 Ting-Yi Lin1
Ting-Yi Lin1 Meng-Chun Tsai1†
Meng-Chun Tsai1† Wei Tu1†
Wei Tu1† Hsin-Chih Yeh1,2,3,4Shu-Chi Wang5Shu-Pin Huang2,4,6,7*
Hsin-Chih Yeh1,2,3,4Shu-Chi Wang5Shu-Pin Huang2,4,6,7* Chia-Yang Li6,7,8*
Chia-Yang Li6,7,8*- 1School of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
- 2Department of Urology, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
- 3Department of Urology, Kaohsiung Municipal Ta-Tung Hospital, Kaohsiung, Taiwan
- 4Department of Urology, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
- 5Department of Medical Laboratory Science and Biotechnology, Kaohsiung Medical University, Kaohsiung, Taiwan
- 6Graduate Institute of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung, Taiwan
- 7Center for Cancer Research, Kaohsiung Medical University, Kaohsiung, Taiwan
- 8Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung, Taiwan
In response to a variety of stresses, mammalian cells activate the inflammasome for targeted caspase-dependent pyroptosis. The research community has recently begun to deduce that the activation of inflammasome is instigated by several known oncogenic stresses and metabolic perturbations; nevertheless, the role of inflammasomes in the context of cancer biology is less understood. In manipulating the expression of inflammasome, researchers have found that NLRP3 serves as a deterministic player in conducting tumor fate decisions. Understanding the mechanistic underpinning of pro-tumorigenic and anti-tumorigenic pathways might elucidate novel therapeutic onco-targets, thereby providing new opportunities to manipulate inflammasome in augmenting the anti-tumorigenic activity to prevent tumor expansion and achieve metastatic control. Accordingly, this review aims to decode the complexity of NLRP3, whereby summarizing and clustering findings into cancer hallmarks and tissue contexts may expedite consensus and underscore the potential of the inflammasome in drug translation.
Introduction
Cancer is a significant public health problem, and cancer deaths are rapidly increasing worldwide (1). In 2000, Drs. Douglas Hanahan and Robert A. Weinberg proposed six hallmarks of cancer, including sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, and activating invasion and metastasis (2). Over the following decade, the authors summarized the latest discoveries into four more hallmarks upon discoveries, such as deregulating cellular energetics, avoiding immune destruction, tumor-promoting inflammation, and genome instability and mutation (3). These universal hallmarks are vital for maintaining tumor development, growth, and adaptation.
Adding a layer of complexity to the oncogenic process, overlapping crosstalk between tumor microenvironment (TME) and local inflammation fuels oncogenic initiation, development and progression. Furthermore, disturbance of the cellular milieu generated by the byproducts of inflammation exacerbates genome instability that promotes the neoplastic disease’s plasticity to expedite tumorigenesis and precipitate multiple hallmarks. Generally poised as the activator of procaspase-1, inflammasomes are cytosolic multi-protein complexes that predominantly engage with innate immunity defense via activation of pro-interleukin (IL)-1β and pro-IL-18 cytokines (4). The active form of IL-18 is essential for interferon-gamma (IFN-γ) production, which augments the cytotoxicity potential of natural killer (NK) cells and T cells (5). Active caspase-1, the pore-forming protein gasdermin D (GSDMD) that disrupts the osmotic potential and results in pyroptosis, is a form of programmed cellular death (6). Inflammasome consists of a danger sensor (nucleotide-binding and oligomerization domain-like receptors, NLR), an adaptor (apoptosis-associated speck-like protein containing a CARD, ASC), and an effector protease (caspase-1) (7). Activation of NLRs with the assembly of the inflammasome requires two sequential activation signals to exert optimal effector response. The cell is initially primed by signal one with the activation of the tumor necrosis factor (TNF), IL-1β, or pathogen-associated molecular patterns (PAMPs) to enable the transcriptional upregulation of NLRP3 inflammasome components and the inactive unspliced cytokines. The inflammasome components are then oligomerized via signal two delivery, consisting of PAMPs or damage-associated molecular patterns (DAMPs). PAMPs are recognized by pattern-recognition receptors (PRRs, including Toll-like receptors, NLRs, RIG-I-like receptors, C-type lectin receptors, etc.), which play a key role in innate immunity for the recognition of pathogens or of cellular injury (8). The DAMPs responsible for NLRP3 activation include a myriad of processes such as perturbation of ion flux, extracellular adenosine triphosphate (ATP), lysosomal degradation, mitochondrial reactive oxygen species (mtROS), and oxidized mitochondrial DNA (ox-mtDNA) (9). The inflammasome cleaves the caspase1-dependent release of the pro-inflammatory cytokines IL-1β, IL-18, and activates pyroptosis (Figure 1) (10).
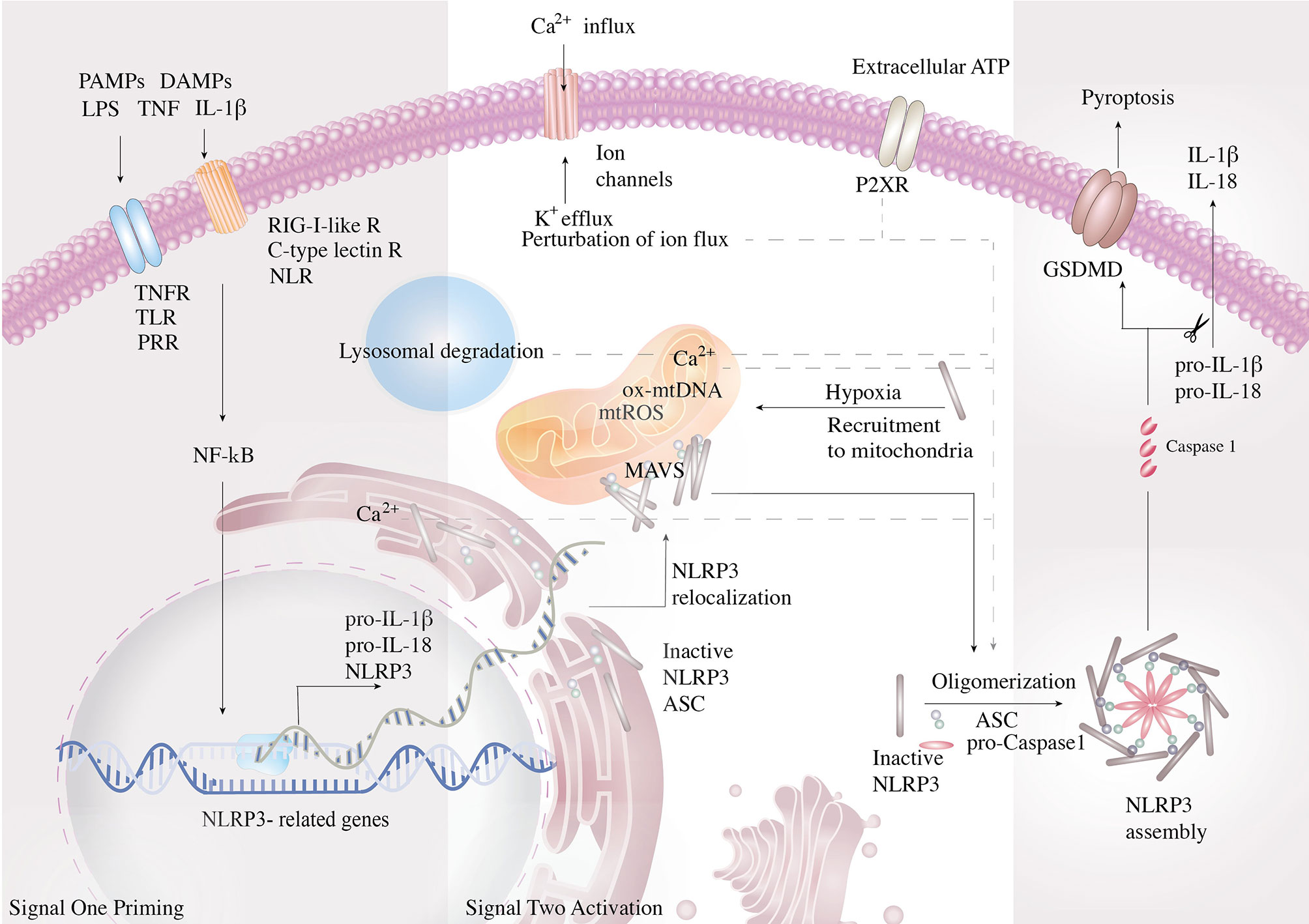
Figure 1 Mechanism and regulation of NLRP3 inflammasome activation. NLRP3 inflammasome is activated in a two-step process. First, NF-κB is initially activated by the pathogen-associated molecular patterns (PAMPs)- and damage-associated molecular pattern (DAMP)-mediated signal cascades and induces the transcription of NLRP3 inflammasome components and the inactive un-spliced cytokines. PAMPs are recognized by pattern-recognition receptors (PRRs, including Toll-like receptors, NLRs, RIG-I-like receptors, C-type lectin receptors). Signal two assembles the inflammasome complex via oligomerization and activates caspase-1, the pore-forming protein gasdermin D (GSDMD), and the inflammatory cytokines. Signal two arises from lysosomal activity. Signal two activating DAMPs consist of perturbation of ion flux, extracellular adenosine triphosphate (ATP), lysosomal degradation, mitochondrial reactive oxygen species (mtROS), and oxidized mitochondrial DNA (ox-mtDNA). To improve efficiency of oligomerization, the organelles work in conjunction to strategically bring the components close to the vital organelles and then together. Resting NLRP3 localizes to endoplasmic reticulum, whereas it is redistributed to mitochondria via mitochondria-associated adaptor molecule (MAV) when activated.
The role of inflammasomes in the context of cancer biology is less understood. NLRP3 recognizes a diverse set of inflammation-inducing stimuli as an intracellular danger sensor, including PAMPs and DAMPs. During cancer expansion, metabolites are actively secreted to the TME as extracellular messengers. ATP, one of the major cancer metabolites and constituents of the TME (11, 12), serves as a key DAMP that activates NLRP3 inflammasome via the purinergic P2X7 receptors (13, 14). The P2XR mediates host–tumor cell interactions and triggers NLRP3 activation via cytosolic Ca2+ influx and K+ efflux, thereby shaping the TME and tumor fate. Besides, hypoxia has been demonstrated to prime the activation of NLRP3 inflammasome (15, 16). Since the TME possesses hypoxic and inflamed sites that accumulate with extracellular ATP, the impacts of NLRP3 inflammasome on the progression of tumor and maintenance of TME have become a critical issue. Herein, we discuss the emerging literature and highlight the role of NLRP3 in oncogenic development by dissecting the functions and justifying each cancer hallmark to gain a comprehensive understanding.
