 Jonas B. Sandbrink
Jonas B. Sandbrink Robin J. Shattock
Robin J. Shattock
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 22 December 2020
Sec. Vaccines and Molecular Therapeutics
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.608460
The COVID-19 pandemic demonstrates the ongoing threat of pandemics caused by novel, previously unrecognized, or mutated pathogens with high transmissibility. Currently, vaccine development is too slow for vaccines to be used in the control of emerging pandemics. RNA-based vaccines might be suitable to meet this challenge. The use of an RNA-based delivery mechanism promises fast vaccine development, clinical approval, and production. The simplicity of in vitro transcription of mRNA suggests potential for fast, scalable, and low-cost manufacture. RNA vaccines are safe in theory and have shown acceptable tolerability in first clinical trials. Immunogenicity of SARS-CoV-2 mRNA vaccines in phase 1 trials looks promising, however induction of cellular immunity needs to be confirmed and optimized. Further optimization of RNA vaccine modification and formulation to this end is needed, which may also enable single injection regimens to be achievable. Self-amplifying RNA vaccines, which show high immunogenicity at low doses, might help to improve potency while keeping manufacturing costs low and speed high. With theoretical properties of RNA vaccines looking promising, their clinical efficacy is the key remaining question with regard to their suitability for tackling emerging pandemics. This question might be answered by ongoing efficacy trials of SARS-CoV-2 mRNA vaccines.
The coronavirus COVID-19 pandemic demonstrates the threat posed by pandemic pathogens. Emerging pandemics are epidemics with potential to spread worldwide caused by potentially novel or emerging pathogens able to rapidly spread in the absence of pre-existing protective immunity. Sociological and ecological factors have favored zoonotic emergence of such pandemics, while biotechnological advances may have increased the danger from human-originated pandemics (1, 2). Indeed, advances in gene synthesis and editing technology may enable actors with malicious intent to synthesize mutated, novel, or previously eradicated pathogens for deliberate release. Vaccines are a powerful tool to counter infectious diseases threats with pandemic potential. However, vaccine development from preclinical phase to licensing takes on average more than 10 years (3). Hence, currently, vaccine development is too slow for vaccines to be available to control an emerging pandemic caused by Disease X.
A growing number of agencies have been engaged in pandemic surveillance and preparedness including agencies such as the WHO, Gavi, BARDA, CDC, and the risk of infectious threats remains high on many national agendas (2). To tackle this issue, the Coalition for Epidemic Preparedness Innovations (CEPI) has set the goal for new vaccines to reach clinical testing within 16 weeks of pathogen detection and for 100,000 doses to be produced within 30 weeks (4). To meet these goals, CEPI has called for the advancement of platform vaccine technologies. This refers to vaccine systems where a universal backbone can be adapted to target different pathogens. RNA vaccines are one such platform which promises fast vaccine development, approval, and manufacturing. Hence, CEPI has funded the development of mRNA vaccines, including the development of SARS-CoV-2 vaccines, by the biotech companies CureVac and Moderna, as well as the development of self-amplifying mRNA (saRNA) vaccine technology by academics at Imperial College London (5–7).
Speed of vaccine development, approval, and manufacturing is not the only characteristic necessary for a vaccine to be suitable for tackling an emerging pandemic. For global availability of a vaccine during a pandemic, scalable and low-cost manufacturing and vaccine thermostability are needed. A given platform should be able to induce both cellular and humoral immunity to be able to tackle novel pathogens in absence of an established correlate of protection, ideally with a single dose. Most importantly, safety, tolerability, and efficacy of a given vaccine need to be shown. This article will evaluate in what ways RNA-based vaccines meet these criteria to date and what steps need to be taken to improve the suitability of RNA vaccines for a pandemic setting. A focus will be laid on vaccines against RNA viruses with particular pandemic potential, such as influenza virus and coronavirus (8). Findings from vaccines targeting other pathogens which provide useful insights into the properties and current state of the RNA vaccine platform will also be considered.
