 Zoe C. Schmiechen1,2
Zoe C. Schmiechen1,2 Ingunn M. Stromnes1,2,3,4*
Ingunn M. Stromnes1,2,3,4*- 1Center for Immunology, University of Minnesota Medical School, Minneapolis, MN, United States
- 2Department of Microbiology and Immunology, University of Minnesota Medical School, Minneapolis, MN, United States
- 3Masonic Cancer Center, University of Minnesota Medical School, Minneapolis, MN, United States
- 4Center for Genome Engineering, University of Minnesota Medical School, Minneapolis, MN, United States
Pancreatic ductal adenocarcinoma (PDA) is a lethal malignancy with an overall 5-year survival rate of 10%. Disease lethality is due to late diagnosis, early metastasis and resistance to therapy, including immunotherapy. PDA creates a robust fibroinflammatory tumor microenvironment that contributes to immunotherapy resistance. While previously considered an immune privileged site, evidence demonstrates that in some cases tumor antigen-specific T cells infiltrate and preferentially accumulate in PDA and are central to tumor cell clearance and long-term remission. Nonetheless, PDA can rapidly evade an adaptive immune response using a myriad of mechanisms. Mounting evidence indicates PDA interferes with T cell differentiation into potent cytolytic effector T cells via deficiencies in naive T cell priming, inducing T cell suppression or promoting T cell exhaustion. Mechanistic research indicates that immunotherapy combinations that change the suppressive tumor microenvironment while engaging antigen-specific T cells is required for treatment of advanced disease. This review focuses on recent advances in understanding mechanisms limiting T cell function and current strategies to overcome immunotherapy resistance in PDA.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is projected to be the 2nd leading cause of cancer related deaths by the year 2030, and has a dismal 5 year survival rate of 10% (1). Disease incidence is on the rise and lethality is attributed to late diagnosis, early metastasis, and therapeutic resistance (2). The median overall survival of patients with metastatic disease is 3 to 6 months, and for patients with locally advanced disease is 8 to 12 months (3, 4). PDA is resectable in less than 20% of cases (5) and often surgery fails to cure (6, 7). Thus, there remains an urgent and unmet need to develop safe and effective therapies for PDA patient treatment.
Immune checkpoint blockade (ICB) is a promising immunotherapy transforming the standard of care for several advanced malignancies (8). ICBs are monoclonal antibodies that interfere with PD-1 and/or CTLA-4 inhibitory proteins expressed on the surface of T cells. Unfortunately, ICB rarely demonstrates clinical responses in PDA (9, 10). A recent phase 2 clinical trial combining anti-CTLA-4 and anti-PD-L1 exhibited an objective response rate of a mere 3.1% (11). Elucidating the reasons for ICB failure is an active area of investigation by our lab and others to inform the design of more effective immune-based treatments for PDA patients.
Mutant Kras is an oncogenic driver in 92% of PDA patients (12), and is sufficient to drive preinvasive disease in murine models (13). The genetically engineered KrasG12D/+;Trp53R172H/+;p48-Cre or Pdx1-Cre (KPC) autochthonous PDA mouse model recapitulates hallmark features of the human disease and has been informative in this regard. Similar to the human disease, KPC PDA originates from precursor histologically defined lesions that are called pancreatic intraepithelial neoplasms (PanINs) (14). At disease inception, these PanINs promote a fibroinflammatory and suppressive tumor microenvironment (TME) (15). The formation of PanIN lesions includes infiltration of suppressive tumor-associated macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), and regulatory T cells (Tregs) which dominate the early immune response and persist to limit T cell activity. In addition to the cellular component, PanINs are surrounded by a dense extracellular matrix (ECM) that contains collagen and hyaluronan. Notably, myeloid-cell induced inflammation is critical for PDA development (16) and limits CD8 T cell anti-tumor responses (17). Moreover, mutant Kras is also required for the maintenance of advanced PDA by promoting the fibroinflammatory stroma and metabolic reprogramming to upregulate glycolytic genes and glucose uptake (18–20).
A hallmark of many cancers including PDA is immune evasion. When there is sufficient immune pressure by tumor-antigen-specific T cells, tumor variants may emerge that are defective in target antigen expression and/or antigen processing and presentation (21–24). Tumors that retain antigen presentation can still avoid T cell recognition by restricting antigen presenting cell differentiation, excluding T cells from tumor nests, immunosuppression, or induction of an altered T cell differentiation state referred to as T cell exhaustion (Figure 1) (25, 26). The particular mechanism(s) driving ICB resistance will impact therapeutic strategies to overcome it. We posit that there are similar mechanisms among multiple patient tumors, yet the hierarchy may depend upon the extent that tumor antigen-specific T cells are engaged. The goal of this review is to discuss major ICB resistance mechanisms and to highlight combination strategies to transform the TME to engage the anti-tumor T cells.
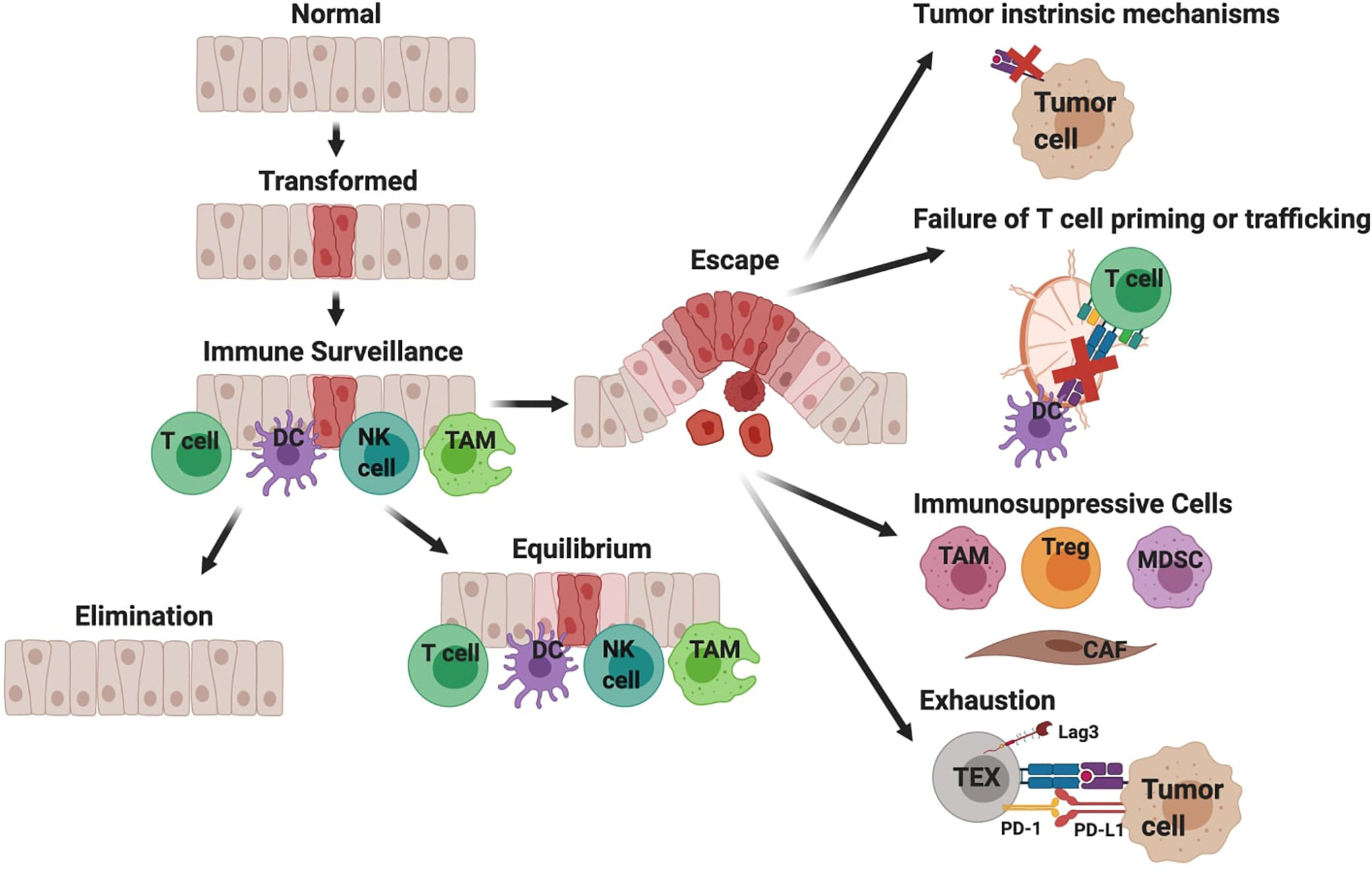
Figure 1 Simplified overview of immune surveillance and tumor evasion in pancreatic ductal adenocarcinoma (PDA). Immune surveillance is the process whereby the immune system surveys the body for malignant or infected cells. Components of immune surveillance include T cells, DCs, NK cells, and macrophages (TAM). Mutations in oncogene KRAS are a driver of PDA. The immune response to transformed tissue can either result in complete elimination of the malignancy, equilibrium to prevent further growth of the malignancy, or escape and development of clinically significant tumors. This figure provides a hypothesized sequence of immune evasion, but this process is likely not linear and instead a dynamic progression. Cancer cells can escape T cell recognition by losing target antigen expression and/or developing defects in antigen processing and presentation. Additionally, defects in T cell priming or trafficking to tumors may limit antigen-specific T cell responses and can be attributed to insufficient mature DCs. When antigen-specific T cells successfully infiltrate tumors, their function may be limited by immunosuppressive cytokines produced by macrophages (TAM, tumor-associated macrophage), regulatory T cells (Treg), myeloid-derived suppressor cells (MDSC), or cancer associated fibroblasts (CAF). Lastly chronic T cell receptor signaling drives T cell exhaustion, resulting in reduced effector function.
Tumor Antigen-Specific T Cells Infiltrate PDA and Are Present in a Subset of Patient Tumors
T cells respond and mediate the anti-tumor effects of most immunotherapies. T cells express T cell receptors (TCRs) that specifically bind peptide:MHC complexes expressed on the cell surface of neighboring cells. During T cell development, most T cells strongly reactive to self-antigens are deleted in the thymus or tolerized in the periphery resulting in a T cell repertoire that is largely tolerant to self-antigens and reactive to foreign antigens (27). The number of nonsynonymous mutations, e.g., tumor mutational burden (TMB), correlates with clinical responses following ICB (28, 29). A high TMB increases the likelihood that a particular mutation will code for a neoepitope, which is a novel tumor-specific antigen recognized by rare antigen-specific T cells. PDAs typically harbor ~25–60 somatic coding mutations (30–32), logs lower than some ICB-responsive cancers such as melanoma (33). A minor fraction of PDAs (<1%) have an abnormally high TMB due to genetic defects in mismatch-repair genes (34). A subset of these patients (5/8) respond to PD-1 blockade, yet even in this setting, responses are often not durable (24, 34). In a long-term PDA survivor cohort, neoepitope similarity to microbial epitopes, rather than neoepitope quantity, correlated with overall patient survival (30). CD8 T cell production of effector molecules IFNγ and Granzyme B are also elevated in long-term survivors (35) and survival is longer when CD8 T cells are proximal to cancer cells (36). Although still ongoing, interim analysis of a combination of a CD40 agonist, anti-PD-1, and standard of care chemotherapy (gemcitabine + abraxane) induced objective responses in over 50% of advanced PDA patients (37). As most preclinical animal studies indicate that CD40 agonist (38–41) or anti-PD-L1 (23) requires T cells for antitumor activity, the apparent success of this ongoing clinical trial supports that endogenous tumor-reactive T cells are present in a subset of PDA patients, despite a relatively low TMB, and can be beneficial for treating advanced disease. Much work remains to determine the independent contributions of CD4 and CD8 T cells, the antigen-specificity of such T cells, and the mechanisms underlying tumor clearance.
Historically, PDA has been considered immunologically “cold” as few T cells were found infiltrating tumor nests (42–44). In both the KPC mouse model and humans, tumor cells are surrounded by a robust fibroinflammatory stroma comprised of cancer-associated fibroblasts (CAFs), TAMs, Tregs, MDSCs, and rare endothelial cells embedded within a complex extracellular matrix (ECM) (Figure 2). In PDA patients, when CD8 T cells are present they are often contained within this stroma and often not directly contacting tumors cells (45, 46). T cell exclusion from tumor nests has been hypothesized to be due to “stromal trapping”, although mechanistically this process is ill-defined in patients (47, 48). Our in situ analysis of resected human PDAs identified marked heterogeneity among the number of T cells among independent tumor samples (45). A proportion of tumors contained few immune cells, a fraction contained abundant T cells within stroma, and a minor fraction of tumors contained abundant T cells within tumor cell nests. These results are consistent with a putative immunogenic subset based on transcriptional profiling (12) and indicate that T cells are present in a fraction of human tumors.
