 Kenneth J. Caldwell
Kenneth J. Caldwell Stephen Gottschalk
Stephen Gottschalk Aimee C. Talleur
Aimee C. Talleur
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 08 January 2021
Sec. Alloimmunity and Transplantation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.618427
This article is part of the Research Topic Moving Towards Allogeneic Cellular Therapies: Opportunities and Challenges View all 14 articles
Adoptive cellular immunotherapy using immune cells expressing chimeric antigen receptors (CARs) has shown promise, particularly for the treatment of hematological malignancies. To date, the majority of clinically evaluated CAR cell products have been derived from autologous immune cells. While this strategy can be effective it also imposes several constraints regarding logistics. This includes i) availability of center to perform leukapheresis, ii) necessity for shipment to and from processing centers, and iii) time requirements for product manufacture and clinical release testing. In addition, previous cytotoxic therapies can negatively impact the effector function of autologous immune cells, which may then affect efficacy and/or durability of resultant CAR products. The use of allogeneic CAR cell products generated using cells from healthy donors has the potential to overcome many of these limitations, including through generation of “off the shelf” products. However, allogeneic CAR cell products come with their own challenges, including potential to induce graft-versus-host-disease, as well as risk of immune-mediated rejection by the host. Here we will review promises and challenges of allogeneic CAR immunotherapies, including those being investigated in preclinical models and/or early phase clinical studies.
Adoptive cellular therapy refers to the isolation of immune cells, followed by ex vivo manipulation and subsequent delivery into patients as a therapeutic intervention. An area of interest is the exploration of cellular or immunotherapeutic approaches for the treatment of oncologic diseases, including using chimeric antigen receptors (CARs) (1–3). CARs combine the specificity of an antibody with signaling domains of effector cells and costimulatory molecules (1–3). When constitutively expressed on the surface of an immune cell through non-viral or viral transduction, CARs enable an effector cell to recognize targets in an antigen-specific manner. CARs designed to target a specific tumor-associated-antigen (TAA) can then be used for anticancer therapy (1–3).
Cell therapy with T cells expressing CARs (CAR T cells) represent a significant advance in the field of cancer immunotherapy and is fueling the development of CAR-based immunotherapies using other immune cells. The most successful CAR cell therapy approach thus far has been the treatment of patients with highly relapsed/refractory CD19-positive hematological malignancies using CD19-CAR T cells derived from autologous T cells. Across numerous institutions, using a variety of CAR constructs and manufacturing strategies, CD19-CAR T cell therapy has been extremely efficacious (4, 5). This success led to the FDA approval of three such products: tisagenlecleucel (Kymriah, Novartis), axicabtagene ciloleucel (Yescarta, Kite Pharmaceuticals), and brexucabtagene autoleucel (Tecartus, Kite Pharmaceuticals) (6–9). Additionally, autologous CAR T cells have shown robust anti-tumor activity for hematological malignancies targeting BCMA, CD20, CD22, and CD30 (10–13).
The autologous (patient-derived) CAR T cell paradigm has also highlighted the limitations of such therapies, including the challenges of leukapheresis, manufacturing and efficacy in an often heavily pre-treated patient population (14). Seeking to overcome these barriers, allogeneic CAR strategies are actively being developed. Significant challenges of using allogeneic cells exist and center upon the inherent immunologic mismatch between donor and recipient. However, despite these challenges, allogeneic CAR strategies hold the potential to offer quicker, more efficacious and more accessible CAR therapies.
In this review, we will discuss a variety of allogeneic CAR cell therapy platforms that are being developed, including the use of different immune cells and/or subtypes, as well as gene-editing techniques (Figure 1). Additionally, we will highlight clinical experiences with allogeneic CAR cell therapies and on-going clinical trials to treat malignancies.
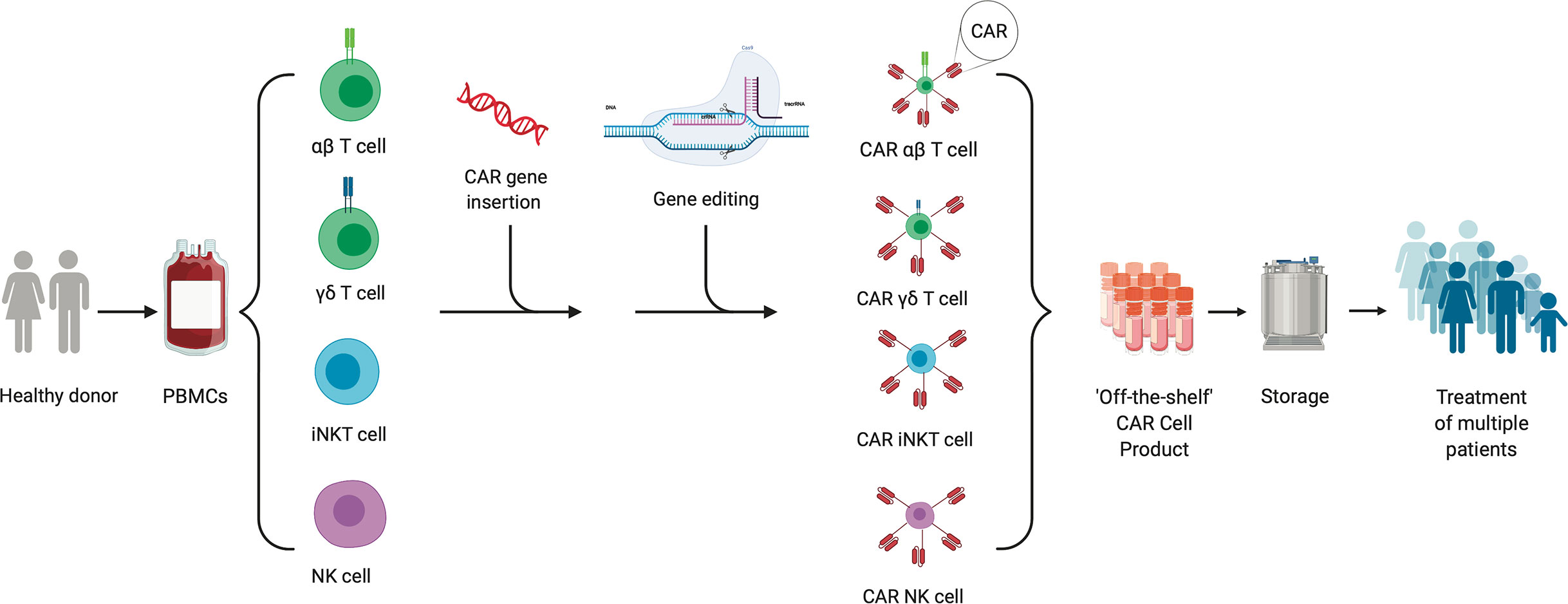
Figure 1 “Off the Shelf” allogeneic cellular therapy production. The production process for an allogeneic cellular therapy starts with collection of peripheral blood mononuclear cells (PBMCs) via leukapheresis from a health donor. Cells can then be sorted and selected for depending on the desired starting cellular material. CAR-encoding genes can be either inserted by non-viral or viral transduction or gene editing into immune cells. Additional gene editing can be performed to knock out genes of interest to mitigate risks such as immunogenicity and/or graft-versus-host-disease. The final product created from a single donor can be expanded, stored and used to treat multiple patients.
Most CAR cell therapies to date, including the FDA approved products, are generated using autologous T cells. This has several important advantages, including infusion of CAR-engineered cell products without immunologic mismatch between donor and recipient. However, the use of autologous immune cells also has clinical and economical disadvantages. Autologous CAR cell production can be long and complicated. The process includes navigating the logistics of performing successful leukapheresis for a patient with relapsed/refractory malignant disease, accessing a manufacturing/treatment facility, and shipping and manufacturing times that commonly take several weeks. This time delay can be significant, particularly in a group of patients with aggressive relapsed/resistant cancers, who are at risk of clinical deterioration which could preclude proceeding with CAR cell therapy. Furthermore, generation of a cell product is not guaranteed and for those whom a product can be successfully generated, a proportion have limited short- or long-term efficacy. This is likely in part due to poor autologous immune cell fitness in cancer patients, particularly following aggressive cancer-directed therapies (15). Earlier collection of T cells may ameliorate some issues related to autologous T cell fitness. However, this strategy would only benefit the subset of patients determined to be at high-risk of needing CAR therapy early on in their disease process. Lastly, autologous cell therapy is performed for individual patients and is associated with significant costs, limiting broader applications of this therapy (16, 17).
The use of immune cells from donors, or allogeneic cell therapies, offers many advantages over autologous cells including the potential to be cost effective, readily available, and provide a higher quality product (Figure 2). Healthy donor cells confer a more uniform starting material, allowing for more predictable manufacturing and performance of generated cell product. Following the single donor to single recipient model, use of a family member would provide an easily accessible and highly motivated allogeneic donor. Furthermore, allogeneic therapies have the potential to provide a ready to use “off the shelf” immunotherapeutic, such that a single manufacturing run would allow dosing for several patients and/or multiple dosing for individual patients. Likewise, by increasing the scale of production and creating an inventory or bank of manufactured CAR immune cells from healthy donors, the cost per patient would decrease while access to product would increase.