RNA vaccines are based on the premise that mRNA, injected for vaccination, when taken up by antigen presenting cells (APCs) and other target cells induces expression of the properly folded and glycosylated antigenic protein (Figure 1) (10). As RNA activates endosomal and cytosolic RNA sensors upon cell entry, these vaccines exhibit self-adjuvanticity and induce both a humoral and cellular immune response against the encoded protein (11, 12). As saRNA vaccines contain a replicon based on alphavirus non-structural proteins, they are able to self-amplify within host cells (13). Hence, saRNA vaccines have the potential to induce higher levels of protein production and immunogenicity relative to the injected dose compared to conventional mRNA vaccines (14).
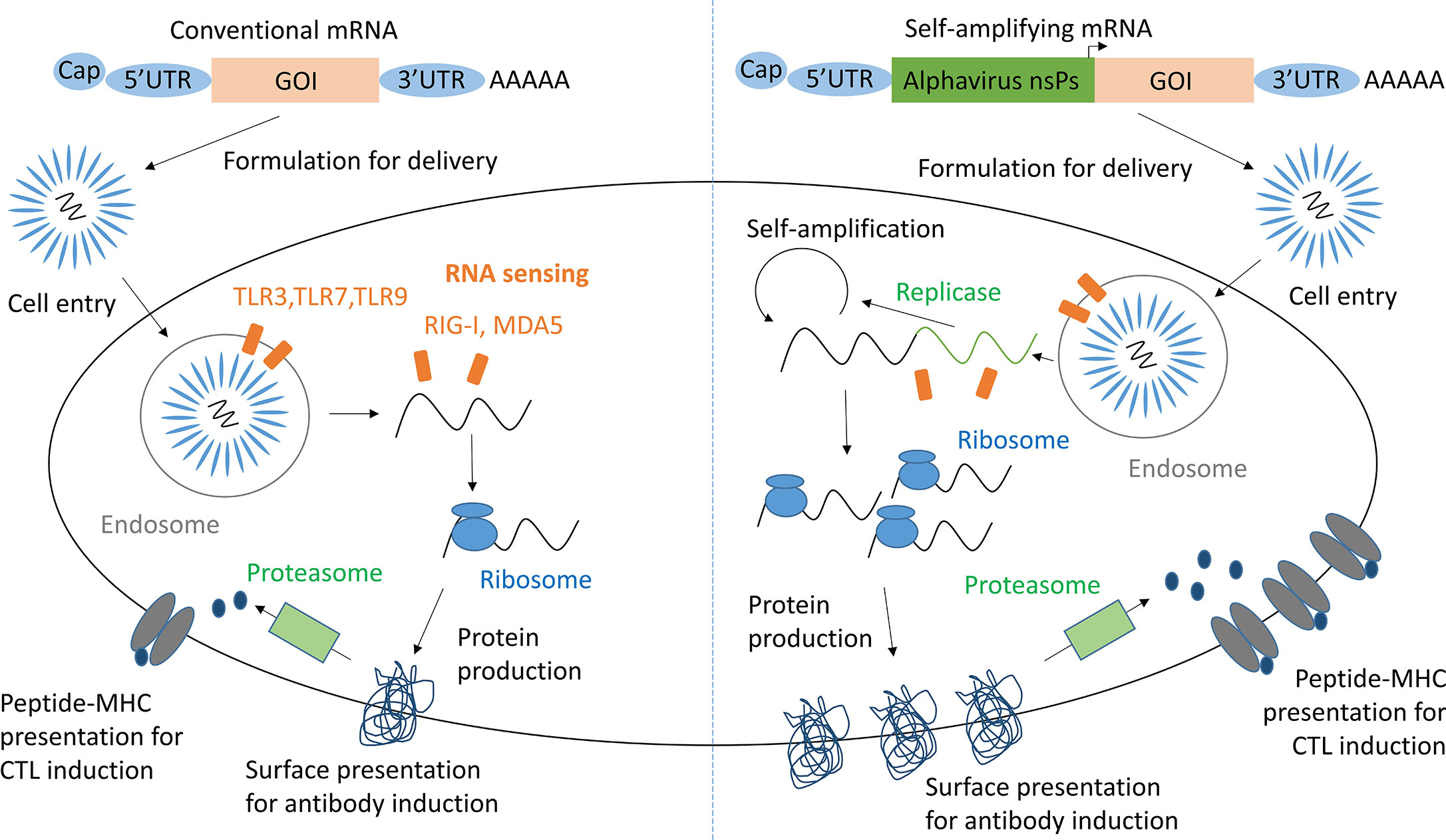
Figure 1 mRNA and saRNA protein production in antigen presenting cells. Adapted from Maruggi et al. (9). GOI, Gene of interest; UTR, Untranslated regions; nsPs, non-structural proteins; CTL, cytotoxic T lymphocyte.
Self-adjuvanticity of RNA may be a double-edged sword. While enabling self-adjuvancy, recognition of foreign RNA by intracellular RNA sensors may also induce its degradation and/or silencing of expression (15). RNA modification strategies such as: the addition of a 5’ cap; the length and structure of a 3’ PolyA tail; use of untranslated regions and the modification of nucleosides, and sequence optimization, have been critical for decreasing the degradation and increasing the immunogenicity of mRNA (16–18). Furthermore, RNA sequence optimization may raise expression efficiency and hence immunogenicity (19, 20). Additionally, RNA may be formulated with a delivery vehicle, such as lipid nanoparticles (LNPs) or cationic polyplexes, which protect RNA from degradation, boost target cell uptake, and increase adjuvancy (21).
RNA vaccines promise fast development as upon sequencing of the target pathogen mRNA candidates can be designed and synthesized very quickly. Additionally, reduced need for optimization and regulatory testing of a new vaccine may further speed up development and the approval process (22). This is because a new RNA vaccine will only differ in the encoded sequence of its target protein, while its formulation and method of delivery will have been optimized and licensed in the past. Indeed, Moderna made news by starting human testing of a SARS-CoV-2 mRNA vaccine only 66 days after viral genome sequencing (23). This unparalleled feat was not only enabled by sequence-informed design and fast manufacturing of the mRNA vaccine for clinical testing, but also by previous work on an mRNA vaccine for MERS-CoV, which informed antigen selection and prefusion stabilization of the SARS-CoV-2 spike protein (23). This supports the notion that in addition to technical advancement of fast-response vaccine platforms applying these platforms to prototype pathogens within viral families of high pandemic potential might be critical for pandemic preparedness (24).
RNA vaccines also have promising properties with regard to manufacturing. Due to similarity between vaccines, a single set of production facilities may be used and production may be swiftly switched to that of a new vaccine for tackling an ongoing outbreak. mRNA is produced by in vitro transcription from a linear DNA template by a bacteriophage RNA polymerase (12). Although current approaches predominantly use DNA templates generated from plasmid DNA produced in bacteria, even this step is likely to be replaced with fully synthetic approaches for generating linear DNA (25). This comparatively simple, high-yield, and virus-free production process enables production facilities to be small and scalable (26). Cell-free production means there is less concern with respect to adventitious agents, microorganisms unintentionally introduced into manufacturing, although similar requirements are needed to ensure endotoxin removal, sterility, and control of any residual reagents. Kis and colleagues have estimated that 1L of saRNA, sufficient for 575,000 doses, could be manufactured in 60h at around $0.30 per dose (27). However, additional cost consideration should consider formulation and distribution costs. Nevertheless, the RNA vaccine platform promises not only fast development but also scalable and low-cost manufacturing.
Safety and tolerability need to be proven for the licensing of any given vaccine. RNA vaccines are comparatively safe. There is no risk of pathogen reactivation and RNA is degraded in vivo with no risk of antigen persistence or integration into the genome (28). Self-replicating RNA could pose additional safety concerns when encoding fusion competent viral glycoproteins, however such theoretical concerns can be mitigated through the use of stabilizing mutations ensuring fusion incompetent structures (29). Although no anti-vector effects have been observed with saRNA, T cell recognition of the replicon proteins necessary for RNA amplification might limit the reusability of a given saRNA-based platform.
As the number of clinical studies of RNA vaccines against infectious diseases is limited, the tolerability data from phase 1 clinical trials to date looks acceptable (30–32). However, the cohort size of clinical trials to date has been small, leaving the potential for rare but potentially severe side effects.
