 Candice Mazewski
Candice Mazewski Ricardo E. Perez1,2
Ricardo E. Perez1,2 Eleanor N. Fish
Eleanor N. Fish Leonidas C. Platanias
Leonidas C. Platanias
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Immunol., 23 November 2020
Sec. Cytokines and Soluble Mediators in Immunity
Volume 11 - 2020 | https://doi.org/10.3389/fimmu.2020.606456
This article is part of the Research TopicStructures, Signaling Mechanisms, and Functions of Types I and III InterferonsView all 9 articles
For several decades there has been accumulating evidence implicating type I interferons (IFNs) as key elements of the immune response. Therapeutic approaches incorporating different recombinant type I IFN proteins have been successfully employed to treat a diverse group of diseases with significant and positive outcomes. The biological activities of type I IFNs are consequences of signaling events occurring in the cytoplasm and nucleus of cells. Biochemical events involving JAK/STAT proteins that control transcriptional activation of IFN-stimulated genes (ISGs) were the first to be identified and are referred to as “canonical” signaling. Subsequent identification of JAK/STAT-independent signaling pathways, critical for ISG transcription and/or mRNA translation, are denoted as “non-canonical” or “non-classical” pathways. In this review, we summarize these signaling cascades and discuss recent developments in the field, specifically as they relate to the biological and clinical implications of engagement of both canonical and non-canonical pathways.
Established cellular signaling pathways have been referred to in the context of canonical or “classical” and non-canonical or “non-classical” signaling cascades that control distinct outcomes in the cell. A canonical pathway indicates the conventional protein signaling, typically considered the main effect or, maybe more appropriately, the first effect discovered and elucidated; non-canonical pathways are alternative pathways to the canonical, but that should not imply less importance (1). Perhaps, the most well-described signaling in terms of canonical and non-canonical pathways is Wnt signaling, specifically the canonical β-catenin pathway (2). Additionally, inflammation and immunoregulatory related pathways such as nuclear factor-κB (NF-κB) and interferon (IFN) signaling are described as canonical and non-canonical (3, 4). Recent discoveries of additional non-canonical pathways, some that interconnect with canonical signaling, add to the complexity surrounding different biological outcomes.
The IFNs are cytokines that can be divided into three groups: type I (IFNα, IFNβ, IFNδ, IFNϵ, IFNκ, IFNτ, IFNω, and IFNζ), type II (IFNγ), and type III (IFNλ) (5). Type I IFNs were first discovered in 1957, followed by type II in 1965, while much more recently, in 2003, type III IFNs were identified (6–8). Type I IFNs have the most family members. The predominant type I IFN subtypes studied are IFNα and IFNβ, partially due to IFNδ, IFNτ, and IFNζ not having human homologs, more specific cellular sources of IFNϵ and IFNκ, mainly female reproductive organs and keratinocytes, respectively, and IFNω being studied more in felines (5, 9, 10). The roles of IFNα and IFNβ in antiviral responses have been most reported, but these type I IFNs also have significant relevance in cancer and autoimmune diseases (11–13).
Production of type I IFNs is induced by pathogen-associated molecular patterns, viral RNA or DNA fragments, and is associated with activation of pattern recognition receptors (11). Once activated, the receptors initiate signal transduction that involves adapter proteins, eventually leading to activation and translocation of IFN regulatory factor 3 (IRF3) and NF-κB, which promote type I IFN production either directly or indirectly through IRF7 (11). IFNα is mainly produced by plasmacytoid dendritic cells (pDCs), whereas IFNβ is ubiquitously produced by immune cells (13).
Following transcriptional activation and mRNA translation, type I IFNs are secreted from immune cells and, on neighboring cells, bind to the two cellular receptor subunits IFNα receptor 1 (IFNAR1) and IFNAR2, which are associated with tyrosine kinases TYK2 and Janus kinase 1 (JAK1), respectively (9). Dimerization of the receptor initiates the autophosphorylation of JAK1, which phosphorylates and activates signal transducers and activators of transcription 1 (STAT1) and STAT2 proteins, which form a complex with IRF9, resulting in a well-characterized complex, IFN-stimulated gene factor 3 (ISGF3). ISGF3 translocates to the nucleus where it binds to IFN-stimulated response elements (ISREs) in the promoters of genes, leading to transcription of IFN stimulated genes (ISG) (14). Additionally, JAKs can phosphorylate and initiate the formation of phosphorylated STAT complexes of STAT1 and STAT3 homodimers, where the STAT1 homodimer is associated with a pro-inflammatory response, mediated by binding to gamma activated sequences (GAS), and the STAT3 homodimer indirectly inhibits inflammatory gene expression, restraining pro-inflammatory responses (15). These JAK/STAT IFN-signaling pathways are considered the canonical pathways. In addition, type I IFNs have also been reported to activate the formation of STAT2:STAT3 heterodimers and a STAT5:CrkL complex, invoking transcriptional activation of ISGs (16, 17).
Non-canonical type I IFN signaling pathways are similarly activated by IFNs binding to the extracellular regions of the dimeric IFNAR1 and IFNAR2 complex, leading to JAK1/TYK2 activation, but diverge from that point, specifically, not involving STAT activation by the JAKs. Evidence points to the regulation of STATs by non-canonical modifiers, with serine phosphorylation of STATs versus the tyrosine phosphorylation by JAK1/TYK2 (18). The main non-canonical IFN pathways identified thus far are the MAP kinase (MAPK) and phosphoinositide 3-kinases (PI3K)/mammalian target of rapamycin (mTOR) pathways, but there are other non-canonical modifiers such as SIRT2 and the Schlafen (SLFN) family (18, 19). MAPK and PI3K/mTOR pathways have been shown to elicit effects on ISG transcription and mRNA translation while also having some interaction with STATs in the canonical cascade (18). Further discoveries on the effectors of these pathways, such as the importance of Unc-51–like kinase (ULK1) in MAPK type I IFN-induced signaling, add to the complexity of type I IFN signaling cascades and demonstrate that the focus cannot be limited to the classical pathways (20). Other non-canonical modifiers include SLFN family members. Type I IFNs upregulate SLFN gene expression, and SLFN5 interaction with STAT1 has been demonstrated, indicating its effect downstream of JAK1 (21). SLFNs have been shown to be involved in antiviral responses, and their high expression in specific human immune cell subsets has been identified, such as elevated SLFN5 in T cells (22). These non-classical IFN-induced effectors have critical roles in ISG transcription, independent of or in conjunction with the canonical pathway, eliciting specific biological responses. A summary of the canonical and non-canonical pathways of type I interferon signaling is shown in Figure 1.
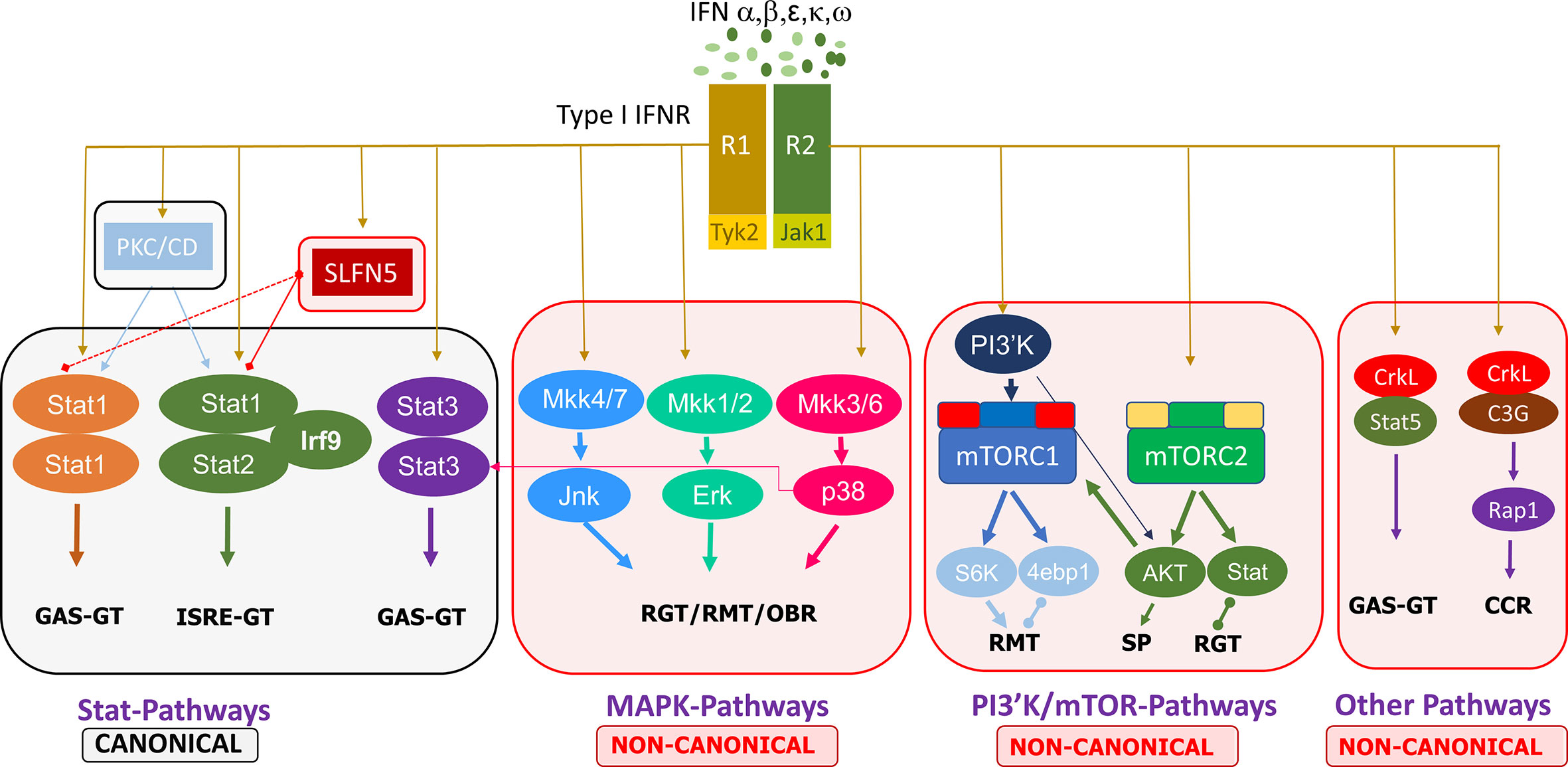
Figure 1 Summary of the canonical and non-canonical pathways involved in type I interferon signaling. 4ebp1, eukaryotic translation initiation factor 4E binding protein 1; CCR, cell cycle regulation; ERK, extracellular signal-regulated kinase; GAS, gamma-activated sequence; GT, gene transcription; IFN, interferon; IFNR, interferon receptor; IRF, interferon regulatory factor; ISRE, interferon-stimulated response element; JAK, janus kinase; Jnk, c-Jun N-terminal kinase; MKK, mitogen activated protein kinase kinase; mTORC, mammalian target of rapamycin complex; PI3′K, phosphoinositide 3-kinase; PKC, protein kinase C; CD, Calmodulin-dependent kinase, R1/R2, receptor 1/2; Rap, Ras-related protein; RGT, regulation of gene transcription; RMT, regulation of mRNA translation; OBR, other biological responses; S6K, ribosomal protein S6 kinase; SLFN, Schlafen; STAT, signal transducer and activator of transcription; SP, survival pathways.
