 Panagiota Bouti1*†
Panagiota Bouti1*† Steven D. S. Webbers1,2*†
Steven D. S. Webbers1,2*† Susanna C. Fagerholm3
Susanna C. Fagerholm3 Ronen Alon4
Ronen Alon4 Markus Moser5
Markus Moser5 Hanke L. Matlung1
Hanke L. Matlung1 Taco W. Kuijpers1,2
Taco W. Kuijpers1,2- 1Sanquin Research and Landsteiner Laboratory, Department of Blood Cell Research, Amsterdam University Medical Center, University of Amsterdam, Amsterdam, Netherlands
- 2Department of Pediatric Immunology, Rheumatology and Infectious Disease, Amsterdam University Medical Center (AUMC), Emma Children’s Hospital, University of Amsterdam, Amsterdam, Netherlands
- 3Research Program of Molecular and Integrative Biosciences, Faculty of Biological and Environmental Sciences, University of Helsinki, Helsinki, Finland
- 4Department of Immunology, The Weizmann Institute of Science, Rehovot, Israel
- 5Institute of Experimental Hematology, School of Medicine, Technical University of Munich, Munich, Germany
Neutrophils are the most prevalent leukocytes in the human body. They have a pivotal role in the innate immune response against invading bacterial and fungal pathogens, while recent emerging evidence also demonstrates their role in cancer progression and anti-tumor responses. The efficient execution of many neutrophil effector responses requires the presence of β2 integrins, in particular CD11a/CD18 or CD11b/CD18 heterodimers. Although extensively studied at the molecular level, the exact signaling cascades downstream of β2 integrins still remain to be fully elucidated. In this review, we focus mainly on inside-out and outside-in signaling of these two β2 integrin members expressed on neutrophils and describe differences between various neutrophil stimuli with respect to integrin activation, integrin ligand binding, and the pertinent differences between mouse and human studies. Last, we discuss how integrin signaling studies could be used to explore the therapeutic potential of targeting β2 integrins and the intracellular signaling cascade in neutrophils in several, among other, inflammatory conditions in which neutrophil activity should be dampened to mitigate disease.
Introduction
Neutrophils
Human neutrophils or polymorphonuclear cells (PMNs) form the frontline of the immune response against bacteria and fungi. Despite their fundamental role in inflammation, recent evidence gives prominence to the controversial role of neutrophils in cancer (1, 2). Whereas recent data using deuterium labeling in vivo suggest that human neutrophils can survive up to 5.4 days in the circulation (3), general belief based on traditional studies still is that the high number of neutrophils emerging from the bone marrow on a daily basis (1011 per day) is balanced with their short half-life in the circulation (up to about 12h) (4). In contrast, the abundancy of neutrophil population in humans, representing up to 70% of circulating leukocytes, is not reflected on mice in which the percentage of neutrophils varies from 10 to 25% of white blood cells (5, 6).
Apart from the daily extravasation to offer that first natural line of defense at the tissue level, neutrophils are able to directionally migrate at larger numbers out of the bloodstream at the site of infection and either clear the pathogen or induce the recruitment of adaptive immune cells by chemokine/cytokine production or antigen presentation (7–9). Neutrophils exit the circulation via trans-endothelial migration, which takes place in sequential steps (tethering, rolling, crawling, cell arrest/firm adhesion/transmigration); all have been thoroughly described (10, 11). At the site of infection, neutrophils exert their killing properties to eliminate the pathogen extracellularly or upon phagocytosis. Neutrophils have the capacity to kill the invaded microorganism by secreting their toxic granular content (degranulation), producing reactive oxygen species (ROS), or releasing their DNA and subsequently forming neutrophil extracellular traps (NETs) in a process of cell death known as NETosis (6, 12). These processes of neutrophil extravasation, migration, and neutrophil effector functions against infectious agents rely to a very large extent on integrins.
Integrins: Expression, Structure and Activation
Integrins are a family of ubiquitously expressed, transmembrane receptors. They anchor cells within their ambient extracellular matrix (ECM) and bind to counter-receptors expressed on other cells (13). Integrins are expressed on the cell surface as heterodimers of non-covalently associated α and β subunits (14, 15). So far, 24 different αβ heterodimers have been described in mammals, which exhibit specific ligand binding properties. The extracellular part of the α subunits defines ligand specificity. It consists of a seven bladed β-propeller module, followed by the thigh and two calf-domains (16–18). The β subunits consist of an N-terminal βA or β-I like domain, a plexin–semaphorin–integrin (PSI) domain, and α hybrid domain, followed by four cysteine-rich epidermal growth factor (EGF) repeats (19). While for most integrins the β-propeller from the α subunit and the N-terminal βA domain from the β subunit form the ligand binding head, a subset of vertebrate integrin α subunits, among them are the β2 integrin family, the collagen, and some laminin-binding integrins, have an additional α-I domain inserted into a loop in the β-propeller domain, which employ ligand binding (20). A hallmark of integrins is their ability to reversibly switch between different ectodomain conformations with distinct affinities for the ligand. It is meanwhile well accepted that integrins can adopt at least three conformations: a low affinity bent conformation, an extended-closed conformation with intermediate affinity, and an extended-open conformation with high ligand affinity (21–23). Notably, the difference in affinity for the ligand between the low and high affinity states varies strongly between different integrins from several hundred to several thousand fold (24–26). The conformational changes are either induced and stabilized by the extracellular ligand (outside-in activation) or are triggered by tightly regulated intracellular signals (inside-out activation); the latter is particularly important for integrins expressed on blood cells and will be described in detail in the following sections.
Both integrin subunits have a single-pass transmembrane helical domain, which are non-covalently associated within the plasma membrane. The two transmembrane domains interact at two interfaces termed the “outer membrane clasp” and the “inner membrane clasp”, which are mediated by helical packing of the N-terminal part and hydrophobic interactions and a salt bridge at the C-terminal part of the transmembrane domains, respectively (27–29). The transmembrane domains are essential for integrin function as they propagate conformational changes across the plasma membrane in both directions. These changes occur upon binding of the ligand to the ectodomain or intracellular adapter proteins to the cytoplasmic domain, which cause the disruption of the transmembrane domain interactions and a separation of the transmembrane and intracellular domains.
The intracellular domains represent the control modules of integrins. These rather short peptides serve as docking stations for numerous intracellular proteins, which regulate the integrin’s activity state and execute their functions. The majority of studies were done on the β integrin tail, which has a length of only 30 to 70 amino acids with the exception of the much longer β4 integrin cytoplasmic domain. Most β-tails contain a membrane proximal NPxY/F and a membrane distal NxxY/F motif flanking a serine/threonine-rich motif (30). Many intracellular proteins interact with these motifs in a phosphorylation dependent or independent manner. The most prominent proteins that bind to these motifs are Talins and Kindlins, which are essential integrin activators and will be described in detail below (31, 32). The cytoplasmic tails of the different α chains show low homology but share a GFFXR sequence in the membrane-proximal region (33). Much less is known about protein interactors, yet the tails of these α chains are important in the regulation of proteins bound to their respective β subunits (34). One of the few proteins known to associate with integrin α chains, including CD11a, is SHARPIN, an integrin negative regulator (35). In addition, CD11a, CD11b, and CD11c cytoplasmic domains have all been reported to be phosphorylated on conserved serine residues, and these phosphorylation sites are important for integrin function (36, 37).
Integrin Activation by Talin and Kindlin
Intracellular signals that eventually trigger changes in integrin affinity for the ligand, also named as integrin inside-out activation, culminate in the binding of Talin and Kindlin to the β integrin tail. While Talin was long thought to be the sole integrin activator, studies in cells and animal models revealed that it requires assistance by Kindlin. Moreover, beside their essential function in integrin activation, both proteins initiate the formation of adhesion complexes that link integrins with the actin cytoskeleton and form signaling hubs that modulate many cellular processes (integrin outside-in signaling) (30, 31, 38, 39).
Talins are large cytoplasmic proteins consisting of an N-terminal head domain, which comprises an atypical FERM domain, and a C-terminal rod domain composed of 13 helical bundles. Talin1 is ubiquitously expressed and the major isoform expressed in hematopoietic cells, while the closely related Talin2 isoform shows a more restricted expression pattern (40). Binding of the Talin head to the membrane proximal NPxY/F motif within the integrin tail destabilizes the transmembrane interactions between the α and β subunits thereby inducing conformational changes of the integrin’s ectodomain (41). Talin is a mechanically regulated protein and provides a direct link with the actin cytoskeleton (42). It contains two actin-binding sites and several binding sites for the actin binding protein Vinculin within its rod (43, 44). Tensile forces generated by the actin-myosin contractile apparatus are transmitted via Talin to the integrin and contribute to full integrin activation. Moreover, the mechanical forces result in stretching and partial unfolding of several bundles within the Talin rod thereby exposing cryptic Vinculin binding sites (45–48). Before Talin is capable to bind and activate integrins it also requires an activation process as intramolecular interactions between the Talin head and rod domains preclude integrin binding (49). Details in the intramolecular interactions were recently provided by the cryo-EM structure of Talin. These structural data also revealed that even in the autoinhibited state the N-terminal F0 and F1 domains are exposed allowing them to bind to activated, membrane-bound Rap1 (50). This recruitment pathway is further promoted by electrostatic interactions between multiple sites within the Talin head and the negatively charged membrane lipids, in particular PIP2 (51). Concurrently, charge repulsions between the plasma membrane and the Talin rod domain interfere with the Talin head–rod interaction and promote the release in autoinhibition (52). Interactions with other proteins such as RIAM, Vinculin, and KANK proteins may further contribute to Talin recruitment and Talin activation and are further discussed below (53–55).
Mammals have three FERMT genes encoding Kindlin protein, which are expressed in a tissue specific manner. While Kindlin-1 is primarily found in epithelial cells, Kindlin-2 is almost ubiquitously expressed, and Kindlin-3 expression is restricted to cells of hematopoietic origin (56). The Kindlin protein structure resembles that of the Talin head and formed by an atypical FERM domain consisting of an N-terminal F0 domain followed by the canonical FERM domains F1, F2, and F3. A striking difference to the Talin FERM domain is an inserted PH domain within the F2 domain of Kindlins (57). The PH domain and a long loop within the F1 domain make contacts with phospholipids of the plasma membrane (58, 59). However, the molecular signals that direct Kindlins to integrin adhesion sites are not known yet. The Kindlin F3 domain binds directly to the membrane distal NxxY/F and the more N-terminally located threonine rich motif of the β integrin cytoplasmic tail (60, 61). Mutations in these motifs abolished Kindlin–integrin interaction resulting in defective integrin activation and function. Whether Kindlins alone, like as it has been shown for Talin, can induce conformational changes of the integrin ectodomain is not known yet. It rather seems that Kindlins support Talin-mediated integrin activation by further destabilizing the interactions between the integrin α and β transmembrane domains, which lead to the fully activated, extended open conformation of integrins (62, 63). Kindlins may also promote integrin activation by clustering integrins and thereby increasing their ligand binding avidity rather than their affinity (64). This model is also supported by structural data, which showed that Kindlin-2 can dimerize (65). Whether dimerization is a general mechanism of all Kindlin members to cluster integrins remains to be shown. Interestingly, recent crystallization studies suggest the formation of trimeric or even higher Kindlin complexes, which are unable to bind integrins (66, 67). Future studies will show whether Kindlin dimer/multimerization does occur and have an impact on integrin activity regulation in vivo and how this is regulated by upstream signals. Irrespective of their crucial role in integrin activity regulation more recent studies began to unravel the important role of Kindlins in adhesion site assembly and stabilization. Meanwhile multiple Kindlin interactors such as Paxillin, Leupaxin, ILK, and actin have been identified, and differences in the interactome or affinities to these and other still unknown interactors may explain the specific features of the individual kindlin members (68–73).
Leukocyte Integrins and the β2 Integrin Family
In leukocytes, αMβ2 or MAC-1 (CD11b/CD18), CR3; αLβ2 or LFA-1 (CD11a/CD18); αDβ2 (CD11d/CD18); αXβ2 (CD11c/CD18); α4β1 or VLA-4 (CD49d/CD29); α5β1 or VLA-5 (CD49e/CD29); α6β1 or VLA-6 (CD49f/CD29); α4β7 (CD49d/HML-1), and αEβ7 (CD103/HML-1) have been described (30, 74). β2 integrins are only found on leukocytes. The four members of this family are expressed on different leukocyte subpopulations and fulfil different roles in immunity. CD11a/CD18 is expressed on all leukocytes, regulating cell–cell communication by binding to ICAM ligands; it has the most restricted ligand repertoire of all the β2 integrins and is structurally more distantly related to the other three. In contrast, only myeloid cells including neutrophils express CD11b/CD18 and CD11c/CD18 on most cells, while macrophages also have additional CD11d/CD18 proteins on their surface, most likely with similar ligand binding properties (75). The most prominent integrin on neutrophils is CD11b/CD18, which has a promiscuous binding capacity to more than 40 ligands, including ICAM, iC3b, fibrinogen, and other ECM proteins (18), indicating its pleiotropic effect on multiple cellular processes.
The complexity of integrin biology is reflected in its intricate mechanism of activation. Recent reviews have proposed how β2 integrins get activated (31, 76). In brief, integrins may exist in three conformational stages, bent-inactive, extended-inactive, and extended-active conformation. Each of them is related to their binding affinity status and the subsequent efficacy of ligand binding. In fact, a recent report on β1 integrin suggests that all of the different integrin conformations can be present on the cell surface in a dynamic equilibrium in order to initiate or preserve cell motility (77). The “switchblade” and the “deadbolt” models have been predicted both describing the chronological order of integrin changes in affinity and ligand binding. However, there is no clear evidence supporting one of these models to date (31).
Focusing on the β2 integrins in leukocytes and, more specifically in neutrophils, neutrophils require the presence of Kindlin-3 protein for all CD11/CD18 integrin-mediated ligand binding (31). The importance of β2 integrin and their protein regulators has been demonstrated by the identification of human leukocyte adhesion deficiency syndromes (LADs) (78). Loss of β2 integrin in humans due to mutations in the gene ITGB2 encoding the β2 subunit causes LAD-I. In contrast, LAD-III (or LAD-I/variant) arises from mutations in FERMT3 encoding the Kindlin-3 protein (79–81) in the presence of normal levels of leukocyte integrins but a lack of high-affinity binding activity upon cell activation. Both syndromes result in similar patient phenotypic characteristics, showing susceptibility to bacterial infections due to the lack of neutrophil migration to the site of infection, poor wound healing, and late detachment of the umbilical cord in most affected children (82). In addition, many LAD-patients suffer from increased IL-17-driven periodontal tissue inflammation, leading to periodontitis, at least in part due to reduced recruitment of neutrophils (83). In LAD-III, the Kindlin-3 mutations also result in a clinical bleeding tendency due to the lack of β3 integrin activity on platelets (84). The role of Talin-1 in initiating intermediate integrin conformation and both Talin-1 and Kindlin-3 in inducing the high affinity state of the CD11a/CD18 integrin was subsequently reported in a murine model (63), very similar to CD11b/CD18 integrin activation by Kindlin-3 (85, 86). After initial activation, integrins engage with their ligands and form nascent clusters. Again, several models of clustering induction and stabilization have been suggested for β1, β2 or β3 integrins, yet the precise mechanism remains to be elucidated (87–90).
Although Talin-1 and Kindlin-3 are the primary proteins associated with β2 integrins in case of both inside-out and outside-in integrin activation, β2 integrins can only function effectively after recruitment of several additional proteins to the β2 integrin cytoplasmic tail, thus regulating or stabilizing its high-affinity state. Although recent technology can identify proteins associating with integrins (91), the heterogeneity among the different β integrin subunits as well as their diverse function in different cell types averts a general statement about integrin signaling and function. Although the proteins that can directly associate with β2 integrins or regulate their affinity and function have been reviewed (92), their contribution to downstream signaling of β2 integrins in the context of neutrophils is limited (38, 93–97). Examples of proteins interacting directly with the cytoplasmic tail of the β2 subunit and having a known function in neutrophils include Cytohesin-1 (98, 99), SYK (100, 101), and Filamin A (97) as we will also discuss below.
Inside-Out Signaling During Endothelial Transmigration
Chemokine Mediated Signaling
For trans-endothelial migration to occur, leukocytes utilize CD11a/CD18 and CD11b/CD18 to adhere to the endothelial wall. As discussed, CD11a/CD18 and CD11b/CD18 are kept in an inactive state, and require activation through recruitment of Talin-1 and Kindlin-3. Activation of these β2 integrins increases their binding affinity, which allows adhesion of leukocytes to several substrates including endothelial membrane proteins ICAM-1, upregulated during inflammation, and ICAM-2. During selectin-mediated rolling and slow rolling, leukocytes engage with chemokines and cytokines presented on the vessel wall with several different receptors, of which most are G-protein coupled receptors (GPCRs), also known as seven transmembrane receptors. Engagement of chemokines, such as CXCL8 (better known as IL-8) and CXCL1 (also known as GRO-α, MGSA, or KC) with their respective receptors causes the activation of CD11a/CD18 and CD11b/CD18 through a common pathway which recruits Talin-1 and Kindlin-3 to the cytoplasmic tail of CD18. Note that Talin-1 can be recruited through both selectin and GPCR signaling.
