
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 16 December 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.606649
This article is part of the Research TopicSteroids and Secosteroids in the Modulation of Inflammation and ImmunityView all 12 articles
 Ya-Jun He1,2†
Ya-Jun He1,2† Ji-Qian Xu1,2†Miao-Miao Sun1,2Xiang-Zhi Fang1,2Zhe-Kang Peng1,2Shang-Wen Pan1,2Ting Zhou1,2Ya-Xin Wang1,2
Ji-Qian Xu1,2†Miao-Miao Sun1,2Xiang-Zhi Fang1,2Zhe-Kang Peng1,2Shang-Wen Pan1,2Ting Zhou1,2Ya-Xin Wang1,2 You Shang1,2*
You Shang1,2*Sepsis is a clinical syndrome that resulting from a dysregulated inflammatory response to infection that leads to organ dysfunction. The dysregulated inflammatory response transitions from a hyper-inflammatory phase to a hypo-inflammatory or immunosuppressive phase. Currently, no phase-specific molecular-based therapies are available for monitoring the complex immune response and treating sepsis due to individual variations in the timing and overlap of the dysregulated immune response in most patients. Glucocorticoid-induced leucine zipper (GILZ), is broadly present in multiple tissues and circumvent glucocorticoid resistance (GCR) or unwanted side effects. Recently, the characteristics of GILZ downregulation during acute hyperinflammation and GILZ upregulation during the immunosuppressive phase in various inflammatory diseases have been well documented, and the protective effects of GILZ have gained attention in the field of sepsis. However, whether GILZ could be a promising candidate biomarker for monitoring and treating septic patients remains unknown. Here, we discuss the effect of GILZ in sepsis and sepsis-induced immunosuppression.
Sepsis is a complex disease that causes life-threatening organ dysfunction due to uncontrolled infection (1). The global burden of sepsis is estimated to be 30 million patient episodes, with a mortality rate approaching 30%–50% annually. Notably, most data are derived from the developed countries, and the true global burden of sepsis is much greater than suggested by these figures (2, 3). The World Health Organization (WHO) rendered sepsis a global health priority in 2017 to improve prevention, diagnosis, and management. Concomitant with early hemodynamic and respiratory support and appropriate antibiotic administration, corticosteroids have been widely used for the management of sepsis as adjuvant therapy to control the immune response to invading pathogens.
Glucocorticoids (GCs) constitute are a class of corticosteroids widely used clinically as anti-inflammatory and anti-shock drugs. The therapeutic glucocorticoids include hydrocortisone, prednisolone and dexamethasone. Several studies and meta-analyses have indicated that low-dose hydrocortisone reduced mortality in patients with septic shock in the intensive care unit (ICU). However, the recent adjunctive corticosteroid treatment in critically ill patients with septic shock (ADRENAL) trial did not show a significantly reduced risk of mortality (4). Mortality is likely associated with the timing of treatment and multiple side effects due to the wide spectrum of effects induced by glucocorticoids (GCs) in addition to the complicated immunopathology of sepsis (5). GCs bind the GC receptor (GR), which belongs to the nuclear receptor superfamily of transcription factors to exert their broad physiological and therapeutic effects.
Glucocorticoid-induced leucine zipper (GILZ or TSC22D3), which is a GC-inducible molecule, has emerged as a GC mediator due to its anti-inflammatory effects and theoretically lacks the side effects of GCs (6). Recently, the potential effect of GILZ in sepsis has gained attention, and GILZ has been recognized as a more promising therapy for polymicrobial sepsis than GCs. Hence, notably, that GILZ may be a checkpoint or even a biomarker of innovative therapies for sepsis-mediated immune responses and is regarded as an actionable target. Here, we review the specific role of GILZ in sepsis.
Sepsis induces a complex immune response and this excessive pro-inflammatory response easily elicits the dysregulation of signaling pathways, resulting in tissue damage and organ dysfunction, and induces an immunosuppressive environment, which could increase susceptibility to secondary infections associated with poor outcomes and subsequent mortality (7), although immunosuppressive responses occur simultaneously upon the initiation of innate immune response (8–10).
Increasing evidence suggests that immune cell apoptosis, autophagy, broad metabolic defects, endotoxin tolerance, T cell exhaustion and epigenetic regulation are all contributors, and aerobic glycolysis is crucial for maintaining the function of the immune system (11). For instance, immune cells, including monocytes and T cells, undergo metabolic reprogramming and shift from oxidative phosphorylation (OXPHOS) to aerobic glycolysis when the host is infected (11). Both glycolysis and oxidative metabolism are apparently defective in leukocytes during sepsis-induced immunosuppression. The phosphorylation of extracellular signal regulated kinase (ERK) promotes B-cell death in sepsis (12). The delayed state of neutrophil apoptosis is the most significant change in innate immunity and contributes to the deficits in of bacterial eradication and ongoing continuous dysfunction (13). In addition, a deficit in the capacity to activate nuclear factor-κB (NF-κB) or deacetylation of p65 is closely related to the macrophage reprogramming (14, 15) and sepsis-induced immunosuppression (16).
Identifying treatment and diagnosis guidelines is essential for survival during sepsis. A rich profile of transcriptional shifts occurs in leukocytes within a few hours of exposure to endotoxins (17). Patients with multiple traumas exhibit an immunosuppressed state with large quantities of danger-associated molecular patterns shortly after trauma (18). Similarly, a reduction HLA-DR in monocytes occurs early and is thought to be a marker of an altered immune state (19). Multiple studies exploring sepsis-associated survival also focused on this immunopathology. For instance, studies have focused on antiapoptotic strategies that could reduce mortality in septic mice (20), and immunomodulation with thymosin α1, IFN-γ, IL-7, GM-CSF, interferon-g and the immune checkpoint inhibitor PD-1 in the clinic (11). However, the timing of treatment and side effects associated with suppressing excessive inflammation or enhancing host immunity have not been fully elucidated. Consequently, more timely, suitable and precise therapy is necessary to reduce cell and tissue damage in sepsis.
In 1976, William Schumer’s study showed that septic shock patients could benefit from glucocorticoid drugs, and thus, GCs have attracted much interest in the treatment of sepsis (21). Glucocorticoids represent a type of steroid hormones secreted by the renicapsule that play key roles in the regulation of reproduction, metabolism, and immunization by binding the GR. To date, more than 37 randomized clinical trials have investigated the treatment effect of steroids in sepsis. While steroid administration reverses shock in some sepsis patients, which patients could benefit from this treatment remains unclear (22, 23). The European Society of Intensive Care Medicine and the Society of Critical Care Medicine suggest that some benefit of using corticosteroids in sepsis occurs only if shock is present (24). Recently, the APPROCHSS trial involving 1241 patients evaluated the effect of hydrocortisone plus fludrocortisone therapy and revealed a lower all-cause 90-day mortality and higher vasopressor-free days in the hydrocortisone plus fludrocortisone group than the placebo group (25). However, the ADRENAL trial, which included 3800 patients, showed that septic patients who were treated with low-dose hydrocortisone did not exhibit reductions in 90-day mortality compared with the patients in the placebo group (4). Given the number of cases included, the difference of these two large-scale trials is likely associated with their broad physiological molecular mechanisms of GCs and sepsis.
Actually, sepsis also involves in neuroendocrine mechanisms (26). The neuroendocrine system is triggered once the body is infected or traumatized to restore dynamic balance and fight noxious stimulation, thus to promote tolerance (27). Cortisol, which is a product of neuroendocrine signaling and a glucocorticoid, is the executor of neuroendocrine responses. The high level of plasma cortisol concentrations may be associated with the severity of sepsis because cortisol is not only an essential response for survival but also mediates endotoxin tolerance (28, 29). Furthermore, the molecules that mediates cortisol clearance including the corticosteroid-binding globulin (CBG), cortisol carrier albumin, A-ring reductases, and 11b-hydroxysteroid dehydrogenase type 2(11β-HSD2), are reduced during in early phase of sepsis (30–32). Notably, when was inhibited by cortisol generation with hypophysectomy, the mortality of lipopolysaccharide (LPS)-induced septic shock was significant increased (27). Thus, functioning endogenous GCs are very important for the regulation of the immune mechanism of sepsis.
