 Jin Kyung Seok
Jin Kyung Seok Han Chang Kang
Han Chang Kang Yong-Yeon Cho
Yong-Yeon Cho Hye Suk Lee
Hye Suk Lee Joo Young Lee
Joo Young Lee- BK21 PLUS Team, College of Pharmacy, The Catholic University of Korea, Bucheon, South Korea
Inflammation is a host protection mechanism that eliminates invasive pathogens from the body. However, chronic inflammation, which occurs repeatedly and continuously over a long period, can directly damage tissues and cause various inflammatory and autoimmune diseases. Pattern recognition receptors (PRRs) respond to exogenous infectious agents called pathogen-associated molecular patterns and endogenous danger signals called danger-associated molecular patterns. Among PRRs, recent advancements in studies of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome have established its significant contribution to the pathology of various inflammatory diseases, including metabolic disorders, immune diseases, cardiovascular diseases, and cancer. The regulation of NLRP3 activation is now considered to be important for the development of potential therapeutic strategies. To this end, there is a need to elucidate the regulatory mechanism of NLRP3 inflammasome activation by multiple signaling pathways, post-translational modifications, and cellular organelles. In this review, we discuss the intracellular signaling events, post-translational modifications, small molecules, and phytochemicals participating in the regulation of NLRP3 inflammasome activation. Understanding how intracellular events and small molecule inhibitors regulate NLRP3 inflammasome activation will provide crucial information for elucidating the associated host defense mechanism and the development of efficient therapeutic strategies for chronic diseases.
Activation of the NLRP3 Inflammasome
NLRP3 is an intracellular sensor in the NLRP3 inflammasome that recognizes the widest range of pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) among NLRP family members. NLRP3 has three domains: an amino-terminal pyrin domain (PYD) that binds to ASC, a NACHT domain with ATPase activity, and an LRR domain that induces autorepression by folding back onto the NACHT domain (1). The ATPase activity of the NACHT domain in particular has been studied as a therapeutic target for NLRP3-related diseases (1).
ASC is an adapter protein that acts as a bridge between NLRP3 and caspase-1. ASC is a bipartite complex consisting of a PYD domain that interacts with NLRP3 and a CARD domain that interacts with caspase-1. The PYD domain of ASC is also required for its self-association as well as its interaction with NLRP3 (2).
Caspase-1 is a cysteine protease that is synthesized as a zymogen and is capable of processing members of the interleukin-1 (IL-1) family, such as IL-1β and IL-18 (3). Full-length caspase-1 has three domains: an amino-terminal CARD, a central large catalytic domain (p20) and a carboxy-terminal small catalytic subunit domain (p10) (4). Caspase-1 clusters on the ASC and is self-cleaving at the linker between p20 and p10, resulting in a complex of p33 (including CARD and p20) and p10. Caspase-1 remains bound to ASC and exhibits proteolytic activity, while additional processing between CARD and p20 releases p20 and p10 from the ASC (5). The released p20–p10 heterotetramer is unstable in the cell and terminates its protease activity (6).
Canonical NLRP3 inflammasome activation requires a priming step. The priming step triggers upregulation of NLRP3 and IL-1β gene as well as post-translational licensing of NLRP3 inflammasome. Priming is initiated by Toll-like receptor (TLR) activation and cytokines such as TNF and IL-1β. Through this process, NF-κB, a transcription factor, is activated and transcriptionally upregulates NLRP3 and pro IL-1β. Moreover, priming sets up NLRP3 to form inflammasome assembly (7) or rescues from degradation (8) by licensing the proteins to form the correct morphology for self-oligomerization and interaction with ASC, through various post-translational modifications (PTM) to NLRP3. PTMs include ubiquitylation, deubiquitination, phosphorylation, and sumoylation of NLRP3 (7). The PTMs of NLRP3 occur during processes such as priming, activation, and resolution. Thus, priming signals regulate NLRP3 inflammasome activation through transcription-dependent pathways and PTMs.
After priming, NLRP3 responds to activating stimuli and assembles the NLRP3 inflammasome complex. These stimuli are derived from PAMPs during pathogen infections or DAMPs released from damaged host cells and include bacterial or viral pathogens, fungi, ATP, pore-forming toxins, crystalline substances, nucleic acids, and hyaluronan. The mechanisms of NLRP3 inflammasome activation that occur in response to various stimuli include the efflux of potassium, the secretion of cathepsin into the cytoplasm following lysosome degradation, the translocation of NLRP3 to the mitochondria, the production of free radicals in the mitochondria, and the secretion of mitochondrial DNA or cardiolipin. Once stimulated, NLRP3 is oligomerized by homotypic interactions in the NACHT domains (9). Subsequently, oligomerized NLRP3 recruits ASC through PYD-PYD interactions and creates multiple helical ASC filaments that combine with ASC speck, a single macromolecule (10). Assembled ASC recruits caspase-1 through CARD-CARD interactions and activates caspase-1 through its self-cleavage (6). Then, the activated NLRP3 inflammasome hydrolyzes inactive pro-caspase-1 to activate caspase-1, and active caspase-1 then induces the production and secretion of inflammasome-specific cytokines such as IL-1β and IL-18 while simultaneously inducing pyroptosis, which is inflammatory cell death (11).
Pyroptosis is an inflammatory form of lytic programmed cell death that is activated by NLRP3 inflammasomes. Interestingly, a recent study showed that GSDMD is a crucial mediator of pyroptosis (12). The amino-terminal cell death domain of GSDMD (GSDMDN-term) possesses a central short linker region and a carboxy-terminal autoinhibition domain (13). Caspase-1 cleaves and releases GSDMD from the carboxyl terminus to overcome intramolecular inhibition, after which GSDMDN-term combines with phosphatidylinositol phosphates and phosphatidylserine in the inner leaflet of the cell membrane and oligomerizes. This oligomer is then inserted into the plasma membrane to form 10-14 nm pores containing 16 symmetrical protomers for cell killing (14, 15). Additionally, GSDMDN-term exhibits bactericidal activities by combining with cardiolipin, which is present in the internal and external bacterial membranes (14, 16). Cardiolipin is also present on the internal and external mitochondrial membranes following NLRP3 activation (17). However, it is unclear whether GSDMDN-term penetrates the mitochondria to combine with mitochondrial cardiolipin.
Full activation of the NLRP3 inflammasome is accomplished by a well-concerted mechanism which includes the priming step and the activation step (Figure 1). The priming is an essential prerequisite to induce expression of NLRP3 and pro-form of IL-1β and IL-18 while the activation step involves an inflammasome assembly in an organized fashion to make pro-caspase-1 activated. The combination of these two events is expected to commonly take place when the host is exposed to PAMPs and DAMPs since PAMPs and DAMPs can activate both priming and activation steps.
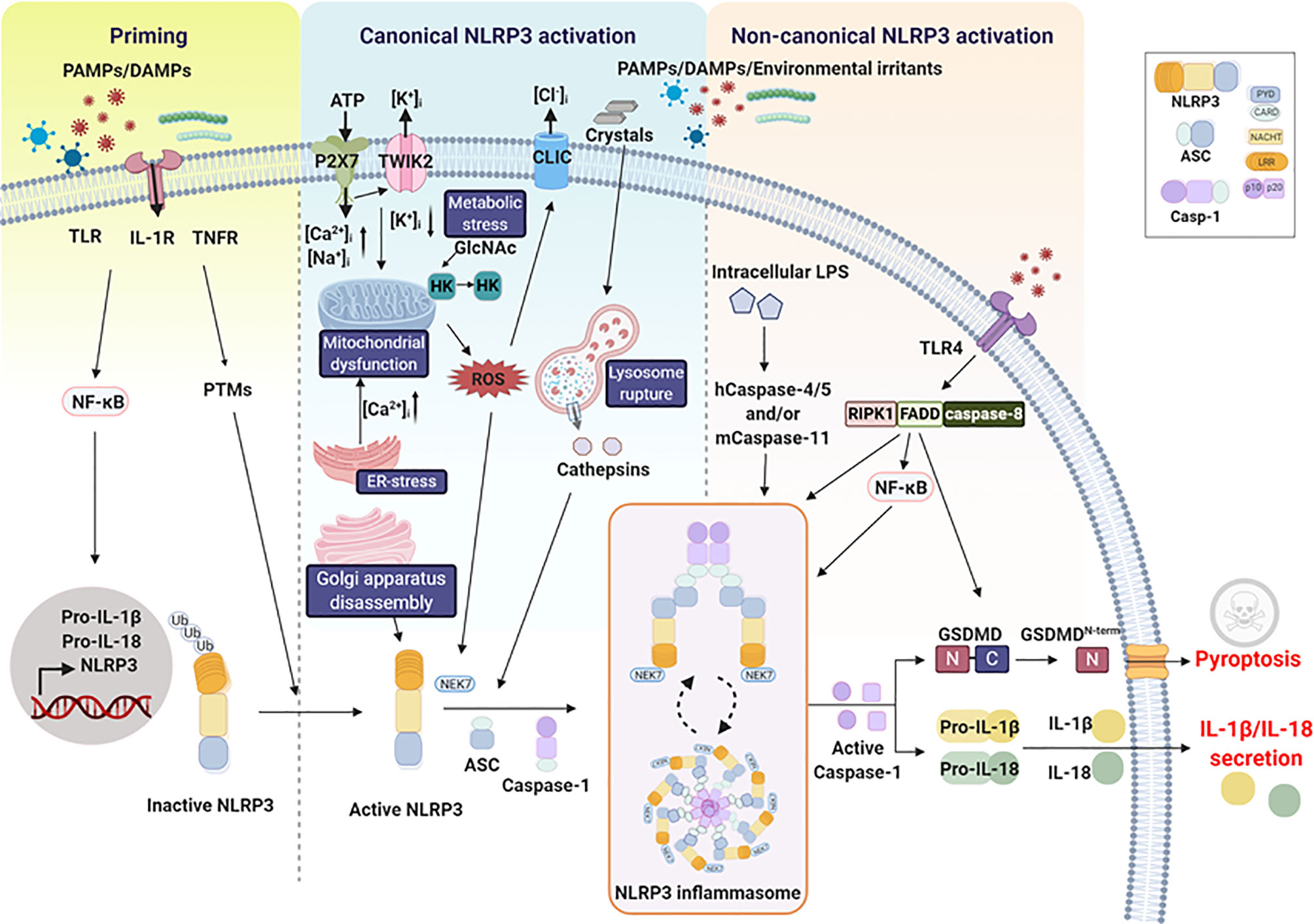
Figure 1 NLRP3 inflammasome pathway. Two steps are required for NLRP3 inflammasome activation. The first step is priming, which is triggered by microbial molecules or endogenous cytokines and is required to induce the expression of pro-interleukin-1β (IL-1β), pro-IL-18, and NLRP3 via the activation of the transcription factor nuclear factor-κB (NF-κB). The second step is NLRP3 inflammasome activation, which includes canonical and noncanonical activation pathways and is induced by a number of PAMPs and DAMPs. The canonical activation pathway involves stimulation-mediated activation signals such as ion fluxes, lysosome rupture, mitochondrial dysfunction, Golgi apparatus disassembly, metabolic stress, and ER stress. Activation of the inflammasome causes caspase-1 activation, leading to the maturation and release of IL-1β/IL-18 and pyroptosis. The noncanonical activation pathway is mediated by human caspase-4, human caspase-5, and mouse caspase-11, indirectly promoting the production of pro-IL-1β or pro-IL-18. Receptor-interacting protein kinase 1 (RIPK1), FAS-associated death domain protein (FADD), and caspase-8 are involved in this pathway by regulating NF-κB activation, leading to NLRP3 inflammasome activation.