This common pathway through GPCRs starts with Gαi and the Gβ/γ subunits, which split after chemokine engagement with the receptor and activate Phospholipase C β2 (PLCβ2) (Figure 1A) (102–104). The signaling then branches into two pathways, as has mostly been supported by gene knockout models in mice. The first is the activation of CalDAG-GEFI (RASGRP2) by PLCβ2 and to a minor extent PLCβ3 through the generation of DAG and a calcium influx by IP3 generation (105, 106), resulting in the activation of Rap1 (107). Activated Rap1 exposes a membrane targeting sequence, and upon interaction with the plasma membrane the N-terminus of Rap1 interacts with RIAM, which contains a Talin-binding sequence (108). A reduced fusion protein consisting of the Rap1 membrane targeting domain and the RIAM-Talin-binding sequence is sufficient to target Talin-1 to the plasma membrane, allowing engagement of Talin-1 with the NPXY motif in the cytoplasmic tail of CD18 (109) and resulting in β2 integrin activation (108). Interestingly, direct Rap1 interactions have also been described with the F0 and F1 domains of Talin-1, which in mice are pivotal for normal overall integrin activation, adhesion, and cell spreading, whereas the RIAM–Talin-1 interaction seems specific for β2 integrins in leukocytes (110–113).
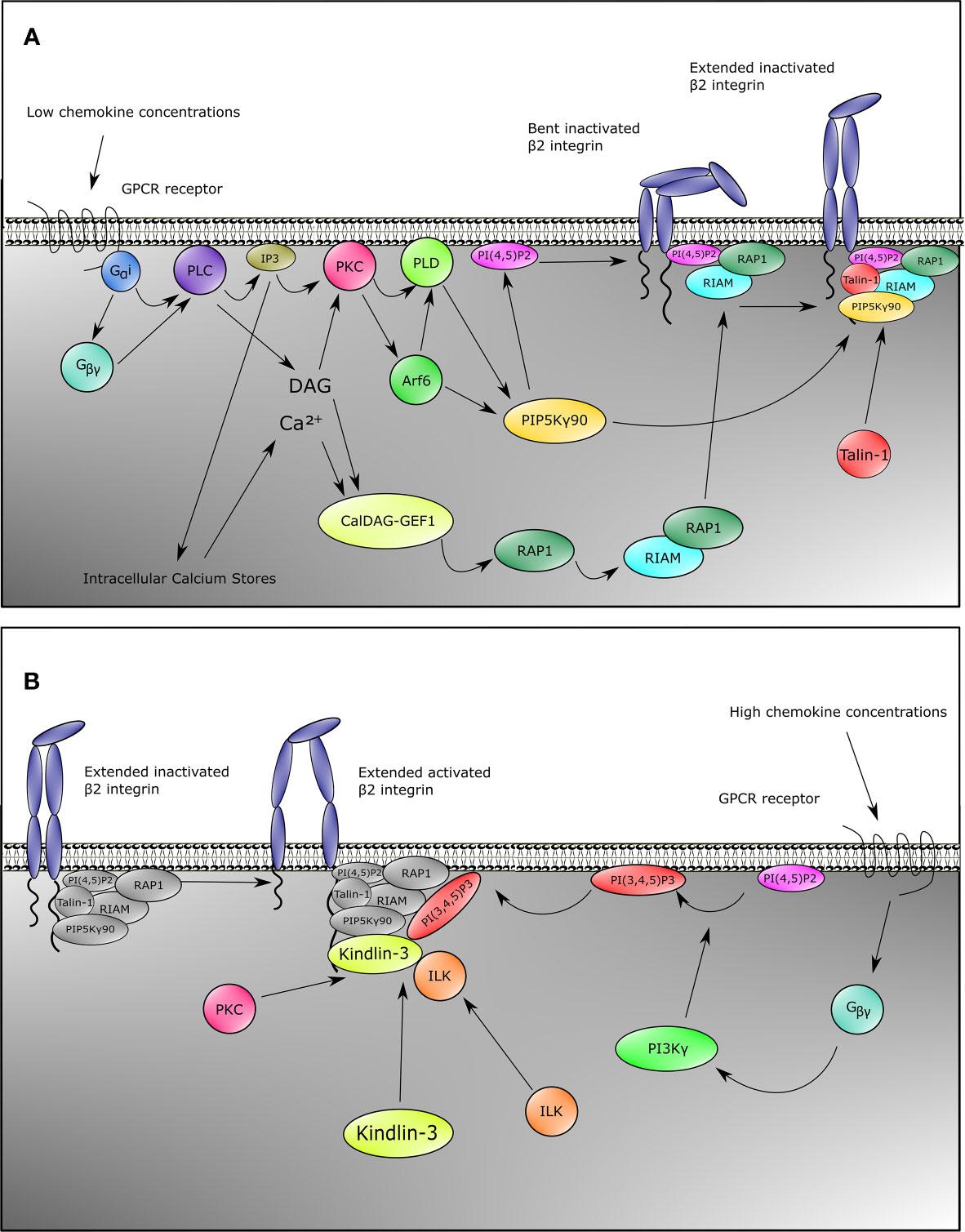
Figure 1 Inside-out signaling recruits Talin-1 and Kindlin-3 to β2 integrin. Upon stimulation of neutrophils with chemokines, both Talin-1 and Kindlin-3 become rapidly recruited to activate β2 integrins. This can be divided into two pathways for Talin-1 and Kindlin-3. For Talin-1 recruitment (A), GPCRs engage with their ligands which causes the heterotrimeric G-proteins to split into Gα and Gβγ. This initiates the activation of PLC by Gαi, and Gβγ. Signaling through the second messengers’ calcium which is released from the endoplasmic reticulum by IP3 and diacylglycerol (DAG) is a critical element in many biological systems. Integration of calcium and DAG signals has been suggested to occur primarily through protein kinase C (PKC) family members, which bind both calcium and DAG. An alternative pathway involves CalDAG-GEF1, which has structural features (calcium-binding EF hands and DAG-binding C1 domains) and functions in calcium and DAG signal integration. This GEF activates RAP1, which then results into activation of RIAM and binding of RIAM to Talin-1. Together with catalyzation of PIP into PI(4,5)P2 by PIP5Kγ90 through the PKC-PLD-Arf6 axis, these events recruit Talin-1 to β2 integrin, thus extending the integrin from the bent conformation. Gβγ is also involved in Kindlin-3 recruitment (B). In addition to activating PLC, Gβγ also activates PI3K, which generates PI(3,4,5)P3. This allows recruitment of Kindlin-3 to β2 integrin. Binding of Kindlin-3 after Talin-1 binding activates extended β2 integrins. Note that the Kindlin-3 recruitment cascade might not be complete.
The second pathway for β2 integrin activation downstream of PLC goes through Protein kinase C (PKC). PKC is activated by both DAG and IP3 generated by PLC. PKC then elicits its function by activation of Phospholipase D (PLD) and Arf6 (Figure 1A) (114, 115). Both Arf6 and PLD subsequently activate PIPKIγ (also known as PIP5K1C) which increases phosphatidylinositol 4,5 (PIP2) levels at the inner plasma membrane (104, 114–117), resulting in the recruitment and activation of Talin-1 via its PIP2 binding F2 domain and subsequent binding to the NPXY motif of the CD18 cytoplasmic tail (Figure 1A) (104, 109, 112, 118). Also taking into account a direct interaction between the C-terminal domain of PIP5K(γ90) and Talin-1, this collectively suggests a feedforward loop of both recruitment of Talin-1 to the β2 integrin tail and activation of Talin-1 through local increase in PIP2 levels close to the β2 integrin (118).
Although multiple studies have shown the importance of Kindlin-3 for β2 integrin activation (31, 63), the signaling pathway essential for activation of Kindlin-3 and recruitment to the CD18 cytoplasmic tail is as yet still less clear compared to what is known about Talin-1 activation (Figure 1B). Notwithstanding, Kindlin-3 recruitment to CD18 temporally precedes the high affinity conformation of β2 integrins (59). Additionally, structure function analyses have indicated that the pleckstrin homology domain of the F2 subdomain of the FERM domain is important for binding phosphatidylinositols at the plasma membrane (119, 120), and that part of this protein population has been suggested to reside in an auto-inhibitory homotrimer (67). Moreover, high chemokine concentrations induce neutrophil arrest, which is mediated through high-affinity β2 integrin by Kindlin-3 associating with the cytoplasmic tail of the β2 subunit and PIP3 increments upon strong activity of PI3K (Figure 1B) (104). Blocking PI3K has been suggested to decrease human neutrophil adhesion to fibrinogen, and adhesion to ICAM-1 is affected by specific blocking of PI3K with wortmannin (103, 121). ILK might also play a role in Kindlin-3 function in mice since genetic abblation resulted in reduced Kindlin-3 phosphorylation and a decrease in high-affinity CD11a/CD18 (122).
Despite these common pathways in β2 integrin activation, some chemokines trigger alternative pathways. For instance, in murine neutrophils, CXCL1 induces a strong calcium influx, which is an important signaling mediator (Figure 1A) for β2 integrin activation (123). This differential signaling effect is substantiated by the knockout of TRPC6 encoding a plasma membrane calcium channel, resulting in lower calcium transient increases, thus leading to reduced Rap1 and β2 integrin activation by CXCL1 (124). Other chemokines remain to be tested for this pathway. Secondly, the actin-binding protein Hematopoietic cell‐specific LYN Substrate 1 (HS1) knockout mice have reduced Rac1 and subsequent Rap1 activation upon CXCL1 stimulation (125), which indicates another integrin activation pathway parallel to the common GPCR pathways (Figure 1A) in at least CXCL1-mediated integrin activation. Additionally, IL-8 also branches towards another pathway via PI3K, which activates the serine/threonine kinases RAF-1 and B-RAF, from which RAF-1 is expressed in human neutrophils. Both RAF-1 and B-RAF can subsequently activate the MEK/ERK/p38-MAPK pathways (126), and blocking MAPK indeed reduces neutrophil adhesion to fibrinogen, indicating a role for this pathway in CD11b/CD18 activation (103).
There are some noticeable differences in the activation of CD11a/CD18 and CD11b/CD18, which can solely be attributed to the presence of either CD11a or CD11b. For example, Cytohesin-1 is known to activate CD11a/CD18 and increases firm adhesion, spreading, and transendothelial migration for mouse neutrophils, whereas the opposite effect was observed for CD11b/CD18 (98, 99, 127–129). Additionally, CD11a Ser1140 and CD11b Ser1126 phosphorylation are critical events in the activation of the respective CD18 heterodimers for binding of ICAM-1, to a lesser extent ICAM-2, but not for the non-cellular ligand inactivated complement 3 fragment b (iC3b) in case of CD11b/CD18 (36, 130). Furthermore, CD11a/CD18 in humans is more rapidly activated than CD11b/CD18, but the activation is more transient, being deactivated after 1 min. In contrast, CD11b/CD18 seems to become fully activated after 1 min and remains activated for up to more than at least 5 min after stimulation with fMLP (131). This may reflect different roles of CD11a/CD18 and CD11b/CD18 in trans-endothelial migration, where CD11a/CD18 facilitates slow rolling and early adhesion, whereas CD11b/CD18 has a more prominent role during firm adhesion and subsequent chemotaxis. These findings are speculative and should be interpreted against background expression levels of these integrins, being much higher for CD11b/CD18 on human but not murine neutrophils. Selective genetic defects for either of the two integrins in gene knockout models of the respective α-chain have not been fully explored and cannot be supported by human ‘experiments of nature’ with genetic mutations in patients as the cause of disease.
Selectin-Mediated Signaling
Aside from GPCRs, other mechanisms of inside-out signaling have been described. Selectin-mediated signaling has been thoroughly described in the past (132), and it is out of the scope of this review, yet we briefly discuss the concept in response to β2 integrin activation. During selectin-mediated rolling, the physical interactions between L-selectins on leukocytes with P- and E-selectins on inflamed endothelium induce recruitment of Talin-1 to the cytoplasmic tail of CD18 (133). Upon engagement of L-selectin with E-selectin, Src-family kinases (SFKs) are recruited and phosphorylate either FcRγ adapters or DAP12 (134) together with ERM proteins Moesin and Ezrin in an ITAM-motif based manner (135). This is followed by phosphorylation of Spleen tyrosine kinase (SYK) (136), which then recruits a complex of SLP76 together with ADAP (137). These two proteins subsequently activate Bruton tyrosine kinase (BTK) to phosphorylate p38-MAPK and also activate PLCγ2 (133) instead of PLCβ2 (as known for chemokine stimulation). Downstream signaling through selectins involves the activation of Rap1a and PIP5K(γ90) (Figure 1A) (104), thus leading to the convergence of selectin-mediated signaling with the common GPCR pathway resulting in the recruitment of Talin-1 to CD11a/CD18 and CD11b/CD18 (110, 111, 138). To date, selectin-mediated CD11a/CD18 activation has only been reported for murine and not human neutrophils, which coincides with a much lower expression of CD11a/CD18 on human neutrophils. The medium avidity state of CD11a/CD18 initiates slow rolling due to transient interactions with ICAM-1 (133, 138). This allows the neutrophil to sample chemokines presented by the endothelial cell wall, thus inducing chemokine mediated inside-out signaling, which leads to further activation of β2 integrins (138). Although it has been found that selectins are sufficient to cause β2 integrin-mediated arrest (139), both the presence of selectins and chemokine signals may be required for full activation of CD11a/CD18 and CD11b/CD18 in human blood vessels (138), as selectins fully activate β2 integrins cooperatively at low chemokine concentrations (104). In human neutrophils, chemokines but not selectins used PI3Kγ in cooperation with Rap1a to mediate integrin-dependent slow rolling (at low chemokine concentrations) and arrest (at high chemokine concentrations). High levels of chemokines activate β2 integrins without the need of selectin signals.
Role of β2 Integrin Downstream Signaling in Slow Rolling, Adhesion, Firm Adhesion, and Crawling
Upon interacting with the vascular wall, neutrophils first decrease their velocity, strengthen their interaction with the endothelium, and finally sample the endothelium by crawling over it to find an appropriate spot to squeeze through and transmigrate into the subendothelial space. In humans, both CD11a/CD18 and CD11b/CD18 have partial redundancy (131), whereas CD11b/CD18 seems redundant in terms of adhesion compared to CD11a/CD18 in mice (140).
For adhesion strengthening, β2 integrins can upregulate their own affinity state. Upon engagement of β2 integrins with their ligands, SFKs are recruited in order to phosphorylate SYK, among other proteins. In mice, the absence of HCK and FGR proteins prevents continued adhesion to ICAM-1 (141), indicating that β2 integrins regulate themselves to maintain a high-affinity conformation. Another example is the binding of 14-3-3ζ isoform to β2 integrins upon phosphorylation of Thr578 of the CD18 cytoplasmic tail (142), although a membrane-proximal site of the cytoplasmic tail which does not require phosphorylation has also been described (143). The binding of 14-3-3ζ to phospho-Thr578 obscures the binding site for Filamin-A (142), a protein which keeps β2 integrins in an inactive state (144). Some of the signals may counter or act to fine-tune the process of adhesion strengthening. This has been indicated in mice by the promoting activity of PKCs (145), whereas hematopoietic progenitor kinase 1 (HPK1) in concert with mammalian actin binding protein 1 (mAbp1) specifically activates CD11a/CD18 (146), thereby negatively affecting intraluminal crawling and spreading of neutrophils.
Aside from regulating their own affinity state, CD11a/CD18 and CD11b/CD18 each also separately clusters with itself. This increases the overall binding avidity of the integrins, which is required for firm adhesion. Integrin clustering also facilitates outside-in signaling by grouping the CD18 cytoplasmic tails close together. Clustering likely occurs due to the linkage of CD11a/CD18 and CD11b/CD18 to the actin cytoskeleton, in which filamentous actin (F-actin) polymerization plays a pivotal role. The initial F-actin polymerization likely is not induced by direct stimulation of CD11a/CD18 or CD11b/CD18, but rather through surface receptors for tumor necrosis factor α (TNFα) and other chemokines (147). Blocking specifically actin polymerization with Cytochalasin D, a strong actin polymerization inhibitor, prevents adhesion strengthening and crawling but does not change CD11a/CD18 or CD11b/CD18 expression in human neutrophils (148). Prior to F-actin polymerization, the SFKs HCK and FGR as well as SYK relocate to the cytoplasmic tail of CD11a/CD18 or CD11b/CD18 (149), and mouse knockout models highlight the relevance of these proteins in spreading and F-actin polymerization (101, 141, 150–152). These studies collectively indicate that F-actin polymerization occurs prior to adhesion strengthening and clustering and that β2 integrins relay signals for actin polymerization. After initial F-actin polymerization, integrins recruit multiple actin binding proteins, including Vinculin and α-actinin (43, 153, 154). Neutrophils express the cytoskeletal protein Vinculin, although they do not form mature focal adhesions as macrophages or fibroblasts do. The role of Vinculin in β2 integrin-dependent neutrophil adhesion, migration, and recruitment was found to be not completely abrogated in knockout mice and more so under static than dynamic flow conditions, suggesting a dispensable ‘mechanosensitive’ function in vivo (155). Other proteins might also promote integrin clustering. The protein 14-3-3ζ, which binds the phosphorylated Thr578 in the CD18 cytoplasmic tail, forms dimers which indicates a role in clustering of β2 integrins (36, 92, 156). Moreover, Cytohesin-1 increases firm adhesion by CD11a/CD18 but decreases adhesion by CD11b/CD18, indicating a role for this protein in the regulation of integrin clustering (98, 99, 127–129). The recruitment of actin binding proteins and CD18 binding proteins ultimately packs the β2 integrins close together due to their collective binding to F-actin filaments close to the membrane, resulting in an increase in binding avidity. It remains to be shown which of these actin-binding proteins are non-redundant in neutrophil behavior. Interesting novel candidates involved in F-actin dynamics might be found in proteomics data from MKL1-deficient patients, which show downregulation of actin-associated proteins AIP1, ARHGAP9 and Profilin-1 (157), regulating actin polymerization and disassembly (AIP1, ARHGAP9), or inhibits polymerization of actin (ARHGAP9, PFN1).
