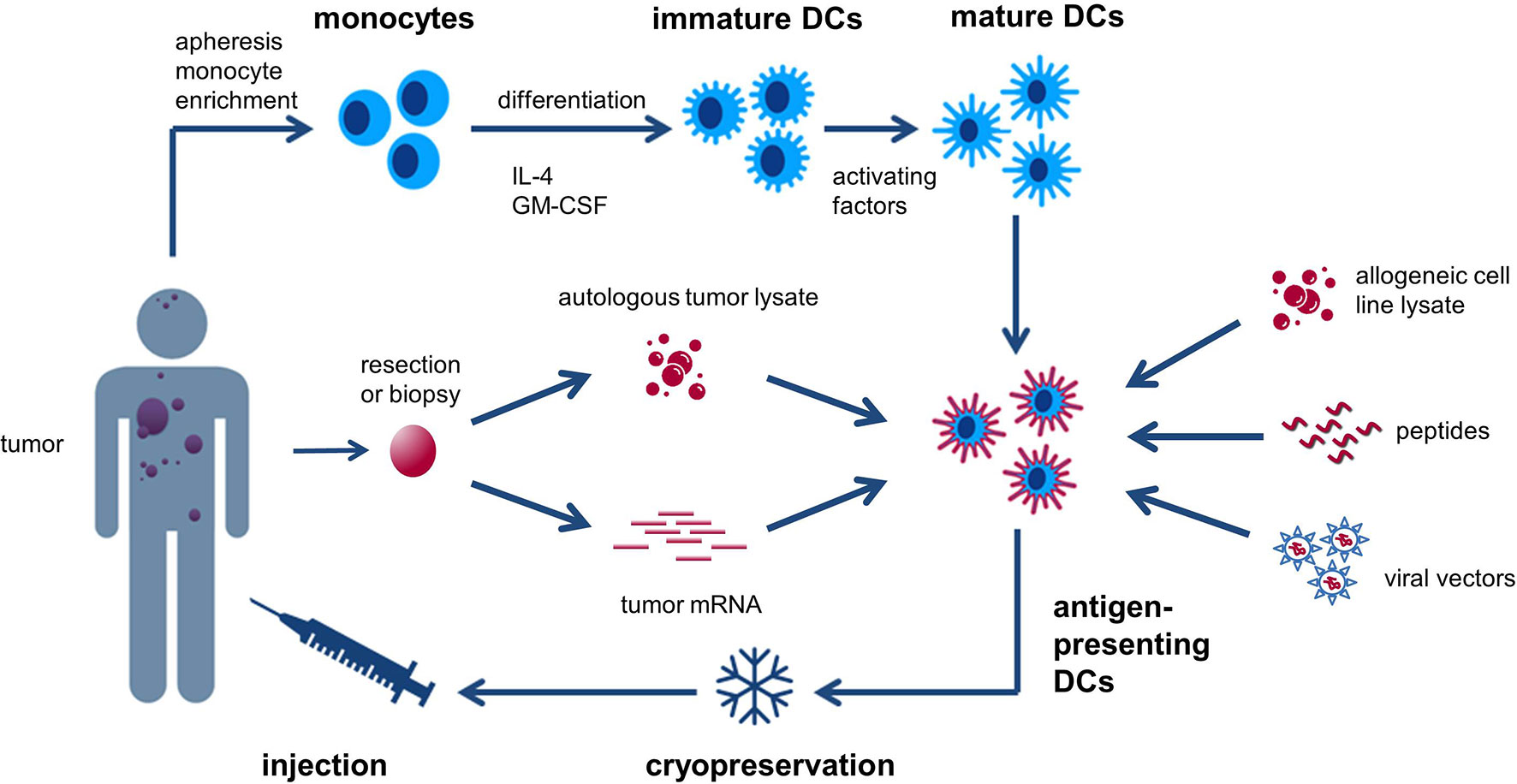
Figure 1 Generic recipe of classical monocyte-derived dendritic cells (DCs). Monocytes are obtained from the patient’s peripheral blood and cultured with IL-4 and GM-CSF to generate immature DCs. These cells are subsequently exposed to activating factors for maturation and loaded with tumor-associated antigens (TAAs). The antigen-loaded DCs are then cryopreserved and injected back into the patient. Different sources of TAAs can be used and include cancer cell line lysate, whole tumor lysate, tumor-derived peptides, (synthetic) protein antigen(s), mRNA(s) encoding selected tumor antigen(s), autologous whole-tumor-derived mRNA, or antigens packaged within viral vectors.
In the field of cancer medicine, DC vaccination has been extensively studied in melanoma patients, as well as in patients with prostate cancer, glioma and renal cell carcinoma, with a favorable safety profile (i.e., no grade 3 or 4 toxicities), but with an ORR that seldom exceeds 15% (32–34). Paradoxically, findings from early-phase trials indicate that DC vaccination might improve survival, advocating implementation of alternative surrogate endpoints to assess the therapeutic effectiveness of DC-based immunotherapy (32).
Still, a major gap exists between the large amount of preclinical data on the exceptional immunogenic power of DCs, and the modest clinical effects in treated cancer patients. The evolving insights into the complex biology of the DC system confront us with a staggering list of parameters that should be adjusted in order to achieve optimal clinical usability. These parameters not only relate to “tweakable” biological properties of the cells, but also to more down-to-earth aspects such as route of administration, dose and frequency of administration, integration into a combinatorial approach, manufacturing, distribution logistics, and costs.
Perhaps one of the most critical factors in DC therapy, yet least systematically investigated is the choice of antigenic targets. This component varies considerably between clinical studies within the same cancer indication, with antigen selections largely made empirically in absence of any solid underlying rationale. Cancer antigens fall into the following different classes: 1) mutated antigens or neo-antigens originating from genomic alterations in cancer cells (single-nucleotide variations and indels), 2) cancer-germline (formerly cancer-testis) antigens whose expression is epigenetically suppressed in normal tissues except for gonadal cells, placenta and many cancers, 3) “differentiation” antigens, which are self-proteins shared between the cancer and the normal tissue from where it originated (e.g., CD20, Melan-A, PSA, CEA), 4) overexpressed shared antigens, which are present in normal tissues and aberrantly overexpressed in tumor cells (e.g., HER2, survivin, WT1), and finally viral oncoproteins, which are expressed in certain virus-induced cancers (e.g., HPV-E6/E7, EBV LMP-1). In addition, some tumor antigens derive their immunogenicity by means of aberrant post-translational modifications, as is the case for the MUC-1 glycoprotein where the tumor-restricted form is strongly hypo-glycosylated. Of all these categories, neo-antigens, cancer-germline antigens and viral oncoproteins are the most attractive targets for DC-based immunotherapy given the highest cancer-restricted expression, and the fact that the natural T cell repertoire has not been tolerized against them. Regardless of the type of antigen used, a major caveat is that studies or databases documenting mRNA expression in a given tumor often do not provide information on protein levels. Moreover, protein expression does not guarantee adequate presentation of antigen-derived peptides on MHC molecules, and if presented, whether these peptides will find a corresponding T cell repertoire with sufficient affinity.
Lung cancer (both NSCLC and SCLC), being a textbook example of a carcinogen-induced tumor, frequently features a high tumor mutational burden, offering opportunities for neoantigen-targeted vaccination approaches. Lung cancers are also rich in cancer-germline antigens [e.g., MAGE-A3 (22)], a number of differentiation antigens (e.g., CEA), and overexpressed shared antigens (e.g., survivin, WT1, MUC1), all being present in variable amounts across different patients. Viral oncoproteins are typically absent in human lung cancers. As we will discuss in the next section, the large majority of completed DC therapy trials in lung cancer made use of shared or tumor-associated antigens (TAAs), a few of the studies also incorporated cancer-germline antigens, and none of the published reports have described a patient-individualized neoantigen approach to date.
In lung cancer, the role of DC-based immunotherapy has yet to be defined. Since the early 2000s, several, mostly non- randomized clinical trials (RCTs) with DC immunotherapy have been conducted, each typically involving a small number of patients and very heterogeneous designs. Over the same timeframe, the lung cancer therapeutic landscape has experienced dramatic changes, with the emergence of oncogene-targeted small molecules, and later immune checkpoint inhibitors. In this review we aim to give an overview of these DC therapy trials which we categorized into four parts: DC therapy in NSCLC, DC/CIK cell therapy in NSCLC, AKT-DC therapy in NSCLC, and DC therapy in SCLC (Tables 1 and 2). We will examine the clinical and immunological outcome as well as safety of DC-based immunotherapy in lung cancer, while also discussing the potential challenges of the different vaccine approaches such as the choice of antigens and DC subset, the use of adjuvants and the route, dose and frequency of administration. Next, we will give some future perspectives in how DCs might be used in clinical practice.
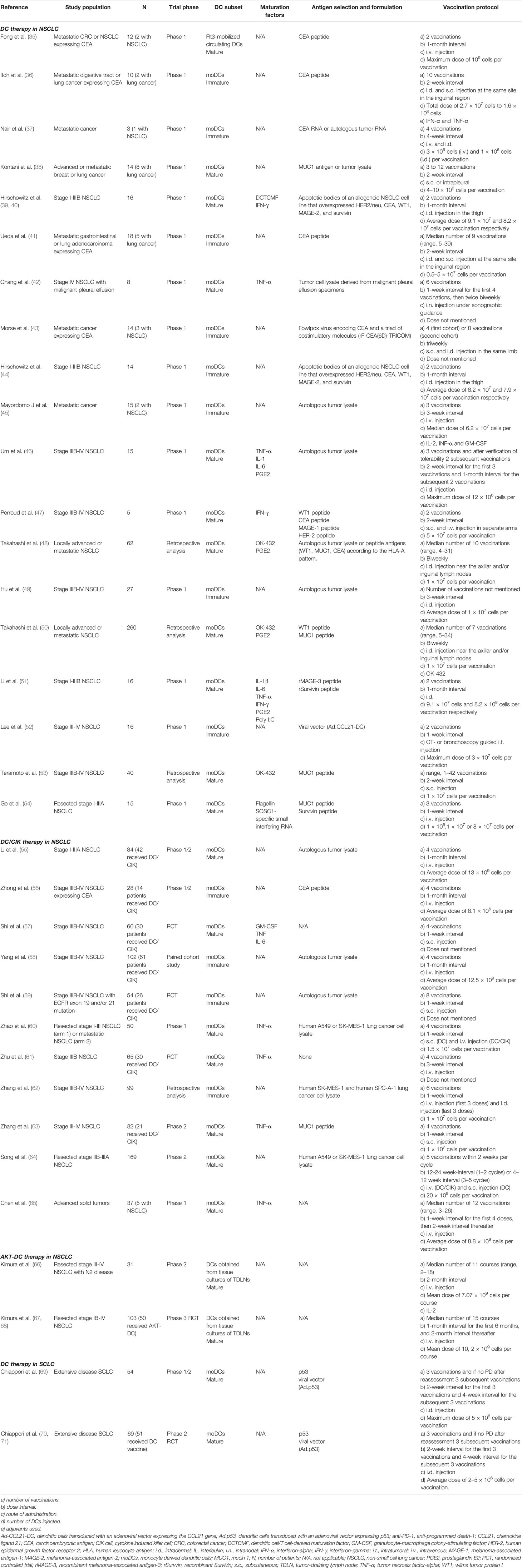
Table 1 Overview of trials with dendritic cell (DC)-based immunotherapy in lung cancer.
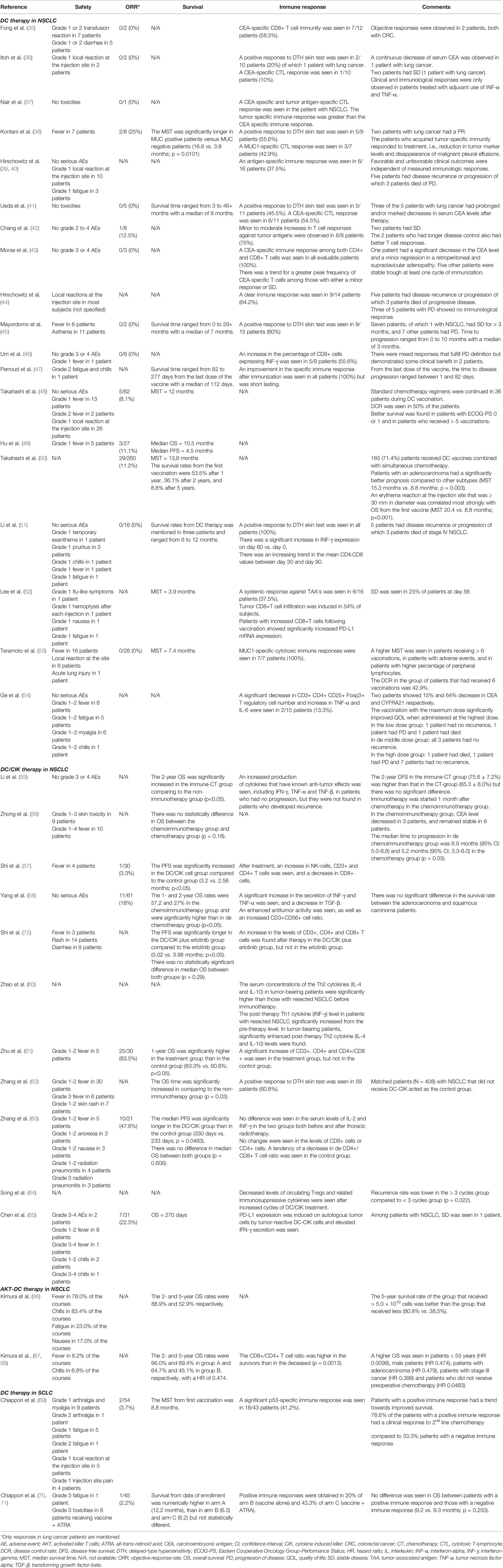
Table 2 Clinical and immunological outcomes of dendritic cell (DC)-based immunotherapy in lung cancer.