Role of NLRP3 in Regulating the Hallmarks of Cancer
Tumor-Promoting Inflammation
Inflammation is frequently observed at the earliest stage of neoplastic initiation and propels oncogenic development in several types of cancer (17). As a danger-sensing protein complex, NLRP3 is activated by numerous oncogenic inducers such as Helicobacter pylori (H. pylori), human papillomavirus (HPV), uric acid, crystals, and reactive oxygen species (ROS) (18). H. pylori infection enhances NLRP3 expression in gastric cancer and triggers the uncontrolled proliferation of epithelial cells (18). Similarly, inflammasome activation in prostate tissue is mediated by uric acid, crystals, and infections increase the risks of prostate gland injury and prostate cancer progression (19). NLRP3-mediated tumorigenesis is observed not only through its downstream activations, but in the upstream root of genetic variants as well. Pontillo et al. discovered that a variant in the NLRP3 gene, rs10754558 (20) was associated with HPV resistance and showed a statistically significant relationship between rs10754558 and cervical cancer development (21). Epistasis analysis revealed that NLRP3 variants together with polymorphisms in inflammasome-related genes modulate both the frequency of inflammasome activation and the process of IL-1β and IL-18 maturation that influence HPV infection outcome and cervical cancer progression (20) (Table 1).
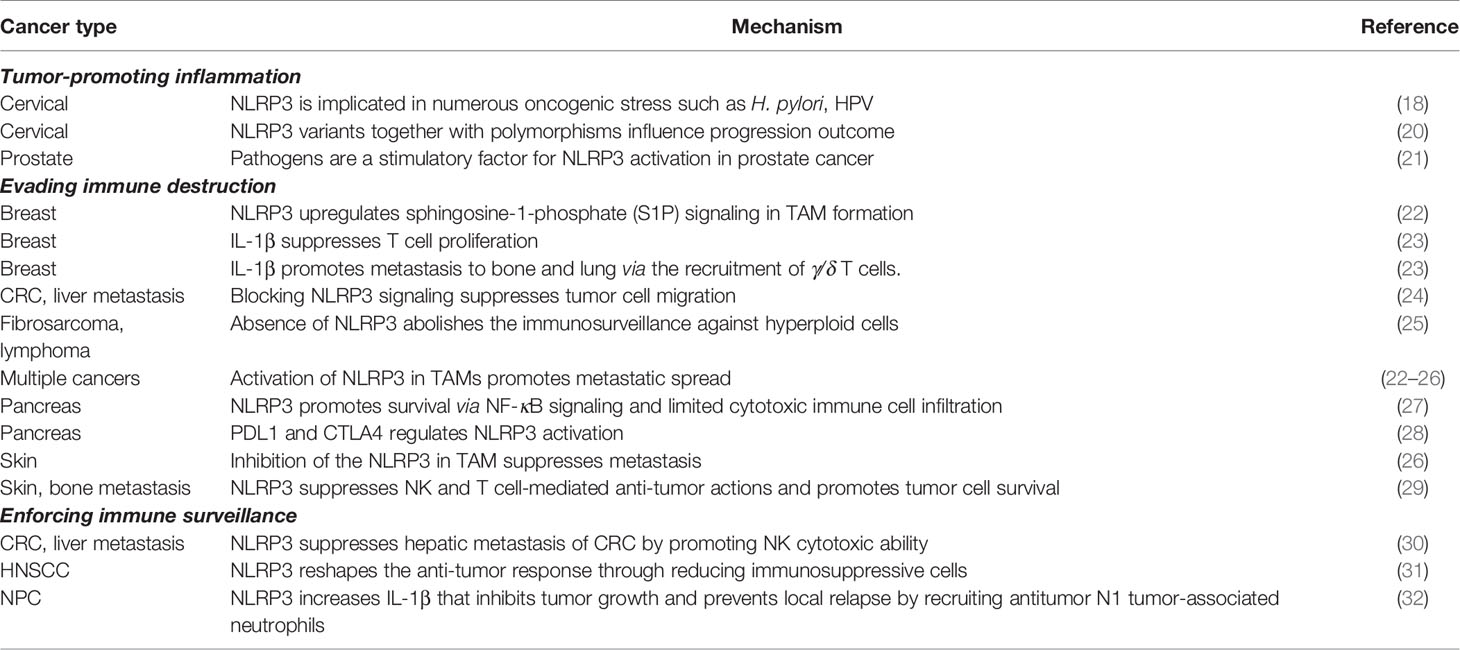
Table 1 Inflammation-associated cancer hallmarks.
Evading Immune Destruction
Immunogenic reprogramming of innate and adaptive immunity within the TME has brought to light the importance of NLRP3 signaling in primary tumors or innate immune cells in mediating immune evasion that not only promotes cancer dissemination but also allows clonogenic survival. Here we discuss the role of NLRP3 activation in immune cells and primary tumor as a failsafe mechanism in inflammation-mediated immune surveillance. Highly immunogenic cancer cells evade immune surveillance by disabling the immune-components or downregulating immunogenic antigens. Recent progress in the action of NLRP3 witnessed remarkable adeptness in paralyzing immune cells dispatched to eliminate cancer by immunosuppressive secretions. The robust immune evasion was emphasized when Aranda et al. demonstrated that the absence of NLRP3 partially abolished immune surveillance against hyperploid tumors (25). Due to the inherent immunogenicity of extra chromosomes, hyperploid cells rarely form tumors or are compromised into slowly growing tumors in vivo (25, 33) (Table 1).
The abundance of immune regulatory subsets of tumor-infiltrating leukocytes in the TME is correlated with disease progression and poor prognosis (34) such as M2 macrophages, myeloid-derived suppressor cells (MDSC), neutrophil subsets, regulatory dendritic cells (DC), and regulatory T cells (Tregs) (35, 36). In sarcoma and metastatic melanoma models, NLRP3 activation enhances MDSC and Treg populations while suppressing both NK and T cell-mediated tumor surveillance. Zhiyu et al. found that NLRP3 silencing resulted in a five-fold reduction of MDSCs in host mice (29). These NLRP3-deficient mice were also presented with less pulmonary metastasis in an orthotopic transplant mouse model of mammary adenocarcinoma (29, 37). NLRP3 expression in tumor-infiltrating macrophages is correlated with survival, lymph node invasion, and metastasis of mammary carcinoma patients (22) (Table 1).
Among tumor-infiltrating immune cells, tumor-associated macrophages (TAMs) take center stage in promoting both metastatic tumor spread and immune evasion (22, 24, 26). NLRP3 mediated macrophage-colorectal cancer (CRC) cell crosstalk and achieved higher migration rates of CRC cells, whereas blocking NLRP3 signaling suppressed CRC cell migration in vitro and liver metastatic ability in vivo (24). In NLRP3- or caspase-1-knockout macrophages, the ability to promote the migration and invasion of melanoma cells was, similarly, greatly diminished and the metastatic potential of melanoma tumor cells was suppressed (26). Mediating immune-suppressive crosstalk, NLRP3 signaling in macrophages has been reported to decrease NK cell activation (21), reduce Th1 cell polarization (27), and attenuate cytotoxic CD8+ T cell activation (27). The suppression of anti-tumor immune responses is accompanied by enhanced differentiation of CD4+ T cells into tumor-promoting cells such as T helper type 2 cells (Th2 cells), Th17 cells, and Treg populations (27). NLRP3 also plays a role in treatment response where chemotherapeutic agents were shown to activate NLRP3 activation in MDSC, leading to immunosuppressive responses through IL-1β production that blunts the anti-tumor therapeutic effect (29, 38) (Table 1).
IL-1β is abundant in the TME, where this cytokine can promote tumor growth and presents anti-tumor activities; thus, alteration of TME from pro-invasive inflammation towards anti-tumor cell immunity against an overwhelming immunosuppressive situation is a strategy for treating cancer (35). Breast cancer-associated fibroblast (CAF)-derived IL-1β in TME drives an immunosuppressive phenotype by promoting TAM’s reprogramming to an M2-like phenotype and restricting the accumulation of T cells in tumors (23). Daley et al. found that inhibition of NLRP3, ASC or caspase-1 protects against pancreatic ductal adenocarcinoma (PDA) via immunogenic reprogramming of innate and adaptive immunity within the TME (27). NLRP3 inflammasome activation in tumor cells can endorse a chronic inflammatory TME that encourages malignant transformation and extinguishes the local immunity provided by NK or T cells (31, 39). IL-1β, has also been reported to recruit and activate γ/δ T cells involved in mediating breast cancer metastasis to bone and lung metastasis of melanoma and breast cancer (23) (Table 1). Not only does the cytokine dampen immune surveillance, but it also blunts the anti-tumor therapeutic effect of chemotherapeutic agents via drug-induced NLRP3 activation in MDSC as well (29, 38) (Table 1).
Immune checkpoint inhibitors targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA4) and programmed cell death protein 1 (PD1) and its ligand-programmed death ligand 1 (PDL1) axis appear to require a pre-existing, primed effector anti-tumor CD8+ T cell infiltration for optimum therapeutic effect (28, 40). Among the known human chemokines, a co-regulated set of four (chemokine (C-C motif) ligand (CCL)-4, CCL-5, chemokine (C-X-C motif) ligand (CXCL)-9, CXCL-10) chemokines is upregulated in primary PDA carcinoma and PDA liver metastasis, which regulates CD8+ T cell infiltration, activates T cells, and promotes NLRP3-mediated T cell priming and enhances anti-tumor CD8+ T cell cytotoxic activity for an effective immune checkpoint therapy response. (Table 1). To reiterate the role of NLRP3 in enhancing immune checkpoint inhibitors, Kaplanov et al. found that although anti-PD-1 reduced tumor growth, the combination of anti-IL-1β plus anti-PD-1 abrogated the tumors completely. These observations support the findings of clinical trials that blockage of IL-1β in cancer might serve as a checkpoint inhibitor (35).