Below we provide an update on type I IFN canonical and non-canonical signaling, related to antiviral responses, antiproliferative effects in cancer, and immune regulation in autoimmune diseases, focusing on studies within the last few years. We address the type I IFN response to SARS-CoV-2 and the potential for therapeutic use for COVID-19.
Type I IFNs have been studied in a wide range of cancers in the last few years, as illustrated in Table 1. These studies have focused exclusively on the IFNα and IFNβ subtypes, demonstrating their clinical relevance over other type I IFN subtypes. The signaling analyses in the last few years have still focused more on the JAK/STAT cascades, specifically STAT1 effects in type I IFN signaling. However, some reports explored the impact of STAT3 versus STAT1, as well as the non-canonical involvement of MAPKs, SIRT2, and SLFN5.
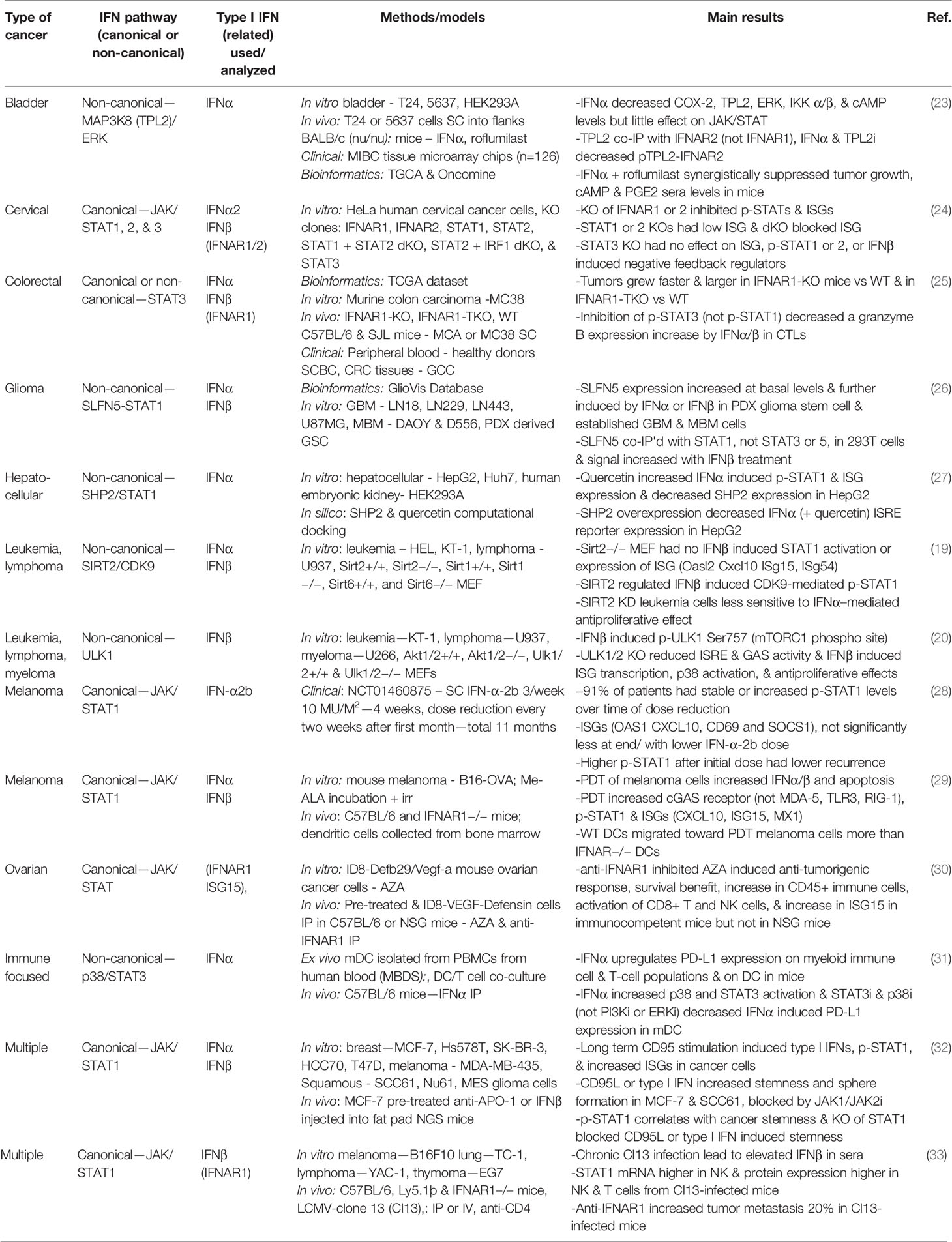
Table 1 Canonical and non-canonical type I interferon signaling in malignancies.
STAT1 phosphorylation and the induced expression of various ISGs such as OASL and ISG15 have commonly been used as indicators of a type I IFN response (19, 27, 28, 34, 35). In a study on cervical cancer, the importance of IFN-inducible activation of STAT1 and STAT2 was demonstrated through the use of STAT1 and STAT2 knockout human HeLa cells, yet the STAT3 knockout did not have any effect on ISGs (24). By contrast, in colorectal cancer, inhibition of p-STAT3 but not p-STAT1 decreased IFNα and IFNβ induced granzyme B expression in cytotoxic T lymphocytes (25). These differences highlight how different effectors activated by type I IFNs are dependent on cell type and disease specificity. Bazhin et al. also explored IFN-activated STAT3 effects, identifying a non-canonical interaction with p38 MAPK on STAT3 phosphorylation in mature DCs (31). ULK1 has been identified as a regulator of p38 MAPK and ISGs, downstream of mTOR, in type I IFN signaling in myeloproliferative neoplasms (20). This demonstrates a connection between both major IFN activated non-canonical signaling pathways. Another MAPK, extracellular signal-regulated kinase (ERK), is involved in non-canonical type I IFN signaling in malignancy, where mitogen-activated protein kinase kinase kinase 8 (MAP3K8) and ERK phosphorylation were decreased upon IFNα treatment in bladder cancer cells (23). Further elucidation is needed on the STAT-dependent and -independent non-canonical functions of the many MAPK pathway proteins.
Additional effects of non-canonical type I IFN-induced signaling in various malignancies have been examined. A glioblastoma study identified SLFN5 as a regulator of STAT1 induction by type I IFNs (26). In leukemia and lymphoma cells, type I IFN induced phosphorylation of STAT1 on serine 727 is mediated by cyclin dependent kinase 9 (CDK9), and this activation is dependent on the deacetylation of CDK9 by SIRT2 (19). Additionally, quercetin, a natural compound, decreases Src Homology Phosphatase 2 (SHP2), a negative regulator of STAT1 (27).
An important issue related to the clinical use of IFNs is toxicity and adverse events. Although approved in 1986 by the FDA for the treatment of malignancies and viral disorders, with demonstrated positive disease outcomes, IFNα is currently not commonly used in cancer treatment due to adverse effects (36, 37). A pilot study looked at the potential of decreasing the dose of IFN-α2b for the treatment of melanoma over the course of an 11-month treatment period. Despite the dose reduction, p-STAT1 levels were induced at comparable levels throughout the 11 months, and the IFN was well-tolerated (28). An alternative strategy has been to stimulate the endogenous type I IFN response in immune cells. Tsuchiya et al. genetically engineered induced pluripotent stem cell (iPSC)-derived proliferation myeloid cells (iPSC-pMCs) to produce IFNα. When injected into mice, these IFN-producing iPSC-pMCs exerted immunomodulatory effects analogous to direct type I IFN administration, yet without adverse effects or hematopoietic stem cell exhaustion (37). Brown et al. studied recombinant poliovirus/rhinovirus chimera PVSRIPO effects in cancer immunosuppression and found PVSRIPO infection of DCs increased IFNβ production and a sustained type I IFN response, as indicated by p-STAT1 and ISG induction (IFIT1, ISG15) (34). In a separate study, the use of photodynamic therapy (PDT) lead to the upregulation of type I IFNs in melanoma cells and DCs co-cultured with the PDT treated cells; the authors proposed this ex vivo strategy of stimulating DCs with the use of PDT as a possible immunotherapy (29).
Distinct from the positive outcomes of type I IFN treatment for malignancies, a number of studies have addressed the potential link of IFN treatment with chemotherapy resistance, immunosuppression, and driving of cancer stemness. Qadir et al. found chronic CD95 activation leading to cancer stemness was driven by IFNα/β-STAT1 canonical signaling (32). They also provided evidence that radio-resistant squamous cancer cells had increased p-STAT1 and ISG expression and that type I IFN treatment of breast and squamous cancer cells increased stemness and sphere formation, which was blocked by JAK inhibition, indicative of the involvement of canonical signaling.
Several studies have evaluated the effects of type I IFN administration in combination with immunotherapy. One group showed that IFNα increased programmed death-ligand 1 (PD-L1) expression on various immune cells through non-canonical p38/STAT3 signaling (31). The inference is that combining immunotherapy with IFNα treatment would limit the immunosuppressive effects of IFN treatment and permit effective growth inhibition. Similarly, another study provided evidence that IFNα-iPSC-pMC treatment increased PD-L1 mRNA, and when combined with a PD-L1 inhibitor, synergistic anti-tumor effects were reported (37). The poliovirus/rhinovirus type I IFN induced response likewise increases PD-L1 expression (34). Additionally, a bioinformatics examination of IFN gene deletions revealed that homozygous deletion of IFN was significantly associated with non-response to anti-CTLA4 treatment among melanoma patients (38). Overall, these studies suggest type I IFNs may have a critical role in immunotherapy strategies, possibly via a combination of type I IFN treatment with PD-L1 inhibition. Moreover, the data suggest that PD-L1 expression may be affected by IFN-induced non-canonical signaling.