The earliest study in this field was performed by Fong and coworkers in patients with metastatic or recurrent cancer who had abnormal or rising serum carcinoembryonic antigen (CEA) levels (35). CEA is a 180-kDa membrane intercellular adhesion glycoprotein that is overexpressed in several malignancies including NSCLC. Twelve patients with either colorectal cancer (CRC) or NSCLC underwent peripheral blood leukapheresis after prior administration of Flt3 ligand, a hematopoietic growth factor known to expand DCs in vivo. DCs were subsequently harvested and loaded with a nonapeptide derived from a human leucocyte antigen (HLA)-A0201-specific peptide of CEA, as well as with keyhole limpet hemocyanin (KLH), a protein with adjuvant properties that also allows to monitor therapy-induced immune responses. Patients were injected intravenously (i.v.) with progressively increasing doses of antigen-exposed DCs with a maximum of 109 cells. Adverse events (AEs) were mild self-limited rigors and fever (7/12), as well as mild diarrhea (5/12). Vaccination elicited a CEA-specific immune response in seven patients. Two out of twelve patients experienced dramatic tumor regression, one patient had a mixed response, and two had stable disease (SD). Clinical responses correlated significantly with the expansion of CD8+ T cells.
A similar CEA-targeted DC vaccination strategy was used by the group of Itoh and Ueda et al. (36, 41). The first study enrolled ten patients with advanced digestive tract or lung cancer expressing CEA (36). PBMCs were harvested from peripheral blood by leukapheresis after five days of priming with granulocyte colony-stimulating factor (G-CSF) and cultured with granulocyte/macrophage colony-stimulating factor (GM-CSF) and interleukin 4 (IL-4) to generate DCs. The DCs showed an immature phenotype and were loaded with CEA652, a nonapeptide restricted to HLA-A24, which is present in 60% of the Japanese population. Patients received repeated intradermal (i.d.) and subcutaneous (s.c.) injections up to a cumulative dose ranging from 2.7 × 107 to 1.6 × 108 DCs. Seven patients also received adjuvant interferon-alpha (IFN-α) and tumor necrosis factor-alpha (TNF-α) twice a week during the vaccination period. No severe toxicity was observed. A positive response to delayed-type hypersensitivity (DTH) skin test was seen in two patients. One of the two demonstrated also a CEA-specific immune response. Two patients, of which one with stage IV lung cancer, had SD for 6 and 9 months respectively, associated with a continuous decrease of serum CEA in the first patient. Clinical and immunological responses were only observed in patients treated with adjuvant use of IFN-α and TNF-α so it is not known whether these responses could be attributed to the DC vaccine. In a follow-up study, 18 patients (five patients with lung cancer) were enrolled and treated using the same immunization protocol, without cytokine adjuvants (41). The vaccine was well tolerated and no toxicity was observed. Although no tumor shrinkage occurred in any patient, long-term SD or marked decreases in the serum CEA level were observed in some subjects. A positive skin response to CEA652-pulsed DCs and a positive in vitro cytotoxic T-lymphocyte (CTL) response to CEA652 peptide after therapy were seen in most of the patients in whom treatment was clinically effective.
Other studies also selected CEA as the antigen of choice for active immunotherapy with DCs. A phase 1 trial included one patient with metastatic lung adenocarcinoma who underwent four monthly immunizations with autologous DCs transfected with CEA-encoding RNA and total tumor RNA (37). Both CEA-specific and tumor-specific CTL immune responses were seen, of which the latter were greater. The authors conclude that RNA-transfected DCs can induce antigen-specific T cell responses in cancer patients with surgically resected malignancies. Morse and coworkers investigated the safety and clinical and immunological efficacy of a DC vaccine modified with a recombinant Fowlpox vector encoding CEA and a triad of stimulatory molecules [rF-CEA(6D)-TRICOM], injected both i.d. and s.c. (43). 14 patients with metastatic CEA-expressing malignancies were enrolled, of which three with NSCLC. There were no treatment-related grade 3 or 4 AEs. One patient had a significant decrease in the CEA level and a minor regression in a retroperitoneal and supraclavicular adenopathy. Five other patients were stable through at least one cycle of immunization. A CEA-specific immune response among both CD4+ and CD8+ T cells was seen in all evaluable patients. There was a trend towards a greater peak frequency of CEA-specific T cells among those with either a minor response or a SD.
Kontani et al. evaluated the clinical effects of a DC vaccine targeting the TAA mucin 1 (MUC1) in 14 patients with advanced or metastatic breast or lung cancer (38). MUC1 is a glycoprotein that is markedly hypoglycosylated in cancer compared to normal tissues, leading to the exposure of immunogenic epitopes (73). PBMCs were collected from peripheral blood samples and supplemented with IL-4 and GM-CSF. Subsequently, DCs were loaded with MUC1 peptides or tumor lysate obtained from malignant pleural effusion specimens of the patients. DCs were then injected s.c. in the supraclavicular region or intrapleurally, at least three times at 2-week intervals. Fever occurred in seven patients. After vaccination, all the evaluable patients with MUC1-positive cancer acquired antigen-specific immunity compared to only one patient with MUC1-negative cancer. Reductions in tumor sizes or tumor marker levels or disappearance of malignant pleural effusion were seen in seven of nine MUC1-positive cancers. The survival of MUC1-positive patients was significantly longer compared to MUC1-negative patients (16.8 vs. 3.8 months; p = 0.0101). The authors conclude in this study that this tumor antigen can elicit a strong immune response and that DC vaccines targeting MUC1, which is expressed in 60% of the lung cancer patients, are a promising immunotherapy in the treatment of cancer (38). Of note, similar signals of clinical efficacy were observed with other MUC1-targeted vaccine approaches in NSCLC (20, 74).
Some important conclusions can be drawn from the different studies mentioned above. First, DC therapy containing one TAA is well tolerated with only minor side effects observed. For CEA-targeted DC vaccination studies, this is reassuring given the severe pulmonary toxicity observed with CEA-specific CAR T cell therapy, which is related to the expression of this antigen on normal pulmonary epithelium (75). Second, this vaccination approach appears to elicit an antigen-specific, T cell-mediated immune response in a substantial fraction of lung cancer patients, despite a number of obstacles: 1) the use of a shared TAA for which high levels of immunological tolerance must be overcome, 2) the injection of immature DCs in some trials, and 3) the climate of systemic immune suppression in the advanced cancer patients enrolled. Yet, clinical responses were rare, possibly explained by the fact that only one tumor antigen was targeted. A limitation of peptide-based formulations is that they are HLA-restricted, which necessitates patient selection. This is not the case for other antigen formulations such as mRNA encoding antigens or tumor lysates that were used in some other trials. A remarkable observation from these studies is also that prior mobilization with Flt3 ligand or G-CSF could expand the number of DCs produced. However, because it was not assessed whether higher DC doses also yielded stronger immune responses, the benefit of mobilization of the donor in the DC manufacturing process remains unclear.
In contrast to the aforementioned trials enrolling different tumor types, the first DC vaccination trial exclusively in NSCLC patients was performed by Hirschowitz et al. (39, 40). In this trial, multiple TAAs were targeted simultaneously. Autologous DC vaccines were delivered to 16 individuals with stage IA to IIIB NSCLC treated with surgery, chemoradiation, or multimodality therapy. DCs were generated from CD14+ precursors and pulsed with apoptotic bodies of an allogeneic NSCLC cell line that overexpressed human epidermal growth factor receptor 2 (HER2/neu), CEA, wilms tumor protein 1 (WT1), and survivin. Interestingly, DCs were only “partially” matured. Patients received two i.d. vaccines with 1 month apart (average dose of 9.1 × 107 and 8.2 × 107 cells per immunization respectively). AEs were limited to a mild skin reaction at the injection site (10/16) and minor fatigue for one to two days after injection (3/16). Of the 16 patients, six showed an antigen-specific response and five showed a tumor-antigen independent response. Five individuals had documented disease recurrence or progression of which three succumbed to the disease. One individual with stage IB NSCLC developed a solitary brain metastasis 2 months following the initial vaccine and had no evidence of disease 15 months following metastasectomy. Two patients with unresectable stage III NSCLC showed no signs of disease progression at 35 and 23 months from chemoradiation, respectively. The aforementioned clinical outcomes were independent of measured immunologic responses. The same group conducted a continuation study with similar inclusion criteria and immunization protocol, using an immature DC vaccine (44). 14 patients were enrolled of which seven had undergone surgical resection, with or without adjuvant therapy, and seven with unresectable stage III who had been treated with chemoradiation. Immunologic responses, measured by IFN-γ enzyme-linked immune absorbent spot (ELISPOT), were seen in 3/7 stage III unresectable, and 6/7 stage I/II surgically resected patients. There were no AEs, except for local reactions in most subjects. The authors conclude that immature DCs pulsed with apoptotic tumor cells have similar biologic activity to a matured DC preparation in a similar clinical protocol (44).
Although clinical outcomes were difficult to interpret, probably due to the heterogeneity of the patient population, and not correlating with immunological responses, the studies of Hirschowitz and colleagues have clearly indicated that DC therapy following surgery, chemoradiation or multimodality treatment is safe and can possibly find its role as an adjuvant treatment. A remarkable observation from these clinical trials was that immature DCs were able to elicit immune responses in almost 2/3 of the patients, since it has been appreciated for a long time that these DC subsets rather induce immune tolerance than immune stimulation. A difference with the previous studies is that an allogeneic tumor cell line was used to produce a multivalent vaccine, targeting several TAAs. Yet, the antigenic make-up of the cell line used may not be representative for each patient’s tumor.
Further exploiting the idea of targeting multiple antigens, Perroud and coworkers assessed the feasibility, safety and immunologic response of a mature, antigen-pulsed autologous DC vaccine loaded with peptides of WT1, CEA, HER2, and Melanoma Antigen 1 (MAGE-1). The trial enrolled five patients with inoperable stage IIIB and IV NSCLC (47). All patients received prior conventional treatment (chemotherapy with or without radiotherapy). PBMCs, obtained after leukapheresis, were cultured in a medium with GM-CSF and IL-4, and subsequently activated with IFN-γ. Patients received two doses of 5 × 107 DCs administered s.c. and i.v. two times at 2-week intervals. One patient developed grade 2 fatigue and chills following the first dose of the vaccine. A lymphoproliferation assay showed an improvement in the specific immune response after immunization in all patients, with a tendency to wane after the second vaccine dose. Survival from the last dose of the vaccine ranged between 82 and 277 days. Three patients had a longer survival time than expected for their tumor, node and metastasis (TNM) classification. The fact that immune responses were not long lasting possibly indicates that multiple doses of the vaccine are required to achieve clinical efficacy.
Li et al. reported the results of a phase 1 trial enrolling 16 patients with stage I to IIIB NSCLC (51). All had no evidence of progression at the time of enrollment and had completed definitive therapy (surgical, medical or multimodal). DC immunotherapy was generated from the patient’s PBMCs and loaded with recombinant survivin and MAGE-3 peptides. To induce DC maturation, a cytokine cocktail consisting of IL-1β, IL-6, TNF-α, IFN-γ, prostaglandin E2 (PGE2), and poly I:C had been added to the culture. A prime immunotherapy (9.1 × 107 cells/dose) and a single boost (8.2 × 107 cells/dose) were administered i.d. 1 month apart. AEs were grade 1 fever, chills and fatigue in one patient, and grade 1 pruritus in three patients. A positive response to DTH skin test was seen in all patients. There was a significant increase in IFN-γ expression on day 60 versus day 0. There was also an increasing trend in the mean CD4:CD8 values between day 30 and day 90; however, the increase was not statistically significant. In total, 5/16 patients experienced disease recurrence or progression, of which three patients succumbed to the disease.
An alternative approach to target multiple antigens simultaneously is to load DCs with autologous tumor cells or cellular lysates. Chang et al. for example conducted a pilot trial using mature DCs pulsed with necrotic tumor cells enriched from malignant pleural effusion specimens (42). Eight patients with advanced NSCLC were injected with antigen-loaded DCs into the inguinal lymph nodes under ultrasound guidance. No major toxicities occurred. Six patients received all six DC injections. Of these, two patients had SD and one patient had a minor response. Minor to moderate increases in T cell responses against tumor antigens were observed after DC vaccination in six of eight patients. The two patients who had a longer disease control also developed better T cell responses. The immunological and clinical effects of a DC vaccine pulsed with autologous tumor lysate was also assessed by the groups of Mayordomo et al. (45) and Um et al. (46). In the first study, 15 patients with metastatic cancer (two with NSCLC) underwent mononuclear cell apheresis after prior mobilization with GM-CSF. PBMCs were cultured with IL-4 and GM-CSF. DCs were then administered i.v. with a median dose of 6.2 × 107 cells per vaccination. In addition, IL-2, IFN-α, and GM-CSF were co-injected s.c. as an adjuvant for several days. A positive response to DTH skin test was noted in 9/15 patients after the first immunization. Seven patients, of whom one with NSCLC, had SD for more than 3 months and seven other patients experienced disease progression. AEs were mild and included fever immediately after DC infusion in six patients and asthenia in eleven patients. The second study enrolled exclusively subjects with stage IIIB and IV NSCLC. DCs were again loaded with autologous tumor lysate by a combination of electroporation and passive loading. Autologous tumor samples were obtained from bronchoscopic biopsies, surgical samples or lymph node biopsies. The antigen-loaded immature DCs were subsequently activated with TNF-α, IL-1, IL-6, and PGE2. In this phase 1 dose-escalation study, 15 patients were assigned to cohorts that received 3, 6, or 12 × 106 DCs by i.d. injection. The maximum dose of the vaccine was shown to be safe with only one patient experiencing low grade fever. In 5/9 patients, the vaccine resulted in an increased IFN-γ production by peripheral blood CD8+ T cells. However, a relationship between the immunological response and the vaccination dose was not seen. Clinical responses were assessed in eight patients. All had PD. Nevertheless, there were mixed responses that fulfilled PD definition but demonstrated some clinical benefit in two patients.