Despite accumulating evidence supporting NLRP3-mediated immune evasion, the inflammasome has also been reported to activate pro-surveillance pathways that heightened anti-tumor control and prevented tumor metastasis. The innate immune system may sense danger signals by the activation of the NLRP3 inflammasome, where downstream crosstalk engages NK cell cytolytic activity, the primarily cytotoxic cell regulating metastatic control. NLRP3 in the primary lesion of cancer cells drives the production of pro-IL-1β, DC maturation, and the secretion of IL-1β to support the evolution of tumor-specific CD8+ T cells (41). Moreover, Dupaul-Chicoine et al. found that the activation of NK cells was IL-18 dependent, but IFN-γ independent. The inflammasome-mediated tumor suppression is proposed to be primed by IL-18, promoting hepatic NK cell maturation, and aims to target FasL-sensitive tumor cells (30). IL-18 also plays an important role in regulating lymphocyte chemoattractant properties and promoting the expression of pro-inflammatory mediators, angiogenesis and adhesion-related factors (42–44). In a separate study, Dagenais et al. found that IL-18-deficient mice reveal a higher metastatic burden than wild-type mice in CRC liver metastasis, suggesting that IL-18 plays an important role in CRC metastatic growth restriction (45). Besides, in an animal model lacking T, B, and NKT cells, Rag1−/− mice exhibit a similar tumor metastatic burden compared to wild-type mice; however, Rag1−/−/IL-18−/− mice presented with an increased metastatic burden, indicating that IL-18-induced tumor cytotoxicity is independent of the major adaptive immunity effectors such as cytotoxic T lymphocytes (45).
In the primary lesion of cancer cells, NLRP3 drives the production of pro-IL-1β, DC maturation, and the secretion of IL-1β to support the differentiation of tumor-specific CD8+ T cells (41). NLRP3 inflammasome has also been reported, in a head and neck squamous carcinoma (HNSC) mice model, to inhibit and delay tumor growth and reshape the anti-tumor response through a decrease in the number of immunosuppressive cells and an enhancement in the function of effector T cells (31). Not only does cancer NLRP3 play a role in adaptive immunity in enforcing immune surveillance, but it also mediates anti-tumor immunity in innate immunity through activation of tumor-associated neutrophils (TAN). Chen et al. observed that irradiated nasopharyngeal carcinoma (NPC) presented with increased NLRP3 signaling and higher levels of tumor-derived IL-1β, and proposed that these cytokines can inhibit tumor growth and prevent local relapse by recruiting antitumor N1 TAN. Correlating with the clinical scenario, Chen et al. found that the presence of TAN significantly promoted survival in NPC patients (32) (Table 1).
Sustaining Proliferative Signaling
The mitogenic signaling in cancer cells is chronically sustained and undamped. It has been proposed that cancer cells achieve such a feat through various approaches: tumor cells can produce growth factor ligands independently, promote receptor’s structural alterations that facilitate ligand-independent activation, and elevate the levels of receptor proteins displayed at the surface resulting in the hyperresponsive reaction under the physiologic growth environment. The current literature has not reported on receptor alterations or dysregulation; nevertheless, NLRP3 has been associated with sending signals to stimulate normal cells within the tumor-associated stroma and supply them with growth factors. The crosstalk results with activation of the downstream constitutive pathway in modulating proliferation and cell cycle and dampening negative feedback: the regular operation to contain uncontrolled proliferation.
NLRP3 downstream IL-1β regulates diverse proliferative processes associated with inflammatory response (21, 42), cell cycle modulation (21, 46), nuclear factor κB (NF-kB)-c-Jun N-terminal kinase (JNK) signaling (21) as well as colony survival (47). The critical role of IL-1β is brought to light in melanoma cells, as the late-stage human melanoma actively secretes IL-1β without exogenous stimulation (48, 49). The spontaneous secretion of IL-1β shows similar characteristics as auto-inflammatory diseases caused by gain-of-function mutations in NLRP3 (48, 49), suggesting that the constitutive secretion of IL-1β may play a role in the underlying pathogenesis of melanoma (48, 49). Another study exploring melanoma found that while late-stage melanoma expresses IL-1β constitutively, intermediate-stage melanoma requires activation of the IL-1 receptor to secrete active IL-1β, and early-stage melanoma requires stimulation of the IL-1 receptor plus the co-stimulant muramyl dipeptide to secrete active IL-1β. These findings reinforce that IL-1-mediated autoinflammation paves the way to human melanoma development and progression (49). In breast cancer, numerous studies have reported a pro-tumorigenic role of IL-1β, whereas blocking IL-1β reduces tumor growth (50–52). Moreover, Voigt et al. indicated a previously unrecognized mechanism by which cancer cells induce IL-22 production from memory CD4+ T cells via activation of the NLRP3 inflammasome in breast cancer. The authors also demonstrated that the IL-1 receptor antagonist anakinra abrogates IL-22 production and reduces tumor growth in a murine breast cancer model (50) (Table 2).

Table 2 Proliferation- and cell death-associated cancer hallmarks.
Varying levels of IL-1β operate distinct cellular signaling pathways and cell fate. Subjected to high levels of IL-1β, cells engage apoptotic pathways, whereas moderate levels of IL-1β peptides stimulate clonal expansion. Roy et al. generated stable IL-1β overexpressed MCF7 cells. In secreting high levels of IL-1β clone, cells underwent apoptosis due to genotoxic stress, whereas moderate IL-1β expressed cells undergoing clonal expansion. These studies support that concentrations of the IL-1β secretion determine its stimulatory or inhibitory signals regulating the growth of tumors that might result in novel preventive strategies (70). NLRP3-mediated IL-1β production activates NF-κB and initiates JNK signaling to cause proliferation in gastric cancer and CRC (21, 39, 71). Similarly, activation of NLRP3 inflammasome in mesothelial cells of lung cancer leads to an inflammatory response that fuels cancer initiation and progression (21) and then activates the NF-κB-signaling pathway in lung cancer, consequently increasing proliferation and inhibiting apoptosis (54). On the other hand, Salaro et al. found that NLRP3 overexpression inhibits cell proliferation and stimulates apoptosis in leukemic cells. We suggest the opposite results in NLRP3-mediated cell proliferation due to different IL-1β levels (57) (Table 2).
Manipulation of NLRP3 downstream components holds potential in determining cell fate. A central component of the inflammasomes, ASC, has been shown to promote tumor development favoring inflammation in infiltrating immune cells (42). Inactivation of NLRP3 inflammasome driven by miR-233-3p has been found to decrease the expression of NLRP3 inflammasome-associated proteins, ASC, IL-1β, and IL-18 in breast cancers and suppress tumor growth. In addition, inactivation of NLRP3 inflammasome has also been found to reduce IL-1β expression and halt development of melanoma (21). Yin et al. found the expression levels of NLRP3, ASC, caspase-1, and IL-1β are highly expressed in high-grade glioma compared to low-grade glioma, indicating an important role for NLRP3 in glioma malignancy. Overexpression of NLRP3 promotes cell proliferation and colony formation in glioma cells, whereas the downregulation of NLRP3 significantly repressed colony formation in glioma cells (56).
Similarly, NLRP3 expression levels are also correlated with the tumor size, lymph node metastatic status and IL-1β expression in oral squamous cell carcinoma (OSCC), and downregulating NLRP3 expression markedly attenuates the proliferation, migration, and invasion of OSCC (47). These results suggest that NLRP3 exerts a vital role in cancer cell proliferation. NLRP3 inflammasomes mediate both suppressions of apoptosis and progression of the cell cycle by leptin-dependent ROS production in breast cancer, which is mediated via estrogen receptor alpha (ERα)/reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase signaling (46). Moreover, NLRP3 downstream, IL-1β, also stimulates the production of ROS that, in turn, induces DNA damage and cancer development in CRC (42) (Table 2).
Evading Growth Suppressors
Sustained proliferation despite the accumulation of endogenous stress and unresolvable metabolic dysregulations requires the cancer cells to circumvent programs that negatively regulate uncontrolled expansion. The replicative machinery intrinsically possesses numerous gatekeepers to maintain homeostasis, and these housekeeping genes integrate diverse signaling inputs to determine if the cell’s condition is suboptimal or normalized for subsequent cell-cycle progression. Previous studies have explored the role of inflammasome activation in the cell cycle mediated by leptin, and the authors found that leptin, a known risk factor for breast cancer, induces cyclin D1 expression (18, 58) and mediates cell cycle progression.
In contrast, physiologic growth becomes abolished when treating with NLRP3 inhibitor or knockout of NLRP3. The decreased population of cells in S- and G2-M phases post-NLRP3 inhibition demonstrates that the activation of NLRP3 inflammasome plays a critical role in leptin-induced cell cycle progression (46). Inflammasome inhibitions were also found to induce cell cycle arrest at the G0/G1 phase and elevated expression levels of negative cell cycle modulators, including p27Kip and p53 (58). Interestingly, activation of NLRP3 inflammasome was only activated during cell cycle interphases by NEK7, mitosis associated with kinase. Known for its role in cellular division, NEK7 mediates centrosome duplication, DNA repair and mitotic spindle activity (72); nevertheless, NEK7 also mediates mitochondrial regulation, and expressions of the NLRP3 inflammasome have been observed. Liu et al. also found that NEK7 is involved in many NLRP3-associated diseases (72). In investigating NEK7’s dual function in regulating cell division and intracellular danger sensor, Shi et al. found that NLRP3 activation requires NEK7 and suggested that NEK7 serves as a cellular switch enforcing exclusive cell fate between inflammasome response and cell division (73) (Table 2).
Enabling Replicative Immortality
A limited number of successive cell division cycles hard-wired into most normal cell lineages safeguard against uncontrolled proliferation. The natural barrier to proliferation, replicative senescence, is the cell’s innate protective measure against oncogenic progression where cells subjected to irreparable stress and uncontrolled growth are forced to undergo cell cycle arrest. Dong et al. found that NLRP3 inhibits senescence and enables replicative immortality through regulating the Wnt/β-catenin pathway via the thioredoxin-interacting protein (TXNIP)/NLRP3 axis (74). A Wnt/β-catenin pathway and NLRP3 was proposed by Xu et al. to be co-regulated by miR-20b, a miRNA targeting TXNIP. TXNIP knockdown or targeting by miR-20b resulted in a pro-tumorigenic phenotype with increased cell proliferation, inhibited cell senescence-reduced cell cycle modulators (p16 and p21), and decreased NLRP3 inflammasome-associated proteins (NLRP3 and cleaved caspase-1). These findings reveal that senescence antagonist miR-20b or TXNIP knockdown resulted in attenuated Wnt/β-catenin pathway via dysregulation of the TXNIP/NLRP3 axis (74) (Table 2).
