 Seppo Meri
Seppo Meri Karita Haapasalo
Karita Haapasalo- 1Department of Bacteriology and Immunology, University of Helsinki, Helsinki, Finland
- 2Department of Bacteriology and Immunology, Translational Immunology Research Program, University of Helsinki, Helsinki, Finland
Complement-mediated inflammation or dysregulation in lipid metabolism are associated with the pathogenesis of several diseases. These include age-related macular degeneration (AMD), C3 glomerulonephritis (C3GN), dense deposit disease (DDD), atherosclerosis, and Alzheimer’s disease (AD). In all these diseases, formation of characteristic lipid-rich deposits is evident. Here, we will discuss molecular mechanisms whereby dysfunction of complement, and especially of its key regulator factor H, could be involved in lipid accumulation and related inflammation. The genetic associations to factor H polymorphisms, the role of factor H in the resolution of inflammation in lipid-rich deposits, modification of macrophage functions, and complement-mediated clearance of apoptotic and damaged cells indicate that the function of factor H is crucial in limiting inflammation in these diseases.
Introduction
A major function of the complement system is to handle invading microbes and clear debris, but without sufficient regulation it can attack and destroy our own cells and tissues. It can be activated through three pathways: the classical, alternative, and lectin pathways. The alternative pathway is constantly active in human plasma and responsible for amplifying all the complement activation cascades. To prevent potentially harmful complement attack toward host tissues the amplification pathway of complement is regulated by membrane inhibitors CD35, CD46, and CD55 and by soluble complement factor H and its alternatively spliced product factor H-like protein (FHL-1) (1). Factor H functions not only in the fluid phase to keep complement activation under control but also on surfaces to prevent attack against host targets. FHL-1 lacks the the ability to discriminate between self and nonself targets.
Factor H recognizes specific host markers directly, such as sialic acids and glycosaminoglycans, or indirectly via C-reactive protein (CRP) present or bound on self cell surfaces or apolipopoprotein E (apoE) on high-density lipoprotein particles (2–4). Binding to CRP is mediated via domains 6-8 and 19–20 of factor H, while domains 5–7 interact with apoE. Factor H usually binds to these structures in the context with surface-deposited C3b and acts as a cofactor for factor I-mediated inactivation of C3b to iC3b (1). From the 20 domains of factor H, the domains 7 and 19–20 mediate surface recognition, while domains 1-4 are required for regulatory activity (Figure 1). Several known mutations and polymorphisms in factor H and anti-factor H antibodies directed against these domains are associated with diseases that can be harmful for the carrier. This indicates that full function of factor H is essential in keeping the spontaneous alternative pathway activation in check and in preventing complement attack against self-structures (5–7).
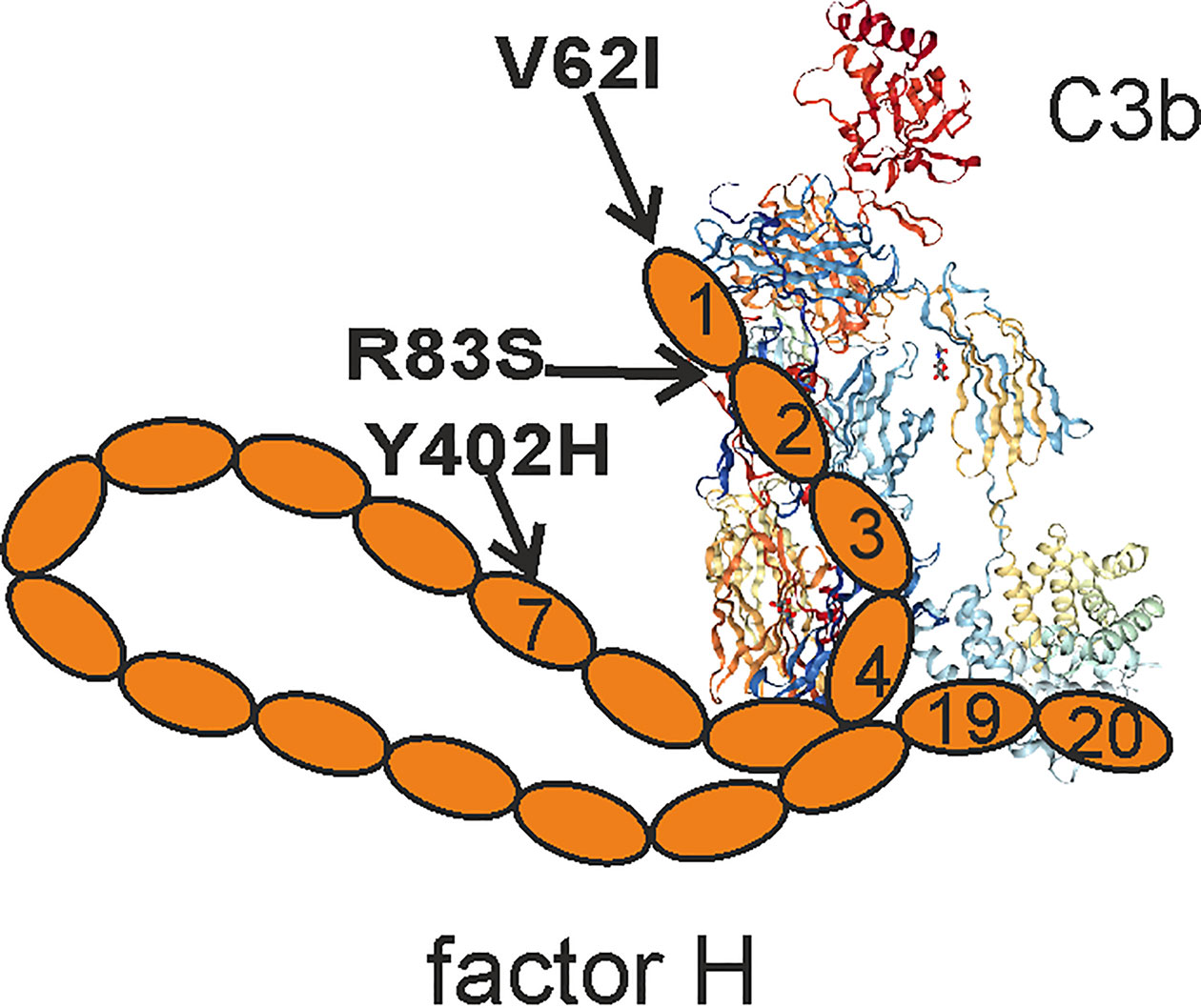
Figure 1 Schematic presentation of factor H domain structure. Factor H domains 1–4 bind to C3b and regulate alternative pathway activation, while domains 19–20 are responsible for surface recognition. The positions of the disease-causing mutations discussed in this manuscript are indicated. C3b crystal structure is from PDB 5FO7.
Due to its ability to bind CRP and control the alternative pathway complement factor H has a central role in the non-inflammatory clearance of extracellular deposits and dying cells in areas of tissue damage (8, 9). Much of the material to be cleared includes various types of phospholipids from cell membranes. Failure in this clearance mechanism may lead to excessive inflammation because of complement activation and overstimulation of macrophages. As a consequence, macrophages can release free radicals that can oxidize lipids and other materials and eventually harm the local tissue. The role of complement is not only restricted to clear microbes and dead cells from human tissues, but it also has an important role in lipid metabolism. For example, complement expression levels are significantly elevated in visceral adipose tissue, and increased expression levels of CFB gene have been suggested to associate positively with triglyceride levels and negatively with high-density lipoprotein (HDL) levels in plasmas of obese individuals. This indicates that complement system is involved in the metabolic consequences associated with increased visceral fat mass (10, 11) and in diseases characterized by the presence of lipid-rich deposits.