Accumulating evidence implicates chronic and persistent type I IFN signaling in systemic inflammation that promotes the pathogenesis of some autoimmune diseases, including systemic lupus erythematosus (SLE), rheumatoid arthritis, multiple sclerosis (MS), type I diabetes (T1D) and Sjögren’s syndrome, among others (13, 39). These conditions are associated with different clinical symptoms and management strategies, yet there are common features related to the underlying inflammatory signaling pathways involved and the dysregulated immune response. Figure 2 summarizes the cell type-specific type I IFN-induced canonical and non-canonical signaling pathways recently implicated in autoimmune diseases.
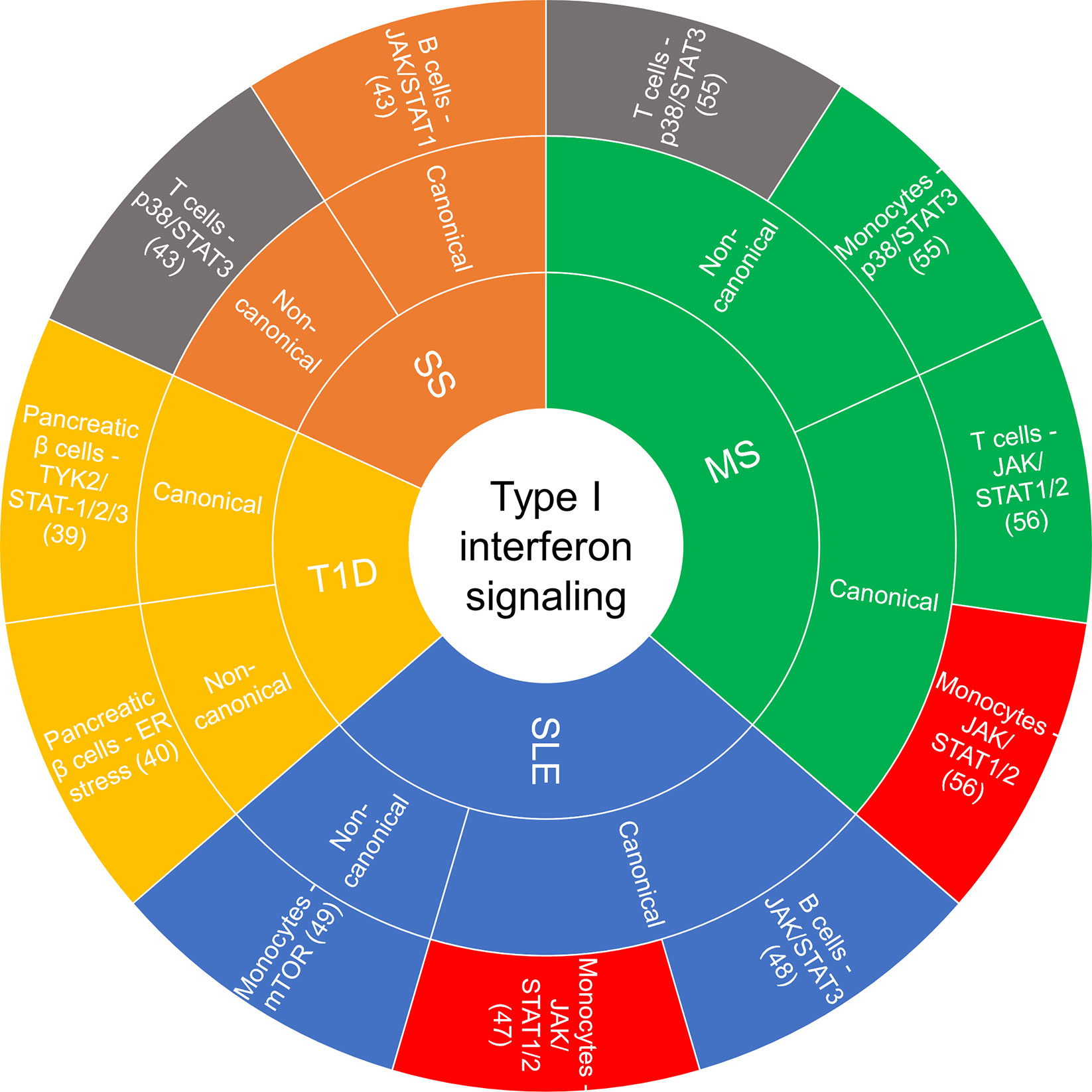
Figure 2 Sunburst chart of cell-specific canonical and non-canonical signaling recently reported on in autoimmune diseases. If there was a commonality in a canonical or non-canonical signaling demonstrated in the same cell type but different disease, they were color coded the same: red—JAK/STAT1/2 in monocytes, gray—p38/STAT3 in T cells. ER, endoplasmic reticulum; JAK, janus kinase; MS, multiple sclerosis; mTOR, mammalian target of rapamycin; SLE, systemic lupus erythematosus; SS, Sjögren’s syndrome; STAT, signal transducer and activator of transcription; T1D, type I diabetes.
IFNα has been shown to impact the onset and progression of T1D, which involves the autoimmune attack of pancreatic β cells (40). One study demonstrated that IFNα activated STAT1, STAT2, and STAT3 in pancreatic β cells through TYK2, and that STAT2 was more critical than STAT1 in mediating the inflammatory and endoplasmic reticulum (ER) stress response (41). Another study likewise reported on IFNα induction of ER stress in pancreatic β cells, leading to the downregulation of insulin production and influence on T1D onset (42). A mouse model study revealed that inhibition of IFNα, but not IFNβ, in the pre-diabetes stage prevented the onset of T1D and blocked autoreactive T cells from entering and killing β cells in the pancreatic islets (43). Notably, patients with neutralizing autoantibodies to type I IFNs, specifically IFNαs, are less likely to develop T1D (44). These studies identify the negative impact of IFNα on the development of T1D.
Sjögren’s syndrome is an autoimmune disease with glandular lymphocyte infiltration leading to symptoms of dry mouth and eyes, where approximately 50% of patients have a type I IFN signature (45, 46). Given that this IFN signature is not present in all patients, one study analyzed the effects of IFN-α2b treatment of peripheral blood mononuclear cells (PBMCs) from patients with Sjögren’s compared with PBMCs from healthy donors, including type I IFN signature-positive and negative patients (45). Baseline effector protein phosphorylation levels differed predominantly in T cells in Sjögren’s patients compared with healthy individuals, with higher p-p38 and p-STAT1 (Y701, S727). Sjögren’s patients also exhibited increased IFNα-inducible JAK phosphorylation of STAT1 (Y701). Further, IFNα-2b treatment of PBMCs upregulated p-STAT1 (Y701) in B cells and downregulated p-STAT3 on S727 in T cells in type I IFN signature-positive patients.
SLE manifestations include organ damage and skin rash (47, 48). There is an IFNα signature in sera of SLE patients. A recent study using inducible IFNα transgenic mice found that upregulation of IFNα alone was capable of inducing an SLE phenotype (47). SLE pathogenesis is characterized by inflammasome overactivation; one study demonstrated that prolonged IFNα treatment increased inflammasome activity, which was eliminated with knockdown of IRF1 in SLE monocytes (49). IFNα treatment increased p-STAT1 and p-STAT2 at tyrosine residues, indicative of a classical JAK/STAT driven response. Another group that analyzed B cells from SLE patients, found increased baseline p-STAT3 (Y705), not p-STAT1, compared to B cells from healthy individuals (50). Additionally, these investigators found that IFNα treatment polarized naïve B cell differentiation towards a lupus-like phenotype, which was reversed by a STAT3 inhibitor and was absent in STAT3-deficient donor naïve B cells. In SLE monocytes, Gkirtzimanaki et al. identified IFNα induced mTOR activity, which promoted oxidative stress, revealing non-canonical IFNα signaling in SLE (51).
Cognizant of the persistent IFNα signature in SLE patients, a phase IIb clinical trial evaluated the effects of vaccination with IFNα kinoid, which produces anti-IFNα antibodies (52). Although the trial did not see a benefit in Based Composite Lupus Assessment (BICLA), the drug did provoke anti-IFN-α2b serum antibodies and decreased the IFN gene signature in 91% of patients. An anti-IFNAR1 monoclonal antibody, anifrolumab, has been evaluated in 11 clinical trials for SLE (9), Sjögren’s (1), and rheumatoid arthritis (1), with encouraging results (53). A recent phase III trial in SLE did not meet its primary endpoint of response, as per the SLE Responder Index; however, the same group conducted another phase III trial using the of British Isles Lupus Assessment Group (BILAG)-BICLA response as the primary endpoint and reported a statistically significant higher percentage of patients having a response as well as seeing a decrease in secondary endpoints, suggesting that a chronic IFNα response in SLE patients may contribute to disease pathogenesis (48).
