 Jiaqi Zou
Jiaqi Zou Clare Thornton2
Clare Thornton2 Emma S. Chambers
Emma S. Chambers Elizabeth C. Rosser
Elizabeth C. Rosser Coziana Ciurtin
Coziana Ciurtin- 1Centre for Rheumatology Research, Division of Medicine, University College London, London, United Kingdom
- 2Department of Rheumatology (Metabolic Bone Diseases), University College London Hospital NHS Foundation Trust, London, United Kingdom
- 3Centre for Immunobiology, Blizard Institute, Queen Mary University of London, London, United Kingdom
- 4Centre for Adolescent Rheumatology Versus Arthritis at University College London, University College London and Great Ormond Street Hospitals, London, United Kingdom
Vitamin D is synthesized in the skin following exposure to UVB radiation or is directly absorbed from the diet. Following hydroxylation in the liver and kidneys, vitamin D becomes its bioactive form, 1,25(OH)2D, which has been described to have potent immunomodulatory capacity. This review will focus on the effect of vitamin D in modulating the dysregulated immune system of autoimmune rheumatic diseases (ARD) patients across age, in particular in arthritis (rheumatoid arthritis and juvenile idiopathic arthritis), and systemic lupus erythematosus (with adult and juvenile onset). As well as delineating the impact of vitamin D on the innate and adaptive immune functions associated with each disease pathology, this review will also summarize and evaluate studies that link vitamin D status with disease prevalence, and supplementation studies that examine the potential benefits of vitamin D on disease outcomes. Exploring this evidence reveals that better designed randomized controlled studies are required to clarify the impact of vitamin D supplementation on ARD outcomes and general health. Considering the accessibility and affordability of vitamin D as a therapeutic option, there is a major unmet need for evidence-based treatment recommendations for the use of vitamin D in this patient population.
Introduction
Vitamin D is an essential micronutrient synthesized in the body or obtained from the diet. In recent decades, vitamin D deficiency (< 50 nmol/L) and insufficiency (50–75 nmol/L) has emerged as a widespread health problem (Table 1) (1, 2). According to available evidence, less than 50% of the global population have adequate vitamin D levels in their blood (3). The predominant bioactive form of vitamin D is cholecalciferol (vitamin D3), which is synthesized in the skin from 7-dehydrocholesterol following exposure to sunlight (UVB radiation). Other forms of vitamin D (mainly as ergocalciferol or vitamin D2) can also be absorbed from various foods e.g., oily fish, red meat, liver, egg yolks, fortified cereals, and specific supplements e.g., fish oil. Both cholecalciferol and ergocalciferol are converted via a two-step activation process (Figure 1). Firstly, cholecalciferol and ergocalciferol are hydroxylated in the liver and converted into the inactive metabolites 25-hydroxyvitamin D [25(OH)D3 (calcifediol) and 25(OH)D2 (ercalcidiol)], respectively. Secondly, calcifediol and ercalcidiol are hydroxylated to the bioactive form 1,25(OH)2D3 (calcitriol) and 1,25(OH)2D2 (ercalcitriol), respectively, in the kidneys. Much like other steroid hormones, both calcitriol and ercalcitriol bind to the vitamin D receptors (VDR) within cells, albeit ercalcitriol with a much lower affinity. VDR then heterodimerise with retinoid X receptors (RXR) and the complex translocates into the nucleus (4). This heterodimer in turn binds to vitamin D receptor elements (VDRE) present in various cell genes, altering gene expression (4). Two types of vitamin D supplementation are available, vitamin D2 and D3.

Table 1 Definition of vitamin D status. 1 nmol/L = 0.4 ng/mL.
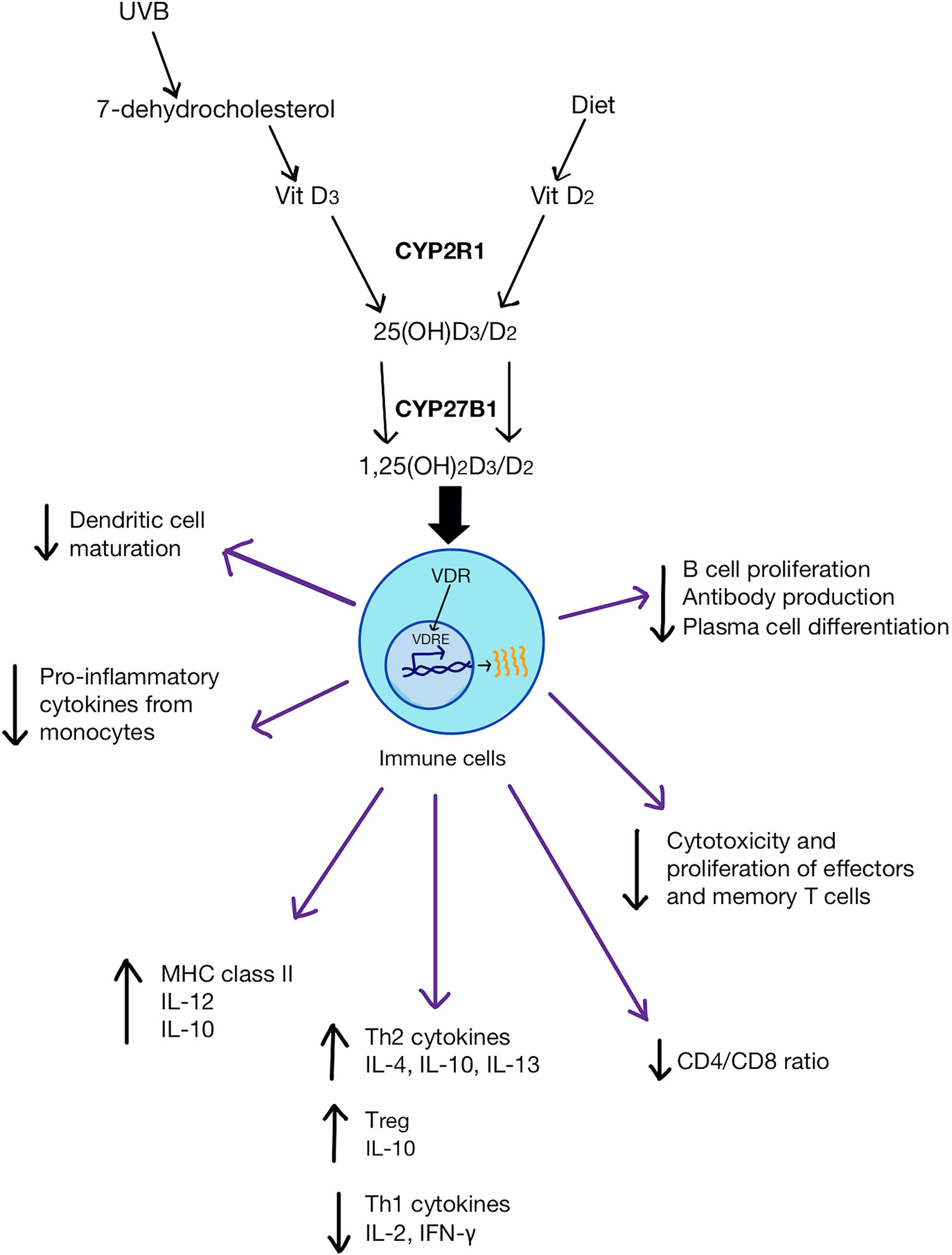
Figure 1 Vitamin D (both D3 and D2) metabolic pathway and its physiological impact on immune cells. D3 is synthesized from either 7-dehydrocholesterol from the skin via UVB activation or through the diet. D2 is obtained from the diet. CYP2R1 enzyme converts D3/D2 to 25(OH)D3/D2 in the liver. CYP27B1 transforms 25(OH)D3/D2 into 1,25(OH)2D3/D2 in the kidneys. 1,25(OH)2D binds to VDR in immune cells which then bind to VDRE on genes to change gene expression. 1,25(OH)2D can inhibit dendritic cell maturation, promote monocyte proliferation and differentiation into macrophages. It can also increase anti-inflammatory cytokines and the frequency of Tregs, as well as reduce pro-inflammatory Th1 cytokines. 1,25(OH)2D can also reduce the ratio of CD4/CD8 and inhibit pro-inflammatory T cell differentiation and suppress antibody production by B cells.
The classical role of bioactive vitamin D is to promote intestinal and renal calcium absorption, maintaining a very precise regulation of calcium transfer between bones and blood stream. Positive calcium balance is characterized by net bone formation, either as bone growth or repair; while in negative calcium balance, vitamin D promotes bone resorption and inhibits bone mineralization to maintain serum calcium levels (5). As a consequence, vitamin D has an active role in the prevention of bone fractures associated with low bone density/osteoporosis, eliciting fracture reduction effect from a minimum serum concentration of 74 nmol/L, which is now widely accepted as lower limit of optimal vitamin D level (Table 1) (6). Following the observation that VDRs are also expressed by immune cells, endothelial cells, and vascular smooth muscle cells (7–9), there is now a wealth of studies demonstrating that vitamin D has an additional immunomodulatory role, as well as implications in cardio-vascular health. Recent research has linked vitamin D deficiency to the pathogenesis of various immune-mediated inflammatory diseases in both children and adults (10, 11), Importantly, there is some evidence that vitamin D deficiency may act as an environmental trigger for autoimmune disease development (12, 13), and in particular autoimmune rheumatic diseases (ARD) (14, 15), with some studies suggesting that there may be some therapeutic benefit of vitamin D supplementation in these diseases (13, 16).
In this review, we will discuss the evidence for an immunomodulatory effect of vitamin D on the two of the most prototypical ARD across age; rheumatoid arthritis (RA) and juvenile idiopathic arthritis (JIA) and juvenile and adult-onset systemic lupus erythematosus (SLE). Although, exploring the immunomodulatory role of vitamin D in the process of atherosclerosis which is a common co-morbidity associated with chronic inflammation as seen in various ARDs is beyond the scope of this review, it is worth mentioning the protective effects of vitamin D on endothelial activation and dysfunction, through inhibition of cyclooxygenase 2 and cellular/platelets adhesion molecules expression, as well as pro-inflammatory cytokines synthesis, therefore minimizing the inflammatory processes contributing to atherosclerosis plaque formation (17, 18).
Immune Dysregulation in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis
Although RA and JIA are completely different diseases, they are both characterized by immune-mediated joint inflammation. In both diseases, autoimmune arthritis is characterized by inflammation of the synovial membrane, which involves the proliferation of synoviocytes and invasion of inflammatory immune cells into the synovium. This leads to synovial membrane thickening, which, if left untreated, ultimately leads to irreversible articular cartilage destruction and bone erosions (19). In adults, RA is one of the most common chronic rheumatic diseases, affecting 0.5–1% of the global population (20). In children, autoimmune inflammatory arthritis is classified under the umbrella term of JIA and is also one of the most common rheumatic diseases in children and adolescents. JIA terminology encompasses many types of arthritis, which are pathologically distinct from RA with the exception of the rheumatoid factor (RF) positive polyarticular JIA phenotype, present in less than 10% JIA patients which is clinically, serologically and genetically similar to the RA phenotype in adults (21, 22). For simplicity, in this review, we will focus on studies in patients with oligo-articular JIA (oligo-JIA), which is the most common phenotype in children, and polyarticular-JIA (poly-JIA), where, as described above, there is some overlap with the pathogenesis of RA (23). Disease activity in RA is measured by using the Disease Activity Score-28 (DAS28) index, which includes swollen and tender joint counts out of 28 designated joints, ESR or CRP levels, and subjective rating of global health measured on a 1–10 visual analogue scale (VAS). For JIA, the most used disease activity measure is the Juvenile Arthritis Disease Activity Score (JADAS), which assesses a variable number of joints (10, 27, or 77), as well as physician and patient/parent VAS rating of disease activity and inflammation (ESR).