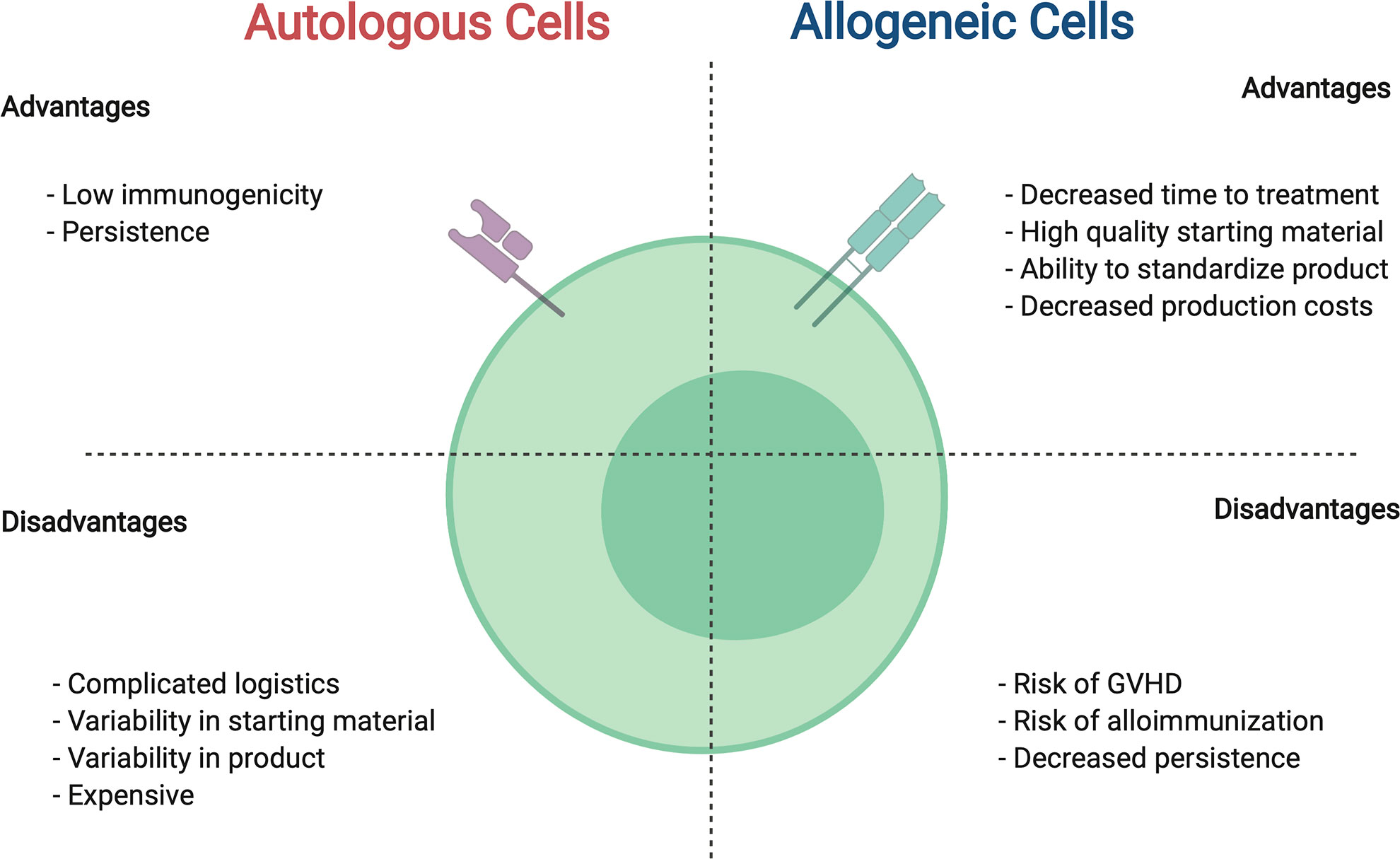
Figure 2 Comparison between autologous and allogeneic cells for use in CAR cell therapies. For details see text.
Despite the recognized potential benefits of allogeneic CAR therapies, they are not without risk and significant challenges must be overcome in order to successfully implement this approach. These challenges stem from the immunologic mismatch between donor and recipient, and the resultant bidirectional risk to the cellular product and to the recipient in vivo. If the administered allogeneic cells recognize and attack healthy recipient tissues, the cellular therapy may cause unwanted graft-versus-host-disease (GVHD). Conversely, if the recipient’s immune system recognizes and reacts against the allogeneic product the cell therapy may be rejected, limiting the therapeutic effect.
Allogeneic hematopoietic cell transplant (HCT) highlights the significant risk of GVHD by adoptive transfer of allogeneic T cells (18). Human leukocyte antigen (HLA) mismatch between donor and recipient leads to donor immune recognition of, and subsequent alloreactivity against, recipient tissues (19–21). Clinically, this is manifested as GVHD. T cells are primarily responsible for causing acute GVHD, triggering tissue cell death via FAS ligand, perforin, granzyme and other signaling pathways (22, 23). The risk of GVHD correlates with increasing donor/recipient HLA-disparity. Most commonly affecting the skin, gastrointestinal tract and liver, GVHD carries a significant risk of post-HCT morbidity and mortality (24, 25). In HCT, the risk of GVHD may be mitigated through donor selection, T cell depletion/selection and/or use of immunosuppressive pharmacologic therapies (26–30). However, some of these strategies are in direct opposition with the goals of allogeneic CAR therapies which depend on highly immunocompetent cells. Therefore, decreasing the risk of GVHD from allogeneic CAR immune cells must balance with the need to retain high levels of immune activity of the effector cell. Such strategies may include T cell-subset selection or gene editing approaches, as well as continued exploration of cell products such as γδ T cells, invariant (i)NKT cells, or allogeneic NK cells that do not induce GVHD.
Expansion and persistence of CAR immune cells are important to achieve both short- and long-term efficacy. While desired duration of persistence may vary based on the malignancy being treated, it has been shown that prolonged remission of acute leukemia correlates with duration of persistence of autologous CAR T cells (31). In the autologous setting, CAR cell longevity can be compromised through immunological rejection of the CARs “foreign” proteins. The use of allogeneic immune cells carries further increased risk of immunogenicity as both the CAR and effector cells are “foreign.” Acutely, this would result in impaired short-term responses as cells are rejected before exerting the intended therapeutic effect. The use of lymphodepleting chemotherapy prior to infusion of allogeneic CAR cellular products should mitigate the risk of acute rejection, augmenting CAR cell persistence (32). However, subsequent recipient immune reconstitution may result in delayed rejection of the adoptively transferred cells, providing an opportunity for malignant relapse. Furthermore, the use of allogeneic cells confers risk of alloimmunization, where the recipient develops donor-specific anti-HLA antibodies (DSAs). Alloimmunization is a well-recognized cause of graft failure and rejection in HCT (33). While desensitization strategies exist, the development of DSAs may preclude a patient from proceeding with HCT in the future or limit re-dosing of the allogeneic CAR product. Genetic modification to remove donor major histocompatibility complex (MHC) molecules or expansion of donor pools to allow for increased HLA-matching may mitigate these risks.
The most widely used CAR platform currently in clinical practice are CAR T cells. These products are largely manufactured using a batched pool of autologous donor T cells collected via peripheral blood leukapheresis and the CAR-T product administered without selection of specific cell types. While this strategy may work in the autologous setting, to mitigate potential risk of using allogeneic immune cells strategies using various T cell subgroups or different immune cell types are being tested in both preclinical and clinical settings. Here we review strategies being explored to make allogeneic CAR immune therapy possible, as well as on-going clinical trials evaluating these strategies (Table 1).
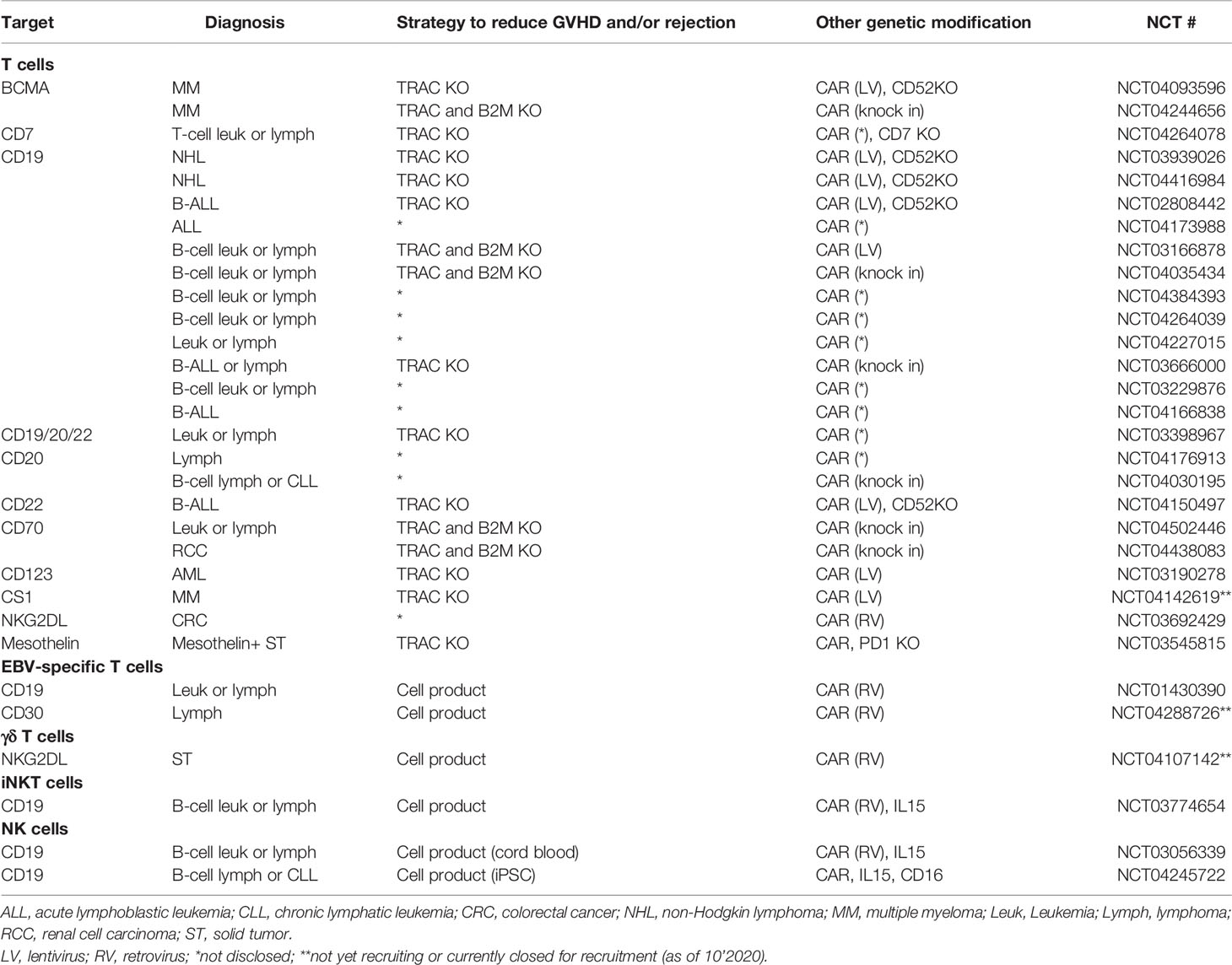
Table 1 Selected clinical studies with allogeneic CAR immune cells.
