
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol. , 24 February 2021
Sec. Immunological Tolerance and Regulation
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.611638
This article is part of the Research Topic Emerging Therapeutics for Immune Tolerance View all 24 articles
 Leila Amini1†
Leila Amini1† Jenny Greig2†
Jenny Greig2† Michael Schmueck-Henneresse1
Michael Schmueck-Henneresse1 Hans-Dieter Volk1
Hans-Dieter Volk1 Séverine Bézie2
Séverine Bézie2 Petra Reinke1
Petra Reinke1 Carole Guillonneau2*‡
Carole Guillonneau2*‡ Dimitrios L. Wagner1*‡
Dimitrios L. Wagner1*‡ Ignacio Anegon2*‡
Ignacio Anegon2*‡Regulatory Tcells (Treg) are essential components of peripheral immune homeostasis. Adoptive Treg cell therapy has shown efficacy in a variety of immune-mediated diseases in preclinical studies and is now moving from phase I/IIa to larger phase II studies aiming to demonstrate efficacy. However, hurdles such as in vivo stability and efficacy remain to be addressed. Nevertheless, preclinical models have shown that Treg function and specificity can be increased by pharmacological substances or gene modifications, and even that conventional T cells can be converted to Treg potentially providing new sources of Treg and facilitating Treg cell therapy. The exponential growth in genetic engineering techniques and their application to T cells coupled to a large body of knowledge on Treg open numerous opportunities to generate Treg with “superpowers”. This review summarizes the genetic engineering techniques available and their applications for the next-generation of Super-Treg with increased function, stability, redirected specificity and survival.
The immune system has developed physiological regulatory mechanisms to avoid excessive intensity or duration of immune responses and inflammation. Undesired immune reactivity needs to be controlled in pathological situations such as autoimmune diseases, solid organ transplantation (SOT), graft-vs.-host disease (GvHD), and immunogenicity of gene therapeutics and biologics. These regulatory mechanisms can be exploited therapeutically to reshape immune responses in subtler ways than conventional immunosuppressors. In fact, conventional immunosuppressors are non-selective, also inhibit protective anti-pathogen immunity and have common off-target toxicities. Although novel treatments dampen immune responses more specifically and induce immune tolerance (1, 2), alternative treatments are needed.
Among the mechanisms that maintain tolerance, both CD4+ and CD8+ FOXP3+ Treg play a central role (3–7). In addition, in both CD4+ and CD8+ compartments FOXP3− Treg are described (8). Treg are multifunctional, adaptable, living drugs, that have the potential to restore/induce durable immune tolerance and thus cure or ameliorate diseases as demonstrated in pathological rodent models (3). Although most Treg used in pre-clinical models have been polyclonal, some were antigen-specific or genetically modified (5, 6, 8–10). Small clinical studies have demonstrated the safety and some efficacy of autologous in vitro expanded polyclonal CD4+ Treg without genetic modifications in a variety of diseases (3).
Genetic modifications hold great potential to enhance their clinical efficacy as previously shown for genetically modified conventional T cells (Tconv) in the cancer field (11). The exponential development of genome engineering approaches enables strategies to generate “Super-Treg.”
This review describes genetic engineering techniques to increase the specificity, functional stability, survival, and suppressive function of Treg, as well as the generation of allogeneic off-the-shelf products, and strategies to eliminate these Super-Treg if necessary.
Targeted genetic manipulation of Treg has surged due to advances in genetic analysis and engineering (12).
The current gold standard for the stable ectopic gene expression by T cells are replication-deficient lenti-/retro-viruses, which insert entire gene expression cassettes into the genome (13) (Figure 1). Multiple studies have demonstrated that Treg from healthy donors and autoimmune patients can be efficiently transduced in vitro (Table 1) (45). Alternatively, transposon-based gene transfer systems allow the random insertion of moderate to large cargo sizes in T cells (46, 47). Random integration of the genetic cargo and insertional mutagenesis are safety concerns requiring long-term monitoring (48), although, so far no leukemic transformation has been reported for virally transduced Tconv (49–51).
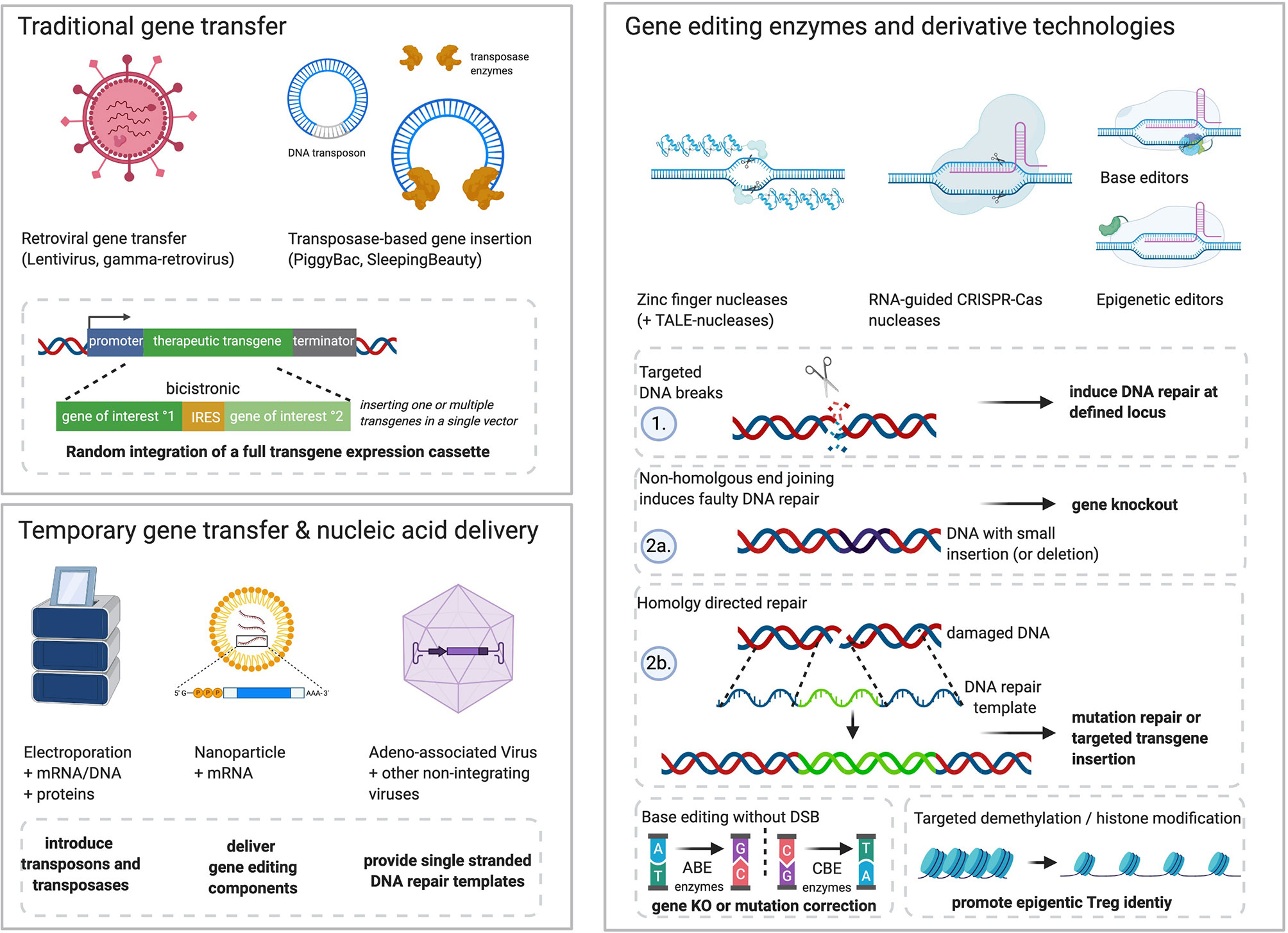
Figure 1 Examples of tools and technologies that allow genetic engineering of Tregs. Traditional gene transfer methods include retroviral transduction or transposase-mediated gene transfer. Random integration of expression cassettes into the Treg genome allow for stable transgene overexpression of one or multiple transgenes connected by internal ribosomal entry sites (IRES) or self-cleaving 2A peptide sequences. Recent advances in the production of plasmids with minimal bacterial backbone (mini-/micro-circles) as well as enhanced transposase enzymes could qualify the use of transposase-modified T cells (52–55), but have not yet been used to generate therapeutic Treg. Gene editing and its derivative technologies allow sequence-specific modification of the human genome. ZFNs and TALE-nucleases bind specific DNA sequences through protein-DNA interaction (zinc-finger arrays, TALE-effectors). Both systems have been used to modify T cell products in preclinical and clinical investigations for HIV or cancer therapy (56–58). CRISPR-Cas ribonucleoprotein complexes can be redirected through small guide RNA (gRNA) and minimal DNA-motif requirements by the Cas enzyme (a.k.a. protospacer adjacent motif, PAM) that are different among Cas variants. After binding of their target sequence, attached or inherent nuclease domains induce DNA double strand breaks (DSB) and subsequently DNA repair. Non-homologous end joining (NHEJ) links the free DNA ends without proofreading, thereby leading to errors like small insertions or deletions that can disrupt genes through frameshifts in their open reading frame. This can be used to knock-out genes and prevent functional protein synthesis. Alternatively, highly activated Treg in S-phase of the cell cycle may also use homology-directed repair (HDR) after DSB. DNA with sequence homology to the cutting site is recognized by the HDR machinery in proximity and used to repair the break via proofreading from the analogous DNA fragment. This can be exploited to correct mutations or introduce new genes. To this end, large amounts of single/double stranded DNA templates including desired changes (e.g., nucleotide changes, transgene inserts) must be delivered into the Treg nucleus (typically by electroporation or non-integrating viruses). Important derivative technologies of programmable nucleases include base editors and epigenetic editing enzymes. Base editors are engineered multi-enzymes complexes typically attached to nuclease-deficient Cas proteins which allow targeted modification of certain bases within the gRNA target sequence. Common variants include adenine base editors (=ABE) which convert adenine to guanine (A:T to G:C) and cytosine base editors (=CBE) which convert cytosine to thymidine (C:G to T:A). Furthermore, targeted changes to the epigenome could be performed through enzymes that interfere with methylation or histone modifications to promote desired epigenetic imprints. While retroviral delivery tools benefit from their ancestors’ capacities to invade T cells naturally, other cargo must be effectively delivered into Treg. Electroporation is a common method to transiently introduce nucleic acids like DNA (transposon technology), but also mRNA encoding gene editing enzymes or even recombinant proteins. Nanoparticles are another alternative for transient delivery of gene engineering tools which are under development for Tconv and Treg. Further, adeno-associated viruses (AAV) and other non-integrating viruses may allow a controlled delivery of DNA templates into Treg nuclei for efficient gene targeting. Figure generated using www.biorender.com.

Table 1 T cell products modified in vitro to treat immune-mediated disease models in vivo or cells from human patients with genetic diseases1.
Zinc finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) enable recognition of a genetic sequence through protein/DNA binding and induce double-strand DNA breaks (DSBs) via dimerization (74–76) (Figure 1). However, the discovery of the CRISPR-Cas system has induced a paradigm shift as it enables easier design of efficient nucleases. Recently, highly efficient optimized Cas9 nuclease variants have been developed (77–79). DSBs at specific sequences are repaired by non-homologous end joining or homology directed repair (HDR) (by providing a DNA repair template) to achieve gene KO or targeted mutation/insertion respectively (Figure 1). This was successfully applied to sorted human Treg in the correction of a pathogenic IL-2Ra in approximately 20% and GFP insertions in up to 40% of CD4+ Treg (80). Targeting multiple genomic loci with site-specific nucleases allows multiplexing of gene knockouts (KOs) in a single intervention. Two recent manuscripts described CRISPR/Cas9 KO screening in Tregs to define genes involved in mouse Treg stability and function (63, 81).
Nucleases without active nuclease domains can be repurposed to shuttle other bioactive cargo to introduce small base changes, modify epigenetic marks or interfere with transcription (82). Nuclease-deficient Cas9 (dCas9) fused to enzymes with different functions, can be used to specifically edit certain bases (83) (Figure 1). Use of base editor proteins for gene multiplexing was successfully achieved with very high efficiency in Tconv (84). Potentially, the newly introduced prime gene editing system could also be applied to insert or replace small gene sequences efficiently without the need for DNA DSBs (85).
Gene editing requires efficient delivery of the respective components into the cells’ nuclei. Gene editing enzymes can be transferred as plasmid, mRNA, or recombinant protein-RNA complexes (RNP). HDR repair templates are required as single- or double-stranded DNA. Electroporation allows the highly efficient transfection of protein, mRNA, or plasmids into T cells. Viral vectors exploit their tropism to deliver their cargo with more control than blunt electroshocks. Adenovirus-associated virus (AAV) serotype 6 has been prominently used to deliver genetic cargo into human Tconv and immunopathology-polyendocrinopathy-enteropathy-X-linked (IPEX) syndrome-patient-derived Tconv to induce Treg (27, 86). Lentiviruses and AAVs can be modified to incorporate nuclease enzymes in their capsids to achieve all-in-one delivery solutions for CRISPR-Cas gene editing and DNA transfer tested in mice and human embryonic kidney cells or lymphoblastoid cell lines (87–90). Combination of transposon-based CAR transfer through an anti-CD3 directed nanoparticle system allowed efficient T cell reprogramming in immunocompetent mice in vivo (52).