Although fundamentally different diseases, oligo-JIA, poly-JIA, and RA share many immunopathogenic features that lead to synovitis, including being characterized by a strong pro-inflammatory cytokine signature. Activated myeloid cells produce increased levels of TNF-α, IL-6, IL-1β at the inflamed site, and this is reflected in the shared therapeutic approaches of blocking these cytokines in both RA and JIA (24–26). There is also an imbalance of T-helper 1 (Th1) and T-helper 17 (Th17) cells compared to T helper 2 (Th2) and regulatory T (Treg cells) (27), and both RA and JIA have been postulated to be mediated by both the type 1 cytokines such as IFN-γ produced and IL-17 (28). Collectively, TNF-α, IL-6, IL-1β, and IL-17 form positive feedback loops increasing the production of these potent inflammatory cytokines, promoting synovial inflammation, and bone resorption, which characterize both diseases. Additionally, in both diseases, B cells secrete autoantibodies, such as rheumatoid factors (RF), anti-cyclic citrullinated peptide (CCP) antibodies, and, in some oligo-JIA and poly-JIA patients, anti-nuclear antibodies (ANAs) (29, 30). B cells can also contribute to inflammation by producing chemokines and cytokines that trigger inflammation and synovial hyperplasia, and by presenting autoantigens to autoreactive T cells (31). It is thought that a complex interplay between genetic and environmental factors leads to this breakdown in immunological tolerance and thus synovial inflammation. Variants within HLA genes have been demonstrated to increase the risk of developing RA and JIA (32, 33), whilst environmental factors such as exposure to tobacco smoke and changes to the gut microbiota have been implicated in disease development (34, 35). In recent years, new evidence has emerged that vitamin D deficiency and insufficiency may also be an important environmental trigger in the onset of disease.
Prevalence of Hypovitaminosis D and Its Impact on the Risk of Developing Rheumatoid Arthritis and Juvenile Idiopathic Arthritis
Numerous studies have investigated the relationship between vitamin D insufficiency/deficiency and autoimmune arthritis, establishing that RA patients present with lower vitamin D levels than healthy controls (36–39). A systematic review with 3,489 people from 24 reports found that RA patients had lower 25(OH)D levels than healthy controls with a mean difference of 16.52 nmol/L(= 6.6 ng/ml) (38). Likewise, the COMORA study measured vitamin D levels of 1,413 RA patients from 15 countries and reported that 63% of patients were either vitamin D insufficient or deficient (36). Similarly, in JIA patients, the evidence for hypovitaminosis D, or low vitamin D levels, is consistent across several studies (40–43). A scoping review in 2018 reported that 32/38 studies showed a high prevalence (84.2%) of suboptimal 25(OH)D levels (< 30ng/ml) in JIA patients (41). Similarly, a meta-analysis revealed widespread vitamin D deficiency status in JIA patients (up to 82%) (42). These data strongly suggest that children and adults with inflammatory arthritis have low concentrations of circulating vitamin D.
Another important consideration is whether individuals with low vitamin D levels are at risk for developing arthritis. There is controversial evidence regarding the impact of vitamin D intake on the risk of developing RA. The Iowa Women’s Health Study recorded the vitamin D intake of 29,368 women without RA at baseline with a self-reported questionnaire and followed them through 11 years (44). They found a negative correlation between vitamin D intake and RA risk (relative risk RR = 0.72). Conversely, the Nurses’ Health Study reported no relationship between vitamin D intake and risk of RA (45). Yet, interestingly, a meta-analysis of the Iowa and NHS cohorts found an overall significant relationship between RA risk and total vitamin D intake (diet and supplements) (RR = 0.758, p = 0.047) (46). Patients with the lowest vitamin D intake had a 24.2% higher risk of RA than those with the highest vitamin D intake. Together, these studies indicate that it is challenging to get an accurate picture of vitamin D intake and RA risk. This brings into question the validity of dietary intake assessments with food frequency questionnaires without measuring serum vitamin D levels. Limitations of these questionnaires include reporting bias and their closed and limited format. There are no similar studies for JIA.
Vitamin D Levels and Disease Activity: Therapeutic Implications for Rheumatoid Arthritis and Juvenile Idiopathic Arthritis
Considering the evidence that both JIA and RA patients have low vitamin D levels, there is growing interest in understanding whether altering vitamin D levels can impact disease activity. For RA, most studies have concluded that serum 25(OH)D levels inversely correlated with the DAS28 index (36–38, 46) (Table 2). From the limited studies available in JIA, it remains controversial whether 25(OH)D levels correlate with disease activity. However, a number of studies found negative correlations between JIA disease activity and serum 25(OH)D levels, although it remains difficult to infer a causal relationship (43, 50, 51), as the majority of studies were cross-sectional. No causal relationship between vitamin D levels and disease activity can be inferred as the evidence from randomized controlled trials (RCTs) is lacking. The evidence for a link between vitamin D supplementation and RA disease activity is inconsistent. Some studies suggested a decrease in DAS28 following supplementation, whereas other studies reported no significant change in DAS28 or flare rate after supplementation (Table 3). Due to the variation in the type of vitamin D supplementation, dose, and duration, it is extremely difficult to compare these studies. The majority of studies supplemented vitamin D deficient patients only (52–55); two studies supplemented patients irrespective of serum levels (57), though in one study, 90% of the patients were vitamin D insufficient (58). For JIA, only one cross-sectional study has investigated the role of vitamin D supplementation in modulating disease activity and did not report any therapeutic benefits (58). Standardizing the method and amount of supplementation would allow more comparison across different studies and cohorts. Further studies are needed to clarify whether vitamin D has any therapeutic benefits for in RA or JIA.
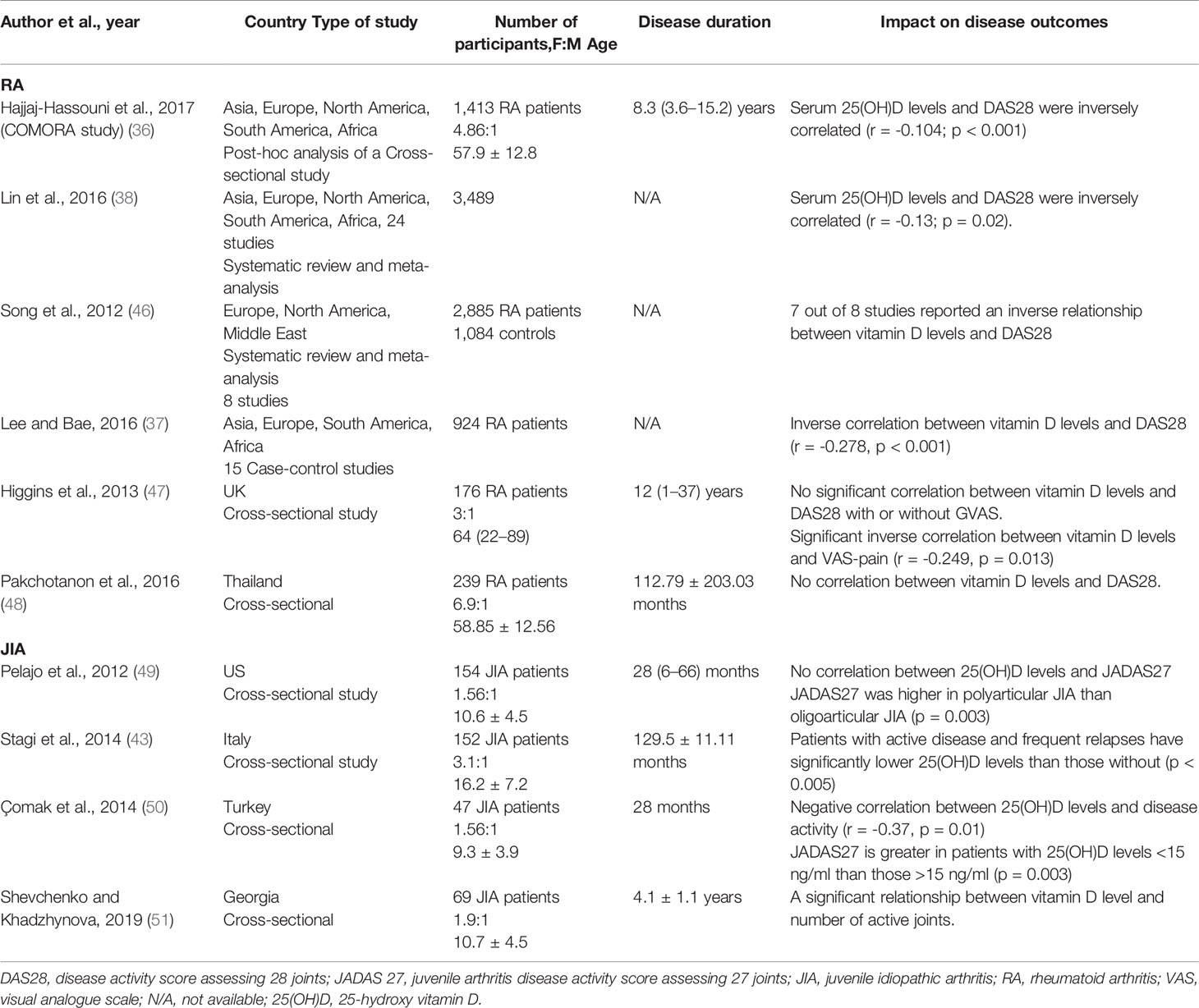
Table 2 Impact of vitamin D concentration on RA and JIA outcomes.
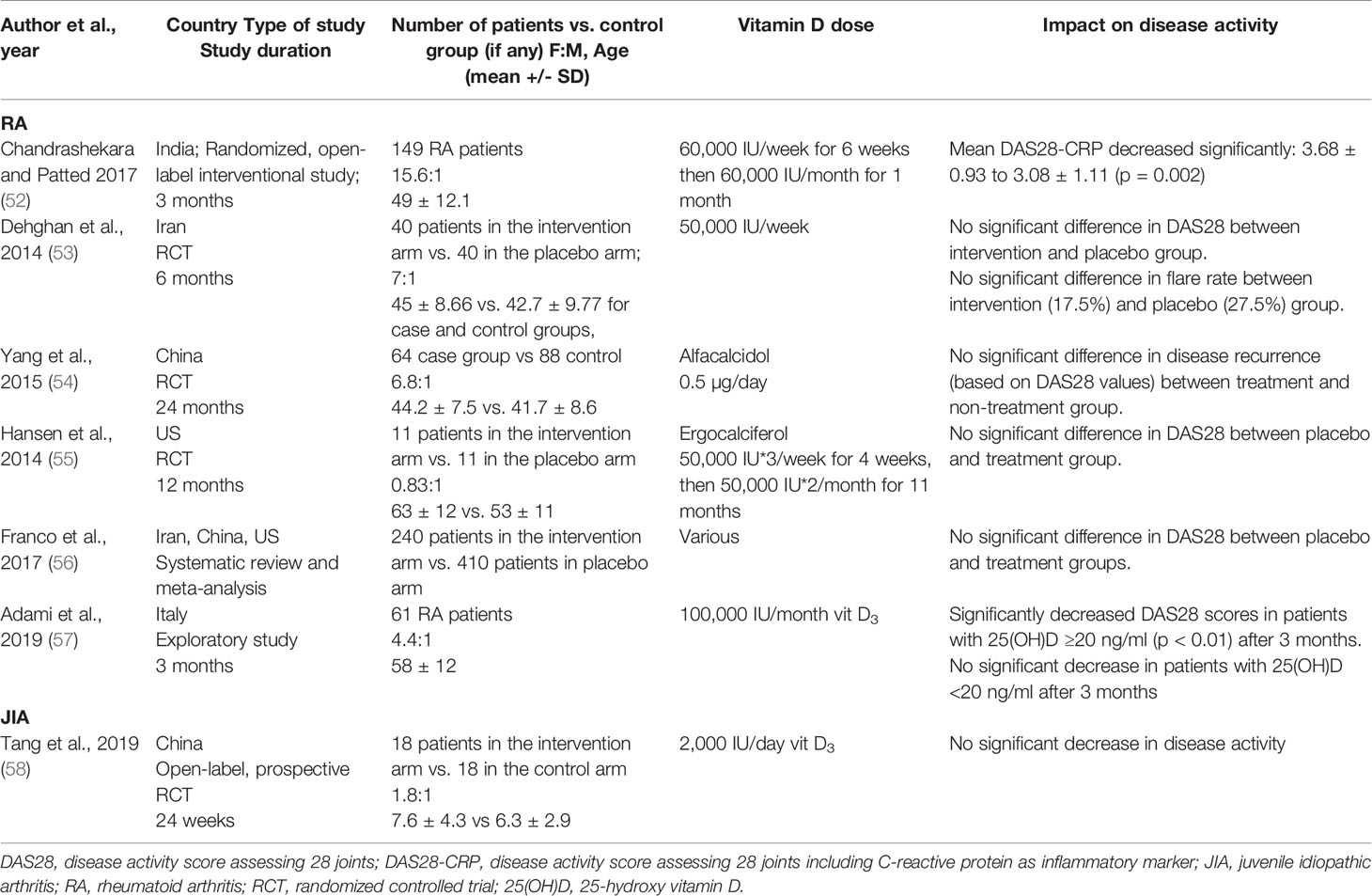
Table 3 Impact of vitamin D supplementation on RA and JIA outcomes.