T cells are a powerful component of the human immune system, providing surveillance for, and protection against, foreign antigen. Antigen recognition occurs via the T cell receptor (TCR), a heterodimer complex composed of two subunits and located on the surface of T cells. In a healthy donor, the majority (> 90%) of circulating T cells have a TCR consisting of an alpha (α) and beta (β) chain, referred to as an αβ T cell (34). The remaining T cells contain a TCR composed of a gamma (γ) and delta (δ) subunit, γδ T cells (35). The interaction between the TCR and antigen triggers a signaling cascade through the TCR which activates the T cell. αβ T cells recognize foreign or non-self-antigen presented through the MHC on antigen presenting cells, while γδ T cells are MHC-independent (36). Furthermore, αβ T cells can be subdivided based on function (i.e., CD4-positive and CD8-positive T cells) and/or degree of differentiation (i.e., naïve and memory T cells) (37, 38). When a T cell is transduced with a CAR, the CAR adds an additional receptor to the T cell without interruption to the native TCR. CAR T cells can expand and contract in response to antigen stimuli via the CAR, allowing for robust responses in the setting of active target recognition, but also potential for memory-surveillance state when an intended target is not currently present. Below, we review different types of T cells used to generate allogeneic CAR T cells.
The use of donor lymphocyte infusion (DLI) after allogeneic HCT is a standard clinical practice. The therapeutic intent of unmodified DLI centers on the properties of donor T cells, such that they can correct mixed donor/recipient chimerism and combat viral infections (39). However, DLIs are not specific for TAAs and therefore have minimal anti-cancer benefit, especially outside the setting of minimal residual disease (39–41). As CAR T cells began to be explored clinically for the treatment of active disease, an initial venture into allogeneic CAR T cell products focused on the post-HCT population using T cells from the HCT donor. Brudno et al. evaluated the use of allogeneic CD19-directed CAR T cells derived from an individual patient’s HCT donor to treat patients with progressive disease after transplant, who had a median donor chimerism of 100%, demonstrating anti-tumor benefits and safety of this approach, including no reports of new-onset GVHD (42). This study exemplified the possibility of increasing the potential for graft-versus-tumor effect of donor-derived T cells, without significantly increasing risk of GVHD.
The use of HCT-donor-derived CAR T cells is limited to post-HCT patients with an available and willing donor, whom are largely treated at facilities with the capability to manufacture clinical grade CAR T cell products. Therefore, this method is innately lacking some of the benefits of an “off the shelf product” and is not widely accessible. However, several benefits of using an allogeneic product are retained, including the use of healthy donor cells, ease of leukapheresis timing and minimal risk of diminished persistence in vivo due to lack of HLA-mismatch. Additionally, this approach allows for exploration into the use of CAR T cells in different disease settings such as prophylaxis post-HCT to reduce relapse in high-risk populations. Data suggest that this strategy is feasible without added toxicity and, in addition to providing leukemic control, may help with control of viral-reactivations post-HCT via native TCR recognition (43, 44).
The adoptive transfer of allogeneic virus-specific T cells (VSTs) has emerged as a safe and effective means of providing antiviral benefit in multiple patient populations (45–48). This has led to the generation of partially HLA-matched banks comprised of libraries of “off the shelf,” purified allogeneic VSTs. Importantly, across numerous clinical studies including in allogeneic HCT populations, the incidence of GVHD has been very minimal. Although the complete mechanism is not fully understood, decreased TCR diversity in VSTs (i.e., memory T cells) is felt to decrease the risk of alloreactivity (i.e., GVHD).
The safety profile seen using allogeneic VSTs created interest in the development of an allogeneic platform using CAR-transduced VSTs. Demonstrating feasibility of VSTs as effector cells, autologous CAR transduced VSTs targeting TAAs (CD30 [Hodgkin lymphoma], HER2 [glioblastoma] and GD2 [neuroblastoma, osteosarcoma]) have successfully been manufactured and infused into patients, with encouraging safety and efficacy profiles (49–51). This strategy has also been explored in the post-HCT patient population using primarily donor-derived VSTs and thus far results are promising. In one study, CD19-redirected VSTs were generated using peripheral blood mononuclear cells (PBMCs) collected from the HCT donor and then infused into patients with B-cell malignancy at escalating doses (52). Manufacturing time for this product was significant, requiring culture for 5 – 6 weeks. Treatment was well tolerated with no GVHD and there was evidence of anti-leukemia activity, as well as retained recognition of viral stimuli. Similar preliminary results have been reported in an on-going trial evaluating allogeneic EBV-specific T cells transduced with a CD19-CAR (NCT 01430390) (53). Notably, donor sources in this trial include the HCT donor or 3rd party donors when the HCT donor is not available, with recipients of the CAR-transduced 3rd party cells also showing encouraging response rates.
Clinical experience to date with allogeneic CAR-transduced VSTs has shown intended anti-tumor effects with minimal GVHD risk. Additional benefit includes the finding that viral-specificity is retained and can trigger CAR T cell expansion in vivo, thereby potentially providing on-going, intermittent stimulus and promoting persistence. A limitation of studies thus far centers on the fact that data are largely confined to CAR-transduced VSTs derived from a patients’ HCT donor, thereby drastically minimizing the challenges of rejection and alloimmunization. Drawing from clinical experiences with unmodified VSTs from 3rd party banks, persistence of VSTs is typically limited to a few months (48); therefore, we would hypothesize that the issue of rejection and limited persistence of CAR-transduced VSTs remains.
When devising strategies for allogeneic cellular therapies the use of memory T cell subsets as effector cells may confer a decreased risk of GVHD. T cell maturation and differentiation inversely correlates with alloreactivity, such that memory T cell subsets are less alloreactive than naïve T cells. Therefore, memory T cells are less likely to cause GVHD in the HLA-mismatched setting (54). Functionally it has also been noted that the effectiveness and persistence of CAR T cells is influenced by the degree of differentiation of the T cell subsets in autologous CAR T cell platforms (55–58). While the use of memory T cells to generate autologous CAR T cells is actively being studied using a variety of CAR constructs (NCT03389230, NCT02146924, NCT02051257, NCT03288493), their use in the allogeneic setting has not yet been evaluated.
T cell subsets can be distinguished through identification of extracellular surface markers, including CD45RO, CD45RA, CD62L, CCR7 and CD27 (37, 38). Several studies have highlighted that generating CAR T cells from central memory (CD45RO+/CD62L+ or CCR7+) T cells or memory stem cells (Tscm) T cell populations is associated with improved CAR T-cell effector function (59–61). Other groups have just focused on utilizing CD45RA-negative T cells, which includes the central memory and effector memory T cell subsets, since these subsets have decreased alloreactive potential (62). After showing promise in animal models, CD45RA-depletion began to be studied in human allogeneic HCT. Clinical studies demonstrated that this approach is feasible and carried a decreased risk of GVHD, both when utilized in primary graft manipulation and post-transplant DLI (63–67). Building upon this clinical experience, the role of memory T cell subsets as effector cells in CAR therapy has been studied pre-clinically. Investigators have shown that CD45RA-negative T cells expressing either a NKG2DL-specific or CD19-CAR have anti-cancer effects and decreased in vivo and in vitro alloreactivity (68–70). Using a similar approach, CD19-CAR-engineered CD27-negative T cells (effector and terminal effector memory subsets) have also shown promise in preclinical models (71). These data suggest that the approach of using allogeneic memory T cells as effector cells in CAR therapy may have merit in the clinical arena; however, additional studies are needed to define the optimal memory T cell subset, which should be used as a source to generate memory CAR T cells with reduced alloreactivity.
Several strategies are being explored to improve allogeneic αβ T cells, which are summarized in Figure 3. Gene editing of T cells to reduce the risk of GVHD and rejection is perhaps the most promising and widespread approach, particularly for the development of an “off the shelf” product. Given that GVHD is driven in large part by TCR recognition of host tissue, gene-editing approaches focused on the native αβ TCR of the effector cell are under investigation. Many groups have explored disrupting the T cell receptor constant alpha chain (TRAC) or beta chain (TRBC). Torikai et al. showed that knocking out the αβ TCR from CD19-CAR T cells did not significantly alter the cells ability to kill CD19-positive targets (72). This initial report in 2012 used zinc finger nuclease mediated knockout of TRAC or TRBC. In recent years, with advancement in gene editing techniques, numerous groups have demonstrated knockout of TRAC using transcription activator like effector nucleases (TALENs) as well as CRISPR/Cas9 (73, 74). Another technique, targeting the CAR to the TRAC locus, was associated with improved anti-tumor activity in one preclinical study (74). In addition to gene-editing approaches, protein-based strategies are being developed to retain the TCR within the Golgi apparatus using an anti-TCR linked to the KDEL motive (75). While these techniques have become increasingly efficient, any remaining T cells that continue to express αβ TCR can be removed magnetically ex vivo using anti- αβTCR antibodies. Stenger et al. showed that TCR knockout of CD19-CAR T cells had high anti-leukemic functionality in the absence of alloreactivity. However, the gene-edited CAR T cells did not persist as long in vivo compared to CAR T cells with endogenous TCR, demonstrating a possible concern with this technique (76). Furthermore, modification to the endogenous TCR does not address the issue of immunogenicity.