Noncanonical and Alternative Pathways for NLRP3 Inflammasome Activation
Recently, caspase-4 and -5 in human and caspase-11 in mouse have been shown to indirectly promote pro-IL-1β or pro-IL-18 activation by inducing NLRP3 inflammasome activation as a noncanonical NLRP3 activation pathway (18, 19). The noncanonical pathway is initiated by direct binding of these caspases to intracellular LPS (iLPS) produced by Gram-negative bacteria, independent of TLR4, the conventional LPS receptor (20). Caspase-11 induces extracellular release of ATP, which in turn activates P2X7 receptor and induces K+ efflux, leading the activation of NLRP3 inflammasome and production of mature IL-1β (21). In addition, activated caspase-4, -5, and -11 cleave gasdermin D (GSDMD), resulting in pyroptosis (13, 16). Caspase-4, -5, and -11 initiate pyroptosis similarly to caspase-1, but they do not directly cleave pro-IL-1β or pro-IL-18 (22).
Caspase-8, the apical activator caspase, provides an alternative pathway of the NLRP3 inflammasome activation, culminating in IL-1β and IL-18 maturation as well as cell death (23, 24). Notably, caspase-8-dependent NLRP3 activation is a species-specific pathway that is present in human and porcine peripheral blood mononuclear cells (PBMCs), but not murine cells (25). TLR4 stimulation by PAMPs and/or DAMPs activates RIPK1-FADD-caspase-8 signaling, which can directly trigger canonical NLRP3 oligomerization and inflammasome assembly as well as facilitate NF-κB transcription (25). In addition, RIPK1-FADD-caspase-8 signaling is involved in TLR3 priming for activation of NLRP3 inflammasome (26). Blockade of TGF-β activated kinase-1 (TAK1) by the Yersinia bacteria leads to cleavage of GSDMD RIPK1- and caspase-8-dependently, promoting the NLRP3 inflammasome activation and IL-1β secretion as well as cell death (27). In contrast, a negative role of caspase-8 in regulation of NLRP3 inflammasome was reported (28). Caspase-8-deficient dendritic cells showed higher production of IL-1β with enhanced activation of NLRP3 inflammasome, which is dependent on the functions of RIPK1 and RIPK3 (28). Although the exact role of caspase-8 in the regulation of NLRP3 inflammasome and inflammatory processes remains to be determined, the studies suggest the possible link between NLRP3 inflammasome, apoptosis, and inflammation mediated by caspase-8.
Considering complexity of in vivo inflammatory processes, noncanonical and alternative pathways for the NLRP3 inflammasome activation in addition to canonical pathway may play differential roles in the pathology of inflammatory diseases in different context.
Intracellular Events Regulating NLRP3 Inflammasome Activation
Ion Fluxes
Most NLRP3 stimuli, including ATP, nigericin, and particulate matter induce K+ efflux (29). The P2X7 receptor, which belongs to the P2X subfamily of ligand-gated ion channels with purine P2 receptors, can be activated by high concentrations of extracellular ATP, leading to the,NLRP3 inflammasome activation (30). The P2X7 receptor plays an important role in mediating the innate immune response by regulating the expression of pro-inflammatory cytokines of the IL-1 family (31). After ATP stimulation, P2X7 promotes Ca2+ and Na+ influx and coordinates with the K+ channel, two-pore domain weakly inward rectifying K+ channel 2 (TWIK2), which mediates K+ efflux (32). Lowering cytoplasmic K+ concentration was sufficient to activate the NLRP3 inflammasome (33). Active caspase-11 led to a decrease of intracellular potassium K+ levels, resulting in the activation of the NLRP3 inflammasome, suggesting the link between noncanonical and canonical NLRP3 activation pathways (33). K+ efflux is also suggested as the mechanism for which cytopathogenic RNA viruses such as vesicular stomatitis virus (VSV) or encephalomyocarditis virus (EMCV) induce activation of the NLRP3 inflammasome (34). These cytopathogenic viruses triggered a lytic cell death, which led to K+ efflux (34).
Na+ influx plays a regulatory role in NLRP3 inflammasome activation, possibly by regulating stimulus-induced K+ efflux. Combination of K+ efflux with Na+ influx is considered necessary for activation of the NLRP3 inflammasome induced by crystalline and lysosomal destabilization (35). Overactivation of the epithelial Na+ channel, ENaC, aggravated NLRP3 inflammasome activation by enhancing Na+ influx and subsequently K+ efflux, culminating in the exacerbated inflammatory responses in cystic fibrosis (36). High salt treatment increased production of IL-1β in monocytes and dendritic cells while treatment with an ENaC inhibitor, amiloride, blocked IL-1β production in these cells (37). Similarly, high salt diet to mice induced increased expression of NLRP3 and pro‐IL-1β in monocytes and dendritic cells whereas treatment with amiloride, an ENaC inhibitor, to mice fed high salt diet showed less expression of NLRP3 and pro‐IL-1β, suggesting an ENaC-dependent activation of NLRP3 inflammasome in response to high salt (37).
The importance of chloride efflux in regulation of NLRP3 inflammasome activation has been noted and the involvement of the volume regulated anion channel (VRAC) was reported (38, 39). Knockout of LRRC8A, a subunit of VRAC, in macrophages resulted in significant impairment of ASC oligomerization, caspase-1 cleavage, and IL-1β processing induced by hypotonicity, showing the critical role of VRAC in the regulation of NLRP3 inflammasome activation (40). Chloride intracellular channels (CLICs) have been reported to regulate NLRP3 inflammasome activation (41). A nonsteroidal anti-inflammatory agent inhibited chloride leakage through VRAC to prevent NLRP3 inflammasome activation (42) and CLIC was suggested to function as a VRAC activator (43). Various Cl- channel inhibitors, including 4,4’-diisothiocyano-2,2’stilbene-disulfonic acid (DIDS) (43), 5-nitro-(3-phenylpropylamino) benzoic acid (NPPB) (38), flufenamic acid, mefenamic acid (42), and indanyloxyacetic acid 94 (IAA94) (43), block the NLRP3 inflammasome but cannot block the NLRC4 or AIM2 inflammasomes. CLICs translocate to the plasma membrane and promote NLRP3-NEK7 interactions, triggering Cl- flux as a downstream event of mitochondrial dysfunction that regulates NLRP3 inflammasome activation (43). Cl- efflux can induce ASC speck formation but does not induce NLRP3 inflammasome activation without K+ efflux (44). Further studies are needed to elucidate how Cl- flux coordinates with other ionic events to trigger NLRP3 inflammasome activation.
Lysosomal Destabilization
The phagocytosis of particulates causes lysosomal rupture, releasing particulates into the cytoplasm. Lysosomal destabilization was first revealed as a pathway mediating the activation of NLRP3 inflammasome by amyloid β, a pathogenic misfolded protein expressed in Alzheimer’s disease (45). The accumulation of crystals such as β-amyloid, monosodium urate (MSU), silica, and asbestos, in the cell destabilizes phagosomes and leads to the release of various components, including proteases, lipases, cathepsins, and Ca 2+ in the cytosol, leading to K+ efflux and the induction of NLRP3 assembly and activation (46). In addition, in Prion disease, the prion protein (PrP) misfolds induced lysosome destabilization and NLRP3 inflammasome activation (47). Lysosome rupture and the release of lysosomal hydrolases, especially cathepsin B, have been shown to be essential for albumin-induced tubulointerstitial inflammation (TI) and fibrosis, suggesting that lysosomal damage is involved in the pathogenesis of chronic kidney disease through NLRP3 inflammasome activation (48). Anti-cancer chemotherapeutic agents such as gemcitabine and 5-fluorouracil, induced activation of the NLRP3 inflammasome, which was dependent on lysosomal permeabilization and the release of cathepsin B, while this activation in myeloid-derived suppressor cells blunted their anticancer efficacy (49). In addition to cathepsin B, multiple cathepsins such as cathepsin L, C, S, and X had been shown to promote both pro-IL-1β synthesis and NLRP3 activation in compensatory and independent manners (50, 51). Lysosome destabilization is linked to ion flux in the process of NLRP3 inflammasome activation (29). The lysosome-destabilizing agonist, Leu-Leu-O-methyl ester (LLME), induced lysosome membrane permeabilization, which correlated with K+ efflux and NLRP3 inflammasome activation in murine dendritic cells (52). However, extensive lysosome destabilization attenuated NLRP3 inflammasome activation with increase of Ca2+ influx, while it potentiated necrosis in murine bone marrow-derived dendritic cells (53).
Lysosomal destabilization-induced activation of the NLRP3 inflammasome is the feature of particulate matter and crystalline, elucidating the pathological mechanisms of the relevant inflammatory diseases, including amyloid β with Alzheimer’s disease, monosodium urate with gout, cholesterol crystalline with atherosclerosis, silica with silicosis, and asbestos with asbestosis.
Mitochondrial Dysfunction
Cardiolipin (1,3-bis(sn-3’-phosphatidyl)-sn-glycerol), a phospholipid constituent of the inner mitochondria membrane, links the mitochondria to the NLRP3 inflammasome activation (17). During mitochondrial stress, cardiolipin is exposed to the outer membrane, where it serves as a binding site during autophagy and apoptosis (54). Cardiolipin independently interacts with NLRP3 and full-length caspase-1, of which event is essential for inflammasome activation. MAVS is a mitochondrial protein that is necessary for the post-stimulus activation of the NLRP3 inflammasome by functioning as an adaptor protein in RNA-sensing pathways associated with RNA virus infections (55, 56). MAVS promotes NLRP3 inflammasome activation by recruiting NLRP3 to the mitochondrial outer membrane (55). Although MAVS is necessary for the RNA virus infection-mediated activation of the NLRP3 inflammasome, it may be dispensable for NLRP3 activation by other stimuli. Mitofusin 2 (Mfn 2), which is present in the outer mitochondrial membrane, endoplasmic reticulum, and contact sites of mitochondria-associated membrane, has been reported to be an essential factor for NLRP3 activation during RNA virus infection (57). During viral infection, Mfn2 forms a complex with MAVS and aids in the localization of NLRP3 in the mitochondria (57).