Role of β2 Integrin Downstream Signaling in Cell Polarization, Spreading, Chemotaxis, and Migration
Once inside-out signaling facilitates β2 integrin intramolecular conformational changes, the outside-in β2 integrin signaling, which is subsequently initiated by adhesion, automatically takes place. Adhesion is followed by polarization of the cell for subsequent directed cell migration or chemotaxis by recognition of chemoattractants such as chemokines, lipid mediators, or chemotactic peptides (158).
The process of chemotaxis can be divided in three separate events: sensing of the chemoattractant, polarization, and cell motility or migration. Here we describe 2D chemotaxis. In fact, chemotaxis in a 3D environment is mechanistically a very different process and can occur even without integrin-mediated adhesion (159). Upon perceiving chemoattractants, leukocytes typically invoke changes in the actin cytoskeleton and activate their β2 integrins as discussed previously. These cytoskeletal rearrangements initiate the second step of cell polarization (160, 161) as initiated by translocation of intracellular and extracellular proteins towards the leading edge of the cell (160, 161). In contrast to tissue cells, leukocytes migrate by forming pseudopods or lamellipodia, actin-filled protrusions important for cell movement at the leading edge of the cell (162). At the trailing end, cells form a uropod which serves as the anchoring point of the cell. For motility to occur, the pseudopod needs to move towards the chemoattractant, adheres with β2 integrins closer to the chemoattractant, and creates a strong interaction between the actin cytoskeleton and the β2 integrins. Subsequently, the uropod pulls the trailing end of the cell towards the new site of adhesion, which requires these strong interactions between the actin cytoskeleton and the β2 integrins. Meanwhile, the uropod also degrades the F-actin at the trailing end after this movement. Concluding, the turnover of β2 integrin interactions at the uropod is critical for proper regulation of motility [also reviewed in (160)].
Following adhesion, activated integrin induces tyrosine phosphorylation of SYK protein by SFKs, such as HCK, FGR, or LYN in neutrophils (101, 163) Activation of SYK results into provisional binding to CD18 molecule, which accordingly facilitates rearrangements of the actin cytoskeleton and subsequent neutrophil spreading (Figure 2A). In mice, neutrophil SYK deficiency resulted in defective adhesion-dependent responses, i.e. spreading, degranulation, and oxidative burst. Yet, neutrophil migration was not impaired in the absence of SYK, indicative of the variation of integrin signaling in each neutrophil response (101).
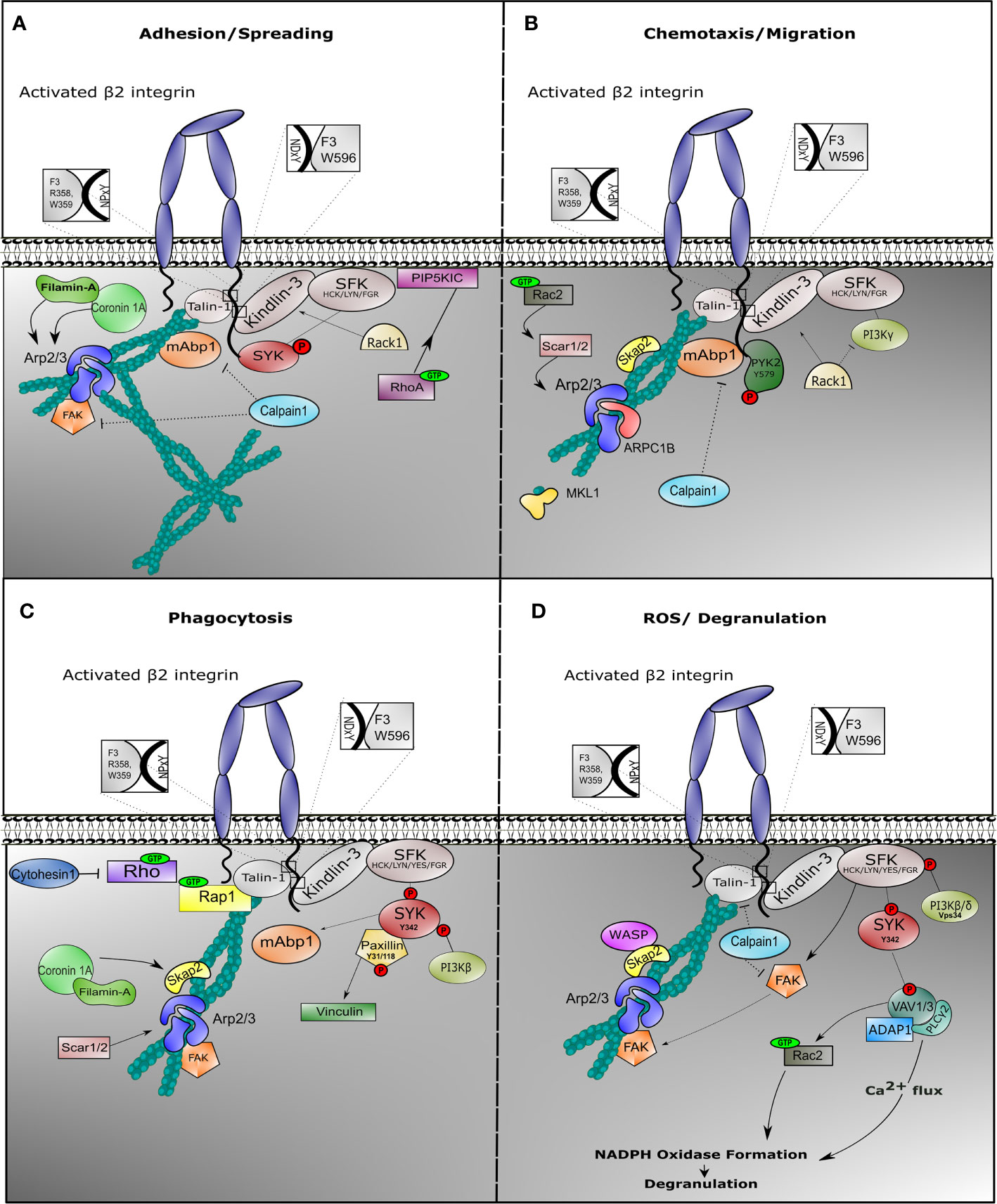
Figure 2 Outside-in signaling involves different protein complexes in response to neutrophil effector function. Once β2 integrin is activated on neutrophils, outside-in signaling is facilitated in order to induce cell effector function. Neutrophils are endowed with multiple mechanism, all achieving target elimination. In all cases, integrin outside-in signaling involves stable interaction with Talin-1 and Kindlin-3, which recruits binding of the actin cytoskeleton and induction of signaling via Sarcoma family kinases (SFKs). During spreading (A), SFKs phosphorylate and recruit SYK to the cytoplasmic tail of the β2 integrin, while Phosphatidylinositol 4-phosphate 5-kinase (PIP5KIC) induces activation of RhoA GTPase. Mitogen-activated binding protein 1 (mAbp1) is involved in stabilization of actin cytoskeleton, while Calpain-1 could play a role to negatively regulate its function. During migration (B), phosphorylated PYK2 is recruited and the Arp2/3 complex is stabilized via SKAP2 and Rac2 GTPase. Filamin A and ARPC1B are important for migration. We do not show WASP protein because deficiency results in impaired neutrophil migration in murine models only. Particularly during phagocytosis (C), phosphorylation of tyrosine protein kinase SYK induces binding of phosphorylated Paxillin with subsequent activation of Vinculin and activation of Phosphoinositide 3-kinase (PI3Kβ) and mAbp1. SCAR1/2 protein (also known as WAVE1/2) recruits Arp2/3 complex to stabilize actin cytoskeleton, while SKAP2 (or SKAP55R) functions as adaptor protein. During ROS production (D), degranulation is facilitated through increase of calcium influx. Phosphorylation of SYK initiates VAV phosphorylation with ADAP1 and PLCγ2 complex formation, promoting oxidative burst. VAV induces Rac1 GTPase activity, which facilitates NAPDH oxidase formation. In this context, Calpain1 could additionally inhibit Talin-1 or Focal adhesion kinase (FAK) function.
During β2-mediated polarization of myeloid HL-60 cells differentiated towards neutrophil-like cells (dHL-60), mAbp1 was enriched towards the cell’s leading edge, suggesting that mAbp1 is a positive regulator under β2 integrin-mediated chemotaxis by counteracting the more adhesion-strengthening signals, thus enabling cell motility (164, 165). HPK1, a protein colocalizing with mAbp1 and actin, was required for CXCL1-induced CD11a/CD18-mediated adhesion in dHL-60 cells, independent of CD11b/CD18 proteins (146). CD11a/CD18 is known to be endo- and exocytosed in a lipid-raft mediated coordination in the presence of chemokines in contrast to CD11b/CD18 (166), again supporting distinctive roles of CD11a/CD18 compared to CD11b/CD18 in the process of cell motility.
Filamin-A was found to be indispensable for dHL-60 spreading but was not essential for neutrophil migration (167). Although Filamin-A is shown to directly interact with Coronin-1A, deficiency of the actin-binding protein Coronin-1A resulted in reduced spreading and impaired chemotaxis, which indicates an independent role of these two proteins during neutrophil motility (96). This might be explained by the role of Coronin-1A in recruiting Arp2/3 complex to the F-actin polymerizing filament (168). Arp2/3 complex is essential to coordinate actin cytoskeleton rearrangement during chemotaxis, while Filamin A may act in more static conditions such as spreading and phagocytosis, and a negative regulator of motility (see below) (Figure 2B).
ARPC1B functions as a subunit of the Arp2/3 complex in hematopoietic cells, and its deficiency in neutrophils resulted in significant defective migration. Additional proteins have been shown to play a role in motility. For instance, human neutrophils lacking Megakaryoblastic leukemia 1 (MKL1) protein which normally sequesters monomeric G-actin, show normal adhesion under static conditions but impaired adhesion under flow conditions and defective chemotaxis (157).
SRC kinase-associated phosphoprotein 2 (SKAP2) and WASP, two adaptor proteins which are also linked to the rearrangements of the actin cytoskeleton, led to impaired neutrophil migration when absent in mice (169). However, neutrophils from WASP-deficient patients show no migratory defect, indicating that murine and human neutrophils may function differently in the context of migration (170). Defects in other cytoskeleton associated proteins also result in impaired neutrophil migratory functions. One such protein is Rac GTPase, a small guanine‐nucleotide binding protein, involved in cytoskeleton rearrangement through signaling via SCAR1/2 (also known as WAVE1/2) and Arp2/3 complex (171). Rac2 deficiency in human neutrophils leads to significantly impaired integrin-dependent chemotaxis (172).
Proline-rich tyrosine kinase 2 (PYK2) has a more complex activity. It is known to interact with cytoskeleton-interacting proteins. It associates with β2 integrin cytoplasmic tail upon fMLP stimulation (173) and was shown to contribute to migration in all-trans retinoic acid (ATRA)-differentiated NB4 myeloid cells (174). Pyk2 deficiency in murine model showed impaired neutrophil migration, with normal phagocytosis and killing (175). PYK2 regulation may be related to differential phosphorylation of PYK2 itself. This was observed for murine CD11a/CD18 during chemotaxis and motility where decreased migration was the result of a defect in the detachment from ICAM-1 at the trailing edge when PYK2 function was inhibited (176).
Although the above mentioned proteins are mainly important to positively regulate β2 integrin during motility, negative regulators have also been identified with the exception of the activated C kinase 1 receptor (RACK1). RACK1, a binding partner of Gβγ heterotrimeric subunits, inhibits PI3Kγ activity (177), and RACK1 deficiency showed enhanced neutrophil chemotaxis (177). Calpain-1 acts as a negative regulator of neutrophil chemotaxis downstream of GPCRs. Even though inhibition of Calpain-1 promoted random migration of resting neutrophils through MAPK and PI3K activation, directed neutrophil chemotaxis was defective in the absence of Calpain-1 due to the lack of directionality (178, 179).
Filamin A has clearly been found to bind to β2 integrin tails at a site which overlaps with Talin and Kindlin-3 (142), and its binding is regulated by phosphorylation of the conserved threonine triplet, where Filamin A can only bind to the unphosphorylated integrin. Filamin A is a large cytoskeletal protein which interacts with a multitude of intracellular partners and has been reported to function as an integrin negative regulator by competing with Talin-1 (180). Indeed, murine neutrophils lacking Filamin A display increased integrin-mediated adhesion of T cells (97), confirming that Filamin A indeed functions as a negative regulator of β2 integrins in these cells. Moreover, neutrophil extravasation in a mouse peritonitis model is affected negatively by both conditional knockout of Filamin A or Talin-1; thus correct interplay between positive and negative regulators is crucial for normal neutrophil function and behavior (63, 181).
Interestingly, we have also found that SHARPIN, which binds to integrin α-tails, can negatively regulate β2 integrin-mediated neutrophil adhesion (Khan, Fagerholm, unpublished observation), indicating that integrin binding proteins indeed can directly regulate integrin function in both positive and negative ways in neutrophils.
It is undeniable that neutrophil function requires a well-balanced equilibrium of positive and negative regulation, yet further proteins that negatively regulate neutrophil chemotaxis in a integrin-dependent manner have not been established (182). Although protein kinases such as SYK and SFKs mediate PMN activation through ITAM signaling domains, SFKs also activate ITIM domains to negatively regulate PMN signal transduction pathways. ITIM phosphorylation by SRC family kinases (HCK, FGR and LYN) recruits SH2 domain-containing phosphatase (SHP) 1 and 2 as well as SHIP 1 and 2 that dephosphorylate substrates of ITAM mediated activating signaling pathways in order to downregulate PMN functional responses. These ITIM containing immunoreceptors may play key roles in negative regulation, including chemotaxis. Well known is the effect of a point mutation in Shp-1 that reduces expression of SHP-1 protein by 90% and displays a hyper-adhesive phenotype of macrophages, contributing to neutrophil inflammation, mostly of the skin, in humans with a spontaneous mutation in the PTPN6 gene encoding SHP-1 (183, 184).
β2 Integrin Signaling in Neutrophil-Mediated Killing and Cytotoxicity
Phagocytosis
Phagocytosis is the uptake of particles by a cell. Here, we describe the uptake of pathogens by professional phagocytes of the immune system (i.e. neutrophils, monocytes, macrophages, and dendritic cells). All phagocytes have the capacity to eliminate invading pathogens by engulfment and degradation (185). Pathogens are often coated with a number of soluble opsonins, which are successfully recognized by phagocytes, including neutrophils (186). Upon recognition of pathogens at the plasma membrane, protrusions encircle the pathogen and draw it into the phagocyte. After engulfing the pathogen, the so-called phagosome fuses with granules in order to degrade the engulfed pathogen. Neutrophil phagocytosis is efficiently initiated upon IgG or iC3b binding to Fcγ receptor or complement receptors, respectively, including CD11b/CD18 (or CR3), while no role for the CD11a/CD18 integrin is described in phagocytosis (187). Earlier studies have primarily focused on macrophage phagocytosis, where it was suggested that Fcγ receptor and CD11b/CD18 integrin-related induction of phagocytosis are two distinct mechanisms (188) (Freeman et al., 2016). Referring to neutrophil phagocytosis, cells from LAD-I patients failed to phagocytose C4b-opsonized sheep erythrocytes or iC3b-opsonized S. aureus due to the specific lack of CD11b/CD18 integrin (189). In fact, CD11b/CD18 integrins and downstream associated proteins are important in both Fc receptor and CR3-induced phagocytosis (190, 191). The extracellular binding site of CD11b/CD18 (CR3) for iC3b is different from the binding site of cellular ligands (REF). In macrophage cell lines the mechanism of phagocytosis by CD11b/CD18 involves Arp2/3 complex formation for an efficient Talin-1-dependent actin-induced internalization of the pathogen. Whether a similar role of Arp2/3 and SYK in phagocytosis applies to human neutrophils is doubted. Neutrophil phagocytosis in patients deficient in ARPC1B or MKL1 is unaffected (192). Also, Arp2/3-inhibited control neutrophils did not show a defect in phagocytosis. We believe that these Arp2/3 processes in macrophages and neutrophils differ. We know that the importance of Rap1 GTPase in phagocytosis is highlighted in murine macrophage cell lines (193, 194) as well as in murine neutrophils, in which disruption of the Rap1/Talin-1 interaction abolishes phagocytosis of serum-opsonized E. coli (111, 195). The complementary function of Talin-1 and Kindlin-3 is also reflected in neutrophil phagocytosis, where LAD-III-derived neutrophils were unable to phagocytize zymosan particles (196) (Figure 2C).
SYK protein initiates integrin inside-out activation through activation of the Rap1/Talin-1 complex, while it is also involved in β2 integrin-induced phagocytosis stabilizing the outside-in signaling (132, 197). Phosphorylated SYK (phospho-Tyr342) colocalizes in macrophage phagosomes with phosphorylated Paxillin (phospho-Tyr31 and phosphor-Tyr118), which leads to the recruitment of Vinculin (198) and subsequent anchorage of F-actin to the membrane to facilitate target internalization. Yet, the above mentioned phosphorylation sites, somehow anchorage points for actin rearrangements, have not yet been verified during neutrophil effector functions.
The process of adhesion strengthening, spreading, or phagocytosis indicates a role of the cytoskeleton in which β2 integrins must provide together with additional proteins anchorage points for membrane rearrangements. In vitro studies have shown localization of Filamin A at the phagocytic cup during phagocytosis (167), but at least murine neutrophil phagocytosis can occur without Filamin A (97). Nonetheless, FLNA mutations identified thus far in humans have not indicated a major impact, suspect of a clear impact and clinical manifestations suggesting a LAD-like phenotype (199).