Interestingly, while IFNα has been implicated in the pathogenesis of various autoimmune diseases, IFNβ has been used to successfully treat MS (54). Employing a mouse model of MS, studies with mice that lack the IFNβ gene revealed that in the absence of IFNβ the mice had a more severe disease with earlier onset and that the lack of IFNβ predisposed the mice to a pro-inflammatory Th17 immunophenotype (55, 56). Given the heterogeneity of the disease, and differing patient responses to IFNβ treatment, the identification of potential biomarkers of response to IFNβ therapy is receiving considerable attention. One study suggested predictors of response could be based on cell type-specific responses to type I IFN signaling, such as higher activation of STAT1, STAT3, and p38, leading to higher TRAIL expression in monocytes of IFN responders (57). Hurtado-Guerrero et al. analyzed monocytes from MS patients ex vivo, either left untreated (baseline) or after short-term IFNβ treatment (58). At baseline, there were no detectable differences in the levels of IFNAR1, IFNAR2, p-STAT1, and p-STAT2 among responders and non-responders, yet following IFNβ treatment, differences were observed. They found a pattern of decreased IFNAR1 and increased IFNAR2, p-STAT1, and p-STAT2 levels representing 68.4% of responder IFNβ-stimulated monocytes. Other groups have employed bioinformatics to uncover gene signatures that determine a response to IFNβ. One study used a feature selection computational method on a longitudinal microarray dataset of relapse-remitting MS (RRMS) patients treated with IFNβ-1b, and found a predictive seven gene signature (CXCL9, IL2RA, CXCR3, AKT1, CSF2, IL2RB, GCA) with 65.08% predictive accuracy (59). Using an alternative method of Elastic net modeling, Fukushima et al. analyzed time-course microarray datasets from PBMCs of MS patients and identified eleven (ZBTB16, ZFP37, HPS5, HOPX, ARFGAP3, CALML5, VPS26A, SLC5A4, MBL2, DLGAP4, CACNA1C) and eight (SMA4, MIR7114_NSMF, LSM8, FLAD1, RRN3P1, RASL10A, IER3IP1, CDH2) genes predictive of an IFNβ response, with 81% and 78% accuracy, respectively, for each dataset (60). A different study employed the GeneRank method to identify monotonically expressed genes (MEGs) that determine a good response (AFTPH, ALOX5, ATG7, MYD88, LILRB1, PRKAB1, PSEN1, VAMP3) and a bad response (AGFG1, CHM, IGLL1, PELI1, PTEN) for responders, and two bad response MEGs for non-responders (NAP1L4, MMS19) in IFNβ treated RRMS patients (61). As an alternative strategy to gene analysis, a logistic regression modeling method was used to examine metabolites from the sera of a cohort of MS patients to predict the production of anti-drug antibodies (ADA) to IFNβ treatment (62). Differences in 29 metabolites were shown to be indicative of ADA production, and the top ten most significant metabolites were lipid related. Another study using a systems immunology approach evaluated ADA production differences in three IFNβ treated cohorts and showed reduced baseline NOTCH2 expression and that a pro-inflammatory phenotype in monocytes was predictive of ADA development (63). Given the preceding, there is a need for further identification and characterization of biomarkers that are reproducibly predictive of an IFNβ response in RRMS patients.
The differences between IFNα and IFNβ in the generation of effects in autoimmune diseases requires additional analysis. Although both type I IFNs bind to and initiate signaling cascades through the dimeric IFNAR, they do differ in primary amino acid sequences and in binding affinity to the receptor which may account for varying impacts of the response on cells (54, 64). Binding affinity for IFNAR1 and IFNAR2 varies among IFNα subunits, with overall higher affinity for IFNAR2 over IFNAR1, and IFNβ has tighter binding to each receptor subunit than any of the IFNα subunits (64, 65). How the induced signaling can differ after the type I IFN ligand binds is not well understood but studies have shown differences further downstream in genes and transcription factor binding sites of IFNα versus IFNβ signaling, such as enrichment of IRF8 binding sites in IFNβ response (54). As previously mentioned, cell-type and disease state lead to variance in type I IFN signaling which is further complicated by the differences invoked by IFNα and IFNβ and requires further studies, especially to understand the protein signaling cascades after binding of type I IFNs to the IFNAR.
IFNs are critical effectors of an antiviral response in mammalian cells. Following viral infection, type I IFNs are produced by immune and non-immune cells, bind to and activate IFNAR, and signal through canonical and non-canonical pathways (66–68). An area of interest has been the involvement of the IFN system in the pathophysiology of Coronavirus Disease 19 (COVID-19).
Since the emergence of severe acute respiratory syndrome coronavirus (SARS-CoV) in 2003 and Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012, therapeutic options for treatment have been limited (69). Type I IFNs are attractive therapeutic candidates because of their ability to clear virus through direct inhibition of viral replication of both DNA and RNA viruses and their effects on the activation of specific immune cell subsets to assist with viral clearance (70). Many viruses, including coronaviruses, evade an IFN antiviral response by inhibiting the production of type I and III IFNs (71–73). Scrutiny of the SARS-CoV genome identified the genes NSP1, NSP3, ORF3b, and ORF6 that are antagonists for type I IFNs, as well as the N protein (74). ORF6 not only inhibits the production of IFN but can also inhibit the expression of ISGs by inhibiting STAT1 nuclear translocation, through disruption of karyopherin-mediated transport. IRF3 is an important transcription factor necessary for IFNβ expression. The papain-like protease (PLpro), conserved in both SARS-CoV and SARS-CoV-2, inhibits the phosphorylation required for IRF3 homodimerization and nuclear translocation leading to its association with CBP/p300 and NF-κB for IFNβ expression (75–77). Comparing the gene sequences between SARS-CoV and SARS-CoV-2 for NSP3, ORF3b, and ORF6, revealed sequence differences that may contribute to the greater sensitivity of SARS-CoV-2 to type I IFNs (77). Konno et al. made the observation that ORF3b inhibits type I IFN induction more so in SARS-CoV-2 than in SARS-CoV, and a naturally arising SARS-CoV-2 variant exerts even greater antagonism of type I IFN induction by ORF3b (78). Accumulating data continue to provide further evidence of a blunted IFN response in COVID-19 cases (79–83).
Recently, data have emerged that indicate that SARS-CoV-2 is sensitive to the antiviral effects of both IFNα and IFNβ in cell culture assays, similar to the sensitivity of SARS-CoV in vitro (84–86). A pilot clinical study during the SARS outbreak of 2003 demonstrated that treatment with an IFNα resulted in reduced disease-associated impaired oxygen saturation and rapid resolution of lung abnormalities (87). The evidence of SARS-CoV-2 sensitivity to IFN treatment and accumulating clinical studies suggest that IFN treatment may have therapeutic benefits for COVID-19 (88). Early on in the pandemic, Zhou et al. provided evidence that treating COVID-19 patients with nebulized IFN-α2b with or without the antiviral drug, arbidol, accelerated viral clearance from the airways of infected patients and also reduced the circulating levels of the inflammatory cytokines, IL-6 and CRP (89). Following up from this exploratory study, there have been several clinical studies evaluating the therapeutic benefit of IFNα and IFNβ treatment for COVID-19 (see Table 2). In vitro studies suggested greater antiviral effectiveness of IFNβ over IFNα against SARS CoV (97). This prompted the WHO SOLIDARITY randomized controlled trial of a combination of lopinavir/ritonavir, ribavirin, and IFNβ-1b versus lopinavir/ritonavir in SARS-CoV-2 (90). The findings suggest that the triple combination treatment was more effective than lopinavir/ritonavir alone, reducing symptom severity and time to viral clearance. Given the emerging evidence that lopinavir/ritonavir treatment may be ineffective against SARS-CoV-2, the ongoing trial had been amended to compare the therapeutic effectiveness of IFNβ with remdesivir, a viral polymerase inhibitor that has demonstrated limited therapeutic efficacy in COVID-19 cases. A prospective observational study was conducted to assess the therapeutic efficacy of IFN-α2b in SARS-CoV-2 patients during the first month after the COVID-19 outbreak began in Cuba. Intramuscular administration of IFN-α2b improved both the rate of recovery and case fatalities (91). However, a retrospective cohort study demonstrated that there is great importance on the timing of administration of IFN-α2b with reduction of in-hospital mortality when administered the first five days of admission but increased mortality and delayed recovery was seen if given later (92). Additionally, inborn errors of type I IFNs and presence of autoantibodies against type I IFNs can be determinants of severity of disease and effectiveness of type I IFN treatment (95, 98). Roughly 10% of COVID-19 patients with severe pneumonia in a cohort of 987 patients had neutralizing autoantibodies against IFNα, IFNω, or both, where patients with no or mild symptoms had no detectable autoantibodies (95). These findings demonstrate that administration of IFNα may not be effective in patients with severe condition and autoantibodies, but since IFNβ autoantibodies were uncommon in the same patients, IFNβ may provide a more beneficial treatment. The same group analyzed a separate cohort of patients with life-threatening pneumonia and found 3.5% had inborn errors in type I IFN related genes, specifically in loci pertaining to TLR3- and IRF7-dependent type I IFN induction (98). This showed a commonality with influenza since similar type I IFN related gene defects have been demonstrated in life-threatening influenza pneumonitis (99).
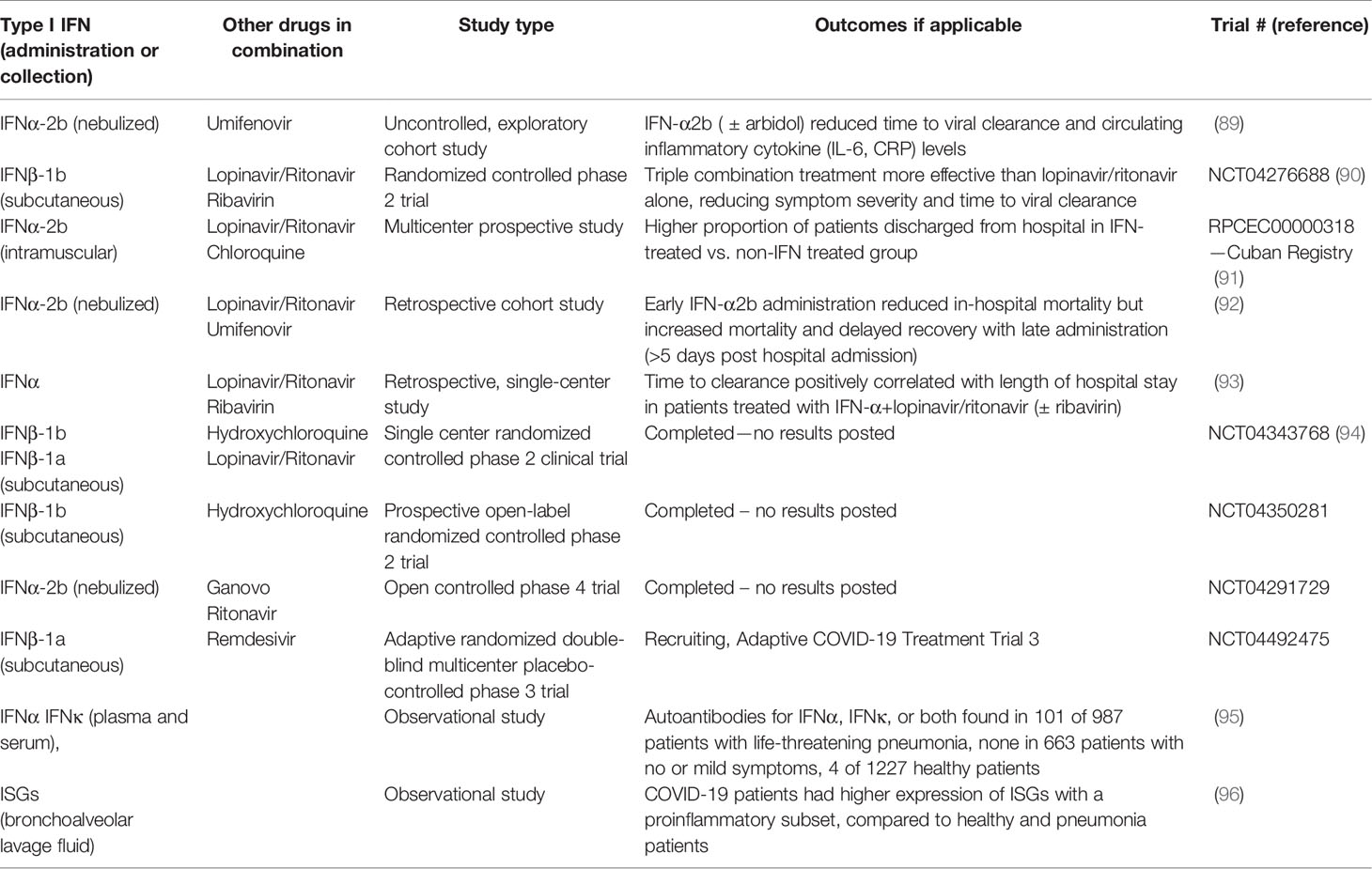
Table 2 Clinical studies involving type I interferons in SARS-CoV-2.