Reactive oxygen species (ROS) production was involved in the activation of NLRP3 inflammasome induced by ATP, MSU crystal, silica, and asbestos, suggesting mitochondrial ROS as a critical mediator for the NLRP3 inflammasome activation (58). Caspase-1 activation and IL-1β production in lipopolysaccharide (LPS)- and ATP-treated macrophages are dependent on mtROS generation and mitochondrial membrane permeability transition (59). The translocation of mitochondrial DNA (mtDNA) to cytosol was correlated with the activation of caspase-1 activation in LPS- and ATP-treated macrophages (59). Furthermore, oxidized mtDNA released during programmed cell death induced by NLRP3 activators such as ATP, bound to and activated the NLRP3 inflammasome (60). The de novo synthesis of mtDNA was induced by TLR signals accompanied with an expression of CMPK2, an enzyme that provides deoxyribonucleotides for mtDNA synthesis (61). CMPK2-dependent mtDNA synthesis resulted in the production of oxidized mtDNA fragments was required for NLRP3 inflammasome activation (61). The role of mtDNA-mediated activation of NLRP3 inflammasome in the development of inflammatory diseases is reported with Type 1 Diabetes (62).
These suggest mitochondria as central regulators of NLRP3 inflammasome activation induced by cellular stress, infections, and the NLRP3 activators, accompanying with mitochondrial dysfunction to promote the activation of NLRP3 inflammasome.
Golgi Apparatus Disassembly
NLRP3 stimuli have been shown to promote the disassembly of the trans-Golgi network into vesicles called the dispersed trans-Golgi network (dTGN) using a cellular reconstitution system. The phospholipid phosphatidylinositol-4-phosphate of dTGN recruits NLRP3 and promotes its aggregation, which is essential for downstream ASC oligomerization and caspase-1 activation (63). The K+ efflux-dependent stimulus (i.e., nigericin) and K+ efflux-independent stimulus (i.e., imiquimod) aid in the formation of dTGN and cause NLRP3 aggregation. However, K+ efflux, is only necessary for the recruitment of NLRP3 and not for dTGN formation (64), indicating that the K+ efflux-dependent and mitochondria-dependent activation of NLRP3 are two separate pathways that converge at the Golgi disassembly stage.
Metabolic Stress
Glucose phosphorylation, the first step in glycolysis, is mediated by hexokinase. During bacterial infection, the decomposition of the bacterial cell wall component peptidoglycan in lysosomes releases N-acetylglucosamine (GlcNAc). Hexokinase, which is located on the mitochondrial membrane, then combines with GlcNAc and promotes its relocalization in the cytosol. This GlcNAc-induced hexokinase relocalization promotes NLRP3 inflammasome activation regardless of K+ efflux (65). Although chemical disruption of glycolysis activates the NLRP3 inflammasome following priming (66), the interpretation of such observations is complicated, as the inhibition of glycolysis during priming results in the inhibition of LPS-induced gene transcription of IL-1β (67).
Saturated fatty acid such as palmitate induced IL-1β secretion in LPS-treated primary macrophages, suggesting the activation of NLRP3 inflammasome by saturated fatty acids, of which mechanism involves the lysosomal rupture and cathepsin B release (68). Saturated fatty acids-induced activation of the NLRP3 inflammasome was mediated by intracellular crystallization accompanied with subsequent lysosomal dysfunction (69). Similarly, oxidized low-density lipoprotein that is recognized by scavenger receptor CD36, causes crystallization to induce the NLRP3 inflammasome activation (70). Oxidized phosphatidylcholine induced activation of the NLRP3 inflammasome mediated by miROS production and mitochondrial destabilization (71). In contrast, unsaturated fatty acids such as oleate and linoleate blocked IL-1β secretion induced by saturated fatty acids, or NLRP3 inducers such as nigericin, alum, and MSU in human monocytes/macrophages (72).
These show the link between the NLRP3 inflammasome and metabolic diseases, suggesting how the metabolic dysfunction leads to augmented inflammation. The activation of the NLRP3 inflammasome by metabolic stress involves a variety of cellular stress pathways including ROS production, ion fluxes, mitochondrial dysfunction, and lysosomal destabilization. The intracellular events involved in the activation of the NLRP3 inflammasome are interconnected and well-correlated to accomplish full activation of the NLRP3 inflammasome.
Collectively, despite of efforts to elucidate the upstream events during NLRP3 activation, a single unifying model has not been proposed, with the evidence obtained to date involving pharmacological inhibition rather than genetic approaches. Thus, it is difficult to analyze the indirect or off-target effects associated with NLRP3 activation. For example, it is difficult to ascertain whether mitochondrial dysfunction and mitochondria-derived ROS (mtROS) are the decisive factors for NLRP3 activation because other events such as ion flux changes and small organ damage, occur simultaneously. The intracellular signaling events may be interconnected or converge to further downstream steps to maximize NLRP3 inflammasome activation.
PTM Regulation of the NLRP3 Inflammasome
Emerging roles have been identified for many intracellular molecules in regulating NLRP3 inflammasome activation. Some of the proteins promote NLRP3 inflammasome activation by binding to the inflammasome components to promote their PTMs, such as phosphorylation, sumoylation, and ubiquitination, while some prevent NLRP3 inflammasome activation via PTM. To manipulate the NLRP3 inflammasome for therapeutic purposes, identifying the intracellular proteins regulating PTM of the NLRP3 inflammasome and understanding their mechanisms of action is of crucial importance (Figure 2).
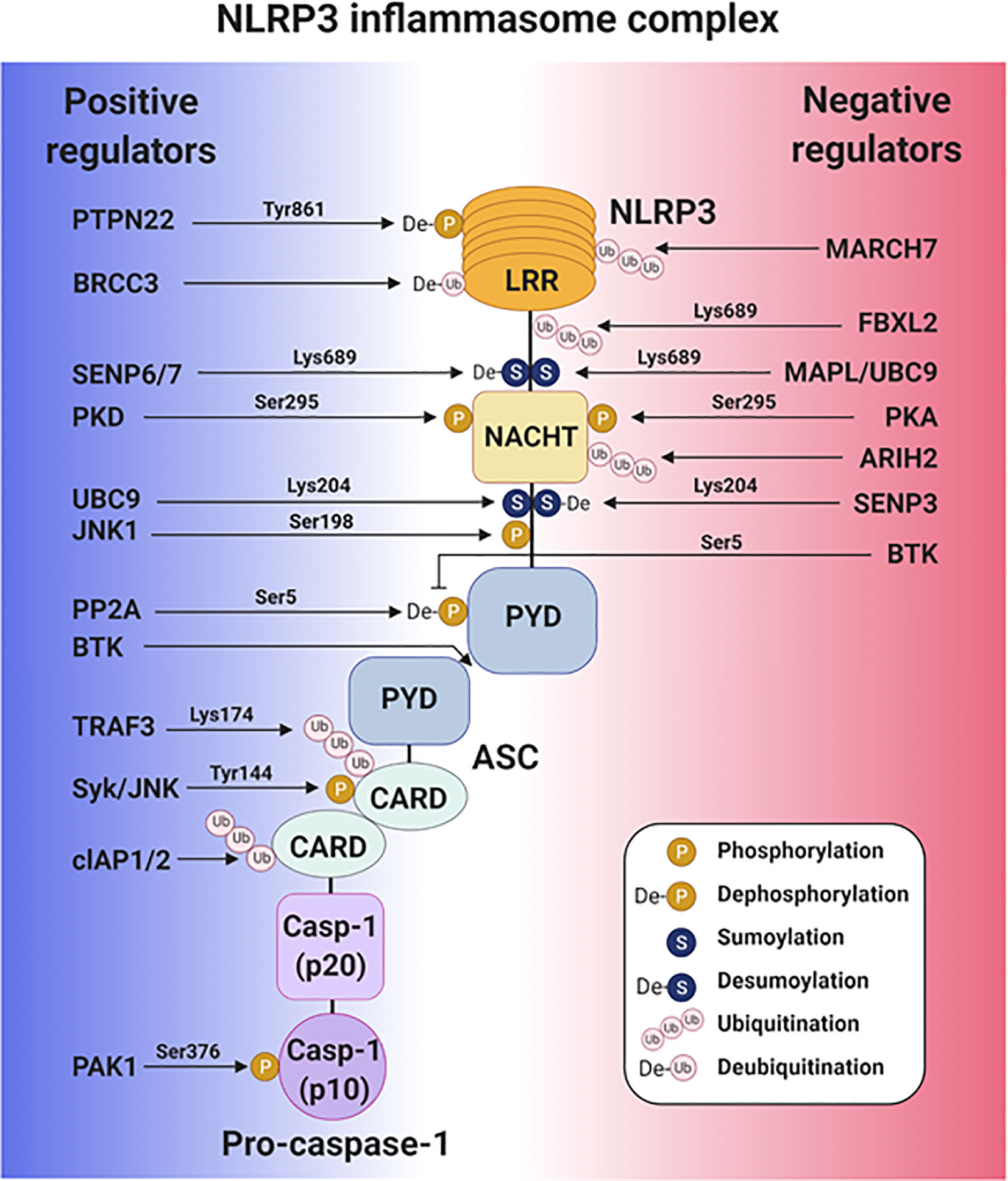
Figure 2 Post-translational regulation of the NLRP3 inflammasome activation. A schematic diagram of the intracellular proteins that positively and negatively regulate NLRP3 inflammasome activity.
Regulators of Phosphorylation
Phosphorylation of NLRP3 at Ser198 by JNK1 is an essential prerequisite event that occurs during the priming step, inducing the self-association of NLRP3 to promote active inflammasome assembly (73). Phosphorylation by JNK1 promotes NLRP3 deubiquitination, while S194A mutation or JNK inhibition interferes with the interaction of NLRP3 with BRCC3 and disrupts NLRP3 deubiquitination (73).
Protein kinase D (PKD) is required for NLRP3 inflammasome activation. PKD phosphorylates human Ser295 (mouse Ser291) in the NACHT domain of NLRP3 at the Golgi to free NLRP3 from mitochondria-associated endoplasmic reticulum membranes (MAMs), resulting in the recruitment of ASC to NLRP3 (74). Blockade of PKD activity led to the suppression of NLRP3 inflammasome activity in macrophages (74).