Immunomodulatory Mechanisms for Vitamin D in Rheumatoid Arthritis and Juvenile Idiopathic Arthritis
There are several mechanisms by which vitamin D could modify inflammatory pathways in autoimmune inflammatory arthritis (Figure 2). For example, 1,25(OH)2D3 can influence myeloid-derived cytokine pathways. An in vitro study demonstrated that the exposure of RA fibroblast-like synviocytes to 1,25(OH)2D3 (in doses ranging from 0.1 to 100 nM, therefore equivalent to sub-physiological and optimal vitamin D serum concentrations) decreased TNF-α and IL-6 expression and at the same time, reduced osteoclastogenic RANKL (receptor activator of nuclear factor kappa-B ligand) levels relative to OPG (osteoprotegerin), an inhibitor of RANKL activity (59). In vivo, 1,25(OH)2D3 treatment at supra-physiological concentration (1 μmol/L) inhibited synoviocyte invasion and reduced IL-1β and matrix metalloprotease-1 (MMP-1) when measured ex vivo (60). Although the mechanism by which this suppression of cytokine production by synoviocytes remains unknown, 1,25(OH)2D3 was detected in the arthritic synovium and VDRs were found in rheumatoid synovial tissue suggesting that there may be direct suppression of cytokine gene expression in these cells (61). Interestingly, it has been suggested that 1,25(OH)2D3 can only act as negative feedback to reduce inflammatory responses when the immune system is activated (62), in which 1,25(OH)2D3 can promote synoviocyte apoptosis but only in the presence of TNF-α (63).
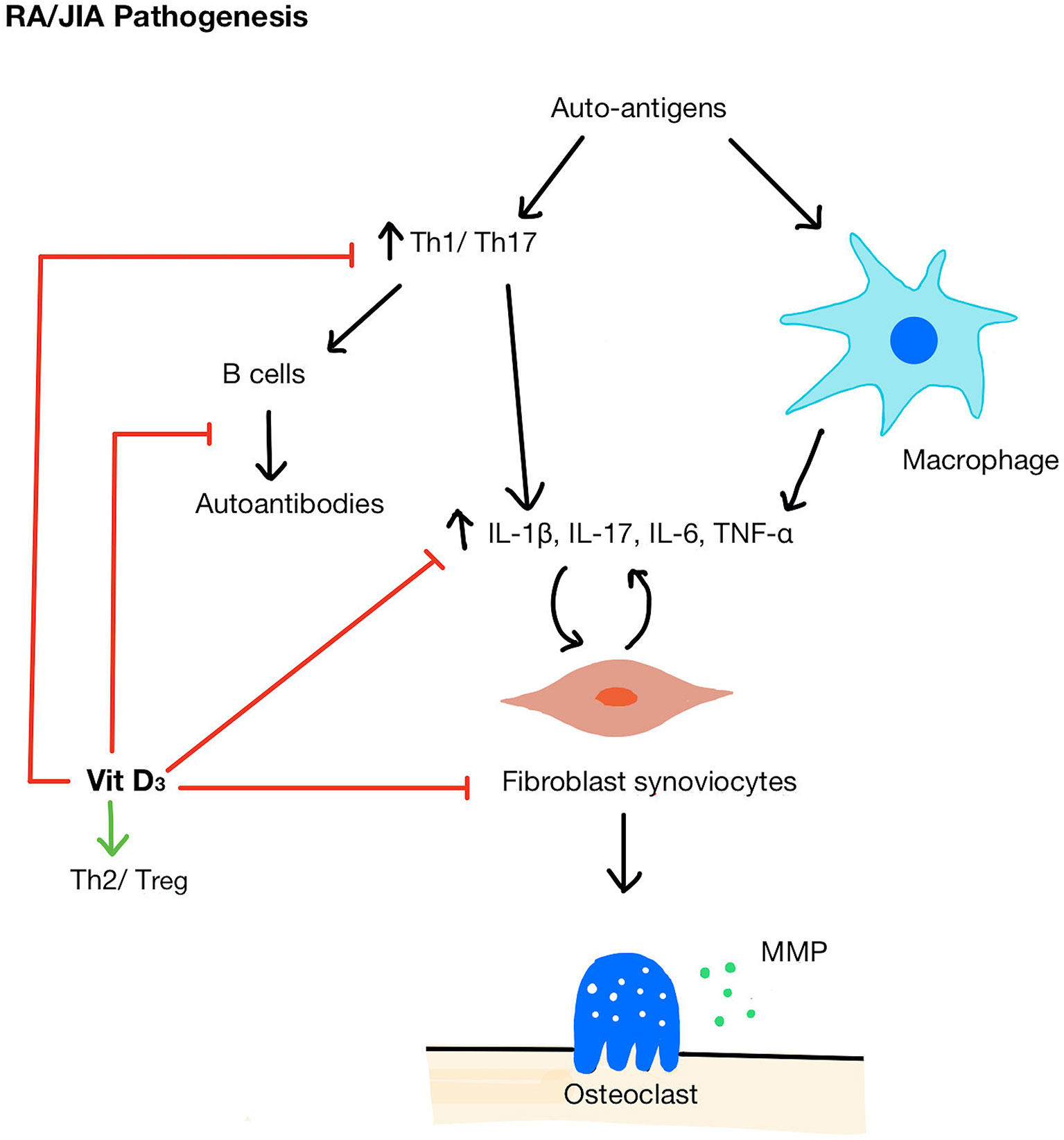
Figure 2 Pathogenesis of RA/JIA and mechanisms of vitamin D acting on immune cells. A breakdown in tolerance in RA/JIA leads to increased Th1/Th17 cells and promotes macrophages to produce pro-inflammatory cytokines (IL-1β, IL-17, IL-6, TNF-α) which in turn stimulates fibroblast synoviocyte proliferation. Synoviocytes then activate osteoclasts and secrete MMPs which break down bone and cartilage. Vitamin D shifts the balance of Th cells from Th1/Th17 to Th2/Treg, inhibit B cells and autoantibody production, reduces pro-inflammatory cytokines, and inhibits synoviocyte proliferation. MMP, matrix metallopeptidase.
1,25(OH)2D3 can also alter T cell differentiation by promoting a shift of Th cells from Th1/Th17 profile (64, 65), which is pathogenic in both RA and JIA, to a Th2/Treg, profile, associated with anti-inflammatory properties (66). 1,25(OH)2D3 stimulates the expression of both IL-10+ and Foxp3+ Treg cells in human T cell culture (67, 68). 1,25(OH)2D3 inhibited cytokines essential for Th1 and Th17 maturation (66) and upregulated the expression of transcription factors that induce Th2 cell development (69). Interestingly, in RA patients, serum 25(OH)D level was inversely correlated with IL-17 levels (70) and in vitro physiological doses of 1,25(OH)2D3 have been shown to decrease the IL-17 production by Th17 cells (71). Considering these complementary actions of immunosuppression, it has been suggested that that vitamin D may be more efficacious at suppressing immunological pathogenic mechanism when acting as an adjunct therapy. For example, Kim et al. found that 25(OH)D sufficient RA patients had greater DAS28 reduction in response to tocilizumab treatment (an IL-6 receptor antagonist) than 25(OH)D deficient patients and the difference persisted for 48 weeks (72). This study also showed that the addition of tocilizumab and 1,25(OH)2D3 in vitro suppressed the IL-17 production synergistically (72). A significant limitation of this study was that only baseline 25(OH)D level was recorded; it did not account for any changes in 25(OH)D levels in patients throughout the follow-up period. Furthermore, in vitro, 1,25(OH)2D3 at an optimal physiological concentration in combination with corticosteroids additively inhibited the TNF-α, IL-17, IL-6, and matrix metallopeptidase (MMP) production by synoviocytes co-cultured with T-cells (73). Notably, TNF-α could only be inhibited by the combination of both 1,25(OH)2D3 and corticosteroid and not by either alone, suggesting that vitamin D was beneficial only in combination with corticosteroid exposure. Although these in vitro experimental setting cannot replicate the pathological synovial environment of RA or JIA, they do provide evidence that vitamin D supplementation could be more efficacious at suppressing inflammatory mediators when given in combination with other therapeutic agents.
It is important to note that one study argued against the use of 1,25(OH)2D3 to treat active RA, by demonstrating that there was a difference in the effect of optimal physiological 1,25(OH)2D3 concentration on Th17 in serum compared to those in the synovial fluid (74). The authors proposed that 1,25(OH)2D3 had a lower inhibitory effect on Th17 in the synovial fluid because 1,25(OH)2D3 is less effective on the more committed T cell phenotypes present in the synovium compared to the naïve T cells in the serum. Thus, they questioned the efficacy of vitamin D as a potential treatment for active RA patients; rather, they proposed it can be used as a prophylactic measure to reduce the recurrence of flares in patients in remission. Future studies should investigate the reason behind the lack of synovial response to 1,25(OH)2D3 and explore ways to improve its sensitivity. At the same time, supplementation studies should be conducted in subjects at risk of RA or in the early stages of RA to explore vitamin D role in preventing abnormal immune activation (62).
Immune Dysregulation in Systemic Lupus Erythematosus With Juvenile and Adult Onset
SLE is a severe autoimmune rheumatic disease that is characterized by chronic, multisystem inflammation (75). It has an unpredictable course, with disease flares alternating with periods of clinical remission (76). The disease is more prevalent in females than males and peaks at the time of puberty in girls. SLE occurs most commonly in patients of 15–44 years of age, although 15–20% of cases start in childhood or adolescence (in which case the disease is called juvenile SLE- JSLE) (77, 78). JSLE can often be more severe, involving life-threatening damage to the kidneys and central nervous system. SLE patients overall require long term and aggressive treatments, and the mortality is increased in JSLE compared with adult-onset SLE. The primary diagnosis for SLE and JSLE is made using a combination of characteristic clinical features and detection of autoimmune antibodies, such as ANA, and in particular, antibodies against double-stranded DNA (dsDNA) which are specific for SLE (79). In clinical settings and experimental studies, SLE disease activity is measured using the SLE disease activity index (SLEDAI) which is a global index comprising 24 clinical and laboratory variables (80).