Wnt signaling is implicated in a multitude of processes such as embryogenesis, carcinogenesis and stem cell pluripotency. In the realm of replicative mortality, Park et al. discovered a novel telomere-independent function of telomerase reverse transcriptase (TERT)/telomerase to amplify signaling by the Wnt pathway. The amplification is achieved by TERT serving as a cofactor of the β-catenin/lymphoid enhancer factor (LEF) transcription factor complex (75). Huang et al. reported that β-catenin promotes NLRP3 inflammasome activation, and silencing of β-catenin impairs NLRP3 activation (76). Nevertheless, Zheng et al. found a reciprocal relationship between NLRP3 and Wnt signaling. NLRP3 agonist induces Wnt/β-catenin activation, whereas inactivation of Wnt/β-catenin results in the inhibition of NLRP3, IL-1β. The authors propose that NLRP3 activation might enhance resistance to gemcitabine via IL-1β/epithelial–mesenchymal transition (EMT)/Wnt/β-catenin signaling (59). Attempting to explore the underlying pathway in colon cancer, Liu et al. indicated that estrogen receptors regulate the Wnt/β-catenin pathway through targeting NLR, and by comparing gene expression between normal and colon cancer tissues, they pointed out that Wnt/β-catenin pathway-associated genes are significantly differentially expressed in ER−/− mice. In addition, selective estrogen receptorα antagonists promote β-catenin degradation via inhibition of NLRP3. Collectively, these results indicate that NLRs are potential cancer markers that mediate estrogen receptor/Wnt/β-catenin signaling axis (61) (Table 2).
Telomeres protect ends of chromosomes and govern the capability of cells to counteract progressive telomere erosions caused by cell divisions. Telomerase elongates specific telomere repeats to the ends of chromosomes, which is absent in non-immortalized cells but overexpressed in immortalized cells and cancer lineages. Employing late generation telomerase knockout mice, Kang et al. found that telomere dysfunction greatly impacts innate immune cells where they present with exaggerated lung inflammation and then increase mortality upon respiratory infection. In investigating the mechanism of dysregulation, telomere dysfunction elicits macrophage mitochondrial abnormality and triggers hyperactivation of the NLRP3 inflammasome. Moreover, Kang et al. demonstrated that the peroxisome proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1α)/estrogen-related receptor alpha (ERRα) axis, controlled by the telomere status, is required not only for maintaining mitochondrial homeostasis but additionally for retaining the TNF-actin interacting protein 3 (AIP3)-dependent machinery during inflammasome activation (56) (Table 2). These results bring to light a previous unexplored crosstalk network between replicative immortality and inflammasome activation.
A previous study indicated that IL-1β stimulates the activation of hypoxic signaling pathways in cancer cells that are critical for regulating and maintaining the characteristics of cancer stem cells (CSCs), including undifferentiated phenotype, self-renewal capacity, and immortality (77). The available evidence suggests that the same sets of transcription factors are implicated in regulating distinct cell fates. These overlapping sets of target genes from developmental genetics and oncogenesis highlight the importance of CSCs in NLRP3-associated carcinogenesis. Inflammasomal production of IL-1β reinforces the cancer stem cell phenotype and contributes to its invasive capacity and survival resiliency (60, 62–65). Huang et al. found that CSC markers are significantly upregulated with enriched CSC population in an IL-1β treated CRC cell line and spherical human primary CRC, and these markers, such as BMI1, nestin, Nanog, and β-catenin, can be used for stemness evaluation. Consistently, Huang et al. demonstrated that cancer expression of stem cell markers BMI1, aldehyde dehydrogenase 1 (ALDH1) and CD44 are associated with NLRP3, and they proposed that inflammasome may regulate CSC (62) (Table 2).
Resisting Cell Death
Programmed cell death by apoptosis serves as a natural barrier against oncogenic development. Despite the major downstream event of NLRP3 inflammation formation of caspase-1 mediated pyroptosis, NLRP3 seems to mediate the dual-function of apoptosis and survival. Herein, elucidation of the NLRP3 signaling circuitry governing the apoptotic program may serve as the identification of the target gene for cell fate determination. When comparing liver cancer biopsies with non-cancerous samples, Wei et al. found that both mRNA and protein levels of NLRP3 are markedly down-regulated in hepatic parenchymal cells derived from liver cancer, suggesting an antitumorigenic role of NLRP3 hepatocellular carcinoma (67). NLRP3 has been associated with suppressing malignant progression by triggering the mitochondrial apoptotic pathway, enhancing immune-cytokine levels in the tumor microenvironment (39), and activating tumor-suppressive protein p53 in epithelial cells (42, 46, 78) (Table 2).
The apoptotic trigger is controlled by counterbalancing pro- and anti-apoptotic commands. Direct inhibition of NLRP3 or its downstream molecules is associated with oncogenic progression and anti-apoptotic effect. NLRP3 inflammasome inactivation, driven by miR-223-3p, increases proliferation, promotes invasion and inhibits apoptosis in breast cancer cells (54). MyD88 knockout mice have been found defective in downstream IL-1β and IL-18 productions, and their downstream signaling reveals an increase in colonic epithelial proliferation and CRC tumorigenesis (66). Furthermore, NLRP3 activation has been proposed to mediate multiple pathways other than that of the caspase-1. NLRP3 inflammasome activation-induced IL-1β and IL-18 in lung cancer may work through mechanisms other than the caspase-1 pathway, indicating that NLRP3 inflammasome can mediate the release of IL-1β and IL-18 through caspase-1-dependent or -independent pathways (69). Moreover, in A549 lung cancer cells, NLRP3 has been demonstrated to associate with sending signals to stimulate normal cells within the tumor-associated stroma, supply them with growth factors, and regulate cell proliferation and migration by releasing IL-1β and IL-18 through an autocrine or paracrine manner (69). Although caspase-1 activation is the major downstream event of NLRP3 inflammasome assembly, recent studies have reported that NLRP3 inflammasome could also be activated by caspase-8 (79). Besides, caspase-8 is also involved in the maturation of IL-1β and IL-18 by inducing a non-canonical process of releasing IL-1β (80) and IL-18 (69, 81) (Table 2).
The attenuation of the NLRP3 downstream pyroptosis pathway promotes apoptosis (54, 68). When attempting to activate pyroptotic signaling in GSDMD-deficient NSCLC, another type of programmed cell death other than pyroptosis was activated. Gao et al. found that in GSDMD-deficient tumor cells, the depletion of GSDMD activated cleavage of caspase-3 and PARP that guided the cell fate towards the intrinsic mitochondrial apoptotic pathway. Furthermore, knockdown of GSDMD attenuates tumor proliferation by promoting apoptosis and inhibiting epidermal growth factor receptor (EGFR)/Akt signaling axis (68). On the other hand, the increase of NLRP3, ASC, caspase-1, and IL-1β proteins in human glioma tissues is significantly correlated with higher World Health Organization grades. Inhibition of NLRP3 suppresses the proliferation, migration and invasion, and promotes apoptosis in glioma cells, while in contrast, increased expression of NLRP3 significantly enhances the proliferation, migration and invasion as well as attenuating apoptosis in glioma cells (56) (Table 2). However, as to how damaged cells decide whether to continue living in a dysfunctional state or die, and how cancer cells escape the inflammasome-induced pyroptosis both remain unclear.
Deregulating Cellular Energetics-Aerobic Glycolysis
Despite its inferiority in ATP production, glycolysis allows diversion of its metabolic products into rewarding intermediates for multiple biosynthetic pathways required for active cellular expansion. An altered metabolic environment and its metabolites provide both the priming signal to induce NLRP3 inflammasome transcription and the trigger signal for inflammasome formation/activation (82). He et al. pointed out that NLRP3 is overexpressed in non-small cell lung cancer (NSCLC) tumor tissues, while in contrast, depletion of NLRP3 reduces cell growth in vitro and in vivo as well as decreases glycolysis while enhancing mitochondrial respiration, where pathways contributing to ATP production and intermediate substrates metabolism serve as the Warburg effect switch (83). Consistently in NPC, Epstein–Barr virus latent membrane protein 1 (LMP1) promotes myeloid-derived suppressor cells MDSC expansion that promotes immune escape in the tumor microenvironment. Cai et al. found that LMP1 promotes the expression of multiple glycolytic genes, including glucose transporter 1 (GLUT1), and upregulates extra-mitochondrial glycolysis pathways in malignant cells. This metabolic reprogramming results in increasing expression of the NLRP3 inflammasome and its downstream effectors, including IL-1β, IL-6, and GM-CSF (84).
In further exploring the underlying mechanism, Moon et al. indicated that activation of the glycolytic phenotypes is intrinsically linked to NLRP3 inflammation; e.g., inhibition of mTOR complex 1 (mTORC1) suppresses NLRP3 inflammasome activation, and its regulatory associated protein of mTOR (Raptor)/mTORC1 downstream HK1-dependent glycolysis is critical for NLRP3 inflammasome activation (85) (Table 3). Previous studies indicated that resting NLRP3 localizes to endoplasmic reticulum structures (89), whereas on inflammasome activation, both NLRP3 and its adaptors redistribute and co-localize with endoplasmic reticulum and mitochondria organelle clusters (90). Mitochondria-associated adaptor molecule (MAVS) is required for optimal NLRP3 inflammasome activity including mediating recruitment of NLRP3 to mitochondria, promoting production of IL-1β, and modulating the pathophysiologic activity of the NLRP3 inflammasome in vivo (91). In addition, Kim et al. found that NLRP3 in renal tubular cells re-localizes from the cytosol to the mitochondria during hypoxia and binds to MAVS, which attenuates mtROS production and depolarization of the mitochondrial membrane potential under hypoxia (92). This evidence illustrates that the activated NLRP3 inflammasome re-localizes within the cell-driven mitochondrial dysfunction in cancer cells, resulting in metabolic reprogramming and cancer progression.

Table 3 Metabolism- and genome instability-associated cancer hallmarks.
A causal mechanism by which obesity promotes the progression of breast cancer via the NLRP3 inflammasome activation has been recently described (42, 93). Wen et al. pointed out that IL-1β and NLRP3 inflammasome regulate lipid and carbohydrate metabolism. Enhanced NLRP3 inflammasome activation and processing of inactive pro-IL-1β to the active mature IL-1β are induced in response to administration of saturated fatty acids. Moreover, NLRP3 downstream IL-1β mRNA induction is required by glycolysis. Both glycolysis and HIF-1α expression are critical for Th17 cell differentiation (86) (Table 3). The AMP-activated protein kinase (AMPK), downregulated in cancer and obesity, has been proposed to mediate survival via its effect on cellular metabolism (94), through oncogenic progression of re-configuring/re-programming its metabolism towards glycolysis. AMPK inhibition blocks autophagy and increases mtROS production, a trigger signal for NLRP3 inflammasome activation (95).
