 Daniela C. Ivan
Daniela C. Ivan Sabrina Walthert
Sabrina Walthert Kristina Berve
Kristina Berve Jasmin Steudler
Jasmin Steudler Giuseppe Locatelli
Giuseppe Locatelli- Theodor Kocher Institute, University Bern, Bern, Switzerland
The central nervous system (CNS) parenchyma is enclosed and protected by a multilayered system of cellular and acellular barriers, functionally separating glia and neurons from peripheral circulation and blood-borne immune cells. Populating these borders as dynamic observers, CNS-resident macrophages contribute to organ homeostasis. Upon autoimmune, traumatic or neurodegenerative inflammation, these phagocytes start playing additional roles as immune regulators contributing to disease evolution. At the same time, pathological CNS conditions drive the migration and recruitment of blood-borne monocyte-derived cells across distinct local gateways. This invasion process drastically increases border complexity and can lead to parenchymal infiltration of blood-borne phagocytes playing a direct role both in damage and in tissue repair. While recent studies and technical advancements have highlighted the extreme heterogeneity of these resident and CNS-invading cells, both the compartment-specific mechanism of invasion and the functional specification of intruding and resident cells remain unclear. This review illustrates the complexity of mononuclear phagocytes at CNS interfaces, indicating how further studies of CNS border dynamics are crucially needed to shed light on local and systemic regulation of CNS functions and dysfunctions.
Introduction
The borders of the central nervous system (CNS) parenchyma are complex structures which maintain organ homeostasis through distinct anatomical specializations. These border areas halt the transit of potentially harmful trespassers contributing to the establishment of a relatively immune-privileged milieu within the CNS parenchyma (1). At the same time, these functional barriers host an extensive variety of yolk sac- and bone marrow-derived myeloid cells, cellular dwellers which are an integral part of the historically overlooked CNS immune capabilities. Altogether, CNS interfaces are fundamental participants in CNS functions and defense mechanisms, as well as contributing to the overall integration of the CNS with the rest of the organism (2–4).
While an increasing body of research is finally dedicating attention to CNS borders and their cellular components, surprisingly much remains to be investigated and understood (5–7).
In this review, we will illustrate the functions and migratory routes of monocyte-derived and tissue-resident macrophages, the immune cells that most densely populate CNS interfaces during homeostasis and upon damage and inflammation (8).
Barrier-Associated Dwellers: Location and Homeostatic Function of CNS Macrophages
CNS borders contain functional barriers separating the CNS parenchyma from peripheral circulation at the level of I- the leptomeningeal/subpial vasculature within the subarachnoid space (SAS), II- the blood-cerebrospinal fluid barrier (BCSFB) of the choroid plexus (ChP) and of the arachnoid mater, and III- the blood-brain/spinal cord barrier (BBB) within parenchymal vessels. As an exception to this rule, circumventricular organs lining the brain ventricles and possessing endocrine functions lack a BBB (9). Furthermore, the CNS parenchyma is protected by the astrocytic glia limitans which envelops perivascular and meningeal surfaces (10) allowing a double layer of separation between parenchymal cells and peripheral circulation (11–13).
CNS interfaces harbor populations of tissue-resident macrophages often referred to as CNS-associated macrophages or barrier-associated macrophages (BAMs, Figure 1) (14–16). Once mistakenly believed to derive from adult bone marrow progenitors (17, 18), most BAMs originate in the yolk sac during embryonic development and stably populate the respective niches by self-renewal throughout adulthood (14), as previously shown for microglia (19). The complex development of BAMs and microglia, deriving from distinct yolk sac-derived progenitor lineages (20), has been extensively reviewed in the last years (7, 21). Sharing high expression of fractalkine receptor (CX3CR1) and a long half-life, compared to circulating monocytes, BAMs and microglia have been often collectively studied as CNS-resident phagocytes (22), at least until the recent discovery of microglia-specific genes and related targeted transgenic approaches (15, 23).
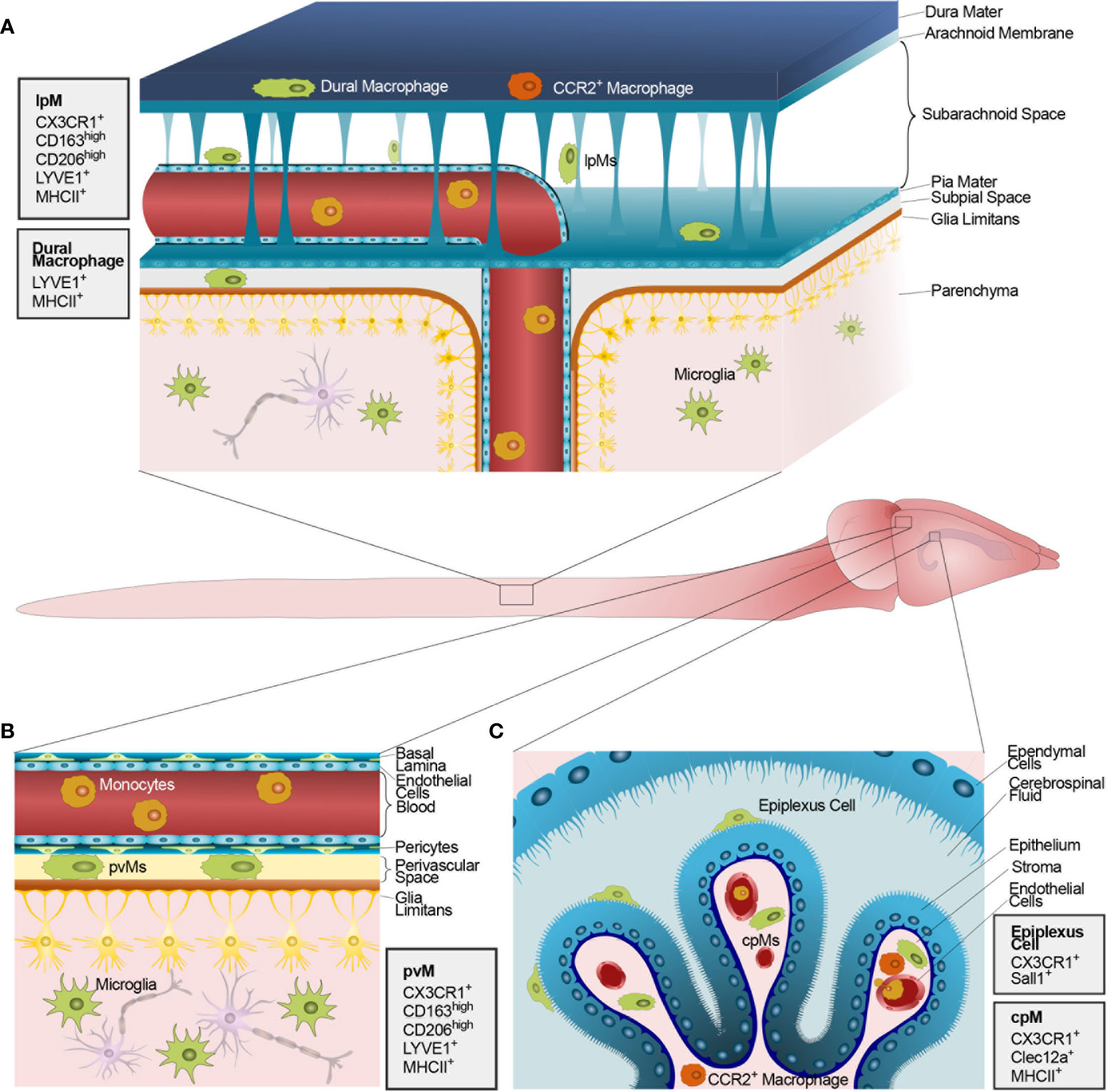
Figure 1 Macrophages populating the CNS barriers and parenchyma at steady state. The figure shows the mouse CNS and, in the magnified inlets, schematic representations of the anatomical CNS interfaces containing functional barriers. (A) The mouse meninges including (top to bottom) the dura mater, the impermeable arachnoid mater, the SAS, the pia mater, the astrocytic glia limitans, and, finally, the CNS parenchyma. The dura mater is populated by both yolk sac-derived (green) and blood-borne CCR2+ macrophages (orange). Conversely, the SAS, the subpial space and the CNS parenchyma host solely long-lived yolk sac-derived lpMs and microglia, respectively. (B) Schematic representation of the perivascular space at the level of post-capillary venules within the CNS parenchyma. The perivascular space hosts yolk sac-derived pvMs between a layer composed of endothelial basal lamina and pericytes and a parenchymal basal lamina. Endothelial cells forming the blood vessel are linked by tight junctions thus constituting a BBB. On the parenchymal side, astrocytic end-feet collectively form the glia limitans vascularis. (C) Schematic representation of the ChP within a CSF-filled brain ventricle lined by ependymal cells. On the apical side of the ChP epithelial cells, resident epiplexus cells are shown. ChP epithelial cells are linked by tight junction thus constituting a functional BCSFB. The ChP stroma hosts a combination of yolk sac-derived cpMs (green) and blood-borne CCR2+ macrophages (orange) extravasated from stromal vessels lacking a BBB. Monocytes circulating within vascular lumens are shown in yellow. Next to each panel, gray boxes illustrate the main protein markers identifying CNS-resident macrophages in their distinct anatomical compartments.
Compared to microglia, BAMs share universally upregulated genes linked to blood vessel development, lipid and cholesterol metabolism, immune response and antigen presentation (16). In addition to the core genes Apoe, Ms4a7, Ms4a6c, Tgfbi and Mrc1 (16), Dab2, F13a1, Mgl2, and Pf4 have been recently proposed as BAM identifiers (24).
Not surprisingly, BAMs also express signature macrophage markers such as integrin αM (CD11b), Aif1 (Iba1), receptor for macrophage-colony stimulating factor (Csf1R), and F4/80 (25), the latter, however, at lower levels compared to activated macrophages and circulating monocytes (26). Expression of the adhesion molecule CD44 is negligible and can thus be used to distinguish BAMs from CD44+ blood-borne macrophages within the CNS (26). Interestingly, some BAMs express the gene encoding for the T cell receptor β, although its function remains unknown (26).
While BAMs at the BBB and within the leptomeninges are solely yolk sac-derived, dura mater, and ChP interfaces harbor a mixed resident population including blood-borne monocyte-derived cells during steady state (14, 16). Novel techniques such as mass cytometry (through CyTOF) and single-cell RNA sequencing (scRNAseq) have indeed revealed a surprising heterogeneity of BAMs (16, 26–28), despite the intrinsic limitations of these approaches due to the use of predefined markers (mass cytometry) and under-representation of lowly expressed genes (scRNAseq) (16, 29).
In the next chapters, we will illustrate how BAM complexity is inherently linked to the different anatomical locations that these cells inhabit (30). A summary of cellular locations, origin and known markers in mice and humans can be found in Tables 1 and 2.

Table 1 The table indicates the main RNA and protein markers described for macrophage populations in the distinct CNS compartments in mice at steady state (homeostasis) and in different disease model.

Table 2 The table indicates the main RNA and protein markers described for macrophage populations in the distinct CNS compartments in human samples at steady state (homeostasis) and upon development of different CNS pathologies.
Resident Perivascular Macrophages
The low pinocytic endothelial cells forming parenchymal CNS vessels possess specialized features constituting the BBB, a relatively impermeable diffusion barrier (81, 82). On the parenchymal side, astrocytic end-feet form the glia limitans to offer a second functional barrier protecting the CNS parenchyma. This astrocytic layer appears impermeable to immune cells (13) but does not form tight junctions during homeostasis and allows movement of low-molecular weight tracers (83). Together, this multilayered border limits trafficking of circulating immune cells and controls the selective exclusion of harmful substances from the CNS parenchyma as well as the intake of water, chemicals, and other molecules (3).
First described in the early 1980s as “granular pericytes” (84), perivascular macrophages (pvMs) reside between the endothelial and glia limitans basement membranes of CNS vessels (excluding capillaries and small arterioles) located in basal ganglia and white matter (85–87). PvM distribution remains, however, controversial, with recent work reporting similar densities of pvMs in peri-arteriolar and peri-venous space of the mouse brain (88).
Given their strategic location, pvMs are proposed to mediate passage of information between the CNS and the periphery (4) and to regulate lymphocyte immunesurveillance (89, 90). Indeed, pvMs express MHC class II and co-stimulatory molecules (35) and secrete cytokines and chemokines, which affect the local microenvironment upon sensing damage or inflammation (8). Moreover, pvMs help to maintain the well-being of the endothelial wall and to contribute to the regulation of vascular permeability (91, 92). In line with the physiological function of the perivascular space (93), PvMs participate in CNS waste clearance (94, 95) displaying a high endocytic rate that can be exploited to mark these cells in vivo (94, 96–98). PvMs can also phagocytose tracers injected in the parenchyma, which demonstrates their ability to sample outflowing CNS interstitial fluids (12). Altogether, given the influence of pvMs on vascular smooth muscle cells (88) and the importance of pvMs on peri-arterial drainage (99), these cells appear key players in CNS fluid dynamics.
Morphologically, pvMs are compact elongated cells displaying continuous movement of cell body and protrusions (14, 100). Homeostatic pvMs are a transcriptionally homogeneous population (27). Compared to monocytes and microglia, pvMs are characterized by high expression of Cd163 (35), a pattern recognition receptor (PRR) recognizing hemoglobin (101), Mrc1 (CD206), a PRR responsible for scavenging circulating glycoproteins (102), and Cd36, a scavenger receptor implicated in efferocytosis (14, 36). Mass cytometry revealed that these cells, similarly to other BAMs, are also highly positive for CD38 (15), an ecto-enzyme with metabolic functions (98).
Earlier reports, likely affected by the technical challenge of distinguishing dendritic cell (DC) from BAMs (15), indicated expression of DC markers such as CD11c and DC-SIGN in pvMs (103). Functionally and ontogenically separate from BAMs, CNS-associated DCs are described elsewhere (16, 27, 28).
Resident Leptomeningeal Macrophages
The cerebrospinal fluid (CSF)-filled SAS regulates CNS fluid, pathogen, and immune cell dynamics (104) and hosts several types of immune and non-immune dwellers including leptomeningeal macrophages (lpMs). The SAS is contained between the tight arachnoid membrane and the pia mater, a thin monolayer of cells linked by desmosomes and gap junctions (104–107). Different collagen-rich trabeculae covered by pial/leptomeningeal cells connect the arachnoid to the pia mater in humans (103). Finally, below the pia mater, the glia limitans functionally separates the SAS from the parenchyma delineating the entire CNS (10, 13).
Importantly, the CSF permeating the SAS also fills the perivascular spaces of parenchymal vessels, with complex exchanges at the level of penetrating arteries surrounded by a layer of pial cells (108). The CSF also collects antigen-rich interstitial fluid from the CNS parenchyma (106), although the extent of this process remains the subject of debate. Accordingly, intra-CNS administration of drugs or tracers [e.g., intra-ventricular injection of clodronate particles (109)] leads to targeting of both lpMs and pvMs (88, 95), an often-overlooked phenomenon in BAM literature.
Altogether, both pvMs and lpMs continuously surveil CSF composition and thus indirectly examine the CNS at a molecular level (12). Given the high local production of immune-regulatory molecules such as TGFβ2 and IL13, the CSF can also influence the phenotype of resident SAS cells (110).
Long-lived lpMs originate in the yolk sac and seed the SAS embrionically (14). Similarly to pvMs, lpMs show an impaired potential for self-renewal following drug-induced inhibition of Csf1r, at least compared to fast-proliferating microglia (16).
LpMs constitute approximately 1/3 of the cells collected from human CSF (111) but are also found in high densities in the subpial layer above the parenchyma (38). Within the SAS, they are often located nearby fibroblast-like leptomeningeal cells (14). Morphologically, lpMs have been described as sessile elongated cells following leptomeningeal vessels (100). Recent intravital observations showed, however, that lpMs are heterogenously able to remain stationary with continuous ameboid movement or to crawl within the SAS (14).
As other BAMs, lpMs are CD163highCD206high sentinels for pathogens and inflammation (33, 34, 49) and important sources of the chemoattractant CXCL12/SDF-1, a key factor in the migration of immune cells and neuronal and oligodendrocyte precursors (112, 113). On a transcriptional level, homeostatic lpMs express high levels of Pf4, Cbr2, Ms4a7, Stab1, Fcrls, and Siglec1, with certain subpopulations expressing Cxcl2 and Nfkbiz (27). A different scRNA-seq study also indicated high expression of P2rx7, Egfl7, Clec4n, Clec10a, Folr2, and Lyve1, with a comparable expression pattern from birth to adulthood (16). Among these, Lyve1, a hyaluronic acid receptor highly expressed in lymphatic vessels (114), has emerged as a marker for MHCIIlow lpMs close to the pia mater (15), as opposed to its low expression in MHCIIhigh lpMs in the SAS (115).
Interestingly, the SAS hosts a small population of CX3CR1lowLyve1+CD38+ lpMs (15, 16) which might have escaped characterization in studies discriminating BAMs based on CX3CR1 positivity (14, 27).
Resident Dural Macrophages
The dura mater is the outermost component of the meninges, containing a high density of collagen and blood vessels that lack a BBB (104, 116). Differentiating this compartment from the rest of the CNS and similar to peripheral organs, the dura displays lymphatics running along major venous sinuses (93) and thus cannot be considered a CNS immune barrier (13, 28). Furthermore, the dura remains delineated from the SAS by a functional BCSFB containing intercellular tight junctions, the impermeable arachnoid membrane (13, 104, 116). While the possible transit of immune cells from the dura to the CNS parenchyma remains unclear, different interchanges between dura and SAS can occur and remain an active area of study (97). Recent investigations have highlighted direct venous connections allowing neutrophils and potentially other myeloid cells to transit between the brain dural vasculature and the skull bone marrow (117, 118).
Dural resident macrophages are characterized as a dense Lyve1lowMHCIIhigh population, with few Lyve1highMHCII+ cells present (15) in a different relative proportion compared to the SAS (119). These cells dynamically surveil the local environment while sensing distal gut biome changes (16). Displaying a mixed embryonic and bone marrow origin (6), dural macrophages account for the vast majority of blood-derived myeloid elements found in CNS preparations during homeostasis, together with CCR2+ macrophages within the ChP stroma (26). During inflammation, further blood-borne monocytes are locally recruited (97), while dural macrophages can regulate lymphoangiogenesis through the release of VEGF-C (120).
Resident Choroid Plexus Macrophages
ChPs are located within the third, fourth, and lateral ventricles of the brain and host a functional barrier for immune cell trafficking, the BCSFB. Separating peripheral circulation from the CSF, this barrier consists of a monolayer of epithelial cells connected through tight and adherens junctions (13) and expressing regulatory factors such as macrophage migration inhibitor factor (MIF) (121). On the basolateral side of this layer, a basement membrane and a thin stroma divide the BCSFB from fenestrated blood vessels (122, 123).
Producing the CSF and maintaining its chemical balance, the ChP has been considered as “the kidney of the CNS”, indispensable for homeostatic equilibrium (124–126). Furthermore, the ChP plays roles in brain development, neurogenesis, metabolism (108, 127, 128) and secretes immunomodulatory microRNAs (129). The CSF itself has mechanical and signaling roles exerted through bioactive molecules and physical/chemical properties such as pH, osmolarity, and flow speed (130).
Different macrophages populate the ChP, albeit at a lower density compared to other CNS interfaces (26). ChP macrophages have been historically described as stromal phagolysosome-rich CD163+MHCII+ antigen presenters (38, 39). Recent studies, however, indicate that the ChP hosts a highly heterogeneous population of yolk sac-derived long-lived stromal macrophages (ChPMs), CCR2+ blood-borne macrophages, and Sall1+ Kolmer/epiplexus cells situated on the apical side of epithelial cells and thus beyond the BCSFB (16).
The dynamic movement of ChP macrophages has been recently described by in vivo two-photon imaging following deep-brain cannula implantation: while epiplexus cells display different kinetic patterns on the apical side of epithelial cells, stromal macrophages continuously surveil ChP vasculature with highly motile processes, efficiently phagocytosing blood-borne fluorescent dextran (131).
Unique among BAMs, epiplexus macrophages share ontogeny, local self-renewal upon depletion, and transcriptome with parenchymal microglia (16). Analysis of the ChP via scRNA-seq identified three macrophage clusters sharing high expression of BAM signature genes Mrc1, Ms4a7, Pf4, Stab1, Cbr2, and Fcrls (27). Another scRNA-seq study also described three ChP clusters sharing signature expression of Lilra5 and Ttr and identified as Cst7+Gm1673+ Clec7a+ epiplexus cells, MHCIIhigh and MHCIIlow ChPMs, the latter two likely corresponding to yolk sac- and bone marrow-derived resident ChPMs, respectively (16). In parallel, mass cytometry indicates equal numbers of Lyve1+MHCII negative, Lyve1negativeMHCII+, and Lyve1+MHCII+ ChP macrophages, in a proportion which differs from the one observed at other CNS barriers (15).