SLE is primarily thought of as a type 1 IFN driven disease. Both JSLE and SLE patients have an increased IFN signature, defined as overexpression of IFN-inducible genes in monocytes and lymphocytes, when compared to healthy controls (81, 82). It is thought that endogenous nucleic acids released by apoptotic cells or historic viral infections lead to the production of IFN-α by plasmacytoid dendritic cells. IFN-α goes on to activate autoreactive B cells, T helper (Th) cells and cytotoxic T cells. These endogenous nucleic acids and apoptotic cells are then bound by anti-nuclear autoantibodies (ANAs), leading to immune complex deposition and epitope spreading. Self-antigens are also presented by mature dendritic cells (non-tolerogenic) (83, 84), which activate pro-inflammatory Th cells (83). Similarly to RA, HLA genetic variants and complement genes have been associated with SLE (85). Environmental factors such as UV light, Epstein-Barr virus infection and obesity could contribute to SLE predisposition. A key feature of lupus is the skin photosensitivity, which leads to many SLE patients avoiding exposure to sunlight, it is perhaps unsurprising that low vitamin D levels have been described in many lupus patients.
Prevalence of Hypovitaminosis D and Its Impact on the Risk of Developing JSLE and Adult-Onset Systemic Lupus Erythematosus
The evidence for inadequate vitamin D levels in SLE is well documented. Recently, a systematic review further confirmed the high prevalence of vitamin D insufficiency in SLE (86). It evaluated 34 case-control studies consisting of 2265 SLE patients and concluded that serum 25(OH)D levels were significantly lower in SLE patients than controls (p < 0.00001). Other cross-sectional studies across Europe, South America, and Asia have reached similar conclusions that the overwhelming majority of SLE patients had insufficient vitamin D levels, which were significantly lower than healthy controls (87–89). As described above, the prevalence of hypovitaminosis D in SLE patients is likely due to the precautions taken by the patients to avoid the sun because of skin photosensitivity, with one study confirming that photosensitivity and photoprotection predicted vitamin D insufficiency and deficiency respectively (87). The use of corticosteroids and antimalarials for SLE treatment can also contribute to reduced vitamin D levels by inhibiting intestinal absorption of cholecalciferol and promoting vitamin D catabolism (90–92). Despite a paucity of studies on JSLE, the existing studies show similar results to adult SLE studies (93–96). Like the adult disease, vitamin D deficiency is prevalent in JSLE patients. This was shown in the APPLE trial which found that 69% of 201 JSLE patients had vitamin D insufficiency and 30% had vitamin D deficiency (93).
As for the relationship between dietary intake and SLE risk, the Nurses’ Health Study (NHS), a prospective cohort study with 186,389 women follow-up for 22 years, reported no relationship between vitamin D intake and risk of SLE (45). Nor was intake in adolescence related to SLE risk in adulthood (97). Similarly, Lourdudoss et al. did not find any protective effect of dietary vitamin D intake on lupus activity in 111 SLE patients during a 2-year period (98).
Vitamin D Levels and Disease Activity: Therapeutic Implications for SLE and JSLE
As with RA, a number of studies have reported an inverse association between serum 25(OH)D levels and disease activity as measured by the SLEDAI score in the majority of studies, and also by the European Consensus Lupus Activity Measurement – ECLAM score, in one study (99). They are summarized in Table 4. In JSLE, one study has also reported a potential inverse relationship between disease activity and 25(OH)D levels (94). Notably, approximately 81% of adult SLE patients also present with fatigue, which remains one of the most troublesome and common symptoms in SLE patients and is inadequately addressed by current treatment strategies (103). Thus, it is important to highlight that two studies have suggested a potentially inverse association between 25(OH)D levels and fatigue scores (87, 102) (Table 4). Like RA, most studies were cross-sectional, thus only correlations were inferred. A causal relationship remains undetermined. Importantly, these studies were conducted in different countries around the world, but all reached the same conclusion which shows that hypovitaminosis D in SLE patients is prevalent. This suggests that changes to geographical sunlight levels does not influence these results.
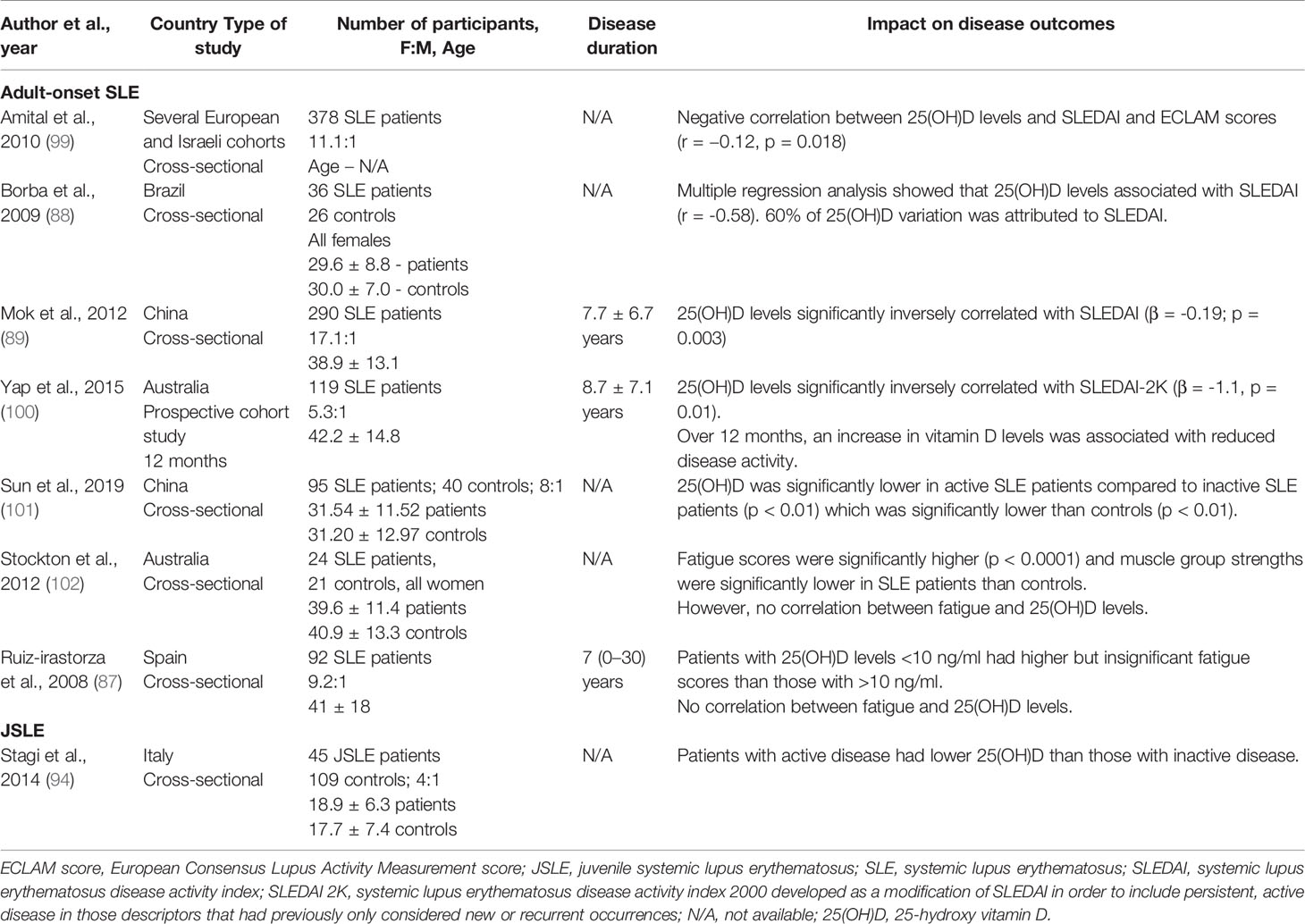
Table 4 Impact of vitamin D concentration on JSLE and adult-onset SLE outcomes.
Regarding the investigation of the vitamin D supplementation role in SLE, most studies reported no significant improvements in disease activity (Table 5). Only one study reported a reduced risk of achieving a moderately-high SLEDAI score (≥ 5) after vitamin D supplementation (106). On the other hand, two JSLE demonstrated decreases in disease activity after supplementation (Table 5). Notably, most studies either solely or mainly supplemented vitamin D insufficient patients (95, 105–109). Two studies treated both vitamin D insufficient and sufficient patients (96, 104).
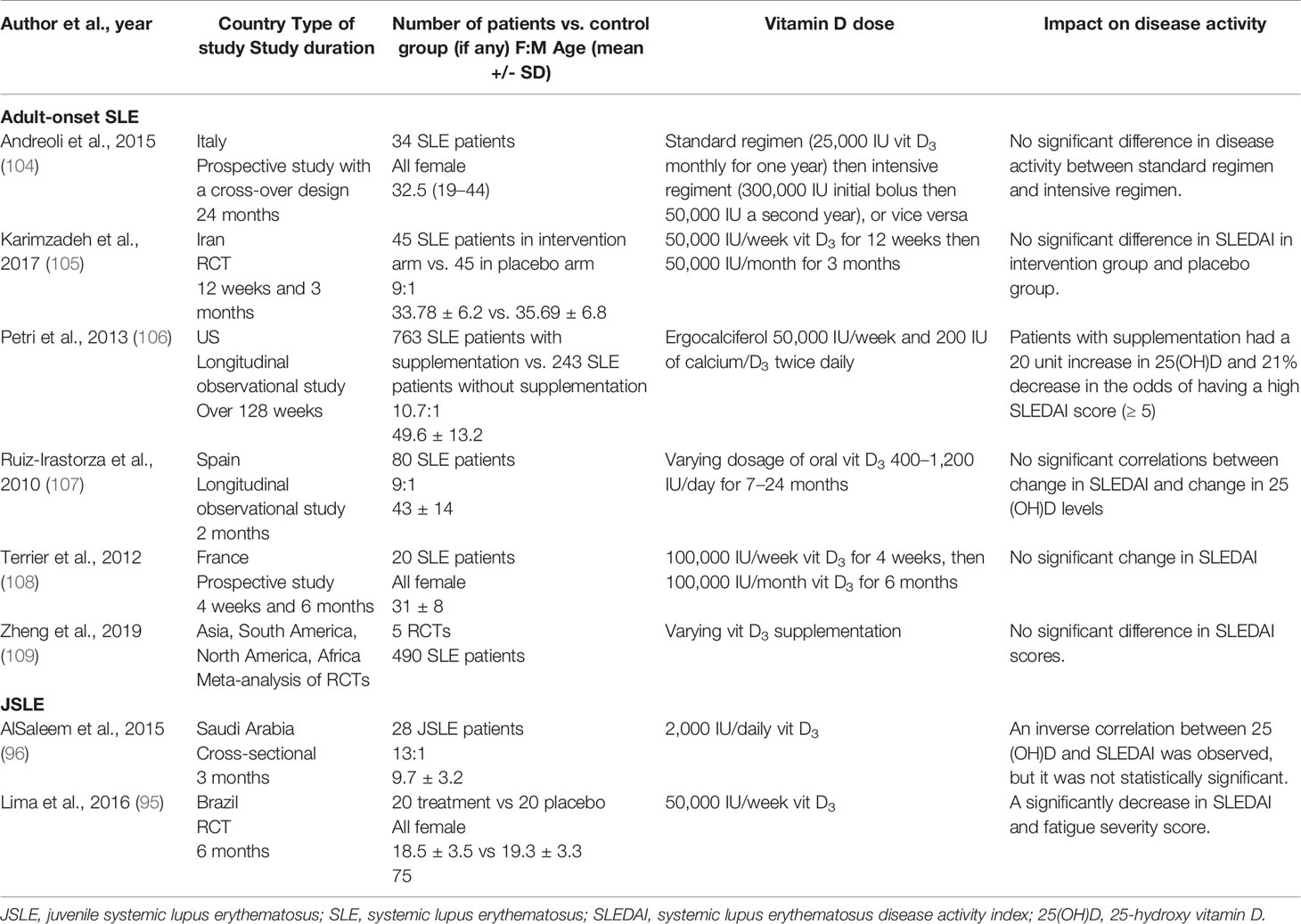
Table 5 Impact of vitamin D supplementation on JSLE and adult-onset SLE outcomes.