NIMA-related kinase 7 (NEK7) is a serine-threonine kinase that may be a crucial component for NLRP3 inflammasome activation. NEK7 interacts with NLRP3 to create ASC specks and induce oligomerization as an essential complex for caspase-1 activation (75). NEK7 can directly bind to NLRP3 protein, leading to NLRP3 inflammasome assembly due to K+ efflux. Moreover, chloride intracellular channel (CLIC)-dependent Cl- efflux can promote NEK7-NLRP3 interactions and subsequent ASC oligomerization (43). However, it remains unclear how intracellular Cl- efflux regulates the NEK7-NLRP3 interaction.
Syk and JNK are responsible for the phosphorylation of ASC in macrophages, with Tyr144 being a putative phosphorylation site (76). Interestingly, Syk has a cell type-specific role in NLRP3 inflammasome activation, as is not essential for this process in dendritic cells (76). The phosphorylation of Tyr144 in mouse ASC was shown to be required for ASC speck formation and caspase-1 activation in NLRP3 inflammasome activation but dispensable for the interaction of ASC with NLRP3 (76).
Protein kinase A (PKA) activation by prostaglandin E2 via the PGE2 receptor E-prostanoid 4 (EP4) leads to inhibitory phosphorylation of NLRP3 at Ser295 in human NLRP3, resulting in the suppression of ATPase activity of NLRP3 (77). In addition, PKA activation by bile acids through the TGR5 bile acid receptor leads to the phosphorylation of mouse NLRP3 at Ser 291, culminating in NLRP3 and ubiquitination the blockade of NLRP3 inflammasome activation (78).
Protein phosphatase (PP2A) participates in NLRP3 activation by dephosphorylating NLRP3 at Ser5 (79). Ser5 is located in the NLRP3 PYD-ASC PYD interaction motif, and its phosphorylation disrupts the association of NLRP3 and ASC (79). However, the kinase that phosphorylates Ser5 of NLRP3 has not been identified.
Protein tyrosine phosphatase nonreceptor type 22 (PTPN22) is required for NLRP3 inflammasome activation since PTPN22 knockdown results in decreased IL-1β secretion (80). PTPN22 promotes NLRP3 inflammasome activation by dephosphorylating Tyr861 in human NLRP3 (80). A Y861C mutation in NLRP3 has been reported in patients with chronic infantile neurologic cutaneous and articular syndrome (CINCA), and the Y861F mutation in NLRP3 has been shown to enhance NLRP3 inflammasome activation compared to wild-type NLRP3 (80).
Src homology 2 (SH2) domain-containing tyrosine phosphatase-2 (SHP2, encoded by the gene PTPN11) is a negative regulator of the NLRP3 inflammasome (81). SHP2 deficiency was shown to lead to excessive NLRP3 inflammasome activation in macrophages and aggravated peritonitis symptoms in a mouse model (81). SHP2 translocates to the mitochondria in response to NLRP3 inflammasome stimulators and binds to and dephosphorylates adenine nucleotide translocase 1 (ANT1), which plays a role in controlling mitochondrial permeability transition. SHP2 prevents the collapse of mitochondrial membrane potential and the subsequent release of mitochondrial DNA and reactive oxygen species, thereby suppressing hyperactivation of the NLRP3 inflammasome (81).
Bruton’s tyrosine kinase (BTK) has been reported to be a positive regulator of the NLRP3 inflammasome (82), as BTK inhibitors and dysfunctional BTK mutants inhibit NLRP3 inflammasome activation in macrophages, and BTK directly interacts with ASC and NLRP3, promoting ASC aggregation (82). The FDA-approved BTK inhibitor ibrutinib was shown to protect against ischemic brain injury by reducing mature IL-1β and caspase-1 activation in infiltrating macrophages and neutrophils in the infarcted area of the ischemic brain (82). Another study showed that a BTK inhibitor or BTK deficiency can impair NLRP3 inflammasome activation (83). BTK is associated with NLRP3 and ASC and promotes ASC speck formation and caspase-1 cleavage (83), with the BTK inhibitor ibrutinib shown to be effective in blocking IL-1β secretion in immune cells derived from Muckle-Wells syndrome patients (83). In contrast, a recent study showed a negative role of BTK in regulating NLRP3 inflammasome activity (84), with BTK deficiency enhancing NLRP3 inflammasome activity in macrophages (84). BTK binds to NLRP3 during the priming step of inflammasome activation, preventing NLRP3 inflammasome assembly induced by NLRP3 activators during the activation phase of inflammasome activation (84). BTK was shown to block PP2A-mediated dephosphorylation of Ser5 in the pyrin domain of NLRP3 to inhibit the NLRP3 inflammasome (84). However, the exact role of BTK in NLRP3 inflammasome regulation and whether BTK has different roles in different contexts needs to be further examined.
TGF-β activated kinase-1 (TAK1) has been suggested to function as a negative regulator to prevent spontaneous NLRP3 activation (85). TAK1 deficiency was shown to lead to spontaneous NLRP3 inflammasome activation in macrophages, suggesting that TAK1 maintains NLRP3 inflammasome quiescence (85).
p21-activated kinase 1 (PAK1) is involved in caspase-1 activation induced by Helicobacter pylori LPS (86) by phosphorylating Ser376 in the p10 subunit of caspase-1 (86).
Regulators of Sumoylation
NLRP3 was conjugated with SUMO-2/-3 and sumoylated at basal state, mediated by interaction between MAPL (also known as MUL1), a SUMO E3 ligase, and NLRP3 (87). Activating signals such as nigericin reduced sumoylated NLRP3, possibly by disrupting NLRP3-MAPL interaction (87). The SUMO E2 ligase, UBC9 interacted with the SUMO consensus motif surrounding K689 of NLRP3 (87). Deficiency of sentrin-specific protease 6 (SENP6) and SENP7, which are SUMO de-conjugating enzymes, reduced NLRP3 inflammasome activation (87). In contrast, SUMO1-catalyzed sumoylation of NLRP3 Lys204 promoted inflammasome activation whereas SENP3, a deSUMOylase, induced NLRP3 deSUMOylation to attenuate inflammasome activation (88). The precise role of sumoylation by different enzymes and at different activation steps needs to be further elucidated.
Regulators of Ubiquitination
Pellino2, an E3 ubiquitin ligase, is required for NLRP3-induced ASC oligomerization and mature IL-1β production (89). LPS induces the interaction of Pellino2 with NLRP3 and Pellino2 FHA, and RING domains facilitate NLRP3 activation by promoting the K63-linked ubiquitination of NLRP3 during the priming step (89). Pellino2-deficient mice and myeloid cells show impaired NLRP3 activation in response to Toll-like receptor priming, NLRP3 stimuli and bacterial challenge (89).
BRCA1-BRCA2 containing complex subunit 3 (BRCC3) plays a role in NLRP3 inflammasome via deubiquitination of the LRR domain of NLRP3 by directly binding to ubiquinated NLRP3 (90). Furthermore, BRCC3 knockdown was shown to reduce the ATP-induced secretion of mature IL-1β by in macrophages (91).
Deficiency of tumor necrosis factor alpha-induced protein 3 (TNFAIP3, also known as A20), a deubiquitinating enzyme, was observed to enhance NLRP3 inflammasome-mediated caspase-1 activation, pyroptosis, and IL-1β secretion in macrophages, while activation of the NLRC4 and AIM2 inflammasomes was not affected (92).
The E3 ubiquitin ligase TRIM31 plays a negative regulatory role in NLRP3 inflammasome activation (93). TRIM31 directly binds to NLRP3, promoting the K48-linked polyubiquitination and proteasomal degradation of NLRP3 (93). Furthermore, TRIM31 deficiency enhances IL-1β secretion in vivo and aggravates alum-induced peritonitis in mice (93).
FBXL2 interacts with Trp73 in NLRP3 and targets Lys689 in NLRP3 for ubiquitin ligation, leading to the degradation of NLRP3 and a reduction in IL-1β and IL-18 secretion in human inflammatory cells (8).
The E3 ubiquitin ligase MARCH7 is involved in the ubiquitination and degradation of NLRP3 in neurotransmitter dopamine-treated macrophages (94). Dopamine negatively regulates the NLRP3 inflammasome via cyclic adenosine monophosphate (cAMP), which binds to NLRP3 and promotes its K48-linked polyubiquitination and degradation through MARCH7 activity (94). In addition, it was demonstrated that the LRR domain of NLRP3 is a key region for MARCH7-mediated ubiquitination and degradation (94).
Ariadne homolog 2 (ARIH2), an E3 ligase, interacts with the NACHT domain of NLRP3, leading to the K48- and K63-linked ubiquitination of NLRP3 and downregulation of NLRP3 inflammasome activation (95). ARIH2 deficiency results in increased NLRP3 inflammasome activation and IL-1β production, while ARIH2 overexpression inhibits NLRP3 inflammasome activation (95).
TNFR-associated factor 3 (TRAF3) is a direct E3 ubiquitin ligase for ASC (96), with ASC ubiquitination at Lys174 being crucial for speck formation and NLRP3 inflammasome activation. TRAF3 deficiency results in impaired ASC ubiquitination and cytosolic aggregate formation, resulting in decreased inflammasome responses during RNA virus infection (96).
HOIL-1L is a component of the linear ubiquitination assembly complex (LUBAC), which consists of HOIL-1L, HOIP, and Sharpin. ASC is linearly ubiquitinated by LUBAC, and HOIL-1L is required for ASC foci formation and NLRP3/ASC inflammasome assembly (97). Furthermore, HOIL-1L-deficient macrophages were observed to be impaired in mature IL-1β secretion upon NLRP3 activation stimuli (97).
Inhibitors of apoptosis proteins (IAPs), such as cIAP1 and cIAP2, have E3 ubiquitin ligase activity. cIAP1 and cIAP2 interact directly with caspase-1 and are required for inflammasome assembly and caspase-1 activation, which is mediated by the K63-linked polyubiquitination of caspase-1 (98).
Collectively, PTMs of the NLRP3 inflammasome show that the NLRP3 inflammasome activity is regulated by various signaling components in a complex and elaborated fashion. It needs to be further elucidated that how each PAMP or DAMP modulates the activity of PTM-related signaling molecules and what the interactions between different PTMs are.