Interestingly, MHCII expression in ChP macrophages is affected by microbiome alterations likely sensed via proximal fenestrated capillaries (16). Unfortunately, the effect of gut flora alterations has not been convincingly investigated in other BAMs.
Circulating Monocytes, Border Trespassers Upon Inflammation
Origin, function and classification of blood monocytes have been reviewed elsewhere (32, 132–134). Briefly, following monopoiesis, monocytes are mobilized by a CCL2-dependent mechanism from the bone marrow and from splenic secondary reservoir (135) and enter the circulation displaying a half-life of approximately 1–2 days in mice and of 1–7 days in humans, depending on the cellular subset (22, 136–138). In the mouse, two major types of blood monocytes can be described as Ly6ChighCCR2+CX3CR1low “classical” inflammatory monocytes and Ly6ClowCCR2lowCX3CR1high “non-classical” patrolling cells (31), with the latter originating from the former both in lymphoid organs and in the periphery (133). While Ly6ChighCCR2+CX3CR1low cells show fast CCR2-mediated recruitment toward inflamed tissues (139), patrolling monocytes mostly participate in endothelial homeostasis within the lumen (137, 140, 141). In humans, a parallel classification exists with “classical” monocytes characterized as CD14highCD16negative, non-classical cells as CD14+CD16high and transitional intermediate monocytes as CD14highCD16+ (32, 60, 61). A more complex categorization of monocyte subtypes is, however, possible and advisable both for mice and human studies (142–145).
Despite their population-specific differences, all circulating monocytes express high levels of Csf1R and the receptor for granulocyte-monocyte colony stimulating factor (GM-CSFR), platelet endothelial cell adhesion molecule 1 (PECAM-1), and β2 and αM integrins, among others (32, 132).
Monocytes sense inflammation and damage via cytokines, chemoattractants, and damage-associated molecular patterns (DAMPs) which contribute to their tissue recruitment (146), with extravasation leading to differentiation to monocyte-derived macrophages (MdMs) (147). Depending on the specific context and highlighting their plastic potential, monocytes can, however, also differentiate into monocyte-derived DCs (148–150) or even to other cellular fates (151).
Dynamic interaction with endothelial cells in the vascular lumen involves a selectin-dependent rolling, a chemokine-dependent arrest and adhesion, and an integrin-mediated crawling eventually resulting into diapedesis (152). Extracellular matrix molecules such as heparane sulfate proteoglycans expressed by the CNS vasculature can also mediate monocyte interaction with endothelial cells (153). Given a differential expression of interaction molecules and chemokine receptors, monocyte subtypes display intrinsic variance in this multistep process (132). Cell deformability through cytoskeletal reorganization and membrane stiffness changes are also regulators of trafficking (154). During trans-endothelial migration, monocytes interact with the endothelial molecules CD99, PECAM1 and CD155 (155) and, following diapedesis, cross the vascular basement membrane and interact with other perivascular cells (144, 156).
Within inflamed tissues, MdMs display substantial differences compared to monocytes. Upregulation of cell differentiation and trafficking genes starts during the first luminal contact with endothelial cells (144, 157, 158), with transmigrated monocytes showing significant changes in metabolism, chemotaxis, survival, inflammatory response (159), and rearrangement in subcellular structures leading to an augmented size (134). Altogether, through the recruitment process, monocytes can acquire distinct pro- or anti-inflammatory polarizations, substantially contributing to pathogen eradication/tissue destruction or to the regulation of inflammation/promotion of tissue regeneration, respectively.
Macrophage Pro- and Anti-Inflammatory Functions
The acquisition of a functional phenotype by tissue macrophages and MdMs is a highly dynamic process which integrates several local cues and thus remains challenging to define in vivo. While these functional adaptations can be modeled and described in high detail in vitro (160) through a variety of techniques (161), the signaling pathways and functional activations observed in vitro and in vivo may diverge significantly depending on the model and the context (162).
Macrophage gene expression displays an inherent plasticity influenced by local signaling, chemical changes and physical confinement (163, 164). While pro-inflammatory macrophages mainly contribute to damage and neurotoxicity by the secretion of chemokines, inflammatory cytokines, and reactive oxygen and nitrogen species, anti-inflammatory cells extensively contribute to neuroprotection by debris scavenging and by releasing tissue regeneration intermediates and growth factors. Functional specifications are also reflected by divergent metabolic adaptations, with pro- and anti-inflammatory polarizations distinctively characterized by differential ATP production and oxygen consumption rates (165). Notably, the acquisition of a specific macrophage phenotype varies substantially also between different mouse strains (166).
To describe the spectrum of macrophage functions, researchers have largely made use of the M1/M2 dichotomy, a jargon introduced in the 1990s to indicate the outcomes of cellular stimulation with IL-4 or lipopolysaccharide (LPS)/IFNγ, respectively (167). Unfortunately, the application of the binary M1/M2 nomenclature to extremely diverse in vitro and in vivo contexts was unable to properly define multifaceted cellular actions (162, 163, 168). The limitations of this dichotomy were also evidenced when studying microglia/macrophage activation in several pathological contexts, including traumatic and neurodegenerative diseases and disease models (165). While some efforts of clarification in macrophage nomenclature have been made (25, 168–170), a generally accepted consensus is still missing. As suggested by experts in the field (170), we support a jargon describing cellular phenotypes via the in vitro stimuli used or, in complex in vivo scenarios, via the observed pro- or anti-inflammatory roles of the described populations.
Besides nomenclature issues, however, differentially polarized macrophage and monocyte subsets from mice and humans possess distinct migratory properties, for example, toward plasminogen (171). Notably, their CNS-invading trajectories and the anatomical site in which they acquire their differential function remain an undeveloped area of study.
Monocyte Trafficking Through the CNS at Steady State
While accumulation of peripheral immune cells at CNS borders is a hallmark of CNS diseases (172), rapid recruitment of monocytes to perivascular CNS spaces is also observed upon peripheral inflammation, such as in endotoxemia (173). While this highlights the potential for active CNS surveillance by blood-borne myeloid cells notwithstanding the absence of local damage, CNS interfaces at steady state host only a limited number of bone marrow-derived immune cells (89, 174, 175). Recruitment of these cells drastically depends on local tissue accessibility, with interfaces such as the dura mater and the ChP hosting fenestrated vessels and a concomitant higher density of monocytes (16).
Importantly, stromal accumulation of blood-borne leukocytes in the ChP might serve as an intermediate step for reaching the CSF by crossing the BCSFB (176, 177). Analysis of human CSF indicates that approximately 1/3 of the cellular compartment comprises monocytes (178), with a vast majority of blood-borne CD16high cells (68). The homeostatic recruitment of these cells, potentially extravasating at the ChP or directly through leptomeningeal vessels as shown upon CNS damage (47, 112), is, however, unknown.
Given the secluded intraventricular location of ChPs, in vitro models have contributed significantly to our understanding of local cell trafficking (179). Using primary ChP mouse epithelial cells, we recently showed that MdMs can migrate through the BCSFB epithelium also in absence of inflammation (37). This transmigration pathway seems possible also for other myeloid cells (180).
Within the CNS parenchyma, basal immunesurveillance is exerted by microglia and pvMs, without apparent contributions by MdMs (14). The ability of MdMs to surveil these border areas at steady state has been historically overestimated due to the absence of tools discriminating yolk sac- and bone marrow-derived myeloid cells and due to the experimental use of chemotherapy or gamma irradiation, artificially increasing BBB permeability and CNS chemokine production (15, 16, 22).
In general, the concept of peripheral immunesurveillance implies that patrolling antigen-presenting cells scan their target organ and, upon infection, move toward secondary lymphoid organs to trigger antigen-specific lymphocyte activation. Key to its relative immune privilege, however, the CNS shows limited afferent routes for cell-mediated antigen drainage (1). Antigen-rich CSF drains to peripheral venous blood via arachnoid villi and granulations and to the lymphatic system along nerve roots and nasal and dura lymphatics (12, 181). Notably, the relative importance of these pathways is still under debate (181, 182). Through these exit routes, CNS antigens can accumulate in peripheral lymph nodes (182, 183), potentially via DCs trafficking from CNS borders to peripheral organs (28, 184). Whether monocytes and MdMs can also participate in this afferent arm of CNS immunity in a comparable manner to that observed in peripheral tissues (185) is, however, unclear (175).
Myeloid Dwellers and Trespassers at CNS Interfaces Upon Auto-Aggressive CNS Inflammation
Macrophages constitute the predominant cell type in the damaged CNS of multiple sclerosis (MS) patients, independently from clinical course (169) and lesion subtype (186, 187). Accordingly, MS disease-modifying therapies strongly affect monocyte/macrophage functions as part of their therapeutic action (169, 188–190).
MS is a chronic inflammatory disease of the CNS with unknown etiology and a heterogeneous pathological course, including relapsing-remitting (RRMS), primary and secondary progressive forms (191). Histopathologically, MS is characterized by multifocal BBB damage and leukocyte infiltration in lesions displaying demyelination and neuronal death (192, 193). To date, whether neurodegeneration is the primary cause or rather the secondary consequence of auto-aggressive inflammation remains debated (194).
Blood monocytes isolated from MS patients show altered expression of microRNAs (195), microvescicle release (196), cytokines (197), norepinephrine (198), and enhanced CCL2-, CCL5-, and CXCL1-driven migration (188, 199) compared to cells from healthy controls. The relative proportions of circulating classical, intermediate, and nonclassical monocytes varies across studies, with some indicating a substantial increase in nonclassical CD14+CD16high monocytes (65), while recent work shows an increase in CD14high and CD16high monocytes specifically in RRMS patients with inactive disease (66).
Within the CNS parenchyma, resident and invading macrophages play complex roles both preclinically and in established lesions (71). Monocyte invasion might, however, vary at different disease stages, with less MdM infiltrates observed in progressive MS compared to RRMS (200).
Inflammatory macrophage functions range from tissue destruction (103) to beneficial roles (201, 202), a continuum reflecting their unique transcriptional plasticity (163, 170). While microglia actions during MS fall in the same context-dependent classification, slowly expanding lesions from progressive MS patients display high density of pro-inflammatory markers in perilesional microglia, showing how these cells can contribute to disease progression (203). In general, however, it remains unclear whether distinct microglia/macrophage actions are preferentially associated with different phases of lesion evolution, or whether they co-exist at every clinical timepoint or even within the same cells (72, 169).
Albeit heterogeneous, the distribution of MS lesions often follows an expected pattern (204), potentially shaped by routes of leukocyte entry and local antigen presentation (205).
To mimic the multifaceted pathological aspects of MS, several inducible and spontaneous animal models have been established. Among these, experimental autoimmune encephalomyelitis (EAE) has been the main tool to study disease mechanisms and to develop and test MS disease-modifying therapies (169), despite its intrinsic limitations as an MS model (206).
As in MS, inflammation in EAE is characterized by a high density of activated macrophages at CNS interfaces and within parenchymal lesions (Figure 2) (44). Given the overlapping expression of key markers including CD11b, Csf1R, CD163, and CD206 (21), the relative pathological contributions of MdMs and resident macrophages has remained unaddressed for decades, but technical advancements finally allow us to define their respective roles (169). Upon induction of EAE, BAMs increase their expression of MHCII, CD44, the immunomodulatory molecule PDL1, CD117 (c-KIT), and Sca-1 (Ly6a) (15). Despite convergent morphological and expression changes, resident macrophages and MdMs remain transcriptionally separate (208) and can be distinguished through mass cytometry (15) and scRNAseq techniques (27). The survival dynamics of recruited MdMs remain, however, unclear, with previous work indicating an inability of invading macrophages to persist as microglia-like cells (209) and recent reports showing the opposite (21, 210, 211).

Figure 2 Macrophages populating CNS barriers and parenchyma during autoimmune CNS inflammation. The figure shows the mouse CNS following induction of EAE and disease development. The magnified inlets illustrate schematic representations of the anatomical CNS interfaces containing functional barriers and blood-borne MdMs. (A) The mouse meninges. Top to bottom, the dura mater hosts yolk sac-derived (green), blood-borne CCR2+ macrophages (orange), and several activated MdMs (red). Different monocytes/MdMs (red) infiltrate the SAS and the subpial space either extravasating at the level of leptomeningeal vessels or crossing the distal ChP BCSFB, thus reaching the CSF. MdMs can invade the CNS parenchyma while yolk sac-derived microglia (green) increase in number. (B) Schematic representation of the perivascular space at the level of post-capillary CNS venules, hosting yolk sac-derived pvMs (green) and invading MdMs (red) collectively forming a perivascular inflammatory cuff. After crossing the glia limitans, MdMs (red) accumulate in parenchymal lesions. (C) Schematic representation of the ChP within a CSF-filled brain ventricle lined by ependymal cells. The ChP stroma is enlarged compared to steady state and hosts a combination of yolk sac-derived cpMs (green) and different populations of blood-borne inflammatory CCR2+ macrophages (orange/red) increasingly extravasated from stromal vessels lacking a BBB. Monocytes circulating within vascular lumens are shown in yellow. The epithelial cells of the BCSFB show decreased density of apical microvilli and are represented as darker and lighter cells to model the ultrastructural alterations previously reported (207). Next to each panel, gray boxes illustrate the main protein markers identifying CNS-resident macrophages in the distinct anatomical compartments, and the main markers commonly expressed by MdMs during CNS inflammation.
During the active phase of EAE, MdMs substantially outnumber BAMs at CNS interfaces (15). Compared to circulating monocytes, CNS-recruited MdMs upregulate glycolytic enzymes and production of inflammatory molecules (44). While experimental MdM removal by apoptosis reduces disease burden (212), MdM accumulation directly correlates with neurodegeneration (208), likely through an increased production of reactive nitrogen and oxygen species (ROS/RNS) (213, 214). Altogether, monocyte infiltration into the CNS parenchyma parallels EAE clinical signs and exerts a significant influence on glial cells (215, 216), at the same time contributing to tissue regeneration (217, 218).
The overall complexity of macrophage phenotypes during EAE is exemplified by the differential expression of the enzymes iNOS and arginase-1 in CCR2+Ly6Chigh MdMs (44). While iNOS+ MdMs revealed higher glycolytic rates, expression of matrix metalloproteinases (MMPs), chemokines (e.g., Ccl5), and pro-inflammatory cytokines, arginase-1+ MdMs showed increased expression of scavenging receptors, complement proteins and oxidative phosphorylation enzymes (44). Notably and beside iNOS+ and arginase-1+ cells, a recent study described two Saa3+ and Cxcl10+ monocyte populations substantially contributing to tissue damage within the CNS parenchyma (43).
Before reaching the white or gray matter, MdMs are increasingly recruited to the ChP stroma (37) and need to cross CNS barriers as shown by accumulation within perivascular cuffs at the BBB (45, 46) or in the CSF, extravasating from the ChP and/or from leptomeningeal vessels. Notably, massive monocyte/macrophage accumulation at CNS borders does not directly lead to clinical symptoms in absence of parenchymal infiltration (219). Passage through these interfaces allows, however, monocytes to adapt to the border microenvironment, acquire distinct functional polarizations and, in turn, regulate the evolution of the disease. Hence, the description of MdM migratory routes and the definition of the parallel role of BAMs represent a fundamental milestone in our understanding of auto-aggressive CNS inflammation.
Macrophages at the BBB During MS and EAE
BAMs efficiently sense the peripheral environment via bidirectional communication with their milieu and in particular with endothelial cells (220), scanning for potential distal danger and reacting to it promptly (100, 221). Experimental evidences from both MS and EAE indeed suggest that pvMs become activated even before development of clinical symptoms and infiltration by peripheral cells (222, 223). Accordingly, pre-clinical CNS lesions observed in marmoset EAE models correlate with increased Iba1+ pvMs (224). CNS inflammation leads to a sudden increase in pvM density and to augmented antigen-presenting capabilities (225). These pvMs also increase in number in EAE (42, 226) via a local proliferation lasting until the chronic disease phase (14). The observed proliferation rate is, however, minor compared to the one described for microglia (27).
Nearby MS lesions, pvMs express CD68, CD64, CD40, CD32, and MHCII, as well as the signature proteins CD163 and CD206 (71, 72). During their activation, pvMs also upregulate expression of interacting molecules such as ICAM-1 and VCAM-1 and chemoattractants such as CCL2/MCP-1 and CCL3/MIP-1α (42). Notably, in both EAE and MS, pvMs appear highly phagocytic and take up substantial amounts of iron, directly linked to demyelination (227, 228).
Surprisingly, however, scRNA-seq analysis indicates that pvMs undergo only mild modifications to their overall transcriptional profile during EAE, compared to their steady state (27). Among the few significantly regulated genes, an increased expression of Ccl5, a T cell chemoattractant, of the MIF receptor Cd74 (41) and a decreased expression of Lyve1 and Ctsd (Cathepsin D, a bactericidal protein) was observed (27).
In parallel, neuroinflammation correlates with massive recruitment of peripheral monocytes which cross the BBB drastically increasing the cellularity of perivascular spaces. Monocyte mobilization from peripheral reservoirs is regulated by several factors including GM-CSF (149), a cytokine playing key roles in both EAE and MS (229).
MdM recruitment results in the formation of perivascular cuffs, a MS pathological hallmark where lymphocytes intersperse with a majority of CD16high myelin-laden MdMs (208, 230). At least in EAE, perivascular MdMs appear morphologically smaller than activated resident pvMs (27). A series of DAMPs/alarmins including HGMB1, IL33, and ATP participate in the recruitment of monocytes (146). In turn, both in vitro (68) and in vivo data (45) indicate that MdM perivascular accumulation enhance recruitment and parenchymal invasion of lymphocytes.
Perivascular inflammatory cell cuff formation is often associated with BBB disruption, a multifaceted concept entailing exogenous and endogenous mechanisms (231). Even though BBB damage aid monocyte extravasation, immune cells can cross endothelial barriers showing intact intercellular tight junctions (3, 37, 190). Monocyte diapedesis is aided by the release of inflammatory molecules such as tissue transglutaminase 2, oncostatin M, histamine, superoxide, GM-CSF, and TNFα (232–235). Contact with endothelial cells involves interactions between monocyte integrins such as VLA-4/LFA-1 and endothelial integrin-binding molecules such as VCAM-1/ICAM-1 (169), homophilic interactions through Ninjurin1 (236) and expression of the constitutively expressed adhesion molecule CD166 (237). Mechanistically, contact of monocytes with the BBB allows release of tissue plasminogen activator from endothelial cells following activation of the NMDA receptor NR1, allowing, in turn, monocyte diapedesis (238, 239). Perivascular cell cuff formation is also dependent on the local expression and accumulation of chemokines, including CCL2 (240), CCL3, CXCL12 (46), and potentially of the constitutively expressed CCL19 (241).
In particular, CCL2 actions have been extensively studied in MS and EAE (242). This chemokine can exert distinct roles depending on its astrocytic or endothelial source (240). CCL2 regulates CCR2+ monocyte adherence and chemotaxis by acting on monocyte integrin conformation and clustering (243–246). Following tissue invasion, CCR2 expression is downregulated contributing to CNS retention of inflammatory MdMs as observed in vivo (44) and in vitro (37). Signaling through CCR1 and CCR5 can similarly regulate monocyte multistep accumulation in the perivascular spaces, collectively guiding disease development (185, 247).
Despite its intrinsic pathological consequences, the accumulation of MdMs in perivascular cuffs also constitutes an efficient checkpoint mechanism ensuring that cells do not indiscriminately enter in contact with neuronal cells. To infiltrate the CNS parenchyma from the perivascular space, immune cells need to additionally cross the parenchymal basement membrane and the glia limitans (45). Notably, during neuroinflammation, BBB disruption induces expression of tight junctions in astrocytic end-feet in both EAE (248) and MS (249). Crossing of this second barrier crucially requires expression of MMPs and of MMP regulatory proteins such as CD147 (169). In this process, MMP2 and MMP9 participate in the lysis of perivascular chemokines such as CXCL12, that increase retention of MdMs preventing parenchymal infiltration (45, 46).