Off-target effects are a concern for the clinical translation of gene editing and careful experimental design as well as thorough off-target analysis are required (91). Transient presence of the components and high-fidelity nucleases reduce the risk of off-targets. Further, unwanted repair outcomes at the edited on-target sites have been observed including large deletions and translocations (92, 93). Translocations are a particular risk when multiplexing loci in a single manipulation (94), and decrease cellular fitness after transfusion (95).
Viral vectors, nuclease systems, and newly introduced transgenes can be immunogenic, potentially decreasing the efficacy of Super-Treg and even posing a potential safety risk as previously seen using Tconv (96, 97). Of note, most human adults have pre-existing adaptive immunity toward Cas9 proteins and enriched Cas9-reactive Tconv can eliminate Cas9-expressing lymphoblastoid cell lines in vitro, which can be reduced by Cas9-specific Treg (98). However, T cells edited using Cas9 delivered by RNP electroporation did not elicit an immune response and persisted, which might be due to the low abundance of Cas9 in the edited final product or defective immune responses in the patients (95).
For TALENs and ZFNs, despite being immunogenic per se, stemming from Xanthomonas, which infects plants, and partially from Flavobacterium okeanokoites, which was isolated from the seabed, there is a low risk for previous exposure to the enzymes. In contrast, Cas9 stems from Streptococcus pyogenes, which is a common human pathogen. However, development of an immune response upon permanent expression may still be relevant.
A limitation of adoptive Treg therapy is that inflammatory conditions can inhibit their function or even switch them to Tconv (99, 100). Therefore, the identification of pathways regulating Treg function and stability are key to define targets for genetically engineering more stable and robust Treg (Figure 2).
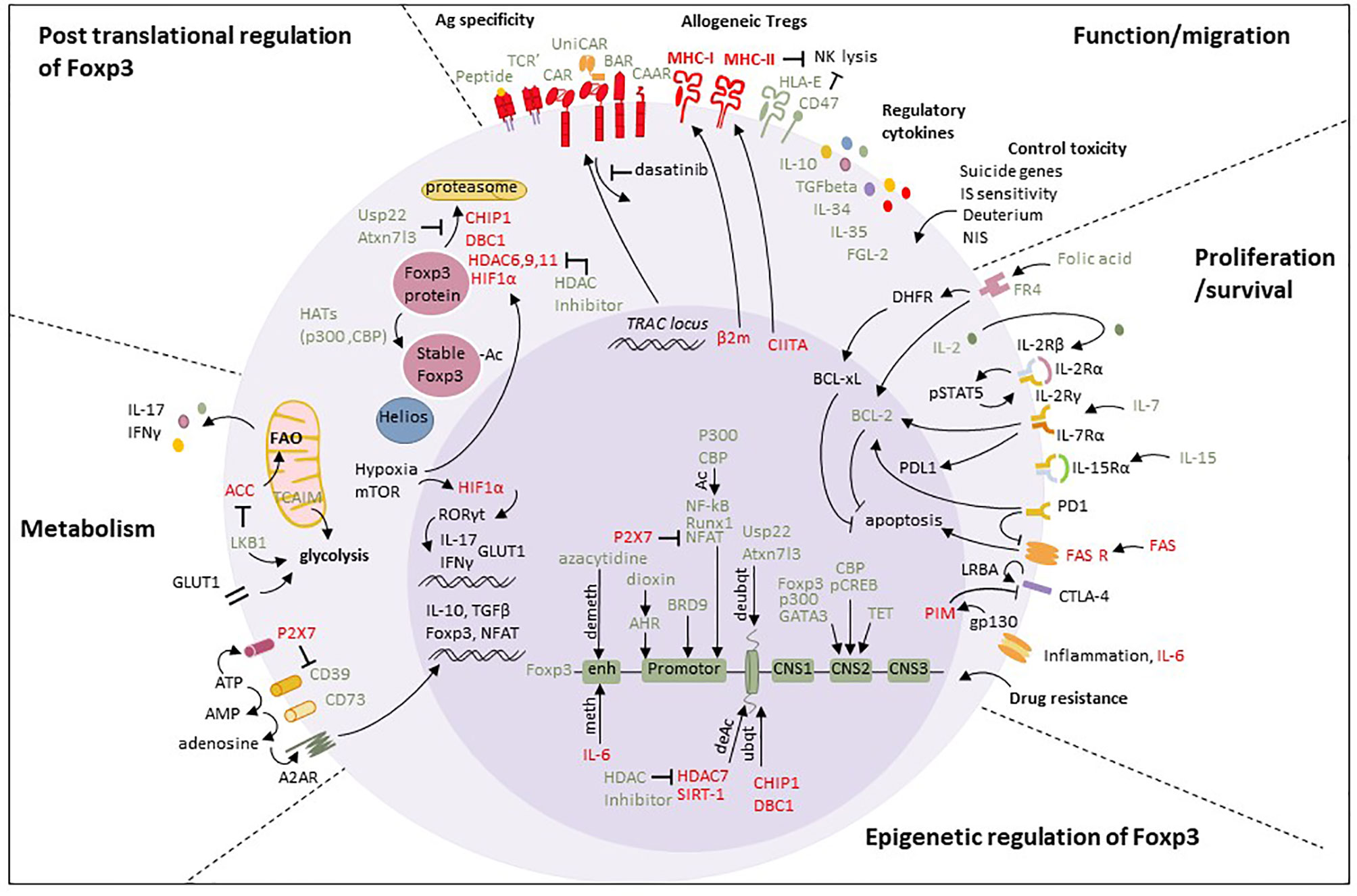
Figure 2 Summary of targets to generate Super-Treg. Foxp3 expression can be modulated at the epigenetic level. Demethylation of CpG dinucleotides at the FOXP3 locus by azacytidine or TET stabilizes FOXP3 expression, while remethylation at the upstream enhancer by IL-6 reduces FOXP3 expression (59). Histone acetylation by HATs (p300, CBP) stabilizes FOXP3 expression by cooperating with key Treg transcription factors that act on the FOXP3 promoter, such as Runx1 and NFAT (60). On the contrary, the histone HDAC SIRT-1 inhibits FOXP3 transcription and its deletion promotes Treg function (61). Treg stability is also maintained in inflammatory environments by CBP and P300 interaction with the Foxp3 CNS2 region, through which CBP is able to regulate pCREB and P300 to regulate expression of GATA3 (60, 62). Histone deubiquitination by Usp22 and Atxn7l3 promotes FOXP3 expression, contrary to CHIP1 and DBC1. Chromatin remodeling by BRD9, a member of the SWI/SNF chromatin remodeling complex, positively regulates FOXP3 as BRD9 depletion reduces the binding of FOXP3 to its enhancers CNS2/CNS0, thereby reducing FOXP3 expression (63). BRD9 also regulates a subset of FOXP3 target genes by promoting both FOXP3 binding at their regulatory element and increasing histone modifications. Knockdown of BRD9 thereby compromises FOXP3 expression and Treg function in vitro and in vivo (63). Thus, BRD9 overexpression could be interesting in Treg. Transcription factor AHR can trigger the differentiation of Treg by the expression of FOXP3 when activated in response to dioxin, whereas carbazole induces Th17 cell development (64). FOXP3 expression can also be regulated at the post translational level. Acetylation by p300 and CBP stabilizes FOXP3 protein, while deacetylation by CHIP1, DBC1, or HDAC7 induces degradation of the protein, which can be inhibited by HDAC inhibitors, Usp22 and Atxn7l3. Glycolysis can be privileged. HIF1α binds to the promoter of RORγt, resulting in expression of IL-17 which drives Th17 cell differentiation (65) and reducing Treg stability through the production of IFN‐γ (66). HIF1α also increases glycolysis by upregulating GLUT1, and promotes FOXP3 ubiquitination. LKB1 and TCAIM (67) promoting glycolysis are promising candidates for consideration. CD39 participates in tolerance induction in kidney grafts (68) and the effector memory Treg subset mainly expressing CD39 is diminished in multiple sclerosis (69). CD39 and CD73 transform ATP in adenosine acting through A2AR, which is limited by ATP uptake by P2X7. Function and migration can be controlled. Antigen specificity of Tregs can be modified by inserting genes encoding for an ectopic TCR or a chimeric antigen receptor preferentially in the TRAC locus. The control of rogue Super-Treg could include potent immunosuppressive drug regimens but they require hours to days for elimination. Recently, the small molecule tyrosine inhibitor dasatinib was shown to be a fast and potent inhibitor of CAR signaling in Tconv and may also be applicable to Treg platforms (72). Allogeneic Tregs present advantages regarding production but allogeneic MHC molecules have to be KO and counterbalanced by HLA-E and CD47 (“do not eat me” molecules). The expression of cytokines known to be responsible for Treg function such as IL-10, TGFβ, IL-34, IL-35, and FGL-2 can be upregulated. Treg-mediated toxicity can be controlled by insertion of suicide genes, such as a truncated version of the epidermal growth factor receptor recognized by the mAb cetuximab (70), or the peptide RQR8 combining epitopes recognized by the mAb rituximab (71) or by dimerization of Casp9 by the small molecule rimiducid (72). Tregs can be tracked using deuterium and NIS. Proliferation and survival can be promoted. Folic acid is suggested to upregulate the anti-apoptotic proteins BCL-2 and BCL-xL via folic acid receptor 4 (FR4) in Treg (73). Hence, increased expression of FR4 or enzymes of this pathway such as dihydrofolate reductase in Treg may preferentially preserve these cells. IL-2, IL-7, and IL-15 signals are important for survival and proliferation of Tregs. To make Treg independent of exogenous IL-2, they could be armed with their own IL-2 for self-supply. PD1 and CTLA-4 have both important roles for Treg function and survival and LRBA, in contrast to PIM, is important for CTLA-4 expression. Drug resistance could also be considered for promoting Treg survival.
Given the key role of FOXP3 to control Treg function and that its expression and function are labile or even lost in patients with mutations in the FOXP3 gene (IPEX), numerous studies have analyzed how to increase or stabilize its expression. Functional CD4+ Treg from IPEX patients could be obtained by ectopic expression of FOXP3 in their Tconv (26), but also with more clinical potential by precise HDR on hematopoietic stem cells (27) (Table 1).
This strategy could also allow high numbers of Treg to be obtained from Tconv. In CD4+ Tconv, ectopic expression of FOXP3 using retroviral vectors (22, 26, 43) or by HDR of a strong promoter upstream of the FOXP3 coding sequences (19) allowed generation of Treg that suppressed CD4+ Tconv not only in vitro but also inhibited GvHD, colitis or dermatitis in animal models (Table 1). Interestingly, tamoxifen-induced but not constitutive FOXP3 expression in CD4+ Tconv resulted in control of autoimmune arthritis by migration into lymph nodes (39). In line with these results, control of type 1 diabetes (T1D) was obtained only with islet-specific CD4+ Tconv homing to lymph nodes and not with polyclonal CD4+ FOXP3-expressing Tconv in a mouse model (33). Transduction of mouse anti-human FVIII T cells with Foxp3 resulted in decrease anti-FVIII antibodies in hemophilia A mice (25). In in vitro studies, human pathogenic synovial Tconv from rheumatoid arthritis patients ectopically expressing FOXP3 showed reduced Tconv responses (101). Other publications with in vitro studies described that ectopic expression of one (102) or both (103) isoforms of FOXP3 in CD4+ Tconv resulted in functional human Treg. Overexpression of the transcription factor HELIOS cooperates with FOXP3 to generate both CD4+ and CD8+ Treg from human Tconv, but particularly CD8+ T cells (20). Similarly, delivery of dCas9 fused to a transcriptional activator and guides recognizing FOXP3 promoter sequences increases FOXP3 expression (104).
Epigenetic regulation of FOXP3 expression is important for the expression of FOXP3 in Treg. Demethylation of CpG dinucleotides at the FOXP3 locus including at regulatory elements in the intronic region, at the proximal promotor and the upstream enhancer stabilizes FOXP3 expression (59, 105, 106). If these Treg-specific demethylated regions (TSDR) are not fully demethylated, such as in the induction of FOXP3 expression by TGF-β, FOXP3 expression can be lost upon restimulation in mouse Treg (107). Epigenetic modifications of FOXP3 for TSDR demethylation in Treg by azacytidine (a DNA methyltransferase inhibitor) induces and stabilizes FOXP3 expression in mouse Treg (107). Partial demethylation of TSDR CNS2 in Treg by catalytically inactive CRISPR-Cas9 (dCas9) fused to the catalytic domain of ten-eleven translocation (TET) protein, which promotes demethylation, resulted in stable FOXP3 expression and increased suppressive activity in vivo in mice (32). However, a similar system using dCas9 fused to TET1 did not increase FOXP3 levels in mouse Treg (108).