Again, clinical outcomes were disappointing. A possible explanation is that most of the patients enrolled in these studies suffered from relapsed or refractory cancer with often bulky disease and a worse performance status, which is shown to be less responsive to DC vaccination. Another potential concern may be the high concentration of suppressive factors released from the tumor cells, which may influence DC functionality. Moreover, in the last study, the autologous tumor samples used for making tumor lysate had been obtained before the initiation of chemotherapy. Changes in the tumor antigenicity during treatment could perhaps explain the low clinical efficacy.
In an attempt to circumvent the limitations of typically small sample sizes in DC vaccination trials, the group of Takahashi and coworkers conducted a pooled retrospective analysis of 62 patients from one center. The patients had previously treated inoperable or postoperatively relapsed NSCLC and received activated DCs pulsed with either autologous tumor lysates or peptide antigens (WT1, MUC1, CEA) matched to their HLA-A type (48). The DCs were activated by in vitro exposure to OK-432, which is a clinically approved lyophilized mixture of group A Streptococcus pyogenes known to promote functional maturation of DCs, including the capacity to secrete IL-12. The vaccines were injected i.d. near the axillar and/or inguinal lymph nodes with a median of 10 immunizations (range, 4–31). Clinical responses were observed in five patients, of which one patient with a complete response (CR). Another 26 patients developed SD. Median survival time (MST) was 12 months from the first DC vaccination. Of note, standard chemotherapy was continued in 36 patients during DC vaccination. A better OS was found in patients who received more than five vaccinations and those with the best performance status. Multivariate analyses also revealed that the use of WT1 peptides significantly affected OS both from initial diagnosis and from the first vaccination. Furthermore, no serious AEs related to the vaccine were observed. In an extended analysis, 260 patients with locally advanced or metastatic NSCLC at six centers were analyzed (50). All had received five or more WT1 and/or MUC1 peptide-pulsed DC vaccinations once every 2 weeks. In some patients, OK-432 was co-administered i.d. as an immunological adjuvant simultaneously with the vaccine. In the majority of the patients (71.4%), DC vaccination was combined with chemotherapy. MST from first vaccination was 13.8 months (95% CI 11.4–16.8) with 8.8% being alive after five years. Patients with an adenocarcinoma had a significantly better prognosis compared with other subtypes (MST 15.3 vs. 8.8 months; p = 0.003). An erythema reaction at the injection site that was ≥ 30 mm in diameter was strongly correlated with OS from the first vaccine (MST 20.4 vs. 8.8 months; p<0.001). Another Japanese group retrospectively analyzed data of 40 patients with MUC1-positive NSCLC treated with a MUC1-targeted and OK-432 activated DC-vaccine, exploring predictive biomarkers for clinical responses. All patients had stage IIIB-IV NSCLC that was refractory to standard anticancer therapies (53). The vaccines were injected s.c. in the axilla or supraclavicular fossa every 2 weeks until disease progression. Low-grade fever occurred in 16 patients and local skin reactions in six individuals. No patients achieved an objective response. The MST after initial vaccination was 7.4 months and the 1-year OS was 25%. Patients who received more than six vaccinations had a longer MST and 1-year OS (9.5 months and 39.3% respectively). Interestingly, in the latter group, patients who developed immune-related AEs had a significantly longer MST compared with patients without those AEs (12.6 vs. 6.7 months; p = 0.042). In addition, longer survival was also seen in patients with > 20% lymphocytes prior to vaccination (12.6 vs. 4.5 months; p = 0.014). All seven patients who had received six vaccinations and were evaluable for immune responses showed an increase in MUC1-specific T cells and a decrease in Tregs.
A major drawback of these studies is however their retrospective design, limiting the interpretation of the results. Since most of the patients also received simultaneous chemotherapy in the first two studies, and no control group was applied, it is difficult to draw definite conclusions regarding the clinical benefit of the vaccine. Furthermore, it was appreciated that patients receiving more DC vaccines also had better survival outcomes, which is of course interesting since the optimal DC dose and frequency of administration is not yet determined. However, this survival benefit could have been possibly attributed to the better performance status of the patients in the group treated with the highest cumulative dose.
Knowledge of negative feedback pathways controlling inflammatory responses can be exploited to re-engineer DCs. Based on this concept, Ge et al. evaluated the safety and efficacy of a DC vaccine activated using the Toll-like receptor (TLR) agonist flagellin, together with siRNA-mediated silencing of the gene encoding for suppressor of cytokine signaling 1 (SOCS1) (54). SOCS1 has been shown to be a negative regulator of DC activation and IL-12 production, thus restricting the DC’s capacity to break immunological tolerance. By analogy to other trials, the DCs were pulsed with peptides of survivin and MUC1. Just as MUC1, survivin is also frequently overexpressed in NSCLC and contributes to oncogenesis. In this phase 1 dose-escalation trial, 15 patients with resected stage I to III NSCLC were i.v. injected with 1 × 106, 1 × 107, or the maximum dose of 1 × 108 DCs at days 7, 14, and 21. The most common AEs were grade 1 flu-like symptoms, which occurred mostly in the group immunized with the maximum dose of the vaccine. A significant decrease in T-regulatory (Treg) cells and increase in TNF-α and IL-6 were seen in two patients. Two patients also showed a 15% and 64% decrease in CEA and CYFRA21, respectively. Interestingly, the patients’ quality of life (QOL) was significantly improved in the high-dose group, compared with the low-dose and middle-dose group after treatment. More importantly, in the long-term follow-up after more than four years, only two patients had died, two patients had progressive disease (PD) and 11 patients had still no recurrence. With the use of SOCS1-silencing, this trial is the first in lung cancer to explore targeted genetic re-engineering of DCs to boost immunogenicity. This manipulation did not translate into increased cytokine-mediated toxicity. Still the added value of SOCS1-silencing in terms of clinical outcome cannot be ascertained from this trial as there was no comparator product treated with a control siRNA.
Based on preclinical evidence pointing to a possible synergistic effect between chemotherapy and vaccination (as discussed below), Hu et al. explored the combination of pemetrexed and DCs pulsed with autologous tumor lysate in 27 patients suffering from stage IIIB or IV lung adenocarcinoma who had failed on maintenance gefitinib or erlotinib treatment after platinum-doublet chemotherapy (49). PBMCs were enriched from a 50-ml blood sample using density gradient centrifugation and subsequently cultured in the presence of IL-4 and GM-CSF. DCs were then given i.d. every 3 weeks at day 8 of each chemotherapy cycle. Grade 1 fever after DC therapy was noted in five patients. Other, mostly hematological, toxicities were attributed to chemotherapy. Three patients (11.1%) experienced a partial response (PR). The median progression-free survival (PFS) was 4.5 months and the median OS 10.5 months, which is better than a previous trial with second line pemetrexed in advanced NSCLC (76). Grade 1 fever after DC therapy was noted in five patients. Other, mostly hematological, toxicities were attributed to chemotherapy. This was the first study to prospectively investigate the added value of DC therapy combined with chemotherapy. However, since there was no control group, the real value of DC vaccination in this setting remains to be confirmed.
In contrast to all the trials using systemic injection of DCs, Lee and coworkers explored the intratumoral injection in terms of feasibility, safety and efficacy. In a phase 1 dose escalation study, autologous DCs were administered intratumorally in 16 subjects with stage IIIB and IV NSCLC (52). Interestingly, the DCs were genetically modified by transduction with an adenoviral (Ad) vector expressing the CCL21 gene (Ad-CCL21-DC). CCL21 is a lymphoid chemokine that strongly attracts effector T cells and DCs and hence facilitates entry into the tumor and in situ vaccination. Endpoints were safety and tumor antigen-specific immune responses. Patients enrolled into a given cohort received the same Ad-CCL21-DC dose (1 × 106, 5 × 106, 1 × 107, or 3 × 107 cells/injection) by CT-guided or bronchoscopic intratumoral injection on days 0 and 7. Three patients developed possibly treatment-related AEs (flu-like syndrome, hemoptysis, nausea and fatigue, all grade 1). Twenty-five percent of the patients had SD at day 56. MST was 3.9 months. A systemic response against TAAs was observed in six of 16 patients by means of an IFN-γ ELISPOT assay. Tumor CD8+ T cell infiltration was induced in 7/13 subjects. Interestingly, intratumoral PD-L1 mRNA expression increased significantly with increased CD8+ T cell infiltration following vaccination. The authors of this study suggest that in situ vaccination itself increases PD-L1 expression as a result of antigen recognition and CD8+ T cell infiltration at the tumor site. In this way, vaccination may be an effective approach to enhance the efficacy of PD-1/PD-L1 checkpoint inhibitors in “cold” tumors with low PD-L1 expression and/or a lack of CD8+ T cell infiltration (52). Still, the major question remains whether DC-induced T cell infiltration and potential priming at one injected site will induce T cells capable of homing into and controlling other metastatic sites.
In recent years, the use of autologous DCs co-cultured with cytokine-induced killer (CIK) cells has been the subject of numerous trials in NSCLC, all of them conducted in the Far-East region (77). CIK cells are a subset of non-MHC restricted natural killer T-lymphocytes with a CD3+ CD56+ phenotype that can proliferate rapidly in vitro and display strong cytolytic activities against malignant cells (59). In DC/CIK therapy, the DCs are derived from mononuclear cells obtained by leukapheresis in typical GM-CSF/IL-4-supplemented medium and loaded with antigens (autologous tumor lysate or peptides). CIK cells are generated by culturing PBMCs in medium supplemented with anti-CD3 antibody, recombinant human IL-1a, IFN-γ, and IL-2 (58).
DC/CIK cell therapy has been evaluated in diverse disease settings: as adjuvant therapy combined with chemotherapy in resectable disease, in stage IIIB and IV patients as first line in combination with chemotherapy, and as a maintenance treatment after first line chemotherapy (55–58, 60, 62, 64, 72). In total, 646 patients were enrolled in these trials. No serious toxicities were observed. Signals of clinical activity were observed in some cases, albeit usually modest and often without statistical significance. Signs of systemic immune activation were reported in patients receiving the experimental arm, including increased numbers of circulating CD8+ and CD4+ T cells, a shift from a Th2 toward a Th1-polarized immune response profile with an increase of the anti-tumoral cytokines IFN-γ, TNF-α, and TNF-β (albeit only in early-stage patients), and a reduction in Tregs after repeated injections of DC/CIK.
A different concept is the combination of DC/CIK cell therapy with thoracic radiotherapy (TRT) or chemoradiotherapy (CRT). The underlying rationale being that radiation-killed tumor cells release tumor antigens and “danger-associated molecular patterns” that can potentially promote DCs to elicit tumor antigen-specific CD8+ T cell responses, which would further consolidate or amplify objective responses and improve survival outcomes (61, 63). The immunogenic effects of radiotherapy are thought to underlie the positive results of MUC1-targeted vaccination and, more convincingly, adjuvant PD-L1 blockade in stage III NSCLC patients treated with chemoradiation (20, 78). In a phase 2 trial, patients with stage III and IV NSCLC received TRT (60 Gy delivered at 2 Gy per fraction) plus MUC1-loaded DC/CIK cell therapy or TRT alone (63). All subjects had previously been treated with two or more cycles of platinum-based doublet chemotherapy without disease progression. Patients that received DC/CIK cells combined with TRT had a longer PFS than those who received TRT alone (330 days vs. 233 days; p<0.05), as well as a better ORR (47.6% vs. 24.6%; p<0.05). Median OS was not significantly different between the two groups. Zhu et al. conducted a RCT in 63 patients with stage IIIB NSCLC (61). Of these, 30 patients were treated with DC/CIK cell therapy combined with platinum-based doublet CRT. DCs were not loaded with tumor antigens. The ORR was significantly higher in the group treated with DC/CIK and CRT than in the group treated with CRT alone (83.3% vs. 54.5%; p = 0.014). One-year survival rate was also better (83.3% vs. 60.6%; p<0.05). These studies suggest that combined treatments with DC/CIK cell therapy and (chemo)radiotherapy can enhance tumor responses and prolong survival.
Recently, an interesting variation on the DC/CIK manufacturing process was reported and evaluated in advanced cancer patients, among them five with NSCLC. In a phase 1 trial by Chen et al., DC/CIK cells were further activated in vitro by incubation with the anti-PD-1 antibody pembrolizumab, and administered i.v. by repeatedly infusions (65). Patients were progressive after at least one previous course of appropriate anti-tumoral treatment. Of note, grade 3 or 4 treatment-related AEs (fever, chills) were noted in two patients. ORR was 22.5% with a median OS and PFS of 270 and 162 days respectively. Still, the actual added value of in vitro activation with anti-PD-1 is not clear from this trial as there was no comparator arm with “standard” DC/CIK infusions.