Diseases with Lipid-Rich Deposits and Factor H Association
Age-related-macular degeneration (AMD), C3 glomerulopathy (C3G) encompassing two different syndromes: C3 glomerulonephritis (C3GN) and dense deposit disease (DDD; previously called membranoproliferative glomerulonephritis type II; MPGN-II), atherosclerosis (AS) and Alzheimer’s disease (AD) have similar histopathological features. They all are associated with accumulation of lipid-rich deposits in the retina (AMD), subendothelially in kidney glomeruli (C3GN), glomerular basement membrane (DDD), arterial intima (AS), or brain (AD). These deposits are called drusen in AMD, dense deposits in DDD, plaques in atherosclerosis, and senile plaques in Alzheimer’s disease. Despite the different names the deposits share common features such as the presence of oxidized lipids and proteins, cholesterol and other lipids and apolipoproteins (12–14) (Figure 2). These diseases are also often affecting the same patient. As an example, individuals with DDD often develop ocular drusen in the macula, whose histopathology is indistinguishable from the drusen in AMD (15). Moreover, patients with atherosclerosis are at higher risk to develop Alzheimer’s disease (16) similarly as AMD patients are at higher risk to develop atherosclerosis (17). These findings indicate that the development of the lipid-rich deposits could share a common pathophysiological mechanism. For example, alterations in vascular glycosaminoglycans that bind the complement inhibitor factor H could lead to both lipid accumulation and complement damage in atherosclerotic lesions (18). Finally, these diseases have been studied for their genetic association with the factor H Y402H polymorphism that is located in the domain seven of this molecule. This polymorphism was first found to associate with AMD (19–21). It confers a five-fold increased risk of developing the disease (22). Interestingly, the AMD risk variant 402H also associates with DDD, and possibly also with Alzheimer’s disease and atherosclerosis, although conflicting results on the latter two have been found in different genetic studies (23–27).
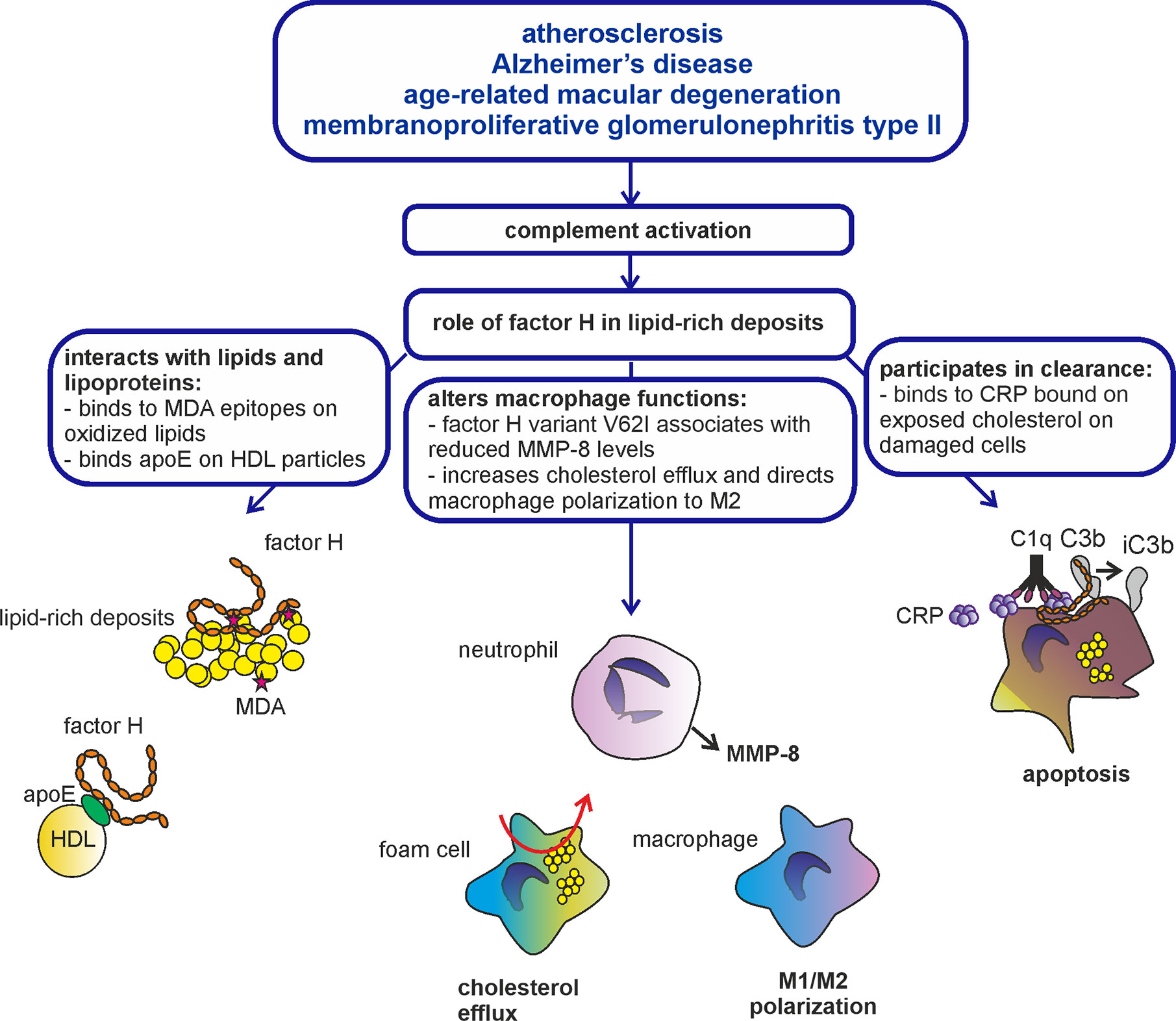
Figure 2 Role of factor H in lipid-rich deposits. Atherosclerosis, Alzheimer’d disease, AMD, and C3G are diseases characterized by formation of lipid-rich deposits and complement-mediated inflammation. Complement regulator factor H is known to bind modified lipids and lipoproteins, interact with macrophages and participate in the clearance of damaged and apoptotic cells through acting as a cofactor for inactivation of C3b to iC3b. Therefore, factor H is likely involved in the resolution of inflammation in lipid-rich deposits accumulated in the arteries, brain, eyes, and kidneys.
Age-Related Macular Degeneration
Age-related macular degeneration (AMD) is the most common cause of visual loss in the elderly people in industrialized countries. Accumulating evidence suggests that a defect in complement regulation by factor H and other abnormalities in the alternative pathway amplification process play a crucial role in the pathogenesis of the disease. The hallmark of early AMD is formation of drusen between the basal surface of the retinal pigmented epithelium (RPE) and Bruch’s membrane. This leads later to necrosis of the RPE cells. Apart from the the genetic background, aging and smoking are the main risk factors for developing AMD (28).