In contrast, the Filamin A-binding protein Coronin-1A was present only during late phagosome formation (167) and most is likely to be redundant and dispensable (97). CD11b/CD18-dependent particle ingestion required the presence of the Arp2/3 protein complex in murine macrophages (200, 201), but human neutrophils from ARPC1B-, MKL-1- or WASP-deficient patients could efficiently phagocytize and kill pathogens in vitro (157, 170, 192), indicating that involvement of Arp2/3 or actin-binding proteins in neutrophil phagocytosis is redundant.
The role of focal adhesion kinase (FAK), also known as cytoplasmic protein tyrosine kinase-2 (PTK2), is interesting in this respect. FAK has been explored in knockout mice, showing impaired phagocytosis of opsonized E. coli (202). FAK deficiency did not result in abnormal cell migration, implicating a role for this protein in stabilization of the phagocytic cup. Human studies on FAK in neutrophils and macrophages are lacking to date. Recently the role of SKAP2 has been described during murine neutrophil phagocytosis, showing impaired phagocytosis of E. coli particles by SKAP2-deficient neutrophils (169). The role of additional proteins in this process still needs to be determined. In this context, signaling molecules such as SYK and PYK2 are important kinases that may contribute to the upstream integrin-mediated signaling required to initiate or stabilize in conjunction with FAK, the actin-based spreading or particle uptake in macrophages or neutrophils.
Last, in contrast to neutrophil chemotaxis, neutrophil phagocytosis is promoted in the presence of Calpain-1 (203). Human neutrophils incubated with Cytohesin-1 inhibitor SecinH3 resulted in significantly increased phagocytosis of unopsonized zymosan (204). Cytohesins are a class of small GEFs for ADP-ribosylation factors (ARFs), which regulate cytoskeletal organization, integrin activation, or integrin signaling, being ubiquitously expressed. In neutrophils, Cytohesin-1, in contrast to the above mentioned proteins, seems to be a negative regulator of phagocytosis, maintaining the equilibrium in integrin regulation.
Antibody-Dependent Cellular Cytotoxicity
As mentioned, the presence of immunoglobulin can initiate Fc receptor activation and facilitate phagocytosis in an integrin-dependent way. When a target is too large to be phagocytized, neutrophils can exploit immunoglobulin binding to perform antibody-dependent cellular cytotoxicity (ADCC) (205). Besides antibody-opsonized bacteria and fungi, neutrophils can also kill antibody-opsonized covered targets such as cancer cells by extracellular toxicity (206, 207). Although Fcγ receptors have been traditionally invoked as the main receptors for antibody binding (208), also Fcα receptors are capable of initiating ADCC in human monocytes and neutrophils (209). Activation of these Fc receptor induces an intracellular signaling cascade with the immunoreceptor tyrosine-based activation motifs (ITAMs) of the Fc tail being phosphorylated, thus recruiting SYK protein binding, which eventually facilitates ADCC response. In ADCC of target cells, cell–cell interactions take place, gearing the response to a certain strength and outcome.
The β2 integrins are involved in ADCC either via inside-out or outside-in integrin regulation (210). CD11b/CD18 can become activated from Fcγ receptor IIa or IIIb through the recruitment and activation of PI3K. To date, the most solid evidence on β2 integrin and FcR involvement during ADCC has been collected from natural killer (NK) cells (211). Moreover, both CD11a/CD18 and CD11b/CD18 ligation to ICAM-1 positively regulates conjugate formation and cytolytic granule release (212, 213), whereas PD-1 signaling impairs outside-in integrin signaling and subsequent granule polarization (214). Yet, the killing effector function of NK cells substantially differs from neutrophils and macrophages, as it involves NK cell granule components.
We and others have clearly implicated the substantial role of CD11b/CD18 on neutrophils in ADCC against cancer cells (215, 216); however, current evidence on the integrin-associated ligands on tumor cells remains very limited. Although in vitro studies have shown that neutrophils adhere to melanoma cells via β2 integrin–ICAM-1 interactions (217), our recent findings have excluded ICAM-1, -2 or -3 as essential ligands for β2 integrin during neutrophil adhesion and cytotoxic activity in the case of breast cancer cells (218), indicating that the neutrophil interactions with different cell targets may vary in the use of counter/receptors or ligands.
ROS Production
Simultaneously with the process of phagosome formation, neutrophils consume an extreme amount of oxygen (respiratory burst), which enables them to produce reactive oxygen species (ROS) (219). The enzymatic machinery responsible for this function is the NADPH oxidase enzyme complex, which converts oxygen to superoxide and consequently hydrogen peroxide (220). As we have previously demonstrated, LAD-III syndrome causes impaired respiratory burst in response to zymosan, indicating a clear correlation between integrin activation and ROS production (196). These data are complementary with CD11b/CD18 deficient cells in mice, which resulted in defective respiratory burst (221). Integrin-mediated ROS induction (169) and Fcγ-receptor-mediated neutrophil activation required members of the VAV guanine nucleotide exchange factor and Rac small GTPase families (VAV1/3 and Rac2, respectively) (222, 223), and the PI3-kinase isoforms PI3Kβ and PI3Kδ instead of PI3Kγ, with a predominant role for PI3Kβ (224–226). SLP76/ADAP1 together with VAV isoforms (VAV1 and VAV3 in neutrophils) forms a complex with PLCγ2 which is also associated with immune complex-mediated signaling (222, 227, 228). VAV protein also functions as a Rac-GEF, enabling activation of Rac2 GTPase and thus inducing NADPH oxidase formation (229).
In an effort to further dissect the integrin-induced signaling cascade leading to NADPH oxidase in murine model systems, apart from the previously reported cascades, an indispensable role of the class III PI3K isoform (Vps34) was established in integrin-related ROS production in response to Escherichia coli or Staphylococcus aureus in neutrophils (Figure 2D). Vps34 is different from the above-mentioned class I PI3K isoforms (Figure 2D), and it is used for successful phagosome maturation and Mycobacterium tuberculosis destruction, while not essential for neutrophil chemotaxis or adhesion (230).
Outside-in signaling is important in adhesion-dependent responses so that neutrophils in suspension will not be able to perform, including degranulation and ROS production in case of TNF-induced effector function (231). Integrin outside-in signaling has been associated with neutrophil effector functions, including adhesion-dependent ROS production, which was dependent on SYK (101). Data on murine or human SFK-deficient neutrophils (lacking all three neutrophil-expressed SFK members, LYN, HCK and FGR) demonstrated the crucial role of SFKs during TNF-α-induced ROS production, accompanied by reduction of VAV protein phosphorylation in mice (232), which is a known regulator of oxidative burst (222).
Therapeutic Approaches
With the emerging role of neutrophils in health and disease and because β2 integrins play such an integral role in the cellular functions of this cell type as discussed above, targeting β2 integrins on neutrophils specifically with small molecules could prove fruitful as a therapy. There are two considerations to use integrin-modulating drugs in the context of neutrophil function. Neutrophil activity could be dampened or enhanced through blockade or induction of integrin activity, respectively. For example, decreasing neutrophil extravasation could provide major benefits to patients suffering from inflammatory bowel disease such as Crohn’s disease (233, 234), whereas LAD-III patients would benefit from an increase in neutrophil extravasation (80, 196, 235). One absolute requirement for such therapy in neutrophils would be the high specificity for the β2 integrin molecule due to the multifunctional nature of β2 integrins, as well as of other proteins involved in its downstream signaling cascade. The way of intervening neutrophil function by altering β2 integrin activity could vary. A large selection of small molecules has been shown to interact with CD11b/CD18, giving rise to various candidates to be further explored as therapeutics (236), and low dosage of such compounds might also already impose a sufficient response with limited side effects on neutrophils and other leukocytes.
Another interesting possibility is the use of antibody-based therapies. A recent study has shown the use of a monoclonal antibody, named anti-M7 that specifically disrupted the interaction between CD11b/CD18 and CD40L and not any other tested ligand. In blocking this CD40L-binding activity of CD11b/CD18, downregulation of pathological inflammation in mice could be accomplished while keeping other functions intact (237). In contrast to general anti-CD11b antibodies (238), this anti-M7 against the ligand-binding I-domain of the CD11b subunit ameliorated instead of aggravated sepsis. Anti-M7 in fact enhanced iC3b binding and phagocytosis while blocking binding to endothelial or platelet CD40L (237). Such ligand-specific antibodies might prove useful during acute inflammatory responses or sepsis. One possible downside to the usage of antibodies is their relatively long half-life and possible off-target effects on other leukocytes, but this might be altered by tempering with the antibody structure, isotype, or using nanobodies which have a much shorter half-life. Humanized monoclonal antibody against CD11a (Efalizumab) has been thoroughly studied in a Phase III clinical trial in psoriasis patients, but the drug has been withdrawn due to a significant risk of developing multifocal leukoencephalopathy (239, 240). Collectively, intervening with β2 integrin-mediated interactions seem to be a promising avenue in both decreasing neutrophil extravasation as well as trying to increase neutrophil phagocytic activity, albeit requiring very specific targeting, dosage, or both, which seems a very unique characteristic to explore in case of the CD40L binding sequence in the I-domain of CD11b (237).
Another possibility in regulating neutrophil integrin function is by modulating other proteins involved in either inside-out or outside-in signaling. This would allow for intervention in integrin activation and thus function, but also modulation of specific processes, such as phagocytosis or ROS production, while excluding or limiting the effects on other cellular processes. For inside-out signaling, Kindlin-3 might be a promising candidate. However, this would require small cell-permeable Kindlin-3-antagonists which specifically interfere with β2 integrin activation while preserving activation of other integrins. This effect might be achieved by obscuring binding between the threonine triplet in the CD18 cytoplasmic tail, since threonine into alanine mutations affect the Kindlin-3 interaction with CD18 in T cells while retaining partial activity (241). Notably, this motive is absent next to the second NXXY motif of other β integrins and could be a specific target for β2 integrins (242), thus avoiding bleeding, a side effect also seen in LAD-III patients. Recent work has also elucidated for the first time the crystal structure of Kindlin-3 (67), which might partially reside as an auto-inhibitory homotrimer. Relaxation or enforcement of this homotrimeric state might be another way in which inside-out signaling can be influenced either positively or negatively, respectively.
Aside from Kindlin-3, Cytohesin-1 is an interesting molecule as a drug target since its inhibition results into neutrophil pre-activation, although its function is also implicated in phagocytosis (204). Since this effect might stem from the relief of negative regulation by Cytohesin-1 on the Arf6 GTPase (98), exploring specifically Arf6 inhibitors or activators could also prove to be a valid therapy as well as expand our current knowledge on its role in integrin activation. Although targeting inside-out signaling shows potential, targeting outside-in signaling proteins solely involved in one cellular process could also be of interest to retain other cellular functions associated with β2 integrins. For example, targeting PI3Kδ could be sufficient to downregulate ROS production and degranulation, although having a minor role compared to PI3Kβ (224–226). Moreover, Pyk2 could be a good target to reduce chemotaxis and migration while keeping other functions intact (175, 176). In conclusion, intracellular proteins regulating integrin activity could be explored as potent therapeutic targets; however, taking into account the multifunctional roles of the proteins in neutrophil response, more studies should be performed to assure maximal efficacy with limited off-target effects.
As mentioned above, Calpain-1 is a negative regulator of integrin-dependent chemotaxis and migration by cleaving a number of integrin-associated proteins such as Talin-1 (243). Calpains are intracellular cysteine proteases with very diverse physiological roles. In vitro studies with human neutrophils and small inhibitory compounds have shown that Calpain-1 is also involved in the β2 integrin signaling cascade of adhesion-dependent TNFα-induced oxidative burst (244). The potential use of therapeutic Calpain-1 inhibitors to counter predictable and unwanted inflammatory reactivity in which neutrophils play a major role, has been explored (244). Yet, aberrant activity of calpains—whether it be over- or under-activation—is often associated with pathological functions and thus with diseases. Effective calpain inhibition may be achieved by peptide and peptidomimetic inhibitors developed to increase the specificity and potency. Since calpains constitute a family of homologous proteins ubiquitously expressed, highly specific inhibition seems problematic. Calpain inhibitors have demonstrated efficacy in animal models of calpain-related diseases, but progression of the inhibitors into clinical trials has been hampered partly due to the lack of calpain isoform selectivity of these inhibitors.
Worth mentioning is the alternative approach for targeting β2 integrins with ICAM-1 blocking antibodies. Studies in patients with partial-thickness burn injury using the anti-ICAM-1 antibody (Enlimomab) have shown promising modulation of the inflammatory response and improved wound healing (245). However, application of the drug has also shown several side effects. Additional ex vivo studies have shown that the concentration of the antibody used in the initial clinical trials could promote complement-dependent neutrophil activation (246). For the treatment of Crohn’s disease or ulcerative colitis, a 20-base antisense oligonucleotide inhibiting ICAM-1 production (Alicarfosen) showed potential clinical benefit (247, 248). Clinical remission rates compared to placebo and whether the response can be maintained in the long-term in larger studies are as yet unknown.
Discussion
We have reviewed and discussed most neutrophil functions requiring signaling through β2 integrins, either inside-out or outside-in integrin-dependent neutrophil activation. Although several proteins downstream of β2 integrins have been identified to play a role in phagocytosis, degranulation, or respiratory burst (Table 1), their exact role and sequential order or interdependence in integrin-mediated outside-in signaling cascade in killing and cytotoxicity remain to be determined. However, additional studies with innovative technologies are needed to develop a full picture of the CD11b/CD18 or CD11a/CD18 signaling cascades and their involvement in neutrophil effector function in humans.
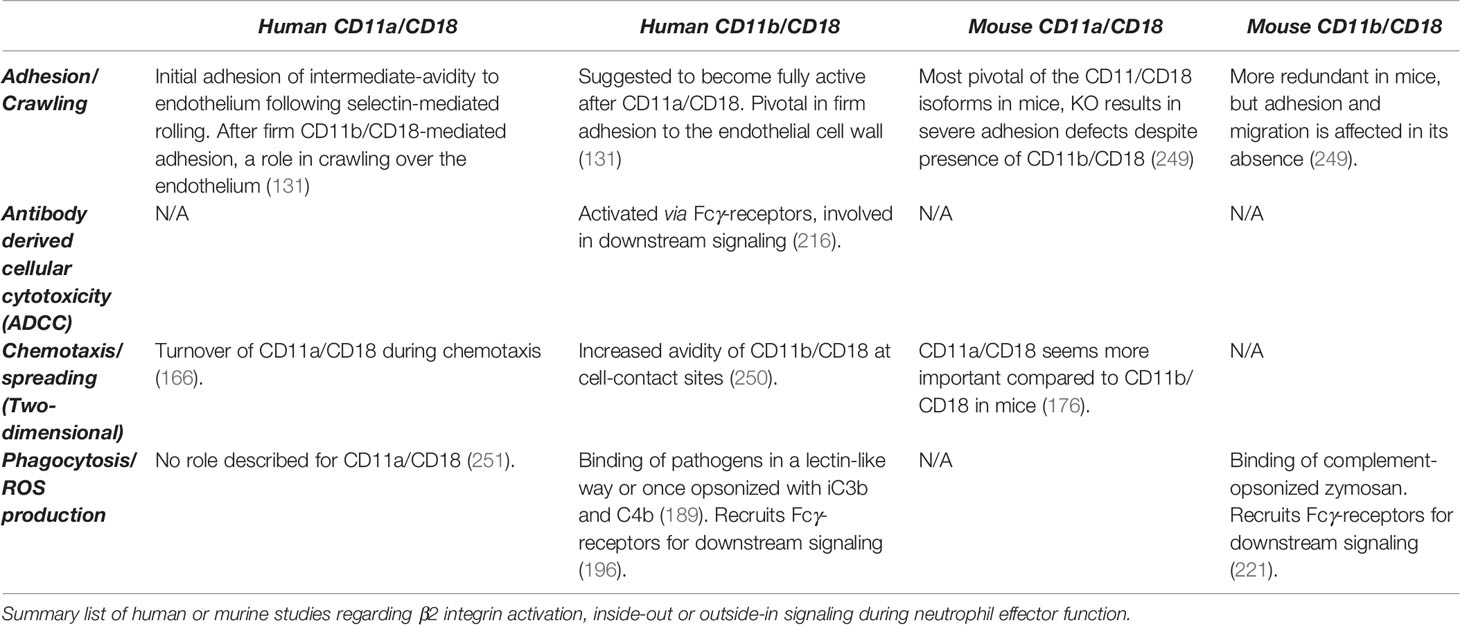
Table 1 Relevance of human and mouse CD11a/CD18 and CD11b/CD18 in neutrophil function.
The last decade, the technology of unbiased OMICS has improved our knowledge in cell biology. From transcriptomics to proteomics, we are now able to characterize RNA and protein expression levels in primary cells with an example of dissecting the composition of neutrophil cytoskeleton and the proteins associated with phagosome maturation (252, 253). In addition, large-scale studies include identification of signaling events and phosphorylation patterns after neutrophil stimulation, all of which have been nicely summarized elsewhere (254). Similar approaches have been followed in case of β1 adhesion receptors or intracellular signaling, where integrin-associated complexes have been identified by proteomic analysis (255–257). Recent methods focusing on interactome mapping based on proximity, such as Enhanced Ascorbate PeroXidase (APEX) or Biotin IDentification (BioID) have been used in order to anticipate function or downstream signaling cascade of β1, β4, and β5 integrins (258–262). Yet the complexity of leukocyte- associated β2 integrin has not allowed the investigation of its pathway with such method to date.