Are perivascular MdMs functionally polarized during anti-CNS responses? While interacting with endothelial cells, transmigrating monocytes encounter several activation signals. GM-CSF, shown to augment monocyte diapedesis at the BBB, can induce a hybrid inflammatory phenotype similar to the one observed in MS tissues (232). GM-CSF can also be released by endothelial cells (250, 251) upon stimulation by Il-1β, an inflammatory cytokine released by invading monocytes and indispensable for their infiltration (251), in a looping mechanism potentiating MdM activation between BBB and parenchyma (252). Furthermore, feedback regulation by reactive species-mediated quorum-sensing might play a key role in phenotype adaptation (30). MdMs within the perivascular space can have an iNOS+ or Arginase-1+ phenotype; furthermore, in vivo imaging indicated that MdMs acquired a pro-inflammatory state immediately following diapedesis and before entering the CNS parenchyma (44). Accordingly, perivascular accumulation of glycolytic phagocytes has been observed in both EAE and MS, with recruited MdMs reducing their rate of glycolysis once in the parenchyma (253). Inhibition of glycolysis and of lactate secretion reduced macrophage invasion, both in vivo and in vitro (253). MdMs in perivascular cuffs were strongly positive for Nrf2, a transcription factor involved in protection against oxidative stress and highly expressed in acute MS lesions (73). Moreover, in both MS and EAE, these cells upregulate expression of the extracellular matrix components lectican versican V1 and chondroitin sulfate glycosaminoglycans, molecules able to enhance MdM migration and secretion of inflammatory cytokine and chemokines (254). Finally, pvMs in active demyelinating MS lesions also show strong reactivity for TGFβ (255), an anti-inflammatory molecule with controversial roles (256).
Taken together, the perivascular space thus appears like a key compartment able to shape the pathological role of recruited monocytes in their migration toward the inflamed parenchyma.
Macrophages in the SAS During MS and EAE
Meningeal inflammation is common in MS clinical manifestations, including primary progressive forms (67, 257), often correlating with neurodegeneration (258). The meninges of MS patients can also host lymphoid follicle-like structures rich in B lymphocytes, potential drivers of disease (259).
However, histological analysis reveals that CD68+ myelin-laden phagocytes are the most represented cell type in the inflamed SAS (67). Collection of CSF via lumbar puncture allows analysis of non-adherent SAS phagocytes and indicates significant variations in CSF cellularity depending on the MS subtype. Compared to healthy donors, the CSF of RRMS patients shows a relative decrease in CD16+ monocytes and an increasing proportion of CCR5highCD64+CD86+CD14+ monocytic cells (68). The described decrease in CD16+ monocytes is not observed in primary progressive patients, potentially reflecting different recruitment mechanisms (68). Other studies have shown an increased presence of monocytes expressing HLA-G, an immunosuppressive non-classical MHC molecule (69). Notably, a recent scRNA-seq analysis of the CSF of MS patients identified a majority of monocytes expressing HLA-DR and the lectin CD33 and a small population of HLA-DRhighCD33midLyve1+ macrophages identified by the authors as “microglia” due to their expression of Trem2 and Olr1 (70). This jargon is, however, misleading, as no evidence of parenchymal microglia crossing the glia limitans toward the CSF exists. In another study, a comparable population (named “Mono2”) showed expression of BAM markers such as Stab1 and Ch25h and of inflammatory genes such as Cd9, Cd163, Egr1, Btg2, C1qa, C1qb, Maf, and Csf1R (62).
Notwithstanding their controversial classification, SAS lpMs clearly play a key role in MS/EAE by producing inflammatory molecules such as CCL5, CXCL9, CXCL10, and CXCL11, in turn, recruiting further leukocytes into the CSF (112, 260).
In EAE, lpMs increase their Iba1 immune reactivity several days before clinical onset of disease, thus indicating a role in the initiation of local inflammation (40). LpMs are highly dynamic dwellers and interact extensively with invading T cells, increasing their proliferation rate during the acute EAE phase (27, 112). This increase in number drops during the chronic phase of disease, paralleled by local appearance of apoptotic lpMs (27). Notably, similar to pvMs, lpM activation in EAE does not lead to dramatic transcriptome changes compared to homeostatic conditions (27).
In MS and EAE, monocytes/MdMs also accumulate in large numbers in the SAS following extravasation from the leptomeningeal vasculature or from the ChP and CSF-filled ventricles. Infiltration through leptomeningeal vessels follows increased intraluminal monocyte crawling and expression of the enzyme tissue transglutaminase 2 (261), known for its involvement in cell adhesion to fibronectin, a glycoprotein released by endothelial cells and pericytes (262).
MdM and lpM dynamics in the SAS during EAE have been extensively explored by intravital imaging (223). Preclinically, lpMs cluster around leptomeningeal vessels following leakage of plasma fibrinogen, which, in turn, triggers ROS production (222). Studies in rats have demonstrated that meningeal phagocytes can present both self and non-self antigens and thus activate infiltrating T cells in a multistep process requiring chemokine signaling (112, 263–265).
Long-lasting contacts seem to occur preferentially between lymphocytes and blood-borne CCR2+ monocyte-derived cells, rather than with resident BAMs (27).
Surprisingly, however, meningeal macrophages do to play an essential role in antigen presentation during EAE. A series of recent reports convincingly demonstrated that expression of MHCII in CD11c+ classic DCs but not in CX3CR1+ macrophages is indispensable for disease induction (28, 266, 267). Nonetheless, the frequency of lpM and MdM contacting lymphocytes suggests the existence of further regulatory roles shaping EAE. In our work, the majority of SAS MdM displayed strong expression of arginase-1+ in striking contrast to the iNOS+ dominated nature of parenchymal lesions, potentially indicating an anti-inflammatory function of lpMs (44). The differential representation of MdM phenotypes in the SAS and in other CNS compartments might also be related to distinct sensitivity toward chemoattractants, as shown in vitro using differentially polarized human cells (268, 269), either as a result of differential chemokine receptor expression or a differential receptor response to transduction.
From the SAS, activated MdMs can reach the CNS parenchyma and participate in the formation of subpial demyelinating lesions, an histological hallmark of progressive MS forms (191). The contribution of MdMs, however, depends on the type of lesion, with so-called leukocortical plaques showing a high number of activated macrophages and purely subpial cortical lesions mostly devoid of inflammatory infiltrates (257). In EAE, subpial white matter demyelination is commonly described in the spinal cord, but cortical gray matter pathology, as observed in MS, is rare. The latter type of lesion can, however, be modeled in mice through cortical injections of TNF and IFNγ (270, 271) or by peripheral injection of β-synuclein-specific T cells (272).
To reach the CNS parenchyma from the SAS, meningeal MdMs need to transverse the pia mater, the parenchyma-associated basement membrane and, eventually, the glia limitans (10). In vivo imaging has shown that cells within the SAS might move toward the parenchyma by crawling on the external surface of leptomeningeal vessels entering the parenchyma (273). However, the permeability of the pia mater to immune cell trafficking remains debated, and the required interaction molecules are unknown (104, 107, 260).
Migration of MdMs from the meninges to the parenchyma can be downregulated by the administration of CXCR7 antagonists, impeding CXCL12 signaling and resulting in meningeal accumulation (274). Retention of phagocytes in the SAS was paralleled by loss of VCAM-1 on astrocytes, thus highlighting a potential role of these cells as interactive partners in the invasion process (275).
To summarize, even though anatomical differences between the meningeal system in rodents and humans impede a fully coherent discussion, several reports have evidenced the central part played by leptomeninges in initiation and evolution of autoimmune CNS inflammation. Nonetheless, many unsolved questions exist regarding macrophage functions and trafficking routes. A detailed anatomical description of these compartments and the creation of transgenic animals allowing visualization of defined meningeal layers (116) remain crucially needed for the progress of the field.
Macrophages in the ChP During MS and EAE
In the context of auto-aggressive CNS inflammation, the ChP has been proposed as the first CNS gateway for autoreactive lymphocytes prior to BBB disruption, subsequently triggering a secondary leukocyte CNS infiltration driving disease progression (40, 260, 276).
Rather than a sealed barrier, the BCSFB is considered an active yet highly regulated exchange surface (108) showing a differential expression of tight junctions compared to the BBB (123).
Immune cell trafficking at the BCSFB seems to be regulated by IFNγ-dependent activation in immune surveillance and repair (277). Both CCL20 and CX3CL1 are constitutively expressed at the ChP and might guide recruitment of CCR6+ (278) and of CX3CR1+ leukocytes, respectively (279). The BCSFB is highly sensitive to systemic inflammation. Thus, peripheral LPS administration leads to local TNF and IL1β secretion, upregulates CXCL1 and CCL2 (280, 281), and triggers release of destabilizing MMP8 and MMP9 (282) and impairment of tight junction barrier properties, an overall reaction suggesting higher trafficking of immune cells.
In the ChP of MS patients, the tight junction protein claudin-3 is downregulated compared to healthy controls (283). Reports of its role in EAE models are, however, controversial, with its deletion increasing numbers of CSF-infiltrated MdMs in one study (283) and to a lack of BCSFB impairment in a recent report (284).
Interestingly, interaction molecules such as ICAM-1 and VCAM-1 are specifically expressed on the apical side of the BCSFB epithelium, facing the CSF (285). During EAE, their increased expression and a de novo apical expression of MAdCAM-1 can be observed (207). Notably, while leukocytes crossing the BCSFB toward the CSF utilize ICAM-1 in the last steps of diapedesis (286), the apical location of these molecules seemingly indicates that leukocytes can also migrate backward from the CSF to the ChP stroma (286, 287).
As shown by 2-photon microscopy, ChP macrophages readily respond to peripheral LPS injections by moving toward nearby vessels, with focal ChP damage leading to spatial reorganization of epiplexus cells around the injury site. In both scenarios not all macrophages responded to the danger stimuli, again highlighting the heterogeneity of ChP dwellers (131).
Compared to steady state, induction of EAE leads to the appearance of disease-associated ChPMs (27). These activated cells show significantly increased expression of antigen presentation molecules, chemokines and cytokines such as Il1b, with one cluster strongly positive for MHCII genes and for Ctss (encoding for Cathepsin S), and the other showing high expression of the antimicrobial products S100a9, S100a8, and Ngp (27). The presence of CCR2+ MdMs in the ChP appears substantially enriched throughout the disease, with a minor proportion of these cells locally expressing iNOS and/or arginase-1 (37).
ChP MdM populations also show high CD74 positivity and can be divided in three different cellular clusters composed of Ly6Chigh monocyte-like, Cd209+ DC-like and MertK+ macrophage-like cells (27).
Beside observations in EAE, not much is known about ChP macrophages in MS. Analysis of human ChP tissue revealed a high density of CD68+MHCII+ macrophages and a minor proportion of MHCIInegative Iba1+ cells, with these cells present within the stroma, intercalated between epithelial cells or lying on the apical side of epithelial cells (63, 64). However, the densities of these cells appeared comparable between progressive MS patients and healthy controls (64).
Do MdMs really access the CSF via the ChP during autoaggressive neuroinflammation? In non-autoimmune disease models, monocytes/macrophages were indeed shown to cross the BCSFB toward the CSF (47, 288, 289). Using in vitro BCSFB models, we could recently confirm that functionally polarized mouse macrophages can actively migrate through the BCSFB monolayer (37). Apparently migrating MHCII+CD68+ macrophages have also been described interspersed between epithelial cells in the ChP of MS patients (63), yet these cells might represent DC surveillants bridging across the BCSFB (290).
To summarize, while in MS the gateway function of the ChP remains unsupported by direct evidence, an active role of the BCSFB in MdM recruitment to the CNS is highly plausable and this CNS interface should become a focus of attention in neuroinflammatory research.
Monocyte/Macrophages at CNS Interfaces in Traumatic CNS Injury
Despite shielding by bones, meninges, and CSF, traumatic damage to the CNS parenchyma is a common pathological occurrence leading to neurodegeneration and to an innate immune response promoting further tissue damage (291, 292). Physical insults can occur to the brain (traumatic brain injury, TBI) or to the spinal cord (spinal cord injury, SCI), with these two compartments showing the evolution of distinct pathologies (293). Depending on their severity, mechanical injuries to the CNS result in local death, DAMP release, activation of BAMs and to different degrees of MdM infiltration (146). Interestingly, compared to brain lesions, physical damage to the spinal cord generally results into a higher activation of CNS macrophages, stronger BBB damage, and denser MdM accumulation (293).
Spinal Cord Injury
Following SCI, perilesional microglia proliferate creating a protective “microglial scar” in concert with astrocytes (294). Communication between microglia and infiltrating MdMs influences their reciprocal polarization as well as lesion evolution (215). While removal of microglia in SCI models proved detrimental, the role of BAMs in SCI was not convincingly addressed. One report showed that pvMs and lpMs do not participate in the disease process, nor do they proliferate extensively in response to SCI (294). The seemingly minor role of BAMs in SCI evolution is supported by observations in demyelinating models: following intra-parenchymal injections of lysophosphatidylcholine, Lyve1+ lpMs and pvMs do not penetrate into demyelinated spinal cord lesions (48). Nonetheless, more research is crucially needed to clarify BAM participation in SCI.
Upon injury, spinal cord endothelial cells upregulate the expression of VCAM-1 and ICAM-1 allowing first neutrophils and later monocytes to accumulate in the damaged region (295). Interestingly, these myeloid cells originate mostly from the spleen reservoir pool rather than from the bone marrow, a pattern likely related to the acuteness of the disease (135, 296). Similar to what is observed in EAE models, sensing of CCL2 and CXCL12 and production of MMP9 are required for monocyte migration from the BBB to the parenchyma upon SCI (297, 298). CCL2 and other chemokines released by glial cells such as astrocytes might also contribute to the acquisition of a functional phenotype by invading and local macrophages (299). Local TNF-α production increases MMP-9 expression highlighting a complex interplay of cytokines and proteases. Once in the parenchyma, MdMs follow a gradient of C5a molecules toward the lesion epicenter in a mechanism regulated by IRF8-purinergic receptor axis, all leading to enhanced tissue repair (300). Accordingly, MdMs in SCI have been described as beneficial (146). Conditional ablation of CD11c+ phagocytes during the first week after SCI resulted in worsened clinical recovery, however, did not affect the pathology when induced 2 weeks following damage (301). The beneficial effect of MdMs was attributed to their expression of anti-inflammatory IL-10 at the lesion margin (301). Notably, a follow-up work showed that while Ly6ChiCX3CR1loCCR2hi monocytes infiltrated the parenchyma in a CCL2-dependent manner via leptomeningeal vessels proximal to the lesion, anti-inflammatory Ly6CloCX3CR1highCCR2lo cells entered the CNS trafficking through the BCSFB (47). In this study, monocyte migration at the ChP relied on VLA4/VCAM-1 interactions as well as on the expression of the adenosine-catalyzing enzyme CD73. Intriguingly, it has been postulated that these polarization differences could be related to the cellular constituents of the two barriers, epithelial cells for the BCSCF and endothelial cells for leptomeningeal vessels (47).
Nonetheless, the net contribution of MdMs to SCI appears time- and location-dependent (302–305). Indeed, one report described locally recruited MdMs as pro-inflammatory players surrounded by anti-inflammatory microglia distal to the injured area (306). Acquisition of an anti-inflammatory phenotype is also affected by the activation status of nearby astroglial cells (299). In another work, inhibition of monocyte infiltration (via splenectomy) accordingly resulted into an ameliorated clinical phenotype (307). Lastly, other studies have observed that MdMs in the lesioned parenchyma show co-expression of pro- and anti-inflammatory markers, once more highlighting the non-binary role of MdMs in CNS injury and indicating complex local functions upon recruitment (308, 309).
Traumatic Brain Injury
Similar to SCI, TBI shows a long-term pathological evolution involving excitotoxicity, cytokine release, ROS/RNS production, and infiltrating myeloid cells with neurotoxic as well as neuroprotective functions (310). Beside the extensively investigated role of microglia (50), an involvement of BAMs upon TBI has been suggested by both human and animal studies.
In TBI patients, CD163+ microglia/macrophages are increased in both the lesion and perivascular spaces indicating a potential participation of CD163+ pvMs to damage evolution (74). CD163+ cells are also increased in a rat TBI model two days post TBI, however, mainly within the lesion (49). Importantly, most CD163+ cells co-expressed heme oxygenase-1 (49), a key enzyme in heme catabolism (311) exerting anti-inflammatory effects (312, 313) likely promoting neuroprotection following TBI. Nonetheless, in these studies, the peripheral or resident nature of pvMs was not convincingly defined.
Blood-borne MdMs play instead a recognized and context-dependent role in TBI (314). Pro-inflammatory macrophages are recruited early in the lesioned area, with CCR2+ MdMs following gradients of chemokines released from activated parenchymal cells (50). Notably, CCR2+ monocytes seem to mediate local ROS/RNS production (315, 316) and might thus constitute important pharmacological targets. Indeed, intravenous injection of immunomodulatory nanoparticles reduced MdM recruitment by affecting monocyte survival and sequestration within the spleen (190), altogether leading to a strong reduction of lesion volume (317).
Conversely, other studies have provided evidence for a beneficial effect of monocyte recruitment after mild TBI, for instance by reducing meningeal vascular damage (318), a pathological hallmark associated to peripheral immune response (319). In the latter study, while “classical” debris-scavenging monocytes were located at the center of meningeal lesions, wound-healing “non-classical” monocytes were localized peri-lesionally and promoted meningeal angiogenesis via expression of MMP-2 (318).
How do MdMs access the CNS parenchyma following TBI? Analysis of patient tissues suggests that CD14+ monocytes initially migrate toward the perilesional perivascular space within the first days following damage and then move toward the parenchyma (75). These MdMs can remain in the perilesional area for weeks (75). In a rat model of TBI, monocytes were instead shown to enter the lesioned CNS parenchyma by crossing SAS microvessels, subsequently accumulating near the injury site (320). Some MdMs appeared to move a short distance along perivascular spaces toward the brain parenchyma (320). This trafficking route utilized by MdMs (and neutrophils) requires cellular interactions that can also be mediated by JAM-A, a junctional adhesion molecule also expressed by macrophages (321, 322).
A potential role of the ChP as a monocyte access gateway in TBI models was suggested by the rapid increase in CCL2 production by the ChP ipsilateral to the lesion. The resulting rise in CCL2 CSF concentration mirrors observations in the CSF of severe TBI patients (323). BCSFB epithelial cultures indicated that CCL2 is secreted across both the apical and basolateral side, a bidirectional production necessary for leukocyte migration at the BCSFB following TBI (288). Accordingly, the blocking of this signaling reduced CCR2+ monocyte infiltration and lesion volume (324) and improved neurological recovery after TBI (315, 323, 325). Interestingly, lack of MdM infiltration correlated with increased astrocyte proliferation and reduced astroglial scar formation, thus suggesting a key role of juxtavascular astrocytes in the interaction with MdMs (326).
Taken together, evidence for a potential involvement of BAMs in traumatic CNS disorders remains sparse. Furthermore, experimental approaches allowing selective investigation of yolk sac-derived macrophages have not yet been utilized in this context. The parallel role of CNS-infiltrating monocytes has been more extensively described and appear extremely dependent on recruitment timing, damage extent and lesion location. A better definition of the role of MdMs would potentially aid the development of novel therapeutics for patients suffering from Sor TBI. and/or TBI.
Myeloid Dwellers and Trespassers at CNS Interfaces in Neurodegenerative Diseases
Parkinson’s Disease
Parkinson’s disease (PD) is a progressive neurodegenerative disorder characterized by neuronal loss in the substantia nigra pars compacta (SNpc) and by chronic CNS inflammation in both patients and animal models (327, 328). Histopathologically, brain samples of PD patients show accumulation of Lewy bodies rich in the neuronal protein α-synuclein (329). Post-mortem and imaging analysis of PD patients revealed detrimental microglial activation (330, 331) accompanied by upregulation of iNOS as well as production of pro-inflammatory cytokines within the parenchyma (332–334). Strong to moderate microglial activation is also observable in the different animal models of PD (327, 335, 336).
However, the role of BAMs in disease progression has rarely been addressed. In a viral model of synucleinopathy, degeneration of neurons in the SNpc coincided with local CD206+ pvM expansion, similar to what has been observed in post-mortem samples of PD patients (52). Depletion of pvMs and lpMs by clodronate liposomes resulted in a significant loss of dopaminergic neurons within the SNpc after two weeks, suggesting that BAMs exert a neuroprotective function in the pathology (52). Notably, clodronate administration increased vascular expression of VCAM-1, enhancing CNS accumulation of lymphocytes (52). Absence of pvMs led to aggravated spreading of misfolded α-synuclein (52), a pathological hallmark of PD (337). On the other hand, in a 6-hydroxydopamine-mediated PD model, no increase in rod-like CD163+ pvMs was found in the striatum as opposed to the recruitment from the blood stream of CD163+ “polygonal” cells (53). Notably, these parenchymal CD163+ cells are also described in the brain of PD patients in association with Aβ deposition and a damaged BBB (77). BBB impairments are commonly observed in both patients and animal models (338).
Several studies have documented an augmented presence of inflammatory cytokines in the CSF of PD patients (338), the increased expression of MHCII in CSF monocytes (76), the presence of α-synuclein at the BCSFB (282, 339) and a potential beneficial role exerted by ChP epithelial cells in transplantation experiments (340). Beyond this sparse evidence of a role of the ChP and in general of BAMs however, the potential contribution of these cells to PD evolution remains unexplored.