Transcription of FOXP3 can be repressed by histone deacetylation in the FOXP3 promoter by histone deacetylase 7 (HDAC7), and HDAC inhibitors increase FOXP3 expression through regulation of both the gene and the protein, and can improve the suppressive action of murine and human Treg (109, 110). A dCas9 fused to the catalytic domain of histone acetyltransferase (HAT) p300 showed that histone acetylation targeted to the promoter locus was able to activate and stabilize FOXP3 levels in mice, even under inflammatory conditions (108).
Lysine acetylation by HATs of both histones in the FOXP3 locus and of FOXP3 itself increases its transcription and reduces its poly-ubiquitination and degradation as well as enhancing FOXP3 chromatin binding in mouse and human Treg (111, 112). Hyperacetylation by HDAC inhibitors or overexpression of HATs can increase FOXP3 levels in mouse and human Treg (112, 113). For example, P300 and CBP HATs acetylate FOXP3, increasing its DNA binding and thereby regulating murine Treg function and stability (60). CBP and P300 affect Treg development through several mechanisms, including promoting FOXP3 production, and by participating in a positive feedback loop that enhances murine Treg stability in inflammatory environments, which could be further exploited through molecular engineering (60).
Ubiquitination of both histones at the FOXP3 locus and of the protein itself is important in the regulation of FOXP3, via members of the deubiquitination module of the SAGA complex, Usp22, and Atxn7l3. Loss of Usp22 in Treg reduces Foxp3 transcript levels, increases FOXP3 ubiquitination and degradation, and reduces suppressive activity in vivo in mice (63, 81). Furthermore, Stub1 (114) and TRAF6 (115) E3 ubiquitin ligases induced by inflammation target the ubiquitination of FOXP3 followed by its degradation in mouse and human Treg and represent interesting targets for genetic ablation in Treg products. In contrast, Hrd1, an E3 ligase critical in suppressing the ER stress response, stabilizes murine FOXP3 expression (116). In terms of kinases and phosphorylation of FOXP3, PIM1 (117), and CDK2 (118) kinases negatively regulate FOXP3 and Treg function.
HIF‐1α reacts to hypoxia by triggering the switch between mitochondrial oxidative phosphorylation and aerobic glycolysis (119), and is also induced by continuous TCR stimulation via mTOR in human T cells (120). In mouse Treg and human embryonic kidney cells, HIF-1α promotes the ubiquitination and proteasomal degradation of FOXP3 (65), and its upregulation in response to hypoxia inhibits FOXP3 expression in mouse T cells (121). HIF-1α also inhibits the development of mouse Treg through increasing glycolysis by upregulating glycolytic proteins (122). HIF-1α deficiency inhibits glycolysis and therefore promotes the differentiation of murine Treg over Th17 cells (123). Deletion of HIF-1α in mice increases FOXP3 expression, and reduces transcription of Th17 cell-related genes (65, 121), suggesting HIF-1α KO as a means to improve Treg stability through metabolic control. Differentiation of induced human Treg is inhibited by IL-1β in a HIF-1α-dependent manner (124). However, in human Jurkat cells, HIF-1α induction increased FOXP3 protein and mRNA levels, which was reversed by knockdown of HIF1α (125, 126). On the same lines, exposure of human PBMCs to hypoxia increased the proportions of FOXP3+ Treg among CD4+ CD25+ T cells and their suppressive potential to inhibit Tconv proliferation, which was also observed in mouse splenocytes (125).
Aryl hydrocarbon receptor (AHR) is a ligand-dependent transcription factor that functions as an environmental sensor and mediates the differentiation of both Th17 cells and FOXP3+ Treg. AHR is highly expressed in peripheral Treg in the gut, and its deletion impairs their function. Conversely, activation of AHR in transgenic mice increases the population and migration of Treg (127). AHR inhibited proinflammatory cytokines (IFNγ and IL-17) and Th1-associated genes, but was dispensable for FOXP3 stability. Ahr activation in a conditional knock-in in Treg in a mouse model of colitis enhances suppressive activity and migration to the inflammatory site, and a reduction in proinflammatory T cells (127). Furthermore, AHR activation was found to promote generation of human induced Treg, producing IL-10 and controlling Tconv via granzyme B, but did not have an effect on thymic-derived human Treg (128, 129).
IL-6 is a proinflammatory cytokine that induces the expression of kinase PIM1 during inflammation, which inhibits expression of Treg markers including CTLA4 and CD25 via phosphorylation of FOXP3 (130). T cell-specific deletion of the IL-6 receptor gp130 in mice reduces IL-6 signaling and promotes the conversion of peripheral Tconv into Treg (131). IL-6 blockade suppresses the immune response in models of autoimmune disease and is used in the treatment of rheumatoid arthritis (132, 133). At the same time, a recent publication showed that IL-6Rα-deficient Treg lost suppression and aggravated experimental glomerulonephritis (134). Therefore, mitigating IL-6 signaling in Treg, which was assumed to be a compelling strategy to enhance their functionality in inflamed tissues and in the presence of high levels of IL-6 needs more investigation and has to be overthought critically.
The purinergic receptor P2X7 induces T cell activation through binding of ATP, pushing the balance toward proinflammatory Th17 cells, and decreasing the viability and suppressive function of mouse Treg (135). In a mouse model of experimental colitis, P2X7 receptor KO resulted in an increase of activated Treg, IL-10, and TGF-β (136). Preventing P2X7 signaling is able to preserve mouse Treg stability by stabilization of nuclear complexes of NFAT and FOXP3, and the resulting downstream transcription of Treg-linked genes (135). CD39 and CD73 expressed by Treg degrade ATP to adenosine and adenosine itself can enhance the expansion and immunosuppressive function of human Treg in vitro by binding to the purinergic P1 adenosine 2A receptor (A2AR) (137, 138). Thus, genetic overexpression of CD39 could be beneficial.
Although Treg produce immunosuppressive cytokines, such as IL-10 (139), TGFβ (140), IL-34 (141, 142), IL-35 (143), and FGL2 (144, 145), their production could be increased by genetic means.
Treg with increased function and stability could therefore be engineered by inhibition of negative regulatory genes (using nucleases), overexpression of positive regulators (using lentiviral vectors), likely giving more precise control than small molecule treatments which may bind multiple members of a family (for example acetylases).
Survival of human naïve CD4+ Treg is mediated by IL-7 signaling, which increases anti-apoptotic BCL-2 (146) while long-term survival of CD4+ Treg is dependent on IL-2 (147) and of CD8+ Treg on IL-15 (4). Low dose IL-2 infusion was shown to increase Treg numbers, FOXP3 expression and lead to a more diverse repertoire of CD4+ and CD8+ Treg in patients (4, 148). Currently, there are different engineered IL-2–based drugs targeting CD25 on Treg, also referred to as IL-2 muteins (149) in clinical trials. Since IL-2 signaling is associated with long-term survival of human Treg, constitutively active STAT5 (150), which is an important signal transducer in this pathway, may improve Treg survival and abolish their dependence on extracellular IL-2 (Figure 2). Moreover, to make Treg independent of exogenous IL-2 that may activate Tconv and NK-cells, they could be armed with their own IL-2 for self-supply. However, ectopic IL-2 expression could compromise the immunosuppressive mechanism of IL-2 deprivation of surrounding Tconv. A mutated IL-2R that only binds an IL-2 mutein and not wild-type IL-2 could be engineered in mouse Treg that are then selectively expanded (151).
A strategy to engineer constitutively active cytokine receptors independent of cytokine availability may also be translated from Tconv to Treg (152), e.g., allowing long-term survival via a constitutively active IL-2 receptor (147). Moreover, chimeric cytokine receptors (CARs) converting pro-inflammatory signals (captured by the extracellular domain of the respective receptor, e.g., IL-6) into Treg-survival signals using the intracellular signal transduction domains (e.g., IL-2 receptor) of pro-survival signals may contribute to improved survival of Treg products as reported previously in a mirroring approach for the support of Tconv (153).
Several pathways inducing Treg apoptosis seem to be dependent on FAS (154, 155) and pro-survival pathways on BCL-2 (73, 146, 154). Thus, disruption of FAS or over-expression of BCL-2 may significantly increase the viability of Treg. An alternative may be to increase the PD1-PDL1 signaling in Treg, since PD1 blockade was reported to lead to downregulation of BCL-2 and increased FAS receptor expression (154). However, mouse Treg lacking PD-1 were shown to be activated and have high suppressive potential (156), which underlines the necessity for further studies of this axis in human Treg. Additionally, human CD4+ Treg express PD-L1 in response to IL-7 (154) and induce apoptosis in PD-1+ Tconv (157) and autoreactive B-cells (158). As CTLA-4 has an important role in Treg function and increased degradation of CTLA-4 as present in LRBA deficiency is associated with high levels of Treg apoptosis, stabilizing strategies for sustained or increased CTLA-4 expression may also improve human Treg survival (159, 160).
Treg and Tconv have different metabolic requirements, as Treg use glycolysis and increase fatty acid oxidation upon activation (161, 162). Acetyl CoA carboxylase (ACC) regulates both biosynthesis and breakdown of long chain fatty acids and ACC1 deficiency induces high levels of FOXP3 expression in mouse and human Treg (163). Therefore, ACC is a potential target for altering the metabolic programming of T cells, as blocking fatty acid synthesis favors Treg induction and prevents Th17 development. Liver kinase 1 (LKB1), a metabolic sensor, is essential for murine Treg stability and suppressive activity by inhibiting expression of pro-inflammatory cytokines and preventing exhaustion (164). LKB1 and its target genes are downregulated in impaired Treg from patients with acute GvHD and Lkb1 deletion in Treg in mice leads to severe autoimmune inflammation, and can aggravate acute GvHD (164, 165). LKB1 also stabilizes FOXP3 expression in Treg and expression levels correlate with Foxp3 expression in human Treg (165). In contrast to Tconv, murine and human Treg do not accumulate lactate and are insensitive to lactate in medium (166). LKB1 increases glycolysis and lactate formation and in mice, abrogation of Lkb1 leads to loss of mitochondrial integrity and to a dramatic reduction of Treg (167). Thus, LKB1 overexpression could be used to stabilize FOXP3 expression, maintain metabolic homeostasis, and avoid exhaustion in Treg.
In several settings, Treg are infused into patients treated with immunosuppressants to inhibit Tconv but also compromise Treg function (168). Hence, making Treg resistant to these drugs may allow their preferential survival. Indeed, strategies aiming to make antiviral T cells resistant to calcineurin inhibitors or glucocorticoids including knockdown (169), knockout (170–172) or introduction of calcineurin-resistant mutants (173), might also be applied to Treg.
Treg-mediated tolerance can be improved by increasing antigen specificity and with the development of gene editing, redirection of Treg specificity became feasible (Figure 2). Indeed, antigen-specific Treg have an increased suppressive ability and a stronger efficacy in the regulation of the immune response and an improved migration to the site of interest compared to polyclonal Treg (9, 10, 174–178). While ex vivo expansion in presence of the antigen of interest, or in vivo by administration of peptides recognized only by Treg (176, 177, 179) is possible, using genome editing would be advantageous as it would confer antigen-specificity to a larger Treg population rather than amplifying a very small subset of antigen-specific Treg, which can be challenging. However, genome editing to redirect specificity would also multiply the danger of contaminating Tconv that may have a proliferative advantage in cytokine-rich medium used during Treg expansion and could overgrow the culture, multiplying their abundance in the final product. Thus, a very pure starting population is required or the undesired cells (such as CD8+ non-Treg) must be depleted at a later time point. However, the latter is challenging for, e.g., CD4+ Tconv, which are not easily distinguishable from Treg after culture, but could also cause detrimental effects. Furthermore, it will be crucial to choose a receptor with appropriate affinity for Treg to exclude the possibility of instabilities. Even pre-selection of more stable Treg subsets or genetic engineering to make them more stable may be required to generate a safe product.
Treg specificity can be redirected by the use of TCRs and CARs (180, 181). Importantly, Treg with a single specificity have been shown to suppress multiple antigens if presented by an APC simultaneously, as shown in autoimmune, GVHD, and SOT models (9, 10, 15, 182).
Redirecting T cell specificity with an engineered TCR was reported as early as 1996 using a chimeric TCRβ chain consisting of a single-chain Fv portion derived from a monoclonal antibody paired with endogenous TCR/CD3 component, thus providing antibody and TCR specificity (183). The use of TCRs has several advantages since it represents a physiological way of activating T cells and allows the targeting of intracellular antigens presented by HLA molecules. In addition, expression of only one antigen per cell is sufficient to activate the TCR-expressing Treg. However, HLA restriction limits coverage to a particular part of the population. Careful identification of a high affinity TCR-α/β is required to ensure that they retain functionality without acquiring a harmful unpredicted specificity when mispaired with the endogenous TCR. To avoid this, disruption of the endogenous TCR using nucleases might be necessary (184). Proofs of concept include human Treg expressing a myelin basic protein-specific TCR derived from a multiple sclerosis patient, which showed in vitro and in vivo efficacy in an EAE model (37). Efficacy was also demonstrated in a mouse model of hemophilia A using human Treg engineered with a factor-VIII-specific TCR isolated from an hemophilia A patient (23). TCRs against autoantigens have also shown in vivo efficacy in models of arthritis (38) (Table 1) or in vitro recognizing islet antigens involved in T1D (182, 185).