One safety concern unspecific to RNA vaccines might be disease enhancement induced by vaccination. For example, the clinical trial of a formalin-inactivated vaccine against RSV in 1967 led to increased mortality in children upon viral infection caused by a type 2 helper CD4+ T cell (Th2) mediated eosinophil invasion of the lungs (33). Lung immunopathology after vaccination has been observed in animal models vaccinated with a range of SARS-CoV and MERS-CoV vaccines (34–36). Hence, concern for disease enhancement might also slow the development of SARS-CoV-2 vaccines. To reduce the risk of lung immunopathology, possible RNA vaccine candidates need to show that they induce a non-Th2 biased immune response. Geall et al. demonstrated in 2012 that an RSV saRNA-LNP vaccine induced twice as high Th1-associated IgG2a titers as Th2-associated IgG1 titers in rats (37). Histological analysis of lung tissue and comparison to a positive and negative control of vaccine-induced RSV exacerbation might have provided additional insight into the safety of this vaccine candidate. In phase 1 clinical studies, mRNA-based SARS-CoV-2 induced Th1-biased immune responses suggesting their safety with regard to disease enhancement (31, 32). Hence, RNA vaccine candidates may be suitable for use against diseases where vaccine-induced disease enhancement is a concern. It is not yet discernable whether RNA-based vaccine approaches universally induce Th1-biased responses and to what degree this depends on the encoded antigen. While classically Th1-polarized responses have been associated with induction of potent cell-mediated immune responses against intracellular pathogens and Th2-polarized responses with humoral immunity against helminthic pathogens, this Th1/Th2 dichotomy has been challenged through the discovery of other CD4+ T cell subsets (38). Almost certainly, different viral pathogens will require the induction of immune responses with different balances of Th1 versus Th2 CD4+ T cell activation.
The fact that no viral culture is required for vaccine development and manufacturing means there are few biosafety concerns around work on RNA vaccines and lack of need for viral modification promises a good biosecurity profile.
Overall, safety and tolerability of RNA vaccines looks promising but needs to be confirmed in larger scale clinical trials. RNA vaccines have the potential to be developed in cases where disease enhancement is a concern; still, thorough testing in these cases will be necessary and might delay vaccine development.
With safety and tolerability looking promising, the key question for whether RNA vaccines will find use in pandemics is whether they will show immunogenicity and efficacy in human trials. Ideally, disease protection after a single dose is needed to ensure fast distribution of vaccines. Additionally, a given platform should be able to induce both cellular and humoral immunity, so generated vaccines are able to induce immunity against a wide range of viral pathogens with different properties and pathology.
Pre-COVID-19, a few human immunogenicity trials using known correlates of protection for evaluation of clinical efficacy had been published, overall showing moderate success (Table 1). In a Moderna H7N9 influenza vaccine trial, the tested mRNA vaccine induced haemagglutinin inhibition assay (HAI) titers of 1 > 1:40 in 96.3% of individuals and virus neutralizing (VN) titers of 1 > 1:20 in 100% of individuals (30). HAI > 1:40 is universally accepted as a correlate of protection for reducing risk of influenza infection by 50%. However, this correlate of protection is largely based on studies of seasonal influenza and has to our knowledge not been confirmed for H7N9 or other potential pandemic subtypes. Additionally, different authors have suggested VN titers between 1:20 and 1:80 as a correlate of protection, complicating the interpretation of the measured VN titers (40). None of the pre-COVID-19 tested rabies or influenza vaccines induced cellular immunity (Table 1).
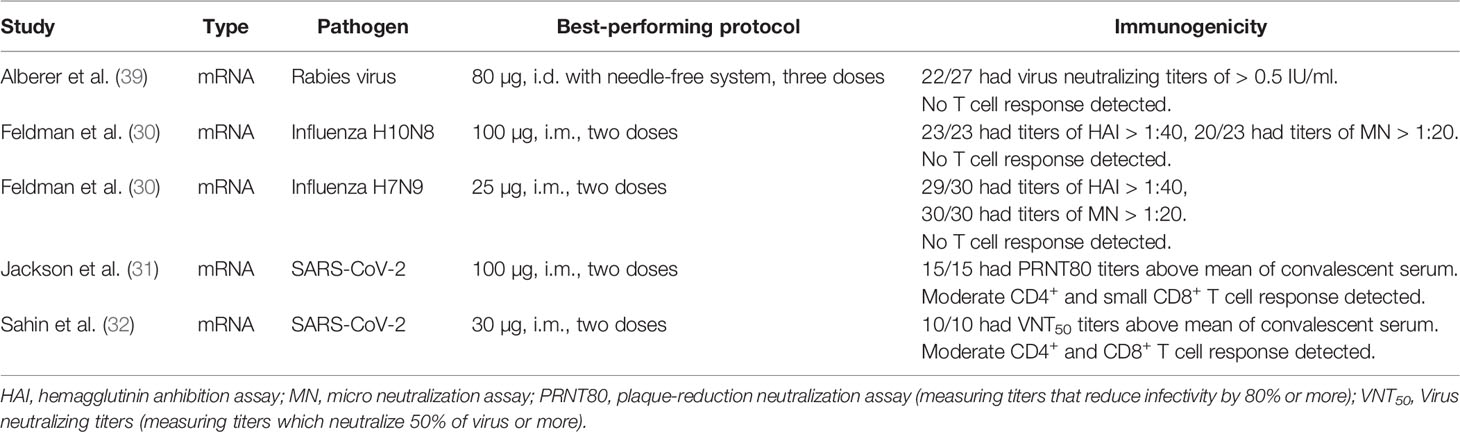
Table 1 Published immunogenicity studies in humans to date.
Evaluating the recently published immunogenicity data of SARS-CoV-2 vaccines is more difficult in absence of a well-established correlate of protection for COVID-19. Phase I clinical data show that Moderna’s mRNA-1273 vaccine was able to induce neutralizing antibody titers similar to those found in convalescent serum samples after two injections (Table 1) (31). The fact that despite rapidly dropping antibody titers in convalescent individuals COVID-19 reinfection is uncommon and cross-reactive T cell responses between coronaviruses seem to confer some degree of protection indicates an important role of cellular immunity in preventing and controlling SARS-CoV-2 infection (41, 42). Hence, the ability of COVID-19 vaccines to induce cellular immune responses might prove critical for vaccine efficacy. While after vaccination with mRNA-1273 a moderate Th1-biased CD4 T cell response was measured, only minimal induction of CD8+ T cell cellular immunity was observed. Pfizer/BioNTech’s vaccine candidate BNT162b1, a LNP-formulated mRNA vaccine encoding the receptor binding domain (RBD) of the SARS-CoV-2 spike protein, induced both CD4+ and CD8+ T cell responses detectable with an ELISpot assay in a majority of trial participants (32). However, a large fraction of these responses also seems very limited in size. Additionally, BNT162b1 induced neutralizing antibodies effective against 17 spike protein variations according to a pseudovirus infection assay. Notably, in all clinically tested mRNA vaccines at least two injections were required for robust immunogenicity.