Similar to SARS-CoV, SARS-CoV-2 interacts with the angiotensin-converting enzyme 2 (ACE2) for cell entry, while MERS-CoV exploits the dipeptidyl peptidase 4 (DPP4) receptor for entry into human cells (100–102). Ziegler et al. demonstrated that nasal secretory cells (goblet cells), type II pneumocytes, and absorptive enterocytes of the ileum are positive for the two critical receptors for SARS-CoV-2 cell entry, ACE2 and the type II transmembrane serine protease, TMPRSS2 (103). Their observation that ACE2 expression is induced by type I IFNs in primary upper airway basal cells and lung tissue is hard to reconcile with IFNs inhibiting infection by SARS-CoV-2, yet recent emerging data suggesting a role for the renin-angiotensin pathway in protection from specific clinical features of COVID-19 would support a role for ACE2 in limiting COVID-19 severity. The inability of mice to uptake SARS-CoV-2 infection through the mouse ortholog of entry receptor ACE2 prompted Israelow et al. to create an adeno associated virus-mediated human ACE2 mouse model that can be utilized to analyze SARS-CoV-2 in mice, and they found increased type I IFN signaling ISGs in the lungs and limited control of SARS-CoV-2 replication by type I IFNs (104). The involvement of canonical versus non-canonical pathways in the induction of IFN-responses against SARS-CoV2 remains to be elucidated.
SARS-CoV-2 and influenza viruses are respiratory infections where disease severity results in lung hyper-inflammation and acute respiratory distress. Findings from clinical studies suggest that the early viral phase of both infections is associated with a blunted IFN response, yet progression to severe disease shows no such failed IFN response, specifically elevated levels of ISGs in PBMCs are observed (105–107). The implications are that the therapeutic benefits of IFN treatment are applicable in the early viral phases of COVID-19 and influenza, but that once the pulmonary phases of both infections progress to hyper-inflammation, IFN treatment is likely to be contra-indicated. Non-canonical effects in type I IFN signaling in influenza have been demonstrated as with p38 MAPK signaling, shown to be important in affecting type I IFN production and signaling in highly pathogenic avian influenza virus infected endothelial cells (108). Additionally, IFN-κ treatment inhibits influenza replication in lung cells, dependent on IFNAR, p38, CHD6, and Fos activation, but not STAT1 (109). Notably, IFNα induced STAT3 activation is crucial for inhibition of influenza viral replication and ISG transcription in mouse embryonic fibroblasts (110).
Although antiretroviral therapy (ART) for Human Immunodeficiency Virus (HIV) infection has transformed this infection from a fatal one to a chronic disease, viral reservoirs complicate efforts for HIV elimination, and a recent review paralleled HIV reservoir persistence to immuno-editing and immune evasion in cancer (111). The roles of type I IFNs in the pathophysiology of HIV infection are not fully understood, but IFNα has been implicated as an adverse factor in the persistence of HIV-1. When circulating levels of IFNα were measured for healthy donors, primary-infected, and chronically-infected patients, higher IFNα levels were associated with higher viral loads and higher expression of the ISG, USP18, which negatively regulates IFNα signaling by displacing JAK2 bound to IFNAR2 (112). Humanized mouse models have provided evidence that whereas type I IFNs suppress early HIV infection, type I IFN signaling induces T cell depletion and impaired functionality during persistent infection. When IFN signaling is blocked in HIV-infected mice or in monkeys receiving ART, this reduces the HIV reservoir, rescues anti-HIV T cells, and reduces HIV-induced inflammation (113–115). Notably, HIV-1 proteins, Vpu and Nef, inhibit ISG expression through canonical IFNα mediated JAK/STAT1 signaling, blocking any antiviral benefits from IFNα (116). Knockout of IFNAR1 in an HIV-induced brain injury mouse model provided memory benefits and neuronal injury protection while suppressing p38 activation, indicating involvement of type I IFN non-canonical signaling in HIV-1–related neurotoxicity (117). Indeed, there is accumulating evidence that sustained type I IFN signaling, surprisingly, can promote viral replication for a number of viruses, mediated by induction of certain ISGs and inhibition of IRFs (14). IFN induced 2′5′-oligoadenylate synthetase-like (OASL) limits RNA virus replication through enhancing RIG-I signaling yet inhibits cGAS and promotes viral replication for DNA viruses such as HSV.
Of late, there are emergent data that SLFN proteins, non-canonical effectors of type I IFN signaling, have a role as antivirals. IFN induced SLFN11 expression controls protein synthesis by regulating tRNA abundance, limiting West Nile virus, dengue virus, and Zika virus replication, all (+) ssRNA viruses, but having little effect on (–) ssRNA viruses (118). Interestingly, SLFN 11 control of HIV-1 infection is independent of type I IFN signaling (119). IFNβ induced SLFN14 exhibits antiviral activity in mouse macrophages, limiting infection with influenza virus or the DNA virus, varicella-zoster virus (120).
Besides the duration of type I IFN signaling influencing whether there is inhibition or enhancement of viral replication (105, 112), cell environmental factors also contribute to a type I IFN response. In a mouse model of vesicular stomatitis virus infection, high salt levels augment type I IFN signaling through the non-canonical p38 pathway (121). In neurons, viral infection may cause pain hypersensitivity; type I IFNs elicit pain sensitization in neurons, by promoting MAPK interacting kinase phosphorylation of eukaryotic initiation translation factor (122).
Though over sixty years have elapsed since the original discovery of IFNs, in recent years, there has been mounting evidence for the critical roles of type I IFNs as immune regulators in multiple biological systems. The mechanisms of induction of type I IFNs and their subsequent biological responses are complex, due in part to the large number of family members, both cell type-dependent and independent biological responses, and varying influences in different disease settings. As identified above, for acute and chronic virus infections, type I IFN signaling can have distinct and sometimes contrasting biological effects. In malignancies, type I IFNs induce antiproliferative and antineoplastic effects but may also upregulate PD-L1 expression, thereby limiting an anti-tumor immune response. In some autoimmune diseases, such as SLE, the persistent exposure of immune cells to endogenous IFNα appears associated with pathogenesis. On the other hand, IFNβ provides therapeutic benefits in MS. Regardless of whether type I IFN associated responses contribute to favorable or poor outcomes, it is clear that both canonical and non-canonical IFN signaling pathways are critical for type I IFN responses. In many cases, both canonical and non-canonical are activated in parallel, but it is possible that in certain cell-type and disease states a given pathway may play a predominant role. With the identification of the roles of non-canonical MAPK and mTOR pathways, the involvement of PKC and SLFN proteins, our understanding of how type I IFN signaling alters the transcriptome to produce proteins that affect changes in biological responses has increased dramatically. The discovery of new non-canonical pathways and effectors has substantially advanced the field, but other non-canonical pathways may have yet to be identified. Understanding how there is connectivity between the classical, canonical JAK/STAT signaling, and non-canonical pathways will provide the basis for further targeting of type I IFN signaling in different diseases.
All authors have contributed in the writing and editing of this review. All authors contributed to the article and approved the submitted version.