Small Molecules and Phytochemicals Regulating the NLRP3 Inflammasome Activation
The NLRP3 inflammasome plays a pivotal role in development and progress of many common inflammatory diseases (99). The NLRP3 inflammasome has been implicated in the pathogenesis of metabolic disorders such as type 2 diabetes (100), atherosclerosis (101), obesity (102), and gout (103). In addition, the role of NLRP3 is noted to contribute to the pathology of central nervous system diseases including Alzheimer’s disease (104) and Parkinson’s disease (105). Abnormal activation of the NLRP3 inflammasome is associated with intestinal cancer and auto-inflammatory diseases such as keratitis/conjunctivitis (106, 107). Therefore, the discovery of pharmacological inhibitors targeting NLRP3 inflammasome components provides a novel strategy for the development of potential therapeutics in a wide range of human diseases (Table 1).
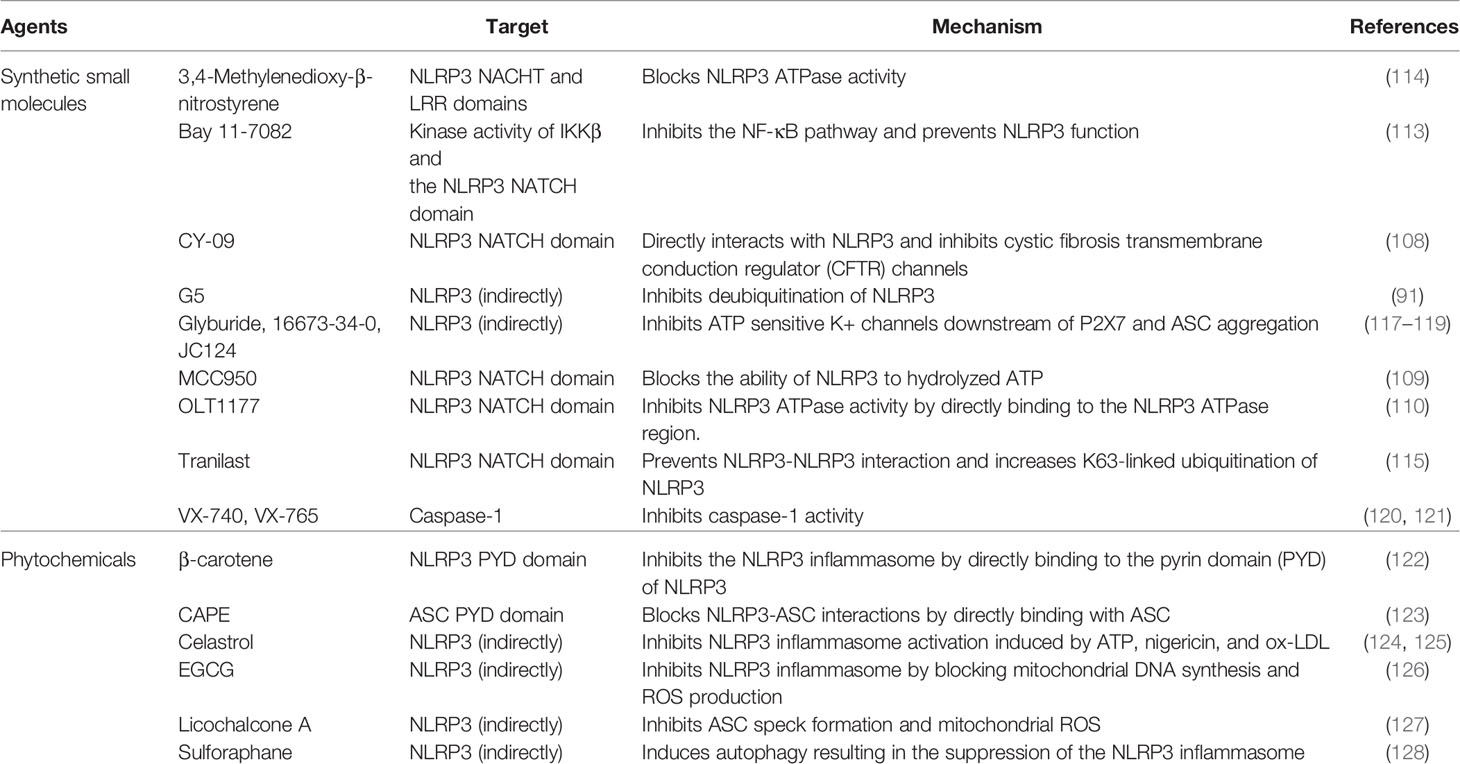
Table 1 Small molecules and phytochemicals regulating the NLRP3 inflammasome activation.
Synthetic Small Molecules
Among the synthetic small molecules, CY-09 (108), MCC950 (109), and OLT1177 (110) bind directly to the NATCH domain of NLRP3 and block NLRP3 ATPase activity. CY-09 is a CFTR (inh)-172 (C172) analog that inhibits cystic fibrosis transmembrane conduction regulator (CFTR) channels by directly interacting with the NLRP3 Walker A motif of NLRP3 NACHT domain and disrupting ATP binding to NLRP3 (108). CY-09 has shown excellent prophylactic and therapeutic properties in mouse models of gout, type 2 diabetes, and Cryopyrin-associated periodic syndrome (CAPS) (108). MCC950 inhibits both canonical and noncanonical NLRP3, but not AIM2, NLRC4 or NLRP1 activation, in mouse macrophages, human monocyte derived macrophages, and human peripheral blood mononuclear cells (111). MCC950 treatment was effective to attenuate inflammatory symptoms of NLRP3-related diseases including experimental autoimmune encephalomyelitis and CAPS (111). MCC950 directly binds to the Walker B motif in the NLRP3 NACHT domain, inhibiting ATPase activity and NLRP3 inflammasome formation (109). OLT1177 is an active β-sulfonylnitrile compound that has passed phase 1 clinical trials for the treatment of degenerative arthritis and is currently being evaluated in phase 2 clinical trials (112). OLT1177 has been shown to block both standard and nonstandard NLRP3 inflammasome activation and ATPase activity by directly binding to NLRP3 (110). Bay 11-7082 binds to NLRP3 through alkylation of cysteine residues in the NLRP3 ATPase region, thereby blocking NLRP3 inflammasome function in addition to its inhibition of the kinase activity of IKKβ (113). 3,4-Methylenedioxy-β-nitrostyrene directly binds to NACHT and LRR domains of NLRP3, blocking its ATPase activity (114). Tranilast, a tryptophan metabolite analog, binds to NACHT domain of NLRP3, preventing NLRP3-NLRP3 interaction and alleviating the symptoms of gouty arthritis, cryopyrin-associated autoinflammatory syndromes, and type 2 diabetes (115). In addition, tranilast increases K63-linked ubiquitination of NLRP3 (116).
Glyburide (99, 117) inhibits NLRP3 inflammasome activity. The small molecule 16673-34-0 (118), an intermediate substrate produced during the synthesis of glyburide and the low-molecular-weight JC124 (119), generated during the structural optimization of glyburide, alleviates the side effects of glyburide.
VX-740 (Pralnacasan) (120) and its analog VX-765 (121) are peptidomimetic inhibitors of caspase-1 and inhibit the proinflammatory cytokines IL-1β and IL-18.
G5 (3,5-bis[(4-Nitrophenyl)methylene]-1,1-dioxide,tetrahydro-4H-thiopyran-4-one), a small molecule inhibitor of deubiquitination, inhibits NLRP3 inflammasome activation induced by ATP and nigericin in LPS-primed macrophages (91). G5 promotes ubiquitination of NLRP3 NACHT and LRR domains with mixed K63 and K48 ubiquitin chains, resulting in the blockade of NLRP3 inflammasome activation (91).
As excessive NLRP3 inflammasome activation has been shown to be closely associated with the pathophysiology of a wide array of disorders, synthetic small molecules that directly or indirectly inhibit the NLRP3 inflammasome have been suggested as promising therapeutic agents (Table 1). The specificity of the target sites with high potency would be the critical prerequisite requirement to develop the new NLRP3 inflammasome inhibitors.
Phytochemicals
Phytochemicals are substances synthesized by medicinal plants and extracted for therapeutic purposes (Katz and Baltz, 2016). Plants are a preferred therapeutic substance because they are easily accessible and cost-effective, and there are many investigations on phytochemicals that modulate the inflammatory response by regulating specific NLRP3 inflammasome components. Phytochemicals that directly or indirectly regulate the NLRP3 inflammasome include β-carotene (122), caffeic acid phenethyl ester (CAPE) (123), sulforaphane (128), celastrol (124, 125), epigallocatechin-3-gallate (EGCG) (126), and licochalcone A (127).
β-Carotene binds directly to pyrin domain (PYD) of NLRP3 (122). Molecular modeling and mutation study showed that β‐carotene interacted with Ala69, Val72, Trp73, Tyr84, and Glu91 in the hydrophobic groove of H5 and H6 (122). β-Carotene inhibits ATP, MSU crystals, and nigericin-induced NLRP3 inflammasome activation in macrophages. In addition, β-carotene was shown to decrease IL-1β secretion by human synovial cells isolated from gout patients, demonstrating the potential inhibitory effect of β-carotene on human gout (122).
CAPE directly binds to PYD of ASC, leading to the disruption of the NLRP3-ASC association as demonstrated by SPR analysis and pull-down experiment (123). In a murine gouty arthritis model, the oral administration of CAPE was shown to attenuate inflammatory symptoms by inhibiting MSU crystal-induced caspase-1 activation and IL-1β production in air pouch exudate and foot tissue (123). To our best knowledge, CAPE is the only phytochemical known to bind directly to ASC.
Sulforaphane (SFN) regulated upstream signaling pathways of AMP-activated protein kinase-autophagy axis to suppress the NLRP3 inflammasome activation. SFN induced autophagosome formation and p62 degradation in hepatocytes, whereas it inhibited the activation of mammalian target of rapamycin (mTOR), a negative regulator of autophagy, suggesting that SFN promotes autophagy in hepatocytes (128). SFN induced phosphorylation of AMP-activated protein kinase and consequent activation of autophagy, resulting in inhibition of the NLRP3 inflammasome in the liver. Oral administration with SFN prevented non-alcoholic fatty liver disease symptoms in mice fed high fat diet by inhibiting the NLRP3 inflammasome in the liver (128). In addition, SFN can effectively alleviate acute gouty inflammation by inhibiting NLRP3 inflammasome activation induced by MSU crystals in in macrophages (129). The inhibition of NLRP3 inflammasome activation by SFN is independent of the reactive oxygen species pathway in macrophages (129). These show that the regulation of autophagy can be used as a beneficial strategy to prevent the NLRP3 inflammasome activation.