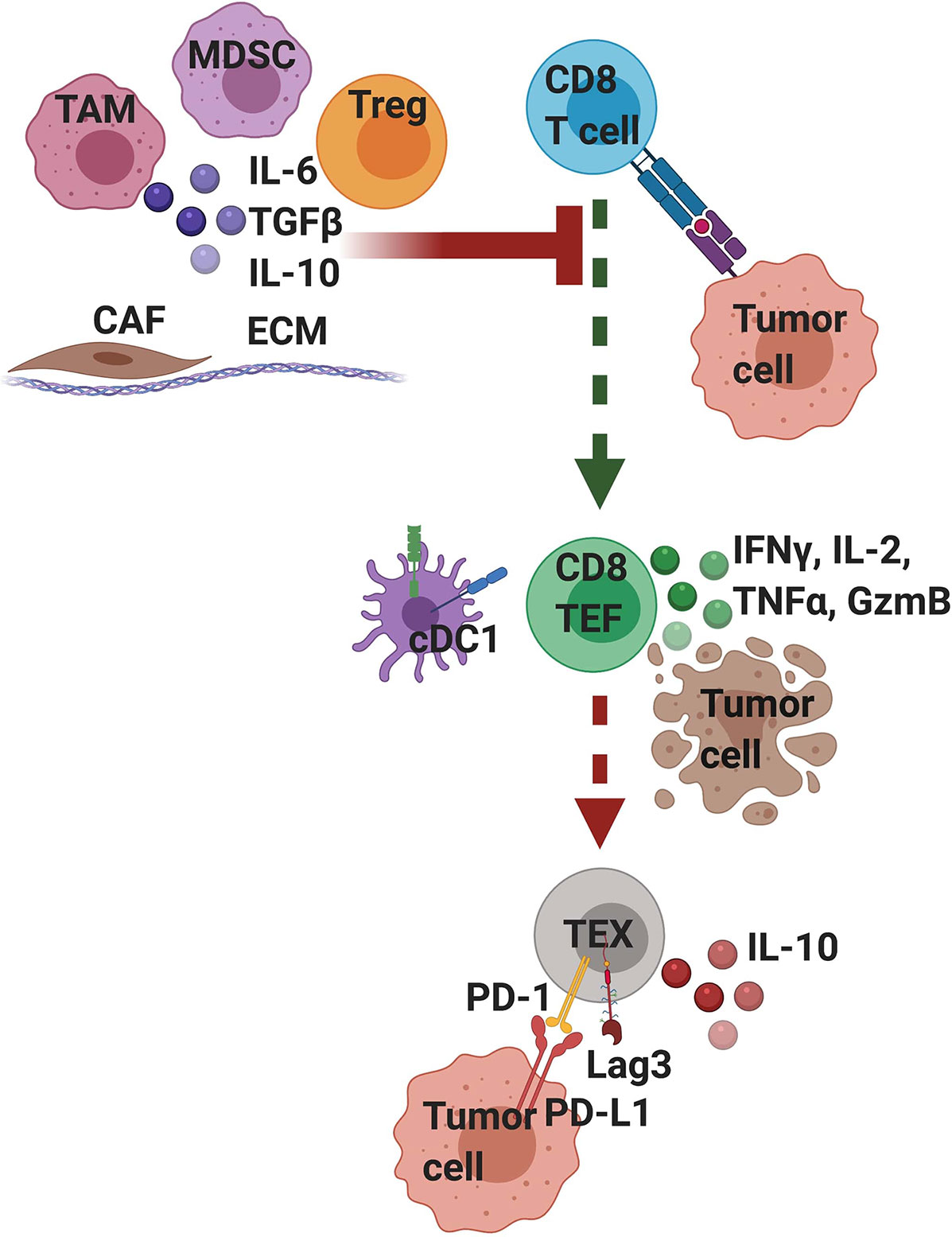
Figure 2 T cell intrinsic and extrinsic factors influence T cell differentiation and functionality in pancreatic cancer. Tumor-infiltrating T cells that strongly recognize tumor antigen mediate transient anti-tumor activity but if the tumor is not cleared, differentiate into exhausted (TEX) T cells, which is driven by persistent T cell receptor (TCR) signaling. Exhausted T cells are often characterized by their expression of PD-1 and Lag3 and are hypofunctional. Tumor cells and other stromal cells can express ligands for these inhibitory receptors, such as PD-L1, a ligand for PD- 1. Signaling through these inhibitory receptors interferes with T cell function and differentiation state. Moreover, exhausted T cells may participate in their own suppression by producing IL-10. A variety of extrinsic cells and factors enriched in the suppressive tumor microenvironment can interfere with TCR signaling and activation. These include tumor associated macrophages (TAM), myeloid-derived suppressor cells (MDSC), regulatory T cells (Tregs), and cancer associated fibroblasts (CAF) all embedded within a dense extracellular matrix (ECM). Immunosuppressive cells secrete cytokines including IL-10 and TGFβ that can suppress T cell activation and TCR signaling. Mechanistically, extrinsic suppression and TCR-driven exhaustion are distinct processes that converge to suppress tumor immunity. We posit that targeting immunosuppression may lead to only transient anti-tumor immune responses because if the tumor is not cleared; T cells will become exhausted. Thus, PDA likely requires combination immunotherapies that target both T cell extrinsic immunosuppression and T cell intrinsic exhaustion.
While murine models of PDA generally recapitulate the immune response seen in human PDA patients, there is a distinction with respect to in T cell infiltration. Although tumors from mouse models exhibit a similar T localization pattern within the stroma rather than direct contact with tumor cells (49, 50), murine models have few T cells in general, without the clear presence of a neoantigen or T cell engagement through immunotherapy (23, 50, 51). The increased heterogeneity in the T cell infiltrate in human compared to murine PDA models has spurred the recent development of mouse models to account for this variability (23, 52). Heterogeneity in T cell infiltration has been reported in murine tumors models in some cases and attributed to differences in tumor intrinsic genetic events (52) or antigenicity (23).
Contrasting with endogenous T cells of unknown antigen specificity that are excluded from PDA, we showed that T cells engineered to express a high affinity mesothelin (Msln)-specific TCR preferentially accumulate in tumors of KPC mice (53). Msln is a self/tumor antigen overexpressed by over 90% of PDAs (53, 54) and due to its low level of expression in normal tissues, is a target for immunotherapy. Thus, antigen-specific effector T cells can overcome stromal barriers and prolong animal survival.
Endogenous T cells specific to native antigens in KPC PDA have not been identified. There are few reported somatic mutations in KPC tumors (55) and as it is a spontaneous model, immunogenic epitopes, when present, will likely be different among individual KPC animals making it challenging to track a reproducible population of antigen-specific T cells. Therefore, we and others have developed tools to study model antigen-specific T cells. Tumor expression of ovalbumin (OVA) in KPC autochthonous mice unexpectedly was reported to accelerate tumor progression (56). In contrast, OVA-expressing KPC cell lines implanted subcutaneously or orthotopically are readily rejected in B6 animals (55) and can prompt outgrowth of OVA-loss variants (23). We developed a KPC orthotopic tumor model by expressing a click beetle red (CB) luciferase in a KPC cell line to monitor tumor growth in real time (23). Unlike the parental KPC CB-negative line that is refractory to anti-PD-1 or anti-PD-L1, CB+ tumors transiently respond to anti-PD-L1. We identified an immunodominant epitope CB101-109:H-2Db, generated a fluorescently-labeled peptide-MHC tetramer and showed that endogenous tumor antigen-specific T cells also preferentially accumulate in PDA (23). This is consistent with our prior study using engineered T cells (53), and contrasts with other studies demonstrating T cells of unknown specificity are excluded from the pancreatic TME (50, 51). As reports often evaluate polyclonal T cells without regard for antigen-specificity, and the polyclonal T cell population is remarkably diverse, the biology of rare tumor antigen-specific T cells can be lost when studying bulk T cells (57).
CD8 T Cell Exhaustion Is Driven by Persistent TCR Signaling
T cell exhaustion was initially identified in models of chronic viral infection, but the full extent to which these findings translate to PDA, remains to be fully investigated. Exhausted T cells are an epigenetically regulated T cell differentiation state that occurs in settings of chronic antigen, is driven by persistent TCR signaling (58–60), and results in defective T cell effector functions (Figure 2). TCR signaling drives expression of the transcription factor Tox, which is required for the survival and persistence of exhausted T cells during chronic LCMV infection as well as in animal models of melanoma and liver cancer (61–63). In PDA animal models, we identified that either engineered or endogenous intratumoral tumor antigen-specific T cells also become defective in their production of IFNγ, TNFα and expression of cytolytic molecules including Granzyme B following specific peptide recognition (23, 53). In contrast, T cells with identical peptide specificity remained largely functional in the spleens within the same tumor-bearing animals, indicating that the tumor and/or the TME drive T cell functional loss. Tox is elevated in both functional effector splenic CD8 T cells as well as intratumoral exhausted CD8 T cells (64). Early during tumor growth, intratumoral tumor-specific T cells co-express both Tox and Granzyme B and produce cytokines following antigen encounter. Over time, however, persisting intratumoral T cells maintain Tox while losing Granzyme B and cytokine production following antigen encounter.
As both PD-1 (23) and Tox appear insufficient to identify exhausted T cells in PDA, we pursued further analysis of these populations. T cell expression of Klrg1 and Lag3 may distinguish these populations in murine PDA (23). Klrg1 is expressed by the majority of tumor-specific T cells in the spleens of tumor-bearing mice. In contrast, it is largely absent on T cells infiltrating PDA. Most Lag3+Klrg1- T cells were exhausted whereas Lag3-Klrg1+ T cells were enriched intratumorally during a productive immunotherapy responses (23, 64) and highly functional. Klrg1 is expressed on NK cells (65) and effector CD8 T cells during acute viral infection (66, 67). It is also expressed by both short-lived (68) or long-lived effector CD8 T cells (69) and can be downregulated during memory T cell transition (70). An inverse relationship between Tox and Klrg1 expression in CD8 T cells during viral infection has been reported (61, 71), and Klrg1 expression correlates with T-bet levels (66). Thus, while further studies are warranted, especially in human samples, Klrg1 may be an intratumoral biomarker for functional antigen-specific T cells.
In a subset of resected human PDAs, intratumoral T cells were enriched for expression of markers of recent or prolonged TCR signaling including 4-1BB and PD-1 (45) suggesting specific antigen recognition. A large fraction of PD-1+ T cells co-expressed putative exhaustion markers Lag3 and Tim-3, similar to exhausted T cells in murine models (23, 53). In the other fraction of tumors, T cells exhibited an effector or effector memory phenotype yet did not express markers indicative of recent antigen encounter. These data suggest that T cells are not specific to tumor antigens, antigen presentation is limiting, and/or immunosuppression is dominant in this fraction (45). Single cell sequencing of human and mouse tumors support an exhausted T cell phenotype based on enrichment of LAG3 and EOMES (72). Further, patients with tumors that expressed high levels of multiple coinhibitory genes (e.g., LAG3, CTLA4, and HAVCR2) had decreased survival compared to patients that expressed low inhibitory gene levels (47). However, many of these markers are also expressed by Tregs, and this data does not definitively identify T cell exhaustion.
The capacity of T cells to self-renew, e.g., stemness, is an emerging concept of cell-based therapy and Tcf7 appears to mark retention of T cell stemness (73). A subpopulation of exhausted T cells that expressed the transcription factor Tcf7 (protein is Tcf1) and intermediate levels of PD-1 proliferate and differentiate into effector T cells following PD-1 blockade during chronic viral infection and melanoma, as opposed to PD1highTcf1- terminally exhausted T cells which fail to proliferate following ICB (74–77). Thus, PD-1 may prevent Tcf1+ T cell differentiation into cytolytic effector T cells (78). As there are many differences between a disseminated chronic viral infection and PDA, we are interested if a similar T cells progenitor subset is present in this malignancy. As Tcf7 is a Tox-responsive gene (79), Tox may promote exhausted T cell survival and persistence during chronic antigen settings via Tcf7. Due to the many differences such as the cytokine milieu, costimulatory/co-inhibitory markers, and antigen presenting cells in PDA, progenitor CD8 T cells may not be found in all situations of chronic antigen stimulation, which may partially explain why some malignancies fail to respond to PD-1/PD-L1 blockade.
We recently identified that intratumoral PD-1+Lag3+Tox+ exhausted tumor antigen-specific T cells not only lose functionality (e.g., IFNγ, TNFα), they progressively express IL-10 specifically in the TME (64). As IL-10 is a regulatory cytokine (80), these data suggest that the TME instructs antigen-specific T cells to actually participate in immune suppression. The T cell intrinsic role of autocrine and paracrine IL-10 on the program of T cell exhaustion and overall antitumor activity of immune based therapies is of current interest. Evaluation of cytokines within the TME point toward IL-27. IL-27 receptor signaling can induce IL-10 (64) and promote a T cell exhaustion program (81). In orthotopic tumor bearing mice, intratumoral myeloid cells including granulocytes, TAMs, and dendritic cells (DC) upregulate IL-27 during tumorigenesis (64). Moreover, Il27ra-/- mice had delayed tumor growth and a lower frequency of intratumoral antigen-specific T cells expressing Tox (64). Overall the data suggest that if endogenous tumor-specific T cells are engaged sufficiently in PDA, IL-27 in combination with chronic TCR signaling promotes tumor growth by inducing T cell exhaustion.
Inflammatory Cytokines Bias CD4 Th17 Differentiation at the Expense of Th1
Antigen-specific CD4 T cells have not been assessed in PDA animal models or humans. Several murine studies have assessed polyclonal CD4 T cells and shown a role in PDA progression and immunotherapy response (40, 41, 82, 83). CD4 T cells express elevated PD-L1 and PD-L1 ligation promotes Th17 at the expense of Th1 polarization (84). Cytokines including IL-27, IL-6, TGFβ, and IL-23 (85) are elevated in PDA and promote Th17 differentiation (56, 86). TAM production of IL-10 also promotes Th2, Th17 or Treg differentiation (87). Pancreatic stellate cell production of IL-6 promotes Th17 differentiation in human PBMCs while decreasing overall T cell proliferation (88). Depletion of immunosuppressive TAMs (89) or genetic ablation of IL-35 or IL-10 (90) increased Th1 cytokine production. Combination of anti-IL-6 plus anti-PD-L1 reduced tumor growth and prolonged mouse survival (91). Mechanistically, this therapy was dependent on CD8 T cells which associated with increased intratumoral Th1 frequency (91). Taken together, the cytokine milieu in PDA biases CD4 T cells away from Th1 and toward Th2, Th17 and Treg which is beneficial for tumor growth.