Interestingly, MHC-I-restricted TCRs have been shown to be functional in human CD4+ Treg, bypassing the need for the CD8 coreceptor, and this was the case for TCRs with low affinity not functional on CD4+ Tconv (186).
A potential future perspective of these studies is the use of TCRs isolated from Treg and not from effector T cells as done until now. Although only very few TCRs and the peptides recognized by Treg TCRs have been identified until now, they do show differences with TCRs from Tconv, e.g., recognizing longer (15aa) peptides or reversed TCR docking modes (176, 177, 187).
Pioneer work by Eshar and colleagues in the autoimmune field allowed the generation of CARs in which antigen recognition signaling domains of antibodies and a TCR-zeta-chain were fused in a single molecule (181). Sequences from co-stimulatory proteins were also fused in cis and the most commonly used ones are the intracellular portions of CD28 or 4-1BB. While the 4-1BB signaling domain used in the CAR construct has been suggested to enhance Tconv persistence and improve the toxicity profile in patients, CD28 was shown to be more beneficial for Treg phenotype and function (18). CAR technology has some advantages over TCRs, the most important one being the absence of HLA restriction. CAR-Treg are less dependent on IL-2 compared to TCR-expressing Treg, potentially due to costimulatory signals received upon activation of the CAR. However, CARs also have several limitations vs. TCRs, as CARs only recognize extracellular antigens. Additionally, CARs require higher expression levels than TCRs (100–10,000 antigens/cell vs. <10, respectively) for sufficient activation although increasing the affinity of CARs can also increase efficacy (188, 189). CAR molecules can be immunogenic, not only due to murine scFv fragments, but also due to the generation of new epitopes in a chimeric molecule, and this impacts the persistence of effector T cells in patients (97, 190). Even induction of anti-CAR antibodies was described for effector T cells and the immune reaction was reported to be able to cause anaphylaxis in a patient repetitively treated with CAR T cells (191). Possibly, anti-CAR immune responses may be less severe if the CAR is expressed in an immunosuppressive Treg compared to expression in pro-inflammatory Tconv.
Human CD4+ and CD8+ CAR-Treg have been used in mouse models of FVIII hemophilia, SOT and GVHD as well as in vitro with CD4+ Treg from IPEX patients (for a complete list see Table 1). Mouse CD4+ CAR-Treg have demonstrated efficacy in mouse models of SOT, GvHD, IPEX, colitis, allergic asthma, rheumatological diseases, and EAE (Table 1).
The importance of internal vs. external antigen targets could orientate toward the generation of a TCR- vs. a CAR-transgenic Treg. There are also new tools developed such as CAR-T cells possessing a TCR-like antibody moiety (TCR-like CAR-T) with a single-chain variable domain specific for a distinct peptide/MHC (192). In an original approach, CD4+ Treg expressing a CAR directed against FITC and ex vivo incubated with FITC-labeled antibodies directed against donor alloantigens inhibited pancreatic islet rejection (17). Similarly, the UniCAR system, in which a universal CAR is indirectly linked to their target cells via a separate targeting module, has been applied to human Treg (193). In a new approach to treat autoantibody-driven diseases, CD4+ Treg have been engineered to express CARs with antigens recognized by B-cells (called BARs, where the scFv fragment is replaced by an antigen) (28). Similarly, Tconv expressing an autoantigen in a chimeric autoantibody receptor (CAAR) mediated killing of the autoreactive B-cells, as shown in pemphigus (44). Also, Tconv expressing anti-CD19 CARs generally used to treat B-cell malignancies were used to treat mice with lupus disease (42) (Table 1).
Genetic engineering of allogeneic Treg as an off-the-shelf product would allow cells from a given donor to treat several patients thereby reducing the cost per dose as well as increasing treatment flexibility (Figure 2). Nevertheless, this approach has the draw-back of allogenicity due to recognition of foreign MHC-I and -II antigens by host T cells. To extend allo-Treg persistence, deletion of β-2 microglobulin and CIITA could be performed to eliminate MHC-I and -II antigens, respectively. Although absence of MHC-I may increase the susceptibility of Treg to NK cell lysis and that could limit therapeutic efficacy, activated Treg may be more resistant in vivo and indeed triple-KO T cells have been found to persist better than HLA-sufficient T cells. In addition, overexpression of HLA-E or CD47, important in NK cell inhibition through inhibitory receptors, could prevent NK cell-mediated lysis (194–196). Preventing an immune response against allogeneic cells is even more important in the Treg setting than in the Tconv setting, as here any pro-inflammatory immune response can be detrimental as opposed to the Tconv setting, in which the goal is to create a pro-inflammatory environment. However, it has also to be considered that Treg have anti-inflammatory properties per se and first applications of 3rd party-derived Treg after umbilical cord stem cell transplantations did not reveal relevant adverse events (197).
Another potential risk using allogeneic T cells is GvHD although the inherent suppressive function of Treg makes this risk less relevant than with allogeneic Tconv. Generating highly specific, allogeneic Treg products also harbors the risk of toxicity in the case of unstable Treg or contaminating Tconv. Strategies such as suicide genes and elimination markers—some already clinically evaluated in Tconv—could be included to shut off adoptively transferred “stealth” Treg in case of toxicity (198) (Figure 2).
Pluripotent stem cells-derived Tconv mainly for cancer use have been described (199, 200) and derivation of Treg would be an important step not only to have an unlimited source of cells but also for generating “stealth” Treg.
It will also be important to better understand the migration of Treg to different anatomical compartments and their survival. To date, Treg infused in patients have only been identified in vivo in T1D patients after labeling CD4+ Treg with deuterium but this strategy is limited to cells in circulation and not in tissues (201). Also, mouse CD4+ Treg have been transduced for the expression of the sodium-iodide symporter (NIS). NIS uptakes into only living cells plasma iodide and other substrates detected using PET or SPECT/CT. NIS-expressing Treg radiolabeled with Technetium-99m pertechnetate were detected in spleen with no effects on cell viability, phenotype, and function (202).
An increasing number of clinical trials employing adoptive Treg transfer are currently ongoing or registered addressing a large variety of applications (3, 5, 6). The generation of Super-Treg with genetic modifications will require the use of improved protocols for the purification and amplification of Treg to prevent contaminating Tconv with putative hazard. Bead-based or flow cytometry-based cell sorters (9, 14–16, 18), either fully closed or open systems, allow for clinical grade CD4+ Treg isolation. For clinical-grade CD8+ Treg isolation flow cytometry-based approaches are used. Typically, in vitro Treg expansion before adoptive transfer takes around 2–3 weeks (9, 14).
Release criteria usually include the classical phenotypic Super-Treg markers (e.g., CD25, FOXP3 for CD4+ Treg) and absence of pro-inflammatory markers, e.g., pro-inflammatory cytokine production and CD45RC for CD8+ Treg. Functional assays or epigenetic assays may be beneficial, however cannot be realized in a timely manner before Treg product infusion.
Super-Treg quality control will require additional control of nuclease delivery and duration of expression as well as maximization of efficacy preferentially using vector-free systems (41). Safety controls will include in silico as well as in vitro analysis of off-target effects of nucleases and careful analysis of the edited loci (41). In this context, a clinical trial using CRISPR/Cas9-genetically modified in cancer patients has been recently published (95).
A Phase I/II clinical trial is approved in UK that plans to apply CAR anti-HLA-A2 CD4+ Treg in kidney transplantation. Academic multi-center consortia (like the ReSHAPE consortium, http://www.reshape-h2020.eu/partnership) aim to generate CD4+ and CD8+ Super-Treg and to apply them to both animal models of immune-mediated disease and clinically in kidney transplanted patients.
The specific challenge of using Treg therapy in general in human pathologies will be to interfere with established autoimmunity, rather than de novo immunizations (SOT, GvHD, gene therapies, biologics), without provoking global immunosuppression.
A future direction is the use of CARs recognizing inflamed or damaged tissues that could direct the Treg to these pathological areas, as shown by preliminary data (203).
The demonstration that Treg can stimulate tissue regeneration (204–208) reveals regenerative medicine as a novel indication for Super-Treg.
As for CAR-Tconv (209), the simultaneous use of both CD4+ and CD8+ CAR-Treg may prove to be superior to each subset alone.
Treg will likely be modified using new T cell engineering strategies, such as synthetic Notch receptors that have an extracellular single-chain antibody and intracellular transcriptional domains that are released and activate expression of target genes (210).
Immune humanized immunodeficient animal models will continue to be useful to address many questions in preclinical studies (211). Moreover, human and/or patient organoids, may gain more importance and are promising candidates for examining Treg function in disease models (212).
Biomarker studies will be important to define not only the effects of Super-Treg therapy but also the timing and doses of their administration.
The knowledge of Treg biology, their success in animal models and early clinical trials as well as the explosion of genome editing techniques are synergistic approaches to treat immune-mediated diseases in the future.
DW, JG, LA, SB, CG, and IA performed bibliography searches and wrote and edited the manuscript. MS-H, H-DV, and PR edited the manuscript. DW generated Figure 1. SB generated Figure 2. LA generated Table 1. IA planned the manuscript. All authors contributed to the article and approved the submitted version.
This work was funded by the ReSHAPE project from the European Union's Horizion 2020 research and innovation programme under grant agreement No 825392. It was also realized in the context and partially funded by the Labex IGO program supported by the National Research Agency via the investment of the future program ANR-11-LABX-0016-01, and a Berlin Institute of Health (BIH) crossfield grant “regulatory T cells.”