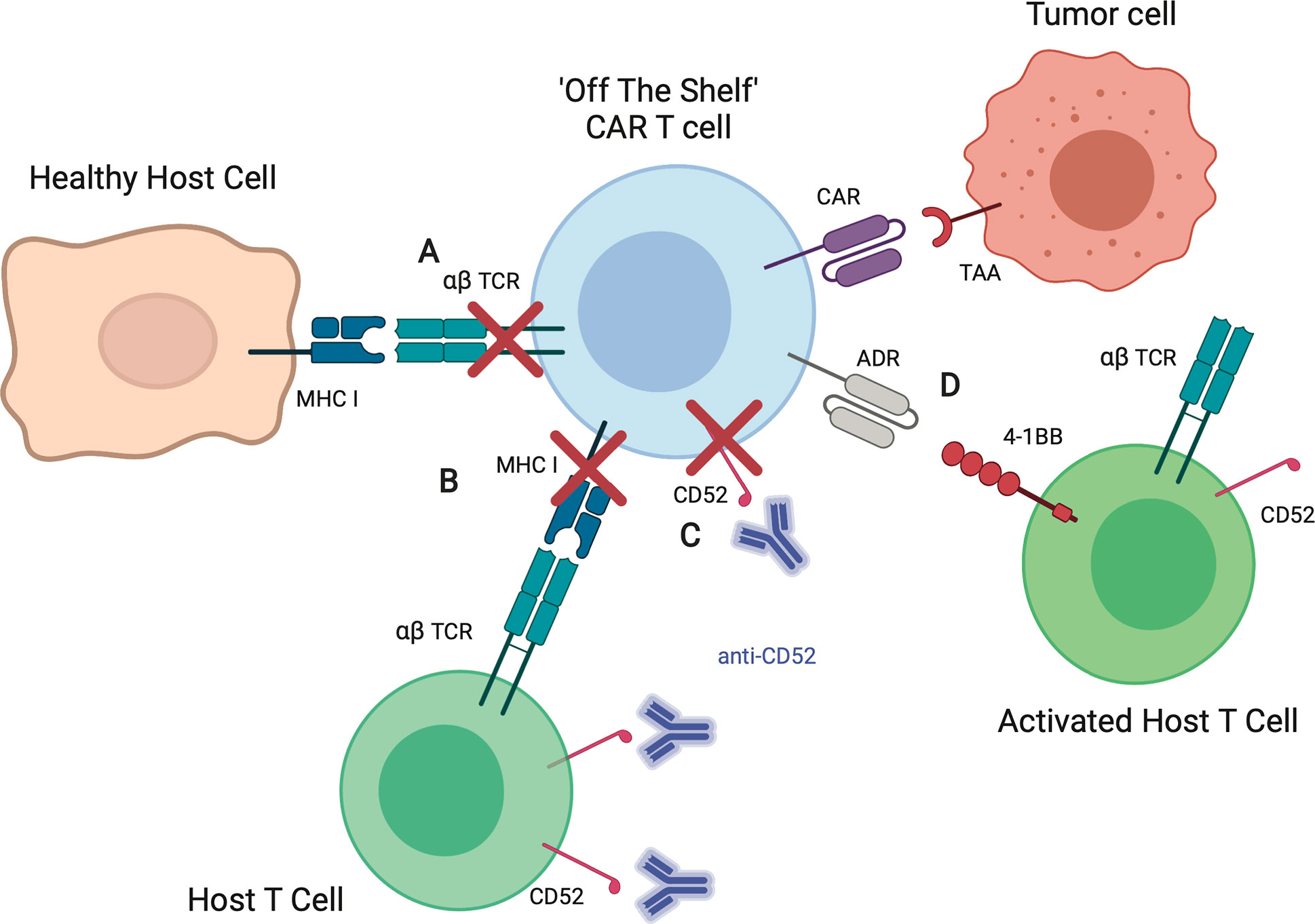
Figure 3 Strategies to improve allogeneic CAR T cells. (A) Elimination of the αβ TCR by knockout of TRAC or retaining TCRs within the Golgi apparatus reduces the risk of allogeneic T cell recognition of healthy host tissues, thereby decreasing risk of graft-versus-host-disease (GVHD). (B) Elimination of MHC I on allogeneic T cells by knockout of β-2 microglobulin (B2M) reduces the risk of host T cell recognition and elimination CAR T cells, increasing likelihood of allogeneic CAR T cell persistence. (C) Knockout of CD52, a T cell marker, allows for the use of CD52 antibody (anti-CD52) for enhanced lymphodepletion of host T cells without affecting infused allogeneic CAR T cells. (D) Expression of alloimmune defense receptor (ADR) destroys alloreactive host T cells targeting 4-1BB, decreasing the risk of rejection.
To decrease immunogenicity, investigators have targeted β-2 microglobulin (B2M), a component of HLA class I molecules that is present on all T cells (77). As recipient T cell recognition of allogeneic CAR T cells can occur via interaction of HLA/MHC, knockout of the B2M gene in CAR T cells may prevent alloantigen presentation by infused the T cells. This thereby offers another strategy to limit host recognition and clearance of infused allogeneic CAR T cells. Ren et al. showed that CAR T cells including knockout of B2M had reduced alloreactivity in vivo (78). Kagoya et al. showed that B2M knockout to eliminate HLA class I as well as knockout of class II major histocompatibility complex transactivator (CIITA) to eliminate HLA class II improved CAR T cells persistence in vitro (79). Other strategies to mitigate immunogenicity rely on depleting resident immune cells. Lymphodepleting chemotherapy prior to cellular infusion by itself should reduce immune-mediated clearance of CAR T cells, however immune responses to components of the CAR have been reported (80–82). To increase immunosuppression post-CAR T cell infusion, investigators have explored the use of the monoclonal antibody, alemtuzumab, which targets the pan-lymphocyte antigen CD52 (83). Since the mean half-life of alemtuzumab is 6.1 days (84), this approach would require the knockout of CD52 on allogeneic CAR T cells to prevent their depletion post infusion (85). Since the use of alemtuzumab is associated with an increased risk of viral reactivation/infection (e.g. cytomegalovirus, adenovirus) after allogeneic HCT (86, 87), close monitoring of recipients of alemtuzumab and CAR T cells is warranted for now since the risk of viral reactivation/infection in the setting of CAR T cell therapy is currently unknown. Lastly, active depletion of alloreactive host T cells is being explored including the expression of so called alloimmune defense receptors (ADRs) on infused allogeneic T cells that selectively recognizes 4-1BB, a cell surface receptor that is temporarily upregulated by activated host T cells (88).
Several clinical trials using gene-edited allogeneic CAR T cells are actively enrolling with some early results presented in abstract format. Many of these products include knockout of both CD52 and TRAC, to address both risk of GVHD and rejection. Pooled data from the CALM (NCT 02746952) and PALL (NCT02808442) studies (cellular product: UCART19) were presented by Allogene Therapeutics and Servier. UCART19 features an anti-CD19 scFv, as well as TRAC and CD52 knockout and was first shown to have clinical activity in two infants with B-ALL who achieved molecular remission after treatment (89). Data presented included 17 patients with relapsed/refractory B-ALL treated with a lymphodepleting regimen including alemtuzamab, 14 of whom had a CR or CRi. Additionally, UCART19 showed overall an acceptable safety profile (90). Neelapu et al. presented early results from the ALLO-501 trial (NCT03939026), which evaluates an allogeneic CAR T cell product targeting CD19 with knock out of TRAC and CD52, and includes a novel CD52 antibody for lymphodepletion prior to CAR T cell infusion. Twenty-two patients had been enrolled, with response seen in 12 of 19 evaluable patients (7 complete responses) (91). Wang et al. presented the first 5 patients treated with TruUCART GC027, an allogeneic anti-CD7 CAR T cell product with knock out of TRAC and CD7 by CRISP/Cas9 gene editing technology to avoid both GVHD and fratricide. Four of the initial 5 treated patients showed a complete response with an acceptable safety profile (92).
Gene editing has emerged as the leading strategy being tested in the clinic as investigators seek to develop an “off the shelf” allogeneic CAR therapy platform. As gene editing techniques advance, this method offers great potential to mitigate potential risks and downsides associated with the use of allogeneic cells. By increasing the number of genetic edits made to a single cell the possibilities increase; however, it will take time to thoroughly investigate the short- and long-term safety of these gene edited products in patients.
Animal models show that γδ T cells play an important role in tissue homeostasis and cancer immunosurveillance (93). Allogeneic γδ T cells have been given to patients with cancer after lymphodepleting chemotherapy and were shown to expand in vivo without causing GVHD (94). γδ T cells recognize cancer through a broad spectrum of receptors, rather than in a single clonally expanded fashion, which may mitigate tumor escape via single antigen loss (95). These T cells are also typically abundant in tissues, which may provide an advantage over αβ T cells when developing therapeutics to treat solid tumors (93). Their ability to recognize targets in an MHC independent manner confers a low risk of alloreactivity and GVHD, thereby increasing the potential of using allogeneic γδ T cells in CAR T cell therapies.
Polyclonal γδT cells transduced with a CD19-CAR have been shown to expand and demonstrated anti-tumor effects in vitro and in in vivo murine models (96). Capsomidis et al. demonstrated CAR dependent antigen specific killing in vitro using GD2-CAR γδ T cells (97). Several companies are now moving forward with clinical trial development using allogeneic CAR-transduced γδ T cells including, Adicet Bio, Cytomed Therapeutics, GammaDelta Therapeutics and TC BioPharm (98). Furthermore, a clinical trial evaluating allogeneic CAR γδ T cells targeting NKG2DL for the treatment of solid tumors has been registered on clinicaltrials.gov, but is not yet recruiting (NCT04107142).
Invariant natural killer T (iNKT) cells are a rare subclass of immune cells that are restricted by CD1d, a glycolipid presenting HLA I like molecule expressed on B cells, antigen presenting cells and some epithelial tissues (99–101). Thus, since iNKT express an invariant TCR, they do not cause GVHD. iNKT cells have been shown to be decreased in number and defective in cancer patients (102–104). iNKT cells also protect from GVHD after allogeneic HCT (105–107). Preclinical studies of iNKT engineered with CARs targeting CD19 and GD2 have been effective in murine models against lymphoma and neuroblastoma, respectively (108–110). CAR-engineered iNKT cells also appear to be safe in humans and are promising for “off the shelf” use given the lack of GVHD. Preliminary results from an ongoing trial (NCT03294954) using autologous GD2-CAR iNKT cells with co-expression of IL15 for the treatment of pediatric patients with neuroblastoma have shown this approach to be feasible and safe (111). Clinical experience of allogeneic CAR iNKT cells is yet to be published so possible adverse events cannot be predicted, however an ongoing clinical trial evaluating allogeneic CAR19-iNKT cells for the treatment of hematological malignancy (NCT03774654) aims to test safety and efficacy.
NK cells, are of great interest in the treatment of cancer as they contribute to graft-versus-tumor effect and do not cause GVHD (112, 113). Endogenous NK cells are part of the innate immune system and can target cancer cells that downregulate HLA class I molecules (112). Tumor cells often downregulate their HLA molecules as an escape mechanism against T cells, making them susceptible to NK cells (114–116). Clinically, adoptively transferred non-CAR engineered allogeneic NK cells have shown to be safe in patients with cancer (117–120). NK cells are a promising alternative to T cells for CAR engineering given the low risk of GVHD and their innate anti-cancer properties. Numerous preclinical studies have shown CAR engineered NK cells to be effective against hematologic malignancy targets (CD19 and CD20), as well as solid tumor targets (WT1 and GD2) (121–126). Notably, Liu et al. published results on 11 patients with lymphoid tumors, treated on an early phase clinical trial using allogeneic (cord blood derived) NK cells transduced with a gene containing a CD19-CAR, IL-15, and an inducible caspase 9 safety switch. In this study, 73% of patients demonstrated anti-tumor response. Inclusion of IL-15 in the CAR construct may have contributed to the persistence of the NK cells, which were shown to expand and persist for at least 12 months. There were no major toxic effects of the therapy (127).