Atherosclerosis
Atherosclerosis is a multifactorial disease driven by inflammation and vascular changes. It is caused by accumulation of lipids, immune cells and fibrous elements in the subendothelium of arteries. The thickening and hardening of the arterial wall may lead to a total obstruction of the blood vessel because of plaque rupture. A major critical site is the coronary arteries of myocardium, where a local infarction may occur. Rupture of coronary artery plaques has been shown to be associated with complement activation in myocardial infarction (29). Similarly, atherosclerosis may lead to cerebral infarction or be involved in the rupture of carotid aneurysms (30). The hallmark of early atherosclerotic lesion is the formation of fatty streaks composed of cholesterol-laden macrophages, which are called foam-cells. The foam cells are formed through macrophage-mediated phagocytosis of oxidized and modified low density lipoproteins (LDLs) that accumulate in the subendothelium of arteries. The local inflammation induces differentiation of monocytes into proinflammatory M1 type macrophages that play a crucial role in the pathogenesis of atherosclerotic plaques (31). It seems like the complement system and especially the alternative pathway of complement has a pro-inflammatory role in in atherosclerosis as it serves as an amplification mechanism for C3b generation (32).
Alzheimer’s Disease
Alzheimer’s disease is characterized by accumulation of amyloid-β, formation of neuronal plaques and neuroinflammation in the brain. Impaired clearance of amyloid-β by microglial cells leads to accumulation of senile plaques in the brain. These can activate the complement system that triggers the development of an inflammatory phenotype in microglia, the phagocytic cells in brain (33). The activated microglial cells trigger astrocytes, which amplify the pro-inflammatory signals and neurotoxic effects (34). Activated microglia are the main source of complement components such as C1q in the brain (35). Interestingly, the classical pathway of complement with its components C1q and C4 appear to contribute to the complement- and microglia-mediated loss of brain synapses in early Alzheimer’s disease (36). Moreover, binding of C1q to apoE has been suggested to reduce C1q-mediated activation of the classical pathway implicating that apoE, the major factor related to Alzheimer’s disease, acts as a complement inhibitor (37). It has, however, been suggested that the alternative pathway of complement could be responsible for the pro-inflammatory effects in the pathogenesis of Alzheimer’s disease (38). Alzheimer’s disease, AMD and atherosclerosis are all degenerative age-dependent diseases, where complement-mediated inflammation most likely plays a crucial role, as several markers of complement activation have been found in the diseased tissues (39, 40).
C3 Glomerulopathy
C3 glomerulopathy (C3G) entails two different diseases, where complement activation, and especially the alternative pathway, has a central role in disease pathogenesis (41). In DDD, C3b deposits accumulate in the glomerular basement membranes and can be seen as dense deposits by electron microscopy. In C3 glomerulonephritis (C3GN), no dense deposits are seen, but C3b accumulation occurs subendothelially and mesangially. In a proportion of cases, C3GN is associated with immunoglobulin paraproteins (42). In DDD, autoantibodies have been detected against against the C3bBb convertase, so called C3 nephritic factors (43) and against the N-terminus of factor H (6). C3GN and DDD are rare chronic nephritic diseases (44). DDD is more common in children. The inflammation in C3G is mediated via hyperactivation of complement that is triggered because of complement dysregulation in the fluid phase. DDD sometimes occurs in association with partial lipodystrophy (PLD). It is characterized by loss of subcutaneous fat from the upper part of the body. It has been suggested that complement-mediated lysis of factor D expressing adipocytes is induced by C3 nephritic factor (C3Nef) in this disease (45). Moreover, in one study, a family with DDD and PLD was found to carry an R83S mutation in factor H in the interface region between domains one and two causing a defect in complement regulation. This indicated a role for alternative pathway dysregulation in the pathogenesis of this disease (46). Several complement components are expressed by adipose tissue including C2, C3, C4, C7, factor B, factor D (D, adipsin), FH, and adiponectin (10, 11, 47, 48). It has been suggested that complement is involved in mediating inflammation in the adipose tissue, but this is maintained only at a reasonable level due to expression of complement regulatory proteins, including factor H (49). Therefore, although not yet shown, a defect in factor H mediated complement regulation could also be involved in the loss of adipose tissue in DDD-related PLD.
Lipid-Rich Deposits
According to current knowledge, inflammation is the key driver in the formation of lipid-rich deposits in AMD, C3G, atherosclerosis, and Alzheimer’s disease. However, the initial trigger has not yet fully been defined except in rare cases, where carrying a mutation can be directly linked to the disease in the family (46). Conversely, lipid deposits may also undergo changes and modifications that convert them into promoters of inflammation. The particles may become proinflammatory themselves or in the context of their microenvironment.
Inflammatory Markers
Accumulation of complement components and inflammatory markers such as CRP around lipid-rich deposits indicate that the deposits have triggered inflammation (50–52). It is known, for example, that surface-exposed cholesterol binds CRP that activates the classical pathway of complement and triggers inflammation in the tissue (53). In atherosclerotic lesions CRP binds to phosphocholine in modified low-density lipoproteins (LDL) and colocalizes with LDL in human atherosclerotic lesions (54). In this context factor H plays a crucial role in limiting alternative complement activation through simultaneous binding to CRP and C3b (8). Inactivation of C3b to iC3b promotes noninflammatory clearance of certain lipid particles and dying cells through interaction with CR3 and CR4 receptors on macrophages (9).
In addition to CRP, there are other pentraxins that have been suggested to play a role in the pathogenesis of at least AMD, atherosclerosis, and Alzheimer’s disease. Pentraxin 3 (PTX3) is expressed by RPE cells and glial cells in vitro and may be involved in oxidative stress-mediated cell injury (55, 56). Moreover, PTX3 is a marker of disease severity in cardiovascular diseases and may exhibit atheroprotective effects (57, 58). Similarly to CRP, PTX3 opsonizes apoptotic and damaged cells and interacts with factor H (59). The involvement of PTX3 also in C3G is possible, because factor H-related protein 5 (FHR5), which is implicated in DDD and FHR5-related glomerulopathy, enhances complement activation by inhibiting binding of factor H to PTX3 (60).
Matrix metalloproteinase 8 (MMP-8) is a pro-inflammatory marker secreted by neutrophils. Elevated plasma and serum levels of MMP-8 associate with conditions such as peritonitis, rheumatoid arthritis and cardiovascular diseases. A recent study showed a significant association between the V62I polymorphism in domain one of factor H and neutrophil MMP-8 levels suggesting that this variation, with increased regulatory activity, could be involved in suppressing complement activation, MMP-8 expression, and inflammation in cardiovascular diseases (61, 62). Interestingly, this same factor H V62I variant is also associated with decreased susceptibility to AMD (63), further strengthening the hypothesis that the function of factor H is crucial in limiting inflammation in these diseases.
Danger-Associated Molecular Patterns
Danger-associated molecular patterns (DAMPs) indicate structures that are normally hidden but exposed during tissue injury. These can be recognized by multiple different types of receptors that often recognize also structures found on microbes. The receptors for DAMPs include Toll-like receptors (TLRs) and Nod-like receptors (NLRs) that can lead to activation of the inflammasome structures inside cells, for example in macrophages. Depending in the nature of interactions, the consequences can be inflammatory or anti-inflammatory. Also, the complement system can be activated on DAMPs or DAMP-like structures, which are generated by tissue injury, protein misfolding. or mislocalization. Such structures include, for example, mitochondria, membrane phospholipids, oxidized lipids in atherosclerosis, amyloid-β in brain, or oxidized bisretinoids on retinal pigment epithelial (RPE) cells in the macular area of the retina (64–66).