Genome Instability
Genomic instability is a characteristic of most cancers, resulting from mutations in DNA repair genes and drives cancer development (96). A recent study indicated that short-wave ultraviolet B (UVB) exposure activates the NLRP3 inflammasome and induces the secretion of IL-1β and IL-18 (97). In addition, a positive correlation has been found between UVB radiation intensity and NLRP3 gene expression (88). Knockdown of NLRP3 suppresses UVB-induced production of IL-1β and attenuates other inflammatory mediators, such as IL-1α, IL-6, TNF-α and PGE2. Inhibition of the DNA repair system by knockdown XPA, a major component of nucleotide excision repair, causes activation of NLRP3 inflammasome. Thus, Hasegawa et al. indicated that accumulating genomic damage due to loss of repair would result in NLRP3 activation (88). ATM-activated autotaxin (ATX) propagates inflammation and accumulates DNA double-strand breaks (DSBs) in epithelial cells. Zheng et al. demonstrated that ATX transfection induces NLRP3 activation and DNA damages. These results suggest that ATX is activated via stress-induced NLRP3-DSB-ataxia telangiectasia-mutated (ATM) response (87) (Table 3).
Inducing Angiogenesis
To satisfy the highly demanding metabolic requirements, cancer switches on aberrant angiogenesis to help sustain expanding neoplastic growth. The role of NLRP3 in tumor-associated neo-vasculature and angiogenesis has been found to regulate angiogenesis in CRC, breast cancer, NSCLC (21, 53). Numerous studies have pointed out the angiogenic role of IL-1β (42, 46, 98). The circuitry of angiogenesis is proposed to be activated through numerous autocrine and paracrine actions. Carmi et al. indicated that IL-1β and vascular endothelial growth factor (VEGF) interact in an autoinduction circuit where these cytokines would result in the positive feedback of the other. The circuitry is proposed to be orchestrated between bone marrow-derived VEGF receptor 1/IL-1 receptor 11 expressed MDSC and tissue endothelial cells (ECs). Inhibition of IL-1β reduces tumor growth in melanoma by attenuating inflammation and promoting MDSC towards M1 macrophage, which subsequently reduces angiogenesis through neutralized VEGF; however, when tumors relapse, the expression of HIF-1α in a paracrine manner has been found to allow angiogenesis to rebound. These findings suggest that IL-1β inhibition acts as an effective anti-tumor therapy in targeting tumor angiogenesis (98) (Table 4).

Table 4 Metastasis-associated cancer hallmarks.
More pathway details were revealed in the study by Voronov et al. where the authors established that IL-1 expression at TME enhances adhesion molecules on EC and facilitates tumor EMT and metastasis (101, 102). Although not regarded as the primary driver of angiogenesis, recombinant interleukin 1 (rIL-1) promotes the proliferation of EC and enhances the expression of adhesion molecules as well as inflammatory mediators and cytokines, which subsequently promote vascular permeability (103, 104). Thus, Voronov et al. aimed to assess the role of IL-1 as a driver of tumor angiogenesis and evaluate its potential as an anti-tumor agent, and by employing IL-1β knockout mice, the authors found that IL-1β is required for in vivo angiogenesis since IL-1β knockout mice failed to recruit blood vessel networks into Matrigel plugs with melanoma tumor cells. The incorporation of IL-1 receptor antagonists into these plugs also inhibited the ingrowth of blood vessel networks in wild-type mice. Similar to melanoma gel models, this phenomenon was consistently found in DA/3 mammary and prostate cancer cell models (99) (Table 4).
Activating Invasion and Metastasis
The invasion and metastasis cascade not only induce in-and-out extravasation of tumor cells into and out of the lymphatic or vascular systems but additionally affect the dissemination and survival in the bloodstream and targeting of metastatic sites while evading immune surveillance. The role of NLRP3 in activating invasion and metastasis seems to be tissue- and context-dependent. Increased activation of the NLRP3 inflammasome promotes migration and invasion activities in gastric cancer cells (24). Consistently, knockdown of NLRP3 induces cell apoptosis in MCF-7 cells and decreases cell migration (54); nevertheless, in other cell-types, NLRP3 inflammasome may pharmacologically repress proliferation and metastasis of hepatic cell carcinoma (HCC) (21) (Table 4).
The role of NLRP3 in promoting invasion has been demonstrated with human endometrial cancer cell lines such as Ishikawa and HEC-1A cells, where knockdown of NLRP3 significantly reduces proliferation, clonogenicity, invasion and migration. In contrast, overexpression of NLRP3 enhances the activities of proliferation, migration and invasion as well as increasing caspase-1 activation and IL-1β secretion in human endometrial cancer cells (55). Liu et al. concluded that the upregulation of NLRP3 expression promotes the progression of endometrial cancer; therefore, NLPR3 inflammasome might be a new therapeutic target for endometrial cancer (55). Unanimously, inhibition of the NLRP3 inflammasome pathway has been suggested to be a promising approach for decreasing tumor cell invasion and survival in HNSC (21) and glioma (56) (Table 4).
The cascade of downstream pathways via activation of the NLRP3-mediated IL-1β axis has been found to activate NF-κB that initiates JNK signaling causing proliferation and invasion in gastric cancer (21). On the other hand, despite the activation of the same downstream pathways, NF-κB, NLRP3 is shown by numerous studies to be anti-oncogenic. MicroRNA-233-3p (miR-223-3p) has been found to directly target NLRP3, which results in diminishing the expression of NLRP3 and its downstream molecules, including ASC, IL-1β and IL-18, suggesting that miR-223-3p might be a dynamic control in determining inflammasome-mediated cell fate (54). Moreover, miR-223 has been demonstrated to increase proliferation, promote invasion and inhibit apoptosis in A549 cells via activation of the NF-κB signaling pathway (105). Adding a layer of complexity into the intracellular crosstalk, NF-κB signaling has also been proposed to act upstream of NLRP3 where NF-κB is required for the upregulation of NLRP3 in transforming growth factor-beta 1 (TGF-β1)- or TNF-α-induced EMT in CRC (100) (Table 4). Whether the complexity of crosstalk remains to be unraveled or whether the role of NLRP3 is cell-specific remains to be elucidated.
It is increasingly apparent that crosstalk between cancer cells and cells of the neoplastic stroma is involved in permitting seeding and successful colonization. The activation of NLRP3 inflammasome in breast CAF promotes tumor metastasis by upregulating the expression of adhesion molecules in EC and promoting the recruitment of MDSCs, suggesting that IL-1β secretion from activated mammary CAFs enhances the expression of adhesion molecules on tumor EC in vivo, thus facilitating cancer cell migration across the blood vessel endothelium (23). IL-1β has also been found to promote metastasis by inducing EMT or increasing matrix metalloproteinases (MMPs) activity in gastric adenocarcinoma, bladder cancer (24) and CRC (42). Moreover, the silencing of NLRP3 significantly decreases the migration and invasion in OSCC cells and reduces EMT-related protein expression. These results suggest that NLRP3 plays an important role in regulating the metastasis of OSCC cells (47) (Table 4).
Discussion
The role of NLRP3 in tumorigenesis may be tissue- or cell-dependent. Conflicting roles have been described where NLRP3 exhibits a protective role against colon cancer and HCC, while NLRP3 also plays a carcinogenic role in gastric and prostate cancers (39, 106). The multitude of cellular impacts demands a comprehensive analysis of tissue-context response, dose-dependent response, and in vivo implications that require further evaluation. Would these complicate benefits in multiple metastatic tumor responses or high-risk patients with cancer susceptibilities? If the drug is to be spread systematically through the oral or intravenous route, what will the overall response be considering immune priming distance from the primary site?
NLRP3 using conditional knockout models and pharmacological activators or inhibitors is needed to decode the precise mechanisms of NLRP3 activity in cancer (39). Awad et al. found that the NLRP3 effector ASC protein plays a differential regulation of NF-κB in different clinical scenarios. In metastatic melanoma, ASC encourages tumorigenesis while suppressing tumorigenesis in primary melanoma. The tissue-specific function of ASC is then confirmed in vivo when ASC acts as a tumor suppressor in keratinocytes and an oncoprotein in myeloid cells (48). Observing that inflammasome pathways serve dual opposing functions, Duan et al. proposed that NLRP3 activation could be enhanced in cells upon stimulated inflammation by LPS and coal tar pitch extract (CTPE) exposure compared to CTPE alone (107). Whether NLRP3 activation may result in proliferation or apoptosis of the cell would depend on the level of stress imposed, and the sensitivity of the cell to produce IL-1β requires elucidation. Would apoptosis be the result from overwhelming stress and pro-tumorigenic response as a consequence of less severe damage, repairable stress? An interesting question that arises would be whether the cell status predicts cell fate and its preposition towards apoptosis and senescence.
While seemingly bizarre with unique cell-dependent phenotypes and crosstalk, NLRP3 at the setting seems to be organized and goal-dependent. Where many uncertainties are regulated and compelled to arrive at one sole cell fate, inhibiting NLRP3 activation with S. muticum extracts has been found to decrease angiogenesis, reduce cancer cell proliferation, and increase apoptosis in human breast cancer cells (53). NLRP3 enhances IL-1β, subsequently activating NF-κB, and initiates JNK signaling to cause proliferation and invasion in gastric cancer (21). In lung cancer, it increases proliferation, promotes invasion and inhibits apoptosis (54). The knockdown of NLRP3 significantly reduces the proliferation, clonogenicity, invasion and migration in both Ishikawa and HEC-1A cells, while in contrast, NLRP3 overexpression enhances the proliferation, migration and invasion in both Ishikawa and HEC-1A cells and furthermore, increases caspase-1 activation and the release of IL-1β in endometrial cancer cells. YVAD-cmk, an inhibitor of caspase-1, decreases the proliferation ability.
Collectively, these results indicate that upregulated NLRP3 expression promotes the progression of endometrial cancer (55). The multifunctional role of NLRP3 in mutually conflicting pathways raises the question of what determines deterministic cell fate decisions in development and oncogenesis. And since inflammasome is a constitutively and physiologically expressed protein in maintaining homeostasis, would the target of NLRP3 inflammasome mitigate malignancy without harming normal neighboring tissue? Addressing these concerns, Bae et al. proposed that significant cell death was observed only when P2X7R and NLRP3 inflammasome were both inhibited by ATP and MCC950, a specific inhibitor of NLRP3 inflammasome (108), and further research into safety manipulation of NLRP3 inflammasome without enhancing significant dose-dependent side effects is required.
It is important not to overlook that EMT, invasion, survival, proliferation, angiogenesis and immune invasion-all traits are implicated in the process of invasion and metastasis (Figure 2). Hence, the distinct crosstalk highlighted in this review implies intertwined processes that are not independently activated and that act synergistically to allow tumor clonogenicity expansion and progression. Given the importance and multifaceted role of inflammasome/inflammation in cancer biology, the complexity of NLRP3 is just beginning to be decoded where inflammasome not only acts as a critical regulator in modulating tumor-associated inflammation but mediates essential roles in regulating cancer hallmarks as well. In response to a variety of stresses, mammalian cells activate the inflammasome for targeted pyroptosis; however, inflammasome has been associated with tumor-promoting effects, raising the interesting question of why inflammasome-mediated pyroptosis fails to outcompete the superior cell fate option in many dysregulated cellular contexts. What are the missing key drivers that will promote the major downstream event of caspase activation and subsequent pyroptosis in mediating tumor control? These recent developments provide new insights and therapeutic opportunities to manipulate inflammasome in augmenting their anti-tumor activity and achieving metastatic control for therapeutic opportunities.