Immunomodulatory Mechanisms of Vitamin D in Systemic Lupus Erythematosus and JSLE
Vitamin D has been demonstrated to influence many immunological pathways that contribute to SLE pathogenesis (Figure 3). For example, 1,25(OH)2D3 has been demonstrated to inhibit of dendritic cell maturation. In one seminal study, VDR knockout mice were shown to have an expansion in mature dendritic cells compared to the wild-type mice (110). In this study, the authors concluded that 1,25(OH)2D3 can physiologically inhibit dendritic cell maturity even when dendritic cells are exposed to stimuli that usually control their maturation. Similar results have been reported in SLE patients (111), where monocyte-derived dendritic cells responded to 1,25(OH)2D3 supplementation at 10 nM, which led to a shift towards a semi-mature, tolerogenic phenotype.
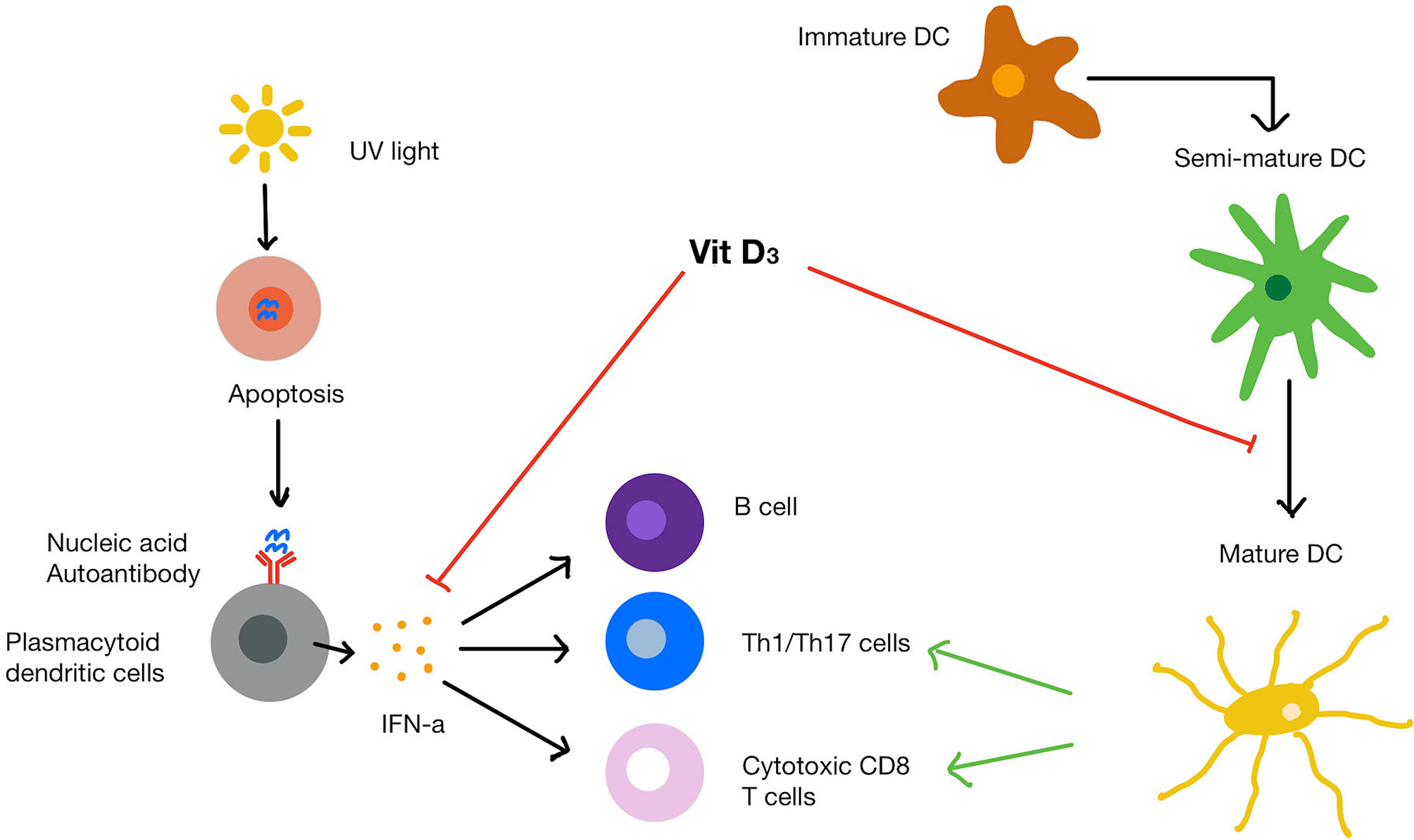
Figure 3 SLE pathogenesis and mechanism of vitamin D on immune cells. UVB light causes apoptosis of cells that release endogenous nucleic acids. The nucleic acids bind to and trigger plasmacytoid dendritic cells to release IFN-α. Mature dendritic cells recruit T helper cells, cytotoxic T cells and B cells. Vitamin D increases protective innate immune response while dampening the over-active immune response. Vitamin D also inhibits the maturation of dendritic cells and suppresses IFN-α production from plasmacytoid dendritic cells as well as IFN-α induced gene expression.
Additionally, there is evidence that 1,25(OH)2D3 can inhibit the expression of IFN-α inducible genes resulting in a reduced IFN signature response (111). A cross-sectional study found that 25(OH)D levels inversely correlated with serum IFN-α levels (r = -0.43, p < 0.0001) and IFN-α gene expression (r = -0.45, p < 0.0001) in SLE patients (112). SLE patients with low serum 25(OH)D levels (≤ 20 ng/ml) overexpressed 2 IFN-α inducible genes compared to those with sufficient serum levels (≥ 20 ng/ml) (16). The expression of these two genes was two-fold lower after cholecalciferol supplementation. However, there are conflicting results as in a subsequent study, Aranow and colleagues did not find any improvement of vitamin D supplementation on IFN signature (113).
1,25(OH)2D3 has also been shown to suppress the proliferation and differentiation of B cells into memory cells or plasma cells and induce anti-inflammatory cytokine production (IL-10) by regulatory B cells (114). Importantly, there is a well-documented imbalance in these B subsets in SLE patients, with an increase in plasma cells, which contribute to autoantibody production, and a defective regulatory B cell compartment (115). Collectively, these data suggest that the ability of 1,25(OH)2D3 to skew B cell differentiation in favour of a regulatory phenotype could be efficacious in suppressing inflammation in SLE patients. Although there are limited studies investigating whether vitamin D affects pathogenic mechanisms in SLE, based on the available evidence in vitro that vitamin D is able to prevent dendritic cell maturation, inhibit IFN-α production, alter B cell phenotype and potentially reduce the IFN signature response it is likely to have a significant impact on the dysregulated immune response. This represents a serious knowledge gap which should be addressed with future studies.
Conclusion
RA, JIA, SLE, and JSLE are ARD driven by a dysregulated innate and adaptive immune system. Current treatments aim to control and relieve the symptoms, as well as prevent long-term damage. Nonsteroidal anti-inflammatory drugs (NSAIDs), corticosteroids, disease-modifying anti-rheumatic drugs (DMARDS), and biologic agents are all part of the therapeutic armamentarium for arthritis and lupus. Importantly, many of these drugs are associated with serious side effects such as an increased risk of infection and malignancy, as well as high socioeconomic costs for health services. While no cure remains for these diseases, a significant unmet need remains for cost-effective therapies with limited side effects.
Hypovitaminosis D is prevalent among patients with inflammatory arthritis and SLE across all ages. The majority of studies have so far provided evidence for an inverse association between vitamin D levels and disease activity in RA and adult SLE. The limited number of studies in JIA and JSLE revealed potential associations between the two, but further research is required to confirm these results. Since most studies were cross-sectional, no causal relationship can be inferred, highlighting the necessity for longitudinal studies that investigate the association between vitamin D levels and disease activity within the same patient over time.
In terms of the therapeutic effect of vitamin D supplementation and suppression of disease activity, randomized controlled trials have yet to provide evidence for its efficacy in both diseases. Notably, the studies included in this review have exposed several limitations of the research on this topic. One prominent problem lies in the high heterogeneity between studies regarding the subject population, vitamin D supplementation dose and frequency of administration, outcome measurements, and even definitions of vitamin D deficiency. Trials have been conducted in over 20 countries in various continents, and many found that the prevalence of low vitamin D levels varied geographically, which could be due to differences related to ethnic background, sun exposure, and clothing style. Developmental status of the country, which significantly influences the quality of diet, and therefore vitamin D intake, will also affect vitamin D levels. In addition, the studies investigating the impact of vitamin D supplementation on the risk of developing arthritis or SLE are limited by the use of self-reported and retrospective data. Furthermore, the lack of consensus in dosage regimens between different studies in both RA and SLE means that there is no conclusive evidence of the role of vitamin D supplementation on disease outcomes.
Nevertheless, immunomodulatory studies have offered promising results. In vitro addition of 1,25(OH)2D3 altered the phenotype of dendritic cells, shifted the balance of T helper cells, inhibited pro-inflammatory cytokines and interferon proteins production, and subsequently affected both T and B cell activity, which could be relevant for the pathogenesis of both RA and SLE. Importantly, the wide-ranging effects of vitamin D on potential pathogenic pathways highlights a potential potent immunosuppressive function in these diseases, if the right therapeutic protocol is identified.
Although the scope of this review was to specifically explore the immunomodulatory role of vitamin D in arthritis and lupus across age, there is evidence of its benefits in other ARDs, such as undifferentiated connective tissue disease (UCTD). A 5-week course of daily alfacalcidol supplementation at various doses administered in two open label trials to patients with UCTD restored the capacity of regulatory T cells to suppress the proliferation of autologous CD4+ T cells implicated in the pathogenesis of the disease (116, 117), providing evidence for a potential therapeutic role of vitamin D in the management of this condition. A large study of 1,029 patients with various ARDs, including RA, SLE, autoimmune myositis, scleroderma, antiphospholipid syndrome, multiple sclerosis, and autoimmune thyroid disease, measured the serum vitamin D levels using an immunoluminometric assays and found suboptimal levels (< 20 ng/ml) in all patients (118). This study found no correlation between vitamin D level and SLE disease activity measured using the ECLAM score. Overcoming the limitations of available research in this field would close the gap between hypothesis and treatment efficacy. Future research should focus on large randomized, controlled trials that investigate different doses of vitamin D supplementation at different frequencies of administration, which should also include adequate stratification of patients based on age, disease activity and background medication. The future calls for better designed research including appropriate collection of metadata which allows confounders to be built into multivariate regression models that allow a better understanding of the relationship between vitamin D levels and disease outcomes. To help reduce the study heterogeneity, a clear definition for vitamin D deficiency/insufficiency should be agreed on for both adults and children. Further recommendations for clinical practice should be guided by good quality evidence of the impact of vitamin D supplementation on disease outcomes, as well as additional health benefits (related or unrelated to the underlying ARD). If beneficial, vitamin D has the advantage of being a relatively cheap and relatively safe treatment option. In conclusion, although the level of evidence for the benefits of vitamin D supplementation on outcome measures in arthritis and SLE across ages is poor, the hypothesis of a potential role of vitamin D as an adjuvant treatment option for these diseases remains promising based on available immunological data, but requires further research.