While early mRNA vaccines for rabies and influenza showed only moderate immunogenicity and did not induce cellular immune responses, recent clinical data on SARS-CoV-2 vaccines look promising. While this improvement likely reflects advances in antigen delivery, the nature of the chosen antigen also influences the quality of the induced immune response. There is need for further optimization of induction of robust cellular immunity with mRNA vaccines, study of immunogenicity in age groups not represented in phase 1 studies to date, and investigation of how single injection protocols may be achieved. Additionally, the promising saRNA technology, which has potential to overcome some of these challenges, needs to be tested in humans. Most importantly, efficacy of RNA vaccines for human protection of disease needs to be shown. The COVID-19 pandemic offers unique opportunities for this, with two phase 2/3 efficacy trials of mRNA vaccines on the way and a saRNA-based vaccine being injected into humans for the first time (43–45).
In absence of human efficacy data, animal data needs to serve as basis for the evaluation of the properties of RNA vaccines and whether this platform will be suitable to tackle pandemics. Recent animal immunogenicity efficacy trials of both mRNA and saRNA vaccines look promising in several regards (Table 2).
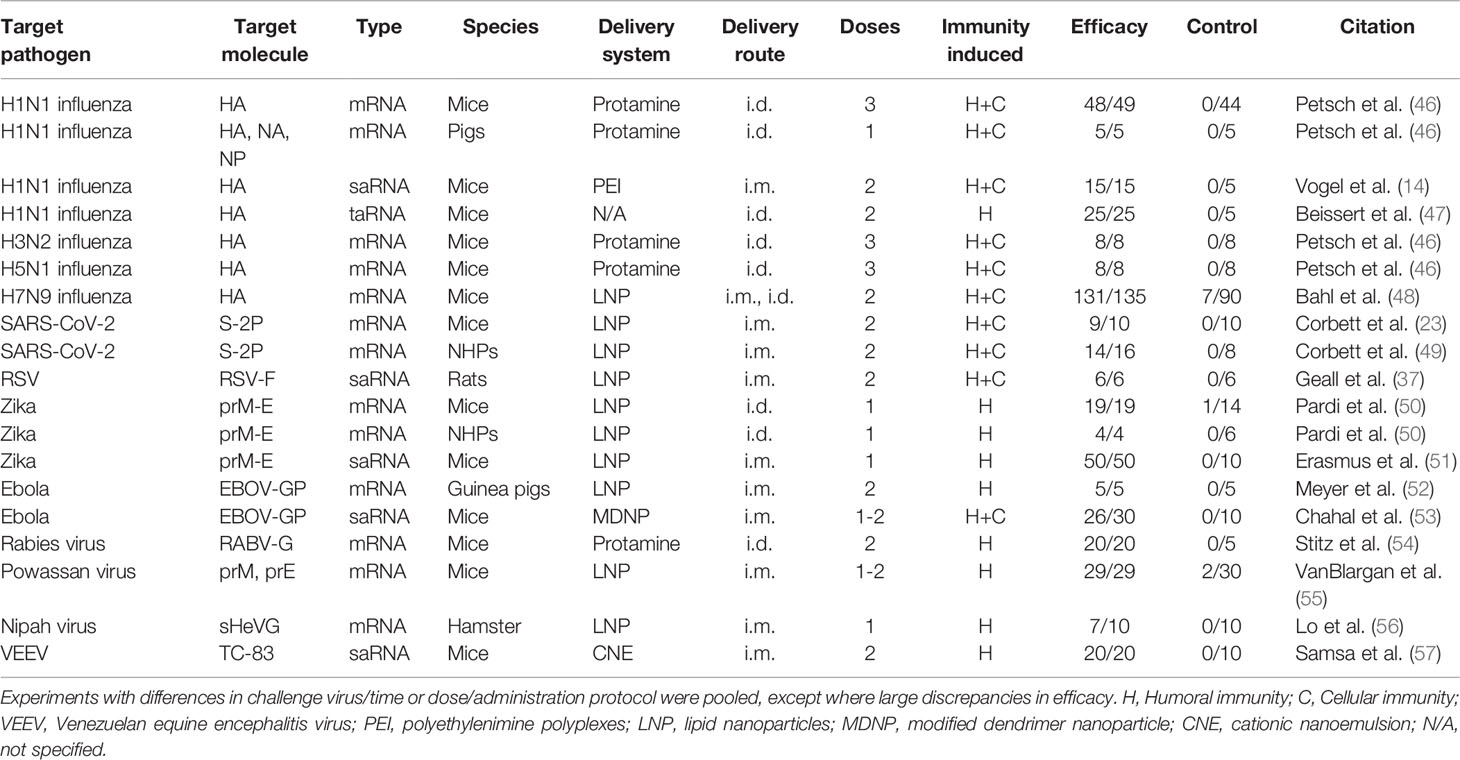
Table 2 Most promising RNA vaccine animal efficacy trials against RNA viruses to date.
Depending on the encoded protein, RNA vaccines are able to induce both cellular and humoral immunity in animals. For example, Vogel et al. showed that two injections of both 1.25 µg saRNA and 80 µg mRNA encoding HA induced both humoral immunity and cellular CD8 T cell responses and protected mice from lethal challenge with H1N1 influenza (14). Limitation of this and similar studies in mice is that induction of strong immune responses in these animals is a lot easier to elicit than in humans. This likely reflects species differences with regards to innate RNA sensing pathways including TLR3, TLR7, and TLR8 (58–60). Additionally, the usefulness of viral challenge efficacy studies in mice may be limited as these animals are not very susceptible to human influenza virus strains; therefore lab- or mouse-adapted viral strains are used in these experiments (61). Nevertheless, an increasing number of RNA vaccines are now reporting success in more relevant non-human primate models (49, 50).
Like all immunogenicity studies in humans, most animal efficacy studies to date have used two or three injections. However, some of the results of single-dose studies look promising (Table 3) (50, 53). For instance, Pardi et al. tested the efficacy of a preM-E encoding Zika virus mRNA vaccine administered with a single injection vaccine in mice and non-human primates (NHPs), which show a very similar disease phenotype and immunity to humans (50). All mice vaccinated with a single 30 µg dose and the four macaques single-dose vaccinated with 200 or 50 µg of mRNA were protected against detectable viraemia after viral challenge.