NK Cells Are Excluded From the Pancreatic TME Yet Have Therapeutic Potential
Often tumors can escape from T cell immune surveillance via selection of MHC class I loss variants. Natural killer (NK) cells can lyse MHC class I low cancer cells (92). Low frequencies of NK cells are typically found in tumors, despite being elevated in the peripheral blood of PDA patients (83, 93). Low intratumoral NK cell frequency in PDA has been attributed to tumor mutant Kras and Myc signaling repressing expression of the type 1 interferon pathway (94). Compared to healthy donors, NK cells from PDA patients have reduced expression of CXCR2, which is expressed on CD56dim cytotoxic NK cells (95), thereby preventing intratumoral NK cell trafficking (93). Circulating NK cells in PDA patients are impaired in cytotoxic degranulation and have reduced IFNγ production, and this impairment was both progressive with disease stage and associated with poor survival (96). Circulating NK cells from PDA patients exhibited reduced expression of NKp30 compared with healthy donors, suggesting decreased cytotoxicity, as CD107a and NKp30 expression were correlated (96). In another study, pancreatic stellate cells reduced NK cell Granzyme B and IFNγ expression, whereas NK cells in the peripheral blood maintained these functions (97).
Immunotherapies improving NK cell infiltration into PDA can induce tumor regression in preclinical models. Type 1 interferon signaling induces intratumoral macrophages to produce CXCL13 and recruit CXCR5+ NK cells (94). A cleavable antibody specifically targeting mesothelin+ tumor cells and containing the chemoattractant CXCL16, which binds CXCR6, improved NK cell infiltration after adoptive transfer, which reduced tumor burden and prolonged survival in orthotopic or metastatic PDA murine models (98). Ex vivo stimulation and expansion of NK cells promoted NK cell-mediated tumor cell lysis in vitro and reduced tumor volume after transfer in vivo (93). Together, these data suggest NK cells may be useful for targeting PDA given sufficient numbers and trafficking signals.
Tumor Cell-Intrinsic Immune Suppressive Mechanisms
Genetic mutations in tumor cells alter cell signaling and subsequently the stromal cells surrounding the tumor. These changes impact the ability of T cells to accumulate and function in PDA. This process was initially identified in melanoma (99) in which tumor β-catenin levels interfered with DC and T cell intratumoral accumulation. PDA is particularly high for β-catenin (100). Other mechanisms often operate through altered cytokines and chemokines. Elevated Myc in tumor cells increases CXCL1 which recruits CXCR2+ suppressive myeloid cells (52). Pancreatic tumor cell production of inflammatory IL-1β activates CAFs to produce IL-6, CCL2 and CCL5 that polarize immunosuppressive TAMs (101). IL-1β stimulation of tumor cells upregulate CXCL13 expression and recruit PD-L1+ regulatory B cells (Bregs) (102). IL-1β blockade combined with anti-PD-1 reduced orthotopic tumor weight and improved CD8 T cell infiltration (101). Knockdown of IL-1β decreased tumor growth and reversed TAM immune suppression while increasing the frequency of polyclonal CD8 T cells and their production of IFNγ and Granzyme B (101, 102). CAF production of CXCL12 also interferes with CD8 T cell infiltration (50, 101), supporting a stromal trapping mechanism. Finally, tumor cells produce Csf1 and IL-34 which recruit immunosuppressive Csf1r+ TAMs that produce IL-6, IL-10, and TGFβ (103, 104), all factors that promote a cycle of immune suppression.
A major tumor-cell intrinsic mechanisms of PDA immune evasion is the disruption of DC homeostasis. A specialized subset of conventional DCs (cDC1s) are efficient at cross-presenting tumor-associated antigen and appear essential for priming tumor-specific T cells (105). cDC1s expand and are necessary participants in mediating tumor control following multiple immunotherapies in PDA mouse models (41, 52, 103, 106). However, there is a scarcity of a cDC1s in the TME, and the DCs that are present have reduced expression of co-stimulatory and maturation markers CD40, CD80, CD86, and MHC class II (56, 107, 108). Tumor-secretion of G-CSF, promotes granulocytes and monocyte differentiation at the detriment of cDC1s (107). IL-6 neutralization in KPC mice increased frequency of cDC1s, which was attributed to reduced apoptosis of cDC1s (108). cDC1 enrichment, either measured by frequency in peripheral blood (107) or gene expression in tumor samples (109), is associated with a better prognosis and immunological response. CD8 T cell infiltration correlates with cDC1 frequency in PDA (52). Thus, once tumor-specific T cells are engaged within the TME, they may produce factors such as GM-CSF (110), Xcl1 (111), and Flt3L (112) that promote DC recruitment.
Tumor Cell-Extrinsic Immune Suppressive Mechanisms
Tumor-Associated Macrophages
Pancreas macrophages are derived from either circulating monocytes or from the embryo during development. Such embryonic-derived tissue resident macrophages are MHC class II low and can promote fibrosis, while monocyte-derived TAMs can be express high levels of MHC class II and promote anti-tumor T cells (113). Tissue resident macrophages are present in autochthonous KPC tumors and orthotopic tumors yet are absent from subcutaneous tumors, underscoring the importance of studying TAM biology within the pancreas (113). TAMs accumulate in PDA and are generally polarized to the immunosuppressive phenotype defined by Csf1R, CD206, and IL-10 expression along with reduced expression of MHC class II and Ly6C (89, 104, 113, 114). While PD-L1 is expressed on a subset of TAMs (45, 115), approximately 50% of intratumoral CD8 T cells also express PD-L1 in both human and murine PDA (84). PD-L1 on T cells ligates PD-1 on macrophages thereby inducing STAT6 signaling, suppressive TAM polarization, and immune tolerance (84). Tumor derived Galectin 9 ligation of Dectin 1 also induces suppressive TAM polarization and promotes polyclonal T cell tolerance (82, 116). Gas6 production by both CAFs and TAMs promote epithelial-mesenchymal transition (EMT) and tumor metastasis, while simultaneously interfering with NK cell proliferation and activation (117). As PDA metastases progress, macrophages are rendered progressively immunosuppressive and suppress CD8 T cell proliferation via production of granulin (118). Microbes that accumulate in PDA polarize immunosuppressive macrophages via TLR signaling and antibiotics reduced tumor weight and improved anti-PD1 therapy (87).
In contrast, anti-tumor macrophages have higher expression of Ly6C and MHC class II (114). In KPC tumors, low CD8 T cells correlated with local and systemic increase in CD11b+ myeloid cells and MDSCs (52). However, T cell infiltration positively correlated with TAMs and CD8+ T cells co-localized with TAMs within the tumor stroma in human samples (45), suggesting potentially a T cell supportive role for TAMs.
Cancer-Associated Fibroblasts
CAFs synthesize collagen and hyaluronan and coordinate ECM signaling, thereby promoting fibroinflammation (72). There are three CAF subsets identified including αSMA+ myofibroblast CAFs that promote stromal remodeling (myCAFs), inflammatory CAFs that have a secretory phenotype and produce IL-6 (iCAFs), and MHC class II+ CAFs that present antigen (apCAFs) (72, 119). myCAFs are generated by TGFβ-signaling and iCAFs by IL-1 signaling (120). After co-culture with tumor organoids, pancreatic stellate cells (PSCs) differentiate into CAFs subtypes (119). CAFs produce IL-6, which is elevated in human and murine PDA serum (88, 91, 108). IL-6 production can promote PDA metastasis to the liver by acting on hepatocytes (121). In vitro co-cultures suggest that human αSMA+ CAFs inhibit T cell function via PGE2 and promote T cell co-inhibitory receptor expression (46). Notably, CAF depletion through genetic ablation or a CXCR4 inhibitor reduced tumor volume in autochthonous KPC mice, and synergized with PD-L1 blockade (50). A CXCR4 inhibitor with PD-1 blockade increased tumor cell apoptosis and CD8 T cell clonal expansion and effector function in a human PDA tumor slice model (48). Cancer cell intrinsic FAK signaling contributes to the dense fibrotic and immunosuppressive TME of PDA by promoting CAF accumulation (122). Pharmacological CAF inhibition in KPC mice significantly decreased PDA fibrosis, as seen by decreased collagen deposition and numbers of αSMA+ fibroblasts (122). However, CAFs may also keep tumors in check as depletion can promote more aggressive PDA in animal models (123, 124). We speculate that how CAFs are targeted, as well as the extent that antigen-specific T cells are engaged, may determine the outcome following CAF targeting.
Myeloid-Derived Suppressor Cells
Myeloid-derived-suppressor-cells (MDSCs) are immature myeloid cells enriched in animal models of PDA (15, 51, 87, 101, 125–127). Both granulocytic and/or monocytic MDSCs suppress naïve T cell proliferation and induce T cell apoptosis (2), which is mediated by ROS and Arg1 production (126). Tumor cell production of GM-CSF and G-CSF (15, 125, 128), microbial signaling through TLR5 (87), and tumor cell production of IL-1β (101) promote MDSC expansion and their suppressive phenotype. The frequency and number of intratumoral CD8 T cells and MDSC negatively correlate in response to therapeutic autophagy inhibition (106), neoadjuvant therapy (129), and at baseline in KPC (52) and KC tumors (51). MDSC depletion with anti-Ly6G increased CD8 T cell infiltration in autochthonous PDA (15). MDSC depletion with anti-Gr-1 also reduced intratumoral Tregs and Tregs promoted MDSC survival (126), suggesting that MDSCs and Tregs sustain each other. Lastly, CXCR2 signaling in MDSCs and neutrophils plays a key role in establishing and maintaining the metastatic niche (130). Treatment of KPC mice with a CXCR2 inhibitor, reduced metastasis and increased survival (130). Moreover, genetic deletion or inhibition of CXCR2 increased T cell infiltration of PDA in KPC mice (130) or mice bearing subcutaneous tumors (131).
Foxp3+ Regulatory T Cells
Tregs restrict T cell cytotoxicity and are barriers to the anti-tumor response in PDA. Tregs are enriched in both human and murine PDA, and progressively increase in frequency from early stages of tumor development (45, 126, 127, 132). PDA Tregs express elevated FOXP3 compared to Tregs in the spleen, suggesting PDA-specific antigen recognition, as FOXP3 can be upregulated after TCR signaling (45, 126). Neoadjuvant therapy reduces the frequency of intratumoral Tregs, while increasing in CD8 T cells (129). In addition to Tregs, intratumoral DCs can induce FOXP3- CD4 T cells that induce peripheral tolerance, and are termed type 1 regulatory cells (Tr1) cells (85). Tr1-related genes correlated with poor prognosis in PDA patients (85). The pleiotropic cytokine TGFβ, produced by Tregs and other immunosuppressive cells, can act directly on effector lymphocytes to impair the anti-tumor immune response. CAFs induce Treg differentiation and IL-10 production (46), while Treg production of TGFβ drives αSMA+ myCAF differentiation (132). Soluble TGFβ1 impairs NK cell function in PDA patients (96) and TGFβ signaling restrains CD8 T cell cytotoxicity, as CD8 T cells deficient in TGFβ signaling were more effective at lysing tumor cells in an adoptive transfer model into KC mice (133). Autochthonous KPC mice treated with TGFβ blockade and anti-PD-1 had increased survival, increased T cell priming, and greater CD8 T cell intratumoral infiltration and Granzyme B production (133).
Treg depletion during early tumor formation accelerated carcinogenesis and accumulated MDSC and immunosuppressive TAMs in the TME without improving CD8 T cell cytotoxicity. In contrast, Treg depletion in an advanced disease led to tumor regression and increased CD8 T cell activation and cytokine production (127, 132). These data indicate both pro-tumor and anti-tumor effects of Tregs depend on disease stage. Tregs can promote tolerance by preventing DC expression of MHC class II, CD40, and CD86 (132). Overall, Tregs both directly limit T cell function and promote immunosuppressive CAFs and myeloid cells that further hinders the T cell anti-tumor response.
Regulatory B Cells
Recent studies suggest that B cells in PDA have regulatory functions. In both PDA patients and mouse models, B cell production of IL-35 correlated with reduced T cell tumor infiltration and increased Tregs (90, 134). Interfering with IL-35 increased T cell infiltration and cytokine production while reducing tumor weight and PanIN lesion formation (90, 102, 134). IL-35 neutralization in combination with anti-PD-1 improved mouse survival while increasing intratumoral CD8 T cells and IFNγ in PDA mouse models (134). Administration of a BTK inhibitor also reduced tumor burden and increased CD8 T cell production of IFNγ in an orthotopic mouse model (135). B cell depletion with a cocktail of anti-CD19, anti-B220, and anti-CD22 reduced the number of preinvasive lesions in KC mice (102). However, B cell depletion with anti-CD20 failed to reduce tumor burden in autochthonous KPC mice, contrasting with above studies performed in an implantable setting (136). Human PDA patients treated with neoadjuvant therapy exhibited a reduction in intratumoral B cells (129), and PDA patients with B cells their tumors had reduced overall survival (102). In contrast, B cells also participate in tertiary lymphoid structure (TLS) formation, which are sites within PDA that contain DCs and participate in T cell priming and regulation of the immune response and are associated with improved survival of PDA patients (45). Thus, the specific role of B cells in PDA is currently unclear and may depend on specific B cell subsets, disease stage, and context which they are studied.