Another form of adoptive immunotherapy involving DCs, although somewhat different from the aforementioned treatments, is a therapy using autologous activated killer T cells and DCs (AKT-DC) obtained from tissue cultures of the tumor-draining lymph nodes of the primary lung tumor. Kimura et al. demonstrated that the tumor-draining lymph nodes of lung cancer patients are a potent source of killer T cells specific to autologous tumor cells, but also of mature DCs, when cultured with low dose IL-2, and that this in vitro expansion of T cells could last for up to 2 months (79). Based on this mechanism, a phase 2 trial was conducted evaluating the safety and feasibility of chemo-immunotherapy using these AKT-DCs in post-surgical N2 NSCLC patients (66). 31 patients were enrolled, of which three subjects eventually dropped out. Four courses of chemotherapy were administered along with AKT-DC immunotherapy every 2 months for 2 years. Fever and chills were the most frequent AEs. The 2- and 5-year OS were 88.9% and 52.9%, respectively.
The same group performed a phase 3 RCT investigating the efficacy of adjuvant chemo-immunotherapy with AKT-DC, targeting residual micrometastases, in 103 patients with resected NSCLC (67, 68). Patients were randomly allocated to receive either chemo-immunotherapy (group A) or chemotherapy alone (group B). Those who were assigned to group A received four courses of platinum-based chemotherapy along with AKT-DC immunotherapy for up to two years after surgery. Almost half of the patients treated with immunotherapy had at least one AE, mostly chills and/or fever. The 2- and 5-year OS rates were 96.0% and 69.4% in group A and 64.7% and 45.1% in group B, respectively, with a hazard ratio (HR) of 0.474. Subgroup analysis also showed that younger patients, male patients, patients with adenocarcinoma, patients with stage III cancer and those who did not receive preoperative chemotherapy had a significantly better OS. This study showed that NSCLC patients could benefit from adoptive cellular immunotherapy as an adjuvant to surgery. However, the heterogeneity of the study population was a major limitation.
The stark differences in biological and clinical behavior of SCLC compared to NSCLC are also reflected at the immunological level. As a demonstration, clinical trials to this date show only limited responses to immune checkpoint inhibition in this aggressive tumor, in contrast to NSCLC (80, 81). Also, DC-based immunotherapy trials in SCLC are scarce. Antonia and Chiappori were the first to test the immunological and clinical effects of a cancer vaccine consisting of DCs transduced with an adenovirus expressing p53 (Ad.p53) in patients with extensive disease SCLC (69, 82). The tumor suppressor gene, p53, plays an important role as a regulator of cell growth and differentiation and is mutated in ≥ 90% of the SCLC cases (82). Hence, it is considered as an ideal TAA. Fifty-four patients were enrolled in this phase 1/2 trial. All patients were treated with conventional chemotherapy prior to the immunizations. PBMCs were obtained after leukapheresis and cultured in media supplemented with GM-CSF and IL-4. At the completion of incubation, DCs were subsequently infected with Ad.p53 at a viral particle to cell ratio of 15,000:1. DCs had a mature phenotype. Patients were scheduled to receive three doses of the vaccine i.d. at 2-week intervals. Those who did not progress after three immunizations underwent a second leukapheresis and received three additional doses of the vaccine, but this time once a month. The number of injected DCs was limited to 5 × 106 cells. p53-specific T cell responses were observed in 18/43 (41.8%) patients by IFN-γ ELISPOT assays. AEs associated with the vaccine were infrequent and mostly mild, with one patient experiencing grade 2 fatigue and one patient grade 2 arthralgia. Two patients achieved a PR and 13 patients had SD. Remarkably, a high rate of ORRs to second line chemotherapy was seen in patients with a positive immune response (78.6%) compared to patients with a negative immune response (33.3%). This is higher than expected based on previous trials with paclitaxel in patients with extensive SCLC (83, 84). Median OS was 8.8 months from the time of first vaccination. Patients with a positive immune response to vaccination had a trend towards an improved survival (MST 12.6 vs. 8.2 months; p = 0.131).
The same group subsequently conducted a randomized phase 2 trial involving 69 patients with extensive SCLC who were responsive to therapy or had non-progressive disease after first-line conventional chemotherapy (70, 71). Subjects were randomized into three arms: arm A (control group), arm B (Ad.p53-DC vaccine only), or arm C (Ad.p53-DC vaccine plus All-trans retinoic acid (ATRA)). The rationale to use ATRA is that it decreases systemic levels of myeloid-derived suppressor cells (MDSCs), which have potent immunosuppressive activity. The same immunization protocol was applied as the previous study. The vaccine was found to be safe with one patient experiencing grade 3 fatigue in arm B and eight patients experiencing grade 3 toxicities in arm C. Positive immune responses were obtained in 3/15 of the patients in arm B and 10/23 patients in arm C. The ORRs to second-line chemotherapy were 15.4%, 16.7%, and 23.8%, respectively for arms A, B and C with no survival differences between the different arms. These ORRs were lower than in the previous studies with the same vaccine. Surprisingly, survival from date of enrollment was numerically higher in the control arm than in de treatment arms (12.2, 6.3, and 6.2 months, respectively). A major limitation of this study was the high dropout rate which prevented patients from completing at least one cycle of salvage chemotherapy. Despite this limitation, some conclusions can also be drawn. First, the safety of the Ad.p53-DC vaccine was confirmed and second, the vaccine was able to elicit a specific cytotoxic T cell response in 20-40% of the patients with extensive SCLC, possibly influenced by the co-administration of ATRA. However, this did not translate into clinical responses, which were poor. The higher-than-expected response rate to second line paclitaxel in the first trial is encouraging and paves the way to combinatorial approaches of chemotherapy with immunotherapy to improve clinical efficacy.
For almost 20 years, long before the introduction of checkpoint inhibitors, DCs have been studied as a form of immunotherapy in lung cancer patients. This was based on a large body of preclinical data demonstrating the power of DCs to elicit de novo cytotoxic T cell responses, and the presence of different classes of TAAs in lung cancer. Evidence, mostly from phase 1 clinical trials, indicates that DC-based immunotherapy is safe and well tolerated with minor side effects depending on the route of administration. Local reactions (e.g., erythema) are a commonly reported AE after cutaneous injection, while systemic side-effects such as fever, chills and fatigue can be triggered as well. These AEs are mostly mild and transient. Severe toxicities rarely occur when DC-based immunotherapy is given solely. This is in contrast to the sometimes serious AEs seen with checkpoint inhibitors. Moreover, even in DC therapies incorporating whole tumor preparations, hence containing a substantial fraction of self-antigens, no clinically significant signs of auto-immunity have been reported so far.
Active immunotherapy involving DCs aims at eliciting cellular immunity in a tumor-specific and robust way. Data from the aforementioned early-phase trials demonstrate that antigen-specific immune responses can be observed in a significant number of patients, even in individuals with metastatic disease. However, positive immune responses as measured by a DTH skin test correlate only imperfectly with clinical outcomes, as shown in other tumor types (85, 86). In addition, these immunological responses tend to abate after the last injected dose.
Despite their proven immunogenicity, DC-based immunotherapy delivers low response rates, with 9.3% (40/432) of the lung cancer patients achieving an objective response. This is comparable to the ORR of second-line docetaxel in metastatic NSCLC (albeit with much less toxicity) and is lower than second-line PD-1/PD-L1 immunotherapy in the same, unselected population (8–11). In SCLC, traditionally considered as a “cold” tumor, the ORR is even lower (3.0%). However, higher ORRs were obtained when DC-based vaccination is combined with CIK cell therapy and/or concurrent chemotherapy (31.2%). Since most of the trials were not designed to assess OS, survival data of DC vaccination in lung cancer patients are scarce and anecdotal. Moreover, a remarkable observation in the DC vaccination field is the disconnect between clinical response and survival, as seen with sipuleucel-T, the FDA-approved DC vaccine for castration-resistant prostate cancer.
A typical limitation of the published studies is the small number of patients and the lack of a control group in almost all clinical trials. Another complicating factor is the huge variability in the methods used. This comprises differences in the type and formulation of TAAs, the DC maturation state at the time of vaccination, different use of co-delivered immunostimulants, as well as variations in the route and frequency of DC injection and dose of the vaccine. Ideally, each of these parameters needs to be optimized in order to improve the clinical efficacy of DC therapy (Figure 2).
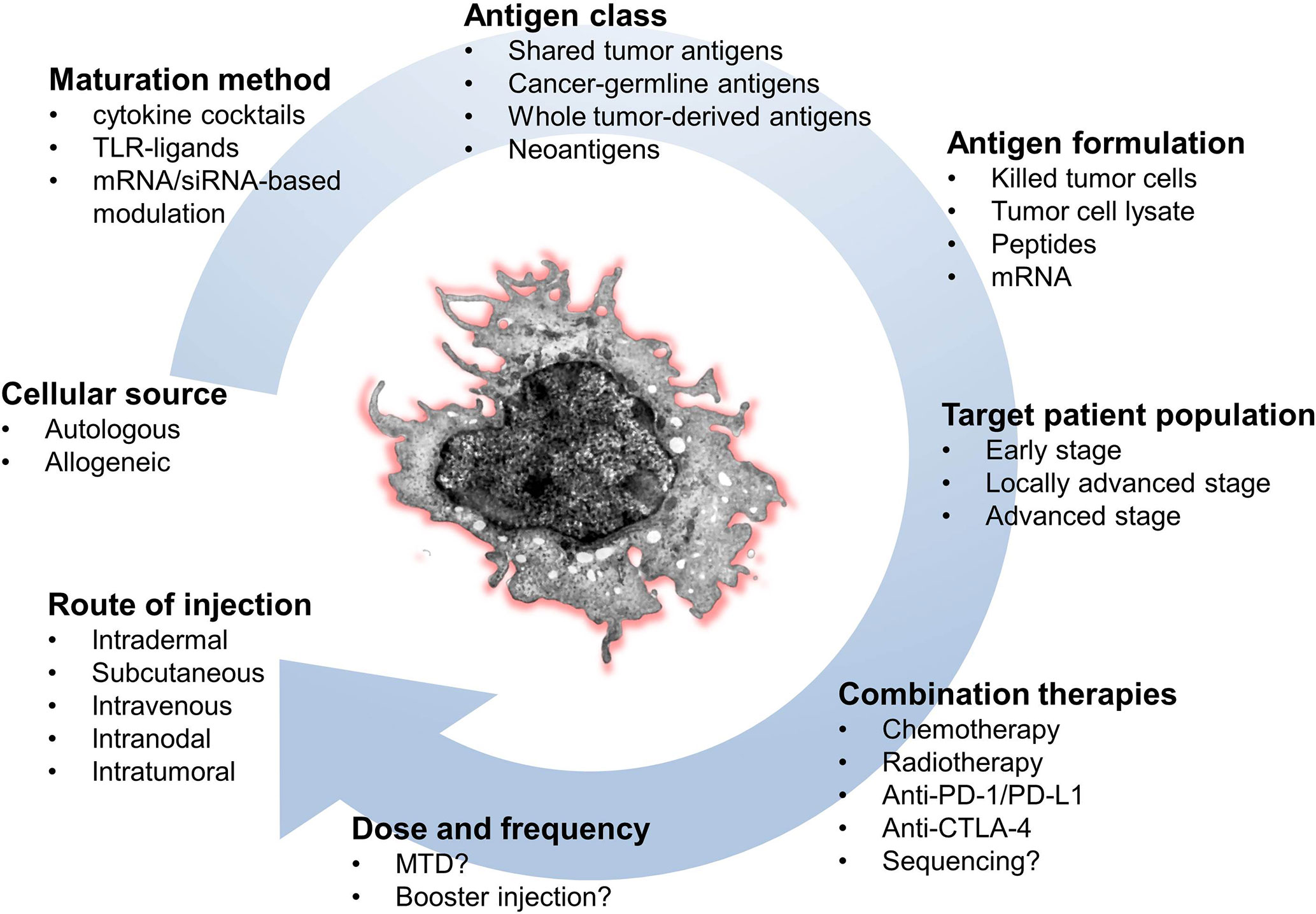
Figure 2 Key parameters to optimize the success of DC-based immunotherapy. CTLA-4, cytotoxic T lymphocyte-associated antigen 4; DC, dendritic cell; MTD, maximum tolerated dose; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1.
Different antigen formulations have been used in DC therapy for lung cancer, with tumor-derived peptides (single or combination) and undefined antigen preparations such as autologous tumor cells or cellular lysates being the most frequently used sources. While the use of peptides imposes restrictions in terms of the HLA-type of the target patient population, this is not the case for antigen preparations which also have the advantage to potentially target a much broader antigenic repertoire corresponding more closely to the patient’s tumor. A limitation in this approach is the often limited access to autologous tumor material for antigen extraction, as is the case in metastatic lung cancer. Furthermore, in some clinical trials, immature DCs were used in the vaccination protocol. While immunological responses were occasionally observed, immature DCs are primarily inducers of immunological tolerance, which is obviously unwanted in the setting of cancer immunotherapy. To achieve maturation, DCs can be exposed to a myriad of molecular combinations. However not all of them can be easily implemented in a clinical-grade production process, either due to stability, toxicity concerns and/or cost. In addition, strong stimuli can lead to the phenomenon of DC “exhaustion” whereby the capacity to produce the type-1 polarizing cytokine IL-12 is lost by the time the cells reach T cells in vivo. Also, inflammatory stimuli can trigger counterregulatory expression of checkpoint molecules such as PD-L1. We have shown that a widely used clinical-grade DC maturation cocktail composed of TNF-α, IL-1β, IL-6, and PGE2 induces high levels of surface PD-L1, which rises even further after cryopreservation and thawing, an effect presumably due to the prostaglandin (87, 88).