Author Contributions
JZ, ER, and CC wrote the manuscript. JZ searched the literature and extracted relevant data. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by a fellowship awarded by the Medical Research Foundation to ECR (MRF-057-0001-RG-ROSS-C0797). ESC was funded by a Barts Charity Lectureship (grant MGU045). CC is supported by NIHR UCLH Biomedical Research Centre (BRC525/III/CC/191350). This work was performed within the Centre for Adolescent Rheumatology Versus Arthritis at UCL, UCLH, and GOSH supported by grants from Versus Arthritis (21593 and 20164), GOSCC, and the NIHR-Biomedical Research Centres at both GOSH and UCLH. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Abbreviations
ANA, Anti-nuclear Antibodies; ARD, Autoimmune Rheumatic Diseases; CCP, anti-Cyclic Citrullinated Peptide; CD4, cluster of differentiation 4; DMARD, disease-modifying anti-rheumatic drug; DAS28, Disease Activity Score-28; ECLAM, European Consensus Lupus Activity Measurement; ESR, Erythrocyte Sedimentation Rate; HLA, Human Leukocyte Antigen; JADAS, Juvenile Arthritis Disease Activity Score; JIA, Juvenile Idiopathic Arthritis; JSLE, Juvenile Systemic Lupus Erythematosus; MMP, Matrix Metalloprotease; NSAID, Nonsteroidal anti-inflammatory drug; OPG, Osteoprotegerin; RA, Rheumatoid Arthritis; RANKL, Receptor Activator of Nuclear factor Kappa-B Ligand; RCT, Randomized Controlled Trial; RF, Rheumatoid Factor; RXR, Retinoid X Receptor; SLE, Systemic Lupus Erythematosus; SLEDAI, SLE disease activity index; Th, T helper; VAS, Visual Analogue Scale; VDR, Vitamin D Receptor; VDRE, Vitamin D Receptor Elements; UCTD, undifferentiated connective tissue disease.
References
1. Lips P. Worldwide status of vitamin D nutrition. J Steroid Biochem Mol Biol (2010) 121(1-2):297–300. doi: 10.1016/j.jsbmb.2010.02.021
3. van Schoor N, Lips P. Global Overview of Vitamin D Status. Endocrinol Metab Clin North Am (2017) 46(4):845–70. doi: 10.1016/j.ecl.2017.07.002
4. Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol (2014) 21(3):319–29. doi: 10.1016/j.chembiol.2013.12.016
5. Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev (2015) 96(1):365–408. doi: 10.1152/physrev.00014.2015
6. Vieth R. The role of vitamin D in the prevention of osteoporosis. Ann Med (2005) 37(4):278–85. doi: 10.1080/07853890510007313
7. Veldman CM, Cantorna MT, DeLuca HF. Expression of 1,25-Dihydroxyvitamin D3 Receptor in the Immune System. Arch Biochem Biophys (2000) 374(2):334–8. doi: 10.1006/abbi.1999.1605
8. Bhalla AK, Amento EP, Clemens TL, Holick MF, Krane SM. Specific High-Affinity Receptors For 1,25-Dihydroxyvitamin D3 In Human Peripheral Blood Mononuclear Cells: Presence In Monocytes And Induction In T Lymphocytes Following Activation. J Clin Endocrinol Metab (1983) 57(6):1308–10. doi: 10.1210/jcem-57-6-1308
9. Kassi E, Adamopoulos C, Basdra EK, Papavassiliou AG. Role of vitamin D in atherosclerosis. Circulation (2013) 128(23):2517–31. doi: 10.1161/CIRCULATIONAHA.113.002654
10. Cantorna MT. Vitamin D and its role in immunology: Multiple sclerosis, and inflammatory bowel disease. Prog Biophys Mol Biol (2006) 92(1):60–4. doi: 10.1016/j.pbiomolbio.2006.02.020
11. Yin K, Agrawal DK. Vitamin D and inflammatory diseases. J Inflamm Res (2014) 7:69–87. doi: 10.2147/JIR.S63898
12. Murdaca G, Tonacci A, Negrini S, Greco M, Borro M, Puppo F, et al. Emerging role of vitamin D in autoimmune diseases: An update on evidence and therapeutic implications. Autoimmun Rev (2019) 18(9):102350. doi: 10.1016/j.autrev.2019.102350
13. Cantorna MT, Mahon BD. Mounting Evidence for Vitamin D as an Environmental Factor Affecting Autoimmune Disease Prevalence. Exp Biol Med (2004) 229(11):1136–42. doi: 10.1177/153537020422901108
14. Arnson Y, Amital H, Shoenfeld Y. Vitamin D and autoimmunity: new aetiological and therapeutic considerations. Ann Rheum Dis (2007) 66(9):1137–42. doi: 10.1136/ard.2007.069831
15. Lemire JM, Ince A, Takashima M. 1,25-Dihydroxyvitamin D3 attenuates the expression of experimental murine lupus of MRL/l mice. Autoimmunity (1992) 12(2):143–8. doi: 10.3109/08916939209150321
16. Aranow C. Vitamin D and the immune system. J Invest Med (2011) 59(6):881–6. doi: 10.2310/JIM.0b013e31821b8755
17. Martinesi M, Bruni S, Stio M, Treves C. 1,25-Dihydroxyvitamin D3 inhibits tumor necrosis factor-alpha-induced adhesion molecule expression in endothelial cells. Cell Biol Int (2006) 30(4):365–75. doi: 10.1016/j.cellbi.2006.01.004
18. Stach K, Kalsch AI, Nguyen XD, Elmas E, Kralev S, Lang S, et al. 1alpha,25-dihydroxyvitamin D3 attenuates platelet activation and the expression of VCAM-1 and MT1-MMP in human endothelial cells. Cardiology (2011) 118(2):107–15. doi: 10.1159/000327547
19. Noss EH, Brenner MB. The role and therapeutic implications of fibroblast-like synoviocytes in inflammation and cartilage erosion in rheumatoid arthritis. Immunol Rev (2008) 223(1):252–70. doi: 10.1111/j.1600-065X.2008.00648.x
20. Alamanos Y, Drosos AA. Epidemiology of adult rheumatoid arthritis. Autoimmun Rev (2005) 4(3):130–6. doi: 10.1016/j.autrev.2004.09.002
21. Hinks A, Marion MC, Cobb J, Comeau ME, Sudman M, Ainsworth HC, et al. Brief Report: The Genetic Profile of Rheumatoid Factor-Positive Polyarticular Juvenile Idiopathic Arthritis Resembles That of Adult Rheumatoid Arthritis. Arthritis Rheumatol (2018) 70(6):957–62. doi: 10.1002/art.40443
22. Peckham H, Cambridge G, Bourke L, Sen D, Radziszewska A, Leandro M, et al. Antibodies to Cyclic Citrullinated Peptides in Patients With Juvenile Idiopathic Arthritis and Patients With Rheumatoid Arthritis: Shared Expression of the Inherently Autoreactive 9G4 Idiotype. Arthritis Rheumatol (2017) 69(7):1387–95. doi: 10.1002/art.40117
23. Lin Y-T, Wang C-T, Gershwin ME, Chiang B-L. The pathogenesis of oligoarticular/polyarticular vs systemic juvenile idiopathic arthritis. Autoimmun Rev (2011) 10(8):482–9. doi: 10.1016/j.autrev.2011.02.001
24. Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth factor Rev (2008) 19(1):41–52. doi: 10.1016/j.cytogfr.2007.10.004
25. Woo P. The cytokine network in juvenile chronic arthritis. Ann Med (1997) 29(2):145–7. doi: 10.3109/07853899709113701
26. de Jager W, Hoppenreijs EP, Wulffraat NM, Wedderburn LR, Kuis W, Prakken BJ. Blood and synovial fluid cytokine signatures in patients with juvenile idiopathic arthritis: a cross-sectional study. Ann Rheum Dis (2007) 66(5):589–98. doi: 10.1136/ard.2006.061853
27. Boissier M-C, Assier E, Falgarone G, Bessis N. Shifting the imbalance from Th1/Th2 to Th17/treg: The changing rheumatoid arthritis paradigm. Joint Bone Spine (2008) 75(4):373–5. doi: 10.1016/j.jbspin.2008.04.005
28. Wedderburn LR, Woo P. Type 1 and type 2 immune responses in children: their relevance in juvenile arthritis. Springer Semin Immunopathol (1999) 21(3):361–74. doi: 10.1007/bf00812262
29. Silverman GJ, Carson DA. Roles of B cells in rheumatoid arthritis. Arthritis Res Ther (2003) 5(4):S1. doi: 10.1186/ar1010
30. Ravelli A, Varnier GC, Oliveira S, Castell E, Arguedas O, Magnani A, et al. Antinuclear antibody–positive patients should be grouped as a separate category in the classification of juvenile idiopathic arthritis. Arthritis Rheum (2011) 63(1):267–75. doi: 10.1002/art.30076
31. Bugatti S, Vitolo B, Caporali R, Montecucco C, Manzo A. B Cells in Rheumatoid Arthritis: From Pathogenic Players to Disease Biomarkers. BioMed Res Int (2014) 2014:681678. doi: 10.1155/2014/681678
32. Padyukov L, Silva C, Stolt P, Alfredsson L, Klareskog L. A gene–environment interaction between smoking and shared epitope genes in HLA–DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum (2004) 50(10):3085–92. doi: 10.1002/art.20553
33. Veit TD, Vianna P, Scheibel I, Brenol C, Brenol JCT, Xavier RM, et al. Association of the HLA-G 14-bp insertion/deletion polymorphism with juvenile idiopathic arthritis and rheumatoid arthritis. Tissue Antigens (2008) 71(5):440–6. doi: 10.1111/j.1399-0039.2008.01019.x
34. Saag KG, Cerhan JR, Kolluri S, Ohashi K, Hunninghake GW, Schwartz DA. Cigarette smoking and rheumatoid arthritis severity. Ann Rheum Dis (1997) 56(8):463–9. doi: 10.1136/ard.56.8.463
35. Horta-Baas G, Romero-Figueroa MDS, Montiel-Jarquín AJ, Pizano-Zárate ML, García-Mena J, Ramírez-Durán N. Intestinal Dysbiosis and Rheumatoid Arthritis: A Link between Gut Microbiota and the Pathogenesis of Rheumatoid Arthritis. J Immunol Res (2017) 2017:4835189. doi: 10.1155/2017/4835189
36. Hajjaj-Hassouni N, Mawani N, Allali F, Rkain H, Hassouni K, Hmamouchi I, et al. Evaluation of Vitamin D Status in Rheumatoid Arthritis and Its Association with Disease Activity across 15 Countries: “the COMORA Study”. Int J Rheumatol (2017) 2017:1–8. doi: 10.1155/2017/5491676
37. Lee YH, Bae SC. Vitamin D level in rheumatoid arthritis and its correlation with the disease activity: A meta-analysis. Clin Exp Rheumatol (2016) 34(5):827–33.