However, inadequate cellular corticosteroid activity has been described as critical illness-related corticosteroid insufficiency (CIRCI) and manifests as insufficient glucocorticoid receptor (GR)-mediated downregulation of proinflammatory transcription factors (33). The following three major pathophysiological theories associated with CIRCI: dysregulation of the hypothalamic–pituitary–adrenal (HPA) axis, altered cortisol metabolism (33) and glucocorticoid resistance (GCR), which limits the activity of endogenous GCs’ and GCs’ therapeutic effects (34) (Figure 1).
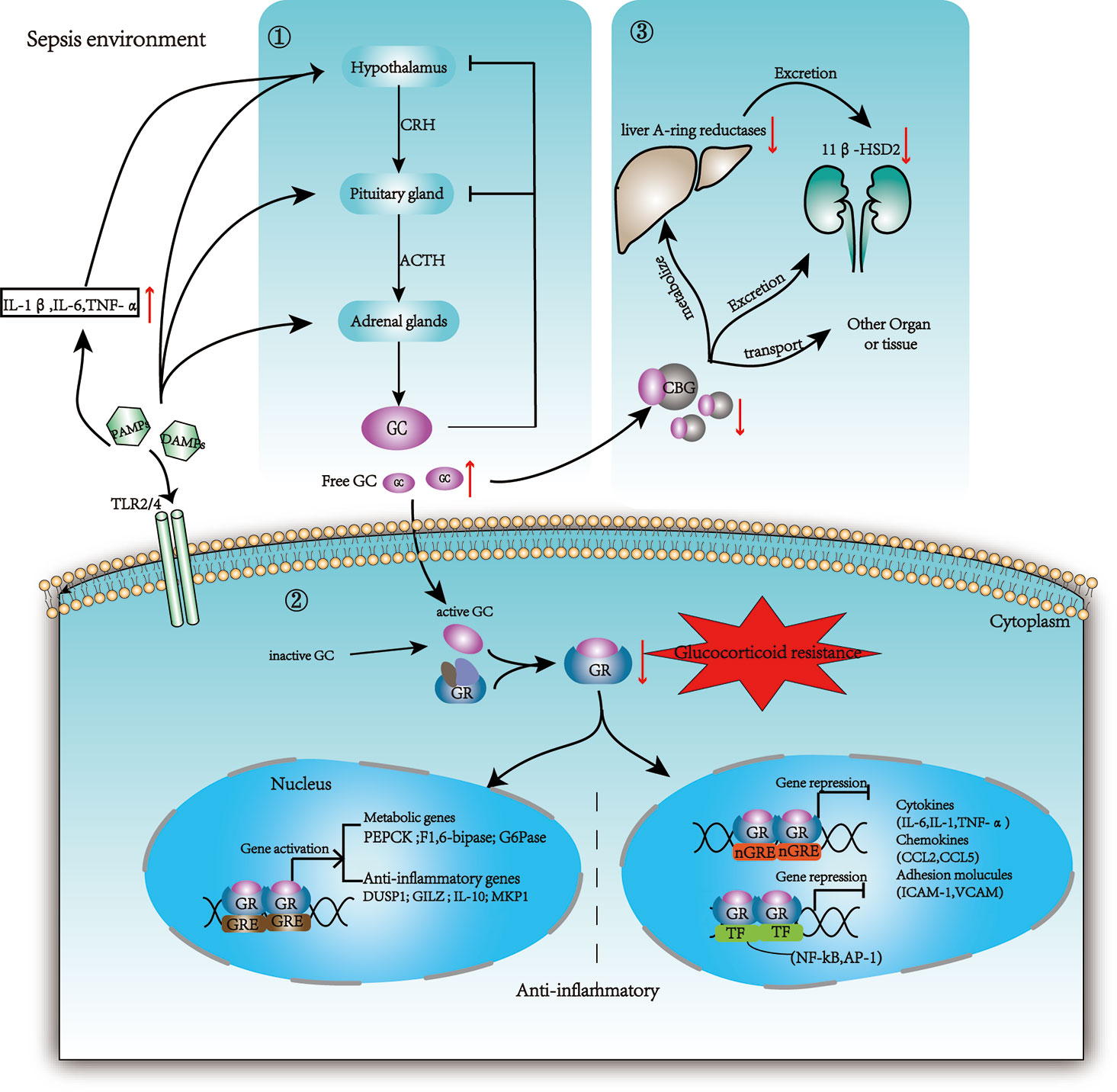
Figure 1 Changes in glucocorticoid production, metabolism, and regulation in sepsis. ① The HPA axis is activated by stress (both physically and mentally), tissue damage, and infection. The paraventricular nucleus of the hypothalamus secretes CRH, and the anterior pituitary gland secretes ACTH or corticotropin; then GC is secreted by the adrenal cortex. Cytokines such as IL-1β, TNF-α, and IL-6 can be projected in the hypothalamus through neuroafferent projections. DAMPs and PAMPs can also directly stimulate adrenocortical cells with toll receptors (TLR), leading to the synthesis of ACTH dependent cortisol. Circulating inflammatory mediators break the brain barrier and act on the hypothalamus. GC exerts negative feedback on both CRH and ACTH production when GC exceeds the threshold. ② Then 5% GC that is free and activated binds the GR and enters the nucleus to influence gene expression. The inactive GC could be reactivated by 11b-HSD1. GR binds promoters to promote metabolic genes and anti-inflammation genes. Additionally, GR binds negative GRE elements (nGRE) on DNA or some transcription factors (TF) to inhibit the expression of target genes. ③75% GC bound to CBG is transported to various tissue. During sepsis, the host protein production is reduced and the CBG in the inflammatory site is cleaved by neutrophil elastases. Cortisol is cleared mainly through A-ring reductases in the liver and through 11β-HSD2 in the kidneys. These enzymes are limited in sepsis resulting in reduced clearance of GC. PAMPs: pathogen-associated molecular patterns; DAMPs: damage-associated molecular patterns; HPA: hypothalamic–pituitary–adrenal axis; CRH: corticotropin-releasing hormone; ACTH: adrenocorticotropic hormone; CBG: corticosteroid-binding globulin; 11β-HSD2: 11b-hydroxysteroid dehydrogenase type 2; 11b-HSD1: 11b-hydroxysteroid dehydrogenase type 1.
The GR is encoded by a single genetic locus, but alternative splicing of the gene product generates the following four distinct messenger RNAs (mRNAs): GRα, GRβ, GRγ, and GR-A. Each of these intracytoplasmic GRα subtypes can bind GCs and, when in the dimer form, can activate gene expression. The GRα protein normally resides in the cytoplasm, and GRα isoforms directly bind GC response elements (GREs) to induce the transcription of multiple genes, including IL-10, IL-1 receptor antagonists, and GILZ (35). GRα expression significantly decreases in patients suffering sepsis, while GRβ is upregulated and cannot bind GCs in septic patients (36). GRβ located in the nucleus could be induced by several proinflammatory cytokines, such as IL-2, IL-4, IL-17A, IL-17F, IL-23, and TNF-α (37, 38),and is thought to be a negative regulator of GC activity. The overexpression of GRβ inhibits GRα-mediated gene transcription (39, 40). Thus, clearly, high GRβ levels seem to be associated with GCR in sepsis.