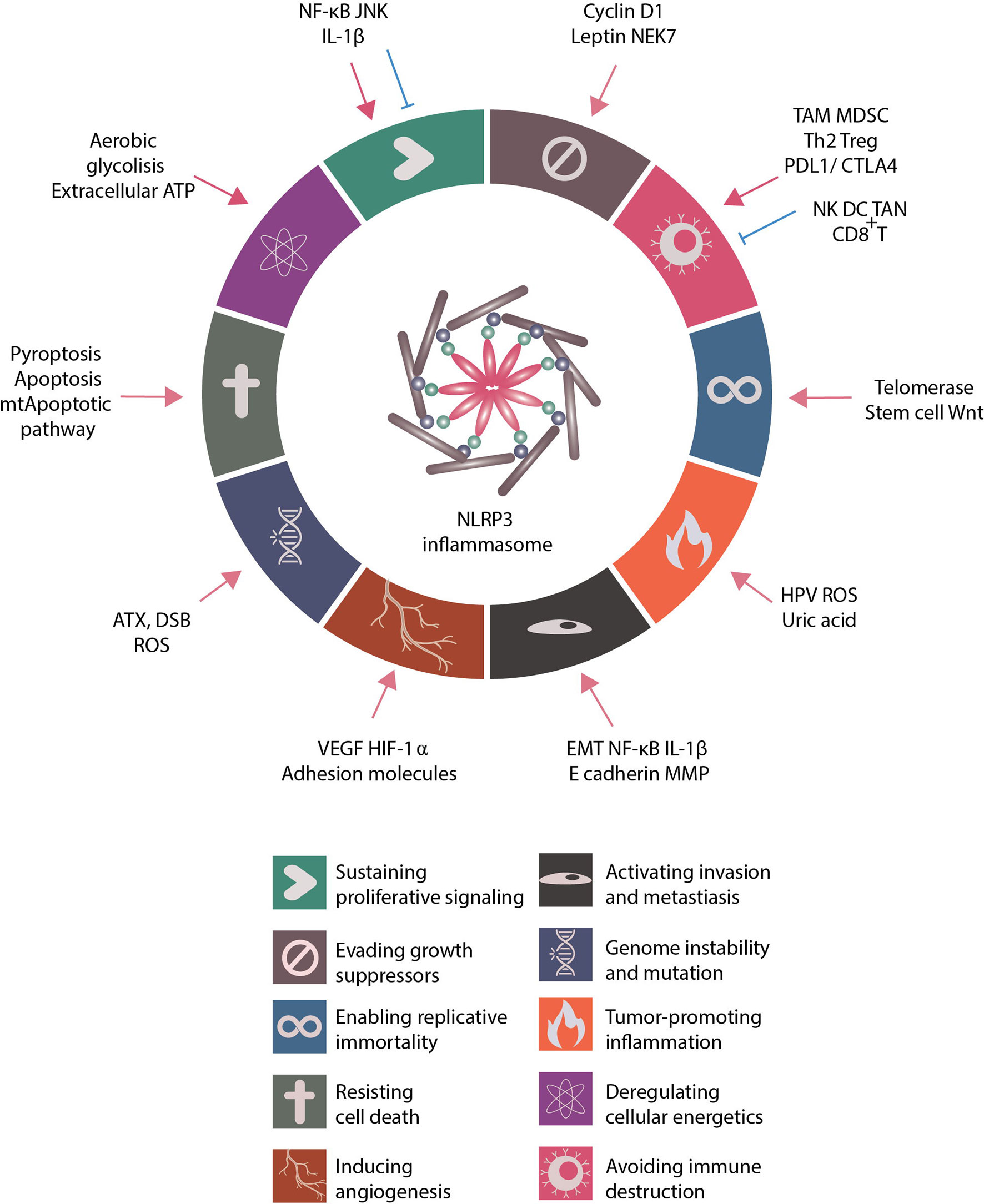
Figure 2 Role of NLRP3 inflammasome in regulating cancer hallmarks. Proposed by Dr. Douglas Hanahan and Dr. Robert A. Weinberg, the cancer hallmarks are universal abilities that cancer cells acquire for survival. These hallmarks include sustaining proliferative signaling, evading growth suppressors, resisting cell death, enabling replicative immortality, inducing angiogenesis, activating invasion and metastasis, deregulating cellular energetics, avoiding immune destruction, tumor-promoting inflammation and genome instability and mutation. The related literature was reviewed and the role of NLRP3 inflammasome in regulating cancer hallmarks summarized. Many NLRP3 inflammasome-related inducers and NLRP3 inflammasome-associated regulations modulate the activities of cancer hallmarks.
Author Contributions
T-YL, S-PH, and C-YL contributed to the conception and design of the review study. T-YL, M-CT, and WT wrote the first draft of the manuscript. T-YL, M-CT, WT, H-CY, S-CW, and C-YL wrote sections of the manuscript. T-YL, S-PH, and C-YL discussed and revised the content of the review article. All authors contributed to the article and approved the submitted version.
Funding
This study was supported in part by grants from the Ministry of Science and Technology, Taiwan, R.O.C. (grant nos. MOST 108-2320-B-037-007, 109-2320-B-037-007-MY3, 109-2314-B-037-108-MY2, 108-2314-B-037-029, and 109-2314-B-037-106-MY3), Kaohsiung Medical University Research Center Grant (KMU-TC108A04), and Kaohsiung Medical University Hospital (grant no: KMUH105-5R42).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin (2020) 70:7–30. doi: 10.3322/caac.21590
2. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell (2000) 100:57–70. doi: 10.1016/j.semcancer.2008.12.002
3. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
4. Huang MY, Tu CE, Wang SC, Hung YL, Su CC, Fang SH, et al. Corylin inhibits LPS-induced inflammatory response and attenuates the activation of NLRP3 inflammasome in microglia. BMC Complement Altern Med (2018) 18:221. doi: 10.1186/s12906-018-2287-5
5. Rex DAB, Agarwal N, Prasad TSK, Kandasamy RK, Subbannayya Y, Pinto SM. A comprehensive pathway map of IL-18-mediated signalling. J Cell Commun Signal (2020) 14:257–66. doi: 10.1007/s12079-019-00544-4
6. Shi J, Gao W, Shao F. Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci (2017) 42:245–54. doi: 10.1016/j.tibs.2016.10.004
7. Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol (2019) 198:477–89. doi: 10.1038/s41577-019-0165-0
8. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev (2009) 22:240–73. doi: 10.1128/CMR.00046-08
9. Yang Y, Wang H, Kouadir M, Song H, Shi F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis (2019) 10:128. doi: 10.1038/s41419-019-1413-8
10. Hung YL, Wang SC, Suzuki K, Fang SH, Chen CS, Cheng WC, et al. Bavachin attenuates LPS-induced inflammatory response and inhibits the activation of NLRP3 inflammasome in macrophages. Phytomedicine (2019) 59:152785. doi: 10.1016/j.phymed.2018.12.008
11. Burnstock G, Di Virgilio F. Purinergic signalling and cancer. Purinergic Signal (2013) 9:491–540. doi: 10.1007/s11302-013-9372-5
12. Di Virgilio F, Sarti AC, Falzoni S, De Marchi E, Adinolfi E, Extracellular ATP. and P2 purinergic signalling in the tumour microenvironment. Nat Rev Cancer (2018) 18:601–18. doi: 10.1038/s41568-018-0037-0
13. Di Virgilio F, Dal Ben D, Sarti AC, Giuliani AL, Falzoni S. The P2X7 receptor in infection and Inflammation. Immunity (2017) 47:15–31. doi: 10.1016/j.immuni.2017.06.020
14. Ferrari D, Pizzirani C, Adinolfi E, Lemoli RM, Curti A, Idzko M, et al. The P2X7 receptor: a key player in IL-1 processing and release. J Immunol (2006) 176:3877–83. doi: 10.4049/jimmunol.176.7.3877
15. Doktor F, Prager P, Wiedemann P, Kohen L, Bringmann A, Hollborn M. Hypoxic expression of NLRP3 and VEGF in cultured retinal pigment epithelial cells: contribution of P2Y2 receptor signaling. Purinergic Signal (2018) 14:471–84. doi: 10.1007/s11302-018-9631-6
16. Panchanathan R, Liu H, Choubey D. Hypoxia primes human normal prostate epithelial cells and cancer cell lines for the NLRP3 and AIM2 inflammasome activation. Oncotarget (2016) 7:28183–94. doi: 10.18632/oncotarget.8594
18. Li S, Liang X, Ma L, Shen L, Li T, Zheng L, et al. MiR-22 sustains NLRP3 expression and attenuates H. pylori-induced gastric carcinogenesis. Oncogene (2018) 37:884–96. doi: 10.1038/onc.2017.381
19. Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol (2008) 9:847–56. doi: 10.1038/ni.1631
20. Pontillo A, Bricher P, Leal VN, Lima S, Souza PR, Crovella S. Role of inflammasome genetics in susceptibility to HPV infection and cervical cancer development. J Med Virol (2016) 88:1646–51. doi: 10.1002/jmv.24514
21. Moossavi M, Parsamanesh N, Bahrami A, Atkin SL, Sahebkar A. Role of the NLRP3 inflammasome in cancer. Mol Cancer (2018) 17:158. doi: 10.1186/s12943-018-0900-3
22. Weichand B, Popp R, Dziumbla S, Mora J, Strack E, Elwakeel E, et al. S1PR1 on tumor-associated macrophages promotes lymphangiogenesis and metastasis via NLRP3/IL-1beta. J Exp Med (2017) 214:2695–713. doi: 10.1084/jem.20160392
23. Ershaid N, Sharon Y, Doron H, Raz Y, Shani O, Cohen N, et al. NLRP3 inflammasome in fibroblasts links tissue damage with inflammation in breast cancer progression and metastasis. Nat Commun (2019) 10:4375. doi: 10.1038/s41467-019-12370-8
24. Deng Q, Geng Y, Zhao L, Li R, Zhang Z, Li K, et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett (2019) 442:21–30. doi: 10.1016/j.canlet.2018.10.030
25. Aranda F, Chaba K, Bloy N, Garcia P, Bordenave C, Martins I, et al. Immune effectors responsible for the elimination of hyperploid cancer cells. Oncoimmunology (2018) 7:e1463947. doi: 10.1080/2162402X.2018.1463947
26. Lee HE, Lee JY, Yang G, Kang HC, Cho YY, Lee HS, et al. Inhibition of NLRP3 inflammasome in tumor microenvironment leads to suppression of metastatic potential of cancer cells. Sci Rep (2019) 9:12277. doi: 10.1038/s41598-019-48794-x
27. Daley D, Mani VR, Mohan N, Akkad N, Pandian G, Savadkar S, et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med (2017) 214:1711–24. doi: 10.1084/jem.20161707
28. Romero JM, Grunwald B, Jang GH, Bavi PP, Jhaveri A, Masoomian M, et al. A four-chemokine signature is associated with a T-cell-inflamed phenotype in primary and metastatic pancreatic cancer. Clin Cancer Res (2020) 26:1997–2010. doi: 10.1158/1078-0432.CCR-19-2803
29. Zhiyu W, Wang N, Wang Q, Peng C, Zhang J, Liu P, et al. The inflammasome: an emerging therapeutic oncotarget for cancer prevention. Oncotarget (2016) 7:50766–80. doi: 10.18632/oncotarget.9391
30. Dupaul-Chicoine J, Arabzadeh A, Dagenais M, Douglas T, Champagne C, Morizot A, et al. The NLRP3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity (2015) 43:751–63. doi: 10.1016/j.immuni.2015.08.013
31. Chen L, Huang CF, Li YC, Deng WW, Mao L, Wu L, et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol Life Sci (2018) 75:2045–58. doi: 10.1007/s00018-017-2720-9
32. Chen LC, Wang LJ, Tsang NM, Ojcius DM, Chen CC, Ouyang CN, et al. Tumour inflammasome-derived IL-1beta recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol Med (2012) 4:1276–93. doi: 10.1002/emmm.201201569
33. Bloy N, Garcia P, Laumont CM, Pitt JM, Sistigu A, Stoll G, et al. Immunogenic stress and death of cancer cells: Contribution of antigenicity vs adjuvanticity to immunosurveillance. Immunol Rev (2017) 280:165–74. doi: 10.1111/imr.12582
34. Mlecnik B, Bindea G, Kirilovsky A, Angell HK, Obenauf AC, Tosolini M, et al. The tumor microenvironment and Immunoscore are critical determinants of dissemination to distant metastasis. Sci Transl Med (2016) 8:327ra326. doi: 10.1126/scitranslmed.aad6352