CG, IA, and SB have patents that have been licensed to TxCell S.A., a Sangamo Company. DW, LA, PR, H-DV, and MS-H have filed patent applications on Treg-related diagnostics and therapeutics. The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Picarda E, Bézie S, Boucault L, Autrusseau E, Kilens S, Meistermann D, et al. Transient antibody targeting of CD45RC induces transplant tolerance and potent antigen-specific regulatory T cells. JCI Insight (2017) 92(3):e90088. doi: 10.1172/jci.insight.90088
2. Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, et al. An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med (2019) 381(7):603–13. doi: 10.1056/NEJMoa1902226
3. Sharabi A, Tsokos MG, Ding Y, Malek TR, Klatzmann D, Tsokos GC. Regulatory T cells in the treatment of disease. Nat Rev Drug Discovery (2018) 17(11):823–44. doi: 10.1038/nrd.2018.148
4. Flippe L, Bézie S, Anegon I, Guillonneau C. Future prospects for CD8+ regulatory T cells in immune tolerance. Immunological Rev (2019) 292(1):209–24. Oct 8 [cited 2019 Nov 6]; Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/imr.12812. doi: 10.1111/imr.12812
5. Romano M, Fanelli G, Albany CJ, Giganti G, Lombardi G. Past, Present, and Future of Regulatory T Cell Therapy in Transplantation and Autoimmunity. Front Immunol (2019) 10:43. doi: 10.3389/fimmu.2019.00043
6. Ferreira LMR, Muller YD, Bluestone JA, Tang Q. Next-generation regulatory T cell therapy. Nat Rev Drug Discovery (2019) 18(10):749–69. doi: 10.1038/s41573-019-0041-4
7. Lees JR. CD8+ T cells: The past and future of immune regulation. Cell Immunol (2020) 357:104212. doi: 10.1016/j.cellimm.2020.104212
8. Gregori S, Roncarolo MG. Engineered T Regulatory Type 1 Cells for Clinical Application. Front Immunol (2018) 9:233. doi: 10.3389/fimmu.2018.00233
9. Bezie S, Charreau B, Vimond N, Lasselin J, Gerard N, Nerriere-Daguin V, et al. Human CD8+ Tregs expressing a MHC-specific CAR display enhanced suppression of human skin rejection and GVHD in NSG mice. Blood Adv (2019) 3(22):3522–38. doi: 10.1182/bloodadvances.2019000411
10. MacDonald KG, Hoeppli RE, Huang Q, Gillies J, Luciani DS, Orban PC, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest (2016) 126(4):1413–24. doi: 10.1172/JCI82771
11. June CH, Sadelain M. Chimeric Antigen Receptor Therapy. N Engl J Med (2018) 379(1):64–73. doi: 10.1056/NEJMra1706169
12. Adli M. The CRISPR tool kit for genome editing and beyond. Nat Commun (2018) 9(1):1911. doi: 10.1038/s41467-018-04252-2
13. Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol (1998) 72(11):8463–71. doi: 10.1128/JVI.72.11.8463-8471.1998
14. Tsang JY-S, Tanriver Y, Jiang S, Xue S-A, Ratnasothy K, Chen D, et al. Conferring indirect allospecificity on CD4+CD25+ Tregs by TCR gene transfer favors transplantation tolerance in mice. J Clin Invest (2008) 118(11):3619–28. doi: 10.1172/JCI33185
15. Boardman DA, Philippeos C, Fruhwirth GO, Ibrahim MA, Hannen RF, Cooper D, et al. Expression of a Chimeric Antigen Receptor Specific for Donor HLA Class I Enhances the Potency of Human Regulatory T Cells in Preventing Human Skin Transplant Rejection. Am J Transplant (2017) 17(4):931–43. 2016/12/28 Ed doi: 10.1111/ajt.14185
16. Noyan F, Zimmermann K, Hardtke-Wolenski M, Knoefel A, Schulde E, Geffers R, et al. Prevention of Allograft Rejection by Use of Regulatory T Cells With an MHC-Specific Chimeric Antigen Receptor. Am J Transplant (2017) 17(4):917–30. doi: 10.1111/ajt.14175
17. Pierini A, Iliopoulou BP, Peiris H, Pérez-Cruz M, Baker J, Hsu K, et al. T cells expressing chimeric antigen receptor promote immune tolerance. JCI Insight (2017) 2(20):e92865. doi: 10.1172/jci.insight.92865
18. Boroughs AC, Larson RC, Choi BD, Bouffard AA, Riley LS, Schiferle E, et al. Chimeric antigen receptor costimulation domains modulate human regulatory T cell function. JCI Insight (2019) 5(8):e126194. doi: 10.1172/jci.insight.126194
19. Honaker Y, Hubbard N, Xiang Y, Fisher L, Hagin D, Sommer K, et al. Gene editing to induce FOXP3 expression in human CD4+ T cells leads to a stable regulatory phenotype and function. Sci Transl Med (2020) 12(546). doi: 10.1126/scitranslmed.aay6422
20. Seng A, Krausz KL, Pei D, Koestler DC, Fischer RT, Yankee TM, et al. Coexpression of FOXP3 and a Helios isoform enhances the effectiveness of human engineered regulatory T cells. Blood Adv (2020) 4(7):1325–39. doi: 10.1182/bloodadvances.2019000965
21. Dawson NAJ, Rosado-Sánchez I, Novakovsky GE, Fung VCW, Huang Q, McIver E, et al. Functional effects of chimeric antigen receptor co-receptor signaling domains in human regulatory T cells. Sci Transl Med (2020) 12(557). doi: 10.1126/scitranslmed.aaz3866
22. Cao J, Chen C, Zeng L, Li L, Li Z, Xu K. Engineered regulatory T cells prevent graft-versus-host disease while sparing the graft-versus-leukemia effect after bone marrow transplantation. Leukemia Res (2010) 34(10):1374–82. doi: 10.1016/j.leukres.2009.11.024
23. Kim YC, Zhang AH, Su Y, Rieder SA, Rossi RJ, Ettinger RA, et al. Engineered antigen-specific human regulatory T cells: immunosuppression of FVIII-specific T- and B-cell responses. Blood (2015) 125(7):1107–15. doi: 10.1182/blood-2014-04-566786
24. Yoon J, Schmidt A, Zhang AH, Konigs C, Kim YC, Scott DW. FVIII-specific human chimeric antigen receptor T-regulatory cells suppress T- and B-cell responses to FVIII. Blood (2017) 129(2):238–45. doi: 10.1182/blood-2016-07-727834
25. Herzog RW, Kuteyeva V, Saboungi R, Terhorst C, Biswas M. Reprogrammed CD4+ T Cells That Express FoxP3+ Control Inhibitory Antibody Formation in Hemophilia A Mice. Front Immunol (2019) 10:274. doi: 10.3389/fimmu.2019.00274
26. Passerini L, Rossi Mel E, Sartirana C, Fousteri G, Bondanza A, Naldini L, et al. CD4+ T cells from IPEX patients convert into functional and stable regulatory T cells by FOXP3 gene transfer. Sci Transl Med (2013) 5(215):215ra174. doi: 10.1126/scitranslmed.3007320
27. Goodwin M, Lee E, Lakshmanan U, Shipp S, Froessl L, Barzaghi F, et al. CRISPR-based gene editing enables FOXP3 gene repair in IPEX patient cells. Sci Adv (2020) 6(19):eaaz0571. doi: 10.1126/sciadv.aaz0571
28. Abdeladhim M, Zhang AH, Kropp LE, Lindrose AR, Venkatesha SH, Mitre E, et al. Engineered ovalbumin-expressing regulatory T cells protect against anaphylaxis in ovalbumin-sensitized mice. Clin Immunol (2019) 207:49–54. doi: 10.1016/j.clim.2019.07.009
29. Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science (2003) 299(5609):1057–61. doi: 10.1126/science.1079490
30. Elinav E, Adam N, Waks T, Eshhar Z. Amelioration of colitis by genetically engineered murine regulatory T cells redirected by antigen-specific chimeric receptor. Gastroenterology (2009) 136(5):1721–31. doi: 10.1053/j.gastro.2009.01.049
31. Blat D, Zigmond E, Alteber Z, Waks T, Eshhar Z. Suppression of Murine Colitis and its Associated Cancer by Carcinoembryonic Antigen-Specific Regulatory T Cells. Mol Ther (2014) 22(5):1018–28. doi: 10.1038/mt.2014.41
32. Someya K, Nakatsukasa H, Ito M, Kondo T, Tateda K-I, Akanuma T, et al. Improvement of Foxp3 stability through CNS2 demethylation by TET enzyme induction and activation. Int Immunol (2017) 29(8):365–75. doi: 10.1093/intimm/dxx049
33. Jaeckel E, von Boehmer H, Manns MP. Antigen-specific FoxP3-transduced T-cells can control established type 1 diabetes. Diabetes (2005) 54(2):306–10. doi: 10.2337/diabetes.54.2.306
34. Tenspolde M, Zimmermann K, Weber LC, Hapke M, Lieber M, Dywicki J, et al. Regulatory T cells engineered with a novel insulin-specific chimeric antigen receptor as a candidate immunotherapy for type 1 diabetes. J Autoimmun (2019) 103. doi: 10.1016/j.jaut.2019.05.017
35. Fransson M, Piras E, Burman J, Nilsson B, Essand M, Lu B, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. (2012) 9:112. doi: 10.1186/1742-2094-9-112
36. Malviya M, Saoudi A, Bauer J, Fillatreau S, Liblau R. Treatment of experimental autoimmune encephalomyelitis with engineered bi-specific Foxp3+ regulatory CD4+ T cells. J Autoimmun (2020) 108:102401. doi: 10.1016/j.jaut.2020.102401
37. Kim YC, Zhang AH, Yoon J, Culp WE, Lees JR, Wucherpfennig KW, et al. Engineered MBP-specific human Tregs ameliorate MOG-induced EAE through IL-2-triggered inhibition of effector T cells. J Autoimmun 2018/06/03 Ed (2018) 92:77–86. doi: 10.1016/j.jaut.2018.05.003
38. Fujio K, Okamoto A, Araki Y, Shoda H, Tahara H, Tsuno NH, et al. Gene Therapy of Arthritis with TCR Isolated from the Inflamed Paw. J Immunol (2006) 177(11):8140. doi: 10.4049/jimmunol.177.11.8140
39. Andersen KG, Butcher T, Betz AG. Specific immunosuppression with inducible Foxp3-transduced polyclonal T cells. PloS Biol (2008) 116(11):e276. doi: 10.1371/journal.pbio.0060276
40. Wright GP, Notley CA, Xue S-A, Bendle GM, Holler A, Schumacher TN, et al. Adoptive therapy with redirected primary regulatory T cells results in antigen-specific suppression of arthritis. Proc Natl Acad Sci USA (2009) 106(45):19078–83. doi: 10.1073/pnas.0907396106
41. Qian Z, Latham KA, Whittington KB, Miller DC, Brand DD, Rosloniec EF. Engineered regulatory T cells coexpressing MHC class II:peptide complexes are efficient inhibitors of autoimmune T cell function and prevent the development of autoimmune arthritis. J Immunol (2013) 190(11):5382–91. doi: 10.4049/jimmunol.1300024
42. Kansal R, Richardson N, Neeli I, Khawaja S, Chamberlain D, Ghani M, et al. Sustained B cell depletion by CD19-targeted CAR T cells is a highly effective treatment for murine lupus. Sci Transl Med (2019) 11(482). doi: 10.1126/scitranslmed.aav1648
43. Loser K, Hansen W, Apelt J, Balkow S, Buer J, Beissert S. In vitro-generated regulatory T cells induced by Foxp3-retrovirus infection control murine contact allergy and systemic autoimmunity. Gene Ther (2005) 12(17):1294–304. doi: 10.1038/sj.gt.3302567
44. Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science (2016) 353(6295):179–84. doi: 10.1126/science.aaf6756
45. Zhou Q, Uhlig KM, Muth A, Kimpel J, Lévy C, Münch RC, et al. Exclusive Transduction of Human CD4 + T Cells upon Systemic Delivery of CD4-Targeted Lentiviral Vectors. JI (2015) 195(5):2493–501. doi: 10.4049/jimmunol.1500956
46. Ivics Z, Hackett PB, Plasterk RH, Izsvák Z. Molecular Reconstruction of Sleeping Beauty, a Tc1-like Transposon from Fish, and Its Transposition in Human Cells. Cell (1997) 91(4):501–10. doi: 10.1016/S0092-8674(00)80436-5
47. Huang X, Wilber AC, Bao L, Tuong D, Tolar J, Orchard PJ, et al. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood (2006) 107(2):483–91. doi: 10.1182/blood-2005-05-2133
48. Hacein-Bey-Abina S. LMO2-Associated Clonal T Cell Proliferation in Two Patients after Gene Therapy for SCID-X1. Science (2003) 302(5644):415–9. doi: 10.1126/science.1088547
49. Scholler J, Brady TL, Binder-Scholl G, Hwang W-T, Plesa G, Hege KM, et al. Decade-Long Safety and Function of Retroviral-Modified Chimeric Antigen Receptor T Cells. Sci Trans Med (2012) 4(132):132ra53–132ra53. doi: 10.1126/scitranslmed.3003761
50. Marcucci KT, Jadlowsky JK, Hwang W-T, Suhoski-Davis M, Gonzalez VE, Kulikovskaya I, et al. Retroviral and Lentiviral Safety Analysis of Gene-Modified T Cell Products and Infused HIV and Oncology Patients. Mol Ther (2018) 26(1):269–79. doi: 10.1016/j.ymthe.2017.10.012
51. Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-Term Follow-up of CD19 CAR Therapy in Acute Lymphoblastic Leukemia. N Engl J Med (2018) 378(5):449–59. doi: 10.1056/NEJMoa1709919
52. Smith TT, Stephan SB, Moffett HF, McKnight LE, Ji W, Reiman D, et al. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat Nanotech (2017) 12(8):813–20. doi: 10.1038/nnano.2017.57
53. Mátés L, Chuah MKL, Belay E, Jerchow B, Manoj N, Acosta-Sanchez A, et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat Genet (2009) 41(6):753–61. doi: 10.1038/ng.343
54. Querques I, Mades A, Zuliani C, Miskey C, Alb M, Grueso E, et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat Biotechnol (2019) 37(12):1502–12. doi: 10.1038/s41587-019-0291-z
55. Clauss J, Obenaus M, Miskey C, Ivics Z, Izsvák Z, Uckert W, et al. Efficient Non-Viral T-Cell Engineering by Sleeping Beauty Minicircles Diminishing DNA Toxicity and miRNAs Silencing the Endogenous T-Cell Receptors. Hum Gene Ther (2018) 29(5):569–84. doi: 10.1089/hum.2017.136
56. Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol (2008) 26(7):808–16. doi: 10.1038/nbt1410
57. Tebas P, Stein D, Tang WW, Frank I, Wang SQ, Lee G, et al. Gene Editing of CCR5 in Autologous CD4 T Cells of Persons Infected with HIV. N Engl J Med (2014) 370(10):901–10. doi: 10.1056/NEJMoa1300662
58. Qasim W, Zhan H, Samarasinghe S, Adams S, Amrolia P, Stafford S, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med (2017) 9(374):eaaj2013. doi: 10.1126/scitranslmed.aaj2013
59. Lal G, Zhang N, van der Touw W, Ding Y, Ju W, Bottinger EP, et al. Epigenetic regulation of Foxp3 expression in regulatory T cells by DNA methylation. J Immunol (2009) 182(1):259–73. doi: 10.4049/jimmunol.182.1.259
60. Liu Y, Wang L, Han R, Beier UH, Akimova T, Bhatti T, et al. Two histone/protein acetyltransferases, CBP and p300, are indispensable for Foxp3+ T-regulatory cell development and function. Mol Cell Biol (2014) 34(21):3993–4007. doi: 10.1128/MCB.00919-14
61. Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, et al. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol Cell Biol (2011) 31(5):1022–9. doi: 10.1128/MCB.01206-10
62. Wohlfert EA, Grainger JR, Bouladoux N, Konkel JE, Oldenhove G, Ribeiro CH, et al. GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J Clin Invest (2011) 121(11):4503–15. doi: 10.1172/JCI57456
63. Loo C-S, Gatchalian J, Liang Y, Leblanc M, Xie M, Ho J, et al. A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function. Immunity 2020 53(1):143–157.e8.