There are other forms of GRs, leading to different variants of GR proteins in addition to GRα and GRβ. GR isoforms and associated subtypes within organs may explain sepsis-induced alterations in GC responses, revealing different clinical responses in septic patients (41). Investigators have identified 27 splice variants of the GR gene and hundreds of single nucleotide polymorphisms (SNPs), insertions and deletions, which could lead to different variants of GR proteins (42). Recently, ANP32E has been shown to correlate with GCR. This protein has been linked to the exchange of H2A. z histone and promotes GR-induced transcription (43). The mechanisms of GC-mediated hypo-responsiveness are heterogeneous due to the various cell types and cytokines involved, and further studies of GR biology may be an important step in promoting GC-based therapies. Hence, supplementing active GCs and activating downstream target molecules are critical for this treatment. Consequently, the identification of a potential treatment that can replace GCs and mediate effective anti-inflammatory effects and endotoxin tolerance without causing GC-associated adverse effects has become the main focus of current studies.
Glucocorticoids function at physiological and pharmacological levels and are mediated by the GR. Following glucocorticoid exposure, the GC/GR complex translocates into the nucleus and directly binds to glucocorticoid response elements (GREs) to promote downstream transcription factors, including GILZ (44). In turn, GILZ binds NF-κB and prevents its nuclear translocation to elicit anti-inflammatory effects. GILZ can also directly bind c-Jun and c-Fos, which are two constituents of AP-1, to inhibit their transcriptional activity and gene expression of proinflammatory molecules (Figure 2). Additionally, GILZ interacts directly with Ras and Raf, thereby inhibiting the downstream activation of mitogen-activated protein kinase 1 (MAPK1), to mediate antiproliferative effects (45, 46).
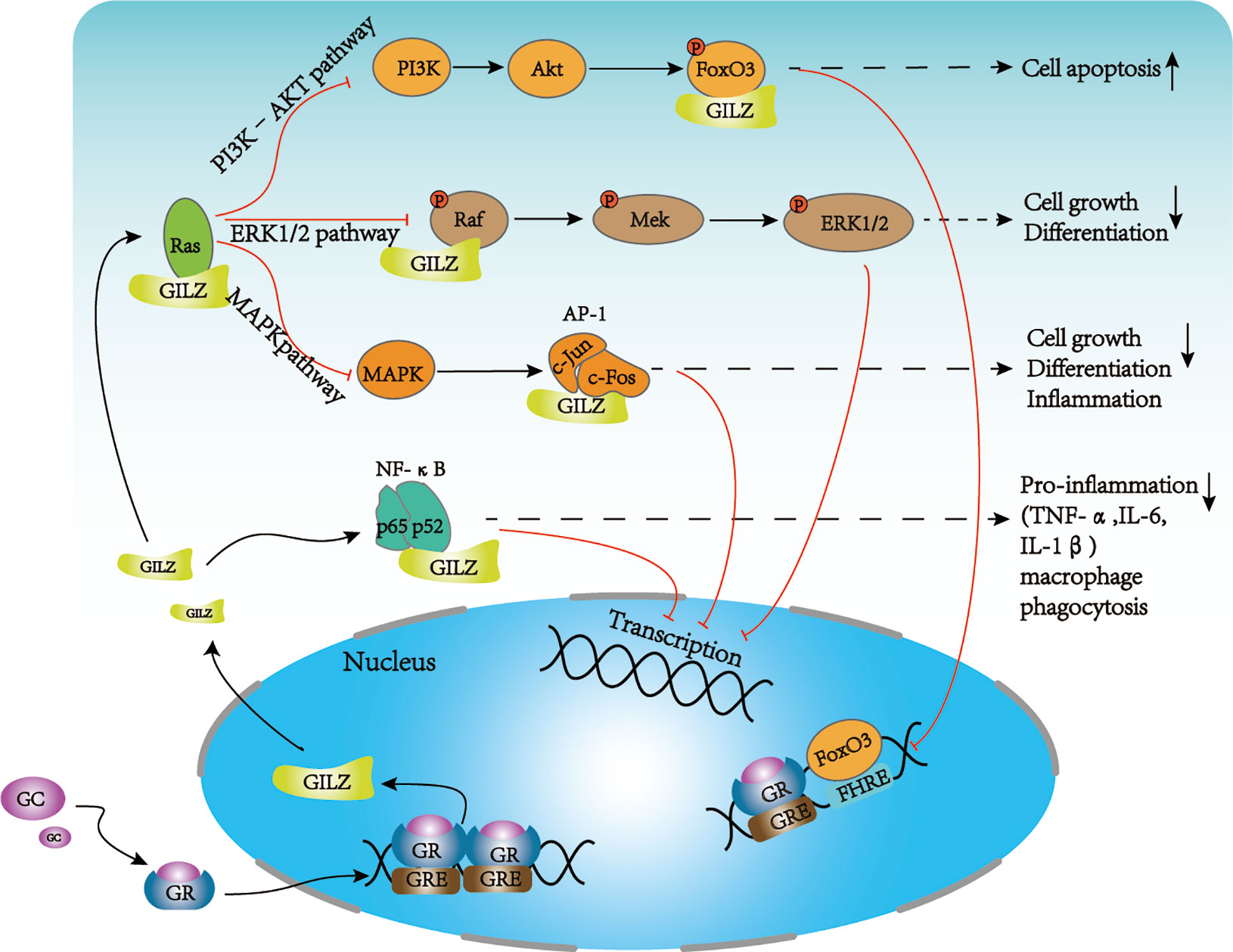
Figure 2 Major signaling pathways of GILZ. GILZ can regulate cell activation, apoptosis, proliferation, and inflammation mainly through several signaling pathways. GILZ directly binds p65 and p52 to inhibit NF-κB signaling to reduce the production of proinflammatory factors and macrophage phagocytosis. GILZ can directly bind Ras and inhibit downstream pathways. (1) inhibits the PI3K–Akt pathway to regulate apoptosis and cell survival; GILZ inhibits FOXO3A-mediated transcription, such as the pro-apoptotic protein, Bim. (2) The direct binding of GILZ to Ras and Raf leads to reduced activation of MEK, ERK, and MAPK, which inhibits cell growth and proliferation. (3) GILZ can directly bind c-Fos and c-Jun to inhibit AP-1 signaling, which prevents cell growth, cell differentiation, and inflammation. NF-κB, nuclear factor κB; AP-1, activator protein 1; FoxO3, forkhead box protein O3; ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; PI3K, phosphoinositide 3-kinase; AKT: Protein kinase B (PKB), also known as Akt.
GILZ, which is widely expressed in various human and mouse organs and is most highly expressed in the lungs (47, 48), was originally identified in 1997 (49). GILZ contains the following three main regions: an N-terminal domain (1–75 amino acids) with a tuberous sclerosis complex (TSC) domain, a leucine zipper (76–97 amino acids) that largely mediates the homodimerization of GILZ, and a proline-rich C-terminal domain (98–137 amino acids) and at least three typical GREs (50) at transcriptional initiation site. GILZ mediates the endogenous and exogenous anti-inflammatory and immunosuppressive effects of GCs in various types of cells, including lymphoid cells, in which it may regulate the activation and apoptosis of cells, which is an important pathophysiological characteristic of the immunopathology of sepsis (6, 51).
GILZ inhibits T lymphocyte activation, apoptosis, and cell proliferation either directly or through antigen-presenting cells (APCs) (52, 53), and inhibit IL-2 withdrawal-induced apoptosis by inhibiting Forkhead Box O3 (FoxO3) transcriptional activity and the proapoptotic gene Bim (54). GILZ also preventsTh-1 and promotes Th-2 differentiation by inhibiting NF-κB activation and nuclear translocation. Transgenic GILZ-overexpressing (GILZ-Tg) mice are less susceptible to Th-1-mediated experimental dinitrobenzene sulfonic acid- (DNBS-) colitis and spinal cord injury. Regulatory T Cells (Tregs) constitute are a subpopulation of T cells that accumulates in the bone marrow and can modulate the immune system, maintain tolerance to self-antigens and control autoimmune disorders. GILZ expression in bone marrow-derived mesenchymal stem cells (BMSCs) and DCs exerts anti-inflammatory effects depending on IL-10-producing Treg and activity. Additionally, GILZ promotes Treg differentiation by activating TGF-β signaling, which is a typical anti-inflammatory factor (55). However, GILZ-KO mice did not have worse collagen-induced arthritis (CIA) compared with WT mice, although the GILZ-KO mice displayed higher T cell proliferation. Furthermore, replenishing GILZ reduces inflammation but does not affect T cell proliferation. Consequently, as a key modulator of Tregs, GILZ plays an anti-inflammatory role in most experimental settings, but the actual role of GILZ in T cells is complex and requires more exploration.