Conversely, several studies addressed monocytes/MdMs functions in PD pathogenesis. The total number of blood monocytes is not affected in PD patients (78), but these cells appear less responsive to activation and more proliferative (341) and show altered phagocytosis (78, 342). Furthermore, monocytes from PD patients display an upregulated FAS/FAS ligand system (78), potentially enhancing myeloid cell recruitment and release of cytokines (343, 344). PD patients show increased CCL2 blood levels and dysregulated CCR2+ monocytes responses (78, 79). Transcriptionally, monocytes from PD patients show a specific expression profile correlating with disease severity and indicating enriched expression of genes related to migration and regulation of inflammation (345).
Blood monocytes with pro-inflammatory features are also increased in PD animal models (346) and seem to infiltrate the degenerating substantia nigra by crossing the BBB (347, 348). Accordingly, CCR2+ MdMs can be found in the brain of PD mouse models early in the disease process, with astrocytes being the main producer of CCL2 (51). Of note, while blocking monocyte recruitment had no effect on dopaminergic neuron survival, overexpression of CCL2 in astrocytes did increase neuronal death and led to augmented infiltration of CCR2+ monocytes, together suggesting that MdMs contribute to neurodegeneration (51). Notably, in the SNpc of a different transgenic PD model, MdMs vastly outnumbered microglia and could be engineered as a “Trojan horse” approach to locally deliver neuroprotective factors (349).
While these studies imply trafficking of monocytes across the BBB during PD, the potential contribution of meningeal and ChP gateways to cell recruitment has never been addressed in the literature. In summary, several evidences point toward an involvement of recruited monocytes to PD pathogenesis, setting the ground for future studies finally testing the importance and the therapeutical value of these cells in PD.
Alzheimer’s Disease
Alzheimer’s disease (AD) is a neurodegenerative disorder characterized by brain atrophy, synaptic loss, extracellular deposition of amyloid-β (Aβ) peptides and intracellular accumulation of neurofibrillary tangles of phosphorylated Tau protein (349–351). The controversial contribution of microglia to AD pathogenesis is discussed by a growing body of literature, with molecules such as CX3CR1, APOE and triggering receptor expressed by myeloid cells 2 (TREM2) showing a critical impact on disease evolution by regulating phagocytosis and anti-inflammatory signaling in macrophages/microglia (352–355).
In parallel, some studies have highlighted the multifactorial contribution of BAMs to AD. Aβ tends to accumulate with age in insoluble depositions limiting drainage along perivascular pathways and typically resulting into cerebral amyloid angiopathy (104, 356). PvMs importantly participate in clearing perivascular Aβ, as shown in different mouse models of AD in which ablation of pvMs resulted in augmented Aβ accumulation (88, 357). PvMs Aβ clearance depended on expression of the scavenger receptor class B type 1 (55) and of CCR2 (56). These pvMs also express CD36 (57), one of the main receptors for Aβ (36). Notably, some studies have demonstrated a CD36-dependent, Nox2-mediated production of ROS in response to Aβ phagomacrocytosis in pvMs, a phenomenon ultimately augmenting vascular pathology and cognitive dysfunctions (358, 359). Taken together, pvMs seem to play both beneficial and detrimental roles in AD models, however, their function in AD patients remains undefined.
The complex equilibrium between fluid drainage and Aβ deposition observed at the perivascular level in AD is also detected within the ChP. Aβ transporter proteins are expressed at both the BBB and BCSFB contributing to a coordinated clearance of Aβ into the peripheral circulation (123, 360, 361). Underscoring disease-specific defects in barrier transport mechanisms, ChP tissue from AD patients shows substantial deposition of Aβ peptide and increased oxidative stress (362). Within the ChP of AD patients, dense fibrillary phosphorylated Tau can also be shown in calcified intracellular inclusion in the proximity of TREM2+ stromal ChPMs (58, 80). These events lead to significant changes in BCSFB permeability (130, 363) and to a reduced ChP expression of trafficking and inflammatory molecules including ICAM-1, VCAM-1, CXCL10, CCL2, and IFNγ, suggesting impaired monocyte migration via the BCSFB and a exacerbatory impact on disease evolution (364).
Concerning meningeal macrophages, one recent report has suggested a contribution of these BAMs to Aβ pathology, yet only within the dura mater and following experimental ablation of lymphatic vessels leading to local accumulation of Aβ and of Iba1+ macrophages (365).
While the role of BAMs in AD remains underinvestigated, the impact of MdM on disease development has been addressed in several studies. In vitro, monocyte trafficking at the BBB drastically increased in the presence of Aβ and was mediated by receptor for advanced glycation end products (RAGE) and PECAM-1 expressed on endothelial cells (366). Furthermore, both mouse and human microglia stimulated with Aβ upregulate their expression of CCL2, CCL3, CCL4, and CXCL2, suggesting a substantial Aβ-dependent recruitment of immune cells to diseased brains (367–369). Accordingly, different CD45highCD11bhighCCR2+ macrophages accumulate in the brain of animal models of AD, with CCR2 deficiency leading to the detrimental accumulation of Aβ (59, 370). These earlier results indicated that MdMs might exert a beneficial effect in the disease process (370, 371). Accordingly, parenchymal CD163+ microglia-like MdMs were described in the brain of AD patients in association with Aβ deposition nearby the damaged BBB (77). Unfortunately, in most of these reports, full body irradiation paradigms were utilized (372), artifactually leading to long-term changes in glia activation and increase in myeloid cell recruitment (56, 373). By shielding the mouse brain during irradiation, it was subsequently shown that CCR2+ MdMs only rarely infiltrate the CNS, thus redimensioning their role in AD (56). This work also contradicted earlier results (367) by showing that microglia accumulation close to Aβ does not strictly depend on the CCL2 system (56).
Nevertheless, monocytes might contribute to Aβ removal without leaving the vascular tree. A recent study has illustrated the role of CX3CR1+Ly6Clow patrolling monocytes in crawling in Aβ+ brain veins and engulfing intraluminal Aβ (54). However, contradictory observations have been made in AD patients, showing that circulating monocytes express reduced TLR levels (374), are defective in Aβ phagocytosis (375, 376) and are more prone to apoptosis compared to monocytes isolated from control patients (376).
In conclusion, in both mouse models and AD patients monocyte/MdM functions remain somewhat elusive. Studies on BAMs have, however, highlighted their complex role in Aβ removal and thus potentially suggested these cells as future targets of therapeutical interventions.
Conclusion and Outlook
Aided by stimulating debates on functional CNS anatomy (13, 97, 106, 182), by technical advancements in reporter tools and imaging (14, 44, 131) and by the recent “single-cell analysis revolution” (16, 27, 28), the study of CNS borders finally bloomed as a research field.
Long-lived dwellers of these CNS interfaces, BAMs mediate systemic communication (4), regulate vascular permeability (91, 92), waste clearance (94), fluid drainage (88), and surveil CSF composition (34). Often ignored and improperly ontogenically and anatomically defined, BAMs can now be precisely identified in contrast to blood-borne myeloid cells (14–16, 20, 22, 23), a necessary advancement allowing to address specific cell functions upon CNS damage. In this context, each CNS interface becomes a complex battleground hosting a myriad of peripherally derived trespassers. Even though the multidimensional interplay between invading monocytes, BAMs and the cellular components of each CNS gateway serves as a key checkpoint in disease evolution, a convincing picture of macrophage dynamics at CNS borders is far from existing.
What is the contribution of BAMs to pathogenesis of CNS disease?
During autoimmune neuroinflammation, BAMs increase in density, contact invading leukocytes and produce inflammatory cytokines and chemokines (42, 112, 225, 260). Surprisingly, however, these cells modify their overall transcriptional profile only mildly compared to steady state (27). Upon TBI/SCI, BAM actions seem beneficial but at the same time negligible (48, 49, 74, 294). The same applies to PD (52, 53, 76), while AD studies conversely indicate a disease-shaping role of BAMs (88, 357, 358, 365). Notably, the discussed results and recent data from brain tumor models (24) seem to indicate a relatively minor contribution of BAMs to the evolution of most CNS dysfunctions.
Conversely, during neuroinflammation, MdMs outnumber BAMs at CNS interfaces and drive disease evolution (15). Molecules such as GM-CSF (149, 229), CCL2 (240, 243–246), and different MMPs (169) primarily orchestrate the overall immigration of these cells into the CNS parenchyma, but the actual route of invasion through different CNS gateways remain remarkably speculative. Monocyte recruitment upon CNS trauma shares similar mechanisms with MS/EAE including the role of CCL2 and the need for MMP production by infiltrating cells (297, 298, 377). Lastly, monocyte trafficking to the CNS during neurodegenerative disorders remain surprisingly understudied, and its dependency on the CCL2-CCR2 axis controversial (51, 56).
To summarize, despite various efforts to understand the functional contribution of myeloid cells to CNS diseases, this review underlines how the study of BAMs and of monocytic invasion pathways is still at its infancy. Recent technical advancements should finally allow understanding whether BAMs, as long-lived dwellers of CNS interfaces, can become relevant therapeutic targets to manipulate CNS dysfunctions. Secondly, a renewed focus on CNS anatomy and barrier functions will hopefully prompt scientists to investigate and describe invading trajectories of monocytes/MdMs in the different disease models. A detailed definition of the infiltration routes and of the polarizing influence of distinct CNS gateways on trespassing monocytes will eventually allow designing targeted strategies to regulate monocyte entry, thus modulating the evolution of CNS pathologies.
Author Contributions
JS designed the figures. DI prepared the tables. DI, SW, KB, and GL wrote the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work is supported by a Swiss Multiple Sclerosis Society grant, Italian MS grant (2019/R-Single/001) and by Scherbarth Foundation funding awarded to GL.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgements
We would like to thank Prof. Britta Engelhardt (University of Bern) and Prof. Nicholas King (Sydney Medical School) for providing helpful comments and critical inputs.
Abbreviations
CNS, central nervous system; SAS, subarachnoid space; BCSFB, blood-cerebrospinal fluid barrier; BBB, blood-brain barrier; ChP, choroid plexus; lpM, resident leptomeningeal macrophage; pvM, resident perivascular macrophage; ChPM, resident stromal choroid plexus macrophage; MdM, monocyte-derived macrophage; BAM, barrier associated macrophage; MS, multiple sclerosis; EAE, experimental autoimmune encephalomyelitis; ROS/RNS, reactive oxygen and nitrogen species; SCI, spinal cord injury; TBI, traumatic brain injury; AD, Alzheimer’s disease; Aβ, amyloid-β; PD, Parkinson’s disease; DAMPs, damage-associated molecular patterns.
References
1. Galea I, Bechmann I, Perry VH. What is immune privilege (not)? Trends Immunol (2007) 28(1):12–8. doi: 10.1016/j.it.2006.11.004
2. Carson MJ, Doose JM, Melchior B, Schmid CD, Ploix CC. CNS immune privilege: hiding in plain sight. Immunol Rev (2006) 213:48–65. doi: 10.1111/j.1600-065X.2006.00441.x
3. Lampron A, Elali A, Rivest S. Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron (2013) 78(2):214–32. doi: 10.1016/j.neuron.2013.04.005
4. Corraliza I. Recruiting specialized macrophages across the borders to restore brain functions. Front Cell Neurosci (2014) 8:262. doi: 10.3389/fncel.2014.00262
5. Kierdorf K, Masuda T, Jordão MJC, Prinz M. Macrophages at CNS interfaces: ontogeny and function in health and disease. Nat Rev Neurosci (2019) 20(9):547–62. doi: 10.1038/s41583-019-0201-x
6. Mastorakos P, McGavern D. The anatomy and immunology of vasculature in the central nervous system. Sci Immunol (2019) 4(37). doi: 10.1126/sciimmunol.aav0492
7. Lopez-Atalaya JP, Askew KE, Sierra A, Gomez-Nicola D. Development and maintenance of the brain’s immune toolkit: Microglia and non-parenchymal brain macrophages. Dev Neurobiol (2018) 78(6):561–79. doi: 10.1002/dneu.22545
8. Kierdorf K, Prinz M, Geissmann F, Gomez Perdiguero E. Development and function of tissue resident macrophages in mice. Semin Immunol (2015) 27(6):369–78. doi: 10.1016/j.smim.2016.03.017
9. Sisó S, Jeffrey M, González L. Sensory circumventricular organs in health and disease. Acta Neuropathol (2010) 120(6):689–705. doi: 10.1007/s00401-010-0743-5
10. Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci (2015) 16(5):249–63. doi: 10.1038/nrn3898
11. Owens T, Bechmann I, Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol (2008) 67(12):1113–21. doi: 10.1097/NEN.0b013e31818f9ca8
12. Engelhardt B, Carare RO, Bechmann I, Flügel A, Laman JD, Vascular RO. glial, and lymphatic immune gateways of the central nervous system. Acta Neuropathol (2016) 132(3):317–38. doi: 10.1007/s00401-016-1606-5
13. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol (2017) 18(2):123–31. doi: 10.1038/ni.3666
14. Goldmann T, Wieghofer P, Jordão MJC, Prutek F, Hagemeyer N, Frenzel K, et al. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat Immunol (2016) 17(7):797–805. doi: 10.1038/ni.3423
15. Mrdjen D, Pavlovic A, Hartmann FJ, Schreiner B, Utz SG, Leung BP, et al. High-Dimensional Single-Cell Mapping of Central Nervous System Immune Cells Reveals Distinct Myeloid Subsets in Health, Aging, and Disease. Immunity (2018) 48(2):380–395.e6. doi: 10.1016/j.immuni.2018.01.011
16. Van Hove H, Martens L, Scheyltjens I, De Vlaminck K, Pombo Antunes AR, De Prijck S, et al. A single-cell atlas of mouse brain macrophages reveals unique transcriptional identities shaped by ontogeny and tissue environment. Nat Neurosci (2019) 22(6):1021–35. doi: 10.1038/s41593-019-0393-4
17. Aguzzi A, Barres BA, Bennett ML. Microglia: scapegoat, saboteur, or something else? Science (2013) 339(6116):156–61. doi: 10.1126/science.1227901
18. Hickey WF, Vass K, Lassmann H. Bone marrow-derived elements in the central nervous system: an immunohistochemical and ultrastructural survey of rat chimeras. J Neuropathol Exp Neurol (1992) 51(3):246–56. doi: 10.1097/00005072-199205000-00002
19. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FMV. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci (2007) 10(12):1538–43. doi: 10.1038/nn2014
20. Utz SG, See P, Mildenberger W, Thion MS, Greter M. Early Fate Defines Microglia and Non-parenchymal Brain Macrophage Development. Cell (2020) 181(3):557–73. doi: 10.1016/j.cell.2020.03.021
21. Brioschi S, Zhou Y, Colonna M. Brain Parenchymal and Extraparenchymal Macrophages in Development, Homeostasis, and Disease. J Immunol (2020) 204(2):294. doi: 10.4049/jimmunol.1900821
22. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity (2013) 38(1):79–91. doi: 10.1016/j.immuni.2012.12.001
23. Buttgereit A, Lelios I, Yu X, Vrohlings M, Krakoski NR, Gautier EL, et al. Sall1 is a transcriptional regulator defining microglia identity and function. Nat Immunol (2016) 17(12):1397–406. doi: 10.1038/ni.3585
24. Ochocka N, Segit P, Walentynowicz K, Wojnicki K, Cyranowski S, Swatler J, et al. Single-cell RNA sequencing reveals functional heterogeneity and sex differences of glioma-associated brain macrophages. BioRxiv (2019).
25. Greter M, Lelios I, Croxford AL. Microglia Versus Myeloid Cell Nomenclature during Brain Inflammation. Front Immunol (2015) 6:249(249). doi: 10.3389/fimmu.2015.00249
26. Korin B, Ben-Shaanan TL, Schiller M, Dubovik T, Azulay-Debby H, Boshnak NT, et al. High-dimensional, single-cell characterization of the brain’s immune compartment. Nat Neurosci (2017) 20(9):1300–9. doi: 10.1038/nn.4610
27. Jordão MJC, Sankowski R, Brendecke SM, Sagar, Locatelli G, Tai Y-H, et al. Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science (2019) 363(6425):eaat7554. doi: 10.1126/science.aat7554
28. Mundt S, Mrdjen D, Utz SG, Greter M, Schreiner B, Becher B. Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci Immunol (2019) 4(31). doi: 10.1126/sciimmunol.aau8380
29. Spitzer MH, Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell (2016) 165(4):780–91. doi: 10.1016/j.cell.2016.04.019
30. Caputa G, Castoldi A, Pearce EJ. Metabolic adaptations of tissue-resident immune cells. Nat Immunol (2019) 20(7):793–801. doi: 10.1038/s41590-019-0407-0
31. Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity (2003) 19(1):71–82. doi: 10.1016/s1074-7613(03)00174-2
32. Mitchell AJ, Roediger B, Weninger W. Monocyte homeostasis and the plasticity of inflammatory monocytes. Cell Immunol (2014) 291(1-2):22–31. doi: 10.1016/j.cellimm.2014.05.010
33. Galea I, Palin K, Newman TA, Van Rooijen N, Perry VH, Boche D. Mannose receptor expression specifically reveals perivascular macrophages in normal, injured, and diseased mouse brain. Glia (2005) 49(3):375–84. doi: 10.1002/glia.20124
34. Herz J, Filiano AJ, Smith A, Yogev N, Kipnis J. Myeloid Cells in the Central Nervous System. Immunity (2017) 46(6):943–56. doi: 10.1016/j.immuni.2017.06.007
35. Fabriek BO, Van Haastert ES, Galea I, Polfliet MMJ, Döpp ED, Van Den Heuvel MM, et al. CD163-positive perivascular macrophages in the human CNS express molecules for antigen recognition and presentation. Glia (2005) 51(4):297–305. doi: 10.1002/glia.20208
36. Silverstein RL, Febbraio M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci Signal (2009) 2(72):re3. doi: 10.1126/scisignal.272re3
37. Ivan DC, Walthert S, Locatelli G. Monocyte recruitment to the inflamed central nervous system: migration pathways and distinct functional polarization. bioRxiv (2020), 2020.04.04.025395. doi: 10.1101/2020.04.04.025395
38. McMenamin PG, Wealthall RJ, Deverall M, Cooper SJ, Griffin B. Macrophages and dendritic cells in the rat meninges and choroid plexus: three-dimensional localisation by environmental scanning electron microscopy and confocal microscopy. Cell Tissue Res (2003) 313(3):259–69. doi: 10.1007/s00441-003-0779-0
39. Strazielle N, Ghersi-Egea JF. Choroid plexus in the central nervous system: biology and physiopathology. J Neuropathol Exp Neurol (2000) 59(7):561–74. doi: 10.1093/jnen/59.7.561
40. Brown DA, Sawchenko PE. Time course and distribution of inflammatory and neurodegenerative events suggest structural bases for the pathogenesis of experimental autoimmune encephalomyelitis. J Comp Neurol (2007) 502(2):236–60. doi: 10.1002/cne.21307
41. Su H, Na N, Zhang X, Zhao Y. The biological function and significance of CD74 in immune diseases. Inflamm Res (2017) 66(3):209–16. doi: 10.1007/s00011-016-0995-1
42. Hofmann N, Lachnit N, Streppel M, Witter B, Neiss WF, Guntinas-Lichius O, et al. Increased expression of ICAM-1, VCAM-1, MCP-1, and MIP-1 alpha by spinal perivascular macrophages during experimental allergic encephalomyelitis in rats. BMC Immunol (2002) 3(11). doi: 10.1186/1471-2172-3-11
43. Giladi A, Wagner LK, Li H, Dörr D, Medaglia C, Paul F, et al. Cxcl10+ monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nat Immunol (2020) 21(5):525–34. doi: 10.1038/s41590-020-0661-1
44. Locatelli G, Theodorou D, Kendirli A, Jordão MJC, Staszewski O, Phulphagar K, et al. Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nat Neurosci (2018) 21(9):1196–208. doi: 10.1038/s41593-018-0212-3
45. Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, et al. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med (2006) 203(4):1007–19. doi: 10.1084/jem.20051342
46. Song J, Wu C, Korpos E, Zhang X, Agrawal SM, Wang Y, et al. Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep (2015) 10(7):1040–54. doi: 10.1016/j.celrep.2015.01.037
47. Shechter R, Miller O, Yovel G, Rosenzweig N, London A, Ruckh J, et al. Recruitment of beneficial M2 macrophages to injured spinal cord is orchestrated by remote brain choroid plexus. Immunity (2013) 38(3):555–69. doi: 10.1016/j.immuni.2013.02.012
48. Plemel JR, Stratton JA, Michaels NJ, Rawji KS, Zhang E, Sinha S, et al. Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Sci Adv (2020) 6(3):eaay6324. doi: 10.1126/sciadv.aay6324
49. Zhang Z, Zhang ZY, Wu Y, Schluesener HJ. Lesional accumulation of CD163+ macrophages/microglia in rat traumatic brain injury. Brain Res (2012) 1461:102–10. doi: 10.1016/j.brainres.2012.04.038
50. Loane DJ, Kumar A. Microglia in the TBI brain: The good, the bad, and the dysregulated. Exp Neurol (2016) 275 Pt 3(0 3):316–27. doi: 10.1016/j.expneurol.2015.08.018
51. Parillaud VR, Lornet G, Monnet Y, Privat A-L, Haddad AT, Brochard V, et al. Analysis of monocyte infiltration in MPTP mice reveals that microglial CX3CR1 protects against neurotoxic over-induction of monocyte-attracting CCL2 by astrocytes. J Neuroinflamm (2017) 14(1):60. doi: 10.1186/s12974-017-0830-9
52. Fuentealba J. Understanding the role of perivascular macrophages in Parkinson’s disease pathophysiology. Doctorate Thesis in Neuroscience, Institut du Cerveau, Paris. Academic Press, 1st Edition. pp. 292. (2017).