Strategies to Enhance the Efficacy of Immunotherapies
Autophagy Inhibition
Pancreatic tumor cells typically express low cell surface MHC class I albeit levels are variable among individual patient tumors and even within the same tumor (45). KPC cell lines are also low for cell surface MHC class I, which can be upregulated by exposure to IFNγ (23). Tumor escape caused by impaired antigen presentation or MHC class I expression on tumor cells is often attributed to mutations in the MHC class I pathway (22, 23, 137). A recent discovery described a novel mechanism whereby cell surface MHC class I molecules are degraded by tumor cells via autophagy (106). Systemic inhibition of autophagy using chloroquine required dual ICB therapy (anti-PD-1 + anti-CTLA-4) for improved T cell infiltration into orthotopic tumors, yielding a 59% response rate (106). Similarly, dual therapy with an ERK inhibitor and chloroquine inhibited tumor growth and improved survival in a PDA xenograft mouse model (138). Interfering with mutant Kras signaling in combination with autophagy inhibition has shown objective responses in advanced human PDA (139) though the relative contribution of the immune response remains to be investigated. Thus, autophagy of MHC class I on tumor cells contributes to tumor escape, and autophagy inhibition renders tumors responsive to immunotherapy.
Agonistic Anti-CD40
CD40 agonist promotes the antitumor activity of a variety of immune-based therapeutic strategies and may do so by engaging DCs, promoting anti-tumor TAMs, and creating a TME that is conducive to anti-tumor cytolytic effector T cells. CD40 agonist altered the cytokines that intratumoral myeloid cells produce, including IL-27, which in turn impacted the intratumoral differentiation of potent cytolytic effector T cells at the expense of exhausted T cells in the TME (64). Agonistic anti-CD40 increased antigen presenting cell antigen processing and presentation to prime and activate antigen-specific T cells, yielding successful anti-tumor responses as a monotherapy or a component of combination approaches (140–142). Treatment of mice bearing subcutaneous tumors with CD40 agonist, anti-PD-1, and anti-CTLA-4 enhanced priming of T cells with unknown antigen-specificity and intratumoral T cell production of TNFα and IFNγ (41).
CCL5 is produced by monocytes, macrophages, cDC2s, and ILC2s to recruit cDC1s to the TME, and intratumoral myeloid cells increased their secretion of CCL5 in response to agonist anti-CD40 therapy (143, 144). Intratumoral CCL5 mediates T cell trafficking to the TME and is required for the efficacy of CD40 agonist + anti-PD-1 + anti-CTLA-4 triple therapy (143). T cells were required for the efficacy of this combination (41, 52). Combination of a TLR4 agonist, agonistic anti-CD40, and anti-PD-1 improved the survival of mice bearing orthotopic tumors, and studies with additional tumor models suggest that this therapy promotes DC priming of naïve T cells and recruitment of antigen-specific T cells to the TME (145). Flt3L administration mobilizes DCs and when combined with agonistic anti-CD40 improved the survival of autochthonous KPPC-OG mice (p48-Cre;KrasLSL-G12D;Trp53fl/fl;R26tm1(LSL-OG)) or mice bearing subcutaneous tumors through the intratumoral enrichment of cDC1s and infiltration of OVA-specific CD8 T cells (56, 108).
A MEK1/2 inhibitor was successful as a single agent in treating mice bearing subcutaneous tumors and synergized with agonistic anti-CD40 to increase mouse survival and prompt rejection in 32% of tumors (146). MEK1/2 inhibitor plus agonistic anti-CD40 dual therapy increased the ratio of intratumoral CD8 T cells to Tregs and increased the frequency of inflammatory anti-tumor macrophages at the loss of immunosuppressive TAMs, whereas agonistic anti-CD40 alone increased CD8 T cell frequency and production of TNFα and IFNγ while also improving DC activation and antigen presentation (146). The MEK1/2 inhibitor alone directly induced tumor cell death, decreased the frequency of intratumoral Tregs and TAMs, yet selectively targeted immunosuppressive TAMs and MDSCs while sparing inflammatory TAMs (146). Similar results were seen with a triple therapy of MEK inhibitor, CDK4/6 inhibitor, and anti-PD-L1 also in a subcutaneous model; triple therapy improved DC antigen presentation, shifted the TME immune landscape to contain more lymphoid cells and fewer myeloid cells as well as enhanced anti-tumor macrophages, CD8 T cells and NK cells (147). These pro-inflammatory anti-tumor TAMs were defined by induction of genes associated with iron metabolism, including Ftl1 and Fth1, as well as genes associated with immune activation including Bnip3l and Ctsd (147). Overall, agonist anti-CD40 improves T cell functionality and synergizes with other immunotherapies to improve the T cell anti-tumor response.
Targeting TAMs
Treatment of orthotopic tumor bearing mice with a CD11b agonist (ADH-503) decreased the total number of intratumoral myeloid cells and remaining TAMs exhibited an inflammatory phenotype that promoted intratumoral antigen-specific T cell recruitment and function (103). Autochthonous KPC mice treated with CD11b agonist combined with anti-PD-1, anti-CTLA-4, and gemcitabine had significantly improved survival and increased intratumoral CD8 T cell infiltration, but the tumors were not rejected as seen with orthotopic tumor bearing mice (103). However, because KPC mice are an autochthonous model, tumor cells are continuously regenerative making curative therapy notoriously difficult. Similarly, genetic ablation of CD11b myeloid cells in an inducible KC mouse model led to a reduction in Tregs and PD-L1 expression along with concurrent activation of CD8 T cells in the TME and delayed PanIN lesion formation (17).
Other strategies to overcome TAM suppression specifically target the signaling pathways contributing to their suppressive nature. Blockade of Csf1/Csf1R, which is expressed by some TAMs, improved mouse survival and reduced tumor weight (104). Csf1/Csf1R blockade reduced collagen deposits, αSMA expression by fibroblasts, and congruently increased CD8 T cell infiltration in PDA mouse models (104, 118). Further, TAM depletion reduced immunosuppressive IL-6 and IL-10 and restored CD8 T cell production of Granzyme B, IFNγ, and perforin, and improved survival of autochthonous KPC mice (104). In a metastatic KPC mouse model, anti-PD-1 and anti-Csf1 synergized to reduce collagen deposits and αSMA+ myofibroblasts, both of which improved CD8 T cell infiltration of PDA (118). Additionally, TAMs in human and murine PDA express PI3Kγ, and genetic ablation or pharmacological inhibition of PI3Kγ reduced tumor weight and metastasis in orthotopic and autochthonous murine models (148). Mechanistically, inhibition of PI3Kγ decreased TAM expression of genes associated with immune suppression and tumor angiogenesis (Arg1, Tgfb, Il1b, Il6, Vegfa), and upregulated expression of genes associated with anti-tumor immunity (Il12 and Ifng), suggesting TAM reprogramming (148).
Engineered T Cell Therapies
A promising immunotherapy is adoptive T cell therapy, which includes the isolation, engineering, and expansion of a defined tumor-reactive T cell population for infusion back into patients (149). This approach may be particularly useful in malignancies where there is a low TMB, and thus a robust endogenous anti-tumor response is lacking. Adoptive transfer of large numbers T cells that express a tumor-reactive TCR have demonstrated proof-of principle for tumor eradication in the clinic in melanoma, leukemia, and some solid tumors (150–160). Such transfer of native or engineered TCR-expressing T cells is distinct from chimeric antigen receptor (CAR)-modified T cells, which recognize cell surface tumor antigens independent of MHC molecules (161). CD19 CAR therapies are effective at eradicating some hematological malignancies (162). However, CAR T cell therapies have not been successful on their own in most solid tumor settings. Phase 1 clinical trials of mesothelin-specific CAR T cells demonstrated safety of the therapy but show limited clinical efficacy and poor persistence (163, 164). Efforts to improve the efficacy of mesothelin-specific CAR T cells include the addition of an oncolytic adenovirus that induced TME expression of TNFα and IL-2 (165). This combination therapy vastly improved in vitro tumor cell lysis and tumor regression in a xenograft NSG mouse model (165). Addition of TNFα and IL-2 enhanced CAR T cell activation as measured by CD69 and CD25 expression along with proliferation, infiltration, and accumulation in tumors of NSG mice (165). Co-infusion of Msln CAR T cells and CD19 CAR T cells successfully depleted B cells yet failed to improve the persistence of mesothelin-specific CAR T cells (166). Prostate stem cell antigen (PSCA)-specific CAR T cells were modified so that the TGFβ receptor was linked to a stimulatory 4-1BB domain and the IL-4 receptor was linked to IL-7 receptor signaling, switching suppressive TGFβ and IL-4 to stimulatory and survival signals (167). Adoptive transfer of these modified CAR T cells yielded complete rejection of subcutaneous tumors in NSG mice, and tumor re-challenge induced CAR T cell expansion and tumor rejection (167).
TCR engineered CD8 T cells specific to mesothelin have yet to be tested in the clinic. The preclinical studies are promising and although engineered T cells are ultimately rendered exhausted in the TME, serial Msln TCR-engineered T cell infusions in concert with a vaccine to expand infused T cells induced objective responses and doubled survival in the aggressive autochthonous KPC animal model (53). We found that administration of the vaccine at the time of T cell infusions was critical for promoting T cell expansion, highlighting the fact that Msln is not presented in an immunogenic manner. However, serial T cell infusions in the clinic are laborious and expensive. Therefore, a major effort by our lab is to identify the factors in the TME that are interfering with engineered T cell function. Notably, accumulating evidence by our lab indicates that strategies to modify the suppressive TME that enhance the activity of endogenous T cells may not yield the same results for infused engineered T cells (23, 53, 114). Engineered T cells are programmed to have a single and defined antigen-specificity and are artificially activated and expanded with anti-CD3/anti-CD28 beads and cytokines in vitro prior to transfer, thereby bypassing the requirement for priming in vivo. Abrogating TAMs using Csf1R blockade had no benefit for engineered T cell accumulation or function (114). In contrast, depleting Csf1R+ TAMs increased endogenous T cell infiltration and responsiveness to PD-1 blockade in an animal model in which antigen-specificity of the T cells is unknown (118). One approach that produced beneficial results with both endogenous (38, 64) and engineered T cells was agonistic anti-CD40 (114). However, while agonist anti-CD40 supported cytotoxic activity of endogenous tumor-specific T cells (64), it failed to rescue cytotoxic function and cytokine production of infused engineered T cells (114). As agonistic anti-CD40 alone is suboptimal in priming and expanding endogenous antigen-specific T cells (64, 168), future combinations with TLR agonists seem promising as this has been shown to dramatically improve vaccination (168). Thus, ICB which was assumed to rescue intratumoral T cell function, may instead act systemically on a new wave of recently activated T cells (23), which are not present in the engineered T cell infused population. Thus, we continue to be cautious to extrapolate results based on endogenous T cell responses to engineered T cell responses.
Of note, the majority of the tumor mass in PDA is stromal cells rather than tumor cells (2). In animal models, MHC class I expression on stromal cells is required for T cell-mediated solid tumor eradication (169). Cross-presentation of tumor antigen by cDC1s that express the transcription factor Batf3 induce priming of rare naïve tumor-reactive T cells (170). Based on data from Batf3-/- mice, cDC1s appear to also be required for adoptively transferred T cells to infiltrate melanoma (171) and for islet infiltration by islet-reactive T cells (172). This suggests a role for cDC1 antigen presentation in the draining lymph node and/or within tumors for T cell migration into tissues and is consistent with clinical data where tumors with high numbers of cDC1s bode better for immunotherapy outcomes (56, 173, 174). Insufficiency of cDC1s and priming of antigen specific T cells is a major road-block to effective immunotherapies, suggesting a need for therapies that draw antigen-specific T cells into the TME and also maintain T cell activation within the TME. cDC1 production of CXCL9 and CXCL10 recruits T cell into the TME (103, 171). Moreover, in a MC38 tumor model, response to anti-PD-1 was dependent on intratumoral cDC1 production of CXCL9 and subsequent signaling through CXCR3 on T cells, which promoted proliferation and production of IFNγ, TNFα, and Granzyme B (175). In a breast cancer model, expansion of adoptively transferred tumor antigen-specific T cells was mediated by DC engagement through a combination therapy of Flt3L, radiotherapy, poly(I:C), and a CD40 agonist (176). Adoptive transfer of CAR T cells engineered to secrete Flt3L and in combination with poly(I:C) and anti-4-1BB enhanced T cell and DC expansion and activation in various tumor models (177). Thus, unlike CARs, TCRs provide T cells the ability to recognize peptide:MHC complexes not only on the tumor cells, but also on antigen presenting cells such as cDC1s, which may be a particular advantage of TCR based approach over CAR-based engineering strategies (53, 114).
Immune Checkpoint Blockade
Similar to PDA patients, anti-PD-L1 or anti-PD-1 monotherapy is ineffective in KPC mouse models and provides minimal benefit in survival or tumor regression even in animal models with a model neoantigen (23, 82, 83, 87, 90, 101, 103, 118, 133, 134). Both TMB and T cell inflamed gene expression profile are biomarkers for response to immune checkpoint blockade (ICB) in human PDA and other cancers (178). Although a low TMB in PDA may partially explain resistance to ICB, it is clear based on trials with MSIhigh PDA that this is not the full story (34).