35. Kaplanov I, Carmi Y, Kornetsky R, Shemesh A, Shurin GV, Shurin MR, et al. Blocking IL-1beta reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc Natl Acad Sci U S A (2019) 116:1361–9. doi: 10.1073/pnas.1812266115
36. Anani W, Shurin MR. Targeting myeloid-derived suppressor cells in cancer. Adv Exp Med Biol (2017) 1036:105–28. doi: 10.1007/978-3-319-67577-0_8
37. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med (2009) 15:1170–8. doi: 10.1038/nm.2028
38. Bruchard M, Mignot G, Derangere V, Chalmin F, Chevriaux A, Vegran F, et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med (2013) 19:57–64. doi: 10.1038/nm.2999
39. Tartey S, Kanneganti TD. Differential role of the NLRP3 inflammasome in infection and tumorigenesis. Immunology (2019) 156:329–38. doi: 10.1111/imm.13046
40. Ino Y, Yamazaki-Itoh R, Shimada K, Iwasaki M, Kosuge T, Kanai Y, et al. Immune cell infiltration as an indicator of the immune microenvironment of pancreatic cancer. Br J Cancer (2013) 108:914–23. doi: 10.1038/bjc.2013.32
41. Emens LA, Middleton G. The interplay of immunotherapy and chemotherapy: harnessing potential synergies. Cancer Immunol Res (2015) 3:436–43. doi: 10.1158/2326-6066.CIR-15-0064
42. Ahechu P, Zozaya G, Marti P, Hernandez-Lizoain JL, Baixauli J, Unamuno X, et al. NLRP3 inflammasome: a possible link between obesity-associated low-grade chronic inflammation and colorectal cancer development. Front Immunol (2018) 9:2918. doi: 10.3389/fimmu.2018.02918
43. Komai-Koma M, Gracie JA, Wei XQ, Xu D, Thomson N, McInnes IB, et al. Chemoattraction of human T cells by IL-18. J Immunol (2003) 170:1084–90. doi: 10.4049/jimmunol.170.2.1084
44. Ye ZB, Ma T, Li H, Jin XL, Xu HM. Expression and significance of intratumoral interleukin-12 and interleukin-18 in human gastric carcinoma. World J Gastroenterol (2007) 13:1747–51. doi: 10.3748/wjg.v13.i11.1747
45. Dagenais M, Saleh M. Linking cancer-induced Nlrp3 inflammasome activation to efficient NK cell-mediated immunosurveillance. Oncoimmunology (2016) 5:e1129484. doi: 10.1080/2162402X.2015.1129484
46. Raut PK, Kim SH, Choi DY, Jeong GS, Park PH. Growth of breast cancer cells by leptin is mediated via activation of the inflammasome: critical roles of estrogen receptor signaling and reactive oxygen species production. Biochem Pharmacol (2019) 161:73–88. doi: 10.1016/j.bcp.2019.01.006
47. Wang H, Luo Q, Feng X, Zhang R, Li J, Chen F. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer (2018) 18:500. doi: 10.1186/s12885-018-4403-9
48. Awad F, Assrawi E, Louvrier C, Jumeau C, Giurgea I, Amselem S, et al. Photoaging and skin cancer: Is the inflammasome the missing link? Mech Ageing Dev (2018) 172:131–7. doi: 10.1016/j.mad.2018.03.003
49. Okamoto M, Liu W, Luo Y, Tanaka A, Cai X, Norris DA, et al. Constitutively active inflammasome in human melanoma cells mediating autoinflammation via caspase-1 processing and secretion of interleukin-1beta. J Biol Chem (2010) 285:6477–88. doi: 10.1074/jbc.M109.064907
50. Voigt C, May P, Gottschlich A, Markota A, Wenk D, Gerlach I, et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc Natl Acad Sci U S A (2017) 114:12994–9. doi: 10.1073/pnas.1705165114
51. Guo B, Fu S, Zhang J, Liu B, Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep (2016) 6:36107. doi: 10.1038/srep36107
52. Holen I, Lefley DV, Francis SE, Rennicks S, Bradbury S, Coleman RE, et al. IL-1 drives breast cancer growth and bone metastasis in vivo. Oncotarget (2016) 7:75571–84. doi: 10.18632/oncotarget.12289
53. Kopalli SR, Kang TB, Lee KH, Koppula S. NLRP3 inflammasome activation inhibitors in inflammation-associated cancer immunotherapy: an update on the recent patents. Recent Pat Anticancer Drug Discovery (2018) 13:106–17. doi: 10.2174/1574892812666171027102627
54. Zhang L, Li H, Zang Y, Wang F. NLRP3 inflammasome inactivation driven by miR2233p reduces tumor growth and increases anticancer immunity in breast cancer. Mol Med Rep (2019) 19:2180–8. doi: 10.3892/mmr.2019.9889
55. Liu SG, Wu XX, Hua T, Xin XY, Feng DL, Chi SQ, et al. NLRP3 inflammasome activation by estrogen promotes the progression of human endometrial cancer. Onco Targets Ther (2019) 12:6927–36. doi: 10.2147/OTT.S218240
56. Yin XF, Zhang Q, Chen ZY, Wang HF, Li X, Wang HX, et al. NLRP3 in human glioma is correlated with increased WHO grade, and regulates cellular proliferation, apoptosis and metastasis via epithelial-mesenchymal transition and the PTEN/AKT signaling pathway. Int J Oncol (2018) 53:973–86. doi: 10.3892/ijo.2018.4480
57. Salaro E, Rambaldi A, Falzoni S, Amoroso FS, Franceschini A, Sarti AC, et al. Involvement of the P2X7-NLRP3 axis in leukemic cell proliferation and death. Sci Rep (2016) 6:26280. doi: 10.1038/srep26280
58. Pham DV, Raut PK, Pandit M, Chang JH, Katila N, Choi DY, et al. Globular adiponectin inhibits breast cancer cell growth through modulation of inflammasome activation: critical role of sestrin2 and AMPK signaling. Cancers (2020) 12:613. doi: 10.3390/cancers12030613
59. Zheng Q, Yao D, Cai Y, Zhou T. NLRP3 augmented resistance to gemcitabine in triple-negative breast cancer cells via EMT/IL-1beta/Wnt/beta-catenin signaling pathway. Biosci Rep (2020) 40:BSR20200730. doi: 10.1042/BSR20200730
60. Arnold KM, Opdenaker LM, Flynn D, Sims-Mourtada J. Wound healing and cancer stem cells: inflammation as a driver of treatment resistance in breast cancer. Cancer Growth Metastasis (2015) 8:1–13. doi: 10.4137/CGM.S11286
61. Liu S, Fan W, Gao X, Huang K, Ding C, Ma G, et al. Estrogen receptor alpha regulates the Wnt/beta-catenin signaling pathway in colon cancer by targeting the NOD-like receptors. Cell Signal (2019) 61:86–92. doi: 10.1016/j.cellsig.2019.05.009
62. Huang CF, Chen L, Li YC, Wu L, Yu GT, Zhang WF, et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res (2017) 36:116. doi: 10.1186/s13046-017-0589-y
63. Li Y, Wang L, Pappan L, Galliher-Beckley A, Shi J. IL-1beta promotes stemness and invasiveness of colon cancer cells through Zeb1 activation. Mol Cancer (2012) 11:87. doi: 10.1186/1476-4598-11-87
64. Wang L, Liu Z, Li Y, Pappan L, Galliher-Beckley A, Shi J. Pro-inflammatory cytokine interleukin-1beta promotes the development of intestinal stem cells. Inflammation Res (2012) 61:1085–92. doi: 10.1007/s00011-012-0501-3
65. Schneider SL, Ross AL, Grichnik JM. Do inflammatory pathways drive melanomagenesis? Exp Dermatol (2015) 24:86–90. doi: 10.1111/exd.12502
66. Perera AP, Sajnani K, Dickinson J, Eri R, Korner H. NLRP3 inflammasome in colitis and colitis-associated colorectal cancer. Mamm Genome (2018) 29:817–30. doi: 10.1007/s00335-018-9783-2
67. Wei Q, Mu K, Li T, Zhang Y, Yang Z, Jia X, et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest (2014) 94:52–62. doi: 10.1038/labinvest.2013.126
68. Gao J, Qiu X, Xi G, Liu H, Zhang F, Lv T, et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol Rep (2018) 40:1971–84. doi: 10.3892/or.2018.6634
69. Wang Y, Kong H, Zeng X, Liu W, Wang Z, Yan X, et al. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol Rep (2016) 35:2053–64. doi: 10.3892/or.2016.4569
70. Roy D, Sarkar S, Felty Q. Levels of IL-1 beta control stimulatory/inhibitory growth of cancer cells. Front Biosci (2006) 11:889–98. doi: 10.2741/1845
71. Hai Ping P, Feng Bo T, Li L, Nan Hui Y, Hong Z. IL-1beta/NF-kb signaling promotes colorectal cancer cell growth through miR-181a/PTEN axis. Arch Biochem Biophys (2016) 604:20–6. doi: 10.1016/j.abb.2016.06.001
72. Liu SH, Shen PC, Chen CY, Hsu AN, Cho YC, Lai YL, et al. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res (2020) 48:D863–70. doi: 10.1093/nar/gkz964
73. Shi H, Wang Y, Li X, Zhan X, Tang M, Fina M, et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol (2016) 17:250–8. doi: 10.1038/ni.3333