There are phytochemicals affecting intracellular events of the NLRP3 inflammasome, including K+ efflux, mitochondrial ROS production, and mitochondrial DNA release. Celastrol suppresses the K+ efflux induced by ATP and nigericin in primary macrophages, resulting in suppression of the NLRP3 inflammasome (124, 125). Celastrol pretreatment reduced the ability of ATP-stimulated macrophages to promote cancer cell migration and invasion (124, 125). The inhibitory effects of licochalcone A and EGCG were mediated through the regulation of mitochondrial dysfunction. Licochalcone A blocked mitochondrial ROS generation induced by Propionibacterium acnes (P. acnes) and rotenone in mouse primary macrophages (127). Similarly, EGCG prevented production of ROS induced by NLRP3 activators such as MSU, ATP, and nigericin and suppressed de novo synthesis of mitochondrial DNAs induced by MSU in primary mouse macrophages (126). The specific target molecules or mechanisms by which these phytochemicals modulate mitochondrial function remain to be further investigated.
There are a few phytochemicals directly binding to the NLRP3 inflammasome components while others work indirectly by modulating upstream signaling pathways of the NLRP3 inflammasome (Table 1). Elucidating the exact action mechanism by which the phytochemicals exert the inhibitory activity would provide an important information on specific targets to be regulated and drug discovery strategies for novel pharmacological inhibitors.
Conclusion
With the lifespan improvements made possible by modern medical developments, the occurrence of metabolic and age-related diseases is increasing. Previous studies have demonstrated that the NLRP3 inflammasome is a key mediator in the development of various metabolic diseases and host inflammatory responses. The discoveries of NLRP3 inflammasome modulators, such as NEK7 and GSDMD, demonstrate significant advances in this field.
The PTM regulation of the NLRP3 inflammasome components can be potential targets for the development of specific drugs or inhibitors of the NLRP3 inflammasome. However, unidentified PTM sites of NLRP3 and PTM-related enzymes remain, and the correlations and interactions between the various types of PTMs of the NLRP3 inflammasome components require further investigation. As research on NLRP3 activation becomes more important, the targeting of NLRP3 as a therapeutic strategy for many diseases is rapidly progressing. The current treatment for NLRP3-related pathologies is to indirectly or directly inhibit the activation of the NLRP3 inflammasome using pharmacological inhibitors. NLRP3 inhibitors function as effective therapeutic agents for many inflammatory disorders by inhibiting the pro-inflammatory cytokines IL-1β and IL-18 and maintaining cellular homeostasis.
In summary, understanding how intracellular proteins, PTMs, and small molecule inhibitors regulate the NLRP3 inflammasome activation will provide crucial information for elucidating the host defense mechanisms in response to pathogen and tissue damage and in constructing effective therapeutic strategies for chronic diseases.
Author Contributions
JS performed the acquisition of the data and information, and mainly wrote a draft of the manuscript. HK and Y-YC provided the data and information and participated in writing a draft of the manuscript. HL critically evaluated the content and participated in writing a draft of the manuscript. JL constructed the idea of the content, performed the acquisition of information, and wrote and finalized the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by grants from the National Research Foundation of Korea (NRF‐2019R1A2C2085739, NRF-2019R1I1A1A01056377, NRF-2020R1A4A2002894), funded by the Korean government (Ministry of Science, ICT and Future Planning).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Figures 1 and 2 were created at Biorender.com.
References
1. Duncan JA, Bergstralh DT, Wang Y, Willingham SB, Ye Z, Zimmermann AG, et al. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc Natl Acad Sci U S A (2007) 104:8041–6. doi: 10.1073/pnas.0611496104
2. Vajjhala PR, Mirams RE, Hill JM. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J Biol Chem (2012) 287:41732–43. doi: 10.1074/jbc.M112.381228
3. Yu HB, Finlay BB. The caspase-1 inflammasome: a pilot of innate immune responses. Cell Host Microbe (2008) 4:198–208. doi: 10.1016/j.chom.2008.08.007
4. Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J Clin Invest (2005) 115:2665–72. doi: 10.1172/JCI26252
5. Broz P, von Moltke J, Jones JW, Vance RE, Monack DM. Differential requirement for Caspase-1 autoproteolysis in pathogen-induced cell death and cytokine processing. Cell Host Microbe (2010) 8:471–83. doi: 10.1016/j.chom.2010.11.007
6. Boucher D, Monteleone M, Coll RC, Chen KW, Ross CM, Teo JL, et al. Caspase-1 self-cleavage is an intrinsic mechanism to terminate inflammasome activity. J Exp Med (2018) 215:827–40. doi: 10.1084/jem.20172222
7. Shim DW, Lee KH. Posttranslational Regulation of the NLR Family Pyrin Domain-Containing 3 Inflammasome. Front Immunol (2018) 9:1054. doi: 10.3389/fimmu.2018.01054
8. Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL, et al. Lipopolysaccharide Primes the NALP3 Inflammasome by Inhibiting Its Ubiquitination and Degradation Mediated by the SCFFBXL2 E3 Ligase. J Biol Chem (2015) 290:18124–33. doi: 10.1074/jbc.M115.645549
9. Schroder K, Tschopp J. The inflammasomes. Cell (2010) 140:821–32. doi: 10.1016/j.cell.2010.01.040
10. Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell (2014) 156:1193–206. doi: 10.1016/j.cell.2014.02.008
11. Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W, et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ (2007) 14:1590–604. doi: 10.1038/sj.cdd.4402194
12. He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res (2015) 25:1285–98. doi: 10.1038/cr.2015.139
13. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature (2015) 526:660–5. doi: 10.1038/nature15514
14. Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature (2016) 535:111–6. doi: 10.1038/nature18590
15. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature (2016) 535:153–8. doi: 10.1038/nature18629
16. Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature (2015) 526:666–71. doi: 10.1038/nature15541
17. Iyer SS, He Q, Janczy JR, Elliott EI, Zhong Z, Olivier AK, et al. Mitochondrial cardiolipin is required for Nlrp3 inflammasome activation. Immunity (2013) 39:311–23. doi: 10.1016/j.immuni.2013.08.001
18. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature (2011) 479:117–21. doi: 10.1038/nature10558
19. Yi YS. Caspase-11 non-canonical inflammasome: a critical sensor of intracellular lipopolysaccharide in macrophage-mediated inflammatory responses. Immunology (2017) 152:207–17. doi: 10.1111/imm.12787
20. Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature (2014) 514:187–92. doi: 10.1038/nature13683
21. Yang D, He Y, Munoz-Planillo R, Liu Q, Nunez G. Caspase-11 Requires the Pannexin-1 Channel and the Purinergic P2X7 Pore to Mediate Pyroptosis and Endotoxic Shock. Immunity (2015) 43:923–32. doi: 10.1016/j.immuni.2015.10.009
22. Zeng C, Wang R, Tan H. Role of Pyroptosis in Cardiovascular Diseases and its Therapeutic Implications. Int J Biol Sci (2019) 15:1345–57. doi: 10.7150/ijbs.33568
23. Antonopoulos C, Russo HM, El Sanadi C, Martin BN, Li X, Kaiser WJ, et al. Caspase-8 as an Effector and Regulator of NLRP3 Inflammasome Signaling. J Biol Chem (2015) 290:20167–84. doi: 10.1074/jbc.M115.652321
24. Chung H, Vilaysane A, Lau A, Stahl M, Morampudi V, Bondzi-Simpson A, et al. NLRP3 regulates a non-canonical platform for caspase-8 activation during epithelial cell apoptosis. Cell Death Differ (2016) 23:1331–46. doi: 10.1038/cdd.2016.14
25. Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity (2016) 44:833–46. doi: 10.1016/j.immuni.2016.01.012
26. Kang S, Fernandes-Alnemri T, Rogers C, Mayes L, Wang Y, Dillon C, et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat Commun (2015) 6:7515. doi: 10.1038/ncomms8515
27. Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science (2018) 362:1064–9. doi: 10.1126/science.aau2818
28. Kang TB, Yang SH, Toth B, Kovalenko A, Wallach D. Caspase-8 blocks kinase RIPK3-mediated activation of the NLRP3 inflammasome. Immunity (2013) 38:27–40. doi: 10.1016/j.immuni.2012.09.015
29. Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity (2013) 38:1142–53. doi: 10.1016/j.immuni.2013.05.016
30. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 Receptor in Infection and Inflammation. Immunity (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
31. Wei L, Syed Mortadza SA, Yan J, Zhang L, Wang L, Yin Y, et al. ATP-activated P2X7 receptor in the pathophysiology of mood disorders and as an emerging target for the development of novel antidepressant therapeutics. Neurosci Biobehav Rev (2018) 87:192–205. doi: 10.1016/j.neubiorev.2018.02.005
32. Di A, Xiong S, Ye Z, Malireddi RKS, Kometani S, Zhong M, et al. The TWIK2 Potassium Efflux Channel in Macrophages Mediates NLRP3 Inflammasome-Induced Inflammation. Immunity (2018) 49:56–65 e4. doi: 10.1016/j.immuni.2018.04.032
33. Ruhl S, Broz P. Caspase-11 activates a canonical NLRP3 inflammasome by promoting K(+) efflux. Eur J Immunol (2015) 45:2927–36. doi: 10.1002/eji.201545772
34. da Costa LS, Outlioua A, Anginot A, Akarid K, Arnoult D. RNA viruses promote activation of the NLRP3 inflammasome through cytopathogenic effect-induced potassium efflux. Cell Death Dis (2019) 10:346. doi: 10.1038/s41419-019-1579-0