The increasing knowledge on the β2 interactome and integrin signaling in the context of neutrophil function allows it to be considered as a target for therapeutic intervention. Due to the short half-life of primary human neutrophils, research on β2 integrin-associated proteins upon activation is very limited. An alternative way to explore β2 integrin signaling is by exploiting neutrophil-like cell lines, such as HL-60 and NB4 cells. However, a considerable disadvantage of such cell lines is that they do not completely recapitulate neutrophil morphology or function, still unable to produce granules or migrate. Studies with these myeloid cell lines are useful for an initial orientation but will not be able to reveal the precise mechanism of signaling events in primary cells (263). Therefore, current efforts are focusing towards the generation of reproducible ways to generate neutrophils derived from induced pluripotent stem cells (iPSCs), being much closer to primary neutrophils (264).
In summary, we have discussed the role and signaling events of β2 integrins during neutrophil effector function. The relevance of this protein in this context is undisputed; however several questions remain unanswered. Although a detailed common pathway for extension of the β2 integrin is known, such a pathway for high-affinity activation of β2 integrins remains elusive; therefore we have summarized the current and most recent findings. We have also described differences regarding proteins involved with β2 integrins during different neutrophil functions such as adhesion, oxidative burst, phagocytosis or ADCC, highlighting the notion that β2 integrin outside-in signaling might include several signaling pathways, each distinctive per neutrophil effector response. Due to short neutrophil half-life, most studies have been focused on murine neutrophils, yet the differences compared to human neutrophils are substantial. To avoid possible confusion and emphasize the potential impact of each study, we have highlighted the data derived from mouse and human neutrophils. To conclude, usage of novel methods for both analysis and generation of neutrophils will be a key to further understanding the β2 integrin signaling. Such studies could lead to new therapeutic targets for modulating β2 integrin mediated neutrophil functions, or even leukocyte function in general.
Author Contributions
HM and TK were the principle investigators that designed the work. SF, RA, MM, HM, and TK supervised the work on behalf of the LADOMICs consortium. PB, SW and MM wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the Dutch Cancer Society [grant number KWF 11537] and eRARE [eRARE, LADOMICs JCT2018/ZonMW 90030376506], Academy of Finland, E-RARE (Academy of Finland), University of Helsinki (HiLife HIPOC), Swedish Cultural Foundation and Liv och Hälsa foundation and a grant of CIDA (Center of ImmunoDeficiencies Amsterdam).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are thankful to Sprenkeler Evelien for her help with the infographic.
Abbreviations
ADCC, Antibody-dependent cellular cytotoxicity; ADCP, Antibody-dependent cellular phagocytosis; EGF, Epidermal growth factor; ECM, Extracellular matrix; F-actin, Filamentous actin; FAK, Focal adhesion kinase; GPCRs, G-protein coupled receptors; HPK1, Hematopoietic progenitor kinase 1; dHL-60, HL-60 cell line differentiated towards neutrophil-like cells; ITAMs, Immunoreceptor tyrosine-based activation motifs; LAD, Leukocyte adhesion deficiency syndrome; fMLP, N-formylmethionine-leucyl-phenylalanine; NK cells, Natural killer cells; NETs, Neutrophil extracellular traps; PSI, Plexin–semaphorin–integrin; PI3K, Phosphoinositide 3-kinase; PMN, Polymorphonuclear; PKC, Protein kinase C; RACK-1, Receptor For Activated C Kinase 1; ROS, Reactive oxygen species; SYK, Spleen tyrosine kinase; TNFα, Tumor necrosis factor α; WASP, Wiskott–Aldrich syndrome protein.
References
1. Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol (2014) 9:181–218. doi: 10.1146/annurev-pathol-020712-164023
2. Sionov RV, Fridlender ZG, Granot Z. The Multifaceted Roles Neutrophils Play in the Tumor Microenvironment. Cancer Microenviron (2015) 8(3):125–58. doi: 10.1007/s12307-014-0147-5
3. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JA, et al. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood (2010) 116(4):625–7. doi: 10.1182/blood-2010-01-259028
4. Lawrence SM, Corriden R, Nizet V. The Ontogeny of a Neutrophil: Mechanisms of Granulopoiesis and Homeostasis. Microbiol Mol Biol Rev (2018) 82(1):e00057–17. doi: 10.1128/MMBR.00057-17
5. Mestas J, Hughes CC. Of mice and not men: differences between mouse and human immunology. J Immunol (2004) 172(5):2731–8. doi: 10.4049/jimmunol.172.5.2731
6. Liew PX, Kubes P. The Neutrophil’s Role During Health and Disease. Physiol Rev (2019) 99(2):1223–48. doi: 10.1152/physrev.00012.2018
7. Li Y, Wang W, Yang F, Xu Y, Feng C, Zhao Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun Signal (2019) 17(1):147. doi: 10.1186/s12964-019-0471-y
8. Papayannopoulos V. Neutrophils Stepping Through (to the Other Side). Immunity (2018) 49(6):992–4. doi: 10.1016/j.immuni.2018.12.006
9. Takashima A, Yao Y. Neutrophil plasticity: acquisition of phenotype and functionality of antigen-presenting cell. J Leukoc Biol (2015) 98(4):489–96. doi: 10.1189/jlb.1MR1014-502R
10. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol (2007) 7(9):678–89. doi: 10.1038/nri2156
11. Sadik CD, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol (2011) 32(10):452–60. doi: 10.1016/j.it.2011.06.008
12. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science (2004) 303(5663):1532–5. doi: 10.1126/science.1092385
13. Calderwood DA, Shattil SJ, Ginsberg MH. Integrins and actin filaments: reciprocal regulation of cell adhesion and signaling. J Biol Chem (2000) 275(30):22607–10. doi: 10.1074/jbc.R900037199
14. Bachmann M, Kukkurainen S, Hytonen VP, Wehrle-Haller B. Cell Adhesion by Integrins. Physiol Rev (2019) 99(4):1655–99. doi: 10.1152/physrev.00036.2018
15. Lenter M, Vestweber D. The integrin chains beta 1 and alpha 6 associate with the chaperone calnexin prior to integrin assembly. J Biol Chem (1994) 269(16):12263–8. doi: 10.1016/S0021-9258(17)32710-2
16. Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol (2011) 3(3):a004994. doi: 10.1101/cshperspect.a004994
17. Gailit J, Ruoslahti E. Regulation of the fibronectin receptor affinity by divalent cations. J Biol Chem (1988) 263(26):12927–32. doi: 10.1016/S0021-9258(18)37650-6
18. Podolnikova NP, Podolnikova AV, Haas TA, Lishko VK, Ugarova TP. Ligand recognition specificity of leukocyte integrin alphaMbeta2 (Mac-1, CD11b/CD18) and its functional consequences. Biochemistry (2015) 54(6):1408–20. doi: 10.1021/bi5013782
19. Lee JO, Bankston LA, Arnaout MA, Liddington RC. Two conformations of the integrin A-domain (I-domain): a pathway for activation? Structure (1995) 3(12):1333–40. doi: 10.1016/S0969-2126(01)00271-4
20. Humphries JD, Byron A, Humphries MJ. Integrin ligands at a glance. J Cell Sci (2006) 119(Pt 19):3901–3. doi: 10.1242/jcs.03098
21. Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell (2008) 32(6):849–61. doi: 10.1016/j.molcel.2008.11.018
22. Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity (2006) 25(4):583–94. doi: 10.1016/j.immuni.2006.07.016
23. Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell (2002) 110(5):599–11. doi: 10.1016/S0092-8674(02)00935-2
24. Li J, Springer TA. Energy landscape differences among integrins establish the framework for understanding activation. J Cell Biol (2018) 217(1):397–412. doi: 10.1083/jcb.201701169
25. Li J, Su Y, Xia W, Qin Y, Humphries J, Vestweber D, et al. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J (2017) 36(5):629–45. doi: 10.15252/embj.201695803
26. Shimaoka M, Xiao T, Liu JH, Yang Y, Dong Y, Jun CD, et al. Structures of the alpha L I domain and its complex with ICAM-1 reveal a shape-shifting pathway for integrin regulation. Cell (2003) 112(1):99–111. doi: 10.1016/S0092-8674(02)01257-6
27. Ginsberg MH. Integrin activation. BMB Rep (2014) 47(12):655–9. doi: 10.5483/BMBRep.2014.47.12.241
28. Lau TL, Kim C, Ginsberg MH, Ulmer TS. The structure of the integrin alphaIIbbeta3 transmembrane complex explains integrin transmembrane signalling. EMBO J (2009) 28(9):1351–61. doi: 10.1038/emboj.2009.63
29. Kim C, Ye F, Ginsberg MH. Regulation of integrin activation. Annu Rev Cell Dev Biol (2011) 27:321–45. doi: 10.1146/annurev-cellbio-100109-104104
30. Legate KR, Fassler R. Mechanisms that regulate adaptor binding to beta-integrin cytoplasmic tails. J Cell Sci (2009) 122(Pt 2):187–98. doi: 10.1242/jcs.041624
31. Moser M, Bauer M, Schmidt S, Ruppert R, Schmidt S, Sixt M, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med (2009) 15(3):300–5. doi: 10.1038/nm.1921
32. Calderwood DA, Campbell ID, Critchley DR. Talins and kindlins: partners in integrin-mediated adhesion. Nat Rev Mol Cell Biol (2013) 14(8):503–17. doi: 10.1038/nrm3624
33. Barczyk M, Carracedo S, Gullberg D. Integrins. Cell Tissue Res (2010) 339(1):269–80. doi: 10.1007/s00441-009-0834-6
34. Vinogradova O, Haas T, Plow EF, Qin J. A structural basis for integrin activation by the cytoplasmic tail of the alpha IIb-subunit. Proc Natl Acad Sci USA (2000) 97(4):1450–5. doi: 10.1073/pnas.040548197
35. Pouwels J, De Franceschi N, Rantakari P, Auvinen K, Karikoski M, Mattila E, et al. SHARPIN regulates uropod detachment in migrating lymphocytes. Cell Rep (2013) 5(3):619–28. doi: 10.1016/j.celrep.2013.10.011
36. Fagerholm SC, Varis M, Stefanidakis M, Hilden TJ, Gahmberg CG. alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood (2006) 108(10):3379–86. doi: 10.1182/blood-2006-03-013557
37. Uotila LM, Aatonen M, Gahmberg CG. Integrin CD11c/CD18 alpha-chain phosphorylation is functionally important. J Biol Chem (2013) 288(46):33494–9. doi: 10.1074/jbc.C113.497446
38. Harburger DS, Calderwood DA. Integrin signalling at a glance. J Cell Sci (2009) 122(Pt 2):159–63. doi: 10.1242/jcs.018093
39. Sun Z, Costell M, Fassler R. Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol (2019) 21(1):25–31. doi: 10.1038/s41556-018-0234-9
40. Klapholz B, Brown NH. Talin - the master of integrin adhesions. J Cell Sci (2017) 130(15):2435–46. doi: 10.1242/jcs.190991
41. Garcia-Alvarez B, de Pereda JM, Calderwood DA, Ulmer TS, Critchley D, Campbell ID, et al. Structural determinants of integrin recognition by talin. Mol Cell (2003) 11(1):49–58. doi: 10.1016/S1097-2765(02)00823-7
42. Goult BT, Yan J, Schwartz MA. Talin as a mechanosensitive signaling hub. J Cell Biol (2018) 217(11):3776–84. doi: 10.1083/jcb.201808061
43. Hemmings L, Rees DJ, Ohanian V, Bolton SJ, Gilmore AP, Patel B, et al. Talin contains three actin-binding sites each of which is adjacent to a vinculin-binding site. J Cell Sci (1996) 109( Pt 11):2715–26.
44. Atherton P, Stutchbury B, Wang DY, Jethwa D, Tsang R, Meiler-Rodriguez E, et al. Vinculin controls talin engagement with the actomyosin machinery. Nat Commun (2015) 6:10038. doi: 10.1038/ncomms10038
45. Kumar A, Ouyang M, Van den Dries K, McGhee EJ, Tanaka K, Anderson MD, et al. Correction: Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J Cell Biol (2016) 214(2):231. doi: 10.1083/jcb.20151001207062016c
46. Kumar A, Ouyang M, Van den Dries K, McGhee EJ, Tanaka K, Anderson MD, et al. Talin tension sensor reveals novel features of focal adhesion force transmission and mechanosensitivity. J Cell Biol (2016) 213(3):371–83. doi: 10.1083/jcb.201510012
47. Yao M, Goult M, Klapholz B, Hu X, Toseland CP, Guo Y, et al. The mechanical response of talin. Nat Commun (2016) 7:11966. doi: 10.1038/ncomms11966
48. del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Stretching single talin rod molecules activates vinculin binding. Science (2009) 323(5914):638–41. doi: 10.1126/science.1162912
49. Goksoy E, Ma YQ, Wang X, Kong X, Perera D, Plow EF, et al. Structural basis for the autoinhibition of talin in regulating integrin activation. Mol Cell (2008) 31(1):124–33. doi: 10.1016/j.molcel.2008.06.011
50. Dedden D, Schumacher S, Kelley CF, Zacharias M, Biertumpfel C, Fassler R, et al. The Architecture of Talin1 Reveals an Autoinhibition Mechanism. Cell (2019) 179(1):120–31.e13. doi: 10.1016/j.cell.2019.08.034
51. Goult BT, Bouaouina M, Elliott PR, Bate N, Patel B, Gingras AR, et al. Structure of a double ubiquitin-like domain in the talin head: a role in integrin activation. EMBO J (2010) 29(6):1069–80. doi: 10.1038/emboj.2010.4
52. Song X, Yang J, Hirbawi J, Ye S, Perera HD, Goksoy E, et al. A novel membrane-dependent on/off switch mechanism of talin FERM domain at sites of cell adhesion. Cell Res (2012) 22(11):1533–45. doi: 10.1038/cr.2012.97
53. Sun Z, Guo SS, Fassler R. Integrin-mediated mechanotransduction. J Cell Biol (2016) 215(4):445–56. doi: 10.1083/jcb.201609037
54. Lee HS, Anekal P, Lim CJ, Liu CC, Ginsberg MH. Two modes of integrin activation form a binary molecular switch in adhesion maturation. Mol Biol Cell (2013) 24(9):1354–62. doi: 10.1091/mbc.e12-09-0695
55. Yang J, Zhu L, Zhang H, Hirbawi J, Fukuda K, Dwivedi P, et al. Conformational activation of talin by RIAM triggers integrin-mediated cell adhesion. Nat Commun (2014) 5:5880. doi: 10.1038/ncomms6880
56. Ussar S, Wang HV, Linder S, Fassler R, Moser M. The Kindlins: subcellular localization and expression during murine development. Exp Cell Res (2006) 312(16):3142–51. doi: 10.1016/j.yexcr.2006.06.030
57. Rognoni E, Ruppert R, Fassler R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci (2016) 129(1):17–27. doi: 10.1242/jcs.161190
58. Bouaouina M, Goult BT, Huet-Calderwood C, Bate N, Brahme NN, Barsukov IL, et al. A conserved lipid-binding loop in the kindlin FERM F1 domain is required for kindlin-mediated alphaIIbbeta3 integrin coactivation. J Biol Chem (2012) 287(10):6979–90. doi: 10.1074/jbc.M111.330845
59. Wen L, Marki A, Roy P, McArdle S, Sun H, Fan Z, et al. Kindlin-3 recruitment to the plasma membrane precedes high affinity beta2 integrin and neutrophil arrest from rolling. Blood (2020). doi: 10.1182/blood.2019003446
60. Ma YQ, Qin J, Wu C, Plow EF. Kindlin-2 (Mig-2): a co-activator of beta3 integrins. J Cell Biol (2008) 181(3):439–46. doi: 10.1083/jcb.200710196
61. Moser M, Nieswandt B, Ussar S, Pozgajova R, Fassler R. Kindlin-3 is essential for integrin activation and platelet aggregation. Nat Med (2008) 14(3):325–30. doi: 10.1038/nm1722
62. Jahed Z, Haydari Z, Rathish A, Mofrad MRK. Kindlin Is Mechanosensitive: Force-Induced Conformational Switch Mediates Cross-Talk among Integrins. Biophys J (2019) 116(6):1011–24. doi: 10.1016/j.bpj.2019.01.038
63. Lefort CT, Rossaint J, Moser M, Petrich BG, Zarbock A, Monkley SJ, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood (2012) 119(18):4275–82. doi: 10.1182/blood-2011-08-373118
64. Ye F, Petrich BG, Anekal P, Lefort CT, Kasirer-Friede A, Shattil SJ, et al. The mechanism of kindlin-mediated activation of integrin alphaIIbbeta3. Curr Biol (2013) 23(22):2288–95. doi: 10.1016/j.cub.2013.09.050
65. Li H, Deng Y, Sun K, Yang H, Liu J, Wang M, et al. Structural basis of kindlin-mediated integrin recognition and activation. Proc Natl Acad Sci U S A (2017) 114(35):9349–54. doi: 10.1073/pnas.1703064114
66. Kadry YA, Maisuria EM, Huet-Calderwood C, Calderwood DA. Differences in self-association between kindlin-2 and kindlin-3 are associated with differential integrin binding. J Biol Chem (2020) 295(32):11161–73. doi: 10.1074/jbc.RA120.013618
67. Bu W, Levitskaya Z, Loh ZY, Jin S, Basu S, Ero R, et al. Structural basis of human full-length kindlin-3 homotrimer in an auto-inhibited state. PloS Biol (2020) 18(7):e3000755. doi: 10.1371/journal.pbio.3000755
68. Theodosiou M, Widmaier M, Bottcher RT, Rognoni E, Veelders M, Bharadwaj M, et al. Kindlin-2 cooperates with talin to activate integrins and induces cell spreading by directly binding paxillin. Elife (2016) 5:e10130. doi: 10.7554/eLife.10130
69. Gao J, Huang M, Lai J, Mao K, Sun P, Cao Z, et al. Kindlin supports platelet integrin alphaIIbbeta3 activation by interacting with paxillin. J Cell Sci (2017) 130(21):3764–75. doi: 10.1242/jcs.205641
70. Klapproth S, Bromberger T, Turk C, Kruger M, Moser M. A kindlin-3-leupaxin-paxillin signaling pathway regulates podosome stability. J Cell Biol (2019) 218(10):3436–54. doi: 10.1083/jcb.201903109
71. Fukuda K, Bledzka K, Yang J, Perera HD, Plow EF, Qin J. Molecular basis of kindlin-2 binding to integrin-linked kinase pseudokinase for regulating cell adhesion. J Biol Chem (2014) 289(41):28363–75. doi: 10.1074/jbc.M114.596692
72. Huet-Calderwood C, Brahme NN, Kumar N, Stiegler AL, Raghavan S, Boggon TJ, et al. Differences in binding to the ILK complex determines kindlin isoform adhesion localization and integrin activation. J Cell Sci (2014) 127(Pt 19):4308–21. doi: 10.1242/jcs.155879
73. Bledzka K, Bialkowska K, Sossey-Alaoui K, Vaynberg J, Pluskota E, Qin J, et al. Kindlin-2 directly binds actin and regulates integrin outside-in signaling. J Cell Biol (2016) 213(1):97–108. doi: 10.1083/jcb.201501006
74. Danen E. An Overview of Structural and Functional Aspects. In: Madame Curie Bioscience Database. Landes Bioscience (2013).