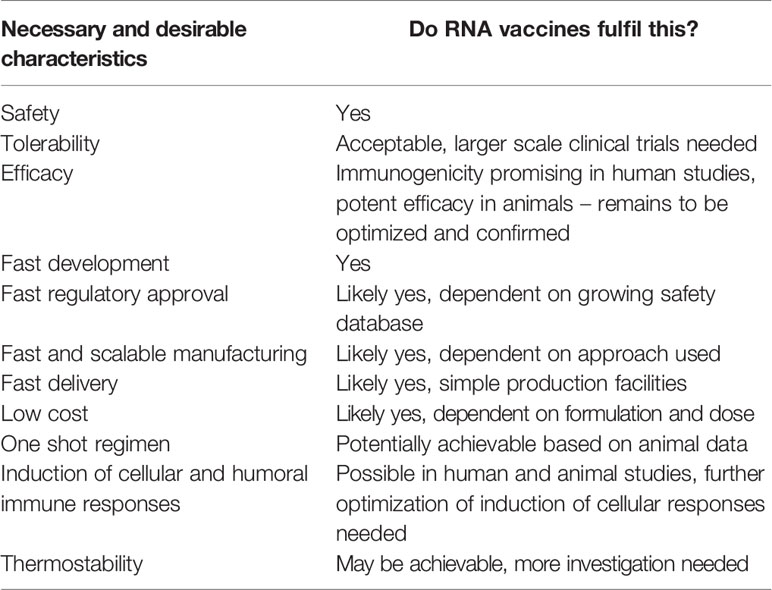
Table 3 Suitability of RNA vaccines for emerging pandemics.
While animal efficacy studies have shown reasonably promising results, translation of these findings to humans is limited with the majority of studies being conducted in small animal models. The extent to which any animal model predicts human response is much debated and highlights the importance of demonstrating proof-of concept in human studies.
Thermostability of a vaccine is critical to reduce the need for a cold chain during vaccine storage and deployment, ensuring global availability of a vaccine in case of a pandemic. RNA is intrinsically unstable, being prone to hydrolysis, and needs to be stored frozen to ensure long-term stability (62). Current RNA vaccines under development for COVID-19 require -80 to -20°C storage during distribution, a significant limitation for global deployment (63, 64). Lyophilisation, freeze-drying, has previously been explored as a method to raise the thermostability of RNA vaccines (54, 65). A freeze-dried mRNA vaccine for rabies was shown to retain immunogenicity and efficacy in mice after 1-month storage at alternating temperatures of 4 and 56°C (54). Freeze-dried versions of the mRNA COVID-19 vaccines might become available eventually. Formulation of RNA with thermostable lipid nanoparticles might contribute to raising the thermostability of RNA vaccines (66). All in all, it seems likely that the thermostability of RNA vaccines will improve in the coming years.
RNA modification and formulation have been critical for raising the immunogenicity of RNA vaccines and will be key to further improve immunogenicity in humans. For example, lipid nanoparticles (LNPs) are one of the most promising methods for the delivery of RNA vaccines. Geall et al. showed that LNP-formulation of saRNA increased IgG titers against RSV 8-fold relative to unformulated saRNA in rats (37). Formulation with LNPs also increased induction of cytotoxic T lymphocyte levels. While the authors found that the properties of the LNP delivery system looked promising, a direct comparison of LNP formulation with other formulation methods would be informative. This reflects a general issue in the field; companies and developers are often reluctant to facilitate head-to-head comparisons. Furthermore, as the field is driven by biotech companies, experiments and even trials are not always published. The COVID-19 pandemic may contribute to resolving some of these issues temporarily. For instance, as most COVID-19 mRNA vaccines encode the spike protein, this may allow direct comparison of immunogenicity of different approaches. Additionally, the urgency of finding a COVID-19 vaccine may continue to result in faster and increased publication of clinical trial results.
Ideally, vaccines against pandemic pathogens should be protective after a single injection. Animal data on single-dose regimens looks promising for certain antigens and vaccine approaches. Additionally, to halt the early phases of an outbreak, neutralizing antibody responses may not need to be very high or durable. Hence, achievement of a single-dose regimen with an RNA vaccine might be possible. RNA modifications aimed at increasing the residual time of RNA may aid achievement of a single-dose regimen. For instance, the use of polyplex-loaded polymeric scaffolds for slow release of mRNA after injection has been explored (67, 68).
Further optimization of immunogenicity of RNA vaccines is needed. However, it will be important not to jeopardize the suitability of RNA vaccines to tackle pandemics through improving immunogenicity at the cost of increased manufacturing complexity and cost. The fact that smaller doses make production cheaper needs to be considered. saRNA vaccines look promising in this regard. As shown by Vogel et al. (14), saRNA vaccines protect mice from lethal influenza virus infection at much lower doses than conventional mRNA vaccines. However, using saRNA increases manufacturing complexity through the need for synthesis of longer RNA constructs. A recent technological advancement may help to overcome this issue. Beissert et al. developed a bipartite vector trans-amplifying RNA (taRNA) system where replicase-encoding RNA is produced in advance and is then added to the on-demand produced transreplicon of the antigen of interest. Using this system, animals were protected from influenza challenge at doses as low as 50 ng (47). Hence, taRNA promises to reduce the amount of RNA needed for immunogenicity, while promising significantly simpler production than saRNA.
The trade-off between improved immunogenicity and increased manufacturing complexity is even more pronounced for vaccine formulations. Vaccine formulations, such as the encapsulation of RNA within lipid nano-particles (LNPs) or vaccine lyophilisation, add further complexity and cost. Modeling by Kis et al. that estimated 575,000 doses of an RNA vaccine could be produced in 60h at around $0.30 per dose assumed a set cost for formulation (27). As the formulation process may make up a large fraction of manufacturing cost, different formulation approaches should be compared for associated expense, in addition to taking into account important considerations around scalability, supply chain logistics of different formulation components, and long-term stability of ready-to-use formulations.
Technological advances may also help to resolve the trade-off between manufacturing complexity and immunogenicity of RNA formulation. Indeed, the immunogenicity of saRNA molecules delivered on the interior or exterior of LNPs is similar (69). Hence, LNPs may be prepared and quality controlled in advance of an outbreak and be combined with RNA later (51).
When optimizing immunogenicity, speed of manufacturing needs to be conserved as much as possible. This needs to be considered when optimizing immunogenicity through increasing doses or vaccine formulation. Technological advances may help to overcome the trade-off between immunogenicity and manufacturing complexity.
RNA vaccines fulfil many of the characteristics necessary to be useful to tackle an emerging pandemic and Disease X (Table 3). They can be rapidly developed and manufactured at low cost. Their safety and tolerability looks acceptable as long as disease enhancement is prevented. The big remaining hurdle is demonstrating the efficacy of RNA vaccines in humans. To this end, clinical efficacy studies are needed, which have been initiated for COVID-19 vaccines. Additionally, current clinical data suggests that cellular immunogenicity of mRNA vaccines may need to be further improved. Here, comparing and combining different RNA modification and formulation approaches may prove critical. At the same time, the cost of RNA modifications, formulations, and supply chain logistics need to be considered. The use of self-amplifying RNA vaccines and additional technological advances might contribute to raising immunogenicity without increasing manufacturing cost and complexity. The coronavirus COVID-19 pandemic is the first field test of whether RNA vaccines can be quickly developed, approved, and produced in face of a pandemic. The race for a COVID-19 vaccine may produce data that will not only answer the question of efficacy of RNA vaccines in humans, but may also advance the RNA vaccine platform to a stage where faster responses to similar future situations are possible.