53. Tentillier N, Etzerodt A, Olesen MN, Rizalar FS, Jacobsen J, Bender D, et al. Anti-Inflammatory Modulation of Microglia via CD163-Targeted Glucocorticoids Protects Dopaminergic Neurons in the 6-OHDA Parkinson’s Disease Model. J Neurosci (2016) 36(36):9375–90. doi: 10.1523/jneurosci.1636-16.2016
54. Michaud JP, Bellavance MA, Préfontaine P, Rivest S. Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell Rep (2013) 5(3):646–53. doi: 10.1016/j.celrep.2013.10.010
55. Thanopoulou K, Fragkouli A, Stylianopoulou F, Georgopoulos S. Scavenger receptor class B type I (SR-BI) regulates perivascular macrophages and modifies amyloid pathology in an Alzheimer mouse model. Proc Natl Acad Sci U S A (2010) 107(48):20816–21. doi: 10.1073/pnas.1005888107
56. Mildner A, Schlevogt B, Kierdorf K, Böttcher C, Erny D, Kummer MP, et al. Distinct and non-redundant roles of microglia and myeloid subsets in mouse models of Alzheimer’s disease. J Neurosci (2011) 31(31):11159–71. doi: 10.1523/jneurosci.6209-10.2011
57. Uekawa K, Park L, Hattori Y, Zhou P, Murphy M, Anrather J, et al. Abstract 149: CD36 in Perivascular Macrophages Contributes to Neurovascular and Cognitive Dysfunction and Amyloid Angiopathy in Mice Overexpressing the Alzheimer Aβ Peptide. Stroke (2018) 49(Suppl_1):A149–9. doi: 10.1161/str.49.suppl_1.149
58. Raha AA, Henderson JW, Stott SR, Vuono R, Foscarin S, Friedland RP, et al. Neuroprotective Effect of TREM-2 in Aging and Alzheimer’s Disease Model. J Alzheimers Dis (2017) 55(1):199–217. doi: 10.3233/jad-160663
59. Simard AR, Soulet D, Gowing G, Julien J-P, Rivest S. Bone Marrow-Derived Microglia Play a Critical Role in Restricting Senile Plaque Formation in Alzheimer’s Disease. Neuron (2006) 49(4):489–502. doi: 10.1016/j.neuron.2006.01.022
60. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood (2010) 116(16):e74–80. doi: 10.1182/blood-2010-02-258558
61. Wong KL, Tai JJ, Wong WC, Han H, Sem X, Yeap WH, et al. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood (2011) 118(5):e16–31. doi: 10.1182/blood-2010-12-326355
62. Schafflick D, Xu CA, Hartlehnert M, Cole M, Schulte-Mecklenbeck A, Lautwein T, et al. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun (2020) 11(1):247. doi: 10.1038/s41467-019-14118-w
63. Vercellino M, Votta B, Condello C, Piacentino C, Romagnolo A, Merola A, et al. Involvement of the choroid plexus in multiple sclerosis autoimmune inflammation: a neuropathological study. J Neuroimmunol (2008) 199(1-2):133–41. doi: 10.1016/j.jneuroim.2008.04.035
64. Rodríguez-Lorenzo S, Konings J, van der Pol S, Kamermans A, Amor S, van Horssen J, et al. Inflammation of the choroid plexus in progressive multiple sclerosis: accumulation of granulocytes and T cells. Acta Neuropathol Commun (2020) 8(1):9. doi: 10.1186/s40478-020-0885-1
65. Gjelstrup MC, Stilund M, Petersen T, Møller HJ, Petersen EL, Christensen T. Subsets of activated monocytes and markers of inflammation in incipient and progressed multiple sclerosis. Immunol Cell Biol (2018) 96(2):160–74. doi: 10.1111/imcb.1025
66. Haschka D, Tymoszuk P, Bsteh G, Petzer V, Berek K, Theurl I, et al. Expansion of Neutrophils and Classical and Nonclassical Monocytes as a Hallmark in Relapsing-Remitting Multiple Sclerosis. Front Immunol (2020) 11:594(594). doi: 10.3389/fimmu.2020.00594
67. Howell OW, Schulz-Trieglaff EK, Carassiti D, Gentleman SM, Nicholas R, Roncaroli F, et al. Extensive grey matter pathology in the cerebellum in multiple sclerosis is linked to inflammation in the subarachnoid space. Neuropathol Appl Neurobiol (2015) 41(6):798–813. doi: 10.1111/nan.12199
68. Waschbisch A, Schröder S, Schraudner D, Sammet L, Weksler B, Melms A, et al. Pivotal Role for CD16+ Monocytes in Immune Surveillance of the Central Nervous System. J Immunol (2016) 196(4):1558–67. doi: 10.4049/jimmunol.1501960
69. Wiendl H, Feger U, Mittelbronn M, Jack C, Schreiner B, Stadelmann C, et al. Expression of the immune-tolerogenic major histocompatibility molecule HLA-G in multiple sclerosis: implications for CNS immunity. Brain (2005) 128(Pt 11):2689–704. doi: 10.1093/brain/awh609
70. Esaulova E, Cantoni C, Shchukina I, Zaitsev K, Bucelli RC, Wu GF, et al. Single-cell RNA-seq analysis of human CSF microglia and myeloid cells in neuroinflammation. Neurol - Neuroimmunol Neuroinflamm (2020) 7(4):e732. doi: 10.1212/NXI.0000000000000732
71. Singh S, Metz I, Amor S, van der Valk P, Stadelmann C, Brück W. Microglial nodules in early multiple sclerosis white matter are associated with degenerating axons. Acta Neuropathol (2013) 125(4):595–608. doi: 10.1007/s00401-013-1082-0
72. Vogel DY, Vereyken EJ, Glim JE, Heijnen PD, Moeton M, van der Valk P, et al. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J Neuroinflamm (2013) 10(35). doi: 10.1186/1742-2094-10-35
73. Licht-Mayer S, Wimmer I, Traffehn S, Metz I, Brück W, Bauer J, et al. Cell type-specific Nrf2 expression in multiple sclerosis lesions. Acta Neuropathol (2015) 130(2):263–77. doi: 10.1007/s00401-015-1452-x
74. Holfelder K, Schittenhelm J, Trautmann K, Haybaeck J, Meyermann R, Beschorner R. De novo expression of the hemoglobin scavenger receptor CD163 by activated microglia is not associated with hemorrhages in human brain lesions. Histol Histopathol (2011) 26(8):1007–17. doi: 10.14670/hh-26.1007
75. Beschorner R, Nguyen TD, Gözalan F, Pedal I, Mattern R, Schluesener HJ, et al. CD14 expression by activated parenchymal microglia/macrophages and infiltrating monocytes following human traumatic brain injury. Acta Neuropathol (2002) 103(6):541–9. doi: 10.1007/s00401-001-0503-7
76. Fiszer U, Mix E, Fredrikson S, Kostulas V, Link H. Parkinson’s disease and immunological abnormalities: increase of HLA-DR expression on monocytes in cerebrospinal fluid and of CD45RO+ T cells in peripheral blood. Acta Neurol Scand (1994) 90(3):160–6. doi: 10.1111/j.1600-0404.1994.tb02699.x
77. Pey P, Pearce RK, Kalaitzakis ME, Griffin WS, Gentleman SM. Phenotypic profile of alternative activation marker CD163 is different in Alzheimer’s and Parkinson’s disease. Acta Neuropathol Commun (2014) 2(21). doi: 10.1186/2051-5960-2-21
78. Grozdanov V, Bliederhaeuser C, Ruf WP, Roth V, Fundel-Clemens K, Zondler L, et al. Inflammatory dysregulation of blood monocytes in Parkinson’s disease patients. Acta Neuropathol (2014) 128(5):651–63. doi: 10.1007/s00401-014-1345-4
79. Funk N, Wieghofer P, Grimm S, Schaefer R, Bühring HJ, Gasser T, et al. Characterization of peripheral hematopoietic stem cells and monocytes in Parkinson’s disease. Mov Disord (2013) 28(3):392–5. doi: 10.1002/mds.25300
80. Raha-Chowdhury R, Henderson JW, Raha AA, Vuono R, Bickerton A, Jones E, et al. Choroid Plexus Acts as Gatekeeper for TREM2, Abnormal Accumulation of ApoE, and Fibrillary Tau in Alzheimer’s Disease and in Down Syndrome Dementia. J Alzheimers Dis (2019) 69(1):91–109. doi: 10.3233/jad-181179
81. Blanchette M, Daneman R. Formation and maintenance of the BBB. Mech Dev (2015) 138:8–16. doi: 10.1016/j.mod.2015.07.007
82. Ballabh P, Braun A, Nedergaard M. The blood-brain barrier: an overview: structure, regulation, and clinical implications. Neurobiol Dis (2004) 16(1):1–13. doi: 10.1016/j.nbd.2003.12.016
83. Nabeshima S, Reese TS, Landis DM, Brightman MW. Junctions in the meninges and marginal glia. J Comp Neurol (1975) 164(2):127–69. doi: 10.1002/cne.901640202
84. Mato M, Ookawara S, Kurihara K. Uptake of exogenous substances and marked infoldings of the fluorescent granular pericyte in cerebral fine vessels. Am J Anat (1980) 157(3):329–32. doi: 10.1002/aja.1001570308
85. Ransohoff RM, Engelhardt B. The anatomical and cellular basis of immune surveillance in the central nervous system. Nat Rev Immunol (2012) 12(9):623–35. doi: 10.1038/nri3265
86. Salzman KL, Osborn AG, House P, Jinkins JR, Ditchfield A, Cooper JA, et al. Giant tumefactive perivascular spaces. AJNR Am J Neuroradiol (2005) 26(2):298–305.
87. Pollock H, Hutchings M, Weller RO, Zhang ET. Perivascular spaces in the basal ganglia of the human brain: their relationship to lacunes. J Anat (1997) 191( Pt 3):337–46. doi: 10.1046/j.1469-7580.1997.19130337.x
88. Yang T, Guo R, Zhang F. Brain perivascular macrophages: Recent advances and implications in health and diseases. CNS Neurosci Ther (2019) 25(12):1318–28. doi: 10.1111/cns.13263
89. Hickey WF. Basic principles of immunological surveillance of the normal central nervous system. Glia (2001) 36(2):118–24. doi: 10.1002/glia.1101
90. Hickey WF, Kimura H. Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science (1988) 239(4837):290–2. doi: 10.1126/science.3276004
91. Willis CL, Garwood CJ, Ray DE. A size selective vascular barrier in the rat area postrema formed by perivascular macrophages and the extracellular matrix. Neuroscience (2007) 150(2):498–509. doi: 10.1016/j.neuroscience.2007.09.023
92. Mendes-Jorge L, Ramos D, Luppo M, Llombart C, Alexandre-Pires G, Nacher V, et al. Scavenger Function of Resident Autofluorescent Perivascular Macrophages and Their Contribution to the Maintenance of the Blood–Retinal Barrier. Invest Ophthalmol Visual Sci (2009) 50(12):5997–6005. doi: 10.1167/iovs.09-3515
93. Wardlaw JM, Benveniste H, Nedergaard M, Zlokovic BV, Mestre H, Lee H, et al. Perivascular spaces in the brain: anatomy, physiology and pathology. Nat Rev Neurol (2020) 16(3):137–53. doi: 10.1038/s41582-020-0312-z
94. Kida S, Steart PV, Zhang ET, Weller RO. Perivascular cells act as scavengers in the cerebral perivascular spaces and remain distinct from pericytes, microglia and macrophages. Acta Neuropathol (1993) 85(6):646–52. doi: 10.1007/bf00334675
95. Faraco G, Park L, Anrather J, Iadecola C. Brain perivascular macrophages: characterization and functional roles in health and disease. J Mol Med (Berlin Germany) (2017) 95(11):1143–52. doi: 10.1007/s00109-017-1573-x
96. Mato M, Ookawara S, Sakamoto A, Aikawa E, Ogawa T, Mitsuhashi U, et al. Involvement of specific macrophage-lineage cells surrounding arterioles in barrier and scavenger function in brain cortex. Proc Natl Acad Sci U S A (1996) 93(8):3269–74. doi: 10.1073/pnas.93.8.3269
97. Coles JA, Myburgh E, Brewer JM, McMenamin PG. Where are we? The anatomy of the murine cortical meninges revisited for intravital imaging, immunology, and clearance of waste from the brain. Prog Neurobiol (2017) 156:107–48. doi: 10.1016/j.pneurobio.2017.05.002
98. Durcanova B, Appleton J, Gurijala N, Belov V, Giffenig P, Moeller E, et al. The Configuration of the Perivascular System Transporting Macromolecules in the CNS. Front Neurosci (2019) 13:511(511). doi: 10.3389/fnins.2019.00511
99. Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, et al. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol (2008) 34(2):131–44. doi: 10.1111/j.1365-2990.2007.00926.x
100. Nayak D, Zinselmeyer BH, Corps KN, McGavern DB. In vivo dynamics of innate immune sentinels in the CNS. Intravital (2012) 1(2):95–106. doi: 10.4161/intv.22823
101. Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal (2013) 18(17):2352–63. doi: 10.1089/ars.2012.4834
102. Lee SJ, Evers S, Roeder D, Parlow AF, Risteli J, Risteli L, et al. Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science (2002) 295(5561):1898–901. doi: 10.1126/science.1069540
103. Bogie JF, Stinissen P, Hendriks JJ. Macrophage subsets and microglia in multiple sclerosis. Acta Neuropathol (2014) 128(2):191–213. doi: 10.1007/s00401-014-1310-2
104. Weller RO, Sharp MM, Christodoulides M, Carare RO, Møllgård K. The meninges as barriers and facilitators for the movement of fluid, cells and pathogens related to the rodent and human CNS. Acta Neuropathol (2018) 135(3):363–85. doi: 10.1007/s00401-018-1809-z
105. Brightman MW, Reese TS. Junctions between intimately apposed cell membranes in the vertebrate brain. J Cell Biol (1969) 40(3):648–77. doi: 10.1083/jcb.40.3.648
106. Abbott NJ, Pizzo ME, Preston JE, Janigro D, Thorne RG. The role of brain barriers in fluid movement in the CNS: is there a ‘glymphatic’ system? Acta Neuropathol (2018) 135(3):387–407. doi: 10.1007/s00401-018-1812-4
107. Adeeb N, Mortazavi MM, Deep A, Griessenauer CJ, Watanabe K, Shoja MM, et al. The pia mater: a comprehensive review of literature. Childs Nerv Syst (2013) 29(10):1803–10. doi: 10.1007/s00381-013-2044-5
108. Ghersi-Egea J-F, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B. Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol (2018) 135(3):337–61. doi: 10.1007/s00401-018-1807-1
109. Polfliet MM, Goede PH, van Kesteren-Hendrikx EM, van Rooijen N, Dijkstra CD, van den Berg TK. A method for the selective depletion of perivascular and meningeal macrophages in the central nervous system. J Neuroimmunol (2001) 116(2):188–95. doi: 10.1016/s0165-5728(01)00282-x
110. Schwartz M, Baruch K. The resolution of neuroinflammation in neurodegeneration: leukocyte recruitment via the choroid plexus. EMBO J (2014) 33(1):7–22. doi: 10.1002/embj.201386609
111. Oehmichen M. Characterization of mononuclear phagocytes in human CSF using membrane markers. Acta Cytol (1976) 20(6):548–52.
112. Schläger C, Körner H, Krueger M, Vidoli S, Haberl M, Mielke D, et al. Effector T-cell trafficking between the leptomeninges and the cerebrospinal fluid. Nature (2016) 530(7590):349–53. doi: 10.1038/nature16939
113. Meiron M, Zohar Y, Anunu R, Wildbaum G, Karin N. CXCL12 (SDF-1alpha) suppresses ongoing experimental autoimmune encephalomyelitis by selecting antigen-specific regulatory T cells. J Exp Med (2008) 205(11):2643–55. doi: 10.1084/jem.20080730
114. Prevo R, Banerji S, Ferguson DJ, Clasper S, Jackson DG. Mouse LYVE-1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J Biol Chem (2001) 276(22):19420–30. doi: 10.1074/jbc.M011004200
115. Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science (2019) 363(6432):eaau0964. doi: 10.1126/science.aau0964
116. Castro Dias M, Mapunda JA, Vladymyrov M, Engelhardt B. Structure and Junctional Complexes of Endothelial, Epithelial and Glial Brain Barriers. Int J Mol Sci (2019) 20(21):5372 . doi: 10.3390/ijms20215372
117. Mecheri B, Paris F, Lübbert H. Histological investigations on the dura mater vascular system of mice. Acta Histochem (2018) 120(8):846–57. doi: 10.1016/j.acthis.2018.09.009
118. Herisson F, Frodermann V, Courties G, Rohde D, Sun Y, Vandoorne K, et al. Direct vascular channels connect skull bone marrow and the brain surface enabling myeloid cell migration. Nat Neurosci (2018) 21(9):1209–17. doi: 10.1038/s41593-018-0213-2
119. Brezovakova V, Jadhav S. Identification of Lyve-1 positive macrophages as resident cells in meninges of rats. J Comp Neurol (2020) 528(12):2021–32. doi: 10.1002/cne.24870
120. Hsu M, Rayasam A, Kijak JA, Choi YH, Harding JS, Marcus SA, et al. Neuroinflammation-induced lymphangiogenesis near the cribriform plate contributes to drainage of CNS-derived antigens and immune cells. Nat Commun (2019) 10(1):229. doi: 10.1038/s41467-018-08163-0
121. Ogata A, Nishihira J, Suzuki T, Nagashima K, Tashiro K. Identification of macrophage migration inhibitory factor mRNA expression in neural cells of the rat brain by in situ hybridization. Neurosci Lett (1998) 246(3):173–7. doi: 10.1016/S0304-3940(98)00203-1
122. Lun MP, Monuki ES, Lehtinen MK. Development and functions of the choroid plexus-cerebrospinal fluid system. Nat Rev Neurosci (2015) 16(8):445–57. doi: 10.1038/nrn3921
123. Kaur C, Rathnasamy G, Ling EA. The Choroid Plexus in Healthy and Diseased Brain. J Neuropathol Exp Neurol (2016) 75(3):198–213. doi: 10.1093/jnen/nlv030
125. Spector R, Johanson CE. The mammalian choroid plexus. Sci Am (1989) 261(5):68–74. doi: 10.1038/scientificamerican1189-68
126. Sathyanesan M, Girgenti MJ, Banasr M, Stone K, Bruce C, Guilchicek E, et al. A molecular characterization of the choroid plexus and stress-induced gene regulation. Transl Psychiatry (2012) 2(7):e139. doi: 10.1038/tp.2012.64
127. Marques F, Sousa JC, Brito MA, Pahnke J, Santos C, Correia-Neves M, et al. The choroid plexus in health and in disease: dialogues into and out of the brain. Neurobiol Dis (2017) 107:32–40. doi: 10.1016/j.nbd.2016.08.011
128. Santos CRA, Duarte AC, Costa AR, Tomás J, Quintela T, Gonçalves I. The senses of the choroid plexus. Prog Neurobiol (2019) 182:101680. doi: 10.1016/j.pneurobio.2019.101680
129. Balusu S, Van Wonterghem E, De Rycke R, Raemdonck K, Stremersch S, Gevaert K, et al. Identification of a novel mechanism of blood-brain communication during peripheral inflammation via choroid plexus-derived extracellular vesicles. EMBO Mol Med (2016) 8(10):1162–83. doi: 10.15252/emmm.201606271
130. Fame RM, Lehtinen MK. Emergence and Developmental Roles of the Cerebrospinal Fluid System. Dev Cell (2020) 52(3):261–75. doi: 10.1016/j.devcel.2020.01.027
131. Shipley F, Dani N, Xu H, Deister C, Cui J, Head J, et al. Tracking Calcium Dynamics and Immune Surveillance at the Choroid Plexus Blood-Cerebrospinal Fluid Interface. SSRN Electronic J (2020) 108(4):623–39. doi: 10.2139/ssrn.3554055
132. Gordon S, Taylor PR. Monocyte and macrophage heterogeneity. Nat Rev Immunol (2005) 5(12):953–64. doi: 10.1038/nri1733
133. Terry RL, Miller SD. Molecular control of monocyte development. Cell Immunol (2014) 291(1-2):16–21. doi: 10.1016/j.cellimm.2014.02.008
134. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol (2017) 17(6):349–62. doi: 10.1038/nri.2017.28
135. Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science (2009) 325(5940):612–6. doi: 10.1126/science.1175202
136. Patel AA, Zhang Y, Fullerton JN, Boelen L, Rongvaux A, Maini AA, et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J Exp Med (2017) 214(7):1913–23. doi: 10.1084/jem.20170355
137. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science (2007) 317(5838):666–70. doi: 10.1126/science.1142883
138. Teh YC, Ding JL, Ng LG, Chong SZ. Capturing the Fantastic Voyage of Monocytes Through Time and Space. Front Immunol (2019) 10:834. doi: 10.3389/fimmu.2019.00834
139. Serbina NV, Pamer EG. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat Immunol (2006) 7(3):311–7. doi: 10.1038/ni1309
140. Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, et al. Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell (2013) 153(2):362–75. doi: 10.1016/j.cell.2013.03.010
141. Rua R, McGavern DB. Elucidation of monocyte/macrophage dynamics and function by intravital imaging. J Leukoc Biol (2015) 98(3):319–32. doi: 10.1189/jlb.4RI0115-006RR
142. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The Heterogeneity of Ly6C(hi) Monocytes Controls Their Differentiation into iNOS(+) Macrophages or Monocyte-Derived Dendritic Cells. Immunity (2016) 45(6):1205–18. doi: 10.1016/j.immuni.2016.12.001
143. Liu Z, Gu Y, Chakarov S, Bleriot C, Kwok I, Chen X, et al. Fate Mapping via Ms4a3-Expression History Traces Monocyte-Derived Cells. Cell (2019) 178(6):1509–25. doi: 10.1016/j.cell.2019.08.009
144. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res (2015) 107(3):321–30. doi: 10.1093/cvr/cvv147
145. Ong SM, Teng K, Newell E, Chen H, Chen J, Loy T, et al. A Novel, Five-Marker Alternative to CD16-CD14 Gating to Identify the Three Human Monocyte Subsets. Front Immunol (2019) 10:1761(1761). doi: 10.3389/fimmu.2019.01761
146. Gadani SP, Walsh JT, Lukens JR, Kipnis J. Dealing with Danger in the CNS: The Response of the Immune System to Injury. Neuron (2015) 87(1):47–62. doi: 10.1016/j.neuron.2015.05.019
147. Ebert RH, Florey HW. The Extravascular Development of the Monocyte Observed In vivo. Br J Exp Pathol (1939) 20(4):342–56.