Allogeneic NK cells can be prepared form numerous sources, including peripheral blood mononuclear cells (PBMCs), cord blood, and pluripotent stem cells (iPSCs) (128). In addition, a NK-cell line, NK-92, genetically modified to express a CAR, is actively being explored in early phase clinical testing (NCT03383978) (129). Current strategies to generate clinical grade NK cells from PBMCs or cord blood rely on the use of irradiated feeder cells, most commonly K562 cells genetically modified to express i) 4-1BBL and membrane-bound (mb) IL15 or ii) mbIL21 and exogenous IL2 (130–132). More recently, exosomes or plasma membrane particles derived from mbIL21 expressing K562 cells have also been successfully used for the ex vivo expansion of NK cells (133). iPSCs present an attractive source for generating NK cells without feeder cells (134, 135), and an early phase clinical study with unmodified iPSC-derived NK cells is in progress (NCT03841110). In addition, iPSC cells can be genetically modified and/or gene edited prior to NK cell differentiation, enabling the provision of an unlimited supply of modified NK cells (136–138).
Autologous CAR T cell therapy has revolutionized the treatment of hematological malignancies, highlighting the therapeutic potential of cellular therapies, as well as opportunities for continued improvement. Subsequently, the number of CAR therapy trials has increased dramatically in recent years, exploring new targets, manufacturing strategies, CAR constructs, patient populations and effector cells. While many of these trials continue to use autologous immune cells, the number using allogeneic CAR products are rapidly increasing. The apparent benefits of allogeneic therapies have spurred a robust interest in developing techniques that counter the predicted limitations, including exploration of various effector cell types and gene editing techniques. One hurdle that will definitively need to be addressed is immunogenicity, which already has emerged as a potential roadblock of autologous CAR T cell therapies, especially when no lymphodepleting chemotherapy is given prior to T cell infusion (139). Thus, recipients of “off the shelf” cell products might require immune-modulation post cell infusion to enable their long-term persistence. However, given the extensive experience with allogeneic HCT and solid organ transplantation, we believe that immunogenicity will not present an unsurmountable barrier. While production of “off the shelf” therapeutic products require increased resources during the development and manufacturing process, the ultimate goal is to develop cell products that have a favorable safety and efficacy profile and are widely accessible and affordable. However, at present it is too early to estimate the cost of an allogeneic cell product; this will depend on the required genetic modifications, which might include not only viral transduction but also gene-editing. Another driving factor of cost will be how many cell doses can be prepared from one lot of “off the shelf” cell products since release testing of genetically-modified cell products is cost intense. Nevertheless, we believe that continued investment in the optimization of these allogeneic strategies is warranted based on the current data and that allogeneic cell products will usher in a new era of cell therapy.
Figures created with BioRender.com.
KC, SG, and AT wrote, reviewed, and edited the manuscript. All authors contributed to the article and approved the submitted version.
The work of the authors is in part supported by the National Cancer Institute/National Institutes of Health grant P30CA021765. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
SG has patent applications in the field of immunotherapy, is a DSMB member of Immatics, and on the scientific advisory board of Tidal.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Eshhar Z, Waks T, Gross G, Schindler DG. Specific activation and targeting of cytotoxic lymphocytes through chimeric single chains consisting of antibody-binding domains and the gamma or zeta subunits of the immunoglobulin and T-cell receptors. Proc Natl Acad Sci USA (1993) 90(2):720–4. doi: 10.1073/pnas.90.2.720
2. Gross G, Gorochov G, Waks T, Eshhar Z. Generation of effector T cells expressing chimeric T cell receptor with antibody type-specificity. Transplant Proc (1989) 21(1 Pt 1):127–30.
3. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci USA (1989) 86(24):10024–8. doi: 10.1073/pnas.86.24.10024
4. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med (2013) 368(16):1509–18. doi: 10.1056/NEJMoa1215134
5. Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med (2011) 365(8):725–33. doi: 10.1056/NEJMoa1103849
6. Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in Children and Young Adults with B-Cell Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):439–48. doi: 10.1056/NEJMoa1709866
7. Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N Engl J Med (2017) 377(26):2531–44. doi: 10.1056/NEJMoa1707447
8. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med (2019) 380(1):45–56. doi: 10.1056/NEJMoa1804980
9. Wang M, Munoz J, Goy A, Locke FL, Jacobson CA, Hill BT, et al. KTE-X19 CAR T-Cell Therapy in Relapsed or Refractory Mantle-Cell Lymphoma. N Engl J Med (2020) 382(14):1331–42. doi: 10.1056/NEJMoa1914347
10. Ramos CA, Grover NS, Beaven AW, Lulla PD, Wu M-F, Ivanova A, et al. Anti-CD30 CAR-T Cell Therapy in Relapsed and Refractory Hodgkin Lymphoma. J Clin Oncol (2020) 38(32):3794–804. doi: 10.1200/JCO.20.01342
11. Raje N, Berdeja J, Lin Y, Siegel D, Jagannath S, Madduri D, et al. Anti-BCMA CAR T-Cell Therapy bb2121 in Relapsed or Refractory Multiple Myeloma. N Engl J Med (2019) 380(18):1726–37. doi: 10.1056/NEJMoa1817226
12. Shah NN, Highfill SL, Shalabi H, Yates B, Jin J, Wolters PL, et al. CD4/CD8 T-Cell Selection Affects Chimeric Antigen Receptor (CAR) T-Cell Potency and Toxicity: Updated Results From a Phase I Anti-CD22 CAR T-Cell Trial. J Clin Oncol (2020) 38(17):1938–50. doi: 10.1200/JCO.19.03279
13. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG, et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood (2012) 119(17):3940–50. doi: 10.1182/blood-2011-10-387969
14. Roddie C, O’Reilly M, Dias Alves Pinto J, Vispute K, Lowdell M. Manufacturing chimeric antigen receptor T cells: issues and challenges. Cytotherapy (2019) 21(3):327–40. doi: 10.1016/j.jcyt.2018.11.009
15. Thommen DS, Schumacher TN. T Cell Dysfunction in Cancer. Cancer Cell (2018) 33(4):547–62. doi: 10.1016/j.ccell.2018.03.012
16. Lin JK, Lerman BJ, Barnes JI, Boursiquot BC, Tan YJ, Robinson AQL, et al. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Relapsed or Refractory Pediatric B-Cell Acute Lymphoblastic Leukemia. J Clin Oncol (2018) 36(32):3192–202. doi: 10.1200/JCO.2018.79.0642
17. Lin JK, Muffly LS, Spinner MA, Barnes JI, Owens DK, Goldhaber-Fiebert JD. Cost Effectiveness of Chimeric Antigen Receptor T-Cell Therapy in Multiply Relapsed or Refractory Adult Large B-Cell Lymphoma. J Clin Oncol (2019) 37(24):2105–19. doi: 10.1200/JCO.18.02079
18. Zeiser R, Blazar BR. Acute Graft-versus-Host Disease - Biologic Process, Prevention, and Therapy. N Engl J Med (2017) 377(22):2167–79. doi: 10.1056/NEJMra1609337
19. Carapito R, Jung N, Kwemou M, Untrau M, Michel S, Pichot A, et al. Matching for the nonconventional MHC-I MICA gene significantly reduces the incidence of acute and chronic GVHD. Blood (2016) 128(15):1979–86. doi: 10.1182/blood-2016-05-719070
20. Martin PJ, Levine DM, Storer BE, Warren EH, Zheng X, Nelson SC, et al. Genome-wide minor histocompatibility matching as related to the risk of graft-versus-host disease. Blood (2017) 129(6):791–8. doi: 10.1182/blood-2016-09-737700
21. Santos N, Rodríguez-Romanos R, Nieto JB, Buño I, Vallejo C, Jiménez-Velasco A, et al. UGT2B17 minor histocompatibility mismatch and clinical outcome after HLA-identical sibling donor stem cell transplantation. Bone Marrow Transplant (2016) 51(1):79–82. doi: 10.1038/bmt.2015.207
22. Baker MB, Altman NH, Podack ER, Levy RB. The role of cell-mediated cytotoxicity in acute GVHD after MHC-matched allogeneic bone marrow transplantation in mice. J Exp Med (1996) 183(6):2645–56. doi: 10.1084/jem.183.6.2645
23. Graubert TA, DiPersio JF, Russell JH, Ley TJ. Perforin/granzyme-dependent and independent mechanisms are both important for the development of graft-versus-host disease after murine bone marrow transplantation. J Clin Invest (1997) 100(4):904–11. doi: 10.1172/JCI119606
24. Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation (1974) 18(4):295–304. doi: 10.1097/00007890-197410000-00001