Polyunsaturated fatty acids are vulnerable to free radical attack caused by activated immune cells or cell apoptosis. As a consequence, oxidation-specific epitopes, such as oxidized phosphocholine, 4-hydroxynonenal, isolevuglandin, and malondialdehyde (MDA), are formed. The roles of oxidized lipids and MDA adducts in the pathogenesis of AMD, atherosclerosis and Alzheimer’s disease have been studied in more detail (67, 68). Interestingly, in AMD, factor H binds to both oxidized lipids and bisretinoids in drusen. Here, the common factor H variant 402Y has a higher affinity for oxidized lipids than the AMD risk allele 402H (69). Also the MDA-epitopes are recognized by factor H. Both the Y402H polymorphism in domain seven, and atypical hemolytic uremic syndrome (aHUS) associated mutations in domains 19–20 impair this interaction (70, 71). Therefore, the found association of factor H Y402H polymorphism with AMD, DDD and possibly also with atherosclerosis and Alzheimer’s disease could be related to the reduced binding of 402H to CRP and oxidized lipids compared to 402Y (19–21, 72). Reduced control of the amplification pathway of complement could thus contribute to the inflammatory pathology in these diseases.
Gangliosides are highly sialylated glycosphingolipids and abundantly expressed in the human body (73). Cells lacking terminal sialic acids, for example, due to oxidative damage, ischemic, senescent, necroptotic or apoptotic cell death become “DAMPs” for the complement system. Therefore, they serve as signals for complement activation and phagocytosis (2, 74). A distictive feature of the alternative pathway of complement is that its activators are structures that lack the ability to bind factor H (75). Thus, nearly any structure without suitable polyanions (sialic acids, glycosaminoglycans, and negatively charged phospholipids) or membrane complement regulators can act as complement activators as long as they provide binding sites for C3b molecules (76).
Because factor H interacts with cell surface sialic acids and surface deposited C3b, it has a crucial role in protecting self cells from complement attack. The presence of anti-ganglioside antibodies in atherosclerosis and Alzheimer’s disease patient sera has been previously observed (77, 78). It is therefore possible, although not shown, that these antibodies could interfere with factor H binding to sialic acids present on gangliosides and thereby contribute to inflammation. The antibodies could have a double-negative effect: They could activate the classical pathway and promote alternative pathway activation by preventing binding of factor H to cell surface gangliosides. A documented role for anti-ganglioside antibodies has been demonstrated in the neurological diseases Guillain-Barré and Miller Fisher syndromes [reviewed recently by Cutillo et al. (73)].
Apolipoprotein E
Apolipoprotein E (ApoE) is the central molecule responsible for cholesterol metabolism in the liver, blood and brain. ApoE has been shown to be involved in amyloid-β clearance in the central nervous system. It promotes anti-atherosclerotic activity by regulating lipoprotein metabolism and promoting cholesterol efflux by the so called reverse cholesterol transport. ApoE also modulates macrophage polarization into the anti-inflammatory M2 phenotype (79, 80). ApoE has three allelic isoforms (apoE2/E3/E4) of which apoE4 is strongly associated with Alzheimer’s disease (81). ApoE4 is also associated with atherosclerosis, nephrotic glomerular disease in children and AMD, of which the latter has the strongest association with FH polymorphism Y402H (26, 81–85).
ApoE is found abundantly in the lipid-rich deposits of AMD, DDD, atherosclerosis and Alzheimer’s disease patients (12, 86–88). However, its function in plaque formation or clearance is not well known. In vivo apoE is mainly associated with lipids, such as HDL particles in plasma or with small HDL-like components in brain. However, a small portion of apoE is found in complex with lipid-free or lipid-poor proteins. Of these, especially the apoE4 variant is likely susceptible for self-aggregation and misfolding (89). Factor H binds both lipid-free and high density lipoprotein (HDL) associated apoE via domains 5–7 (4). On HDL particles factor H regulates the alternative pathway of complement but its role in binding to lipid-free apoE is not known. The single amino acid difference in residues 112 and 158 between the apoE isoforms is responsible for the structural differences between these proteins (90). Moreover, knowing that FH interacts with apoE via the domain 7, where the Y402H polymorphism is located, it is possible that these genetic variations could also affect binding between apoE and FH.
Phagocytic Cells
Phagocytic cells are closely associated with disease progression in AMD, DDD, atherosclerosis, and Alzheimer’s disease. In atherosclerosis, macrophages play a crucial role in the phagocytosis of modified LDL particles and cholesterol efflux, while microglia are involved in amyloid-β phagocytosis. Increased intake of cholesterol by macrophages and reduced cholesterol efflux capacity leads to formation of foam cells in the arterial intima, while an increased intake of amyloid-β by microglial cells induces microglial activation and neurotoxicity (91). In microglia, the increase in free radical generation has been suggested to be related to the binding of amyloid-β to complement receptor type 3, CR3. CR3 is a phagocytic receptor that is involved in complement-mediated clearance of iC3b-coated particles and suppression of inflammation. It interacts with several different ligands, including factor H. Factor H interacts with CR3 on several cell types (92) and possibly has a direct effect on phagocytic functions. In cholesterol-loaded macrophages factor H has been shown to simultaneously promote cholesterol efflux, reduce transcription of proinflammatory genes and increase transcription of antiatherogenic genes such as ABCA1 and PPAR-α (93). PPAR-α transcription factor is known to induce the expression ABCA1, the intracellular ATP binding cassette transporter that regulates cellular cholesterol homeostasis (94). Binding of factor H to CR3 has been shown to reduce acute subretinal inflammation in mice indicating that factor H could be involved in suppressing inflammation in human AMD as well (95).
Necrotic and Apoptotic Cells
Formation of necrotic and apoptotic cells is involved in the pathogenesis of AMD, C3G, atherosclerosis and Alzheimer’s disease. As mentioned earlier, complement system is involved in the non-inflammatory clearance of apoptotic and necrotic cells that is initiated through recognition of CRP by C1q that activates the classical pathway of complement (96, 97). Here, binding of factor H to CRP is crucial, as it is involved in suppressing activation of the alternative and terminal pathways at the site of tissue damage and during local inflammation (8, 98, 99).
While CR3 is the receptor of iC3b, complement receptor type 1 (CR1 and CD35) is the receptor that has a higher affinity toward C3b (100, 101). CR1 is a cell surface complement regulator that acts as a cofactor for factor I in the inactivation of C3b (similarly to factor H) and further in the cleavage of iC3b to C3c and C3dg. In addition to phagocytic cells CR1 is also found on red blood cells, where it participates in the clearance of immune complexes by transporting them for elimination in the spleen or liver (102, 103). Factor H blocks binding of CR1 to C3b (104) as it competes for the same binding site with CR1 on C3b. This competitive binding leads to decreased C3c/C3dg formation as only CR1, but not factor H, acts as a cofactor for factor I in the cleavage of iC3b to C3c and C3dg. As C3d/C3dg is recognized by CR2 on B-cells and thus links the innate and adaptive immunity, it is possible that inhibition of C3d formation by factor H could have consequences for the adaptive immunity and formation of autoantibodies that has been described at least in atherosclerosis and C3G (46, 105–107).