The impact of the route of DC injection has also been insufficiently addressed so far. Many trials have used the i.d. or s.c. route of injection, as it is very safe and feasible. However, a disadvantage of this route of administration is that the majority of DCs remain stuck at the injection site and will fail to migrate to the T cell rich areas within draining lymph nodes (89). Direct intranodal administration of DCs has been tested in melanoma, however it is technically challenging, while being not superior in terms of evoked immune responses (90). A much more predictable biodistribution can be achieved by i.v. injection, where the totality of the dose encounters the pulmonary vascular bed first, with subsequent distribution to the spleen and liver, as shown in a unique imaging study on human subjects (91). Preclinical experiments have shown that the “lung vascular filter” acts as a site where robust T cell-mediated immune responses can be efficiently evoked (92). The fact that the human lung represents a reservoir of around 10 billion resident T cells (93) raises the likelihood of productive interactions with antigen-carrying DCs injected intravenously. In addition, the route of DC injection can have an impact on the trafficking pattern of primed T cells, as shown in preclinical studies (94). Whereas an s.c. injection will program elicited T cells to home towards the skin, i.v. injection of DCs induces splenic CD8+ T cells capable of homing towards hematogenic metastases, which is especially relevant for lung cancer. Interestingly, in a therapeutic DC vaccination phase 1 trial in advanced melanoma comparing several ratios of i.v. versus i.d. injection, the data suggest that the i.v. rather than the i.d. injection route could be pivotal for the exceptional objective responses observed (95). Importantly, despite the potential of activated DCs to secrete large amounts of inflammatory cytokines and chemokines, none of the trials using i.v. injection have observed life-threatening toxicity events. A different strategy is the intratumoral injection of DCs, the idea being that relevant tumor antigens are present in abundance, and that T cells can then be primed in situ. One study using CCL21 gene-modified DCs delivered into lung tumors documented systemic antigen-specific CD8+ T cell responses in a fraction of the patients (52). This is surprising considering the strongly immunosuppressive microenvironment in lung tumors, shown to corrupt the function of DCs and T cells alike (96). Also, intratumoral injection is technically challenging and not feasible in every patient.
Finally, the optimal DC dose and injection schedule has not been determined yet. Given the complex and indirect mechanism of action of DC therapy and the imperfect nature of immune responses as surrogate for clinical responses, accurate modelling of a dose-effect relationship has not been achieved yet. What is clear, however, from all the early-phase trials, is that no dose-limiting AEs have been observed to date. Often the maximum dose delivered is practically limited by the production capacity of autologous DC vaccines. Still, one study suggested a better survival in NSCLC patients receiving the highest dose of DCs (54). Two other studies demonstrated a better OS in patients receiving respectively five and six or more vaccinations (48, 53). A bias may be present in these retrospective studies as the group of patients that received fewer vaccinations generally had a worse performance status.
The choice of target antigens for loading onto DCs is crucial to maximize the likelihood of eliciting a strong and tumor-directed immune response. Ideally, the antigen should meet different criteria: tumor-specific (expressed by cancer cells only), highly immunogenic, and necessary for cancer survival (97).
To date, most of the DC vaccination trials in lung cancer have targeted TAAs, which are self-antigens that are abnormally expressed by cancer cells, but may be present in normal cells as well. Since TAAs are shared with normal tissues, they can display limited immunogenicity due to central and peripheral tolerance, hence affecting the clinical efficacy of the vaccine. This can be partly circumvented by targeting more than one cancer antigen which will induce a broader immune response (98), as was the case in several of the abovementioned trials. However, the detectable immune responses that were evoked by the DC vaccine in these trials were often not powerful enough to translate into clinical effectiveness.
Development of personalized cancer vaccines based on neoantigens has become a new approach in cancer immunotherapy (99, 100). Neoantigens are tumor specific antigens that arise as a consequence of non-synonymous somatic mutations in the tumor cell genome (100). As their expression is tumor-restricted, in contrast to TAAs, these antigens are not subject to central tolerance and are potentially recognized by high avidity T cells. Hence, these antigens are ideal targets for DC vaccines. Neoantigens can be identified and selected using whole exome sequencing of tumor and blood cell DNA and bioinformatics algorithms. In a murine lung carcinoma model, neoantigen-pulsed DC vaccines were superior to neoantigen-adjuvant vaccines in activating immune responses and inhibiting tumor growth (101). A first demonstration of this approach in human subjects was provided in a phase 1 trial in metastatic melanoma, showing a remarkable induction of de novo T cell responses after vaccination with personalized tumor neoantigen-loaded DCs. Several phase 1 clinical trials are currently exploring neoantigen-targeted DC vaccines in lung cancer (Table 3), including a study from our group in surgically resected NSCLC (MIDRIXNEO-LUNG/NCT04078269), as well as in lung cancer patients who failed on standard anticancer therapies (NCT03871205). A major drawback for neoantigen-targeted vaccination is the lengthy and complex process leading up to the identification of potential neo-epitopes, which precludes implementation in patients with advanced or progressing disease. Also, the lack of standardization of in silico neo-epitope identification pipelines, with different algorithms producing diverging target lists, is a concern. One workaround consists of harvesting and profiling the “real” HLA-bound mutanome-derived peptides from tumor cells using advanced mass-spectrometric methods (102). This however requires access to large tumor samples which is a challenge in some clinical settings.
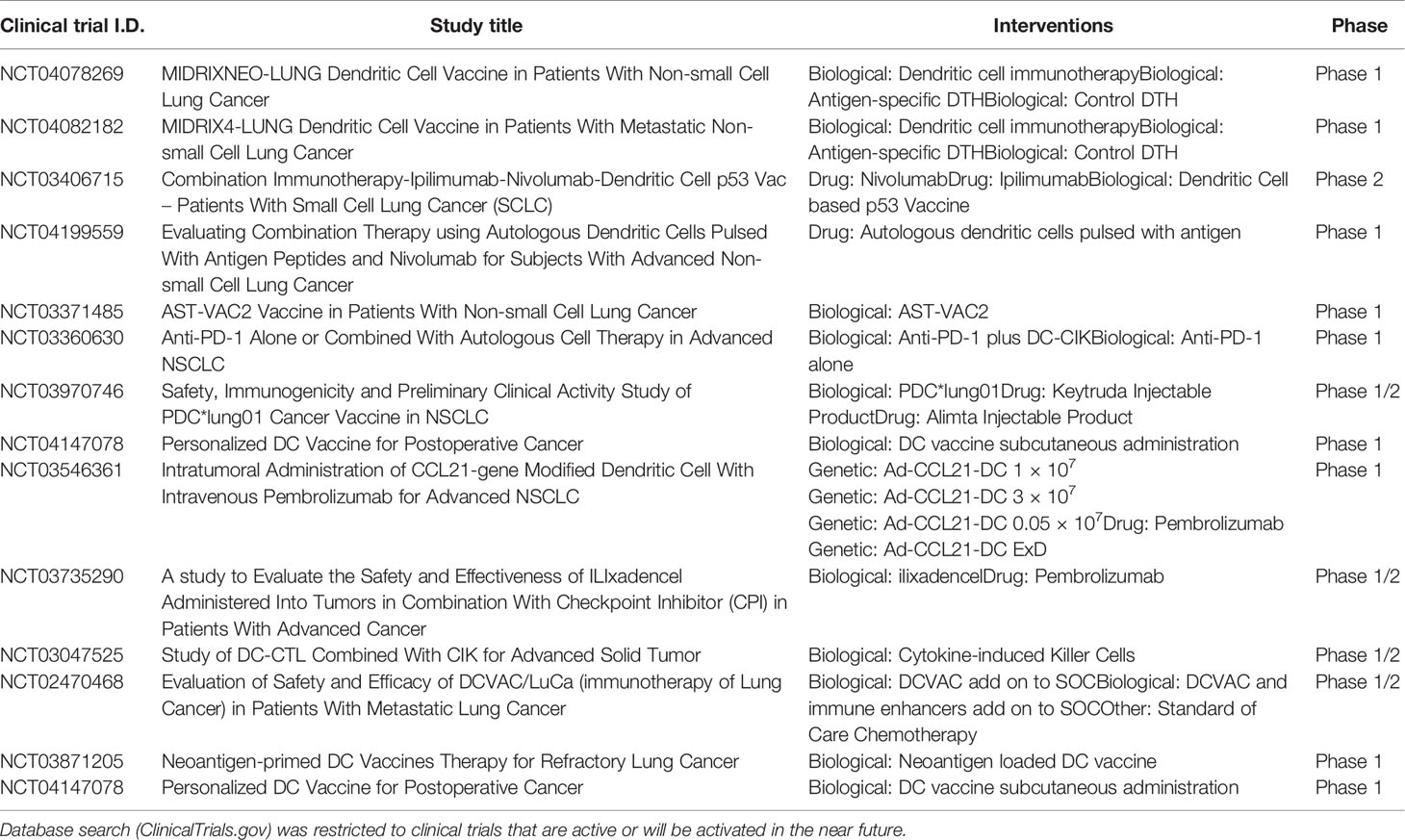
Table 3 Current clinical trials exploring dendritic cell (DC)-based immunotherapy in lung cancer.
Autologous cell therapies such as DC-based immunotherapies are labor-intensive and expensive to produce, and scaling-out to address a large patient population is difficult. Affordability of these therapies will be an important issue and challenge for both manufacturers and healthcare providers (103). Hence, a crucial question is which patients will derive most clinical benefit from these treatments. In early stage and locally advanced disease, treatable with curative intent (low tumor burden), the goal of DC-based immunotherapy is primarily to induce immunological memory to prevent later disease relapse (prophylactic vaccination). However, to show any therapeutic efficacy in this patient population, large and lengthy randomized trials are needed. In patients with metastatic disease on the other hand, DC-based immunotherapy actually aims to control the existing tumor (therapeutic vaccination). Considering the delayed antitumor effect and the systemic immunosuppression that is proportionate to the tumor load, patients with rapidly progressive or bulky tumors are unlikely to be appropriate candidates for DC vaccination, unless combinations with other systemic therapies are applied. In order to make DC therapy a viable option in clinical practice, biomarkers to enrich for responders/exclude non-responders upfront must imperatively be developed. Matching the targets loaded into the vaccine with the antigen expression pattern in the tumor is an obvious step. In addition, one can envision to exclude patients whose tumor biopsies harbor immune escape features such as loss-of-function or truncating mutations in Janus kinase (JAK) 1/2 or β2-microglobulin respectively, as vaccine-elicited T lymphocytes will fail to recognize and destroy the escape variants.
Combination with other treatment modalities, such as chemotherapy, radiotherapy and especially immune checkpoint inhibition, may be the key to the success of DC-based immunotherapy and is currently the subject of several clinical trials (Table 3). In principle, all combinatorial strategies aiming to potentiate cancer vaccines in general are applicable to DC therapy in particular (see (18) for an extensive review).
The combination of DC therapy with chemotherapy may seem less suitable due to the immunosuppressive effects of the latter. However, it has become clear that cytotoxic drugs do also have several immune-potentiating effects, not only by inducing immunogenic cell death (104), but also by some ancillary effects on both cancer cells and immune cells present within the tumor3 microenvironment (TME) (105). Chemotherapy can for instance reduce systemic levels of MDSCs and Tregs, which are important factors of lymphocyte suppression in metastatic cancer patients. It was previously shown that vaccination in between platinum-containing chemotherapy cycles can indeed boost antigen-specific T cell responses (106), which is attributed to the MDSC-depleting effect of myelotoxic platinum salts. In addition, chemotherapeutics can also promote antitumor immune responses by upregulating the expression of tumor antigens and MHC class I molecules on the tumor, thereby increasing the capacity for antigen presentation (105). As such, chemotherapy could improve the efficacy of DC-based immunotherapy by rendering tumor cells more susceptible for immune-mediated killing elicited by the DC vaccine (107). Finally, different classes of chemotherapeutics can directly affect DC biology, resulting in upregulated costimulatory molecule expression and increased antigen presentation. For some chemotherapeutics such as taxanes, the effects are mediated by TLR triggering (108). To date, combinatorial approaches of chemotherapy and DC-based immunotherapy (mostly DC/CIK cell therapy) have been investigated only sporadically in lung cancer. A relevant and very recent study in NSCLC is SLU01, a phase 1/2 randomized, open-label, multicenter trial evaluating the clinical efficacy and safety of DCVAC/LuCa added to standard first-line chemotherapy (paclitaxel and carboplatin) and immune-enhancers (IFN-α and hydroxychloroquine) in patients with stage IV NSCLC (NCT02470468). Preliminary results, presented in abstract form (109), demonstrated a better OS in patients receiving the combination therapy versus chemotherapy alone (15.5 vs. 11.8 months, HR 0.55, 95% CI 0.33–0.93; p = 0.0232).
An emerging research topic is the complex interplay between radiotherapy (RT) and the immune system, since it was appreciated that RT can mediate tumor regression outside the radiation field. This phenomenon, called the “abscopal effect”, is shown to be the result of an immune-mediated mechanism (110, 111). The advent of immunotherapies, especially immune checkpoint inhibitors, has created special interest in strategies that combine RT with immunotherapeutic agents. RT can enhance systemic antitumor immune responses by several immunomodulatory mechanisms (112), which were already briefly mentioned earlier in this review. In this way, RT could act synergistically with DC-vaccination and thus improve clinical outcomes. Preclinical tumor models have indeed shown potent local and systemic antitumor responses when direct intratumoral administration of DCs was combined with RT (113, 114). The first modest signs of clinical efficacy in humans were demonstrated in small clinical trials involving patients with digestive tumors and high-risk soft tissue sarcomas (115–117), whereas evidence in lung cancer is limited only to some studies with DC/CIK cell therapy.