The research of LP is supported by grants CA77816 and CA121192 by the NIH. REP is supported in part by NIH/NCI training grant T32 CA070085.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Platanias LC. Interferon Signals: What Is Classical and What Is Nonclassical? J Interferon Cytokine Res (2005) 25:732. doi: 10.1089/jir.2005.25.732
2. Komiya Y, Habas R. Wnt signal transduction pathways. Organogenesis (2008) 4(2):68–75. doi: 10.4161/org.4.2.5851
3. Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol (2017) 17(9):545–58. doi: 10.1038/nri.2017.52
4. Majoros A, Platanitis E, Kernbauer-Holzl E, Rosebrock F, Muller M, Decker T. Canonical and Non-Canonical Aspects of JAK-STAT Signaling: Lessons from Interferons for Cytokine Responses. Front Immunol (2017) 8:29. doi: 10.3389/fimmu.2017.00029
5. Li SF, Gong MJ, Zhao FR, Shao JJ, Xie YL, Zhang YG, et al. Type I Interferons: Distinct Biological Activities and Current Applications for Viral Infection. Cell Physiol Biochem (2018) 51(5):2377–96. doi: 10.1159/000495897
6. Isaacs A, Lindenmann J. Virus interference. I. The interferon. Proc R Soc Lond B (1957) 147:258–67. doi: 10.1098/rspb.1957.0048
7. Wheelock EF. Interferon-Like Virus-Inhibitor Induced in Human Leukocytes by Phytohemagglutinin. Science (1965) 149:310–11. doi: 10.1126/science.149.3681.310
8. Kotenko SV, Gallagher G, Baurin VV, Lewis-Antes A, Shen M, Shah NK, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol (2003) 4(1):69–77. doi: 10.1038/ni875
9. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5(5):375–86. doi: 10.1038/nri1604
10. Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol (2005) 23:307–36. doi: 10.1146/annurev.immunol.23.021704.115843
11. Acosta PL, Byrne AB, Hijano DR, Talarico LB. Human Type I Interferon Antiviral Effects in Respiratory and Reemerging Viral Infections. J Immunol Res (2020) 2020:1372494. doi: 10.1155/2020/1372494
12. Borden EC. Interferons alpha and beta in cancer: therapeutic opportunities from new insights. Nat Rev Drug Discovery (2019) 18(3):219–34. doi: 10.1038/s41573-018-0011-2
13. Jiang J, Zhao M, Chang C, Wu H, Lu Q. Type I Interferons in the Pathogenesis and Treatment of Autoimmune Diseases. Clin Rev Allergy Immunol (2020) 59(2):248–72. doi: 10.1007/s12016-020-08798-2
14. Lukhele S, Boukhaled GM, Brooks DG. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin Immunol (2019) 43:101277. doi: 10.1016/j.smim.2019.05.001
15. Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol (2014) 14:36–49. doi: 10.1038/nri3581
16. Brierley MM, Fish EN. Functional relevance of the conserved DNA-binding domain of STAT2. J Biol Chem (2005) 280(13):13029–36. doi: 10.1074/jbc.M500426200
17. Grumbach IM, Mayer I, Uddin S, Lekmine F, Majchrzak B, Yamauchi H, et al. Engagement of the CrkL adapter in interferon α signaling in BCR-ABL expressing cells. Br J Haematol (2001) 112:327–36. doi: 10.1046/j.1365-2141.2001.02556.x
18. Saleiro D, Platanias LC. Interferon signaling in cancer. Non-canonical pathways and control of intracellular immune checkpoints. Semin Immunol (2019) 43:101299. doi: 10.1016/j.smim.2019.101299
19. Kosciuczuk EM, Mehrotra S, Saleiro D, Kroczynska B, Majchrzak-Kita B, Lisowski P, et al. Sirtuin 2-mediated deacetylation of cyclin-dependent kinase 9 promotes STAT1 signaling in type I interferon responses. J Biol Chem (2019) 294(3):827–37. doi: 10.1074/jbc.RA118.005956
20. Saleiro D, Mehrotra S, Kroczynska B, Beauchamp EM, Lisowski P, Majchrzak-Kita B, et al. Central role of ULK1 in type I interferon signaling. Cell Rep (2015) 11(4):605–17. doi: 10.1016/j.celrep.2015.03.056
21. Mavrommatis E, Fish EN, Platanias LC. The schlafen family of proteins and their regulation by interferons. J Interferon Cytokine Res (2013) 33(4):206–10. doi: 10.1089/jir.2012.0133
22. Puck A, Aigner R, Modak M, Cejka P, Blaas D, Stockl J. Expression and regulation of Schlafen (SLFN) family members in primary human monocytes, monocyte-derived dendritic cells and T cells. Results Immunol (2015) 5:23–32. doi: 10.1016/j.rinim.2015.10.001
23. Qiang Z, Zhou ZY, Peng T, Jiang PZ, Shi N, Njoya EM, et al. Inhibition of TPL2 by interferon-alpha suppresses bladder cancer through activation of PDE4D. J Exp Clin Cancer Res (2018) 37(1):288. doi: 10.1186/s13046-018-0971-4
24. Urin V, Shemesh M, Schreiber G. CRISPR/Cas9-based Knockout Strategy Elucidates Components Essential for Type 1 Interferon Signaling in Human HeLa Cells. J Mol Biol (2019) 431(17):3324–38. doi: 10.1016/j.jmb.2019.06.007
25. Lu C, Klement JD, Ibrahim ML, Xiao W, Redd PS, Nayak-Kapoor A, et al. Type I interferon suppresses tumor growth through activating the STAT3-granzyme B pathway in tumor-infiltrating cytotoxic T lymphocytes. J Immunother Cancer (2019) 7(1):157. doi: 10.1186/s40425-019-0635-8
26. Arslan AD, Sassano A, Saleiro D, Lisowski P, Kosciuczuk EM, Fischietti M, et al. Human SLFN5 is a transcriptional co-repressor of STAT1-mediated interferon responses and promotes the malignant phenotype in glioblastoma. Oncogene (2017) 36(43):6006–19. doi: 10.1038/onc.2017.205
27. Igbe I, Liu WL, Shen X-F, Liu HW, Jiao W, Zhang G-L, et al. Dietary quercetin potentiates the antiproliferative effect of interferon-α in hepatocellular carcinoma cells through activation of JAK/STAT pathway signaling by inhibition of SHP2 phosphatase. Oncotarget (2017) 8(69):113734–48. doi: 10.18632/oncotarget.22556
28. Suarez-Kelly LP, Levine KM, Olencki TE, Del Campo SEM, Streacker EA, Brooks TR, et al. A pilot study of interferon-alpha-2b dose reduction in the adjuvant therapy of high-risk melanoma. Cancer Immunol Immunother (2019) 68(4):619–29. doi: 10.1007/s00262-019-02308-w
29. Lamberti MJ, Mentucci FM, Roselli E, Araya P, Rivarola VA, Rumie Vittar NB, et al. Photodynamic Modulation of Type 1 Interferon Pathway on Melanoma Cells Promotes Dendritic Cell Activation. Front Immunol (2019) 10:2614. doi: 10.3389/fimmu.2019.02614
30. Stone ML, Chiappinelli KB, Li H, Murphy LM, Travers ME, Topper MJ, et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc Natl Acad Sci USA (2017) 114(51):E10981–E90. doi: 10.1073/pnas.1712514114
31. Bazhin AV, von Ahn K, Fritz J, Werner J, Karakhanova S. Interferon-alpha Up-Regulates the Expression of PD-L1 Molecules on Immune Cells Through STAT3 and p38 Signaling. Front Immunol (2018) 9:2129. doi: 10.3389/fimmu.2018.02129
32. Qadir AS, Ceppi P, Brockway S, Law C, Mu L, Khodarev NN, et al. CD95/Fas Increases Stemness in Cancer Cells by Inducing a STAT1-Dependent Type I Interferon Response. Cell Rep (2017) 18(10):2373–86. doi: 10.1016/j.celrep.2017.02.037
33. Oh JH, Kim MJ, Choi SJ, Ban YH, Lee HK, Shin EC, et al. Sustained Type I Interferon Reinforces NK Cell-Mediated Cancer Immunosurveillance during Chronic Virus Infection. Cancer Immunol Res (2019) 7(4):584–99. doi: 10.1158/2326-6066.CIR-18-0403
34. Brown MC, Holl EK, Boczkowski D, Dobrikova E, Mosaheb M, Chandramohan V, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen–specific CTLs. Sci Trans Med (2017) 9. doi: 10.1126/scitranslmed.aan4220
35. Izquierdo-Bouldstridge A, Bustillos A, Bonet-Costa C, Aribau-Miralbes P, Garcia-Gomis D, Dabad M, et al. Histone H1 depletion triggers an interferon response in cancer cells via activation of heterochromatic repeats. Nucleic Acids Res (2017) 45(20):11622–42. doi: 10.1093/nar/gkx746
36. Gutterman JU. Cytokine Therapeutics: Lessons from Interferon α. Proc Natl Acad Sci (1994) 91:1198–205. doi: 10.1073/pnas.91.4.1198
37. Tsuchiya N, Zhang R, Iwama T, Ueda N, Liu T, Tatsumi M, et al. Type I Interferon Delivery by iPSC-Derived Myeloid Cells Elicits Antitumor Immunity via XCR1(+) Dendritic Cells. Cell Rep (2019) 29(1):162–75.e9. doi: 10.1016/j.celrep.2019.08.086
38. Ye Z, Dong H, Li Y, Ma T, Huang H, Leong HS, et al. Prevalent Homozygous Deletions of Type I Interferon and Defensin Genes in Human Cancers Associate with Immunotherapy Resistance. Clin Cancer Res (2018) 24(14):3299–308. doi: 10.1158/1078-0432.CCR-17-3008
39. Muskardin TLW, Niewold TB. Type I interferon in rheumatic diseases. Nat Rev Rheumatol (2018) 14(4):214–28. doi: 10.1038/nrrheum.2018.31
40. Lombardi A, Tsomos E, Hammerstad SS, Tomer Y. Interferon alpha: The key trigger of type 1 diabetes. J Autoimmun (2018) 94:7–15. doi: 10.1016/j.jaut.2018.08.003
41. Marroqui L, Dos Santos RS, Op de Beeck A, Coomans de Brachene A, Marselli L, Marchetti P, et al. Interferon-alpha mediates human beta cell HLA class I overexpression, endoplasmic reticulum stress and apoptosis, three hallmarks of early human type 1 diabetes. Diabetologia (2017) 60(4):656–67. doi: 10.1007/s00125-016-4201-3
42. Lombardi A, Tomer Y. Interferon alpha impairs insulin production in human beta cells via endoplasmic reticulum stress. J Autoimmun (2017) 80:48–55. doi: 10.1016/j.jaut.2017.02.002
43. Marro BS, Ware BC, Zak J, de la Torre JC, Rosen H, Oldstone MB. Progression of type 1 diabetes from the prediabetic stage is controlled by interferon-alpha signaling. Proc Natl Acad Sci U S A (2017) 114(14):3708–13. doi: 10.1073/pnas.1700878114