While anti-PD-1 therapy alone is not successful at treating PDA, it synergizes with many other immunotherapies in preclinical studies and thus we are hopeful that in the near future combinations will prove efficacious in the clinic (37). We identified that anti-PD-1 plus anti-PD-L1 treatment of mice bearing orthotopic tumors improved mouse survival and induced systemic expansion of tumor-specific T cells that were then recruited to the TME and further differentiated into central and tissue resident memory T cells following orthotopic tumor clearance (23). Importantly, dual therapy promoted Klrg1+ effector cells producing IFNγ and TNFα and decreased the frequency of PD-1 and Lag3 expressing T cells due to recruitment of new effector cells and not reinvigoration of intratumoral exhausted T cell (23). These results raise the question if reinvigoration of intratumoral T cells is possible in PDA. Dual therapy with anti-PD-1 + anti-OX40 led to complete response and long-term survival along with recall response to re-challenge in an orthotopic tumor model, while autochthonous KPC mice had improved survival by 40 days (83). This dual therapy required CD4 T cells for efficacy and memory response, and led to intratumoral enrichment of total CD4 T cells, CD8 and CD4 memory T cells, and a reduced frequency of Tregs and PD-1+ T cells (83). Similarly, anti-PD-1 + anti-OX40 improved the efficacy of PancVAX, a neoantigen targeted vaccine with a STING adjuvant, and induced a durable response in mice bearing subcutaneous tumors from a Panc02 cells line and that correlated with reduced frequency of intratumoral T cells co-expressing PD-1 and Lag3 (179). However this dual therapy was not tested without the vaccine and may have been the main therapeutic driver (179). In orthotopic but not subcutaneous mouse models, IL-33 activates ILC2s, which in turn produce CCL5 to recruit CD103+ cDC1s to the TME which then prime and activate CD8 T cells to infiltrate the tumor and exert an anti-tumor immune response (144). Dual therapy with recombinant IL-33 plus anti-PD-1 engaged ILC2s to accumulate in orthotopic KPC tumors and improved T cell infiltration through DC priming, allowing for improved mouse survival and decreased tumor volume (144). Overall, anti-PD-1 therapy synergizes with many other immunotherapy drugs to improve the anti-tumor immune response of T cells in PDA mouse models. The challenge ahead is to identify the approaches that can be safely used in patients and that elicit durable responses.
Challenges to Modeling the Immune Response in PDA Preclinical Studies
Successful immunotherapy for targeting PDA will likely require multiple agents used in combination and sequenced appropriately. Target identification as well as safety and efficacy testing require rational immune-competent preclinical animal models. There are many labs utilizing variations of the KPC animal model. How this model is used to interrogate immunotherapy response is quite important, however. In the autochthonous setting, high-resolution ultrasound imaging should be performed to locate and measure a defined tumor mass within the pancreas to ensure that mice have invasive disease before enrolling for treatments or else regimens will be tested in animals with only preinvasive disease. Implanted KPC cell lines under the skin of wild type animals will likely not fully recapitulate the TME of the native organ as most of the myeloid cells in the skin tumors will be of blood origin and such tumors will lack pancreas tissue resident macrophages (113). Orthotopic KPC tumor implantation is a better alternative to subcutaneous tumors. As discussed, a major limitation is the lack of identified native antigens in the KPC model to interrogate endogenous tumor-specific T cells. While Ova can be used, as tumors that express Ova are rejected, it remains questionable the extent this models a T cell response in PDA. We target mesothelin with engineered T cells, but we also showed that most endogenous T cells reactive to Msln are deleted during T cell development (53). Thus, while we developed a neoantigen model by engineering the tumors to express CB as discussed above, the epitope binds MHC with high affinity and thus models a highly immunogenic epitope. Consequently, there remains a major knowledge gap regarding the specificity of the CD4 and CD8 T cells that are engaged during combination therapies and the extent that the epitopes are immunogenic on their own.
Summary
Mechanistic studies support that priming and expanding systemic T cells, rather than rescuing or reinvigorating exhausted T cells, is necessary for tumor rejection. This is in opposition to the preponderance of the literature describing T cell exhaustion during chronic viral infection. There are striking differences between chronic viral infection and PDA including the location of the target antigen and the surrounding environment. Chronic LCMV results in a systemic infection where antigen and antigen presentation are not limiting, and the biology of the T cells is almost always assessed from the spleen. In contrast, many studies support that antigen and/or antigen presentation is limiting in PDA and thus T cell differentiation toward the identical exhausted T cell state is unlikely, and lack of T cell function may be due to the myriad of suppressive mechanisms described above.
PDA is characterized by a dense desmoplastic stroma and enrichment of immunosuppressive cells. While antigen-specific lymphocytes can infiltrate the TME, proliferate and mediate some anti-tumor activity transiently, they lose function either through progressive exhaustion due to chronic TCR signaling and/or immunosuppressive cells and cytokines that limit their activation and cytotoxicity. PDA is resistant to single agent T cell-directed immunotherapies, and combination therapies targeting lymphocytes and reprograming the TME are necessary to induce a productive anti-tumor immune response. While there are many combination therapies that induce a partial or complete response in murine models, clinical trials have yet to successfully identify an effective immunotherapy to treat PDA patients. As such, further investigation into the individual mechanisms that limit T cell function in PDA is required. Moreover, these individual mechanisms likely form a hierarchy of suppression. By elucidating the key mechanism(s) of suppression we can identify the best targets for PDA immunotherapy under specific conditions of T cell activation, suppression or exhaustion (Figure 3). For example, immune checkpoint blockade may complement approaches that can engage tumor-specific T cells while simultaneously modifying the immunosuppressive TME. A deficiency in T cell priming can may be addressed by approaches that expand and activate DCs or adoptive T cell therapy. However, once a sufficient T cell response is engaged, if tumor cells are not eradicated, tumor escape either through defects in target antigen and/or antigen presentation, or T cell exhaustion due to chronic antigen encounter may prevail. Thus, identifying strategies to anticipate and overcome escape following immunotherapy may be critical for durable responses in the clinic.
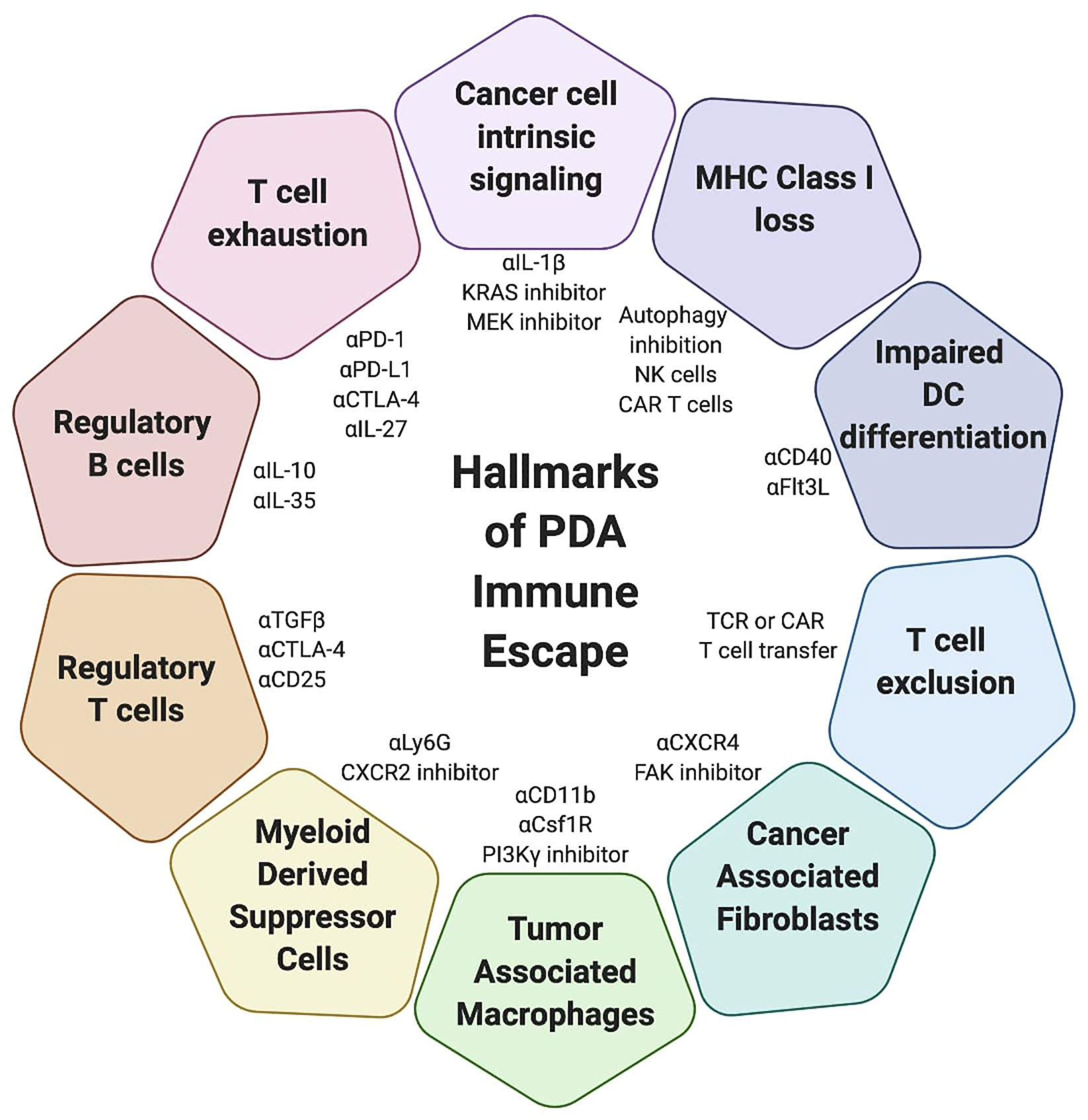
Figure 3 Mechanisms of immune escape in pancreatic cancer. Simplified overview of mechanisms by which pancreatic ductal adenocarcinoma (PDA) evades the immune response and examples of potential therapeutic strategies to target these mechanisms. Some strategies may obviate multiple tumor evasion mechanisms. Combination strategies may be necessary to promote synergy and overcome tumor evasion from the immune system.
Author Contributions
All authors contributed to the article and approved the submitted version.
Funding
IS is supported by an AACR Pancreatic Cancer Action Network Career Development Award (17-20-25-STRO), AACR Pancreatic Cancer Action Network Catalyst Award (19-35-STRO), an American Cancer Society Institutional Research Grant (124166-IRG-58-001-55-IRG65), and pilot awards from the Masonic Cancer Center and Cancer Research Translational Initiative, (University of Minnesota Medical School) and NIH U54-CA-210190.
Conflict of Interest
IS serves on the scientific advisory board for Luminary Therapeutics and Immunogenesis.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Adam Burrack for editorial feedback. The figures were created using birender.com with the help of Meagan Rollins.
References
1. Howlader N, Noone A, Krapcho M, Miller D, Brest A, Yu M, et al. SEER Cancer Statistics Review, 1975-2016. Bethesda, MD: National Cancer Institute (2019). Available at: https://seer.cancer.gov/csr/1975_2016/.