64. Quintana FJ, Basso AS, Iglesias AH, Korn T, Farez MF, Bettelli E, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature (2008) 453(7191):65–71. doi: 10.1038/nature06880
65. Dang EV, Barbi J, Yang H-Y, Jinasena D, Yu H, Zheng Y, et al. Control of T(H)17/T(reg) balance by hypoxia-inducible factor 1. Cell (2011) 146(5):772–84. doi: 10.1016/j.cell.2011.07.033
66. Overacre-Delgoffe AE, Chikina M, Dadey RE, Yano H, Brunazzi EA, Shayan G, et al. Interferon-γ Drives Treg Fragility to Promote Anti-tumor Immunity. Cell (2017) 169(6):1130–41.e11. doi: 10.1016/j.cell.2017.05.005
67. Schumann J, Stanko K, Woertge S, Appelt C, Schumann M, Kuhl AA, et al. The mitochondrial protein TCAIM regulates activation of T cells and thereby promotes tolerance induction of allogeneic transplants. Am J Transplant 2014/11/05 Ed (2014) 14(12):2723–35. doi: 10.1111/ajt.12941
68. Durand M, Dubois F, Dejou C, Durand E, Danger R, Chesneau M, et al. Increased degradation of ATP is driven by memory regulatory T cells in kidney transplantation tolerance. Kidney Int (2018) 93(5):1154–64. doi: 10.1016/j.kint.2017.12.004
69. Álvarez-Sánchez N, Cruz-Chamorro I, Díaz-Sánchez M, Lardone PJ, Guerrero JM, Carrillo-Vico A. Peripheral CD39-expressing T regulatory cells are increased and associated with relapsing-remitting multiple sclerosis in relapsing patients. Sci Rep (2019) 9(1):2302. doi: 10.1038/s41598-019-38897-w
70. Wang X, Chang W-C, Wong CW, Colcher D, Sherman M, Ostberg JR, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood (2011) 118(5):1255–63. doi: 10.1182/blood-2011-02-337360
71. Philip B, Kokalaki E, Mekkaoui L, Thomas S, Straathof K, Flutter B, et al. A highly compact epitope-based marker/suicide gene for easier and safer T-cell therapy. Blood (2014) 124(8):1277–87. doi: 10.1182/blood-2014-01-545020
72. Mestermann K, Giavridis T, Weber J, Rydzek J, Frenz S, Nerreter T, et al. The tyrosine kinase inhibitor dasatinib acts as a pharmacologic on/off switch for CAR T cells. Sci Transl Med (2019) 11(499). doi: 10.1126/scitranslmed.aau5907
73. Kinoshita M, Kayama H, Kusu T, Yamaguchi T, Kunisawa J, Kiyono H, et al. Dietary folic acid promotes survival of Foxp3+ regulatory T cells in the colon. J Immunol (2012) 189(6):2869–78. doi: 10.4049/jimmunol.1200420
74. Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci (1996) 93(3):1156–60. doi: 10.1073/pnas.93.3.1156
75. Porteus MH. Chimeric Nucleases Stimulate Gene Targeting in Human Cells. Science (2003) 300(5620):763–3. doi: 10.1126/science.1078395
76. Urnov FD, Miller JC, Lee Y-L, Beausejour CM, Rock JM, Augustus S, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature (2005) 435(7042):646–51. doi: 10.1038/nature03556
77. Nishimasu H, Shi X, Ishiguro S, Gao L, Hirano S, Okazaki S, et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science (2018) 361(6408):1259–62. doi: 10.1126/science.aas9129
78. Huang TP, Zhao KT, Miller SM, Gaudelli NM, Oakes BL, Fellmann C, et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat Biotechnol (2019) 37(6):626–31. doi: 10.1038/s41587-019-0134-y
79. Walton RT, Christie KA, Whittaker MN, Kleinstiver BP. Unconstrained genome targeting with near-PAMless engineered CRISPR-Cas9 variants. Science (2020) 368(6488):290–6. doi: 10.1126/science.aba8853
80. Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature (2018) 559(7714):405–9. doi: 10.1038/s41586-018-0326-5
81. Cortez JT, Montauti E, Shifrut E, Gatchalian J, Zhang Y, Shaked O, et al. CRISPR screen in regulatory T cells reveals modulators of Foxp3. Nature 2020 582(7812):416–20. doi: 10.1038/s41586-020-2246-4.
82. Broeders M, Herrero-Hernandez P, Ernst MPT, van der Ploeg AT, Pijnappel WWMP. Sharpening the Molecular Scissors: Advances in Gene-Editing Technology. iScience (2020) 23(1):100789. doi: 10.1016/j.isci.2019.100789
83. Rees HA, Liu DR. Base editing: precision chemistry on the genome and transcriptome of living cells. Nat Rev Genet (2018) 19(12):770–88. doi: 10.1038/s41576-018-0059-1
84. Webber BR, Lonetree C, Kluesner MG, Johnson MJ, Pomeroy EJ, Diers MD, et al. Highly efficient multiplex human T cell engineering without double-strand breaks using Cas9 base editors. Nat Commun (2019) 10(1):5222. doi: 10.1038/s41467-019-13778-y
85. Anzalone AV, Randolph PB, Davis JR, Sousa AA, Koblan LW, Levy JM, et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature (2019) 576(7785):149–57. doi: 10.1038/s41586-019-1711-4
86. Bak RO, Porteus MH. CRISPR-Mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cell Reports (2017) 20(3):750–6. doi: 10.1016/j.celrep.2017.06.064
87. Ibraheim R, Song C-Q, Mir A, Amrani N, Xue W, Sontheimer EJ. All-in-one adeno-associated virus delivery and genome editing by Neisseria meningitidis Cas9 in vivo. Genome Biol (2018) 19(1):137. doi: 10.1186/s13059-018-1515-0
88. Krooss SA, Dai Z, Schmidt F, Rovai A, Fakhiri J, Dhingra A, et al. Ex Vivo/In vivo Gene Editing in Hepatocytes Using “All-in-One” CRISPR-Adeno-Associated Virus Vectors with a Self-Linearizing Repair Template. iScience (2020) 23(1):100764. doi: 10.1016/j.isci.2019.100764
89. Ortinski PI, O’Donovan B, Dong X, Kantor B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol Ther - Methods Clin Dev (2017) 5:153–64. doi: 10.1016/j.omtm.2017.04.002
90. Lyu P, Javidi-Parsijani P, Atala A, Lu B. Delivering Cas9/sgRNA ribonucleoprotein (RNP) by lentiviral capsid-based bionanoparticles for efficient ‘hit-and-run’ genome editing. Nucleic Acids Res (2019) 47(17):e99–9. doi: 10.1093/nar/gkz605
91. Lee CM, Cradick TJ, Fine EJ, Bao G. Nuclease Target Site Selection for Maximizing On-target Activity and Minimizing Off-target Effects in Genome Editing. Mol Ther (2016) 24(3):475–87. doi: 10.1038/mt.2016.1
92. Kosicki M, Tomberg K, Bradley A. Repair of double-strand breaks induced by CRISPR–Cas9 leads to large deletions and complex rearrangements. Nat Biotechnol (2018) 36(8):765–71. doi: 10.1038/nbt.4192
93. Cullot G, Boutin J, Toutain J, Prat F, Pennamen P, Rooryck C, et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat Commun (2019) 10(1):1136. doi: 10.1038/s41467-019-09006-2
94. Ren J, Zhang X, Liu X, Fang C, Jiang S, June CH, et al. A versatile system for rapid multiplex genome-edited CAR T cell generation. Oncotarget (2017) 8(10):17002–11. doi: 10.18632/oncotarget.15218
95. Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR-engineered T cells in patients with refractory cancer. Science (2020) 367(6481):eaba7365. doi: 10.1126/science.aba7365
96. Gorovits B, Koren E. Immunogenicity of Chimeric Antigen Receptor T-Cell Therapeutics. BioDrugs (2019) 33(3):275–84. doi: 10.1007/s40259-019-00354-5
97. Hege KM, Bergsland EK, Fisher GA, Nemunaitis JJ, Warren RS, McArthur JG, et al. Safety, tumor trafficking and immunogenicity of chimeric antigen receptor (CAR)-T cells specific for TAG-72 in colorectal cancer. J Immunotherapy cancer (2017) 5(1):22. doi: 10.1186/s40425-017-0222-9
98. Wagner DL, Amini L, Wendering DJ, Burkhardt L-M, Akyüz L, Reinke P, et al. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat Med (2019) 25(2):242–8. doi: 10.1038/s41591-018-0204-6
99. Hoffmann P, Boeld TJ, Eder R, Huehn J, Floess S, Wieczorek G, et al. Loss of FOXP3 expression in natural human CD4+CD25+ regulatory T cells upon repetitive in vitro stimulation. Eur J Immunol (2009) 39(4):1088–97. doi: 10.1002/eji.200838904
100. Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martínez-Llordella M, Ashby M, et al. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat Immunol (2009) 10(9):1000–7. doi: 10.1038/ni.1774
101. Beavis PA, Gregory B, Green P, Cribbs AP, Kennedy A, Amjadi P, et al. Resistance to regulatory T cell-mediated suppression in rheumatoid arthritis can be bypassed by ectopic foxp3 expression in pathogenic synovial T cells. Proc Natl Acad Sci USA (2011) 108(40):16717–22. doi: 10.1073/pnas.1112722108
102. Allan SE, Alstad AN, Merindol N, Crellin NK, Amendola M, Bacchetta R, et al. Generation of potent and stable human CD4+ T regulatory cells by activation-independent expression of FOXP3. Mol Ther (2008) 16(1):194–202. doi: 10.1038/sj.mt.6300341
103. Aarts-Riemens T, Emmelot ME, Verdonck LF, Mutis T. Forced overexpression of either of the two common human Foxp3 isoforms can induce regulatory T cells from CD4(+)CD25(-) cells. Eur J Immunol (2008) 38(5):1381–90. doi: 10.1002/eji.200737590
104. Forstnerič V, Oven I, Ogorevc J, Lainšček D, Praznik A, Lebar T, et al. CRISPRa-mediated FOXP3 gene upregulation in mammalian cells. Cell Biosci (2019) 9:93. doi: 10.1186/s13578-019-0357-0
105. Kim H-P, Leonard WJ. CREB/ATF-dependent T cell receptor-induced FoxP3 gene expression: a role for DNA methylation. J Exp Med (2007) 204(7):1543–51. doi: 10.1084/jem.20070109
106. Janson PCJ, Winerdal ME, Marits P, Thörn M, Ohlsson R, Winqvist O. FOXP3 promoter demethylation reveals the committed Treg population in humans. PloS One (2008) 3(2):e1612. doi: 10.1371/journal.pone.0001612
107. Polansky JK, Kretschmer K, Freyer J, Floess S, Garbe A, Baron U, et al. DNA methylation controls Foxp3 gene expression. Eur J Immunol (2008) 38(6):1654–63. doi: 10.1002/eji.200838105
108. Okada M, Kanamori M, Someya K, Nakatsukasa H, Yoshimura A. Stabilization of Foxp3 expression by CRISPR-dCas9-based epigenome editing in mouse primary T cells. Epigenet Chromatin (2017) 10:24. doi: 10.1186/s13072-017-0129-1
109. Tao R, de Zoeten EF, Ozkaynak E, Wang L, Li B, Greene MI, et al. Histone deacetylase inhibitors and transplantation. Curr Opin Immunol (2007) 19(5):589–95. doi: 10.1016/j.coi.2007.07.015
110. Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat Med (2007) 13(11):1299–307. doi: 10.1038/nm1652
111. Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, et al. TGF-beta and IL-6 signals modulate chromatin binding and promoter occupancy by acetylated FOXP3. Proc Natl Acad Sci USA (2008) 105(37):14023–7. doi: 10.1073/pnas.0806726105
112. van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YYJ, Beekman JM, van Beekum O, et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood (2010) 115(5):965–74. doi: 10.1182/blood-2009-02-207118
113. Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Histone acetyltransferase mediated regulation of FOXP3 acetylation and Treg function. Curr Opin Immunol (2010) 22(5):583–91. doi: 10.1016/j.coi.2010.08.013
114. Chen Z, Barbi J, Bu S, Yang H-Y, Li Z, Gao Y, et al. The ubiquitin ligase Stub1 negatively modulates regulatory T cell suppressive activity by promoting degradation of the transcription factor Foxp3. Immunity (2013) 39(2):272–85. doi: 10.1016/j.immuni.2013.08.006
115. Ni X, Kou W, Gu J, Wei P, Wu X, Peng H, et al. TRAF6 directs FOXP3 localization and facilitates regulatory T-cell function through K63-linked ubiquitination. EMBO J (2019) 38(9). doi: 10.15252/embj.201899766
116. Xu Y, Melo-Cardenas J, Zhang Y, Gau I, Wei J, Montauti E, et al. The E3 ligase Hrd1 stabilizes Tregs by antagonizing inflammatory cytokine-induced ER stress response. JCI Insight (2019) 4(5). doi: 10.1172/jci.insight.121887
117. Deng G, Nagai Y, Xiao Y, Li Z, Dai S, Ohtani T, et al. Pim-2 Kinase Influences Regulatory T Cell Function and Stability by Mediating Foxp3 Protein N-terminal Phosphorylation. J Biol Chem (2015) 290(33):20211–20. doi: 10.1074/jbc.M115.638221
118. Morawski PA, Mehra P, Chen C, Bhatti T, Wells AD. Foxp3 protein stability is regulated by cyclin-dependent kinase 2. J Biol Chem (2013) 288(34):24494–502. doi: 10.1074/jbc.M113.467704
119. Semenza GL. Hypoxia-inducible factor 1 (HIF-1) pathway. Sci STKE (2007) 2007(407):cm8. doi: 10.1126/stke.4072007cm8
120. Nakamura H, Makino Y, Okamoto K, Poellinger L, Ohnuma K, Morimoto C, et al. TCR engagement increases hypoxia-inducible factor-1 alpha protein synthesis via rapamycin-sensitive pathway under hypoxic conditions in human peripheral T cells. J Immunol (2005) 174(12):7592–9. doi: 10.4049/jimmunol.174.12.7592
121. Shehade H, Acolty V, Moser M, Oldenhove G. Cutting Edge: Hypoxia-Inducible Factor 1 Negatively Regulates Th1 Function. J Immunol (2015) 195(4):1372–6. doi: 10.4049/jimmunol.1402552
122. Lee JH, Elly C, Park Y, Liu Y-C. E3 ubiquitin ligase VHL regulates hypoxia-inducible factor-1α to maintain regulatory T cell stability and suppressive capacity. Immunity (2015) 42(6):1062–74.
123. Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, et al. HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med (2011) 208(7):1367–76. doi: 10.1084/jem.20110278
124. Feldhoff LM, Rueda CM, Moreno-Fernandez ME, Sauer J, Jackson CM, Chougnet CA, et al. IL-1β induced HIF-1α inhibits the differentiation of human FOXP3+ T cells. Sci Rep (2017) 7(1):465.
125. Ben-Shoshan J, Schwartz S, Luboshits G, Maysel-Auslender S, Barzelay A, Polak-Charcon S, et al. Constitutive expression of HIF-1alpha and HIF-2alpha in bone marrow stromal cells differentially promotes their proangiogenic properties. Stem Cells Dayt Ohio (2008) 26(10):2634–43.
126. Clambey ET, McNamee EN, Westrich JA, Glover LE, Campbell EL, Jedlicka P, et al. Hypoxia-inducible factor-1 alpha-dependent induction of FoxP3 drives regulatory T-cell abundance and function during inflammatory hypoxia of the mucosa. Proc Natl Acad Sci USA (2012) 109(41):E2784–2793.
127. Ye J, Qiu J, Bostick JW, Ueda A, Schjerven H, Li S, et al. The Aryl Hydrocarbon Receptor Preferentially Marks and Promotes Gut Regulatory T Cells. Cell Rep (2017) 21(8):2277–90. doi: 10.1016/j.celrep.2017.10.114
128. Gandhi R, Kumar D, Burns EJ, Nadeau M, Dake B, Laroni A, et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat Immunol (2010) 11(9):846–53.
129. Zamali I, Rekik R, Belhadj Hmida N, Ben Hmid A, Kammoun O, Barbouche M-R, et al. An endogenous aryl hydrocarbon receptor ligand enhances de novo generation of regulatory T cells in humans. J Leukoc Biol (2019) 105(2):291–5.
130. Li Z, Lin F, Zhuo C, Deng G, Chen Z, Yin S, et al. PIM1 kinase phosphorylates the human transcription factor FOXP3 at serine 422 to negatively regulate its activity under inflammation. J Biol Chem (2014) 289(39):26872–81. doi: 10.1074/jbc.M114.586651
131. Korn T, Mitsdoerffer M, Croxford AL, Awasthi A, Dardalhon VA, Galileos G, et al. IL-6 controls Th17 immunity in vivo by inhibiting the conversion of conventional T cells into Foxp3+ regulatory T cells. Proc Natl Acad Sci USA (2008) 105(47):18460–5. doi: 10.1073/pnas.0809850105
132. Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, et al. Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheumatol (2008) 58(12):3710–9. doi: 10.1002/art.24126
133. Jones SA, Scheller J, Rose-John S. Therapeutic strategies for the clinical blockade of IL-6/gp130 signaling. J Clin Invest (2011) 121(9):3375–83. doi: 10.1172/JCI57158
134. Hagenstein J, Melderis S, Nosko A, Warkotsch MT, Richter JV, Ramcke T, et al. A Novel Role for IL-6 Receptor Classic Signaling: Induction of ROR γ t + Foxp3 + Tregs with Enhanced Suppressive Capacity. JASN (2019) 30(8):1439–53. doi: 10.1681/ASN.2019020118
135. Schenk U, Frascoli M, Proietti M, Geffers R, Traggiai E, Buer J, et al. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci Signal (2011) 4(162):ra12. doi: 10.1126/scisignal.2001270
136. Figliuolo VR, Savio LEB, Safya H, Nanini H, Bernardazzi C, Abalo A, et al. P2X7 receptor promotes intestinal inflammation in chemically induced colitis and triggers death of mucosal regulatory T cells. Biochim Biophys Acta Mol Basis Dis (2017) 1863(6):1183–94. doi: 10.1016/j.bbadis.2017.03.004
137. Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood (1997) 90(4):1600–10. doi: 10.1182/blood.V90.4.1600
138. Ohta A, Kini R, Ohta A, Subramanian M, Madasu M, Sitkovsky M. The development and immunosuppressive functions of CD4(+) CD25(+) FoxP3(+) regulatory T cells are under influence of the adenosine-A2A adenosine receptor pathway. Front Immunol (2012) 3:190. doi: 10.3389/fimmu.2012.00190
139. Saraiva M, Vieira P, O’Garra A. Biology and therapeutic potential of interleukin-10. J Exp Med (2020) 217(1). doi: 10.1084/jem.20190418
140. Levings MK, Sangregorio R, Sartirana C, Moschin AL, Battaglia M, Orban PC, et al. Human CD25+CD4+ T suppressor cell clones produce transforming growth factor beta, but not interleukin 10, and are distinct from type 1 T regulatory cells. J Exp Med (2002) 196(10):1335–46. doi: 10.1084/jem.20021139
141. Bézie S, Picarda E, Ossart J, Tesson L, Usal C, Renaudin K, et al. IL-34 is a Treg-specific cytokine and mediates transplant tolerance. J Clin Invest (2015) 125(10):3952–64. doi: 10.1172/JCI81227
142. Guillonneau C, Bézie S, Anegon I. Immunoregulatory properties of the cytokine IL-34. Cell Mol Life Sci (2017) 74(14):2569–86. doi: 10.1007/s00018-017-2482-4
143. Sawant DV, Hamilton K, Vignali DAA. Interleukin-35: Expanding Its Job Profile. J Interferon Cytokine Res (2015) 35(7):499–512. doi: 10.1089/jir.2015.0015
144. Bézie S, Picarda E, Tesson L, Renaudin K, Durand J, Ménoret S, et al. Fibrinogen-Like Protein 2/Fibroleukin Induces Long-Term Allograft Survival in a Rat Model through Regulatory B Cells. Chatenoud L, editor. PloS One (2015) 10(3):e0119686. doi: 10.1371/journal.pone.0119686
145. Hu J, Yan J, Rao G, Latha K, Overwijk WW, Heimberger AB, et al. The Duality of Fgl2 - Secreted Immune Checkpoint Regulator Versus Membrane-Associated Procoagulant: Therapeutic Potential and Implications. Int Rev Immunol (2016) 35(4):325–39. doi: 10.3109/08830185.2014.956360
146. Silva SL, Albuquerque AS, Serra-Caetano A, Foxall RB, Pires AR, Matoso P, et al. Human naive regulatory T-cells feature high steady-state turnover and are maintained by IL-7. Oncotarget (2016) 7(11):12163–75. doi: 10.18632/oncotarget.7512
147. Fan MY, Low JS, Tanimine N, Finn KK, Priyadharshini B, Germana SK, et al. Differential Roles of IL-2 Signaling in Developing versus Mature Tregs. Cell Rep (2018) 25(5):1204–13.e4. doi: 10.1016/j.celrep.2018.10.002
148. Whangbo JS, Kim HT, Nikiforow S, Koreth J, Alho AC, Falahee B, et al. Functional analysis of clinical response to low-dose IL-2 in patients with refractory chronic graft-versus-host disease. Blood Adv (2019) 3(7):984–94. doi: 10.1182/bloodadvances.2018027474
149. Khoryati L, Kumari S, Nguyet Pham TM, Bogdani M, Gavin M. Selectively Agonizing Treg in Type 1 Diabetes with IL-2 Muteins. Diabetes (2018) 67(Supplement 1). doi: 10.2337/db18-314-OR
150. Passerini L, Allan SE, Battaglia M, Di Nunzio S, Alstad AN, Levings MK, et al. STAT5-signaling cytokines regulate the expression of FOXP3 in CD4+CD25+ regulatory T cells and CD4+CD25- effector T cells. Int Immunol (2008) 20(3):421–31. doi: 10.1093/intimm/dxn002
151. Sockolosky JT, Trotta E, Parisi G, Picton L, Su LL, Le AC, et al. Selective targeting of engineered T cells using orthogonal IL-2 cytokine-receptor complexes. Science (2018) 359(6379):1037–42. doi: 10.1126/science.aar3246
152. Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, et al. Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discovery (2017) 7(11):1238–47. doi: 10.1158/2159-8290.CD-17-0538
153. Leen AM, Sukumaran S, Watanabe N, Mohammed S, Keirnan J, Yanagisawa R, et al. Reversal of tumor immune inhibition using a chimeric cytokine receptor. Mol Ther (2014) 22(6):1211–20. doi: 10.1038/mt.2014.47
154. Asano T, Meguri Y, Yoshioka T, Kishi Y, Iwamoto M, Nakamura M, et al. PD-1 modulates regulatory T-cell homeostasis during low-dose interleukin-2 therapy. Blood (2017) 129(15):2186–97. doi: 10.1182/blood-2016-09-741629
155. Li Q, Wang Y, Li H, Shen G, Hu S. Ox-LDL influences peripheral Th17/Treg balance by modulating Treg apoptosis and Th17 proliferation in atherosclerotic cerebral infarction. Cell Physiol Biochem (2014) 33(6):1849–62. doi: 10.1159/000362963
156. Tan CL, Kuchroo JR, Sage PT, Liang D, Francisco LM, Buck J, et al. PD-1 restraint of regulatory T cell suppressive activity is critical for immune tolerance. J Exp Med (2021) 218(1).
157. Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med (2002) 8(8):793–800. doi: 10.1038/nm730
158. Gotot J, Dhana E, Yagita H, Kaiser R, Ludwig-Portugall I, Kurts C. Antigen-specific Helios(-) , Neuropilin-1(-) Tregs induce apoptosis of autoreactive B cells via PD-L1. Immunol Cell Biol (2018) 96(8):852–62. doi: 10.1111/imcb.12053
159. Charbonnier L-M, Janssen E, Chou J, Ohsumi TK, Keles S, Hsu JT, et al. Regulatory T-cell deficiency and immune dysregulation, polyendocrinopathy, enteropathy, X-linked–like disorder caused by loss-of-function mutations in LRBA. J Allergy Clin Immunol (2015) 135(1):217–27.e9. doi: 10.1016/j.jaci.2014.10.019
160. Lo B, Zhang K, Lu W, Zheng L, Zhang Q, Kanellopoulou C, et al. AUTOIMMUNE DISEASE. Patients with LRBA deficiency show CTLA4 loss and immune dysregulation responsive to abatacept therapy. Science (2015) 349(6246):436–40. doi: 10.1126/science.aaa1663
161. Howie D, Cobbold SP, Adams E, Ten Bokum A, Necula AS, Zhang W, et al. Foxp3 drives oxidative phosphorylation and protection from lipotoxicity. JCI Insight (2017) 2(3):e89160. doi: 10.1172/jci.insight.89160
162. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) 186(6):3299–303. doi: 10.4049/jimmunol.1003613
163. Berod L, Friedrich C, Nandan A, Freitag J, Hagemann S, Harmrolfs K, et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat Med (2014) 20(11):1327–33. doi: 10.1038/nm.3704
164. Timilshina M, You Z, Lacher SM, Acharya S, Jiang L, Kang Y, et al. Activation of Mevalonate Pathway via LKB1 Is Essential for Stability of Treg Cells. Cell Rep (2019) 27(10):2948–61. doi: 10.1016/j.celrep.2019.05.020
165. Su X, Wang Q, Guo W, Pei X, Niu Q, Liu M, et al. Loss of Lkb1 impairs Treg function and stability to aggravate graft-versus-host disease after bone marrow transplantation. Cell Mol Immunol (2020) 17(5):483–95. doi: 10.1038/s41423-019-0312-3
166. Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab (2017) 25(6):1282–93.e7. doi: 10.1016/j.cmet.2016.12.018
167. He N, Fan W, Henriquez B, Yu RT, Atkins AR, Liddle C, et al. Metabolic control of regulatory T cell (Treg) survival and function by Lkb1. Proc Natl Acad Sci USA (2017) 114(47):12542–7. doi: 10.1073/pnas.1715363114
168. San Segundo D, Ruiz JC, Fernandez-Fresnedo G, Izquierdo M, Gomez-Alamillo C, Cacho E, et al. Calcineurin inhibitors affect circulating regulatory T cells in stable renal transplant recipients. Transplant Proc 2006/11/14 Ed (2006) 38(8):2391–3. doi: 10.1016/j.transproceed.2006.08.081
169. De Angelis B, Dotti G, Quintarelli C, Huye LE, Zhang L, Zhang M, et al. Generation of Epstein-Barr virus-specific cytotoxic T lymphocytes resistant to the immunosuppressive drug tacrolimus (FK506). Blood (2009) 114(23):4784–91. doi: 10.1182/blood-2009-07-230482
170. Amini L, Wagner DL, Rössler U, Zarrinrad G, Wagner LF, Vollmer T, et al. CRISPR-Cas9-Edited Tacrolimus-Resistant Antiviral T Cells for Advanced Adoptive Immunotherapy in Transplant Recipients. Mol Ther (2021) 29(1):32–46. doi: 10.1016/j.ymthe.2020.09.011
171. Basar R, Daher M, Uprety N, Gokdemir E, Alsuliman A, Ensley E, et al. Large-scale GMP-compliant CRISPR-Cas9–mediated deletion of the glucocorticoid receptor in multivirus-specific T cells. Blood Adv (2020) 4(14):3357–67. doi: 10.1182/bloodadvances.2020001977
172. Kaeuferle T, Deisenberger L, Jablonowski L, Stief TA, Blaeschke F, Willier S, et al. CRISPR-Cas9-Mediated Glucocorticoid Resistance in Virus-Specific T Cells for Adoptive T Cell Therapy Posttransplantation. Mol Ther (2020) 28(9):1965–73. doi: 10.1016/j.ymthe.2020.06.002
173. Brewin J, Mancao C, Straathof K, Karlsson H, Samarasinghe S, Amrolia PJ, et al. Generation of EBV-specific cytotoxic T cells that are resistant to calcineurin inhibitors for the treatment of posttransplantation lymphoproliferative disease. Blood (2009) 114(23):4792–803. doi: 10.1182/blood-2009-07-228387
174. Mahic M, Henjum K, Yaqub S, Bjørnbeth BA, Torgersen KM, Taskén K, et al. Generation of highly suppressive adaptive CD8+CD25+FOXP3+ regulatory T cells by continuous antigen stimulation. Eur J Immunol (2008) 38(3):640–6. doi: 10.1002/eji.200737529
175. Zheng J, Liu Y, Qin G, Chan P-L, Mao H, Lam K-T, et al. Efficient Induction and Expansion of Human Alloantigen-Specific CD8 Regulatory T Cells from Naive Precursors by CD40-Activated B Cells. J Immunol (2009) 183(6):3742–50. doi: 10.4049/jimmunol.0901329
176. Picarda E, Bézie S, Venturi V, Echasserieau K, Mérieau E, Delhumeau A, et al. MHC-derived allopeptide activates TCR-biased CD8+ Tregs and suppresses organ rejection. J Clin Invest (2014) 124(6):2497–512. doi: 10.1172/JCI71533
177. Picarda E, Bézie S, Usero L, Ossart J, Besnard M, Halim H, et al. Cross-Reactive Donor-Specific CD8+ Tregs Efficiently Prevent Transplant Rejection. Cell Reports (2019) 29(13):4245–55.e6. doi: 10.1016/j.celrep.2019.11.106
178. Tang Q, Henriksen KJ, Bi M, Finger EB, Szot G, Ye J, et al. In vitro-expanded antigen-specific regulatory T cells suppress autoimmune diabetes. J Exp Med (2004) 199(11):1455–65. doi: 10.1084/jem.20040139
179. Bézie S, Meistermann D, Boucault L, Kilens S, Zoppi J, Autrusseau E, et al. Ex Vivo Expanded Human Non-Cytotoxic CD8+CD45RClow/– Tregs Efficiently Delay Skin Graft Rejection and GVHD in Humanized Mice. Front Immunol (2018) 8:2014. doi: 10.3389/fimmu.2017.02014
180. Adair PR, Kim YC, Zhang A-H, Yoon J, Scott DW. Human Tregs Made Antigen Specific by Gene Modification: The Power to Treat Autoimmunity and Antidrug Antibodies with Precision. Front Immunol (2017) 8:1117. doi: 10.3389/fimmu.2017.01117
181. Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci (1989) 86(24):10024–8. doi: 10.1073/pnas.86.24.10024
182. Yeh W-I, Seay HR, Newby B, Posgai AL, Moniz FB, Michels A, et al. Avidity and Bystander Suppressive Capacity of Human Regulatory T Cells Expressing De Novo Autoreactive T-Cell Receptors in Type 1 Diabetes. Front Immunol (2017) 8:1313. doi: 10.3389/fimmu.2017.01313
183. Brocker T, Riedinger M, Karjalainen K. Redirecting the complete T cell receptor/CD3 signaling machinery towards native antigen via modified T cell receptor. Eur J Immunol (1996) 26(8):1770–4. doi: 10.1002/eji.1830260816
184. Provasi E, Genovese P, Lombardo A, Magnani Z, Liu P-Q, Reik A, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med (2012) 18(5):807–15. doi: 10.1038/nm.2700
185. Hull CM, Nickolay LE, Estorninho M, Richardson MW, Riley JL, Peakman M, et al. Generation of human islet-specific regulatory T cells by TCR gene transfer. J Autoimmun (2017) 79:63–73. doi: 10.1016/j.jaut.2017.01.001
186. Plesa G, Zheng L, Medvec A, Wilson CB, Robles-Oteiza C, Liddy N, et al. TCR affinity and specificity requirements for human regulatory T-cell function. Blood (2012) 119(15):3420–30. doi: 10.1182/blood-2011-09-377051
187. Beringer DX, Kleijwegt FS, Wiede F, van der Slik AR, Loh KL, Petersen J, et al. T cell receptor reversed polarity recognition of a self-antigen major histocompatibility complex. Nat Immunol (2015) 16(11):1153–61. doi: 10.1038/ni.3271
188. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res (2015) 75(17):3596–607. doi: 10.1158/0008-5472.CAN-15-0159
189. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res (2015) 75(17):3505–18. doi: 10.1158/0008-5472.CAN-15-0139
190. Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest (2016) 126(6):2123–38.
191. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X, et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer Immunol Res (2013) 1:26–31.
192. Akatsuka Y. TCR-Like CAR-T Cells Targeting MHC-Bound Minor Histocompatibility Antigens. Front Immunol (2020) 11:257. doi: 10.3389/fimmu.2020.00257
193. Koristka S, Kegler A, Bergmann R, Arndt C, Feldmann A, Albert S, et al. Engrafting human regulatory T cells with a flexible modular chimeric antigen receptor technology. J Autoimmun 2018/03/06 Ed (2018) 90:116–31. doi: 10.1016/j.jaut.2018.02.006
194. Gornalusse GG, Hirata RK, Funk SE, Riolobos L, Lopes VS, Manske G, et al. HLA-E-expressing pluripotent stem cells escape allogeneic responses and lysis by NK cells. Nat Biotechnol (2017) 35(8):765–72. doi: 10.1038/nbt.3860
195. Deuse T, Hu X, Gravina A, Wang D, Tediashvili G, De C, et al. Hypoimmunogenic derivatives of induced pluripotent stem cells evade immune rejection in fully immunocompetent allogeneic recipients. Nat Biotechnol (2019) 37(3):252–8. doi: 10.1038/s41587-019-0016-3
196. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell (2009) 138(2):271–85. doi: 10.1016/j.cell.2009.05.046
197. Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: kinetics, toxicity profile, and clinical effect. Blood (2016) 127(8):1044–51.
198. Brandt LJB, Barnkob MB, Michaels YS, Heiselberg J, Barington T. Emerging Approaches for Regulation and Control of CAR T Cells: A Mini Review. Front Immunol (2020) 11:326. doi: 10.3389/fimmu.2020.00326
199. Timmermans F, Velghe I, Vanwalleghem L, De Smedt M, Van Coppernolle S, Taghon T, et al. Generation of T cells from human embryonic stem cell-derived hematopoietic zones. J Immunol (2009) 182(11):6879–88. doi: 10.4049/jimmunol.0803670
200. Themeli M, Kloss CC, Ciriello G, Fedorov VD, Perna F, Gonen M, et al. Generation of tumor-targeted human T lymphocytes from induced pluripotent stem cells for cancer therapy. Nat Biotechnol (2013) 31(10):928–33. doi: 10.1038/nbt.2678
201. Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, et al. Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med (2015) 7(315):315ra189. doi: 10.1126/scitranslmed.aad4134
202. Sharif-Paghaleh E, Sunassee K, Tavaré R, Ratnasothy K, Koers A, Ali N, et al. In Vivo SPECT Reporter Gene Imaging of Regulatory T Cells. Zimmer J, editor. PloS One (2011) 6(10):e25857. doi: 10.1371/journal.pone.0025857
203. Raffin C, Zhou Y, Piccoli L, Lanzavecchia A, Sadelain M, Tareen SU, et al. Development of citrullinated-vimentin-specific CAR for targeting Tregs to treat autoimmune rheumatoid arthritis. J Immunol (2018) 200(1 Supplement):176.17.
204. Burzyn D, Kuswanto W, Kolodin D, Shadrach JL, Cerletti M, Jang Y, et al. A Special Population of Regulatory T Cells Potentiates Muscle Repair. Cell (2013) 155(6):1282–95. doi: 10.1016/j.cell.2013.10.054
205. Dombrowski Y, O’Hagan T, Dittmer M, Penalva R, Mayoral SR, Bankhead P, et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat Neurosci (2017) 20(5):674–80. doi: 10.1038/nn.4528
206. Nosbaum A, Prevel N, Truong H-A, Mehta P, Ettinger M, Scharschmidt TC, et al. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. JI (2016) 196(5):2010–4. doi: 10.4049/jimmunol.1502139
207. Zacchigna S, Martinelli V, Moimas S, Colliva A, Anzini M, Nordio A, et al. Paracrine effect of regulatory T cells promotes cardiomyocyte proliferation during pregnancy and after myocardial infarction. Nat Commun (2018) 9(1):2432. doi: 10.1038/s41467-018-04908-z
208. Schlundt C, Reinke S, Geissler S, Bucher CH, Giannini C, Märdian S, et al. Individual Effector/Regulator T Cell Ratios Impact Bone Regeneration. Front Immunol (2019) 10:1954. doi: 10.3389/fimmu.2019.01954
209. Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia (2016) 30(2):492–500. doi: 10.1038/leu.2015.247
210. Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, et al. Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell (2016) 167(2):419–432.e16. doi: 10.1016/j.cell.2016.09.011
211. Adigbli G, Ménoret S, Cross AR, Hester J, Issa F, Anegon I. Humanization of Immunodeficient Animals for the Modeling of Transplantation, Graft Versus Host Disease and Regenerative Medicine. Transplantation (2020) 104(11):2290–306. doi: 10.1097/TP.0000000000003177
Keywords: immune tolerance, transplantation, autoimmunity, genome editing, CAR, cell therapy, immune regulation, regulatory T cells
Citation: Amini L, Greig J, Schmueck-Henneresse M, Volk H-D, Bézie S, Reinke P, Guillonneau C, Wagner DL and Anegon I (2021) Super-Treg: Toward a New Era of Adoptive Treg Therapy Enabled by Genetic Modifications. Front. Immunol. 11:611638. doi: 10.3389/fimmu.2020.611638
Received: 29 September 2020; Accepted: 24 December 2020;
Published: 24 February 2021.
Edited by:
James Moon, University of Michigan, United StatesCopyright © 2021 Amini, Greig, Schmueck-Henneresse, Volk, Bézie, Reinke, Guillonneau, Wagner and Anegon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ignacio Anegon, aWFuZWdvbkBuYW50ZXMuaW5zZXJtLmZy; Dimitrios L. Wagner, RGltaXRyaW9zLUwuV2FnbmVyQGNoYXJpdGUuZGU=; Carole Guillonneau, Q2Fyb2xlLkd1aWxsb25uZWF1QHVuaXYtbmFudGVzLmZy
†These authors have contributed equally to this work
‡These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.