35. Katsnelson M, Dubyak G. Cytosolic K+ and extracellular Na+ as regulators of NLRP3 inflammasome activation and the IL-1β secretion response of macrophages to crystalline stimuli. FASEB J (2013) 27:138.8–.8. doi: 10.1096/fasebj.27.1_supplement.138.8
36. Scambler T, Jarosz-Griffiths HH, Lara-Reyna S, Pathak S, Wong C, Holbrook J, et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife (2019) 8:e49248. doi: 10.7554/eLife.49248
37. Pitzer AL, Barbaro NR, Aden L, Ray E, Kleyman T, Kirabo A. High salt activates NLRP3 inflammasome in antigen presenting cells via ENaC to promote salt-sensitive hypertension. FASEB J (2020) 34:1–. doi: 10.1096/fasebj.2020.34.s1.05532
38. Compan V, Baroja-Mazo A, Lopez-Castejon G, Gomez AI, Martinez CM, Angosto D, et al. Cell volume regulation modulates NLRP3 inflammasome activation. Immunity (2012) 37:487–500. doi: 10.1016/j.immuni.2012.06.013
39. Verhoef PA, Kertesy SB, Lundberg K, Kahlenberg JM, Dubyak GR. Inhibitory effects of chloride on the activation of caspase-1, IL-1beta secretion, and cytolysis by the P2X7 receptor. J Immunol (2005) 175:7623–34. doi: 10.4049/jimmunol.175.11.7623
40. Green JP, Swanton T, Morris LV, El-Sharkawy LY, Cook J, Yu S, et al. LRRC8A is essential for hypotonicity-, but not for DAMP-induced NLRP3 inflammasome activation. eLife (2020) 9:e59704. doi: 10.7554/eLife.59704
41. Domingo-Fernandez R, Coll RC, Kearney J, Breit S, O’Neill LAJ. The intracellular chloride channel proteins CLIC1 and CLIC4 induce IL-1beta transcription and activate the NLRP3 inflammasome. J Biol Chem (2017) 292:12077–87. doi: 10.1074/jbc.M117.797126
42. Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun (2016) 7:12504. doi: 10.1038/ncomms12504
43. Tang T, Lang X, Xu C, Wang X, Gong T, Yang Y, et al. CLICs-dependent chloride efflux is an essential and proximal upstream event for NLRP3 inflammasome activation. Nat Commun (2017) 8:202. doi: 10.1038/s41467-017-00227-x
44. Green JP, Yu S, Martin-Sanchez F, Pelegrin P, Lopez-Castejon G, Lawrence CB, et al. Chloride regulates dynamic NLRP3-dependent ASC oligomerization and inflammasome priming. Proc Natl Acad Sci U S A (2018) 115:E9371–E80. doi: 10.1073/pnas.1812744115
45. Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, et al. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol (2008) 9:857–65. doi: 10.1038/ni.1636
46. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
47. Hafner-Bratkovič I, Benčina M, Fitzgerald KA, Golenbock D, Jerala R. NLRP3 inflammasome activation in macrophage cell lines by prion protein fibrils as the source of IL-1β and neuronal toxicity. Cell Mol Life Sci CMLS (2012) 69:4215–28. doi: 10.1007/s00018-012-1140-0
48. Liu D, Wen Y, Tang TT, Lv LL, Tang RN, Liu H, et al. Megalin/Cubulin-Lysosome-mediated Albumin Reabsorption Is Involved in the Tubular Cell Activation of NLRP3 Inflammasome and Tubulointerstitial Inflammation. J Biol Chem (2015) 290:18018–28. doi: 10.1074/jbc.M115.662064
49. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med (2013) 19:57–64. doi: 10.1038/nm.2999
50. Orlowski GM, Colbert JD, Sharma S, Bogyo M, Robertson SA, Rock KL. Multiple Cathepsins Promote Pro-IL-1beta Synthesis and NLRP3-Mediated IL-1beta Activation. J Immunol (2015) 195:1685–97. doi: 10.4049/jimmunol.1500509
51. Campden RI, Zhang Y. The role of lysosomal cysteine cathepsins in NLRP3 inflammasome activation. Arch Biochem Biophys (2019) 670:32–42. doi: 10.1016/j.abb.2019.02.015
52. Katsnelson MA, Rucker LG, Russo HM, Dubyak GR. K+ efflux agonists induce NLRP3 inflammasome activation independently of Ca2+ signaling. J Immunol (2015) 194:3937–52. doi: 10.4049/jimmunol.1402658
53. Katsnelson MA, Lozada-Soto KM, Russo HM, Miller BA, Dubyak GR. NLRP3 inflammasome signaling is activated by low-level lysosome disruption but inhibited by extensive lysosome disruption: roles for K+ efflux and Ca2+ influx. Am J Physiol Cell Physiol (2016) 311:C83–C100. doi: 10.1152/ajpcell.00298.2015
54. Dudek J. Role of Cardiolipin in Mitochondrial Signaling Pathways. Front Cell Dev Biol (2017) 5:90. doi: 10.3389/fcell.2017.00090
55. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi: 10.1016/j.cell.2013.02.054
56. Park S, Juliana C, Hong S, Datta P, Hwang I, Fernandes-Alnemri T, et al. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J Immunol (2013) 191:4358–66. doi: 10.4049/jimmunol.1301170
57. Ichinohe T, Yamazaki T, Koshiba T, Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc Natl Acad Sci U S A (2013) 110:17963–8. doi: 10.1073/pnas.1312571110
58. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
59. Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol (2011) 12:222–30. doi: 10.1038/ni.1980
60. Shimada K, Crother TR, Karlin J, Dagvadorj J, Chiba N, Chen S, et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity (2012) 36:401–14. doi: 10.1016/j.immuni.2012.01.009
61. Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature (2018) 560:198–203. doi: 10.1038/s41586-018-0372-z
62. Pereira CA, Carlos D, Ferreira NS, Silva JF, Zanotto CZ, Zamboni DS, et al. Mitochondrial DNA Promotes NLRP3 Inflammasome Activation and Contributes to Endothelial Dysfunction and Inflammation in Type 1 Diabetes. Front Physiol (2019) 10:1557. doi: 10.3389/fphys.2019.01557
63. Chen J, Chen ZJ. PtdIns4P on dispersed trans-Golgi network mediates NLRP3 inflammasome activation. Nature (2018) 564:71–6. doi: 10.1038/s41586-018-0761-3
64. Xu B, Jiang M, Chu Y, Wang W, Chen D, Li X, et al. Gasdermin D plays a key role as a pyroptosis executor of non-alcoholic steatohepatitis in humans and mice. J Hepatol (2018) 68:773–82. doi: 10.1016/j.jhep.2017.11.040
65. Wolf AJ, Reyes CN, Liang W, Becker C, Shimada K, Wheeler ML, et al. Hexokinase Is an Innate Immune Receptor for the Detection of Bacterial Peptidoglycan. Cell (2016) 166:624–36. doi: 10.1016/j.cell.2016.05.076
66. Sanman LE, Qian Y, Eisele NA, Ng TM, van der Linden WA, Monack DM, et al. Disruption of glycolytic flux is a signal for inflammasome signaling and pyroptotic cell death. Elife (2016) 5:e13663. doi: 10.7554/eLife.13663
67. Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, et al. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature (2013) 496:238–42. doi: 10.1038/nature11986
68. Weber K, Schilling JD. Lysosomes integrate metabolic-inflammatory cross-talk in primary macrophage inflammasome activation. J Biol Chem (2014) 289:9158–71. doi: 10.1074/jbc.M113.531202
69. Karasawa T, Kawashima A, Usui-Kawanishi F, Watanabe S, Kimura H, Kamata R, et al. Saturated Fatty Acids Undergo Intracellular Crystallization and Activate the NLRP3 Inflammasome in Macrophages. Arterioscler Thromb Vasc Biol (2018) 38:744–56. doi: 10.1161/ATVBAHA.117.310581
70. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol (2013) 14:812–20. doi: 10.1038/ni.2639
71. Yeon SH, Yang G, Lee HE, Lee JY. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J Leukoc Biol (2017) 101:205–15. doi: 10.1189/jlb.3VMA1215-579RR
72. L’Homme L, Esser N, Riva L, Scheen A, Paquot N, Piette J, et al. Unsaturated fatty acids prevent activation of NLRP3 inflammasome in human monocytes/macrophages. J Lipid Res (2013) 54:2998–3008. doi: 10.1194/jlr.M037861
73. Song N, Liu ZS, Xue W, Bai ZF, Wang QY, Dai J, et al. NLRP3 Phosphorylation Is an Essential Priming Event for Inflammasome Activation. Mol Cell (2017) 68:185–97.e6. doi: 10.1016/j.molcel.2017.08.017
74. Zhang Z, Meszaros G, He WT, Xu Y, de Fatima Magliarelli H, Mailly L, et al. Protein kinase D at the Golgi controls NLRP3 inflammasome activation. J Exp Med (2017) 214:2671–93. doi: 10.1084/jem.20162040
75. Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature (2019) 570:338–43. doi: 10.1038/s41586-019-1295-z
76. Hara H, Tsuchiya K, Kawamura I, Fang R, Hernandez-Cuellar E, Shen Y, et al. Phosphorylation of the adaptor ASC acts as a molecular switch that controls the formation of speck-like aggregates and inflammasome activity. Nat Immunol (2013) 14:1247–55. doi: 10.1038/ni.2749
77. Mortimer L, Moreau F, MacDonald JA, Chadee K. NLRP3 inflammasome inhibition is disrupted in a group of auto-inflammatory disease CAPS mutations. Nat Immunol (2016) 17:1176–86. doi: 10.1038/ni.3538
78. Guo C, Xie S, Chi Z, Zhang J, Liu Y, Zhang L, et al. Bile Acids Control Inflammation and Metabolic Disorder through Inhibition of NLRP3 Inflammasome. Immunity (2016) 45:944. doi: 10.1016/j.immuni.2016.10.009
79. Stutz A, Kolbe CC, Stahl R, Horvath GL, Franklin BS, van Ray O, et al. NLRP3 inflammasome assembly is regulated by phosphorylation of the pyrin domain. J Exp Med (2017) 214:1725–36. doi: 10.1084/jem.20160933
80. Spalinger MR, Kasper S, Gottier C, Lang S, Atrott K, Vavricka SR, et al. NLRP3 tyrosine phosphorylation is controlled by protein tyrosine phosphatase PTPN22. J Clin Invest (2016) 126:4388. doi: 10.1172/JCI90897
81. Guo W, Liu W, Chen Z, Gu Y, Peng S, Shen L, et al. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat Commun (2017) 8:2168. doi: 10.1038/s41467-017-02351-0
82. Ito M, Shichita T, Okada M, Komine R, Noguchi Y, Yoshimura A, et al. Bruton’s tyrosine kinase is essential for NLRP3 inflammasome activation and contributes to ischaemic brain injury. Nat Commun (2015) 6:7360. doi: 10.1038/ncomms8360
83. Liu X, Pichulik T, Wolz OO, Dang TM, Stutz A, Dillen C, et al. Human NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) inflammasome activity is regulated by and potentially targetable through Bruton tyrosine kinase. J Allergy Clin Immunol (2017) 140:1054–67.e10. doi: 10.1016/j.jaci.2017.01.017