GILZ exerts suppressive effects on B cells by inhibiting cell activation, proliferation, differentiation, apoptosis and IgG production (56). The accumulation of B cell precursors in the bone marrow and peripheral lymphoid organs with elevated Bcl-2 and NF-κB activity has been shown in GILZ-deficient mice (57). The deletion of GILZ increased IFN-γ and AP-1 activity in B cells in a colitis mouse model (58). Furthermore, the above effects were all reversed by the compensation of the GILZ protein, demonstrating the potential therapeutic role of GILZ in regulating B cell-dependent diseases (58).
GILZ not only plays an important role in adaptive immunity, but also inhibits the activity of the innate immune system. In DCs, GILZ and GCs are critical for balancing the anti-inflammatory response and tolerance phenotypes (59, 60), which are closely related to sepsis-induced immunosuppression. GILZ could mediate the downregulation of MHC class II molecules, costimulatory factors, and Treg cell generation (61). Some pro-inflammatory factors including IL-1α, IL-1β, IL-6, and IL-23, were strongly increased in GILZ-KO bone marrow-derived DCs (BMDCs) following upon TLR4 and TLR7 stimulation (62, 63). GILZ prevents DCs from activating the antigen-specific T lymphocyte response. GILZ-/- DCs increased IFN-γ and IL-17 secretion in CD4+ T cells and CD8+ T cells (64, 65). Moreover, GILZ could regulate antigen capture and cross-presentation by DCs and limits antigen internalization in DCs from GILZ-/- mice (62, 66). Dexamethasone enhanced antigen capture by DCs (67, 68), which does not different from the roles of GILZ. Additionally, GILZ can inhibit DC maturation, which could increase the production of IL-10 and promote the development of tolerant DC phenotypes (65, 69).
The GILZ protein and mRNA levels were obviously decreased in alveolar macrophages (AMs) and THP-1 cells exposed to LPS (6, 46, 70). GILZ could decrease macrophage sensitivity to LPS and proinflammatory cytokines expression (71). GILZ deficiency enhances NF-κB pathway-mediated macrophage phagocytosis. Consistent with this result, GILZ-KO macrophages were observed to have increased NO production (45). Interestingly, GC exerts the opposite effect to promote macrophage phagocytosis (72).
GILZ also inhibits neutrophil migration to inflammatory sites via annexin A1 (73) and alleviates the proinflammatory response by inhibiting reactive oxygen species (ROS) generation and the accumulation of leukocytes at the site of inflammation or inducing neutrophil apoptosis (57, 74). Neutrophils derived from GILZ-KO mice showed a stronger capacity to clear pathogens in a candida albicans intraperitoneal infection model. Although the role of GILZ is extensive, it has not been deeply studied in sepsis patients. Furthermore, few studies investigated about the immune response associated with GILZ in myeloid-derived suppressor cells, which perform potent immunosuppressive functions and act on both innate and adaptive immunity.
GILZ has been studied in septic patients and CLP models (Table 1). During the hyperinflammatory stage of sepsis, GILZ regulation is contrary to inflammatory release. GILZ mRNA was reduced by 50% in peripheral polymorphonuclear cells from critically ill patients (75, 80). In an experimental model of CLP, the GILZ expression level was also downregulated in both blood cells and the liver after two hours of the CLP procedure (75). Human alveolar macrophages (AMs) were treated with LPS, upon TLR4 activation, both the mRNA and protein levels of GILZ rapidly decreased, while the TNF and IL-6 mRNA levels increased (70, 77).
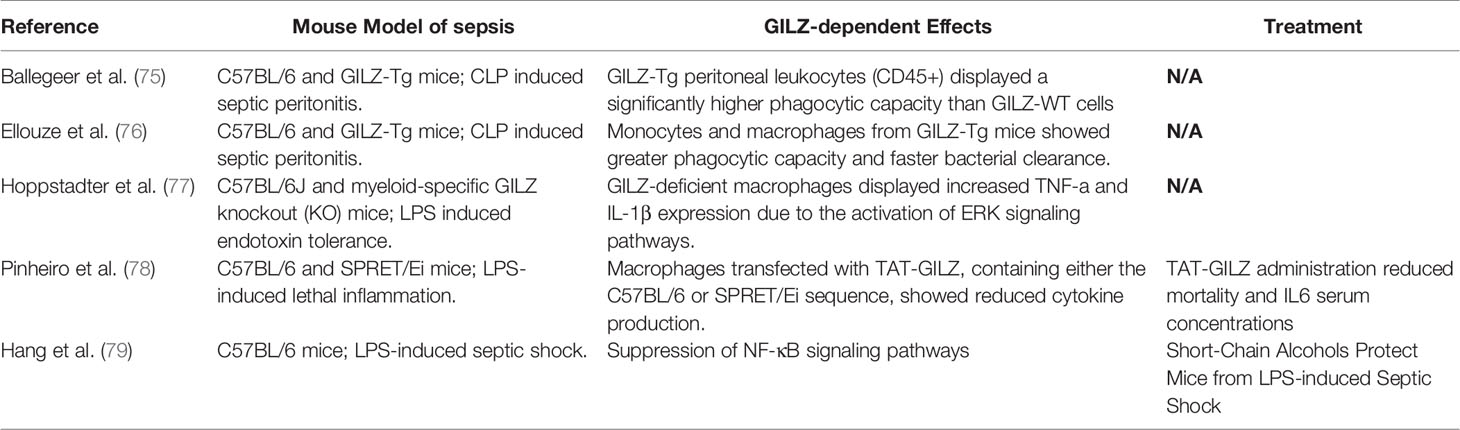
Table 1 A Summary of Preclinical Evidence that revealing the Involvement of glucocorticoid-induced leucine zipper (GILZ) in the pathogenesis of sepsis.
The inbred mouse strain SPRET/Ei has been shown to exhibit marked resistance to LPS that depending at least partially on the GR levels and GILZ mRNA was higher than that in C57BL/6 mice (78, 81). In addition, the TAT-GILZ fusion protein, which is a synthetic fusion construct containing the TAT peptide followed by the GILZ cDNA sequence from C57BL/6 and SPRET/Ei, has been shown to reduce mortality [58]. The recombinant GILZ protein was first demonstrated to attenuate colon inflammation in DNBS-induced colitis in 2009 (82). The administration of the TAT-GILZ fusion protein could reduce tissue edema and increase perfusion of the ischemic/reperfused kidney in mice with acute kidney injury (AKI), and improve the disruption of mitochondrial membrane potential and cell death in vitro (83, 84). GILZ-Tg mice had a lower mortality and blood bacterial load than GILZ-WT mice in a CLP model (75). Peritoneal leukocytes in GILZ-Tg mice are more likely have a higher phagocytic capacity, which may be mediated in inflammatory resolution due to neutrophils apoptosis or M2−type macrophages polarization (74, 85, 86) In addition, mice with GILZ overexpress mononuclear macrophages (M/M) exhibited an increased survival rate and reduced levels of plasma inflammatory cytokines and blood bacterial load (76). It is likely that overexpression of GILZ elicits an increase in macrophages phagocytosis. A recent study found that the management of short-chain alcohols protects mice from LPS septic shock by increasing peripheral blood GILZ in a dose-dependent manner and suppressing IκB phosphorylation, which is a mechanisms that promoting immune tolerance (16, 79).