JS wrote the manuscript. RS revised the manuscript. All authors contributed to the article and approved the submitted version.
RS is funded by the Department of Health and Social Care using UK Aid funding and is managed by the Engineering and Physical Sciences Research Council (EPSRC, grant number: EP/R013764/1, note: the views expressed in this publication are those of the author (s) and not necessarily those of the Department of Health and Social Care).
RS is a co-founder of VacEquity Global Health, a Social Business focused on the development of a self-amplifying RNA vaccine against CoV2.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Sarah Gilbert for advice.
1. Cascio A, Bosilkovski M, Rodriguez-Morales AJ, Pappas G. The socio-ecology of zoonotic infections. Clin Microbiol Infect (2011) 17:336–42. doi: 10.1111/j.1469-0691.2010.03451.x
2. Bloom DE, Cadarette D. Infectious disease threats in the twenty-first century: Strengthening the global response. Front Immunol (2019) 10:549. doi: 10.3389/fimmu.2019.00549
3. Pronker ES, Weenen TC, Commandeur H, Claassen EHJHM, Osterhaus ADME. Risk in Vaccine Research and Development Quantified. PloS One (2013) 8:e57755. doi: 10.1371/journal.pone.0057755
4. Coalition for Epidemic Preparedness Innovations (CEPI). Coalition for Epidemic Preparedness Innovations (CEPI) Call for proposals (CfP) Topic: Platform technologies to enable rapid vaccine development for epidemic prone infections (2017). Available at: https://www.nodoka.co/apc-aa-files/73d01f29fa83c3ff16b52972bc6a4a3c/call-for-proposals.pdf (Accessed March 7, 2020).
5. CEPI announces three programmes to develop coronavirus vaccine. Available at: https://www.drugtargetreview.com/news/55371/cepi-announces-three-programmes-to-develop-coronavirus-vaccine/ (Accessed March 10, 2020).
6. CEPI partners with Imperial College to develop transformative rapid-response technology to create vaccines against emerging infectious diseases – CEPI. Available at: https://cepi.net/news_cepi/cepi-partners-with-imperial-college-to-develop-transformative-rapid-response-technology-to-create-vaccines-against-emerging-infectious-diseases/ (Accessed March 7, 2020).
7. CureVac and CEPI extend their Cooperation to Develop a Vaccine against Coronavirus nCoV-2019 | English. Available at: https://www.curevac.com/news/curevac-and-cepi-extend-their-cooperation-to-develop-a-vaccine-against-coronavirus-ncov-2019 (Accessed March 10, 2020).
8. Adalja AA, Watson M, Toner ES, Cicero A, Inglesby TV. The Characteristics of Pandemic Pathogens. (2018).
9. Maruggi G, Zhang C, Li J, Ulmer JB, Yu D. mRNA as a Transformative Technology for Vaccine Development toControl Infectious Diseases. Mol Ther (2019) 27:757–72. doi: 10.1016/j.ymthe.2019.01.020
10. Lindsay KE, Bhosle SM, Zurla C, Beyersdorf J, Rogers KA, Vanover D, et al. Visualization of early events in mRNA vaccine delivery in non-human primates via PET-CT and near-infrared imaging. Nat BioMed Eng (2019) 3:371–80. doi: 10.1038/s41551-019-0378-3
11. Pepini T, Pulichino A-M, Carsillo T, Carlson AL, Sari-Sarraf F, Ramsauer K, et al. Induction of an IFN-Mediated Antiviral Response by a Self-Amplifying RNA Vaccine: Implications for Vaccine Design. J Immunol (2017) 198:4012–24. doi: 10.4049/jimmunol.1601877
12. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines-a new era in vaccinology. Nat Rev Drug Discovery (2018) 17:261–79. doi: 10.1038/nrd.2017.243
13. Perri S, Greer CE, Thudium K, Doe B, Legg H, Liu H, et al. An Alphavirus Replicon Particle Chimera Derived from Venezuelan Equine Encephalitis and Sindbis Viruses Is a Potent Gene-Based Vaccine Delivery Vector. J Virol (2003) 77:10394–403. doi: 10.1128/jvi.77.19.10394-10403.2003
14. Vogel AB, Lambert L, Kinnear E, Busse D, Erbar S, Reuter KC, et al. Self-Amplifying RNA Vaccines Give Equivalent Protection against Influenza to mRNA Vaccines but at Much Lower Doses. Mol Ther (2018) 26:446–55. doi: 10.1016/j.ymthe.2017.11.017
15. Chakrabarti A, Jha BK, Silverman RH. New insights into the role of RNase L in innate immunity. J Interf Cytokine Res (2011) 31:49–57. doi: 10.1089/jir.2010.0120
16. Malone RW, Felgner PL, Verma IM. Cationic liposome-mediated RNA transfection. Proc Natl Acad Sci USA (1989) 86:6077–81. doi: 10.1073/pnas.86.16.6077
17. Holtkamp S, Kreiter S, Selmi A, Simon P, Koslowski M, Huber C, et al. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood (2006) 108:4009–17. doi: 10.1182/blood-2006-04-015024
18. Andries O, Mc Cafferty S, De Smedt SC, Weiss R, Sanders NN, Kitada T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J Control Release (2015) 217:337–44. doi: 10.1016/j.jconrel.2015.08.051
19. Kudla G, Lipinski L, Caffin F, Helwak A, Zylicz M. High guanine and cytosine content increases mRNA levels in mammalian cells. PloS Biol (2006) 4:0933–42. doi: 10.1371/journal.pbio.0040180
20. Richner JM, Himansu S, Dowd KA, Butler SL, Salazar V, Fox JM, et al. Modified mRNA Vaccines Protect against Zika Virus Infection.[Erratum appears in Cell. 2017 Mar 23;169(1):176; PMID: 28340344]. Cell (2017) 168:1114–25.e10. doi: 10.1016/j.cell.2017.02.017