84. Mao L, Kitani A, Hiejima E, Montgomery-Recht K, Zhou W, Fuss I, et al. Bruton tyrosine kinase deficiency augments NLRP3 inflammasome activation and causes IL-1beta-mediated colitis. J Clin Invest (2020) 130:1793–807. doi: 10.1172/JCI128322
85. Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med (2018) 215:1023–34. doi: 10.1084/jem.20171922
86. Basak C, Pathak SK, Bhattacharyya A, Mandal D, Pathak S, Kundu M. NF-kappaB- and C/EBPbeta-driven interleukin-1beta gene expression and PAK1-mediated caspase-1 activation play essential roles in interleukin-1beta release from Helicobacter pylori lipopolysaccharide-stimulated macrophages. J Biol Chem (2005) 280:4279–88. doi: 10.1074/jbc.M412820200
87. Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun (2018) 9:3001. doi: 10.1038/s41467-018-05321-2
88. Shao L, Liu Y, Wang W, Li A, Wan P, Liu W, et al. SUMO1 SUMOylates and SENP3 deSUMOylates NLRP3 to orchestrate the inflammasome activation. FASEB J (2020) 34:1497–515. doi: 10.1096/fj.201901653R
89. Humphries F, Bergin R, Jackson R, Delagic N, Wang B, Yang S, et al. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat Commun (2018) 9:1560. doi: 10.1038/s41467-018-03669-z
90. Ma Q, Chen S, Hu Q, Feng H, Zhang JH, Tang J. NLRP3 inflammasome contributes to inflammation after intracerebral hemorrhage. Ann Neurol (2014) 75:209–19. doi: 10.1002/ana.24070
91. Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell (2013) 49:331–8. doi: 10.1016/j.molcel.2012.11.009
92. Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P, et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature (2014) 512:69–73. doi: 10.1038/nature13322
93. Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun (2016) 7:13727. doi: 10.1038/ncomms13727
94. Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell (2015) 160:62–73. doi: 10.1016/j.cell.2014.11.047
95. Kawashima A, Karasawa T, Tago K, Kimura H, Kamata R, Usui-Kawanishi F, et al. ARIH2 Ubiquitinates NLRP3 and Negatively Regulates NLRP3 Inflammasome Activation in Macrophages. J Immunol (2017) 199:3614–22. doi: 10.4049/jimmunol.1700184
96. Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, et al. MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. J Immunol (2015) 194:4880–90. doi: 10.4049/jimmunol.1402851
97. Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med (2014) 211:1333–47. doi: 10.1084/jem.20132486
98. Labbe K, McIntire CR, Doiron K, Leblanc PM, Saleh M. Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase-1 activation by the inflammasome. Immunity (2011) 35:897–907. doi: 10.1016/j.immuni.2011.10.016
99. Mangan MSJ, Olhava EJ, Roush WR, Seidel HM, Glick GD, Latz E. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov (2018) 17:588–606. doi: 10.1038/nrd.2018.97
100. Yaribeygi H, Mohammadi MT, Rezaee R, Sahebkar A. Fenofibrate improves renal function by amelioration of NOX-4, IL-18, and p53 expression in an experimental model of diabetic nephropathy. J Cell Biochem (2018) 119:7458–69. doi: 10.1002/jcb.27055
101. Hoseini Z, Sepahvand F, Rashidi B, Sahebkar A, Masoudifar A, Mirzaei H. NLRP3 inflammasome: Its regulation and involvement in atherosclerosis. J Cell Physiol (2018) 233:2116–32. doi: 10.1002/jcp.25930
102. Wu KK, Cheung SW, Cheng KK. NLRP3 Inflammasome Activation in Adipose Tissues and Its Implications on Metabolic Diseases. Int J Mol Sci (2020) 21:4184. doi: 10.3390/ijms21114184
103. Kim HY, Lee HJ, Chang YJ, Pichavant M, Shore SA, Fitzgerald KA, et al. Interleukin-17-producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity-associated airway hyperreactivity. Nat Med (2014) 20:54–61. doi: 10.1038/nm.3423
104. Ising C, Venegas C, Zhang S, Scheiblich H, Schmidt SV, Vieira-Saecker A, et al. NLRP3 inflammasome activation drives tau pathology. Nature (2019) 575:669–73. doi: 10.1038/s41586-019-1769-z
105. Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int Immunopharmacol (2019) 67:458–64. doi: 10.1016/j.intimp.2018.12.019
106. Yang X, Zhao G, Yan J, Xu R, Che C, Zheng H, et al. Pannexin 1 Channels Contribute to IL-1beta Expression via NLRP3/Caspase-1 Inflammasome in Aspergillus Fumigatus Keratitis. Curr Eye Res (2019) 44:716–25. doi: 10.1080/02713683.2019.1584321
107. de Torre-Minguela C, Mesa Del Castillo P, Pelegrin P. The NLRP3 and Pyrin Inflammasomes: Implications in the Pathophysiology of Autoinflammatory Diseases. Front Immunol (2017) 8:43. doi: 10.3389/fimmu.2017.00043
108. Jiang H, He H, Chen Y, Huang W, Cheng J, Ye J, et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med (2017) 214:3219–38. doi: 10.1084/jem.20171419
109. Coll RC, Hill JR, Day CJ, Zamoshnikova A, Boucher D, Massey NL, et al. MCC950 directly targets the NLRP3 ATP-hydrolysis motif for inflammasome inhibition. Nat Chem Biol (2019) 15:556–9. doi: 10.1038/s41589-019-0277-7
110. Marchetti C, Swartzwelter B, Gamboni F, Neff CP, Richter K, Azam T, et al. OLT1177, a beta-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc Natl Acad Sci U S A (2018) 115:E1530–E9. doi: 10.1073/pnas.1716095115
111. Coll RC, Robertson AA, Chae JJ, Higgins SC, Munoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med (2015) 21:248–55. doi: 10.1038/nm.3806
112. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
113. Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW, et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem (2010) 285:9792–802. doi: 10.1074/jbc.M109.082305
114. He Y, Varadarajan S, Munoz-Planillo R, Burberry A, Nakamura Y, Nunez G. 3,4-methylenedioxy-beta-nitrostyrene inhibits NLRP3 inflammasome activation by blocking assembly of the inflammasome. J Biol Chem (2014) 289:1142–50. doi: 10.1074/jbc.M113.515080
115. Huang Y, Jiang H, Chen Y, Wang X, Yang Y, Tao J, et al. Tranilast directly targets NLRP3 to treat inflammasome-driven diseases. EMBO Mol Med (2018) 10:e8689. doi: 10.15252/emmm.201708689
116. Chen S, Wang Y, Pan Y, Liu Y, Zheng S, Ding K, et al. Novel Role for Tranilast in Regulating NLRP3 Ubiquitination, Vascular Inflammation, and Atherosclerosis. J Am Heart Assoc (2020) 9:e015513. doi: 10.1161/JAHA.119.015513
117. Riddle MC. Editorial: sulfonylureas differ in effects on ischemic preconditioning–is it time to retire glyburide? J Clin Endocrinol Metab (2003) 88:528–30. doi: 10.1210/jc.2002-021971
118. Marchetti C, Chojnacki J, Toldo S, Mezzaroma E, Tranchida N, Rose SW, et al. A novel pharmacologic inhibitor of the NLRP3 inflammasome limits myocardial injury after ischemia-reperfusion in the mouse. J Cardiovasc Pharmacol (2014) 63:316–22. doi: 10.1097/FJC.0000000000000053
119. Fulp J, He L, Toldo S, Jiang Y, Boice A, Guo C, et al. Structural Insights of Benzenesulfonamide Analogues as NLRP3 Inflammasome Inhibitors: Design, Synthesis, and Biological Characterization. J Med Chem (2018) 61:5412–23. doi: 10.1021/acs.jmedchem.8b00733
120. Rudolphi K, Gerwin N, Verzijl N, van der Kraan P, van den Berg W. Pralnacasan, an inhibitor of interleukin-1beta converting enzyme, reduces joint damage in two murine models of osteoarthritis. Osteoarthritis Cartilage (2003) 11:738-46. doi: 10.1016/s1063-4584(03)00153-5
121. Audia JP, Yang XM, Crockett ES, Housley N, Haq EU, O’Donnell K, et al. Caspase-1 inhibition by VX-765 administered at reperfusion in P2Y12 receptor antagonist-treated rats provides long-term reduction in myocardial infarct size and preservation of ventricular function. Basic Res Cardiol (2018) 113:32. doi: 10.1007/s00395-018-0692-z
122. Yang G, Lee HE, Moon SJ, Ko KM, Koh JH, Seok JK, et al. Direct Binding to NLRP3 Pyrin Domain as a Novel Strategy to Prevent NLRP3-Driven Inflammation and Gouty Arthritis. Arthritis Rheumatol (2020) 72:1192–202. doi: 10.1002/art.41245
123. Lee HE, Yang G, Kim ND, Jeong S, Jung Y, Choi JY, et al. Targeting ASC in NLRP3 inflammasome by caffeic acid phenethyl ester: a novel strategy to treat acute gout. Sci Rep (2016) 6:38622. doi: 10.1038/srep38622
124. Lee HE, Lee JY, Yang G, Kang HC, Cho YY, Lee HS, et al. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci Rep (2019) 9:12277. doi: 10.1038/s41598-019-48794-x
125. Sun Z, Li Y, Qian Y, Wu M, Huang S, Zhang A, et al. Celastrol attenuates ox-LDL-induced mesangial cell proliferation via suppressing NLRP3 inflammasome activation. Cell Death Discov (2019) 5:114. doi: 10.1038/s41420-019-0196-0
126. Lee HE, Yang G, Park YB, Kang HC, Cho YY, Lee HS, et al. Epigallocatechin-3-Gallate Prevents Acute Gout by Suppressing NLRP3 Inflammasome Activation and Mitochondrial DNA Synthesis. Molecules (2019) 24:2138. doi: 10.3390/molecules24112138
127. Yang G, Lee HE, Yeon SH, Kang HC, Cho YY, Lee HS, et al. Licochalcone A attenuates acne symptoms mediated by suppression of NLRP3 inflammasome. Phytother Res (2018) 32:2551–9. doi: 10.1002/ptr.6195
128. Yang G, Lee HE, Lee JY. A pharmacological inhibitor of NLRP3 inflammasome prevents non-alcoholic fatty liver disease in a mouse model induced by high fat diet. Sci Rep (2016) 6:24399. doi: 10.1038/srep24399
Keywords: innate immunity, pattern-recognition receptors, cell signaling, inflammation, pharmacological inhibitor
Citation: Seok JK, Kang HC, Cho Y-Y, Lee HS and Lee JY (2021) Regulation of the NLRP3 Inflammasome by Post-Translational Modifications and Small Molecules. Front. Immunol. 11:618231. doi: 10.3389/fimmu.2020.618231
Received: 16 October 2020; Accepted: 29 December 2020;
Published: 02 February 2021.
Edited by:
Jong-Seok Moon, Soonchunhyang University, South KoreaReviewed by:
Eun-Kyeong Jo, Chungnam National University, South KoreaIva Hafner Bratkovic, National Institute of Chemistry Slovenia, Slovenia
Copyright © 2021 Seok, Kang, Cho, Lee and Lee. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joo Young Lee, am9vbGVlQGNhdGhvbGljLmFjLmty