Although the upregulation of GILZ can alleviate inflammatory storms and improve survival during the early stage of sepsis, but not endotoxin tolerance (77), the downregulation of GILZ expression independent of GR activation abrogated LPS tolerance and increased responsiveness to LPS by enhancing ERK activity and rescuing MAPK signals, which was independent of GR activation (77). Meanwhile, Wang et al. (87) found that the elevated level of GCs or GILZ is related to late stage inflammation in sepsis, and artesunate can inhibit the upregulation of GILZ mRNA and increase bacterial clearance of hydrocortisone-induced immunosuppression peritoneal macrophages.
Immune phenotypes of infection, especially in sepsis, have many similarities with those in cancer (88). GILZ is a crucial immunosuppressive molecule mediated by GCs in the immunosuppressive tumor environment. Recently, GILZ was shown to be highly expressed in the immunosuppressive tumor microenvironment, which was sufficient to abolish the therapeutic control of tumors (89). DC-specific GILZ deletion or GR antagonists could reverse these negative effects. An online survival analysis suggested that the GILZ level was negatively correlated with prognosis in lung cancer patients (90, 91). These seemingly conflicting data may indicate the cell-specific nature and different stages of the regulation of GILZ in sepsis, which is similar to GCs. As mentioned above, GILZ may play a pivotal role in the immune tolerance of sepsis, although only a few related researches. Of note is, GCs are responsible for osteoporosis though osteoblast formation after long-term GC treatment. In contrast, GILZ increases osteogenic differentiation and inhibits adipocyte formation in mesenchymal stem cells (MSCs) (92). Consistently, cell-specific GILZ overexpression in osteoblasts induced high bone mass and increased bone formation and the osteoblast number (93). Whether GILZ exerts the opposite effect on other GC side effects remains unclear, although the latest published data indicate that GILZ is a critical mediator leading to statin-induced myopathy (90).
Collectively, the potential significance of GILZ in the potential diagnosis, treatment, and prediction of sepsis is worth discussing. To date, few studies investigated the mechanism and therapeutic applications of GILZ in inflammatory diseases and sepsis, and more studies are needed to determine its efficacy and safety.
Sepsis and septic shock are both severe diseases that should be treated and resuscitated immediately. Evidence increasingly suggests that the early treatment of sepsis increases the chances of survival, and no study has shown that treatment works better when applied later (94). Although the immune system changes as sepsis progresses, GILZ expression is stable during the late stage of inflammation, especially during immunosuppression, which is probably due to GILZ acting as a downstream molecule and regulation by endogenous cortisol in vivo. Hence, GILZ might become a potential monitoring molecule under the regulation of endogenous glucocorticoids or guide treatment with glucocorticoids. Currently, studies are exploring the key role of GILZ in sepsis and whether it can be a critical marker, such as the Rapid Recognition of Corticosteroid Resistant or Sensitive Sepsis (RECORDS) trial (NCT04280497) (95).
According to the current study, GILZ overexpression mice or using GILZ peptides all simulated the effect of early anti-inflammatory agents in acute sepsis. Recombinant GILZ, which couples GILZ to membrane-penetrating sequences, is an attractive prospect, although further studies are needed to demonstrate the feasibility and safety of GILZ in clinical setting. Alternatively, the targeted activation of GILZ expression using the CRISPR/dCas9 activator complex specifically in macrophages could be used (96). It should be noted that a peptide targeting the C-terminal region of GILZ 115-137 aa in the mouse GILZ sequence has demonstrated a potential therapeutic effect in experimental autoimmune encephalitis (EAE) (97), which suggesting that diverse regions of GILZ can bind different partner proteins. It may be necessary to further determine the optional GILZ peptide that binds specific targets.
Sepsis is a syndrome associated with an immune system disorder in response to infection. Currently, the mechanism underlying sepsis development is unclear, further limiting treatment. Clinical studies have shown that the earlier sepsis is resolved, the more likely the patient is to survive. GILZ, which is downstream of the GR, has powerful anti-inflammatory effects similar to GCs and might circumvent GCR in sepsis. Proof-of-concept studies have shown that GILZ has significant therapeutic effects in CLP-induced septic models. Additionally, cell-specific targeting of GILZ may circumvent side effects, which could be of particular interest in sepsis models or other experimental models. Cortisol is essential for inflammatory responses, and endogenous and exogenous GCs induce GILZ to change dynamically. Detection of GILZ expression over time can guide the use of GCs and evaluate the timeline of sepsis, which is of great importance for the treatment of sepsis. As a potential alternative treatment to GCs, further research concerning the role of GILZ in sepsis is needed to identify its powerful regulatory role and molecular mechanisms.
Y-JH, J-QX, and YS designed the review. Y-JH and J-QX wrote the manuscript with supervision of YS. All authors contributed to the article and approved the submitted version.
This study was supported by National Natural Science Foundation of China (No. 82002026, 81971818, and 81772047).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to all authors who could not be cited due to space constraints.
1. Cecconi M, Evans L, Levy M, Rhodes A. Sepsis and septic shock. Lancet (Lond Engl) (2018) 392(10141):75–87. doi: 10.1016/S0140-6736(18)30696-2
2. Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, et al. Global, regional, and national sepsis incidence and mortality, 1990-2017: analysis for the Global Burden of Disease Study. Lancet (2020) 395(10219):200–11. doi: 10.1016/S0140-6736(19)32989-7
3. Reinhart K, Daniels R, Kissoon N, Machado FR, Schachter RD, Finfer S. Recognizing Sepsis as a Global Health Priority - A WHO Resolution. N Engl J Med (2017) 377(5):414–7. doi: 10.1056/NEJMp1707170
4. Venkatesh B, Finfer S, Cohen J, Rajbhandari D, Arabi Y, Bellomo R, et al. Adjunctive Glucocorticoid Therapy in Patients with Septic Shock. N Engl J Med (2018) 378(9):797–808. doi: 10.1056/NEJMoa1705835
5. Seymour CW, Gesten F, Prescott HC, Friedrich ME, Iwashyna TJ, Phillips GS, et al. Time to Treatment and Mortality during Mandated Emergency Care for Sepsis. N Engl J Med (2017) 376(23):2235–44. doi: 10.1056/NEJMoa1703058
6. Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J (2009) 23(11):3649–58. doi: 10.1096/fj.09-134684