Conclusions
AMD, C3G, atherosclerosis and Alzheimer’s disease seem to be unrelated to each other because of the different locations of the affected tissues. However, current knowledge on the role of complement-mediated inflammation in the development of these diseases indicates that they partially share common pathophysiology. Current genetic and biochemical data indicate that complement regulator factor H participates in the modification of both complement activation and cell responses and that defects in complement regulation by factor H play an important role in the pathogenesis of these diseases. More knowledge is, however, needed to understand the exact molecular mechanisms whereby factor H protects the eyes, the kidneys, the brain, and arteries from inflammation that eventually leads to formation of lipid-rich deposits in AMD, C3G, Alzheimer’s disease, and atherosclerosis.
Author Contributions
SM and KH have been involved with original research leading to concepts described in this work. They both wrote the review. All authors contributed to the article and approved the submitted version.
Funding
The study was supported by the Jane and Aatos Erkko Foundation (15032019), Finnish Foundation for Cardiovascular Research (08042019), Academy of Finland (331108), and Helsinki University Hospital Funds (VTR).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Walport MJ. Complement. First of two parts. N Engl J Med (2001) 344:1058–66. doi: 10.1056/NEJM200104053441406
2. Kajander T, Lehtinen MJ, Hyvarinen S, Bhattacharjee A, Leung E, Isenman DE, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc Natl Acad Sci USA (2011) 108:2897–902. doi: 10.1073/pnas.1017087108
3. Giannakis E, Jokiranta TS, Male DA, Ranganathan S, Ormsby RJ, Fischetti VA, et al. A common site within factor H SCR 7 responsible for binding heparin, C-reactive protein and streptococcal M protein. Eur J Immunol (2003) 33:962–9. doi: 10.1002/eji.200323541
4. Haapasalo K, van Kessel K, Nissila E, Metso J, Johansson T, Miettinen S, et al. Complement Factor H Binds to Human Serum Apolipoprotein E and Mediates Complement Regulation on High Density Lipoprotein Particles. J Biol Chem (2015) 290:28977–87. doi: 10.1074/jbc.M115.669226
5. Pangburn MK, Schreiber RD, Müller-Eberhard HJ. Formation of the initial C3 convertase of the alternative complement pathway. Acquisition of C3b-like activities by spontaneous hydrolysis of the putative thioester in native C3. J Exp Med (1981) 154:856–67. doi: 10.1084/jem.154.3.856
6. Meri S, Koistinen V, Miettinen A, Tornroth T, Seppala IJ. Activation of the alternative pathway of complement by monoclonal lambda light chains in membranoproliferative glomerulonephritis. J Exp Med (1992) 175:939–50. doi: 10.1084/jem.175.4.939
7. Jokiranta TS, Solomon A, Pangburn MK, Zipfel PF, Meri S. Nephritogenic lambda light chain dimer: a unique human miniautoantibody against complement factor H. J Immunol (1999) 163:4590–6. doi: 10.2210/pdb2loi/pdb
8. Jarva H, Jokiranta TS, Hellwage J, Zipfel PF, Meri S. Regulation of complement activation by C-reactive protein: targeting the complement inhibitory activity of factor H by an interaction with short consensus repeat domains 7 and 8-11. J Immunol (1999) 163:3957–62. doi: 10.4049/jimmunol.176.12.7612
9. Gershov D, Kim S, Brot N, Elkon KB. C-Reactive protein binds to apoptotic cells, protects the cells from assembly of the terminal complement components, and sustains an antiinflammatory innate immune response: implications for systemic autoimmunity. J Exp Med (2000) 192:1353–64. doi: 10.1084/jem.192.9.1353
10. Gabrielsson BG, Johansson JM, Lonn M, Jernas M, Olbers T, Peltonen M, et al. High expression of complement components in omental adipose tissue in obese men. Obes Res (2003) 11:699–708. doi: 10.1038/oby.2003.100
11. Moreno-Navarrete JM, Martinez-Barricarte R, Catalan V, Sabater M, Gomez-Ambrosi J, Ortega FJ, et al. Complement factor H is expressed in adipose tissue in association with insulin resistance. Diabetes (2010) 59:200–9. doi: 10.2337/db09-0700
12. Sethi S, Gamez JD, Vrana JA, Theis JD, Bergen HR 3rd, Zipfel PF, et al. Glomeruli of Dense Deposit Disease contain components of the alternative and terminal complement pathway. Kidney Int (2009) 75:952–60. doi: 10.1038/ki.2008.657
13. Johnson LV, Forest DL, Banna CD, Radeke CM, Maloney MA, Hu J, et al. Cell culture model that mimics drusen formation and triggers complement activation associated with age-related macular degeneration. Proc Natl Acad Sci USA (2011) 108:18277–82. doi: 10.1073/pnas.1109703108
14. Kiskis J, Fink H, Nyberg L, Thyr J, Li JY, Enejder A. Plaque-associated lipids in Alzheimer’s diseased brain tissue visualized by nonlinear microscopy. Sci Rep (2015) 5:13489. doi: 10.1038/srep13489
15. Mullins RF, Aptsiauri N, Hageman GS. Structure and composition of drusen associated with glomerulonephritis: implications for the role of complement activation in drusen biogenesis. Eye (Lond) (2001) 15:390–5. doi: 10.1038/eye.2001.142
16. Hofman A, Ott A, Breteler MM, Bots ML, Slooter AJ, van Harskamp F, et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet (1997) 349:151–4. doi: 10.1016/S0140-6736(96)09328-2
17. Vingerling JR, Dielemans I, Bots ML, Hofman A, Grobbee DE, de Jong PT. Age-related macular degeneration is associated with atherosclerosis. The Rotterdam Study. Am J Epidemiol (1995) 142:404–9. doi: 10.1093/oxfordjournals.aje.a117648
18. Oksjoki R, Jarva H, Kovanen PT, Laine P, Meri S, Pentikainen MO. Association between complement factor H and proteoglycans in early human coronary atherosclerotic lesions: implications for local regulation of complement activation. Arterioscler Thromb Vasc Biol (2003) 23:630–6. doi: 10.1161/01.ATV.0000057808.91263.A4
19. Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, et al. Complement factor H polymorphism in age-related macular degeneration. Science (2005) 308:385–9. doi: 10.1126/science.1109557
20. Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA, et al. Complement factor H polymorphism and age-related macular degeneration. Science (2005) 308:421–4. doi: 10.1126/science.1110189
21. Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, et al. Complement factor H variant increases the risk of age-related macular degeneration. Science (2005) 308:419–21. doi: 10.1126/science.1110359
22. Schramm EC, Clark SJ, Triebwasser MP, Raychaudhuri S, Seddon J, Atkinson JP. Genetic variants in the complement system predisposing to age-related macular degeneration: a review. Mol Immunol (2014) 61:118–25. doi: 10.1016/j.molimm.2014.06.032
23. Volcik KA, Ballantyne CM, Braun MC, Coresh J, Mosley TH, Boerwinkle E. Association of the complement factor H Y402H polymorphism with cardiovascular disease is dependent upon hypertension status: The ARIC study. Am J Hypertens (2008) 21:533–8. doi: 10.1038/ajh.2007.81
24. Abrera-Abeleda MA, Nishimura C, Smith JL, Sethi S, McRae JL, Murphy BF, et al. Variations in the complement regulatory genes factor H (CFH) and factor H related 5 (CFHR5) are associated with membranoproliferative glomerulonephritis type II (dense deposit disease). J Med Genet (2006) 43:582–9. doi: 10.1136/jmg.2005.038315
25. Sofat R, Casas JP, Kumari M, Talmud PJ, Ireland H, Kivimaki M, et al. Genetic variation in complement factor H and risk of coronary heart disease: eight new studies and a meta-analysis of around 48,000 individuals. Atherosclerosis (2010) 213:184–90. doi: 10.1016/j.atherosclerosis.2010.07.021
26. Zetterberg M, Landgren S, Andersson ME, Palmer MS, Gustafson DR, Skoog I, et al. Association of complement factor H Y402H gene polymorphism with Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet (2008) 147B:720–6. doi: 10.1002/ajmg.b.30668
27. Le Fur I, Laumet G, Richard F, Fievet N, Berr C, Rouaud O, et al. Association study of the CFH Y402H polymorphism with Alzheimer’s disease. Neurobiol Aging (2010) 31:165–6. doi: 10.1016/j.neurobiolaging.2008.03.003
28. Xu Q, Cao S, Rajapakse S, Matsubara JA. Understanding AMD by analogy: systematic review of lipid-related common pathogenic mechanisms in AMD, AD, AS and GN. Lipids Health Dis (2018) 17:3. doi: 10.1186/s12944-017-0647-7
29. Laine P, Pentikainen MO, Wurzner R, Penttila A, Paavonen T, Meri S, et al. Evidence for complement activation in ruptured coronary plaques in acute myocardial infarction. Am J Cardiol (2002) 90:404–8. doi: 10.1016/S0002-9149(02)02498-0
30. Tulamo R, Frosen J, Junnikkala S, Paetau A, Pitkaniemi J, Kangasniemi M, et al. Complement activation associates with saccular cerebral artery aneurysm wall degeneration and rupture. Neurosurgery (2006) 59:1069–76; discussion 1076-7. doi: 10.1227/01.NEU.0000245598.84698.26
31. Hansson GK, Robertson AK, Soderberg-Naucler C. Inflammation and atherosclerosis. Annu Rev Pathol (2006) 1:297–329. doi: 10.1146/annurev.pathol.1.110304.100100
32. Malik TH, Cortini A, Carassiti D, Boyle JJ, Haskard DO, Botto M. The alternative pathway is critical for pathogenic complement activation in endotoxin- and diet-induced atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation (2010) 122:1948–56. doi: 10.1161/CIRCULATIONAHA.110.981365
33. Shen Y, Meri S. Yin and Yang: complement activation and regulation in Alzheimer’s disease. Prog Neurobiol (2003) 70:463–72. doi: 10.1016/j.pneurobio.2003.08.001
34. Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature (2017) 541:481–7. doi: 10.1038/nature21029
35. Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, et al. Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J Neuroinflam (2017) 14:48. doi: 10.1186/s12974-017-0814-9
36. Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science (2016) 352:712–6. doi: 10.1126/science.aad8373
37. Yin C, Ackermann S, Ma Z, Mohanta SK, Zhang C, Li Y, et al. ApoE attenuates unresolvable inflammation by complex formation with activated C1q. Nat Med (2019) 25:496–506. doi: 10.1038/s41591-018-0336-8
38. Fonseca MI, Chu SH, Berci AM, Benoit ME, Peters DG, Kimura Y, et al. Contribution of complement activation pathways to neuropathology differs among mouse models of Alzheimer’s disease. J Neuroinflam (2011) 8:4. doi: 10.1186/1742-2094-8-4
39. Vlaicu SI, Tatomir A, Rus V, Mekala AP, Mircea PA, Niculescu F, et al. The role of complement activation in atherogenesis: the first 40 years. Immunol Res (2016) 64:1–13. doi: 10.1007/s12026-015-8669-6
40. Zanjani H, Finch CE, Kemper C, Atkinson J, McKeel D, Morris JC, et al. Complement activation in very early Alzheimer disease. Alzheimer Dis Assoc Disord (2005) 19:55–66. doi: 10.1097/01.wad.0000165506.60370.94
41. Pickering MC, D’Agati VD, Nester CM, Smith RJ, Haas M, Appel GB, et al. C3 glomerulopathy: consensus report. Kidney Int (2013) 84:1079–89. doi: 10.1038/ki.2013.377
42. Sethi S, Rajkumar SV. Monoclonal gammopathy-associated proliferative glomerulonephritis. Mayo Clin Proc (2013) 88:1284–93. doi: 10.1016/j.mayocp.2013.08.002
43. Daha MR, Austen KF, Fearon DT. The incorporation of C3 nephritic factor (C3NeF) into a stabilized C3 convertase, C3bBb(C3NeF), and its release after decay of convertase function. J Immunol (1977) 119:812–7.
44. Smith RJH, Appel GB, Blom AM, Cook HT, D’Agati VD, Fakhouri F, et al. C3 glomerulopathy - understanding a rare complement-driven renal disease. Nat Rev Nephrol (2019) 15:129–43. doi: 10.1038/s41581-018-0107-2
45. Mathieson PW, Wurzner R, Oliveria DB, Lachmann PJ, Peters DK. Complement-mediated adipocyte lysis by nephritic factor sera. J Exp Med (1993) 177:1827–31. doi: 10.1084/jem.177.6.1827
46. Wong EK, Anderson HE, Herbert AP, Challis RC, Brown P, Reis GS, et al. Characterization of a factor H mutation that perturbs the alternative pathway of complement in a family with membranoproliferative GN. J Am Soc Nephrol (2014) 25:2425–33. doi: 10.1681/ASN.2013070732
47. Choy LN, Rosen BS, Spiegelman BM. Adipsin and an endogenous pathway of complement from adipose cells. J Biol Chem (1992) 267:12736–41.
48. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem (1995) 270:26746–9. doi: 10.1074/jbc.270.45.26746
49. Kaye S, Lokki AI, Hanttu A, Nissila E, Heinonen S, Hakkarainen A, et al. Upregulation of Early and Downregulation of Terminal Pathway Complement Genes in Subcutaneous Adipose Tissue and Adipocytes in Acquired Obesity. Front Immunol (2017) 8:545. doi: 10.3389/fimmu.2017.00545
50. McGeer EG, Yasojima K, Schwab C, McGeer PL. The pentraxins: possible role in Alzheimer’s disease and other innate inflammatory diseases. Neurobiol Aging (2001) 22:843–8. doi: 10.1016/S0197-4580(01)00288-3
51. Montecucco F, Mach F. New evidences for C-reactive protein (CRP) deposits in the arterial intima as a cardiovascular risk factor. Clin Interv Aging (2008) 3:341–9. doi: 10.2147/CIA.S2706
52. Molins B, Romero-Vazquez S, Fuentes-Prior P, Adan A, Dick AD. C-Reactive Protein as a Therapeutic Target in Age-Related Macular Degeneration. Front Immunol (2018) 9:808. doi: 10.3389/fimmu.2018.00808
53. Taskinen S, Hyvonen M, Kovanen PT, Meri S, Pentikainen MO. C-reactive protein binds to the 3beta-OH group of cholesterol in LDL particles. Biochem Biophys Res Commun (2005) 329:1208–16. doi: 10.1016/j.bbrc.2005.02.091
54. Chang MK, Binder CJ, Torzewski M, Witztum JL. C-reactive protein binds to both oxidized LDL and apoptotic cells through recognition of a common ligand: Phosphorylcholine of oxidized phospholipids. Proc Natl Acad Sci USA (2002) 99:13043–8. doi: 10.1073/pnas.192399699
55. Hwang N, Kwon MY, Woo JM, Chung SW. Oxidative Stress-Induced Pentraxin 3 Expression Human Retinal Pigment Epithelial Cells is Involved in the Pathogenesis of Age-Related Macular Degeneration. Int J Mol Sci (2019) 20:1–12. doi: 10.3390/ijms20236028
56. Jeon H, Lee S, Lee WH, Suk K. Analysis of glial secretome: the long pentraxin PTX3 modulates phagocytic activity of microglia. J Neuroimmunol (2010) 229:63–72. doi: 10.1016/j.jneuroim.2010.07.001
57. Norata GD, Marchesi P, Pulakazhi Venu VK, Pasqualini F, Anselmo A, Moalli F, et al. Deficiency of the long pentraxin PTX3 promotes vascular inflammation and atherosclerosis. Circulation (2009) 120:699–708. doi: 10.1161/CIRCULATIONAHA.108.806547
58. Deban L, Jaillon S, Garlanda C, Bottazzi B, Mantovani A. Pentraxins in innate immunity: lessons from PTX3. Cell Tissue Res (2011) 343:237–49. doi: 10.1007/s00441-010-1018-0
59. Kopp A, Strobel S, Tortajada A, Rodriguez de Cordoba S, Sanchez-Corral P, Prohaszka Z, et al. Atypical hemolytic uremic syndrome-associated variants and autoantibodies impair binding of factor h and factor h-related protein 1 to pentraxin 3. J Immunol (2012) 189:1858–67. doi: 10.4049/jimmunol.1200357
60. Csincsi AI, Kopp A, Zoldi M, Banlaki Z, Uzonyi B, Hebecker M, et al. Factor H-related protein 5 interacts with pentraxin 3 and the extracellular matrix and modulates complement activation. J Immunol (2015) 194:4963–73. doi: 10.4049/jimmunol.1403121
61. Reiner AP, Hartiala J, Zeller T, Bis JC, Dupuis J, Fornage M, et al. Genome-wide and gene-centric analyses of circulating myeloperoxidase levels in the charge and care consortia. Hum Mol Genet (2013) 22:3381–93. doi: 10.1093/hmg/ddt189
62. Salminen A, Vlachopoulou E, Havulinna AS, Tervahartiala T, Sattler W, Lokki ML, et al. Genetic Variants Contributing to Circulating Matrix Metalloproteinase 8 Levels and Their Association With Cardiovascular Diseases: A Genome-Wide Analysis. Circ Cardiovasc Genet (2017) 10. doi: 10.1161/CIRCGENETICS.117.001731
63. Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci USA (2005) 102:7227–32. doi: 10.1073/pnas.0501536102
64. Radu RA, Hu J, Jiang Z, Bok D. Bisretinoid-mediated complement activation on retinal pigment epithelial cells is dependent on complement factor H haplotype. J Biol Chem (2014) 289:9113–20. doi: 10.1074/jbc.M114.548669
65. Eikelenboom P, Hack CE, Rozemuller JM, Stam FC. Complement activation in amyloid plaques in Alzheimer’s dementia. Virchows Arch B Cell Pathol Incl Mol Pathol (1989) 56:259–62. doi: 10.1007/BF02890024
66. Berliner JA, Subbanagounder G, Leitinger N, Watson AD, Vora D. Evidence for a role of phospholipid oxidation products in atherogenesis. Trends Cardiovasc Med (2001) 11:142–7. doi: 10.1016/S1050-1738(01)00098-6
67. Sultana R, Perluigi M, Butterfield DA. Lipid peroxidation triggers neurodegeneration: a redox proteomics view into the Alzheimer disease brain. Free Radic Biol Med (2013) 62:157–69. doi: 10.1016/j.freeradbiomed.2012.09.027
68. Holvoet P, Perez G, Zhao Z, Brouwers E, Bernar H, Collen D. Malondialdehyde-modified low density lipoproteins in patients with atherosclerotic disease. J Clin Invest (1995) 95:2611–9. doi: 10.1172/JCI117963
69. Shaw PX, Zhang L, Zhang M, Du H, Zhao L, Lee C, et al. Complement factor H genotypes impact risk of age-related macular degeneration by interaction with oxidized phospholipids. Proc Natl Acad Sci USA (2012) 109:13757–62. doi: 10.1073/pnas.1121309109
70. Weismann D, Hartvigsen K, Lauer N, Bennett KL, Scholl HP, Charbel Issa P, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature (2011) 478:76–81. doi: 10.1038/nature10449
71. Hyvarinen S, Uchida K, Varjosalo M, Jokela R, Jokiranta TS. Recognition of malondialdehyde-modified proteins by the C terminus of complement factor H is mediated via the polyanion binding site and impaired by mutations found in atypical hemolytic uremic syndrome. J Biol Chem (2014) 289:4295–306. doi: 10.1074/jbc.M113.527416
72. Laine M, Jarva H, Seitsonen S, Haapasalo K, Lehtinen MJ, Lindeman N, et al. Y402H polymorphism of complement factor H affects binding affinity to C-reactive protein. J Immunol (2007) 178:3831–6. doi: 10.4049/jimmunol.178.6.3831
73. Cutillo G, Saariaho AH, Meri S. Physiology of gangliosides and the role of antiganglioside antibodies in human diseases. Cell Mol Immunol (2020) 17:313–22. doi: 10.1038/s41423-020-0388-9
74. Meesmann HM, Fehr EM, Kierschke S, Herrmann M, Bilyy R, Heyder P, et al. Decrease of sialic acid residues as an eat-me signal on the surface of apoptotic lymphocytes. J Cell Sci (2010) 123:3347–56. doi: 10.1242/jcs.066696
75. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA (1990) 87:3982–6. doi: 10.1073/pnas.87.10.3982
76. Meri S. Self-nonself discrimination by the complement system. FEBS Lett (2016) 590:2418–34. doi: 10.1002/1873-3468.12284
77. Koutsouraki E, Hatzifilippou E, Michmizos D, Banaki T, Costa V, Baloyannis S. The probable auto-antigenic role of lipids (anti-ganglioside antibodies) in the pathogenesis of Alzheimer’s disease. J Alzheimers Dis (2014) 42 Suppl 3:S163–6. doi: 10.3233/JAD-132633
78. Golovanova NK, Gracheva EV, Basharova LA, Kozlov SG, Lyakishev AA, Prokazova NV, et al. Autoantibodies to gangliosides in sera of atherosclerotic patients. Clin Chim Acta (1998) 272:197–207. doi: 10.1016/S0009-8981(98)00013-8
79. Terwel D, Steffensen KR, Verghese PB, Kummer MP, Gustafsson JA, Holtzman DM, et al. Critical role of astroglial apolipoprotein E and liver X receptor-alpha expression for microglial Abeta phagocytosis. J Neurosci (2011) 31:7049–59. doi: 10.1523/JNEUROSCI.6546-10.2011
80. Braesch-Andersen S, Paulie S, Smedman C, Mia S, Kumagai-Braesch M. ApoE production in human monocytes and its regulation by inflammatory cytokines. PloS One (2013) 8:e79908. doi: 10.1371/journal.pone.0079908
81. Saunders AM, Strittmatter WJ, Schmechel D, George-Hyslop PH, Pericak-Vance MA, Joo SH, et al. Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology (1993) 43:1467–72. doi: 10.1212/WNL.43.8.1467
82. Fritsche LG, Chen W, Schu M, Yaspan BL, Yu Y, Thorleifsson G, et al. Seven new loci associated with age-related macular degeneration. Nat Genet (2013) 45:433–9439e1–2. doi: 10.1038/ng.2578
83. Klaver CC, Kliffen M, van Duijn CM, Hofman A, Cruts M, Grobbee DE, et al. Genetic association of apolipoprotein E with age-related macular degeneration. Am J Hum Genet (1998) 63:200–6. doi: 10.1086/301901
84. Asami T, Ciomartan T, Hayakawa H, Uchiyama M, Tomisawa S. Apolipoprotein E epsilon 4 allele and nephrotic glomerular diseases in children. Pediatr Nephrol (1999) 13:233–6. doi: 10.1007/s004670050599
85. Marais AD. Apolipoprotein E in lipoprotein metabolism, health and cardiovascular disease. Pathology (2019) 51:165–76. doi: 10.1016/j.pathol.2018.11.002
86. Malek G, Li CM, Guidry C, Medeiros NE, Curcio CA. Apolipoprotein B in cholesterol-containing drusen and basal deposits of human eyes with age-related maculopathy. Am J Pathol (2003) 162:413–25. doi: 10.1016/S0002-9440(10)63836-9
87. O’Brien KD, Deeb SS, Ferguson M, McDonald TO, Allen MD, Alpers CE, et al. Apolipoprotein E localization in human coronary atherosclerotic plaques by in situ hybridization and immunohistochemistry and comparison with lipoprotein lipase. Am J Pathol (1994) 144:538–48.