21. Guan S, Rosenecker J. Nanotechnologies in delivery of mRNA therapeutics using nonviral vector-based delivery systems. Gene Ther (2017) 24:133–43. doi: 10.1038/gt.2017.5
22. Graham BS, Mascola JR, Fauci AS. Novel vaccine technologies: Essential components of an adequate response to emerging viral diseases. JAMA J Am Med Assoc (2018) 319:1431–2. doi: 10.1001/jama.2018.0345
23. Corbett KS, Edwards DK, Leist SR, Abiona OM, Boyoglu-Barnum S, Gillespie RA, et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature (2020) 586:1–8. doi: 10.1038/s41586-020-2622-0
24. Graham BS, Corbett KS. Prototype pathogen approach for pandemic preparedness: world on fire. J Clin Invest (2020) 130:3348–9. doi: 10.1172/JCI139601
25. Walters AA, Kinnear E, Shattock RJ, Mcdonald JU, Caproni LJ, Porter N, et al. Comparative analysis of enzymatically produced novel linear DNA constructs with plasmids for use as DNA vaccines. Gene Ther (2014) 21:645–52. doi: 10.1038/gt.2014.37
26. Kis Z, Shattock R, Shah N, Kontoravdi C. Emerging Technologies for Low-Cost, Rapid Vaccine Manufacture. Biotechnol J (2019) 14:1800376. doi: 10.1002/biot.201800376
27. Kis Z, Papathanasiou M, Calvo-Serrano R, Kontoravdi C, Shah N. A model-based quantification of the impact of new manufacturing technologies on developing country vaccine supply chain performance: A Kenyan case study. J Adv Manuf Process (2019) 1:e10025. doi: 10.1002/amp2.10025
28. Karikó K, Muramatsu H, Welsh FA, Ludwig J, Kato H, Akira S, et al. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol Ther (2008) 16:1833–40. doi: 10.1038/mt.2008.200
29. Rolls MM, Webster P, Balba NH, Rose JK. Novel infectious particles generated by expression of the vesicular stomatitis virus glycoprotein from a self-replicating RNA. Cell (1994) 79:497–506. doi: 10.1016/0092-8674(94)90258-5
30. Feldman RA, Fuhr R, Smolenov I, (Mick)Ribeiro A, Panther L, Watson M, et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine (2019) 37:3326–34. doi: 10.1016/j.vaccine.2019.04.074
31. Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, et al. An mRNA Vaccine against SARS-CoV-2 — Preliminary Report. N Engl J Med (2020) 383:1920–31. doi: 10.1056/nejmoa2022483
32. Sahin U, Muik A, Derhovanessian E, Vogler I, Kranz LM, Baum A, et al. Concurrent human antibody and TH1 type T-cell responses elicited by a COVID-19 RNA vaccine. medRxiv (2020) 586:2020.07.17.20140533. doi: 10.1101/2020.07.17.20140533
33. Acosta PL, Caballero MT, Polack FP. Brief History and Characterization of Enhanced Respiratory Syncytial Virus Disease. Clin Vaccine Immunol (2016) 23:189–95. doi: 10.1128/CVI.00609-15
34. Tseng C-T, Sbrana E, Iwata-Yoshikawa N, Newman PC, Garron T, Atmar RL, et al. Immunization with SARS Coronavirus Vaccines Leads to Pulmonary Immunopathology on Challenge with the SARS Virus. PloS One (2012) 7:e35421. doi: 10.1371/journal.pone.0035421
35. Agrawal AS, Tao X, Algaissi A, Garron T, Narayanan K, Peng BH, et al. Immunization with inactivated Middle East Respiratory Syndrome coronavirus vaccine leads to lung immunopathology on challenge with live virus. Hum Vaccines Immunother (2016) 12:2351–6. doi: 10.1080/21645515.2016.1177688
36. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H, et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight (2019) 4:e123158. doi: 10.1172/jci.insight.123158
37. Geall AJ, Verma A, Otten GR, Shaw CA, Hekele A, Banerjee K, et al. Nonviral delivery of self-amplifying RNA vaccines. Proc Natl Acad Sci USA (2012) 109:14604–9. doi: 10.1073/pnas.1209367109
38. Jankovic D, Feng CG. CD4+ T Cell Differentiation in Infection: Amendments to the Th1/Th2 Axiom. Front Immunol (2015) 6:198. doi: 10.3389/fimmu.2015.00198
39. Alberer M, Gnad-Vogt U, Hong HS, Mehr KT, Backert L, Finak G, et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: an open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet (2017) 390:1511–20. doi: 10.1016/S0140-6736(17)31665-3
40. Trombetta CM, Montomoli E. Influenza immunology evaluation and correlates of protection: a focus on vaccines. Expert Rev Vaccines (2016) 15:967–76. doi: 10.1586/14760584.2016.1164046
41. Long QX, Tang XJ, Shi QL, Li Q, Deng HJ, Yuan J, et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat Med (2020) 26:1–5. doi: 10.1038/s41591-020-0965-6
42. Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature (2020) 584:1–10. doi: 10.1038/s41586-020-2550-z
43. Phase 3 Clinical Trial of Investigational Vaccine for COVID-19 Begins | NIH: National Institute of Allergy and Infectious Diseases. Available at: https://www.niaid.nih.gov/news-events/phase-3-clinical-trial-investigational-vaccine-covid-19-begins (Accessed August 5, 2020).
44. Pfizer and BioNTech Choose Lead mRNA Vaccine Candidate Against COVID-19 and Commence Pivotal Phase 2/3 Global Study | BioNTech. Available at: https://investors.biontech.de/news-releases/news-release-details/pfizer-and-biontech-choose-lead-mrna-vaccine-candidate-against (Accessed August 5, 2020).
45. First volunteers receive Imperial COVID-19 vaccine | Imperial News | Imperial College London. Available at: https://www.imperial.ac.uk/news/198533/first-volunteers-receive-imperial-covid-19-vaccine/ (Accessed August 5, 2020).