7. Ding R, Meng Y, Ma X. The Central Role of the Inflammatory Response in Understanding the Heterogeneity of Sepsis-3. BioMed Res Int (2018) 2018:5086516. doi: 10.1155/2018/5086516
8. Venet F, Monneret G. Advances in the understanding and treatment of sepsis-induced immunosuppression. Nat Rev Nephrol (2018) 14(2):121–37. doi: 10.1038/nrneph.2017.165
9. Maslove DM, Wong HR. Gene expression profiling in sepsis: timing, tissue, and translational considerations. Trends Mol Med (2014) 20(4):204–13. doi: 10.1016/j.molmed.2014.01.006
10. Tang BM, Huang SJ, McLean AS. Genome-wide transcription profiling of human sepsis: a systematic review. Crit Care (2010) 14(6):R237. doi: 10.1186/cc9392
11. Cheng SC, Scicluna BP, Arts RJ, Gresnigt MS, Lachmandas E, Giamarellos-Bourboulis EJ, et al. Broad defects in the energy metabolism of leukocytes underlie immunoparalysis in sepsis. Nat Immunol (2016) 17(4):406–13. doi: 10.1038/ni.3398
12. Shankar-Hari M, Fear D, Lavender P, Mare T, Beale R, Swanson C, et al. Activation-Associated Accelerated Apoptosis of Memory B Cells in Critically Ill Patients With Sepsis. Crit Care Med (2017) 45(5):875–82. doi: 10.1097/CCM.0000000000002380
13. Hotchkiss RS, Nicholson DW. Apoptosis and caspases regulate death and inflammation in sepsis. Nat Rev Immunol (2006) 6(11):813–22. doi: 10.1038/nri1943
14. Rackov G, Hernandez-Jimenez E, Shokri R, Carmona-Rodriguez L, Manes S, Alvarez-Mon M, et al. p21 mediates macrophage reprogramming through regulation of p50-p50 NF-kappaB and IFN-beta. J Clin Invest (2016) 126(8):3089–103. doi: 10.1172/JCI83404
15. Qin K, Han C, Zhang H, Li T, Li N, Cao X. NAD + dependent deacetylase Sirtuin 5 rescues the innate inflammatory response of endotoxin tolerant macrophages by promoting acetylation of p65. J Autoimmun (2017) 81:120–9. doi: 10.1016/j.jaut.2017.04.006
16. Hoogendijk AJ, Garcia-Laorden MI, van Vught LA, Wiewel MA, Belkasim-Bohoudi H, Duitman J, et al. Sepsis Patients Display a Reduced Capacity to Activate Nuclear Factor-kappaB in Multiple Cell Types. Crit Care Med (2017) 45(5):e524–31. doi: 10.1097/CCM.0000000000002294
17. Talwar S, Munson PJ, Barb J, Fiuza C, Cintron AP, Logun C, et al. Gene expression profiles of peripheral blood leukocytes after endotoxin challenge in humans. Physiol Genomics (2006) 25(2):203–15. doi: 10.1152/physiolgenomics.00192.2005
18. Timmermans K, Kox M, Vaneker M, van den Berg M, John A, van Laarhoven A, et al. Plasma levels of danger-associated molecular patterns are associated with immune suppression in trauma patients. Intensive Care Med (2016) 42(4):551–61. doi: 10.1007/s00134-015-4205-3
19. Kim OY, Monsel A, Bertrand M, Coriat P, Cavaillon JM, Adib-Conquy M. Differential down-regulation of HLA-DR on monocyte subpopulations during systemic inflammation. Crit Care (2010) 14(2):R61. doi: 10.1186/cc8959
20. Hotchkiss RS, Coopersmith CM, Karl IE. Prevention of lymphocyte apoptosis–a potential treatment of sepsis? Clin Infect Dis (2005) 41 Suppl 7:S465–9. doi: 10.1086/431998
21. Patel GP, Balk RA. Systemic Steroids in Severe Sepsis and Septic Shock. Am J Resp Crit Care (2012) 185(2):133–9. doi: 10.1164/rccm.201011-1897CI
22. Steigbigel NH. Effect of treatment with low doses of hydrocortisone and fludrocortisone on mortality in patients with septic shock. Curr Infect Dis Rep (2003) 5(5):363–4. doi: 10.1007/s11908-003-0014-x
23. Minneci PC, Deans KJ, Natanson C. Corticosteroid therapy for severe sepsis and septic shock. JAMA (2009) 302(15):1643, 1644–5. doi: 10.1001/jama.2009.1480
24. Pastores SM, Annane D, Rochwerg B. Guidelines for the diagnosis and management of critical illness-related corticosteroid insufficiency (CIRCI) in critically ill patients (Part II): Society of Critical Care Medicine (SCCM) and European Society of Intensive Care Medicine (ESICM) 2017. Intensive Care Med (2018) 44(4):474–7. doi: 10.1007/s00134-017-4951-5
25. Annane D, Renault A, Brun-Buisson C, Megarbane B, Quenot JP, Siami S, et al. Hydrocortisone plus Fludrocortisone for Adults with Septic Shock. N Engl J Med (2018) 378(9):809–18. doi: 10.1056/NEJMoa1705716
26. Jara EL, Munoz-Durango N, Llanos C, Fardella C, Gonzalez PA, Bueno SM, et al. Modulating the function of the immune system by thyroid hormones and thyrotropin. Immunol Lett (2017) 184:76–83. doi: 10.1016/j.imlet.2017.02.010
27. Vandermosten L, Vanhorebeek I, De Bosscher K, Opdenakker G, Van den Steen PE. Critical Roles of Endogenous Glucocorticoids for Disease Tolerance in Malaria. Trends Parasitol (2019) 35(11):918–30. doi: 10.1016/j.pt.2019.08.007
28. Jenniskens M, Weckx R, Dufour T, Vander Perre S, Pauwels L, Derde S, et al. The Hepatic Glucocorticoid Receptor Is Crucial for Cortisol Homeostasis and Sepsis Survival in Humans and Male Mice. Endocrinology (2018) 159(7):2790–802. doi: 10.1210/en.2018-00344
29. Beishuizen A, Thijs LG, Vermes I. Patterns of corticosteroid-binding globulin and the free cortisol index during septic shock and multitrauma. Intensive Care Med (2001) 27(10):1584–91. doi: 10.1007/s001340101073
30. Boonen E, Vervenne H, Meersseman P, Andrew R, Mortier L, Declercq PE, et al. Reduced cortisol metabolism during critical illness. N Engl J Med (2013) 368(16):1477–88. doi: 10.1056/NEJMoa1214969
31. Thomas Z, Fraser GL. An update on the diagnosis of adrenal insufficiency and the use of corticotherapy in critical illness. Ann Pharmacother (2007) 41(9):1456–65. doi: 10.1345/aph.1K082
32. Annane D. The Role of ACTH and Corticosteroids for Sepsis and Septic Shock: An Update. Front Endocrinol (Lausanne) (2016) 7:70. doi: 10.3389/fendo.2016.00070
33. Annane D, Pastores SM, Arlt W, Balk RA, Beishuizen A, Briegel J, et al. Critical Illness-Related Corticosteroid Insufficiency (CIRCI). Crit Care Med (2017) 45(12):2089–98. doi: 10.1097/CCM.0000000000002724
34. Marik PE, Pastores SM, Annane D, Meduri GU, Sprung CL, Arlt W, et al. Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. Crit Care Med (2008) 36(6):1937–49. doi: 10.1097/CCM.0b013e31817603ba
35. Flynn JK, Dankers W, Morand EF. Could GILZ Be the Answer to Glucocorticoid Toxicity in Lupus? Front Immunol (2019) 10:1684. doi: 10.3389/fimmu.2019.01684
36. Guerrero J, Gatica HA, Rodriguez M, Estay R, Goecke IA. Septic serum induces glucocorticoid resistance and modifies the expression of glucocorticoid isoforms receptors: a prospective cohort study and in vitro experimental assay. Crit Care (2013) 17(3):R107. doi: 10.1186/cc12774
37. Vazquez-Tello A, Halwani R, Hamid Q, Al-Muhsen S. Glucocorticoid receptor-beta up-regulation and steroid resistance induction by IL-17 and IL-23 cytokine stimulation in peripheral mononuclear cells. J Clin Immunol (2013) 33(2):466–78. doi: 10.1007/s10875-012-9828-3
38. Webster JC, Oakley RH, Jewell CM, Cidlowski JA. Proinflammatory cytokines regulate human glucocorticoid receptor gene expression and lead to the accumulation of the dominant negative beta isoform: a mechanism for the generation of glucocorticoid resistance. ProcNatl Acad Sci U S A (2001) 98(12):6865–70. doi: 10.1073/pnas.121455098
39. Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, McKee DD, et al. Crystal structure of the glucocorticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell (2002) 110(1):93–105. doi: 10.1016/s0092-8674(02)00817-6
40. Li LB, Leung D, Hall CF, Goleva E. Divergent expression and function of glucocorticoid receptor beta in human monocytes and T cells. J Leukoc Biol (2006) 79(4):818–27. doi: 10.1189/jlb.0805466
41. Abraham MN, Jimenez DM, Fernandes TD, Deutschman CS. Cecal Ligation and Puncture Alters Glucocorticoid Receptor Expression. Crit Care Med (2018) 46(8):e797–804. doi: 10.1097/CCM.0000000000003201
42. Leventhal SM, Lim D, Green TL, Cantrell AE, Cho K, Greenhalgh DG. Uncovering a multitude of human glucocorticoid receptor variants: an expansive survey of a single gene. BMC Genet (2019) 20(1):16. doi: 10.1186/s12863-019-0718-z
43. Loren V, Garcia-Jaraquemada A, Naves JE, Carmona X, Manosa M, Aransay AM, et al. ANP32E, a Protein Involved in Steroid-Refractoriness in Ulcerative Colitis, Identified by a Systems Biology Approach. J Crohns Colitis (2019) 13(3):351–61. doi: 10.1093/ecco-jcc/jjy171
44. Pérez LMSA. Glucocorticoids and Glucocorticoid-Induced-Leucine-Zipper (GILZ) in Psoriasis. Front Immunol (2019) 10:2220. doi: 10.3389/fimmu.2019.02220
45. Hoppstadter J, Diesel B, Linnenberger R, Hachenthal N, Flamini S, Minet M, et al. Amplified Host Defense by Toll-Like Receptor-Mediated Downregulation of the Glucocorticoid-Induced Leucine Zipper (GILZ) in Macrophages. Front Immunol (2018) 9:3111. doi: 10.3389/fimmu.2018.03111
46. Ricci E, Ronchetti S, Gabrielli E, Pericolini E, Gentili M, Roselletti E, et al. GILZ restrains neutrophil activation by inhibiting the MAPK pathway. J Leukoc Biol (2019) 105(1):187–94. doi: 10.1002/JLB.3AB0718-255R
47. Ayyar VS, Almon RR, Jusko WJ, DuBois DC. Quantitative tissue-specific dynamics of in vivo GILZ mRNA expression and regulation by endogenous and exogenous glucocorticoids. Physiol Rep (2015) 3(6):e12382. doi: 10.14814/phy2.12382
48. Cari L, Ricci E, Gentili M, Petrillo MG, Ayroldi E, Ronchetti S, et al. A focused Real Time PCR strategy to determine GILZ expression in mouse tissues. Results Immunol (2015) 5:37–42. doi: 10.1016/j.rinim.2015.10.003
49. D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, et al. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity (1997) 7(6):803–12. doi: 10.1016/s1074-7613(00)80398-2
50. Asselin-Labat ML, David M, Biola-Vidamment A, Lecoeuche D, Zennaro MC, Bertoglio J, et al. GILZ, a new target for the transcription factor FoxO3, protects T lymphocytes from interleukin-2 withdrawal-induced apoptosis. Blood (2004) 104(1):215–23. doi: 10.1182/blood-2003-12-4295
51. Bereshchenko O, Migliorati G, Bruscoli S, Riccardi C. Glucocorticoid-Induced Leucine Zipper: A Novel Anti-inflammatory Molecule. Front Pharmacol (2019) 10(3):308–18. doi: 10.3389/fphar.2019.00308
52. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, et al. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood (2001) 98(3):743–53. doi: 10.1182/blood.v98.3.743
53. Cannarile L, Delfino DV, Adorisio S, Riccardi C, Ayroldi E. Implicating the Role of GILZ in Glucocorticoid Modulation of T-Cell Activation. Front Immunol (2019) 10:1823. doi: 10.3389/fimmu.2019.01823
54. Latre DLP, Pepin A, Assaf-Vandecasteele H, Espinasse C, Nicolas V, Asselin-Labat ML, et al. Glucocorticoid-induced leucine zipper (GILZ) promotes the nuclear exclusion of FOXO3 in a Crm1-dependent manner. J Biol Chem (2010) 285(8):5594–605. doi: 10.1074/jbc.M109.068346
55. Bereshchenko O, Coppo M, Bruscoli S, Biagioli M, Cimino M, Frammartino T, et al. GILZ promotes production of peripherally induced Treg cells and mediates the crosstalk between glucocorticoids and TGF-beta signaling. Cell Rep (2014) 7(2):464–75. doi: 10.1016/j.celrep.2014.03.004
56. Gruver-Yates AL, Quinn MA, Cidlowski JA. Analysis of glucocorticoid receptors and their apoptotic response to dexamethasone in male murine B cells during development. Endocrinology (2014) 155(2):463–74. doi: 10.1210/en.2013-1473
57. Bruscoli S, Biagioli M, Sorcini D, Frammartino T, Cimino M, Sportoletti P, et al. Lack of glucocorticoid-induced leucine zipper (GILZ) deregulates B-cell survival and results in B-cell lymphocytosis in mice. Blood (2015) 126(15):1790–801. doi: 10.1182/blood-2015-03-631580
58. Bruscoli S, Sorcini D, Flamini S, Gagliardi A, Adamo F, Ronchetti S, et al. Glucocorticoid-Induced Leucine Zipper Inhibits Interferon-Gamma Production in B Cells and Suppresses Colitis in Mice. Front Immunol (2018) 9:1720. doi: 10.3389/fimmu.2018.01720
59. Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, et al. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood (2007) 110(1):211–9. doi: 10.1182/blood-2006-10-052506
60. Futterleib JS, Feng H, Tigelaar RE, Choi J, Edelson RL. Activation of GILZ gene by photoactivated 8-methoxypsoralen: potential role of immunoregulatory dendritic cells in extracorporeal photochemotherapy. Transfus Apher Sci (2014) 50(3):379–87. doi: 10.1016/j.transci.2013.10.003
61. Calmette J, Ellouze M, Tran T, Karaki S, Ronin E, Capel F, et al. Glucocorticoid-induced leucine zipper enhanced expression in dendritic cells is sufficient to drive regulatory T cells expansion in vivo. J Immunol (2014) 193(12):5863–72. doi: 10.4049/jimmunol.1400758
62. Calmette J, Bertrand M, Vetillard M, Ellouze M, Flint S, Nicolas V, et al. Glucocorticoid-Induced Leucine Zipper Protein Controls Macropinocytosis in Dendritic Cells. J Immunol (2016) 197(11):4247–56. doi: 10.4049/jimmunol.1600561
63. Jones SA, Perera DN, Fan H, Russ BE, Harris J, Morand EF. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J Autoimmun (2015) 61:73–80. doi: 10.1016/j.jaut.2015.05.010
64. Cathelin D, Met O, Svane IM. Silencing of the glucocorticoid-induced leucine zipper improves the immunogenicity of clinical-grade dendritic cells. Cytotherapy (2013) 15(6):740–9. doi: 10.1016/j.jcyt.2013.02.005
65. Hackstein H, Thomson AW. Dendritic cells: emerging pharmacological targets of immunosuppressive drugs. Nat Rev Immunol (2004) 4(1):24–34. doi: 10.1038/nri1256
66. Lebson L, Wang T, Jiang Q, Whartenby KA. Induction of the glucocorticoid-induced leucine zipper gene limits the efficacy of dendritic cell vaccines. Cancer Gene Ther (2011) 18(8):563–70. doi: 10.1038/cgt.2011.23
67. Maggi J, Schinnerling K, Pesce B, Hilkens CM, Catalán D, Aguillón JC. Dexamethasone and Monophosphoryl Lipid A-Modulated Dendritic Cells Promote Antigen-Specific Tolerogenic Properties on Naive and Memory CD4(+) T Cells. Front Immunol (2016) 7:359. doi: 10.3389/fimmu.2016.00359
68. Piemonti L, Monti P, Allavena P, Leone BE, Caputo A, Di Carlo V. Glucocorticoids increase the endocytic activity of human dendritic cells. Int Immunol (1999) 11(9):1519–26. doi: 10.1093/intimm/11.9.1519
69. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, et al. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood (2006) 107(5):2037–44. doi: 10.1182/blood-2005-07-2760
70. Hoppstadter J, Diesel B, Eifler LK, Schmid T, Brune B, Kiemer AK. Glucocorticoid-induced leucine zipper is downregulated in human alveolar macrophages upon Toll-like receptor activation. Eur J Immunol (2012) 42(5):1282–93. doi: 10.1002/eji.201142081
71. Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, et al. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood (2003) 101(2):729–38. doi: 10.1182/blood-2002-02-0538
72. Giles KM, Ross K, Rossi AG, Hotchin NA, Haslett C, Dransfield I. Glucocorticoid augmentation of macrophage capacity for phagocytosis of apoptotic cells is associated with reduced p130Cas expression, loss of paxillin/pyk2 phosphorylation, and high levels of active Rac. J Immunol (2001) 167(2):976–86. doi: 10.4049/jimmunol.167.2.976
73. Ricci E, Ronchetti S, Pericolini E, Gabrielli E, Cari L, Gentili M, et al. Role of the glucocorticoid-induced leucine zipper gene in dexamethasone-induced inhibition of mouse neutrophil migrationvia control of annexin A1 expression. FASEB J (2017) 31(7):3054–65. doi: 10.1096/fj.201601315R
74. Espinasse MA, Pepin A, Virault-Rocroy P, Szely N, Chollet-Martin S, Pallardy M, et al. Glucocorticoid-Induced Leucine Zipper Is Expressed in Human Neutrophils and Promotes Apoptosis through Mcl-1 Down-Regulation. J Innate Immun (2016) 8(1):81–96. doi: 10.1159/000439052
75. Ballegeer M, Vandewalle J, Eggermont M, Van Isterdael G, Dejager L, De Bus L, et al. Overexpression of Gilz Protects Mice Against Lethal Septic Peritonitis. Shock (2019) 52(2):208–14. doi: 10.1097/SHK.0000000000001252
76. Ellouze M, Vigouroux L, Tcherakian C, Woerther PL, Guguin A, Robert O, et al. Overexpression of GILZ in macrophages limits systemic inflammation while increasing bacterial clearance in sepsis in mice. Eur J Immunol (2019) 50(4):589–602. doi: 10.1002/eji.201948278
77. Hoppstadter J, Kessler SM, Bruscoli S, Huwer H, Riccardi C, Kiemer AK. Glucocorticoid-induced leucine zipper: a critical factor in macrophage endotoxin tolerance. J Immunol (2015) 194(12):6057–67. doi: 10.4049/jimmunol.1403207
78. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimège L, Mahieu T, et al. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med (2013) 5(3):456–70. doi: 10.1002/emmm.201201683
79. Ng HP, Jennings S, Nelson S, Wang G. Short-Chain Alcohols Upregulate GILZ Gene Expression and Attenuate LPS-Induced Septic Immune Response. Front Immunol (2020) 11(3):53–65. doi: 10.3389/fimmu.2020.00053
80. Vassiliou AG, Stamogiannos G, Jahaj E, Botoula E, Floros G, Vassiliadi DA, et al. Longitudinal evaluation of glucocorticoid receptor alpha/beta expression and signalling, adrenocortical function and cytokines in critically ill steroid-free patients. Mol Cell Endocrinol (2020) 501:110656. doi: 10.1016/j.mce.2019.110656
81. Dejager L, Pinheiro I, Bogaert P, Huys L, Libert C. Role for neutrophils in host immune responses and genetic factors that modulate resistance to Salmonella enterica serovar typhimurium in the inbred mouse strai SPRET/Ei. Infect Immun (2010) 78(9):3848–60. doi: 10.1128/IAI.00044-10
82. Cannarile L, Cuzzocrea S, Santucci L, Agostini M, Mazzon E, Esposito E, et al. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology (2009) 136(2):530–41. doi: 10.1053/j.gastro.2008.09.024
83. Baban B, Marchetti C, Khodadadi H, Malik A, Emami G, Lin PC, et al. Glucocorticoid-Induced Leucine Zipper Promotes Neutrophil and T-Cell Polarization with Protective Effects in Acute Kidney Injury. J Pharmacol Exp Ther (2018) 367(3):483–93. doi: 10.1124/jpet.118.251371
84. Baban B, Khodadadi H, Vaibhav K, Marchetti C, Riccardi C, Mozaffari MS. Regulation of Innate Lymphoid Cells in Acute Kidney Injury: Crosstalk between Cannabidiol and GILZ. J Immunol Res (2020) 2020:1–10. doi: 10.1155/2020/6056373
85. Vago JP, Tavares LP, Garcia CC, Lima KM, Perucci LO, Vieira EL, et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J Immunol (2015) 194(10):4940–50. doi: 10.4049/jimmunol.1401722
86. Dalli J, Jones CP, Cavalcanti DM, Farsky SH, Perretti M, Rankin SM. Annexin A1 regulates neutrophil clearance by macrophages in the mouse bone marrow. FASEB J (2012) 26(1):387–96. doi: 10.1096/fj.11-182089
87. Wang Y, Liao M, Zhang Y, Deng F, Luo J, Wang N, et al. Artesunate protects immunosuppression mice induced by glucocorticoids via enhancing pro-inflammatory cytokines release and bacterial clearance. Eur J Pharmacol (2020), 173630. doi: 10.1016/j.ejphar.2020.173630
88. Hotchkiss RS, Moldawer LL. Parallels between Cancer and Infectious Disease. N Engl J Med (2014) 371(4):380–3. doi: 10.1056/NEJMcibr1404664
89. Yang H, Xia L, Chen J, Zhang S, Martin V, Li Q, et al. Stress–glucocorticoid–TSC22D3 axis compromises therapy-induced antitumor immunity. Nat Med (2019) 25(9):1428–41. doi: 10.1038/s41591-019-0566-4
90. Hoppstadter J, Valbuena PJ, Linnenberger R, Dahlem C, Legroux TM, Hecksteden A, et al. The glucocorticoid-induced leucine zipper mediates statin-induced muscle damage. FASEB J (2020) 34(3):4684–701. doi: 10.1096/fj.201902557RRR
91. Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One (2013) 8(12):e82241. doi: 10.1371/journal.pone.0082241
92. Zhang W, Yang N, Shi XM. Regulation of mesenchymal stem cell osteogenic differentiation by glucocorticoid-induced leucine zipper (GILZ). J Biol Chem (2008) 283(8):4723–9. doi: 10.1074/jbc.M704147200
93. Pan G, Cao J, Yang N, Ding K, Fan C, Xiong WC, et al. Role of glucocorticoid-induced leucine zipper (GILZ) in bone acquisition. J Biol Chem (2014) 289(28):19373–82. doi: 10.1074/jbc.M113.535237
94. Levy MM, Evans LE, Rhodes A. The Surviving Sepsis Campaign Bundle: 2018 update. Intensive Care Med (2018) 44(6):925–8. doi: 10.1007/s00134-018-5085-0
95. Rapid Recognition of Corticosteroid Resistant or Sensitive Sepsis. Issue 03 NCT04280497. (2020). Available at: https://clinicaltrials.gov/show/NCT04280497
96. Vandewalle J, Libert C. GILZ in sepsis: “Poor is the pupil who does not surpass his master”. Eur J Immunol (2020) 50(4):490–3. doi: 10.1002/eji.202048582
Keywords: sepsis, glucocorticoid-induced leucine zipper, glucocorticoids, sepsis-induced immunosuppression, anti-inflammatory
Citation: He Y-J, Xu J-Q, Sun M-M, Fang X-Z, Peng Z-K, Pan S-W, Zhou T, Wang Y-X and Shang Y (2020) Glucocorticoid-Induced Leucine Zipper: A Promising Marker for Monitoring and Treating Sepsis. Front. Immunol. 11:606649. doi: 10.3389/fimmu.2020.606649
Received: 15 September 2020; Accepted: 13 November 2020;
Published: 16 December 2020.
Edited by:
Bidesh Mahata, University of Cambridge, United KingdomReviewed by:
Peter A. Ward, University of Michigan, United StatesCopyright © 2020 He, Xu, Sun, Fang, Peng, Pan, Zhou, Wang and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: You Shang, eW91X3NoYW5naHVzdEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.