74. Dong F, Dong S, Liang Y, Wang K, Qin Y, Zhao X. miR20b inhibits the senescence of human umbilical vein endothelial cells through regulating the Wnt/betacatenin pathway via the TXNIP/NLRP3 axis. Int J Mol Med (2020) 45:847–57. doi: 10.3892/ijmm.2020.4457
75. Park JI, Venteicher AS, Hong JY, Choi J, Jun S, Shkreli M, et al. Telomerase modulates Wnt signalling by association with target gene chromatin. Nature (2009) 460:66–72. doi: 10.1038/nature08137
76. Huang L, Luo R, Li J, Wang D, Zhang Y, Liu L, et al. beta-catenin promotes NLRP3 inflammasome activation via increasing the association between NLRP3 and ASC. Mol Immunol (2020) 121:186–94. doi: 10.1016/j.molimm.2020.02.017
77. Tanno T, Matsui W. Development and maintenance of cancer stem cells under chronic inflammation. J Nippon Med Sch (2011) 78:138–45. doi: 10.1272/jnms.78.138
78. Drexler SK, Yazdi AS. Complex roles of inflammasomes in carcinogenesis. Cancer J (2013) 19:468–72. doi: 10.1097/PPO.0000000000000004
79. Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ (2013) 20:1149–60. doi: 10.1038/cdd.2013.37
80. Antonopoulos C, El Sanadi C, Kaiser WJ, Mocarski ES, Dubyak GR. Proapoptotic chemotherapeutic drugs induce noncanonical processing and release of IL-1beta via caspase-8 in dendritic cells. J Immunol (2013) 191:4789–803. doi: 10.4049/jimmunol.1300645
81. Li S, Sun R, Chen Y, Wei H, Tian Z. TLR2 limits development of hepatocellular carcinoma by reducing IL18-mediated immunosuppression. Cancer Res (2015) 75:986–95. doi: 10.1158/0008-5472.CAN-14-2371
82. Haneklaus M, O’Neill LA. NLRP3 at the interface of metabolism and inflammation. Immunol Rev (2015) 265:53–62. doi: 10.1111/imr.12285
83. He ZF, Jin XR, Lin JJ, Zhang X, Liu Y, Xu HL, et al. NALP3 orchestrates cellular bioenergetics to facilitate non-small cell lung cancer cell growth. Life Sci (2020) 241:117165. doi: 10.1016/j.lfs.2019.117165
84. Cai TT, Ye SB, Liu YN, He J, Chen QY, Mai HQ, et al. LMP1-mediated glycolysis induces myeloid-derived suppressor cell expansion in nasopharyngeal carcinoma. PloS Pathog (2017) 13:e1006503. doi: 10.1371/journal.ppat.1006503
85. Moon JS, Hisata S, Park MA, DeNicola GM, Ryter SW, Nakahira K, et al. mTORC1-induced HK1-dependent glycolysis regulates NLRP3 inflammasome activation. Cell Rep (2015) 12:102–15. doi: 10.1016/j.celrep.2015.05.046
86. Wen H, Ting JP, O’Neill LA. A role for the NLRP3 inflammasome in metabolic diseases–did Warburg miss inflammation? Nat Immunol (2012) 13:352–57. doi: 10.1038/ni.2228
87. Zheng H, Hogberg J, Stenius U. ATM-activated autotaxin (ATX) propagates inflammation and DNA damage in lung epithelial cells: a new mode of action for silica-induced DNA damage? Carcinogenesis (2017) 38:1196–206. doi: 10.1093/carcin/bgx100
88. Hasegawa T, Nakashima M, Suzuki Y. Nuclear DNA damage-triggered NLRP3 inflammasome activation promotes UVB-induced inflammatory responses in human keratinocytes. Biochem Biophys Res Commun (2016) 477:329–35. doi: 10.1016/j.bbrc.2016.06.106
89. Wang W, Hu D, Wu C, Feng Y, Li A, Liu W, et al. STING promotes NLRP3 localization in ER and facilitates NLRP3 deubiquitination to activate the inflammasome upon HSV-1 infection. PloS Pathog (2020) 16:e1008335. doi: 10.1371/journal.ppat.1008335
90. Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature (2011) 469:221–5. doi: 10.1038/nature09663
91. Subramanian N, Natarajan K, Clatworthy MR, Wang Z, Germain RN. The adaptor MAVS promotes NLRP3 mitochondrial localization and inflammasome activation. Cell (2013) 153:348–61. doi: 10.1016/j.cell.2013.02.054
92. Kim SM, Kim YG, Kim DJ, Park SH, Jeong KH, Lee YH, et al. Inflammasome-independent role of NLRP3 mediates mitochondrial regulation in renal injury. Front Immunol (2018) 9:2563. doi: 10.3389/fimmu.2018.02563
93. Kolb R, Phan L, Borcherding N, Liu Y, Yuan F, Janowski AM, et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun (2016) 7:13007. doi: 10.1038/ncomms13007
94. Li W, Saud SM, Young MR, Chen G, Hua B. Targeting AMPK for cancer prevention and treatment. Oncotarget (2015) 6:7365–78. doi: 10.18632/oncotarget.3629
95. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol (2011) 12:408–15. doi: 10.1038/ni.2022
96. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability–an evolving hallmark of cancer. Nat Rev Mol Cell Biol (2010) 11:220–28. doi: 10.1038/nrm2858
97. Korhonen E, Bisevac J, Hyttinen JMT, Piippo N, Hytti M, Kaarniranta K, et al. UV-B-induced inflammasome activation can be prevented by cis-urocanic acid in human corneal epithelial cells. Invest Ophthalmol Vis Sci (2020) 61:7. doi: 10.1167/iovs.61.4.7
98. Carmi Y, Dotan S, Rider P, Kaplanov I, White MR, Baron R, et al. The role of IL-1beta in the early tumor cell-induced angiogenic response. J Immunol (2013) 190:3500–9. doi: 10.4049/jimmunol.1202769
99. Voronov E, Shouval DS, Krelin Y, Cagnano E, Benharroch D, Iwakura Y, et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A (2003) 100:2645–50. doi: 10.1073/pnas.0437939100
100. Wang H, Wang Y, Du Q, Lu P, Fan H, Lu J, et al. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp Cell Res (2016) 342:184–92. doi: 10.1016/j.yexcr.2016.03.009
101. Chirivi RG, Chiodoni C, Musiani P, Garofalo A, Bernasconi S, Colombo MP, et al. IL-1alpha gene-transfected human melanoma cells increase tumor-cell adhesion to endothelial cells and their retention in the lung of nude mice. Int J Cancer (1996) 67:856–63. doi: 10.1002/(SICI)1097-0215(19960917)67:6<856::AID-IJC16>3.0.CO;2-
102. Vidal-Vanaclocha F, Alvarez A, Asumendi A, Urcelay B, Tonino P, Dinarello CA. Interleukin 1 (IL-1)-dependent melanoma hepatic metastasis in vivo; increased endothelial adherence by IL-1-induced mannose receptors and growth factor production in vitro. J Natl Cancer Inst (1996) 88:198–205. doi: 10.1093/jnci/88.3-4.198
103. Dinarello CA. Biologic basis for interleukin-1 in disease. Blood (1996) 87:2095–147. doi: 10.1182/blood.V87.6.2095.bloodjournal8762095
104. Mantovani A, Dejana E. Cytokines as communication signals between leukocytes and endothelial cells. Immunol Today (1989) 10:370–75. doi: 10.1016/0167-5699(89)90270-3
105. Huang L, Li F, Deng P, Hu C. microRNA-223 promotes tumor progression in lung cancer A549 cells via activation of the NF-kappaB signaling pathway. Oncol Res (2016) 24:405–13. doi: 10.3727/096504016X14685034103437
106. Xu Y, Li H, Chen W, Yao X, Xing Y, Wang X, et al. Mycoplasma hyorhinis activates the NLRP3 inflammasome and promotes migration and invasion of gastric cancer cells. PloS One (2013) 8:e77955. doi: 10.1371/journal.pone.0077955
107. Duan S, Wang N, Huang L, Shao H, Zhang P, Wang W, et al. NLRP3 inflammasome activation involved in LPS and coal tar pitch extract-induced malignant transformation of human bronchial epithelial cells. Environ Toxicol (2019) 34:585–93. doi: 10.1002/tox.22725
Keywords: NLRP3 inflammasome, cancer hallmarks, tumor microenvironment, pyroptosis, interleukin-1β
Citation: Lin T-Y, Tsai M-C, Tu W, Yeh H-C, Wang S-C, Huang S-P and Li C-Y (2021) Role of the NLRP3 Inflammasome: Insights Into Cancer Hallmarks. Front. Immunol. 11:610492. doi: 10.3389/fimmu.2020.610492
Received: 26 September 2020; Accepted: 16 December 2020;
Published: 03 February 2021.
Edited by:
Kuo-Feng Hua, National Ilan University, TaiwanReviewed by:
Francesco Di Virgilio, University of Ferrara, ItalyMary A. Markiewicz, University of Kansas Medical Center, United States
Copyright © 2021 Lin, Tsai, Tu, Yeh, Wang, Huang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shu-Pin Huang, c2hwaWh1NzNAZ21haWwuY29t; Chia-Yang Li, Y2hpYXlhbmdsaUBrbXUuZWR1LnR3
†These authors have contributed equally to this work and share second authorship