38. Lin J, Liu J, Davies ML, Chen W. Serum Vitamin D level and rheumatoid arthritis disease activity: Review and meta-analysis. PLoS One (2016) 11(1):1–17. doi: 10.1371/journal.pone.0146351
39. Mouyis M, Ostor AJK, Crisp AJ, Ginawi A, Halsall DJ, Shenker N, et al. Hypovitaminosis D among rheumatology outpatients in clinical practice. Rheumatology (2008) 47(9):1348–51. doi: 10.1093/rheumatology/ken203
40. Dağdeviren-Çakır A, Arvas A, Barut K, Gür E, Kasapçopur Ö. Serum vitamin D levels during activation and remission periods of patients with juvenile idiopathic arthritis and familial Mediterranean fever. Turkish J Pediatr (2016) 58(2):125–31. doi: 10.24953/turkjped.2016.02.001
41. Finch SL, Rosenberg AM, Vatanparast H. Vitamin D and juvenile idiopathic arthritis. Pediatr Rheumatol (2018) 16(1):34–4. doi: 10.1186/s12969-018-0250-0
42. Nisar MK, Masood F, Cookson P, Sansome A, Östör AJ. What do we know about juvenile idiopathic arthritis and vitamin D? A systematic literature review and meta-analysis of current evidence. Clin Rheumatol (2013) 32(6):729–34. doi: 10.1007/s10067-012-2159-1
43. Stagi S, Bertini F, Cavalli L, Matucci-Cerinic M, Brandi ML, Falcini F. Determinants of vitamin D levels in children, adolescents, and young adults with juvenile idiopathic arthritis. J Rheumatol (2014) 41(9):1884–92. doi: 10.3899/jrheum.131421
44. Merlino LA, Curtis J, Mikuls TR, Cerhan JR, Criswell LA, Saag KG. Vitamin D Intake Is Inversely Associated With Rheumatoid Arthritis: Results From the Iowa Women’s Health Study. Arthritis Rheum (2004) 50(1):72–7. doi: 10.1002/art.11434
45. Costenbader KH, Feskanich D, Holmes M, Karlson EW, Benito-Garcia E. Vitamin D intake and risks of systemic lupus erythematosus and rheumatoid arthritis in women. Ann Rheumatic Dis (2008) 67(4):530–5. doi: 10.1136/ard.2007.072736
46. Song GG, Bae SC, Lee YH. Association between vitamin D intake and the risk of rheumatoid arthritis: A meta-analysis. Clin Rheumatol (2012) 31(12):1733–9. doi: 10.1007/s10067-012-2080-7
47. Higgins M, Mackie S, Thalayasingam N, Bingham S, Hamilton J, Kelly C. The effect of vitamin D levels on the assessment of disease activity in rheumatoid arthritis. Clin Rheumatol (2013) 32(6):863–7. doi: 10.1007/s10067-013-2174-x
48. Pakchotanon R, Chaiamnuay S, Narongroeknawin P, Asavatanabodee P. The association between serum vitamin D Level and disease activity in Thai rheumatoid arthritis patients. Int J Rheum Dis (2016) 19(4):355–61. doi: 10.1111/1756-185X.12222
49. Pelajo CF, Lopez-Benitez JM, Kent DM, Price L L, Miller LC, Dawson-Hughes B. 25-Hydroxyvitamin D levels and juvenile idiopathic arthritis: Is there an association with disease activity? Rheumatol Int (2012) 32(12):3923–9. doi: 10.1007/s00296-011-2287-y
50. Çomak E, Doğan ÇS, Uslu-Gökçeoğlu A, Akbaş H, Özdem S, Koyun M, et al. Association between vitamin D deficiency and disease activity in juvenile idiopathic arthritis. Turkish J Pediatr (2014) 56(6):626–31.
51. Shevchenko N, Khadzhynova Y. Juvenile idiopathic arthritis and vitamin D status in Ukrainian patients. Georgian Med News (2019) 294):88–91.
52. Chandrashekara S, Patted A. Role of vitamin D supplementation in improving disease activity in rheumatoid arthritis: An exploratory study. Int J Rheumatic Dis (2017) 20(7):825–31. doi: 10.1111/1756-185X.12770
53. Dehghan A, Rahimpour S, Soleymani-Salehabadi H, Owlia MB. Role of vitamin D in flare ups of rheumatoid arthritis. Z fur Rheumatologie (2014) 73(5):461–4. doi: 10.1007/s00393-013-1297-4
54. Yang J, Liu L, Zhang Q, Li M, Wang J. Effect of vitamin D on the recurrence rate of rheumatoid arthritis. Exp Ther Med (2015) 10(5):1812–6. doi: 10.3892/etm.2015.2747
55. Hansen KE, Bartels CM, Gangnon RE, Jones AN, Gogineni J. An evaluation of high-dose vitamin d for rheumatoid arthritis. J Clin Rheumatol (2014) 20(2):112–4. doi: 10.1097/RHU.0000000000000072
56. Franco AS, Freitas TQ, Bernardo WM, Pereira RMR. Vitamin D supplementation and disease activity in patients with immune-mediated rheumatic diseases. Medicine 96(23). doi: 10.1097/MD.0000000000007024
57. Adami G, Rossini M, Bogliolo L, Cantatore FP, Varenna M, Malavolta N, et al. An exploratory study on the role of vitamin D supplementation in improving pain and disease activity in rheumatoid arthritis. Modern Rheumatol (2019) 29(6):1059–62. doi: 10.1080/14397595.2018.1532622
58. Tang T, Zhang Y, Luo C, Liu M, Xu L, Tang X. Adjunctive vitamin D for the treatment of active juvenile idiopathic arthritis: An open−label, prospective, randomized controlled trial. Exp Ther Med (2019) 18(6):4921–6. doi: 10.3892/etm.2019.8133
59. Feng X, Lv C, Wang F, Gan K, Zhang M, Tan W. Modulatory effect of 1,25-dihydroxyvitamin D3 on IL1 β -induced RANKL, OPG, TNF α, and IL-6 expression in human rheumatoid synoviocyte MH7A. Clin Dev Immunol (2013) 2013:1–8. doi: 10.1155/2013/160123
60. Laragione T, Shah A, Gulko PS. The vitamin D receptor regulates rheumatoid arthritis synovial fibroblast invasion and morphology. Mol Med (2012) 18(2):194–200. doi: 10.2119/molmed.2011.00410
61. Nagpal S, Na S, Rathnachalam R. Noncalcemic actions of vitamin D receptor ligands. Endocr Rev (2005) 26(5):662–87. doi: 10.1210/er.2004-0002
62. Harrison SR, Li D, Jeffery LE, Raza K, Hewison M. Vitamin D, Autoimmune Disease and Rheumatoid Arthritis. Calcified Tissue Int (2020) 106(1):58–75. doi: 10.1007/s00223-019-00577-2
63. Gu X, Gu B, Lv X, Yu Z, Wang R, Zhou X, et al. 1, 25-dihydroxy-vitamin D3 with tumor necrosis factor-alpha protects against rheumatoid arthritis by promoting p53 acetylation-mediated apoptosis via sirt1 in synoviocytes. Cell Death Dis (2016) 7(10):1–17. doi: 10.1038/cddis.2016.300
64. Nanzer AM, Chambers ES, Ryanna K, Richards DF, Black C, Timms PM, et al. Enhanced production of IL-17A in patients with severe asthma is inhibited by 1α,25-dihydroxyvitamin D3 in a glucocorticoid-independent fashion. J Allergy Clin Immunol (2013) 132(2):297–304.e3. doi: 10.1016/j.jaci.2013.03.037
65. Chambers ES, Nanzer AM, Pfeffer PE, Richards DF, Timms PM, Martineau AR, et al. Distinct endotypes of steroid-resistant asthma characterized by IL-17A(high) and IFN-γ(high) immunophenotypes: Potential benefits of calcitriol. J Allergy Clin Immunol (2015) 136(3):628–37.e4. doi: 10.1016/j.jaci.2015.01.026
66. Daniel C, Sartory NA, Zahn N, Radeke HH, Stein JM. Immune modulatory treatment of trinitrobenzene sulfonic acid colitis with calcitriol is associated with a change of a T helper (Th) 1/Th17 to a Th2 and regulatory T cell profile. J Pharmacol Exp Ther (2008) 324(1):23–33. doi: 10.1124/jpet.107.127209
67. Urry Z, Chambers ES, Xystrakis E, Dimeloe S, Richards DF, Gabryšová L, et al. The role of 1α,25-dihydroxyvitamin D3 and cytokines in the promotion of distinct Foxp3+ and IL-10+ CD4+ T cells. Eur J Immunol (2012) 42(10):2697–708. doi: 10.1002/eji.201242370
68. Jeffery LE, Burke F, Mura M, Zheng Y, Qureshi OS, Hewison M, et al. 1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell production of inflammatory cytokines and promote development of regulatory T cells expressing CTLA-4 and FoxP3. J Immunol (2009) 183(9):5458–67. doi: 10.4049/jimmunol.0803217
69. Boonstra A, Barrat FJ, Crain C, Heath VL, Savelkoul HFJ, O’Garra A. 1α,25-Dihydroxyvitamin D3 Has a Direct Effect on Naive CD4 + T Cells to Enhance the Development of Th2 Cells. J Immunol (2001) 167(9):4974–80. doi: 10.4049/jimmunol.167.9.4974
70. Ranganathan P, Khalatbari S, Yalavarthi S, Marder W, Brook R, Kaplan MJ. Vitamin D Deficiency, Interleukin 17, and Vascular Function in Rheumatoid Arthritis HHS Public Access. J Rheumatol (2013) 40(9):1529–34. doi: 10.3899/jrheum.130012
71. Colin EM, Asmawidjaja PS, Van Hamburg JP, Mus AMC, Van Driel M, Hazes JMW, et al. 1,25-Dihydroxyvitamin D3 modulates Th17 polarization and interleukin-22 expression by memory T cells from patients with early rheumatoid arthritis. Arthritis Rheum (2010) 62(1):132–42. doi: 10.1002/art.25043
72. Kim H, Baek S, Hong SM, Lee J, Jung SM, Lee J, et al. 1,25-dihydroxy Vitamin D3 and Interleukin-6 Blockade Synergistically Regulate Rheumatoid Arthritis by Suppressing Interleukin-17 Production and Osteoclastogenesis. J Korean Med Sci (2020) 35(6):1–10. doi: 10.3346/jkms.2020.35.e40
73. Dankers W, González-Leal C, Davelaar N, Asmawidjaja PS, Mus AMC, Hazes JMW, et al. 1,25(OH) 2 D 3 and dexamethasone additively suppress synovial fibroblast activation by CCR6 + T helper memory cells and enhance the effect of tumor necrosis factor alpha blockade. Arthritis Res Ther (2018) 20(1):1–10. doi: 10.1186/s13075-018-1706-9
74. Jeffery LE, Henley P, Marium N, Filer A, Sansom DM, Hewison M, et al. Decreased sensitivity to 1,25-dihydroxyvitamin D3 in T cells from the rheumatoid joint. J Autoimmun (2018) 88:50–60. doi: 10.1016/j.jaut.2017.10.001
75. Kaul A, Gordon C, Crow MK, Touma Z, Urowitz MB, Van Vollenhoven R, et al. Systemic lupus erythematosus. Nat Rev Dis Primers (2016) 2(1):1–21. doi: 10.1038/nrdp.2016.39
76. Petri M, Buyon J, Kim M. Classification and definition of major flares in SLE clinical trials. Thousand Oaks, CA: Sage PublicationsSage CA (1999) p. 685–91.