75. Tan SM. The leucocyte beta2 (CD18) integrins: the structure, functional regulation and signalling properties. Biosci Rep (2012) 32(3):241–69. doi: 10.1042/BSR20110101
76. Shattil SJ, Kim C, Ginsberg MH. The final steps of integrin activation: the end game. Nat Rev Mol Cell Biol (2010) 11(4):288–300. doi: 10.1038/nrm2871
77. Spiess M, Hernandez-Varas P, Olofsson A, Oddone H, Blom H, Waithe D, et al. Active and inactive beta1 integrins segregate into distinct nanoclusters in focal adhesions. J Cell Biol (2018) 217(6):1929–40. doi: 10.1083/jcb.201707075
78. van de Vijver E, van den Berg TK, Kuijpers TW. Leukocyte adhesion deficiencies. Hematol Oncol Clin North Am (2013) 27(1):101–16. doi: 10.1016/j.hoc.2012.10.001
79. Malinin NL, Zhang L, Choi J, Ciocea A, Razorenova O, Ma YQ, et al. A point mutation in KINDLIN3 ablates activation of three integrin subfamilies in humans. Nat Med (2009) 15(3):313–8. doi: 10.1038/nm.1917
80. Kuijpers TW, van de Vijver E, Weterman MA, de Boer M, Tool AT, van den Berg TK, et al. LAD-1/variant syndrome is caused by mutations in FERMT3. Blood (2009) 113(19):4740–6. doi: 10.1182/blood-2008-10-182154
81. Svensson L, Howarth K, McDowall A, Patzak I, Evans R, Ussar S, et al. Leukocyte adhesion deficiency-III is caused by mutations in KINDLIN3 affecting integrin activation. Nat Med (2009) 15(3):306–12. doi: 10.1038/nm.1931
82. van de Vijver E, Maddalena A, Sanal O, Holland SM, Uzel G, Madkaikar M, et al. Hematologically important mutations: leukocyte adhesion deficiency (first update). Blood Cells Mol Dis (2012) 48(1):53–61. doi: 10.1016/j.bcmd.2011.10.004
83. Moutsopoulos NM, Konkel J, Sarmadi M, Eskan MA, Wild T, Dutzan N, et al. Defective neutrophil recruitment in leukocyte adhesion deficiency type I disease causes local IL-17-driven inflammatory bone loss. Sci Transl Med (2014) 6(229):229ra40. doi: 10.1126/scitranslmed.3007696
84. Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol (2009) 27:339–62. doi: 10.1146/annurev.immunol.021908.132554
85. Kahner BN, Kato H, Banno A, Ginsberg MH, Shattil SJ, Ye F. Kindlins, integrin activation and the regulation of talin recruitment to alphaIIbbeta3. PloS One (2012) 7(3):e34056. doi: 10.1371/journal.pone.0034056
86. Hyduk SJ, Rullo J, Cano AP, Xiao H, Chen M, Moser M, et al. Talin-1 and kindlin-3 regulate alpha4beta1 integrin-mediated adhesion stabilization, but not G protein-coupled receptor-induced affinity upregulation. J Immunol (2011) 187(8):4360–8. doi: 10.4049/jimmunol.1003725
87. Kadry YA, Calderwood DA. Chapter 22: Structural and signaling functions of integrins. Biochim Biophys Acta Biomembr (2020) 1862(5):183206. doi: 10.1016/j.bbamem.2020.183206
88. Paszek MJ, Boettiger D, Weaver VM, Hammer DA. Integrin clustering is driven by mechanical resistance from the glycocalyx and the substrate. PloS Comput Biol (2009) 5(12):e1000604. doi: 10.1371/journal.pcbi.1000604
89. Roca-Cusachs P, Gauthier NC, Del Rio A, Sheetz MP. Clustering of alpha(5)beta(1) integrins determines adhesion strength whereas alpha(v)beta(3) and talin enable mechanotransduction. Proc Natl Acad Sci USA (2009) 106(38):16245–50. doi: 10.1073/pnas.0902818106
90. Welf ES, Naik UP, Ogunnaike BA. A spatial model for integrin clustering as a result of feedback between integrin activation and integrin binding. Biophys J (2012) 103(6):1379–89. doi: 10.1016/j.bpj.2012.08.021
91. Dong JM, Tay FP, Swa HL, Gunaratne J, Leung T, Burke B, et al. Proximity biotinylation provides insight into the molecular composition of focal adhesions at the nanometer scale. Sci Signal (2016) 9(432):rs4. doi: 10.1126/scisignal.aaf3572
92. Thome S, Begandt D, Pick R, Salvermoser M, Walzog B. Intracellular beta2 integrin (CD11/CD18) interacting partners in neutrophil trafficking. Eur J Clin Invest (2018) 48 Suppl 2:e12966. doi: 10.1111/eci.12966
93. Sun H, Fan Z, Gingras AR, Lopez-Ramirez MA, Ginsberg MH, Ley K. Frontline Science: A flexible kink in the transmembrane domain impairs beta2 integrin extension and cell arrest from rolling. J Leukoc Biol (2020) 107(2):175–83. doi: 10.1002/JLB.1HI0219-073RR
94. Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front Immunol (2019) 10:254. doi: 10.3389/fimmu.2019.00254
95. Green HJ, Brown NH. Integrin intracellular machinery in action. Exp Cell Res (2019) 378(2):226–31. doi: 10.1016/j.yexcr.2019.03.011
96. Pick R, Begandt D, Stocker TJ, Salvermoser M, Thome S, Bottcher RT, et al. Coronin 1A, a novel player in integrin biology, controls neutrophil trafficking in innate immunity. Blood (2017) 130(7):847–58. doi: 10.1182/blood-2016-11-749622
97. Uotila LM, Guenther C, Savinko T, Lehti TA, Fagerholm SC. Filamin A Regulates Neutrophil Adhesion, Production of Reactive Oxygen Species, and Neutrophil Extracellular Trap Release. J Immunol (2017) 199(10):3644–53. doi: 10.4049/jimmunol.1700087
98. Weber KS, Weber C, Ostermann G, Dierks H, Nagel W, Kolanus W. Cytohesin-1 is a dynamic regulator of distinct LFA-1 functions in leukocyte arrest and transmigration triggered by chemokines. Curr Biol (2001) 11(24):1969–74. doi: 10.1016/S0960-9822(01)00597-8
99. El azreq MA, Bourgoin SG. Cytohesin-1 regulates human blood neutrophil adhesion to endothelial cells through beta2 integrin activation. Mol Immunol (2011) 48(12-13):1408–16. doi: 10.1016/j.molimm.2011.03.018
100. Woodside DG, Obergfell A, Talapatra A, Calderwood DA, Shattil SJ, Ginsberg MH. The N-terminal SH2 domains of Syk and ZAP-70 mediate phosphotyrosine-independent binding to integrin beta cytoplasmic domains. J Biol Chem (2002) 277(42):39401–8. doi: 10.1074/jbc.M207657200
101. Mocsai A, Zhou M, Meng F, Tybulewicz VL, Lowell CA. Syk is required for integrin signaling in neutrophils. Immunity (2002) 16(4):547–58. doi: 10.1016/S1074-7613(02)00303-5
102. Wu D, LaRosa GJ, Simon MI. G protein-coupled signal transduction pathways for interleukin-8. Science (1993) 261(5117):101–3. doi: 10.1126/science.8316840
103. Takami M, Terry V, Petruzzelli L. Signaling pathways involved in IL-8-dependent activation of adhesion through Mac-1. J Immunol (2002) 168(9):4559–66. doi: 10.4049/jimmunol.168.9.4559
104. Yago T, Zhang N, Zhao L, Abrams CS, McEver RP. Selectins and chemokines use shared and distinct signals to activate beta2 integrins in neutrophils. Blood Adv (2018) 2(7):731–44. doi: 10.1182/bloodadvances.2017015602
105. Cook AA, Deng W, Ren J, Sondek J, Bergmeier W. Calcium-induced structural rearrangements release autoinhibition in the Rap-GEF CalDAG-GEFI. J Biol Chem (2018) 293(22):8521–9. doi: 10.1074/jbc.RA118.002712
106. Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science (2000) 287(5455):1046–9. doi: 10.1126/science.287.5455.1046
107. L’Heureux GP, Bourgoin S, Jean N, McColl SR, Naccache PH. Diverging signal transduction pathways activated by interleukin-8 and related chemokines in human neutrophils: interleukin-8, but not NAP-2 or GRO alpha, stimulates phospholipase D activity. Blood (1995) 85(2):522–31. doi: 10.1182/blood.V85.2.522.bloodjournal852522
108. Lee HS, Lim CJ, Puzon-McLaughlin W, Shattil SJ, Ginsberg MH. RIAM activates integrins by linking talin to ras GTPase membrane-targeting sequences. J Biol Chem (2009) 284(8):5119–27. doi: 10.1074/jbc.M807117200
109. Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science (2003) 302(5642):103–6. doi: 10.1126/science.1086652
110. Medrano-Fernandez I, Reyes R, Olazabal I, Rodriguez E, Sanchez-Madrid F, Boussiotis VA, et al. RIAM (Rap1-interacting adaptor molecule) regulates complement-dependent phagocytosis. Cell Mol Life Sci (2013) 70(13):2395–410. doi: 10.1007/s00018-013-1268-6
111. Bromberger T, Klapproth S, Rohwedder I, Zhu L, Mittmann L, Reichel CA, et al. Direct Rap1/Talin1 interaction regulates platelet and neutrophil integrin activity in mice. Blood (2018) 132(26):2754–62. doi: 10.1182/blood-2018-04-846766
112. Bromberger T, Zhu L, Klapproth S, Qin J, Moser M. Rap1 and membrane lipids cooperatively recruit talin to trigger integrin activation. J Cell Sci (2019) 132(21):2754–62. doi: 10.1242/jcs.235531
113. Zhu L, Yang J, Bromberger T, Holly A, Lu F, Liu H, et al. Structure of Rap1b bound to talin reveals a pathway for triggering integrin activation. Nat Commun (2017) 8(1):1744. doi: 10.1038/s41467-017-01822-8
114. Chang LC, Huang TH, Chang CS, Tsai YR, Lin RH, Lee PW, et al. Signaling mechanisms of inhibition of phospholipase D activation by CHS-111 in formyl peptide-stimulated neutrophils. Biochem Pharmacol (2011) 81(2):269–78. doi: 10.1016/j.bcp.2010.10.007
115. Olson SC, Lambeth JD. Biochemistry and cell biology of phospholipase D in human neutrophils. Chem Phys Lipids (1996) 80(1-2):3–19. doi: 10.1016/0009-3084(96)02541-8
116. Honda A, Nogami M, Yokozeki T, Yamazaki M, Nakamura H, Watanabe H, et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell (1999) 99(5):521–32. doi: 10.1016/S0092-8674(00)81540-8
117. Iyer SS, Agrawal RS, Thompson CR, Thompson S, Barton JA, Kusner DJ. Phospholipase D1 regulates phagocyte adhesion. J Immunol (2006) 176(6):3686–96. doi: 10.4049/jimmunol.176.6.3686
118. Ye X, McLean MA, Sligar SG. Phosphatidylinositol 4,5-Bisphosphate Modulates the Affinity of Talin-1 for Phospholipid Bilayers and Activates Its Autoinhibited Form. Biochemistry (2016) 55(36):5038–48. doi: 10.1021/acs.biochem.6b00497
119. Hart R, Stanley P, Chakravarty P, Hogg N. The kindlin 3 pleckstrin homology domain has an essential role in lymphocyte function-associated antigen 1 (LFA-1) integrin-mediated B cell adhesion and migration. J Biol Chem (2013) 288(21):14852–62. doi: 10.1074/jbc.M112.434621
120. Ni T, Kalli AC, Naughton FB, Yates LA, Naneh O, Kozorog M, et al. Structure and lipid-binding properties of the kindlin-3 pleckstrin homology domain. Biochem J (2017) 474(4):539–56. doi: 10.1042/BCJ20160791
121. Knall C, Worthen GS, Johnson GL. Interleukin 8-stimulated phosphatidylinositol-3-kinase activity regulates the migration of human neutrophils independent of extracellular signal-regulated kinase and p38 mitogen-activated protein kinases. Proc Natl Acad Sci USA (1997) 94(7):3052–7. doi: 10.1073/pnas.94.7.3052
122. Margraf A, Germena G, Drexler HCA, Rossaint J, Ludwig N, Prystaj B, et al. The integrin linked kinase is required for chemokine-triggered highaffinity conformation of neutrophil beta2-integrin LFA1. Blood (2020) 136(19):2200–5. doi: 10.1182/blood.2020004948
123. Damaj BB, McColl SR, Neote K, Hebert CA, Naccache PH. Diverging signal transduction pathways activated by interleukin 8 (IL-8) and related chemokines in human neutrophils. IL-8 and Gro-alpha differentially stimulate calcium influx through IL-8 receptors A and B. J Biol Chem (1996) 271(34):20540–4. doi: 10.1074/jbc.271.34.20540
124. Lindemann O, Rossaint J, Najder K, Schimmelpfennig S, Hofschroer V, Walte M, et al. Intravascular adhesion and recruitment of neutrophils in response to CXCL1 depends on their TRPC6 channels. J Mol Med (Berl) (2020) 98(3):349–60. doi: 10.1007/s00109-020-01872-4
125. Latasiewicz J, Artz A, Jing D, Blanco MP, Currie SM, Avila MV, et al. HS1 deficiency impairs neutrophil recruitment in vivo and activation of the small GTPases Rac1 and Rap1. J Leukoc Biol (2017) 101(5):1133–42. doi: 10.1189/jlb.1A0416-195R
126. Knall C, Young S, Nick JA, Buhl AM, Worthen GS, Johnson GL. Interleukin-8 regulation of the Ras/Raf/mitogen-activated protein kinase pathway in human neutrophils. J Biol Chem (1996) 271(5):2832–8. doi: 10.1074/jbc.271.5.2832
127. Geiger C, Nagel W, Boehm T, van Kooyk Y, Figdor CG, Kremmer E, et al. Cytohesin-1 regulates beta-2 integrin-mediated adhesion through both ARF-GEF function and interaction with LFA-1. EMBO J (2000) 19(11):2525–36. doi: 10.1093/emboj/19.11.2525
128. Kolanus W, Nagel W, Schiller B, Zeitlmann L, Godar S, Stockinger H, et al. Alpha L beta 2 integrin/LFA-1 binding to ICAM-1 induced by cytohesin-1, a cytoplasmic regulatory molecule. Cell (1996) 86(2):233–42. doi: 10.1016/S0092-8674(00)80095-1
129. Hmama Z, Knutson KL, Herrera-Velit P, Nandan D, Reiner NE. Monocyte adherence induced by lipopolysaccharide involves CD14, LFA-1, and cytohesin-1. Regulation by Rho and phosphatidylinositol 3-kinase. J Biol Chem (1999) 274(2):1050–7. doi: 10.1074/jbc.274.2.1050
130. Fagerholm SC, Hilden TJ, Nurmi SM, Gahmberg CG. Specific integrin alpha and beta chain phosphorylations regulate LFA-1 activation through affinity-dependent and -independent mechanisms. J Cell Biol (2005) 171(4):705–15. doi: 10.1083/jcb.200504016
131. Neelamegham S, Taylor AD, Burns AR, Smith CW, Simon SI. Hydrodynamic shear shows distinct roles for LFA-1 and Mac-1 in neutrophil adhesion to intercellular adhesion molecule-1. Blood (1998) 92(5):1626–38. doi: 10.1182/blood.V92.5.1626.417a02_1626_1638
132. Futosi K, Fodor S, Mocsai A. Neutrophil cell surface receptors and their intracellular signal transduction pathways. Int Immunopharmacol (2013) 17(3):638–50. doi: 10.1016/j.intimp.2013.06.034
133. Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood (2010) 116(3):485–94. doi: 10.1182/blood-2009-12-259556
134. Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med (2008) 205(10):2339–47. doi: 10.1084/jem.20072660
135. Urzainqui A, Serrador JM, Viedma F, Yanez-Mo M, Rodriguez A, Corbi AL, et al. ITAM-based interaction of ERM proteins with Syk mediates signaling by the leukocyte adhesion receptor PSGL-1. Immunity (2002) 17(4):401–12. doi: 10.1016/S1074-7613(02)00420-X
136. Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity (2007) 26(6):773–83. doi: 10.1016/j.immuni.2007.04.011
137. Block H, Herter JM, Rossaint J, Stadtmann A, Kliche S, Lowell CA, et al. Crucial role of SLP-76 and ADAP for neutrophil recruitment in mouse kidney ischemia-reperfusion injury. J Exp Med (2012) 209(2):407–21. doi: 10.1084/jem.20111493
138. Ma YQ, Plow EF, Geng JG. P-selectin binding to P-selectin glycoprotein ligand-1 induces an intermediate state of alphaMbeta2 activation and acts cooperatively with extracellular stimuli to support maximal adhesion of human neutrophils. Blood (2004) 104(8):2549–56. doi: 10.1182/blood-2004-03-1108
139. Green CE, Pearson DN, Camphausen RT, Staunton DE, Simon SI. Shear-dependent capping of L-selectin and P-selectin glycoprotein ligand 1 by E-selectin signals activation of high-avidity beta2-integrin on neutrophils. J Immunol (2004) 172(12):7780–90. doi: 10.4049/jimmunol.172.12.7780