88. Shao Y, Gearing M, Mirra SS. Astrocyte-apolipoprotein E associations in senile plaques in Alzheimer disease and vascular lesions: a regional immunohistochemical study. J Neuropathol Exp Neurol (1997) 56:376–81. doi: 10.1097/00005072-199704000-00006
89. Hatters DM, Zhong N, Rutenber E, Weisgraber KH. Amino-terminal domain stability mediates apolipoprotein E aggregation into neurotoxic fibrils. J Mol Biol (2006) 361:932–44. doi: 10.1016/j.jmb.2006.06.080
90. Frieden C, Garai K. Structural differences between apoE3 and apoE4 may be useful in developing therapeutic agents for Alzheimer’s disease. Proc Natl Acad Sci USA (2012) 109:8913–8. doi: 10.1073/pnas.1207022109
91. Zhang D, Hu X, Qian L, Chen SH, Zhou H, Wilson B, et al. Microglial MAC1 receptor and PI3K are essential in mediating beta-amyloid peptide-induced microglial activation and subsequent neurotoxicity. J Neuroinflam (2011) 8:3. doi: 10.1186/1742-2094-8-3
92. Avery VM, Gordon DL. Characterization of factor H binding to human polymorphonuclear leukocytes. J Immunol (1993) 151:5545–53.
93. Nissila E, Hakala P, Leskinen K, Roig A, Syed S, Van Kessel KPM, et al. Complement Factor H and Apolipoprotein E Participate in Regulation of Inflammation in THP-1 Macrophages. Front Immunol (2018) 9:2701. doi: 10.3389/fimmu.2018.02701
94. Chinetti G, Lestavel S, Bocher V, Remaley AT, Neve B, Torra IP, et al. PPAR-alpha and PPAR-gamma activators induce cholesterol removal from human macrophage foam cells through stimulation of the ABCA1 pathway. Nat Med (2001) 7:53–8. doi: 10.1038/83348
95. Calippe B, Augustin S, Beguier F, Charles-Messance H, Poupel L, Conart JB, et al. Complement Factor H Inhibits CD47-Mediated Resolution of Inflammation. Immunity (2017) 46:261–72. doi: 10.1016/j.immuni.2017.01.006
96. Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol (2010) 28:157–83. doi: 10.1146/annurev-immunol-030409-101305
97. Volanakis JE, Narkates AJ. Interaction of C-reactive protein with artificial phosphatidylcholine bilayers and complement. J Immunol (1981) 126:1820–5.
98. Mihlan M, Stippa S, Jozsi M, Zipfel PF. Monomeric CRP contributes to complement control in fluid phase and on cellular surfaces and increases phagocytosis by recruiting factor H. Cell Death Differ (2009) 16:1630–40. doi: 10.1038/cdd.2009.103
99. Haapasalo K, Meri S. Regulation of the Complement System by Pentraxins. Front Immunol (2019) 10:1–11. doi: 10.3389/fimmu.2019.01750
100. Ross GD, Jarowski CI, Rabellino EM, Winchester RJ. The sequential appearance of Ia-like antigens and two different complement receptors during the maturation of human neutrophils. J Exp Med (1978) 147:730–44. doi: 10.1084/jem.147.3.730
101. Todd RF3. The continuing saga of complement receptor type 3 (CR3). J Clin Invest (1996) 98:1–2. doi: 10.1172/JCI118752
102. Schifferli JA, Ng YC, Peters DK. The role of complement and its receptor in the elimination of immune complexes. N Engl J Med (1986) 315:488–95. doi: 10.1056/NEJM198608213150805
103. Fearon DT. Regulation of the amplification C3 convertase of human complement by an inhibitory protein isolated from human erythrocyte membrane. Proc Natl Acad Sci USA (1979) 76:5867–71. doi: 10.1073/pnas.76.11.5867
104. Amdahl H, Haapasalo K, Tan L, Meri T, Kuusela PI, van Strijp JA, et al. Staphylococcal protein Ecb impairs complement receptor-1 mediated recognition of opsonized bacteria. PloS One (2017) 12:e0172675. doi: 10.1371/journal.pone.0172675
105. Dempsey PW, Allison ME, Akkaraju S, Goodnow CC, Fearon DT. C3d of complement as a molecular adjuvant: bridging innate and acquired immunity. Science (1996) 271:348–50. doi: 10.1126/science.271.5247.348
106. Fearon DT, Locksley RM. The instructive role of innate immunity in the acquired immune response. Science (1996) 272:50–3. doi: 10.1126/science.272.5258.50
Keywords: apoE, C-reactive protein, adiponectin, HDL, amyloid-beta- protein
Citation: Meri S and Haapasalo K (2020) Function and Dysfunction of Complement Factor H During Formation of Lipid-Rich Deposits. Front. Immunol. 11:611830. doi: 10.3389/fimmu.2020.611830
Received: 29 September 2020; Accepted: 09 November 2020;
Published: 08 December 2020.
Edited by:
Robert Braidwood Sim, University of Oxford, United KingdomReviewed by:
Deborah Fraser, California State University, Long Beach, United StatesPéter Gál, Hungarian Academy of Sciences (MTA), Hungary
Copyright © 2020 Meri and Haapasalo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karita Haapasalo, a2FyaXRhLmhhYXBhc2Fsb0BoZWxzaW5raS5maQ==