2. Stromnes IM, DelGiorno KE, Greenberg PD, Hingorani SR. Stromal reengineering to treat pancreas cancer. Carcinogenesis (2014) 35:1451–60. doi: 10.1093/carcin/bgu115
3. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. N Engl J Med (2014) 371:1039–49. doi: 10.1056/NEJMra1404198
4. Huang L, Jansen L, Balavarca Y, van der Geest L, Lemmens L, Van Eycken L, et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: a large, international population-based study Lei. BMC Med (2018) 16. doi: 10.1186/s12916-018-1120-9
5. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM, et al. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the united states. Cancer Res (2014) 74:2913–21. doi: 10.1158/0008-5472.CAN-14-0155
6. Chin V, Nagrial A, Sjoquist K, O’Connor CA, Chantrill L, Biankin AV, et al. Chemotherapy and radiotherapy for advanced pancreatic cancer. Cochrane Database Syst Rev (2018) 3. doi: 10.1002/14651858.CD011044.pub2
7. Rahman SH, Urquhart R, Molinari M. Neoadjuvant therapy for resectable pancreatic cancer. World J Gastrointest. Oncol (2017) 9:457–65. doi: 10.4251/wjgo.v9.i12.457
8. Hu-Lieskovan S, Ribas A. New combination strategies using PD-1/ L1 checkpoint inhibitors as a backbone. Cancer J (2017) 23:10–22. doi: 10.1097/PPO.0000000000000246
9. Royal RE, Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 Trial of Single Agent Ipilimumab (Anti-CTLA-4) for Locally Advanced or Metastatic Pancreatic Adenocarcinoma Richard. J Immunother (2010) 33:828–33. doi: 10.1097/CJI.0b013e3181eec14c
10. Brahmer JR, Tykodi SS, Chow LQM, Hwu WJ, Topalian SL, Hwu P, et al. Safety and Activity of Anti-PD-L1 Antibody in Patients with Advanced Cancer. N Engl J Med (2012) 366:2455–65. doi: 10.1056/NEJMoa1200694
11. O’Reilly EM, Oh DY, Dhani N, Renouf DJ, Lee MA, Sun W, et al. Durvalumab with or Without Tremelimumab for Patients with Metastatic Pancreatic Ductal Adenocarcinoma: A Phase 2 Randomized Clinical Trial. JAMA Oncol (2019) 5:1431–8. doi: 10.1001/jamaoncol.2019.1588
12. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531:47–52. doi: 10.1038/nature16965
13. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell (2003) 4:437–50. doi: 10.1016/S1535-6108(03)00309-X
14. Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell (2005) 7:469–83. doi: 10.1016/j.ccr.2005.04.023
15. Stromnes IM, Brockenbrough JS, Izeradjene K, Carlson MA, Cuevas C, Simmons RM, et al. Targeted depletion of an MDSC subset unmasks pancreatic ductal adenocarcinoma to adaptive immunity. Gut (2014) 63:1769–81. doi: 10.1136/gutjnl-2013-306271
16. Guerra C, Schuhmacher AJ, Cañamero M, Grippo PJ, Verdaguer L, Pérez-Gallego L, et al. Chronic Pancreatitis Is Essential for Induction of Pancreatic Ductal Adenocarcinoma by K-Ras Oncogenes in Adult Mice. Cancer Cell (2007) 11:291–302. doi: 10.1016/j.ccr.2007.01.012
17. Zhang Y, Velez-Delgado A, Mathew E, Li D, Mendez FM, Flannagan K, et al. Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer. Gut (2017) 66:124–36. doi: 10.1136/gutjnl-2016-312078
18. Dey P, Li J, Zhang J, Chaurasiya S, Strom A, Wang H, et al. Oncogenic KRAS-driven metabolic reprogramming in pancreatic cancer cells utilizes cytokines from the tumor microenvironment. Cancer Discovery (2020) 10:608–25. doi: 10.1158/2159-8290.CD-19-0297
19. Collins MA, Bednar F, Zhang Y, Brisset JC, Galbán S, Galbán CJ, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. (2012) 122:639–53. doi: 10.1172/JCI59227
20. Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, et al. Oncogenic kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell (2012) 149:656–70. doi: 10.1016/j.cell.2012.01.058
21. Pandha H, Rigg A, John J, Lemoine N. Loss of expression of antigen-presenting molecules in human pancreatic cancer and pancreatic cancer cell lines. Clin Exp Immunol (2007) 148:127–35. doi: 10.1111/j.1365-2249.2006.03289.x
22. Garrido F, Aptsiauri N, Doorduijn EM, Lora AMG, Hall T. Van. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol (2016) 39:44–51. doi: 10.1016/j.coi.2015.12.007
23. Burrack AL, Spartz EJ, Raynor JF, Wang I, Olson M, Stromnes IM. Combination PD-1 and PD-L1 Blockade Promotes Durable Neoantigen-Specific T Cell-Mediated Immunity in Pancreatic Ductal Adenocarcinoma. Cell Rep (2019) 28:2140–2155.e6. doi: 10.1016/j.celrep.2019.07.059
24. Hu Z II, Hellmann MD, Wolchok JD, Vyas M, Shia J, Stadler ZK, et al. Acquired resistance to immunotherapy in MMR-D pancreatic cancer. J Immunother. Cancer (2018) 6:1–6. doi: 10.1186/s40425-018-0448-1
25. Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: a case for synergistic therapies. Cancer Cell (2017) 31:311–25. doi: 10.1016/j.ccell.2017.02.008
26. Thommen DS. & Schumacher, T. N. T Cell Dysfunction in Cancer. Cancer Cell (2018) 33:547–62. doi: 10.1016/j.ccell.2018.03.012
27. Klein L, Kyewski B, Allen PM, Hogquist KA. Positive and negative selection of the T cell repertoire: what thymocytes see and don’t see. Nat Rev Immunol (2014) 14:377–91. doi: 10.1038/nri3667
28. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
29. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Mutational landscape determines sensitivity to PD-1 blockade in non–small cell lung cancer. Science (2015) 348:124–8. doi: 10.1126/science.aaa1348
30. Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral A, Remark R, et al. Identification of unique neoantigen qualities in long term pancreatic cancer survivors. Nature (2018) 551:512–6. doi: 10.1038/nature24462
31. Biankin AV, Waddell N, Kassahn KS, Gingras MC, Muthuswamy LB, Johns AL, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature (2012) 491:399–405. doi: 10.1038/nature11547
32. Bailey P, Chang DK, Forget MA, Lucas FAS, Alvarez HA, Haymaker C, et al. Exploiting the neoantigen landscape for immunotherapy of pancreatic ductal adenocarcinoma. Sci Rep (2016) 6:1–8. doi: 10.1038/srep35848
33. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499:214–8. doi: 10.1038/nature12213
34. Hu Z II, Shia J, Stadler ZK, Varghese AM, Capanu M, Salo-Mullen E, et al. Evaluating mismatch repair deficiency in pancreatic adenocarcinoma: Challenges and recommendations. Clin Cancer Res (2018) 24:1326–36. doi: 10.1158/1078-0432.CCR-17-3099
35. Riquelme E, Zhang Y, Zhang L, Montiel M, Zoltan M, Dong W, et al. Tumor Microbiome Diversity and Composition Influence Pancreatic Cancer Outcomes. Cell (2019) 178:795–806.e12. doi: 10.1016/j.cell.2019.07.008
36. Carstens JL, De Sampaio PC, Yang D, Barua S, Wang H, Rao A, et al. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat Commun (2017) 8. doi: 10.1038/ncomms15095
37. Vonderheide RH. CD40 Agonist Antibodies in Cancer Immunotherapy. Annu Rev Med (2020) 71:47–58. doi: 10.1146/annurev-med-062518-045435
38. Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer ARL, Bajor DL, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res (2015) 3:399–411. doi: 10.1158/2326-6066.CIR-14-0215
39. Luheshi NM, Coates-Ulrichsen J, Harper J, Mullins S, Sulikowski MG, Martin P, et al. Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget (2016) 7:18508–20. doi: 10.18632/oncotarget.7610
40. Ma HS, Poudel B, Torres ER, Sidhom JW, Robinson TM, Christmas B, et al. A CD40 Agonist and PD-1 Antagonist Antibody Reprogram the Microenvironment of Nonimmunogenic Tumors to Allow T-cell–Mediated Anticancer Activity. Cancer Immunol Res (2019) 7:428–42. doi: 10.1158/2326-6066.CIR-18-0061
41. Morrison AH, Diamond MS, Hay CA, Byrne KT, Vonderheide RH. Sufficiency of CD40 activation and immune checkpoint blockade for T cell priming and tumor immunity. Proc Natl Acad Sci (2020), 201918971. doi: 10.1073/pnas.1918971117
42. Von Bernstorff W, Voss M, Freichel S, Schmid A, Vogel I, Jöhnk C, et al. Systemic and local immunosuppression in pancreatic cancer patients. Clin Cancer Res (2001) 7:925–32.
43. Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology (2013) 145:1121–32. doi: 10.1053/j.gastro.2013.07.025
44. Hartmann N, Giese NA, Giese T, Poschke I, Offringa R, Werner J, Ryschich E. Prevailing role of contact guidance in intrastromal T-cell trapping in human pancreatic cancer. Clin Cancer Res (2014) 20:3422–33. doi: 10.1158/1078-0432.CCR-13-2972
45. Stromnes IM, Hulbert A, Pierce RH, Greenberg PD. & Hingorani, S. R. T-cell localization, activation, and clonal expansion in human pancreatic ductal adenocarcinoma. Cancer Immunol Res (2017) 5:978–91. doi: 10.1158/2326-6066.CIR-16-0322
46. Gorchs L, Moro CF, Bankhead P, Kern KP, Sadeak I, Meng Q, et al. Human pancreatic carcinoma-associated fibroblasts promote expression of co-inhibitory markers on CD4+ and CD8+ T-cells. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00847
47. Blando J, Sharma A, Higa MG, Zhao H, Vence L, Yadav SS, et al. Comparison of immune infiltrates in melanoma and pancreatic cancer highlights VISTA as a potential target in pancreatic cancer. Proc Natl Acad Sci U. S. A. (2019) 116:1692–7. doi: 10.1073/pnas.1811067116
48. Seo YD, Jiang X, Sullivan KM, Jalikis FG, Smythe KS, Abbasi A, et al. Mobilization of CD8+ T cells via CXCR4 blockade facilitates PD-1 checkpoint therapy in human pancreatic cancer. Clin Cancer Res (2019) 25:3934–45. doi: 10.1158/1078-0432.CCR-19-0081
49. Beatty GL, Winograd R, Evans RA, Long KB, Luque SL, Lee JW, et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice is Regulated by Ly6Clow F4/80+ Extra-tumor Macrophages. Gastroenterology (2015) 149:201–10. doi: 10.1053/j.gastro.2015.04.010
50. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U. S. A. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
51. Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH, et al. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res (2007) 67:9518–27. doi: 10.1158/0008-5472.CAN-07-0175
52. Li J, Byrne KT, Yan F, Yamazoe T, Chen Z, Baslan T, et al. Tumor Cell-Intrinsic Factors Underlie Heterogeneity of Immune Cell Infiltration and Response to Immunotherapy. Immunity (2018) 49:178–193.e7. doi: 10.1016/j.immuni.2018.06.006
53. Stromnes IM, Schmitt TM, Hulbert A, Brockenbrough JS, Nguyen H, Cuevas C, et al. T cells engineered against a native antigen can surmount immunologic and physical barriers to treat pancreatic ductal adenocarcinoma Ingunn. Cancer Cell (2015) 28:638–52. doi: 10.1016/j.ccell.2015.09.022
54. Argani P, Iacobuzio-Donahue C, Ryu B, Rosty C, Goggins M, Wilentz RE, et al. Mesothelin is overexpressed in the vast majority of ductal adenocarcinomas of the pancreas: Identification of a new pancreatic cancer marker by serial analysis of gene expression (SAGE). Clin Cancer Res (2001) 7:3862–8.
55. Evans RA, Diamond MS, Rech AJ, Chao T, Richardson MW, Lin JH, et al. Lack of immunoediting in murine pancreatic cancer reversed with neoantigen. JCI Insight (2016) 1. doi: 10.1172/jci.insight.88328
56. Hegde S, Krisnawan VE, Herzog BH, Zuo C, Breden MA, Knolhoff BL, et al. Dendritic Cell Paucity Leads to Dysfunctional Immune Surveillance in Pancreatic Cancer. Cancer Cell (2020) 37:289–307.e9. doi: 10.1016/j.ccell.2020.02.008
57. Jenkins MK, Moon JJ. The Role of Naive T Cell Precursor Frequency and Recruitment in Dictating Immune Response Magnitude. J Immunol (2012) 188:4135–40. doi: 10.4049/jimmunol.1102661
58. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD-1 blockade. Science (2016) 354:1160–5. doi: 10.1126/science.aaf2807
59. Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity (2016) 45:389–401. doi: 10.1016/j.immuni.2016.07.011
60. Martinez GJ, Pereira RM, Aijo T, Kim EY, Marangoni F, Pipkin ME, et al. The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity (2015) 42:265–78. doi: 10.1016/j.immuni.2015.01.006
61. Khan O, Giles JR, McDonald S, Manne S, Ngiow SF, Patel KP, et al. TOX transcriptionally and epigenetically programs CD8+ T cell exhaustion. Nature (2019) 571:211–8. doi: 10.1038/s41586-019-1325-x
62. Scott AC, Dündar F, Zumbo P, Chandran SS, Klebanoff CA, Shakiba M, et al. TOX is a critical regulator of tumour-specific T cell differentiation. Nature (2019) 571:270–4. doi: 10.1038/s41586-019-1324-y
63. Alfei F, Kanev K, Hofmann M, Wu M, Ghoneim HE, Roelli P, et al. TOX reinforces the phenotype and longevity of exhausted T cells in chronic viral infection. Nature (2019) 571:265–9. doi: 10.1038/s41586-019-1326-9
64. Burrack AL, Rollins MR, Spartz EJ, Mesojednik TD, Schmiechen ZC, Raynor JF, et al. Agonist anti-CD40 overcomes T cell exhuastion induced by chronic myeloid cell IL-27 production in a preclinical model of pancreatic cancer. J Immunol (2020). (in press).