148. Davison AM, King NJC. Accelerated Dendritic Cell Differentiation from Migrating Ly6C<sup<lo</sup< Bone Marrow Monocytes in Early Dermal West Nile Virus Infection. J Immunol (2011) 186(4):2382. doi: 10.4049/jimmunol.1002682
149. King IL, Dickendesher TL, Segal BM. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood (2009) 113(14):3190–7. doi: 10.1182/blood-2008-07-168575
150. Ifergan I, Kébir H, Bernard M, Wosik K, Dodelet-Devillers A, Cayrol R, et al. The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain (2008) 131(Pt 3):785–99. doi: 10.1093/brain/awm295
151. Tso C, Rye KA, Barter P. Phenotypic and functional changes in blood monocytes following adherence to endothelium. PLoS One (2012) 7(5):e37091. doi: 10.1371/journal.pone.0037091
152. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity (2014) 41(5):694–707. doi: 10.1016/j.immuni.2014.10.008
153. Floris S, van den Born J, Van der Pol SMA, Dijkstra CD, de Vries HE. Heparan Sulfate Proteoglycans Modulate Monocyte Migration across Cerebral Endothelium. J Neuropathol Exp Neurol (2003) 62(7):780–90. doi: 10.1093/jnen/62.7.780
154. Bashant KR, Toepfner N, Day CJ, Mehta NN, Kaplan MJ, Summers C, et al. The mechanics of myeloid cells. Biol Cell (2020) 112(4):103–12. doi: 10.1111/boc.201900084
155. Muller WA. How endothelial cells regulate transmigration of leukocytes in the inflammatory response. Am J Pathol (2014) 184(4):886–96. doi: 10.1016/j.ajpath.2013.12.033
156. Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol (2010) 11(5):366–78. doi: 10.1038/nrm2889
157. Njock MS, Cheng HS, Dang LT, Nazari-Jahantigh M, Lau AC, Boudreau E, et al. Endothelial cells suppress monocyte activation through secretion of extracellular vesicles containing antiinflammatory microRNAs. Blood (2015) 125(20):3202–12. doi: 10.1182/blood-2014-11-611046
158. Thomas-Ecker S, Lindecke A, Hatzmann W, Kaltschmidt C, Zänker KS, Dittmar T. Alteration in the gene expression pattern of primary monocytes after adhesion to endothelial cells. Proc Natl Acad Sci U S A (2007) 104(13):5539–44. doi: 10.1073/pnas.0700732104
159. Williams MR, Sakurai Y, Zughaier SM, Eskin SG, McIntire LV. Transmigration across activated endothelium induces transcriptional changes, inhibits apoptosis, and decreases antimicrobial protein expression in human monocytes. J Leukoc Biol (2009) 86(6):1331–43. doi: 10.1189/jlb.0209062
160. Seim GL, Britt EC, John SV, Yeo FJ, Johnson AR, Eisenstein RS, et al. Two-stage metabolic remodelling in macrophages in response to lipopolysaccharide and interferon-γ stimulation. Nat Metab (2019) 1(7):731–42. doi: 10.1038/s42255-019-0083-2
161. Bertani FR, Mozetic P, Fioramonti M, Iuliani M, Ribelli G, Pantano F, et al. Classification of M1/M2-polarized human macrophages by label-free hyperspectral reflectance confocal microscopy and multivariate analysis. Sci Rep (2017) 7(1):8965. doi: 10.1038/s41598-017-08121-8
162. Orecchioni M, Ghosheh Y, Pramod AB, Ley K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol (2019) 10:1084(1084). doi: 10.3389/fimmu.2019.01084
163. Murray PJ, Wynn TA. Obstacles and opportunities for understanding macrophage polarization. J Leukoc Biol (2011) 89(4):557–63. doi: 10.1189/jlb.0710409
164. Meli VS, Veerasubramanian PK, Atcha H, Reitz Z, Downing TL, Liu WF. Biophysical regulation of macrophages in health and disease. J Leukoc Biol (2019) 106(2):283–99. doi: 10.1002/jlb.Mr0318-126r
165. Devanney NA, Stewart AN, Gensel JC. Microglia and macrophage metabolism in CNS injury and disease: The role of immunometabolism in neurodegeneration and neurotrauma. Exp Neurol (2020) 329:113310. doi: 10.1016/j.expneurol.2020.113310
166. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm.0022-1767 (Print)).
167. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep (2014) 6(13). doi: 10.12703/p6-13
168. Ransohoff RM. A polarizing question: do M1 and M2 microglia exist? Nat Neurosci (2016) 19(8):987–91. doi: 10.1038/nn.4338
169. Mishra MK, Yong VW. Myeloid cells - targets of medication in multiple sclerosis. Nat Rev Neurol (2016) 12(9):539–51. doi: 10.1038/nrneurol.2016.110
170. Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity (2014) 41(1):14–20. doi: 10.1016/j.immuni.2014.06.008
171. Thaler B, Baik N, Hohensinner PJ, Baumgartner J, Panzenböck A, Stojkovic S, et al. Differential expression of Plg-R(KT) and its effects on migration of proinflammatory monocyte and macrophage subsets. Blood (2019) 134(6):561–7. doi: 10.1182/blood.2018850420
172. Mottahedin A, Joakim Ek C, Truvé K, Hagberg H, Mallard C. Choroid plexus transcriptome and ultrastructure analysis reveals a TLR2-specific chemotaxis signature and cytoskeleton remodeling in leukocyte trafficking. Brain Behav Immun (2019) 79:216–27. doi: 10.1016/j.bbi.2019.02.004
173. Audoy-Rémus J, Richard JF, Soulet D, Zhou H, Kubes P, Vallières L. Rod-Shaped monocytes patrol the brain vasculature and give rise to perivascular macrophages under the influence of proinflammatory cytokines and angiopoietin-2. J Neurosci (2008) 28(41):10187–99. doi: 10.1523/jneurosci.3510-08.2008
174. Ousman SS, Kubes P. Immune surveillance in the central nervous system. Nat Neurosci (2012) 15(8):1096–101. doi: 10.1038/nn.3161
175. Harrison-Brown M, Liu G-J, Banati R. Checkpoints to the Brain: Directing Myeloid Cell Migration to the Central Nervous System. Int J Mol Sci (2016) 17(12):2030. doi: 10.3390/ijms17122030
176. Ransohoff RM, Kivisäkk P, Kidd G. Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol (2003) 3(7):569–81. doi: 10.1038/nri1130
177. Ling EA, Kaur C, Origin J. nature, and some functional considerations of intraventricular macrophages, with special reference to the epiplexus cells. Microsc Res Tech (1998) 41(1):43–56. doi: 10.1002/(sici)1097-0029(19980401)41:1<43::Aid-jemt5>3.0.Co;2-v
178. Seehusen DA, Reeves MM, Fomin DA. Cerebrospinal fluid analysis. Am Fam Physician (2003) 68(6):1103–8.
179. Lazarevic I, Engelhardt B. Modeling immune functions of the mouse blood-cerebrospinal fluid barrier in vitro: primary rather than immortalized mouse choroid plexus epithelial cells are suited to study immune cell migration across this brain barrier. Fluids Barriers CNS (2016) 13(2). doi: 10.1186/s12987-016-0027-0
180. Petersen N, Torz L, Jensen KHR, Hjortø GM, Spiess K, Rosenkilde MM. Three-Dimensional Explant Platform for Studies on Choroid Plexus Epithelium. Front Cell Neurosci (2020) 14:108(108). doi: 10.3389/fncel.2020.00108
181. Ma Q, Ineichen BV, Detmar M, Proulx ST. Outflow of cerebrospinal fluid is predominantly through lymphatic vessels and is reduced in aged mice. Nat Commun (2017) 8(1):1434. doi: 10.1038/s41467-017-01484-6
182. Alves de Lima K, Rustenhoven J, Kipnis J. Meningeal Immunity and Its Function in Maintenance of the Central Nervous System in Health and Disease. Annu Rev Immunol (2020) 38(1):597–620. doi: 10.1146/annurev-immunol-102319-103410
183. Locatelli G, Wörtge S, Buch T, Ingold B, Frommer F, Sobottka B, et al. Primary oligodendrocyte death does not elicit anti-CNS immunity. Nat Neurosci (2012) 15(4):543–50. doi: 10.1038/nn.3062
184. Papadopoulos Z, Herz J, Kipnis J. Meningeal Lymphatics: From Anatomy to Central Nervous System Immune Surveillance. J Immunol (2020) 204(2):286–93. doi: 10.4049/jimmunol.1900838
185. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol (2011) 11(11):762–74. doi: 10.1038/nri3070
186. Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H. Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol (2000) 47(6):707–17. doi: 10.1002/1531-8249(200006)47:6<707::aid-ana3>3.0.co;2-q
187. Brück W, Sommermeier N, Bergmann M, Zettl U, Goebel HH, Kretzschmar HA, et al. CHAPTER 14 - Macrophages in Multiple Sclerosis. Immunobiology (1996) 195(4):588–600. doi: 10.1016/S0171-2985(96)80024-6
188. Fischer HJ, Finck TLK, Pellkofer HL, Reichardt HM, Lühder F. Glucocorticoid Therapy of Multiple Sclerosis Patients Induces Anti-inflammatory Polarization and Increased Chemotaxis of Monocytes. Front Immunol (2019) 10:1200(1200). doi: 10.3389/fimmu.2019.01200
189. Fiedler SE, George JD, Love HN, Kim E, Spain R, Bourdette D, et al. Analysis of IL-6, IL-1β and TNF-α production in monocytes isolated from multiple sclerosis patients treated with disease modifying drugs. J Syst Integr Neurosci (2017) 3(3). doi: 10.15761/jsin.1000166
190. Getts DR, Terry RL, Getts MT, Deffrasnes C, Müller M, van Vreden C, et al. Therapeutic inflammatory monocyte modulation using immune-modifying microparticles. Sci Trans Med (2014) 6(219):219ra7–7. doi: 10.1126/scitranslmed.3007563
191. Lassmann H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front Immunol (2018) 9:3116(3116). doi: 10.3389/fimmu.2018.03116
192. Frischer JM, Weigand SD, Guo Y, Kale N, Parisi JE, Pirko I, et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann Neurol (2015) 78(5):710–21. doi: 10.1002/ana.24497
193. Calabrese M, Favaretto A, Martini V, Gallo P. Grey matter lesions in MS: from histology to clinical implications. Prion (2013) 7(1):20–7. doi: 10.4161/pri.22580
194. Lassmann H. What drives disease in multiple sclerosis: Inflammation or neurodegeneration? Clin Exp Neuroimmunol (2010) 1(1):2–11. doi: 10.1111/j.1759-1961.2009.00003.x
195. Amoruso A, Blonda M, Gironi M, Grasso R, Di Francescantonio V, Scaroni F, et al. Immune and central nervous system-related miRNAs expression profiling in monocytes of multiple sclerosis patients. Sci Rep (2020) 10(1):6125. doi: 10.1038/s41598-020-63282-3
196. Dalla Costa G, Finardi A, Garzetti L, Carandini T, Comi G, Martinelli V, et al. Disease-modifying treatments modulate myeloid cells in multiple sclerosis patients. Neurological Sci (2018) 39(2):373–6. doi: 10.1007/s10072-017-3176-2
197. Kouwenhoven M, Teleshova N, Ozenci V, Press R, Link H. Monocytes in multiple sclerosis: phenotype and cytokine profile. J Neuroimmunol (2001) 112(1-2):197–205. doi: 10.1016/s0165-5728(00)00396-9
198. Shaked I, Hanna RN, Shaked H, Chodaczek G, Nowyhed HN, Tweet G, et al. Transcription factor Nr4a1 couples sympathetic and inflammatory cues in CNS-recruited macrophages to limit neuroinflammation. Nat Immunol (2015) 16(12):1228–34. doi: 10.1038/ni.3321
199. George JD, Kim E, Spain R, Bourdette D, Salinthone S. Effects of lipoic acid on migration of human B cells and monocyte-enriched peripheral blood mononuclear cells in relapsing remitting multiple sclerosis. J Neuroimmunol (2018) 315:24–7. doi: 10.1016/j.jneuroim.2017.12.009
200. Böttcher C, van der Poel M, Fernández-Zapata C, Schlickeiser S, Leman JKH, Hsiao C-C, et al. Single-cell mass cytometry reveals complex myeloid cell composition in active lesions of progressive multiple sclerosis. Acta Neuropathol Commun (2020) 8(1):136. doi: 10.1186/s40478-020-01010-8
201. Boven LA, Van Meurs M, Van Zwam M, Wierenga-Wolf A, Hintzen RQ, Boot RG, et al. Myelin-laden macrophages are anti-inflammatory, consistent with foam cells in multiple sclerosis. Brain (2006) 129(2):517–26. doi: 10.1093/brain/awh707
202. Grajchen E, Hendriks JJA, Bogie JFJ. The physiology of foamy phagocytes in multiple sclerosis. Acta Neuropathol Commun (2018) 6(1):124. doi: 10.1186/s40478-018-0628-8
203. Jäckle K, Zeis T, Schaeren-Wiemers N, Junker A, van der Meer F, Kramann N, et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis.1460-2156 (Electronic)).
204. Oppenheimer DR. The cervical cord in multiple sclerosis. Neuropathol Appl Neurobiol (1978) 4(2):151–62. doi: 10.1111/j.1365-2990.1978.tb00555.x
205. Gross CC, Schulte-Mecklenbeck A, Hanning U, Posevitz-Fejfár A, Korsukewitz C, Schwab N, et al. Distinct pattern of lesion distribution in multiple sclerosis is associated with different circulating T-helper and helper-like innate lymphoid cell subsets. Mult Scler (2017) 23(7):1025–30. doi: 10.1177/1352458516662726
206. Lassmann H, Bradl M. Multiple sclerosis: experimental models and reality. Acta Neuropathol (2017) 133(2):223–44. doi: 10.1007/s00401-016-1631-4
207. Engelhardt B, Wolburg-Buchholz K, Wolburg H. Involvement of the choroid plexus in central nervous system inflammation. Microsc Res Tech (2001) 52(1):112–29. doi: 10.1002/1097-0029(20010101)52:1<112::Aid-jemt13>3.0.Co;2-5
208. Yamasaki R, Lu H, Butovsky O, Ohno N, Rietsch AM, Cialic R, et al. Differential roles of microglia and monocytes in the inflamed central nervous system. J Exp Med (2014) 211(8):1533–49. doi: 10.1084/jem.20132477
209. Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FMV. Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci (2011) 14(9):1142–9. doi: 10.1038/nn.2887
210. Cronk JC, Filiano AJ, Louveau A, Marin I, Marsh R, Ji E, et al. Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. J Exp Med (2018) 215(6):1627–47. doi: 10.1084/jem.20180247
211. Lund H, Pieber M, Parsa R, Han J, Grommisch D, Ewing E, et al. Competitive repopulation of an empty microglial niche yields functionally distinct subsets of microglia-like cells. Nat Commun (2018) 9(1):4845. doi: 10.1038/s41467-018-07295-7
212. Halder SK, Milner R. Chronic mild hypoxia accelerates recovery from preexisting EAE by enhancing vascular integrity and apoptosis of infiltrated monocytes. Proc Natl Acad Sci U S A (2020) 117(20):11126–35. doi: 10.1073/pnas.1920935117
213. van Horssen J, Witte ME, Schreibelt G, de Vries HE. Radical changes in multiple sclerosis pathogenesis. Biochim Biophys Acta (2011) 1812(2):141–50. doi: 10.1016/j.bbadis.2010.06.011
214. Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM, et al. A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med (2011) 17(4):495–9. doi: 10.1038/nm.2324
215. Greenhalgh AD, Zarruk JG, Healy LM, Baskar Jesudasan SJ, Jhelum P, Salmon CK, et al. Peripherally derived macrophages modulate microglial function to reduce inflammation after CNS injury. PLoS Biol (2018) 16(10):e2005264. doi: 10.1371/journal.pbio.2005264
216. Greenhalgh AD, David S, Bennett FC. Immune cell regulation of glia during CNS injury and disease. Nat Rev Neurosci (2020) 21(3):139–52. doi: 10.1038/s41583-020-0263-9
217. Weber MS, Prod’homme T, Youssef S, Dunn SE, Rundle CD, Lee L, et al. Type II monocytes modulate T cell-mediated central nervous system autoimmune disease. Nat Med (2007) 13(8):935–43. doi: 10.1038/nm1620
218. Miron VE, Boyd A, Zhao JW, Yuen TJ, Ruckh JM, Shadrach JL, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci (2013) 16(9):1211–8. doi: 10.1038/nn.3469
219. Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T. Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol (1998) 161(7):3767–75.
220. Perry VH, Teeling J. Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration. Semin Immunopathol (2013) 35(5):601–12. doi: 10.1007/s00281-013-0382-8
221. Elmquist JK, Breder CD, Sherin JE, Scammell TE, Hickey WF, Dewitt D, et al. Intravenous lipopolysaccharide induces cyclooxygenase 2-like immunoreactivity in rat brain perivascular microglia and meningeal macrophages. J Comp Neurol (1997) 381(2):119–29. doi: 10.1002/(sici)1096-9861(19970505)381:2<119::aid-cne1>3.0.co;2-6
222. Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, et al. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun (2012) 3(1227). doi: 10.1038/ncomms2230
223. Borjini N, Paouri E, Tognatta R, Akassoglou K, Davalos D. Imaging the dynamic interactions between immune cells and the neurovascular interface in the spinal cord. Exp Neurol (2019) 322(113046). doi: 10.1016/j.expneurol.2019.113046
224. Maggi P, Sati P, Massacesi L. Magnetic resonance imaging of experimental autoimmune encephalomyelitis in the common marmoset. J Neuroimmunol (2017) 304:86–92. doi: 10.1016/j.jneuroim.2016.09.016
225. Zhang Z, Zhang ZY, Schittenhelm J, Wu Y, Meyermann R, Schluesener HJ. Parenchymal accumulation of CD163+ macrophages/microglia in multiple sclerosis brains. J Neuroimmunol (2011) 237(1-2):73–9. doi: 10.1016/j.jneuroim.2011.06.006
226. Polfliet MMJ, van de Veerdonk F, Döpp EA, van Kesteren-Hendrikx EML, van Rooijen N, Dijkstra CD, et al. The role of perivascular and meningeal macrophages in experimental allergic encephalomyelitis. J Neuroimmunol (2002) 122(1):1–8. doi: 10.1016/S0165-5728(01)00445-3
227. Lee NJ, Ha SK, Sati P, Absinta M, Nair G, Luciano NJ, et al. Potential role of iron in repair of inflammatory demyelinating lesions. J Clin Invest (2019) 129(10):4365–76. doi: 10.1172/jci126809
228. Schuh C, Wimmer I, Hametner S, Haider L, Van Dam AM, Liblau RS, et al. Oxidative tissue injury in multiple sclerosis is only partly reflected in experimental disease models. Acta Neuropathol (2014) 128(2):247–66. doi: 10.1007/s00401-014-1263-5
229. Monaghan KL, Wan ECK. The Role of Granulocyte-Macrophage Colony-Stimulating Factor in Murine Models of Multiple Sclerosis. Cells (2020) 9(3):611. doi: 10.3390/cells9030611
230. Tanaka R, Iwasaki Y, Koprowski H. Ultrastructural studies of perivascular cuffing cells in multiple sclerosis brain. Am J Pathol (1975) 81(3):467–78.