Given the spectacular emergence of immune checkpoint blockers (ICBs) in the lung cancer therapeutic landscape, questions inevitably arise as to the role of DC vaccination in this setting. Although ICBs, more specifically PD-1/PD-L1 inhibitors, can sometimes trigger dramatic durable responses, the majority of lung cancer patients still experiences disease progression within a year of treatment. This is not surprising given the fact that anti-PD-1 antibodies, the most commonly used ICBs in lung cancer, only “fix” one specific step in the cancer immunity cycle, which is alleviating T cell paralysis in the TME. Upstream of this, a whole sequence of events leading up to the induction of tumor-homing cytotoxic T cells is operated by DCs, which are known to be dysfunctional within cancer-bearing hosts. Hence, adoptive transfer of fully functional ex vivo generated DCs can be seen as an ideal complement to checkpoint inhibition, as a fitting illustration of “pushing the gas pedal” while also “releasing the brakes” (118). ICB failure is often a manifestation of an “immune cold” tumor, i.e., a phenotype characterized by a lack of T cell priming against tumor antigens and consequently an absence of tumor-infiltrating T cells. DC therapy can turn a “cold” into a “hot” tumor through its capacity to prime and generate a de novo tumor antigen-specific T cell population. In addition, expanding insights into the mechanisms of action of ICBs could help to design better DC-based therapeutic approaches. The emerging knowledge that exhausted T cells consist of a multi-stage and dynamic group of lymphocytes is extremely relevant in this context. Differences in abundance and distribution of these T cell subsets could underlie differential responsiveness to ICBs, as only “progenitor exhausted” T cells can be expanded by this therapy (119). It raises the question whether DC vaccination may replenish the immune system with the type of progenitor T cells that is amenable to rescue by anti-PD-1 blockade. New insights in the mechanism of action of anti-PD-L1 ICBs are also emerging, mostly diverting the traditional focus from T cell/cancer cell interactions in the TME. As recently reported, anti-PD-L1 ICBs may achieve much of its effect by blocking PD-L1/PD-1 interactions in lymph node-resident DC-T cell clusters, rather than at the level of the tumor (120). Also, adoptively transferred activated DCs express variable levels of surface PD-L1, such that the anti-PD-L1 combination partner must be judiciously chosen. On one hand the combination can indeed result in boosting of T cell responses. However PD-L1-blockade may also be detrimental to DC-mediated T cell priming as PD-L1 protects DCs from cytotoxic T cells during antigen-specific cognate interactions (121). At worse, an anti-PD-L1 ICB with a specific IgG subtype could in theory trigger elimination of the injected DCs through antibody-dependent cytotoxicity. Finally, although CTLA-4 blockade as such is not part of the standard-of-care in lung cancer, its capacity to boost T cell priming could make it an ideal partner in a DC-based combinatorial approach. Clinical evidence for this type of combination was already provided in a phase 1 trial in advanced melanoma patients, where a DC vaccine combined with ipilimumab resulted in remarkably high ORRs (122).
Clinical data supporting the combination of DC therapy and ICBs is not yet available in lung cancer, however several early-phase trials are already addressing this issue (NCT03406715, NCT03360630, NCT03970746, NCT03546361, and NCT03970746). Of these, PDC-LUNG-101 (NCT03970746) seems promising, evaluating the safety, clinical efficacy and immunogenicity of PDC*lung01, a peptide-pulsed allogeneic plasmacytoid DC line in combination with pembrolizumab in patients with metastatic NSCLC. Still many questions remain unanswered such as which class of immune checkpoint inhibition is most suited (anti-PD-1/PD-L1/CTLA4)?. Choosing the correct sequencing could also be critical as preclinical data suggest that PD-1 inhibition can induce a population of T cells that are refractory to subsequent stimulation by a vaccine (123). Additional factors may come into play as our understanding of ICB mechanism of action grows. Of note, accumulating data around the role of the gut microbiome in shaping responses to ICBs (124) may sooner or later impact the way we design cancer vaccination combinatorial studies, including DC immunotherapy.
DC-based immunotherapy is safe and well-tolerated and can elicit antitumor immune responses in many patients with lung cancer, with occasional yet remarkable objective responses despite the predominant immunosuppressive climate in the metastatic setting. Combining DC-based immunotherapy with other anticancer therapies, such as chemotherapy, radiotherapy and/or checkpoint inhibition, can potentially improve their effectiveness. Alternatively, a choice of antigens based on neoepitopes with proven expression by the tumor cells may not merely induce immune responses but could result in clinical responses. Clinical trials to prove these hypotheses are underway and the results are eagerly awaited. Additional challenges for the future of DC therapy are determining the adequate dose, frequency, and duration of treatment, improving the choice of target antigens, and finding biomarkers to select potential responders upfront. Finally, identifying the most synergistic combinatorial regimen can hold the real key to long term disease control and survival in this lethal disease.
DS and KV wrote the manuscript. JI, SVL and BV read and corrected where needed. DS and KV took part in the discussion leading up to the manuscript. All authors contributed to the article and approved the submitted version.
KV is supported by an FWO Senior Clinical Investigator Grant. JI received a University BOF (Bijzonder Onderzoeksfonds) grant.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Ad, adenovirus; AE, adverse event; AKT, activated killer T cell; APC, antigen presenting cell; ATRA, all-trans retinoic acid; CEA, carcinoembryonic antigen; CIK, cytokine-induced killer cell; CR, complete response; CRC, colorectal cancer; CRT, chemoradiotherapy; CTA, cancer-testis antigen; CTL, cytotoxic T lymphocyte; CTLA-4, cytotoxic T lymphocyte-associated antigen 4; DC, dendritic cell; DCTCMF, dendritic cell/T cell-derived maturation factor; DTH, delayed-type hypersensitivity; ELISPOT, enzyme-linked immune absorbent spot; G-CSF, granulocyte colony-stimulating factor; GM-CSF, granulocyte/macrophage colony-stimulating factor; HR, hazard ratio; HER2, human epidermal growth factor receptor 2; HLA, human leucocyte antigen; ICB, immune checkpoint blocker/blockade; IFN, interferon; IL, interleukin; i.d., intradermal; i.v., intravenous; JAK, Janus kinase; KHL, keyhole limpet hemocyanin; MAGE, melanoma Antigen; MDSC, myeloid-derived suppressor cell; MHC, major histocompatibility complex; MST, median survival time; MUC1, mucin 1; NSCLC, non-small cell lung cancer; ORR, objective response rate; OS, overall survival; PBMC, peripheral blood monocyte; PD, progressive disease; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PFS, progression-free survival; PGE2, prostaglandin E2; PR, partial response; QOL, quality of life; RCT, randomized controlled trial; RT, radiotherapy; s.c., subcutaneous; SCLC, small cell lung cancer; SD, stable disease; SOCS1, suppressor of cytokine signaling 1; TAA, tumor-associated antigens; TLR, toll-like receptor; TME, tumor microenvironment; TNF, tumor necrosis factor; TNM, tumor node metastasis; Treg, t-regulatory; TRT, thoracic radiotherapy; WT1, wilms tumor protein 1.
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin (2018) 68:394–424. doi: 10.3322/caac.21492
2. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA: A Cancer J Clin (2018) 68:7–30. doi: 10.3322/caac.21442
3. Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med (2016) 375:1823–33. doi: 10.1056/NEJMoa1606774
4. Gandhi L, Rodríguez-Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, et al. Pembrolizumab plus Chemotherapy in Metastatic Non–Small-Cell Lung Cancer. N Engl J Med (2018) 378:2078–92. doi: 10.1056/NEJMoa1801005
5. Horn L, Mansfield AS, Szczęsna A, Havel L, Krzakowski M, Hochmair MJ, et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N Engl J Med (2018) 379:2220–9. doi: 10.1056/NEJMoa1809064
6. Paz-Ares L, Luft A, Vicente D, Tafreshi A, Gümüş M, Mazières J, et al. Pembrolizumab plus Chemotherapy for Squamous Non–Small-Cell Lung Cancer. N Engl J Med (2018) 379:2040–51. doi: 10.1056/NEJMoa1810865
7. Paz-Ares L, Dvorkin M, Chen Y, Reinmuth N, Hotta K, Trukhin D, et al. Durvalumab plus platinum-etoposide versus platinum-etoposide in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): a randomised, controlled, open-label, phase 3 trial. Lancet (2019) 394:1929–39. doi: 10.1016/S0140-6736(19)32222-6
8. Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:1627–39. doi: 10.1056/NEJMoa1507643
9. Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med (2015) 373:123–35. doi: 10.1056/NEJMoa1504627
10. Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet (2016) 387:1540–50. doi: 10.1016/S0140-6736(15)01281-7
11. Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, Von Pawel J, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet (2017) 389:255–65. doi: 10.1016/S0140-6736(16)32517-X
12. Muenst S, Laubli H, Soysal SD, Zippelius A, Tzankov A, Hoeller S. The immune system and cancer evasion strategies: therapeutic concepts. J Intern Med (2016) 279:541–62. doi: 10.1111/joim.12470
13. O’Donnell JS, Teng MWL, Smyth MJ. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat Rev Clin Oncol (2019) 16:151–67. doi: 10.1038/s41571-018-0142-8
14. van der Burg SH, Arens R, Ossendorp F, van Hall T, Melief CJ. Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nat Rev Cancer (2016) 16:219–33. doi: 10.1038/nrc.2016.16
15. Villanueva N, Bazhenova L. New strategies in immunotherapy for lung cancer: beyond PD-1/PD-L1. Ther Adv Respir Dis (2018) 12:1–29. doi: 10.1177/1753466618794133
16. Albright C, Garst J. Vaccine therapy in non—small-cell lung cancer. Curr Oncol Rep (2007) 9:241–6. doi: 10.1007/s11912-007-0029-9
17. Kalinski P, Urban J, Narang R, Berk E, Wieckowski E, Muthuswamy R. Dendritic cell-based therapeutic cancer vaccines: what we have and what we need. Future Oncol (2009) 5:379–90. doi: 10.2217/fon.09.6
18. Vermaelen K. Vaccine Strategies to Improve Anti-cancer Cellular Immune Responses. Front Immunol (2019) 10:8. doi: 10.3389/fimmu.2019.00008
19. Brunsvig PF, Kyte JA, Kersten C, Sundstrom S, Moller M, Nyakas M, et al. Telomerase Peptide Vaccination in NSCLC: A Phase II Trial in Stage III Patients Vaccinated after Chemoradiotherapy and an 8-Year Update on a Phase I/II Trial. Clin Cancer Res (2011) 17:6847–57. doi: 10.1158/1078-0432.CCR-11-1385
20. Butts C, Socinski MA, Mitchell PL, Thatcher N, Havel L, Krzakowski M, et al. Tecemotide (L-BLP25) versus placebo after chemoradiotherapy for stage III non-small-cell lung cancer (START): a randomised, double-blind, phase 3 trial. Lancet Oncol (2014) 15:59–68. doi: 10.1016/S1470-2045(13)70510-2
21. Nemunaitis J, Jahan T, Ross H, Sterman D, Richards D, Fox B, et al. Phase 1/2 trial of autologous tumor mixed with an allogeneic GVAX® vaccine in advanced-stage non-small-cell lung cancer. Cancer Gene Ther (2006) 13:555–62. doi: 10.1038/sj.cgt.7700922
22. Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol (2016) 17:822–35. doi: 10.1016/S1470-2045(16)00099-1
23. Thomas A, Giaccone G. Why has active immunotherapy not worked in lung cancer? Ann Oncol (2015) 26:2213–20. doi: 10.1093/annonc/mdv323
24. Steinman RM, Cohn ZA. Identification of a novel cell type in peripheral lymphoid organs of mice. J Exp Med (1973) 137:1142–62. doi: 10.1084/jem.137.5.1142
25. Melief CJM. Cancer Immunotherapy by Dendritic Cells. Immunity (2008) 29:372–83. doi: 10.1016/j.immuni.2008.08.004
26. Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature (2007) 449:419–26. doi: 10.1038/nature06175
27. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer (2012) 12:265–77. doi: 10.1038/nrc3258
28. Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol (2003) 15:138–47. doi: 10.1016/s0952-7915(03)00015-3
29. Cranmer LD, Trevor KT, Hersh EM. Clinical applications of dendritic cell vaccination in the treatment of cancer. Cancer Immunol Immunother (2004) 53:275–306. doi: 10.1007/s00262-003-0432-5