46. Petsch B, Schnee M, Vogel AB, Lange E, Hoffmann B, Voss D, et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat Biotechnol (2012) 30:1210–6. doi: 10.1038/nbt.2436
47. Beissert T, Perkovic M, Vogel A, Erbar S, Walzer KC, Hempel T, et al. A Trans-amplifying RNA Vaccine Strategy for Induction of Potent Protective Immunity. Mol Ther (2019) 28:119–28. doi: 10.1016/j.ymthe.2019.09.009
48. Bahl K, Senn JJ, Yuzhakov O, Bulychev A, Brito LA, Hassett KJ, et al. Preclinical and Clinical Demonstration of Immunogenicity by mRNA Vaccines against H10N8 and H7N9 Influenza Viruses. Mol Ther (2017) 25:1316–27. doi: 10.1016/j.ymthe.2017.03.035
49. Corbett KS, Flynn B, Foulds KE, Francica JR, Boyoglu-Barnum S, Werner AP, et al. Evaluation of the mRNA-1273 Vaccine against SARS-CoV-2 in Nonhuman Primates. N Engl J Med (2020) 383:1544–55. doi: 10.1056/NEJMoa2024671
50. Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, et al. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature (2017) 543:248–51. doi: 10.1038/nature21428
51. Erasmus JH, Khandhar AP, Guderian J, Granger B, Archer J, Archer M, et al. A Nanostructured Lipid Carrier for Delivery of a Replicating Viral RNA Provides Single, Low-Dose Protection against Zika. Mol Ther (2018) 26(10):2507–22. doi: 10.1016/j.ymthe.2018.07.010
52. Meyer M, Huang E, Yuzhakov O, Ramanathan P, Ciaramella G, Bukreyev A. Modified mRNA-Based Vaccines Elicit Robust Immune Responses and Protect Guinea Pigs From Ebola Virus Disease. J Infect Dis J Infect Dis ® (2018) 451:451–6. doi: 10.1093/infdis/jix592
53. Chahal JS, Khan OF, Cooper CL, McPartlan JS, Tsosie JK, Tilley LD, et al. Dendrimer-RNA nanoparticles generate protective immunity against lethal ebola, H1N1 influenza, and Toxoplasma gondii challenges with a single dose. Proc Natl Acad Sci USA (2016) 113:E4133–42. doi: 10.1073/pnas.1600299113
54. Stitz L, Vogel A, Schnee M, Voss D, Rauch S, Mutzke T, et al. A thermostable messenger RNA based vaccine against rabies. PloS Negl Trop Dis (2017) 11:e0006108. doi: 10.1371/journal.pntd.0006108
55. VanBlargan LA, Himansu S, Foreman BM, Ebel GD, Pierson TC, Diamond MS. An mRNA Vaccine Protects Mice against Multiple Tick-Transmitted Flavivirus Infections. Cell Rep (2018) 25:3382–3392.e3. doi: 10.1016/j.celrep.2018.11.082
56. Lo MK, Spengler JR, Welch SR, Harmon JR, Coleman-Mccray JD, Scholte FEM, et al. Evaluation of a Single-Dose Nucleoside-Modified Messenger RNA Vaccine Encoding Hendra Virus-Soluble Glycoprotein Against Lethal Nipah virus Challenge in Syrian Hamsters. J Infect Dis (2019) 221:S493–8. doi: 10.1093/infdis/jiz553
57. Samsa MM, Dupuy LC, Beard CW, Six CM, Schmaljohn CS, Mason PW, et al. Self-Amplifying RNA Vaccines for Venezuelan Equine Encephalitis Virus Induce Robust Protective Immunogenicity in Mice. Mol Ther (2019) 27:850–65. doi: 10.1016/j.ymthe.2018.12.013
58. Mestas J, Hughes CCW. Of Mice and Not Men: Differences between Mouse and Human Immunology. J Immunol (2004) 172:2731–8. doi: 10.4049/jimmunol.172.5.2731
59. Lundberg AM, Drexler SK, Monaco C, Williams LM, Sacre SM, Feldmann M, et al. Key differences in TLR3/poly I:C signaling and cytokine induction by human primary cells: A phenomenon absent from murine cell systems. Blood (2007) 110:3245–52. doi: 10.1182/blood-2007-02-072934
60. Barchet W, Wimmenauer V, Schlee M, Hartmann G. Accessing the therapeutic potential of immunostimulatory nucleic acids. Curr Opin Immunol (2008) 20:389–95. doi: 10.1016/j.coi.2008.07.007
61. Thangavel RR, Bouvier NM. Animal models for influenza virus pathogenesis, transmission, and immunology. J Immunol Methods (2014) 410:60–79. doi: 10.1016/j.jim.2014.03.023
62. Kaukinen U, Lyytikäinen S, Mikkola S, Lönnberg H. The reactivity of phosphodiester bonds within linear single-stranded oligoribonucleotides in strongly dependent on the base sequence. Nucleic Acids Res (2002) 30:468–74. doi: 10.1093/nar/30.2.468
63. Moderna. mRNA-1273 Vaccine Against COVID-19 Phase 1 Interim Analysis of Older Adult Cohorts (ages 56-70 and 71+) (2020). Available at: https://investors.modernatx.com/static-files/1dc3deaf-4a1b-46f8-8aed-33bfd7b2c979 (Accessed August 28, 2020).
64. Mast J. Will supply chain demands freeze Pfizer and BioNTech out of a big chunk of the Covid-19 market? – Endpoints News, in: Endpoints News (2020). Available at: https://endpts.com/will-supply-chain-demands-freeze-pfizer-and-biontech-out-of-a-big-chunk-of-the-covid-19-market/ (Accessed August 28, 2020).
65. Zhang N-N, Li X-F, Deng Y-Q, Zhao H, Huang Y-J, Yang G, et al. A thermostable mRNA vaccine against COVID-19. Cell (2020) 182:1271–83.e16. doi: 10.1016/j.cell.2020.07.024
66. Lball R, Bajaj P, Whitehead KA. Achieving long-term stability of lipid nanoparticles: Examining the effect of pH, temperature, and lyophilization. Int J Nanomed (2017) 12:305–15. doi: 10.2147/IJN.S123062
67. Chen R, Zhang H, Yan J, Bryers JD. Scaffold-mediated delivery for non-viral mRNA vaccines. Gene Ther (2018) 25:556–67. doi: 10.1038/s41434-018-0040-9
68. Yan J, Chen R, Zhang H, Bryers JD. Injectable Biodegradable Chitosan-Alginate 3D Porous Gel Scaffold for mRNA Vaccine Delivery. Macromol Biosci (2019) 19:e1800242. doi: 10.1002/mabi.201800242
Keywords: mRNA vaccine, infectious disease, pandemics, outbreak, vaccine development, vaccine platform, self-amplifying RNA
Citation: Sandbrink JB and Shattock RJ (2020) RNA Vaccines: A Suitable Platform for Tackling Emerging Pandemics? Front. Immunol. 11:608460. doi: 10.3389/fimmu.2020.608460
Received: 20 September 2020; Accepted: 18 November 2020;
Published: 22 December 2020.
Edited by:
Anke Huckriede, University Medical Center Groningen, NetherlandsReviewed by:
Axel T. Lehrer, University of Hawaii at Manoa, United StatesCopyright © 2020 Sandbrink and Shattock. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jonas B. Sandbrink, am9uYXMuc2FuZGJyaW5rQGd0Yy5veC5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.