140. Ding ZM, Babensee JF, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. (0022-1767 (Print)).
141. Giagulli C, Ottoboni L, Caveggion E, Rossi B, Lowell C, Constantin G, et al. The Src family kinases Hck and Fgr are dispensable for inside-out, chemoattractant-induced signaling regulating beta 2 integrin affinity and valency in neutrophils, but are required for beta 2 integrin-mediated outside-in signaling involved in sustained adhesion. J Immunol (2006) 177(1):604–11. doi: 10.4049/jimmunol.177.1.604
142. Takala H, Nurminen E, Nurmi SM, Aatonen M, Strandin T, Takatalo M, et al. Beta2 integrin phosphorylation on Thr758 acts as a molecular switch to regulate 14-3-3 and filamin binding. Blood (2008) 112(5):1853–62. doi: 10.1182/blood-2007-12-127795
143. Bonet R, Vakonakis I, Campbell ID. Characterization of 14-3-3-zeta Interactions with integrin tails. J Mol Biol (2013) 425(17):3060–72. doi: 10.1016/j.jmb.2013.05.024
144. Das M, Ithychanda SS, Plow EF. Migfilin and filamin as regulators of integrin activation in endothelial cells and neutrophils. PloS One (2011) 6(10):e26355. doi: 10.1371/journal.pone.0026355
145. Bertram A, Zhang H, von Vietinghoff S, de Pablo C, Haller H, Shushakova N, et al. Protein kinase C-theta is required for murine neutrophil recruitment and adhesion strengthening under flow. J Immunol (2012) 188(8):4043–51. doi: 10.4049/jimmunol.1101651
146. Jakob SM, Pick R, Brechtefeld D, Nussbaum C, Kiefer F, Sperandio M, et al. Hematopoietic progenitor kinase 1 (HPK1) is required for LFA-1-mediated neutrophil recruitment during the acute inflammatory response. Blood (2013) 121(20):4184–94. doi: 10.1182/blood-2012-08-451385
147. Norgauer J, Krutmann J, Dobos GJ, Traynor-Kaplan AE, Oades ZG, Schraufstatter IU. Actin polymerization, calcium-transients, and phospholipid metabolism in human neutrophils after stimulation with interleukin-8 and N-formyl peptide. J Invest Dermatol (1994) 102(3):310–4. doi: 10.1111/1523-1747.ep12371788
148. Silveira AAA, Dominical VM, Almeida CB, Chweih H, Ferreira WA Jr, Vicente CP, et al. TNF induces neutrophil adhesion via formin-dependent cytoskeletal reorganization and activation of beta-integrin function. J Leukoc Biol (2018) 103(1):87–98.
149. Yan SR, Berton G. Antibody-induced engagement of beta2 integrins in human neutrophils causes a rapid redistribution of cytoskeletal proteins, Src-family tyrosine kinases, and p72syk that precedes de novo actin polymerization. J Leukoc Biol (1998) 64(3):401–8. doi: 10.1002/jlb.64.3.401
150. Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci U.S.A. (2003) 100(23):13298–302. doi: 10.1073/pnas.2336149100
151. Berton G, Mocsai A, Lowell CA. Src and Syk kinases: key regulators of phagocytic cell activation. Trends Immunol (2005) 26(4):208–14. doi: 10.1016/j.it.2005.02.002
152. Frommhold D, Mannigel I, Schymeinsky J, Mocsai A, Poeschl J, Walzog B, et al. Spleen tyrosine kinase Syk is critical for sustained leukocyte adhesion during inflammation in vivo. BMC Immunol (2007) 8:31. doi: 10.1186/1471-2172-8-31
153. Gilmore AP, Burridge K. Regulation of vinculin binding to talin and actin by phosphatidyl-inositol-4-5-bisphosphate. Nature (1996) 381(6582):531–5. doi: 10.1038/381531a0
154. Sampath R, Gallagher PJ, Pavalko FM. Cytoskeletal interactions with the leukocyte integrin beta2 cytoplasmic tail. Activation-dependent regulation of associations with talin and alpha-actinin. J Biol Chem (1998) 273(50):33588–94. doi: 10.1074/jbc.273.50.33588
155. Wilson ZS, Witt H, Hazlett L, Harman M, Neumann BM, Whitman A, et al. Context-Dependent Role of Vinculin in Neutrophil Adhesion, Motility and Trafficking. Sci Rep (2020) 10(1):2142. doi: 10.1038/s41598-020-58882-y
156. Murata K, Yoshitomi H, Furu M, Ishikawa M, Shibuya H, Ito H, et al. MicroRNA-451 down-regulates neutrophil chemotaxis via p38 MAPK. Arthritis Rheumatol (2014) 66(3):549–59. doi: 10.1002/art.38269
157. Sprenkeler EGG, Henriet SSV, Tool ATJ, Kreft IC, van der Bijl I, Aarts CEM, et al. MKL1 deficiency results in a severe neutrophil motility defect due to impaired actin polymerization. Blood (2020) 135(24):2171–81. doi: 10.1182/blood.2019002633
158. Petri B, Sanz MJ. Neutrophil chemotaxis. Cell Tissue Res (2018) 371(3):425–36. doi: 10.1007/s00441-017-2776-8
159. Lammermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Soldner R, Hirsch K, et al. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature (2008) 453(7191):51–5. doi: 10.1038/nature06887
160. Hind LE, Vincent WJ, Huttenlocher A. Leading from the Back: The Role of the Uropod in Neutrophil Polarization and Migration. Dev Cell (2016) 38(2):161–9. doi: 10.1016/j.devcel.2016.06.031
161. Ren C, Yuan Q, Braun M, Zhang X, Petri B, Zhang J, et al. Leukocyte Cytoskeleton Polarization Is Initiated by Plasma Membrane Curvature from Cell Attachment. Dev Cell (2019) 49p. 206–219(2):e7. doi: 10.1016/j.devcel.2019.02.023
162. Fritz-Laylin LK, Riel-Mehan M, Chen BC, Lord SJ, Goddard TD, Ferrin TE, et al. Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. Elife (2017) 6:e26990. doi: 10.7554/eLife.26990
163. Yan SR, Fumagalli L, Berton G. Activation of p58c-fgr and p53/56lyn in adherent human neutrophils: evidence for a role of divalent cations in regulating neutrophil adhesion and protein tyrosine kinase activities. J Inflammation (1995) 45(4):297–311.
164. Hepper I, Schymeinsky J, Weckbach LT, Jakob SM, Frommhold D, Sixt M, et al. The mammalian actin-binding protein 1 is critical for spreading and intraluminal crawling of neutrophils under flow conditions. J Immunol (2012) 188(9):4590–601. doi: 10.4049/jimmunol.1100878
165. Schymeinsky J, Sperandio M, Walzog B. The mammalian actin-binding protein 1 (mAbp1): a novel molecular player in leukocyte biology. Trends Cell Biol (2011) 21(4):247–55. doi: 10.1016/j.tcb.2010.12.001
166. Fabbri M, Di Meglio S, Gagliani MC, Consonni E, Molteni R, Bender JR, et al. Dynamic partitioning into lipid rafts controls the endo-exocytic cycle of the alphaL/beta2 integrin, LFA-1, during leukocyte chemotaxis. Mol Biol Cell (2005) 16(12):5793–803. doi: 10.1091/mbc.e05-05-0413
167. Roth H, Samereier M, Begandt D, Pick R, Salvermoser M, Brechtefeld D, et al. Filamin A promotes efficient migration and phagocytosis of neutrophil-like HL-60 cells. Eur J Cell Biol (2017) 96(6):553–66. doi: 10.1016/j.ejcb.2017.05.004
168. Gandhi M, Goode BL. Coronin: the double-edged sword of actin dynamics. Subcell Biochem (2008) 48:72–87. doi: 10.1007/978-0-387-09595-0_7
169. Boras M, Volmering S, Bokemeyer A, Rossaint J, Block H, Bardel B, et al. Skap2 is required for beta2 integrin-mediated neutrophil recruitment and functions. J Exp Med (2017) 214(3):851–74. doi: 10.1084/jem.20160647
170. Cooper MD, Chae HP, Lowman JT, Krivit W, Good RA. Wiskott-Aldrich syndrome. An immunologic deficiency disease involving the afferent limb of immunity. Am J Med (1968) 44(4):499–513. doi: 10.1016/0002-9343(68)90051-X
171. Pantarelli C, Welch HCE. Rac-GTPases and Rac-GEFs in neutrophil adhesion, migration and recruitment. Eur J Clin Invest (2018) 48 Suppl 2:e12939. doi: 10.1111/eci.12939
172. Williams DA, Tao W, Yang F, Kim C, Gu Y, Mansfield P, et al. Dominant negative mutation of the hematopoietic-specific Rho GTPase, Rac2, is associated with a human phagocyte immunodeficiency. Blood (2000) 96(5):1646–54.
173. Miura Y, Tohyama Y, Hishita T, Lala A, De Nardin E, Yoshida Y, et al. Pyk2 and Syk participate in functional activation of granulocytic HL-60 cells in a different manner. Blood (2000) 96(5):1733–9. doi: 10.1182/blood.V96.5.1733.h8001733_1733_1739
174. Ovcharenko A, Granot G, Shpilberg O, Raanani P. Retinoic acid induces adhesion and migration in NB4 cells through Pyk2 signaling. Leuk Res (2013) 37(8):956–62. doi: 10.1016/j.leukres.2013.03.010
175. Kamen LA, Schlessinger J, Lowell CA. Pyk2 is required for neutrophil degranulation and host defense responses to bacterial infection. J Immunol (2011) 186(3):1656–65. doi: 10.4049/jimmunol.1002093
176. Cheung SM, Ostergaard HL. Pyk2 Controls Integrin-Dependent CTL Migration through Regulation of De-Adhesion. J Immunol (2016) 197(5):1945–56. doi: 10.4049/jimmunol.1501505
177. Chen S, Lin F, Shin ME, Wang F, Shen L, Hamm HE. RACK1 regulates directional cell migration by acting on G betagamma at the interface with its effectors PLC beta and PI3K gamma. Mol Biol Cell (2008) 19(9):3909–22. doi: 10.1091/mbc.e08-04-0433
178. Katsube M, Kato T, Kitagawa M, Noma H, Fujita H, Kitagawa S. Calpain-mediated regulation of the distinct signaling pathways and cell migration in human neutrophils. J Leukoc Biol (2008) 84(1):255–63. doi: 10.1189/jlb.0907664
179. Lokuta MA, Nuzzi PA, Huttenlocher A. Calpain regulates neutrophil chemotaxis. Proc Natl Acad Sci U S A (2003) 100(7):4006–11. doi: 10.1073/pnas.0636533100
180. Kiema T, Lad Y, Jiang T, Oxley CL, Baldassarre M, Wegener KL, et al. The molecular basis of filamin binding to integrins and competition with talin. Mol Cell (2006) 21(3):337–47. doi: 10.1016/j.molcel.2006.01.011
181. Sun C, Forster C, Nakamura F, Glogauer M. Filamin-A regulates neutrophil uropod retraction through RhoA during chemotaxis. PloS One (2013) 8(10):e79009. doi: 10.1371/journal.pone.0079009
182. Azcutia V, Parkos CA, Brazil JC. Role of negative regulation of immune signaling pathways in neutrophil function. J Leukoc Biol (2017). doi: 10.1002/JLB.3MIR0917-374R
183. Nesterovitch AB, Gyorfy Z, Hoffman MD, Moore EC, Elbuluk N, Tryniszewska B, et al. Alteration in the gene encoding protein tyrosine phosphatase nonreceptor type 6 (PTPN6/SHP1) may contribute to neutrophilic dermatoses. Am J Pathol (2011) 178(4):1434–41. doi: 10.1016/j.ajpath.2010.12.035
184. Speir M, Nowell CJ, Chen AA, O'Donnell JA, Shamie IS, Lakin PR, et al. Ptpn6 inhibits caspase-8- and Ripk3/Mlkl-dependent inflammation. Nat Immunol (2020) 21(1):54–64. doi: 10.1038/s41590-019-0550-7
185. Gordon S. Phagocytosis: An Immunobiologic Process. Immunity (2016) 44(3):463–75. doi: 10.1016/j.immuni.2016.02.026
186. Rosales C, Uribe-Querol E. Phagocytosis: A Fundamental Process in Immunity. BioMed Res Int (2017) 2017:9042851. doi: 10.1155/2017/9042851
187. Gazendam RP, van de Geer A, Roos D, van den Berg TK, Kuijpers TW. How neutrophils kill fungi. Immunol Rev (2016) 273(1):299–311. doi: 10.1111/imr.12454
188. Freeman SA, Goyette J, Furuya W, Woods EC, Bertozzi CR, Bergmeier W, et al. Integrins Form an Expanding Diffusional Barrier that Coordinates Phagocytosis. Cell (2016) 164(1-2):128–40. doi: 10.1016/j.cell.2015.11.048
189. Gresham HD, Graham IL, Anderson DC, Brown EJ. Leukocyte adhesion-deficient neutrophils fail to amplify phagocytic function in response to stimulation. Evidence for CD11b/CD18-dependent and -independent mechanisms of phagocytosis. J Clin Invest (1991) 88(2):588–97. doi: 10.1172/JCI115343
190. Patel PC, Harrison RE. Membrane ruffles capture C3bi-opsonized particles in activated macrophages. Mol Biol Cell (2008) 19(11):4628–39. doi: 10.1091/mbc.e08-02-0223
191. Le Cabec V, Carreno S, Moisand A, Bordier C, Maridonneau-Parini I. Complement receptor 3 (CD11b/CD18) mediates type I and type II phagocytosis during nonopsonic and opsonic phagocytosis, respectively. J Immunol (2002) 169(4):2003–9. doi: 10.4049/jimmunol.169.4.2003
192. Kuijpers TW, Tool ATJ, van der Bijl I, de Boer M, van Houdt M, de Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol (2017) 140(1):273–77.e10. doi: 10.1016/j.jaci.2016.09.061
193. Caron E, Self AJ, Hall A. The GTPase Rap1 controls functional activation of macrophage integrin alphaMbeta2 by LPS and other inflammatory mediators. Curr Biol (2000) 10(16):974–8. doi: 10.1016/S0960-9822(00)00641-2
194. Dupuy AG, Caron E. Integrin-dependent phagocytosis: spreading from microadhesion to new concepts. J Cell Sci (2008) 121(11):1773–83. doi: 10.1242/jcs.018036
195. Lim J, Wiedemann A, Tzircotis G, Monkley SJ, Critchley DR, Caron E. An essential role for talin during alpha(M)beta(2)-mediated phagocytosis. Mol Biol Cell (2007) 18(3):976–85.