231. Erickson MA, Banks WA. Neuroimmune Axes of the Blood-Brain Barriers and Blood-Brain Interfaces: Bases for Physiological Regulation, Disease States, and Pharmacological Interventions. Pharmacol Rev (2018) 70(2):278–314. doi: 10.1124/pr.117.014647
232. Vogel DY, Kooij G, Heijnen PD, Breur M, Peferoen LA, van der Valk P, et al. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur J Immunol (2015) 45(6):1808–19. doi: 10.1002/eji.201444960
233. Van der Goes A, Wouters D, Van Der Pol SM, Huizinga R, Ronken E, Adamson P, et al. Reactive oxygen species enhance the migration of monocytes across the blood-brain barrier in vitro. FASEB J (2001) 15(10):1852–4. doi: 10.1096/fj.00-0881fje
234. Larochelle C, Alvarez JI, Prat A. How do immune cells overcome the blood-brain barrier in multiple sclerosis? FEBS Lett (2011) 585(23):3770–80. doi: 10.1016/j.febslet.2011.04.066
235. van Strien ME, de Vries HE, Chrobok NL, Bol J, Breve JJP, van der Pol SMP, et al. Tissue Transglutaminase contributes to experimental multiple sclerosis pathogenesis and clinical outcome by promoting macrophage migration. Brain Behav Immun (2015) 50:141–54 doi: 10.1016/j.bbi.2015.06.023
236. Ifergan I, Kebir H, Terouz S, Alvarez JI, Lécuyer MA, Gendron S, et al. Role of Ninjurin-1 in the migration of myeloid cells to central nervous system inflammatory lesions. Ann Neurol (2011) 70(5):751–63. doi: 10.1002/ana.22519
237. Lyck R, Lécuyer MA, Abadier M, Wyss CB, Matti C, Rosito M, et al. ALCAM (CD166) is involved in extravasation of monocytes rather than T cells across the blood-brain barrier. J Cereb Blood Flow Metab (2017) 37(8):2894–909. doi: 10.1177/0271678x16678639
238. Reijerkerk A, Kooij G, van der Pol SM, Leyen T, van Het Hof B, Couraud PO, et al. Tissue-type plasminogen activator is a regulator of monocyte diapedesis through the brain endothelial barrier. J Immunol (2008) 181(5):3567–74. doi: 10.4049/jimmunol.181.5.3567
239. Reijerkerk A, Kooij G, van der Pol SM, Leyen T, Lakeman K, van Het Hof B, et al. The NR1 subunit of NMDA receptor regulates monocyte transmigration through the brain endothelial cell barrier. J Neurochem (2010) 113(2):447–53. doi: 10.1111/j.1471-4159.2010.06598.x
240. Paul D, Ge S, Lemire Y, Jellison ER, Serwanski DR, Ruddle NH, et al. Cell-selective knockout and 3D confocal image analysis reveals separate roles for astrocyte-and endothelial-derived CCL2 in neuroinflammation. J Neuroinflamm (2014) 11(10). doi: 10.1186/1742-2094-11-10
241. Krumbholz M, Theil D, Steinmeyer F, Cepok S, Hemmer B, Hofbauer M, et al. CCL19 is constitutively expressed in the CNS, up-regulated in neuroinflammation, active and also inactive multiple sclerosis lesions. J Neuroimmunol (2007) 190(1-2):72–9. doi: 10.1016/j.jneuroim.2007.07.024
242. Mahad DJ, Ransohoff RM. The role of MCP-1 (CCL2) and CCR2 in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Semin Immunol (2003) 15(1):23–32. doi: 10.1016/s1044-5323(02)00125-2
243. Greenwood J, Heasman SJ, Alvarez JI, Prat A, Lyck R, Engelhardt B. Review: leucocyte-endothelial cell crosstalk at the blood-brain barrier: a prerequisite for successful immune cell entry to the brain. Neuropathol Appl Neurobiol (2011) 37(1):24–39. doi: 10.1111/j.1365-2990.2010.01140.x
244. Subileau EA, Rezaie P, Davies HA, Colyer FM, Greenwood J, Male DK, et al. Expression of chemokines and their receptors by human brain endothelium: implications for multiple sclerosis. J Neuropathol Exp Neurol (2009) 68(3):227–40. doi: 10.1097/NEN.0b013e318197eca7
245. Gerszten RE, Garcia-Zepeda EA, Lim YC, Yoshida M, Ding HA, Gimbrone MA Jr., et al. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature (1999) 398(6729):718–23. doi: 10.1038/19546
246. Gschwandtner M, Derler R, Midwood KS. More Than Just Attractive: How CCL2 Influences Myeloid Cell Behavior Beyond Chemotaxis. Front Immunol (2019) 10:2759(2759). doi: 10.3389/fimmu.2019.02759
247. Weber C, Weber KS, Klier C, Gu S, Wank R, Horuk R, et al. Specialized roles of the chemokine receptors CCR1 and CCR5 in the recruitment of monocytes and T(H)1-like/CD45RO(+) T cells. Blood (2001) 97(4):1144–6. doi: 10.1182/blood.v97.4.1144
248. Horng S, Therattil A, Moyon S, Gordon A, Kim K, Argaw AT, et al. Astrocytic tight junctions control inflammatory CNS lesion pathogenesis. J Clin Invest (2017) 127(8):3136–51. doi: 10.1172/jci91301
249. Hollier P-L, Guimbal S, Mora P, Diop A, Cornuault L, Couffinhal T, et al. Genetic disruption of the Blood Brain Barrier leads to protective barrier formation at the Glia Limitans. bioRxiv (2020), 2020.03.13.990762. doi: 10.1101/2020.03.13.990762
250. Paré A, Mailhot B, Lévesque SA, Juzwik C, Ignatius Arokia Doss PM, Lécuyer MA, et al. IL-1β enables CNS access to CCR2(hi) monocytes and the generation of pathogenic cells through GM-CSF released by CNS endothelial cells. Proc Natl Acad Sci U S A (2018) 115(6):E1194–e1203. doi: 10.1073/pnas.1714948115
251. Lévesque SA, Paré A, Mailhot B, Bellver-Landete V, Kébir H, Lécuyer MA, et al. Myeloid cell transmigration across the CNS vasculature triggers IL-1β-driven neuroinflammation during autoimmune encephalomyelitis in mice. J Exp Med (2016) 213(6):929–49. doi: 10.1084/jem.20151437
252. Jost MM, Ninci E, Meder B, Kempf C, Van Royen N, Hua J, et al. Divergent effects of GM-CSF and TGFbeta1 on bone marrow-derived macrophage arginase-1 activity, MCP-1 expression, and matrix metalloproteinase-12: a potential role during arteriogenesis. FASEB J (2003) 17(15):2281–3. doi: 10.1096/fj.03-0071fje
253. Kaushik DK, Bhattacharya A, Mirzaei R, Rawji KS, Ahn Y, Rho JM, et al. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J Clin Invest (2019) 129(8):3277–92. doi: 10.1172/jci124012
254. Stephenson EL, Mishra MK, Moussienko D, Laflamme N, Rivest S, Ling CC, et al. Chondroitin sulfate proteoglycans as novel drivers of leucocyte infiltration in multiple sclerosis. Brain (2018) 141(4):1094–110. doi: 10.1093/brain/awy033
255. De Groot CJ, Montagne L, Barten AD, Sminia P, Van Der Valk P. Expression of transforming growth factor (TGF)-beta1, -beta2, and -beta3 isoforms and TGF-beta type I and type II receptors in multiple sclerosis lesions and human adult astrocyte cultures. J Neuropathol Exp Neurol (1999) 58(2):174–87. doi: 10.1097/00005072-199902000-00007
256. Derada Troletti C, de Goede P, Kamermans A, de Vries HE. Molecular alterations of the blood-brain barrier under inflammatory conditions: The role of endothelial to mesenchymal transition. Biochim Biophys Acta (2016) 1862(3):452–60. doi: 10.1016/j.bbadis.2015.10.010
257. Popescu BF, Lucchinetti CF. Meningeal and cortical grey matter pathology in multiple sclerosis. BMC Neurol (2012) 12(11). doi: 10.1186/1471-2377-12-11
258. Bevan RJ, Evans R, Griffiths L, Watkins LM, Rees MI, Magliozzi R, et al. Meningeal inflammation and cortical demyelination in acute multiple sclerosis. Ann Neurol (2018) 84(6):829–42. doi: 10.1002/ana.25365
259. Michel L, Touil H, Pikor NB, Gommerman JL, Prat A, Bar-Or A. B Cells in the Multiple Sclerosis Central Nervous System: Trafficking and Contribution to CNS-Compartmentalized Inflammation. Front Immunol (2015) 6:636(636). doi: 10.3389/fimmu.2015.00636
260. Russi AE, Brown MA. The meninges: new therapeutic targets for multiple sclerosis. Transl Res (2015) 165(2):255–69. doi: 10.1016/j.trsl.2014.08.005
261. Chrobok NL, Jaouen A, Fenrich KK, Bol JGJM, Wilhelmus MMM, Drukarch B, et al. Monocyte behaviour and tissue transglutaminase expression during experimental autoimmune encephalomyelitis in transgenic CX3CR1gfp/gfp mice. Amino Acids (2017) 49(3):643–58. doi: 10.1007/s00726-016-2359-0
262. Akimov SS, Belkin AM. Cell surface tissue transglutaminase is involved in adhesion and migration of monocytic cells on fibronectin. Blood (2001) 98(5):1567–76. doi: 10.1182/blood.v98.5.1567
263. Bartholomäus I, Kawakami N, Odoardi F, Schläger C, Miljkovic D, Ellwart JW, et al. Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature (2009) 462(7269):94–8. doi: 10.1038/nature08478
264. Mues M, Bartholomäus I, Thestrup T, Griesbeck O, Wekerle H, Kawakami N, et al. Real-time in vivo analysis of T cell activation in the central nervous system using a genetically encoded calcium indicator. Nat Med (2013) 19(6):778–83. doi: 10.1038/nm.3180
265. Kyratsous NI, Bauer IJ, Zhang G, Pesic M, Bartholomäus I, Mues M, et al. Visualizing context-dependent calcium signaling in encephalitogenic T cells in vivo by two-photon microscopy. Proc Natl Acad Sci U S A (2017) 114(31):E6381–e6389. doi: 10.1073/pnas.1701806114
266. Wolf Y, Shemer A, Levy-Efrati L, Gross M, Kim JS, Engel A, et al. Microglial MHC class II is dispensable for experimental autoimmune encephalomyelitis and cuprizone-induced demyelination. Eur J Immunol (2018) 48(8):1308–18. doi: 10.1002/eji.201847540
267. Giles DA, Duncker PC, Wilkinson NM, Washnock-Schmid JM, Segal BM. CNS-resident classical DCs play a critical role in CNS autoimmune disease. J Clin Invest (2018) 128(12):5322–34. doi: 10.1172/jci123708
268. Vogel DY, Heijnen PD, Breur M, de Vries HE, Tool AT, Amor S, et al. Macrophages migrate in an activation-dependent manner to chemokines involved in neuroinflammation. J Neuroinflamm (2014) 11(23). doi: 10.1186/1742-2094-11-23
269. Müller J, von Bernstorff W, Heidecke CD, Schulze T. Differential S1P Receptor Profiles on M1- and M2-Polarized Macrophages Affect Macrophage Cytokine Production and Migration. BioMed Res Int (2017) 2017(7584621). doi: 10.1155/2017/7584621
270. Merkler D, Ernsting T, Kerschensteiner M, Brück W, Stadelmann C. A new focal EAE model of cortical demyelination: multiple sclerosis-like lesions with rapid resolution of inflammation and extensive remyelination. Brain (2006) 129(Pt 8):1972–83. doi: 10.1093/brain/awl135
271. James RE, Schalks R, Browne E, Eleftheriadou I, Munoz CP, Mazarakis ND, et al. Persistent elevation of intrathecal pro-inflammatory cytokines leads to multiple sclerosis-like cortical demyelination and neurodegeneration. Acta Neuropathol Commun (2020) 8(1):66. doi: 10.1186/s40478-020-00938-1
272. Lodygin D, Hermann M, Schweingruber N, Flügel-Koch C, Watanabe T, Schlosser C, et al. β-Synuclein-reactive T cells induce autoimmune CNS grey matter degeneration. Nature (2019) 566(7745):503–8. doi: 10.1038/s41586-019-0964-2
273. Caravagna C, Jaouën A, Desplat-Jégo S, Fenrich KK, Bergot E, Luche H, et al. Diversity of innate immune cell subsets across spatial and temporal scales in an EAE mouse model. Sci Rep (2018) 8(1):5146. doi: 10.1038/s41598-018-22872-y
274. Cruz-Orengo L, Holman DW, Dorsey D, Zhou L, Zhang P, Wright M, et al. CXCR7 influences leukocyte entry into the CNS parenchyma by controlling abluminal CXCL12 abundance during autoimmunity. J Exp Med (2011) 208(2):327–39. doi: 10.1084/jem.20102010
275. Gimenez MA, Sim JE, Russell JH. TNFR1-dependent VCAM-1 expression by astrocytes exposes the CNS to destructive inflammation. J Neuroimmunol (2004) 151(1-2):116–25. doi: 10.1016/j.jneuroim.2004.02.012
276. Lopes Pinheiro MA, Kooij G, Mizee MR, Kamermans A, Enzmann G, Lyck R, et al. Immune cell trafficking across the barriers of the central nervous system in multiple sclerosis and stroke. Biochim Biophys Acta (2016) 1862(3):461–71. doi: 10.1016/j.bbadis.2015.10.018
277. Kunis G, Baruch K, Rosenzweig N, Kertser A, Miller O, Berkutzki T, et al. IFN-γ-dependent activation of the brain’s choroid plexus for CNS immune surveillance and repair. Brain (2013) 136(Pt 11):3427–40. doi: 10.1093/brain/awt259
278. Reboldi A, Coisne C, Baumjohann D, Benvenuto F, Bottinelli D, Lira S, et al. C-C chemokine receptor 6-regulated entry of TH-17 cells into the CNS through the choroid plexus is required for the initiation of EAE. Nat Immunol (2009) 10(5):514–23. doi: 10.1038/ni.1716
279. Mills JH, Alabanza LM, Mahamed DA, Bynoe MS. Extracellular adenosine signaling induces CX3CL1 expression in the brain to promote experimental autoimmune encephalomyelitis. J Neuroinflamm (2012) 9(1):193. doi: 10.1186/1742-2094-9-193
280. Marques F, Sousa JC, Correia-Neves M, Oliveira P, Sousa N, Palha JA. The choroid plexus response to peripheral inflammatory stimulus. Neuroscience (2007) 144(2):424–30. doi: 10.1016/j.neuroscience.2006.09.029
281. Marques F, Sousa JC, Coppola G, Falcao AM, Rodrigues AJ, Geschwind DH, et al. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. J Cereb Blood Flow Metab (2009) 29(5):921–32. doi: 10.1038/jcbfm.2009.15
282. Solár P, Zamani A, Kubíčková L, Dubový P, Joukal M. Choroid plexus and the blood–cerebrospinal fluid barrier in disease. Fluids Barriers CNS (2020) 17(1):35. doi: 10.1186/s12987-020-00196-2
283. Kooij G, Kopplin K, Blasig R, Stuiver M, Koning N, Goverse G, et al. Disturbed function of the blood-cerebrospinal fluid barrier aggravates neuro-inflammation. Acta Neuropathol (2014) 128(2):267–77. doi: 10.1007/s00401-013-1227-1
284. Castro Dias M, Coisne C, Lazarevic I, Baden P, Hata M, Iwamoto N, et al. Claudin-3-deficient C57BL/6J mice display intact brain barriers. Sci Rep (2019) 9(1):203. doi: 10.1038/s41598-018-36731-3
285. Wolburg K, Gerhardt H, Schulz M, Wolburg H, Engelhardt B. Ultrastructural localization of adhesion molecules in the healthy. Cell Tissue Res (1999) 296(2):259–69. doi: 10.1007/s004410051287
286. Nishihara H, Soldati S, Mossu A, Rosito M, Rudolph H, Muller WA, et al. Human CD4(+) T cell subsets differ in their abilities to cross endothelial and epithelial brain barriers in vitro. Fluids Barriers CNS (2020) 17(1):3. doi: 10.1186/s12987-019-0165-2
287. Strominger I, Elyahu Y, Berner O, Reckhow J, Mittal K, Nemirovsky A, et al. The Choroid Plexus Functions as a Niche for T-Cell Stimulation Within the Central Nervous System. Front Immunol (2018) 9:1066(1066). doi: 10.3389/fimmu.2018.01066
288. Szmydynger-Chodobska J, Strazielle N, Gandy JR, Keefe TH, Zink BJ, Ghersi-Egea JF, et al. Posttraumatic invasion of monocytes across the blood-cerebrospinal fluid barrier. J Cereb Blood Flow Metab (2012) 32(1):93–104. doi: 10.1038/jcbfm.2011.111
289. Meeker RB, Bragg DC, Poulton W, Hudson L. Transmigration of macrophages across the choroid plexus epithelium in response to the feline immunodeficiency virus. Cell Tissue Res (2012) 347(2):443–55. doi: 10.1007/s00441-011-1301-8
290. Serot JM, Foliguet B, Béné MC, Faure GC. Ultrastructural and immunohistological evidence for dendritic-like cells within human choroid plexus epithelium. Neuroreport (1997) 8(8):1995–8. doi: 10.1097/00001756-199705260-00039
291. Huber-Lang M, Lambris JD, Ward PA. Innate immune responses to trauma. Nat Immunol (2018) 19(4):327–41. doi: 10.1038/s41590-018-0064-8
292. Farooqui AA. “Chapter 10 - Summary, Perspective, and Direction for Future Research on Neurotraumatic Diseases”. In: Farooqui AA, editor. Ischemic and Traumatic Brain and Spinal Cord Injuries. Academic Press (2018). p. 419–38.