77. Danchenko N, Satia JA, Anthony MS. Epidemiology of systemic lupus erythematosus: A comparison of worldwide disease burden. Lupus (2006) 15(5):308–18. doi: 10.1191/0961203306lu2305xx
78. Stagi S, Rigante D. Vitamin D and juvenile systemic lupus erythematosus: Lights, shadows and still unresolved issues. Autoimmun Rev (2018) 17(3):290–300. doi: 10.1016/j.autrev.2018.01.004
79. Wichainun R, Kasitanon N, Wangkaew S, Hongsongkiat S, Sukitawut W, Louthrenoo W. Sensitivity and specificity of ANA and anti-dsDNA in the diagnosis of systemic lupus erythematosus: A comparison using control sera obtained from healthy individuals and patients with multiple medical problems. Asian Pacific J Allergy Immunol (2013) 31(4):292–8. doi: 10.12932/AP0272.31.4.2013
80. Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH, Austin A, et al. Derivation of the sledai. A disease activity index for lupus patients. Arthritis Rheum (1992) 35(6):630–40. doi: 10.1002/art.1780350606
81. Rönnblom L, Pascual V. The innate immune system in SLE: Type I interferons and dendritic cells. Lupus (2008) 17(5 SPEC. ISS.):394–9. doi: 10.1177/0961203308090020
82. Wahadat MJ, Bodewes ILA, Maria NI, van Helden-Meeuwsen CG, van Dijk-Hummelman A, Steenwijk EC, et al. Type I IFN signature in childhood-onset systemic lupus erythematosus: a conspiracy of DNA- and RNA-sensing receptors? Arthritis Res Ther (2018) 20(1):4. doi: 10.1186/s13075-017-1501-z
83. Lutz MB, Schuler G. Immature, semi-mature and fully mature dendritic cells: Which signals induce tolerance or immunity? Trends Immunol (2002) 23(9):445–9. doi: 10.1016/S1471-4906(02)02281-0
84. Morel PA, Turner MS. Dendritic cells and the maintenance of self-tolerance. Immunol Res (2011) 50(2–3):124–9. doi: 10.1007/s12026-011-8217-y
85. Tsao BP. The genetics of human systemic lupus erythematosus. Trends Immunol (2003) 24(11):595–602. doi: 10.1016/j.it.2003.09.006
86. Islam MA, Khandker SS, Alam SS, Kotyla P, Hassan R. Vitamin D status in patients with systemic lupus erythematosus (SLE): A systematic review and meta-analysis. Autoimmun Rev (2019) 18(11):1–19. doi: 10.1016/j.autrev.2019.102392
87. Ruiz-irastorza G, Egurbide MV, Olivares N, Martinez-Berriotxoa A, Aguirre C. Vitamin D deficiency in systemic lupus erythematosus: Prevalence, predictors and clinical consequences. Rheumatology (2008) 47(6):920–3. doi: 10.1093/rheumatology/ken121
88. Borba VZC, Vieira JGH, Kasamatsu T, Radominski SC, Sato EI, Lazaretti-Castro M. Vitamin D deficiency in patients with active systemic lupus erythematosus. Osteoporosis Int (2009) 20(3):427–33. doi: 10.1007/s00198-008-0676-1
89. Mok CC, Birmingham DJ, Leung HW, Hebert LA, Song H, Rovin BH. Vitamin D levels in Chinese patients with systemic lupus erythematosus: Relationship with disease activity, vascular risk factors and atherosclerosis. Rheumatology (2012) 51(4):644–52. doi: 10.1093/rheumatology/ker212
90. Hahn TJ, Halstead LR, Baran DT. Effects of Short Term Glucocorticoid Administration on Intestinal Calcium Absorption and Circulating Vitamin D Metabolite Concentrations in Man*. J Clin Endocrinol Metab (1981) 52(1):111–5. doi: 10.1210/jcem-52-1-111
91. Chaiamnuay S, Chailurkit L-O, Narongroeknawin P, Asavatanabodee P, Laohajaroensombat S, Chaiamnuay P. Current Daily Glucocorticoid Use and Serum Creatinine Levels Are Associated With Lower 25(OH) Vitamin D Levels in Thai Patients With Systemic Lupus Erythematosus. JCR: J Clin Rheumatol (2013) 19(3):121–5. doi: 10.1097/RHU.0b013e318289bd16
92. Barré PE, Gascon-Barré M, Meakins JL, Goltzman D. Hydroxychloroquine treatment of hypercalcemia in a patient with sarcoidosis undergoing hemodialysis. Am J Med (1987) 82(6):1259–62. doi: 10.1016/0002-9343(87)90237-3
93. Robinson AB, Tangpricha V, Yow E, Gurion R, Schanberg LE, McComsey GA, et al. Vitamin D status is a determinant of atorvastatin effect on carotid intima medial thickening progression rate in children with lupus: An Atherosclerosis Prevention in Pediatric Lupus Erythematosus (APPLE) substudy. Lupus Sci Med (2014) 1(1):e000037–e000037. doi: 10.1136/lupus-2014-000037
94. Stagi S, Cavalli L, Bertini F, De Martino M, Cerinic MM, Brandi ML, et al. Vitamin D levels in children, adolescents, and young adults with juvenile-onset systemic lupus erythematosus: A cross-sectional study. Lupus (2014) 23(10):1059–65. doi: 10.1177/0961203314532564
95. Lima GL, Paupitz J, Aikawa NE, Takayama L, Bonfa E, Pereira RMR. Vitamin D Supplementation in Adolescents and Young Adults With Juvenile Systemic Lupus Erythematosus for Improvement in Disease Activity and Fatigue Scores: A Randomized, Double-Blind, Placebo-Controlled Trial. Arthritis Care Res (2016) 68(1):91–8. doi: 10.1002/acr.22621
96. AlSaleem A, AlE’ed A, AlSaghier A, Al-Mayouf SM. Vitamin D status in children with systemic lupus erythematosus and its association with clinical and laboratory parameters. Clin Rheumatol (2015) 34(1):81–4. doi: 10.1007/s10067-014-2811-z
97. Hiraki LT, Munger KL, Costenbader KH, Karlson EW. Dietary intake of vitamin D during adolescence and risk of adult-onset systemic lupus erythematosus and rheumatoid arthritis. Arthritis Care Res (2012) 64(12):1829–36. doi: 10.1002/acr.21776
98. Lourdudoss C, Hafström I, Frostegård J, van Vollenhoven R. The association between diet and glucocorticoid treatment in patients with SLE. Lupus Sci Med (2016) 3(1):e000135. doi: 10.1136/lupus-2015-000135
99. Amital H, Szekanecz Z, Szucs G, Danko K, Nagy E, Csepany T, et al. Serum concentrations of 25-OH vitamin D in patients with systemic lupus erythematosus (SLE) are inversely related to disease activity: is it time to routinely supplement patients with SLE with vitamin D? Ann Rheum Dis (2010) 69(6):1155–7. doi: 10.1136/ard.2009.120329
100. Yap KS, Northcott M, Hoi AB-Y, Morad EF, Nikpour M. Association of low vitamin D with high disease activity in an Australian systemic lupus erythematosus cohort. Lupus Sci Med (2015) 2(1). doi: 10.1136/lupus-2014-000064
101. Sun J, Liu C, Zhang C, Yi B, Gui M, Zhang W, et al. Vitamin D receptor expression in peripheral blood mononuclear cells is inversely associated with disease activity and inflammation in lupus patients. Clin Rheumatol (2019) 38(9):2509–18. doi: 10.1007/s10067-019-04594-2
102. Stockton KA, Kandiah DA, Paratz JD, Bennell KL. Fatigue, muscle strength and vitamin D status in women with systemic lupus erythematosus compared with healthy controls. Lupus (2012) 21(3):271–8. doi: 10.1177/0961203311425530
103. Tench CM, McCurdie I, White PD, D’Cruz DP. The prevalence and associations of fatigue in systemic lupus erythematosus. Rheumatology (2000) 39(11):1249–54. doi: 10.1093/rheumatology/39.11.1249
104. Andreoli L, Dall’Ara F, Piantoni S, Zanola A, Piva N, Cutolo M, et al. A 24-month prospective study on the efficacy and safety of two different monthly regimens of Vitamin D supplementation in pre-menopausal women with systemic lupus erythematosus. Lupus (2015) 24(4-5):499–506. doi: 10.1177/0961203314559089
105. Karimzadeh H, Shirzadi M, Karimifar M. The effect of Vitamin D supplementation in disease activity of systemic lupus erythematosus patients with Vitamin D deficiency: A randomized clinical trial. J Res Med Sci (2017) 22(1):1–6. doi: 10.4103/1735-1995.199089
106. Petri M, Bello KJ, Fang H, Magder LS. Vitamin D in systemic lupus erythematosus: Modest association with disease activity and the urine protein-to-creatinine ratio. Arthritis Rheum (2013) 65(7):1865–71. doi: 10.1002/art.37953
107. Ruiz-Irastorza G, Gordo S, Olivares N, Egurbide MV, Aguirre C. Changes in vitamin D levels in patients with systemic lupus erythematosus: Effects on fatigue, disease activity, and damage. Arthritis Care Res (2010) 62(8):1160–5. doi: 10.1002/acr.20186
108. Terrier B, Derian N, Schoindre Y, Chaara W, Geri G, Zahr N, et al. Restoration of regulatory and effector T cell balance and B cell homeostasis in systemic lupus erythematosus patients through vitamin D supplementation. Arthritis Res Ther (2012) 14(5):1–10. doi: 10.1186/ar4060
109. Zheng R, Gonzalez A, Yue J, Wu X, Qiu M, Gui L, et al. Efficacy and Safety of Vitamin D Supplementation in Patients With Systemic Lupus Erythematosus: A Meta-analysis of Randomized Controlled Trials. Am J Med Sci (2019) 358(2):104–14. doi: 10.1016/j.amjms.2019.04.020
110. Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1α,25 dihydroxyvitamin D3 and its analogs: A vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci U S A (2001) 98(12):6800–5. doi: 10.1073/pnas.121172198
111. Ben-Zvi I, Aranow C, Mackay M, Stanevsky A, Kamen DL, Marinescu LM, et al. The impact of vitamin D on dendritic cell function in patients with systemic lupus erythematosus. PLoS One (2010) 5(2):1–8. doi: 10.1371/journal.pone.0009193
112. Mandal M, Tripathy R, Panda AK, Pattanaik SS, Dakua S, Pradhan AK, et al. Vitamin D levels in Indian systemic lupus erythematosus patients: Association with disease activity index and interferon alpha. Arthritis Res Ther (2014) 16(1):1–8. doi: 10.1186/ar4479
113. Aranow C, Kamen DL, Dall’Era M, Massarotti EM, MacKay MC, Koumpouras F, et al. Randomized, double-blind, placebo-controlled trial of the effect of vitamin D3 on the interferon signature in patients with systemic lupus erythematosus. Arthritis Rheumatol (2015) 67(7):1848–57. doi: 10.1002/art.39108
114. Heine G, Niesner U, Chang HD, Steinmeyer A, Zügel U, Zuberbier T, et al. 1,25-dihydroxyvitamin D3 promotes IL-10 production in human B cells. Eur J Immunol (2008) 38(8):2210–8. doi: 10.1002/eji.200838216
115. Blair PA, Noreña LY, Flores-Borja F, Rawlings DJ, Isenberg DA, Ehrenstein MR, et al. CD19+CD24hiCD38hi B Cells Exhibit Regulatory Capacity in Healthy Individuals but Are Functionally Impaired in Systemic Lupus Erythematosus Patients. Immunity (2010) 32(1):129–40. doi: 10.1016/j.immuni.2009.11.009
116. Zold E, Szodoray P, Nakken B, Barath S, Kappelmayer J, Csathy L, et al. Alfacalcidol treatment restores derailed immune-regulation in patients with undifferentiated connective tissue disease. Autoimmun Rev (2011) 10(3):155–62. doi: 10.1016/j.autrev.2010.09.018
117. Zold E, Szodoray P, Kappelmayer J, Gaal J, Csathy L, Barath S, et al. Impaired regulatory T-cell homeostasis due to vitamin D deficiency in undifferentiated connective tissue disease. Scand J Rheumatol (2010) 39(6):490–7. doi: 10.3109/03009741003781951
Keywords: vitamin D, rheumatoid arthritis, juvenile idiopathic arthritis, systemic lupus erythematosus, immunomodulatory, autoimmunity
Citation: Zou J, Thornton C, Chambers ES, Rosser EC and Ciurtin C (2021) Exploring the Evidence for an Immunomodulatory Role of Vitamin D in Juvenile and Adult Rheumatic Disease. Front. Immunol. 11:616483. doi: 10.3389/fimmu.2020.616483
Received: 12 October 2020; Accepted: 22 December 2020;
Published: 18 February 2021.
Edited by:
Attila Mócsai, Semmelweis University, HungaryReviewed by:
Zoltan Szekanecz, University of Debrecen, HungaryOzgur Kasapcopur, Istanbul University-Cerrahpasa, Turkey
Copyright © 2021 Zou, Thornton, Chambers, Rosser and Ciurtin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Elizabeth C. Rosser, ZS5yb3NzZXJAdWNsLmFjLnVr; Coziana Ciurtin, Yy5jaXVydGluQHVjbC5hYy51aw==
†These authors have contributed equally to this work and share senior authorship