44. Meyer S, Woodward M, Hertel C, Vlaicu P, Haque Y, Karner J, et al. AIRE-Deficient Patients Harbor Unique High-Affinity Disease-Ameliorating Autoantibodies. Cell (2016) 166(3):582–95. doi: 10.1016/j.cell.2016.06.024
45. Davies R, Hammenfors D, Bergum B, Vogelsang P, Gavasso S, Brun JG, et al. Aberrant cell signalling in PBMCs upon IFN-alpha stimulation in primary Sjogren’s syndrome patients associates with type I interferon signature. Eur J Immunol (2018) 48(7):1217–27. doi: 10.1002/eji.201747213
46. Brkic Z, Maria NI, van Helden-Meeuwsen CG, van de Merwe JP, van Daele PL, Dalm VA, et al. Prevalence of interferon type I signature in CD14 monocytes of patients with Sjogren’s syndrome and association with disease activity and BAFF gene expression. Ann Rheum Dis (2013) 72(5):728–35. doi: 10.1136/annrheumdis-2012-201381
47. Akiyama C, Tsumiyama K, Uchimura C, Honda E, Miyazaki Y, Sakurai K, et al. Conditional Upregulation of IFN-alpha Alone Is Sufficient to Induce Systemic Lupus Erythematosus. J Immunol (2019) 203(4):835–43. doi: 10.4049/jimmunol.1801617
48. Morand EF, Furie R, Tanaka Y, Bruce IN, Askanase AD, Richez C, et al. Trial of Anifrolumab in Active Systemic Lupus Erythematosus. N Engl J Med (2020) 382(3):211–21. doi: 10.1056/NEJMoa1912196
49. Liu J, Berthier CC, Kahlenberg JM. Enhanced Inflammasome Activity in Systemic Lupus Erythematosus Is Mediated via Type I Interferon-Induced Up-Regulation of Interferon Regulatory Factor 1. Arthritis Rheumatol (2017) 69(9):1840–9. doi: 10.1002/art.40166
50. De Groof A, Ducreux J, Aleva F, Long AJ, Ferster A, van der Ven A, et al. STAT3 phosphorylation mediates the stimulatory effects of interferon alpha on B cell differentiation and activation in SLE. Rheumatol (Oxford) (2020) 59(3):668–77. doi: 10.1093/rheumatology/kez354
51. Gkirtzimanaki K, Kabrani E, Nikoleri D, Polyzos A, Blanas A, Sidiropoulos P, et al. IFNalpha Impairs Autophagic Degradation of mtDNA Promoting Autoreactivity of SLE Monocytes in a STING-Dependent Fashion. Cell Rep (2018) 25(4):921–33 e5. doi: 10.1016/j.celrep.2018.09.001
52. Houssiau FA, Thanou A, Mazur M, Ramiterre E, Gomez Mora DA, Misterska-Skora M, et al. IFN-alpha kinoid in systemic lupus erythematosus: results from a phase IIb, randomised, placebo-controlled study. Ann Rheum Dis (2020) 79(3):347–55. doi: 10.1136/annrheumdis-2019-216379
53. Felten R, Scher F, Sagez F, Chasset F, Arnaud L. Spotlight on anifrolumab and its potential for the treatment of moderate-to-severe systemic lupus erythematosus: evidence to date. Drug Des Devel Ther (2019) 13:1535–43. doi: 10.2147/DDDT.S170969
54. de Jong TD, Vosslamber S, Mantel E, de Ridder S, Wesseling JG, van der Pouw Kraan TC, et al. Physiological evidence for diversification of IFNalpha- and IFNbeta-mediated response programs in different autoimmune diseases. Arthritis Res Ther (2016) 18:49. doi: 10.1186/s13075-016-0946-9
55. Galligan CL, Pennell LM, Murooka TT, Baig E, Majchrzak-Kita B, Rahbar R, et al. Interferon-beta is a key regulator of proinflammatory events in experimental autoimmune encephalomyelitis. Mult Scler (2010) 16(12):1458–73. doi: 10.1177/1352458510381259
56. Pennell LM, Fish EN. Immunoregulatory Effects of Interferon-β in Suppression of Th17 cells. J Interferon Cytokine Res (2014) 34(5):330–41. doi: 10.1089/jir.2013.0088
57. Zula JA, Green HC, Ransohoff RM, Rudick RA, Stark GR, van Boxel-Dezaire AH. The role of cell type-specific responses in IFN-beta therapy of multiple sclerosis. Proc Natl Acad Sci U S A (2011) 108(49):19689–94. doi: 10.1073/pnas.1117347108
58. Hurtado-Guerrero I, Pinto-Medel MJ, Urbaneja P, Rodriguez-Bada JL, Leon A, Guerrero M, et al. Activation of the JAK-STAT Signaling Pathway after In Vitro Stimulation with IFNss in Multiple Sclerosis Patients According to the Therapeutic Response to IFNss. PloS One (2017) 12(1):e0170031. doi: 10.1371/journal.pone.0170031
59. Jin T, Wang C, Tian S. Feature selection based on differentially correlated gene pairs reveals the mechanism of IFN-beta therapy for multiple sclerosis. Peer J (2020) 8:e8812. doi: 10.7717/peerj.8812
60. Fukushima A, Sugimoto M, Hiwa S, Hiroyasu T. Elastic net-based prediction of IFN-beta treatment response of patients with multiple sclerosis using time series microarray gene expression profiles. Sci Rep (2019) 9(1):1822. doi: 10.1038/s41598-018-38441-2
61. Tian S, Zhang L. Identification of Monotonically Differentially Expressed Genes for IFN-beta-Treated Multiple Sclerosis Patients. BioMed Res Int (2019) 2019:5647902. doi: 10.1155/2019/5647902
62. Waddington KE, Papadaki A, Coelewij L, Adriani M, Nytrova P, Kubala Havrdova E, et al. Using Serum Metabolomics to Predict Development of Anti-drug Antibodies in Multiple Sclerosis Patients Treated With IFNbeta. Front Immunol (2020) 11:1527. doi: 10.3389/fimmu.2020.01527
63. Adriani M, Nytrova P, Mbogning C, Hassler S, Medek K, Jensen PEH, et al. Monocyte NOTCH2 expression predicts IFN-beta immunogenicity in multiple sclerosis patients. JCI Insight (2018) 3(11). doi: 10.1172/jci.insight.99274
64. Schreiber G. The molecular basis for differential type I interferon signaling. J Biol Chem (2017) 292(18):7285–94. doi: 10.1074/jbc.R116.774562
65. Lavoie TB, Kalie E, Crisafulli-Cabatu S, Abramovich R, DiGioia G, Moolchan K, et al. Binding and activity of all human alpha interferon subtypes. Cytokine (2011) 56(2):282–9. doi: 10.1016/j.cyto.2011.07.019
66. Nan Y, Wu C, Zhang YJ. Interplay between Janus Kinase/Signal Transducer and Activator of Transcription Signaling Activated by Type I Interferons and Viral Antagonism. Front Immunol (2017) 8:1758. doi: 10.3389/fimmu.2017.01758
67. Randall RE, Goodbourn S. Interferons and viruses: an interplay between induction, signalling, antiviral responses and virus countermeasures. J Gen Virol (2008) 89(Pt 1):1–47. doi: 10.1099/vir.0.83391-0
68. Harrison AR, Moseley GW. The dynamic interface of viruses with STATs. J Virol (2020) 94(22):e00856-20. doi: 10.1128/JVI.00856-20
69. Yu C, Kang L, Chen J, Zang N. Evaluation of safety, efficacy, tolerability, and treatment-related outcomes of type I interferons for human coronaviruses (HCoVs) infection in clinical practice: An updated critical systematic review and meta-analysis. Int Immunopharmacol (2020) 86:106740. doi: 10.1016/j.intimp.2020.106740
70. Wang BX, Fish EN. Global virus outbreaks: Interferons as 1st responders. Semin Immunol (2019) 43:101300. doi: 10.1016/j.smim.2019.101300
71. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J Virol (2009) 83(13):6689–705. doi: 10.1128/JVI.02220-08
72. Kopecky-Bromberg SA, Martinez-Sobrido L, Frieman M, Baric RA, Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J Virol (2007) 81(2):548–57. doi: 10.1128/JVI.01782-06
73. Menachery VD, Eisfeld AJ, Schafer A, Josset L, Sims AC, Proll S, et al. Pathogenic influenza viruses and coronaviruses utilize similar and contrasting approaches to control interferon-stimulated gene responses. mBio (2014) 5(3):e01174–14. doi: 10.1128/mBio.01174-14
74. Totura AL, Baric RS. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr Opin Virol (2012) 2(3):264–75. doi: 10.1016/j.coviro.2012.04.004
75. Yoneyama M, Suhara W, Fujita T. Control of IRF-3 activation by phosphorylation. J Interferon Cytokine Res (2002) 22(1):73–6. doi: 10.1089/107999002753452674
76. Devaraj SG, Wang N, Chen Z, Chen Z, Tseng M, Barretto N, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem (2007) 282(44):32208–21. doi: 10.1074/jbc.M704870200
77. Lokugamage KG, Hage A, Schindewolf C, Rajsbaum R, Menachery VD. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv (2020) 2020.03.07.982264. doi: 10.1101/2020.03.07.982264
78. Konno Y, Kimura I, Uriu K, Fukushi M, Irie T, Koyanagi Y, et al. SARS-CoV-2 ORF3b is a potent interferon antagonist whose activity is increased by a naturally occurring elongation variant. Cell Rep (2020) 32(12):108185. doi: 10.1101/2020.05.11.088179
79. Li JY, Liao CH, Wang Q, Tan YJ, Luo R, Qiu Y, et al. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res (2020) 286:198074. doi: 10.1016/j.virusres.2020.198074
80. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Moller R, et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell (2020) 181(5):1036–45.e9. doi: 10.1016/j.cell.2020.04.026
81. Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y, et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study With Implications for the Pathogenesis of COVID-19. Clin Infect Dis (2020) 71(6):1400–9. doi: 10.1093/cid/ciaa410
82. Bost P, Giladi A, Liu Y, Bendjelal Y, Xu G, David E, et al. Host-Viral Infection Maps Reveal Signatures of Severe COVID-19 Patients. Cell (2020) 181(7):1475–88 e12. doi: 10.1016/j.cell.2020.05.006
83. Hadjadj J, Yatim N, Barnabei L, Corneau A, Boussier J, Smith N, et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science (2020) 369:718–24. doi: 10.1126/science.abc6027