293. Schnell L, Fearn S, Klassen H, Schwab ME, Perry VH. Acute inflammatory responses to mechanical lesions in the CNS: differences between brain and spinal cord. Eur J Neurosci (1999) 11(10):3648–58. doi: 10.1046/j.1460-9568.1999.00792.x
294. Bellver-Landete V, Bretheau F, Mailhot B, Vallières N, Lessard M, Janelle M-E, et al. Microglia are an essential component of the neuroprotective scar that forms after spinal cord injury. Nat Commun (2019) 10(1):518. doi: 10.1038/s41467-019-08446-0
295. Carlson SL, Parrish ME, Springer JE, Doty K, Dossett L. Acute inflammatory response in spinal cord following impact injury. Exp Neurol (1998) 151(1):77–88. doi: 10.1006/exnr.1998.6785
296. Milich LM, Ryan CB, Lee JK. The origin, fate, and contribution of macrophages to spinal cord injury pathology. Acta Neuropathol (2019) 137(5):785–97. doi: 10.1007/s00401-019-01992-3
297. Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB. Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res (2002) 68(6):691–702. doi: 10.1002/jnr.10269
298. Zhang H, Trivedi A, Lee JU, Lohela M, Lee SM, Fandel TM, et al. Matrix metalloproteinase-9 and stromal cell-derived factor-1 act synergistically to support migration of blood-borne monocytes into the injured spinal cord. J Neurosci (2011) 31(44):15894–903. doi: 10.1523/jneurosci.3943-11.2011
299. Li L, Ni L, Heary RF, Elkabes S. Astroglial TLR9 antagonism promotes chemotaxis and alternative activation of macrophages via modulation of astrocyte-derived signals: implications for spinal cord injury. J Neuroinflamm (2020) 17(1):73. doi: 10.1186/s12974-020-01748-x
300. Kobayakawa K, Ohkawa Y, Yoshizaki S, Tamaru T, Saito T, Kijima K, et al. Macrophage centripetal migration drives spontaneous healing process after spinal cord injury. Sci Adv (2019) 5(5):eaav5086. doi: 10.1126/sciadv.aav5086
301. Shechter R, London A, Varol C, Raposo C, Cusimano M, Yovel G, et al. Infiltrating blood-derived macrophages are vital cells playing an anti-inflammatory role in recovery from spinal cord injury in mice. PLoS Med (2009) 6(7):e1000113. doi: 10.1371/journal.pmed.1000113
302. Jeong SJ, Cooper JG, Ifergan I, McGuire TL, Xu D, Hunter Z, et al. Intravenous immune-modifying nanoparticles as a therapy for spinal cord injury in mice. Neurobiol Dis (2017) 108:14947–54. doi: 10.1016/j.nbd.2017.08.006
303. Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT. Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol (1999) 158(2):351–65. doi: 10.1006/exnr.1999.7118
304. Iannotti CA, Clark M, Horn KP, van Rooijen N, Silver J, Steinmetz MP. A combination immunomodulatory treatment promotes neuroprotection and locomotor recovery after contusion SCI. Exp Neurol (2011) 230(1):3–15. doi: 10.1016/j.expneurol.2010.03.010
305. Norden DM, Faw TD, McKim DB, Deibert RJ, Fisher LC, Sheridan JF, et al. Bone Marrow-Derived Monocytes Drive the Inflammatory Microenvironment in Local and Remote Regions after Thoracic Spinal Cord Injury. J Neurotrauma (2019) 36(6):937–49. doi: 10.1089/neu.2018.5806
306. Nakajima H, Honjoh K, Watanabe S, Kubota A, Matsumine A. Distribution and polarization of microglia and macrophages at injured sites and the lumbar enlargement after spinal cord injury. Neurosci Lett (2020) 737:135152–2. doi: 10.1016/j.neulet.2020.135152
307. Blomster LV, Brennan FH, Lao HW, Harle DW, Harvey AR, Ruitenberg MJ. Mobilisation of the splenic monocyte reservoir and peripheral CX3;CR1 deficiency adversely affects recovery from spinal cord injury. Exp Neurol (2013) 247:226–40. doi: 10.1016/j.expneurol.2013.05.002
308. Kigerl KA, Gensel JC, Ankeny DP, Alexander JK, Donnelly DJ, Popovich PG. Identification of two distinct macrophage subsets with divergent effects causing either neurotoxicity or regeneration in the injured mouse spinal cord. J Neurosci (2009) 29(43):13435–44. doi: 10.1523/jneurosci.3257-09.2009
309. Hsieh CL, Kim CC, Ryba BE, Niemi EC, Bando JK, Locksley RM, et al. Traumatic brain injury induces macrophage subsets in the brain. Eur J Immunol (2013) 43(8):2010–22. doi: 10.1002/eji.201243084
310. Bramlett HM, Dietrich WD. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J neurotrauma (2015) 32(23):1834–48. doi: 10.1089/neu.2014.3352
311. Shibahara S, Kitamuro T, Takahashi K. Heme degradation and human disease: diversity is the soul of life. Antioxid Redox Signal (2002) 4(4):593–602. doi: 10.1089/15230860260220094
312. Jazwa A, Cuadrado A. Targeting heme oxygenase-1 for neuroprotection and neuroinflammation in neurodegenerative diseases. Curr Drug Targets (2010) 11(12):1517–31. doi: 10.2174/1389450111009011517
313. Paine A, Eiz-Vesper B, Blasczyk R, Immenschuh S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem Pharmacol (2010) 80(12):1895–903. doi: 10.1016/j.bcp.2010.07.014
314. Fux M, van Rooijen N, Owens T. Macrophage-independent T cell infiltration to the site of injury-induced brain inflammation. J Neuroimmunol (2008) 203(1):64–72. doi: 10.1016/j.jneuroim.2008.06.025
315. Morganti JM, Jopson TD, Liu S, Riparip LK, Guandique CK, Gupta N, et al. CCR2 antagonism alters brain macrophage polarization and ameliorates cognitive dysfunction induced by traumatic brain injury. J Neurosci (2015) 35(2):748–60. doi: 10.1523/jneurosci.2405-14.2015
316. Kumar A, Barrett JP, Alvarez-Croda DM, Stoica BA, Faden AI, Loane DJ. NOX2 drives M1-like microglial/macrophage activation and neurodegeneration following experimental traumatic brain injury. Brain Behav Immun (2016) 58:291–309. doi: 10.1016/j.bbi.2016.07.158
317. Sharma S, Ifergan I, Kurz JE, Linsenmeier RA, Xu D, Cooper JG, et al. Intravenous Immunomodulatory Nanoparticle Treatment for Traumatic Brain Injury. Ann Neurol (2020) 87(3):442–55. doi: 10.1002/ana.25675
318. Russo MV, Latour LL, McGavern DB. Distinct myeloid cell subsets promote meningeal remodeling and vascular repair after mild traumatic brain injury. Nat Immunol (2018) 19(5):442–52. doi: 10.1038/s41590-018-0086-2
319. Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature (2014) 505(7482):223–8. doi: 10.1038/nature12808
320. Szmydynger-Chodobska J, Shan R, Thomasian N, Chodobski A. The Involvement of Pial Microvessels in Leukocyte Invasion after Mild Traumatic Brain Injury. PLoS One (2016) 11(12):e0167677. doi: 10.1371/journal.pone.0167677
321. Cera MR, Fabbri M, Molendini C, Corada M, Orsenigo F, Rehberg M, et al. JAM-A promotes neutrophil chemotaxis by controlling integrin internalization and recycling. J Cell Sci (2009) 122(Pt 2):268–77. doi: 10.1242/jcs.037127
322. Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, Rademakers T, et al. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation (2014) 129(1):66–76. doi: 10.1161/circulationaha.113.004149
323. Semple BD, Bye N, Rancan M, Ziebell JM, Morganti-Kossmann MC. Role of CCL2 (MCP-1) in traumatic brain injury (TBI): evidence from severe TBI patients and CCL2-/- mice. J Cereb Blood Flow Metab (2010) 30(4):769–82. doi: 10.1038/jcbfm.2009.262
324. Gyoneva S, Kim D, Katsumoto A, Kokiko-Cochran ON, Lamb BT, Ransohoff RM. Ccr2 deletion dissociates cavity size and tau pathology after mild traumatic brain injury. J Neuroinflamm (2015) 12(228). doi: 10.1186/s12974-015-0443-0
325. Hsieh CL, Niemi EC, Wang SH, Lee CC, Bingham D, Zhang J, et al. CCR2 deficiency impairs macrophage infiltration and improves cognitive function after traumatic brain injury. J Neurotrauma (2014) 31(20):1677–88. doi: 10.1089/neu.2013.3252
326. Frik J, Merl-Pham J, Plesnila N, Mattugini N, Kjell J, Kraska J, et al. Cross-talk between monocyte invasion and astrocyte proliferation regulates scarring in brain injury. EMBO Rep (2018) 19(5). doi: 10.15252/embr.201745294
327. Chen Z, Chen S, Liu J. The role of T cells in the pathogenesis of Parkinson’s disease. Prog Neurobiol (2018) 169:1–23. doi: 10.1016/j.pneurobio.2018.08.002
328. Brochard V, Combadière B, Prigent A, Laouar Y, Perrin A, Beray-Berthat V, et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest (2009) 119(1):182–92. doi: 10.1172/jci36470
329. Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature (1997) 388(6645):839–40. doi: 10.1038/42166
330. Gerhard A, Pavese N, Hotton G, Turkheimer F, Es M, Hammers A, et al. In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. Neurobiol Dis (2006) 21(2):404–12. doi: 10.1016/j.nbd.2005.08.002
331. Imamura K, Hishikawa N, Sawada M, Nagatsu T, Yoshida M, Hashizume Y. Distribution of major histocompatibility complex class II-positive microglia and cytokine profile of Parkinson’s disease brains. Acta Neuropathol (2003) 106(6):518–26. doi: 10.1007/s00401-003-0766-2
332. Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC. Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett (1994) 172(1-2):151–4. doi: 10.1016/0304-3940(94)90684-x
333. Hunot S, Boissière F, Faucheux B, Brugg B, Mouatt-Prigent A, Agid Y, et al. Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience (1996) 72(2):355–63. doi: 10.1016/0306-4522(95)00578-1
334. Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. J Neural Transm Suppl (2000) (58):143–51.
335. Kin K, Yasuhara T, Kameda M, Date I. Animal Models for Parkinson’s Disease Research: Trends in the 2000s. Int J Mol Sci (2019) 20(21):5402. doi: 10.3390/ijms20215402
336. Ambrosi G, Kustrimovic N, Siani F, Rasini E, Cerri S, Ghezzi C, et al. Complex Changes in the Innate and Adaptive Immunity Accompany Progressive Degeneration of the Nigrostriatal Pathway Induced by Intrastriatal Injection of 6-Hydroxydopamine in the Rat. Neurotoxicity Res (2017) 32(1):71–81. doi: 10.1007/s12640-017-9712-2
337. Wong YC, Krainc D. α-synuclein toxicity in neurodegeneration: mechanism and therapeutic strategies. Nat Med (2017) 23(2):1–13. doi: 10.1038/nm.4269
338. Fuzzati-Armentero MT, Cerri S, Blandini F. Peripheral-Central Neuroimmune Crosstalk in Parkinson’s Disease: What Do Patients and Animal Models Tell Us? Front Neurol (2019) 10:232(232). doi: 10.3389/fneur.2019.00232
339. Bates CA, Fu S, Ysselstein D, Rochet JC, Zheng W. Expression and Transport of α-Synuclein at the Blood-Cerebrospinal Fluid Barrier and Effects of Manganese Exposure. Admet dmpk (2015) 3(1):15–33. doi: 10.5599/admet.3.1.159
340. Luo XM, Lin H, Wang W, Geaney MS, Law L, Wynyard S, et al. Recovery of neurological functions in non-human primate model of Parkinson’s disease by transplantation of encapsulated neonatal porcine choroid plexus cells. J Parkinsons Dis (2013) 3(3):275–91. doi: 10.3233/jpd-130214
341. Nissen SK, Shrivastava K, Schulte C, Otzen DE, Goldeck D, Berg D, et al. Alterations in Blood Monocyte Functions in Parkinson’s Disease. Mov Disord (2019) 34(11):1711–21. doi: 10.1002/mds.27815
342. Wijeyekoon RS, Kronenberg-Versteeg D, Scott KM, Hayat S, Jones JL, Clatworthy MR, et al. Monocyte Function in Parkinson’s Disease and the Impact of Autologous Serum on Phagocytosis. Front Neurol (2018) 9:870(870). doi: 10.3389/fneur.2018.00870
343. Park DR, Thomsen AR, Frevert CW, Pham U, Skerrett SJ, Kiener PA, et al. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J Immunol (2003) 170(12):6209–16. doi: 10.4049/jimmunol.170.12.6209
344. Letellier E, Kumar S, Sancho-Martinez I, Krauth S, Funke-Kaiser A, Laudenklos S, et al. CD95-ligand on peripheral myeloid cells activates Syk kinase to trigger their recruitment to the inflammatory site. Immunity (2010) 32(2):240–52. doi: 10.1016/j.immuni.2010.01.011
345. Schlachetzki JCM, Prots I, Tao J, Chun HB, Saijo K, Gosselin D, et al. A monocyte gene expression signature in the early clinical course of Parkinson’s disease. Sci Rep (2018) 8(1):10757. doi: 10.1038/s41598-018-28986-7
346. Côté M, Poirier AA, Aubé B, Jobin C, Lacroix S, Soulet D. Partial depletion of the proinflammatory monocyte population is neuroprotective in the myenteric plexus but not in the basal ganglia in a MPTP mouse model of Parkinson’s disease. Brain Behav Immun (2015) 46:154–67. doi: 10.1016/j.bbi.2015.01.009
347. Kokovay E, Cunningham LA. Bone marrow-derived microglia contribute to the neuroinflammatory response and express iNOS in the MPTP mouse model of Parkinson’s disease. Neurobiol Dis (2005) 19(3):471–8. doi: 10.1016/j.nbd.2005.01.023
348. Depboylu C, Stricker S, Ghobril J-P, Oertel WH, Priller J, Höglinger GU. Brain-resident microglia predominate over infiltrating myeloid cells in activation, phagocytosis and interaction with T-lymphocytes in the MPTP mouse model of Parkinson disease. Exp Neurol (2012) 238(2):183–91. doi: 10.1016/j.expneurol.2012.08.020
349. Chen C, Li X, Ge G, Liu J, Biju KC, Laing SD, et al. GDNF-expressing macrophages mitigate loss of dopamine neurons and improve Parkinsonian symptoms in MitoPark mice. Sci Rep (2018) 8(1):5460. doi: 10.1038/s41598-018-23795-4
350. Mammana S, Fagone P, Cavalli E, Basile MS, Petralia MC, Nicoletti F, et al. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int J Mol Sci (2018) 19(3):831. doi: 10.3390/ijms19030831
351. Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med (2011) 1(1):a006189. doi: 10.1101/cshperspect.a006189
352. Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol (2018) 217(2):459–72. doi: 10.1083/jcb.201709069
353. Hemonnot A-L, Hua J, Ulmann L, Hirbec H. Microglia in Alzheimer Disease: Well-Known Targets and New Opportunities. Front Aging Neurosci (2019) 11:233(233). doi: 10.3389/fnagi.2019.00233
354. Gratuze M, Leyns CEG, Holtzman DM. New insights into the role of TREM2 in Alzheimer’s disease. Mol Neurodegener (2018) 13(1):66. doi: 10.1186/s13024-018-0298-9
355. Jay TR, Hirsch AM, Broihier ML, Miller CM, Neilson LE, Ransohoff RM, et al. Disease Progression-Dependent Effects of TREM2 Deficiency in a Mouse Model of Alzheimer’s Disease. J Neurosci (2017) 37(3):637–47. doi: 10.1523/jneurosci.2110-16.2016
356. Ramirez J, Berezuk C, McNeely AA, Gao F, McLaurin J, Black SE. Imaging the Perivascular Space as a Potential Biomarker of Neurovascular and Neurodegenerative Diseases. Cell Mol Neurobiol (2016) 36(2):289–99. doi: 10.1007/s10571-016-0343-6
357. Hawkes CA, McLaurin J. Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A (2009) 106(4):1261–6. doi: 10.1073/pnas.0805453106
358. Park L, Uekawa K, Garcia-Bonilla L, Koizumi K, Murphy M, Pistik R, et al. Brain Perivascular Macrophages Initiate the Neurovascular Dysfunction of Alzheimer Aβ Peptides. Circ Res (2017) 121(3):258–69. doi: 10.1161/circresaha.117.311054
359. Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci (2004) 5(5):347–60. doi: 10.1038/nrn1387
360. Alvira-Botero X, Carro EM. Clearance of amyloid-β peptide across the choroid plexus in Alzheimer’s disease. Curr Aging Sci (2010) 3(3):219–29. doi: 10.2174/1874609811003030219
361. Shen X, Xia L, Liu L, Jiang H, Shannahan J, Du Y, et al. Altered clearance of beta-amyloid from the cerebrospinal fluid following subchronic lead exposure in rats: Roles of RAGE and LRP1 in the choroid plexus. J Trace Elements Med Biol (2020) 61(126520). doi: 10.1016/j.jtemb.2020.126520
362. Perez-Gracia E, Blanco R, Carmona M, Carro E, Ferrer I. Oxidative stress damage and oxidative stress responses in the choroid plexus in Alzheimer’s disease. Acta Neuropathol (2009) 118(4):497–504. doi: 10.1007/s00401-009-0574-4
363. Bergen AA, Kaing S, ten Brink JB, Gorgels TG, Janssen SF, The Netherlands Brain, B. Gene expression and functional annotation of human choroid plexus epithelium failure in Alzheimer’s disease. BMC Genomics (2015) 16(1):956. doi: 10.1186/s12864-015-2159-z
364. Baruch K, Rosenzweig N, Kertser A, Deczkowska A, Sharif AM, Spinrad A, et al. Breaking immune tolerance by targeting Foxp3+ regulatory T cells mitigates Alzheimer’s disease pathology. Nat Commun (2015) 6(1):7967. doi: 10.1038/ncomms8967
365. Da Mesquita S, Louveau A, Vaccari A, Smirnov I, Cornelison RC, Kingsmore KM, et al. Functional aspects of meningeal lymphatics in ageing and Alzheimer’s disease. Nature (2018) 560(7717):185–91. doi: 10.1038/s41586-018-0368-8
366. Giri R, Shen Y, Stins M, Du Yan S, Schmidt AM, Stern D, et al. beta-amyloid-induced migration of monocytes across human brain endothelial cells involves RAGE and PECAM-1. Am J Physiol Cell Physiol (2000) 279(6):C1772–81. doi: 10.1152/ajpcell.2000.279.6.C1772
367. El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, et al. CD36 mediates the innate host response to beta-amyloid. J Exp Med (2003) 197(12):1657–66. doi: 10.1084/jem.20021546
368. Walker DG, Lue LF, Beach TG. Gene expression profiling of amyloid beta peptide-stimulated human post-mortem brain microglia. Neurobiol Aging (2001) 22(6):957–66. doi: 10.1016/s0197-4580(01)00306-2
369. Ishizuka K, Kimura T, Igata-yi R, Katsuragi S, Takamatsu J, Miyakawa T. Identification of monocyte chemoattractant protein-1 in senile plaques and reactive microglia of Alzheimer’s disease. Psychiatry Clin Neurosci (1997) 51(3):135–8. doi: 10.1111/j.1440-1819.1997.tb02375.x
370. Hickman SE, El Khoury J. Mechanisms of mononuclear phagocyte recruitment in Alzheimer’s disease. CNS Neurol Disord Drug Targets (2010) 9(2):168–73. doi: 10.2174/187152710791011982
371. Naert G, Rivest S. A deficiency in CCR2+ monocytes: the hidden side of Alzheimer’s disease. J Mol Cell Biol (2013) 5(5):284–93. doi: 10.1093/jmcb/mjt028
372. Malm TM, Koistinaho M, Pärepalo M, Vatanen T, Ooka A, Karlsson S, et al. Bone-marrow-derived cells contribute to the recruitment of microglial cells in response to beta-amyloid deposition in APP/PS1 double transgenic Alzheimer mice. Neurobiol Dis (2005) 18(1):134–42. doi: 10.1016/j.nbd.2004.09.009
373. Malm T, Koistinaho M, Muona A, Magga J, Koistinaho J. The role and therapeutic potential of monocytic cells in Alzheimer’s disease. Glia (2010) 58(8):889–900. doi: 10.1002/glia.20973
374. Fiala M, Liu PT, Espinosa-Jeffrey A, Rosenthal MJ, Bernard G, Ringman JM, et al. Innate immunity and transcription of MGAT-III and Toll-like receptors in Alzheimer’s disease patients are improved by bisdemethoxycurcumin. Proc Natl Acad Sci U S A (2007) 104(31):12849–54. doi: 10.1073/pnas.0701267104
375. Fiala M, Lin J, Ringman J, Kermani-Arab V, Tsao G, Patel A, et al. Ineffective phagocytosis of amyloid-beta by macrophages of Alzheimer’s disease patients. J Alzheimers Dis (2005) 7(3):221–32; discussion 255-62. doi: 10.3233/jad-2005-7304
376. Zaghi J, Goldenson B, Inayathullah M, Lossinsky AS, Masoumi A, Avagyan H, et al. Alzheimer disease macrophages shuttle amyloid-beta from neurons to vessels, contributing to amyloid angiopathy. Acta Neuropathol (2009) 117(2):111–24. doi: 10.1007/s00401-008-0481-0
Keywords: macrophage cell, meninges, CNS inflammation, cell trafficking, choroid plexus
Citation: Ivan DC, Walthert S, Berve K, Steudler J and Locatelli G (2021) Dwellers and Trespassers: Mononuclear Phagocytes at the Borders of the Central Nervous System. Front. Immunol. 11:609921. doi: 10.3389/fimmu.2020.609921
Received: 24 September 2020; Accepted: 29 December 2020;
Published: 05 March 2021.
Edited by:
Bruno Stankoff, Sorbonne Universités, FranceReviewed by:
Ulises Gomez-Pinedo, Instituto de Investigación Sanitaria del Hospital Clínico San Carlos, SpainVioletta Zujovic, Institut National de la Santé et de la Recherche Médicale (INSERM), France
Kuti Baruch, ImmunoBrain Checkpoint Ltd., Israel
Copyright © 2021 Ivan, Walthert, Berve, Steudler and Locatelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Giuseppe Locatelli, Z2l1c2VwcGUubG9jYXRlbGxpQHRraS51bmliZS5jaA==