196. Kuijpers TW, van Bruggen R, Kamerbeek N, Tool AT, Hicsonmez G, Gurgey A, et al. Natural history and early diagnosis of LAD-1/variant syndrome. Blood (2007) 109(8):3529–37. doi: 10.1182/blood-2006-05-021402
197. Reyes-Reyes M, Mora N, Gonzalez G, Rosales C. beta1 and beta2 integrins activate different signalling pathways in monocytes. Biochem J (2002) 363(Pt 2):273–80. doi: 10.1042/bj3630273
198. Jaumouille V, Cartagena-Rivera AX, Waterman CM. Coupling of beta2 integrins to actin by a mechanosensitive molecular clutch drives complement receptor-mediated phagocytosis. Nat Cell Biol (2019) 21(11):1357–69. doi: 10.1038/s41556-019-0414-2
199. Wade EM, Halliday EM, Jenkins ZA, O'Neill AC, Robertson SP. The X-linked filaminopathies: Synergistic insights from clinical and molecular analysis. Hum Mutat (2020) 41(5):865–83. doi: 10.1002/humu.24002
200. May RC, Caron E, Hall A, Machesky LM. Involvement of the Arp2/3 complex in phagocytosis mediated by FcgammaR or CR3. Nat Cell Biol (2000) 2(4):246–8. doi: 10.1038/35008673
201. Pollard TD. Regulation of actin filament assembly by Arp2/3 complex and formins. Annu Rev Biophys Biomol Struct (2007) 36:451–77. doi: 10.1146/annurev.biophys.35.040405.101936
202. Kasorn A, Alcaide P, Jia Y, Subramanian KK, Sarraj B, Li Y, et al. Focal adhesion kinase regulates pathogen-killing capability and life span of neutrophils via mediating both adhesion-dependent and -independent cellular signals. J Immunol (2009) 183(2):1032–43. doi: 10.4049/jimmunol.0802984
203. Kumar V, Everingham S, Hall C, Greer PA, Craig AW. Calpains promote neutrophil recruitment and bacterial clearance in an acute bacterial peritonitis model. Eur J Immunol (2014) 44(3):831–41. doi: 10.1002/eji.201343757
204. El Azreq MA, Garceau V, Bourgoin SG. Cytohesin-1 regulates fMLF-mediated activation and functions of the beta2 integrin Mac-1 in human neutrophils. J Leukoc Biol (2011) 89(6):823–36. doi: 10.1189/jlb.0410222
205. Gómez Román VR, Murray JC, Weiner LM. Antibody-Dependent Cellular Cytotoxicity (ADCC). (2014) p:1–27. doi: 10.1016/B978-0-12-394802-1.00001-7
206. Heemskerk N, van Egmond M. Monoclonal antibody-mediated killing of tumour cells by neutrophils. Eur J Clin Invest (2018) 48 Suppl 2:e12962. doi: 10.1111/eci.12962
207. Worley MJ, Fei K, Lopez-Denman AJ, Kelleher AD, Kent SJ, Chung AW, et al. Neutrophils mediate HIV-specific antibody-dependent phagocytosis and ADCC. J Immunol Methods (2018) 457:41–52. doi: 10.1016/j.jim.2018.03.007
208. Bruhns P, Jonsson F. Mouse and human FcR effector functions. Immunol Rev (2015) 268(1):25–51. doi: 10.1111/imr.12350
209. Zwick A, Bernhard M, Knoerck A, Linxweiler M, Schick B, Heinzelmann J, et al. Monitoring kinetics reveals critical parameters of IgA-dependent granulocyte-mediated anti-tumor cell cytotoxicity. J Immunol Methods (2019) 473:112644. doi: 10.1016/j.jim.2019.112644
210. Ortiz-Stern A, Rosales C. Cross-talk between Fc receptors and integrins. Immunol Lett (2003) 90(2-3):137–43. doi: 10.1016/j.imlet.2003.08.004
212. Steblyanko M, Anikeeva N, Campbell KS, Keen JH, Sykulev Y. Integrins Influence the Size and Dynamics of Signaling Microclusters in a Pyk2-dependent Manner. J Biol Chem (2015) 290(19):11833–42. doi: 10.1074/jbc.M114.614719
213. Liu D, Bryceson YT, Meckel T, Vasiliver-Shamis G, Dustin ML, Long EO. Integrin-dependent organization and bidirectional vesicular traffic at cytotoxic immune synapses. Immunity (2009) 31(1):99–109. doi: 10.1016/j.immuni.2009.05.009
214. Huang Y, Chen Z, Jang JH, Baig MS, Bertolet G, Schroeder C, et al. PD-1 blocks lytic granule polarization with concomitant impairment of integrin outside-in signaling in the natural killer cell immunological synapse. J Allergy Clin Immunol (2018) 142(4):1311–1321 e8. doi: 10.1016/j.jaci.2018.02.050
215. van Spriel AB, Leusen JH, van Egmond M, Dijkman HB, Assmann KJ, Mayadas TN, et al. Mac-1 (CD11b/CD18) is essential for Fc receptor-mediated neutrophil cytotoxicity and immunologic synapse formation. Blood (2001) 97(8):2478–86. doi: 10.1182/blood.V97.8.2478
216. Matlung HL, Babes L, Zhao XW, van Houdt M, Treffers LW, van Rees DJ, et al. Neutrophils Kill Antibody-Opsonized Cancer Cells by Trogoptosis. Cell Rep (2018) 23(13):3946–59.e6. doi: 10.1016/j.celrep.2018.05.082
217. Liang S, Slattery MJ, Dong C. Shear stress and shear rate differentially affect the multi-step process of leukocyte-facilitated melanoma adhesion. Exp Cell Res (2005) 310(2):282–92. doi: 10.1016/j.yexcr.2005.07.028
218. Bouti P, Zhao XW, Verkuijlen P, Tool AT, van Houdt M, Koker N, et al. Kindlin3-dependent 1270 CD11b/CD18-integrin activation is required for potentiation of neutrophil cytotoxicity by CD47-1271 SIRPalpha checkpoint disruption. Cancer Immunol Res (2020).
219. Lehman HK, Segal BH. The role of neutrophils in host defense and disease. J Allergy Clin Immunol (2020) 145(6):1535–44. doi: 10.1016/j.jaci.2020.02.038
220. Dahlgren C, Karlsson A, Bylund J. Intracellular Neutrophil Oxidants: From Laboratory Curiosity to Clinical Reality. J Immunol (2019) 202(11):3127–34. doi: 10.4049/jimmunol.1900235
221. Husemann J, Obstfeld A, Febbraio M, Kodama T, Silverstein SC. CD11b/CD18 mediates production of reactive oxygen species by mouse and human macrophages adherent to matrixes containing oxidized LDL. Arterioscler Thromb Vasc Biol (2001) 21(8):1301–5. doi: 10.1161/hq0801.095150
222. Utomo A, Cullere X, Glogauer M, Swat W, Mayadas TN. Vav proteins in neutrophils are required for FcgammaR-mediated signaling to Rac GTPases and nicotinamide adenine dinucleotide phosphate oxidase component p40(phox). J Immunol (2006) 177(9):6388–97. doi: 10.4049/jimmunol.177.9.6388
223. Hall AB, Gakidis MA, Glogauer M, Wilsbacher JL, Gao S, Swat W, et al. Requirements for Vav guanine nucleotide exchange factors and Rho GTPases in FcgammaR- and complement-mediated phagocytosis. Immunity (2006) 24(3):305–16. doi: 10.1016/j.immuni.2006.02.005
224. Anderson KE, Boyle KB, Davidson K, Chessa TA, Kulkarni S, Jarvis GE, et al. CD18-dependent activation of the neutrophil NADPH oxidase during phagocytosis of Escherichia coli or Staphylococcus aureus is regulated by class III but not class I or II PI3Ks. Blood (2008) 112(13):5202–11. doi: 10.1182/blood-2008-04-149450
225. Houslay DM, Anderson KE, Chessa T, Kulkarni S, Fritsch R, Downward J, et al. Coincident signals from GPCRs and receptor tyrosine kinases are uniquely transduced by PI3Kbeta in myeloid cells. Sci Signal (2016) 9(441):ra82. doi: 10.1126/scisignal.aae0453
226. Kulkarni S, Sitaru C, Jakus Z, Anderson KE, Damoulakis G, Davidson K, et al. PI3Kbeta plays a critical role in neutrophil activation by immune complexes. Sci Signal (2011) 4(168):ra23. doi: 10.1126/scisignal.2001617
227. Jakus Z, Simon E, Frommhold D, Sperandio M, Mocsai A. Critical role of phospholipase Cgamma2 in integrin and Fc receptor-mediated neutrophil functions and the effector phase of autoimmune arthritis. J Exp Med (2009) 206(3):577–93. doi: 10.1084/jem.20081859
228. Newbrough SA, Mocsai A, Clemens RA, Wu JN, Silverman MA, Singer AL, et al. SLP-76 regulates Fcgamma receptor and integrin signaling in neutrophils. Immunity (2003) 19(5):761–9. doi: 10.1016/S1074-7613(03)00305-4
229. Fernandez-Espartero CH, Ramel D, Farago M, Malartre M, Luque CM, Limanovich S, et al. GTP exchange factor Vav regulates guided cell migration by coupling guidance receptor signalling to local Rac activation. J Cell Sci (2013) 126(Pt 10):2285–93. doi: 10.1242/jcs.124438
230. Fratti RA, Backer JM, Gruenberg J, Corvera S, Deretic V. Role of phosphatidylinositol 3-kinase and Rab5 effectors in phagosomal biogenesis and mycobacterial phagosome maturation arrest. J Cell Biol (2001) 154(3):631–44. doi: 10.1083/jcb.200106049
231. Nguyen GT, Green ER, Mecsas J. Neutrophils to the ROScue: Mechanisms of NADPH Oxidase Activation and Bacterial Resistance. Front Cell Infect Microbiol (2017) 7:373. doi: 10.3389/fcimb.2017.00373
232. Fumagalli L, Campa CC, Germena G, Lowell CA, Hirsch E, Berton G. Class I phosphoinositide-3-kinases and SRC kinases play a nonredundant role in regulation of adhesion-independent and -dependent neutrophil reactive oxygen species generation. J Immunol (2013) 190(7):3648–60. doi: 10.4049/jimmunol.1201951
233. Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discovery (2016) 15(3):173–83. doi: 10.1038/nrd.2015.10
234. McLean LP, Cross RK. Integrin antagonists as potential therapeutic options for the treatment of Crohn’s disease. Expert Opin Investig Drugs (2016) 25(3):263–73. doi: 10.1517/13543784.2016.1148137
235. Bednarczyk M, Stege H, Grabbe S, Bros M. beta2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int J Mol Sci (2020) 21(4):1402. doi: 10.3390/ijms21041402
236. Faridi MH, Maiguel D, Brown BT, Suyama E, Barth CJ, Hedrick M, et al. High-throughput screening based identification of small molecule antagonists of integrin CD11b/CD18 ligand binding. Biochem Biophys Res Commun (2010) 394(1):194–9. doi: 10.1016/j.bbrc.2010.02.151
237. Wolf D, Anto-Michel N, Blankenbach H, Wiedemann A, Buscher K, Hohmann JD, et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat Commun (2018) 9(1):525. doi: 10.1161/atvb.38.suppl_1.031
238. Yonekawa K, Harlan JM. Targeting leukocyte integrins in human diseases. J Leukoc Biol (2005) 77(2):129–40. doi: 10.1189/jlb.0804460
239. Dedrick RL, Walicke P, Garovoy M. Anti-adhesion antibodies: Efalizumab, a humanized anti-CD11a monoclonal antibody. Transplant Immunol (2002) 9(2):181–6. doi: 10.1016/S0966-3274(02)00029-1
240. Johnson-Huang LM, Pensabene CA, Shah KR, Pierson KC, Kikuchi T, Lentini T, et al. Post-therapeutic relapse of psoriasis after CD11a blockade is associated with T cells and inflammatory myeloid DCs. PLoS One (2012) 7(2):e30308. doi: 10.1371/journal.pone.0030308
241. Morrison VL, MacPherson M, Savinko T, Lek HS, Prescott A, Fagerholm SC. The beta2 integrin-kindlin-3 interaction is essential for T-cell homing but dispensable for T-cell activation in vivo. Blood (2013) 122(8):1428–36. doi: 10.1182/blood-2013-02-484998
242. Filardo EJ, Brooks PC, Deming SL, Damsky C, Cheresh DA. Requirement of the NPXY motif in the integrin beta 3 subunit cytoplasmic tail for melanoma cell migration in vitro and in vivo. J Cell Biol (1995) 130(2):441–50. doi: 10.1083/jcb.130.2.441
243. Inomata M, Hayashi M, Ohno-Iwashita Y, Tsubuki S, Saido TC, Kawashima S, et al. Involvement of calpain in integrin-mediated signal transduction. Arch Biochem Biophys (1996) 328(1):129–34. doi: 10.1006/abbi.1996.0152
244. Wiemer AJ, Lokuta MA, Surfus JC, Wernimont SA, Huttenlocher A. Calpain inhibition impairs TNF-alpha-mediated neutrophil adhesion, arrest and oxidative burst. Mol Immunol (2010) 47(4):894–902. doi: 10.1016/j.molimm.2009.10.002
245. Mileski WJ, Burkhart D, Hunt JL, Kagan RJ, Saffle JR, Herndon DN, et al. Clinical effects of inhibiting leukocyte adhesion with monoclonal antibody to intercellular adhesion molecule-1 (enlimomab) in the treatment of partial-thickness burn injury. J Trauma (2003) 54(5):950–8. doi: 10.1097/01.TA.0000030626.84680.11
246. Vuorte J, Lindsberg PJ, Kaste M, Meri S, Jansson S-E, Rothlein R, et al. Anti-ICAM-1 Monoclonal Antibody R6.5 (Enlimomab) Promotes Activation of Neutrophils in Whole Blood. J Immunol (1999) 162(4):2353.
247. van Deventer SJ, Tami JA, Wedel MK. A randomised, controlled, double blind, escalating dose study of alicaforsen enema in active ulcerative colitis. (0017-5749 (Print)).
248. Greuter T, Biedermann L, Rogler G, Sauter B, Seibold F, et al. Alicaforsen, an antisense inhibitor of ICAM-1, as treatment for chronic refractory pouchitis after proctocolectomy: A case series. (2050-6406 (Print)).
249. Burlak C, Whitney AR, Mead DJ, Hackstadt T, Deleo FR, et al. Maturation of human neutrophil phagosomes includes incorporation of molecular chaperones and endoplasmic reticulum quality control machinery. Mol Cell Proteomics (2006) 5(4):620–34. doi: 10.1074/mcp.M500336-MCP200
250. Xu P, Crawford M, Way M, Godovac-Zimmermann J, Segal AW, Radulovic M. Subproteome analysis of the neutrophil cytoskeleton. Proteomics (2009) 9(7):2037–49. doi: 10.1002/pmic.200800674
251. McLeish KR, Merchant ML, Klein JB, Ward RA. Technical note: proteomic approaches to fundamental questions about neutrophil biology. J Leukoc Biol (2013) 94(4):683–92. doi: 10.1189/jlb.1112591
252. Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Fremillon A, Robertson J, et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol (2015) 17(12):1577–87. doi: 10.1038/ncb3257
253. Humphries JD, Byron A, Bass MD, Craig SE, Pinney JW, Knight D, et al. Proteomic analysis of integrin-associated complexes identifies RCC2 as a dual regulator of Rac1 and Arf6. Sci Signal (2009) 2(87):ra51. doi: 10.1126/scisignal.2000396
254. Byron A. Proteomic Profiling of Integrin Adhesion Complex Assembly. Methods Mol Biol (2018) 1764:193–236. doi: 10.1007/978-1-4939-7759-8_13
255. Myllymaki SM, Kamarainen UR, Liu X, Cruz SP, Miettinen S, Vuorela M, et al. Assembly of the beta4-Integrin Interactome Based on Proximal Biotinylation in the Presence and Absence of Heterodimerization. Mol Cell Proteomics (2019) 18(2):277–93. doi: 10.1074/mcp.RA118.001095
256. Sahgal P, Alanko J, Icha J, Paatero I, Hamidi H, Arjonen A, et al. GGA2 and RAB13 promote activity-dependent beta1-integrin recycling. J Cell Sci (2019) 132(11):jcs233387. doi: 10.1242/jcs.233387
257. Trinkle-Mulcahy L. Recent advances in proximity-based labeling methods for interactome mapping. F1000Res (2019) 8(31):F1000. doi: 10.12688/f1000research.16903.1
258. Zuidema A, Wang W, Kreft M, Te Molder L, Hoekman L, Bleijerveld OB, et al. Mechanisms of integrin alphaVbeta5 clustering in flat clathrin lattices. J Cell Sci (2018) 131(21):jcs221317. doi: 10.1242/jcs.221317
259. Chastney MR, Lawless C, Humphries JD, Warwood S, Jones MC, Knight D, et al. Topological features of integrin adhesion complexes revealed by multiplexed proximity biotinylation. J Cell Biol (2020) 219(8):e202003038. doi: 10.1083/jcb.202003038
260. Rincon E, Rocha-Gregg BL, Collins SR. A map of gene expression in neutrophil-like cell lines. BMC Genomics (2018) 19(1):573. doi: 10.1186/s12864-018-4957-6
261. Lachmann N, Ackermann M, Frenzel E, Liebhaber S, Brennig S, Happle C, et al. Large-scale hematopoietic differentiation of human induced pluripotent stem cells provides granulocytes or macrophages for cell replacement therapies. Stem Cell Rep (2015) 4(2):282–96. doi: 10.1016/j.stemcr.2015.01.005
262. Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol (1999) 163(9):5029–38.
263. Rochon YP, Kavanagh TJ, Harlan JM. Analysis of integrin (CD11b/CD18) movement during neutrophil adhesion and migration on endothelial cells. J Microsc (2000) 197(Pt 1):15–24. doi: 10.1046/j.1365-2818.2000.00645.x
Keywords: CD11b/CD18 integrin, β2 integrin signaling, neutrophils, neutrophil function, therapeutic targets
Citation: Bouti P, Webbers SDS, Fagerholm SC, Alon R, Moser M, Matlung HL and Kuijpers TW (2021) β2 Integrin Signaling Cascade in Neutrophils: More Than a Single Function. Front. Immunol. 11:619925. doi: 10.3389/fimmu.2020.619925
Received: 21 October 2020; Accepted: 31 December 2020;
Published: 18 February 2021.
Edited by:
Vicky L. Morrison, University of Glasgow, United KingdomReviewed by:
Vineet Gupta, Rush University Medical Center, United StatesKeehoon Jung, Seoul National University, South Korea
Copyright © 2021 Bouti, Webbers, Fagerholm, Alon, Moser, Matlung and Kuijpers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Panagiota Bouti, p.bouti@sanquin.nl; Steven D. S. Webbers, s.webbers@sanquin.nl
†These authors have contributed equally to this work