25. Harris AC, Young R, Devine S, Hogan WJ, Ayuk F, Bunworasate U, et al. International, Multicenter Standardization of Acute Graft-versus-Host Disease Clinical Data Collection: A Report from the Mount Sinai Acute GVHD International Consortium. Biol Blood Marrow Transplant (2016) 22(1):4–10. doi: 10.1016/j.bbmt.2015.09.001
26. Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest (2015) 125(7):2677–89. doi: 10.1172/JCI81229
27. Kröger N, Solano C, Wolschke C, Bandini G, Patriarca F, Pini M, et al. Antilymphocyte Globulin for Prevention of Chronic Graft-versus-Host Disease. N Engl J Med (2016) 374(1):43–53. doi: 10.1056/NEJMoa1506002
28. Lang P, Feuchtinger T, Teltschik HM, Schwinger W, Schlegel P, Pfeiffer M, et al. Improved immune recovery after transplantation of TCRαβ/CD19-depleted allografts from haploidentical donors in pediatric patients. Bone Marrow Transplant (2015) 50(Suppl 2):S6–10. doi: 10.1038/bmt.2015.87
29. Urbano-Ispizua A, Rozman C, Martínez C, Marín P, Briones J, Rovira M, et al. Rapid engraftment without significant graft-versus-host disease after allogeneic transplantation of CD34+ selected cells from peripheral blood. Blood (1997) 89(11):3967–73. doi: 10.1182/blood.V89.11.3967
30. Zeiser R, Leveson-Gower DB, Zambricki EA, Kambham N, Beilhack A, Loh J, et al. Differential impact of mammalian target of rapamycin inhibition on CD4+CD25+Foxp3+ regulatory T cells compared with conventional CD4+ T cells. Blood (2008) 111(1):453–62. doi: 10.1182/blood-2007-06-094482
31. Finney OC, Brakke HM, Rawlings-Rhea S, Hicks R, Doolittle D, Lopez M, et al. CD19 CAR T cell product and disease attributes predict leukemia remission durability. J Clin Invest (2019) 129(5):2123–32. doi: 10.1172/JCI125423
32. Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol (2005) 26(2):111–7. doi: 10.1016/j.it.2004.12.003
33. Ciurea SO, Cao K, Fernandez-Vina M, Kongtim P, Malki MA, Fuchs E, et al. The European Society for Blood and Marrow Transplantation (EBMT) Consensus Guidelines for the Detection and Treatment of Donor-specific Anti-HLA Antibodies (DSA) in Haploidentical Hematopoietic Cell Transplantation. Bone Marrow Transplant (2018) 53(5):521–34. doi: 10.1038/s41409-017-0062-8
34. Kreslavsky T, Gleimer M, Garbe AI, von Boehmer H. αβ versus γδ fate choice: counting the T-cell lineages at the branch point. Immunol Rev (2010) 238(1):169–81. doi: 10.1111/j.1600-065X.2010.00947.x
35. Pauza CD, Liou ML, Lahusen T, Xiao L, Lapidus RG, Cairo C, et al. Gamma Delta T Cell Therapy for Cancer: It Is Good to be Local. Front Immunol (2018) 9:1305. doi: 10.3389/fimmu.2018.01305
36. Legut M, Cole DK, Sewell AK. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell Mol Immunol (2015) 12(6):656–68. doi: 10.1038/cmi.2015.28
37. Koch S, Larbi A, Derhovanessian E, Ozcelik D, Naumova E, Pawelec G. Multiparameter flow cytometric analysis of CD4 and CD8 T cell subsets in young and old people. Immun Ageing (2008) 5:6. doi: 10.1186/1742-4933-5-6
38. van den Broek T, Borghans JAM, van Wijk F. The full spectrum of human naive T cells. Nat Rev Immunol (2018) 18(6):363–73. doi: 10.1038/s41577-018-0001-y
39. Deol A, Lum LG. Role of donor lymphocyte infusions in relapsed hematological malignancies after stem cell transplantation revisited. Cancer Treat Rev (2010) 36(7):528–38. doi: 10.1016/j.ctrv.2010.03.004
40. Kolb HJ, Mittermüller J, Clemm C, Holler E, Ledderose G, Brehm G, et al. Donor leukocyte transfusions for treatment of recurrent chronic myelogenous leukemia in marrow transplant patients. Blood (1990) 76(12):2462–5. doi: 10.1182/blood.V76.12.2462.2462
41. Pati AR, Godder K, Lamb L, Gee A, Henslee-Downey PJ. Immunotherapy with donor leukocyte infusions for patients with relapsed acute myeloid leukemia following partially mismatched related donor bone marrow transplantation. Bone Marrow Transpl (1995) 15(6):979–81.
42. Brudno JN, Somerville RP, Shi V, Rose JJ, Halverson DC, Fowler DH, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol (2016) 34(10):1112–21. doi: 10.1200/JCO.2015.64.5929
43. Kebriaei P, Singh H, Huls MH, Figliola MJ, Bassett R, Olivares S, et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J Clin Invest (2016) 126(9):3363–76. doi: 10.1172/JCI86721
44. Kebriaei P, Ciurea SO, Huls MH, Singh H, Olivares S, Su S, et al. Pre-Emptive Donor Lymphocyte Infusion with CD19-Directed, CAR-Modified T Cells Infused after Allogeneic Hematopoietic Cell Transplantation for Patients with Advanced CD19+ Malignancies. Blood (2015) 126(23):862–. doi: 10.1182/blood.V126.23.862.862
45. Tzannou I, Papadopoulou A, Naik S, Leung K, Martinez CA, Ramos CA, et al. Off-the-Shelf Virus-Specific T Cells to Treat BK Virus, Human Herpesvirus 6, Cytomegalovirus, Epstein-Barr Virus, and Adenovirus Infections After Allogeneic Hematopoietic Stem-Cell Transplantation. J Clin Oncol (2017) 35(31):3547–57. doi: 10.1200/JCO.2017.73.0655
46. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virus-specific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood (2013) 121(26):5113–23. doi: 10.1182/blood-2013-02-486324
47. Melenhorst JJ, Leen AM, Bollard CM, Quigley MF, Price DA, Rooney CM, et al. Allogeneic virus-specific T cells with HLA alloreactivity do not produce GVHD in human subjects. Blood (2010) 116(22):4700–2. doi: 10.1182/blood-2010-06-289991
48. O’Reilly RJ, Prockop S, Hasan AN, Koehne G, Doubrovina E. Virus-specific T-cell banks for ‘off the shelf’ adoptive therapy of refractory infections. Bone Marrow Transpl (2016) 51(9):1163–72. doi: 10.1038/bmt.2016.17
49. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med (2008) 14(11):1264–70. doi: 10.1038/nm.1882
50. Savoldo B, Rooney CM, Di Stasi A, Abken H, Hombach A, Foster AE, et al. Epstein Barr virus–specific cytotoxic T lymphocytes expressing the anti-CD30ζ artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood (2007) 110(7):2620–30. doi: 10.1182/blood-2006-11-059139
51. Ahmed N, Brawley V, Hegde M, Bielamowicz K, Kalra M, Landi D, et al. Specific Chimeric Antigen Receptor-Modified Virus-Specific T Cells for Progressive Glioblastoma: A Phase 1 Dose-Escalation Trial. HER2-JAMA Oncol (2017) 3(8):1094–101. doi: 10.1001/jamaoncol.2017.0184
52. Cruz CR, Micklethwaite KP, Savoldo B, Ramos CA, Lam S, Ku S, et al. Infusion of donor-derived CD19-redirected virus-specific T cells for B-cell malignancies relapsed after allogeneic stem cell transplant: a phase 1 study. Blood (2013) 122(17):2965–73. doi: 10.1182/blood-2013-06-506741
53. Curran KJ, Sauter CS, Kernan NA, Prockop SE, Boulad F, Perales M, et al. Durable Remission Following “Off-the-Shelf” Chimeric Antigen Receptor (CAR) T-Cells in Patients with Relapse/Refractory (R/R) B-Cell Malignancies. Biol Blood Marrow Transplant (2020) 26(3, Supplement):S89. doi: 10.1016/j.bbmt.2019.12.590
54. Anderson BE, McNiff J, Yan J, Doyle H, Mamula M, Shlomchik MJ, et al. Memory CD4+ T cells do not induce graft-versus-host disease. J Clin Invest (2003) 112(1):101–8. doi: 10.1172/JCI17601
55. Golubovskaya V, Wu L. Different Subsets of T Cells, Memory, Effector Functions, and CAR-T Immunotherapy. Cancers (Basel) (2016) 8(3):36. doi: 10.3390/cancers8030036
56. Riddell SR, Sommermeyer D, Berger C, Liu LS, Balakrishnan A, Salter A, et al. Adoptive therapy with chimeric antigen receptor-modified T cells of defined subset composition. Cancer J (2014) 20(2):141–4. doi: 10.1097/PPO.0000000000000036
57. Singh N, Perazzelli J, Grupp SA, Barrett DM. Early memory phenotypes drive T cell proliferation in patients with pediatric malignancies. Sci Transl Med (2016) 8(320):320ra3. doi: 10.1126/scitranslmed.aad5222
58. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O, et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR.CD19-T cells and are preserved by IL-7 and IL-15. Blood (2014) 123(24):3750–9. doi: 10.1182/blood-2014-01-552174
59. Priesner C, Aleksandrova K, Esser R, Mockel-Tenbrinck N, Leise J, Drechsel K, et al. Automated Enrichment, Transduction, and Expansion of Clinical-Scale CD62L+ T Cells for Manufacturing of Gene Therapy Medicinal Products. Hum Gene Ther (2016) 27(10):860–9. doi: 10.1089/hum.2016.091
60. Sabatino M, Hu J, Sommariva M, Gautam S, Fellowes V, Hocker JD, et al. Generation of clinical-grade CD19-specific CAR-modified CD8+ memory stem cells for the treatment of human B-cell malignancies. Blood (2016) 128(4):519–28. doi: 10.1182/blood-2015-11-683847
61. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi: 10.1038/leu.2015.247
62. Sallusto F, Geginat J, Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu Rev Immunol (2004) 22:745–63. doi: 10.1146/annurev.immunol.22.012703.104702
63. Maschan M, Blagov S, Shelikhova L, Shekhovtsova Z, Balashov D, Starichkova J, et al. Low-dose donor memory T-cell infusion after TCR alpha/beta depleted unrelated and haploidentical transplantation: results of a pilot trial. Bone Marrow Transplant (2018) 53(3):264–73. doi: 10.1038/s41409-017-0035-y