30. Wimmers F, Schreibelt G, Skãld AE, Figdor CG, De Vries IJM. Paradigm Shift in Dendritic Cell-Based Immunotherapy: From in vitro Generated Monocyte-Derived DCs to Naturally Circulating DC Subsets. Front Immunol (2014) 5:165. doi: 10.3389/fimmu.2014.00165
31. Huber A, Dammeijer F, Aerts JGJV, Vroman H. Current State of Dendritic Cell-Based Immunotherapy: Opportunities for in vitro Antigen Loading of Different DC Subsets? Front Immunol (2018) 9:2804. doi: 10.3389/fimmu.2018.02804
32. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol (2014) 15:e257–67. doi: 10.1016/S1470-2045(13)70585-0
33. Carreno BM, Magrini V, Becker-Hapak M, Kaabinejadian S, Hundal J, Petti AA, et al. A dendritic cell vaccine increases the breadth and diversity of melanoma neoantigen-specific T cells. Science (2015) 348:803–8. doi: 10.1126/science.aaa3828
34. Draube A, Klein-González N, Mattheus S, Brillant C, Hellmich M, Engert A, et al. Dendritic Cell Based Tumor Vaccination in Prostate and Renal Cell Cancer: A Systematic Review and Meta-Analysis. PLoS One (2011) 6:e18801. doi: 10.1371/journal.pone.0018801
35. Fong L, Hou Y, Rivas A, Benike C, Yuen A, Fisher GA, et al. Altered peptide ligand vaccination with Flt3 ligand expanded dendritic cells for tumor immunotherapy. Proc Natl Acad Sci (2001) 98:8809–14. doi: 10.1073/pnas.141226398
36. Itoh T, Ueda Y, Kawashima I, Nukaya I, Fujiwara H, Fuji N, et al. Immunotherapy of solid cancer using dendritic cells pulsed with the HLA-A24-restricted peptide of carcinoembryonic antigen. Cancer Immunol Immunother (2002) 51:99–106. doi: 10.1007/s00262-001-0257-z
37. Nair SK, Morse M, Boczkowski D, Ian Cumming R, Vasovic L, Gilboa E, et al. Induction of Tumor-Specific Cytotoxic T Lymphocytes in Cancer Patients by Autologous Tumor RNA-Transfected Dendritic Cells. Ann Surg (2002) 235:540–9. doi: 10.1097/00000658-200204000-00013
38. Kontani K, Taguchi O, Ozaki Y, Hanaoka J, Sawai S, Inoue S, et al. Dendritic cell vaccine immunotherapy of cancer targeting MUC1 mucin. Int J Mol Med (2003) 12:493–502. doi: 10.3892/ijmm.12.4.493
39. Hirschowitz EA. Autologous Dendritic Cell Vaccines for Non-Small-Cell Lung Cancer. J Clin Oncol (2004) 22:2808–15. doi: 10.1200/JCO.2004.01.074
40. Yannelli JR, Sturgill J, Foody T, Hirschowitz E. The large scale generation of dendritic cells for the immunization of patients with non-small cell lung cancer (NSCLC). Lung Cancer (2005) 47:337–50. doi: 10.1016/j.lungcan.2004.08.008
41. Ueda Y, Itoh T, Nukaya I, Kawashima I, Okugawa K, Yano Y, et al. Dendritic cell-based immunotherapy of cancer with carcinoembryonic antigen-derived, HLA-A24-restricted CTL epitope: Clinical outcomes of 18 patients with metastatic gastrointestinal or lung adenocarcinomas. Int J Oncol (2004) 24:909–17. doi: 10.3892/ijo.24.4.909
42. Chang G-C, Lan H-C, Juang S-H, Wu Y-C, Lee H-C, Hung Y-M, et al. A pilot clinical trial of vaccination with dendritic cells pulsed with autologous tumor cells derived from malignant pleural effusion in patients with late-stage lung carcinoma. Cancer (2005) 103:763–71. doi: 10.1002/cncr.20843
43. Morse MA. Phase I Study of Immunization with Dendritic Cells Modified with Fowlpox Encoding Carcinoembryonic Antigen and Costimulatory Molecules. Clin Cancer Res (2005) 11:3017–24. doi: 10.1158/1078-0432.CCR-04-2172
44. Hirschowitz EA, Foody T, Hidalgo GE, Yannelli JR. Immunization of NSCLC patients with antigen-pulsed immature autologous dendritic cells. Lung Cancer (2007) 57:365–72. doi: 10.1016/j.lungcan.2007.04.002
45. Mayordomo JI, Andres R, Isla MD, Murillo L, Cajal R, Yubero A, et al. Results of a pilot trial of immunotherapy with dendritic cells pulsed with autologous tumor lysates in patients with advanced cancer. Tumori (2007) 93:26–30. doi: 10.1177/030089160709300106
46. Um S-J, Choi YJ, Shin H-J, Son CH, Park Y-S, Roh MS, et al. Phase I study of autologous dendritic cell tumor vaccine in patients with non-small cell lung cancer. Lung Cancer (2010) 70:188–94. doi: 10.1016/j.lungcan.2010.02.006
47. Perroud MW, Honma HN, Barbeiro AS, Gilli SC, Almeida MT, Vassallo J, et al. Mature autologous dendritic cell vaccines in advanced non-small cell lung cancer: a phase I pilot study. Journal of Experimental & Clinical Cancer Research (2011) 30:65. doi: 10.1186/1756-9966-30-65
48. Takahashi H, Okamoto M, Shimodaira S, Tsujitani S-I, Nagaya M, Ishidao T, et al. Impact of dendritic cell vaccines pulsed with Wilms’ tumour-1 peptide antigen on the survival of patients with advanced non-small cell lung cancers. Eur J Cancer (2013) 49:852–9. doi: 10.1016/j.ejca.2012.11.005
49. Hu RH, Shi SB, Qi JL, Tian J, Tang XY, Liu GF, et al. Pemetrexed plus dendritic cells as second-line treatment for patients with stage IIIB/IV non-small cell lung cancer who had treatment with TKI. Med Oncol (2014) 31:63. doi: 10.1007/s12032-014-0063-z
50. Takahashi H, Shimodaira S, Ogasawara M, Ota S, Kobayashi M, Abe H, et al. Lung adenocarcinoma may be a more susceptive subtype to a dendritic cell-based cancer vaccine than other subtypes of non-small cell lung cancers: a multicenter retrospective analysis. Cancer Immunol Immunother (2016) 65:1099–111. doi: 10.1007/s00262-016-1872-z
51. Li D, He S. MAGE3 and Survivin activated dendritic cell immunotherapy for the treatment of non-small cell lung cancer. Oncol Lett (2018) 15:8777–83. doi: 10.3892/ol.2018.8362
52. Lee JM, Lee M-H, Garon E, Goldman JW, Salehi-Rad R, Baratelli FE, et al. Phase I Trial of Intratumoral Injection ofCCL21Gene–Modified Dendritic Cells in Lung Cancer Elicits Tumor-Specific Immune Responses and CD8+T-cell Infiltration. Clin Cancer Res (2017) 23:4556–68. doi: 10.1158/1078-0432.CCR-16-2821
53. Teramoto K, Ozaki Y, Hanaoka J, Sawai S, Tezuka N, Fujino S, et al. Predictive biomarkers and effectiveness of MUC1-targeted dendritic-cell-based vaccine in patients with refractory non-small cell lung cancer. Ther Adv Med Oncol (2017) 9:147–57. doi: 10.1177/1758834016678375
54. Ge C, Li R, Song H, Geng T, Yang J, Tan Q, et al. Phase I clinical trial of a novel autologous modified-DC vaccine in patients with resected NSCLC. BMC Cancer (2017) 17:884. doi: 10.1186/s12885-017-3859-3
55. Li H, Wang C, Yu J, Cao S, Wei F, Zhang W, et al. Dendritic cell-activated cytokine-induced killer cells enhance the anti-tumor effect of chemotherapy on non-small cell lung cancer in patients after surgery. Cytotherapy (2009) 11:1076–83. doi: 10.3109/14653240903121252
56. Zhong R, Teng J, Han B, Zhong H. Dendritic cells combining with cytokine-induced killer cells synergize chemotherapy in patients with late-stage non-small cell lung cancer. Cancer Immunol Immunother (2011) 60:1497–502. doi: 10.1007/s00262-011-1060-0
57. Shi SB, Ma TH, Li CH, Tang XY. Effect of maintenance therapy with dendritic cells: cytokine-induced killer cells in patients with advanced non-small cell lung cancer. Tumori (2012) 98:314–9. doi: 10.1700/1125.12398
58. Yang L, Ren B, Li H, Yu J, Cao S, Hao X, et al. Enhanced antitumor effects of DC-activated CIKs to chemotherapy treatment in a single cohort of advanced non-small-cell lung cancer patients. Cancer Immunol Immunother (2013) 62:65–73. doi: 10.1007/s00262-012-1311-8
59. Gao X, Mi Y, Guo N, Xu H, Xu L, Gou X, et al. Cytokine-Induced Killer Cells As Pharmacological Tools for Cancer Immunotherapy. Front Immunol (2017) 8:774. doi: 10.3389/fimmu.2017.00774
60. Zhao P, Bu X, Wei X, Sun W, Xie X, Li C, et al. Dendritic cell immunotherapy combined with cytokine-induced killer cells promotes skewing toward Th2 cytokine profile in patients with metastatic non-small cell lung cancer. Int Immunopharmacol (2015) 25:450–6. doi: 10.1016/j.intimp.2015.02.010
61. Zhu XP, Xu YH, Zhou J, Pan XF. A clinical study evaluating dendritic and cytokine-induced killer cells combined with concurrent radiochemotherapy for stage IIIB non-small cell lung cancer. Genet Mol Res (2015) 14:10228–35. doi: 10.4238/2015.August.28.6
62. Zhang L, Yang X, Sun Z, Li J, Zhu H, Li J, et al. Dendritic cell vaccine and cytokine-induced killer cell therapy for the treatment of advanced non-small cell lung cancer. Oncol Lett (2016) 11:2605–10. doi: 10.3892/ol.2016.4273
63. Zhang L, Xu Y, Shen J, He F, Zhang D, Chen Z, et al. Feasibility study of DCs/CIKs combined with thoracic radiotherapy for patients with locally advanced or metastatic non-small-cell lung cancer. Radiat Oncol (2016) 11:60. doi: 10.1186/s13014-016-0635-5
64. Song H, Liu S, Zhao Z, Sun W, Wei X, Ma X, et al. Increased cycles of DC/CIK immunotherapy decreases frequency of Tregs in patients with resected NSCLC. Int Immunopharmacol (2017) 52:97–202. doi: 10.1016/j.intimp.2017.09.014
65. Chen C-L, Pan Q-Z, Weng D-S, Xie C-M, Zhao J-J, Chen M-S, et al. Safety and activity of PD-1 blockade-activated DC-CIK cells in patients with advanced solid tumors. OncoImmunology (2018) 7:e1417721. doi: 10.1080/2162402X.2017.1417721
66. Kimura H, Iizasa T, Ishikawa A, Shingyouji M, Yoshino M, Kimura M, et al. Prospective phase II study of post-surgical adjuvant chemo-immunotherapy using autologous dendritic cells and activated killer cells from tissue culture of tumor-draining lymph nodes in primary lung cancer patients. Anticancer Res (2008) 28:1229–38.
67. Kimura H, Matsui Y, Ishikawa A, Nakajima T, Iizasa T. Randomized controlled phase III trial of adjuvant chemoimmunotherapy with activated cytotoxic T cells and dendritic cells from regional lymph nodes of patients with lung cancer. Cancer Immunol Immunother (2018) 67:1231–8. doi: 10.1007/s00262-018-2180-6
68. Kimura H, Matsui Y, Ishikawa A, Nakajima T, Yoshino M, Sakairi Y. Randomized controlled phase III trial of adjuvant chemo-immunotherapy with activated killer T cells and dendritic cells in patients with resected primary lung cancer. Cancer Immunol Immunother (2015) 64:51–9. doi: 10.1007/s00262-014-1613-0
69. Chiappori AA, Soliman H, Janssen WE, Antonia SJ, Gabrilovich DI. INGN-225: a dendritic cell-based p53 vaccine (Ad.p53-DC) in small cell lung cancer: observed association between immune response and enhanced chemotherapy effect. Expert Opin Biol Ther (2010) 10:983–91. doi: 10.1517/14712598.2010.484801
70. Chiappori AA, Williams CC, Gray JE, Tanvetyanon T, Haura EB, Creelan BC, et al. Randomized-controlled phase II trial of salvage chemotherapy after immunization with a TP53-transfected dendritic cell-based vaccine (Ad.p53-DC) in patients with recurrent small cell lung cancer. Cancer Immunol Immunother (2019) 68:517–27. doi: 10.1007/s00262-018-2287-9
71. Iclozan C, Antonia S, Chiappori A, Chen D-T, Gabrilovich D. Therapeutic regulation of myeloid-derived suppressor cells and immune response to cancer vaccine in patients with extensive stage small cell lung cancer. Cancer Immunol Immunother (2013) 62:909–18. doi: 10.1007/s00262-013-1396-8
72. Shi SB, Tang XY, Tian J, Chang CX, Li P, Qi JL. Efficacy of erlotinib plus dendritic cells and cytokine-induced killer cells in maintenance therapy of advanced non-small cell lung cancer. J Immunother (2014) 37:250–5. doi: 10.1097/CJI.0000000000000015
73. Cascio S, Finn O. Intra- and Extra-Cellular Events Related to Altered Glycosylation of MUC1 Promote Chronic Inflammation, Tumor Progression, Invasion, and Metastasis. Biomolecules (2016) 6:39. doi: 10.3390/biom6040039
74. Quoix E, Lena H, Losonczy G, Forget F, Chouaid C, Papai Z, et al. TG4010 immunotherapy and first-line chemotherapy for advanced non-small-cell lung cancer (TIME): results from the phase 2b part of a randomised, double-blind, placebo-controlled, phase 2b/3 trial. Lancet Oncol (2016) 17:212–23. doi: 10.1016/S1470-2045(15)00483-0
75. Thistlethwaite FC, Gilham DE, Guest RD, Rothwell DG, Pillai M, Burt DJ, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother (2017) 66:1425–36. doi: 10.1007/s00262-017-2034-7
76. Hanna N, Shepherd FA, Fossella FV, Pereira JR, De Marinis F, von Pawel J, et al. Randomized phase III trial of pemetrexed versus docetaxel in patients with non-small-cell lung cancer previously treated with chemotherapy. J Clin Oncol (2004) 22:1589–97. doi: 10.1200/JCO.2004.08.163
77. Wang S, Wang Z. Efficacy and safety of dendritic cells co-cultured with cytokine-induced killer cells immunotherapy for non-small-cell lung cancer. Int Immunopharmacol (2015) 28:22–8. doi: 10.1016/j.intimp.2015.05.021
78. Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med (2018) 379:2342–50. doi: 10.1056/NEJMoa1809697
79. Kimura H, Dobrenkov K, Iida T, Suzuki M, Ando S, Yamamoto N. Tumor-draining lymph nodes of primary lung cancer patients: a potent source of tumor-specific killer cells and dendritic cells. Anticancer Res (2005) 25:85–94.