65. Corral L, Hanke T, Vance RE, Cado D, Raulet DH. NK cell expression of the killer cell lectin-like receptor G1 (KLRG1), the mouse homolog of MAFA, is modulated by MHC class I molecules. Eur J Immunol (2000) 30:920–30. doi: 10.1002/1521-4141(200003)30:3<920::AID-IMMU920>3.0.CO;2-P
66. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, et al. Inflammation Directs Memory Precursor and Short-Lived Effector CD8(+) T Cell Fates via the Graded Expression of T-bet Transcription Factor. Immunity (2007) 27:281–95. doi: 10.1016/j.immuni.2007.07.010
67. Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, et al. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity (2007) 27:670–84. doi: 10.1016/j.immuni.2007.09.006
68. Kaech SM, Wherry EJ. Heterogeneity and Cell-Fate Decisions in Effector and Memory CD8+ T Cell Differentiation during Viral Infection. Immunity (2007) 27:393–405. doi: 10.1016/j.immuni.2007.08.007
69. Olson JA, McDonald-Hyman C, Jameson SC, Hamilton SE. Effector-like CD8+ T Cells in the Memory Population Mediate Potent Protective Immunity. Immunity (2013) 38:1250–60. doi: 10.1016/j.immuni.2013.05.009
70. Herndler-Brandstetter D, Ishigame H, Shinnakasu R, Plajer V, Stecher C, Zhao J, et al. KLRG1+ Effector CD8+ T Cells Lose KLRG1, Differentiate into All Memory T Cell Lineages, and Convey Enhanced Protective Immunity. Immunity (2018) 48:716–729.e8. doi: 10.1016/j.immuni.2018.03.015
71. Yao C, Sun H, Lacey NE, Ji Y, Moseman EA, Shih H, et al. Single-cell RNA-seq reveals TOX as a key regulator of CD8+ T cell persistence in chronic infection. Nat Immunol (2019) 20:890–901. doi: 10.1038/s41590-019-0403-4
72. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA, et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discovery (2019) 9:1102–23. doi: 10.1158/2159-8290.CD-19-0094
73. Sade-Feldman M, Yizhak K, Bjorgaard SL, Ray JP, de Boer CG, Jenkins RW, et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell (2018) 175:998–1013.e20. doi: 10.1016/j.cell.2018.10.038
74. Miller BC, Sen DR, Abosy R, Al Bi K, Yamini V, Lafleur MW, et al. Subsets of exhausted CD8+ T cells differentially mediate tumor control and respond to checkpoint blockade. Nat Immunol (2019) 20:326–36. doi: 10.1038/s41590-019-0312-6
75. McLane LM, Abdel-Hakeen MS, Wherry EJ. CD8 T Cell Exhaustion During Chronic Viral Infection and Cancer. Annu Rev Immunol (2019) 37:457–95. doi: 10.1146/annurev-immunol-041015-055318
76. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature (2016) 537:417–21. doi: 10.1038/nature19330
77. Wang X, He Q, Shen H, Xia A, Tian W, Yu W, et al. TOX promotes the exhaustion of antitumor CD8+ T cells by preventing PD1 degradation in hepatocellular carcinoma. J Hepatol (2019) 71:731–41. doi: 10.1016/j.jhep.2019.05.015
78. Chen Z, Ji Z, Ngiow SF, Manne S, Cai Z, Huang AC, et al. TCF-1-Centered Transcriptional Network Drives an Effector versus Exhausted CD8 T Cell-Fate Decision. Immunity (2019) 51:840–855.e5. doi: 10.1016/j.immuni.2019.09.013
79. Page N, Klimek B, De Roo M, Steinbach K, Soldati H, Lemeille S, et al. Expression of the DNA-Binding Factor TOX Promotes the Encephalitogenic Potential of Microbe-Induced Autoreactive CD8+ T Cells. Immunity (2018) 48:937–950.e8. doi: 10.1016/j.immuni.2018.04.005
80. Sato T, Terai M, Tamura Y, Alexeev V, Mastrangelo MJ, Selvan SR. Interleukin 10 in the tumor microenvironment: A target for anticancer immunotherapy. Immunol Res (2011) 51:170–82. doi: 10.1007/s12026-011-8262-6
81. Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, et al. Induction and Transcriptional Regulation of the Co-inhibitory Gene Module in T Cell. Nature (2018) 558:454–9. doi: 10.1038/s41586-018-0206-z
82. Daley D, Mani VR, Mohan N, Akkad N, Ochi A, Heindel DW, et al. Dectin 1 activation on macrophages by galectin 9 promotes pancreatic carcinoma and peritumoral immune tolerance. Nat Med (2017) 23:556–67. doi: 10.1038/nm.4314
83. Ma Y, et al. Combination of PD1 Inhibitor and OX40 Agonist Induces Tumor Rejection and Immune Memory in Mouse Models of Pancreatic Cancer. Gastroenterology (2020), 1–14. doi: 10.1053/j.gastro.2020.03.018
84. Diskin B, Adam S, Cassini MF, Sanchez G, Liria M, Aykut B, et al. PD-L1 engagement on T cells promotes self-tolerance and suppression of neighboring macrophages and effector T cells in cancer. Nat Immunol (2020) 21:442–54. doi: 10.1038/s41590-020-0620-x
85. Barilla RM, Diskin B, Caso RC, Lee KB, Mohan N, Buttar C, et al. Specialized dendritic cells induce tumor-promoting IL-10 + IL-17 + FoxP3 neg regulatory CD4 + T cells in pancreatic carcinoma. Nat Commun (2019) 10. doi: 10.1038/s41467-019-09416-2
86. McAllister F, Bailey JM, Alsina J, Nirschl CJ, Sharma R, Fan H, et al. Oncogenic kras activates a hematopoietic-to-epithelial IL-17 signaling axis in preinvasive pancreatic neoplasia. Cancer Cell (2014) 25:621–37. doi: 10.1016/j.ccr.2014.03.014
87. Pushalkar S, Hundeyin M, Daley D, Zambirinis CP, Kurz E, Mishra A, et al. The pancreatic cancer microbiome promotes oncogenesis by induction of innate and adaptive immune suppression. Cancer Discovery (2018) 8:403–16. doi: 10.1158/2159-8290.CD-17-1134
88. Delitto D, Delitto AE, DiVita BB, Pham K, Han S, Hartlage ER, et al. Human pancreatic cancer cells induce a MyD88-dependent stromal response to promote a tumor-tolerant immune microenvironment. Cancer Res (2017) 77:672–83. doi: 10.1158/0008-5472.CAN-16-1765
89. Daley D, Mani VR, Mohan N, Akkad N, Balasubramania Pandian GSD, Savadkar S, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med (2017) 214:1711–24. doi: 10.1084/jem.20161707
90. Mirlekar B, Michaud D, Searcy R, Greene K, Pylayeva-Gupta Y. IL35 Hinders endogenous antitumor T-cell immunity and responsiveness to immunotherapy in pancreatic cancer. Cancer Immunol Res (2018) 6:1014–24. doi: 10.1158/2326-6066.CIR-17-0710
91. Mace TA, Shakya R, Pitarresi JR, Swanson B, McQuinn CW, Loftus S, et al. IL-6 and PD-L1 antibody blockade combination therapy reduces tumour progression in murine models of pancreatic cancer. Gut (2018) 67:320–32. doi: 10.1136/gutjnl-2016-311585
92. Karre K, Ljunggren HG, Piontek G, Kiessling R. Selective rejection of H–2-deficient lymphoma variants suggests alternative immune defence strategy. Nature (1986) 319:675–8. doi: 10.1038/319675a0
93. Lim SA, Kim J, Jeon S, Shin MH, Kwon J, Kim TJ, et al. Defective localization with impaired tumor cytotoxicity contributes to the immune escape of NK cells in pancreatic cancer patients. Front Immunol (2019) 10. doi: 10.3389/fimmu.2019.00496
94. Muthalagu N, Monteverde T, Raffo-Iraolagoitia X, Wiesheu R, Whyte D, Hedley A, et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discovery (2020) 10:872–87. doi: 10.1158/2159-8290.CD-19-0620
95. Lima M, Leander M, Santos M, Santos AH, Lau C, Queirós ML, et al. Chemokine Receptor Expression on Normal Blood CD56+ NK-Cells Elucidates Cell Partners That Comigrate during the Innate and Adaptive Immune Responses and Identifies a Transitional NK-Cell Population. J Immunol Res (2015) 2015. doi: 10.1155/2015/839684
96. Jun E, Song AY, Choi JW, Lee HH, Kim MY, Ko DH, et al. Progressive impairment of NK cell cytotoxic degranulation is associated with TGF-β1 deregulation and disease progression in pancreatic cancer. Front Immunol (2019) 10:1–14. doi: 10.3389/fimmu.2019.01354
97. Huang Q, Huang M, Meng F, Sun R. Activated pancreatic stellate cells inhibit NK cell function in the human pancreatic cancer microenvironment. Cell Mol Immunol (2019) 16:98–100. doi: 10.1038/s41423-018-0014-2
98. Lee J, Kang TH, Yoo W, Choi H, Jo S, Kong K, et al. An antibody designed to improve adoptive NK-cell therapy inhibits pancreatic cancer progression in a murine model. Cancer Immunol Res (2019) 7:219–29. doi: 10.1158/2326-6066.CIR-18-0317
99. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature (2015) 523:231–5. doi: 10.1038/nature14404
100. Zeng G, Germinaro M, Micsenyi A, Monga NK, Bell A, Sood A, et al. Aberrant Wnt/β-catenin signaling in pancreatic adenocarcinoma. Neoplasia (2006) 8:279–89. doi: 10.1593/neo.05607
101. Das S, Shapiro B, Vucic EA, Vogt S, Bar-Sagi D. Tumor cell-derived IL1β promotes desmoplasia and immune suppression in pancreatic cancer. Cancer Res (2020) 80:1088–101. doi: 10.1158/0008-5472.CAN-19-2080
102. Takahashi R, et al. Interleukin-1 β -induced pancreatitis promotes pancreatic ductal adenocarcinoma via B lymphocyte – mediated immune suppression. Gut (2020), 1–12. doi: 10.1136/gutjnl-2019-319912
103. Panni RZ, Herndon JM, Zuo C, Hegde S, Hogg GD, Knolhoff BL, et al. Agonism of CD11b reprograms innate immunity to sensitize pancreatic cancer to immunotherapies. Sci Transl Med (2019) 11. doi: 10.1126/scitranslmed.aau9240
104. Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R+ Macrophages Sustain Pancreatic Tumor Growth through T Cell Suppression and Maintenance of Key Gene Programs that Define the Squamous Subtype. Cell Rep (2018) 23:1448–60. doi: 10.1016/j.celrep.2018.03.131
105. Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 deficiency reveals a critical role for CD8α+ dendritic cells in cytotoxic T cell immunity. Science (2008) 322:1097–100. doi: 10.1126/science.1164206
106. Yamamoto K, Venida A, Yano J, Biancur DE, Kakiuchi M, Gupta S, et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature (2020) 1–6. doi: 10.1038/s41586-020-2229-5
107. Meyer MA, Baer JM, Knolhoff BL, Nywening TM, Panni RZ, Su X, et al. Breast and pancreatic cancer interrupt IRF8-dependent dendritic cell development to overcome immune surveillance. Nat Commun (2018) 9. doi: 10.1038/s41467-018-03600-6
108. Lin JH, Huffman AP, Wattenberg MM, Walter DM, Carpenter EL, Feldser DM, et al. Type 1 conventional dendritic cells are systemically dysregulated early in pancreatic carcinogenesis. JEM (2020) 217:1–20. doi: 10.1084/jem.20190673
109. Romero JM, Grünwald B, Jang GH, Bavi PP, Jhaveri A, Masoomian M, et al. A four-chemokine signature is associated with a T-cell- inflamed phenotype in primary and metastatic pancreatic cancer. Clin Cancer Res (2020) 26:1997–2010. doi: 10.1158/1078-0432.CCR-19-2803
110. Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T Cells Is Increased in Multiple Sclerosis and Suppressed by IFN-β Therapy. J Immunol (2015) 194:5085–93. doi: 10.4049/jimmunol.1403243
111. Brewitz A, Eickhoff S, Dähling S, Quast T, Bedoui S, Kroczek RA, et al. CD8+ T Cells Orchestrate pDC-XCR1+ Dendritic Cell Spatial and Functional Cooperativity to Optimize Priming. Immunity (2017) 46:205–19. doi: 10.1016/j.immuni.2017.01.003
112. Saito Y, Boddupalli CS, Borsotti C, Manz MG. Dendritic cell homeostasis is maintained by nonhematopoietic and T-cell-produced Flt3-ligand in steady state and during immune responses. Eur J Immunol (2013) 43:1651–8. doi: 10.1002/eji.201243163
113. Zhu Y, Herndon JM, Sojka DK, Kim KW, Knolhoff BL, Zuo C, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity (2017) 47:323–338.e6. doi: 10.1016/j.immuni.2017.07.014
114. Stromnes IM, Burrack AL, Hulbert A, Bonson P, Black C, Brockenbrough JS, et al. Differential effects of depleting versus programming tumor-associated macrophages on engineered T cells in pancreatic ductal adenocarcinoma. Cancer Immunol Res (2019) 7:977–89. doi: 10.1158/2326-6066.CIR-18-0448
115. Rahn S, Krüger S, Mennrich R, Goebel L, Wesch D, Oberg HH, et al. POLE Score: A comprehensive profiling of programmed death 1 ligand 1 expression in pancreatic ductal adenocarcinoma. Oncotarget (2019) 10:1572–88. doi: 10.18632/oncotarget.26705
116. Seifert AM, Reiche C, Heiduk M, Tannert A, Meinecke AC, Baier S, et al. Detection of pancreatic ductal adenocarcinoma with galectin-9 serum levels. Oncogene (2020) 39:3102–13. doi: 10.1038/s41388-020-1186-7
117. Ireland L, Luckett T, Schmid MC, Mielgo A. Blockade of Stromal Gas6 Alters Cancer Cell Plasticity, Activates NK Cells, and Inhibits Pancreatic Cancer Metastasis. Front Immunol (2020) 11:1–16. doi: 10.3389/fimmu.2020.00297
118. Quaranta V, Rainer C, Nielsen SR, Raymant ML, Ahmed MS, Engle DD, et al. Macrophage-derived granulin drives resistance to immune checkpoint inhibition in metastatic pancreatic cancer. Cancer Res (2018) 78:4253–69. doi: 10.1158/0008-5472.CAN-17-3876
119. Öhlund D, Handly-Santana A, Biffi G, Elyada E, Almeida AS, Ponz-Sarvise M, et al. Distinct populations of inflammatory fibroblasts and myofibroblasts in pancreatic cancer. J Exp Med (2017) 214:579–96. doi: 10.1084/jem.20162024
120. Biffi G, Oni TE, Spielman B, Hao Y, Elyada E, Park Y, et al. Il1-induced Jak/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discovery (2019) 9:282–301. doi: 10.1158/2159-8290.CD-18-0710
121. Lee JW, Stone ML, Porrett PM, Thomas SK, Komar CA, Li JH, et al. Hepatocytes direct the formation of a pro-metastatic niche in the liver. Nature (2019) 567:249–52. doi: 10.1038/s41586-019-1004-y
122. Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting Focal Adhesion Kinase Renders Pancreatic Cancers Responsive to Checkpoint Immunotherapy. Nat Med (2016) 22:851–60. doi: 10.1038/nm.4123
123. Ozdemir BC, Pentcheva-Hoang T, Carstens JL, Xiofeng Z, Wu C-C, Simpson T, et al. Depletion of Carcinoma-Associated Fibroblasts andFibrosis Induces Immunosuppression and Accelerates Pancreas Cancer with Reduced Survival. Cancer Cell (2014) 25:719–34. doi: 10.1016/j.ccr.2014.04.005
124. Rhim AD, Oberstein PE, Thomas DL, Mirek ET, Palermo CF, Sastra SA, et al. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell (2014) 25:735–47. doi: 10.1016/j.ccr.2014.04.021
125. Bayne LJ, Beatty GL, Jhala N, Clark CE, Rhim AD, Stanger BZ, et al. Tumor-Derived Granulocyte-Macrophage Colony-Stimulating Factor Regulates Myeloid Inflammation and T Cell Immunity in Pancreatic Cancer. Cancer Cell (2012) 21:822–35. doi: 10.1016/j.ccr.2012.04.025
126. Siret C, Collignon A, Silvy F, Robert S, Cheyrol T, Andrá P, et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front Immunol (2020) 10:1–16. doi: 10.3389/fimmu.2019.03070
127. Zhang Y, Lazarus J, Steele NG, Yan W, Lee HJ, Nwosu ZC, et al. Regulatory T-cell depletion alters the tumor microenvironment and accelerates pancreatic carcinogenesis. Cancer Discovery (2020) 10:422–39. doi: 10.1158/2159-8290.CD-19-0958
128. Pylayeva-Gupta Y, Lee KE, Hajdu CH, Miller G, Bar-Sagi D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell (2012) 21:836–47. doi: 10.1016/j.ccr.2012.04.024
129. Reyes CM, Teller S, Muckenhuber A, Konukiewitz B, Safak O, Weichert W, et al. Neoadjuvant therapy remodels the pancreatic cancer microenvironment via depletion of protumorigenic immune cells. Clin Cancer Res (2020) 26:220–31. doi: 10.1158/1078-0432.CCR-19-1864
130. Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. CXCR2 Inhibition Profoundly Suppresses Metastases and Augments Immunotherapy in Pancreatic Ductal Adenocarcinoma. Cancer Cell (2016) 29:832–45. doi: 10.1016/j.ccell.2016.04.014
131. Chao T, Furth EE, Vonderheide RH. CXCR2-dependent accumulation of tumor-associated neutrophils regulates T-cell immunity in pancreatic ductal adenocarcinoma. Cancer Immunol Res (2016) 4:968–82. doi: 10.1158/2326-6066.CIR-16-0188
132. Jang JE, Hajdu CH, Liot C, Miller G, Dustin ML, Bar-Sagi D. Crosstalk between Regulatory T Cells and Tumor-Associated Dendritic Cells Negates Anti-tumor Immunity in Pancreatic Cancer. Cell Rep (2017) 20:558–71. doi: 10.1016/j.celrep.2017.06.062
133. Principe DR, Park A, Dorman MJ, Kumar S, Viswakarma N, Rubin J, et al. TGFb blockade augments PD-1 inhibition to promote T-cell–mediated regression of pancreatic cancer. Mol Cancer Ther (2019) 18:613–20. doi: 10.1158/1535-7163.MCT-18-0850
134. Mirlekar B, Michaud D, Lee SJ, Kren NP, Harris C, Greene K, et al. B cell-Derived IL35 Drives STAT3-Dependent CD8+ T-cell Exclusion in Pancreatic Cancer. Cancer Immunol Res (2020) 8:292–308. doi: 10.1158/2326-6066.CIR-19-0349
135. Das S, Bar-Sagi D. BTK signaling drives CD1d hi CD5 + regulatory B-cell differentiation to promote pancreatic carcinogenesis. Oncogene (2019) 38:3316–24. doi: 10.1038/s41388-018-0668-3
136. Spear S, Candido JB, McDermott JR, Ghirelli C, Maniati E, Beers SA, et al. Discrepancies in the tumor microenvironment of spontaneous and orthotopic murine models of pancreatic cancer uncover a new immunostimulatory phenotype for B cells. Front Immunol (2019) 10:1–18. doi: 10.3389/fimmu.2019.00542
137. Garrido F, Festenstein H, Schirrmacher V. Further evidence for derepression of H-2 and Ia like specificities of foreign haplotypes in mouse tumor cell lines. Nature (1976) 261:228–30. doi: 10.1038/259228a0
138. Bryant KL, Stalnecker CA, Zeitouni D, Klomp JE, Peng S, Tikunov AP, et al. Combination of ERK and autophagy inhibition as a treatment approach for pancreatic cancer. Nat Med (2019) 25:628–40. doi: 10.1038/s41591-019-0368-8
139. Kinsey CG, Camolotto SA, Boespflug AM, Guillen KP, Foth M, Truong A, et al. Protective autophagy elicited by RAF→MEK→ERK inhibition suggests a treatment strategy for RAS-driven cancers. Nat Med (2019) 25:620–7. doi: 10.1038/s41591-019-0367-9
140. French RR, Chan HTC, Tutt AL, Glennie MJ. CD40 antibody evokes a cytotoxic T-cell response that eradicates lymphoma and bypasses T-cell help. Nat Med (1999) 5:548–53. doi: 10.1038/8426
141. Diehl L, Den Boer AT, Schoenberger SP, Van Der Voort EIH, Schumacher TNM, Melief CJM, et al. CD40 activation in vivo overcomes peptide-induced peripheral cytotoxic T-lymphocyte tolerance and augments anti-tumor vaccine efficacy. Nat Med (1999) 5:774–9. doi: 10.1038/10495
142. Sotomayor EM, Borrello I, Tubb E, Rattis FM, Bien H, Lu Z, et al. Conversion of tumor-specific CD4+ T-cell tolerance to T-cell priming through in vivo ligation of cd40. Nat Med (1999) 5:780–7. doi: 10.1038/10503
143. Huffman AP, Lin JH, Kim S II, Byrne KT, Vonderheide RH. CCL5 mediates CD40-driven CD4+ T-cell tumor infiltration and immunity. JCI Insight (2020). doi: 10.1172/jci.insight.137263
144. Moral JA, Leung J, Rojas LA, Ruan J, Zhao J, Sethna Z, et al. ILC2s amplify PD-1 blockade by activating tissue-specific cancer immunity. Nature (2020) 579:130–5. doi: 10.1038/s41586-020-2015-4
145. Khalil DN, Wolchok JD, Merghoub T. In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J Clin Invest (2019) 129:3435–47. doi: 10.1172/JCI128562
146. Baumann D, Hägele T, Mochayedi J, Drebant J, Vent C, Blobner S, et al. Proimmunogenic impact of MEK inhibition synergizes with agonist anti-CD40 immunostimulatory antibodies in tumor therapy. Nat Commun (2020) 11:1–18. doi: 10.1038/s41467-020-15979-2
147. Knudsen E, Kumarasamy V, Chung S, Ruiz A, Vail P, Tzetzo S, et al. Targeting dual signaling pathways in concert with immune checkpoints for the treatment of pancreatic cancer. Gut (2020) 0:1–12. doi: 10.1136/gutjnl-2020-321000
148. Kaneda MM, Cappello P, Nguyen AV, Ralainirina N, Hardamon CR, Foubert P, et al. Macrophage PI3Kγ drives pancreatic ductal adenocarcinoma progression. Cancer Discovery (2016) 6:870–85. doi: 10.1158/2159-8290.CD-15-1346
149. Stromnes IM, Schmitt TM, Chapuis AG, Hingorani SR, Greenberg PD. Re-adapting T cells for cancer therapy: from mouse models to clinical trials. Immunol Rev (2014) 257:145–64. doi: 10.1111/imr.12141
150. Tran E, Turcotte S, Gros A, Robbins PF, Lu Y, Dudley ME, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T cells in a Patient with Epithelial Cancer. Science (2014) 344:641–5. doi: 10.1126/science.1251102
151. Tran E, Robbins PF, Lu YC, Prickett TD, Gartner JJ, Jia L, et al. T-cell transfer therapy targeting mutant KRAS in cancer. N Engl J Med (2016) 375:2255–62. doi: 10.1056/NEJMoa1609279
152. Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: In vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci U.S.A. (2002) 99:16168–73. doi: 10.1073/pnas.242600099
153. Zacharakis N, Chinnasamy H, Black M, Xu H, Lu YC, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med (2018) 24:724–30. doi: 10.1038/s41591-018-0040-8
154. Johnson LA, Morgan RA, Dudley ME, Cassard L, Yang JC, Hughes MS, et al. Gene therapy with human and mouse T-cell receptors mediates cancer regression and targets normal tissues expressing cognate antigen. Blood (2009) 114:535–46. doi: 10.1182/blood-2009-03-211714
155. Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science (2006) 314:126–9. doi: 10.1126/science.1129003
156. Chapuis AG, Ragnarsson GB, Nguyen HN, Chaney CN, Pufnock JS, Schmitt TM, et al. Transferred WT1-reactive CD8+ T cells can mediate antileukemic activity and persist in post-transplant patients. Sci Transl Med (2013) 5. doi: 10.1126/scitranslmed.3004916
157. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
158. Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: Long-term follow-up and correlates with response. Clin Cancer Res (2015) 21:1019–27. doi: 10.1158/1078-0432.CCR-14-2708
159. Rapoport AP, Stadtmauer EA, Binder-Scholl GK, Goloubeva O, Vogl DT, Lacey SF, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med (2015) 21:914–21. doi: 10.1038/nm.3910
160. Kageyama S, Ikeda H, Miyahara Y, Imai N, Ishihara M, Saito K, et al. Adoptive transfer of MAGE-A4 T-cell receptor gene-transduced lymphocytes in patients with recurrent esophageal cancer. Clin Cancer Res (2015) 21:2268–77. doi: 10.1158/1078-0432.CCR-14-1559
161. Martinez M, Moon EK. CAR T cells for solid tumors: New strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol (2019) 10:1–21. doi: 10.3389/fimmu.2019.00128
162. Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell L, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med (2013) 5. doi: 10.1126/scitranslmed.3005930
163. Haas AR, Tanyi JL, O’Hara MH, Gladney WL, Lacey SF, Torigian DA, et al. Phase I Study of Lentiviral-Transduced Chimeric Antigen Receptor-Modified T Cells Recognizing Mesothelin in Advanced Solid Cancers. Mol Ther (2019) 27:1919–29. doi: 10.1016/j.ymthe.2019.07.015
164. Beatty GL, O’Hara MH, Lacey SF, Torigian DA, Nazimuddin F, Chen F, et al. Activity of Mesothelin-Specific Chimeric Antigen Receptor T Cells Against Pancreatic Carcinoma Metastases in a Phase 1 Trial. Gastroenterology (2018) 155:29–32. doi: 10.1053/j.gastro.2018.03.029
165. Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI insight (2018) 3. doi: 10.1172/jci.insight.99573
166. Ko AH, Jordan AC, Tooker E, Lacey SF, Chang RB, Li Y, et al. Dual targeting of mesothelin and CD19 with chimeric antigen receptor-modified T cells in patients with metastatic pancreatic ductal adenocarcinoma. Mol Ther (2020) 28:1–12. doi: 10.1016/j.ymthe.2020.07.017
167. Sukumaran S, Watanabe N, Bajgain P, Raja K, Mohammed S, Fisher WE, et al. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discovery (2018) 8:972–87. doi: 10.1158/2159-8290.CD-17-1298
168. Ahonen CL, Doxsee CL, McGurran SM, Riter TR, Wade WF, Barth RJ, et al. Combined TLR and CD40 Triggering Induces Potent CD8+ T Cell Expansion with Variable Dependence on Type I IFN. J Exp Med (2004) 199:775–84. doi: 10.1084/jem.20031591
169. Spiotto MT, Rowley DA, Schreiber H. Bystander elimination of antigen loss variants in established tumors. Nat Med (2004) 10:294–8. doi: 10.1038/nm999
170. Cruz FM, Colbert JD, Merino E, Kriegsman BA, Rock KL. The biology and underlying mechanisms of cross-presentation of exogenous antigens on MHC-I molecules. Annu Rev Immunol (2017) 35:149–76. doi: 10.1146/annurev-immunol-041015-055254
171. Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell (2017) 31:711–723.e4. doi: 10.1016/j.ccell.2017.04.003
172. Ferris ST, Carrero JA, Mohan JF, Calderon B, Murphy KM, Unanue ER, et al. A Minor Subset of Batf3-Dependent Antigen-Presenting Cells in Islets of Langerhans Is Essential for the Development of Autoimmune Diabetes. Immunity (2014) 41:657–69. doi: 10.1016/j.immuni.2014.09.012
173. Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell (2014) 26:638–52. doi: 10.1016/j.ccell.2014.09.007
174. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103+ /CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell (2016) 30:324–36. doi: 10.1016/j.ccell.2016.06.003
175. Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti- PD-1 therapy. Immunity (2019) 50:1498–512. doi: 10.1016/j.immuni.2019.04.010
176. Oba T, Long MD, Keler T, Marsh HC, Minderman H, Abrams SI, et al. Overcoming primary and acquired resistance to anti-PD-L1 therapy by induction and activation of tumor-residing cDC1s. Nat Commun (2020) 11. doi: 10.1038/s41467-020-19192-z
177. Lai J, Mardiana S, House IG, Sek K, Henderson MA, Giuffrida L, et al. Adoptive cellular therapy with T cells expressing the dendritic cell growth factor Flt3L drives epitope spreading and antitumor immunity. Nat Immunol (2020) 21:914–26. doi: 10.1038/s41590-020-0676-7
178. Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science (2018) 362. doi: 10.1126/science.aar3593
Keywords: pancreatic cancer, PD-1, PD-L1, pancreatic ductal adenocarcinoma, T cell, exhaustion, immunosuppression, immunotherapy
Citation: Schmiechen ZC and Stromnes IM (2021) Mechanisms Governing Immunotherapy Resistance in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 11:613815. doi: 10.3389/fimmu.2020.613815
Received: 03 October 2020; Accepted: 10 December 2020;
Published: 28 January 2021.
Edited by:
Bryon Johnson, Medical College of Wisconsin, United StatesReviewed by:
Robert Vonderheide, University of Pennsylvania, United StatesMichael Dwinell, Medical College of Wisconsin, United States
Copyright © 2021 Schmiechen and Stromnes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ingunn M. Stromnes, aW5ndW5uQHVtbi5lZHU=