84. Dahl H, Linde A, Strannegard O. In vitro inhibition of SARS virus replication by human interferons. Scand J Infect Dis (2004) 36(11-12):829–31. doi: 10.1080/00365540410021144
85. Moriguchi H, Sato C. Treatment of SARS with human interferons. Lancet (2003) 362(9390):1159. doi: 10.1016/S0140-6736(03)14484-4
86. Mantlo E, Bukreyeva N, Maruyama J, Paessler S, Huang C. Antiviral activities of type I interferons to SARS-CoV-2 infection. Antiviral Res (2020) 179:104811. doi: 10.1016/j.antiviral.2020.104811
87. Loutfy MR, Blatt L, Siminovitch K, Ward S, Wolff B, Lho H, et al. Therapeutic potential of interferon alfacon-1 plus corticosteroids in severe acute respiratory syndrome: a preliminary study. J Am Med Assoc (2003) 290(24):3222–8. doi: 10.1001/jama.290.24.3222
88. Sallard E, Lescure FX, Yazdanpanah Y, Mentre F, Peiffer-Smadja N. Type 1 interferons as a potential treatment against COVID-19. Antiviral Res (2020) 178:104791. doi: 10.1016/j.antiviral.2020.104791
89. Zhou Q, Chen V, Shannon CP, Wei XS, Xiang X, Wang X, et al. Interferon-alpha2b Treatment for COVID-19. Front Immunol (2020) 11:1061. doi: 10.3389/fimmu.2020.615275
90. Hung IF-N, Lung K-C, Tso EY-K, Liu R, Chung TW-H, Chu M-Y, et al. Triple combination of interferon beta-1b, lopinavir–ritonavir, and ribavirin in the treatment of patients admitted to hospital with COVID-19: an open-label, randomised, phase 2 trial. Lancet (2020) 395(10238):1695–704. doi: 10.1016/S0140-6736(20)31042-4
91. Pereda R, González D, Rivero HB, Rivero JC, Pérez A, López LR, et al. Therapeutic effectiveness of interferon-α2b against COVID-19: the Cuban experience. J Interferon Cytokine Res (2020) 40(9):438–42. doi: 10.1089/jir.2020.0124
92. Wang N, Zhan Y, Zhu L, Hou Z, Liu F, Song P, et al. Retrospective Multicenter Cohort Study Shows Early Interferon Therapy Is Associated with Favorable Clinical Responses in COVID-19 Patients. Cell Host Microbe (2020) 28(3):455–64.e2. doi: 10.1016/j.chom.2020.07.005
93. Yuan J, Zou R, Zeng L, Kou S, Lan J, Li X, et al. The correlation between viral clearance and biochemical outcomes of 94 COVID-19 infected discharged patients. Inflammation Res (2020) 69(6):599–606. doi: 10.1007/s00011-020-01342-0
94. Irvani SSN, Golmohammadi M, Pourhoseingholi MA, Shokouhi S, Darazam IA. Effectiveness of Interferon Beta 1a, compared to Interferon Beta 1b and the usual therapeutic regimen to treat adults with moderate to severe COVID-19: structured summary of a study protocol for a randomized controlled trial. Trials (2020) 21(1):473. doi: 10.1186/s13063-020-04382-3
95. Bastard P, Rosen LB, Zhang Q, Michailidis E, Hoffmann HH, Zhang Y, et al. Auto-antibodies against type I IFNs in patients with lifethreatening COVID-19. Sci (American Assoc Advancement Sci (2020) 370(6515):eabd4585. doi: 10.1126/science.abd4585
96. Zhou Z, Ren L, Zhang L, Zhong J, Xiao Y, Jia Z, et al. Heightened Innate Immune Responses in the Respiratory Tract of COVID-19 Patients. Cell Host Microbe (2020) 27(6):883–90.e2. doi: 10.1016/j.chom.2020.04.017
97. Cinatl J, Morgenstern B, Bauer G, Chandra P, Rabenau H, Doerr HW. Treatment of SARS with human interferons. Lancet (2003) 362(9380):293–4. doi: 10.1016/S0140-6736(03)13973-6
98. Zhang Q, Bastard P, Liu Z, Le Pen J, Moncada-Velez M, Chen J, et al. Inborn Errors of Type I IFN Immunity in Patients with Life-threatening COVID-19. Science (2020) 370(6515):eabd4570. doi: 10.1126/science.abd4570
99. Zhang Q. Human genetics of life-threatening influenza pneumonitis. Hum Genet (2020) 139(6-7):941–8. doi: 10.1007/s00439-019-02108-3
100. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature (2003) 426(6965):450–4. doi: 10.1038/nature02145
101. Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol (2020) 5(4):562–9. doi: 10.1038/s41564-020-0688-y
102. Raj VS, Mou H, Smits SL, Dekkers DH, Muller MA, Dijkman R, et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature (2013) 495(7440):251–4. doi: 10.1038/nature12005
103. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell (2020) 181(5):1016–35.e19. doi: 10.1016/j.cell.2020.04.035
104. Israelow B, Song E, Mao T, Lu P, Meir A, Liu F, et al. Mouse model of SARS-CoV-2 reveals inflammatory role of type I interferon signaling. J Exp Med (2020) 217(12):e20201241. doi: 10.1084/jem.20201241
105. Lee JS, Park S, Jeong HW, Ahn JY, Choi SJ, Lee H, et al. Immunophenotyping of COVID-19 and influenza highlights the role of type I interferons in development of severe COVID-19. Sci Immunol (2020) 5(49):eabd1554. doi: 10.1126/sciimmunol.abd1554
106. Wu W, Metcalf JP. The Role of Type I IFNs in Influenza: Antiviral Superheroes or Immunopathogenic Villains? J Innate Immun (2020) 19:1–11. doi: 10.1159/000508379
107. Lucas C, Wong P, Klein J, Castro TBR, Silva J, Sundaram M, et al. Longitudinal analyses reveal immunological misfiring in severe COVID-19. Nature (2020) 584(7821):463–9. doi: 10.1038/s41586-020-2588-y
108. Borgeling Y, Schmolke M, Viemann D, Nordhoff C, Roth J, Ludwig S. Inhibition of p38 mitogen-activated protein kinase impairs influenza virus-induced primary and secondary host gene responses and protects mice from lethal H5N1 infection. J Biol Chem (2014) 289(1):13–27. doi: 10.1074/jbc.M113.469239
109. He Y, Fu W, Cao K, He Q, Ding X, Chen J, et al. IFN-K suppresses the replication of influenza A viruses through the IFNAR-MAPK-Fos-CHD6 axis. Sci Signaling (2020) 13(626):eaaz3381. doi: 10.1126/scisignal.aaz3381
110. Mahony R, Gargan S, Roberts KL, Bourke N, Keating SE, Bowie AG, et al. A novel anti-viral role for STAT3 in IFN-alpha signalling responses. Cell Mol Life Sci (2017) 74(9):1755–64. doi: 10.1007/s00018-016-2435-3
111. Huang SH, McCann CD, Mota TM, Wang C, Lipkin SM, Jones RB. Have Cells Harboring the HIV Reservoir Been Immunoedited? Front Immunol (2019) 10:1842. doi: 10.3389/fimmu.2019.01842
112. Dagenais-Lussier X, Loucif H, Cadorel H, Blumberger J, Isnard S, Bego MG, et al. USP18 is a significant driver of memory CD4 T-cell reduced viability caused by type I IFN signaling during primary HIV-1 infection. PloS Pathog (2019) 15(10):e1008060. doi: 10.1371/journal.ppat.1008060
113. Cheng L, Ma J, Li J, Li D, Li G, Li F. Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J Clin Invest (2017) 127(1):269–79. doi: 10.1172/JCI90745
114. Zhen A, Rezek V, Youn C, Lam B, Chang N, Rick J. Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J Clin Invest (2017) 127(1):260–8. doi: 10.1172/JCI89488
115. Su L. Pathogenic Role of Type I Interferons in HIV-Induced Immune Impairments in Humanized Mice. Curr HIV/AIDS Rep (2019) 16(3):224–9. doi: 10.1007/s11904-019-00444-7
116. Nguyen NV, Tran JT, Sanchez DJ. HIV blocks Type I IFN signaling through disruption of STAT1 phosphorylation. Innate Immun (2018) 24(8):490–500. doi: 10.1177/1753425918803674
117. Singh H, Ojeda-Juarez D, Maung R, Shah R, Roberts AJ, Kaul M. A pivotal role for Interferon-alpha receptor-1 in neuronal injury induced by HIV-1. J Neuroinflamm (2020) 17(1):226. doi: 10.1186/s12974-020-01894-2
118. Valdez F, Salvador J, Palermo PM, Mohl JE, Hanley KA, Watts D, et al. Schlafen 11 Restricts Flavivirus Replication. J Virol (2019) 93(15):e00104-19. doi: 10.1128/JVI.00104-19
119. Borrego AR, Corona-Ayala C, Salvador JC, Valdez FC, Llano M. Gene Expression Regulation of the Type I Interferon-Induced Protein Schlafen 11. J Fed Am Societies Exp Biol (2020) 34(S1). doi: 10.1096/fasebj.2020.34.s1.00603
120. Seong RK, Seo SW, Kim JA, Fletcher SJ, Morgan NV, Kumar M, et al. Schlafen 14 (SLFN14) is a novel antiviral factor involved in the control of viral replication. Immunobiology (2017) 222(11):979–88. doi: 10.1016/j.imbio.2017.07.002
121. Zhang W-C, Du L-J, Zheng X-J, Chen X-Q, Shi C, Chen B-Y, et al. Elevated sodium chloride drives type I interferon signaling in macrophages and increases antiviral resistance. J Biol Chem (2018) 293(3):1030–9. doi: 10.1074/jbc.M117.805093
122. Barragan-Iglesias P, Franco-Enzastiga U, Jeevakumar V, Shiers S, Wangzhou A, Granados-Soto V, et al. Type I Interferons Act Directly on Nociceptors to Produce Pain Sensitization: Implications for Viral Infection-Induced Pain. J Neurosci (2020) 40(18):3517–32. doi: 10.1523/JNEUROSCI.3055-19.2020
Keywords: interferon, signaling, MAP kinase signaling, signal transducer and activator of transcription, mammalian target of rapamycin, mRNA translation, SARS-CoV-2, COVID-19
Citation: Mazewski C, Perez RE, Fish EN and Platanias LC (2020) Type I Interferon (IFN)-Regulated Activation of Canonical and Non-Canonical Signaling Pathways. Front. Immunol. 11:606456. doi: 10.3389/fimmu.2020.606456
Received: 15 September 2020; Accepted: 26 October 2020;
Published: 23 November 2020.
Edited by:
Mark R. Walter, University of Alabama at Birmingham, United StatesReviewed by:
Howard M. Johnson, University of Florida, United StatesCopyright © 2020 Mazewski, Perez, Fish and Platanias. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leonidas C. Platanias, bC1wbGF0YW5pYXNAbm9ydGh3ZXN0ZXJuLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.