64. Muffly L, Sheehan K, Armstrong R, Jensen K, Tate K, Rezvani AR, et al. Infusion of donor-derived CD8(+) memory T cells for relapse following allogeneic hematopoietic cell transplantation. Blood Adv (2018) 2(6):681–90. doi: 10.1182/bloodadvances.2017012104
65. Shook DR, Triplett BM, Eldridge PW, Kang G, Srinivasan A, Leung W. Haploidentical stem cell transplantation augmented by CD45RA negative lymphocytes provides rapid engraftment and excellent tolerability. Pediatr Blood Cancer (2015) 62(4):666–73. doi: 10.1002/pbc.25352
66. Triplett BM, Shook DR, Eldridge P, Li Y, Kang G, Dallas M, et al. Rapid memory T-cell reconstitution recapitulating CD45RA-depleted haploidentical transplant graft content in patients with hematologic malignancies. Bone Marrow Transplant (2015) 50(7):1012. doi: 10.1038/bmt.2015.139
67. Talleur AC, Li Y, Akel S, Sharma A, Qudeimat A, Srinivasan A, et al. Haploidentical CD45RA-Negative Donor Lymphocyte Infusions Are Feasible, Safe and Associated with Clinical Benefit. Biol Blood Marrow Transplant (2020) 26(3, Supplement):S268. doi: 10.1016/j.bbmt.2019.12.435
68. Chan WK, Suwannasaen D, Throm RE, Li Y, Eldridge PW, Houston J, et al. Chimeric antigen receptor-redirected CD45RA-negative T cells have potent antileukemia and pathogen memory response without graft-versus-host activity. Leukemia (2015) 29(2):387–95. doi: 10.1038/leu.2014.174
69. Fernández L, Fernández A, Mirones I, Escudero A, Cardoso L, Vela M, et al. GMP-Compliant Manufacturing of NKG2D CAR Memory T Cells Using CliniMACS Prodigy. Front Immunol (2019) 10:2361. doi: 10.3389/fimmu.2019.02361
70. Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood (2012) 119(1):72–82. doi: 10.1182/blood-2011-07-366419
71. Talleur AC, Métais J-Y, Li Y, Throm RE, Tillman H, Rooney B, et al. Allogeneic CD27-depleted cells in adoptive cell therapy. Adv Cell Gene Ther (2019) 2(2):e45. doi: 10.1002/acg2.45
72. Torikai H, Reik A, Liu PQ, Zhou Y, Zhang L, Maiti S, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood (2012) 119(24):5697–705. doi: 10.1182/blood-2012-01-405365
73. Osborn MJ, Webber BR, Knipping F, Lonetree CL, Tennis N, DeFeo AP, et al. Evaluation of TCR Gene Editing Achieved by TALENs, CRISPR/Cas9, and megaTAL Nucleases. Mol Ther (2016) 24(3):570–81. doi: 10.1038/mt.2015.197
74. Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature (2017) 543(7643):113–7. doi: 10.1038/nature21405
75. Maciocia P, Wawrzyniecka P, Kassimatis L, Pule M. A Protein-Based Method to Develop Allogeneic Chimeric Antigen Receptor T-Cells. Blood (2018) 132(Supplement 1):700–. doi: 10.1182/blood-2018-99-112544
76. Stenger D, Stief TA, Kaeuferle T, Willier S, Rataj F, Schober K, et al. Endogenous TCR promotes in vivo persistence of CD19-CAR-T cells compared to a CRISPR/Cas9-mediated TCR knockout CAR. Blood (2020) 136(12):1407–18. doi: 10.1182/blood.2020005185
77. Wang D, Quan Y, Yan Q, Morales JE, Wetsel RA. Targeted Disruption of the β2-Microglobulin Gene Minimizes the Immunogenicity of Human Embryonic Stem Cells. Stem Cells Transl Med (2015) 4(10):1234–45. doi: 10.5966/sctm.2015-0049
78. Ren J, Liu X, Fang C, Jiang S, June CH, Zhao Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin Cancer Res (2017) 23(9):2255–66. doi: 10.1158/1078-0432.CCR-16-1300
79. Kagoya Y, Guo T, Yeung B, Saso K, Anczurowski M, Wang CH, et al. Genetic Ablation of HLA Class I, Class II, and the T-cell Receptor Enables Allogeneic T Cells to Be Used for Adoptive T-cell Therapy. Cancer Immunol Res (2020) 8(7):926–36. doi: 10.1158/2326-6066.CIR-18-0508
80. Brentjens RJ, Rivière I, Park JH, Davila ML, Wang X, Stefanski J, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood (2011) 118(18):4817–28. doi: 10.1182/blood-2011-04-348540
81. Hay KA, Turtle CJ. Chimeric Antigen Receptor (CAR) T Cells: Lessons Learned from Targeting of CD19 in B-Cell Malignancies. Drugs (2017) 77(3):237–45. doi: 10.1007/s40265-017-0690-8
82. Jensen MC, Popplewell L, Cooper LJ, DiGiusto D, Kalos M, Ostberg JR, et al. Antitransgene rejection responses contribute to attenuated persistence of adoptively transferred CD20/CD19-specific chimeric antigen receptor redirected T cells in humans. Biol Blood Marrow Transplant (2010) 16(9):1245–56. doi: 10.1016/j.bbmt.2010.03.014
83. Levitsky J, Leventhal JR, Miller J, Huang X, Chen L, Chandrasekaran D, et al. Favorable effects of alemtuzumab on allospecific regulatory T-cell generation. Hum Immunol (2012) 73(2):141–9. doi: 10.1016/j.humimm.2011.11.008
84. Fernandez O. Alemtuzumab in the treatment of multiple sclerosis. J Inflammation Res (2014) 7:19–27. doi: 10.2147/JIR.S38079
85. Poirot L, Philip B, Schiffer-Mannioui C, Le Clerre D, Chion-Sotinel I, Derniame S, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res (2015) 75(18):3853–64. doi: 10.1158/0008-5472.CAN-14-3321
86. Chakrabarti S, Mackinnon S, Chopra R, Kottaridis PD, Peggs K, O’Gorman P, et al. High incidence of cytomegalovirus infection after nonmyeloablative stem cell transplantation: potential role of Campath-1H in delaying immune reconstitution. Blood (2002) 99(12):4357–63. doi: 10.1182/blood.V99.12.4357
87. Chakrabarti S, Mautner V, Osman H, Collingham KE, Fegan CD, Klapper PE, et al. Adenovirus infections following allogeneic stem cell transplantation: incidence and outcome in relation to graft manipulation, immunosuppression, and immune recovery. Blood (2002) 100(5):1619–27. doi: 10.1182/blood-2002-02-0377
88. Mo F, Watanabe N, McKenna MK, Hicks MJ, Srinivasan M, Gomes-Silva D, et al. Engineered off-the-shelf therapeutic T cells resist host immune rejection. Nat Biotechnol (2020). doi: 10.1038/s41587-020-0601-5
89. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med (2017) 9(374). doi: 10.1126/scitranslmed.aaj2013
90. Benjamin R, Graham C, Yallop D, Jozwik A, Ciocarlie O, Jain N, et al. Preliminary Data on Safety, Cellular Kinetics and Anti-Leukemic Activity of UCART19, an Allogeneic Anti-CD19 CAR T-Cell Product, in a Pool of Adult and Pediatric Patients with High-Risk CD19+ Relapsed/Refractory B-Cell Acute Lymphoblastic Leukemia. Blood (2018) 132(Supplement 1):896–. doi: 10.1182/blood-2018-99-111356
91. Neelapu SS, Munoz J, Locke FL, Miklos DB, Brown R, McDevitt JT, et al. First-in-human data of ALLO-501 and ALLO-647 in relapsed/refractory large B-cell or follicular lymphoma (R/R LBCL/FL): ALPHA study. J Clin Oncol (2020) 38(15_suppl):8002–. doi: 10.1200/JCO.2020.38.15_suppl.8002
92. Wang X, Li S, Gao L, Yuan Z, Wu K, Liu L, et al. Abstract CT052: Clinical safety and efficacy study of TruUCAR™ GC027: The first-in-human, universal CAR-T therapy for adult relapsed/refractory T-cell acute lymphoblastic leukemia (r/r T-ALL). Cancer Res (2020) 80(16 Supplement):CT052–CT. doi: 10.1158/1538-7445.AM2020-CT052
93. Khairallah C, Chu TH, Sheridan BS. Tissue Adaptations of Memory and Tissue-Resident Gamma Delta T Cells. Front Immunol (2018) 9:2636. doi: 10.3389/fimmu.2018.02636
94. Wilhelm M, Smetak M, Schaefer-Eckart K, Kimmel B, Birkmann J, Einsele H, et al. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J Transl Med (2014) 12:45. doi: 10.1186/1479-5876-12-45
95. Scheper W, Sebestyen Z, Kuball J. Cancer Immunotherapy Using γδT Cells: Dealing with Diversity. Front Immunol (2014) 5:601. doi: 10.3389/fimmu.2014.00601
96. Deniger DC, Switzer K, Mi T, Maiti S, Hurton L, Singh H, et al. Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol Ther (2013) 21(3):638–47. doi: 10.1038/mt.2012.267
97. Capsomidis A, Benthall G, Van Acker HH, Fisher J, Kramer AM, Abeln Z, et al. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol Ther (2018) 26(2):354–65. doi: 10.1016/j.ymthe.2017.12.001
98. Kabelitz D, Serrano R, Kouakanou L, Peters C, Kalyan S. Cancer immunotherapy with γδ T cells: many paths ahead of us. Cell Mol Immunol (2020) 17(9):925–39. doi: 10.1038/s41423-020-0504-x
99. Bendelac A, Savage PB, Teyton L. The biology of NKT cells. Annu Rev Immunol (2007) 25:297–336. doi: 10.1146/annurev.immunol.25.022106.141711
100. Exley M, Garcia J, Wilson SB, Spada F, Gerdes D, Tahir SM, et al. CD1d structure and regulation on human thymocytes, peripheral blood T cells, B cells and monocytes. Immunology (2000) 100(1):37–47. doi: 10.1046/j.1365-2567.2000.00001.x
101. Salio M, Silk JD, Jones EY, Cerundolo V. Biology of CD1- and MR1-restricted T cells. Annu Rev Immunol (2014) 32:323–66. doi: 10.1146/annurev-immunol-032713-120243
102. Fujii S, Shimizu K, Klimek V, Geller MD, Nimer SD, Dhodapkar MV. Severe and selective deficiency of interferon-gamma-producing invariant natural killer T cells in patients with myelodysplastic syndromes. Br J Haematol (2003) 122(4):617–22. doi: 10.1046/j.1365-2141.2003.04465.x
103. Molling JW, Kölgen W, van der Vliet HJ, Boomsma MF, Kruizenga H, Smorenburg CH, et al. Peripheral blood IFN-gamma-secreting Valpha24+Vbeta11+ NKT cell numbers are decreased in cancer patients independent of tumor type or tumor load. Int J Cancer (2005) 116(1):87–93. doi: 10.1002/ijc.20998