80. Antonia SJ, López-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol (2016) 17:883–95. doi: 10.1016/S1470-2045(16)30098-5
81. Chung HC, Piha-Paul SA, Lopez-Martin J, Schellens JHM, Kao S, Miller WH Jr, et al. Pembrolizumab After Two or More Lines of Previous Therapy in Patients With Recurrent or Metastatic SCLC: Results From the KEYNOTE-028 and KEYNOTE-158 Studies. J Thorac Oncol (2020) 15:618–27. doi: 10.1016/j.jtho.2019.12.109
82. Antonia SJ, Mirza N, Fricke I, Chiappori A, Thompson P, Williams N, et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res (2006) 12:878–87. doi: 10.1158/1078-0432.CCR-05-2013
83. Smit EF, Fokkema E, Biesma B, Groen HJ, Snoek W, Postmus PE. A phase II study of paclitaxel in heavily pretreated patients with small-cell lung cancer. Br J Cancer (1998) 77:347–51. doi: 10.1038/bjc.1998.54
84. Yamamoto N, Tsurutani J, Yoshimura N, Asai G, Moriyama A, Nakagawa K, et al. Phase II study of weekly paclitaxel for relapsed and refractory small cell lung cancer. Anticancer Res (2006) 26:777–81.
85. Aarntzen EHJG, Figdor CG, Adema GJ, Punt CJA, De Vries IJM. Dendritic cell vaccination and immune monitoring. Cancer Immunol Immunother (2008) 57:1559–68. doi: 10.1007/s00262-008-0553-y
86. Lesterhuis WJ, De Vries IJM, Schuurhuis DH, Boullart ACI, Jacobs JFM, De Boer AJ, et al. Vaccination of colorectal cancer patients with CEA-loaded dendritic cells: antigen-specific T cell responses in DTH skin tests. Ann Oncol (2006) 17:974–80. doi: 10.1093/annonc/mdl072
87. Brabants E, Heyns K, De Smet S, Devreker P, Ingels J, De Cabooter N, et al. An accelerated, clinical-grade protocol to generate high yields of type 1-polarizing messenger RNA-loaded dendritic cells for cancer vaccination. Cytotherapy (2018) 20:1164–81. doi: 10.1016/j.jcyt.2018.06.006
88. Prima V, Kaliberova LN, Kaliberov S, Curiel DT, Kusmartsev S. COX2/mPGES1/PGE2pathway regulates PD-L1 expression in tumor-associated macrophages and myeloid-derived suppressor cells. Proc Natl Acad Sci (2017) 114:1117–22. doi: 10.1073/pnas.1424355112
89. Verdijk P, Aarntzen EH, Lesterhuis WJ, Boullart AC, Kok E, van Rossum MM, et al. Limited amounts of dendritic cells migrate into the T-cell area of lymph nodes but have high immune activating potential in melanoma patients. Clin Cancer Res (2009) 15:2531–40. doi: 10.1158/1078-0432.CCR-08-2729
90. Bol KF, Figdor CG, Aarntzen EH, Welzen ME, Van Rossum MM, Blokx WA, et al. Intranodal vaccination with mRNA-optimized dendritic cells in metastatic melanoma patients. OncoImmunology (2015) 4:e1019197. doi: 10.1080/2162402X.2015.1019197
91. Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res (1999) 59:56–8.
92. Willart MA, Jan de Heer H, Hammad H, Soullie T, Deswarte K, Clausen BE, et al. The lung vascular filter as a site of immune induction for T cell responses to large embolic antigen. J Exp Med (2009) 206:2823–35. doi: 10.1084/jem.20082401
93. Purwar R, Campbell J, Murphy G, Richards WG, Clark RA, Kupper TS. Resident memory T cells (T(RM)) are abundant in human lung: diversity, function, and antigen specificity. PLoS One (2011) 6:e16245. doi: 10.1371/journal.pone.0016245
94. Mullins DW, Sheasley SL, Ream RM, Bullock TN, Fu YX, Engelhard VH. Route of immunization with peptide-pulsed dendritic cells controls the distribution of memory and effector T cells in lymphoid tissues and determines the pattern of regional tumor control. J Exp Med (2003) 198:1023–34. doi: 10.1084/jem.20021348
95. Wilgenhof S, Van Nuffel AM, Benteyn D, Corthals J, Aerts C, Heirman C, et al. A phase IB study on intravenous synthetic mRNA electroporated dendritic cell immunotherapy in pretreated advanced melanoma patients. Ann Oncol (2013) 24:2686–93. doi: 10.1093/annonc/mdt245
96. Pyfferoen L, Brabants E, Everaert C, De Cabooter N, Heyns K, Deswarte K, et al. The transcriptome of lung tumor-infiltrating dendritic cells reveals a tumor-supporting phenotype and a microRNA signature with negative impact on clinical outcome. Oncoimmunology (2017) 6:e1253655. doi: 10.1080/2162402X.2016.1253655
97. Hollingsworth RE, Jansen K. Turning the corner on therapeutic cancer vaccines. NPJ Vaccines (2019) 4:7. doi: 10.1038/s41541-019-0103-y
98. Lybaert L, Vermaelen K, De Geest BG, Nuhn L. Immunoengineering through cancer vaccines - A personalized and multi-step vaccine approach towards precise cancer immunity. J Control Rel (2018) 289:125–45. doi: 10.1016/j.jconrel.2018.09.009
99. Mastelic-Gavillet B, Balint K, Boudousquie C, Gannon PO, Kandalaft LE. Personalized Dendritic Cell Vaccines—Recent Breakthroughs and Encouraging Clinical Results. Front Immunol (2019) 10:766. doi: 10.3389/fimmu.2019.00766
100. Peng M, Mo Y, Wang Y, Wu P, Zhang Y, Xiong F, et al. Neoantigen vaccine: an emerging tumor immunotherapy. Mol Cancer (2019) 18:128. doi: 10.1186/s12943-019-1055-6
101. Zhang R, Yuan F, Shu Y, Tian Y, Zhou B, Yi L, et al. Personalized neoantigen-pulsed dendritic cell vaccines show superior immunogenicity to neoantigen-adjuvant vaccines in mouse tumor models. Cancer Immunol Immunother (2020) 69:135–45. doi: 10.1007/s00262-019-02448-z
102. Ebrahimi-Nik H, Michaux J, Corwin WL, Keller GL, Shcheglova T, Pak H, et al. Mass spectrometry driven exploration reveals nuances of neoepitope-driven tumor rejection. JCI Insight (2019) 5:e129152. doi: 10.1172/jci.insight.129152
103. Lopes AG SA, Frohlich B. Cost Analysis of Cell Therapy Manufacture: Autologous Cell Therapies, Part 1. BioProcess Int (2018) 16:S3–8.
104. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi: 10.1038/nm.2028
105. Chen G, Emens LA. Chemoimmunotherapy: reengineering tumor immunity. Cancer Immunol Immunother (2013) 62:203–16. doi: 10.1007/s00262-012-1388-0
106. Welters MJ, van der Sluis TC, van Meir H, Loof NM, van Ham VJ, van Duikeren S, et al. Vaccination during myeloid cell depletion by cancer chemotherapy fosters robust T cell responses. Sci Transl Med (2016) 8:334ra52. doi: 10.1126/scitranslmed.aad8307
107. van Gulijk M, Dammeijer F, Aerts J, Vroman H. Combination Strategies to Optimize Efficacy of Dendritic Cell-Based Immunotherapy. Front Immunol (2018) 9:2759. doi: 10.3389/fimmu.2018.02759
108. Truxova I, Hensler M, Skapa P, Halaska MJ, Laco J, Ryska A, et al. Rationale for the Combination of Dendritic Cell-Based Vaccination Approaches With Chemotherapy Agents. Int Rev Cell Mol Biol (2017) 330:115–56. doi: 10.1016/bs.ircmb.2016.09.003
109. Havel L, Kolek V, Pesek M, Cernovská M, Bartunkova J, Spisek R, et al. Dendritic-cell vaccine (DCVAC) with first-line chemotherapy in patients with stage IV NSCLC: Final analysis of phase II, open label, randomized, multicenter trial. J Clin Oncol (2019) 37:9039. doi: 10.1200/JCO.2019.37.15_suppl.9039
110. Demaria S, Formenti SC. The abscopal effect 67 years later: from a side story to center stage. Br J Radiol (2020) 93:20200042. doi: 10.1259/bjr.20200042
111. Demaria S, Ng B, Devitt ML, Babb JS, Kawashima N, Liebes L, et al. Ionizing radiation inhibition of distant untreated tumors (abscopal effect) is immune mediated. Int J Radiat Oncol Biol Phys (2004) 58:862–70. doi: 10.1016/j.ijrobp.2003.09.012
112. Ferrara TA, Hodge JW, Gulley JL. Combining radiation and immunotherapy for synergistic antitumor therapy. Curr Opin Mol Ther (2009) 11:37–42.
113. Akutsu Y, Matsubara H, Urashima T, Komatsu A, Sakata H, Nishimori T, et al. Combination of direct intratumoral administration of dendritic cells and irradiation induces strong systemic antitumor effect mediated by GRP94/gp96 against squamous cell carcinoma in mice. Int J Oncol (2007) 31:509–15.
114. Teitz-Tennenbaum S, Li Q, Rynkiewicz S, Ito F, Davis MA, McGinn CJ, et al. Radiotherapy potentiates the therapeutic efficacy of intratumoral dendritic cell administration. Cancer Res (2003) 63:8466–75.
115. Chi KH, Liu SJ, Li CP, Kuo HP, Wang YS, Chao Y, et al. Combination of conformal radiotherapy and intratumoral injection of adoptive dendritic cell immunotherapy in refractory hepatoma. J Immunother (2005) 28:129–35. doi: 10.1097/01.cji.0000154248.74383.5e
116. Finkelstein SE, Iclozan C, Bui MM, Cotter MJ, Ramakrishnan R, Ahmed J, et al. Combination of External Beam Radiotherapy (EBRT) With Intratumoral Injection of Dendritic Cells as Neo-Adjuvant Treatment of High-Risk Soft Tissue Sarcoma Patients. Int J Radiat OncologyBiologyPhysics (2012) 82:924–32. doi: 10.1016/j.ijrobp.2010.12.068
117. Wang C, Pu J, Yu H, Liu Y, Yan H, He Z, et al. A Dendritic Cell Vaccine Combined With Radiotherapy Activates the Specific Immune Response in Patients With Esophageal Cancer. J Immunother (2017) 40:71–6. doi: 10.1097/CJI.0000000000000155
118. Versteven M, Van den Bergh JMJ, Marcq E, Smits ELJ, Van Tendeloo VFI, Hobo W, et al. Dendritic Cells and Programmed Death-1 Blockade: A Joint Venture to Combat Cancer. Front Immunol (2018) 9:394. doi: 10.3389/fimmu.2018.00394
119. Beltra JC, Manne S, Abdel-Hakeem MS, Kurachi M, Giles JR, Chen Z, et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity (2020) 52:825–41.e8. doi: 10.1016/j.immuni.2020.04.014
120. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The PD-1/PD-L1-Checkpoint Restrains T cell Immunity in Tumor-Draining Lymph Nodes. Cancer Cell (2020) 38:685–700.e8. doi: 10.1016/j.ccell.2020.09.001
121. Mohsenzadegan M, Peng RW, Roudi R. Dendritic cell/cytokine-induced killer cell-based immunotherapy in lung cancer: What we know and future landscape. J Cell Physiol (2020) 235:74–86. doi: 10.1002/jcp.28977
122. Wilgenhof S, Corthals J, Heirman C, van Baren N, Lucas S, Kvistborg P, et al. Phase II Study of Autologous Monocyte-Derived mRNA Electroporated Dendritic Cells (TriMixDC-MEL) Plus Ipilimumab in Patients With Pretreated Advanced Melanoma. J Clin Oncol (2016) 34:1330–8. doi: 10.1200/JCO.2015.63.4121
123. Verma V, Shrimali RK, Ahmad S, Dai W, Wang H, Lu S, et al. PD-1 blockade in subprimed CD8 cells induces dysfunctional PD-1(+)CD38(hi) cells and anti-PD-1 resistance. Nat Immunol (2019) 20:1231–43. doi: 10.1038/s41590-019-0441-y
Keywords: dendritic cell, cancer vaccine, lung cancer, immunotherapy, tumor antigen, immune checkpoint blockade
Citation: Stevens D, Ingels J, Van Lint S, Vandekerckhove B and Vermaelen K (2021) Dendritic Cell-Based Immunotherapy in Lung Cancer. Front. Immunol. 11:620374. doi: 10.3389/fimmu.2020.620374
Received: 22 October 2020; Accepted: 29 December 2020;
Published: 12 February 2021.
Edited by:
Stephanie E. B. McArdle, Nottingham Trent University, United KingdomCopyright © 2021 Stevens, Ingels, Van Lint, Vandekerckhove and Vermaelen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karim Vermaelen, a2FyaW0udmVybWFlbGVuQHVnZW50LmJl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.