104. Molling JW, Langius JA, Langendijk JA, Leemans CR, Bontkes HJ, van der Vliet HJ, et al. Low levels of circulating invariant natural killer T cells predict poor clinical outcome in patients with head and neck squamous cell carcinoma. J Clin Oncol (2007) 25(7):862–8. doi: 10.1200/JCO.2006.08.5787
105. Chaidos A, Patterson S, Szydlo R, Chaudhry MS, Dazzi F, Kanfer E, et al. Graft invariant natural killer T-cell dose predicts risk of acute graft-versus-host disease in allogeneic hematopoietic stem cell transplantation. Blood (2012) 119(21):5030–6. doi: 10.1182/blood-2011-11-389304
106. Leveson-Gower DB, Olson JA, Sega EI, Luong RH, Baker J, Zeiser R, et al. Low doses of natural killer T cells provide protection from acute graft-versus-host disease via an IL-4-dependent mechanism. Blood (2011) 117(11):3220–9. doi: 10.1182/blood-2010-08-303008
107. Rubio MT, Bouillié M, Bouazza N, Coman T, Trebeden-Nègre H, Gomez A, et al. Pre-transplant donor CD4(-) invariant NKT cell expansion capacity predicts the occurrence of acute graft-versus-host disease. Leukemia (2017) 31(4):903–12. doi: 10.1038/leu.2016.281
108. Heczey A, Liu D, Tian G, Courtney AN, Wei J, Marinova E, et al. Invariant NKT cells with chimeric antigen receptor provide a novel platform for safe and effective cancer immunotherapy. Blood (2014) 124(18):2824–33. doi: 10.1182/blood-2013-11-541235
109. Tian G, Courtney AN, Jena B, Heczey A, Liu D, Marinova E, et al. CD62L+ NKT cells have prolonged persistence and antitumor activity in vivo. J Clin Invest (2016) 126(6):2341–55. doi: 10.1172/JCI83476
110. Rotolo A, Caputo VS, Holubova M, Baxan N, Dubois O, Chaudhry MS, et al. Enhanced Anti-lymphoma Activity of CAR19-iNKT Cells Underpinned by Dual CD19 and CD1d Targeting. Cancer Cell (2018) 34(4):596–610.e11. doi: 10.1016/j.ccell.2018.08.017
111. Heczey A, Courtney AN, Montalbano A, Robinson S, Liu K, Li M, et al. Anti-GD2 CAR-NKT cells in patients with relapsed or refractory neuroblastoma: an interim analysis. Nat Med (2020) 26(11):1686–90. doi: 10.1038/s41591-020-1074-2
112. Wu J, Lanier LL. Natural killer cells and cancer. Adv Cancer Res (2003) 90:127–56. doi: 10.1016/S0065-230X(03)90004-2
113. Olson JA, Leveson-Gower DB, Gill S, Baker J, Beilhack A, Negrin RS. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood (2010) 115(21):4293–301. doi: 10.1182/blood-2009-05-222190
114. Algarra I, García-Lora A, Cabrera T, Ruiz-Cabello F, Garrido F. The selection of tumor variants with altered expression of classical and nonclassical MHC class I molecules: implications for tumor immune escape. Cancer Immunol Immunother (2004) 53(10):904–10. doi: 10.1007/s00262-004-0517-9
115. Costello RT, Gastaut JA, Olive D. Tumor escape from immune surveillance. Arch Immunol Ther Exp (Warsz) (1999) 47(2):83–8.
116. Mehta RS, Rezvani K. Chimeric Antigen Receptor Expressing Natural Killer Cells for the Immunotherapy of Cancer. Front Immunol (2018) 9:283. doi: 10.3389/fimmu.2018.00283
117. Nguyen R, Wu H, Pounds S, Inaba H, Ribeiro RC, Cullins D, et al. A phase II clinical trial of adoptive transfer of haploidentical natural killer cells for consolidation therapy of pediatric acute myeloid leukemia. J Immunother Cancer (2019) 7(1):81. doi: 10.1186/s40425-019-0564-6
118. Rubnitz JE, Inaba H, Kang G, Gan K, Hartford C, Triplett BM, et al. Natural killer cell therapy in children with relapsed leukemia. Pediatr Blood Cancer (2015) 62(8):1468–72. doi: 10.1002/pbc.25555
119. Rubnitz JE, Inaba H, Ribeiro RC, Pounds S, Rooney B, Bell T, et al. NKAML: a pilot study to determine the safety and feasibility of haploidentical natural killer cell transplantation in childhood acute myeloid leukemia. J Clin Oncol (2010) 28(6):955–9. doi: 10.1200/JCO.2009.24.4590
120. Geller MA, Cooley S, Judson PL, Ghebre R, Carson LF, Argenta PA, et al. A phase II study of allogeneic natural killer cell therapy to treat patients with recurrent ovarian and breast cancer. Cytotherapy (2011) 13(1):98–107. doi: 10.3109/14653249.2010.515582
121. Zhao Q, Ahmed M, Tassev DV, Hasan A, Kuo TY, Guo HF, et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia (2015) 29(11):2238–47. doi: 10.1038/leu.2015.125
122. Esser R, Müller T, Stefes D, Kloess S, Seidel D, Gillies SD, et al. NK cells engineered to express a GD2 -specific antigen receptor display built-in ADCC-like activity against tumour cells of neuroectodermal origin. J Cell Mol Med (2012) 16(3):569–81. doi: 10.1111/j.1582-4934.2011.01343.x
123. Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood (2005) 106(1):376–83. doi: 10.1182/blood-2004-12-4797
124. Shimasaki N, Fujisaki H, Cho D, Masselli M, Lockey T, Eldridge P, et al. A clinically adaptable method to enhance the cytotoxicity of natural killer cells against B-cell malignancies. Cytotherapy (2012) 14(7):830–40. doi: 10.3109/14653249.2012.671519
125. Chu Y, Hochberg J, Yahr A, Ayello J, van de Ven C, Barth M, et al. Targeting CD20+ Aggressive B-cell Non-Hodgkin Lymphoma by Anti-CD20 CAR mRNA-Modified Expanded Natural Killer Cells In Vitro and in NSG Mice. Cancer Immunol Res (2015) 3(4):333–44. doi: 10.1158/2326-6066.CIR-14-0114
126. Müller T, Uherek C, Maki G, Chow KU, Schimpf A, Klingemann HG, et al. Expression of a CD20-specific chimeric antigen receptor enhances cytotoxic activity of NK cells and overcomes NK-resistance of lymphoma and leukemia cells. Cancer Immunol Immunother (2008) 57(3):411–23. doi: 10.1007/s00262-007-0383-3
127. Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med (2020) 382(6):545–53. doi: 10.1056/NEJMoa1910607
128. Veluchamy JP, Kok N, van der Vliet HJ, Verheul HMW, de Gruijl TD, Spanholtz J. The Rise of Allogeneic Natural Killer Cells As a Platform for Cancer Immunotherapy: Recent Innovations and Future Developments. Front Immunol (2017) 8(631). doi: 10.3389/fimmu.2017.00631
129. Tang X, Yang L, Li Z, Nalin AP, Dai H, Xu T, et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am J Cancer Res (2018) 8(6):1083–9.
130. Fujisaki H, Kakuda H, Shimasaki N, Imai C, Ma J, Lockey T, et al. Expansion of Highly Cytotoxic Human Natural Killer Cells for Cancer Cell Therapy. Cancer Res (2009) 69(9):4010–7. doi: 10.1158/0008-5472.CAN-08-3712
131. Lee DA. Cellular therapy: Adoptive immunotherapy with expanded natural killer cells. Immunol Rev (2019) 290(1):85–99. doi: 10.1111/imr.12793
132. Shimasaki N, Coustan-Smith E, Kamiya T, Campana D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy (2016) 18(11):1422–34. doi: 10.1016/j.jcyt.2016.06.013
133. Oyer J, Gitto SB, Khederzadeh S, Shaver K, Lee DA, Altomare D, et al. Assessment of antitumor function of NK cells expanded with exosomes from K562.mb21 cells. J Clin Oncol (2017) 35(7_suppl):132–. doi: 10.1200/JCO.2017.35.7_suppl.132
134. Ueda T, Kumagai A, Iriguchi S, Yasui Y, Miyasaka T, Nakagoshi K, et al. Non-clinical efficacy, safety and stable clinical cell processing of induced pluripotent stem cell-derived anti-glypican-3 chimeric antigen receptor-expressing natural killer/innate lymphoid cells. Cancer Sci (2020) 111(5):1478–90. doi: 10.1111/cas.14374
135. Cichocki F, Bjordahl R, Gaidarova S, Mahmood S, Abujarour R, Wang H, et al. iPSC-derived NK cells maintain high cytotoxicity and enhance in vivo tumor control in concert with T cells and anti–PD-1 therapy. Sci Trans Med (2020) 12(568):eaaz5618. doi: 10.1126/scitranslmed.aaz5618
136. Zhu H, Blum RH, Bernareggi D, Ask EH, Wu Z, Hoel HJ, et al. Metabolic Reprograming via Deletion of CISH in Human iPSC-Derived NK Cells Promotes In Vivo Persistence and Enhances Anti-tumor Activity. Cell Stem Cell (2020) 27(2):224–37.e6. doi: 10.1016/j.stem.2020.05.008
137. Li Y, Hermanson DL, Moriarity BS, Kaufman DS. Human iPSC-Derived Natural Killer Cells Engineered with Chimeric Antigen Receptors Enhance Anti-tumor Activity. Cell Stem Cell (2018) 23(2):181–92.e5. doi: 10.1016/j.stem.2018.06.002
138. Shankar K, Capitini CM, Saha K. Genome engineering of induced pluripotent stem cells to manufacture natural killer cell therapies. Stem Cell Res Ther (2020) 11(1):234–. doi: 10.1186/s13287-020-01741-4
Keywords: allogeneic, CAR, cell therapy, immunotherapy, cancer
Citation: Caldwell KJ, Gottschalk S and Talleur AC (2021) Allogeneic CAR Cell Therapy—More Than a Pipe Dream. Front. Immunol. 11:618427. doi: 10.3389/fimmu.2020.618427
Received: 16 October 2020; Accepted: 30 November 2020;
Published: 08 January 2021.
Edited by:
Alice Bertaina, Stanford University, United StatesReviewed by:
Kara Davis, Stanford University, United StatesCopyright © 2021 Caldwell, Gottschalk and Talleur. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Aimee C. Talleur, YWltZWUudGFsbGV1ckBzdGp1ZGUub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.